
H FOURATI, M BOUGAMRA, L BEN MANSOUR*, H KETATA, K ISSA*, E DAOUD, M HACHICHA*, Z MNIF - SFAX - TUNISIE,
*Service de pédiatrie, CHU Hédi Chaker
Service de Radiologie, CHU Habib Bourguiba
Service de Radiologie, CHU Hédi Chaker
Sfax Tunisie


Les malformations broncho-pulmonaires (MBP) résultent d’accidents de développement du système broncho-pulmonaire.
Ce sont des affections rares et polymorphes.
Bien que largement décrites en littérature, il n’existe pas une classification étiologique des mécanismes pathogéniques universellement admise.
Elles constituent une urgence diagnostique et thérapeutique devant le risque de détresse respiratoire et d’infection incitant à mieux connaître cette pathologie.

Les malformations broncho-pulmonaires de l’enfant
comprennent :
L’emphysème lobaire congénital
La malformation adénomatoïde kystique
La séquestration pulmonaire
Le kyste bronchogénique
L’agénésie, l’aplasie et l’hypoplasie pulmonaires
L’étude des MBP a largement bénéficié des
différents moyens d’imagerie particulièrement
anténatale dont l’échographie et l’IRM fœtale.


Etude rétrospective réalisée entre 1989 et 2011 de 23 observations: 9 filles et 14 garçons dont l’âge varie entre j0 de vie et 5 ans
Une radiographie de thorax a été réalisée dans tous les cas;
Une tomodensitométrie thoracique était réalisée chez 20 patients;
L’échographie et/ou doppler a été réalisée pour 3 enfants (dont deux échographies anténatales)
La scintigraphie pulmonaire dans 3 cas.


Les patients de notre série étaient répartis
comme suit:
Emphysème lobaire géant: n=12
Malformation adénomatoïde kystique: n=2
Agénésie ou hypoplasie pulmonaire : n=5
Séquestration pulmonaire n=3
Kyste bronchogénique n=1


Les malformations broncho-pulmonaires sont des affections rares.
Il s’agit d’accidents de développement sans facteur génétique connu hormis quelques exceptions.
Elles peuvent être mises en évidence par le dépistage anténatal.
Ailleurs, elles peuvent être: asymptomatiques découvertes à l’occasion d’un cliché
standard systématique ou fait pour une pathologie intercurrente ou
symptomatiques.
Elles peuvent être évoqués sur des radiographies standards, mais leur diagnostic précis requiert souvent des examens plus invasifs et parfois le diagnostic n’est retenu qu’après la chirurgie.


L’emphysème lobaire géant dit aussi congénital (ELG) est caractérisé par une hyperinflation du parenchyme pulmonaire d’un lobe, consécutive à une obstruction en amont.
Son caractère néonatal, la fréquence d’une malformation associée, le font qualifier de
« congénital ».
Il est Considéré comme l’aboutissement d’anomalies de développement pulmonaire responsable d’«air trapping»

Les causes de l’obstruction sont variables:
une immaturité du cartilage bronchique (67%
des cas);
une obstruction intrinsèque: bouchon
muqueux, une atrésie bronchique ou un repli
muqueux;
une obstruction extrinsèque: arc vasculaire ou
masse médiastinale;
Aucune cause apparente n’est retrouvée dans
40% des cas.

L’ELG est rare et représente 3 à 15% des malformations pulmonaires.
Une prédominance masculine est généralement retrouvée.
Quelques cas familiaux d’ELG ont été décrits bien que le facteur héréditaire n’est pas retenu.
Le diagnostic anténatal est rarement fait

Cette malformation se révèle habituellement dans les premiers jours ou mois de vie.
En effet, les manifestations cliniques apparaissent à la naissance dans 33 % des cas et avant l’âge d’un mois dans 50 % des cas.
La dyspnée est le signe clinique le plus fréquent, elle est souvent progressive, évoluant dans un contexte d’apyrexie faisant penser à une origine malformative.

La radiographie met en évidence une distension progressive d’un lobe avec compression et déplacement des autres lobes entraînant une détresse respiratoire.
Permet de poser le diagnostic d’ELG et précise la topographie du lobe atteint.
Elle montre une:
une hyperclarté homogène avec diminution de la vascularisation pulmonaire,
une hernie pulmonaire transmédiastinale controlatérale
une distension de l’hémithorax avec élargissement des espaces intercostaux, abaissement de la coupole diaphragmatique homolatérale et déviation du médiastin vers le côté controlatéral

Plus sensible que la radiographie pour démontrer la distension et l’aspect caractéristique des structures vasculaires étirés et grêles en cas d’ELG.
Elle permet de mieux localiser l’emphysème, de
chercher une éventuelle étiologie et de le distinguer de l’emphysème compensateur en particulier.
L’aspect de l’emphysème est celui d’une hyperclarté avec une hypovascularisation du lobe atteint, associé à une hernie transmédiastinale et un refoulement du médiastin vers le côté controlatéral.

