
Histiozytosen
G. Ratzingera*, W. Burgdorfb und B. ZelgeraaUniversitätsklinik f€ur Dematologie und Venerologie, Medizinische Universität, Innsbruck, ÖsterreichbUniversitätsklinik f€ur Dematologie und Allergologie, Ludwig Maximilian Universität, M€unchen, Deutschland
1 Makrophagen- und Langerhans-Zell-Erkrankungen
Unter dem Begriff der Histiozytosen werden Erkrankungen zusammenfasst, die sich von Makrophagenoder dendritischen Zellen ableiten. Beide Zelltypen finden ihren Ursprung im Knochenmark. Makro-phagen entwickeln sich €uber Blutmonozyten durch Einwanderung in verschiedene Gewebe zu Makro-phagen. Phagozytose verschiedener Materialien und Reifung beeinflussen die Gestalt dieser Zellen, diedadurch morphologisch sehr heterogen werden. Dendritische Zellen stammen ebenso vom Knochenmarkab und finden sich danach in der Haut, in den Lymphknoten und anderen Organen. Langerhans-Zellensind die dendritischen Zellen der Epidermis. Sie sind der Prototyp einer Antigen-präsentierenden Zelle,die Antigen in der Epidermis aufnimmt, prozessiert und dieses anschließend nach Migration zumLymphknoten ebendort präsentiert und damit eine Immunantwort initiiert. Langerhans-Zellen (CD1a+,Langerin (CD207)+, S100+) und Makrophagen (CD68+) sind zumeist immunhistochemisch leicht zuunterscheiden. Expressionsprofile können sich jedoch im Rahmen der Zellreifung ändern, und auchÜberlappungen kommen vor (Tab. 1). Daraus ergibt sich ein dritter Zelltyp, der im Zusammenhang mitHistiozytosen Erwähnung findet, die sog. indeterminierte Zelle, deren Eigenständigkeit kontroversdiskutiert wird. Wahrscheinlich handelt es sich dabei um Makrophagen oder dendritische Zellen, derenExpressionsprofil von der Norm abweicht (S100+ Makrophagen, CD207- Langerhans-Zellen). DieEinteilung der Histiozytosen kann primär nach Zelltyp, sowie sekundär nach dem klinischen Befundund der Histologie erfolgen (Übersicht „Klassifikation der Histiozytosen“).
Klassifikation der HistiozytosenLangerhans-Zell-Erkrankungen
– Akut disseminierte Langerhans-Zell-Histiozytose (Abt-Letterer-Siwe-Syndrom) (ICD-O: 9754/3)– Chronisch multifokale oder disseminierte Langerhans-Zell-Histiozytose (Hand-Sch€uller-
Christian Syndrom) (ICD-O: 9753/1)– Kongenitale selbstheilende Retikulohistiozytose (Hashimoto-Pritzker Syndrom) (ICD-O: 9751/1)– Chronisch fokale Langerhans-Zell-Histiozytose (Eosinophiles Granulom) (ICD-O: 9752/1)
Makrophagenerkrankungen
– Xanthogranulom-Familie– Xanthome
(Fortsetzung)
*E-Mail: [email protected]
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 1 von 23
Sonderformen
– Histiozytose der indeterminierten Zelle (ICD-O: 9657/3)– Rosai-Dorfman Krankheit (RDD)– Erdheim-Chester-Krankheit– Maligne Proliferationen von Makrophagen und Langerhans-Zellen (ICD-O: 9756/3)
2 Langerhans-Zell-Histiozytose (LZH)
Definition und klinisches Bild Schon 1953 fasste Lichtenstein die klassischen Vertreter (Abt-Letterer-Siwe-Syndrom, Hand-Sch€uller-Christian-Syndrom und das eosinophile Granulom) unter dem BegriffHistiozytosis X zusammen. Gemeinsam ist den Erkrankungen dieser Gruppe die Pathogenese, nämlichdie klonale Proliferation der Ursprungszelle einerseits und eine starke entz€undliche Komponente ande-rerseits. Verbindend ist weiterhin die einheitliche Histopathologie. Das klinische Bild ist heterogen undumfasst eine Reihe von verschiedenen Ausprägungen, wobei Überlappungen die Regel sind. Die klini-schen Verläufe reichen von gutartigen Einzelläsionen bis zu fatalem Multisystembefall.
Immer noch wird die epidermale Langerhans-Zelle als Ausgangszelle f€ur die LZH angesehen, obwohldiesbez€uglich Diskussionen bestehen. Es gibt Hinweise, dass andere dendritische Zellen, wiez. B. unreife oder fehldifferenzierte myeloide dendritische Zellen involviert sind. Neben vielen Gemein-samkeiten mit der epidermalen Langerhans-Zelle (CD1a, Langerin-CD207, Birbeck Granula) gibt eswichtige Unterschiede: der LZH-Zelle fehlen morphologisch Dendriten und funktionell die Migrations-fähigkeit. Sie zeigt auch ein unterschiedliches Genexpressionsprofil. Eine aktivierte BRAFV600EMuta-tion, die in> 50 % aller LZH-Läsionen gefunden wird, unterstreicht weiter die neoplastische Genese, hatwahrscheinlich auch funktionelle Bedeutung und könnte möglicherweise auch therapeutische Optioneneröffnen.
Die Erkrankung kann verschiedene Organe befallen, wobei Hautbeteiligung bei den meisten Formenhäufig ist und als Primärmanifestation oft diagnostisch im Vordergrund steht. Die Subtypen unterscheidensich durch den Befall verschiedener Organe und die daraus resultierende unterschiedliche Symptomatikund Prognose. Überlappungen sind häufig. Man unterscheidet disseminierte von fokalen Formen.
Varianten des KrankheitsbildesDisseminierte Formen
Akute disseminierte Langerhans-Zell-Histiozytose Die akute disseminierte Langerhans-Zell-Histiozytose (Abt-Letterer-Siwe-Syndrom) ist die aggressivste Verlausform. Sie tritt bevorzugt bei Klein-kindern zwischen dem 6. und dem 24. Lebensmonat auf, kann aber grundsätzlich in jedemAlter auftreten.In 80 % findet man Hautläsionen in Form von dicht ausgesäten, rötlich-braunen, schuppigen oder erosiv-
Tab. 1 Markerprofile
CD1a CD207-Langerin S100-Protein CD68
Langerhans-Zell-Erkrankung + + + -
Makrophagen-Erkrankung - - 95 % -5 % +
+
Rosai-Dorfman-Syndrom - - + +
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 2 von 23

krustigen Papeln bis Knoten, manchmal begleitet von petechialen Blutungen, die vor allem an denHandflächen als prognostisch ung€unstig gelten. Läsionen können auch vesikulär, ulzerös, urtikariell oderxanthomatös imponieren. Prädilektionsstellen sind Kopf, oberer Rumpf sowie intertriginöse und seborr-hoische Areale. Bei älteren Menschen findet sich vergleichsweise oft anogenitaler Befall. Die Schweredes Hautbefalls ist direkt proportional zu Multiorganbefall. Fieber, Anämie, Thrombozytopenie, Hepato-splenomegalie, Lymphadenopathie und Lungenbeteiligung (letzteres als prognostisch ung€unstiges Merk-mal) sind charakteristisch. Patienten sollten deshalb immer einer Staginguntersuchung unterzogenwerden. Seltene osteolytische Läsionen zeigen sich bevorzugt im Mastoid, imitieren klinisch eine Otitismedia und sind prognostisch ung€unstig.
Chronische multifokale oder disseminierte Langerhans-Zell-Histiozytose Die chronische multifokaleoder disseminierte Langerhans-Zell-Histiozytose (Hand-Sch€uller-Christian-Syndrom) tritt vor allem beiKleinkindern, seltener bei älteren Kindern und Jugendlichen auf. Überlappungen mit der akuten disse-minierten LZH sind häufig. Hautbefall findet sich in 30 % der Fälle. Am häufigsten sind axilläre,anogenitale oder orale Knoten und Plaques, die im Verlauf ulzerieren können. Weniger häufig zeigensich disseminierte schuppig-krustige Papeln ähnlich der akuten disseminierten LZH, zumeist jedochweniger intensiv und mit geringerer hämorrhagischer Komponente. Selten findet man Xanthome. DieOrganbeteiligung umfasst typischerweise die klassische Trias von Diabetes insipidus, osteolytischenKnochendefekten und Exophthalmus. Granulomatöse Infiltrationen im Bereich des Hypothalamus-Hypophysenstiels, im Bereich von Knochen, besonders im Schädeldach, sowie retrobulbär f€uhren zuden genannten Symptomen. Die Kardinalsymptome sind sehr variabel und können zum Teil oder auch zurGänze fehlen. Zusätzlich können granulomatöse Infiltrationen im Bereich von Leber, Milz, Lunge,Lymphknoten oder Mastoid auftreten.
Kongenitale selbstheilende Retikulohistiozytose Die kongenitale selbstheilende Retikulohistiozytose(Hashimoto-Pritzker-Syndrom) ist benigne und heilt zumeist, aber nicht immer, innerhalb des erstenLebensjahres spontan ab. Sie ist entweder bei der Geburt schon vorhanden oder tritt in den erstenLebenstagen bis Wochen zutage. Die Hautmanifestation steht im Vordergrund und zeigt sich als isolierteoder locker disseminierte rötlich-braune bis bläuliche Papeln und Knoten, z. T. vesikulär oder exsudativ,die in Folge verkrusten. Prädilektionsstellen sind Kopf und Gesicht. In 25 % handelt es sich um Einzel-läsionen, zumeist findet man wenige und nur in Ausnahmenfällen zahlreiche Läsionen am gesamtenKörper. Selten kann es zu einem Rezidiv mit Knochenbeteiligung bzw. zu einem Rezidiv mit Progressionin eine akute disseminierte LZH kommen.
Die disseminierten Formen der LZH f€uhren zu 10 % zum Tod, zu 30 % in die Spontanheilung und zu60 % in einen chronisch-undulierenden Verlauf. Die Prognose der Erkrankung wird durch das Alter desPatienten, die Anzahl der befallenen Organe und das Ausmaß der Organbeteiligung bestimmt. Beteili-gung von Knochenmark, Milz, Leber oder Lunge sind prognostisch ung€unstig. Hautbeteiligung bei derGeburt ist g€unstig (kongenitale selbstheilende Retikulohistiozytose), vor dem 2. Lebensjahr allerdingsung€unstig (akute disseminierte Langerhans-Zell-Histiozytose). F€ur Erwachsene ist ein erhöhtes Risiko f€urhämatologische oder pulmonale Neoplasien beschrieben, zusätzlich kann es sekundär zu einer reaktivenhämophagozytischen Lymphohistiozytose kommen, die zumeist letal ist.
Fokale Form
Chronische fokale Langerhans-Zell-Histiozytose Die chronische fokale Langerhans-Zell-Histiozytose(eosinophiles Granulom) zeichnet sich durch einen chronisch-benignen Verlauf aus. Sie tritt vor allem beiKleinkindern auf. Gelegentlich kommt es zur Spontanheilung. Hautveränderungen stehen imHintergrund
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 3 von 23

