
1
Hypothetical Potential Energy Surface
Chapter 15 (26): Computational Chemistry
stationary pointsall ν s >0 :
minimum
one ν im:transition statesaddle point
multiple ν im:hilltop
2
Ethane conformationsHartree-Fock theory, 6-31G(d) basis set
Source: Hyperchem calculationHF 6-31G(d)geometry relaxed at each angle
Chapter 15 (26): Computational Chemistry
staggered
eclipsed
3
Potential Energy Surface for F( 2P3/2,
2P1/2 ) + CH4 → FH + CH3
Chapter 15 (26): Computational Chemistry
MP2 with 6-311+G(2df,2pd) basis setSource: Cipriano Rángel, et al., J. Phys. Chem. A, 109 (7), 1441 -1448, 2005. 4
Thermodynamic versus kinetic control
Engel's Figures 15.4,5,6
∆E relates to thermodynamic control. Ea relates to kinetic control.
Chapter 15 (26): Computational Chemistry
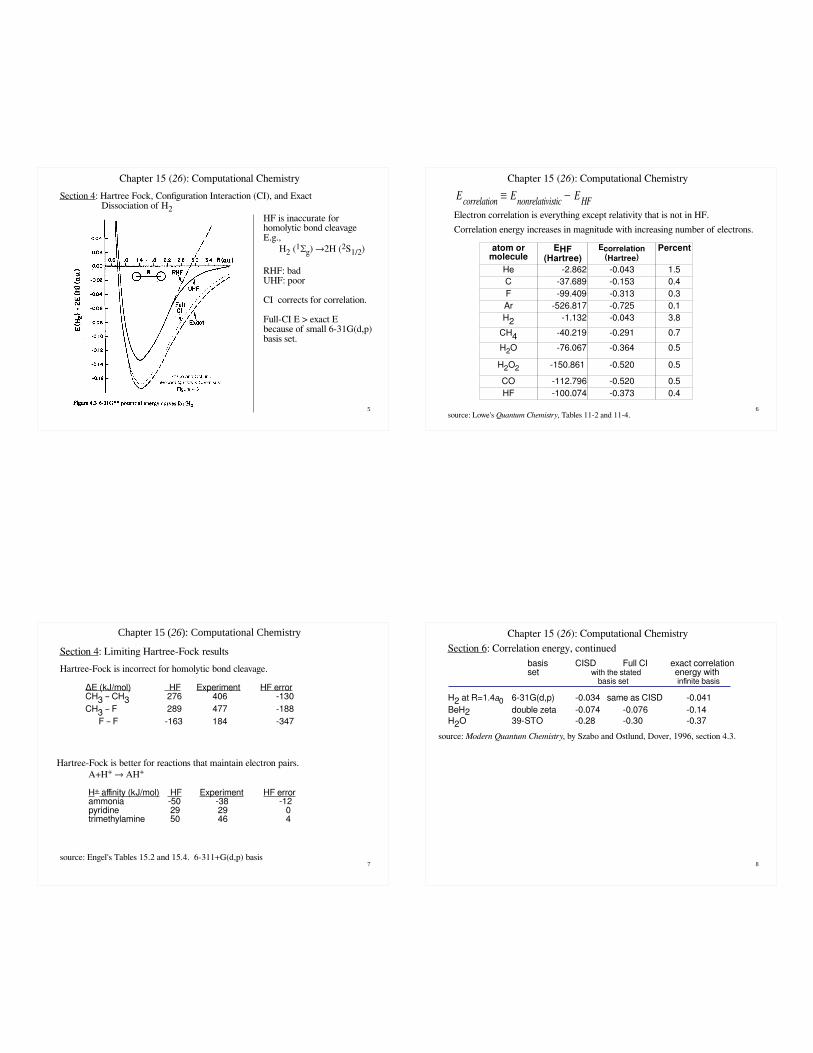
5
Section 4: Hartree Fock, Configuration Interaction (CI), and Exact Dissociation of H2
Chapter 15 (26): Computational Chemistry
HF is inaccurate for homolytic bond cleavageE.g.,
H2 (1Σg) →2H (2S1/2)
RHF: badUHF: poor
CI corrects for correlation.
Full-CI E > exact E because of small 6-31G(d,p) basis set.
