
Supporting Information
© Wiley-VCH 2007
69451 Weinheim, Germany

1
Annularly-fused Hexapyrrolohexaazacoronenes: A Multiple InteriorNitrogen-containing π-System with Stable Oxidation States
Masayoshi Takase, Volker Enkelmann, Daniel Sebastiani, Martin Baumgarten, and
Klaus Müllen*
Max-Planck-Institute for Polymer Research
Ackermannweg 10, D-55128 Mainz, Germany
General Methods1H NMR and 13C NMR spectra were recorded in deuterated solvents such as CD2Cl2,
CDCl3, C2D2Cl4 on a Bruker DPX 250 and Bruker DRX 500 spectrometer with use of
the solvent proton or carbon signal as an internal standard. Field desoption (FD) mass
spectra were obtained on a VG Instruments ZAB 2-SE-FPD. High resolution MALDI
mass spectra were recorded on a Bruker Reflex II-TOF Spectrometer using a 337 nm
nitrogen laser with TCNQ as matrix. ��Melting points were measured� on Büchi B-545 and
not corrected. Thermogravimetric analysis (TGA) were carried out by a Mettler TG 50
thermogravimetric analyzer. UV/Vis/NIR spectra were recorded at room temperature
on a Perkin-Elmer Lambda 9 spectrophotometer. �Fluorescence spectra were recorded
on a SPEX-Fluorolog II (212) spectrometer. Electron spin resonance spectroscopy
(ESR) were recorded on CW X-band ESP 300E equipped with an NMR gauss meter
(Brucker ER035), a frequency counter (Brucker ER 041 XK) and a variable
temperature control continues flow nitrogen cryostat (Brucker B-VT 2000). All reactions
were carried out under a nitrogen atmosphere. Dry CH2Cl2 and CD2Cl2 for the
absorption and NMR spectra were dried over CaH2 and distilled.
Synthetic procedures and spectral data of 1b, 1c, 2b, and 2c.
Hexakis(3,4-dibromopyrrolyl)benzene 1b. To a DMF (20 mL) solution of sodium
methoxide (285 mg, 5.28 mmol), a DMF (10 mL) solution of N-(triisopropylsilyl)-3,4-
dibromopyrrole1 1.83 g (4.80 mmol) was added dropwise at 0 °C over 10 min. The
reaction mixture was stirred for an additional 30 min at room temperature, and then

2
hexafluorobenzene (74 mg, 0.40 mmol) was added. The reaction mixture was stirred
for an additional 1 h at the same temperature before it was poured into water (150 mL).
The aqueous phase was extracted with diethylether. After evaporation of the organic
phase, the residue was subjected to the silica gel column chromatography (eluent:
hexane:EtOAc = 1:1) to give 1b as a white solid (460 mg, 81%). dec. > 338 °C; 1HNMR (CDCl3) δ 6.13; 13C NMR (CDCl3) δ 133.3,120.4,104.4; FD MS calcd for M+,
C30H12N6Br12: 1415.12; found 1415.4.
Hexakis(3,4-dibromopyrrolo)[2,1,5-bc:2',1',5'-ef:2'',1'',5''-hi:2''',1''',5'''-
kl:2'''',1'''',5''''-no:2''''',1''''',5'''''-qr][2a,4a,6a,8a,10a,12a]hexaazacoronene 2b.To a CH2Cl2 (200 mL) solution of 1b (200 mg, 0.1413 mmol), a CH3NO2 (3 mL) solution
of FeCl3 (458 mg, 2.826 mmol) was dropwise at room temperature over 10 min. After
stirring for 1 h, hydrazine (2 mL) was added and stirred for 5 min. Adding MeOH and
filtration gave 2b as a mixture with mono- and di-debrominated compounds as an
orange solid (165 mg, 83%). dec > 360 °C (TGA); MALDI-TOF-MS (TCNQ matrix)
calcd for M+, C30N6Br12: 1403.03; found 1405 (100%), C30HN6Br11: 1325.12; found 1325
(73%), C30H2N6Br10: 1245.21; found 1247 (9.7%).
Hexakis(3,4-bis(4-(trifluoromethyl)phenyl)pyrrolyl)benzene 1c. To a DMF (3 mL)
solution of NaH (60% oil dispersion, 45 mg, 1.13 mmol), 380 mg (1.07 mmol) of 3,4-
di-(4-trifluoromethylphenyl)pyrrolyl2 was added at 0 °C. After the liberation of H2 gas
had ceased, the reaction mixture was stirred for an additional 30 min at the same
temperature, and then a DMF (1 mL) solution of hexafluorobenzene (16 mg, 0.089
mmol) was added. The reaction mixture was stirred for an additional 6 h at 50 °C
before it was poured into ice-water (20 mL). The white precipitate was filtered off and
washed with water. The filtrate was subjected to the silica gel column chromatography
(eluent: hexane:CH2Cl2 = 3:2) to give 1c as a white solid. (144 mg, 61%). dec. >
350 °C; 1H NMR (CD2Cl2) δ 7.63 (d, J = 7.5 Hz, 24H), 7.21 (d, J = 7.5 Hz, 24H), 6.72 (s,
12H); 13C NMR (C2D2Cl4, 373K) δ 137.0,133.7, 129.6 (q, J = 129.8 Hz), 128.2, 125.5
(br), 122.8, 121.2; FD MS calcd for M+, C114H60F36N6: 2197.43; found 2200.1.
Hexakis(3,4-bis(4-(trifluoromethyl)phenyl)pyrrolo)[2,1,5-bc:2',1',5'-ef:2'',1'',5''-hi:2''',1''',5'''-kl:2'''',1'''',5''''-no:2''''',1''''',5'''''-

