
Page 1
Chemoselective sp2-sp3 Cross-Couplings: Iron-Catalyzed Alkyl Transfer to Dihaloaromatics
Sushant Malhotra,* Pamela S. Seng, Stefan G. Koenig, Alan J. Deese, and Kevin A. Ford
SUPPORTING INFORMATION

Page 2
General Information:
All reactions were performed under a nitrogen atmosphere. All solvents and Grignard reagents were used without further purification
or further analyses. All dihaloaromatics were either commercially-available or prepared using literature known methods. All
reactions were monitored using a X-Select CSH Phenyl Hexyl reverse phase column or an SL Only Monolithic C18 column using a
mobile phase containing mixtures of 0.05% aqueous TFA and acetonitrile. Silica gel chromatography was performed using RediSep®
pre-packed silica gel columns commercially-available from Teledyne Isco (Catalog 69-2203). Proton nuclear magnetic resonance (1H
NMR) spectra were acquired on either a Bruker 600 MHz or Bruker 300 MHz spectrometer. Carbon nuclear magnetic resonance (1H
NMR) spectra were acquired on a Bruker 151 MHz spectrometer. All NMR spectra are referenced internally according to residual
solvent signals. Data for 1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s, singlet; d, doublet; t, triplet; q,
quartet; m, multiplet), integration, coupling constant (Hz). Data for 13C NMR are reported in terms of chemical shift (δ, ppm).
Melting points are uncorrected and were measured on a Büchi B-540 melting apparatus. All IR spectra were recorded on a Thermo
Electron Corporation Nicolet 6700 FT-IR instrument using a SMART Performer sampling attachment and Ge ATR crystal.
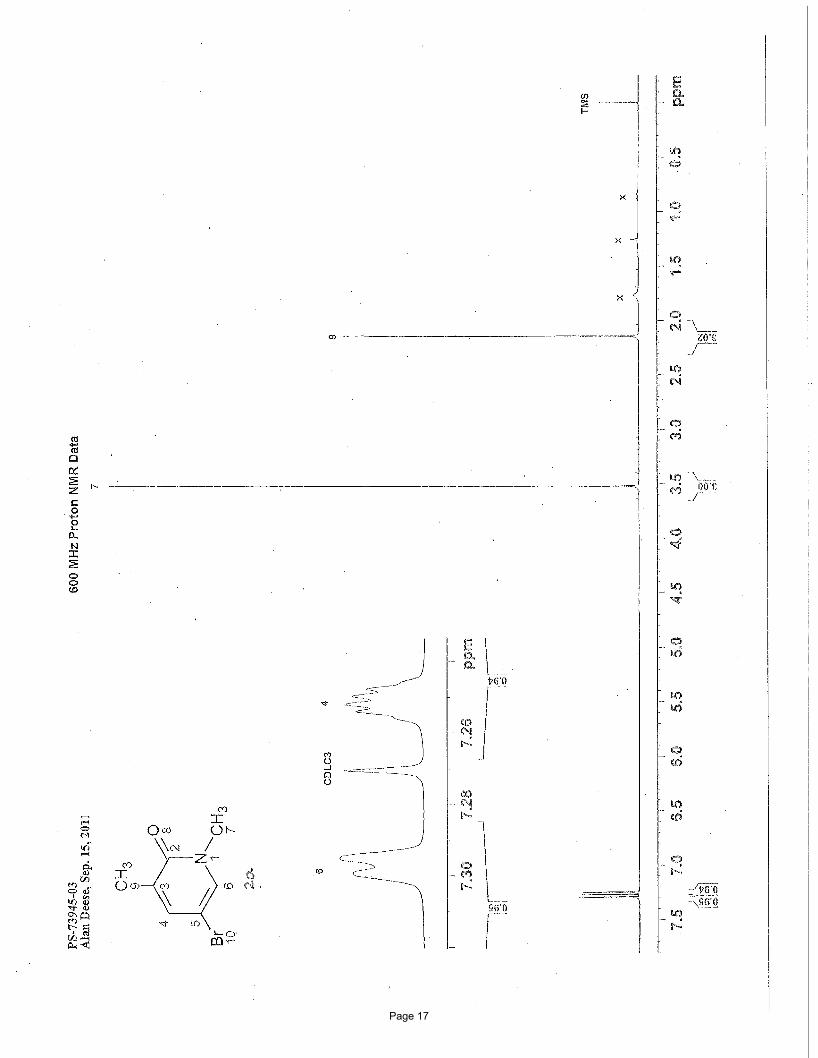
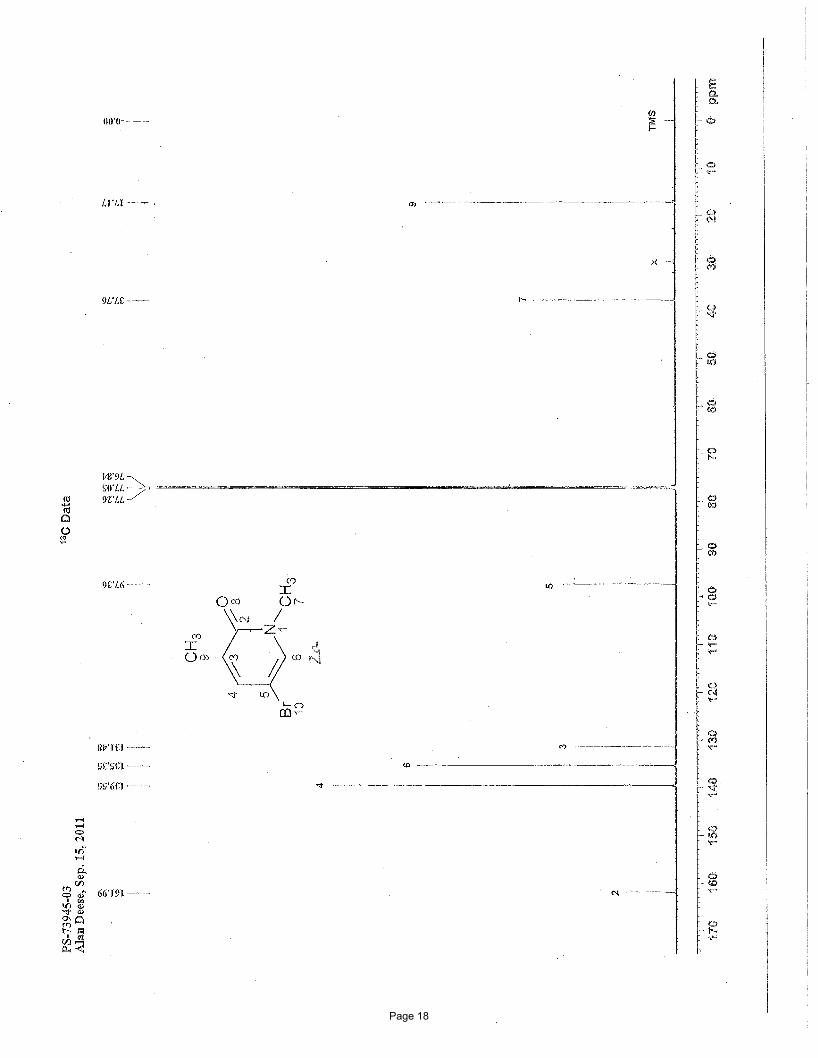
5-bromo-1,3-dimethylpyridin-2(1H)-one (2a)
N Me
OMe
BrN Me
O
Br
Br
MeMgBr
3.75 mol % Fe(acac)3
To a vial was added 3,5-dibromo-1-methylpyridin-2(1H)-one (200 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375
equiv). The vial was sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (5.0 mL). The
reaction mixture was treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.86
mmol, 1.15 equiv) at 0 ºC. After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3 x 5
mL). The organic layers were combined and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 65%
EtOAc/hexanes afforded 124 mg (82%) of the desired product as a white solid. This compound has been previously reported.i m.p. =
104.0 – 105.4 ºC (lit. 106-107 ºC)i; TLC Rf = 0.33 (50% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 7.30 (d, J = 2.4 Hz, 1H),
7.25 (m, 1H), 3.52 (s, 3H), 2.13 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 162.0, 139.6, 135.4, 131.5, 97.4, 37.8, 17.2 ppm; IR
(neat): υmax 1649, 1590, 1556, 1423, 1229, 1221, 892, 761 cm-1.
Structural elucidation:
N Me
OMe
Br
2.17 (s, 3H)
3.52 (s, 3H)
N Me
OMe
Br
2.13 (s, 3H)
3.54 (s, 3H)
J. Org. Chem. 1974, 39, 2116. This work
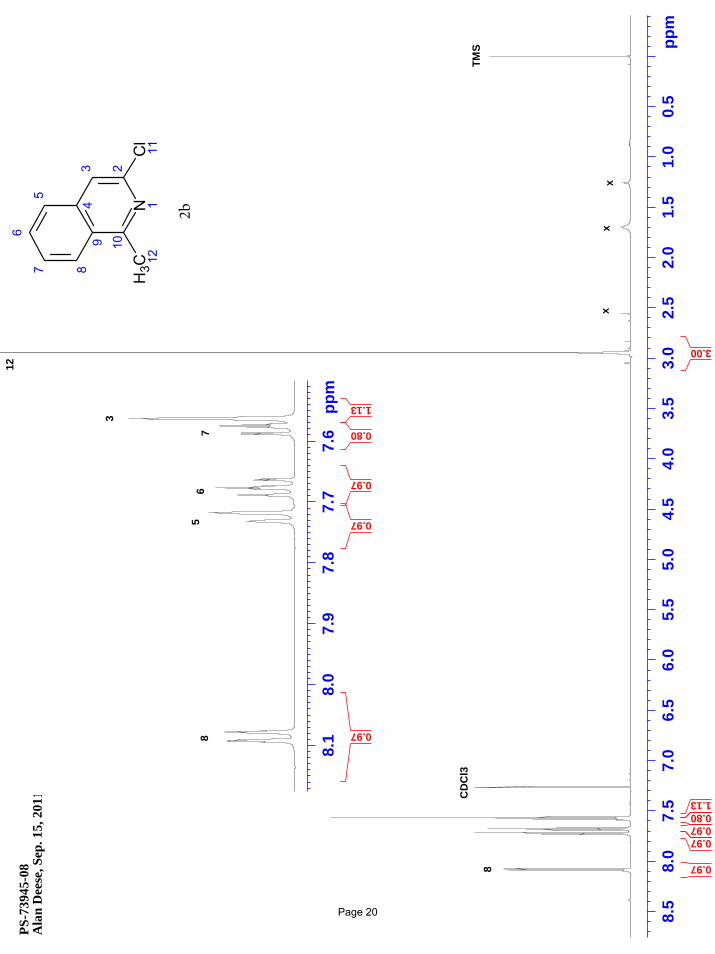
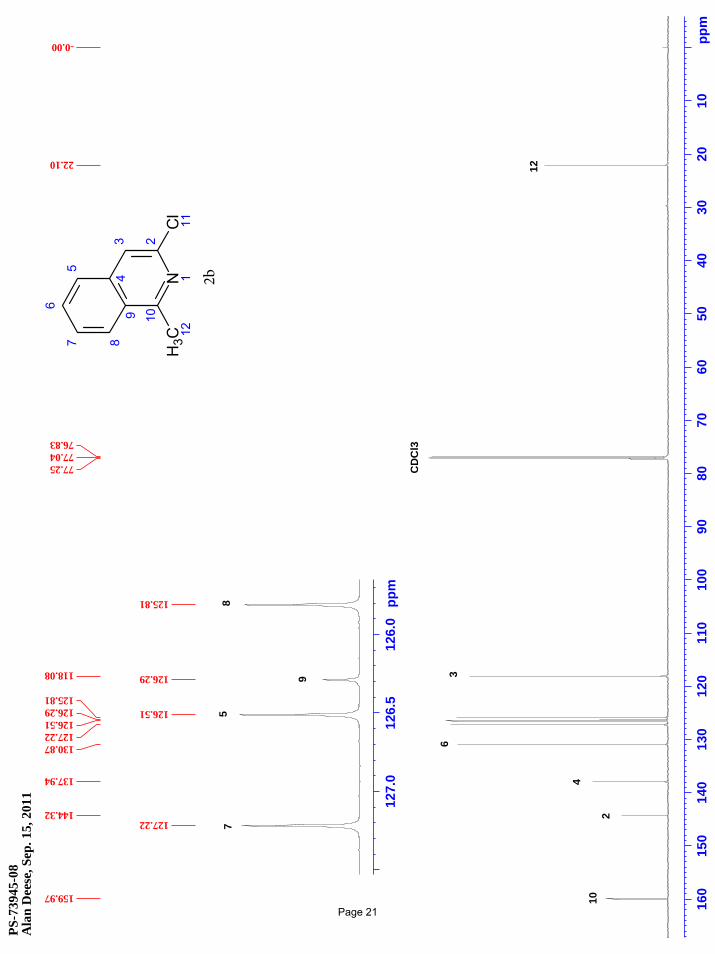
3-chloro-1-methylisoquinoline (2b)
N ClMeN ClClMeMgBr
3.75 mol % Fe(acac)3
To a vial was added 1,3-dichloroisoquinoline (148 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (5.0 mL). The reaction mixture was
treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.86 mmol, 1.15 equiv) at 0 ºC.
After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3 x 5 mL). The organic layers
were combined and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 35% EtOAc/hexanes afforded 121 mg
(91%) of the desired product as a pale pink solid. m.p. = 58.3 – 59.1 ºC; TLC Rf = 0.49 (50% EtOAc/hexanes); 1H NMR (600 MHz,
CDCl3): δ 8.09 (m, 1H), 7.73 (m, 1H), 7.68 (m, 1H), 7.59 (m, 1H), 7.56 (d, J = 1.0 Hz, 1H), 2.94 (s, 3H) ppm; 13C NMR (151 MHz,
CDCl3): δ 160.0, 144.3, 137.9, 130.9, 127.2, 126.5, 126.3, 125.8, 118.1, 22.1 ppm; IR (neat): υmax 1616, 1555, 1425, 1397, 1317, 1265,
1163, 1101, 968 cm-1.

Page 3
Structural elucidation:
N ClMe N MeCl
2.95 (s, 3H) 2.65 (s, 3H)
H H
7.56 (d, J = 1.0 Hz, 1H) 7.25 (s, 1H)
W.O. Patent 03099274, 2003.This work
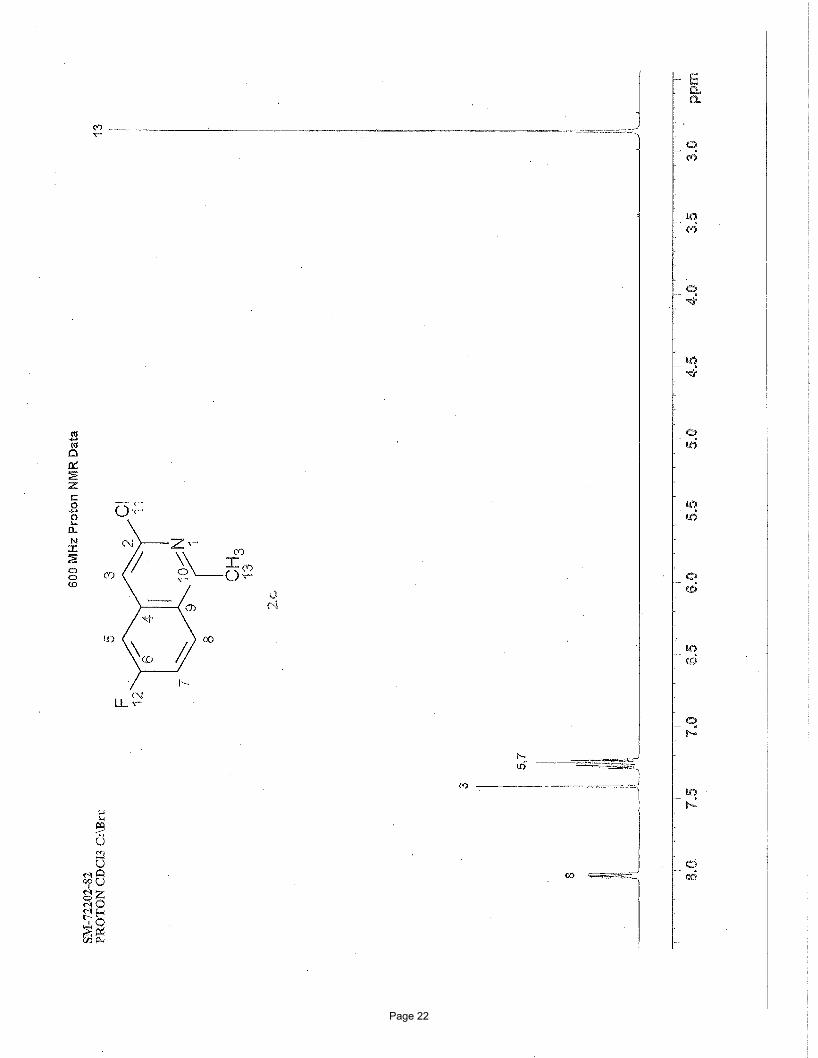
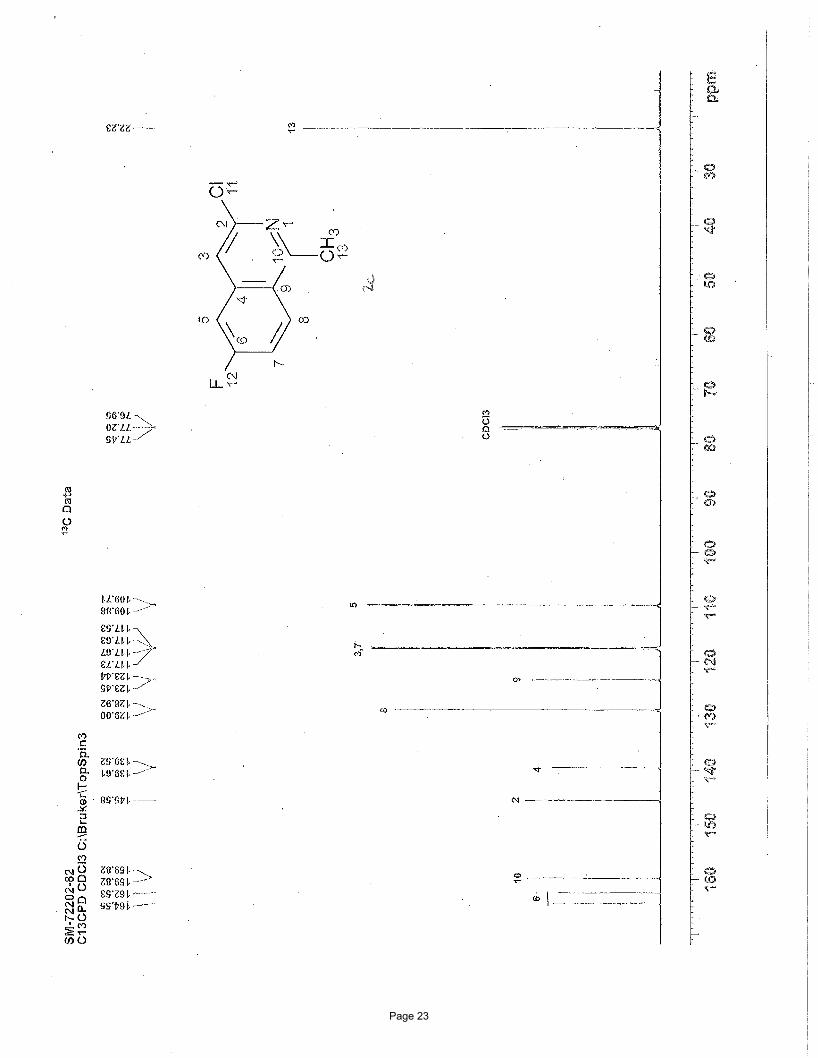
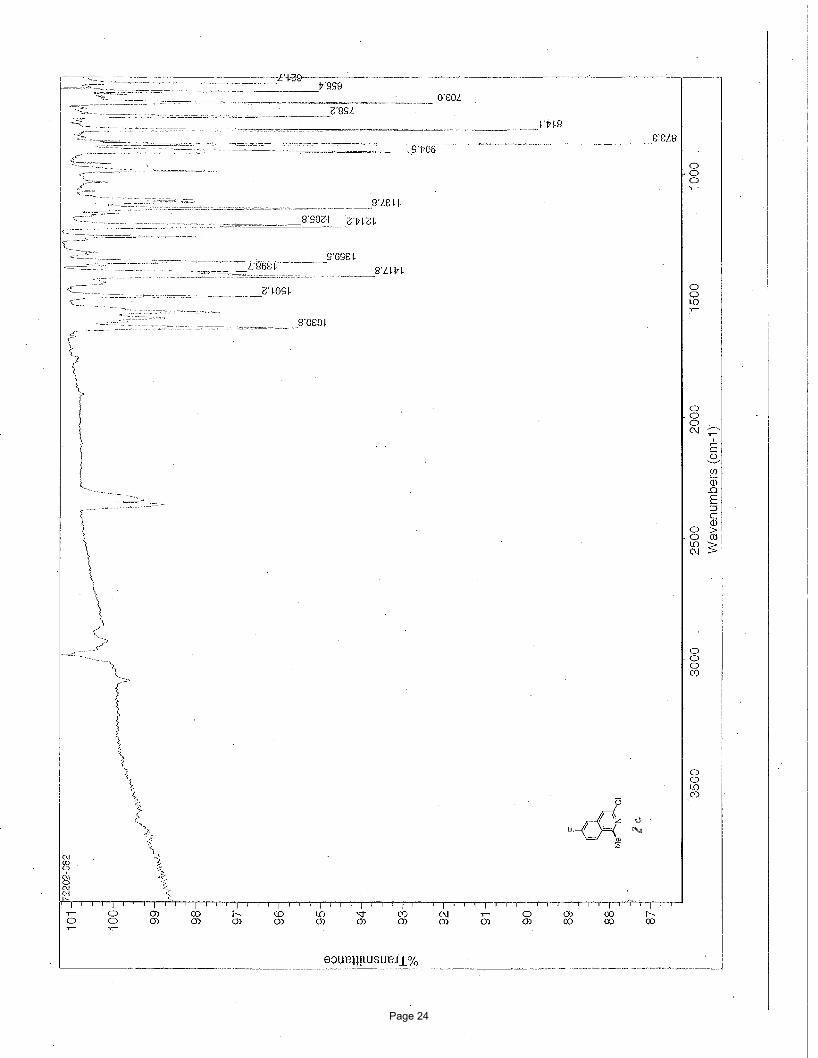
3-chloro-6-fluoro-1-methylisoquinoline (2c)
N ClMe
F
N Cl
F
ClMeMgBr
3.75 mol % Fe(acac)3
To a vial was added 1,3-dichloro-6-fluoroisoquinoline (147 mg, 0.74 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The
vial was sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL). The reaction mixture
was treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.86 mmol, 1.15 equiv) at
ambient temperature. After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3 x 5 mL).
The organic layers were combined and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 50%
EtOAc/hexanes afforded 135 mg (92%) of the desired product as a white solid. m.p. = 94.2 – 95.7 ºC; TLC Rf = 0.68 (20%
EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.04 (dd, J = 9.11, 5.33(F) Hz, 1H), 7.42 (s, 1H), 7.28 (dd, J = 7.24(F), 2.38 Hz, 1H),
7.24 (dd, J = 9.11, 7.24(F) Hz, 1H), 2.13 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 164.6-162.5, 159.8, 145.6, 139.6-139.5, 129.0-
128.9, 123.5-123.4, 117.7-117.6, 117.7-117.5, 109.9-109.7, 22.2 ppm; IR (neat): υmax 1631, 1501, 1418, 1360, 1214, 1138, 905, 873,
814, 703 cm-1.
Structural elucidation: Structure assigned based on analogy to product obtained from 1,3-dichloroisoquinoline.
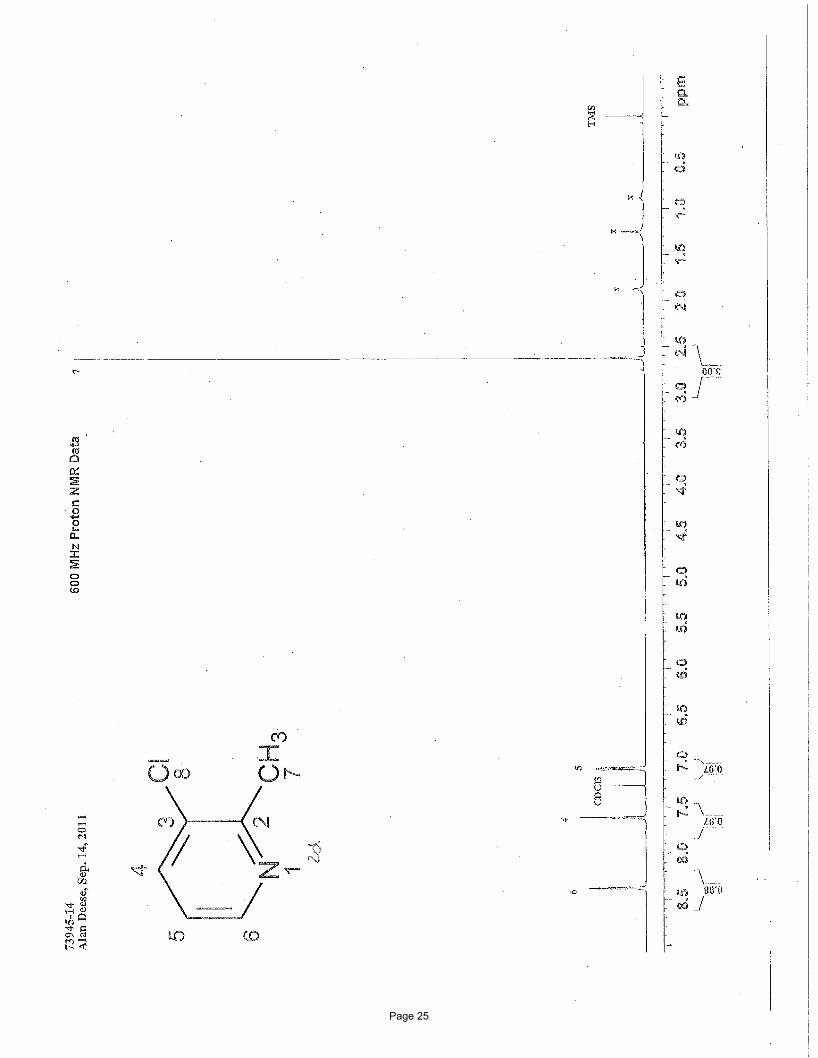
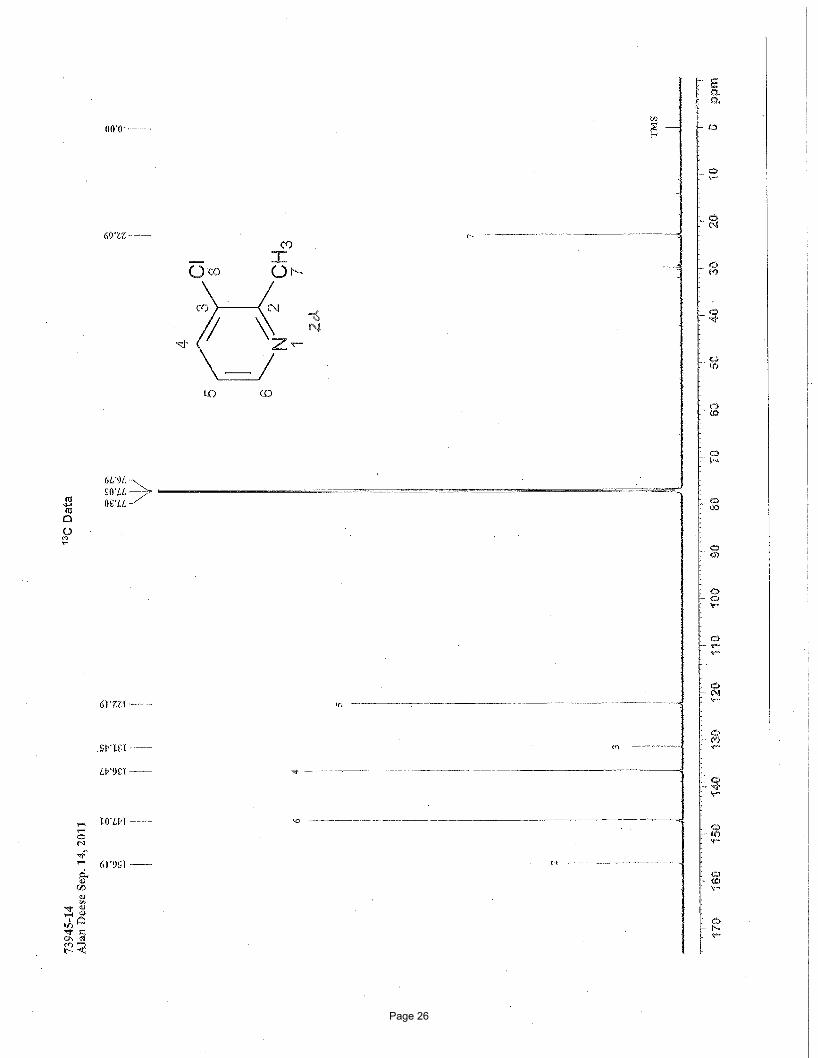
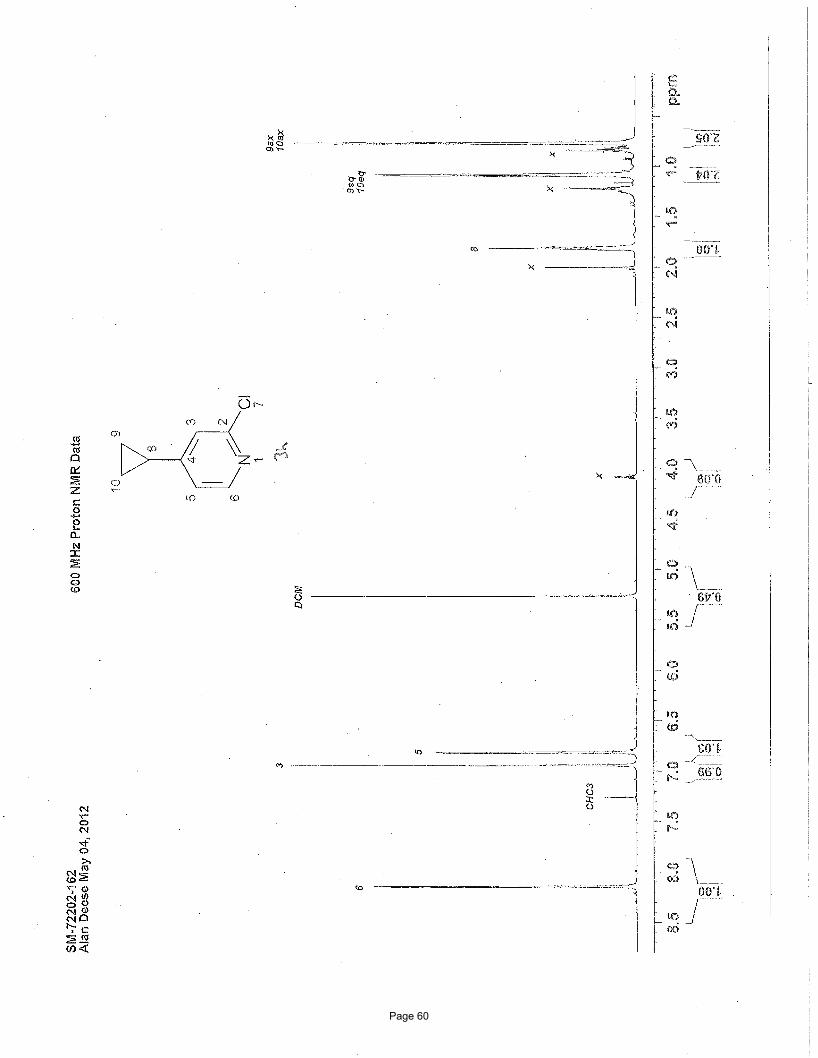
3-chloro-2-methylpyridine (2d)
N
Cl
MeN
Cl
Cl MeMgBr
3.75 mol % Fe(acac)3
To a vial was added 2,3-dichloropyridine (200 mg, 1.35 mmol) and Fe(acac)3 (18 mg, 0.051 mmo, 0.0375 equiv). The vial was sealed
with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (5.0 mL) and NMP (1.0 mL). The reaction
mixture was treated dropwise with methylmagnesium bromide (1.10 mL of a 1.4 M solution in THF:toluene (1:3), 1.55 mmol, 1.15
equiv) at ambient temperature. After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3
x 5 mL). The organic layers were combined and concentrated in vacuo. Silica gel chromatography using a gradient of 20% – 50%
MTBE/hexanes afforded 133 mg (77%) of the desired product as a colorless oil. TLC Rf = 0.39 (50% EtOAc/hexanes); Spectral data
matches literature known valuesii: 1H NMR (600 MHz, CDCl3): δ 8.39 (d, J = 3.8 Hz, 1H), 7.62 (dd, J = 8.0, 1.2 Hz, 1H); 7.09 (dd, J
= 8.0, 4.8 Hz, 1H), 2.63 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 156.2, 147.0, 136.5, 131.5, 122.2, 22.7 ppm.
Structural elucidation:
N Me
H
2.63 (s, 3H)
7.10 (dd, J = 8.0, 4.8 Hz, 1H)
7.63 (dd, J = 8.0, 1.2 Hz, 1H)
8.39 (d, J = 3.7 Hz, 1H)
H Cl
H
N Me
H
2.63 (s, 3H)
7.09 (dd, J = 8.0, 4.8 Hz, 1H)7.62 (dd, J = 8.0, 1.2 Hz, 1H)
8.39 (d, J = 3.8 Hz, 1H)
H Cl
H
This Work
N Cl
H2.39 (s, 3H)7.05 (dd, J = 7.0, 5.0 Hz, 1H)
8.1 (d, J = 5.0, 2.0 Hz, 1H)
H Me
H
Org. Lett. 2004, 6, 3517.
Synth. Commun. 2004, 34, 1097.

