
Data processing and analysis of genetic variation using next-
generation sequencing
Mark DePristo Dec. 1st - 2nd, 2010
Manager, Medical and Population Genetics Analysis Broad Institute of MIT and Harvard

Acknowledgments!Genome sequencing and
analysis group (MPG)!Eric Banks (Development lead)!
Ryan Poplin!Guillermo del Angel!Menachem Fromer!
Kiran Garimella (Analysis Lead)!Corin Boyko!Chris Hartl!Matt Hanna!
Khalid Shakir!Aaron McKenna!
Broad postdocs, staff, and faculty!
Anthony Philippakis!Vineeta Agarwala!
Manny Rivas!Jared Maguire!
Carrie Sougnez!David Jaffe!
Nick Patterson!Steve Schaffner!Shamil Sunyaev!Paul de Bakker!
1000 Genomes project!In general but notably:!
Matt Hurles!Philip Awadalla!Richard Durbin!
Goncalo Abecasis!Richard Gibbs!Gabor Marth!
Thomas Keane!Gil McVean!
Gerton Lunter!Heng Li!
Copy number group!Bob Handsaker
Jim Nemesh Josh Korn!
Steve McCarroll !
Cancer genome analysis!
Kristian Cibulskis!Andrey Sivachenko!
Gad Getz!
Integrative Genomics Viewer (IGV)!Jim Robinson!
Helga Thorvaldsdottir!
Genome Sequencing Platform!In general but notably:!
Lauren Ambrogio!Illumina Production Team!
Tim Fennell!Kathleen Tibbetts!
Alec Wysoker!Toby Bloom! MPG directorship!
Stacey Gabriel!David Altshuler!
Mark Daly!

Self-introduction: who am I? • Currently the manager of Medical and
Population Genetics Analysis in MPG – Run Genome Sequencing and Analysis (GSA)
group • Before this I was:
– A business consultant at a local consulting firm – A Damon Runyon fellow doing experimental
bacterial evolution at Harvard – A Marshall Fellow in Biochemistry at Cambridge – Mathematics and Computer Science major at
Northwestern

Outline • Goal: understand how we can turn raw NGS
into reliable information about genetic variation in our samples
• Next-generation DNA sequencing (NGS) machines
• Processing of NGS to discover and genotype – SNPs – Indels – CNVs
• Quality control

SNPs
Indels
Structural variation (SV)
Rawindels
RawSVs
Typically by lane Typically multiple samples simultaneously but can be single sample alone
Input
Output
Mapping
Local realignment
Duplicate marking
Base quality recalibration
Analysis-ready reads
Raw reads Sample 1 reads
Raw variants
RawSNPs
Genotype refinement
Variant quality recalibration
Analysis-ready variants
Pedigrees Known variation
Known genotypes
Population structure
Phase 1: NGS data processing Phase 2: Variant discovery and genotyping Phase 3: Integrative analysis
Sample N reads
External data

NGS TECHNOLOGIES AND TERMINOLOGY

Sequencing Generations
= =
= = Next, Next Generation
??
= + +
454 Illumina SOLiD
ABI 377 ABI 3730
Helicos
Stacey Gabriel

Tota
l Gb
prod
uced
0
10
20
30
40
50
60
70
0
500
1000
1500
2000
2500
3000
2006 2007 2008
Tota
l Gb
prod
uced
0
20000
40000
60000
80000
100000
120000
2008 2009 2010
Tota
l Gb
prod
uced
Illumina
90
454 3730XL
2 14
SOLiD
8
Helicos
1
Prototype Instruments
Production Instruments
Lots of Bases….
5
…2011 = 500,000 Gb
Stacey Gabriel
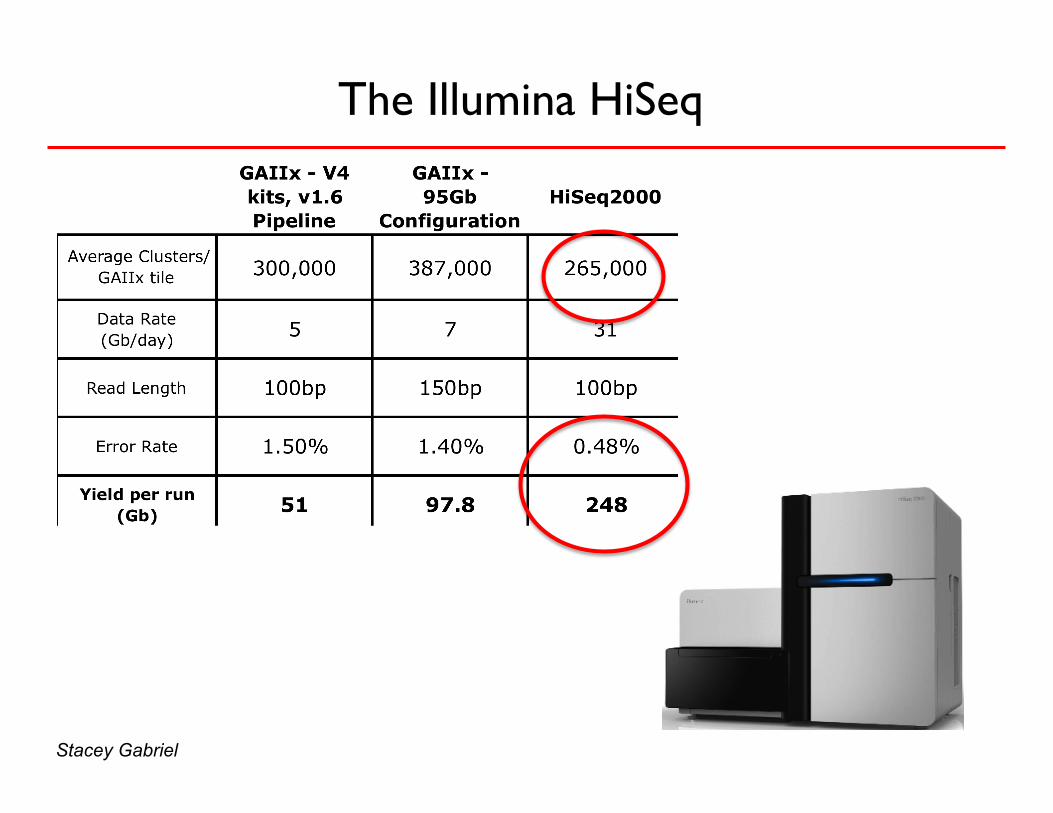
The Illumina HiSeq
Stacey Gabriel

Libraries, lanes, and flowcells
Each reaction produces a unique library of DNA fragments for
sequencing.
Each NGS machine processes a single flowcell containing several independent lanes during a single
sequencing run
Illumina
Lanes
Flowcell

Reads and fragments
Depth of coverage
Individual reads aligned to the genome
Clean C/T heterozygote
Details about specific read
First and second read from the same fragment
Reference genome
Non-reference bases are colored; reference bases are grey
IGV screenshot

