
DOI: 10.1002/zaac.200600360
Synthese und Kristallstrukturen von �- und β-Ba3(PS4)2 sowie von Ba3(PSe4)2
Synthesis and Crystal Structures of �-, β-Ba3(PS4)2 and Ba3(PSe4)2
Stefan Jörgens und Albrecht Mewis*
Düsseldorf, Institut für Anorganische Chemie und Strukturchemie II der Heinrich-Heine-Universität
Bei der Redaktion eingegangen am 19. Dezember 2006.
Abstract. Ba3(PS4)2 and Ba3(PSe4)2 were prepared by heating mix-tures of the elements at 800 °C for 25 h. Both compounds wereinvestigated by single crystal X-ray methods. The thiophosphate isdimorphic and undergoes a displacive phase transition at about75 °C. Both modifications crystallize in new structure types. In theroom temperature phase (�-Ba3(PS4)2: P21/a; a � 11.649(3), b �
6.610(1), c � 17.299(2) A, β � 90.26(3)°; Z � 4) three crystallogra-phically independent Ba atoms are surrounded by ten sulfur atomsforming distorted polyhedra. The arrangement of the PS4 tetrahe-dra, isolated from each other, is comparable with the formation ofthe SO4
2� ions of β-K2SO4. In β-Ba3(PS4)2 (C2/m; a � 11.597(2),
Einleitung
Durch die Umsetzung von Metallen (A) mit Phosphor undSchwefel bzw. Selen entstehen in vielen Fällen Chalcogeno-phosphate. Deren Formeltyp kann durchaus von der La-dung des Metallkations bestimmt werden: Einerseits existie-ren zahlreiche Verbindungen (AI)3PX4 sowie AIIIPX4 (X: S,Se), andererseits werden mit zweiwertigen Kationen vorran-gig Chalcogenohypodiphosphate des Typs (AII)2P2X6
gebildet [1]. Demzufolge lassen sich die Hexathio- undHexaselenohypodiphosphate der Erdalkalimetalle problem-los darstellen, deren Kristallstrukturen auf der Basis vonEinkristalldaten bereits beschrieben wurden [2�4]. DieSynthese der entsprechenden Orthophosphate ist dagegenentweder bislang nicht gelungen oder aber recht mühsamund erfordert mehrwöchiges Tempern, wie die nur Raman-spektroskopisch charakterisierten Thiophosphate Ca3(PS4)2
und Ba3(PS4)2 [5] belegen. Wir haben nun eine Möglichkeiterarbeitet, von Ba3(PS4)2 Einkristalle zu züchten, um dieKristallstruktur zu bestimmen. Dies ist uns auch beiBa3(PSe4)2 gelungen. Über die strukturelle Charakterisie-
* Prof. Dr. A. MewisInstitut für Anorganische Chemie und Strukturchemie II der Hein-rich-Heine-UniversitätUniversitätsstr. 1D-40225 DüsseldorfFax: (0211) 81 14146e-mail: [email protected]
570 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim Z. Anorg. Allg. Chem. 2007, 633, 570�574
b � 6.727(1), c � 8.704(2) A; β � 90.00(3)°; Z � 2) the PS4 tetrahe-dra are no more tilted along [001], but oriented parallel to eachother inducing less distorted tetrahedra and polyhedra around theBa atoms, respectively. Ba3(PSe4)2 (P21/a; a � 12.282(2), b �
6.906(1), c � 18.061(4) A; β � 90.23(3)°; Z � 4) is isotypic to �-Ba3(PS4)2 and no phase transition could be detected up to about550 °C.
Keywords: Chalcogenophosphates; Barium; Phase transition;Crystal structure
rung dieser beiden Verbindungen wird im folgenden berich-tet.
