homogeneous and heterogeneous metallocene/mao-catalyzed polymerization of trialkylsilyl-protected...
TRANSCRIPT

Macromol. Rapid Commun. 19,511-5I5 (1998) 511
~omogeneous and heterogeneous m e ~ l l o c e n ~ ~ O - c a t a ~ y z e d polymerization of trialkylsilyl-protected alcohols
Ralf Goretzki, Gerhard Fink*
Max-Planck-Institut fur Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mulheim a. d. Ruhr, Gemany
(Received: June 18,1998)
SUMMARY We investigated the ethylene co~lymerization by utilizing M e z S i ( I n d ) ~ ~ l ~ A O and Me&( Ind)zZrCl~~AO/SiO~ with 1 O-undecene- 1 -0xytrimethylsilane and 1 O-undecene- 1 -0xytriisopropylsi- lane and the ethylene copolymerization by using 'Pr(Cp1nd)ZrClJMAO and 'Pr(CpInd)ZrC12/MAO/SiOz with 5-norbornene-2-methyleneoxytrimethylsilane and 5-norbornene-2-methyleneoxytriisopropylsilane. The tri- methylsilyl (TMS) protecting group could not prevent the catalyst deactivation caused by the addition of the polar comonomer. In contrast to that, good catalyst activities and comonomer contents were obtained with the triisopropylsilyl (TIPS) protected monomers. The homopolymerization of 10-undecene- 1 -0TIPS was car- ried out with MezSi(Ind)zZrCl&IAO.
Introduction On account of the Lewis acidity of Ziegler-catalysts direct polymerization of polar monomers, which are able to coordinate at the cationic polymerization active center, is not possible',*). Revious attempts to produce funct- ionalized polyolefins with Ziegler catalysts can essen- tially be divided into two main approaches:
The first approach is the polymerization of polar mono- mers which are precomplexed with alumini~ma&yls~-~) and the polymerization of monomers with sterically hin- dered The other way is the polymerization of Lewis acidic bor- ane monorner~~. '~), which can be transformed to a variety of polar groups in a post polymerization step.
In this report we present results in respect to the ethy- lene copolymerization with trimethyl- and triisopropylsi- lyl protected alcohols.
or with silyl protected polar
Experimental part
Materials
'Pr(Cp1nd)ZrClz was obtained from Hoechst, rac- MezSi(Ind)2ZrCl,, MA0 (10 wt.-% in toluene) and SiOJ MA0 were kindly supplied by Witco. Toluene was purified by distillation under argon over NaA1(Et)4, triisobutylalumi- nium (Tiba) was produced in the technical department of our institute. Supported metallocenes were prepared as follows: A toluene solution of the metallocene was added to a stirred suspension of SiOmAO in toluene and stirred for 3 h at 25 "C. The supported catalyst was filtered off, washed thor- oughly with toluene and dried in vacuum. The zirconium content was measured by ICP analysis.
Synthesis of 10-undecene-1 -oxytrimethylsilane: In a two- necked flask 40.9 mL (0.2 mol) of 10-undecene-1-01 was introduced and 50 mL (0.2 mol) of ~,U-bis(trimethylsily1)-
acetamide and 0.5 mL of trimethylsilyl chloride were added under stirring and ice cooling (under argon). After stirring over night (25 "C) and filtration (crist. N-trimethylsilylacet- amide was removed) the product was distilled (bp = 73°C 6 mbar).
'H NMR (CDC13): 6 = 0.0 (s, 9H, TMS), 1.88-1.98 (m,
4.78-4.92 (m, 2H, --CH=C€Jz), 5.59-5.79 (m, lH,
Synthesis of 5-norbornene-2-~eth~~eneo~tr~m~thylsi~ane: In a two-necked flask 30.6 g (0.25 mol) of 5-norbornene-2- methanol (81% endo) was introduced and 40.0 g (0.28 mol) of ~-methyl-N-trimethyls~lylacetamide and 0.5 mL. of tri- methylsilyl chloride were added under stirring and ice cool- ing (under argon). After stirring over night (25 "C), crystalli- zation of N-methylacetamide by cooling down and filtration, the product was distilled (bp = 36"C, 0.05 mbar).