EXPERIMENTAL DESIGNS

Experimental parameters
Number of samples
Depth per
sample
Target size
~100 genes
Whole genome
Whole exome
Low-pass multi-sample (1000 Genomes, T2D GO)
Deep single genomes (Cancer)
Exome ARRA projects (ESP, Autism, T2D)
Candidate gene follow-up (FHS, Pfizer)

“Pond” Sheared whole genome library
“Baits” Biotinylated oligo
Sequence
“Catch” Hybrid-selected
targets
Hybridize and capture on bead
Hybrid Selection : an Approach to “Fish” for Exons
Stacey Gabriel

Genotyping vs. pooling Genotyping
• Known relationship between reads and samples
• Analysis algorithms empowered to by using this information – Helps control errors
• E.g., multi-sample SNP calling
Pooling
• Unknown relationship between reads and samples – Samples mixed together
before sequencing
• Complicates analysis algorithms – Harder to control errors – Less reliable output
Sample 1!
Variants!
Sample 2!Samples 1 and 2!
Het Hom Het Rare Rare Common

Shearing (50 uL)
End Repair
A-Base Addition
Adapter Ligation
2.2x SPRI (no transfer)
Modified 2.2x SPRI w/ SPRI Buffer
Modified 1x SPRI w/ SPRI Buffer
Limited Pond PCR
v 4.2
Modified 2.2x SPRI w/ SPRI Buffer
Hybridization
Capture on Bead Direct Catch PCR off Bead
Modified 1x SPRI w/ SPRI Buffer
1 x 50 ul reaction 20 cycles
Sequencing
Input: 3 ug
1 x 50 ul reactions 6 cycles
Blocking Oligos
July
Barcode Addition
Pooling Option 1 After Selection
Pooling Option 2 Before Selection
Indexed Hybrid Selection
DNA fragment
add adapters
PCR with universal oligos
8-base index
SP1
SP2’
P5
P7
P5 P7 SP1
SP2
Barcode
InP
Sheila Fisher

Single vs. multi-sample analysis
Deep single-sample
• Higher sensitivity for variants in the sample
• More accurate genotyping per sample
• Cost: no information about other samples
Shallower multi-sample
Sample 1!
Variants!
Fixed sequencing budget
Found 3 variants total! Found 5 variants total!
Sample 1!
Sample 2!
Sample 3!
Sample 4!
• Sensitivity dependent on frequency of variation
• Worse genotyping • More total variants
discovered

High-pass sequencing design
Exon I! Exon II!Intron I!Inter-genic! Inter-genic!
Targeted bases! ~3 Gb!
Coverage! Avg. 30x!
# sequenced bases! 100 Gb!
# lanes of HiSeq! ~8 lanes!
Variants found per sample! ~3-5M!
Percent of variation in genome! >99%!
Pr{singleton discovery}! >99%!
Pr{common allele discovery}! >99%!
Data requirements per sample! Variant detection among multiple samples!
~30x reads!
Variant site!
Excellent sensitivity for hetero- and homozygotes!
High depth allows excellent genotype calling!
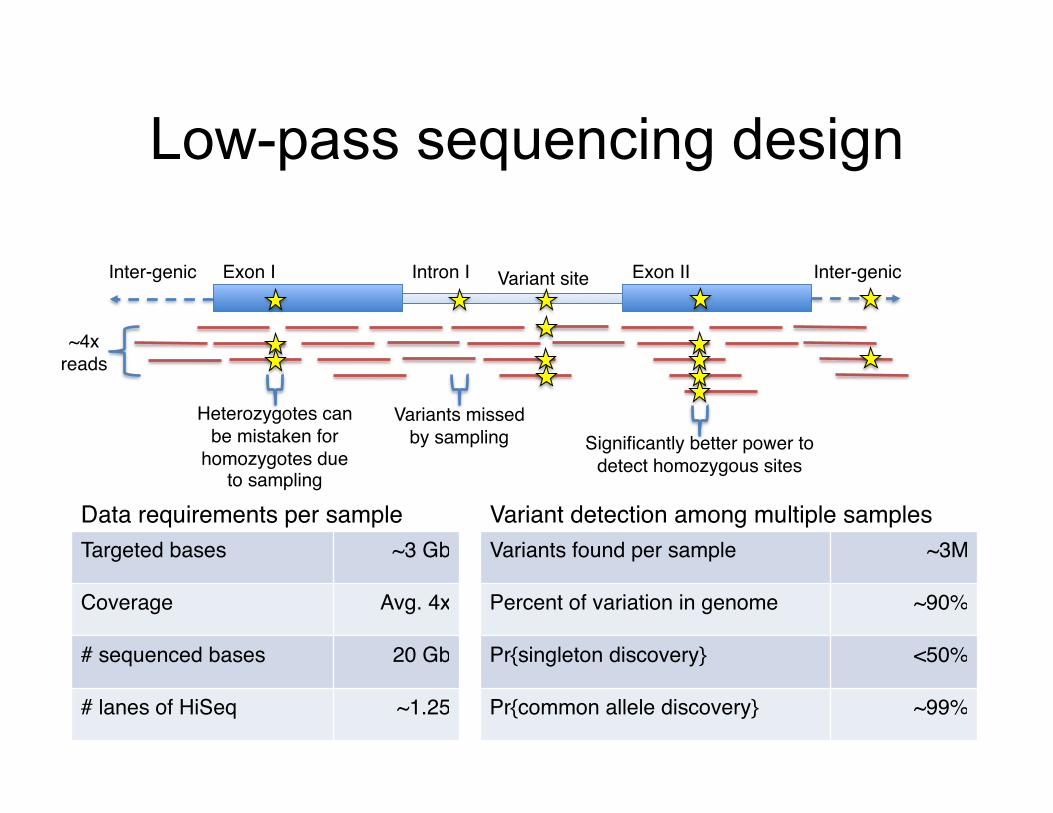
Low-pass sequencing design
Exon I! Exon II!Intron I!Inter-genic! Inter-genic!
Targeted bases! ~3 Gb!
Coverage! Avg. 4x!
# sequenced bases! 20 Gb!
# lanes of HiSeq! ~1.25!
Variants found per sample! ~3M!
Percent of variation in genome! ~90%!
Pr{singleton discovery}! <50%!
Pr{common allele discovery}! ~99%!
Data requirements per sample! Variant detection among multiple samples!
~4x reads!
Variant site!
Variants missed by sampling!
Heterozygotes can be mistaken for
homozygotes due to sampling!
Significantly better power to detect homozygous sites!

Exome capture sequencing design
Exon I! Exon II!Intron I!Inter-genic! Inter-genic!
Targeted bases! ~32Mb!
Coverage! >80% 20x!
# sequenced bases! 5 Gb!
# lanes of HiSeq! ~0.33!
Variants found per sample! ~20K!
Percent of variation in genome! 0.5%!
Pr{singleton discovery}! ~95%!
Pr{common allele discovery}! ~95%!
Data requirements per sample! Variant detection among multiple samples!
150x reads! Little off-target
coverage!
Variant site!