Ergebnisse und Diskussion
Von Ba3(PS4)2 konnten transparente schwach rosa Einkri-stalle erhalten werden. Da die Verbindung dimorph ist(s.u.), wird die bei Raumtemperatur stabile Modifikationals �- und die Hochtemperaturform als β-Ba3(PS4)2 be-zeichnet. Einkristalle von Ba3(PSe4)2 sind ebenfalls transpa-rent, aber gelb. Die Bestimmung der Elementarzellen wurdein beiden Fällen durch das Vorliegen von drei Kristall-domänen erschwert, die längs [001] um je 120° gegeneinan-der verdreht sind, was zunächst hexagonale Symmetrie vor-täuschte. Beide Chalcogenophosphate kristallisieren jedochmonoklin und sind isotyp. Das Auslöschungssymbol ist2/mP � 21/c, welches zur Raumgruppe P21/c führt. Für ei-nen besseren Vergleich mit der Struktur von β-Ba3(PS4)2
wurde die Aufstellung P21/a gewählt. Die Kristallstruktu-ren des Thio- und Selenophosphats konnten unter anisotro-per Aufspaltung der Auslenkungsparameter zu den in denTabellen 1 � 3 aufgeführten Ergebnissen verfeinert werden.Da infolge der vorliegenden Domänen in vielen Reflexendie Intensität von zwei oder drei Kristallindividuen enthal-ten ist, dürfte dies der Grund für das nicht restlos zufriedenstellende Resultat sein.
�-Ba3(PS4)2 und Ba3(PSe4)2 kristallisieren in einem neuenStrukturtyp, der im folgenden an Hand des Thiophosphatsbeschrieben wird. Er wird von isolierten PS4-Tetraedern ge-

�- und β-Ba3(PS4)2 sowie Ba3(PSe4)2
Tabelle 1 Kristallographische Daten und ihre Bestimmung
�-Ba3(PS4)2 β-Ba3(PS4)2 Ba3(PSe4)2
Raumgruppe P21/a C2/m P21/aMessgerät IPDS IPDS AED2Messtemperatur 25 °C ca. 100 °C 25 °CGitterparameter a � 11,649(3) A a � 11,597(2) A a � 12,282(2) A
b � 6,610(1) A b � 6,727(1) A b � 6,906(1) Ac � 17,299(2) A c � 8,704(2) A c � 18,061(4) Aβ � 90,26(3)° β � 90,00(3)° β � 90,23(3)°
Zellvolumen/A3 1332,0(4) 679,0(2) 1531,9(5)Formeleinheiten/Zelle Z � 4 Z � 2 Z � 4Röntg. Dichte/g·cm�3 3,642 3,573 4,794Messbereich 3°�2θ�52° 3°�2θ�54° 3°�2θ�60°
�14�h�14 �14�h�14 �17�h�0�8�k�8 �8�k�8 �9�k�9�21�l�21 �11�l�11 0�l�25
Anzahl der Reflexe 13621 4705 4964symmetrieunabh. 2610 758 2401mit I � 2σ (I) 2108 715 1158Rint 0,205 0,336 0,229R1 0,093 0,076 0,085wR2 (alle Reflexe) 0,231 0,193 0,194
