cardiac sodium channels as targets for new inotropic agents
TRANSCRIPT

Cardiac Sodium Channels as Targets forNew Inotropic Agents
Steinberg et al.Na
1Channels as Targets for Inotropic Agents
Mitchell I. Steinberg, Eileen McCall, Hans-JürgenMest, Achim Raap, and Theressa WrightLilly Research Laboratories, Indianapolis, Indiana U.S.A. andBeiersdorf-Lilly GmbH, Hamburg, Germany
Abstract. Congestive heart failure (CHF) is the most fre-
quent cause of hospitalization in patients over 65. Hospital-
ized patients with severe CHF could bene~t from new
agents that directly support myocardial contractility and
peripheral hemodynamics. The few classes of drugs avail-
able for short-term use (beta agonists, digoxin, and phos-
phodiesterase inhibitors) all act via cyclic AMP and have
signi~cant limitations, including tolerance, tachyarrhyth-
mias, and excessive vasodilation, especially in late-stage
disease. Newer agents are in development that increase
contractility by novel mechanisms, including calcium sensi-
tizers and ion channel modulators. Among the latter, so-
dium-channel modulators (e.g., DPI 201-106 and LY333612)
interfere directly with inactivation of the rapid sodium
channel. We review the structure and function of the human
sodium channel and discuss the role of the sodium–calcium
exchange mechanism in modulating the amount of calcium
available for use in myocardial contraction. Sodium channel
enhancers markedly increase contractility independent of
cAMP in papillary muscle strips from patients with ad-
vanced CHF that are refractory to standard agents. More-
over, the unfavorable systolic and diastolic force–frequency
relationships of isolated papillary muscle in advanced CHF
are improved by some agents in this class. In ischemic CHF
animal models, stroke volume and output are enhanced in
the absence of positive chronotropic or arrhythmogenic ac-
tivity. Additional studies are suggested to help determine
the ultimate role for these agents in the therapy of end-
stage CHF.
Progressive left ventricular dysfunction occurs even inthe absence of symptoms [1]. Eventually, the adaptivehemodynamic and neurohormonal mechanisms respon-sible for sustaining the circulation fail and the signsand symptoms of congestive heart failure (CHF) de-velop. Of the over 4 million people in the United Stateswith heart failure, about 10% have NYHA class IVsymptoms [2–4]. At this stage, both hospital admissionrates and medical costs escalate rapidly [5,6]. In itsadvanced stages, CHF has the highest hospital read-mission rate of any medical diagnosis and representsthe largest diagnostic related group in the UnitedStates [6], accounting for over 2 million hospital admis-sions per year [7]. Roughly twice as much is spent byMedicare on hospitalized patients with CHF than forpatients with either cancer or myocardial infarction.
The increasing costs and morbidity associated withCHF result from a general aging of the population [8],the postponement of mortality with the use of neuro-hormonal antagonists in earlier stages of heart failure[9,10], and improved drug therapy for coronary heartdisease and hypertension. Moreover, advances in non-pharmacological treatments such as cardiac transplan-tation have led to a nearly sevenfold increase in candi-dates and a 30% increase in waiting times. Whileawaiting de~nitive therapy, most patients receive addi-tional drug and mechanical advanced life supportmeasures, further contributing to increased costs[11,12].
In advanced stages of CHF, intravenous inotropictherapy still represents the primary pharmacologic op-tion for maintaining adequate pump function [13]. Thedevelopment of inotropic agents has largely remainedstatic since the introduction of dobutamine anddopamine over two decades ago (see [14]). Dobutaminehas proven useful not only for CHF and general post-surgical support but also for the diagnosis and riskstrati~cation of ischemic heart disease [15]. Whilethere is widespread agreement that inotropic agentsare useful to improve the quality of life in patients withadvanced CHF, there is as yet little evidence to sup-port the long-term ability of these agents to alter thenatural course of the disorder and reduce mortality[13,16].
Prior attempts at introducing phosphodiesterase in-hibitors (PDEI) and oral b-agonists were based on thebelief that the bene~ts of acute inotropic support couldbe extended to the outpatient setting, resulting insymptom relief as well as decreased mortality [17,18].Unfortunately, all such attempts have resulted in anincreased risk of premature death [19]. However, it isunclear whether longer-term inotropic support per seis undesirable or whether the means by which long-term inotropic support is achieved is more important.For instance, a marked and sustained increase in ejec-tion fraction resulting from the slowing or reversal of
Heart Failure Reviews 1998;2:173–193
© Kluwer Academic Publishers, Boston. Printed in U.S.A.
173
Address for correspondence: Dr. Mitchell I. Steinberg, Lilly Re-search Laboratories, DC-0524, Dept. MC 304, Lilly CorporateCenter, Indianapolis, IN 46285, U.S.A.

ventricular remodeling is a consistent response thataccompanies the reduction in mortality seen with long-term b-blockade [20].
Inotropic Drug Classi~cation
Attempting to classify inotropic agents is dif~cult due tothe variety of overlapping mechanisms by which suchdrugs can act ([21]; see Figure 1). However, a simple andpractical classi~cation distinguishes between agentswhose primary mechanism is mediated by 1) increasedintracellular cyclic adenosine monophosphate (cAMP),e.g., b-agonists and phosphodiesterase inhibitors(PDEI), and 2) increased intracellular calcium (Ca21)availability to myo~brils. The second group includesagents that affect sodium (Na1) movement, sensitizethe contractile elements to endogenous Ca21, or in-crease Ca21 availability (Table 1). Unfortunately, agents
in the second group are often rather nonspeci~c, andmost also increase levels of cAMP by inhibiting PDE. Itshould be understood that primary increases in cAMPwill necessarily modify subsequent Ca21 transients, butthis categorization emphasizes that some agents canmodify contractility entirely independent of cAMP. Thisdistinction is important because, other than the recent(Digital Investigation Group) DIG trial [22] (vide infra),there have never been any long-term trials of inotropicagents that do not either directly or re_exly activate theb-receptor/cAMP system.
Agents acting via cAMP
Most agents that are used clinically for acute inotropicsupport activate the b-receptor/cAMP pathway and in-clude both inotropic catecholamines and PDEIs. CyclicAMP-dependent phosphorylation reactions enhancein_ux of Ca21 through the voltage-sensitive Ca21 chan-nel, promote relaxation by enhancing uptake of Ca21 by
Fig. 1. Schematic representation of a myocardial cell showing major sites of action of known classes of inotropic agents and their sec-ond messenger systems. The sodium channel is depicted in close apposition to the Na1–Ca21 exchanger mechanism, functionally link-ing the two. Thus, sodium channel enhancers may modulate contractile activity in the absence of effects on cAMP.