DATA PROCESSING

SNPs
Indels
Structural variation (SV)
Rawindels
RawSVs
Typically by lane Typically multiple samples simultaneously but can be single sample alone
Input
Output
Mapping
Local realignment
Duplicate marking
Base quality recalibration
Analysis-ready reads
Raw reads Sample 1 reads
Raw variants
RawSNPs
Genotype refinement
Variant quality recalibration
Analysis-ready variants
Pedigrees Known variation
Known genotypes
Population structure
Phase 1: NGS data processing Phase 2: Variant discovery and genotyping Phase 3: Integrative analysis
Sample N reads
External data

Commonly used tools • MAQ/BWA
– Mapping (Illumina, 454, SOLID)
• BFAST – Mapping (SOLiD)
• SSAHA – Mapping (454)
• Mosaik – Mapping (Illumina, 454,
SOLID) – Variant calling
• SOAP – Mapping (Illumina) – SNP calling
• IGV – Data visualization
• Samtools – Data processing – Variant calling
• glfTools – SNP calling
• QCALL – SNP calling
• GATK – Data processing – Variant calling – Variant manipulation – Variant QC
• DINDEL – Indel calling

The GATK is a general toolkit for developing NGS data processing algorithms!
Local realignment! SNP calling!Base quality score recalibration!
Variant evaluation!
http://www.broadinstitute.org/gsa/wiki/!
Popular GATK tools!
Indel calling! Phasing and imputation!
Genome Analysis Toolkit (GATK)! SAM/BAM format!
• NGS standard: technology agnostic, binary, indexed, portable and extensible file format for NGS reads!
• Used throughput Broad production pipeline!
http://samtools.sourceforge.net/!
VCF format!• Standard and accessible
format for storing variation and genotypes!
• SNPs, indels, SV!• Many supporters: 1000
Genomes, dbSNP, GAP!
• Open-source map/reduce programming framework for developing analysis tools for next-gen sequencing data!
• Easy-to-use, CPU and memory efficient, automatically parallelizing Java engine!
http://vcftools.sourceforge.net/ !
GATKʼs Queue!
• Simple, distributable pipeline engine!• Underlies MPG-Firehose data processing!

The BAM format facilitates sequencer-agnostic analyses
SLX1:1:127:63:4 … 1 10052169 … 23M6N10M … GAAGATACTGGTTTTTTTCTTATGAGACGGAGT 768832'48::::::;;:/78$88818099897 SM:Z:JPTGBMN01 …!
BAM file!Read name!
Locus!
Alignment gap information!
Read sequence!
Quality scores (fastq format)!
Meta data!
Data processing
and analysis!
BAM file allows us to represent the data of any sequencer. Analyses can then be conducted largely agnostic to the particular sequencer used.!
Bases, quality scores, (optionally) alignments, and meta data!
Kiran Garimella

BAM headers: an optional (but essential) part of a BAM file
@HD VN:1.0 GO:none SO:coordinate @SQ SN:chrM LN:16571 @SQ SN:chr1 LN:247249719 @SQ SN:chr2 LN:242951149 [cut for clarity] @SQ SN:chr9 LN:140273252 @SQ SN:chr10 LN:135374737 @SQ SN:chr11 LN:134452384 [cut for clarity] @SQ SN:chr22 LN:49691432 @SQ SN:chrX LN:154913754 @SQ SN:chrY LN:57772954 @RG ID:20FUK.1 PL:illumina PU:20FUKAAXX100202.1 LB:Solexa-18483 SM:NA12878 CN:BI @RG ID:20FUK.2 PL:illumina PU:20FUKAAXX100202.2 LB:Solexa-18484 SM:NA12878 CN:BI @RG ID:20FUK.3 PL:illumina PU:20FUKAAXX100202.3 LB:Solexa-18483 SM:NA12878 CN:BI @RG ID:20FUK.4 PL:illumina PU:20FUKAAXX100202.4 LB:Solexa-18484 SM:NA12878 CN:BI @RG ID:20FUK.5 PL:illumina PU:20FUKAAXX100202.5 LB:Solexa-18483 SM:NA12878 CN:BI @RG ID:20FUK.6 PL:illumina PU:20FUKAAXX100202.6 LB:Solexa-18484 SM:NA12878 CN:BI @RG ID:20FUK.7 PL:illumina PU:20FUKAAXX100202.7 LB:Solexa-18483 SM:NA12878 CN:BI @RG ID:20FUK.8 PL:illumina PU:20FUKAAXX100202.8 LB:Solexa-18484 SM:NA12878 CN:BI @PG ID:BWA VN:0.5.7 CL:tk @PG ID:GATK TableRecalibration VN:1.0.2864 20FUKAAXX100202:1:1:12730:189900 163 chrM 1 60 101M = 282 381
GATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTA…[more bases] ?BA@A>BBBBACBBAC@BBCBBCBC@BC@CAC@:BBCBBCACAACBABCBCCAB…[more quals] RG:Z:20FUK.1 NM:i:1 SM:i:37 AM:i:37 MD:Z:72G28 MQ:i:60 PG:Z:BWA UQ:i:33
Required: Standard header
Essential: contigs of aligned reference
sequence. Should be in karotypic order.
Essential: read groups. Carries platform (PL), library
(LB), and sample (SM) information. Each read is
associated with a read group
Useful: Data processing tools applied to the reads

Finding the true origin of each read is a computationally demanding first step
Mapping!
GATK local realignment!
Duplicate marking!
Raw reads!
Analysis-ready reads!
Input!
Output!
Phase 1: Picard!NGS data processing!
GATK base quality recal!
----- By lane/sample ----!
Region 1
Enormous pile of short reads from
NGS
Detects correct read origin and flags them
with high certainty
Detects ambiguity in the origin of reads and
flags them as uncertain
Reference genome
Region 2 Region 3
For more information see:
Li and Homer (2010). A survey of sequence alignment algorithms for next-generation sequencing. Briefings in Bioinformatics.
Mapping and alignment algorithms