und ähneln denen der chronischen disseminierten LZH. Am häufigsten finden sich einzelne oder einigewenige Knochenläsionen in Form vonOsteolysen, die asymptomatisch sind oder je nach Lage Schmerzenbzw. Spontanfrakturen nach sich ziehen können. Bevorzugt sind Schädel (Kiefer mit Zahnlockerung),Schulter- und Beckeng€urtel, Rippen und lange Röhrenknochen befallen. Selten kommt es zu einemÜbergang in eine multifokale oder disseminierte Erkrankung.
Histologie und Immunhistologie Die typische LZH-Zelle besitzt Merkmale der epidermalenLangerhans-Zelle. Sie hat einen großen gelappten oder nierenförmigen Kern (Abb. 1). Das Zytoplasmaist hell und leicht eosinophil, die Nukleoli sind prominent. Ultrastrukturell können Tennisschläger-förmige Birbeck Granula nachgewiesen werden. Immunhistochemisch färben S100-Protein, CD1a undLangerin-CD207 (Abb. 2) positiv. Die Umgebungsreaktion kann proliferativ oder granulomatös sowieseltener xanthomatös sein, wobei eine klare Beziehung zum klinischen Subtyp der Erkrankung besteht.
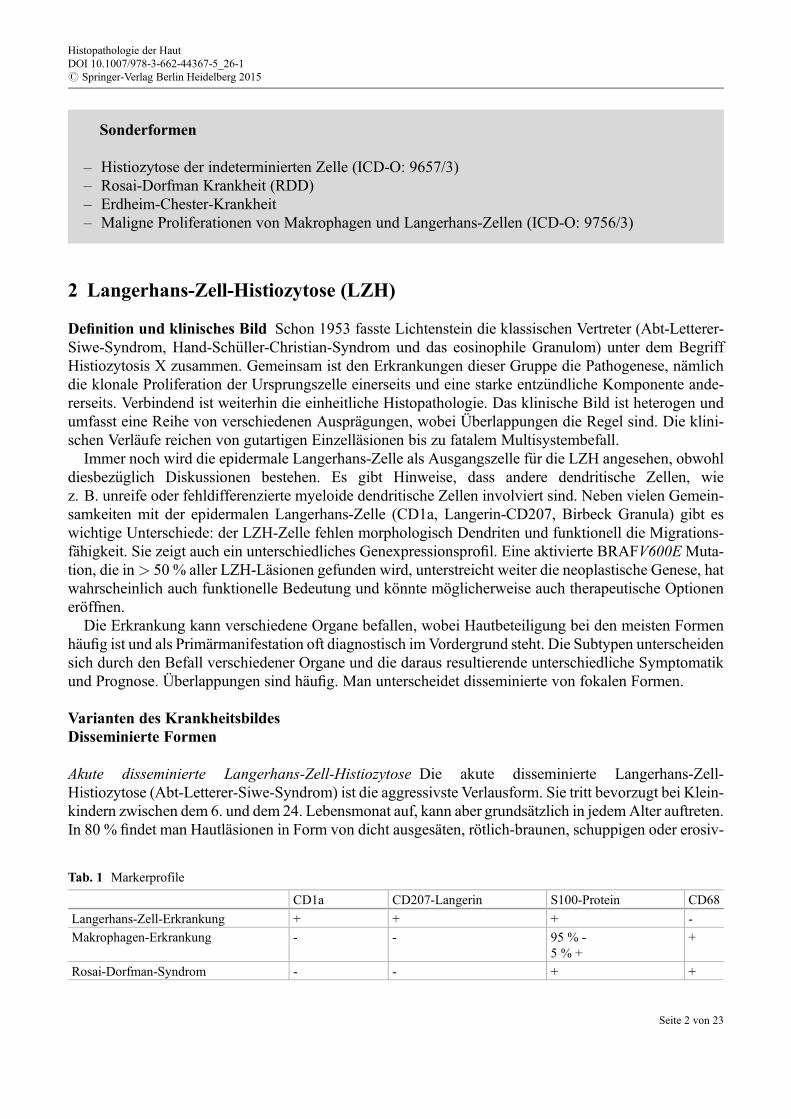
Die proliferative Reaktion, die oft sehr mild sein kann, findet sich typischerweise in der akutendisseminierten LZH. Sie ist charakterisiert durch ein ausgedehntes, fast reines Infiltrat von LZH-Zellen,die knapp subepidermal oder auch epidermal (sog. Epidermotropismus) liegen, was in Folge zu Ulze-
Abb. 1 LZH (HE 40x): Subepidermal bis intraepidermal große Zellen mit nierenförmigem Kern
Abb. 2 LZH – Langerin (40x): kräftige Reaktivität auf Langerin (CD207)
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 4 von 23
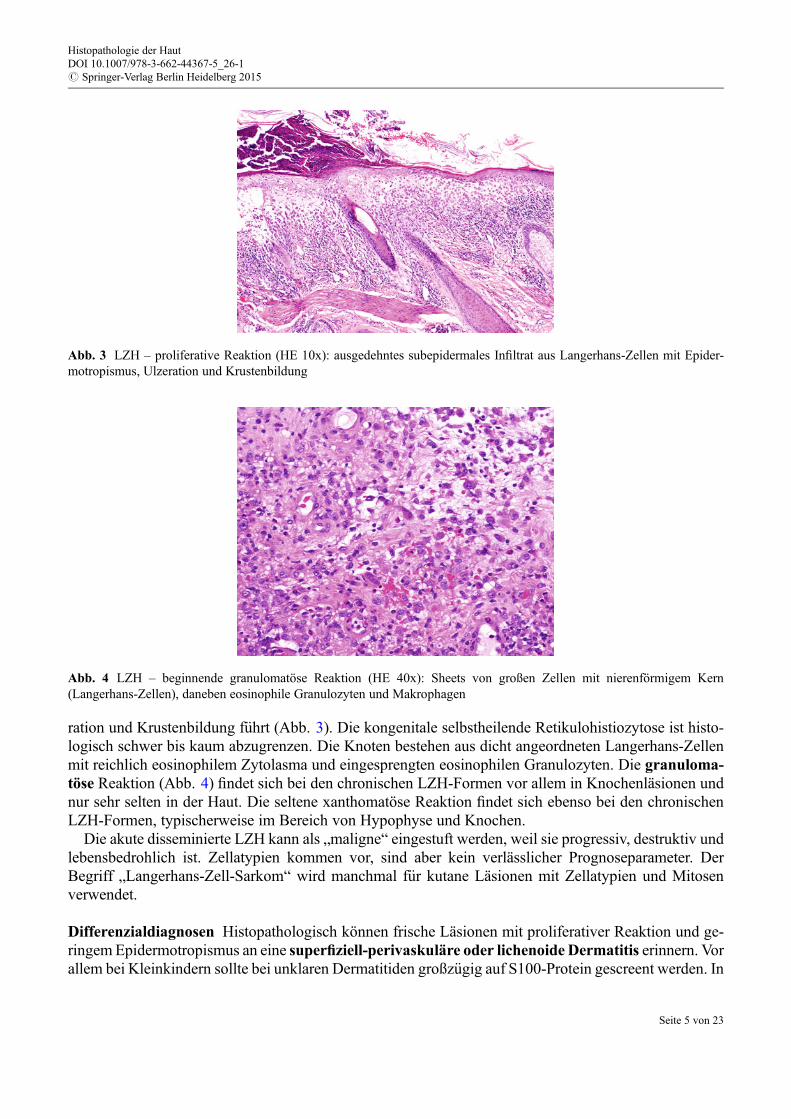
ration und Krustenbildung f€uhrt (Abb. 3). Die kongenitale selbstheilende Retikulohistiozytose ist histo-logisch schwer bis kaum abzugrenzen. Die Knoten bestehen aus dicht angeordneten Langerhans-Zellenmit reichlich eosinophilem Zytolasma und eingesprengten eosinophilen Granulozyten. Die granuloma-töse Reaktion (Abb. 4) findet sich bei den chronischen LZH-Formen vor allem in Knochenläsionen undnur sehr selten in der Haut. Die seltene xanthomatöse Reaktion findet sich ebenso bei den chronischenLZH-Formen, typischerweise im Bereich von Hypophyse und Knochen.
Die akute disseminierte LZH kann als „maligne“ eingestuft werden, weil sie progressiv, destruktiv undlebensbedrohlich ist. Zellatypien kommen vor, sind aber kein verlässlicher Prognoseparameter. DerBegriff „Langerhans-Zell-Sarkom“ wird manchmal f€ur kutane Läsionen mit Zellatypien und Mitosenverwendet.
Differenzialdiagnosen Histopathologisch können frische Läsionen mit proliferativer Reaktion und ge-ringem Epidermotropismus an eine superfiziell-perivaskuläre oder lichenoide Dermatitis erinnern. Vorallem bei Kleinkindern sollte bei unklaren Dermatitiden großz€ugig auf S100-Protein gescreent werden. In
Abb. 3 LZH – proliferative Reaktion (HE 10x): ausgedehntes subepidermales Infiltrat aus Langerhans-Zellen mit Epider-motropismus, Ulzeration und Krustenbildung
Abb. 4 LZH – beginnende granulomatöse Reaktion (HE 40x): Sheets von großen Zellen mit nierenförmigem Kern(Langerhans-Zellen), daneben eosinophile Granulozyten und Makrophagen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 5 von 23