3
qr][2a,4a,6a,8a,10a,12a]hexaazacoronene 2c.To a CH2Cl2 (100 mL) solution of 1c (95 mg, 0.0432 mmol), a CH3NO2 (2 mL) solution
of FeCl3 (252 mg, 1.556 mmol) was dropwise at room temperature over 5 min. After
stirring for 19 h at the same temperature, hydrazine (5 mL) was added and stirred for 5
min. Organic phase was washed with water and evaporated. The residue was
subjected to the silica gel column chromatography (eluent: hexane:CH2Cl2 = 3:2) to
give 2c as an orange solid (50 mg, 53%). dec > 535 °C (TGA); 1H NMR (CDCl3) δ 6.81
(d, J = 8.4 Hz, 24H), 6.57 (d, J = 8.4 Hz, 24H); 13C NMR (C2D2Cl4, 413K) δ 136.4, 130.6,
129.3 (br), 125.0, 124.0, 118.4, 107.8 (One peak for the carbon atom bonding to the
nitrogen atom is not observed probably due to quadrupolar relaxation); MALDI-TOF-
MS calcd for M+, C114H48F36N6: 2185.34; found 2185.51.
Reference(1) Bray, B. L.; Mathies, P. H.; Naef, R.; Solas, D. R.; Tidwell, T. T.; Artis, D. R.;
Muchowski, J. M. J Org. Chem. 1990, 55, 6317-6328.
(2) Fujii, K,; Yahashi, Y.; Nakano, T.; Imanishi, S.; Baldia, S. F.; Harada, K. Tetrahedron,
2002, 58, 6873-6879.
4
0
20
40
60
80
100
500 1000 1500 2000 2500 3000 3500