rs28782535!
rs28783181! rs28788974! rs34877486! rs28788974!
1,000 Genomes Pilot 2 data, raw MAQ alignments! 1,000 Genomes Pilot 2 data, after MSA!
HiSeq data, raw BWA alignments! HiSeq data, after MSA!
Effect of MSA on alignments!NA12878, chr1:1,510,530-1,510,589!
Dealing with indels: local realignment and per-base-alignment qualities (BAQ)
28
Mapping!
GATK local realignment!
Duplicate marking!
Raw reads!
Analysis-ready reads!
Input!
Output!
Phase 1: Picard!NGS data processing!
GATK base quality recal!
----- By lane/sample ----!
DePristo, M., Banks, E., Poplin, R. et. al, A framework for variation discovery and genotyping using next-generation DNA sequencing data. Manuscript in review.!
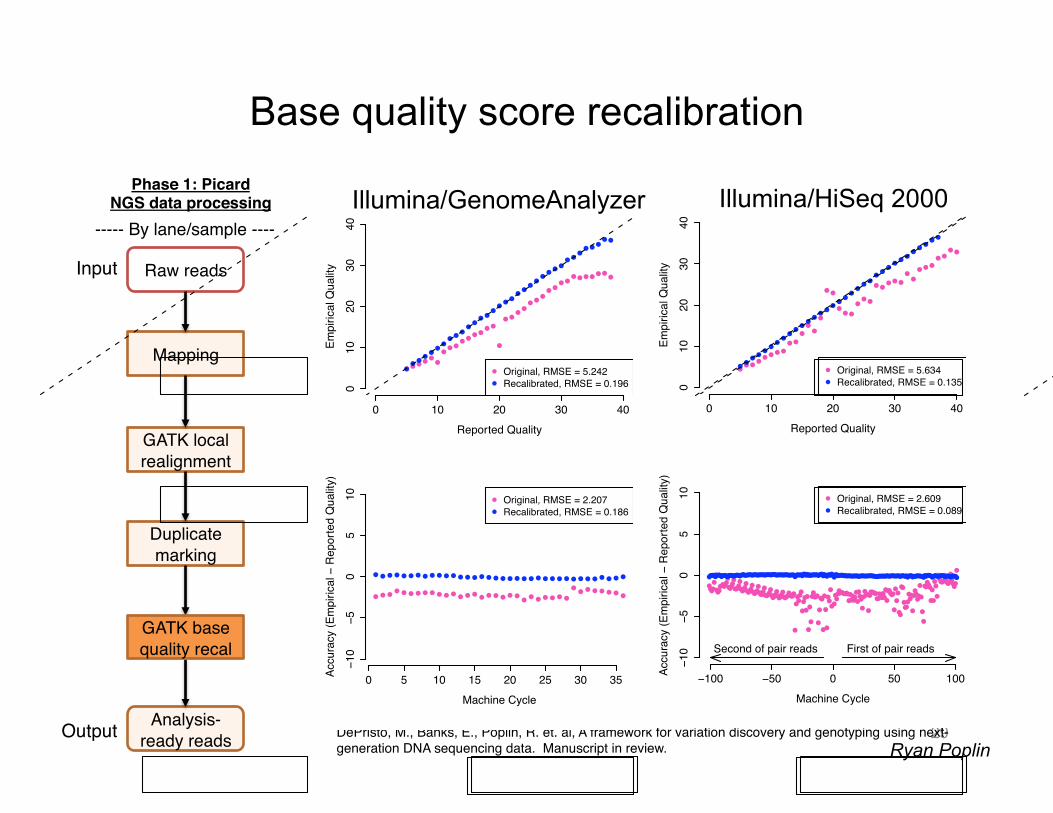
Base quality score recalibration
29
Mapping!
GATK local realignment!
Duplicate marking!
Raw reads!
Analysis-ready reads!
Input!
Output!
Phase 1: Picard!NGS data processing!
GATK base quality recal!
----- By lane/sample ----!
DePristo, M., Banks, E., Poplin, R. et. al, A framework for variation discovery and genotyping using next-generation DNA sequencing data. Manuscript in review.!
!!!!!!
!!!
!!!!
!!
!
!
!!
!!
!!
!!
!!!!!! !
!!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!!!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!!
!
!
Original, RMSE = 5.242Recalibrated, RMSE = 0.196
!!
!!
!!!
!!
!!
!!
!!
!
!!!!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!
!
!
Original, RMSE = 2.556Recalibrated, RMSE = 0.213 !!!
!
!
!!!
!!!
!!
!!
!!
!!!!
!!
!!
!
!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!!!!!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!!!
!!!!
!
!
Original, RMSE = 1.215Recalibrated, RMSE = 0.756
!!!
!!!!!
!!
! !!
!
!
!
!
!!
!!
!
!
!!
!!
!!!!
!!
!!
!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!!!!!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!
!
Original, RMSE = 5.634Recalibrated, RMSE = 0.135
!!!!!!!!!!!!!!! !! !! !! !
! !! !!
!
!
!! !!!!!
0 5 10 15 20 25 30 35
!1
0!
50
51
0
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!!!!!! !! !! !! !! !! !! !! !! !!!! !
!
!
Original, RMSE = 2.207Recalibrated, RMSE = 0.186
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!!
!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!
!!!
!!!!!!!! !!!!!!!! !!!!!!!! !!
!!!!!! !!!!!!!!
!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!! !!!! !!!
!!!!! !!!!
!!!!!!
!!!!!!!!!!
!!
!
!
!
!!
!!
!!
0 50 100 150 200
!1
0!
50
51
0
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!! !!!! !!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!!!!!!!!!! !!!!!!!!!!!!!!!! !!!! !!!!!!!! !!!! !!!!!!!!!!!!!!!!
!!!
!!!!!
!
!
Original, RMSE = 1.784Recalibrated, RMSE = 0.136
!!
!
!
!
!
!
!
!
!
!!
!
!!!
!!
! !!!
! !!!
!
!!
!!
!!
!!! !!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!!!!!
!30 !20 !10 0 10 20 30
!1
0!
50
51
0
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!! !!! !! !! !!!! !! !! !! !! !! !! !! !! !! !! !! !! !! !! !! !
! !! !! ! !! !!!!!
! !! ! !!!!!
Second of pair reads First of pair reads
!
!
Original, RMSE = 1.688Recalibrated, RMSE = 0.213
!
!!!!!!!!!!!
!!!!!!!!
!!!!
!
!!
!
!!!!!!!!!!!!
!
!!!!!
! !
!
!!
!!
!!
!!
!!
!!!!
!!!
!
!
!
!!
!!!!
!
!
!
!
!
!!!
!
!
!
!
!!!!!!!! !
!
!!
!!
!
!!!
!!
!
!!!!
!!
!
!
!
!!!!
!
!
!!
!
!
!
!
!!
!
!
!!!! !!!! !!!
!
!!!! !
!!!!!!!!!!!!!!!
!
!!!!
!
!!!!
!!!!!
! !!!!!!!!
!!!!
!!!!
!!
!!!
!100 !50 0 50 100!
10
!5
05
10
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!! !!!! !!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!! !!!! !!!!!!!!!!!!!!!!!!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!!!!!!!
Second of pair reads First of pair reads
!
!
Original, RMSE = 2.609Recalibrated, RMSE = 0.089
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!
!!
!
!!!!!!!!!
!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 2.598Recalibrated, RMSE = 0.052
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!
!!!!!
!
!!!!!!!
!!
!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 2.169Recalibrated, RMSE = 0.135
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!
!!!!
!!!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 1.656Recalibrated, RMSE = 0.088
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!
!!
!!!!
!!!!!
!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 2.469Recalibrated, RMSE = 0.083
Illumina/GenomeAnalyzer Roche/454 Life/SOLiD Illumina/HiSeq 2000
!!!!!!
!!!
!!!!
!!
!
!
!!
!!
!!
!!
!!!!!! !
!!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!!!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!!
!
!
Original, RMSE = 5.242Recalibrated, RMSE = 0.196
!!
!!
!!!
!!
!!
!!
!!
!
!!!!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!
!
!
Original, RMSE = 2.556Recalibrated, RMSE = 0.213 !!!
!
!
!!!
!!!
!!
!!
!!
!!!!
!!
!!
!
!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!!!!!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!!!
!!!!
!
!
Original, RMSE = 1.215Recalibrated, RMSE = 0.756
!!!
!!!!!
!!
! !!
!
!
!
!
!!
!!
!
!
!!
!!
!!!!
!!
!!
!
0 10 20 30 40
01
02
03
04
0
Reported Quality
Em
pir
ica
l Qu
alit
y
!!!!!!!!!!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!!
!
!
Original, RMSE = 5.634Recalibrated, RMSE = 0.135
!!!!!!!!!!!!!!! !! !! !! !
! !! !!
!
!
!! !!!!!
0 5 10 15 20 25 30 35
!1
0!
50
51
0
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!!!!!! !! !! !! !! !! !! !! !! !!!! !
!
!
Original, RMSE = 2.207Recalibrated, RMSE = 0.186
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!!
!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!
!!!
!!!!!!!! !!!!!!!! !!!!!!!! !!
!!!!!! !!!!!!!!
!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!! !!!! !!!
!!!!! !!!!
!!!!!!
!!!!!!!!!!
!!
!
!
!
!!
!!
!!
0 50 100 150 200!
10
!5
05
10
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!! !!!! !!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!!!!!!!!!! !!!!!!!!!!!!!!!! !!!! !!!!!!!! !!!! !!!!!!!!!!!!!!!!
!!!
!!!!!
!
!
Original, RMSE = 1.784Recalibrated, RMSE = 0.136
!!
!
!
!
!
!
!
!
!
!!
!
!!!
!!
! !!!
! !!!
!
!!
!!
!!
!!! !!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!
!!!!!
!30 !20 !10 0 10 20 30
!1
0!
50
51
0
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!! !!! !! !! !!!! !! !! !! !! !! !! !! !! !! !! !! !! !! !! !! !
! !! !! ! !! !!!!!
! !! ! !!!!!
Second of pair reads First of pair reads
!
!
Original, RMSE = 1.688Recalibrated, RMSE = 0.213
!
!!!!!!!!!!!
!!!!!!!!
!!!!
!
!!
!
!!!!!!!!!!!!
!
!!!!!
! !
!
!!
!!
!!
!!
!!
!!!!
!!!
!
!
!
!!
!!!!
!
!
!
!
!
!!!
!
!
!
!
!!!!!!!! !
!
!!
!!
!
!!!
!!
!
!!!!
!!
!
!
!
!!!!
!
!
!!
!
!
!
!
!!
!
!
!!!! !!!! !!!
!
!!!! !
!!!!!!!!!!!!!!!
!
!!!!
!
!!!!
!!!!!
! !!!!!!!!
!!!!
!!!!
!!
!!!
!100 !50 0 50 100
!1
0!
50
51
0
Machine Cycle
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!! !!!! !!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!! !!!! !!!! !!!! !!!!!!!!!!!!!!!!!!!!!!!! !!!!!!!! !!!!!!!! !!!!!!!!!!!!!
Second of pair reads First of pair reads
!
!
Original, RMSE = 2.609Recalibrated, RMSE = 0.089
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!
!!
!
!!!!!!!!!
!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 2.598Recalibrated, RMSE = 0.052
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!
!!!!!
!
!!!!!!!
!!
!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 2.169Recalibrated, RMSE = 0.135
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!!!!!!
!!!!
!!!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 1.656Recalibrated, RMSE = 0.088
!1
0!
50
51
0
Dinucleotide
Acc
ura
cy (
Em
pir
ica
l ! R
ep
ort
ed
Qu
alit
y)
!!!!!
!!
!!!!
!!!!!
!!!!!!!!!!!!!!!!
AA AG CA CG GA GG TA TG
Original, RMSE = 2.469Recalibrated, RMSE = 0.083
Illumina/GenomeAnalyzer Roche/454 Life/SOLiD Illumina/HiSeq 2000
Ryan Poplin