6
atom ormolecule
EHF (Hartree)
Ecorrelation(Hartree)
Percent
He -2.862 -0.043 1.5C -37.689 -0.153 0.4F -99.409 -0.313 0.3Ar -526.817 -0.725 0.1H2 -1.132 -0.043 3.8
CH4 -40.219 -0.291 0.7
H2O -76.067 -0.364 0.5
H2O2 -150.861 -0.520 0.5
CO -112.796 -0.520 0.5HF -100.074 -0.373 0.4
source: Lowe's Quantum Chemistry, Tables 11-2 and 11-4.
Electron correlation is everything except relativity that is not in HF.
Correlation energy increases in magnitude with increasing number of electrons.
Chapter 15 (26): Computational Chemistry
E correlation ≡ E nonrelativistic − E HF
7
Hartree-Fock is incorrect for homolytic bond cleavage.
ΔE (kJ/mol) HF Experiment HF errorCH3 – CH3 276 406 -130
CH3 – F 289 477 -188
F – F -163 184 -347
Hartree-Fock is better for reactions that maintain electron pairs. A+H+ → AH+
source: Engel's Tables 15.2 and 15.4. 6-311+G(d,p) basis
H+ affinity (kJ/mol) HF Experiment HF errorammonia -50 -38 -12pyridine 29 29 0trimethylamine 50 46 4
Chapter 15 (26): Computational Chemistry
Section 4: Limiting Hartree-Fock results
8
Section 6: Correlation energy, continued
Chapter 15 (26): Computational Chemistry
source: Modern Quantum Chemistry, by Szabo and Ostlund, Dover, 1996, section 4.3.
basis CISD Full CI exact correlationset with the stated energy with
basis set infinite basis
H2 at R=1.4a0 6-31G(d,p) -0.034 same as CISD -0.041BeH2 double zeta -0.074 -0.076 -0.14H2O 39-STO -0.28 -0.30 -0.37

9
CI-Singles and Doubles is not size consistent.Example He monomer and dimer with {1s,2s} basis.
CISD for He monomer
CISD for HeaHeb dimer
CISD for separated Hea Heb monomers includes four additional double excitations.
Figures are from Engel's chapter 15.
CIS and full CI are size consistent. 10
DFT focuses on ρ(x,y,z) rather than on Ψ(all electrons' x,y,z)▪ Walter Kohn (1923-2016) and Pierre Hohenberg theoretical basis, 1963
▪ Walter Kohn and Lu Sham, 1964expanded ρ(x,y,z) in electron orbitals (like MO theory)Kohn-Sham equations for orbitals and their energies (SCF)
Chapter 15 (26): Computational ChemistryDensity Functional Theory (DFT)
Walter KohnNobel 1998www.nobelprize.org
[− 12
∇2− ∑
nuclei A
Z A
|⃗r− R⃗A|+∫
ρ( r⃗ ' )|⃗r − r⃗ '| d r⃗ ' + v xc ]ψi
KS( r⃗ ) = ϵi
KSψi
KS( r⃗ )
11
Kohn-Sham and Hartree-Fock orbitals for ethene
HOMO-LUMO0.24 Eh
HOMO-LUMO0.55 Eh
source: GAMESS, images from wxMacMolPlt.12
Chapter 15 (26): Computational Chemistry
Density Functional Theory, results
mean absolute errors
Bond Lengths1 Atomization2 Dipole3
Method (Angstrom) (kJ/mol) (Debye)HF 0.022 310 0.21B3LYP 0.004 9.2 0.03
1. for 32 molecules containing only first-row atoms. Basis set 6-311G(d,p). Cramer, Essentials of Computational Chemistry, Table 8.5.2. Atomization energies were calculated for the G2 set: 55 molecules. Basis 6-31G(d). Cramer, Essentials of Computational Chemistry, Table 8.1.3. Average error in Debye for CO, H2O, H2S, NH3, PH3 and SO Cohen, Chemical Physics Letters, 299, 465-472, 1999.