HPyBδ (TCNQ)
m/e
0
20
40
60
80
100
2180 2182 2184 2186 2188 2190 2192 2194
HPyBδ (TCNQ)
m/e
55.566.57
ppm
N
NN
N
NN
F3CF3C
F3C
F3C
F3CF3C CF3
CF3
CF3
CF3
CF3CF3
Pyrrole -NH
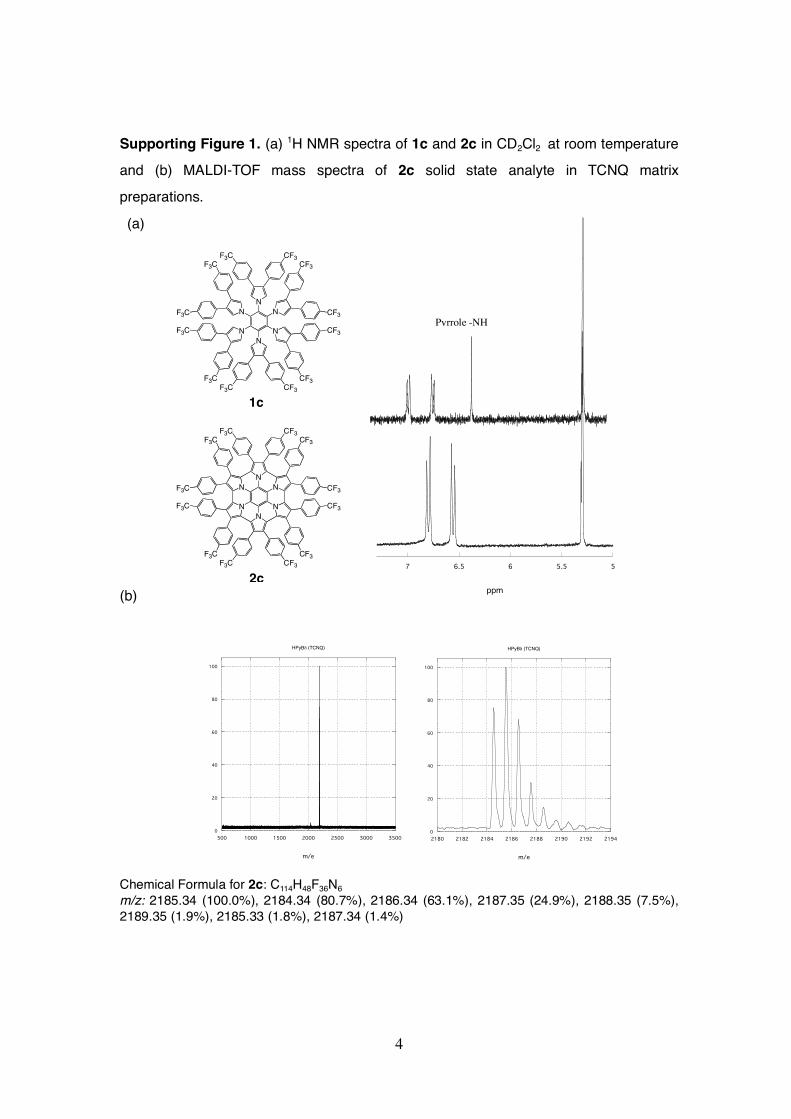
Supporting Figure 1. (a) 1H NMR spectra of 1c and 2c in CD2Cl2 at room temperature
and (b) MALDI-TOF mass spectra of 2c solid state analyte in TCNQ matrix
preparations.
(a)
(b)
Chemical Formula for 2c: C114H48F36N6m/z: 2185.34 (100.0%), 2184.34 (80.7%), 2186.34 (63.1%), 2187.35 (24.9%), 2188.35 (7.5%),2189.35 (1.9%), 2185.33 (1.8%), 2187.34 (1.4%)
N
NN
N
NN
F3CF3C
F3C
F3C
F3CF3C CF3
CF3
CF3
CF3
CF3CF3
1c
2c
5
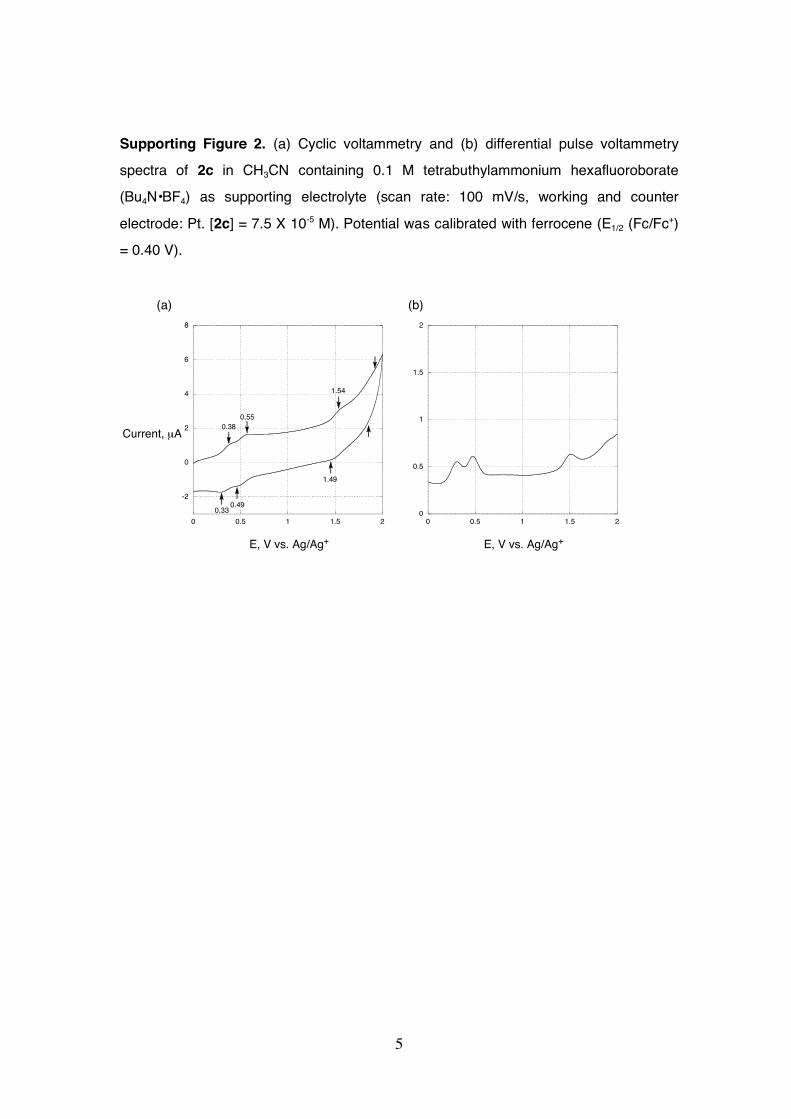
Supporting Figure 2. (a) Cyclic voltammetry and (b) differential pulse voltammetry
spectra of 2c in CH3CN containing 0.1 M tetrabuthylammonium hexafluoroborate
(Bu4N•BF4) as supporting electrolyte (scan rate: 100 mV/s, working and counter
electrode: Pt. [2c] = 7.5 X 10-5 M). Potential was calibrated with ferrocene (E1/2 (Fc/Fc+)
= 0.40 V).
-2
0
2
4
6
8
0 0.5 1 1.5 20
0.5
1
1.5
2
0 0.5 1 1.5 2
E, V vs. Ag/Ag+
Current, µA
E, V vs. Ag/Ag+
(a) (b)
0.55
0.49
0.38
0.33
1.54
1.49