Tabelle 2 Lage- und äquivalente Auslenkungsparameter/pm2 von�-Ba3(PS4)2 und Ba3(PSe4)2 (besetzte Punktlage: 4e (x, y, z)
Atom x y z Ueq
�-Ba3(PS4)2:Ba1 0,02185(9) 0,9618(2) 0,25089(5) 261(4)Ba2 0,16900(8) 0,4908(1) 0,42805(6) 226(4)Ba3 0,33921(8) 0,9949(1) 0,07225(6) 237(4)P1 0,1617(3) 0,4738(5) 0,1406(2) 166(8)P2 0,3288(3) 0,9852(5) 0,3599(3) 172(8)S1 0,1119(4) 0,4503(8) 0,2511(2) 385(10)S2 0,8313(3) 0,0248(6) 0,1154(3) 267(9)S3 0,0927(3) 0,2328(6) 0,0827(2) 252(8)S4 0,0939(3) 0,7303(6) 0,0921(2) 231(8)S5 0,3032(3) 0,9075(8) 0,2489(2) 388(10)S6 0,9072(3) 0,2491(6) 0,3852(2) 270(9)S7 0,9223(3) 0,7377(6) 0,4115(2) 273(9)S8 0,1730(3) 0,9916(5) 0,4139(2) 203(9)
Ba3(PSe4)2:Ba1 0,0274(2) 0,9368(3) 0,2534(2) 197(8)Ba2 0,1734(2) 0,4864(3) 0,4237(2) 229(10)Ba3 0,3501(2) 0,9926(3) 0,0777(2) 272(9)P1 0,1653(8) 0,4568(12) 0,1381(5) 110(30)P2 0,3334(8) 0,9798(12) 0,3619(5) 120(30)Se1 0,0913(4) 0,4185(5) 0,2489(2) 242(14)Se2 0,8438(3) 0,0439(5) 0,1232(3) 235(15)Se3 0,1017(4) 0,2169(5) 0,0712(3) 258(15)Se4 0,1047(3) 0,7299(5) 0,0899(2) 227(14)Se5 0,2990(4) 0,8598(5) 0,2507(2) 231(13)Se6 0,9076(3) 0,2348(5) 0,3783(2) 222(14)Se7 0,9342(3) 0,7336(5) 0,4201(2) 215(13)Se8 0,1737(3) 0,9914(5) 0,4191(2) 171(12)
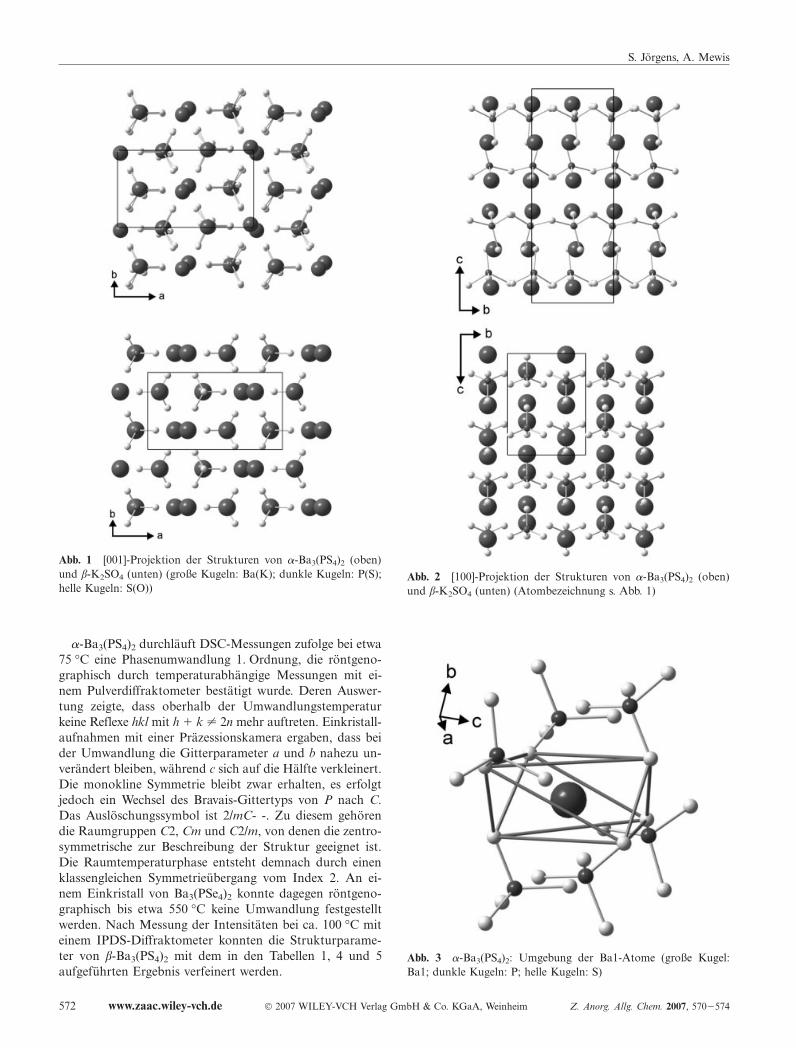
prägt, innerhalb derer die P-S-Abstände mit 2,01 � 2,05 Ain dem für Orthothiophosphate üblichen Bereich liegen undauf einen Doppelbindungsanteil schließen lassen. DieTetraeder sind insbesondere durch die Vergrößerung einesWinkels auf 119° verzerrt, während die übrigen Winkel von105° bis 109° reichen. Für die Beschreibung der Tetraeder-anordnung als strukturbildendes Element kann ein Ver-gleich mit der β-K2SO4-Struktur (Pcmn; a � 10,07, b �5,77, c � 7,48 A) herangezogen werden. Ein Blick entlang[001] zeigt, dass die Orientierung der Tetraeder in beidenFällen nahezu identisch ist und die Polyeder bei dem Thio-phosphat lediglich etwas gegeneinander verdreht sind (s.Abb. 1). Während die Anordnung auch entlang [010] sehr
Z. Anorg. Allg. Chem. 2007, 570�574 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.zaac.wiley-vch.de 571