Bébé Y.A âgé de 6 mois consulte pour une dyspnée progressive
Radiographie du thorax:
Hyperclarté et distension de l’hémi-champ pulmonaire
droit, élargissement des espaces intercostaux
homolatéraux , déviation du médiastin à gauche
TDM thoracique: coupes
axiales (a),
recnstructions
multiplanaires dans les
plans coronal (b) et
sagittal (c): distension du
lobe moyen avec
importante
hypoatténuation,
raréfaction de la
vascularisation en son
sein, hernie trans-
médiastinale antérieure,
déviation du médiastin
a c b

Radiographie de thorax de face
Hyperclarté du champ pulmonaire
gauche, une hernie transmediastinale et
aspect rétracté du champ pulmonaire
controlatéral
Scanner thoracique coupe axiale fenêtre
parenchymateux:
Distension du lobe supérieur gauche avec
aspect grêle et étiré des vaisseaux ,
déviation du médiastin
Bébé âgé de 5 mois consulte pour cyanose, tachycardie et détresse respiratoire.

Scanner initial Scanner de contrôle
Sur le premier scanner: discrète distention du poumon gauche avec déviation
médiastinale vers le côté droit, pas de signe évident d’ELG.
Sur le scanner de contrôle: accentuation de l’hyperinflation du culmen, hypodense
avec une importante raréfaction vasculaire en son sein: aspect évocateur d’ELG.
Observation N°3: Nouveau-né Y.T se présente à la naissance avec une
dyspnée paroxystique, une radio et un scanner thoraciques ont été réalisés,
il ya eu ensuite une aggravation progressive de la symptomatologie clinique
ayant motivé la réalisation d’un 2ème scanner à l’âge de 1 mois.

La scintigraphie pulmonaire de ventilation
permet de visualiser au niveau du lobe
emphysémateux une lacune à l’inspiration et
un foyer radioactif isolé à l’expiration et en fin
du rinçage.
La perfusion est diminuée au niveau du lobe
atteint vu la compression des vaisseaux
sanguins par l’emphysème qui les entoure.

La bronchoscopie a un intérêt étiologique et
thérapeutique mais peut être grave risquant
d’augmenter l’hyperdistension.
Elle permet d’éliminer la présence d’un corps
étranger intra-bronchique, un bouchon
muqueux ou de rechercher une anomalie
bronchique pouvant être responsable de
l’emphysème.


La séquestration pulmonaire est définie comme un territoire non fonctionnel du parenchyme pulmonaire séparé de ses connexions bronchiques et vasculaires normales ; sa vascularisation est assurée par une artère systémique.
Il existe deux formes: la forme intra-lobaire et la forme extra-lobaire.
Elle représente 0,15 % à 1,7% de toutes les anomalies pulmonaires congénitales.
Le segment postéro basal est atteint dans 97 % des cas, elle siège à gauche dans 60 % des cas.
Cliniquement, elle se manifeste le plus souvent par des infections récurrentes dans le même territoire, une toux et expectoration chronique. Par ailleurs, elle peut se révéler par une insuffisance cardiaque néonatale.