'H NMR (CDC13): 6 = 0.0 (d, 9H, TMS), 5.80-6.05 (m, 2H, -CH=€H-); GC: 98.7% (81.4% endo, 17.3% exo).
Synthe~~s of 5-norbo~nene-~-methyleneoxytriisopropyl- silune"': A two-necked flask was charged with 160 mL of methylene chloride, 0.16 mol of 5-norbornene-2-methanol and 0.40 rnol of 2.6-dimethylpyridine. Under stirring and ice cooling 0.2 1 mol of triisoprop~lsilyl trifluoromethanesulfo- nate was slowly added and stirred over night at 25°C. The silylating agent was destroyed with 100 mL of water and the organic layer was washed with a KHC03 solution. After dry- ing over NazS04, methylene chloride was removed and the product distilled in two steps: 30°C 0.05 mbar to remove dimethylpyridine and triisopropylsilane and 69 "C, 2 + lO-' mbar for the main fraction,
28.21, 28.83 (C3), 41.21, 41.44 (C4), 41.48, 41.86 (C2), 42.99, 43.37 (Cl), 44.48, 48.97 (C7), 66.46 (endo <HZ- 0-), 67.35 (ex0 -CH2+), 132.28 (C6), 136.43 (C5).
2H, -C&.-CH*--O-), 3.43-3.50 (t, 2H, I J H z U ) ,
-CE*H,).
I3C NMR (CDC13, 50.323 MHz): 6 = 11.98, 17.68 (TIPS),
GC: 80.7% endo, 16.9% exo. Synthesis of 10-undecene-1 -oxytriisopropylsilane: A two-
necked flask was charged with 200 mL of methylene chlor- ide, 0.2 mol of 10-undecene-1-01 and 0.50 mol of 2.6-
Macromol. Rapid Commun. 19, No. 10 0 WILEY-VCH Verlag GmbH, D-69451 Weinheim 1998 1022- 1336/98/101o-O5 11 $1 7.50+.50/0
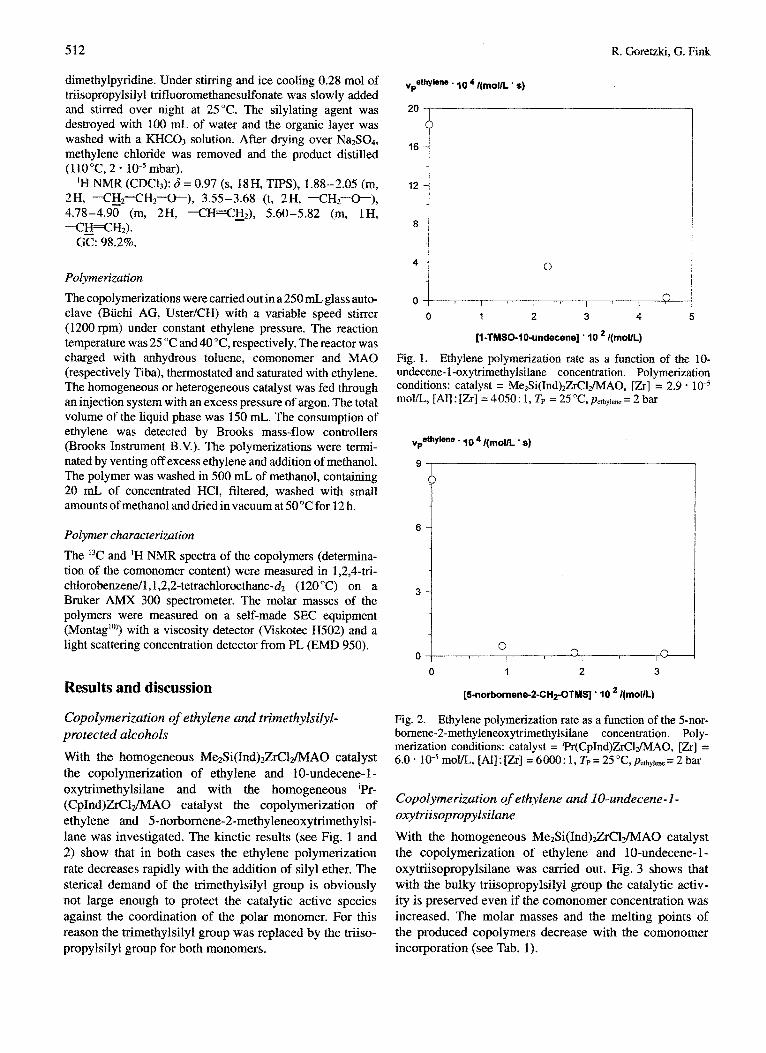
512
12 -
8 -
4 -
R. Goretzki, G. Fink
dimethylpyridine. Under stirring and ice cooling 0.28 mol of ~isopropylsilyl ~fluorometh~esulfonate was slowly added and stirred over night at 25°C. The silylating agent was destroyed with 100 mL of water and the organic layer was washed with a KHCO3 solution. After drying over NazS04, methylene chloride was removed and the product distilled (llO"C, 2 mbar). 'H NMR (CDCI3): 6 = 0.97 (s, l8H, TIPS), 1.88-2.05 (m,
4.78-4.90 (m, 2H, -CH=CEz), 5.60-5.82 (m, lH, 2H, --CBz*Hz+), 3.55-3.68 (t, 2H, -CHz+),
--C€J=CH2). GC: 98.2%.
Polymerization The copolymerizations were carried out in a250 mL glass auto- clave (Buchi AG, UsteriCH) with a variable speed stirrer (1200 rpm) under constant ethylene pressure. The reaction temperature was 25 "C and 40 "C, respectively. The reactor was charged with anhydrous toluene, comonomer and MA0 (respectively Tiba), thermostated and saturated with ethylene. The homogeneous or heterogeneous catalyst was fed through an injection system with an excess pressure of argon. The total volume of the liquid phase was 150 mL. The consumption of ethylene was detected by Brooks mass-flow controllers (Brooks I n s ~ m e n t B.V.). The po~yme~zations were termi- nated by venting off excess ethylene and addition of methanol. The polymer was washed in 500 mL of methanol, containing 20 mL of concentrated HCI, filtered, washed with small amounts of methanol and dried in vacuum at 50 "C for 12 h.