L(G | D) = P(G)P(D |G) = P(b |G)b∈ good _ bases{ }∏
SNP calling I: genotype likelihoods per sample
• Genotype likelihoods describe the probability of the reads for each genotype (AA, AC, …, GT, TT) at each locus
• Likelihood of data computed using pileup of bases and associated quality scores at given locus
• Only “good bases” are included: those satisfying minimum base quality, mapping read quality, pair mapping quality, NQS
• P(b | G) uses platform-specific confusion matrices
Prior for the genotype!
Likelihood for the genotype!
Likelihood of the data given the genotype!
Bayesian model
Independent base model!
See http://www.broadinstitute.org/gsa/wiki/index.php/Unified_genotyper for more information 30

• Simultaneous estimation of: – Allele frequency (AF) spectrum Pr{AF = i | D} – The probability that a SNP exists Pr{AF > 0 | D} – Assignment of genotypes to each sample
Individual 1!
Sample-associated reads!
Individual 2!
Individual N!
Genotype likelihoods!
Joint estimate across samples!
Genotype frequencies!
Allele frequency!
SNPs!
Excellent mathematical treatment at http://lh3lh3.users.sourceforge.net/download/samtools.pdf
SNP calling II: multi-sample SNP calling

Variant Quality Score Recalibration: training on highly confident known sites to determine the probability that other sites are true
The HapMap3 sites from NA12878 HiSeq!calls are used to train the GMM. Shown!here is the 2D plot of strand bias vs. the!variant quality / depth for those sites.!
Variants are scored based on their!fit to the Gaussians. The variants!(here just the novels) clearly!separate into good and bad clusters.!
Eric Banks, Ryan Poplin

Dindel: an algorithm by Kees Albers for calling small (~50 bp) indels in NGS Illumina data
33
Raw SNPs!
Raw variants!
Filtered variants!
Sample 1 reads!
Sample N reads!
….!
Phase 2: MPG-Firehose!Variant discovery and genotyping!
---- Multiple samples simultaneously ----!
*!Raw indels!
Figure and work by Kees Albers (http://sites.google.com/site/keesalbers/soft/dindel). A version of this is being implemented in the GATK by Guillermo del Angel with permission from Kees.!
The GATK version of this algorithm is still under development. We use it here experimentally for its genotyping facility. For initial discovery, we rely on sites emitted by IndelGenotyperV2, written by Andrey Sivachenko.!
Guillermo del Angel, Kiran Garimella