Masse hyperéchogène, globalement homogène
ovalaire , bien limitée, inférieure droite, centrée
par une artère nourricière systémique provenant
de l’aorte.
Cliché échographique, coupes longitudinales et
transversale
Radiographie de thorax de face:
opacité basale paracardiaque
droite.
Une échographie morphologique a été réalisée pour une femme enceinte à 22
SA, montrant chez le fœtus une formation hyperéchogène de la gouttière
costovertébrale droite présentant une vascularisation d’origine systémique
visualisée grâce au doppler couleur.
Une échographie thoracique réalisée après la naissance
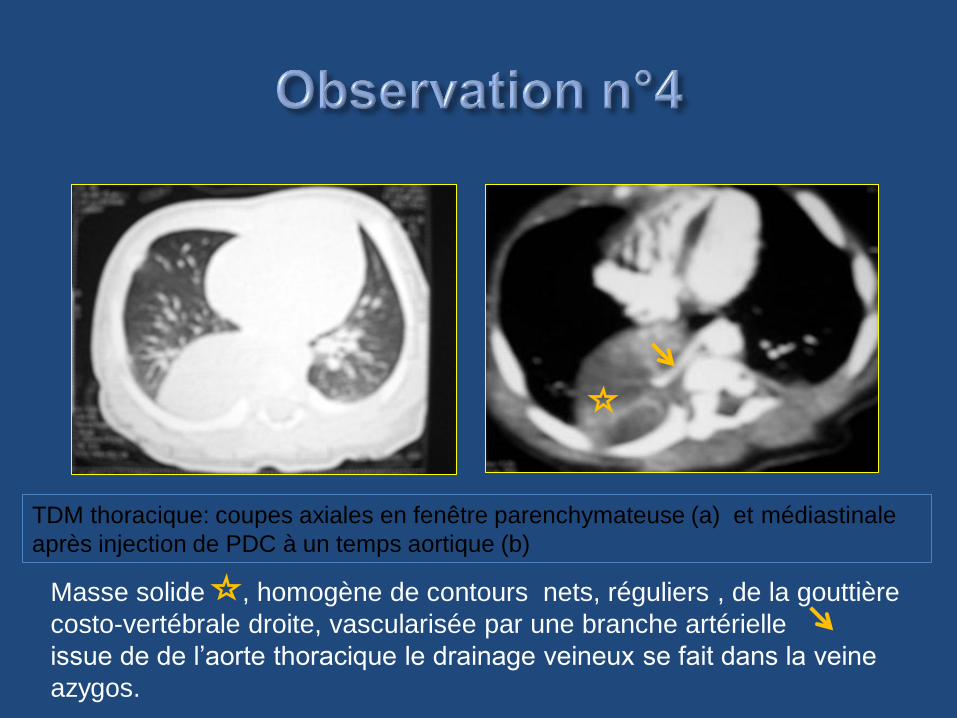
TDM thoracique: coupes axiales en fenêtre parenchymateuse (a) et médiastinale
après injection de PDC à un temps aortique (b)
Masse solide , homogène de contours nets, réguliers , de la gouttière
costo-vertébrale droite, vascularisée par une branche artérielle
issue de de l’aorte thoracique le drainage veineux se fait dans la veine
azygos.
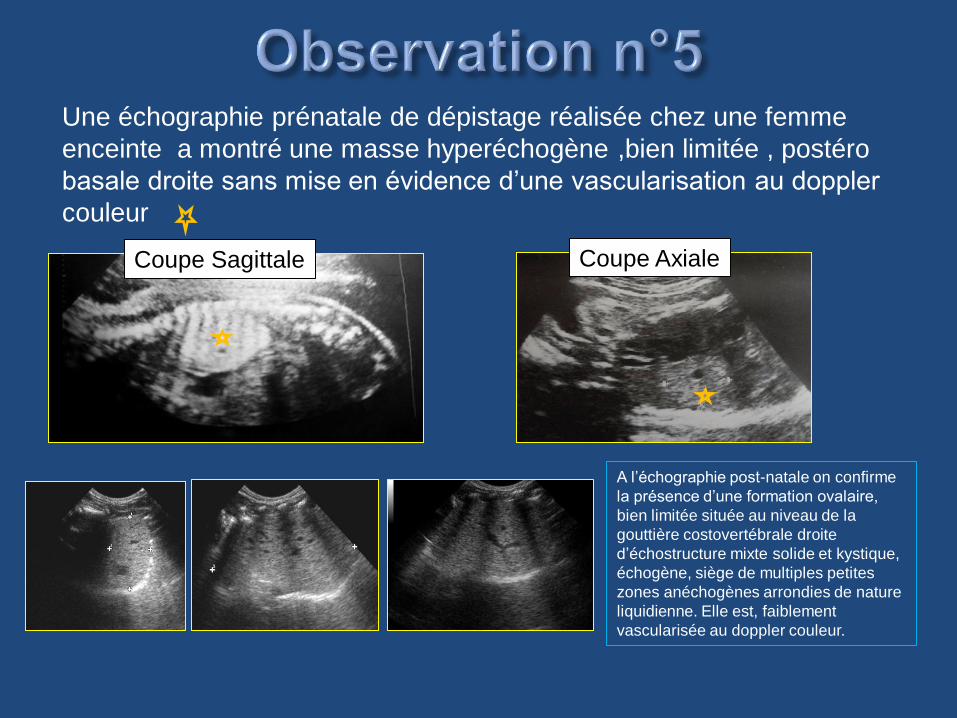
Une échographie prénatale de dépistage réalisée chez une femme
enceinte a montré une masse hyperéchogène ,bien limitée , postéro
basale droite sans mise en évidence d’une vascularisation au doppler
couleur
Coupe Sagittale Coupe Axiale
A l’échographie post-natale on confirme
la présence d’une formation ovalaire,
bien limitée située au niveau de la
gouttière costovertébrale droite
d’échostructure mixte solide et kystique,
échogène, siège de multiples petites
zones anéchogènes arrondies de nature
liquidienne. Elle est, faiblement
vascularisée au doppler couleur.
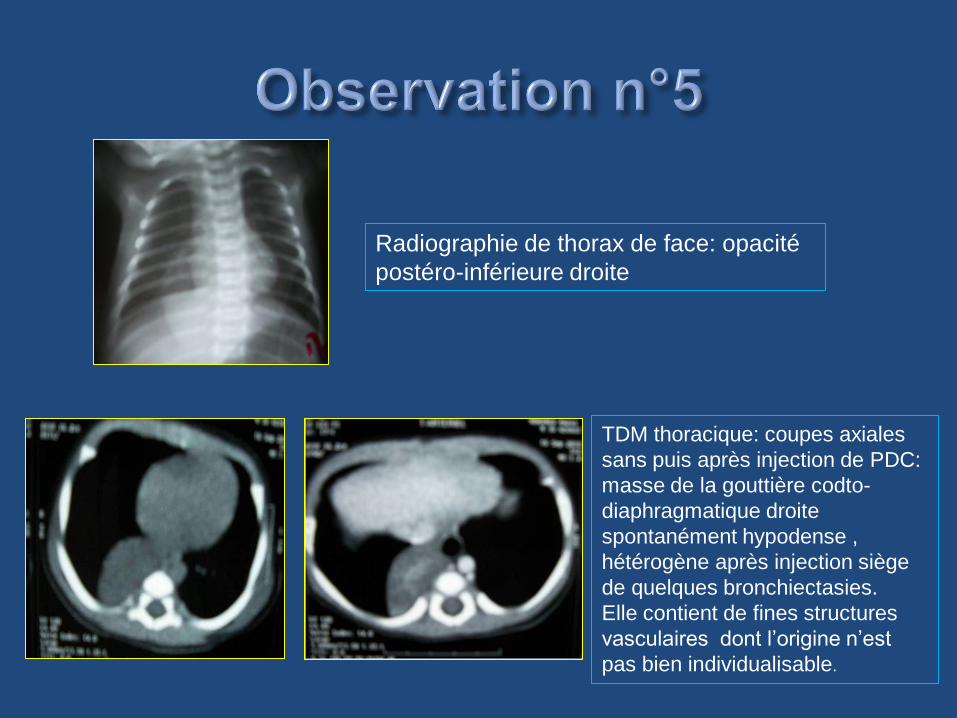
TDM thoracique: coupes axiales
sans puis après injection de PDC:
masse de la gouttière codto-
diaphragmatique droite
spontanément hypodense ,
hétérogène après injection siège
de quelques bronchiectasies.
Elle contient de fines structures
vasculaires dont l’origine n’est
pas bien individualisable.
Radiographie de thorax de face: opacité
postéro-inférieure droite