6
Supporting Figure 3. The stepwise oxidation of 2c (1.0 X 10-5 M) with incremental
addition of 0 to 6.7 equiv (-) and 6.7 to 15 equiv (-) of SbCl5 (1.0 X 10-3 M) in
dichloromethane at room temperature.
0
0.5
1
1.5
2
400 800 1200 1600 2000
HPHAC(1X10-5M)1eq2eq2.67eq3eq3.33eq4eq4.675.33eq6.67eq10eq13.316.7eq20eq23.3eq26.7eq30eq33.3eq
nm
0
0.1
0.2
0.3
0.4
0.5
400 800 1200 1600 2000
HPHAC(1X10-5M)1eq2eq2.67eq3eq3.33eq4eq4.675.33eq6.67eq10eq13.316.7eq20eq23.3eq26.7eq30eq33.3eq
nm
0
0.1
0.2
0.3
0.4
0.5
400 800 1200 1600 2000
HPHAC(1X10-5M)1eq2eq2.67eq3eq3.33eq4eq4.675.33eq6.67eq10eq13.316.7eq20eq23.3eq26.7eq30eq33.3eq
nm
(a) 250 ~ 2200 nm
(b) 380 ~ 2200 nm

7
Supporting Figure 4. ESR spectra of (a) monocation 2c+• in the presence of 1.2 equiv.
of NOSbCl6 and (b) dication 2c2+ in the presence of ca. 5 equiv. of NOSbCl6 in dry
CH2Cl2 at room temperature ([2c] = 1.0 X 10-3 M).
-3000
-2000
-1000
0
1000
2000
3000
334 334.5 335 335.5 336 336.5
mT
-3000
-2000
-1000
0
1000
2000
3000
334 334.5 335 335.5 336 336.5
mT
(a) (b)
g = 2.0023
Δ H = 0.298 mT