NGS processing requires significant but consistent CPU and storage costs
●●●
●
●
●
●
●
●●
●●
●
●
●●
●
020
4060
8010
0
CPU time required to process a project
Number of samples
CPU
tim
e (d
ays)
0 50 100 150
y = 0.74x + 0.79
MCKD1Amish_Rare_CFSC Ashuldin_mennonite
BMI_Hirschhorn_NHGRI
Autism_Walsh
Autism_Daly_WholeExome_Batch1
Autism_Daly_WholeExome_Batch2
Autism_Daly_WholeExome_Batch3
Ashuldin_MennoniteMCKD1_20exomes
Autism_Daly_WholeExome_Batch4Autism_Daly_WholeExome_Batch5
Autism_Daly_WholeExome_Batch6
Height_Hirschhorn_NHGRI
EOMI_Kathiresan_NHGRIHIV_deBakker_WholeExome_48Exomes
SCZ_Sklar
Data type Target Storage
Per-sample
Single WEx 32 Mb 30-50 Gb
Deep WGS 2.85 Gb 250 Gb
Shallow WGS 2.85 Gb 25-50 Gb
Complete Project
700 EOMI exomes 32 Mb 35 Tb
1000G Pilot 2.85 Gb 9 Tb
1000G Phase I 2.85 Gb 60 Tb
Example Storage requires
Misc. mathematical equations
Mark DePristo
November 29, 2010
1 Storage requirements
storage ≈ 2bytes
bp× targeted area (1)
1
Kiran Garimella

QUALITY CONTROL

Evaluating SNP call quality Expected number of calls?
• The number of SNP calls should be close to the average human heterozygosity of 1 variant per 1000 bases
• Only detects gross under/over calling
What fraction of my calls are already known?
• dbSNP catalogs most common variation, so most of the true variants found will be in dbSNP
• For single sample calls, ~90 of variants should be in dbSNP
• Need to adjust expectation when considering calls across samples
Concordance with genotype chip calls?
• Often we have genotype chip data that indicates the hom-ref, het, hom-var status at millions of sites
• Good SNP calls should be >99.5% consistent these chip results, and >99% of the variable sites should be found
• The chip sites are in the better parts of the genome, and so are not representative of the difficulties at novel sites
Transition to transversion ratio (Ti/Tv)?
• Transitions are twice as frequent as transversions (see Ebersberger, 2002)
• Validated human SNP data suggests that the Ti/Tv should be ~2.1 genome-wide and ~2.8 in exons
• FP SNPs should has Ti/Tv around 0.5 • Ti/Tv is a good metric for assessing SNP
call quality
A
G T
C
transversions transitions

How many calls should I expect?
Sample ethnicity
Target (L) Heterozygosity (θ) Expected number of variants
Single sample => 2N = 2 European Whole genome 0.78 x 10-3 ~3.3M European 32Mb exome 0.42 x 10-3 ~20K African Whole genome 1.00 x 10-3 ~4.3M African 32Mb exome 0.48 x 10-3 ~23K 100 samples => 2N = 200 European Whole genome 0.78 x 10-3 ~13M

05
10
15
20
70 80 90 100 110 120 130
0
20
40
60
80
100
NA12878 (CEU)
NA19240 (YRI)
Including the 1000 Genomes Pilot, ~92-95% of all SNPs discovered in whole genome
sequencing projects will be already known
% of novel SNPs discovered in whole-genome sequencing
Number of SNPs in dbSNP (Millions)
dbSNP build ID
1000 Genomes
pilot 1
70 80 90 100 110 120 130
0
20
40
60
80
100
NA12878 (CEU)
NA19240 (YRI)
* NA12878 and NA19240 from 1KG Pilot 2 deep whole genome sequencing

Genotype sensitivity and concordance
• Genotype chips (Illumina 1M, e.g.) reliably identify genotypes at many sites – >99% accuracy at known polymorphic sites
• Sensitivity measure: what fraction of variant sites in sample on chip did I found?
• Genotype quality: how concordance are the genotype calls from NGS with the chip?
• Caveats: not comprehensive, only at easy sites, does not apply to novel calls

• Blue elements / red area elements
• Measures fraction of sites called variant (A/B or B/B) in comparison that are also called variant in evaluation data
• Penalizes hom-ref and no-calls at variant sites
Non-reference sensitivity Genotype of comparison calls
Gen
otyp
e of
eva
luat
ion
calls
A is ref allele, B is alt allele, . is no call
(6+7+10+11) / (2+3+6+7+10+11+14+15)
= 34/ 68
A/A A/B B/B ./.
A/A 1 2 3 4
A/B 5 6 7 8
B/B 9 10 11 12
./. 13 14 15 16

• Blue elements / red area elements
• Measures accuracy of genotype calls at sites called by both sets
– Excludes concordant A/A genotypes since these are often large in number and easier to get correct
• Good metric for assaying accuracy of genotype calls
Non-reference discrepancy rate Genotype of comparison calls
Gen
otyp
e of
eva
luat
ion
calls
A is ref allele, B is alt allele, . is no call
(2+3+5+7+9+10) / (2+3+5+6+7+9+10+11)
= 36 / 53
A/A A/B B/B ./.
A/A 1 2 3 4
A/B 5 6 7 8
B/B 9 10 11 12
./. 13 14 15 16
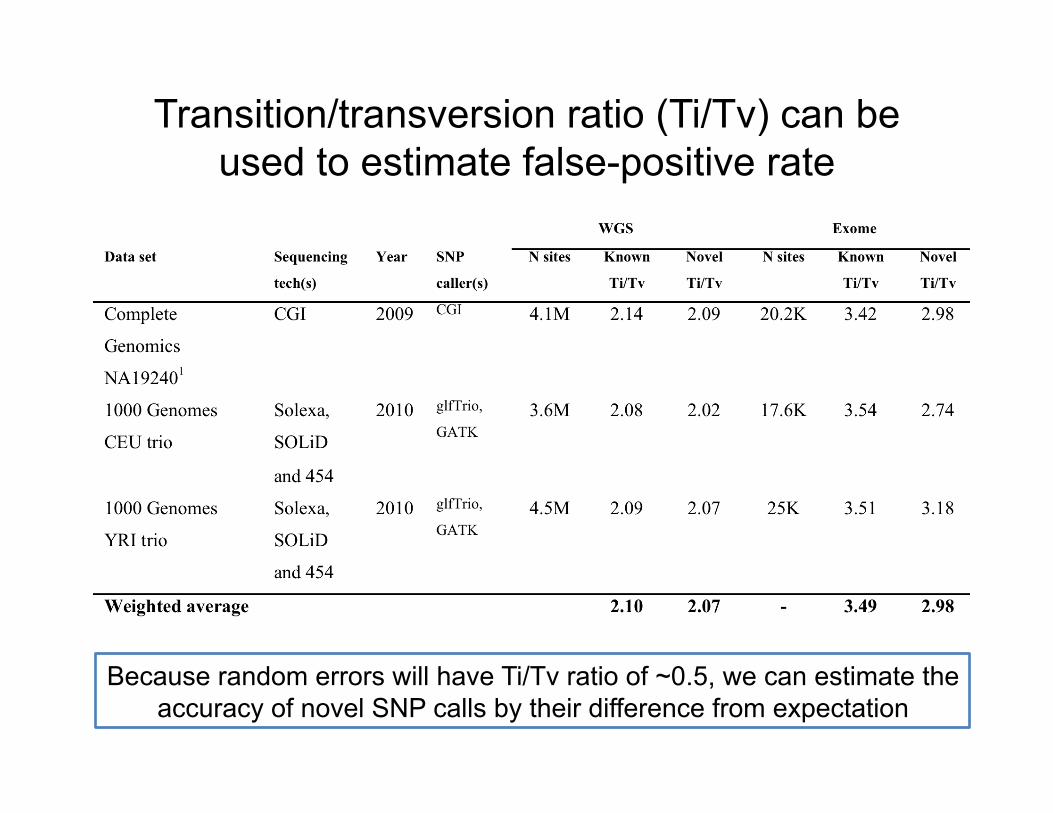
Transition/transversion ratio (Ti/Tv) can be used to estimate false-positive rate
Because random errors will have Ti/Tv ratio of ~0.5, we can estimate the accuracy of novel SNP calls by their difference from expectation