der Abgrenzung zu Xanthomen kann die Positivität auf S100-Protein und CD1a im umgebenden Infiltrathilfreich sein, nachdem die Schaumzellen selbst oft nur schwach positiv oder negativ sind. Klinisch kanndie selbstheilende multizentrische Retikulohistiozytose mit einem Blueberry-Muffin-Syndrom, einerMastozytose oder kongenitalen leukämischen Infiltraten verwechselt werden.
3 Xanthogranulom-Familie
Definition Diese Erkrankungen findet man auch unter dem NamenNicht-Langerhans-Zell-Histiozyto-sen, da CD1a und Birbeck-Granula fehlen. Als Ausgangszellen werden Makrophagen oder in zweiterLinie dendritische Zellen mit Makrophagenfunktion angenommen. Der zugrunde liegende pathologischeProzess ist eine noduläre bis diffuse Dermatitis mit Makrophagen als vorherrschendem Zelltyp. DasXanthogranulom ist der Archetyp in dieser Familie, die anderen Entitäten sind Variationen dazu.
Die Xanthogranulom-Zellen können morphologisch vielfältig sein: xanthomatisiert (helles, schaum-iges Zytoplasma), scalloped (mild eosinophiles Zytoplasma mit sternförmigen Ausziehungen), onkozytär(epithelioide Zellen mit stark eosinophilem Zytoplasma), spindelig oder vakuolisiert (helles Zytoplasmamit Vakuolen) sein (Abb. 5).
Manche Varianten können durch Reifung ineinander €ubergehen. Während der vakuolisierte Zelltyphauptsächlich in frischen Läsionen vorkommt, spricht Lipideinlagerung (Xanthomatisierung) f€ur späteLäsionen bzw. terminale Differenzierung. Es besteht keine Assoziation zu Hyperlipidämien. Fast immerfinden sich in einer Läsion mehrere morphologische Makrophagenvarianten. Bei Fehlen eines vor-herrschenden Zelltyps spricht man von einer polymorphen Form, bei Vorhandensein eines vorherrsch-
Abb. 5 Vereinheitlichendes Schema der Makrophagenerkrankungen – Typ Xanthogranulome. Mod. nach Zelger und Burg-dorf (2013)
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 6 von 23

enden Zelltyps spricht man von einer monomorphen Form. In beiden Fällen gibt es lokalisierte undgeneralisierte Verlaufsformen. Pigmentinkontinenz und nachfolgende Pigmentaufnahme in Makropha-gen ist als Xanthosiderohistiozytose bezeichnet worden, eine Manifestationsform, die in chronischenVerläufen aller Varianten gesehen werden kann. Der Großteil der Läsionen bleibt dauerhaft bestehen. Nursehr fr€uhe Läsionen aus vakuolisierten Zellen bei benigner zephaler Histiozytose oder generalisierter,eruptiver Histiozytose sowie auch die meisten juvenilen Xanthogranulome bei Kindern neigen zuSpontanregression.
Entsprechend dem vereinheitlichendem Schema der Xanthogranulome wird das folgende Kapitelzunächst in polymorphe und monomorphe Formen untergliedert (Abb. 5). Die monomorphen Formenwerden nach Zelltyp behandelt, wobei jeweils zuerst die lokalisierte Form und danach die generalisierteForm besprochen wird.
3.1 Polymorphe Form: Juveniles Xanthogranulom (JXG)
Klinisches Bild Dies ist eine nicht seltene, benigne, Erkrankung, die zumeist im ersten Lebensjahr inErscheinung tritt oder bei der Geburt bereits vorhanden ist (20 %). Die Läsionen wachsen rasch, bildensich aber fast immer innerhalb eines Jahres unter Hinterlassung von milder Atrophie und Hyperpigmen-tierung spontan zur€uck. Prädilektionsstellen sind Kopf und Streckseiten der Extremitäten. Läsionen beiErwachsenen kommen vor und neigen eher zu Persistenz. Es finden sich einzelne bis zahlreiche rot-gelbePapeln bis Knoten. Das solitäre Riesenxanthogranulom kann größer als 5 cm sein. Lichenoide Papeln,flächige Plaques oder auch destruierende Läsionen mit Verunstaltung z. B. der Nase (Cyrano-Typ)kommen vor. Es wurden auch subkutane oder tiefe JXG beschrieben.
Okuläre Läsionen sind selten (<10 %), Glaukom und Einblutung in die vordere Augenkammer sindmögliche Komplikationen. Orale Läsionen, seltene Assoziationen mit Cafe-au-lait Flecken, Neurofi-bromatose I und juveniler chronisch myeloischer Leukämie sowie Organbefall in ZNS, Niere, Lunge,Leber, Hoden und Perikard wurden beschrieben. Nur 5 % der Kinder zeigen systemischen Befall, diesekönnen in weiterer Folge Infektionen oder ein Makrophagenaktivierungssyndrom erleiden. Zumeistweisen Kinder mit Organbefall keine Hautbeteiligung auf. Bei Kindern mit kutanen Xanthogranulomenist deshalb eine Staginguntersuchung als Routinemaßnahme nicht nötig.
Histologie In der Übersicht findet man einen umschriebenen, manchmal exophytischen Knoten in derpapillären bis retikulären Dermis (Abb. 6a). Verschiedene morphologische Varianten von Makrophagensind nebeneinander vorhanden (Abb. 6b). Fr€uhe Läsionen bestehen vorwiegend aus vakuolisierten Zellenmit nur geringer Fettspeicherung sowie geringer Beimengung von Eosinophilen und Lymphozyten. Involl ausgeprägten Läsionen findet sich meist ein granulomatöses Entz€undungsinfiltrat mit Schaumzellen,Fremdkörperriesenzellen und Touton-Riesenzellen neben Lymphozyten und Eosinophilen. In regredien-ten Läsionen ersetzt Fibrose teilweise das entz€undliche Infiltrat. Touton Riesenzellen mit einemKranz ausKernen, umgeben von schaumigem Zytoplasma sind typisch, aber nicht diagnostisch f€ur JXG, sie könnenz. B. auch bei melanozytären Nävi vorkommen. Bei der mononukleären Variante des JXG fehlenSchaumzellen und Riesenzellen, der vorherrschende Makrophagentyp ist vakuolisiert. Idente Läsionenfinden sich auch bei der benignen zephalen Histiozytose und bei der generalisierten eruptiven Histio-zytose. Manchmal können Touton Riesenzellen auch in voll ausgeprägten Läsionen fehlen. Manchmalfinden sich Mitosen, die aber klinisch nicht bedeutsam sind und nie in Schaumzellen auftreten, da dieseterminal differenziert sind.
Differenzialdiagnose Klinisch muss man an melanozytäre Nävi Typ Spitz, Mastozytome, Dermatofi-brome denken. Histologisch ist das Bild eindeutig abgesehen von den vielen Varianten der
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 7 von 23

Xanthogranulom-Familie. JXG können auch bei LZH als xanthogranulomatöse Reaktion auf die Ent-z€undung auftreten.
3.2 Monomorphe Formen
3.2.1 Xanthomatisierte FormenPapulöses Xanthom (PX)Klinisches Bild
Es finden sich einzelne bis viele kleine gelbe bis rot-braune Papeln in regelloser Anordnung. OraleLäsionen kommen vor. PX kommen auch bei der progressiven nodulären Histiozytose vor. Betroffen sindv. a. Erwachsene, der Verlauf ist benigne.
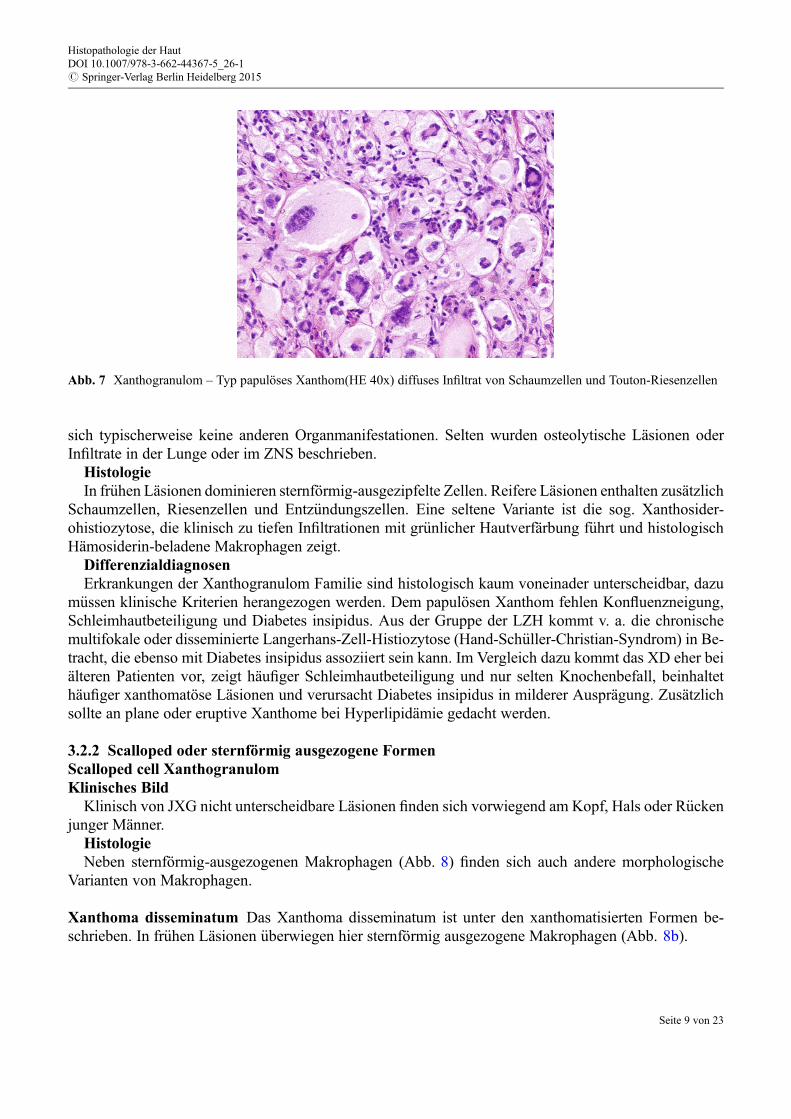
HistologieKnoten aus Schaumzellen und Touton-Riesenzellen finden sich in der Dermis (Abb. 7). Extrazelluläre
Lipide fehlen. Xanthomatöse Schaumzellen €uberwiegen sowohl in fr€uhen als auch in voll ausgeprägtenLäsionen.
DifferenzialdiagnoseXanthome im Rahmen von Hyperlipidämie oder als Paraneoplasie sind abzugrenzen. Einzelläsionen
können mit Dermatofibromen oder melanozytären Nävi Typ Spitz verwechselt werden.
Xanthoma disseminatum (XD)Klinisches Bild
Die meisten Fälle beginnen in der Kindheit, können grundsätzlich aber jedes Alter betreffen. Es bestehteine Androtropie (2:1). An der Haut sieht man zahlreiche, disseminierte, aber symmetrisch angeordnete,zur Konfluenz neigende orange bis gelb-braune Papeln und Knoten. Prädilektionsstellen sind Hals, großeBeugen, der Stamm und periokulär. Schleimhautbefall findet sich in ca. 40 %, besonders oral, nasal undokulär. Pharyngeale und laryngeale Läsionen f€uhren zu Heiserkeit und Atemnot. Es werden drei Ver-laufsformen unterschieden. Am häufigsten persistieren die Läsionen €uber Jahre. Nur selten kommt es zuSpontanregression. Sehr selten kommt es zu Progression mit Organbeteiligung. Am häufigsten findet sichein Diabetes insipidus in 40% der Fälle, bedingt durch Infiltration der Hypophyse. In diesen Fällen finden
Abb. 6 a, b Juveniles Xanthogranulom. a (HE 4x) Umschriebener Knoten mit collarette-artiger Einfassung durch dieEpidermis; b (HE 40x) mononukleäre und mehrkernige Makrophagen mit Xanthomatisierung in Form eines Schaumkranzes(Touton-Riesenzellen), daneben auch spindelförmige und sternförmig ausgezogene Makrophagen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 8 von 23
sich typischerweise keine anderen Organmanifestationen. Selten wurden osteolytische Läsionen oderInfiltrate in der Lunge oder im ZNS beschrieben.
HistologieIn fr€uhen Läsionen dominieren sternförmig-ausgezipfelte Zellen. Reifere Läsionen enthalten zusätzlich
Schaumzellen, Riesenzellen und Entz€undungszellen. Eine seltene Variante ist die sog. Xanthosider-ohistiozytose, die klinisch zu tiefen Infiltrationen mit gr€unlicher Hautverfärbung f€uhrt und histologischHämosiderin-beladene Makrophagen zeigt.
DifferenzialdiagnosenErkrankungen der Xanthogranulom Familie sind histologisch kaum voneinader unterscheidbar, dazu
m€ussen klinische Kriterien herangezogen werden. Dem papulösen Xanthom fehlen Konfluenzneigung,Schleimhautbeteiligung und Diabetes insipidus. Aus der Gruppe der LZH kommt v. a. die chronischemultifokale oder disseminierte Langerhans-Zell-Histiozytose (Hand-Sch€uller-Christian-Syndrom) in Be-tracht, die ebenso mit Diabetes insipidus assoziiert sein kann. Im Vergleich dazu kommt das XD eher beiälteren Patienten vor, zeigt häufiger Schleimhautbeteiligung und nur selten Knochenbefall, beinhaltethäufiger xanthomatöse Läsionen und verursacht Diabetes insipidus in milderer Ausprägung. Zusätzlichsollte an plane oder eruptive Xanthome bei Hyperlipidämie gedacht werden.
3.2.2 Scalloped oder sternförmig ausgezogene FormenScalloped cell XanthogranulomKlinisches Bild
Klinisch von JXG nicht unterscheidbare Läsionen finden sich vorwiegend am Kopf, Hals oder R€uckenjunger Männer.
HistologieNeben sternförmig-ausgezogenen Makrophagen (Abb. 8) finden sich auch andere morphologische
Varianten von Makrophagen.
Xanthoma disseminatum Das Xanthoma disseminatum ist unter den xanthomatisierten Formen be-schrieben. In fr€uhen Läsionen €uberwiegen hier sternförmig ausgezogene Makrophagen (Abb. 8b).
Abb. 7 Xanthogranulom – Typ papulöses Xanthom(HE 40x) diffuses Infiltrat von Schaumzellen und Touton-Riesenzellen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 9 von 23