8
Supporting Figure 5. Packing structures (side views) of a) HPHAC 2c and b) itsdication salt 2c2+ (2c2+[SbCl6-]2).
(a)
(b)

9
CH2Cl2
CH
8 7.5 7 6.5 6 5.5 5δ / nm
145 140 135 130 125 120 115δ / nm
* *
*
°°CH
HPHAC C(sp2)°CF3
C(sp2)CF3
Supporting Figure 6. (a) 1H (500 MHz) and (b) 13C (125 MHz) NMR spectra of 2c2+ inCD2Cl2 at RT. A suspension of 2c (3.3 X 10-3 mmol) and NOSbCl6 (3.3 X 10-2 mmol) indry CD2Cl2 (0.65 mL) was shaken for 10 min. After decantation, the supernatant wasanalyzed by NMR spectroscopy. The signal “°” indicates the inner sp2 carbons ofHPHAC core. Assignments for peripheral 4-trifluoromethylphenyl groups’ carbons arebased on the comparison of the starting materials and spin-echo experiment. Asteriskindicates impurities from the solvent used.
(b)
(a)

10
Supporting Figure 7. Three-dimensional NICS maps of a) neutral form 2a and b)dication form 2a2+ and possible resonance forms for them.
a) b)
NN
NN
N
N
NN
NN
N
N

11
Supporting Figure 8. Isosurfaces of the induced electronic current density of 2a for anexternal magnetic field perpendicular to the molecular plane. The arrows indicate theorientation of the currents (extracted from the cartesian components of the currentdensity, not shown).
The origin of this difference can be explained using the electronic current density
pattern (often referred to as “ring currents”). The current density is a three-dimensional
vector field, and can therefore not be fully explained with simple arguments. However,
we shall attempt a partial interpretation of the patterns. In both molecules, there is a
competition between differently oriented currents on the outer rim versus those inside
the individual rings. In the neutral form (a), the intensities of these two opposite
currents is about the same, leading to an almost complete canceling of their induced
magnetic fields. In contrast to this, there is a clear winner in the dication (b): the outer
rim appears significantly stronger, yielding a highly aromatic behavior.
All calculations of NICS maps were carried out with the DFT program package CPMD[Computer code CPMD, Version 3.12, Copyright IBM Corp. and MPI-FKF Stuttgart,http://www.cpmd.org] using plane waves with 60Ry energy cutoff as basis set, the BLYPfunctional and normconserving pseudopotentials [Becke, A. D.; Phys. Rev. A 1988: 38, 3098and Lee, C; Yang, W; Parr, R. G.; Phys. Rev. B 1988: 37, 785-789 and Goedecker, S.; Teter, M.;Hutter, J.; Phys. Rev. B 1996: 54, 1703]. The geometries of the molecules were optimized priorto the NICS calculations using the same computational setup. For bothmolecules, large supercells (35A x 35A x 30A) were used to isolate the molecules from theirperiodic images and to avoid that the NICS fields overlay each other. The graphicalvisualizations were done with the Molekel program and postprocessed with the GIMP package[P. Flukiger, H. P. Luthi, S. Portmann, J. Weber, Molecular visualization program Molekel,version 4.0. Swiss Center for Scientific Computing, Manno (Switzerland), 2000; S. Kimball, P.Mattis, GNU Image Manipulation Program GIMP, http://www.gimp.org].
(a) neutral 2a (b) dication 2a2+