Page 4
7-chloro-4-methylquinoline (2e)
MeMgBr
3.75 mol % Fe(acac)3
NCl
Cl
NCl
Me
To a vial was added 4,7-dichloroquinoline (150 mg, 0.76 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.87
mmol, 1.15 equiv) at ambient temperature. After stirring for 4 h, the reaction was quenched by adding water (5 mL) and extracted
with EtOAc (3 x 5 mL). The organic layers were combined and concentrated in vacuo. Silica gel chromatography using a gradient of
0% – 50% EtOAc/hexanes afforded 121 mg (93%) of the desired product as a colorless oil. TLC Rf = 0.22 (20% EtOAc/hexanes); 1H
NMR (600 MHz, CDCl3): δ 8.65 (d, J = 4.39 Hz, 1H), 7.98 (d, J = 2.2 Hz, 1H), 7.75 (d, J = 8.9 Hz, 1H), 7.35 (dd, J = 8.9, 2.2 Hz,
1H), 7.08 (d, J = 4.4, 1H), 2.55 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 151.2, 148.5, 144.4, 134.9, 128.9, 127.2, 126.7, 125.3,
122.1, 18.6 ppm; IR (neat): υmax 1605, 1591, 1497, 1092, 886, 844, 831, 766 cm-1.
Structural elucidation
N
Me
Cl
2.70 (s, 3H)
7.28 (d, J = 4.2 Hz, 1H)7.55 (d, J = 9.0, 1.8 Hz, 1H)
7.95 (d, J = 9.0, 1.8 Hz, 1H)
8.14 (d, J = 1.8 Hz, 1H)
8.75 (d, J = 4.2 Hz, 1H) 8.65 (d, J = 4.4 Hz, 1H)
2.55 (s, 3H)
7.98 (d, J = 2.2 Hz, 1H)
7.08 (d, J = 4.4, 1H)7.35 (dd, J = 8.92, 2.2 Hz, 1H)
7.75 (d, J = 8.92 Hz, 1H)
Bose, D. S.; Kumar, R. K. Heterocycles, 2006, 68, 549. This work
N
H HH
•• •HMBCHMBC
Cl
N
Me
Cl
Note: The 1H NMR data for the product isolated from the Fe-catalyzed coupling does not match literature reported
data. The assignment is based on HMBC data.
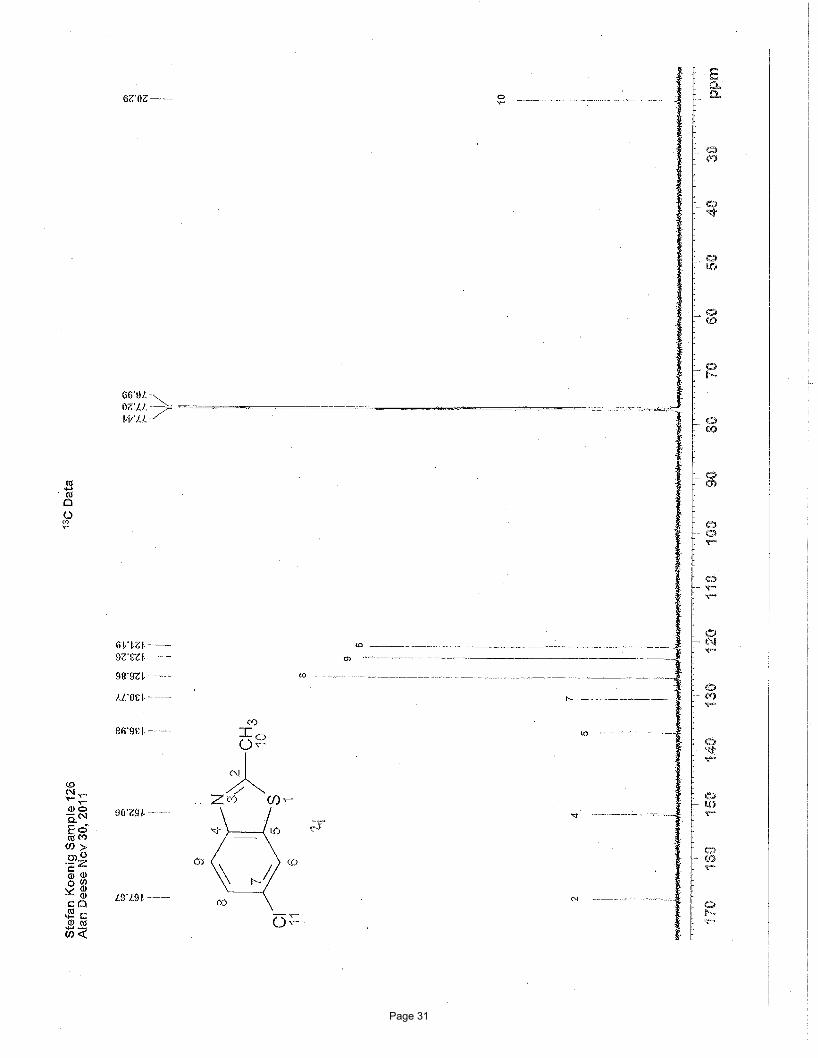
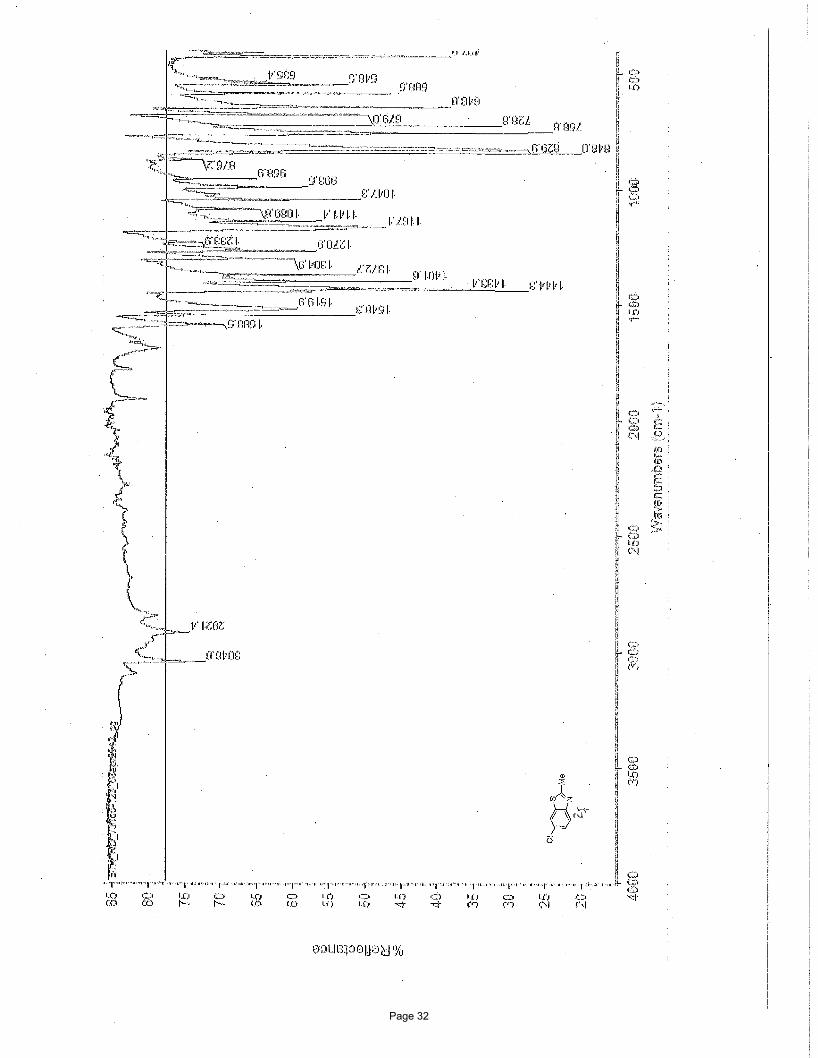
6-chloro-2-methylbenzo[d]thiazole (2f)
MeMgBr
3.75 mol % Fe(acac)3
N
SMe
Cl
N
SClCl
To a vial was added 2,6-dichlorobenzo[d]thiazole (155 mg, 0.757 mmol) and Fe(acac)3 (10 mg, 0.028 mmol, 0.0375 equiv). The vial
was sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4 mL) and NMP (1 mL). The
reaction mixture was treated dropwise with methylmagnesium bromide (0.619 mL of a 1.4 M solution in THF:toluene (1:3), 0.87
mmol, 1.15 equiv) at ambient temperature. The reaction was monitored by HPLC, which showed partial conversion after 1h.
Additional MeMgBr (0.12 mL, 0.17 mmol, 0.22 equiv) was added dropwise. After stirring for 16 h, the reaction was quenched by
adding water (5 mL) and extracted with EtOAc (3 x 5 mL). The organic layers were combined and concentrated in vacuo. The residue
was purified by silica gel chromatography using a gradient of 0% to 30% EtOAc/hexanes, affording 0.090 g (65%) of the desired
product as a colored solid. m.p. = 82 – 83 ºC (lit 84 – 86 ºC); TLC Rf = 0.82 (50% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ
7.84 (d, J = 8.7 Hz, 1H), 7.79 (d, J = 2.1 Hz, 1H), 7.40 (dd, J = 8.7, 2.1 Hz, 1H), 2.82 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ
167.7, 152.1, 137.0, 130.8, 126.9, 123.3, 121.2, 20.3 ppm; IR (neat): υmax 3049, 2921, 1589, 1548, 1520, 1444, 1433, 1402, 1373,
1305, 1271, 1167 cm-1.
Structure Elucidation:
Literature data for 6-chloro-2-methylbenzothazole: Huang, X.; Tang, J. Tetrahedron 2003, 59, 4851: 1H NMR (CDCl3, 400 MHz): δ
7.83 (s, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.38-7.41 (dd, J2 = 8.6 Hz, J1 = 2.0 Hz, 1H), 2.82 (s, 3H) ppm; IR (neat): υmax 1548, 1523, 1446,
1305, 1271 cm-1. For regioisomeric product see Cressier, D.; Prouillac C.; Hernandez, P.; Amourette, C.; Diserbo, M.; Lion, C.;
Rima, G. Bioorg. Med. Chem. 2009, 17, 5275: m.p. = 50 – 52 ºC; 1H NMR (CDCl3, 300.1 MHz): δ 7.01-7.81 (m, 4H), 2.32 (s, 3H)
ppm; 13C NMR (CDCl3, 75.4 MHz, CDCl3): δ 172.0, 152.0, 149.0, 136.0, 128.2, 122.3, 120.8, 21.5 ppm.

