Download - Intro CompChem

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 1/79
Materials Simulation Center presents:
Introduction toComputational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 2/79
Course contents
• verv ew: a s ompu a ona em s ry
• 1: ab initio method
• 2: Density Functional Theory
• 3: Molecular mechanics method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 3/79
Overview: What is Computational Chemistry
Computational Chemistry uses the results of theoreticalchemistry, incorporated into efficient computer programs, tocalculate the structures and properties of molecules and solids.
Computational chemistry methods range from highly accurate(i.e. Ab initio methods and DFT) to less accurate, (i.e. semi-empirical method), to very approximate (i.e. Molecular
, - .
It can deal with system of a single molecule, a group of molecules, a liquid or solid.
It calculates properties such as properties are structure, relativeenergies, charge distributions, dipoles and multipole moments,
,quantities, etc.
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 4/79
Simulation method for length and time scale
ContinuumContinuumContinuumContinuum
-3
100
TIME
/s
Mesoscale methodsAtomistic
e o s
-3
100
TIME
/s
Mesoscale methodsAtomistic
e o s
-3
100
TIME
/s
Mesoscale methodsAtomistic
e o s
-3
100
TIME
/s
Mesoscale methodsAtomistic
e o s
-9
10-6(μs)
mu a onMethods
Semi-empirical
Lattice Monte Carlo
Brownian dynamicsDissipative particle dyn
-9
10-6(μs)
mu a onMethods
Semi-empirical
Lattice Monte Carlo
Brownian dynamicsDissipative particle dyn
-9
10-6(μs)
mu a onMethods
Semi-empirical
Lattice Monte Carlo
Brownian dynamicsDissipative particle dyn
-9
10-6(μs)
mu a onMethods
Semi-empirical
Lattice Monte Carlo
Brownian dynamicsDissipative particle dyn
-15
10-12(ps)
me o s
Ab initio
methods
Monte Carlo
molecular dynamics
tight-bindingMNDO, INDO/S-15
10-12(ps)
me o s
Ab initio
methods
Monte Carlo
molecular dynamics
tight-bindingMNDO, INDO/S-15
10-12(ps)
me o s
Ab initio
methods
Monte Carlo
molecular dynamics
tight-bindingMNDO, INDO/S-15
10-12(ps)
me o s
Ab initio
methods
Monte Carlo
molecular dynamics
tight-bindingMNDO, INDO/S
10-10 10-9 10-8 10-7 10-6 10-5 10-4
(nm) (μm)
LENGTH
10-10 10-9 10-8 10-7 10-6 10-5 10-4
(nm) (μm)
LENGTH
10-10 10-9 10-8 10-7 10-6 10-5 10-4
(nm) (μm)
LENGTH
10-10 10-9 10-8 10-7 10-6 10-5 10-4
(nm) (μm)
LENGTH
/meters /meters
Figure 1. Schematic illustration of the simulation
/meters /meters
Figure 1. Schematic illustration of the simulation
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 5/79
Methods
• n t o quantum c em stry met o s
• Density functional theory (DFT) methods
• Semi-empirical quantum chemistry method
• Molecular mechanics method
• Specially treatment for periodic boundary condition
• Plane waves implementation
• Ewald summation for long-range interaction
• Molecular dynamics and Monte Carlo simulation
• Hybrid quantum mechanics/molecular mechanics method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 6/79
Ab initio quantum chemistry methods
Ab initio quantum chemistry solves electronic Schrödinger equation frommathematical principles. The term ab initio indicates that thecalculation is derived from first principles and does not rely on anyemp r ca a a.
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 7/79
Commonly used methods
Hartree-Fock methods
o Hartree-Fock (HF)o Restricted Open-shell Hartree-Fock (ROHF)o Unrestricted Hartree-Fock (UHF)
Post-Hartree-Fock methods
-o Configuration interaction (CI)o Coupled cluster (CC)
o Quadratic configuration interaction (QCI)o uan um c em s ry compos e me o s , ,
Multi-reference methods
o Multi-configurational self-consistent field (MCSCF)o Multi-reference configuration interaction (MRCI)o Complete Active Space Perturbation Theory (CASPTn)
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 8/79
Electron atomic and molecular orbitals
HOMO LUMO
HOMO-1 LUMO+1
HOMO-2
LUMO+2
-
+
Introduction to Computational Chemistry
HOMO-4 LUMO+4