EXPECTED OUTPUTS

! "#$%!&#'()*%+, -)./0+#')1!$)!2345676!*0+#01$'
! 2)8!)9!"2:' ;#<;* =>?!()1()+&01(% 4@A!()1()+&01(%
-0BB!'%$! 3BB! @1)C1 2)*%B &D"2:E @1)C1 2)*%B 2F!
'%1'#$#*#$,!
2FG!+0$% 2F!
'%1'#$#*#$,!
2FG!+0$%
=#"%H!
I#10B!(0BB!'%$! ?8J5>! ?857>! ?K6@! LM8?7! 584K! 58M7! LL8?5! M8M6! L7867! M844!
N)CO/0''! ! ! ! ! ! ! ! ! ! !
P0+#01$!+%(0B#D+0$%&!
(0BB!'%$!
6847>! Q8LK>! 585Q>! 758QK! 5847! 48L4! R$%.#S%&!D%B)C!
"0./B%!*0+#01$!(0BB'!9)+!2345676!)1B,!
TU).%!(0/$V+%! ! ! ! ! ! ! ! ! ! !
How do indel realignment and base quality recalibration affect SNP calling?
http://www.broadinstitute.org/gsa/wiki/index.php/Base_quality_score_recalibration
http://www.broadinstitute.org/gsa/wiki/index.php/Local_realignment_around_indels
6.5% of calls on raw reads are likely false positives due to indels!
The process doesnʼt remove real SNPs!
Eric Banks

•
•
•
•
•
•
•
•
•
•
! "#$%!&#'()*%+, -)./0+#')1!$)!2345676!*0+#01$'
! 2)8!)9!"2:' ;#<;* =>?!()1()+&01(% 4@A!()1()+&01(%
-0BB!'%$! 3BB! @1)C1 2)*%B &D"2:E @1)C1 2)*%B 2F!
'%1'#$#*#$,!
2FG!+0$% 2F!
'%1'#$#*#$,!
2FG!+0$%
How well does SNP calling work?
Germline Somatic False Positive
Hard Filtered 49 952 1304 Variant Recalibrated 49 929 85 Complete Genomics 47 886 255
93.5% reduction in
FPs
Validation results of de novo mutation calls on NA12878 !
We get more calls with better
metrics
The old approach!
The new approach!
NA12878!
Eric Banks

Variant Quality Score Recalibration: enables one to choose the tranche that suits the needs of the task at hand, targeting
sensitivity or specificity
http://www.broadinstitute.org/gsa/wiki/index.php/Variant_quality_score_recalibration
Number of Novel Variants (1000s)0 500 1000 1500 2000
Ti/Tv FDR
10
5
1
0.1
1.91
1.99
2.06
2.07
0 100 200 300 400
Cumulative TPsTranch!specific TPsTranch!specific FPsCumulative FPs
Ti/Tv FDR
10
2
1
0.1
1.92
2.04
2.05
2.07
0.0 0.2 0.4 0.6 0.8
Ti/Tv FDR
10
2
1
0.1
2.79
2.96
2.98
3.01
Evaluating novel variants
More Bias
Less Bias
HiSeq: training on HapMap
More Bias
Less Bias
Likely dbSNP errors
Gaussian mixture model fits
A HM3 1KG Trio
99.5
99.5
99.5
99.5
98.2
98.5
98.4
98.6
NR sensitivity (%):
HiSeq C
HiSeq
88.0
89.7
88.0
93.8
HM3
96.0
96.8
96.0
98.3
D
65.1
66.2
65.3
66.9 86.5 With imputation
82.3
82.8
82.4
96.7 NGS only 83.0
B
Heterozygous variants Homozygous
variants
Exome
Low-pass E
Analysis tranche
Analysis tranche
HiSeq: evaluating novel variants
HiSeq HM3
Analysis tranche
Eric Banks
Number of Novel Variants

0.0
0.2
0.4
0.6
0.8
1.0
1 ! Specificity vs. HiSeq calls
Sensitiv
ity v
s. H
iSeq c
alls
Sensitivity after imputation with all samples
Fraction of sites with at least one alternate allele in NA12878
Fraction of sites with at least one alternate allele in any sample
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
0.00 0.05 0.10 0.15 0.20
N=0
N=1
N=2
N=3
N=4
N=5
N=10
N=20
N=30
N=40
N=50
N=60
Discovery of HiSeq sites for NA12878 + N other samples
We hit our target here.
Eric Banks, Ryan Poplin