Le tissu pulmonaire anormal est anatomiquement enfermé dans sa propre plèvre en dehors de la plèvre viscérale.
90% des SEL se situent à gauche.
Elle se présente le plus souvent comme une masse unilatérale en continuité avec l’hémi-diaphragme gauche soit dans le thorax soit dans l’abdomen supérieur.
Quand elle n’est pas associée à une communication avec l’intestin primitif, elle se manifeste rarement par une infection pulmonaire.
Elle peut s’associer à d’autres malformations congénitales: éventration ou paralysie diaphragmatique ipsilatérale, hernie diaphragmatique, hypoplasie pulmonaire ipsilatérale, Malformation adénomatoïde kystique, cardiopathies congénitales.

Elle est partiellement ou totalement supplée par une vascularisation systémique.
Radio thorax: opacité ronde ou triangulaire rétro-cardiaque adjacente à l’hémi-diaphragme gauche.
Moins fréquemment: topographie médiastinale, paravertébrale, rétro-péritonéale.
TDM: lésion homogène, bien circonscrite siège occasionnellement de zones kystiques. L’apport vasculaire systémique est rarement démontré.

Le tissu pulmonaire repose à l‘intérieur de la plèvre viscérale pulmonaire normale.
L’étiologie de ce type de séquestration est encore controversée, certains auteurs la considèrent comme une anomalie acquise et d’autres comme congénitale.
Cette séquestration se manifeste soit précocement par des infections pulmonaires à répétition, occasionnellement par une hémoptysie ou par insuffisance cardiaque secondaire au shunt à travers le segment séquestré
Rarement, elle est découverte fortuitement sur une radiographie thoracique.

Histologie: non spécifique, les zones kystiques peuvent
ressembler à des bronches dilatées, des remaniements
inflammatoires sont fréquemment notés.
Rarement s’associe à d’autres malformations: hernie
diaphragmatique, anomalies squelettiques ou cardiaques.
Radiothorax: opacité para-spinale du lobe inférieur.
TDM: masse tissulaire homogène ou mixte solide et kystique.
Le scanner montre fréquemment la vascularisation systémique.


La malformation adénomatoïde kystique congénitale (MAKC) est due à l’hyperplasie des structures terminales bronchiolaires qui prennent un aspect pseudo glandulaires.
C’est une malformation hamartomateuse segmentaire habituellement unilatérale et secondaire à un arrêt de maturation des voies de conduction au niveau des bronchioles sans formation de tissu alvéolaire.
Sa vascularisation est d’origine pulmonaire.
Elle représente 25 % des malformations broncho-pulmonaires
C’est la malformation broncho-pulmonaire la plus fréquemment diagnostiquée en anténatal.