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 9/79
Molecular Orbital Theory & Hartree-Fock Method
)2()2()2(
)1()1()1(
1
N21
φφφ
φφφ
L
L
N
) N() N() N(! N
N21 φφφ
=ψ
L
LLLL
χ==μ
μμii c
1
LCAO: Linear Combination of Atomic Orbitals
N
Hartree-Fock (HF)
=μμμii
1
Variation Principle
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 10/79
Post-Hartree-Fock Method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 11/79
Basis Set
STO
∑ χ=φ=
μμ N
ii c
1
GTO
r • Minimal basis sets: STO-3G• Split-valence basis sets: 3-21G, 6-31G
• Polarisation functions are denoted with * (3-21G*, 6-31G*)
• use unc ons are eno e y + - +
* The selection of basis set is a key factor to the accuracy of the numeric solution.
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 12/79
Level of Theory
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 13/79
Cost vs. Accuracy
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 14/79
Methods
• n t o quantum c em stry met o s
• Density functional theory (DFT) methods
• Semi-empirical quantum chemistry method
• Molecular mechanics method
• Specially treatment for periodic boundary condition
• Plane waves implementation
• Ewald summation for long-range interaction
• Molecular dynamics and Monte Carlo simulation
• Hybrid quantum mechanics/molecular mechanics method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 15/79
Density functional theory methods
• -
― All properties of the many-body system are determined by the
― Each property is a functional of the ground state density n0(r) which
―
A functional f [n0] maps a function to a result: n0(r)→
f
• Kohn-Sham ansatz (1965)
• Replace original many-body problem with an independent electron
pro em – a can e so ve• The ground state density is required to be the same as the exact
density
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 16/79
Exchange-Correlation Functional
• Local Density Approximation - LDA
Exc can be determined usin homo eneous electron as model or using quantum Monte Carlo methods
• -
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 17/79
Correlation-Exchange Functional
LDAlocal density
l c o s t
VWN5
BLYP
GGAgradient corrected
p u t a t i o n
BP86
Meta-GGAkinetic energy density
included t y a n d c oTPSS
M06-L
Hybrid“exact” HF exchange
s i n g q u a l i
B3LYPB97/2MPW1K
Hybrid-meta-GGA I n c r e a
MPWB1KM06
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 18/79
Software supporting DFT
Introduction to Computational Chemistry
http://en.wikipedia.org/wiki/Density_functional_theory

