
Secondary structure assignment Secondary structure assignment and prediction and prediction
Eran EyalMay 2011
Talk overview
•Why to predict secondary structures in proteins
• Methods to predict secondary structures in proteins
• Performance and evaluation
• Machine learning approaches
• Detailed description of several specific programs (PHD)
•Secondary structure assignment
Automatic assignment of secondary structures to a set of protein coordinates
Assignment of secondary structures to known secondary structures is a relatively simple bioinformatics task.
Given exact definitions for secondary structures, all we need todo is to see which part of the structure falls within each definition
α-helix
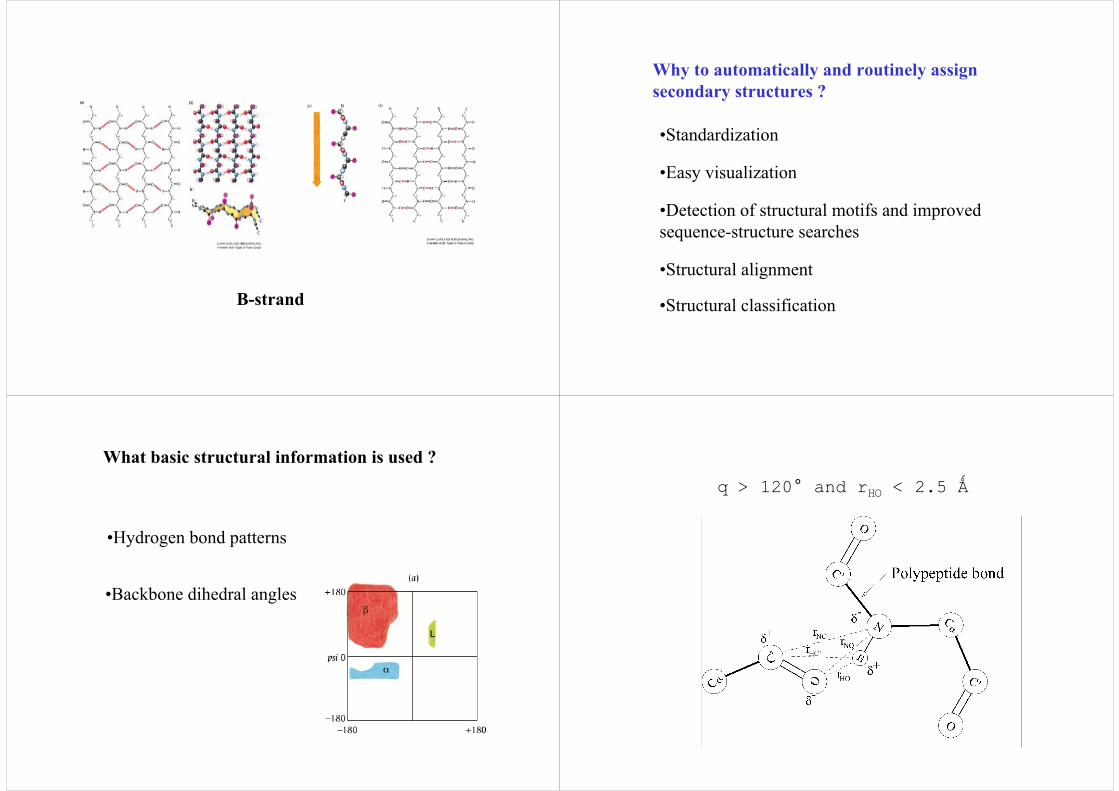
Β-strand
Why to automatically and routinely assign secondary structures ?
•Standardization
•Easy visualization
•Detection of structural motifs and improved sequence-structure searches
•Structural alignment
•Structural classification
What basic structural information is used ?
•Hydrogen bond patterns
•Backbone dihedral angles
q > 120° and rHO < 2.5 Ǻ