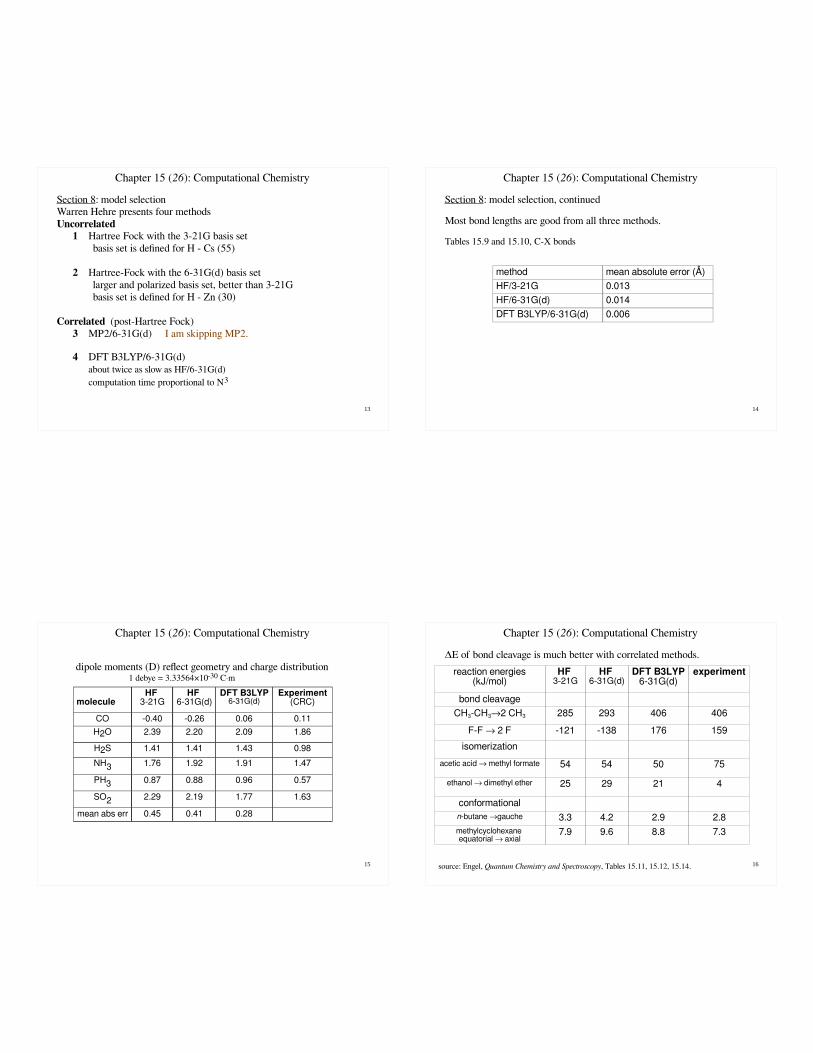
13
Chapter 15 (26): Computational Chemistry
Section 8: model selectionWarren Hehre presents four methodsUncorrelated
1 Hartree Fock with the 3-21G basis setbasis set is defined for H - Cs (55)
2 Hartree-Fock with the 6-31G(d) basis setlarger and polarized basis set, better than 3-21Gbasis set is defined for H - Zn (30)
Correlated (post-Hartree Fock)3 MP2/6-31G(d) I am skipping MP2.
4 DFT B3LYP/6-31G(d)about twice as slow as HF/6-31G(d)computation time proportional to N3
14
Chapter 15 (26): Computational Chemistry
Section 8: model selection, continued
Most bond lengths are good from all three methods.
Tables 15.9 and 15.10, C-X bonds
method mean absolute error (Å)
HF/3-21G 0.013
HF/6-31G(d) 0.014
DFT B3LYP/6-31G(d) 0.006
15
dipole moments (D) reflect geometry and charge distribution 1 debye = 3.33564×10-30 C·m
moleculeHF
3-21GHF
6-31G(d)DFT B3LYP
6-31G(d)Experiment
(CRC)
CO -0.40 -0.26 0.06 0.11
H2O 2.39 2.20 2.09 1.86
H2S 1.41 1.41 1.43 0.98
NH3 1.76 1.92 1.91 1.47
PH3 0.87 0.88 0.96 0.57
SO2 2.29 2.19 1.77 1.63
mean abs err 0.45 0.41 0.28
Chapter 15 (26): Computational Chemistry
16
ΔE of bond cleavage is much better with correlated methods.
reaction energies (kJ/mol)
HF 3-21G
HF 6-31G(d)
DFT B3LYP6-31G(d)
experiment
bond cleavage
CH3-CH3→2 CH3 285 293 406 406
F-F → 2 F -121 -138 176 159
isomerization
acetic acid → methyl formate 54 54 50 75
ethanol → dimethyl ether 25 29 21 4
conformationaln-butane →gauche 3.3 4.2 2.9 2.8methylcyclohexane equatorial → axial
7.9 9.6 8.8 7.3
Chapter 15 (26): Computational Chemistry
source: Engel, Quantum Chemistry and Spectroscopy, Tables 15.11, 15.12, 15.14.