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 19/79
Methods
• n t o quantum c em stry met o s
• Density functional theory (DFT) methods
• Semi-empirical quantum chemistry method
• Molecular mechanics method
• Specially treatment for periodic boundary condition
• Plane waves implementation
• Ewald summation for long-range interaction
• Molecular dynamics and Monte Carlo simulation
• Hybrid quantum mechanics/molecular mechanics method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 20/79
Semi-empirical quantum chemistry method
CNDO (1965,CNDO (1965, PoplePople et al)et al)
MINDO (1975, Dewar )MINDO (1975, Dewar )
STOSTO--basis (/Sbasis (/S--spectra,/2 dspectra,/2 d--orbitalsorbitals))
//1/2/3, organics1/2/3, organics
MNDO (1977,MNDO (1977, ThielThiel)) /d, organics, transition metals/d, organics, transition metals
INDO (1967,INDO (1967, PoplePople et al)et al)
ZINDOZINDO
rgan csrgan cs
Electronic spectra, transition metalsElectronic spectra, transition metals
SINDO1SINDO1 11--3 row binding energies,3 row binding energies,
photochemistry and transition metalsphotochemistry and transition metals
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 21/79
Further improvement
AM1 AM1 Modified nuclear repulsion terms model to account for Modified nuclear repulsion terms model to account for
-- ,,
Widely used today (transition metals,Widely used today (transition metals, inorganicsinorganics))
PM3 (1989, Stewart)PM3 (1989, Stewart)
Larger data set for parameterization compared to AM1Larger data set for parameterization compared to AM1 Widely used today (transition metals,Widely used today (transition metals, inorganicsinorganics))
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 22/79
Methods
• n t o quantum c em stry met o s
• Density functional theory (DFT) methods
• Semi-empirical quantum chemistry method
• Molecular mechanics method
• Specially treatment for periodic boundary condition
• Plane waves implementation
• Ewald summation for long-range interaction
• Molecular dynamics and Monte Carlo simulation
• Hybrid quantum mechanics/molecular mechanics method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 23/79
Molecular mechanics method
Interactions between atoms (Potential Energy Function) are representedby functions of distance, angle or dihedral
Collection of empirical parameters and potential functions is known as aforce field.
i.e. AMBER
force field:
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 24/79
Commonly used force field
AmberDNA, proteins and lipids. Generalized Amber Force Field (GAFF) covers most organicspace.
OPLSOrganic molecules in the liquid phase. Available as all-atom (AA) or united-atom (UA)form.
CHARMM DNA, proteins, lipids, sugars.
GROMOS General purpose. Organic and biochemical space.
Dreiding General purpose. Includes some parameters for metals and main group elements.
MM2/MM3General purpose. MM2 used for hydrocarbons. MM3 includes most of organic space plussome other main rou and metal atom t es.
Water Rigid and flexible models available. TIP3P widely used in biological simulation.
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 25/79
Commonly used softwares
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 26/79
Common tasks
• Sampling conformational spaces (Structure and dynamics)
•
• Structural refinement (based on NMR data)
• Reaction path (Transition Path Sampling)
• Molecular interaction (Ligand-Protein)
• Investigate mutational effects
• etc.
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 27/79
Applications
• Speed of method opens up a wide range of systems
Amorphous condensed phase – liquids, liquid crystals, glasses
Biological systems – Biopolymers, membranes
Nanotubes and molecular machines
• Fast calculations allow large numbers of conformers to be screened
• Drug docking analysis
• Global minimum searches
• Statistical data can be collected
•
• Simulation of finite temperature behaviour and phase changes
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 28/79
Methods
• n t o quantum c em stry met o s
• Density functional theory (DFT) methods
• Semi-empirical quantum chemistry method
• Molecular mechanics method
• Specially treatment for periodic boundary condition
― Periodic Electronic Structure Calculations
― Ewald summation for long-range interaction
• Molecular dynamics and Monte Carlo simulation
• Hybrid quantum mechanics/molecular mechanics method
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 29/79
Systems with periodic boundary condition
• , ,(polymers) and 0D (molecule))
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 30/79
Bloch’s Theorem
e e ec ron c wave unc on n a per o c po en a can eexpressed as
.ex r r ik cr =
r unk
k is wavevector within the first Brillouin zone
nk n
Introduction to Computational Chemistry
7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 31/79
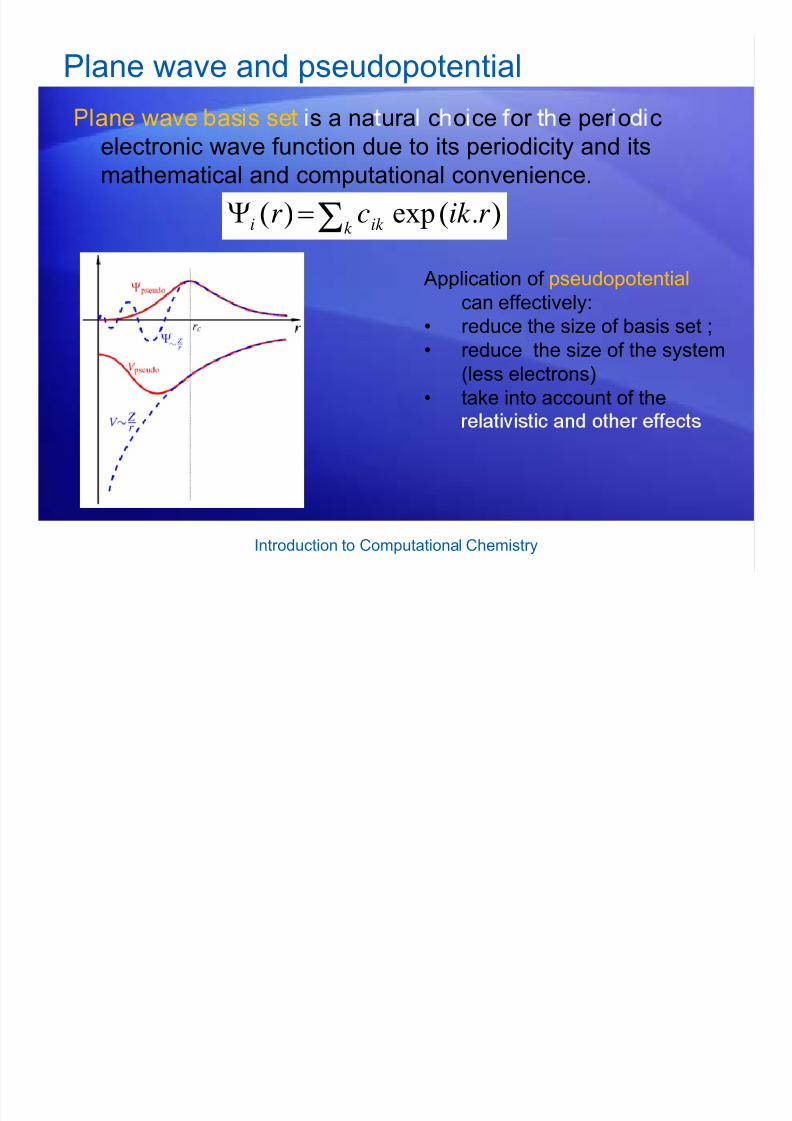
Plane wave and pseudopotential
ane wave as s se s a na ura c o ce or e per o celectronic wave function due to its periodicity and itsmathematical and computational convenience.
).(exp)( r ik cr ik k i Σ=Ψ
Application of pseudopotentialcan effectively:
• reduce the size of basis set ;• reduce the size of the system
(less electrons)
• take into account of the
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 32/79
Long-range interaction in Molecular Mechanics
Ewald O(N 3/2 ) Ewald, 1921
summation
Fast MultipoleMethod
O(N) Greengard, 1987
Particle Mesh O N lo N Darden, 1993
EwaldMulti-gridsummation
O(N) Brandt et al., 1990
Skeel et al., 2002
Izaguirre et al., 2003
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 33/79
Computational Chemistry Codes on Clusters
• ― GAMESS― GAUSSIAN―
― NWCHEM― ORCA―
― SPARTAN― TURBOMOLE
• Quantum chemistry package for solids/surface― ― ―
• Molecular Mechanics Packa es
― CPMD
― NWCHEM
― AMBER― CHARMM― DL_POLY
― GROMACS― LAMMPS― NAMD
― XPLOR
• Others― 3D-DOCK― AUTODOCK
― Molden― Pymol
― VMD
Introduction to Computational Chemistry
― HADDOCK ― Rosetta

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 34/79
Molecular Quantum Chemistry Packages
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 35/79
Quantum Chemistry package on clusters
― GAMESS (US) ― QCHEM
―
―NWCHEM
―
―TURBOMOLE
― ORCA
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 36/79
GAMESS
• General Atomic and Molecular Electronic Structure System
― Primary focus is on ab initio quantum chemistry calculations
― Also includes DFT semi-em irical AM1 PM3 and QM/MMcapabilities
• Early version of GAMESS is available under /usr/global/gamess
― scripts need to execute the program
Introduction to Computational Chemistry