DSSP algorithm
•The so-called “Dictionary of Secondary Structure of Proteins”(DSSP) by Kabsch and Sander makes its sheet and helix assignments solely on the basis of backbone-backbone hydrogen bonds.
•The DSSP method defines a hydrogen bond when the bond energy is below -0.5 kcal/mol from a Coulomb approximation of the hydrogen bond energy.
•The structural assignments are defined such that visually appealing and unbroken structures result.
• In case of overlaps, alpha-helix is given first priority.
•The helix definition does not include the terminal residue having the initial and final hydrogen bonds in the helix.
•A minimal size helix is set to have two consecutive hydrogen bonds in the helix, leaving out single helix hydrogen bonds, which are assigned as turns (state 'T').
•beta-sheet residues (state 'E') are defined as either having two hydrogen bonds in the sheet, or being surrounded by two hydrogen bonds in the sheet.
•The minimal sheet consists of two residues at each partner segment.
STRIDE
•The secondary STRuctural IDEntification method by Frishman and Argos uses an empirically derived hydrogen bond energy and phi-psi torsion angle criteria to assign secondary structure.
•Torsion angles are given alpha-helix and beta-sheet propensities according to how close they are to their regions in Ramachandranplots.
•The parameters are optimized to mirror visual assignments made by crystallographers for a set of proteins.
•By construction, the STRIDE assignments agreed better with the expert assignments than DSSP, at least for the data set used to optimize the free parameters.

•Like DSSP, STRIDE assigns the shortest alpha-helix ('H') if it contains at least two consecutive i - i+4 hydrogen bonds.
•In contrast to DSSP, helices are elongated to comprise one or both edge residues if they have acceptable phi-psi angles, similarly a short helix can be vetoed.
•hydrogen bond patterns may be ignored if the phi-psi angles are unfavorable.
•The sheet category does not distinguish between parallel and anti-parallel sheets. The minimal sheet ('E') is composed of two residues.
•The dihedral angles are incorporated into the final sheetassignment criterion as was done for the alpha-helix.
DEFINE
•An algorithm by Richards and Kundrot which assigns secondary structures by matching Cα-coordinates with a linear distance mask of the ideal secondary structures.
•First, strict matches are found, which subsequently are elongated and/or joined allowing moderate irregularities or curvature.
•The algorithm locates the starts and ends of α- and 310-helices, beta-sheets, turns and loops. With these classifications the authors are able to assign 90-95% of all residues to at least one of the given secondary structure classes.

•Prediction of tertiary structures based on the amino acid sequence is still a very difficult task.
•Prediction of more local structural properties is easier
•Prediction of secondary structures and solvent accessibility (SAS) is important and more feasible
Secondary structure prediction
•Prediction of secondary structures is a “bridge” between the linear information and the 3D structure
•Programs in this field often employ different types of machine learning approaches
A-C-H-Y-T-T-E-K-R-G-G-S-G-T-K-K-R-E-A
A-C-H-Y-T-T-E-K-R-G-G-S-G-T-K-K-R-E-AH-H-H-H-H-H-H-H-O-O-O-O-O-S-S-S-S-S-S
Many degrees of freedomLong search. Pruned to errors
Few degrees of freedomFast search
The importance and the need of predicting the secondary structures in proteins
The information might give clues concerning the function of the protein and the existing of specific structural motifs
Intermediate step toward construction of a complete 3D model from the sequence.
Secondary structure content also allows us to classify a protein to the basic levels of structure type based on its sequence alone.
Generations in algorithm development
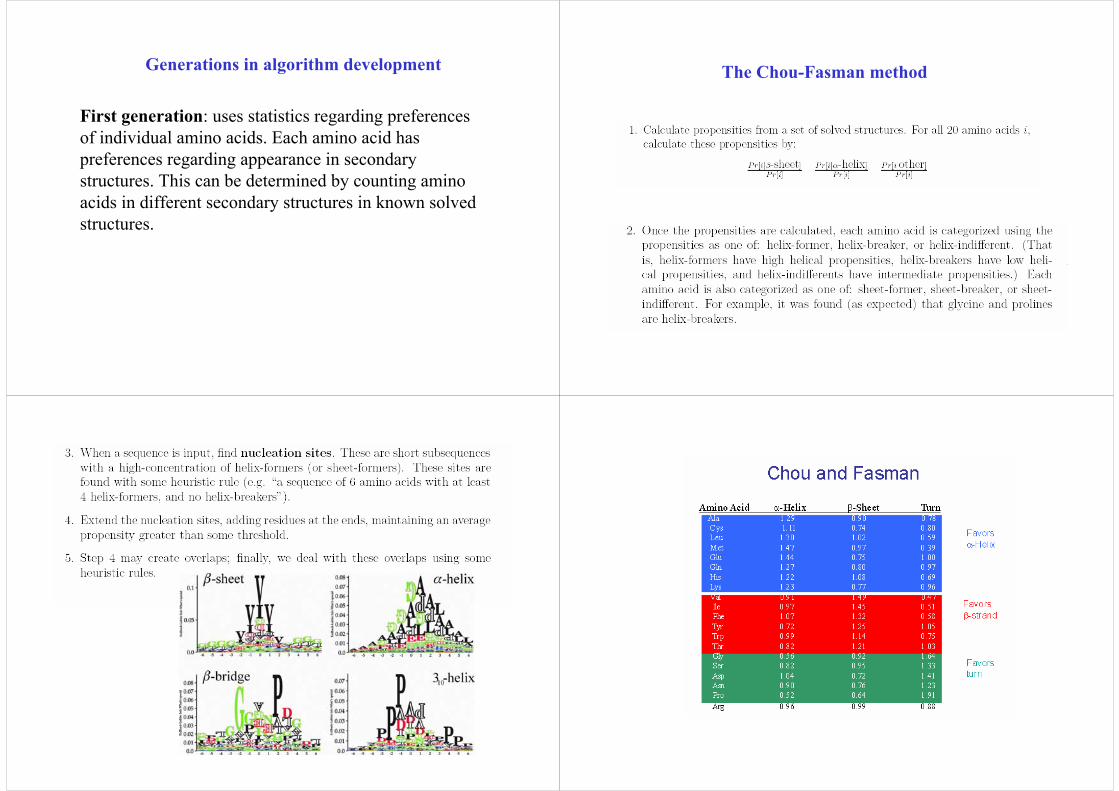
First generation: uses statistics regarding preferences of individual amino acids. Each amino acid has preferences regarding appearance in secondary structures. This can be determined by counting amino acids in different secondary structures in known solved structures.
The Chou-Fasman method