Polymer characteriza~~o~
The I3C and 'H NMR spectra of the copolymers (determina- tion of the comonomer content) were measured in 1,2,4-tri- chlorobenzene/l,l,2,2-tetrachloroethane-~~ (120°C) on a Bruker AMX 300 spectrometer. The molar masses of the polymers were measured on a self-made SEC equipment (MontaglO)) with a viscosity detector (Viskotec H502) and a light scattering concentration detector from PL (EMD 950).
Results and discussion
~ o p o l y m e ~ ~ a t ~ o n a~ethy~ene and t ~ ~ e t h y ~ s i l y l - protected alcohols
With the homogeneous M e * S i ( I n d ) ~ ~ l ~ ~ A O catalyst the copolymerization of ethylene and 1 O-undecene- 1 - oxytrimethylsilane and with the homogeneous 'Pr- ( C p I n d ~ ~ l * ~ A O catalyst the copolyme~zation of ethylene and 5-norbornene-2-methyleneoxytrimethylsi- lane was investigated. The kinetic results (see Fig. 1 and 2) show that in both cases the ethylene polyme~zation rate decreases rapidly with the addition of silyl ether. The sterical demand of the trimethylsilyl group is obviously not large enough to protect the catalytic active species against the coordination of the polar monomer. For this reason the ~ m e t h y l s i l ~ l group was replaced by the triiso-
16
0
0 1 2 3 4 5
[I-TMSO-10-undecene] ' 10 I(mollL)
Fig. 1. Ethylene ~ lyme~zat ion rate as a function of the 10- undecene- 1 -oxytrimethylsilane concentration. Polymerization conditions: catalyst = MezSi(Ind)2ZrCI&fAO, [Zr] = 2.9 - mom, [All : [Zr] = 4050: 1, Tp = 25 "C, pe&yie.e = 2 bar
[S-norbornene-2-CH24TMSJ * 10 * /(moUL)
Fig. 2. Ethylene ~ lyme~zat ion rate as a function of the 5-nor- bornene-2-methyleneoxytrimethylsilane concentration. Poly- merization conditions: catalyst = 'Pr(CpInd)ZrClz/MAO, [Zr] = 6.0 - mom, [All : [a] = 6000: 1, TP = 25 "C, pe&.,kE= 2 bar
Copolymerization of ethylene and 20-undecene-1- oxytrdisopropy lsilane With the homogeneous MezSi(Ind)zZrC12/MA0 catalyst the copolyme~zation of ethylene and 10-undecene- 1 - oxytriisopropylsilane was carried out. Fig. 3 shows that with the bulky triisopropylsilyl group the catalytic activ- ity is preserved even if the comonomer concentration was increased. The molar masses and the melting points of the produced copolymers decrease with the comonomer
propylsilyl group for both monomers. incorporation (see Tab. 1).