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 37/79
GAMESS Input file
$CONTRL SCFTYP=RHF RUNTYP=OPTI MI ZE COORD=CART
NZVAR=0 MULT=1 I CHARG=0 $END
SYSTEM TI MLI M=20000 MEMORY=10000000 END
$STATPT NSTEP=1000 $END
$BASI S GBASI S=STO NGAUSS=3 $END
$GUESS GUESS=HUCKEL $END
$DATA
Test . . . HCHO mol ecul e - RHF/ STO- 3G ( a comment l i ne)
Cn 1
C 6. 0 0. 6084782705 - 0. 0000011694 0. 000000000000
O 8. 0 - 0. 6082418894 - 0. 0000002093 0. 000000000000
H 1. 0 1. 2040919862 - 0. 9264398115 0. 000000000000
H 1. 0 1. 2040973125 0. 9264340484 0. 00000000000$END
Introduction to Computational Chemistry
GAUSSIAN

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 38/79
GAUSSIAN
Gaussian 03 is a general electronic structure programs. Gaussian 03 isused by chemists, chemical engineers, biochemists, physicists andothers for research in established and emerging areas of chemicaln eres .
Introduction to Computational Chemistry
G i I t Fil St t

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 39/79
Gaussian Input File Structures
Route section (# lines): Specify calculation type, model chemistry and other optionsTitle section: Brief description of the calculationMolecule specification: Specify molecular system to be studied
%NPr ocShar ed=2 Link 0 section (#of processors for SMP)
Optional additional sections: Additional input needed for specific job types
=%chk=h2o_opt . chk# RHF/ 6- 31g** OPT
(checkpoint file name and location)
Route section
- g op m za on
0 1O
Molecule Specification section (charge, multiplicity)Structure representation in Z-matrix format
r
H 1 r1 2 t ha1
r 1 1. 000
Introduction to Computational Chemistry
a .
R t S ti K d

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 40/79
Route Section: Keywords
ADMP ExtendedHuckel MaxDisk Scan
AM1 External MINDO3 SCF
Amber ExtraBasis MM SCRF
Archive Frozen Core Options MNDO SP
B3LYP Field MP* Keywords Sparse
BD FMM Name Stable
BOMD Force NMR Symmetry
CASSCF Frequency ONIOM TD
*
CBSExtrapolate Gen Output Test
CCD Geom OVGF TestMO
Charge GFInput PBC TrackIO
ChkBasis GFPrint PM3 Transformationuess o ar
CIS GVB Population Units
CNDO Hartree-Fock Pressure Volume
Complex Huckel Prop W1U
Constants INDO Pseudo Zindo
Counterpoise Integral Punch Link 0 Commands
CPHF IOp QCISD Non-Standard Routes
Density IRC ReArchive Program Development Keywords
DensityFit IRCMax SAC-CIDensity Functional Methods
Introduction to Computational Chemistry
K d (J b t )

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 41/79
Keywords (Job types)
• ng e po nt energy.•Opt Geometry optimization.
•Freq Frequency and thermochemical analysis.
•IRC Reaction path following.
•IRCMax Find the maximum energy along a specific reaction path.
•Scan Potential energy surface scan.
•Polar Polarizabilities and hyperpolarizabilities.
•ADMP and BOMD Direct dynamics trajectory calculation.
• .
•Volume Compute molecular volume.
Introduction to Computational Chemistry
Keywords (Molecular properties)

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 42/79
Keywords (Molecular properties)
, ,o Electron affinities and Ionization potentials via propagator methods: OVGF
o Electron density: cubegen
o Electronic circular dichroism: TD
,o Electrostatic-potential derived charges: Pop=Chelp, ChelpG or MK
o Frequency-dependent polarizabilities/hyperpolarizabilities: Polar CPHF=RdFreqo High accuracy energies: CBS-QB3, G2, G3, W1U
o Hyperfine spectra tensors (incl. g tensors): Freq=(VCD, VibRot[, Anharmonic])
o Hyperpolarizabilities: Freq, Polar
o IR and Raman spectra: Freq
= o NMR shielding and chemical shifts: NMR
o NMR spin-spin coupling constants: NMR=SpinSpin
o Optical rotations: Polar =OptRot CPHF=RdFreq
,
o Thermochemical analysis: Freqo UV/Visible spectra: CIS, Zindo, TD
o Vibration-rotation coupling: Freq=VibRot
=
Introduction to Computational Chemistry
Available model calculations