174 Steinberg et al.

the sarcoplasmic reticulum (SR), and decrease the sen-sitivity of contractile proteins [23] (see Figure 1).
Although highly effective for maintaining contrac-tility in a variety of acute situations, drugs actingthrough this mechanism may also possess distinct clini-cal disadvantages [24]. First, these agents increaseheart rate and ventricular automaticity, either directlyor re_exly, especially at higher doses. This outcome isparticularly disadvantageous for patients with heartfailure, since heart rate is a major determinant of myo-cardial oxygen consumption (MVO2) [25]. Second, inadvanced CHF, b-receptors are downregulated, andthe maximum inotropic responses to both b-agonistsand PDEIs tend to be blunted [26,27]. Third, activationof the b-receptor/cAMP pathway is associated with avariety of cellular responses that could, at least theo-retically, contribute to direct myocyte toxicity [28,29].For instance, high levels of inotropic catecholaminesmay cause Ca21 overload and generate superoxide an-ion, which could contribute to the initiation of necrosisand/or programmed cell death [30–33]. Elevation ofintracellular cAMP increases not only the rate of con-traction but also the rate of relaxation, further contrib-uting to an increase in MVO2. The decrease in cAMP inheart failure [34] and the downregulation of b-recep-tors could thus be seen as compensatory efforts by thefailing heart to protect against excessive MVO2. Theadverse effects of cAMP-mediated inotropy could havecontributed, at least in part, to the adverse risk ofmortality seen in longer-term clinical trials with eitheroral or i.v. agents in this class [5,35].
Agents acting independently of cAMP
Agents acting independently of cAMP either directlymodify sarcolemmal ionic transport mechanisms or en-hance the sensitivity of the contractile machinery to
Ca21 (Figure 1). Included in this class is digitalis, themost widely used and oldest inotropic agent. The roleof digitalis in CHF patients with normal sinus rhythmhas been debated ever since Withering ~rst describedthe actions of the foxglove extract over 200 years ago[36]. Intravenous digoxin has limited utility in the set-ting of acute CHF and has its main utility for chronicmaintenance therapy [37]. Recent studies have conclu-sively shown that digoxin is effective in relieving con-gestive symptoms and decreasing hospital readmis-sions for worsening CHF [38,39]. Because of itsbene~ts for symptom relief, its use is likely to continueeven though the recent DIG trial did not show reduc-tions in overall mortality [22].
Digitalis remains the only inotropic agent forchronic use that does not increase the risk of death. Itsrelatively benign pro~le may derive from its mild inot-ropic actions along with favorable hemodynamic andautonomic effects; digitalis neither increases heart ratenor excessively increases MVO2 [37]. The ability ofdigitalis to sensitize the activity of impaired barorecep-tors in patients with CHF appears particularly rele-vant. Unfortunately, digoxin blood levels must remainwithin a relatively narrow range (recent studies sug-gest levels not much above 1 ng/mL). It has been esti-mated that perhaps 25% of hospitalized patients ad-ministered digitalis show some signs of drug toxicitythat can be dif~cult to distinguish from the underlyingdisease state [36]. Most of its untoward actions relateclosely to its mechanism of action, i.e., inhibiting mem-brane-bound Na1–K1 adenosine triphosphatase (AT-Pase). Of particular concern are the numerous druginteractions that can alter serum levels of digoxin andthe potentiation of digitalis toxicity by electrolyte de-pletion (especially in conjunction with intense diuretictherapy). Because of these limitations, attempts are
Table 1. Classi~cation of inotropic agents
Agents Acting via cAMP Agents Acting Independent of cAMP
b-agonists Digitalis derivativesDobutamine, dopexamine Digoxin, digitoxin, ouabainDopamine, ibopamine Na1–K1ATPase isoform inhibitorsIsoproterenol, norepinephrine Sodium channel activatorsXamoterol, prenalterol Racemates
Phosphodiesterase (PDEI) III inhibitors DPI 201-106Milrinone, amrinone, enoximone Carsatrin (RWJ 24517)Imazodan, piroximone LY333612 (BDF 9146)
LY348395 (BDF 9198)Mixed action Single enantiomers
Myo~lament sensitization and PDEI LY366634 (BDF 9196)Sulmazol, pimobendan LY368052Levosmendan, EMD 57033, MCI-154 Calcium agonists
Ion channel modulation and PDEI SelectiveVesnarinone, OPC 18790, OPC 8490 BAY K 86466, BAY 5959
Calcium sensitizingCGP 48506
Na1 Channels as Targets for Inotropic Agents 175

being made to develop selective inhibitors of the car-diac isoform of Na1–K1-ATPase that might have agreater therapeutic index [40].
Inotropic drugs are also available that act by modu-lating Ca21 or Na1 transport independently of ATPaseinhibition. Included in this group are agents that in-crease the responsiveness of the contractile elementsto Ca21 or increase their mechanical ef~ciency [21], aswell as those that stereospeci~cally enhance Ca21
in_ux through L-type Ca21 channels [41]. The proto-type of the Ca21 channel activators is the (S)-enan-tiomer of BAY K 8644, a dihydropyridine analoguethat increases Ca21 transport in both cardiac andsmooth muscle. Recently, newer cardioselective ana-logues such as BAY Y 5959 have been reported toincrease contractility without altering blood pressurein an animal model of heart failure [42]. However, byincreasing diastolic Ca21, such drugs have the poten-tial to impair relaxation and promote myocyte Ca21
overload [43].Several Ca21 sensitizing agents have been studied
in man, including levosimendan and pimobendan. Al-though initially promising, pimobendan has beenshown recently in a phase III clinical trial to increasemortality. Pimobendan not only increases the sensitiv-ity of troponin C to Ca21 but also inhibits PDE andincreases cAMP in cardiac and smooth muscle [44–46].Levosimendan (perhaps the most potent Ca21 sensi-tizer acting directly at the level of troponin C [46])likewise possesses signi~cant PDE inhibitory activity,resulting in vasodilatation, re_ex activation of thesympathetic nervous system, and increased heart rate[47,48]. Recently, selective enantiomeric Ca21 sensitiz-ers have been reported, including EMD 570033 andCGP 48506. However, EMD 570033 still retains PDEIactivity [49]. Although CGP 48506 increases contractil-ity independent of cAMP in human failed myocardium[50], it markedly prolongs relaxation. Nevertheless,CGP 48506 is the only sensitizer to date that does notseem to affect PDE.