Second generation: the improvements comparing to the first were the uses of better statistics and statistical methods, and by looking on a set of adjacent amino acids on the sequence (usually windows of 11-21 amino acids) rather than on individual amino acids
The new statistics determined what is the probability of an amino acid to be in a particular secondary structure given that it is in the middle of a local sequence segment.Other segments similar to the given segments might also assist in the prediction. Different methods tried to correspond the segments to other segments in the 3D database by sequence alignments and other methods.
The GOR method
Helix tableStrand table

General problems of methods in generations I,II
Overall prediction rate was rather low:
• Overall prediction: 60%• B-strands prediction: ~35%• Predictions included small secondary elements, with disability to integrate them to longer structures such as those found in protein structures.
Third generation: the improvement of the programs in the third generation was mainly due to incorporation of evolutionary information. This was done by looking at the multiple alignment which included sequences similar to the sequence we wish to predict.
Such information presented as MSA or by other way include plenty of information which can not be obtained from evolutionary sequences:• Which regions are more conserved• which substitution are allowed in each position• Information regarding interacting sites
SAARDFFRT--HAAGRFFTFTSAARDFFRS--GTRAKFFTFTTAARDFFRF-GKAA-KFFTFTSAARRFFRTGDHAALDFFTFTSAARRFFRWHGLAAIDFFTFT
Comparison of many sequences of protein families helps to detect conserved regions
AAARDFFRTGGHAAGRFFTFTAAARDFFRSGGHAAGKFFTFTAAARDFFRTGGHAAGKFFTFTAAARRFFRTGAHAAGDFYTFSAAARRFFRTGGHAAGDFFTFT
Comparison of many sequences of protein families helps to detect interactions in space