Elle constitue des lésions sporadiques non
héréditaires ou peut s’associer à des
syndromes génétiques comme la trisomie 18
ou la dysplasie rénale héréditaire.
Elle est actuellement, de plus en plus
fréquemment rapportée dans la littérature
grâce aux performances de l’échographie
prénatale avec un diagnostic anténatal qui se
fait dès la 16ème semaine de grossesse.

Il existe une prédominance masculine.
Dans 80 à 95 % des cas, la MAK ne touche
qu’un lobe, avec un certain tropisme pour le
poumon droit.
L’extension à plusieurs lobes peut se
rencontrer.
L’atteinte bilatérale est extrêmement rare.

Chez la plupart des patients, la MAKC est
suspectée sur des critères cliniques et des
images radiologiques. Dans de rares cas, elle
demeure asymptomatique et va être de
découverte radiologique fortuite.
Le diagnostic différentiel radiologique est celui
d’une hyperclarté circonscrite : hernie
diaphragmatique, emphysème lobaire géant,
kyste bronchogénique ou dysembryonnaire,
pneumatocèle post-traumatique ou infectieuse.

La tomodensitométrie thoracique révèle de multiples kystes aériques connectés aux bronches et bien démarquées du parenchyme pulmonaire normal.
L’angioscanner est parfois utile dans le diagnostic différentiel permettant de rechercher une vascularisation systémique qui serait en faveur d’une séquestration pulmonaire.
L’imagerie par résonance magnétique a peu d’intérêt dans cette pathologie.
Seule l’étude histologique confirme le diagnostic et élimine les autres malformations kystiques pulmonaires.

Une classification (Stocker) selon la taille des kystes selon
des données histologiques et cliniques a été proposée et
actuellement la plus employée :
Type I : multiples kystes de diamètre supérieur à 2 cm ou kyste
volumineux unique avec des petits kystes qui l’entourent
contenant de l’air et/ou du liquide. Ce type représente 50 % des
cas et possède le meilleur pronostic.
Type II : multiples kystes de moins de 1 cm de taille associés dans
70% des cas à d’autres anomalies congénitales particulièrement
uro-génitales. Il représente 40 % des cas.
Type III : petits kystes de moins de 0,5 cm de diamètre se présentant
comme une masse d’aspect solide. Il représente 10% des cas et il
est de plus mauvais pronostic

.
Diagramme des trois types de la malformation adénomatoïde
kystique:
Type I: larges kystes de taille variable.
Type II: Kystes de petite taille.
Type III: Structures pseudo-glandulaires
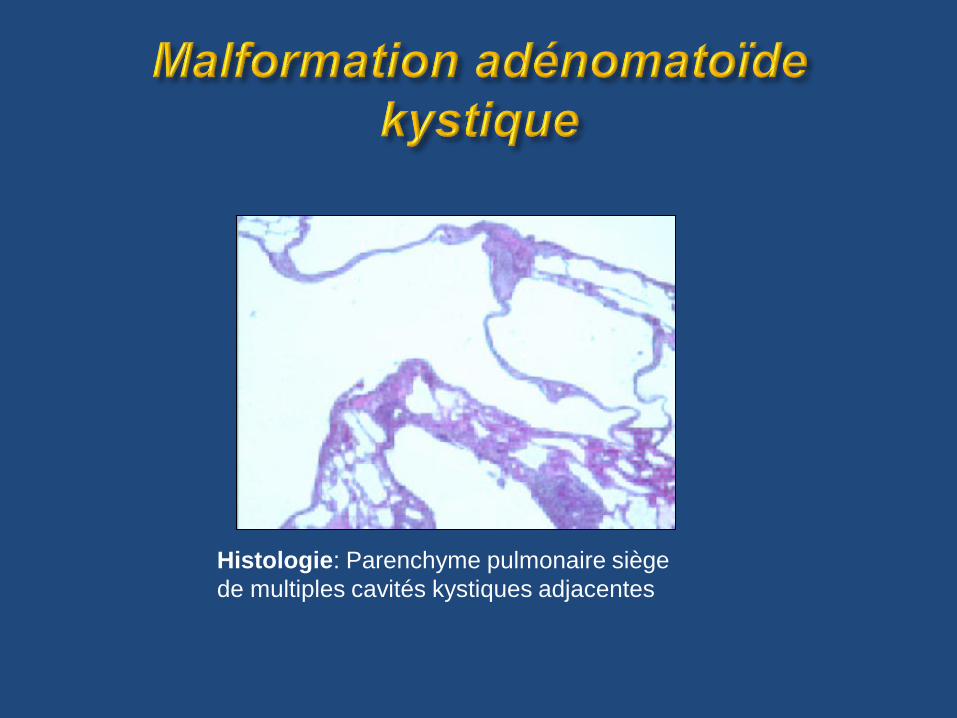
Histologie: Parenchyme pulmonaire siège
de multiples cavités kystiques adjacentes

En général, les MAKC sont isolées.
Cependant, elles s’associent à d’autres
malformations dans 18 % des cas, soit
broncho-pulmonaires (séquestration
pulmonaire, bronche trachéale, hypoplasie
pulmonaire), soit digestives (hernie
diaphragmatique), soit rénales (polykystose,
dysgénésie), soit cardiaque ou autre.