An ion channel modulator–PDEI that has gener-ated intense clinical interest is vesnarinone (OPC8212). This inotropic agent was originally reported todramatically increase survival in chronic congestiveheart failure in doses of 60 mg, while larger dosesmarkedly decreased survival [51]. The results ap-peared to support the notion that small increases incontractility might be bene~cial, while “whipping theheart” with large doses are detrimental. Moreover,some investigators postulated that low doses of ves-narinone might possess, “noninotropic actions,” e.g.,cytokine inhibition, that might mediate the agent’sbene~cial effects. However, additional studies using 60and 30 mg/day of vesnarinone have shown, in contrastto the original report, that even low doses of this agentare associated with an adverse risk of mortality(mainly due to increases in sudden cardiac death), andclinical trials with this drug have now been suspended.These disappointing results once again raise the issue
of whether there exists any dose of an inotropic drugthat can be safely administered to heart failure pa-tients during chronic therapy [19]. Vesnarinone and itsquinolinone derivatives (OPC 8490 and OPC 18790)have other cellular effects that could potentially con-tribute to an adverse outcome in chronic therapy.These include effects on potassium conductance [52],direct activation of Ca21 transport, and potent inhibi-tion of PDE [53–55]. Thus, it is possible that the ad-verse effects of vesnarinone on mortality are related toactivation of the cAMP pathway and/or subtle adverseelectrophysiological effects and not to its “inotropic”actions.
Recently, a series of synthetic, lipid-soluble com-pounds that enhance Na1 transport while retaininghigh af~nity for cardiac tissues have been described[56,57]. Such agents exhibit profound inotropic activityin the absence of heart rate change and hence are ofpotential interest as a new class of positive inotropicagent that acts independently of the cAMP pathway(for reviews, see [58,59]). While compounds in thiscategory have little or no propensity to affect cAMP,their inotropic mechanisms will likely include morethan direct effects on the Na1 channel [56,60,61]. For amore complete understanding of the mechanism of ac-tion of these agents and their potential role in thetherapy of CHF, the current view of the structure,molecular biology, and function of the cardiac Na1
channel is discussed below.
Sodium Channel Structure andFunction
Voltage-gated Na1 channels are essential for the initia-tion and spread of action potentials in excitable tissue.The ionic basis of the action potential and the centralrole of Na1 were ~rst demonstrated in Hodgkin andHuxley’s classic voltage-clamp study of squid giant ax-ons (for a review, see [62]). Many insights at the mo-lecular level have been gained more recently usingsingle-channel electrophysiology, antipeptide antibod-ies, genetic mapping, and site-directed mutagenesis[63]. Na1 channels are thought to have been derivedfrom potassium (K1) channels after gene duplication[63], although the population is not as diverse as thatof K1 channels [64]. Structural homology is also appar-ent between the tetrodotoxin (TTX)-sensitive Na1
channel and the a1-subunit of the L-type Ca21 channel.
A topological model of the sodium
channel
The currently accepted schematic of the cardiac humanheart Na1 channel, hH1, is shown in Figure 2. In 1992Gellens et al. [65] cloned, sequenced, and functionallyexpressed the major human cardiac Na1 channel gene(SCN5A) consisting of 2016 amino acid residues com-prising four domains (D), each domain consisting of sixmembrane-spanning segments (S1–6). All four do-
176 Steinberg et al.

mains and the DIII–DIV loop are necessary for thechannel to function and are highly conserved [66]. Thechannel consists of two subunits; a large a-subunit andup to four smaller b-subunits. The a-subunits containall the necessary structures for voltage-dependent gat-ing and ion conduction, while the b-subunits modulatethese properties and may be required for optimal fastinactivation [67]. Thus, a-subunits expressed aloneshow slower inactivation and more positive voltagedependence than when the subunits are coexpressed.The a-subunit has a molecular weight of about 260 kDa,and consists of the four-domains (DI–DIV) and theirsix imbedded segments (Figure 2A). These segmentsare most likely antiparallel alpha-helices connected bynonconserved, hydrophilic linker sections. The aminoand carboxyl terminal and linker segments are intra-cellular, while the hydrophilic segments bridging thehelices form loops that are exposed to the aqueousenvironment and may be extracellular (S1–2, S3–4, andS5–6) or intracellular (S2–3 and S4–5). The b1-subunitis an intrinsic membrane protein with a molecularweight of 36 kDa. It is linked to the a-subunit withnoncovalent bonds and may have an extracellular sur-face, since it can be labeled by extracellular neurotox-ins [68]. The small family of b1-subunits share the char-acteristics of four glycosylation sites, a single a-helicalmembrane-spanning area, and a very small intracellu-lar domain. These subunits may speci~cally associatewith particular a-subunits [69].
The cardiac Na1 current. Sodium carries the pri-mary depolarizing current in most cardiac tissues ex-cept the AV node. From studies in cardiac Purkinje~bers [70], it was established that the cardiac actionpotential upstroke resembled that of neurons, and thatthe resultant Na1 current (INa) was distinct from theCa21 current (ICa) [71,72]. The cardiac INa can, however,be differentiated from neuronal INa both kinetically andpharmacologically. Cardiac INa is slower than neuronalcurrents and is insensitive to saxitoxin and TTX butcan be blocked by some divalent ions, e.g., cadmiumand zinc.
At resting membrane potentials (-80 to -90 mV), theNa1 channel is normally closed. It opens in response todepolarization, allowing Na1 in_ux, with energy de-rived from existing electrochemical gradients. As themembrane potential continues to fall, more Na1 chan-nels open due to positive feedback. INa flows longitudi-nally along cells, depolarizing the membranes of adja-cent cells at a rate of 100–500 V/sec and generating moreINa to activate these cells [73]. Depolarization also in-duces inactivation of the channel so that Na1 channelsare normally open only transiently. The Na1 channelcycle can be thought of as a three-state Markov chainmodel, with the states being 1) closed, nonconducting,but excitable (also known as the resting state), 2) open(or conducting), and 3) inactive, (nonconducting), thetransitions among them being time and voltage depend-ent. An important difference between the resting and
inactivated states is that the latter cannot conduct cur-rent until it recycles back through the resting state.These differences are important, since most drugs act-ing at the Na1 channel are state dependent.
Na1 channel structure differs among tissues, andvarious Na1 channel isoforms may be expressed evenin the same tissue (see [74] and below). At least ~vedifferent cardiac Na1 channel phenotypes are nowknown [75–77]. The predominant isoform is voltage de-pendent and TTX insensitive. Slow TTX- and Mn21-in-sensitive Na1 channels have been found in myocardialmembranes from cardiomyopathic hamsters [78,79]. Inthe future, each Na1 channel isoform may serve as anovel therapeutic target as more becomes knownabout their pathophysiological roles.