Page 5
Structure elucidation
N
SMe
Cl
N
SMeCl
Bioorg. Med. Chem.17, 5275, 2003.
This work &Tetrahedron 2003
59, 4851.
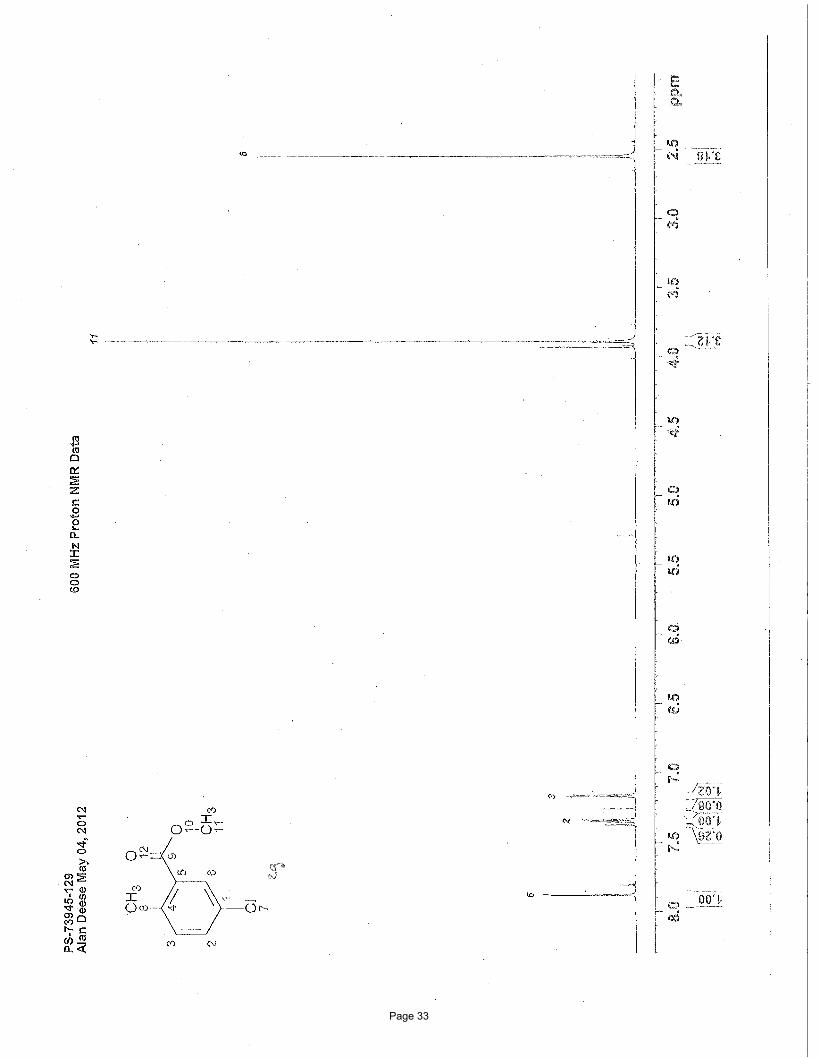
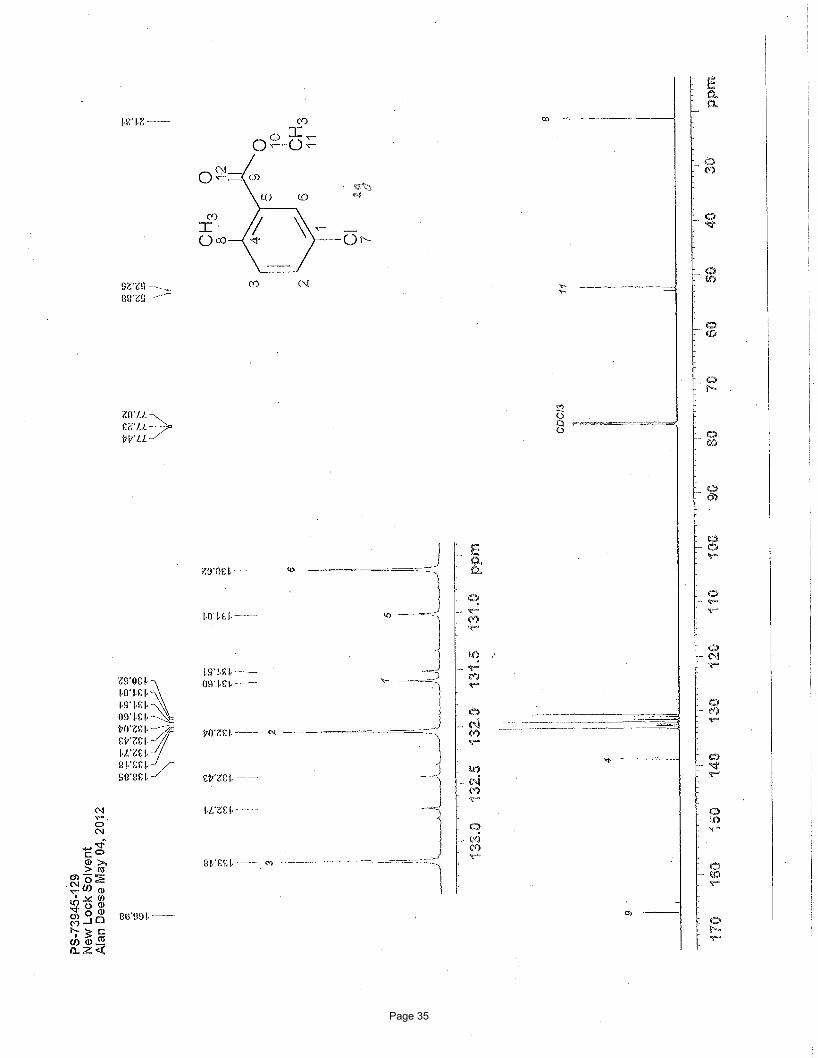
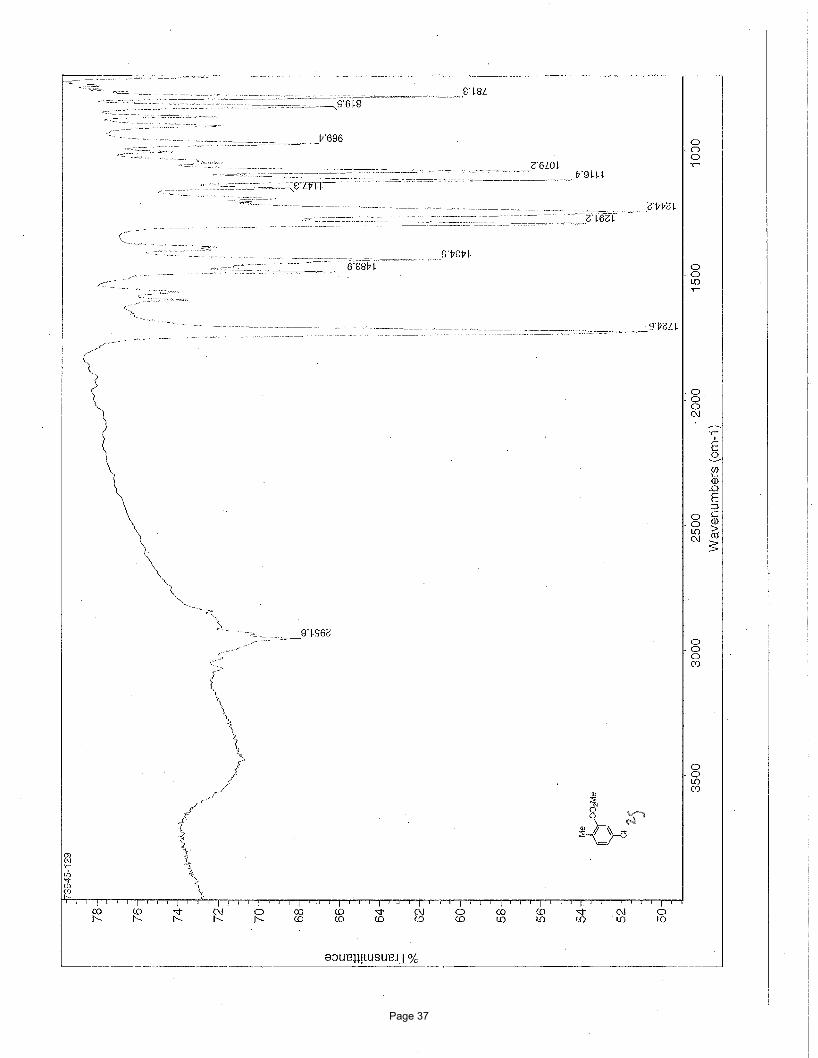
methyl 5-chloro-2-methylbenzoate (2g)
MeMgBr
3.75 mol % Fe(acac)3Me
CO2Me
Cl
CO2Me
Cl
Cl
To a vial was added methyl 2,5-dichlorobenzoate (155 mg, 0.76 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial
was sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4 mL) and NMP (1 mL). The
reaction mixture was treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.87
mmol, 1.15 equiv) at ambient temperature. After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted
with EtOAc (3 x 5 mL). The organic layers were combined and concentrated in vacuo. Preparative thin layer chromatography using
5% EtOAc/hexanes to afforded 76 mg (54%) of the desired product as a clear oil. TLC Rf = 0.66 (20% EtOAc/hexanes); 1H NMR
(600 MHz, CDCl3): δ 7.89 (d, J = 2.4 Hz, 1H), 7.36 (dd, J = 8.2, 2.4 Hz, 1H), 7.18 (d, J = 8.2 Hz, 1H), 3.90 (s, 3H), 2.56 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 167.0, 138.9, 133.2, 132.0, 131.6, 131.0, 130.6, 52.3, 21.3 ppm; IR (neat): υmax 2952, 1725, 1484,
1435, 1291, 1244, 1116, 1079 cm-1.
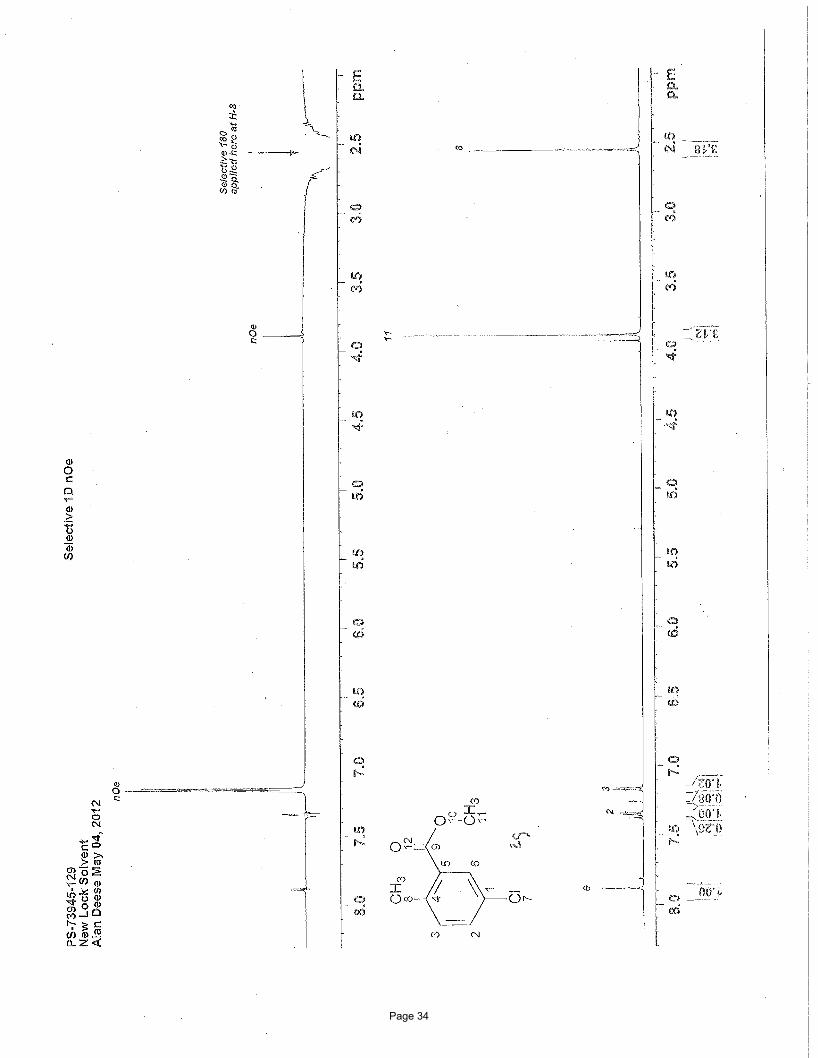
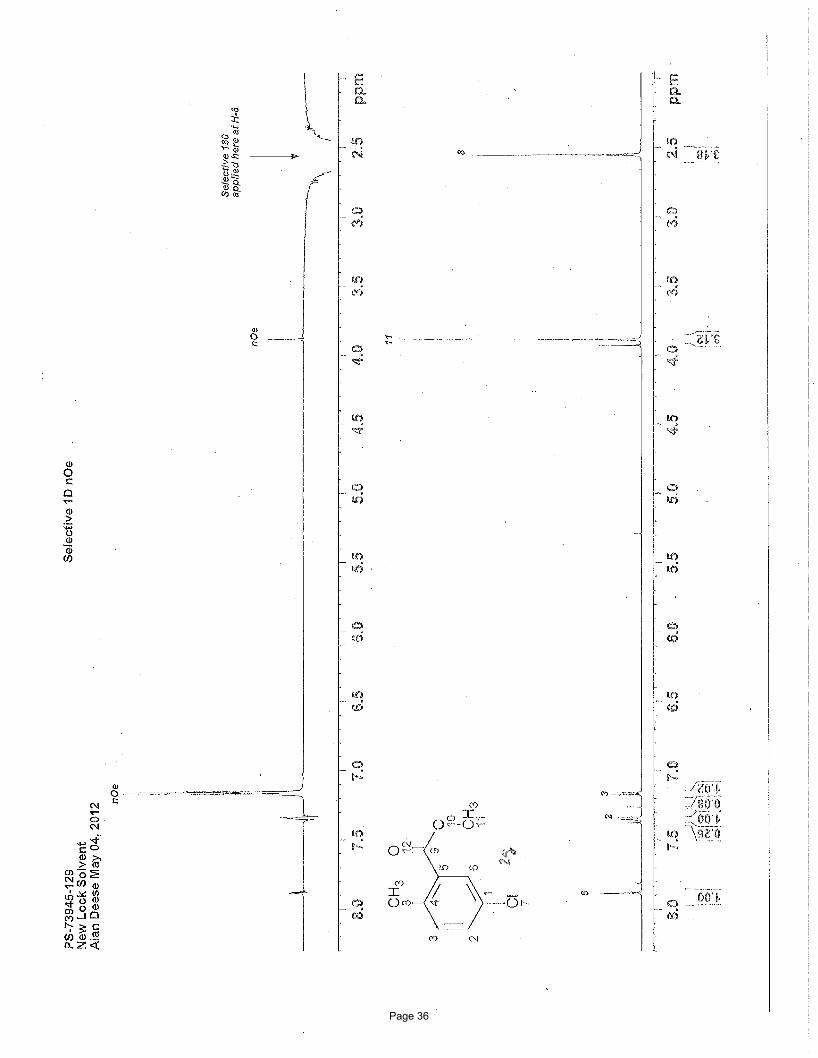
Structural elucidation
CO2Me
Cl
H HH
H7.18 (d, J = 8.2 Hz, 1H)
nOe
7.36 (dd, J = 8.2, 2.4 Hz, 1H) 7.89 (d, J = 2.4 Hz, 1H)H H
Preparation of isomeric methyl 2-chloro-5-methylbenzoate to determine site selectivity:
CO2MeCl
CO2HCl
MeOH
H2SO4 (cat)Me Me
To a flask containing a solution of 2-chloro-5-methylbenzoic acid (500 mg, 2.93 mmol) in methanol (15 mL) was added 1 drop of
concentrated sulfuric acid. The mixture was heated to reflux for 18h and the solvent was evaporated by distillation. The resulting oil
was dissolved in EtOAc (15 mL), the mixture was washed with saturated aq. NaHCO3 (15 mL), dried (Na2SO4), and concentrated
under reduced pressure to afford 497 mg (92%) of the desired product as a lightly colored oil. 1H NMR (300 MHz, CDCl3): δ 7.63 (d,
J = 2.1 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 7.20 (d, J = 8.2, 2.1 Hz, 1H), 3.92 (s, 3H), 2.35 (s, 3H) ppm. This compound has been
reported previously.iii
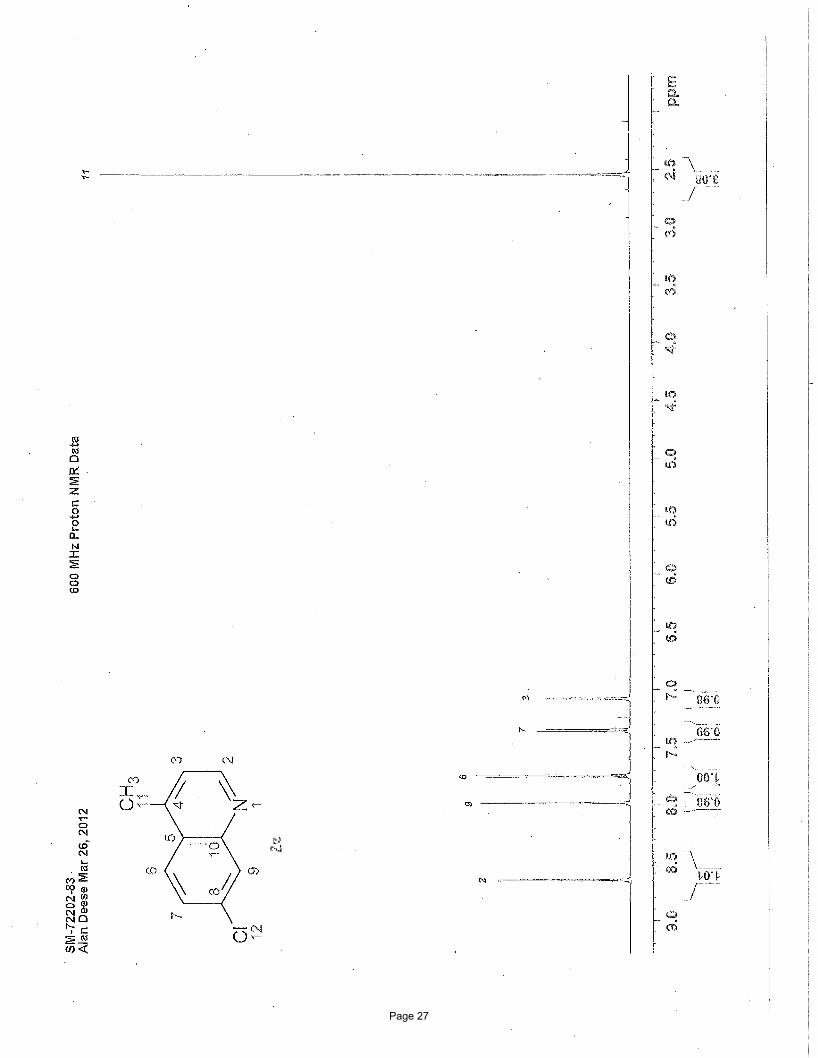
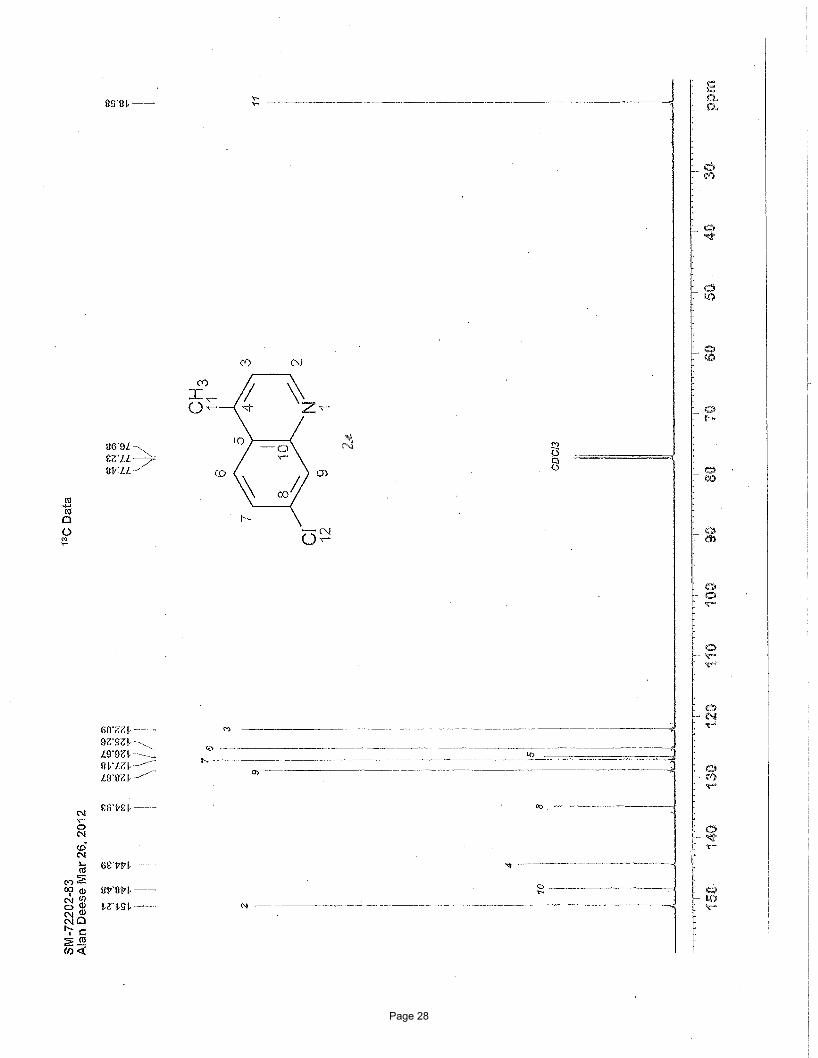
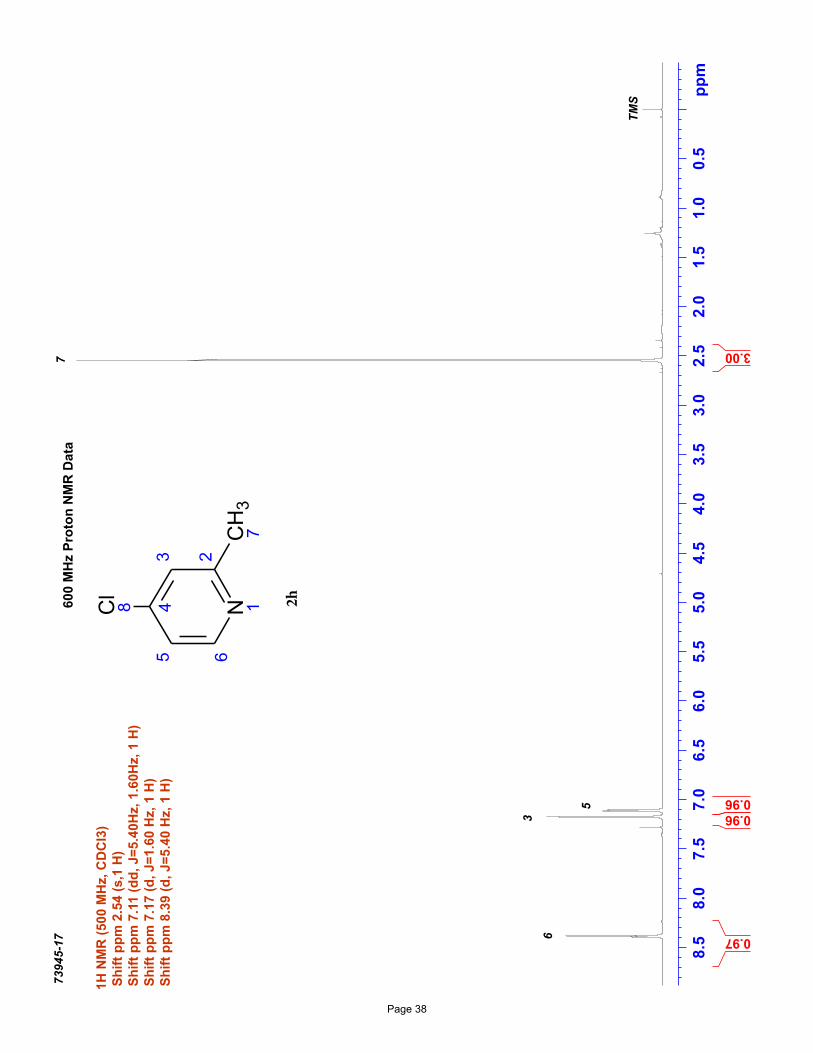
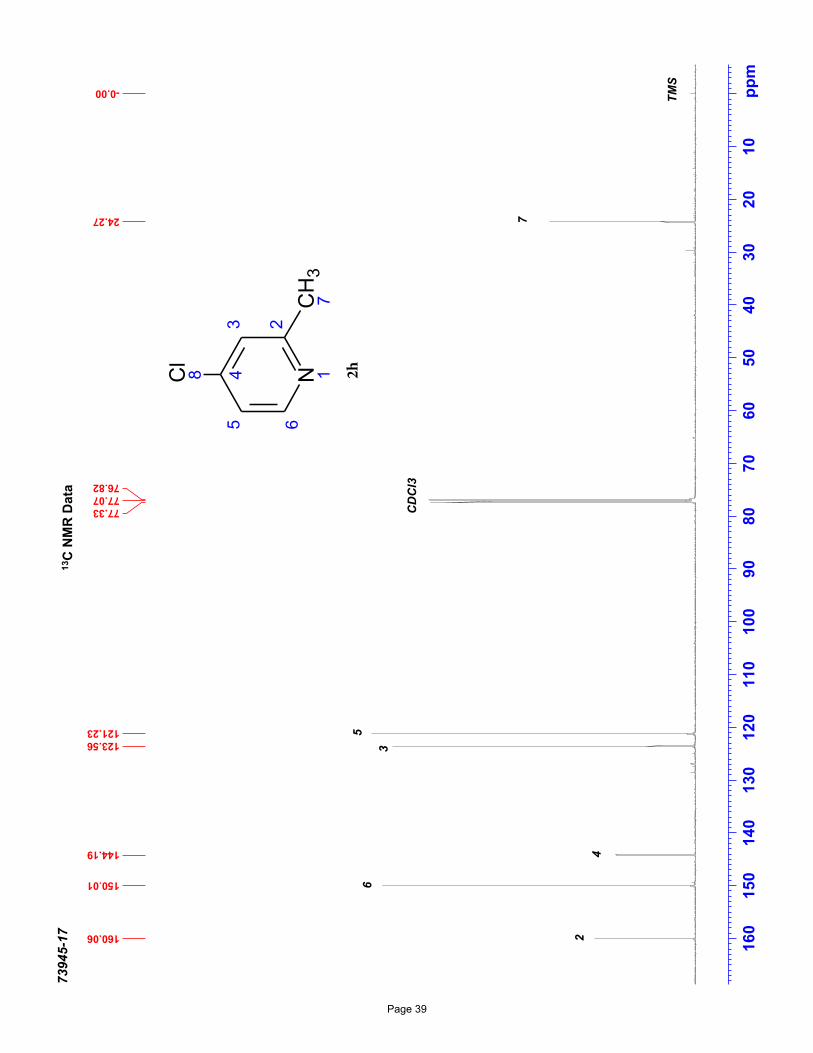
4-chloro-2-methylpyridine (2h)
MeMgBr
3.75 mol % Fe(acac)3
N Me
Cl
N Cl
Cl
4:1 THF: NMP To a vial was added 2,4-dichloropyridine (111 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4 mL) and NMP (1 mL). The reaction
mixture was treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.86 mmol, 1.15
equiv) at ambient temperature. After stirring for 4 h, the reaction was quenched by adding water (5 mL) and extracted with MTBE (2