Tabelle 3 Atomabstände/A und Bindungswinkel/° von �-Ba3(PS4)2 und Ba3(PSe4)2
�-Ba3(PS4)2:Ba1 - 1 S1 3,395(5) Ba2 - 1 S1 3,141(4)
1 S1 3,540(5) 1 S6 3,284(4)1 S2 3,248(5) 1 S6 3,519(4)1 S3 3,518(4) 1 S6 3,757(4)1 S4 3,257(4) 1 S7 3,316(4)1 S5 3,297(4) 1 S7 3,338(4)1 S5 3,528(5) 1 S7 3,466(4)1 S6 3,288(4) 1 S8 3,289(4)1 S7 3,360(4) 1 S8 3,309(3)1 S8 3,324(4) 1 S8 3,320(3)
Ba3 - 1 S2 3,263(4) P1 - 1 S1 2,006(6)1 S2 3,517(4) 1 S2 2,025(6)1 S2 3,821(4) 1 S3 2,045(5)1 S3 3,280(4) 1 S4 2,048(5)1 S3 3,291(4) �S1-P1-S2 119,5(3)1 S3 3,462(4) �S1-P1-S3 107,0(2)1 S4 3,336(4) �S1-P1-S4 110,0(2)1 S4 3,337(4) �S2-P1-S3 106,2(2)1 S4 3,369(4) �S2-P1-S4 106,4(2)1 S5 3,141(4) �S3-P1-S4 107,1(2)
P2 - 1 S5 2,008(6) �S5-P2-S6 119,5(3)1 S6 2,027(5) �S5-P2-S7 108,1(2)1 S7 2,036(5) �S5-P2-S8 108,3(2)1 S8 2,045(5) �S6-P2-S7 107,0(3)
�S6-P2-S8 106,5(2)�S7-P2-S8 106,8(2)
Ba3(PSe4)2:Ba1 - 1 Se1 3,420(4) Ba2 - 1 Se1 3,343(6)
1 Se1 3,665(4) 1 Se6 3,362(4)1 Se2 3,334(6) 1 Se6 3,784(5)1 Se3 3,929(6) 1 Se7 3,398(5)1 Se4 3,420(4) 1 Se7 3,471(4)1 Se5 3,378(5) 1 Se7 3,743(5)1 Se5 3,473(5) 1 Se8 3,399(6)1 Se6 3,393(4) 1 Se8 3,419(4)1 Se7 3,516(4) 1 Se8 3,489(4)1 Se8 3,504(6)
Ba3 - 1 Se2 3,306(4) P1 - 1 Se1 2,216(8)1 Se2 3,796(4) 1 Se2 2,210(9)1 Se3 3,350(5) 1 Se3 2,193(10)1 Se3 3,422(5) 1 Se4 2,205(10)1 Se3 3,687(5) �Se1-P1-Se2 121,4(6)1 Se4 3,488(5) �Se1-P1-Se3 105,1(3)1 Se4 3,490(5) �Se1-P1-Se4 108,6(3)1 Se4 3,525(5) �Se2-P1-Se3 106,4(3)1 Se5 3,318(5) �Se2-P1-Se4 106,7(3)
�Se3-P1-Se4 108,0(5)
P2 - 1 Se5 2,213(11) �Se5-P2-Se6 122,5(4)1 Se6 2,191(10) �Se5-P2-Se7 106,8(4)1 Se7 2,190(10) �Se5-P2-Se8 105,6(5)1 Se8 2,222(7) �Se6-P2-Se7 107,9(5)
�Se6-P2-Se8 105,8(3)�Se7-P2-Se8 107,5(4)
ähnlich ist, wird der wesentliche Unterschied in der Aus-richtung der Tetraeder bei einem Blick längs der a-Achsebesonders deutlich: Bei dem Sulfat sind die Tetraeder ent-lang [001] jeweils gleich ausgerichtet, entlang [010] aber um180° verdreht. Letzteres ist bei dem Thiophosphat nicht derFall, die Tetraeder stehen statt dessen entlang [001] Spitzeauf Spitze und sind zudem gegeneinander leicht verkippt (s.Abb. 2).