A functional model of the cardiac
sodium channel
For more than a decade, the Na1 channel has beenviewed as having a dilated region at either end (withthe outer dilatation being the TTX receptor), a centralselectivity ~lter, and two gates at its inner end (for areview, see [80]). This model was developed with sev-eral constraints: 1) TTX acts externally and does notaffect gating; 2) the selectivity ~lter is 3 Å by 5 Å, and3) the inactivation site is only accessible when the acti-vation gate is partly open. Developments in molecularbiology and modeling techniques now have allowed amore sophisticated model of the Na1 channel to beformulated, as outlined below. Differences betweencardiac and other channels are noted where appropri-ate.
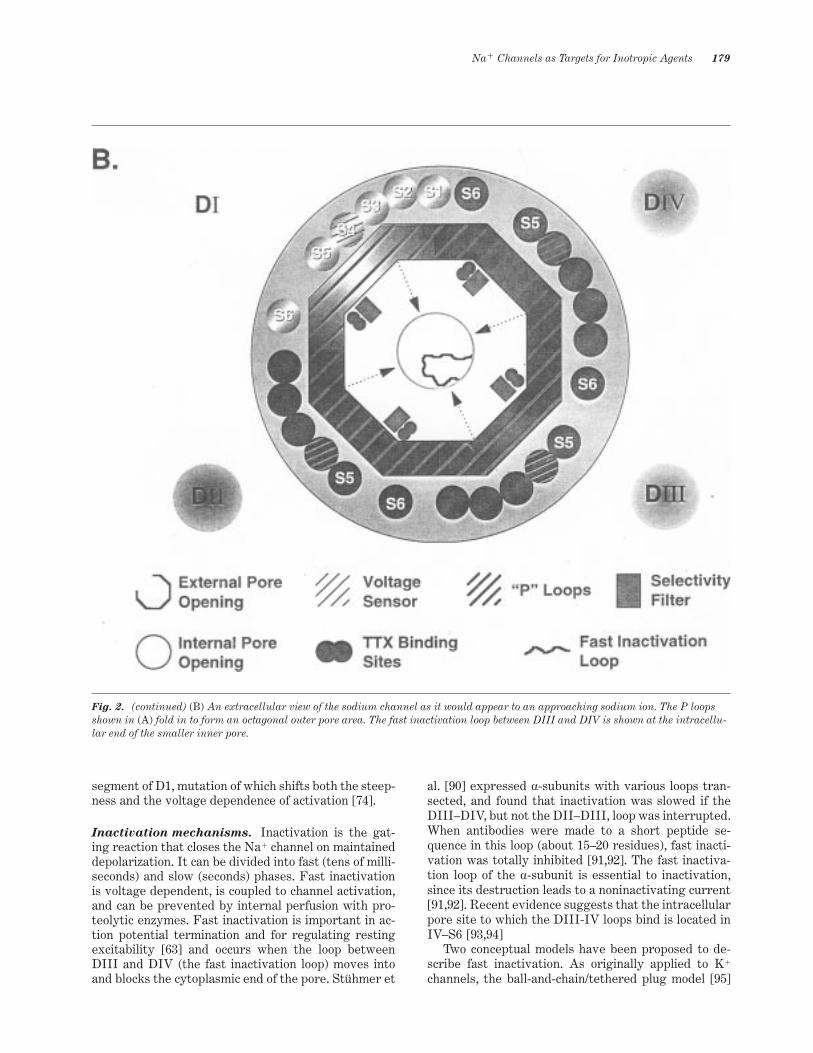
The ion conductance pathway. The pore is the ionconductance pathway and is the binding site for mostinhibitory toxins and local anesthetic agents. The fourextracellular loops between S5 and S6 in each domainof the a-subunit contain the amino acid sequences thatline the pore. These four loops (known as P loops) foldback on themselves and into the membrane to form anoctagonal pore (Figure 2B). Glycosylation occursmainly on the P loop of domain I. Tetrodotoxin andsaxitoxin are potent water soluble blockers of the Na1
ion conductance pathway and antagonize the effect ofall known Na1 channel activator substances [81]. Thesetoxins thus serve as critical tools to demonstrate therole of Na1 channels in the mechanism of action ofchannel activators. In contrast to these natural toxins,most synthetic compounds that interact with Na1
channels inhibit channel function [82], have highaf~nity for cardiac tissue, and are used therapeuticallyas local anesthetics and in the treatment of cardiacarrhythmias [76].
Site-directed mutagenesis [83,84] has shown thatTTX binding is controlled by two rings of carboxylresidues around the external mouth of the pore andthat both the charge and the nature of the constituentamino acids confer speci~city to TTX binding (Figure
Na1 Channels as Targets for Inotropic Agents 177

2). The difference in TTX sensitivity between cardiacand neuronal channels is thought to be due to the sub-stitution of a cysteine (in cardiac channels) for tyrosineor phenyalanine at a critical position proximal to thecarboxyl rings. This cysteine is essential for the high-af~nity cadmium block of cardiac Na1 channels [85–87],since substitution leads to cadmium insensitivity. Thissite is probably critical in determining cardiac andneuronal Na1 channel susceptibility to inhibitory tox-ins and divalent cations. The intracellular surface ofthe pore can be phosphorylated by multiple proteinkinases [88].
To be selective for Na1, the channel must attractmonovalent cations but exclude K1 and divalent cat-ions, especially Ca21. In 1994, Lipkind and Fozzard [89]envisioned a conical pore containing the TTX site thatnarrowed to form the ion selectivity ~lter (Figure 2B).The constriction is formed by four speci~c residues(DEKA), one from each of the four domains, and muta-tions in at least three of these residues affect selectiv-ity [88]. Lysine 1422 and alanine 1714 are critical, withmutations of these residues causing the channel to be-
come highly Ca21 selective, showing high-af~nity Ca21
binding and block of monovalent ion conductance [87].Lysine 1422 is also important in selecting Na1 over K1
as the permeant ion.
Voltage sensing and activation. Sodium channelactivation occurs through a voltage-driven conforma-tional change in the membrane, causing the pore toopen. The S4 helix in each domain of the a-subunit isthe voltage sensor, containing repeated motifs of onepositive then two hydrophobic residues (see Figure2A). Gating charge arises from the movement of sixpositive residues through the membrane. Individualresidues can stabilize the channel in either an activatedor a closed state, depending on their position. The twomodels of charge movement are 1) the sliding he-lix/helical screw model, in which the S4 helix movesacross the membrane and exchanges ion pairs with thesurrounding transmembrane segments, and 2) thepropagating helix model, in which charged residues aremoved due to a-helix/b-sheet transition. Activation de-pends on four positively charged residues in the S4
Fig. 2. Topological depiction of the human cardiac sodium channel. (A) Cross sectional view, illustrating the arrangement of the sixindividual segments composing each domain. Shown are the critical residues involved in activation, inactivation, and ligand recogni-tion.