3.2.3 Onkozytäre FormenRiesenzellretikulohistiozytom und multizentrische RetikulohistiozytoseKlinisches Bild
Das Riesenzellretikulohistiozytom ist eine benigne, von einem JXG klinisch nicht unterscheidbarepapulöse bis knotige Läsion, die fast ausschließlich bei Erwachsenen auftritt. In>90% handelt es sich umsolitäre Läsionen. Auch bei Auftreten multipler Läsionen fehlt eine Systembeteiligung. Die multizentri-sche Retikulohistiozytose (MRH), erstbeschrieben von Goltz und Laymon (1954), tritt ebenso haupt-sächlich bei Erwachsenen zwischen der 5. und 6. Lebensdekade auf und zeigt Gynäkotropie. Klinischzeigt sich eine Aussaat von gelblich-braunen Papeln und Knoten mit Betonung der Extremitäten (hierv. a. periartikulär) und des Gesichtes. Konfluierende Knoten im Gesicht können zu einer Facies leoninaf€uhren. Kleine Papeln am Nagelfalz bilden das sog. coral-bead-Zeichen. Schleimhautbefall (oral odernasal) findet sich in 50 %, Xanthelasmen finden sich in 25 % der Fälle. Bis zu 60 % erleiden einePolyarthropathie, die dem Hautbefall vorausgehen oder folgen kann. Sie wird durch synoviale Infiltrationdurch epithelioide Makrophagen initiiert und kann infolge von Knorpelzerstörung bis zur Gelenksde-struktion gehen. Eine Assoziation mit Hyperlipidämien (30–50 %) oder mit Autoimmunerkrankungen(5–15 %) wurde beschrieben. Bei 20 % der Patienten treten allerdings assoziiert interne Neoplasien auf.
HistologieTypisch sind onkozytäre Makrophagen mit mehrkernigen, irregulären Riesenzellen mit reichlich
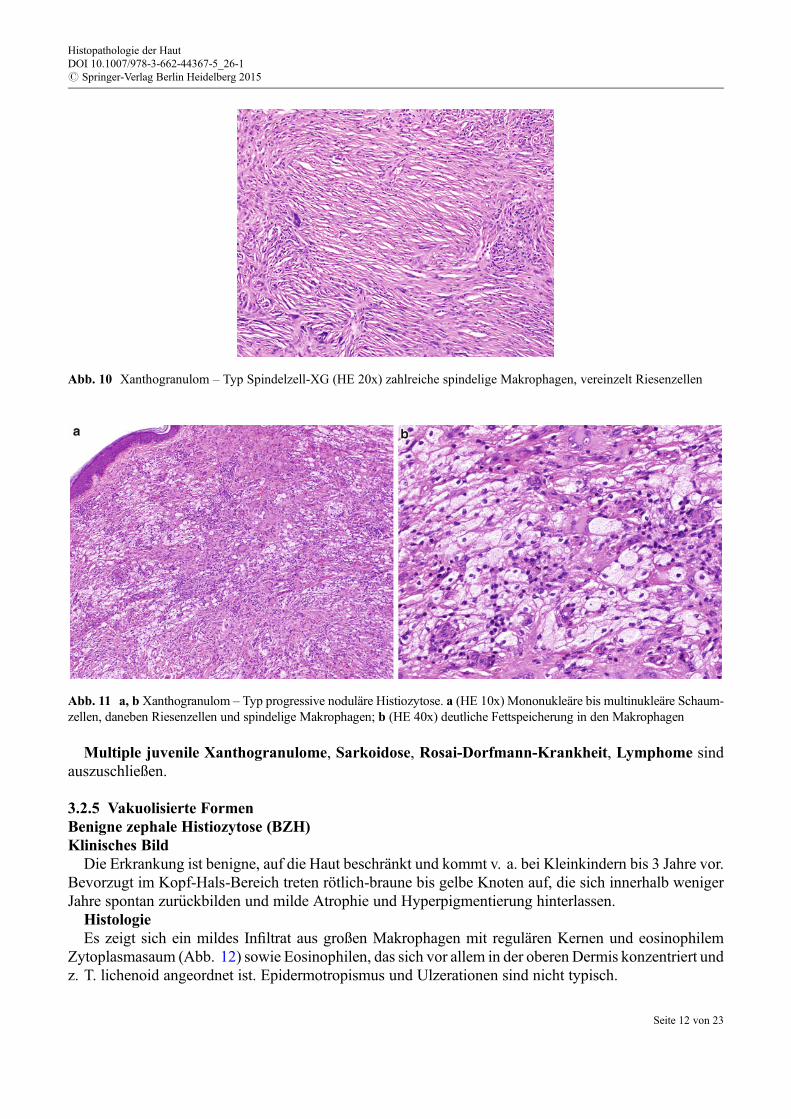
eosinophilem und feingranulärem Zytoplasma, das von den Erstbeschreibern auch als Milchglas-artigbezeichnet wurde (Abb. 9). Ultrastrukturell korreliert dies mit Reichtum an Mitochondrien und Lysoso-men. In reiferen Läsionen €uberwiegen Riesenzellen und Fibrose.
DifferenzialdiagnosenKlinisch sollten Gicht, rheumatoide Arthritis, Sarkoidose, Dermatomyositis und Lepra ausgeschlossen
werden. Auch die Varianten der Xanthogranulom-Familie m€ussen klinisch abgegrenzt werden. Mehrker-
Abb. 8 a, b Xanthogranulom – Typ Scalloped cell Xanthogranulom; a (HE 10x) papulöse Läsion aus eosinophilen Makro-phagen und eingestreuten Lymphozyten; b (HE 40x) sternförmig ausgezogene Makrophagen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 10 von 23