Die drei kristallographisch unabhängigen Ba-Atome wer-den von jeweils zehn S-Atomen mit einem mittleren Ab-stand von 3,37 A bzw. 3,38 A umgeben, was etwas über derRadiensumme von 3,26 A liegt (Ba: Atomradius (KZ 12);S: Kovalenzradius) [6]. Für die Ba1-Atome ergibt sich dabeiein verzerrtes trigonales Antiprisma, bei dem vier derDreieckflächen überkappt sind (s. Abb. 3). Die Polyeder umdie beiden anderen Ba-Atome sind dagegen unregelmäßig.
S. Jörgens, A. Mewis
Abb. 1 [001]-Projektion der Strukturen von �-Ba3(PS4)2 (oben)und β-K2SO4 (unten) (große Kugeln: Ba(K); dunkle Kugeln: P(S);helle Kugeln: S(O))
�-Ba3(PS4)2 durchläuft DSC-Messungen zufolge bei etwa75 °C eine Phasenumwandlung 1. Ordnung, die röntgeno-graphisch durch temperaturabhängige Messungen mit ei-nem Pulverdiffraktometer bestätigt wurde. Deren Auswer-tung zeigte, dass oberhalb der Umwandlungstemperaturkeine Reflexe hkl mit h � k � 2n mehr auftreten. Einkristall-aufnahmen mit einer Präzessionskamera ergaben, dass beider Umwandlung die Gitterparameter a und b nahezu un-verändert bleiben, während c sich auf die Hälfte verkleinert.Die monokline Symmetrie bleibt zwar erhalten, es erfolgtjedoch ein Wechsel des Bravais-Gittertyps von P nach C.Das Auslöschungssymbol ist 2/mC- -. Zu diesem gehörendie Raumgruppen C2, Cm und C2/m, von denen die zentro-symmetrische zur Beschreibung der Struktur geeignet ist.Die Raumtemperaturphase entsteht demnach durch einenklassengleichen Symmetrieübergang vom Index 2. An ei-nem Einkristall von Ba3(PSe4)2 konnte dagegen röntgeno-graphisch bis etwa 550 °C keine Umwandlung festgestelltwerden. Nach Messung der Intensitäten bei ca. 100 °C miteinem IPDS-Diffraktometer konnten die Strukturparame-ter von β-Ba3(PS4)2 mit dem in den Tabellen 1, 4 und 5aufgeführten Ergebnis verfeinert werden.
www.zaac.wiley-vch.de 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim Z. Anorg. Allg. Chem. 2007, 570�574572
Abb. 2 [100]-Projektion der Strukturen von �-Ba3(PS4)2 (oben)und β-K2SO4 (unten) (Atombezeichnung s. Abb. 1)
Abb. 3 �-Ba3(PS4)2: Umgebung der Ba1-Atome (große Kugel:Ba1; dunkle Kugeln: P; helle Kugeln: S)

�- und β-Ba3(PS4)2 sowie Ba3(PSe4)2
Tabell 4 Lage- und äquivalente Auslenkungsparameter/pm2 vonβ-Ba3(PS4)2
Atom Lage x y z Ueq
Ba1 2a 0 0 0 653(7)Ba2 4i 0,33315(8) 0 0,3638(1) 379(5)P 4i 0,3335(3) 0 0,7814(4) 322(8)S1 8j 0,0854(3) 0,2564(3) 0,3077(4) 450(9)S2 4i 0,1714(3) 0 0,6922(5) 443(11)S3 4i 0,3354(12) 0 0,0101(7) 1260(40)
Tabelle 5 Atomabstände/A und Bindungswinkel/° von β-Ba3(PS4)2
Ba1 - 4 S1 3,336(3) Ba2 - 2 S1 3,387(3)2 S2 3,336(5) 2 S1 3,388(3)6 S3 3,880(4) 2 S1 3,429(3)