La malformation adénomatoïde kystique est
une malformation pulmonaire nécessitant un
traitement chirurgical. Cette malformation est
rarement révélée par un pneumothorax
spontané.
Les données tomodensitométriques sont bien
corrélées avec les lésions histologiques.
Le pronostic peut être réservé nécessitant une
chirurgie urgente.
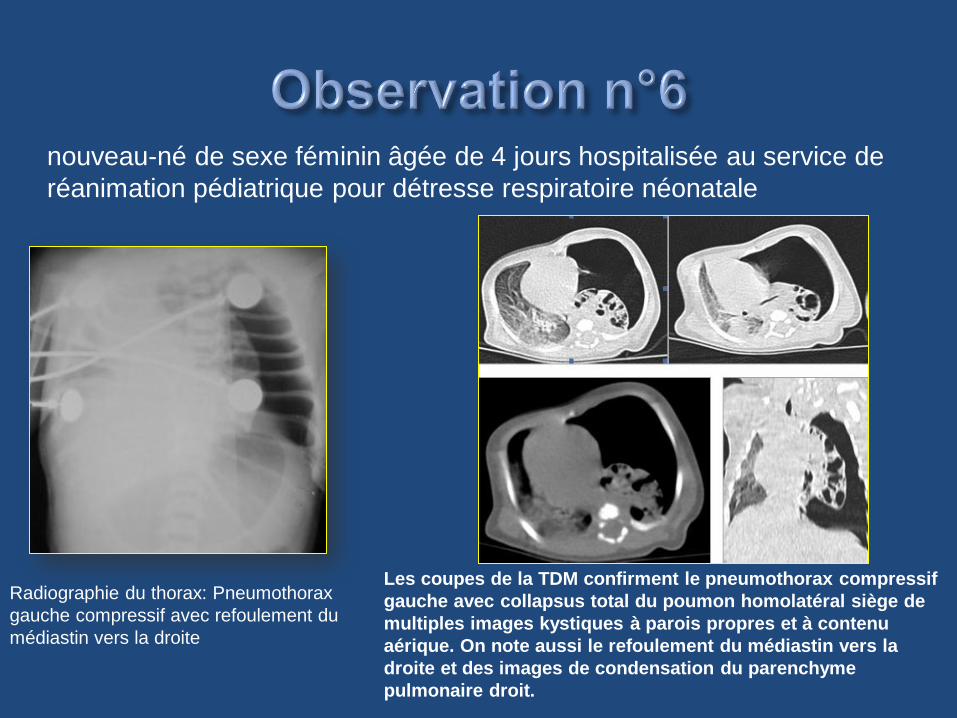
Radiographie du thorax: Pneumothorax
gauche compressif avec refoulement du
médiastin vers la droite
Les coupes de la TDM confirment le pneumothorax compressif
gauche avec collapsus total du poumon homolatéral siège de
multiples images kystiques à parois propres et à contenu
aérique. On note aussi le refoulement du médiastin vers la
droite et des images de condensation du parenchyme
pulmonaire droit.
nouveau-né de sexe féminin âgée de 4 jours hospitalisée au service de
réanimation pédiatrique pour détresse respiratoire néonatale


L’agénésie est définie par une absence
complète du parenchyme pulmonaire, de sa
vascularisation et de tout tissu en aval de la
bifurcation trachéale.
L’aplasie est similaire, mais s’en distingue par
la présence d’un bronche souche rudimentaire
et borgne.
Une agénésie ou aplasie bilatérale est
extrêmement rare et létale.

Cliniquement, une agénésie pulmonaire unilatérale est souvent inapparente, découverte à l’occasion d’exploration d’autres anomalies associées.
Les cas d’agénésie pulmonaire sont également répartis entre le côté droit et le côté gauche, cependant le pronostic est meilleur dans les agénésies gauches.
L’agénésie droite peut être associée à une fistule oeso-trachéale.
Des malformations associées des vertèbres, des membres, cardio-vasculaires, gastro-intestinales ou génito-urinaires sont fréquentes.

L’agénésie droite peut être associée à une
fistule oeso-trachéale.
Des malformations associées des vertèbres,
des membres, cardio-vasculaires, gastro-
intestinales ou génito-urinaires sont fréquentes.
La prise en charge est basée sur la
surveillance et la préservation du poumon
restant.