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 43/79
Available model calculations
Introduction to Computational Chemistry
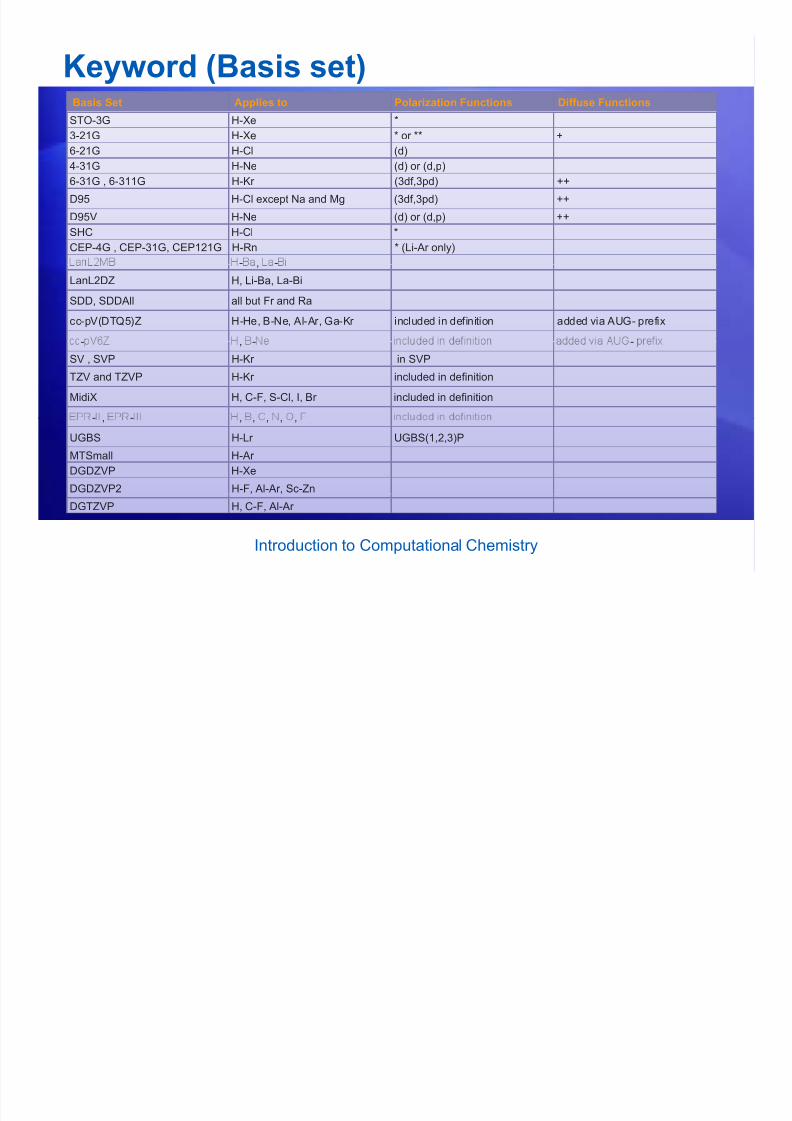
Keyword (Basis set)
7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 44/79
Keyword (Basis set)Basis Set Applies to Polarization Functions Diffuse Functions
STO-3G H-Xe *3-21G H-Xe * or ** +
6-21G H-Cl (d)
4-31G H-Ne (d) or (d,p)
6-31G , 6-311G H-Kr (3df,3pd) ++
D95 H-Cl except Na and Mg (3df,3pd) ++
D95V H-Ne (d) or (d,p) ++
SHC H-Cl *
CEP-4G , CEP-31G, CEP121G H-Rn * (Li-Ar only)
- -,
LanL2DZ H, Li-Ba, La-Bi
SDD, SDDAll all but Fr and Ra
cc-pV(DTQ5)Z H-He, B-Ne, Al-Ar, Ga-Kr included in definition added via AUG- prefix
- , - -
SV , SVP H-Kr in SVP
TZV and TZVP H-Kr included in definition
MidiX H, C-F, S-Cl, I, Br included in definition
- -- , - , , , , ,
UGBS H-Lr UGBS(1,2,3)PMTSmall H-Ar
DGDZVP H-Xe
DGDZVP2 H-F, Al-Ar, Sc-Zn
Introduction to Computational Chemistry
DGTZVP H, C-F, Al-Ar
Molecular Specification

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 45/79
Molecular SpecificationMolecules can be s ecified b Z- matrix or Cartesian coordinate.
The Z-matrix is a way to represent asystem u t o atoms. t prov es a
description of each atom in a moleculein terms of its atomic number, bond, ,
angle, the so-called internal
coordinates.
Some softwares provide sketcher and fragment library to build molecules with’
Molecular builders
, . . , , ,
Molecular Editor, HyperChem, Gabedit, etc.
Some softwares provide interface to write out z-matrix or Cartesian
Introduction to Computational Chemistry
coordinates, i.e. Molden
NWCHEM

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 46/79
NWCHEM
NWChem is a computational chemistry package with many capabilities,including:
― Molecular electronic structure calculations
― Pseudopotential plane-wave electronic structure calaculations
― Ab intio and classical molecular dynamics
―
Introduction to Computational Chemistry
NWCHEM Input file format

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 47/79
NWCHEM Input file format
s ar wa er
Ti t l e “H2o ener gy”
•Job name determines namesfor temporary files
•
geomet r y uni t s auO 0. 00000 0. 000000 0. 000000
-
change settings for futurecalculations
H 0. 00000 - 1. 43042809 - 1. 10715266
End
•task directive triggers
calculation
• - -Basi s
* l i br ar y 3- 21G
End
bottom (only settings above a
task have an impact)
MP2; f r eeze at omi c; END
Task mp2 ener gy
•Python procedures for morecomplicated structures
Introduction to Computational Chemistry
Quantum chemistry package for solids/surface