nige Riesenzellen können manchmal auch Lymphozyten enthalten. In solchen Fällen sollte man dif-ferenzialdiagnostisch an die Rosai-Dorfmann-Erkrankung denken.
3.2.4 Spindelige FormenSpindelzellxanthogranulomKlinisches Bild
Von JXG oder Dermatofibromen klinisch nicht unterscheidbare Läsionen im Kopf-Halsbereich jungerErwachsener. Tiefe Spindelzellxanthogranulome können diagnostisch schwierig sein und auch mit JXGgemeinsam auftreten.
HistologieVerwechslungen mit Dermatofibromen oder melanozytären Nävi Typ blau/Tieche sind häufig. Die
reaktive Verbreiterung der Epidermis oder seitliche Abgrenzung durch Kollagen (wie bei Dermatofi-bromen typisch) oder auch Melanin fehlen. Die spindeligen Makrophagen (Abb. 10) zeigen nebentypischen Makrophagenmarkern, wie Ki-M1p, CD163 und CD68, auch Faktor XIIIa und Aktin.
Progressive noduläre Histiozytose (PNH)Klinisches Bild
Typischerweise finden sich hunderte rotbraune Papeln und Knoten, die schubweise auftreten. Es zeigtsich ein Nebeneinander von einerseits oberflächlichen, xanthomatösen Papeln und Knoten von bis zu10 mm sowie andererseits tiefen fibrösen Knoten und Tumoren von bis zu 3 cm Größe. Letztere könnendurch Konfluenz im Gesicht zu einer Facies leonina f€uhren. Größere Läsionen zeigen häufig Einblutun-gen und folglich Hämosiderin-beladene Makrophagen (Xanthosiderohistiozytose), auch Ulzerationenkommen vor. Schleimhautbefall (Konjunktiven, Mund, Larynx) und Organbefall sind selten. Spontanre-gression kommt kaum vor. Die Erkrankung ist selten, benigne und zumeist auf die Haut beschränkt.
HistologieDie kleinen Läsionen entsprechen papulösen Xanthomen aus Schaumzellen und Touton-Riesenzellen
(Abb. 11). Die größeren Läsionen entsprechen Spindelzellxanthogranulomen aus vorwiegend spindeli-gen Makrophagen.
Differenzialdiagnose
Abb. 9 Xanthogranulom – Typ Retikulohistiozytom (HE 40x) vorwiegend onkozytäre Makrophagen sowie milchglasartigeRiesenzellen, daneben spindelige oder sternförmig ausgezogene Makrophagen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 11 von 23
Multiple juvenile Xanthogranulome, Sarkoidose, Rosai-Dorfmann-Krankheit, Lymphome sindauszuschließen.
3.2.5 Vakuolisierte FormenBenigne zephale Histiozytose (BZH)Klinisches Bild
Die Erkrankung ist benigne, auf die Haut beschränkt und kommt v. a. bei Kleinkindern bis 3 Jahre vor.Bevorzugt im Kopf-Hals-Bereich treten rötlich-braune bis gelbe Knoten auf, die sich innerhalb wenigerJahre spontan zur€uckbilden und milde Atrophie und Hyperpigmentierung hinterlassen.
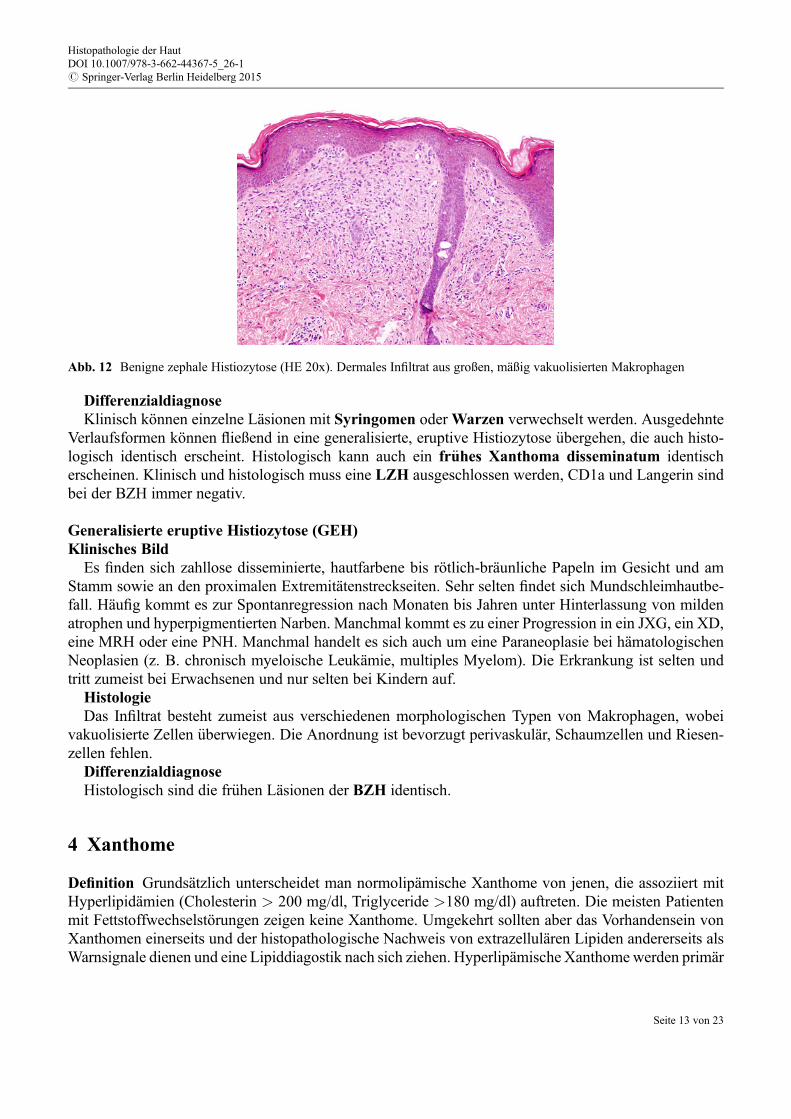
HistologieEs zeigt sich ein mildes Infiltrat aus großen Makrophagen mit regulären Kernen und eosinophilem
Zytoplasmasaum (Abb. 12) sowie Eosinophilen, das sich vor allem in der oberen Dermis konzentriert undz. T. lichenoid angeordnet ist. Epidermotropismus und Ulzerationen sind nicht typisch.
Abb. 11 a, b Xanthogranulom – Typ progressive noduläre Histiozytose. a (HE 10x) Mononukleäre bis multinukleäre Schaum-zellen, daneben Riesenzellen und spindelige Makrophagen; b (HE 40x) deutliche Fettspeicherung in den Makrophagen
Abb. 10 Xanthogranulom – Typ Spindelzell-XG (HE 20x) zahlreiche spindelige Makrophagen, vereinzelt Riesenzellen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 12 von 23
DifferenzialdiagnoseKlinisch können einzelne Läsionen mit Syringomen oderWarzen verwechselt werden. Ausgedehnte
Verlaufsformen können fließend in eine generalisierte, eruptive Histiozytose €ubergehen, die auch histo-logisch identisch erscheint. Histologisch kann auch ein fr€uhes Xanthoma disseminatum identischerscheinen. Klinisch und histologisch muss eine LZH ausgeschlossen werden, CD1a und Langerin sindbei der BZH immer negativ.
Generalisierte eruptive Histiozytose (GEH)Klinisches Bild
Es finden sich zahllose disseminierte, hautfarbene bis rötlich-bräunliche Papeln im Gesicht und amStamm sowie an den proximalen Extremitätenstreckseiten. Sehr selten findet sich Mundschleimhautbe-fall. Häufig kommt es zur Spontanregression nach Monaten bis Jahren unter Hinterlassung von mildenatrophen und hyperpigmentierten Narben. Manchmal kommt es zu einer Progression in ein JXG, ein XD,eine MRH oder eine PNH. Manchmal handelt es sich auch um eine Paraneoplasie bei hämatologischenNeoplasien (z. B. chronisch myeloische Leukämie, multiples Myelom). Die Erkrankung ist selten undtritt zumeist bei Erwachsenen und nur selten bei Kindern auf.
HistologieDas Infiltrat besteht zumeist aus verschiedenen morphologischen Typen von Makrophagen, wobei
vakuolisierte Zellen €uberwiegen. Die Anordnung ist bevorzugt perivaskulär, Schaumzellen und Riesen-zellen fehlen.
DifferenzialdiagnoseHistologisch sind die fr€uhen Läsionen der BZH identisch.
4 Xanthome
Definition Grundsätzlich unterscheidet man normolipämische Xanthome von jenen, die assoziiert mitHyperlipidämien (Cholesterin > 200 mg/dl, Triglyceride >180 mg/dl) auftreten. Die meisten Patientenmit Fettstoffwechselstörungen zeigen keine Xanthome. Umgekehrt sollten aber das Vorhandensein vonXanthomen einerseits und der histopathologische Nachweis von extrazellulären Lipiden andererseits alsWarnsignale dienen und eine Lipiddiagostik nach sich ziehen. Hyperlipämische Xanthome werden primär
Abb. 12 Benigne zephale Histiozytose (HE 20x). Dermales Infiltrat aus großen, mäßig vakuolisierten Makrophagen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 13 von 23

klinisch diagnostiziert. Übergänge zwischen den klinischen Varianten oder auch zu normolipämischenXanthomen sind häufig.
4.1 Xanthome assoziiert mit Hyperlipidämie
Klinisches Bild
Eruptive Xanthome kommen vor allem bei sekundären Hyperlipidämien vor und sind fast immer mitChylomikronämie assoziiert. Es zeigen sich kleine, weiche, gelbe Papeln am Gesäß und den dorsalenOberschenkeln, die in Abhängigkeit vom Chylomikronspiegel kommen und gehen. Die eingelagertenLipide sind hautsächlich Triglyceride.
Tuberose Xanthome oder tubero-eruptive Xanthome kommen bevorzugt bei erhöhten LDL und VLDLvor. Es zeigen sich große Knoten oder Plaques an den Ellbogen, den Knien, den Fingern oder am Gesäß.Das eingelagerte Lipid ist hauptsächlich Cholesterin.
Sehnenscheidenxanthome kommen vor allem bei Patienten mit stark erhöhtem LDL vor, wie z. B. beifamiliärer Hypercholesterinämie oder familiärem Apolipoprotein B-100 Defekt sowie auch bei Phytos-terolämie oder Cholestanolämie (zerebrotendinöse Xanthomatose). Bevorzugt betroffen sind Achilles-sehne und Fingersehnen.
Plane Xanthome finden sich vor allem in Hautfalten. Das Auftreten in den Handlinien ist diagnostisch f€urDysbetalipoproteinämie. Bei homozygoter, familiärer Hypercholesterinämie finden sie sich vor allem inden großen Beugen. Palmare Xanthome treten auch bei Cholestase (bei primär-biliärer Zirrhose oderbiliärer Atresie) auf. Diffuse plane Xanthome können auch ohne Hyperlipidämie im Rahmen vonParaproteinämie, Lymphomen oder Leukämien auftreten und erscheinen als gruppierte gelbliche Papelnund Plaques.
Xanthelasmen sind flach erhabene, gelbliche Plaques an den Augenlidern. Sie sind sehr häufig, aber auchsehr unspezifisch. Zumeist treten sie ohne Hyperlipidämie auf. Im Fall von Hyperlipidämie sind sie oft mitanderen hyperlipämischen Xanthomvarianten assoziiert. Sie verschwinden nicht mit lipidsenkendenMaßnahmen. Manche Autoren sehen Xanthelasmen als Variante des Xanthogranuloms.
Histologie Die verschiedenen klinischen Varianten zeigen histopathologisch ein recht einheitliches Bild.Makrophagen mit Lipidspeicherung, sog. Schaumzellen, sind typisch. Mehrkernige Riesenzellen, v. a.Touton-Typ, mit kranzförmig angeordneten Kernen, sind eingestreut. Wichtig ist das Vorhandensein vonfreien, noch nicht phagozytierten Lipiden als Ausdruck der Fettstoffwechselstörung. Fett kann durchScharlachrot oder Sudanrot hervorgehoben werden. Eine begleitende Fibrose kann in variabler Aus-prägung, besonders bei chronischen Läsionen, vorhanden sein. Xanthome zeigen regelmäßig Fixations-artefakte, weil Lipide durch Formalin und Paraffin herausgelöst werden und Schatten bzw. Spalten, sog.Clefts hinterlassen. Weiterhin sind die doppelbrechenden Eigenschaften von Cholesterolestern zu erwäh-nen. Unter polarisiertem Licht könnenmanchmal, bei nicht vollständiger Lyse der Cholesterin-Kristalle inFormalin, Cholesterol-reiche Sehnenscheidenxanthome oder auch tuberöse Xanthome von Xanthomen,die andere Lipide speichern, unterschieden werden. Die verschiedenen klinischen Erscheinungsformenzeigen auch histopathologisch Besonderheiten.
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 14 von 23