178 Steinberg et al.
segment of D1, mutation of which shifts both the steep-ness and the voltage dependence of activation [74].
Inactivation mechanisms. Inactivation is the gat-ing reaction that closes the Na1 channel on maintaineddepolarization. It can be divided into fast (tens of milli-seconds) and slow (seconds) phases. Fast inactivationis voltage dependent, is coupled to channel activation,and can be prevented by internal perfusion with pro-teolytic enzymes. Fast inactivation is important in ac-tion potential termination and for regulating restingexcitability [63] and occurs when the loop betweenDIII and DIV (the fast inactivation loop) moves intoand blocks the cytoplasmic end of the pore. Stühmer et
al. [90] expressed a-subunits with various loops tran-sected, and found that inactivation was slowed if theDIII–DIV, but not the DII–DIII, loop was interrupted.When antibodies were made to a short peptide se-quence in this loop (about 15–20 residues), fast inacti-vation was totally inhibited [91,92]. The fast inactiva-tion loop of the a-subunit is essential to inactivation,since its destruction leads to a noninactivating current[91,92]. Recent evidence suggests that the intracellularpore site to which the DIII-IV loops bind is located inIV–S6 [93,94]
Two conceptual models have been proposed to de-scribe fast inactivation. As originally applied to K1
channels, the ball-and-chain/tethered plug model [95]
Fig. 2. (continued) (B) An extracellular view of the sodium channel as it would appear to an approaching sodium ion. The P loopsshown in (A) fold in to form an octagonal outer pore area. The fast inactivation loop between DIII and DIV is shown at the intracellu-lar end of the smaller inner pore.
Na1 Channels as Targets for Inotropic Agents 179

envisions a short, positively charged peptide sequence,loosely attached to the fast inactivation loop, that be-comes bound to a negatively charged receptor site atthe inner mouth of the pore. The receptor site (presum-ably within DIV–S6) develops a high af~nity for theloop residues on channel activation, providing the linkbetween activation and inactivation [88]. In thehinged-lid model [96], based on allosteric enzyme inac-tivation, the lid is represented by the fast inactivationloop and would have the _exibility to pivot to place thepeptide sequence close enough to the mouth of the poreto allow binding.
The peptide sequence of the fast inactivation loopis highly conserved. A cluster of hydrophobic resi-dues, designated IFM (isoleucine, phenylalanine,methionine), control the binding of the DIII–DIV loopto the internal pore area (DIV–S6), thereby servinga latchlike function in preventing the inactivation gatefrom reopening (Figure 2A). Mutation of the IFMcluster in rat brain Na1 channels completely blocksinactivation [97]. In hH1 Na1 channels, simultaneousmutation of IFM to I1484Q, F1485Q, and M1486Qcompletely inhibits fast inactivation from both openand closed channels. The F1485Q mutation alonecauses inactivation that is tightly coupled to the openstate and leads to repetitive cycling between restingand open states at depolarized potentials [98]. Themechanism is proposed to be due to the destabilizationof the channel in the inactivated state and to a re-duction in the association rate of the open channel andthe inactivation gate.
Slow inactivation is apparently involved in diastolicregulation. It remains intact after internal proteolysis.Recent evidence in wild-type channels suggests thatfast and slow inactivation may compete, with fast inac-tivation being predominant [99]. The topological re-gion(s) mediating slow inactivation in Na1 channels arepoorly de~ned. However, it is known that mutation ofamino acid residues in the S6 segment of voltage-gatedCa21 channels impairs slow inactivation [100]. In ratskeletal muscle Na1 channels, this segment is also in-volved with slow inactivation [101]. In addition to Na1
and Ca21 channels, K1 channels also exhibit slow inac-tivation [102], suggesting that cation channels in gen-eral might share analogous regions for slow inactiva-tion.
Clinical consequences of Na1 channel dysfunc-
tion. An improved understanding of the structureand function of Na1 channels has led to the recognitionof a variety of gain-of-function genetic mutations ofskeletal and cardiac muscle [103,104]. In cardiac mus-cle, the major phenotypic expression of abnormal Na1
channel gene expression is the long QT syndrome. Thissyndrome was the ~rst genetic cardiac ion channel dis-ease recognized [105] and is characterized by delayedventricular repolarization (prolonged QT interval),ventricular arrhythmia (predominantly torsades despointes), syncope, and an increased risk of premature
death. Various investigators [106–109] have identi~edfour chromosomal loci for K1 and Na1 channel genesassociated with long QT syndrome.
LQT3 is due to a mutation of the fast cardiac Na1
channel gene SCN5A on chromosome 3 that causesin-frame deletions destabilizing the inactivation gate.Mutations so far identi~ed result in the deletion ofthree amino acids, KPQ, in the DIII–DIV loop (Figure2A) so that the Na1 channel repeatedly reopens duringdepolarization, shifting between normal and persistentopenings to prolong the APD [105]. Single residue sub-stitutions in KPQ mediate late, persistent Na1 current,thereby delaying repolarization [107]. Priori et al. [110]modeled the genetic abnormalities of long QT syn-drome in isolated guinea pig myocytes using pharma-cological probes to mimic the phenotype of LQT3 (an-thopleurin A). In the case of the LQT3 model, theprolonged action potentials were shortened by mex-ilitine, rapid pacing, and isoproterenol, interventionsthat also are useful in patients with prolonged QT syn-drome linked to mutations in SCN5A [111]. Thus, cel-lular models of abnormal Na1 channel activation maybe useful for determining the potential arrhythmo-genicity or antiarrhythmic effects of new drugs thatmight interact with the cardiac Na1 channel.
Pharmacology of Na1 ChannelEnhancers
Sodium Channel LigandsIt has been long known that a variety of naturallyoccurring neural toxins act to prolong the open state ofNa1 channels in excitable tissue [81,112]. These includethe water-soluble, polypeptide toxins, e.g., alpha scor-pion toxin, goniopora toxin from salt water coral [113],and the sea anemone toxin, ATX-II, and a diversegroup of lipid-soluble, Na1 channel activator toxins,e.g., veratridine, batrachotoxin, aconitine, and the py-rethroids. These toxins are generally more speci~c forneural and skeletal muscle Na1 channels than for car-diac tissue—hence their extreme toxicity in man.