Information obtained from MSA might help in the prediction. Because the fold of all members of the family is identical, every sequence can contribute the structure prediction of other given sequence in the family
The best MSA for this purpose is one which includes many sequences of the family but being not too close one to another
Neurons cells are the basic components of the nerve system
Every neuron gets information from several other neurons by the dendrites
Introduction to neural networks
The information is being processed and the neuron makes binary decision if to transfer a signal to other neurons
The information is transferred by the axon
Computational tasks that the nerve system executes:
•Representation of data•Holding data•Learning procedures•Decision making•Pattern recognition
Neural networks - properties
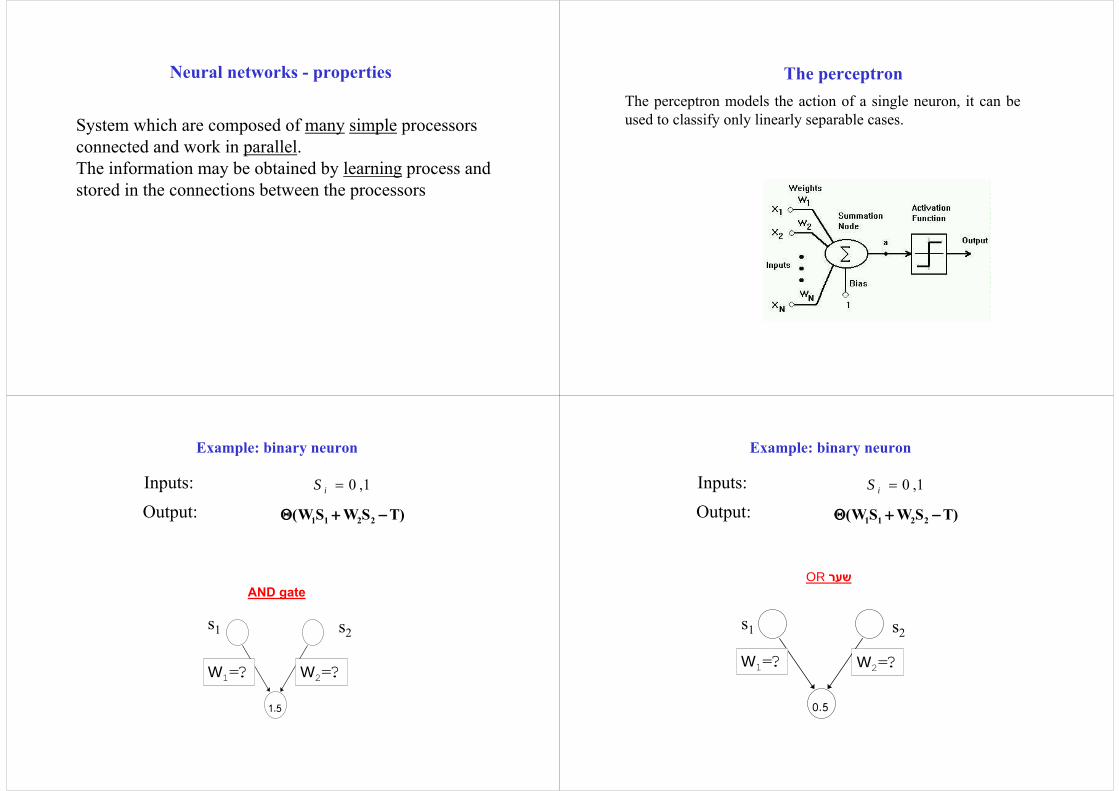
System which are composed of many simple processors connected and work in parallel. The information may be obtained by learning process and stored in the connections between the processors
The perceptronThe perceptron models the action of a single neuron, it can be used to classify only linearly separable cases.
Example: binary neuron
S i = 0 1,
)TSWSW( 2211 −+Θ
1.5
1 1
AND gate
W1=? W2=?
Inputs:
s1 s2
Output:
0.5
1 1
ORשער
W1=? W2=?
Example: binary neuron
S i = 0 1,
)TSWSW( 2211 −+Θ
Inputs:Output:
s1 s2
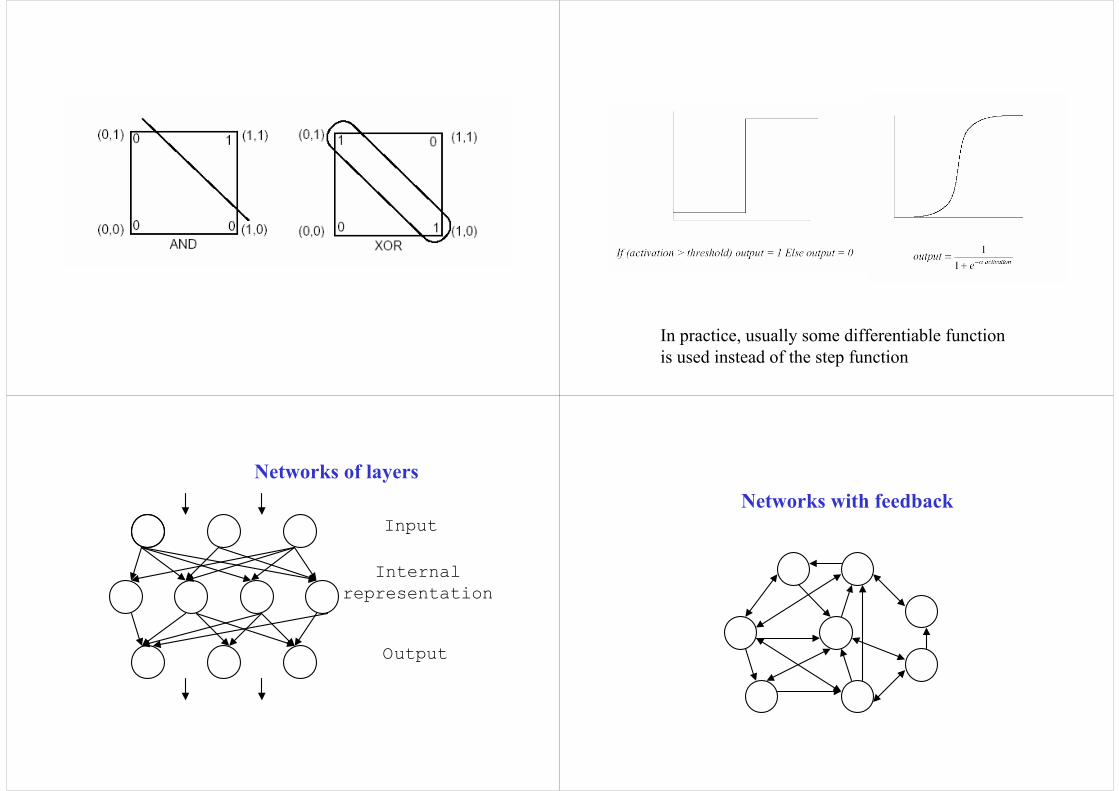
In practice, usually some differentiable function is used instead of the step function
Networks of layers
Input
Internal representation
Output
Networks with feedback