Eruptive Xanthome sind oft reich an vakuolisierten Makrophagen, Lymphozyten und Neutrophilen.Extrazelluläre Lipide sind häufig. Mit der Bestandsdauer der Läsionen nehmen Schaumzellen zu.
Tuberose Xanthome zeigen kleine und große Ansammlungen von Schaumzellen (Abb. 13). Fr€uheLäsionen enthalten zusätzlich vakuolisierte Makrophagen, Lymphozyten und Neutrophile, die mit derReifung verschwinden. Schließlich werden Zellen durch Kollagen ersetzt (Abb. 13a).
Sehnenscheidenxanthome sind ähnlich, oft größer und enthalten oft keine Hautanteile.
Plane Xanthome sind nur in palmarer Lokalisation durch epidermale Hyperkeratose und ein Stratumlucidum unterscheidbar.
Xanthelasmen unterscheiden sich durch die sehr oberflächliche Lokalisation und das Fehlen von Fibrose(Abb. 14). Oberflächliche Anteile von quergestreiften Muskelb€undeln, Vellushaare und die sehr d€unneEpidermis charakterisieren die Lokalisation an den Lidern. Es finden sich Riesenzellen, aber kaum jeextrazelluläre Lipide.
Differenzialdiagnosen Verschiedene Xanthomvarianten, Gicht (bei Nachweis von Uratkristallen),Artefakte (Filler), primäre Adenokarzinome von Talgdr€usen oder Siegelringzellen, Metastasen vonAdenokarzinomen (Mammakarzinom) m€ussen ausgeschlossen werden.
4.2 Normolipämische Xanthome
Definition Diese Gruppe von Xanthomen tritt ohne assoziierte Hypercholesterinämie oder Hypertrigly-ceridämie auf. Tuberöse Xanthome und Sehnenscheidenxanthome können aber auch auf einem Über-schuss von anderen Sterolen, wie z. B. Cholestanol (bei zerebrotendinöser Xanthomatose) oder Sitosterol(bei Phytosterolämie) beruhen. Xanthome, v. a. diffuse plane Xanthome und das nekrobiotische Xantho-granulom, können mit lymphoproliferativen Erkrankungen assoziiert sein oder auch posttraumatisch bzw.idiopathisch auftreten. Manche normolipämischen Xanthome werden der Xanthogranulomfamilie
Abb. 13 a, b Tuberöses Xanthom. a (HE 4x) Diffuses Infiltrat aus Schaumzellen in fibrosklerotischer Umgebung; b (HE 40x)schaumige Makrophagen sowie extrazelluläre, freie Lipideinlagerungen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 15 von 23

(Xanthoma disseminatum und papulöses Xanthom) oder sehr selten den Langerhans-Zell-Histiozytosenzugerechnet. Überlappungen zwischen den klinischen Varianten sind sehr häufig.
Klinisches Bild
Nekrobiotisches Xanthogranulom mit Paraproteinämie Charakteristisch sind große, gelbliche, derbePlaques mit Atrophie, Teleangiektasien und gelegentlicher Ulzeration v. a. periorbital oder auch thorakal.Kardiomyopathien mit Riesenzellinfiltraten kommen assoziiert vor. Zumeist findet sich in der Serum-elektrophorese eine monoklonale Gammopathie in IgG mit Leichtkettenrestriktion. Die Behandlung derhämatologischen Grunderkrankung beeinflusst die Hautläsionen nicht. Spirochäten konnten in einigenLäsionen nachgewiesen werden, eine pathogenetische Rolle oder eine Triggerfunktion von Borreliaburgdorferi wird diskutiert.
Diffuse normolipämische plane Xanthome (DNPX) Es finden sich gelb-orange Papeln, Plaques oderflächige Verfärbungen v. a. periorbital oder thorakal mit initialer Ähnlichkeit zu Xanthelasmen ohneR€uckbildungstendenz. Lipidstoffwechselstörungen fehlen. In 50 % sind myeloproliferative Erkrankun-gen assoziiert, die den Xanthomen zeitlich oft bis zu Jahren nachhinken
Verruziformes Xanthom (VX) Es finden sich zumeist solitäre, hyperkeratotische, papillomatöse Knotenan der Mundschleimhaut. An der Haut können sie im Rahmen eines epidermalen Nävus (CHILD Nävus)oder auch mechanisch getriggert v. a. perioral und genitoanal auftreten. Weiters wurde das Auftreten ansonnengeschädigter Haut oder sekundär zu entz€undlichen Dermatosen (Lichen ruber, Lupus erythema-tosus) berichtet. Ätiologisch wird ein Zusammenhang mit Viruswarzen diskutiert.
Abb. 14 Xanthelasma (HE 10x) unter einer d€unnen Epidermis Infiltrat aus Schaumzellen, in der Tiefe ein Vellushaar undMuskelb€undel
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 16 von 23
Histologie
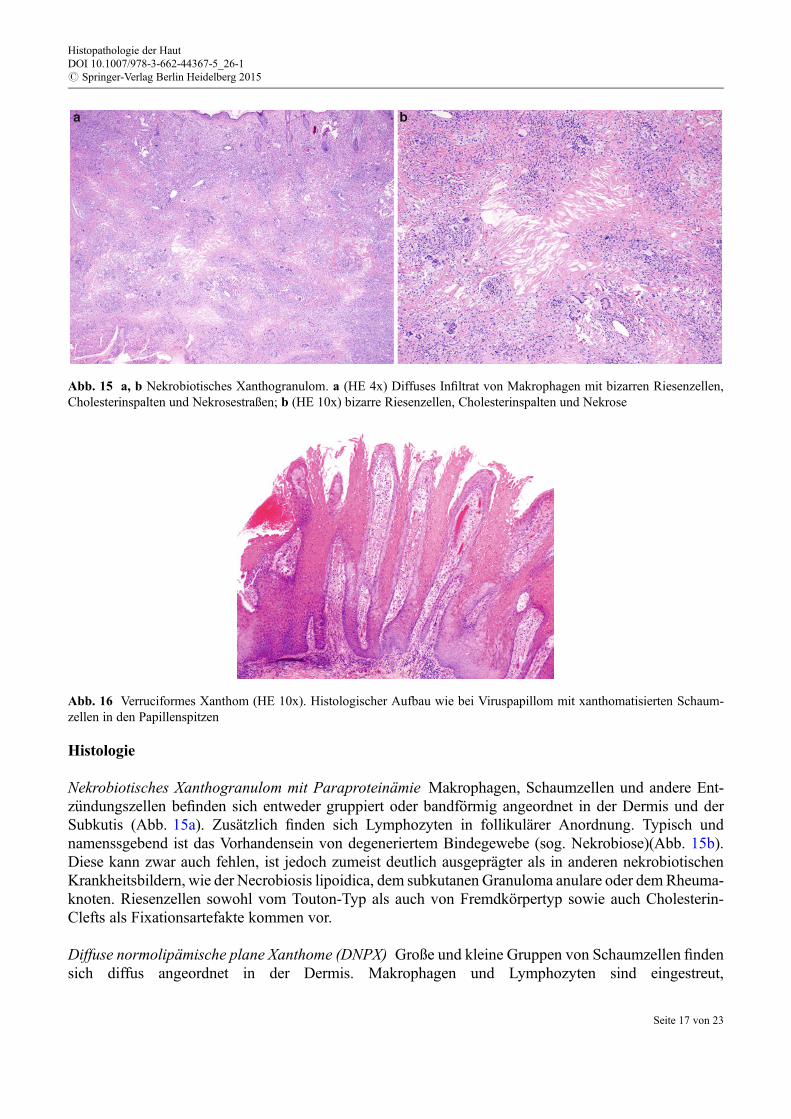
Nekrobiotisches Xanthogranulom mit Paraproteinämie Makrophagen, Schaumzellen und andere Ent-z€undungszellen befinden sich entweder gruppiert oder bandförmig angeordnet in der Dermis und derSubkutis (Abb. 15a). Zusätzlich finden sich Lymphozyten in follikulärer Anordnung. Typisch undnamenssgebend ist das Vorhandensein von degeneriertem Bindegewebe (sog. Nekrobiose)(Abb. 15b).Diese kann zwar auch fehlen, ist jedoch zumeist deutlich ausgeprägter als in anderen nekrobiotischenKrankheitsbildern, wie der Necrobiosis lipoidica, dem subkutanen Granuloma anulare oder demRheuma-knoten. Riesenzellen sowohl vom Touton-Typ als auch von Fremdkörpertyp sowie auch Cholesterin-Clefts als Fixationsartefakte kommen vor.
Diffuse normolipämische plane Xanthome (DNPX) Große und kleine Gruppen von Schaumzellen findensich diffus angeordnet in der Dermis. Makrophagen und Lymphozyten sind eingestreut,
Abb. 15 a, b Nekrobiotisches Xanthogranulom. a (HE 4x) Diffuses Infiltrat von Makrophagen mit bizarren Riesenzellen,Cholesterinspalten und Nekrosestraßen; b (HE 10x) bizarre Riesenzellen, Cholesterinspalten und Nekrose
Abb. 16 Verruciformes Xanthom (HE 10x). Histologischer Aufbau wie bei Viruspapillom mit xanthomatisierten Schaum-zellen in den Papillenspitzen
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 17 von 23