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 48/79
Quantum chemistry package for solids/surface
― ABINIT ― CASTEP
― ADF/Band
― CPMD
― NWCHEM
― VASP
Introduction to Computational Chemistry
VASP (Vienna Ab-initio Simulation Package)

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 49/79
VASP (Vienna Ab initio Simulation Package)
VASP is a complex package for performing ab-initio quantum-mechanicalmolecular dynamics (MD) simulations using pseudopotentials or theprojector-augmented wave method and a plane wave basis set.
• The approach implemented in VASP is based on the (finite-temperature) local-density approximation with the free energy asvariational quantity and an exact evaluation of the instantaneouselectronic ground state at each MD time step. VASP uses efficient
matrix diagonalisation schemes and an efficient Pulay/Broyden chargeens y m x ng.
• The interaction between ions and electrons is described by ultra-softVanderbilt pseudopotentials (US-PP) or by the projector-augmentedwave me o .
• Forces and the full stress tensor can be calculated with VASP andused to relax atoms into their instantaneous ground-state.
Introduction to Computational Chemistry
VASP Input

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 50/79
VASP Input
INPUT Files
POSCAR
OUPUT Files
OUTCAR
KPOINTS CONTCAR
WAVECAR
EIGENVALPROCAR
XDATCARSTOPCAR is used to
LOCPOTDOSCAR
signal stopping VASPexecution
Introduction to Computational Chemistry
Structure Input File

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 51/79
Structure Input File
Introduction to Computational Chemistry
Pseudopotential File

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 52/79
Pseudopotential File
Introduction to Computational Chemistry
KPOINT Sampling

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 53/79
p g
Introduction to Computational Chemistry
CONTROL File

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 54/79
Introduction to Computational Chemistry
Result analysis

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 55/79
y
Introduction to Computational Chemistry
CPMD

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 56/79
e co e s a p ane wave pseu opotent a mp ementat on oDensity Functional Theory, particularly designed for ab-initiomolecular dynamics.
―
isolated systems and system with periodic boundary conditions; k-points― molecular and crystal symmetry
― wavefunction optimization: direct minimization and diagonalization
― geometry optimization: local optimization and simulated annealing
― molecular dynamics: constant energy, constant temperature and constantpressure
― path integral MD
― response functions
― exc e s a es
―many electronic properties
― time-dependent DFT (excitations, molecular dynamics in excited states)
Introduction to Computational Chemistry
CPMD Input file 1 (Wave Function)

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 57/79
&CPMD 120.0 0.998611043 1.609250000 7.210402167
OPTIMIZE WAVEFUNCTIONCONVERGENCE ORBITALS
1.0e-8
MAXSTEP
200
CHARGE0.0
&END
&ATOMS
*O-q6.psp
LMAX=P
20
0.261992993 1.609250000 1.089966007
RHOOUT
ELECTROSTATIC POTENTIAL
ELF PARAMETERS
0.0 0.0
*K-q9.psp
LMAX=P
4
3.014818112 1.609250000 14.986109219
. . - .
1.769401965 1.609250000 2.032139989
3.428690989 1.609250000 3.491585919
1.059363053 1.609250000 17.051501595
1.636507007 4.827750000 10.326966007
&END
&DFT
FUNCTIONAL LDA
GC-CUTOFF
. . .
0.782181888 4.827750000 3.487890781
1.116318112 1.609250000 12.724890781
*Ti-q12.psp
LMAX=D
. . .
0.129098035 4.827750000 11.269139989
2.266809011 4.827750000 12.728585919
0.839136947 4.827750000 7.814501595
3.535007007 4.827750000 17.384033993
1.256806888 4.827750000 18.6217920075.0e-6
&END
&SYSTEM
SYMMETRY
4
1.249212986 1.609250000 0.568999197
0.649287014 4.827750000 9.805999197
2.547787014 4.827750000 17.905000803
3.147712986 1.609250000 8.668000803
2.027598035 4.827750000 16.441860011
0.368309011 4.827750000 14.982414081
2.737636947 4.827750000 19.896498405
2.160492993 1.609250000 8.147033993
0.641693112 1.609250000 9.384792007
POINT GROUP
AUTO
ANGSTROM
CELL
*Ta-q13.psp
LMAX=D
4
2.897111043 1.609250000 2.026597833
. . .
1.530190989 1.609250000 5.745414081
2.957863053 1.609250000 10.659498405
&END
. . . . . .
CUTOFF
. . .
0.899888957 4.827750000 16.447402167
Introduction to Computational Chemistry
CPMD Input file 2 (NMR Calculation)