Homogeneous and heterogeneous metallocene~A0-catalyzed polymerization of ~alkylsilyl-p~tected alcohols 513
Tab. 1. Co~lymerization of ethylene and 10-undecene- 1-oxy~isopropylsilane with ~ e 2 S ~ ( I n d ) z Z ~ l ~ ~ A 0 a)
[ 10-undecene- 1 -0TIPSl Content of polar monomer Mw/( g/mol) Md(glrno1) TJ OC in feed x 10' in molL in polymer in wt.-% (mol-%)
0 - 662 000 22 1 000 134.6 3 1.4 (0.15) 354 000 121 000 126.1 6 11.8 (1.25) 245 000 95 000 117.9
12 20.6 (2.18) 203 000 87 000 104.1
Polymenzation conditions: [Zr] = 7.4 * lo4 mom, [All : [Zr] = 4800: 1, TP = 2 5 O C , p = ~ ~ ~ ~ = 2 bar.
Tab. 2. Copolyme~zation of ethylene and 10-undeeene- 1 -oxy~isopropylsilane with M e 2 S i ( I n d ) a Z ~ l ~ A O / S i ~ a)
pethydbar [ 10-undecene- I-OTIPS] Content of polar Activity in M,l(g/rnol) MW/@" T,IT Tg/"C in feed x lo2 in moVL monomer in polymer
in wt.-% (mol-%) kg/(mol Zr - h)
2.0 0.0 2.0 12 0.6 12
- 1 240 418000 2.8 134.5 - 3.3 133.8 - 3.49 (0.31) 1190 246 000
66.2 (14.4) 58 67 OOO 1.6 125.2 -53.5
Polyme~za~on conditions: catalyst: [All = 23.88, [Al]:[Zr] = 359: 1, silica: Grace SD 3216-30; 2.0 bar ethylene: [Zr] = 3.2 * mom, [Tiba] : [Zr] = 300: I, Tp = 40°C; 0.6 bar ethylene: [Zr] = 7.2 * mom, [Tiba] : [Zr] = 249: 1.
Tab. 3. Homopolymerization of 10-undecene- 1 -oxytriisopropylsilane with MeaSi(Ind)2ZrC1&IA0 a)
[ 10-undecene-1 -0TIPSl Activity in Conversion M,J(g/mol) M,l(g/rnol) Tg /T T J T in feed x 102 in m o m in % kg/(mol Zr * h)
0.23 42 77 23 300 17 900 42 .7 -
a) Polymerization c o n ~ ~ o n s : [Zr] = 7.1 * mom, [All : [Zrl= 3 100: 1, Tp= 25 "C, tp = 20 h.
i 0 4- !
2 4 6 8 10 12 0 - t - I 1 I ' I ' I
0
[~-TlPSO-lO-undecene] ' 10 ' /(mol/L)
Fig. 3. Ethylene ~lymenzation rate as a function of the 10- undecene- 1 -ox ytriisopropylsil~e concentration. P o l ~ e ~ z a t i o n conditions: catalyst = M~Si(Ind)zZrCl~AO, [Zr] = 7.4 * lo4 mom, [All : [Zr] = 4 800 : 1, Tp = 25 "C, pelhylene = 2 bar
oxytriisopropylsilane in the copolymer is significantly lower than in the case of the homogeneous catalysis (see Tab. 2). This may be due to the diffusion restriction for the polar monomer by the crystalline polyethylene layer around the catalyst particle. When on the other hand the ethylene concentration was reduced to an extent (0.6 bar ethylene pressure) that the formation of an amorphous copolymer layer was possible, the comonomer was well i n c o ~ r a t e d (Tab. 2).