Interest in synthetic cardiospeci~c inotropic Na1
channel modulators was initially stimulated by the de-scription of the novel piperazinylindole derivative, DPI201-106 [56,57,106,114–123]. Agents in this class exerta positive inotropic effect independent of cAMP byenhancing Na1 in_ux through voltage-dependent Na1
channels. The consequent increase in intracellular Na1
activates the Na1 2 Ca21 exchange mechanism work-ing in reverse mode to increase intracellular Ca21
[57,118,119,124]. DPI 201-106 is a fairly potent ligandat the phenylalkylamine [125,126] and dihydropyridine[127] sites of the Ca21 channel, as well as a nonstereose-lective antagonist of the Ca21 current in cardiac muscle[116,127] and vascular smooth muscle [128].
More potent and speci~c agents such as LY333612
180 Steinberg et al.
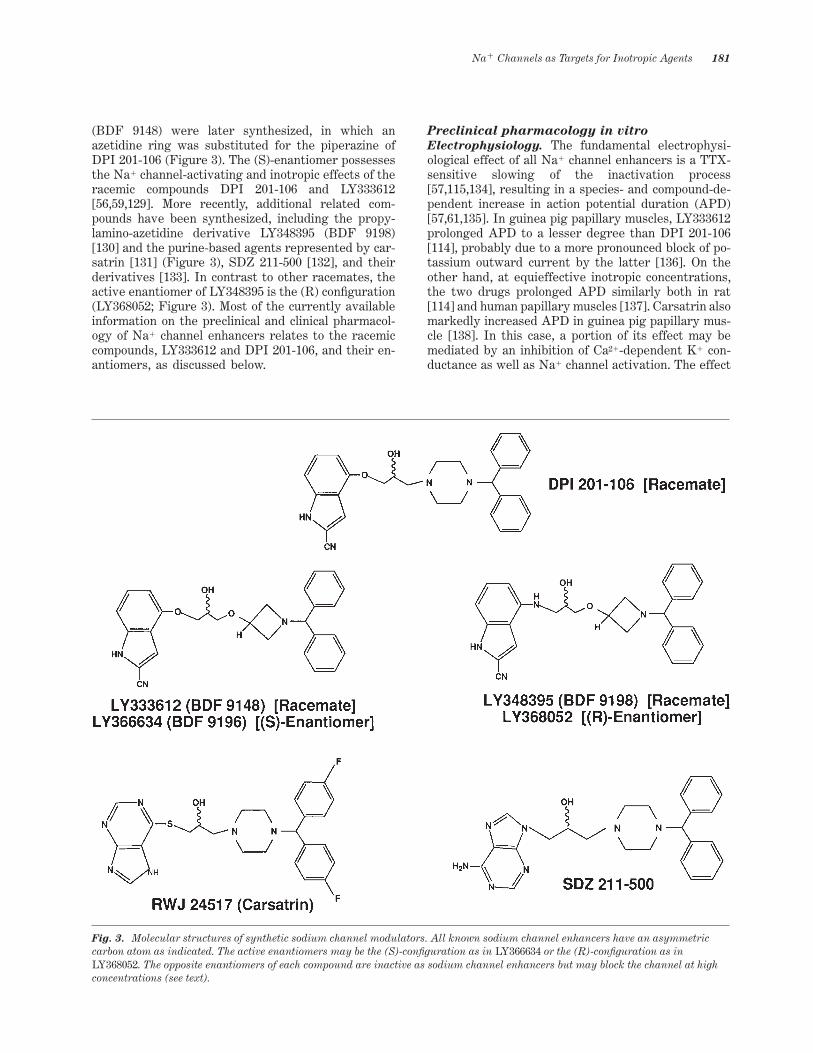
(BDF 9148) were later synthesized, in which anazetidine ring was substituted for the piperazine ofDPI 201-106 (Figure 3). The (S)-enantiomer possessesthe Na1 channel-activating and inotropic effects of theracemic compounds DPI 201-106 and LY333612[56,59,129]. More recently, additional related com-pounds have been synthesized, including the propy-lamino-azetidine derivative LY348395 (BDF 9198)[130] and the purine-based agents represented by car-satrin [131] (Figure 3), SDZ 211-500 [132], and theirderivatives [133]. In contrast to other racemates, theactive enantiomer of LY348395 is the (R) con~guration(LY368052; Figure 3). Most of the currently availableinformation on the preclinical and clinical pharmacol-ogy of Na1 channel enhancers relates to the racemiccompounds, LY333612 and DPI 201-106, and their en-antiomers, as discussed below.
Preclinical pharmacology in vitro
Electrophysiology. The fundamental electrophysi-ological effect of all Na1 channel enhancers is a TTX-sensitive slowing of the inactivation process[57,115,134], resulting in a species- and compound-de-pendent increase in action potential duration (APD)[57,61,135]. In guinea pig papillary muscles, LY333612prolonged APD to a lesser degree than DPI 201-106[114], probably due to a more pronounced block of po-tassium outward current by the latter [136]. On theother hand, at equieffective inotropic concentrations,the two drugs prolonged APD similarly both in rat[114] and human papillary muscles [137]. Carsatrin alsomarkedly increased APD in guinea pig papillary mus-cle [138]. In this case, a portion of its effect may bemediated by an inhibition of Ca21-dependent K1 con-ductance as well as Na1 channel activation. The effect
Fig. 3. Molecular structures of synthetic sodium channel modulators. All known sodium channel enhancers have an asymmetriccarbon atom as indicated. The active enantiomers may be the (S)-con~guration as in LY366634 or the (R)-con~guration as inLY368052. The opposite enantiomers of each compound are inactive as sodium channel enhancers but may block the channel at highconcentrations (see text).