Page 6
x 5 mL). The organic layers were combined, washed with water (10 mL), and concentrated in vacuo. Silica gel chromatography
using a gradient of 20%-50% MTBE/hexanes afforded 72 mg (76%) of the desired product as a colorless oil. TLC Rf = 0.38 (50%
EtOAc/hexanes); Spectral data matches literature known valuesiv: 1H NMR (600 MHz, CDCl3): δ 8.38 (d, J = 5.4 Hz, 1H), 7.18 (d, J =
1.2 Hz, 1H), 7.11 (dd, J = 5.4, 1.0 Hz, 1H), 2.54 (s, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 160.1, 150.0, 144.2, 123.6, 121.2, 24.3
ppm.
Structural elucidation
N
Me
ClN Me
ClH H
H
H
H
H
Eur. J. Org. Chem. 2004, 2004, 3477.
8.22 (s, 1H)
Reaction with THF:NMP(major)
8.22 (s, 1H)N
Me
Cl
H
H
H
8.38 (s, 1H)
Reaction with THF:NMP(minor)
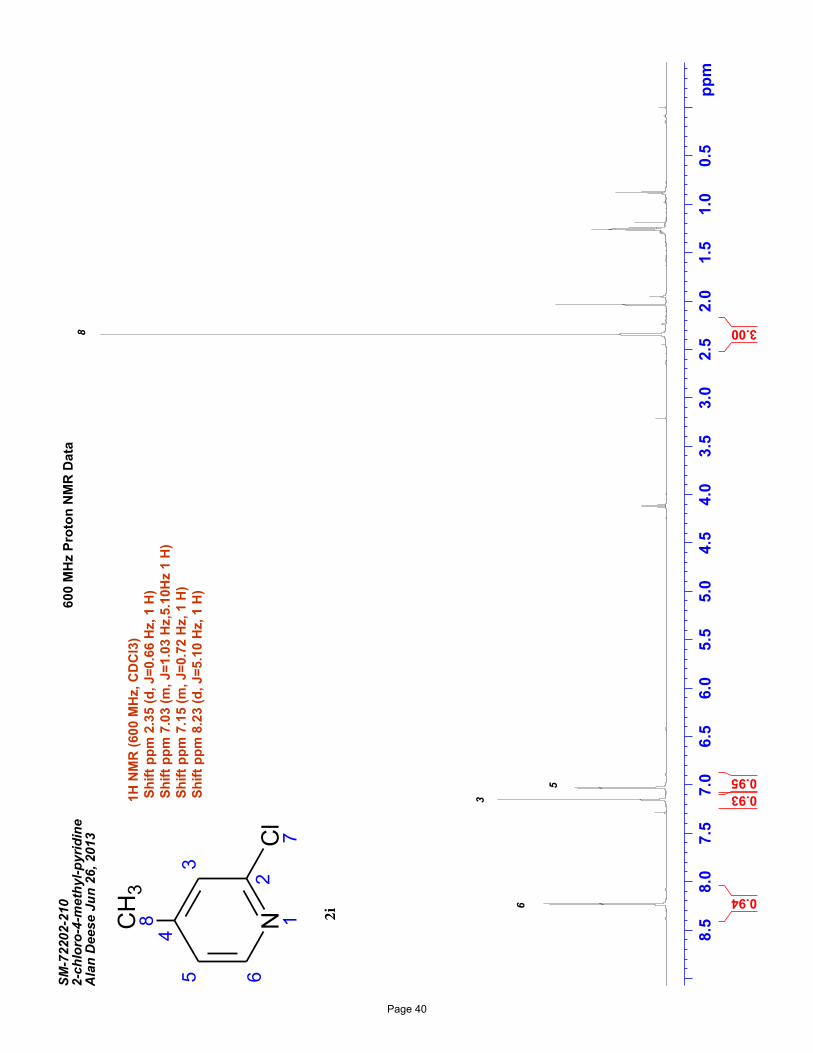
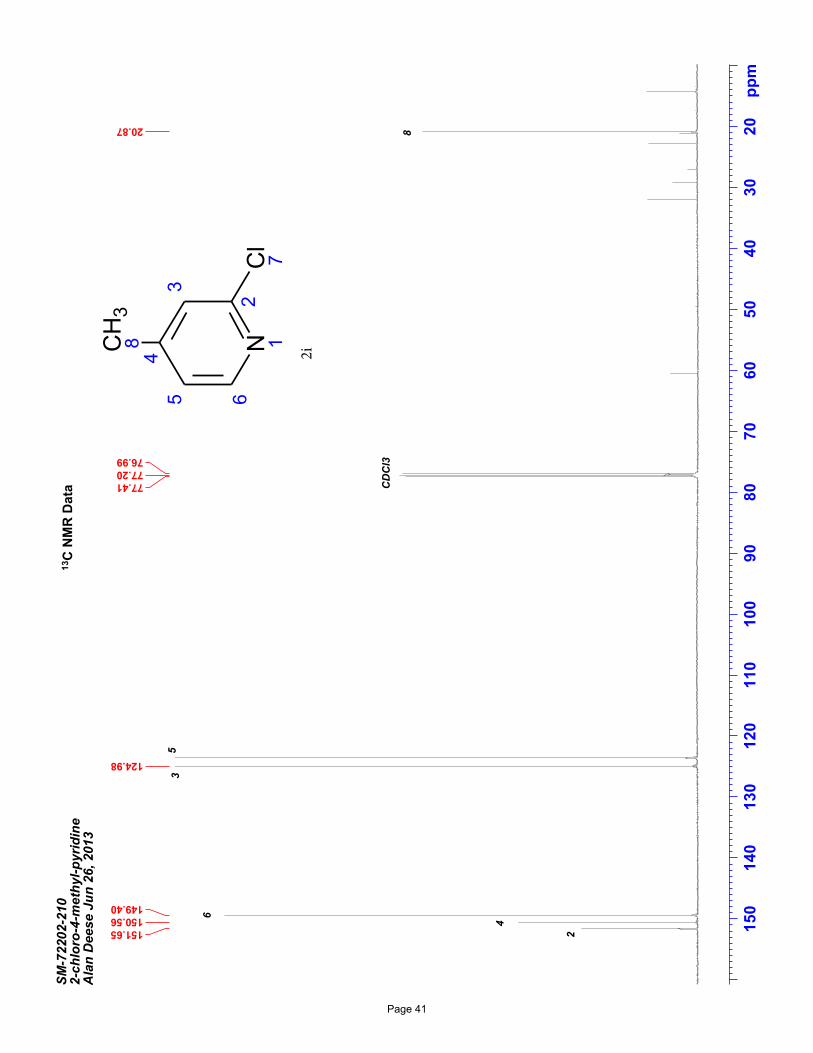
2-chloro-4-methylpyridine (2i)
MeMgBr
3.75 mol % Fe(acac)3
N Cl
Me
N Cl
Cl
THF To a vial was added 2,4-dichloropyridine (200 mg, 1.35 mmol) and Fe(acac)3 (17.9 mg, 0.051 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (9 mL). The reaction mixture was
treated dropwise with methylmagnesium bromide (1.1 mL of a 1.4 M solution in THF:toluene (1:3), 1.55 mmol, 1.15 equiv) at
ambient temperature. After stirring for 2 h, the reaction was quenched by adding satd. NH4Cl (5 mL) and extracted with EtOAc (2 x 5
mL). The organic layers were combined and concentrated in vacuo. Silica gel chromatography using a gradient of 0%-50%
EtOAc/hexanes afforded 86 mg (50%) of the desired product as a colorless oil (potential loss in yield may be due to compound
volatility). Spectral data matches literature known valuesiv: 1H NMR (600 MHz, CDCl3): δ 8.23 (d, J = 5.1 Hz, 1H), 7.15 (m, J = 1.0
Hz, 1H), 7.03 (m, J = 5.1, 1.0 Hz, 1H), 2.35 (s, 3H) ppm. Spectral data shows the presence of residual EtOAc – special care was taken
to minimize product loss due to compound volatility.
N
Me
ClN Me
ClH H
H
H
H
H
Eur. J. Org. Chem. 2004, 2004, 3477.
8.22 (s, 1H)
Reaction with just THF(minor)
8.22 (s, 1H)N
Me
Cl
H
H
H
8.38 (s, 1H)
Reaction with just THF(major)

Page 7
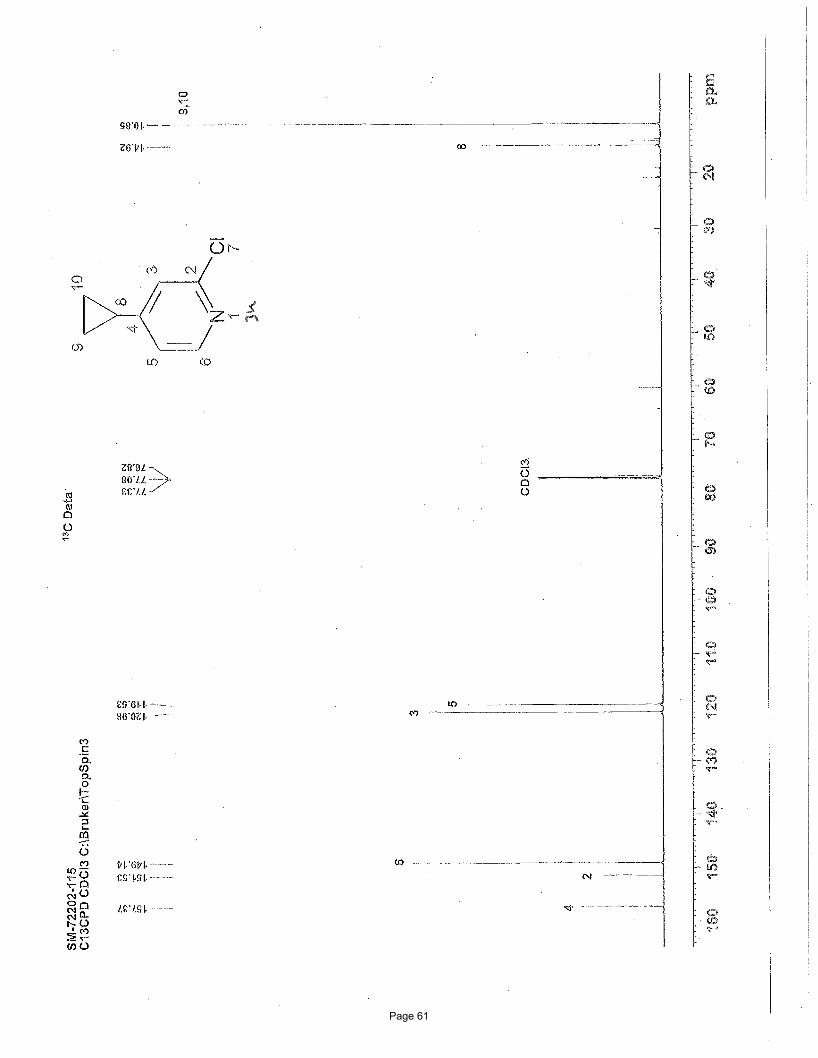
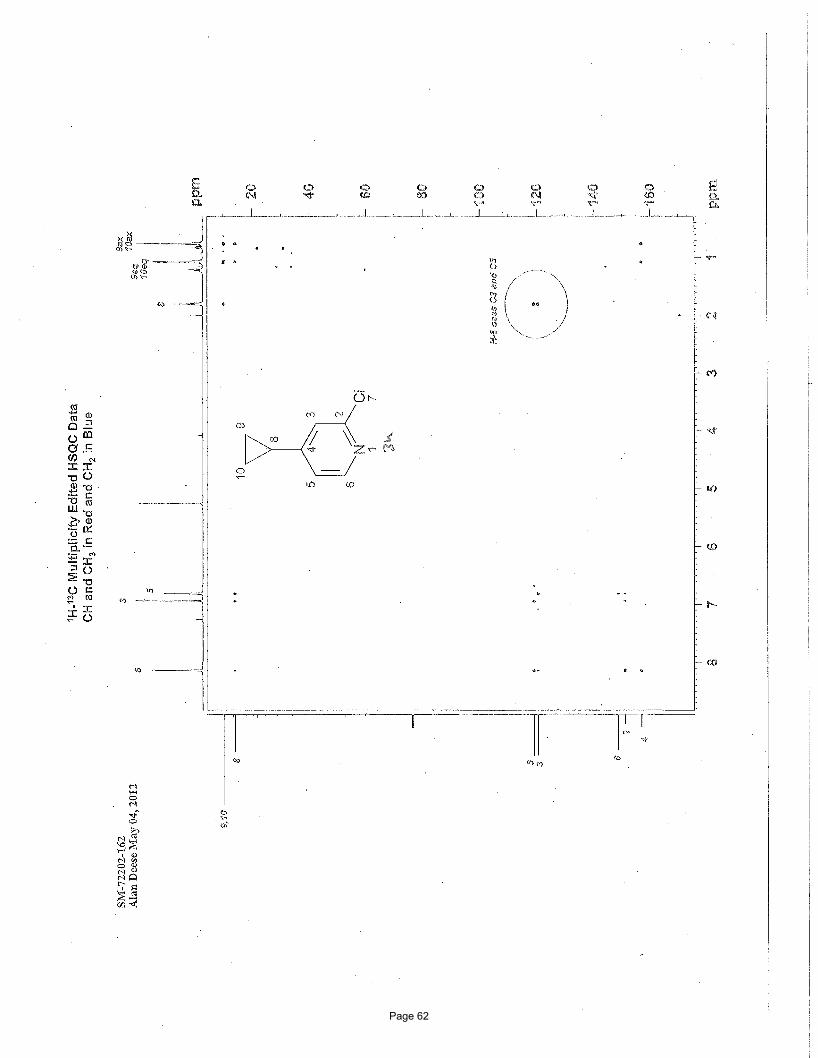
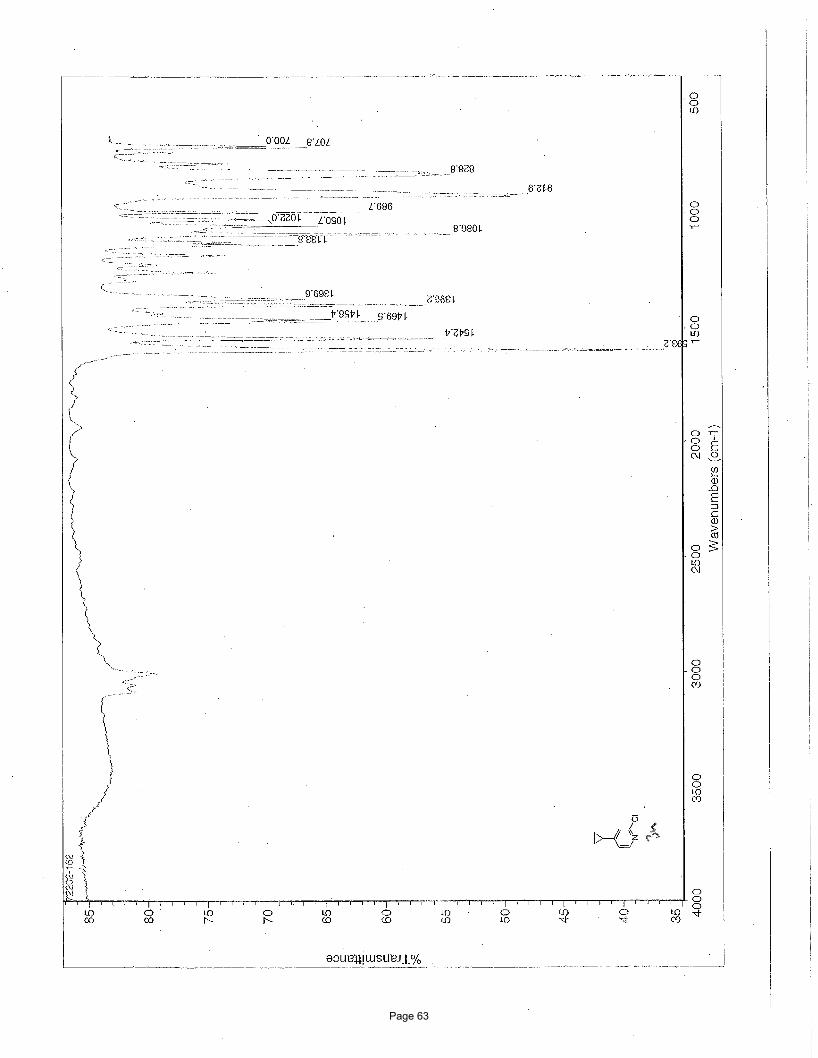
5-bromo-3-cyclopropyl-1-methylpyridin-2(1H)-one (3a)
N Me
O
BrN Me
O
Br
Br3.75 mol % Fe(acac)3
MgBr
To a vial was added 3,5-dibromo-1-methylpyridin-2(1H)-one (200 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375
equiv). The vial was sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP
(1.0 mL). The reaction mixture was treated with cyclopropylmagnesium bromide (2.48 mL of a 0.5 M solution in THF, 1.12 mmol,
1.50 equiv, added over 7.5 min using a syringe pump) at ambient temperature. After stirring for 2 h, the reaction was quenched with
saturated ammonium chloride, filtered through celite, and the resulting solids were washed with EtOAc (10 mL). The mixture was
poured into a separatory funnel, the top layer was collected and the bottom aqueous layer was extracted with EtOAc (3 x 5 mL). The
organic layers were concentrated, washed with brine (2 X 25 mL), dried (Na2SO4), and concentrated in vacuo. Silica gel
chromatography using a gradient of 0% – 50% EtOAc/hexanes to afforded 76 mg (50%) of the desired product as a light yellow solid.
m.p. = 91.9 – 93.9 ºC; TLC Rf = 0.1 (20% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 7.20, (d, J = 2.6 Hz, 1H), 6.82 (d, J = 2.6
Hz, 1H), 3.48 (s, 3H), 2.08 (m, 1H), 0.91 (m, 2H), 0.57 (m, 2H) ppm; 13C NMR (151 MHz, CDCl3): δ 162.1,137.1, 134.7, 134.6, 97.7,
37.9, 11.0, 8.25; IR (neat): υmax 1646, 1589, 1554, 1423, 1218, 1015, 906, 888, 771, 751 cm-1. Structure assigned based on analogy to
structure obtained for compound 2a.
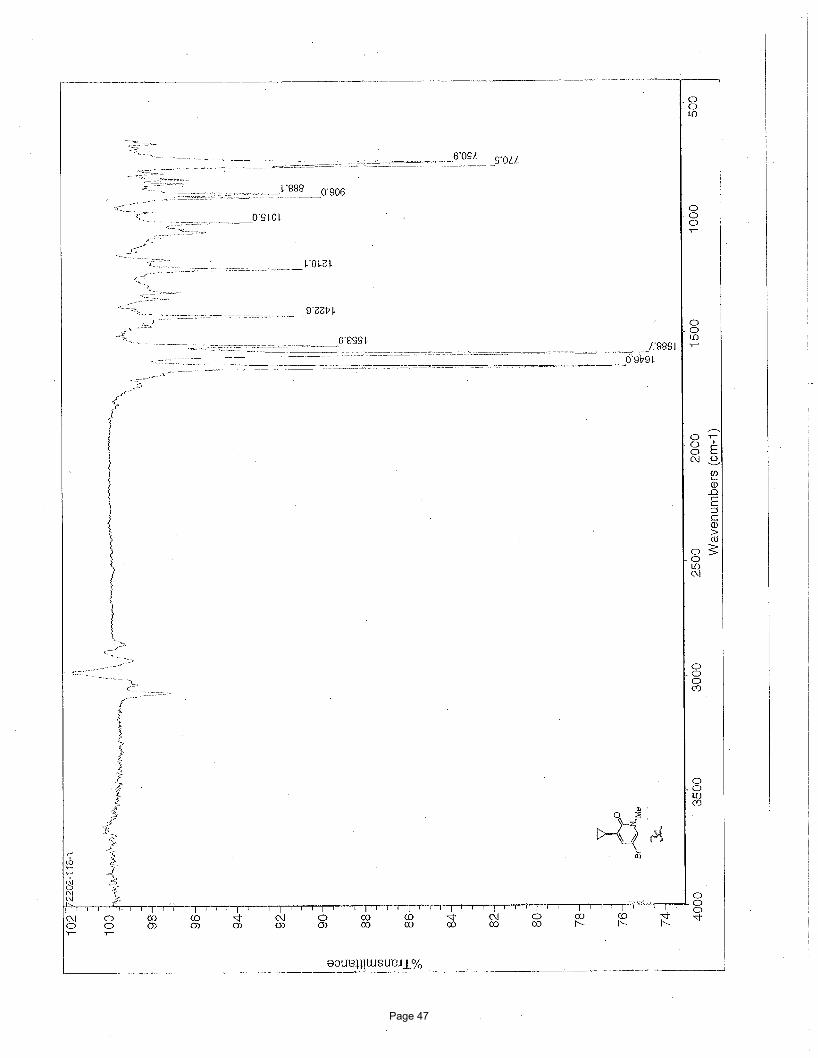
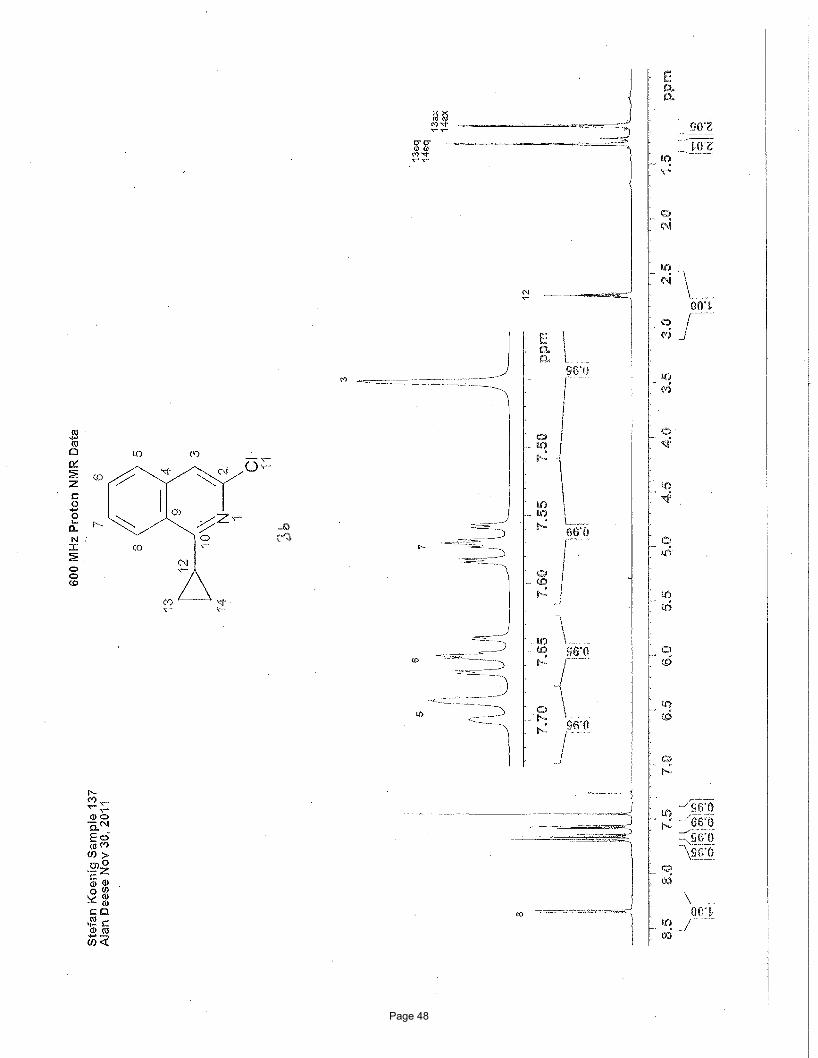
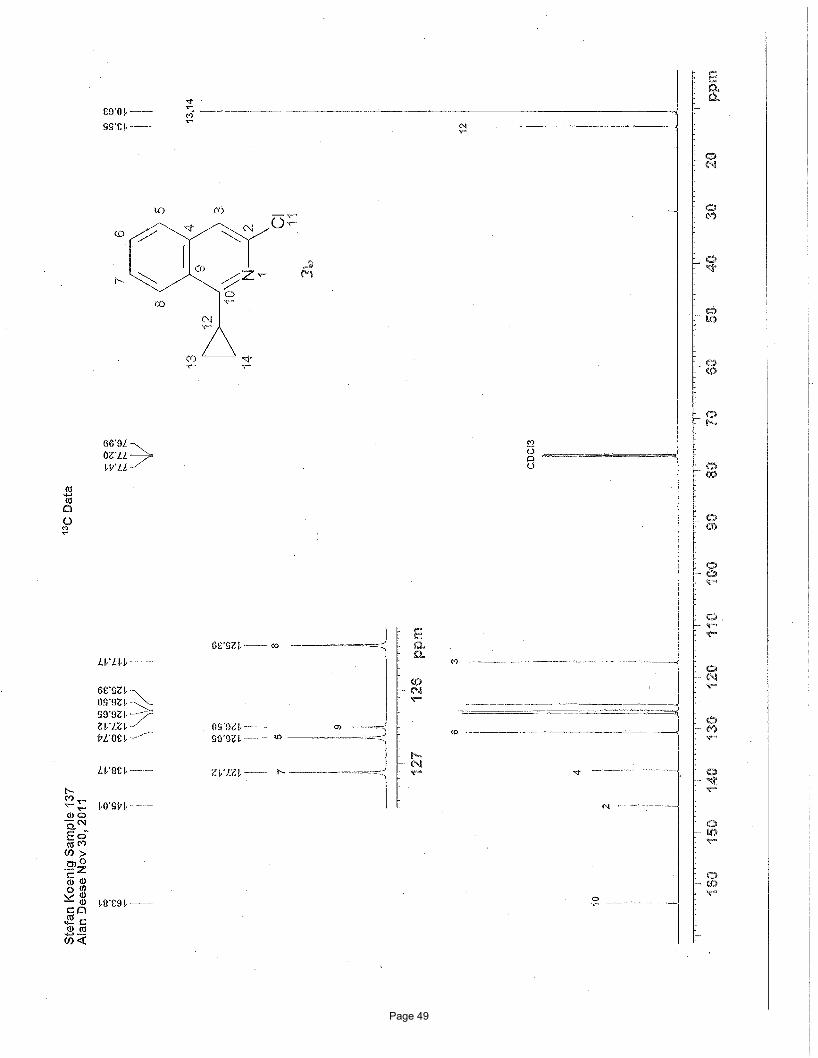
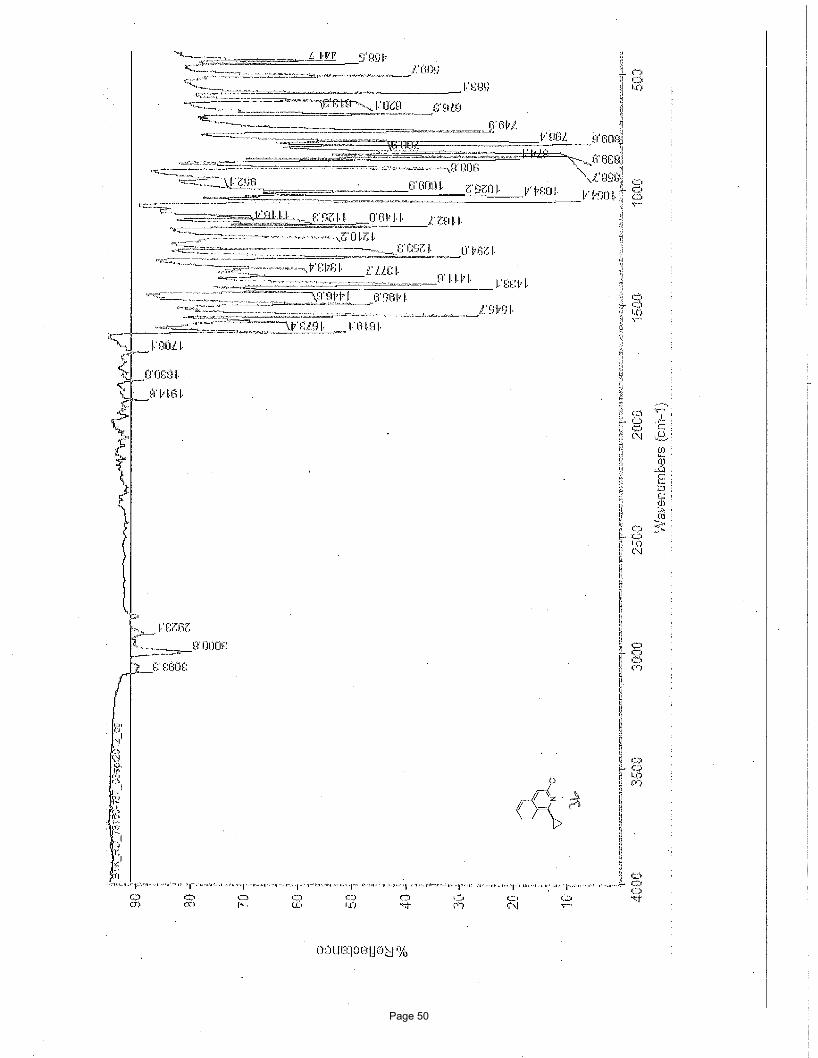
3-chloro-1-cyclopropylisoquinoline (3b)
N ClN ClCl
3.75 mol % Fe(acac)3
MgBr
To a vial was added 1,3-dichloroisoquinoline (150 mg, 0.757 mmol) and Fe(acac)3 (10 mg, 0.0283 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (5 mL). The reaction mixture was
treated dropwise with cyclopropylmagnesium bromide (3.47 mL of a 0.5 M solution in THF, 1.741 mmol, 2.30 equiv) at ambient
temperature. After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3 x 5 mL). The
organic layers were combined and concentrated in vacuo. The residue was purified by silica gel chromatography using a gradient of
0% to 10% EtOAc/hexanes, affording 0.108 g (70%) of the desired product as a lightly colored semi-solid. TLC Rf = 0.73 (20%
EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.35 (d, J = 8.52 Hz, 1H), 7.70 (d, J = 8.26 Hz, 1H), 7.65 (m, 1H), 7.57 (m, 1H),
7.45 (s, 1H), 2.70 (m, 1H), 1.31 (m, 2H), 1.14 (m, 2H) ppm; 13C NMR (151 MHz, CDCl3): δ 163.8, 145.0, 138.2, 130.7, 127.1, 126.7,
126.5, 125.4, 117.1, 13.6, 10.6; IR (neat): υmax 3093, 3001, 2923, 1619, 1573, 1546, 1433, 1412, 1294, 1163 cm-1.
Structural elucidation
N Cl
H 7.45 (s, 1H)
This work
N
H
Cl
7.55 (s, 1H)
W. O. Patent 03099274, 2003
1.04 - 1.00 (m, 4H)2.18 - 2.11 (m, 1H)
HH2.70 (m, 1H)
1.31 (m, 2H) and1.14 (m, 2H)
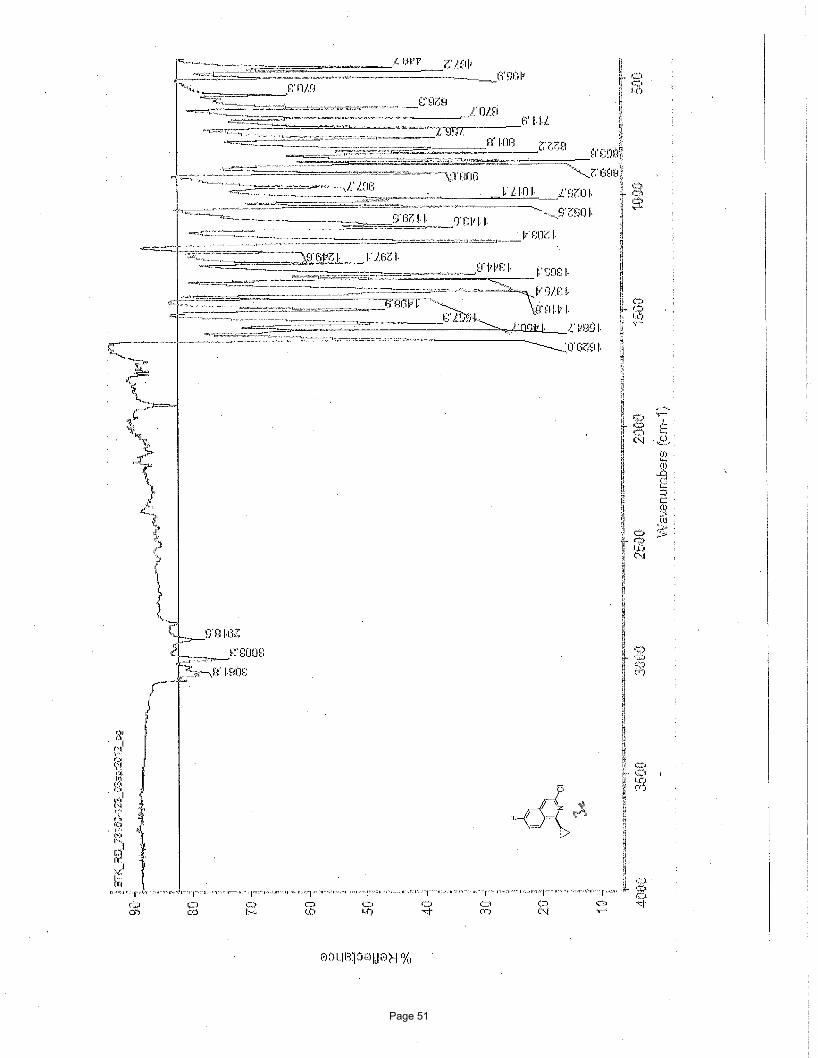
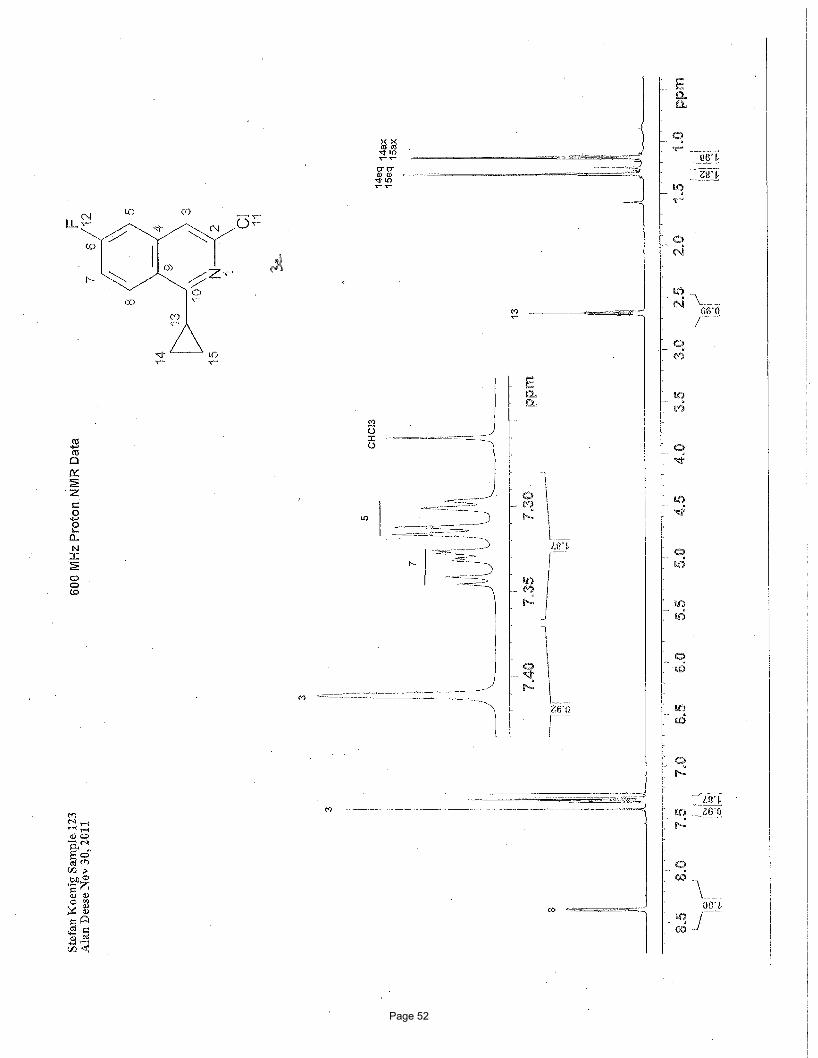
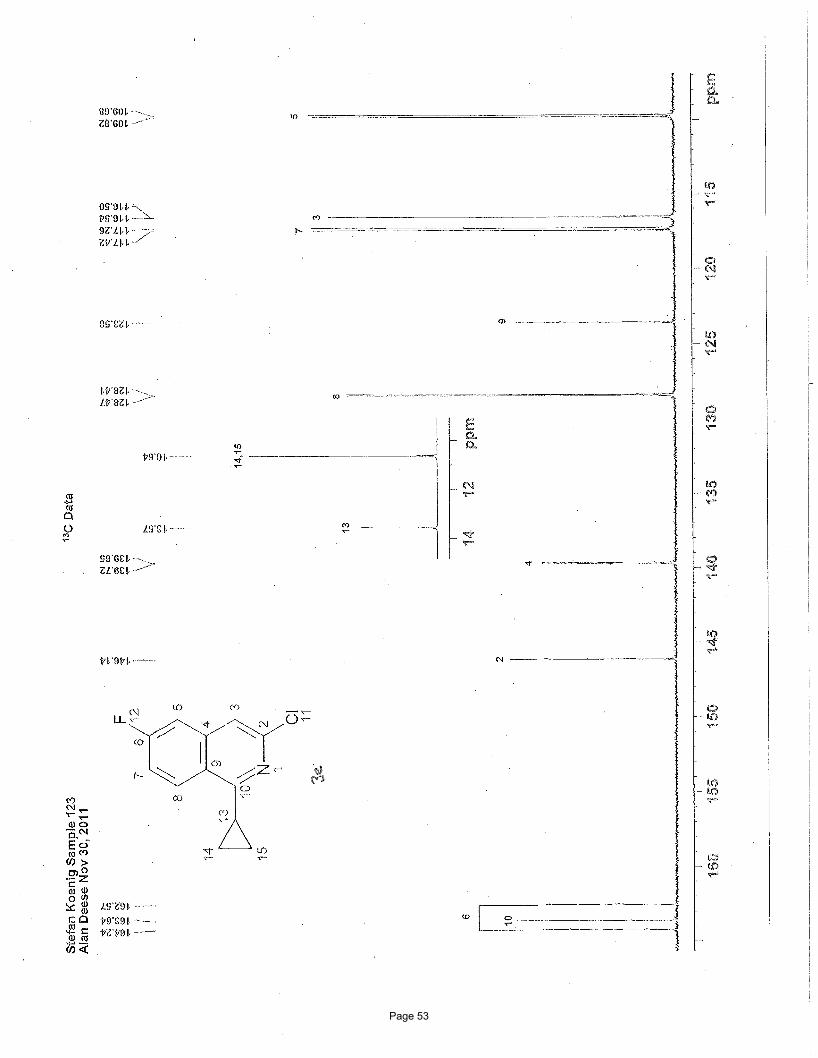
3-chloro-1-cyclopropyl-6-fluoroisoquinoline (3c)
N ClN ClCl
3.75 mol % Fe(acac)3
FF
MgBr
To a vial was added 1,3-dichloro-6-fluoroisoquinoline (163 mg, 0.757 mmol) and Fe(acac)3 (10 mg, 0.0283 mmol, 0.0375 equiv). The
vial was sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (5 mL). The reaction mixture
was treated dropwise with cyclopropylmagnesium bromide (3.47 mL of a 0.5 M solution in THF, 1.74 mmol, 2.30 equiv) at ambient
temperature. After stirring for 2 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3 x 5 mL). The