Homopolymerization of 10-undecene-I - oxytriisopropylsilane With the homogeneous Me2Si(Ind)2ZrC12/MA0 catalyst the homopolymerization of 10-undecene- 1 -0xytriisopro- pylsilane was carried out with a 77% conversion of the monomer (see Tab. 3).
Copolymerization of ethylene and 5-norbornene-2- methyleneoxytriisopropylsilane With the homogeneous 'Pr(CpInd)ZrCl2/MA0 and the heterogeneous 'Pr(CpInd)ZrC12/MAO/Si02 catalyst the copolymerization of ethylene and 5-norbornene-2-methy- leneoxytriisopropylsilane was investigated. Fig. 4 shows
In the case of the heterogeneous catalysis performed with the Me2Si(Ind)zZrC12/MAO/SiOz system at 2.0 bar ethylene pressure, the incorporation of 10-undecene- 1 -

5 14 R. Goretzki. G. Fink
6 1 I
4 0 P 4 1 O
0
0 2 4 6 8 10 12
[S-norbornene-2CH~-OTips~ ' 10 /(moUL)
Fig. 4. Ethylene polymerization rate as a function of the 5-nor- bornene-2-methyleneoxytriisopropylsilane concentration. Poly- merization conditions: (0) catalyst = 'Pr(CpInd)ZrCldMAO, [Zr] = 6.0 - lo-' mom, [Al]:[Zr] = 6000: 1, TP = 25"C, Peihyienc = 2 bar; (0) catalyst = 'Pr(CpInd)ZrClz/MAO/Si02, 0.25% Zr, [All: [Zr] = 320: 1, silica: PQ MS 3040; [Zr] = 7.3 * mom, [Tiba] : [Zr] = 277 : 1, Tp = 40 "C, = 2 bar
Tab. 4. Copolymerization of ethylene and 5-norbornene-2- methyleneoxytriisopropylsilane with 'Pr(CpInd)ZrC1zIMAO a)
pethy~en&ar [5-norbornene-2- Content of Tmb)/ "C CHz-OTIPS] in feed polar monomer
in polymer in wt.-% (mol-%)
x lo2 in mom
2.0 0.0 - 11 5.W127.5 2.0 1.5 13.2 (1.5) 108.01124.8 2.0 3 19.8 (2.4) 89.4/124.1 2.0 6 37.7 (5.7) 78N122.8
*) Polymerization conditions: [Zrl = 6.0 * mom, [All : [Zr]
b, A11 samples have a bimodal molar mass dis~bution, main fraction (ca. 97 wt.-%): a,.., = 16000 gfmol, minor fraction: M,, = ca. 1 * lo6 g/moI.
=6000:1, Tpz25OC.
(CpInd)ZxC12/MA0 catalyst", The lower comonomer incorporation under the heterogeneous catalysis condi- tions can be extremely improved by the reduction of the ethylene concentration (see Tab. 5).
The cleavage of the TIPS groups to obtain the free hydroxyl groups is not as easily accomplished as in syn-
Tab. 5. Copolymerization of ethylene and 5-norbornene-2-methyleneoxytriisopropylsi~~e with 'P~CpInd)ZrCl~AO/SiO~ a)
PethyleneJbar [5-norbornene-2-CH2-OTIPS] Content of polar monomer in Tmb)/ "C Tgl "C in feed x lo2 in mom polymer in wt.-% (mol-%)
2.0 2.0 2.0 2.0 2.0 0.8
0.0 1 3 6
12 12
116.2/125.7 . 0.91 (0.09) 1 15.3h25.5 8.59 (0.93) 113.4/132.3
16.4 (1.92) 108.1/125.5 37.3 (5.6) 106.6/125.5 65.4 (15.9) 127.8 66.8
-
*) Polymerization conditions: catalyst: 0.25% Zr, [All : [Zr] = 320: 1, silica: PQ MS 3 040; [Zr] = 7.3 * mom, [Tiba] : [Zr] =
b, All samples have a bimodal molar mass distribution, main fraction (ca. 99.5 wt.-%): aw = 16000 g/mol(O.8 bar ethylene: aw = 40000 g/mol), minor fraction: gp = ca. 1 - lo6 g/mol.