Training
Preparation of a large training set
The neural network gets the input and random initial values for the parameters (weights)
The network tries to maximize the number of correctly predicted cases by changes in the values of the parameters (weights)
To test the net we evaluate its performance on a collection of solved examples (test set)
The test set should be independent of the training set. The first interaction of the net with this set should be done during evaluation
The test set should be large and representative. It is better to use test set already used for evaluation of other programs designed to solve similar task

PHD – a third generation program the uses neural networks.
PhD is the most popular secondary structure prediction program, although other programs reach the same accuracy, it is still very popular today
The versions of this program implement and demonstrate the recent elements which are considered the most important for prediction accuracy
Demonstrates the use of machine learning approaches in this field
Input: sequence of amino acids. Using data base sequence alignment, similar alignments are found and MSA is built
The composition of this alignment is the input to the neural network which is the core of the program
Every position in the input sequence is expressed by 21 parameters: the prectage of each amino acid in that position and another character which indicate the start/end of the sequence
In addition the input for each position includes global information about the protein composition and the sequence distance between the predicted region to the start/end positions

Important of variability in the input sequences
Good alignment!
Output: the secondary structure with the highest score is the final prediction for that position
The neural network includes several layers:
Input layer: sequence -> structure
Intermediate layer: structure -> structure
Output system: summation of several networks