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 58/79
&CPMD &END 2.798388957 4.827750000 11.263597833
LINEAR RESPONSERESTART ALL LATEST
CONVERGENCE ORBITALS
1.0d-7
STRUCTURE BONDS ANGLES
&ATOMS
ISOTOPES
39.00
48.00
0.899888957 4.827750000 16.4474021670.998611043 1.609250000 7.210402167
*O-q6.psp
LMAX=P
&RESP
NMR
FULL
.
16.00
*K-q9.psp
LMAX=P
0.261992993 1.609250000 1.089966007
2.540193112 1.609250000 -0.147792007
1.769401965 1.609250000 2.032139989
3.428690989 1.609250000 3.491585919
&DFT
FUNCTIONAL LDA
GC-CUTOFF
1.0e-6
3.014818112 1.609250000 14.986109219
2.680681888 4.827750000 5.749109219
0.782181888 4.827750000 3.487890781
1.116318112 1.609250000 12.724890781
. . .
1.636507007 4.827750000 10.326966007
3.155306888 4.827750000 9.089207993
0.129098035 4.827750000 11.269139989
2.266809011 4.827750000 12.728585919
0.839136947 4.827750000 7.814501595&END
&SYSTEM
SYMMETRY
8
*Ti-q12.psp
LMAX=D
4
1.249212986 1.609250000 0.568999197
0.649287014 4.827750000 9.805999197
3.535007007 4.827750000 17.384033993
1.256806888 4.827750000 18.621792007
2.027598035 4.827750000 16.441860011
0.368309011 4.827750000 14.982414081
2.737636947 4.827750000 19.896498405
CELL
3.797 1.69528575 4.86542 0.0 0.0 0.0
CUTOFF
120.0
. . .
3.147712986 1.609250000 8.668000803
*Ta-q13.psp
LMAX=D
. . .
0.641693112 1.609250000 9.384792007
3.667901965 1.609250000 7.204860011
1.530190989 1.609250000 5.745414081
2.957863053 1.609250000 10.659498405
0.0 2.897111043 1.609250000 2.026597833
Introduction to Computational Chemistry
ABINIT

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 59/79
ABINIT is a package whose main program allows one to find the totalenergy, charge density and electronic structure of systems made of electrons and nuclei (molecules and periodic solids) within Density
unc ona eory , us ng pseu opo en a s an a p anewave
basis.
Introduction to Computational Chemistry
ABINIT Input

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 60/79
To run abinis you need four things:(1) Access to the executable, abinis/abinip.
2 An in ut file.
(3) A files file (list of file names in a file).
(4) A pseudopotential input file for each kind of element in the unit cell.
Introduction to Computational Chemistry
Sample file of filelist

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 61/79
t par al _1. i n
tparal_1.filelist
par a _ . out par al _1it par al _1o
_ 82pb. 4. hgh
- The main output filename
- The root of input files
- The root of output files
- The root of temporary files
- The pseudopotential file name
Introduction to Computational Chemistry
Sample input control file

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 62/79
#
# Lead crystal
kptopt 1
nshiftk 4
typat 1
xred
_ .
#
# Simulation parameters
shiftk
0.5 0.5 0.50.5 0.0 0.0
0.000 0.000 0.000
nband 4
ecut 30.0
acell 10.0 10.0 10.0
rprim
0.0 0.5 0.0
0.0 0.0 0.5
occopt 7
# SCF procedure
nstep 3
tolvrs 1.0d-100.0 0.5 0.5
0.5 0.0 0.5
0.5 0.5 0.0
tsmear 0.01
# System description
# This line added whendefaults were changed(v5.3) to keep the
# K-points
ngkpt 8 8 8
ntypat 1znucl 82
natom 1
,
iscf 5
Introduction to Computational Chemistry
Sample pseudopotential file

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 63/79
Har t wi gsen- Goedecker - Hut t er psp f or Pb, f r om PRB58, 3641 ( 1998)
82pb.4.hgh
za om, z on, psp a
3 1 2 0 2001 0 pspcod, pspxc, l max, l l oc, mmax, r 2wel l
0. 617500 0. 753143 0. 000000 0. 000000 0. 000000 r l oc, c1, c2, c3, c40. 705259 1. 979927 - 0. 164960 - 0. 806060 r s, h11s, h22s, h33s
0. 846641 0. 864420 - 0. 540969 0. 000000 r p, h11p, h22p, h33p
0. 207711 0. 012948 0. 000000 k11p, k22p, k33p
0. 971939 0. 374967 0. 000000 0. 000000 r d, h11d, h22d, h33d
0. 029256 0. 000000 0. 000000 k11d, k22d, k33d
0. 000000 0. 000000 0. 000000 0. 000000 r f , h11f , h22f , h33f
. . . , ,
Introduction to Computational Chemistry
Sample PBS Job script

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 64/79
#PBS -l nodes=1:ppn=2#PBS -l walltime=4:00:00
#PBS - oe
#PBS -N test_abinitp
#
_ _
#
mpirun /usr/global/abinit/5.4/bin/abinip < tparal_1.files > tparal_1.log
exit
Introduction to Computational Chemistry
Molecular Mechanics Packages

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 65/79
― AMBER―
― GROMACS―
― XPLOR―
― DL_POLY ― NAMD
Introduction to Computational Chemistry
AMBER (Assisted Model Building with Energy Refinement)

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 66/79
• Classical molecular dynamics simulations (NVT, NPT, etc)• Force field for biomolecular simulations (proteins, nucleic acids,
carbohydrates and organic molecules)
• Combined Quantum Mechanics/Molecular Mechanics (QM/MM)
implementation• Parallelized d namics codes
• Explicit Solvent Models with particle-mesh Ewald sum (PME), ImplicitSolvent models with Poisson-Boltzmann and Generalized Born
a roach• Enhanced sampling (replica exchange MD, Locally Enhanced
Sampling)
• , .
• Structural and trajectory analysis• ……
Introduction to Computational Chemistry
Normal procedure to set up a simulation