Page 8
organic layers were combined and concentrated in vacuo. The residue was purified by silica gel chromatography using a gradient of
0% to 15% EtOAc/hexanes, affording 0.118 g (70%) of the desired product as a lightly colored semi-solid. TLC Rf = 0.69 (20%
EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.38 (dd, J = 9.2, 5.41(F) Hz, 1H), 7.41 (s, 1H), 7.34 (m, 1H), 7.31 (dd, J = 9.2, 2.5
(F) Hz, 1H), 2.65 (m, 1H), 1.31 (m, 2H), 1.15 (m, 2H) ppm; 13C NMR (151 MHz, CDCl3): δ 163.6, 163.4 (d, J = 253.4 Hz, C-6 C-F),
146.1, 139.7 (d, J = 10.68 Hz, C-4a), 128.4 (d, J = 9.85 Hz, C-8), 123.6, 117.3 (d, J = 25.29 Hz, C-7), 116.5 (d, J = 5.15 Hz, C-4),
109.8 (d, J = 21.05 Hz, C-5), 13.6, 10.6 ppm; IR (neat): υmax 3062, 3003, 2919, 1629, 1585, 1557, 1451, 1365, 1345, 1203 cm-1.
Structure assigned based on analogy to structure obtained for compound 3b.
3-chloro-2-cyclopropylpyridine (3d)
N
Cl
N
Cl
Cl
3.75 mol % Fe(acac)3
MgBr
To a vial was added 2,3-dichloropyridine (111 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated with cyclopropylmagnesium bromide (1.73 mL of a 0.5 M solution in THF, 0.86 mmol, 1.15 equiv,
added over 7.5 min using a syringe pump) at ambient temperature. After stirring for 1h, the reaction mixture was treated with
additional cyclopropylmagnesium bromide (0.86 mL of a 0.5 M solution in THF, 0.43 mmol, 0.58 equiv, added over 7.5 min using a
syringe pump) at ambient temperature. After stirring for 2h, the reaction was quenched with saturated ammonium chloride, filtered
through celite, and the resulting solids were washed with EtOAc (10 mL). The mixture was poured into a separatory funnel, the top
layer was collected and the bottom aqueous layer was extracted with EtOAc (3 x 5 mL). The organic layers were concentrated,
washed with brine (2 X 10 mL), dried (Na2SO4), and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 50%
EtOAc/hexanes to afforded 81 mg (70%) of the desired product as a light yellow oil. TLC Rf = 0.72 (20% EtOAc/hexanes); 1H NMR
(600 MHz, CDCl3): δ 8.28 (dd, J = 4.6, 1.19 Hz, 1H), 7.54 (dd, J = 8.0, 1.49, 1H), 6.93 (dd, J = 8.0, 4.6 Hz, 1H), 2.48 (m, 1H), 1.07
(m, 2H), 0.99 (m, 2H) ppm; 13C NMR (X MHz, CDCl3): δ 159.6, 147.3, 136.3, 131.1, 121.0, 13.7, 10.1; IR (neat): υmax 1576, 1448,
1132, 1053, 1024, 903, 790, 755 cm-1. Structure assigned based on analogy to structure obtained for compound 2d.
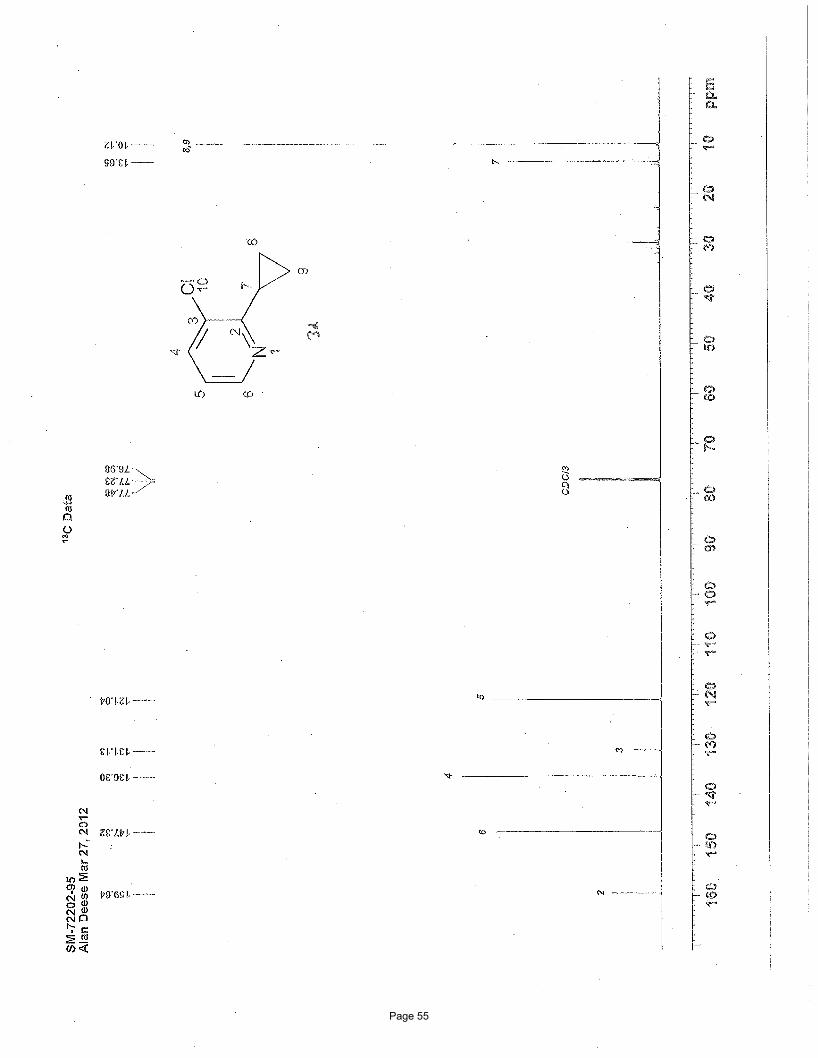
7-chloro-4-cyclopropylquinoline (3e)
NClNCl
Cl3.75 mol % Fe(acac)3
MgBr
To a vial was added 4,7-dichloroquinoline (149 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated with cyclopropylmagnesium bromide (2.26 mL of a 0.5 M solution in THF, 1.13 mmol, 1.5 equiv, added
over 7.5 min using a syringe pump) at ambient temperature. After stirring for 2h, the reaction was quenched with saturated
ammonium chloride and extracted with EtOAc (3 x 5 mL). The organic layers were combined, washed with brine, and concentrated
in vacuo. Silica gel chromatography using a gradient of 0% – 40% EtOAc/hexanes to afforded 138 mg (90%) of the desired product
as a clear oil. TLC Rf = 0.37 (20% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.72 (d, J = 4.6 Hz, 1H), 8.17 (d, J = 9.0 Hz, 1H),
8.06 (d, J = 2.1 Hz, 1H), 7.45 (dd, J = 9.0, 2.1 Hz, 1H), 6.94 (d, J = 4.6 Hz, 1H), 2.3 (m, 1H), 1.13 (m, 2H), 0.80 (m, 2H) ppm; 13C
NMR (151 MHz, CDCl3): δ 149.3, 148.0, 146.4, 133.0, 126.8, 125.2, 125.2, 123.5, 115.3, 10.0, 6.2 ppm; IR (neat): υmax 1605, 1588,
1422, 1306, 1073, 871, 820 cm-1. Structure assigned based on analogy to structure obtained for compound 2e.
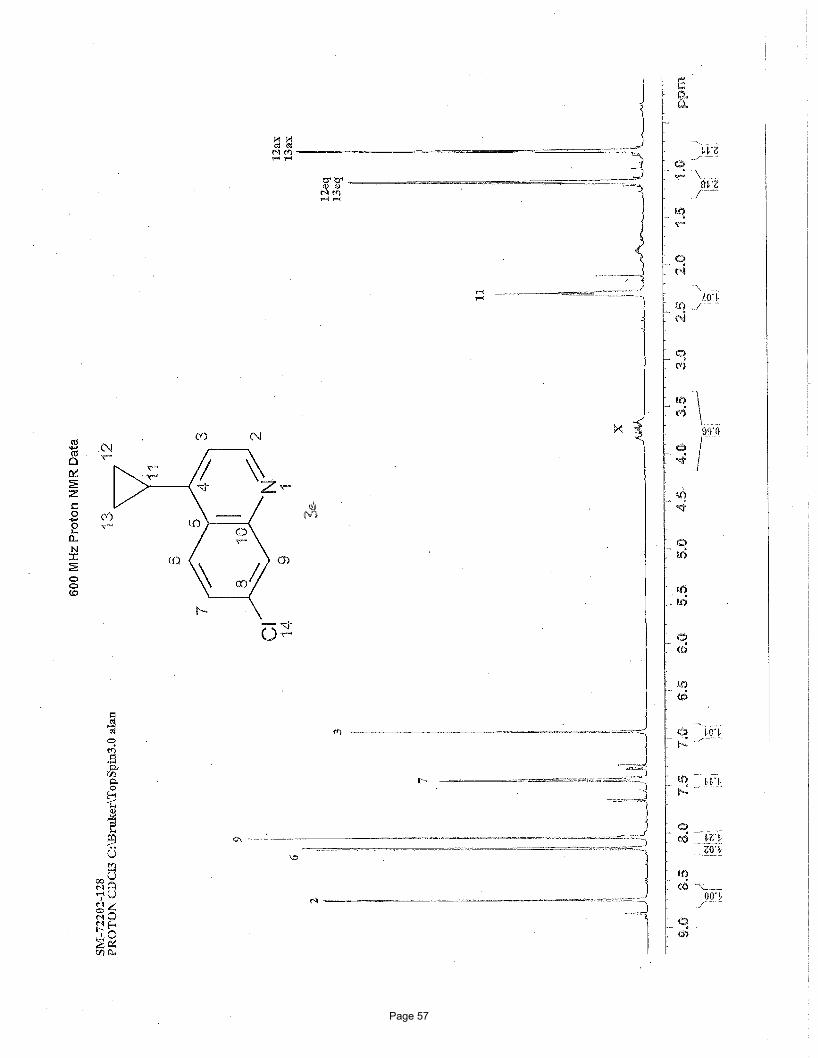
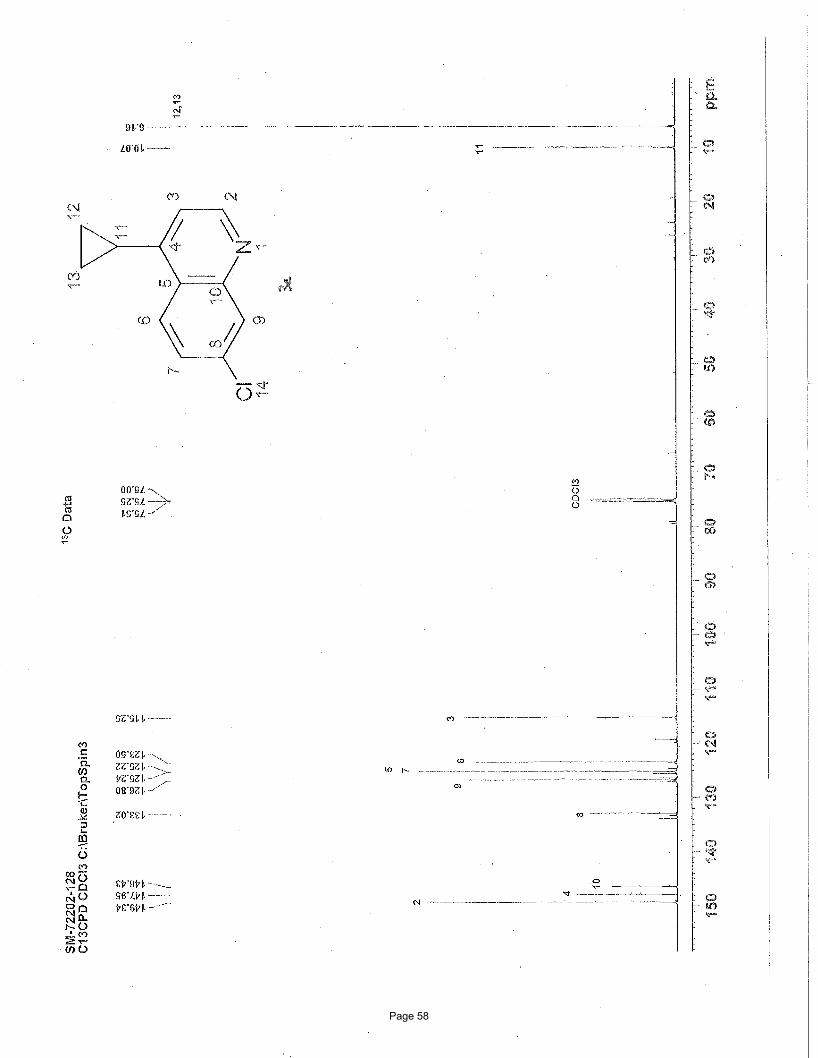
2-chloro-4-cyclopropylpyridine (3h)
N ClN Cl
Cl3.75 mol % Fe(acac)3
MgBr
To a vial was added 2,4-dichloropyridine (111 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The