Touton-Riesenzellen sind selten. Manchmal lagern sich die Schaumzellen strichförmig an dieKollagenfaserb€undel an.
Verruziformes Xanthom (VX) In der Übersicht zeigt sich das Bild einer Verruca vulgaris. In den ausge-zogenen dermalen Papillen finden sich jedoch Schaumzellen (Abb. 16).
5 Sonderformen
5.1 Histiozytose der indeterminierten Zelle
Definition und klinisches Bild Wir vermuten, dass diese Krankheitsbilder zumeist von Makrophagenausgehen, wobei sich die vorherrschende Zelle von einem fr€uheren oder aberranten Entwicklungsstadiumableitet. Andere Fälle, die der kongenitalen selbstheilenden Retikulohistiozytose ähneln, sind Variantender LZH. Es handelt sich aus unserer Sicht also nicht um eine eigene Entität, sondern um einenSchmelztopf von einerseits Makrophagenerkrankungen, die S100-Protein exprimieren und anderer-seits Langerhans-Zell-Erkrankungen, die Birbeck Granula vermissen. Die meisten Formen sindwahrscheinlich den Xanthogranulomen zuzurechnen. Die Patienten zeigen zahlreiche, rotbraune Papelnbis Knoten, die zur Konfluenz neigen und persistieren.
Histologie Zumeist besteht das Infiltrat aus vakuolisierten Zellen mit eingestreuten Eosinophilen, Rie-senzellen und Schaumzellen. Die Zellen zeigen S100-Protein, CD68 und fokal CD1a als Hintergrund-reaktion. Starke Reaktivität auf S100-Protein ist mit Eosinophilenreichtum assoziiert. Eine Variante zeigthistologische Kriterien der LZH, allerdings ohne Langerin oder Birbeck Granula. Kutane und nodaleLäsionen wurden beschrieben.
5.2 Rosai-Dorfman-Krankheit (RDD)
Definition und klinisches Bild Destombes (1965) hat die Erkrankung erstmals beschrieben, Rosai undDorfman (1969) prägten 4 Jahre später den Namen Sinushistiozytose mit massiver Lymphadenopathie.Die vorherrschenden Zellen sind aktivierte Makrophagen, die neben Makrophagenmarkern und Fascinauch Positivität f€ur S100-Protein zeigen. Es findet sich regelmäßig eine ausgeprägte, zumeist zervikale,schmerzlose, bilaterale Lymphadenopathie. Diagnostisch steht die Lymphknotenbiopsie im Vordergrund.Zusätzliche Haut- oder Bindegewebsbeteiligung in Form von solitären oder multiplen Papeln oderKnoten betrifft 10 % der Patienten. Sie betrifft v. a. junge Erwachsene und ist häufiger bei Weißen undDunkelhäutigen. Auch isolierter Hautbefall kommt vor, in diesem Fall erfolgt die Diagnose €uber denDermatopathologen. Zumeist kommt es zur Spontanheilung, seltener zu Persistenz und nur in Aus-nahmen zu letalem Verlauf.
Histologie In der Übersicht findet man das Bild des „Lymphknotens in der Haut“ (Abb. 17a). Es zeigtsich ein polymorphes Infiltrat aus Lymphozyten und epithelioiden Makrophagen mit blass-schaumigembis eosinophilem Zytoplasma. Gelegentlich können Makrophagen auch mehrkernig sein. Das typischeMerkmal ist die Emperipolesis (Abb. 17b). Intakte Lymphozyten oder seltener Erythrozyten finden sichintrazellulär in Makrophagen. Oft sind zusätzlich IgG4-positive Plasmazellen mit kristallinen Immung-lobulinablagerungen zu sehen. Die betroffenen Lymphknoten zeigen ausgeweitete Sinus, die von Ent-z€undungszellen, insbesonders Makrophagen mit reichlich schaumigem Zytoplasma gef€ullt sind. Auchhier findet sich Emperipolesis. Makrophagenmarker und S100-Protein sind positiv.
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 18 von 23

5.3 Erdheim-Chester-Krankheit
Definition, klinisches und histologisches Bild Die Erkrankung ist charakterisiert durch osteoskleroti-sche Läsionen in den langen Röhrenknochen. Zumeist sind Erwachsene betroffen, selten wurden Fälle beiKindern beschrieben. In 50 % kommt es zu extrasklettalem Befall mit Beteiligung von Nieren, Retro-peritoneum, Haut, Lunge und ZNS. An der Haut finden sich z. B. Xanthelasmen oder rötlich-braunePapeln und Knoten, die an JXG erinnern, jedoch mehr Fibrose und auch Hämosiderineinlagerungenaufweisen. Histologisch zeigen sich Makrophagen in diffuser oder granulomatöser Anordnung, diexanthomatisiert sein können und Makrophagenmarker aufweisen. Touton-Riesenzellen sind manchmaleingestreut. Die Prognose ist bei extrasklettalem Befall schlecht, die Patienten sterben innerhalb wenigerJahre an Organversagen. Koexsistenz von LHZ und Erdheim-Chester-Krankheit wurde beschrieben.
5.4 Maligne Proliferationen von Makrophagen und Langerhans-Zellen
Definition, klinisches und histologisches Bild Viele Erkrankungen, die fr€uher den malignen Histiozy-tosen zugeordnet wurden, sind heute als B- oder T-Zell-Lymphome klassifiziert. Nur wenige maligneErkrankungen bleiben €ubrig, die sich tatsächlich von Makrophagen oder sehr selten von Langerhans-Zellen ableiten. Myelo-monozytäre Leukämien zeigen gelegentlich Haut- und Schleimhautbefall in Formvon rasch auftretenden lividen bis hämorrhagischen Papeln bis Knoten, manchmal auch als Primär-manifestation. Andere Formen maligner Makrophagen-Erkrankungen sind sehr selten. Im Falle eineszytologisch malignen Infiltrates sollten zunächst Leukämien und Lymphome ausgeschlossen werden. Inder Immunhistochemie zeigen sich dann die Zellen CD68 positiv und in 90 % positiv f€ur Lysozym. S100-Protein kann positiv sein. Ultrastrukturell finden sich Lysosomen, aber keine Birbeck-Granula oderDesmosomen. Die Prognose hängt von der Ausprägung ab. Umschriebene, extranodale Herde könnengeheilt werden, zumeist kommt es jedoch zu einem aggressiven Verlauf. Langerhans-Zell-Leukämiensowie auch primär maligne Hautinfiltrate mit Langerhans-Zell-Charakter sind extrem selten.
Abb. 17 a, b Rosai-Dorfmann-Krankheit. a (HE 2x) Knotiges dermales Infiltrat – „Lymphknoten in der Haut“; b (HE 40x)diffuses Infiltrat aus blass eosinophilen Makrophagen umgeben von Lymphozyten und Plasmazellen, außerdem Phagozytosevon Lymphozyten durch Makrophagen – Emperiopolesis (Pfeile)
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 19 von 23

Weiterführende Literatur
Weiterführende Literatur zu Makrophagen- und Langerhans-Zell-ErkrankungenJaffe R, Chikwava KR (2012) Disorders of histiocytes. In: Hsi ED (Hrsg) Hematopathology, Bd 19.
Saunders, Philadelphia, S 3–40Satter EK, HighWA (2008) Langerhans cell histiocytosis: a review of the current recommendations of the
Histiocyte Society. Pediatr Dermatol 25:291–295Swerdlow SH, Campo E, Harris NL et al. (Hrsg) (2008)WHO classification of tumours of haematopoietic
and lymphoid tissue. IARC, LyonWeitzman S, Egeler RM (2005) Histiocytic disorders of children and adults. Cambridge University Press,
Cambridge
Weiterführende Literatur zur Langerhans-Zell-Histiozytose (LZH)Allen CE, Li L, Peters TL et al (2010) Cell-specific gene expression in Langerhans cell histiocytosis
lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol 184:4557–4567Badalian-Very G, Vergilio JA, Degar BA et al (2012) Recent advances in the understanding of Langerhans
cell histiocytosis. Br J Haematol 156:163–172Battistella M, Fraitag S, Teillac DH et al (2010) Neonatal and early infantile cutaneous Langerhans cell
histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol146:149–156
Berres ML, Allen CE, Merad M (2013) Pathological consequence of misguided dendritic cell differen-tiation in histiocytic diseases. Adv Immunol 120:127–161
Hashimoto K, PritzkerMS (1973) Electron microscopic study of reticulohistiocytoma. An unusual case ofcongenital, self-healing reticulohistiocytosis. Arch Dermatol 107:263–270
Haupt R, Minkov M, Astigarraga I et al (2013) Langerhans cell histiocytosis (LCH): guidelines fordiagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer60:175–184
Kapur P, Erickson C, Rakheja D et al (2007) Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol56:290–294
Lichtenstein L, Histiocytosis X (1953) Integration of eosinophilic granuloma, Letterer-Siwe disease andHand-Sch€uller-Christian disease as related manifestations of a single nosologic entity. Arch Pathol56:84–102
Lieberman PH, Jones CR, Steinman RM et al (1996) Langerhans cell (eosinophilic) granulomatosis.A clinicopathologic study encompassing 50 years. Am J Surg Pathol 20:519–552
Pileri SA, Grogan TM, Harris NL et al (2002) Tumours of histiocytes and accessory dendritic cells: animmunohistochemical approach to classification from the International Lymphoma Study Group basedon 61 cases. Histopathology 41:1–29
SagranskyMJ, Deng AC,Magro CM (2013) Primary cutaneous Langerhans cell sarcoma: a report of fourcases and review of the literature. Am J Dermatopathol 35:196–204
Weiterführende Literatur zur Xanthogranulom-FamilieBattaglini J, Olsen TG (1984) Disseminated xanthosiderohistiocytosis, a variant of xanthoma dissemi-
natum, in a patient with a plasma cell dyscrasia. J Am Acad Dermatol 11:750–755Breier F, Zelger B, Reiter H et al (2002) Papular xanthoma: a clinicopathological study of 10 cases.
J Cutan Pathol 29:200–206
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 20 von 23