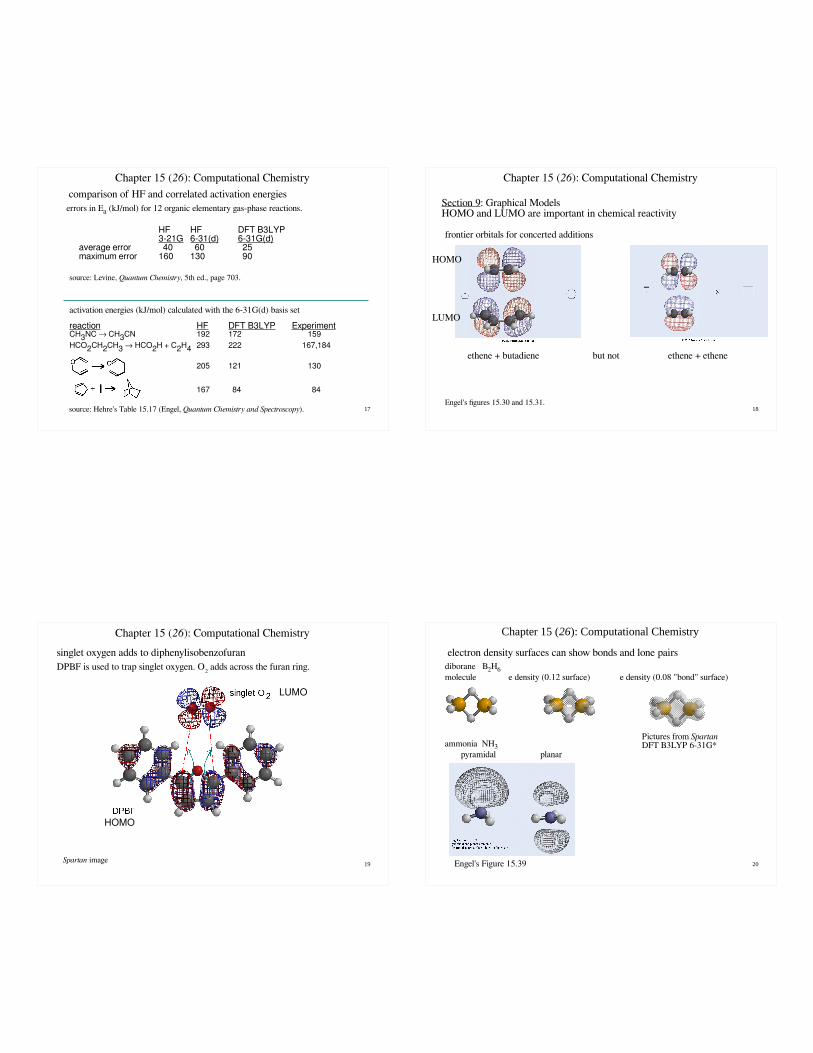
17
reaction HF DFT B3LYP ExperimentCH3NC → CH3CN 192 172 159
HCO2CH2CH3 → HCO2H + C2H4 293 222 167,184
205 121 130
167 84 84
comparison of HF and correlated activation energies
source: Hehre's Table 15.17 (Engel, Quantum Chemistry and Spectroscopy).
activation energies (kJ/mol) calculated with the 6-31G(d) basis set
errors in Ea (kJ/mol) for 12 organic elementary gas-phase reactions.
source: Levine, Quantum Chemistry, 5th ed., page 703.
HF HF DFT B3LYP3-21G 6-31(d) 6-31G(d)
average error 40 60 25maximum error 160 130 90
Chapter 15 (26): Computational Chemistry
18
Section 9: Graphical ModelsHOMO and LUMO are important in chemical reactivity
ethene + butadiene but not ethene + ethene
frontier orbitals for concerted additions
Engel's figures 15.30 and 15.31.
Chapter 15 (26): Computational Chemistry
HOMO
LUMO
19
singlet oxygen adds to diphenylisobenzofuranDPBF is used to trap singlet oxygen. O
2 adds across the furan ring.
Spartan image
HOMO
LUMO
Chapter 15 (26): Computational Chemistry
20
electron density surfaces can show bonds and lone pairsdiborane B2H6molecule e density (0.12 surface) e density (0.08 "bond" surface)
ammonia NH3pyramidal planar
Engel's Figure 15.39
Chapter 15 (26): Computational Chemistry
Pictures from SpartanDFT B3LYP 6-31G*

21
electrostatic potential near H correlates with acidity
Spartan. PM3 geometry. HF 6-31G(d) electrostatic potential.
Chapter 15 (26): Computational Chemistry