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 67/79
Step 1, obtain and edit initial structure
Step 2, prepare input parameter and topology file
Step 3, run simulations and save production trajectory
Step 4, analyze output and trajectory files
Preparatory
Pro rams
Simulation
Programs AnalysisLEaP
antechamber
Sander
PMEMD
rograms
PtrajMM/PBSA
Introduction to Computational Chemistry
Preparation using Leap

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 68/79
Leap command script
source leaprc.ff03
oxy = loadPdb oxyt.pdb
on oxy. . oxy. .
charge oxy
check oxysaveAmberParm oxy oxy_vac.top oxy_vac.crd
solvateOct oxy TIP3PBOX 9.0
. .
quit
Introduction to Computational Chemistry
Sander Input

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 69/79
Introduction to Computational Chemistry
Analysis using ptraj

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 70/79
RMSD analysis (ptraj 1qs4A.top ptraj.input2)t r aj i n 1qs4A_md01_nowat . mdcr d
r ef er ence 1qs4A. cr d_ _ .
: 1- 154go
-t r aj i n 1qs4A_md01_nowat . mdcr d
r ms f i r st out 1qs4A_md01_CA. dat : 1-154@CA
a om c uc ou qs _m . ac orbyat om bf act or
go
Introduction to Computational Chemistry
Other Packages

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 71/79
― 3D-DOCK ― Pymol
― AUTODOCK
―
― Rosetta
―
― HADDOCK
―o en
Introduction to Computational Chemistry
Quantum mechanics/molecular mechanics method

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 72/79
•
o able to model large molecules by defining two or three layers within the structure that are treated atdifferent levels of accuracy;
o applicable in many other areas, including enzyme,
reactions, photochemical processes, substituenteffects and reactivity of organic and organometalliccompounds, and homogeneous catalysis.
• Q-Chem/CHARMM, Gaussian/Tinker, ChemShell, NWChem, etc.
special treatment at the bourdary
Introduction to Computational Chemistry
Gaussian – ONIOM Input-

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 73/79
# ONIOM(mp2/6-311g**:b3lyp/6-31g*:hf/3-21g)
specification
# oniom(blyp/3-21g:amber)
3-layer ONIOM
0 1 0 1 0 1 0 1 0 1 0 1 0 1C -0.006049274275 0.000000000000 0.066754956170 H
O 0.011403425950 0.000000000000 1.308239478983 H
geom=connect v ty# oniom(blyp/3-21g/dga1:amber) geom=connectivity opt=loose
blyp/3-21g:amber with density fitting from Luecke
3 1 1 1
H 0.944762558657 0.000000000000 -0.507359536461 H
C -1.307562483867 0.000000000000 -0.766510748030 M H 1 0.723886 0.723886 0.723886
C -1.047480751885 0.000000000000 -2.301387120377 L H 4 0.723886 0.723886 0.723886
H -1.903669606697 -0.885256630266 -0.468844831106 M
H -1.903669606697 0.885256630266 -0.468844831106 M
N-N3-0.181200 22.076181 24.563316 -13.077047 L
C-CT-0.003400 20.736756 24.514869 -13.699982 L
C-C-0.616300 20.734957 25.313129 -14.988472 L
O-O--0.572200 20.731825 24.680955 -16.030293 L
……
C-CT--0.043900 19.775830 32.473200 7.920456 L
H -1.988817319373 0.000000000000 -2.842389774687 L
H -0.482972255230 0.881286097766 -2.591806824941 L
H -0.482972255230 -0.881286097766 -2.591806824941 L
- -- . . . . - . .
C-CT--0.082400 17.966350 32.942557 6.232525 H
C-CT-0.377886 16.928731 33.900731 5.689445 H
N-N2--0.693824 17.521649 35.160530 5.191031 H
N-N--0.415700 22.301376 31.316157 8.955475 L
……
H-HW-0.417000 10.154733 46.125705 25.662719 L
Three-Layer ONIOM specification
# ONIOM m 2/6-311 **:b3l /6-31 *:hf/3-21
H-HW-0.417000 11.339041 47.039598 25.433539 L
H-H-0.274700 21.866560 34.137178 7.953109 L
H-H-0.377886 17.847763 35.188083 4.238902 H
H-H-0.341200 18.066775 49.503351 -1.731201 L
H-HO-0.474700 15.931300 31.668829 17.328019 L
Introduction to Computational Chemistry
1 2 1.0 1770 1.0 1771 1.0 1772 1.0
2 3 1.0 5 1.0 1769 1.0
3 4 2.0 8 2.0
4
5 6 1.0 7 1.0 1764 1.0
6 1768 1.0
Visualization tools

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 74/79
Molecules• Gaussview
Periodic Systems• Materials Studio• Cr stal Maker
• o en
• Molekel•
• VMD
• ECCE
• ArgusLab
• VMD
• VegaZZ
• Discovery Studio• JMol
Introduction to Computational Chemistry
MOLDEN

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 75/79
Introduction to Computational Chemistry
Gaussview

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 76/79
Introduction to Computational Chemistry
VMD

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 77/79
Introduction to Computational Chemistry
ArgusLab

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 78/79
Introduction to Computational Chemistry
Acknowledgement

7/29/2019 Intro CompChem
http://slidepdf.com/reader/full/intro-compchem 79/79
graphics from other authors.
Introduction to Computational Chemistry