BurgdorfWH, Zelger B (2004) JXG, NF1, and JMML: alphabet soup or a clinical issue? Pediatr Dermatol21:174–176
Burgdorf WH, Kusch SL, Nix TE Jr et al (1981) Progressive nodular histiocytoma. Arch Dermatol117:644–649
Caputo R, Grimalt R, Gelmetti C et al (1993) Unusual aspects of juvenile xanthogranuloma. J Am AcadDermatol 29:868–870
Caputo R, Veraldi S, Grimalt R et al (1995) The various clinical patterns of xanthoma disseminatum.Considerations on seven cases and review of the literature. Dermatology 190:19–24
Chang MW, Frieden IJ, Good W (1996) The risk of intraocular juvenile xanthogranuloma: survey ofcurrent practices and assessment of risk. J Am Acad Dermatol 34:445–449
Dehner LP (2003) Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 27:579–593
Fett N, Liu RH (2011) Multicentric reticulohistiocytosis with dermatomyositis-like features: a morecommon disease presentation than previously thought. Dermatology 222:102–108
Gianotti F, Caputo R, Ermacora E et al (1986) Benign cephalic histiocytosis. Arch Dermatol122:1038–1043
Goltz RW, Laymon CW (1954) Multicentric reticulohistiocytosis of the skin and synovia. Arch Dermatol69:717–730
Hilker O, Kovneristy A, Varga R et al (2013) Progressive nodular histiocytosis. J Dtsch Dermatol Ges11:301–307
Janssen D, Harms D (2005) Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologicstudy of 129 patients from the Kiel pediatric tumor registry. Am J Surg Pathol 29:21–28
Jih DM, Salcedo SL, Jaworsky C (2002) Benign cephalic histiocytosis: a case report and review. J AmAcad Dermatol 47:908–913
Miettinen M, Fetsch JF (2006) Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopatho-logic and immunohistochemical study of 44 cases. Am J Surg Pathol 30:521–528
Mowbray M, Schofield OM (2007) Juvenile xanthogranuloma en plaque. Pediatr Dermatol 24:670–671Ngendahayo P, de Saint AN (2012) Mitotically active xanthogranuloma: a case report with review of the
literature. Am J Dermatopathol 34:e27–e30Sanchez Yus E, Requena L, Villegas C et al (1995) Subcutaneous juvenile xanthogranuloma. J Cutan
Pathol 22:460–465Seward JL, Malone JC, Callen JP (2004) Generalized eruptive histiocytosis. J Am Acad Dermatol
50:116–120Shapiro PE, Silvers DN, Treiber RK et al (1991) Juvenile xanthogranulomas with inconspicuous or absent
foam cells and giant cells. J Am Acad Dermatol 24:1005–1009Skinner M, Briant M, Morgan MB (2011) Erdheim-Chester disease: a histiocytic disorder more than skin
deep. Am J Dermatopathol 33:e24–e26Tajirian AL, Malik MK, Robinson-Bostom L et al (2006) Multicentric reticulohistiocytosis. Clin Der-
matol 24:486–492Torrelo A, Juarez A, Hernandez A et al (2009) Multiple lichenoid juvenile xanthogranuloma. Pediatr
Dermatol 26:238–240Tran DT, Wolgamot GM, Olerud J et al (2008) An ‘eruptive’ variant of juvenile xanthogranuloma
associated with Langerhans cell histiocytosis. J Cutan Pathol 35(Suppl 1):50–54Winkelmann RK, Muller SA (1963) Generalized eruptive histiocytoma. Arch Dermatol 88:586–596Zelger B, BurgdorfW (2013) In: Plewig G, Landthaler M, BurgdorfW, Hertl M, Ruzicka T (Hrsg) Braun-
Falco's Dermatologie, Venerologie und Allergologie. Springer, Heidelberg, S 1818
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 21 von 23

Zelger BW, Cerio R (2001) Xanthogranuloma is the archetype of non-Langerhans cell histiocytoses. BrJ Dermatol 145:369–371
Zelger B, Cerio R, Orchard G et al (1992) Histologic and immunohistochemical study comparingxanthoma disseminatum and histiocytosis X. Arch Dermatol 128:1207–1212
Zelger B, Cerio R, Soyer HP et al (1994) Reticulohistiocytoma and multicentric reticulohistiocytosis.Histopathologic and immunophenotypic distinct entities. Am J Dermatopathol 16:577–584
Zelger BG, Zelger B, Steiner H et al (1995a) Solitary giant xanthogranuloma and benign cephalichistiocytosis–variants of juvenile xanthogranuloma. Br J Dermatol 133:598–604
Zelger BW, Staudacher C, Orchard G et al (1995b) Solitary and generalized variants of spindle cellxanthogranuloma (progressive nodular histiocytosis). Histopathology 27:11–19
Zelger BW, Sidoroff A, Orchard G et al (1996) Non-Langerhans cell histiocytoses. A new unifyingconcept. Am J Dermatopathol 18:490–504
Zelger BG, Orchard G, Rudolph P et al (1998) Scalloped cell xanthogranuloma. Histopathology32:368–374
Weiterführende Literatur zu XanthomenAltmann J, Winkelmann RK (1962) Diffuse normolipemic plane xanthoma. Arch Dermatol 85:633–640Blankenship DW, Zech L, Mirzabeigi M et al (2013) Verruciform xanthoma of the upper-extremity in the
absence of chronic skin disease or syndrome: a case report and review of the literature. J Cutan Pathol40:745–752
Braun-Falco O, Eckert F (1991) Macroscopic and microscopic structure of xanthomatous eruptions. CurrProbl Dermatol 20:54–62
Cruz PD Jr, East C, Bergstresser PR (1988) Dermal, subcutaneous, and tendon xanthomas: diagnosticmarkers for specific lipoprotein disorders. J Am Acad Dermatol 19:95–111
Cumberland L, Dana A, Resh B et al (2010) Verruciform xanthoma in the setting of cutaneous trauma andchronic inflammation: report of a patient and a brief review of the literature. J Cutan Pathol 37:895–900
Ferrara G, Palombi N, Lipizzi A et al (2007) Nonnecrobiotic necrobiotic xanthogranuloma. AmJ Dermatopathol 29:306–308
Finan MC, Winkelmann RK (1987) Histopathology of necrobiotic xanthogranuloma with paraproteine-mia. J Cutan Pathol 14:92–99
Kossard S, Winkelmann RK (1980) Necrobiotic xanthogranuloma with paraproteinemia. J Am AcadDermatol 3:257–270
Marcoval J, Moreno A, Bordas X et al (1998) Diffuse plane xanthoma: clinicopathologic study of 8 cases.J Am Acad Dermatol 39:439–442
Mohsin SK, Lee MW, Amin MB et al (1998) Cutaneous verruciform xanthoma: a report of five casesinvestigating the etiology and nature of xanthomatous cells. Am J Surg Pathol 22:479–487
Shafer WG (1971) Verruciform xanthoma. Oral Surg Oral Med Oral Pathol 31:784–789Wood AJ, Wagner MV, Abbott JJ et al (2009) Necrobiotic xanthogranuloma: a review of 17 cases with
emphasis on clinical and pathologic correlation. Arch Dermatol 145:279–284Zelger B, Eisendle K, Mensing C et al (2007) Detection of spirochetal micro-organisms by focus-floating
microscopy in necrobiotic xanthogranuloma. J Am Acad Dermatol 57:1026–1030
Weiterführende Literatur zu den SonderformenDestombes P (1965) Adenites avec surcharge lipidique, de l'enfant ou de l'adulte jeune, observees aux
Antilles et au Mali. (Quatre observations) [Adenitis with lipid excess, in children or young adults, seenin the Antilles and in Mali. (4 cases)]. Bull Soc Pathol Exot Filiales 58:1169–1175
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 22 von 23

Foucar E, Rosai J, Dorfman R (1990) Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 7:19–73
Frater JL, Maddox JS, Obadiah JM et al (2006) Cutaneous Rosai-Dorfman disease: comprehensivereview of cases reported in the medical literature since 1990 and presentation of an illustrative case.J Cutan Med Surg 10:281–290
Kuo TT, Chen TC, Lee LYet al (2009) IgG4-positive plasma cells in cutaneous Rosai-Dorfman disease:an additional immunohistochemical feature and possible relationship to IgG4-related sclerosing disea-se. J Cutan Pathol 36:1069–1073
Ratzinger G, Burgdorf WH, Metze D et al (2005) Indeterminate cell histiocytosis: fact or fiction? J CutanPathol 32:552–560
Rezk SA, Spagnolo DV, Brynes RK et al (2008) Indeterminate cell tumor: a rare dendritic neoplasm. AmJ Surg Pathol 32:1868–1876
Rosai J, Dorfman RF (1969) Sinus histiocytosis with massive lymphadenopathy. A newly recognizedbenign clinicopathological entity. Arch Pathol 87:63–70
Shamburek RD, Brewer HB Jr, Gochuico BR (2001) Erdheim-Chester disease: a rare multisystemhistiocytic disorder associated with interstitial lung disease. Am J Med Sci 321:66–75
Sidoroff A, Zelger B, Steiner H et al (1996) Indeterminate cell histiocytosis–a clinicopathological entitywith features of both X- and non-X histiocytosis. Br J Dermatol 134:525–532
Tsai JW, Tsou JH, Hung LY et al (2010) Combined Erdheim-Chester disease and Langerhans cellhistiocytosis of skin are both monoclonal: a rare case with human androgen-receptor gene analysis.J Am Acad Dermatol 63:284–291
Volpicelli ER, Doyle L, Annes JP et al (2011) Erdheim-Chester disease presenting with cutaneousinvolvement: a case report and literature review. J Cutan Pathol 38:280–285
Wang KH, Chen WY, Liu HN et al (2006) Cutaneous Rosai-Dorfman disease: clinicopathologicalprofiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol 154:277–286
Wood GS, Hu CH, Beckstead JH et al (1985) The indeterminate cell proliferative disorder: report of a casemanifesting as an unusual cutaneous histiocytosis. J Dermatol Surg Oncol 11:1111–1119
Histopathologie der HautDOI 10.1007/978-3-662-44367-5_26-1# Springer-Verlag Berlin Heidelberg 2015
Seite 23 von 23