277:1,Tp=4OoC.
the polymerization rate curves of ethylene as a function of the silyl ether concentration. The ethylene polymeriza- tion rate indicates that the acceptance of the comonomer is higher in the heterogeneous than in the homogeneous case, but in comparison to the trimethylsilyl protected 5- norbornene-%methanol the activity of the homogeneous catalyst could be preserved.
As in the case for the ethylene/lO-undecene-1-OTIPS copolymerization, the silyl ether incorporation in the copolymer is higher under homogeneous catalyst condi- tions. In the case of the heterogeneous catalysis the diffu- sion problem obviously also exists for the polar monomer in comparison to the smaller ethylene (see Tab. 4 and 5). A11 copolymers produced have two melting points, which are caused by the bimodal working method of the 'Pr-
thetic organic chemistry, where acetic acid or hydrochlo- ric acid are commonly used cleaving agents13), because the polymer must be in solution to remove all protection groups. This can be completely achieved, for example, in trichlorobenzene and trifluoroacetic acid.
If U. Gi~n in i , G. Briickner, E. Pellino, A. Cassata, J. Polym. Sci., Part C 22, 157 (1968)
2, J. Boor, Jr., Ziegler-Natta-Catalysts and Polymerizations, Academic Press, New York 1979
3, DE-0s 1947109 (1969) Mitsui, Chem. Abstr: 7 2 134045~; EP 0014822 (1980) Bayer AG, Chem. Abstr: 93: 168864d; USP 4,987,200 (1991) Exxon, Chem. Abstr: 110: 173976r
4, S. Datta, G. Ver Strate, E. N. Kresge, Polym Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 33,899 (1992)

Homogeneous and heterogeneous m e t a l l ~ e n e ~ A 0 - c a t ~ y z e d ~lymenzation of t~aIkylsily1-protected alcohols 515
P. Aaltonen, B. Lofgren, Macromolecules 28,5353 (1995); P. Aaltonen, G. Fink, B. Lofgren, J. Seppala, Macromolecules 29,5255 (1996)
6, M. R. Kesti, G. W. Coates, R. M. Waymouth, J. Am. Chem. SOC. 114,9679 (1992)
7, U. M. Stehling, K. M. Stein, M. R. Kesti, R. M. Waymouth, Macromolecules 31,2019 (1998)
8, M. J. Schneider, R. Sch2fer, R. Mulhaupt, Polymer 38, 2455 ( 1997)
9, T. C. Chung, Macromolecules 21,865 (1988); S. Ramakrish- nan, E. Berluche, T. C. Chung, Macromolecules 23, 378 (1990); T. C. Chung, H. L. Lu, C. L. Li, Macromolecules 27, 7533 (1994); T. C. Chung, Polym. PrepE (Am. Chem. SOC.,
Div. Polym. Chenz) 35, 674 (1994); C. Chung, W. Janvikul, R. Bernard, G. J. Jiang, Macromolecules 27, 26 (1994); T. C. Chung, H. L. Lu, J. Mol. Cabal. A 115, 115 (1997); T. C. Chung, H. L. Lu, J. Polym. Sci., Pard A: Polym. Chem. 35, 575 (1997)
lo) P. Montag, Thesis, University of Diisseldorf 1995 11) E. J. Corey, C. Riicker, Tetrahedron Lett. 22,3455 (1981) 12) D. Ruchatz, Thesis, University of Dusseldorf 1997 13) a) K. K. Ogilvie, K. L. Sadana, E. A. Thompson, M. A. Quil-
liam, Y. B. Coestmore, Tetrahedron Lett. 33, 2861 (1974); b) K. K. Ogilvie, E. A. Thompson, M. A. Quilliam, Y. B. Coest- more, Tetrahedron Lett. 33,2865 (1974)