Radiographie thoracique: Déviation du cœur et des structures médiastinales vers
le côté atteint complètement opaque et rétracté.
Une partie du poumon contro-latéral distendu est souvent herniée de l'autre côté de la ligne médiane. Ceci peut rendre le diagnostic difficile.
Sur le cliché standard la clé du diagnostic est l'absence de bronche souche du côté atteint, sur les clichés de médiastin filtré par un examen attentif de la région carinaire.

Angio-scanner thoracique:
Peut éliminer une compression vasculaire des
bronches proximales
Peut montrer une artère pulmonaire
rudimentaire ou absente
Peut différencier l’aplasie de l’agénésie
pulmonaire

Nouveau-né M.A se présente pour détresse respiratoire néonatale.
Radiographie thoracique:
Hémithorax droit opaque et rétracté. Pincement des
espaces intercostaux à droite.
Déviation du médiastin à droite (sonde naso-gastrique
déviée)
Hernie parenchymateuse trans-médiastinale.
Une malformation costale est associée
Le diagnostic a été confirmé par un angioscanner.

Observation n°8
Radiographie de thorax de face
Rétraction de l’hémichamps pulmonaire
droit, déviation médiastinale vers le côté
droit, hyperinflation du poumon controlatéral
Nourrisson de 5 mois consulte pour dyspnée progressive
Le cathétérisme cardiaque a
révélé une agénésie de l’artère
pulmonaire droite

Hypoplasie pulmonaire

Elle se définit comme une diminution du nombre des bronches
périphériques et des acini.
Elle peut être uni ou bilatérale, et elle est souvent associée à
d'autres anomalies comme l'agénésie rénale, la hernie
diaphragmatique.
Elle peut parfois être acquise à la suite d'infections pulmonaires.
L'artère pulmonaire du côté atteint est habituellement petite.
Dans l'hypoplasie pulmonaire l'hémi-thorax affecté est en général
petit, il y a un déplacement du médiastin du côté atteint.
les opacités hilaires sont petites et le poumon est hyperclair.
Observation 9: Nourisson, découverte fortuite
sur une radiographie thoracique d’une asymétrie
entre les deux champs pulmonaires.
Scanner: confirmation d’une hypoplasie du
poumon droit.


Les kystes bronchogéniques peuvent être considérés comme des dérivés de bourgeons anormaux issus de l'oesophage primitif ou de l'ébauche trachéobronchique et n'ayant pas abouti à la différenciation alvéolaire.
Le kyste bronchogénique résulte d'un bourgeon aberrant qui se détache de l'ébauche trachéobronchique à un moment précoce de son évolution pour les kystes médiastinaux et tardifs pour les kystes intraparenchymateux.

Les kystes bronchogéniques ont une paroi de type bronchique, sécrétante, ce qui explique le contenu habituellement liquidien.
Les kystes bronchogéniques ne sont pas associés à des malformations vertébrales ce qui les oppose aux kystes médiastinaux à revêtement digestif (kyste œsophagien ou kyste neuro-entérique).
Le diagnostic exact ne peut être qu'anatomo-pathologique.

En anténatal, l'échographie montre une image
kystique bien limitée, de taille variable, en
général uniloculaire, à contenu anéchogène ou
finement échogène. Parfois le contenu
mucoïde peut être échogène pouvant faire
discuter une formation solide. L'IRM permet de
préciser la lésion.
Ces kystes peuvent être compressifs mais
l'évolution est bonne en général.

A la naissance, ils peuvent être asymptomatiques, se surinfecter ou peuvent entraîner une compression médiastinale.
Le bilan radiologique post natal est fonction de l'état clinique de l'enfant: la radiographie pulmonaire, l'échographie et le transit oesophagien sont les examens de base. La tomodensitométrie ou l'IRM offrent une analyse sémiologique plus fine.
Habituellement : ils se présntent comme des formations liquidiennes homogènes arrondies ou ovalaires rarement polylobées. Il peut apparaître des niveaux hydro-aériques soit par communication congénitale avec les bronches ou acquise lors d'une surinfection.
La paroi du kyste peut être calcifiée au cours de complications inflammatoires.
Le contenu du kyste est variable expliquant que les densités analysées en tomodensitométrie varient de 0 à 120 unités Hounsfield.

L'échographie est une méthode de choix pour affirmer le caractère
liquidien de la masse.
Les KB sont de plus en plus diagnostiqués en anténatal lors de
l’échographie prénatale de dépistage.
Observation 10: Radiographie thoracique (cliché
de profil) chez un enfant présentant des infections
broncho-pulmonaires intercurrentes: Image
arrondie siège d’un niveau hydro-aérique.
Le diagnostic d’abcès a été évoqué. Stabilité de
la masse sous antibiothérapie
Diagnostic post opératoire: kyste
bronchogénique.