Na1 Channels as Targets for Inotropic Agents 181
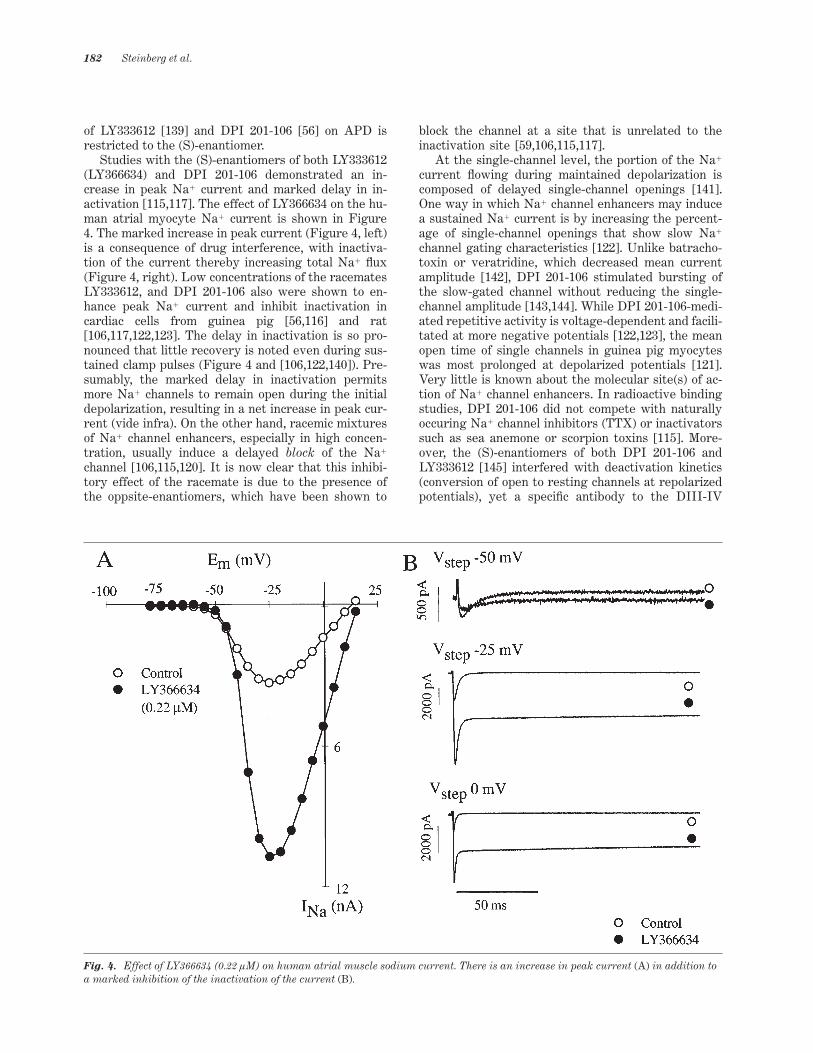
of LY333612 [139] and DPI 201-106 [56] on APD isrestricted to the (S)-enantiomer.
Studies with the (S)-enantiomers of both LY333612(LY366634) and DPI 201-106 demonstrated an in-crease in peak Na1 current and marked delay in in-activation [115,117]. The effect of LY366634 on the hu-man atrial myocyte Na1 current is shown in Figure4. The marked increase in peak current (Figure 4, left)is a consequence of drug interference, with inactiva-tion of the current thereby increasing total Na1 _ux(Figure 4, right). Low concentrations of the racematesLY333612, and DPI 201-106 also were shown to en-hance peak Na1 current and inhibit inactivation incardiac cells from guinea pig [56,116] and rat[106,117,122,123]. The delay in inactivation is so pro-nounced that little recovery is noted even during sus-tained clamp pulses (Figure 4 and [106,122,140]). Pre-sumably, the marked delay in inactivation permitsmore Na1 channels to remain open during the initialdepolarization, resulting in a net increase in peak cur-rent (vide infra). On the other hand, racemic mixturesof Na1 channel enhancers, especially in high concen-tration, usually induce a delayed block of the Na1
channel [106,115,120]. It is now clear that this inhibi-tory effect of the racemate is due to the presence ofthe oppsite-enantiomers, which have been shown to
block the channel at a site that is unrelated to theinactivation site [59,106,115,117].
At the single-channel level, the portion of the Na1
current _owing during maintained depolarization iscomposed of delayed single-channel openings [141].One way in which Na1 channel enhancers may inducea sustained Na1 current is by increasing the percent-age of single-channel openings that show slow Na1
channel gating characteristics [122]. Unlike batracho-toxin or veratridine, which decreased mean currentamplitude [142], DPI 201-106 stimulated bursting ofthe slow-gated channel without reducing the single-channel amplitude [143,144]. While DPI 201-106-medi-ated repetitive activity is voltage-dependent and facili-tated at more negative potentials [122,123], the meanopen time of single channels in guinea pig myocyteswas most prolonged at depolarized potentials [121].Very little is known about the molecular site(s) of ac-tion of Na1 channel enhancers. In radioactive bindingstudies, DPI 201-106 did not compete with naturallyoccuring Na1 channel inhibitors (TTX) or inactivatorssuch as sea anemone or scorpion toxins [115]. More-over, the (S)-enantiomers of both DPI 201-106 andLY333612 [145] interfered with deactivation kinetics(conversion of open to resting channels at repolarizedpotentials), yet a speci~c antibody to the DIII-IV
Fig. 4. Effect of LY366634 (0.22 lM) on human atrial muscle sodium current. There is an increase in peak current (A) in addition toa marked inhibition of the inactivation of the current (B).
182 Steinberg et al.

linker region modi~ed only inactivation. This findingsuggests that additional sites, beside the known inacti-vation linker region, are involved in the action of thesedrugs [140].
Inotropy: nonhuman myocardium. In contrast tob-adrenergic drugs or PDEIs, Na1 channel enhancersincrease contractility without altering levels of cAMP[56,130,139,146,147]. Unlike cardiac glycosides, Na1
channel enhancers did not inhibit guinea pig myocar-dial Na1–K1-ATPase [135,139,147]. Moreover, additionof propranolol, cimetidine, or prazosin did not abolishtheir positive inotropic effect in guinea pig papillarymuscles [135,139] or guinea pig atria [56]. Thus, thepositive inotropic effect is not mediated by a, b, orhistaminergic receptors. However, DPI 201-106 wasreported to possess Ca21 - sensitizing effects in ferretand guinea pig papillary muscles [60] and in porcinecardiac trabeculae [56]. TTX, however, abolished thepositive inotropic response of LY333612[116,135,139,148], LY348395 [130], and DPI 201-106[60], supporting the idea that the inotropic action iscausally related to the increase in Na1 in_ux. The in-crease in intracellular Na1 inhibits the Na1–Ca21 ex-change mechanism [57,116] resulting in a decrease inCa21 ef_ux [149]. There is also strong evidence that theexchanger also serves as a source for Ca21 in_ux whenoperating in reverse mode [149–153].
The inotropic activity of DPI 201–106, LY333612, andits (S)-enantiomer (LY366634) has been described incardiac tissues from various species, including rat, rab-bit, and guinea pig atrial and ventricular tissue[56,57,116,129,135,148,154–158] (Figure 5). The rank or-der of potency in increasing force of contraction of iso-lated electrically stimulated guinea pig papillary mus-cles is as follows: LY348395 @ LY366634 . LY366612 .DPI 201–106 [130,139] (Figure 5). In left atria from theguinea pig [129,139,158] and rat [129], LY333612 waslikewise 3- to 10-fold more potent than DPI 201–106.The basis for the enhanced potency of LY333612 com-pared to DPI 201–106 is likely related to the greaterpotency of the former in prolonging the Na1 current[114]. The effect of LY368052 (the (R)-enantiomer ofLY348395; see Figure 3) on the contractility of an iso-lated rat ventricular myocyte obtained from a geneticCHF model [159] is shown in Figure 6. In the absence ofdrug, force of contraction increased with increasing fre-quency (in nonfailing rats, the interval–force relation isnegative [160]). LY368052 (2 nM) potently increasedcontractility, with the greatest effect occurring atslower frequencies. The increase in contraction oc-curred despite no change in the Ca21 content of the SR,suggesting that in the presence of LY368052 a greaterfraction of Ca21 is released per stimulus [161]. Themechanism underlying the frequency-dependent ef-fects of this agent is as yet unclear.