http://www.embl-heidelberg.de/predictprotein/
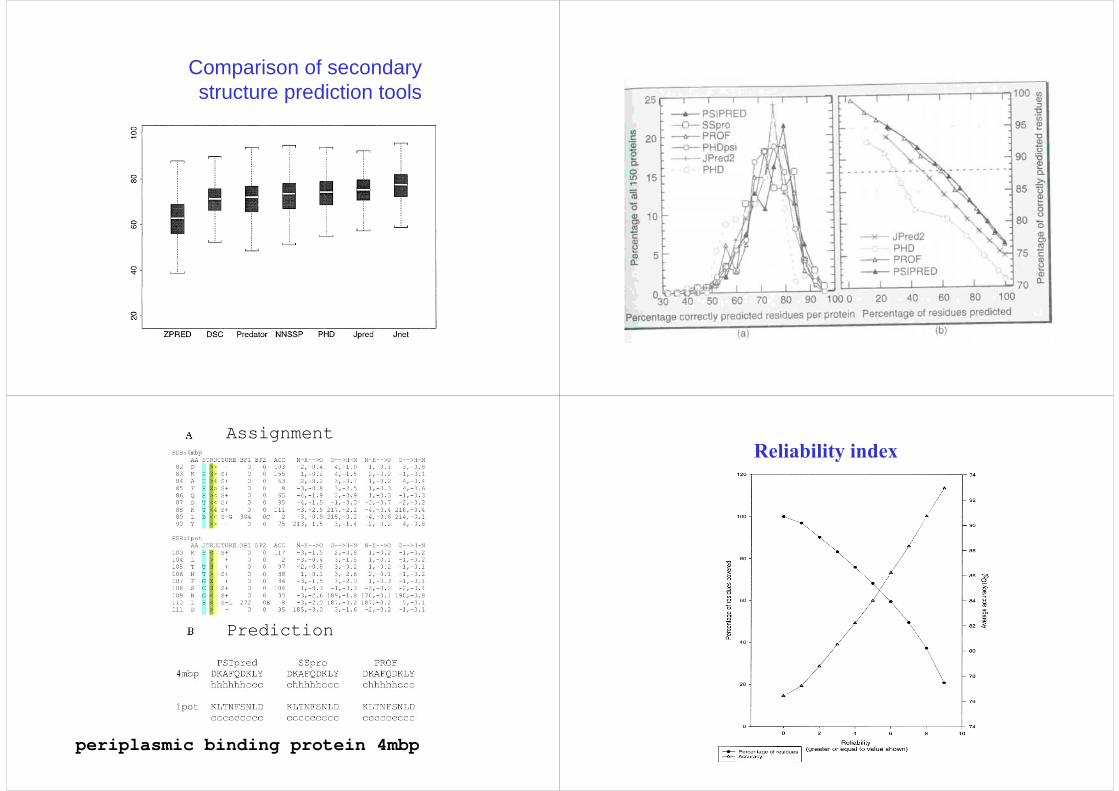
Comparison of secondary structure prediction tools
periplasmic binding protein 4mbp
Assignment
Prediction
Reliability index

Reliability index -PHDCombination of different prediction methods
Every method has errors which can be classified to 2 general types:
1.Systematic errors2.Non-systematic errors
Several methods can be therefore combined to increase the prediction accuracy
The basic condition to successful combination is that the source of error of each individual method is not only systematical
Several new methods exploit this fact and train independently several neural networks and predict based on average prediction of all the networks.
Another method (Jpred) gets as input results of several existing methods and predict based on that.

Many web-server available ….
http://www.compbio.dundee.ac.uk/www-jpred/
http://bmerc-www.bu.edu/psa/
To understand some of the sequence signals that might be used we can consider the basic biochemistry of secondary structures
α-helix for example has a periodicity of 3.6 amino acids. Helices on the protein surface are expected to posses some signal in this periodicity for positions occupied by hydrophilic and hydrophobic side chains.
Finding hydrophobic amino acids in positions i,i+3,i+7, i+10 for example is a strong indication for a helix

α-helix in Myoglobin
β-strand of CD8
Similarly, in surface B-strands, there is preferences for “Zigzag’ pattern. For example, hydrophilic side chain at positions i, i+2, i+4... and hydrophobic side chains at positions i+1, i+3, i+5…
Related topics
•Prediction secondary structures of membrane proteins
•Prediction of solvent accessibility
Rel. Acc. (%)
PSIBLAST (%)
HMMER2 (%)
Combined [change] (%)
25% 75.0 74.2 76.25% 79.0 78.8 79.80% 86.6 86.3 86.5
Average prediction accuracies from (based on the 480 protein set) for 2-state Solvent Accessibility