Page 9
reaction mixture was treated with cyclopropylmagnesium bromide (2.25 mL of a 0.5 M solution in THF, 1.13 mmol, 1.5 equiv, added
over 7.5 min using a syringe pump) at 0 ºC. After stirring for 30 min, the reaction mixture was treated with additional
cyclopropylmagnesium bromide (0.8 mL of a 0.5 M solution in THF, 0.375 mmol, 0.5 equiv, added over 7.5 min using a syringe
pump) at 0 ºC. After stirring for 2h, the reaction was quenched with saturated ammonium chloride and extracted with EtOAc (3 x 5
mL). The organic layers were combined, washed with brine, and concentrated in vacuo. Silica gel chromatography using a gradient
of 0% – 40% EtOAc/hexanes to afforded 88 mg (76%) of the desired product as a clear oil. TLC Rf = 0.5 (20% EtOAc/hexanes); 1H
NMR (600 MHz, CDCl3): δ 8.17 (d, J = 5.2 Hz, 1H), 6.97 (d, J = 1.0 Hz, 1H), 6.84 (dd, J = 5.2, 1.5 Hz, 1H), 1.84 (m, 1H), 1.11 (m,
1H), 0.8 (m, 1H) ppm; 13C NMR (151 MHz, CDCl3): δ 159.6, 147.3, 136.3, 131.13, 121.0, 13.7, 10.1 ppm; IR (neat): υmax 1593, 1542,
1470, 1458, 1396, 10887, 913, 827cm-1.
Structural elucidation
N Cl
H
• •• HMBCHMBC
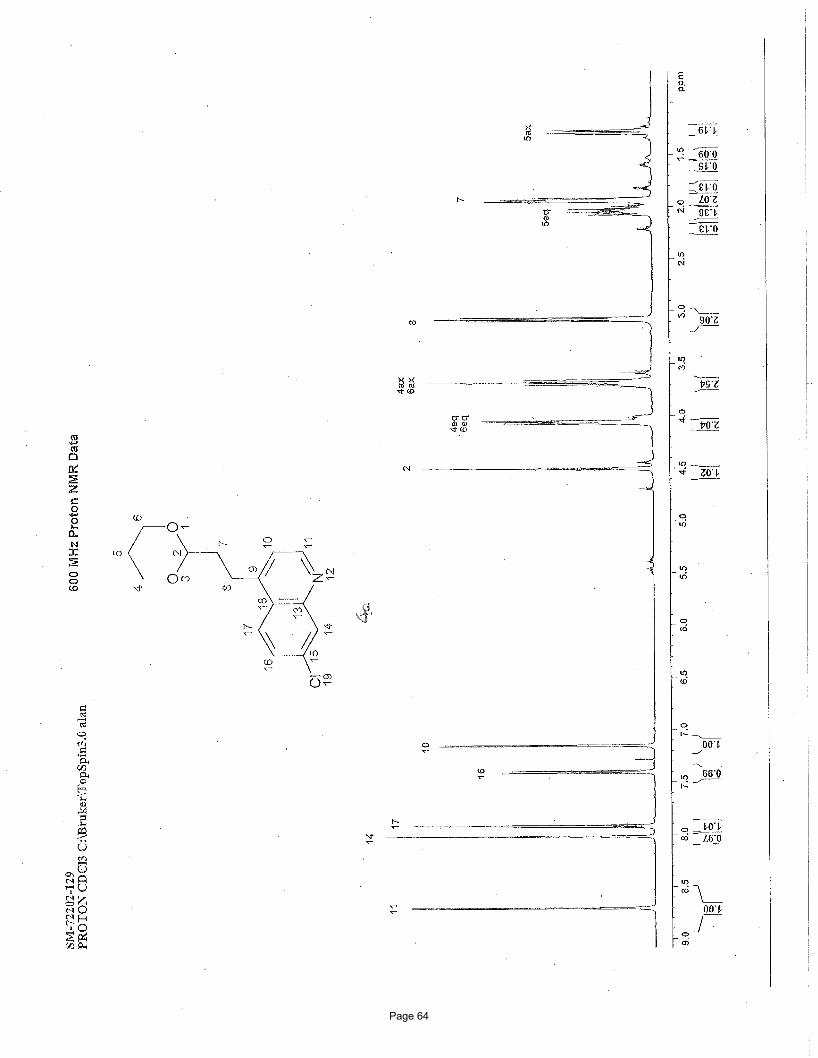
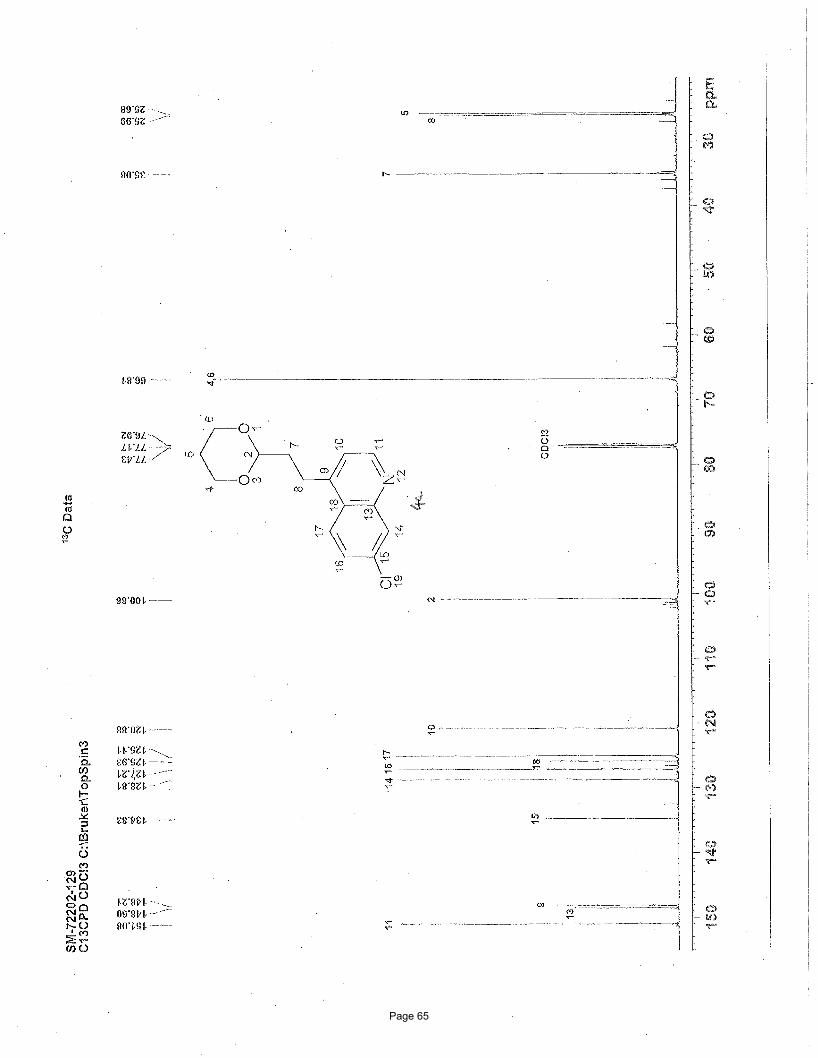
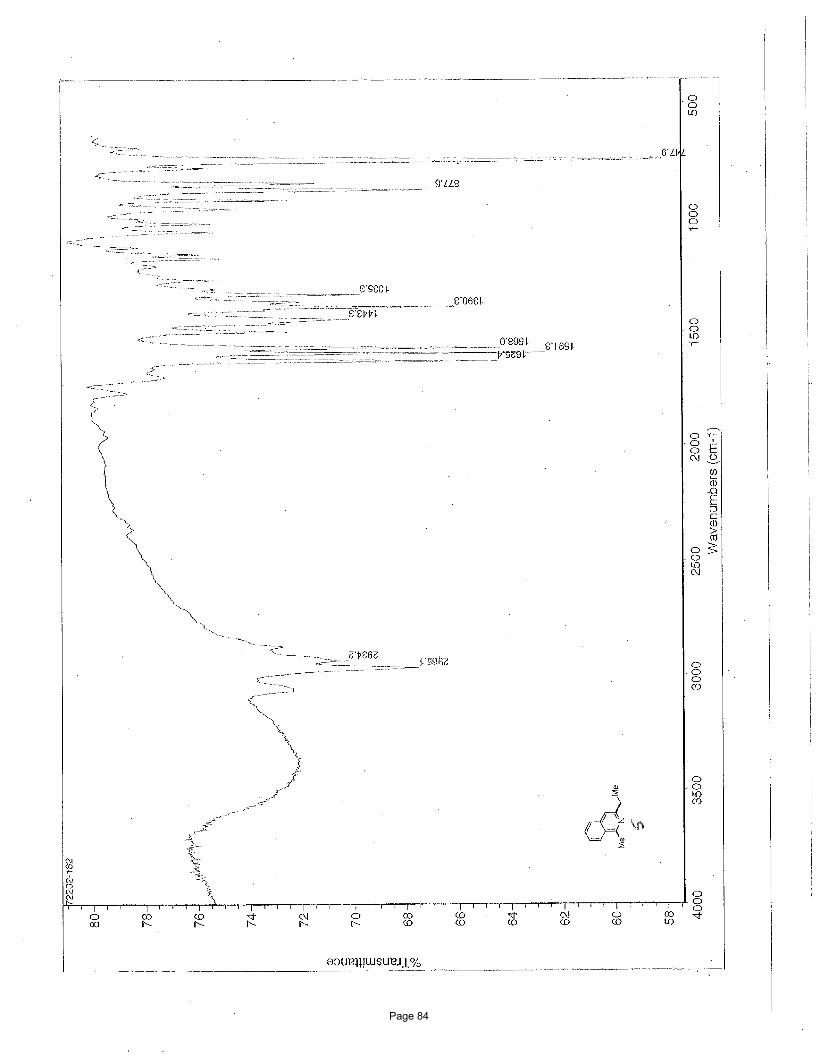
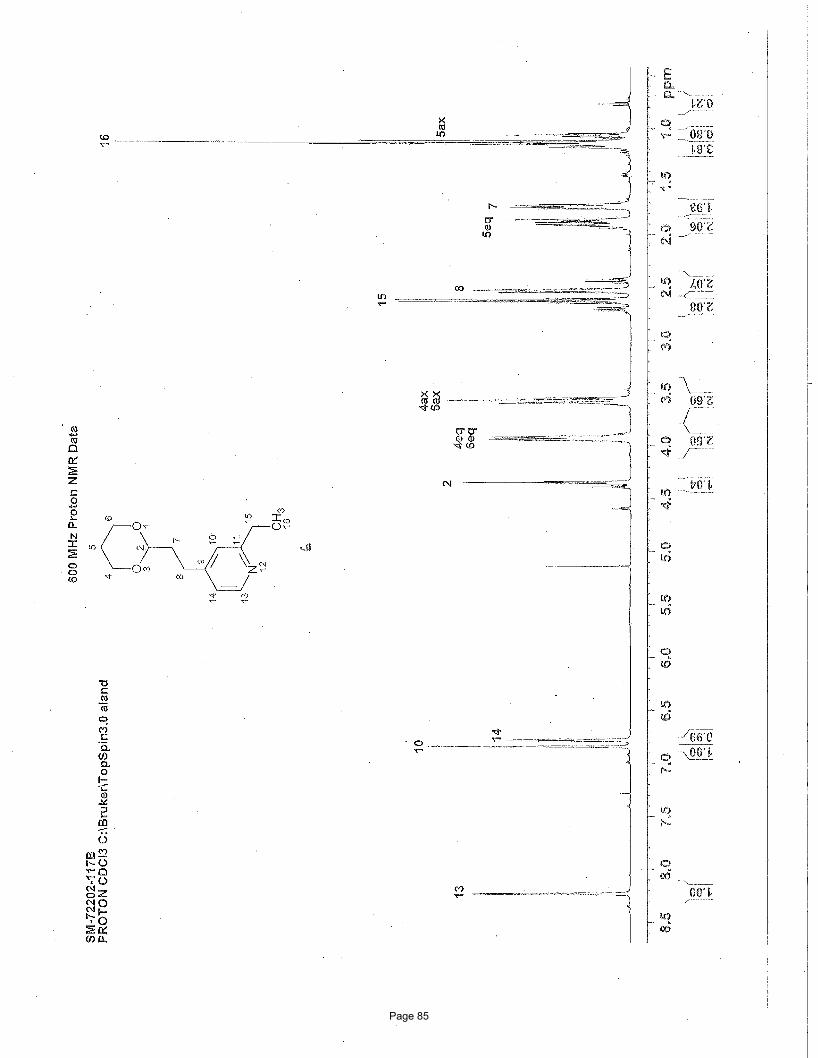
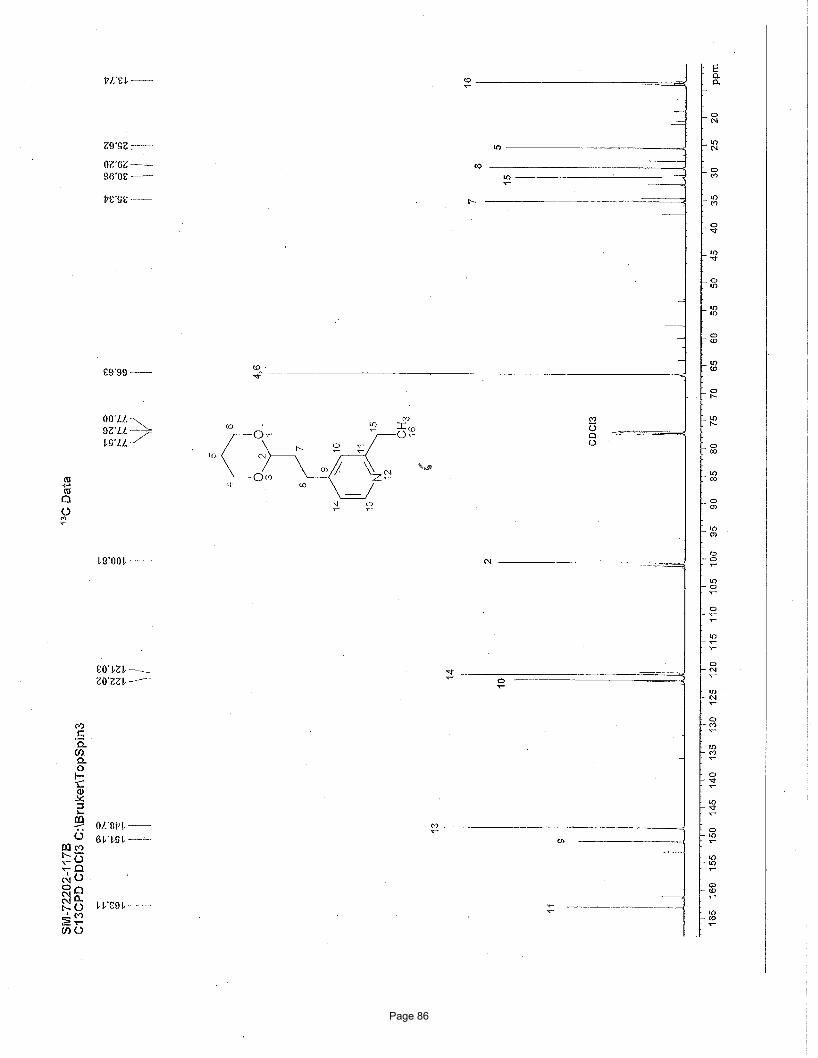
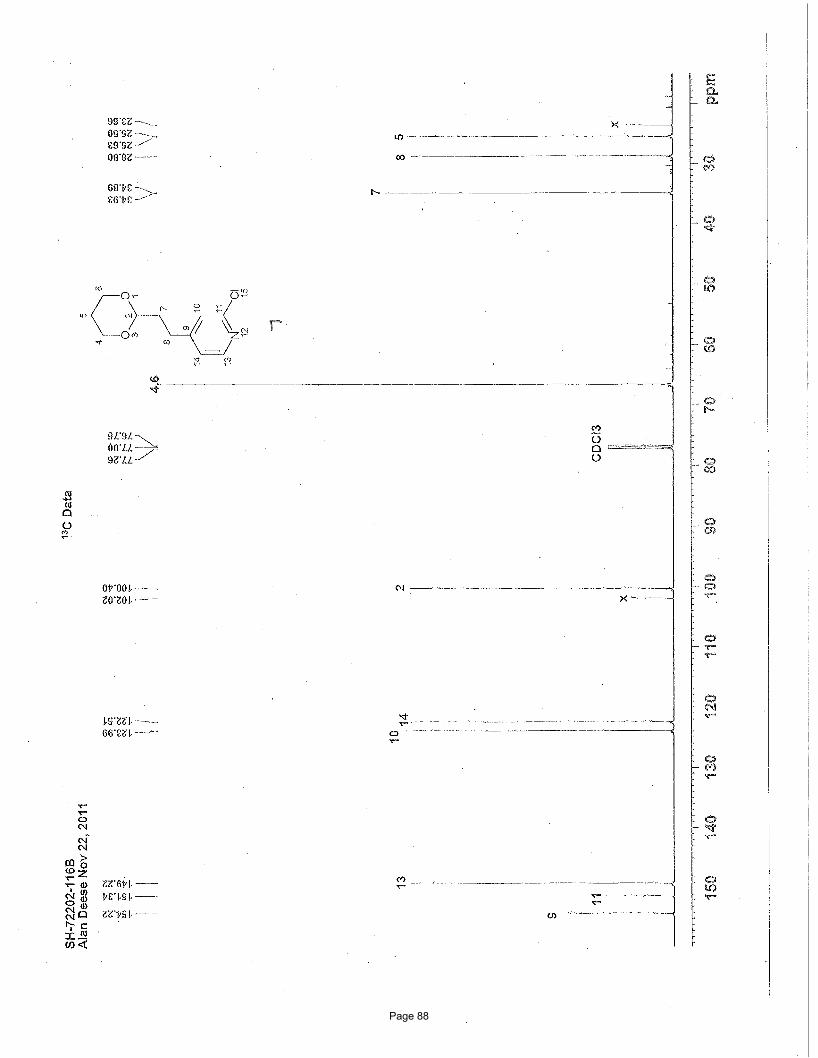
4-(2-(1,3-dioxan-2-yl)ethyl)-7-chloroquinoline (4a)
3.75 mol % Fe(acac)3
NCl
Cl
NCl
O
O
O
OBrMg
To a vial was added 4,7-dichloroquinoline (150 mg, 0.76 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated dropwise with (1,3-dioxan-2-ylethyl)magnesium bromide (2.26 mL of a 0.5 M solution in THF, 1.5
equiv) at ambient temperature. After stirring for 4 h, the reaction was quenched by adding water (5 mL) and extracted with EtOAc (3
x 5 mL). The organic layers were combined and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 50%
EtOAc/hexanes afforded 188 mg (90%) of the desired product as a colorless oil. TLC Rf = 0.12 (20% EtOAc/hexanes); 1H NMR (600
MHz, CDCl3): δ 8.71 (d, J = 4.6 Hz, 1H), 8.03 (d, J = 2.1 Hz, 1H), 7.93 (d, J = 9.0 Hz, 1H), 7.41 (dd, J = 9.0, 2.1 Hz, 1H), 7.16 (d, J=
4.6 Hz, 1H), 4.51 (t, J = 4.84 Hz, 1H), 4.06 (m, 2H), 3.69 (m, 2H), 3.08 (t, J = 16.0, 8.0 Hz, 2H), 2.04 (m, 1H), 1.94 (m, 2H), 1.29 (d,
J = 13.4 Hz, 1H) ppm; 13C NMR (151 MHz, CDCl3): δ 151.1, 148.6, 148.2, 134.8, 128.8, 127.2, 125.9, 125.1, 120.9, 110.9, 66.8, 35.1,
26.0, 25.7 ppm; IR (neat): υmax 2961, 2852, 1591, 1241, 1145, 1131, 887 cm-1. Structure assigned based on analogy to structure
obtained for compound 2e.
7-chloro-4-propylquinoline (4b)
3.75 mol % Fe(acac)3
NCl
Cl
NCl
Me
MeBrMg
To a vial was added 4,7-dichloroquinoline (150 mg, 0.76 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated dropwise with n-propylmagnesium bromide (0.43 mL of a 2.0 M solution in THF, 1.15 equiv) at ambient
temperature. After stirring for 75 min, the reaction was quenched by adding saturated aqueous NH4Cl (3 mL) and extracted with
MTBE (15 mL). The organic layer was concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 50%
EtOAc/hexanes afforded 120 mg (78%) of the desired product as a colorless oil. 1H NMR (600 MHz, CDCl3): δ 8.72 (d, J = 4.4 Hz,
1H), 8.04 (d, J = 2.2 Hz, 1H), 7.86 (d, J = 9.0 Hz, 1H), 7.40 (dd, J = 9.0, 2.2 Hz, 1H), 7.12 (d, J = 4.4 Hz, 1H), 2.95 (m, 2H), 1.71 (m,
2H), 0.95 (t, J = 7.5 Hz, 3H) ppm; 13C NMR (151 MHz, CDCl3): δ 151.2, 148.8, 148.7, 134.8, 129.0, 127.2, 126.1, 125.1, 121.0, 34.0,
23.2, 14.1 ppm; IR (neat): υmax 2860, 2931, 2871, 1605, 1589, 1497, 881, 833 cm-1. Structure assigned based on analogy to structure
obtained for compound 2e.