Consistency across samples
●
1000
2000
3000
Sample
Nov
el v
aria
nts
(not
in d
bSN
P)
●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●
●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●●
●●●●●●●●●● ●
5481
.11
PAR
C_9
85PA
RC
_106
9PA
RC
_121
6AC
TG_2
837
PAR
C_1
67AC
TG_1
079
PAR
C_4
4355
45.4
ACTG
_146
162
30.3
PAR
C_1
09_r
esen
dAC
TG_1
031
PAR
C_0
61_r
esen
dAC
TG_4
5255
86.4
ACTG
_274
8PA
RC
_085
ACTG
_407
PAR
C_6
9056
61.6
PAR
C_1
3333
51.5
PAR
C_7
4254
94.5
ACTG
_385
PAR
C_1
102
5657
.4AC
TG_1
220
PAR
C_8
4331
00.6
5624
.355
87.2
6277
.7PA
RC
_005
PAR
C_1
94PA
RC
_117
0AC
TG_1
778
PAR
C_6
25PA
RC
_734
PAR
C_1
84AC
TG_1
249
PAR
C_1
56_r
esen
d55
47.3
ACTG
_161
7AC
TG_3
000
ACTG
_224
8PA
RC
_404
ACTG
_746
ACTG
_285
877
86.4
ACTG
_508
PAR
C_7
0555
91.4
7779
.250
75.7
7780
.463
09.2
5466
.4AC
TG_5
55PA
RC
_876
_res
end
5859
.537
59.5
PAR
C_6
4661
09.4
PAR
C_0
2234
87.3
PAR
C_7
0255
82.2
ACTG
_169
4PA
RC
_006
PAR
C_9
6562
09.2
5474
.8PA
RC
_108
4AC
TG_1
280
5543
.9PA
RC
_449
PAR
C_1
078
6200
.261
25.3
5505
.678
03.4
PAR
C_1
026
3999
.663
07.5
3641
.550
02.1
256
01.3
5567
.656
45.4
PAR
C_1
024
5565
.11
ACTG
_123
956
56.4
7843
.1PA
RC
_920
5599
.3PA
RC
_215
PAR
C_4
33PA
RC
_437
PAR
C_1
089
PAR
C_6
91PA
RC
_442
PAR
C_4
22PA
RC
_455
PAR
C_0
19PA
RC
_432
PAR
C_4
25PA
RC
_445
ACTG
_302
PAR
C_4
24PA
RC
_423
PAR
C_4
31PA
RC
_447
PAR
C_4
14AC
TG_8
6338
70.4
PAR
C_0
02PA
RC
_028
7852
.4PA
RC
_434
● QC+ sampleQC− sample
●
1.0
2.0
3.0
Sample
Ti/T
v of
nov
el v
aria
nts
(not
in d
bSN
P) ●
●●
●
●●
●●●
●
●
●●●
●
●
●
●
●
●●
●
●●●●●
●●●
●
●●●
●
●
●●
●●
●●●
●
●●●●
●
●
●●●●
●
●●
●
●
●●●
●●●
●
●●●●
●
●●●●●
●
●●●●●●●●
●●●
●
●●●
●●●●
●
●
●●
●●
● ●
5481
.11
PAR
C_9
85PA
RC
_106
9PA
RC
_121
6AC
TG_2
837
PAR
C_1
67AC
TG_1
079
PAR
C_4
4355
45.4
ACTG
_146
162
30.3
PAR
C_1
09_r
esen
dAC
TG_1
031
PAR
C_0
61_r
esen
dAC
TG_4
5255
86.4
ACTG
_274
8PA
RC
_085
ACTG
_407
PAR
C_6
9056
61.6
PAR
C_1
3333
51.5
PAR
C_7
4254
94.5
ACTG
_385
PAR
C_1
102
5657
.4AC
TG_1
220
PAR
C_8
4331
00.6
5624
.355
87.2
6277
.7PA
RC
_005
PAR
C_1
94PA
RC
_117
0AC
TG_1
778
PAR
C_6
25PA
RC
_734
PAR
C_1
84AC
TG_1
249
PAR
C_1
56_r
esen
d55
47.3
ACTG
_161
7AC
TG_3
000
ACTG
_224
8PA
RC
_404
ACTG
_746
ACTG
_285
877
86.4
ACTG
_508
PAR
C_7
0555
91.4
7779
.250
75.7
7780
.463
09.2
5466
.4AC
TG_5
55PA
RC
_876
_res
end
5859
.537
59.5
PAR
C_6
4661
09.4
PAR
C_0
2234
87.3
PAR
C_7
0255
82.2
ACTG
_169
4PA
RC
_006
PAR
C_9
6562
09.2
5474
.8PA
RC
_108
4AC
TG_1
280
5543
.9PA
RC
_449
PAR
C_1
078
6200
.261
25.3
5505
.678
03.4
PAR
C_1
026
3999
.663
07.5
3641
.550
02.1
256
01.3
5567
.656
45.4
PAR
C_1
024
5565
.11
ACTG
_123
956
56.4
7843
.1PA
RC
_920
5599
.3PA
RC
_215
PAR
C_4
33PA
RC
_437
PAR
C_1
089
PAR
C_6
91PA
RC
_442
PAR
C_4
22PA
RC
_455
PAR
C_0
19PA
RC
_432
PAR
C_4
25PA
RC
_445
ACTG
_302
PAR
C_4
24PA
RC
_423
PAR
C_4
31PA
RC
_447
PAR
C_4
14AC
TG_8
6338
70.4
PAR
C_0
02PA
RC
_028
7852
.4PA
RC
_434
● QC+ sampleQC− sample
All samples (QC+ and QC-):!
SNPs Ti/Tv! All: 123K 2.7!Known: 57K 3.4!Novel: 65K 2.3!
Good samples (QC+ only):!
SNPs Ti/Tv! All: 96K 3.2!Known: 50K 3.5!Novel: 45K 3.0!
✗
✓
Misbehaving samples: unusually high number of novel variants, correlated low Ti/Tv ratio. !
48 Kiran Garimella

Excellent overall concordance to the HIV GWAS data – one outlier (possible swapped sample)
!!!!!!!!!!
!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!
!!!!!!!!!!!!
!!!!!!!!!!!!!!!!!!
!!!!!!!!!!!!!!!!
!!!!!!!!
!!!!!!
!!
0.5
1.0
2.0
5.0
10.0
20.0
50.0
Comparison of HIV exome data to HIV GWAS data
Perc
ent non!
refe
rence d
iscre
pancy r
ate
NA
12878
5543.9
5657.4
3999.6
5075.7
5661.6
5591.4
5565.1
16309.2
5645.4
5599.3
3759.5
5587.2
5582.2
PA
RC
_005
5859.5
5505.6
5656.4
6109.4
7780.4
3641.5
6230.3
5002.1
25466.4
3487.3
PA
RC
_433
5567.6
7843.1
5586.4
5601.3
PA
RC
_133
7786.4
AC
TG
_385
6277.7
5474.8
5624.3
7779.2
6125.3
5494.5
PA
RC
_184
PA
RC
_167
PA
RC
_1026
7803.4
PA
RC
_742
6200.2
PA
RC
_404
AC
TG
_452
PA
RC
_1170
AC
TG
_1220
AC
TG
_2837
AC
TG
_2248
PA
RC
_449
PA
RC
_1089
PA
RC
_156_re
send
3100.6
5545.4
PA
RC
_1102
PA
RC
_1024
PA
RC
_006
PA
RC
_646
PA
RC
_702
PA
RC
_705
6209.2
PA
RC
_061_re
send
AC
TG
_746
AC
TG
_1461
PA
RC
_194
5547.3
AC
TG
_3000
PA
RC
_1084
AC
TG
_2858
PA
RC
_437
AC
TG
_1617
PA
RC
_843
PA
RC
_734
AC
TG
_555
PA
RC
_1078
AC
TG
_1694
AC
TG
_1079
PA
RC
_1216
AC
TG
_2748
PA
RC
_625
PA
RC
_920
PA
RC
_109_re
send
AC
TG
_1778
AC
TG
_508
PA
RC
_085
3351.5
PA
RC
_442
PA
RC
_1069
PA
RC
_690
AC
TG
_1249
PA
RC
_022
AC
TG
_407
AC
TG
_1239
PA
RC
_876_re
send
AC
TG
_1280
PA
RC
_691
PA
RC
_985
AC
TG
_1031
allS
am
ple
sPA
RC
_215
PA
RC
_019
PA
RC
_965
PA
RC
_443
6307.5
! QC+ samplesQC! samples
Sample 6307.5 has ~70% discordance with the variants in the GWAS data, but did not show up in other QC checks. !
Potentially a sample mix-up.!
49
820K sites in the whole-genome GWAS data.!17K sites (sites falling within the exome) used for evaluation.!
Kiran Garimella

Summary
• Next-generation sequencing is a maturing technology – Multiple robust NGS technologies – Several canonical experimental designs
• Sophisticated data processing algorithms • Reliable statistical approaches for SNP
and indel discovery and genotyping • Suite of quality-control metrics to assess
the reliability of your experimental results