Les malformations pulmonaires sont fréquentes et très variées.
Elles sont souvent bien expliquées par un trouble de l'organogénèse. Un certain nombre d’entre elles peuvent engager le pronostic vital d’où la nécessité d’en connaître l’existence
L’exploration radiologique des malformations pulmonaires à largement bénéficié de l’apport des techniques d’imagerie particulièrement en prénatal dont l’échographie obstétricale et l’IRM fœtale.
Bien que le diagnostic prénatal précis n’est pas toujours possible, l’évaluation et la prise en charge sont cmmunes à la plupart des lésions

Le diagnostic prénatal permet surtout d’évaluer
le caractère isolé ou non de la malformation,
son pronostic, de surveiller l’évolution et
d’optimiser la prise en charge péri-natale.
L’apport de la radiographie et de la TDM
thoracique en post-natal est également
important pour préciser, confirmer et parfois
redresser certains diagnostics.

TDM thoracique:
coupes axiales en
fenêtre médiastinale
et parenchymateuse
Enfant M. B âgée de 5 mois est explorée pour infections broncho-
pulmonaires à répétition
Radiographie de thorax de face
en position couchée

A la radiographie thoracique:
A. il existe un pneumothorax droit.
B. Masse kystique occupant l’hémi-champ
pulmonaire droit.
C. il y a une déviation du médiastin vers le côté
gauche.
D. on retrouve un foyer de condensation
parenchymateux à gauche.
C. on retrouve un épanchement pleural de grande
abondance à droite.

Réponses: B-C
Cliché de radiographie de thorax réalisé en
position couchée: formation de tonalité aérique
occupant l’hémichamps pulmonaire droit
avec compression médiastinale vers le côté
gauche

Sur le scanner, on retrouve:
A. Une formation de nature kystique pure.
B. Il existe un niveau-hydro-aérqiue au sein de
cette formation.
C. La formation sus décrite est non compressive.
D. Elle s’étend à la région sous carénaire.
E. Elle présente une paroi.

Sur le scanner, on retrouve:
A. Une formation de nature kystique pure.
B. Il existe un niveau-hydro-aérqiue au sein de
cette formation
C. La formation sus décrite est non compressive.
D. Elle s’étend à la région sous carénaire
E. Elle présente une paroi
Réponses: B. D. E

A. Kyste bronchogénique.
B. Malformation adénomatoïde kystique.
C. duplication oesophagienne.
D. Séquestration pulmonaire.
E. abcès pulmonaire

Quel est votre diagnostic ?
A. Kyste bronchogénique.
B. Malformation adénomatoïde kystique.
C. duplication œsophagienne.
D. Séquestration pulmonaire.
E. abcès pulmonaire
Réponse: A, B, D
L’examen anatomo-pathologique a révélé l’association de ces trois malformations.
Le diagnostic d’abcès pulmonaire a été évoqué sur ces examens parce que la patiente
se présentait pour des infections broncho-pulmonaires intercurrents parfois dans
un contexte fébrile

Affections rares et polymorphes.
Résultats d’accidents de développement du système broncho-
pulmonaire
Urgence diagnostique et thérapeutique: risque de détresse
respiratoire et d’infection.
Apport de l’imagerie en anténatal +++: dépistage, pronostic,
planifier la prise en charge.
La radiographie thoracique permet de poser le diagnostic.
Angio-scanner, scintigraphie, bronchoscopie en post-natal pour
mieux étayer le diagnostic
Le diagnostic définitif anatomopathologique.

[1] Stocker JT : Congenital and developmental diseases. In: Dail DH, Hammar SP. Editors. Pulmonary pathology. Berlin. Springer-Verlag ; 1994 : 155-81. [2] Belkheiri M, Elidrissi F, Bouslamti T, Eddaniaoui M, Banani A, Khabouze S, Aghoutane M, Zerhouni H, Ettayebi F, Kharbach A, Chaoui A : Malformation adénomatoïde kystique congénitale du poumon : diagnostic anténatal à propos d’un cas. Rev Pneumol Clin 2001 ; 57 : 356-60. [3] Oh BJ, Lee JS, Kim JS, Lim CM, Koh Y : Congenital cystic adenomatoid malformation of the lung in adults: clinical and CT evaluation of seven patients. Respirology 2006 ; 11 : 496-501. [4]Bentur L, Canny G, Thorner P, Superina R, Babyn P, Levison H. Sponatneous pneumothorax in cystic adenomatoid malformation. Unusual clinical and histologic features. Chest 1991; 99: 1292-3. [5]Adirim TA, King R, Klein BL. Radiological case of the mounth: congenital cystic adenomatoid malformation of the lung and pulmonary blastoma. Arch Pediatr Adolesc Med 1997; 151: 1053-4. [6] Stocker JT, Madewell JE, Drake RM : Congenital cystic adenomatoid malformation of the lung: classification and morphologic spectrum. Hum Pathol 1977 ; 8 : 155-71.