Page 10
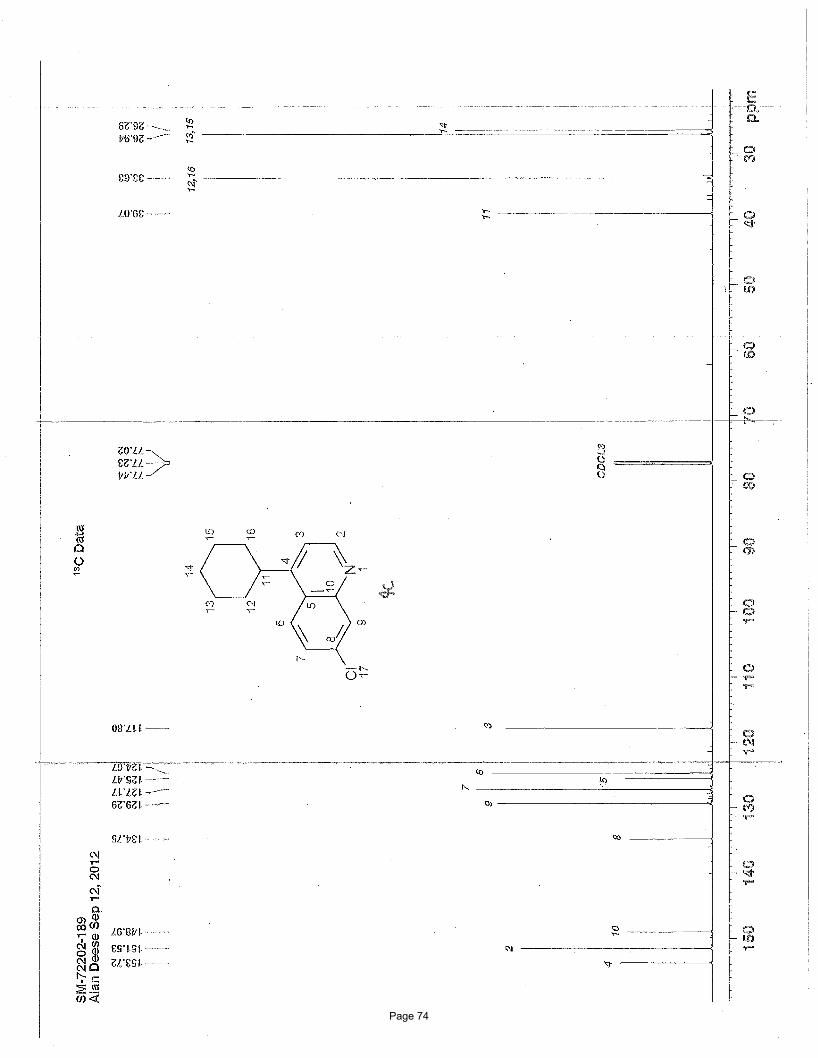
7-chloro-4-cyclohexylquinoline (4c)
3.75 mol % Fe(acac)3
NCl
Cl
NClBrMg
To a vial was added 4,7-dichloroquinoline (150 mg, 0.76 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated dropwise with cyclohexylmagnesium bromide (0.95 mL of a 17% solution in THF g, ~1.0 M, 1.15 equiv)
at ambient temperature. After stirring for 75 min, the reaction was quenched by adding saturated aqueous NH4Cl (3 mL) and extracted
with MTBE (15 mL). The organic layer was concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 50%
EtOAc/hexanes afforded 93 mg (68%) of the desired product as a colorless oil. 1H NMR (600 MHz, CDCl3): δ 8.81 (d, J = 4.6 Hz,
1H), 8.09 (d, J = 2.3 Hz, 1H), 7.99 (d, J = 9.1 Hz, 1H), 7.46 (dd, J = 9.1, 2.3 Hz, 1H), 7.24 (d, J = 4.6 Hz, 1H), 3.24 (m, 1H), 1.94-
1.32 (m, 10H) ppm; 13C NMR (151 MHz, CDCl3): δ 153.7, 151.5, 149.0, 134.8, 129.3, 127.2, 125.5, 124.7, 117.8, 39.0, 33.6, 27.0,
26.3 ppm; IR (neat): υmax 2925, 2851, 1603, 1586, 1497, 1448, 881, 860, 822 cm-1. Structure assigned based on analogy to structure
obtained for compound 2e.
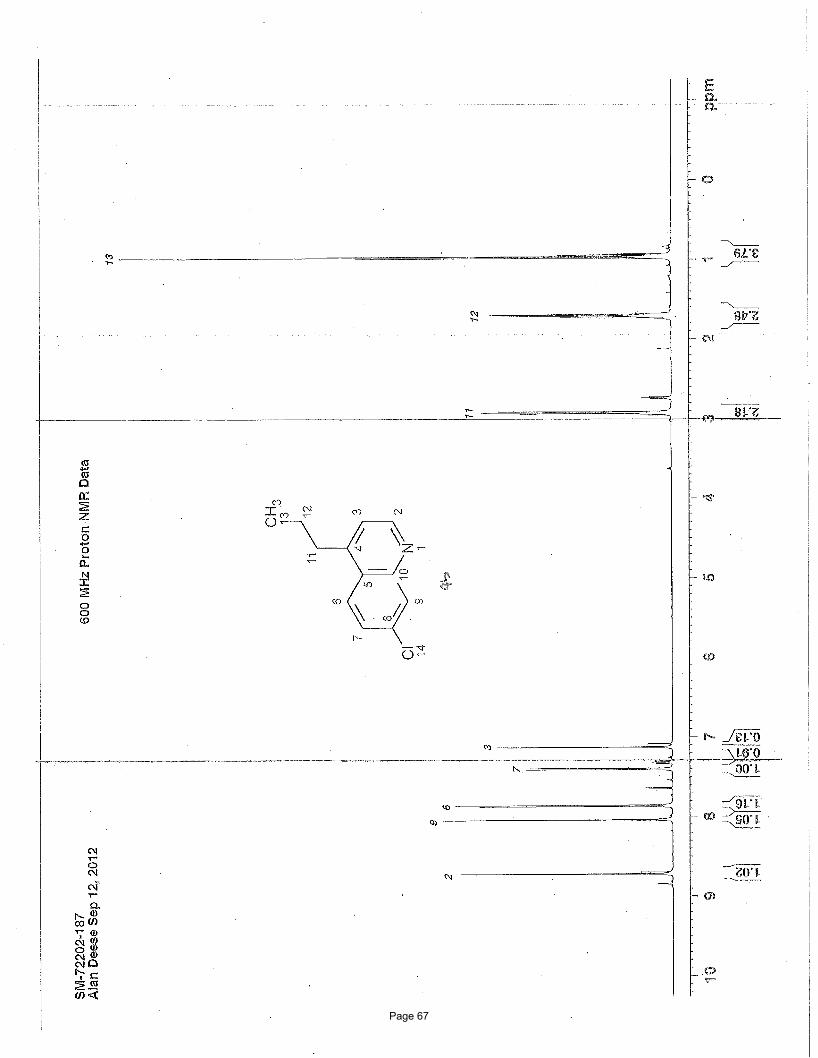
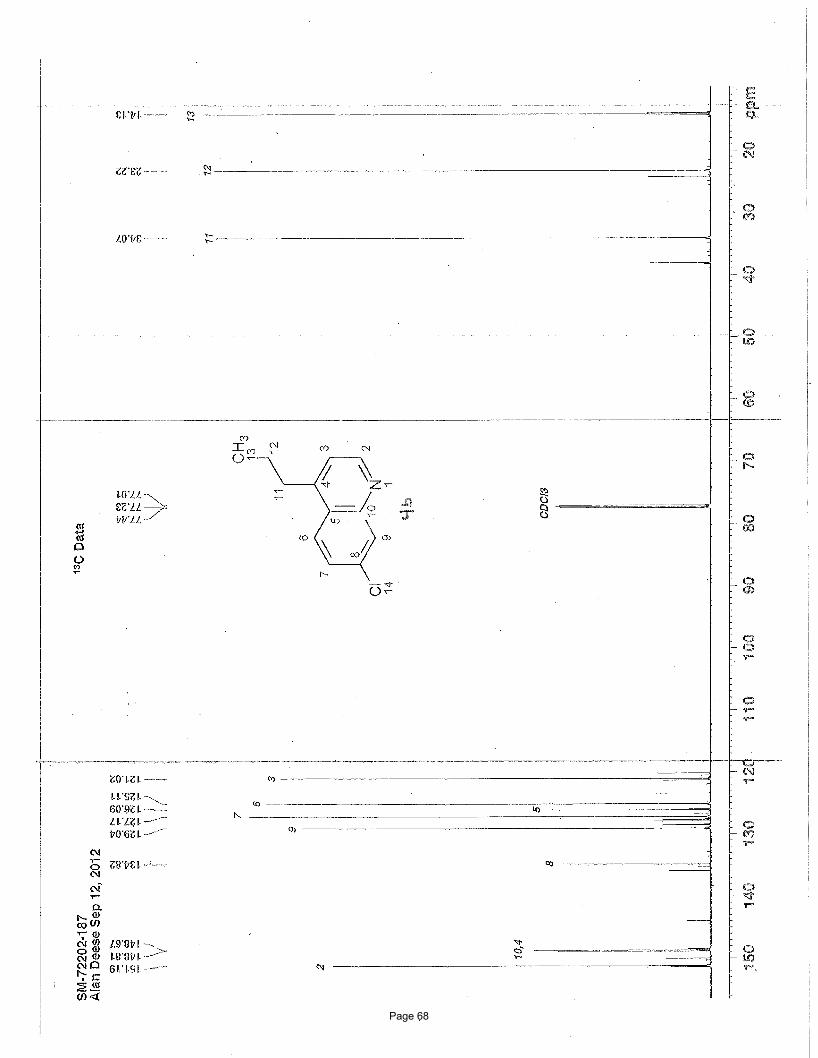
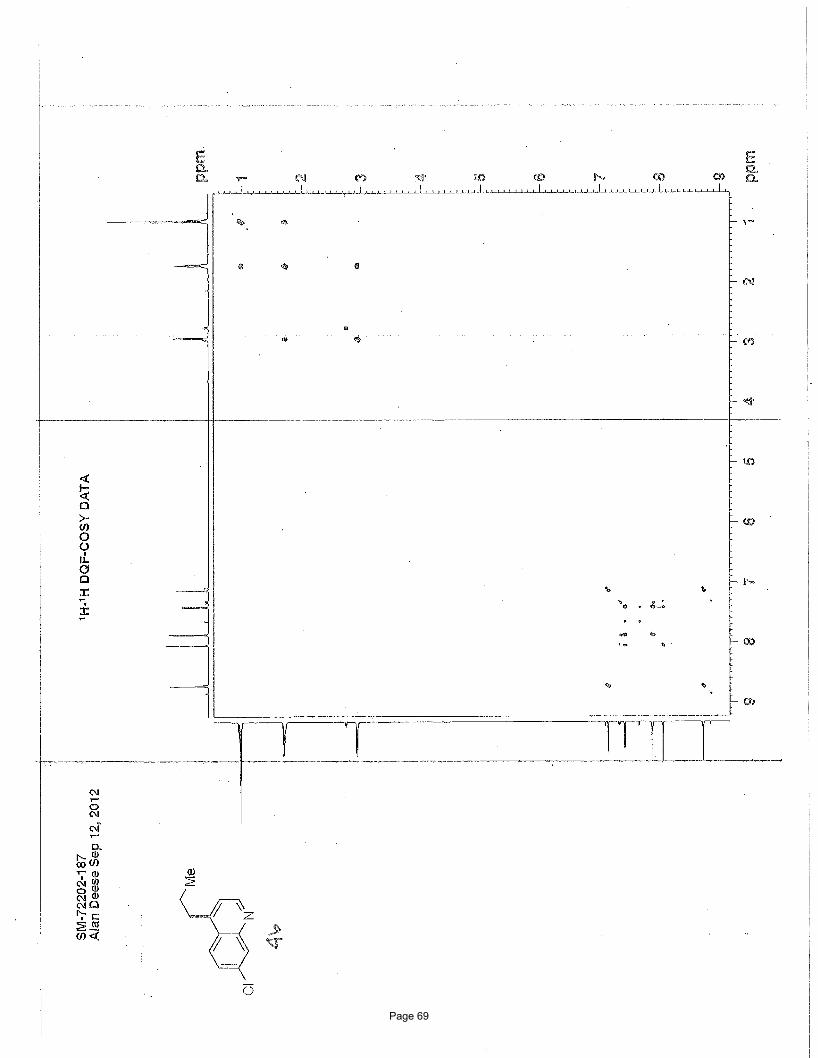
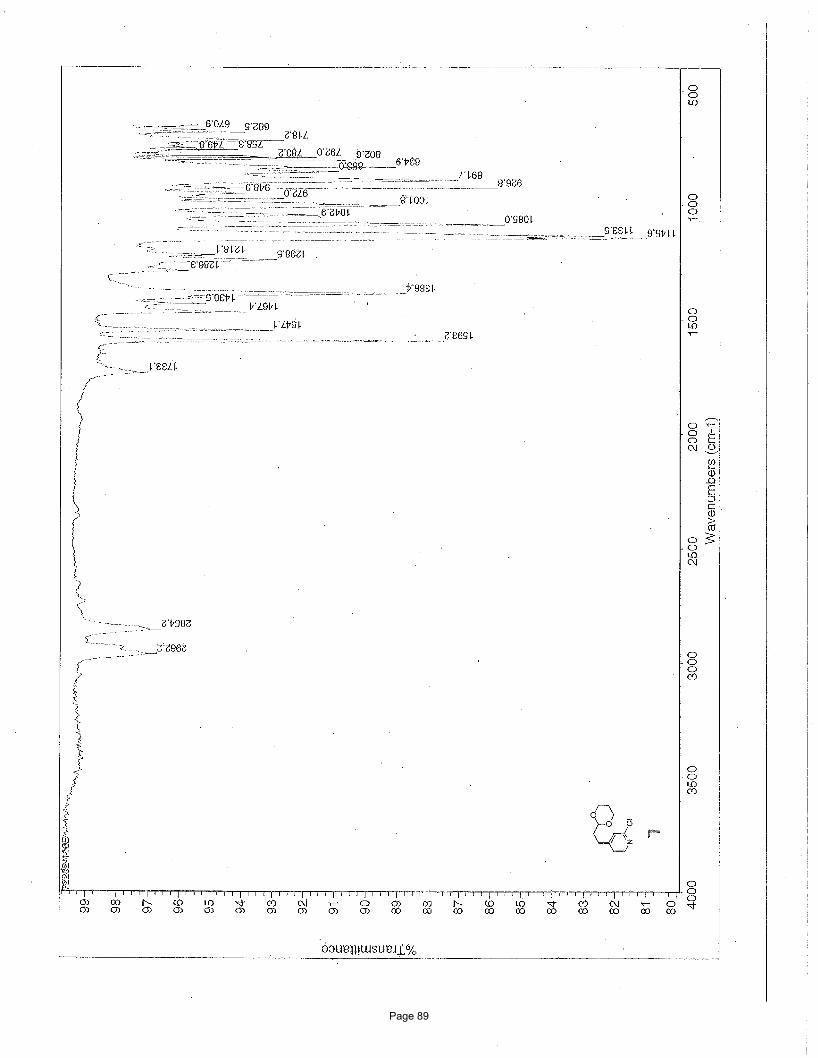
3-ethyl-1-methylisoquinoline (5)
NMeN ClCl
3.75 mol % Fe(acac)3
MeMgBr, THF; then3.75 mol % Fe(acac)3
NMP, EtMgBr
Me
To a vial was added 1,3-dichloroisoquinoline (149 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (5.0 mL). The reaction mixture was
treated dropwise with methylmagnesium bromide (0.62 mL of a 1.4 M solution in THF:toluene (1:3), 0.86 mmol, 1.15 equiv) at
ambient temperature. After stirring for 2h (reaction monitored by HPLC for complete conversion), a solution of Fe(acac)3 (9.9 mg,
0.028 mmol, 0.0375 equiv) in NMP (1 mL) and a solution of ethylmagnesium bromide (1.5 mL of a 1.0 M solution in THF, 2.0 equiv)
was added over 7.5 min at ambient temperature using a syringe pump. After stirring for 2h, the reaction was quenched by adding
saturated aqueous NH4Cl (10 mL), filtered through celite, and poured into a separatory funnel. The top organic layer was collected
and the aqueous layer was extracted with EtOAc (3 x 5 mL). The organic layers were combined and washed with brine (2 x 25 mL),
dried (Na2SO4), and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 100% EtOAc/hexanes afforded 107
mg (84%) of the desired product as a light yellow oil. TLC Rf = 0.38 (20% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.03 (d, J
= 8.5 Hz, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.59 (m, 1H), 7.47 (m, 1H), 7.30 (s, 3H), 2.95 (q, J = 7.7 Hz, 1H), 2.93 (s, 3H), 1.39 (t, J = 7.7
Hz, 1H) ppm; 13C NMR (151 MHz, CDCl3): δ 157.8, 155.4, 136.6, 129.6, 126.6, 125.9, 125.7, 125.3, 30.9, 22.0, 14.0 ppm; IR (neat):
υmax 2965, 2934, 1625, 1591, 1568, 1443, 1390, 1335, 878, 748 cm-1.
Structural elucidation
• HMBCHMBCN
Me
H
H
•
4-(2-(1,3-dioxan-2-yl)ethyl)-2-ethylpyridine (6)
N
Et
N
ClCl
3.75 mol % Fe(acac)3
THF:NMP (4:1); then
3.75 mol % Fe(acac)3NMP, EtMgBr
O
OMgBrO
O

Page 11
To a vial was added 2,4-dichloropyridine (222 mg, 1.50 mmol) and Fe(acac)3 (19.9 mg, 0.056 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (8.0 mL) and NMP (2.0 mL). The
reaction mixture was treated dropwise with (1,3-dioxan-2-ylethyl)magnesium bromide (4.5 mL of a 0.5 M solution in THF, 1.5 equiv,
added over 7.5 minutes by syringe pump) at ambient temperature. After stirring for 1h (reaction monitored by HPLC for complete
conversion), a solution of Fe(acac)3 (19.9 mg, 0.056 mmol, 0.0375 equiv) in NMP (1 mL) and a solution of ethylmagnesium bromide
(3.02 mL of a 1.0 M solution in THF, 2.0 equiv) was added over 10 min at ambient temperature. After stirring for 2h, the reaction was
quenched by adding saturated aqueous NH4Cl (10 mL), filtered through celite, and poured into a separatory funnel. The top organic
layer was collected and the aqueous layer was extracted with EtOAc (3 x 5 mL). The organic layers were combined and washed with
brine (2 x 25 mL), dried (Na2SO4), and concentrated in vacuo. Silica gel chromatography using a gradient of 0% – 50%
EtOAc/hexanes afforded 286 mg (86%) of the desired product as a mixture of inseparable regioisomeric products. TLC Rf = 0.1 (20%
EtOAc/hexanes); 1H NMR (600 MHz, CDCl3, major isomer): δ 8.23 (d, J = 5.1 Hz, 1H), 6.84 (s, 1H), 6.79 (d, J = 5.1 Hz, 1H), 4.35 (t,
J = 5.10, 1H), 3.92 (m, 2H), 2.57 (m, 2H), 2.63 (q, J = 7.5 Hz, 2H), 2.53 (m, 2H), 1.90 (m, 1H), 1.73 (m, 2H), 1.13 (t, J = 7.5 Hz, 3H),
1.06 (m, 1H) ppm; 13C NMR (151 MHz, CDCl3, major isomer): δ 163.1, 151.2, 148.7, 122.0, 121.0, 100.8, 66.6, 35.3, 31.0, 29.2, 25.6,
13.7 ppm.
Structural elucidation
N Et• •• HMBCHMBC
OO
HH
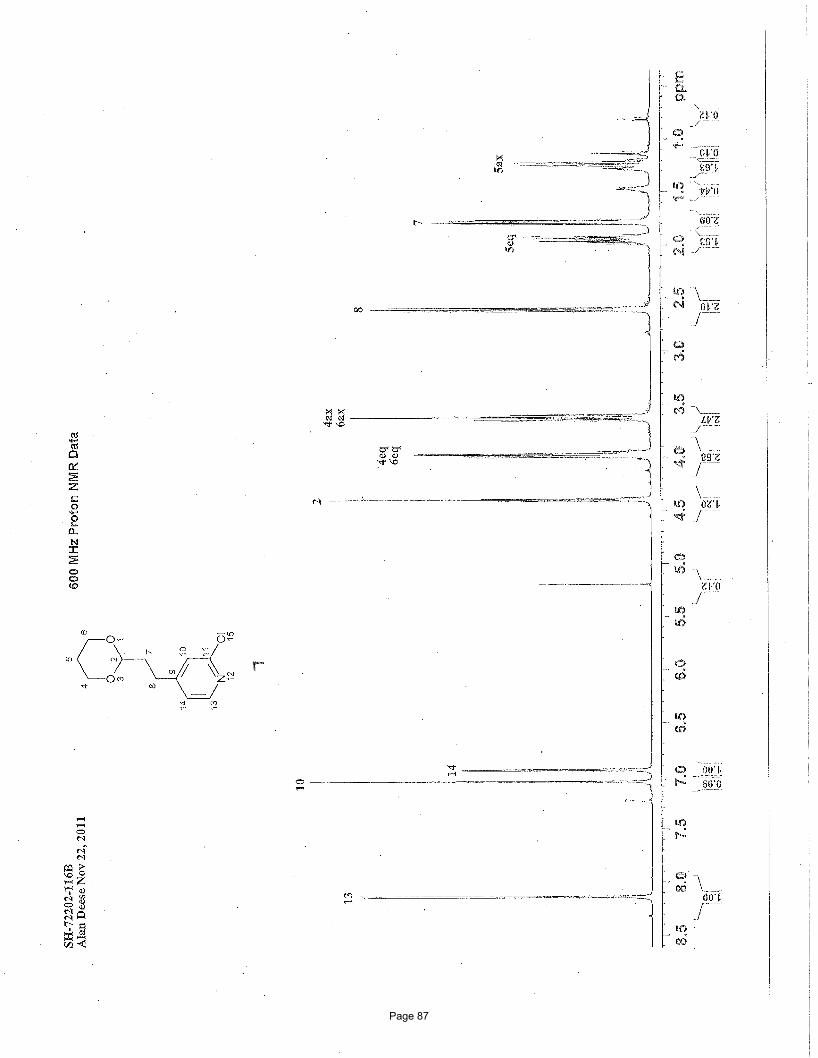
4-(2-(1,3-dioxan-2-yl)ethyl)-2-chloropyridine (7)
3.75 mol % Fe(acac)3
N
Cl
N
O
O
O
OBrMgCl Cl
To a vial was added 2,4-dichloropyridine (111 mg, 0.75 mmol) and Fe(acac)3 (9.9 mg, 0.028 mmol, 0.0375 equiv). The vial was
sealed with a septum cap, evacuated, purged with nitrogen and the mixture dissolved in THF (4.0 mL) and NMP (1.0 mL). The
reaction mixture was treated dropwise with (1,3-dioxan-2-ylethyl)magnesium bromide (2.25 mL of a 0.5 M solution in THF, 1.5
equiv) at ambient temperature. After stirring for 1h, additional (1,3-dioxan-2-ylethyl)magnesium bromide (0.75 mL of a 0.5 M
solution in THF, 0.5 equiv) at ambient temperature. After stirring for 4h, the reaction was quenched by adding water (5 mL) and
extracted with EtOAc (3 x 5 mL). The organic layers were combined and concentrated in vacuo. Analysis of the crude reaction
mixture by 1H NMR showed that the reaction had gone to roughly 80% conversion (selectivity was determined to be 4.8:1 based on 1H
NMR analysis of the crude reaction mixture. Silica gel chromatography using a gradient of 0% – 50% EtOAc/hexanes afforded 127
mg (75%) of the desired product as a colorless oil. TLC Rf = 0.17 (20% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.19 (d, J =
5.1 Hz, 1H), 7.10 (s, 1H), 7.00 (d, J = 5.10 Hz, 1H), 4.45 (t, J = 5.00 Hz, 1H), 4.03 (m, 2H), 3.68 (m, 2H), 2.66 (m, 2H), 1,99 (m, 1H),
1.83 (m, 2H), 1.28 (m, 1H) ppm; 13C NMR (151 MHz, CDCl3): δ 154.2, 151.3, 149.2, 124.0, 122.5, 100.4, 66.8, 34.9, 28.8, 25.6 ppm;
IR (neat): υmax 2962, 2854, 1593, 1387, 1239, 1146, 1134, 1085, 1002, 892 cm-1. Structure assigned based on analogy to structure
obtained for compound 3j and 7.
Trace metal analysis for Fe(acac)3 used for the entire study:
Co = < 3 ppm
Cu = < 3 ppm
Mn = 8 ppm
Pd = 81 ppm
Ni = 6 ppm