2 S2 3,399(1)P - 2 S1 2,043(4) 1 S2 3,419(5)
1 S2 2,033(5) 1 S3 3,078(6)1 S3 1,991(7)
�S1-P-S1 106,7(3) �S1-P-S2 106,3(2) 2x�S1-P-S3 112,0(3) 2x �S2-P-S3 113,1(5)
Die Phasenumwandlung ist displaziv, d.h. die Kristall-struktur von β-Ba3(PS4)2 entspricht im wesentlichen dervon �-Ba3(PS4)2. Dadurch, dass die S1- und S5-Atome von�-Ba3(PS4)2 nun als S3-Atome auf der Spiegelebene in y �0 liegen, sind die PS4-Tetraeder entlang [001] nicht mehrverkippt, sondern parallel zueinander ausgerichtet, was zueiner Halbierung der c-Achse führt. Außerdem ist derenVerzerrung deutlich geringer, was sich an den von 106,3°bis 113,1° reichenden Winkeln ablesen lässt. Auch das Ba1-Atom bewegt sich im Zuge der Phasenumwandlung. Es ver-lässt seine allgemeine Lage (x � 0,02; y � 0,96) und sitztnun im Ursprung der neuen Zelle (s. Abb. 4). Die Verände-rungen der Atompositionen wirken sich auch auf die Um-gebung der Ba-Atome aus. Das Ba1-Atom wird von sechsS-Atomen mit einem Abstand von 3,34 A in Form einestrigonalen Antiprismas umgeben. Die Koordinationszahlist demnach kleiner als bei vergleichbaren Thiophosphaten,die z.B. 9 bei KBaPS4 [7] und Ba2P2S6 [3] bzw. 8 und 9 beiBa3Ln2(PS4)4 (Ln � Gd-Er) [8] beträgt. Allerdings befindensich über den Seitenflächen noch sechs S3-Atome, jedochin einem signifikant größeren Abstand von 3,88 A. Werdendiese berücksichtigt, ergibt sich die Koordinationszahl 12und ein mittlerer Ba1-S-Abstand von 3,61 A. Dabei fälltauf, dass die S3-Atome bei den Lageparametern relativhohe Standardabweichungen und zudem sehr große Auslen-kungsparameter aufweisen (U11 � 1810 pm2; U22 �1680 pm2). Das S3-Atom gehört zu den Koordinationspoly-edern von einem Ba2-Atom (Abstand: 3,08 A) sowie vondrei Ba1-Atomen (3,88 A). Es wäre denkbar, dass die S3-Atome in der a,b-Ebene fehlgeordnet sind und dadurch dieBa1-Atome in Wirklichkeit mit kürzeren Abständen umge-ben; dies ließ sich durch entsprechende Rechnungen abernicht verifizieren. Bei der Phasenumwandlung in die �-Form und der damit verbundenen Verkippung der PS4-Te-
Z. Anorg. Allg. Chem. 2007, 570�574 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.zaac.wiley-vch.de 573
Abb. 4 β-Ba3(PS4)2: Projektionen entlang [001], [010] und [100](große Kugeln: Ba; dunkle Kugeln: P; helle Kugeln: S)
traeder verliert das Ba1-Atom zwar den Kontakt zu zwei S-Nachbarn, gleichzeitig sinkt aber sein mittlerer Ba-S-Ab-stand von 3,61 A in der β- auf 3,38 A in der �-Phase. DieKoordinationssphäre des Ba2-Atoms wird von zehn S-Ato-men (mittlerer Abstand: 3,37 A) gebildet. Sechs von ihnenbilden einen Ring, über bzw. unter diesem befinden sich einbzw. 3 weitere S-Atome (s. Abb. 5).