Page 12
Ag = 22 ppm
Zn = 3 ppm

Page 13
Table 1. Crystal data and structure refinement for gscb015.
X-ray ID gscb015
Sample/notebook ID 76003-64-01
Empirical formula C10 H8 Cl N
Formula weight 177.62
Temperature 100(2) K
Wavelength 1.54178 Å
Crystal system Monoclinic
Space group P2(1)/c
Unit cell dimensions a = 6.7894(2) Å α= 90°.
b = 18.0944(5) Å β= 110.484(2)°.
c = 7.2390(2) Å γ = 90°.
Volume 833.08(4) Å3
Z 4
Density (calculated) 1.416 Mg/m3
Absorption coefficient 3.513 mm-1
F(000) 368
Crystal size 0.05 x 0.05 x 0.02 mm3
Crystal color/habit colorless needle
Theta range for data collection 4.89 to 67.69°.
Index ranges -8<=h<=8, -21<=k<=21, -8<=l<=6
Reflections collected 10339
Independent reflections 1472 [R(int) = 0.0308]
Completeness to theta = 67.00° 97.8 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.9331 and 0.8439
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 1472 / 0 / 110
Goodness-of-fit on F2 1.075
Final R indices [I>2sigma(I)] R1 = 0.0326, wR2 = 0.0847
R indices (all data) R1 = 0.0369, wR2 = 0.0879
Largest diff. peak and hole 0.326 and -0.139 e.Å-3
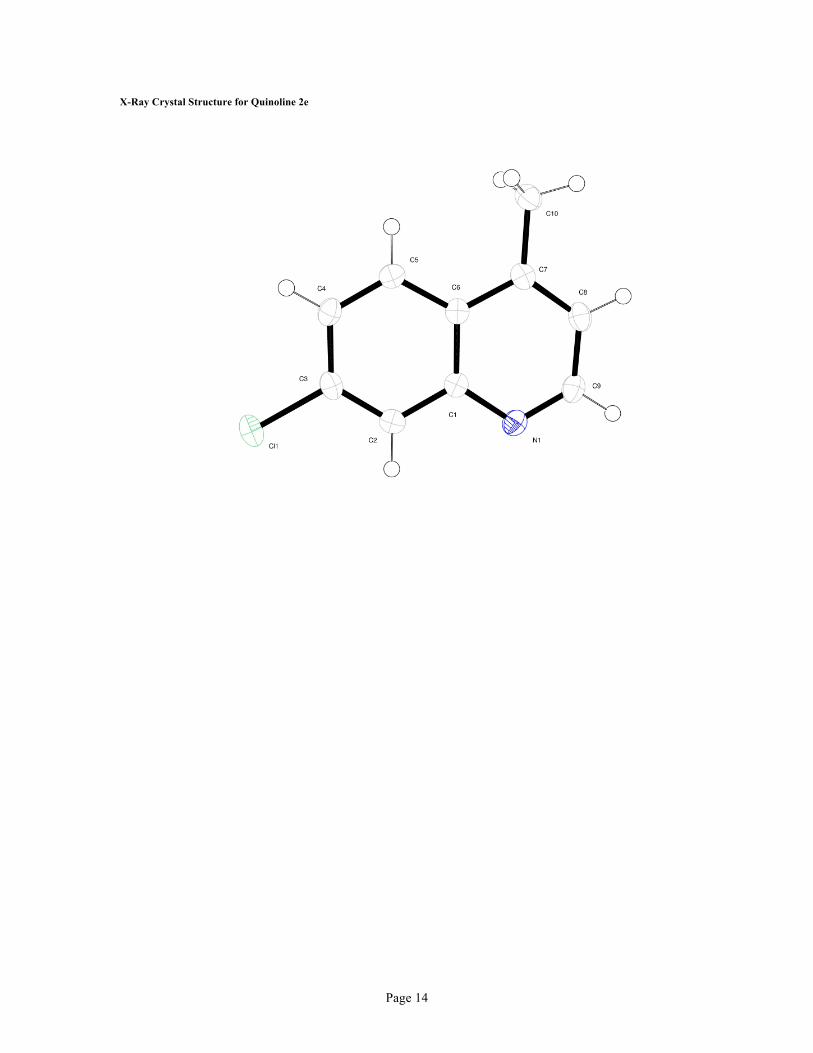
Page 14
X-Ray Crystal Structure for Quinoline 2e

Page 15
References:
i (a) Ashton, W. T.; Sisco, R. M.; Kieczykowski, G. R.; Yang, Y. T.; Yudkovitz, J. B.; Cui, J.; Mount, G.R.; Ren, R. N.; Wu, T.-J.; Shen, X.; Lyons, K. A.; Mao, A.-H.; Carlin, J. R.; Karanam, B. V.; Vincent, S. H.; Cheng, K.; Goulet, M. T. Bioorg. Med. Chem. Lett. 2001, 11, 2597. (b) Buzuya, M.; Noguchi, A.; Mano, E.; Okuda, T. Bull. Chem. Soc. Jpn. 1985, 58, 1149. (c) Comins, D.L.; Lyle, R.E.; J. Org. Chem. 1976, 41, 2065. (d) Morrow, C. J.; Rapoport, H. J. Org. Chem. 1974, 39, 2116. ii (a) Spectral data for 3-chloro-2-methyl pyridine: Lemire, A.; Grenon, M.; Pourashraf, M.; Charette, A. B. Org. Lett. 2004, 6, 3517. (a) Spectral data for 2-chloro-3-methyl pyridine: Narendar, P.; Gangadasu, B.; Ramesh, Ch. Raju, B. C.; Rao, V. J. Synth. Commun. 2004, 34, 1097. iii Universite, L.; Attardo, G.; Tripathy, S.; Gagnon, M. Methyl Sulfanyl Pyrimidines Useful as Antiinflammatories, Analgesics, and Antiepileptics. WIPO Pat. Appl. WO2010/132999 A1, 2010. iv Connon, S. J.; Hegarty, A. F. Eur. J. Org. Chem. 2004, 2004, 3477.

Page 16
Page 17
Page 18

Page 19
8.5
8.0
7.5
7.0
6.5
6.0
5.5
5.0
4.5
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
ppm
3.00
1.130.800.970.97
0.97
PS-7
3945
-08
Ala
n D
eese
, Sep
. 15,
201
17 8
94
5
6
10
N 1
23
CH
312
Cl
11
7.6
7.7
7.8
7.9
8.0
8.1
ppm
1.13
0.80
0.97
0.97
0.978
56
7
3
12
TMS
xx
x
CD
Cl3
8
2b
Page 20
160
150
140
130
120
110
100
9080
7060
5040
3020
10pp
m
-0.00
22.10
76.8377.0477.25
118.08
125.81126.29126.51127.22130.87
137.94
144.32
159.97
PS-7
3945
-08
Ala
n D
eese
, Sep
. 15,
201
1
7 89
4
5
6
10
N 1
23
CH
312
Cl
11
126.
012
6.5
127.
0pp
m
125.81
126.29
126.51
127.22
10
12
CD
Cl3
2
4
63
75
9
82b
Page 21
Page 22
Page 23
Page 24
Page 25
Page 26
Page 27
Page 28

Page 29

Page 30
Page 31
Page 32
Page 33
Page 34
Page 35
Page 36
Page 37
600
MH
z Pr
oton
NM
R D
ata
8.5
8.0
7.5
7.0
6.5
6.0
5.5
5.0
4.5
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
ppm
3.00
0.960.96
0.97
3 2
4 N 1
5 6
Cl8
CH3
7
6
5
3
7
TMS
73945-17
1H N
MR
(500
MH
z, C
DC
l3)
Shi
ft pp
m 2
.54
(s,1
H)
Shi
ft pp
m 7
.11
(dd,
J=5
.40H
z, 1
.60H
z, 1
H)
Shi
ft pp
m 7
.17
(d, J
=1.6
0 H
z, 1
H)
Shi
ft pp
m 8
.39
(d, J
=5.4
0 H
z, 1
H)
2h
Page 38
13C
NM
R D
ata
160
150
140
130
120
110
100
9080
7060
5040
3020
10ppm
-0.00
24.27
76.8277.0777.33
121.23123.56
144.19
150.01
160.06
73945-17
3 2
4 N 1
5 6
Cl8
CH3
7
2
6
4
3
5
CDCl3
7
TMS
2h
Page 39
600
MH
z Pr
oton
NM
R D
ata
8.5
8.0
7.5
7.0
6.5
6.0
5.5
5.0
4.5
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
ppm
3.00
0.950.93
0.94
SM-7
2202
-210
2-ch
loro
-4-m
ethy
l-pyr
idin
eA
lan
Dee
se J
un 2
6, 2
013
5 6N 1
2
34CH3
8
Cl7
6
5
3
8
1H N
MR
(600
MH
z, C
DC
l3)
Shi
ft pp
m 2
.35
(d, J
=0.6
6 H
z, 1
H)
Shi
ft pp
m 7
.03
(m, J
=1.0
3 H
z,5.
10H
z 1
H)
Shi
ft pp
m 7
.15
(m, J
=0.7
2 H
z, 1
H)
Shi
ft pp
m 8
.23
(d, J
=5.1
0 H
z, 1
H)
2i
Page 40
13C
NM
R D
ata
2030
4050
6070
8090
100
110
120
130
140
150
ppm
20.87
76.9977.2077.41
124.98
149.40150.56151.65
SM-7
2202
-210
2-ch
loro
-4-m
ethy
l-pyr
idin
eA
lan
Dee
se J
un 2
6, 2
013
5 6N 1
2
34CH3
8
Cl7
2
4
6
35
CD
Cl3
8
2i
Page 41
1 H-1
H D
QF-
CO
SY D
ATA
ppm
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
ppm
1 2 3 4 5 6 7 8
SM-7
2202
-210
2-ch
loro
-4-m
ethy
l-pyr
idin
eA
lan
Dee
se J
un 2
6, 2
013
Page 42
1 H-1
3 C M
ultip
licity
Edi
ted
HSQ
C D
ata
CH
and
CH
3 in
Red
and
CH
2 in
Blu
e
ppm
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
ppm20 40 60 80 100
120
140
SM-7
2202
-210
2-ch
loro
-4-m
ethy
l-pyr
idin
eA
lan
Dee
se J
un 2
6, 2
013
Page 43
1 H-1
3 C H
MB
C D
ata
ppm
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
8.5
ppm20 30 40 50 60 70 80 90 100
110
120
130
140
150
SM-7
2202
-210
2-ch
loro
-4-m
ethy
l-pyr
idin
eA
lan
Dee
se J
un 2
6, 2
013
Page 44
Page 45
Page 46
Page 47
Page 48
Page 49
Page 50
Page 51
Page 52
Page 53
Page 54
Page 55

Page 56
Page 57
Page 58

Page 59
Page 60
Page 61
Page 62
Page 63
Page 64
Page 65

Page 66
Page 67
Page 68
Page 69
Page 70
Page 71
Page 72
Page 73
Page 74
Page 75
Page 76
Page 77
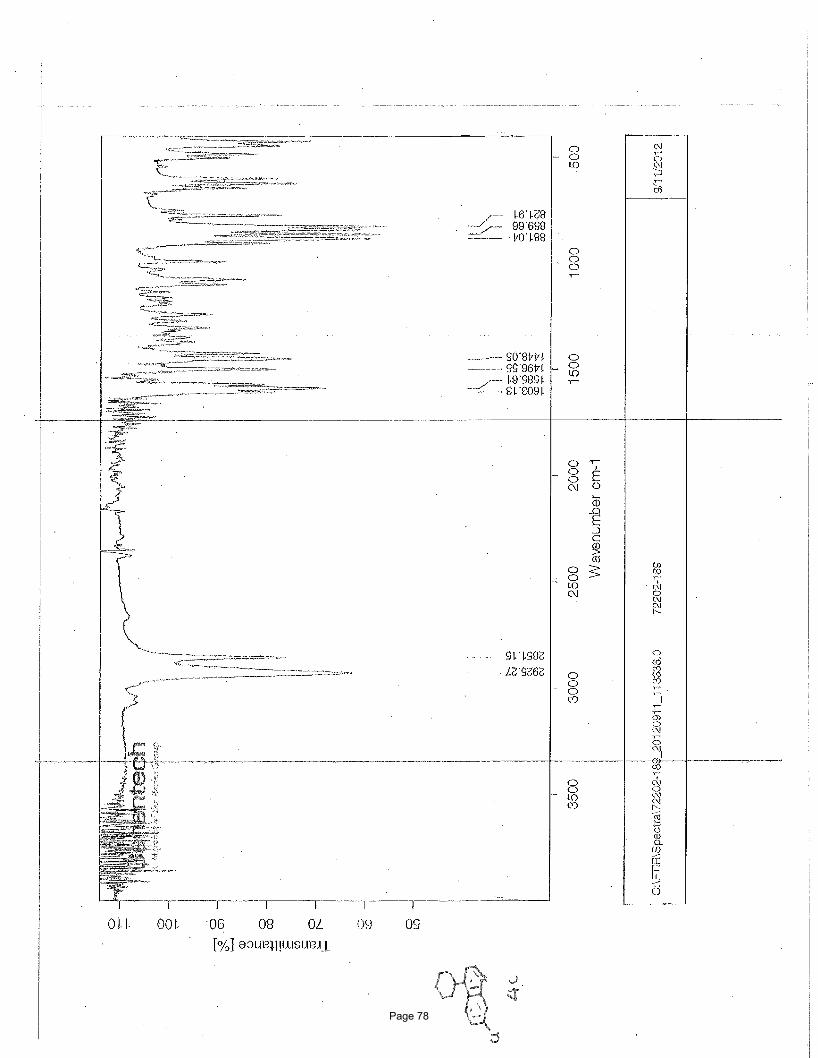
Page 78
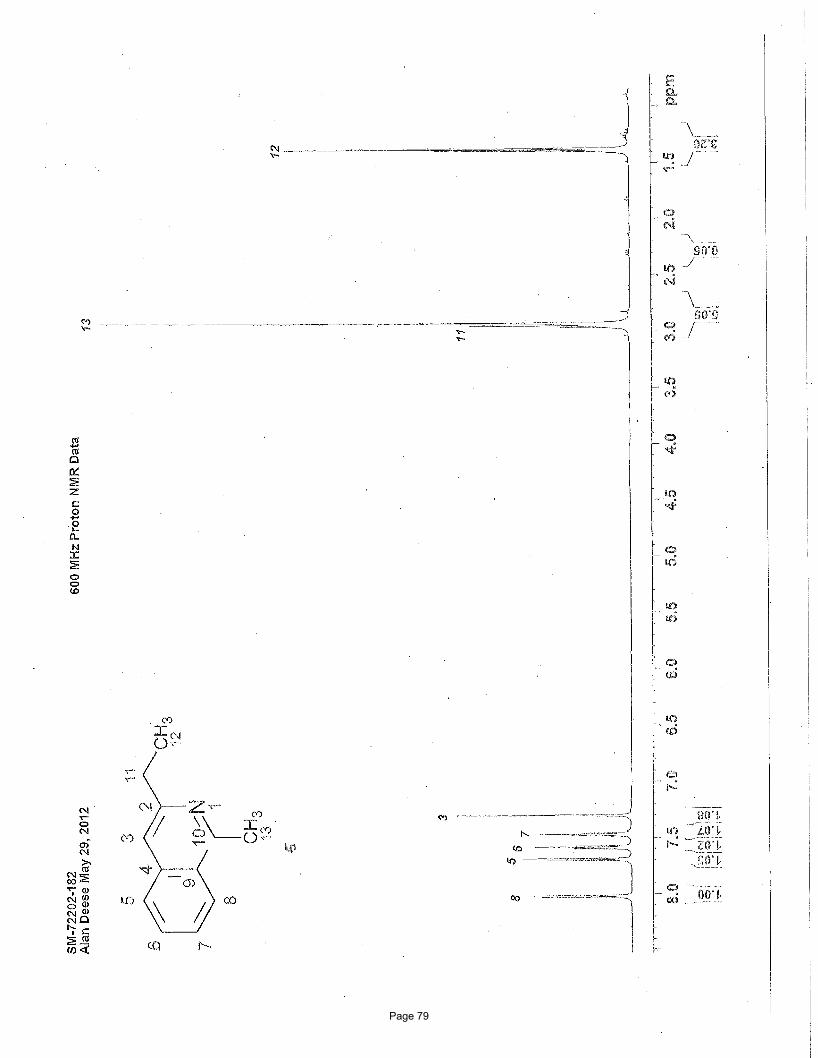
Page 79
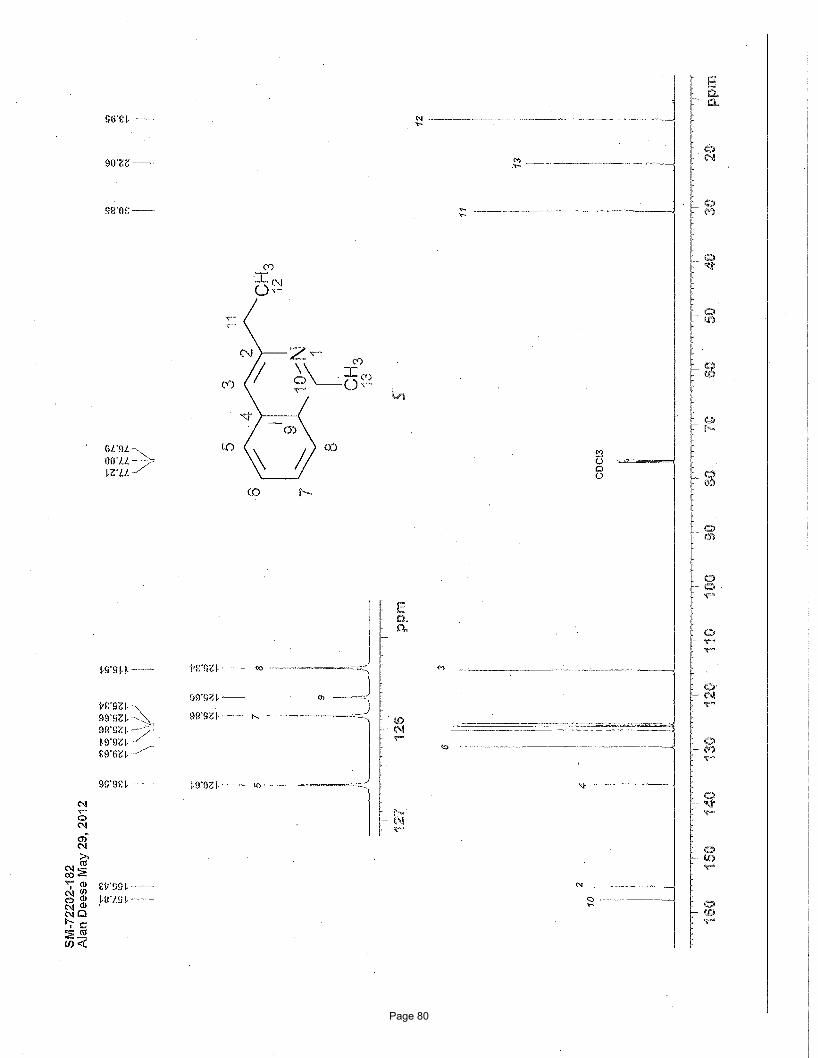
Page 80

Page 81

Page 82
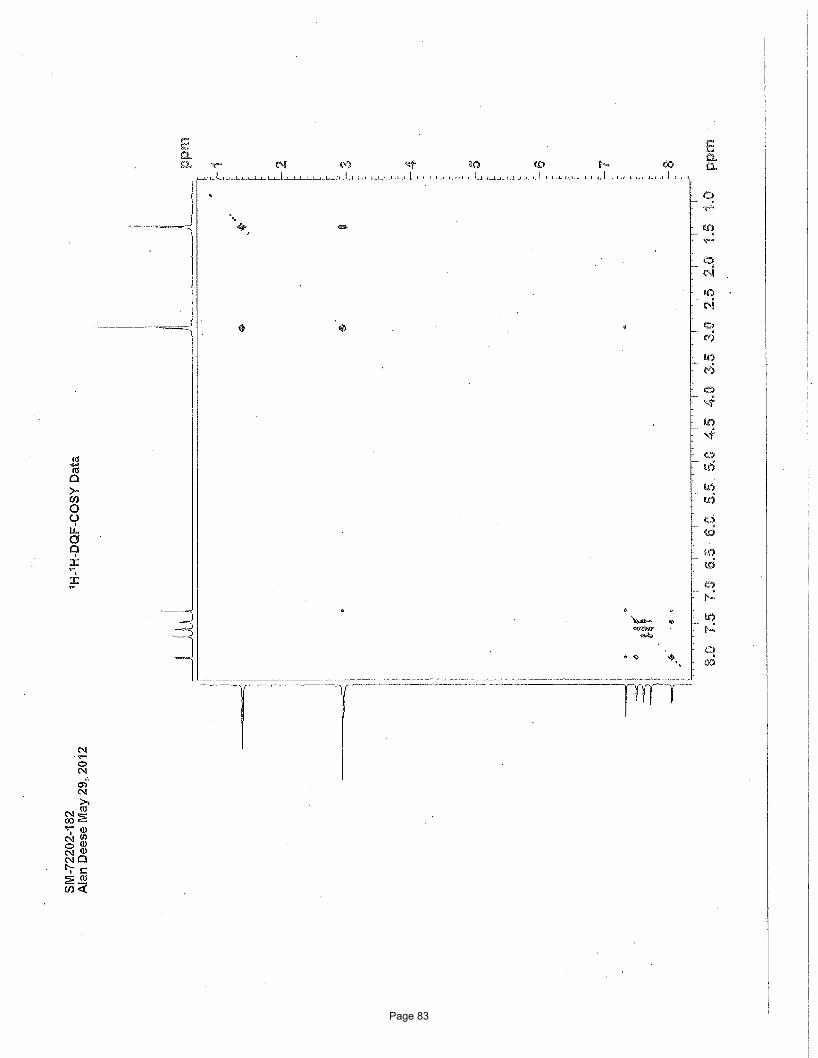
Page 83
Page 84
Page 85
Page 86
Page 87
Page 88
Page 89