S. Jörgens, A. Mewis
Abb. 5 β-Ba3(PS4)2: Umgebung der Ba2-Atome (große Kugel:Ba2; dunkle Kugeln: P; helle Kugeln: S)
Experimentelle Angaben
Zur Darstellung von Ba3(PS4)2 und Ba3(PSe4)2 wurden die Ele-mente in einem der Formel entsprechenden Verhältnis in evakuier-ten Quarzglasampullen erhitzt. Im Fall von Ba3(PS4)2 betrug dieReaktionszeit 10 � 15 h bei 800 °C, die Ampullen wurden anschlie-ßend abgeschreckt. Dabei bildeten sich kleine schwach rosa Kri-stallite der Zielverbindung im oberen Teil der Ampulle, währendsich im unteren Teil farbloses Ba2P2S6 [3] als Nebenprodukt be-fand. Ba3(PS4)2 wurde anschließend mit einem AgI-Überschuss ineiner Quarzglasampulle für 100 h auf 800 °C erhitzt und mit10 °/h abgekühlt, um Kristalle mit einer für die Röntgenstruktur-analyse geeigneten Größe zu züchten. Gelbe transparente Kristallevon Ba3(PSe4)2 wurden durch Umsetzung der Elemente bei 800 °Cüber einen Zeitraum von 25 h erhalten. Als Nebenprodukte fielen
www.zaac.wiley-vch.de 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim Z. Anorg. Allg. Chem. 2007, 570�574574
dabei glasige Phosphorselenide sowie Ba2P2Se6 [3] an. Beide Chal-cogenophosphate können ohne weiteres an der Luft gehandhabtwerden.
Pulverdiffraktogramme bei höheren Temperaturen wurden mit ei-nem Gerät X�Pert PRO (Fa. Philips Analytical) aufgenommen, fürdie entsprechende Vermessung der Einkristalle kam eine selbst an-gefertigte Heizvorrichtung zum Einsatz. Einkristalle der beidenVerbindungen wurden nach einer Voruntersuchung (Präzessions-Kamera) auf einem Diffraktometer (IPDS, AED2, Fa. Stoe) ver-messen (MoKα; Graphitmonochromator), wobei Absorptionsein-flüsse bei Ba3(PSe4)2 mit Hilfe von ψ-scans korrigiert wurden. ZurStrukturlösung und der anschließenden Optimierung freier Para-meter wurde das Programmsystem SHELXL-97 [9] herangezogen.Weitere Einzelheiten zur Strukturbestimmung können beim Fach-informationszentrum Karlsruhe, D-76344 Eggenstein-Leopolds-hafen, unter Angabe der CSD-Hinterlegungsnummern 417421(�-Ba3(PS4)2), 417422 (β-Ba3(PS4)2) sowie 417423 (Ba3(PSe4)2) an-gefordert werden.
Literatur
[1] P. Villars, L. D. Calvert, Pearson’s Handbook of CrystallographicData for Intermetallic Compounds, 2nd ed. 1991 and desk edition1997, ASM, Metals Park, Oh 44073.
[2] C. Hadenfeldt, D. Hoedel, Z. Anorg. Allg. Chem. 1996, 622,1495.
[3] S. Jörgens, A. Mewis, R.-D. Hoffmann, R. Pöttgen, B. D.Mosel, Z. Anorg. Allg. Chem. 2003, 629, 429.
[4] S. Jörgens, A. Mewis, Z. Anorg. Allg. Chem. 2004, 630, 51.[5] W. Brockner, U. Pätzmann, Z. Naturforsch. 1987, 42a, 513.[6] L. Pauling, Die Natur der chemischen Bindung, 3. Auflage,
Verlag Chemie, Weinheim 1976, S. 379.[7] S. Jörgens, A. Mewis, Z. Naturforsch. 2005, 60b, 431.[8] Y. Klawitter, W. Bensch, C. Wickleder, Chem. Mater. 2006, 18,
187.[9] G. M. Sheldrick, SHELXL-97, Univ. Göttingen (1997).



![2121 21 SPARCO BÅ3 21 C] -07 BA3 2103, (K 2103, 2107 ( c BA3 BA3 2101—07 2101 2102, 2103, 21 C 7 ( 21 C I BPT (I-LAO 21 C I BPT (I-LAO 7 BA3 ruo ruo NED EurcEx M3ÅT3-2 …](https://cdn.vdocuments.site/doc/165x107/610a4c5a32cfb84fad3ebd8c/2121-21-sparco-b3-21-c-07-ba3-2103-k-2103-2107-c-ba3-ba3-2101a07-2101.jpg)