In guinea pig papillary muscles, LY333612 and DPI201–106 decreased the time to maximum force develop-ment, but only DPI 201–106 prolonged the time for
relaxation [114]. Somewhat different results in guineapig papillary muscles were reported by others, wheretime to peak tension was either unaffected [135,139] orprolonged [56]. Both compounds prolong relaxationtime [56,60,61,135,139], although the increase with DPI201–106 was greater [135] and more sustained [139].Functional refractory period also increases in the pres-
Fig. 5. Cumulative concentration response curves illustratingthe effect of sodium channel enhancers on guinea pig papillarymuscle force of contraction. LY348395 (n=7), LY366634 (n=10),LY333612 (n=6), (R)-LY333612 (n=6), and racemic DPI 201-106(n=6). The racemic propylamino-derivative, LY348395, is themost potent agent. Baseline tension values were LY348395 (4.326 0.39 mN), LY366634 (5.2 6 0.64 mN), LY333612 (5.6 6 0.8mN), (R)- LY333612 (4.48 6 0.77 mN), and DPI 201-106 (5.366 0.43 mN). Data are mean 6 SEM.
Fig. 6. The effect of LY368052 (2 nM) (the (R)-enantiomer ofthe racemate LY348395) on the contraction–frequency relationin a ventricular myocyte isolated from a chronic heart failure(SHHF) rat. LY368052 increases peak contractility without af-fecting diastolic length. Note the inverse frequency-dependenceof contractility in the control situation and its reversal byLY368052 (see text).
Na1 Channels as Targets for Inotropic Agents 183

ence of Na1 channel enhancers in rat [147] and guineapig cardiac muscle [56,139].
The positive inotropic effects of the racematesLY333612 and DPI 201–106 are due entirely to theactivity of the (S)-enantiomers, whereas the (R)-enan-tiomers are ineffective as positive inotropic agents[56,129,139] (Figure 5). Moreover, no evidence for in-teraction between the (R)- and (S)-enantiomers wasseen [129]. The slight negative inotropic effect of DPI201–106 and LY333612 at high concentrations [135,139]is likely due to the presence of the (R)-enantiomersthat block the Na1 channel and possibly the effect ofthe active enantiomer (vide supra).
Inotropy: human myocardium. Failing myocar-dium is characterized by a downregulation of the b1-ad-renergic pathway [26,27,152]. Thus, in end-stage heartfailure, drugs such as isoproterenol or dobutaminetend to be less effective [26,162–164]. In contrast,LY333612 [53,162,164] and its (S)-enantiomerLY366634 [163,165] remained effective and in fact weremore potent in increasing the force of contraction infailing compared to nonfailing papillary muscle (Table2) [166]. An analogous increase in sensitivity of failingmyocardium to ouabain was also reported [166]. Re-cent studies demonstrated that the mRNA and proteinlevels of the Na1–Ca21 exchanger are increased in hu-man cardiomyopathy [146,163]. While it may be postu-lated that this upregulation is an attempt to extrudeexcessive Ca21 from the myocyte, it is possible that theexchanger is activated to function at least part of thetime in reverse mode to resupply Ca21 to the sacroplas-mic reticulum [57,163,166]. This could help explain whyfailing heart tissues may be more sensitive than non-failing heart to Ca21 loading by the exchange mecha-nism [146,165,167]. As in animal tissues, the propy-lamino-derivative LY348395 exerted about a 10-foldhigher potency in increasing systolic tension in isolatedhuman ventricular muscle strips (NYHA IV) com-
pared to LY333612 and LY366634 (Figure 7) ([168] andR.W. Schwinger, personal communication).
A speci~c effect of Na1 channel enhancers on Na1
channels is supported by the following observations: 1)3H-ouabain binding was not in_uenced by LY333612[162,164]; 2) carbachol, adenosine [162,165], andforskolin [53] failed to affect the positive inotropic ef-fects of Na1 channel enhancers in vitro; 3) LY333612mediated an increase in intracellular Na1 in failingheart tissues [137]; 4) potency was maintained in myo-cardium from end-stage CHF [137,163–166,168]; and 5)
Table 2. Inotropic actions of the Na1 -channel activators LY333612, LY366634, (R)-LY333612, and Ca21 in human nonfailing andfailing myocardium
Compound n Basal FOC (mN) Max PIE (mN) EC50 (lM; 95%-CL)
Nonfailing
LY333612 5 1.5 6 0.4 5.3 6 1.2 1.54; 0.95–2.45LY366634 4 2.7 6 0.9 8.5 6 0.9 0.92; 0.68–1.35(R)-LY333612 5 2.0 6 0.5 21.0 6 0.4 —Calcium 9 1.6 6 0.2 6.4 6 0.4 7.36 mM; 5.94–9.12
NYHA IV
LY333612 11 2.9 6 0.3 6.2 6 0.5 0.36; 0.24–0.55LY366634 9 2.6 6 0.5 7.7 6 1.0 0.25; 0.12–0.51(R)-LY333612 9 2.5 6 0.3 21.3 6 0.5 —Calcium 16 1.9 6 0.2 6.5 6 0.5 6.22 mM; 4.19–9.23
Abbreviations: Basal FOC, basal force of contraction in mN; Max PIE, maximal positive inotropic effect; n, number of preparations studied; EC50,half maximal concentration; 95%-CL, con~dence interval for p,0.05; NYHA IV, New York Heart Association Classi~cation IV. Values shown asmean 6 S.E.From Schwinger et al., J Pharmacol Exp Ther 1996; 276: 1180–1188, with permission.
Fig. 7. Concentration response curves for the effects ofLY348395, LY366634, LY333612, and (R)-LY333612 on force ofcontraction in electrically driven (1 Hz, 378 C) papillary mus-cle strips from failing human myocardium. The most potentagent is the racemic propylamino-analogue, LY348395. Dataare mean 6 SEM. Basal force of contraction and number of ex-periments are indicated in Table 2.
184 Steinberg et al.
TTX blocked the contractile effect [61]. While initialreports suggested a Ca21 sensitizing effect for DPI201–106 in myopathic right ventricular myocardium[169], LY333612 and DPI 201–106 were each reportedrecently to be without direct effect in human chemi-cally skinned ~bers from terminally failing human leftventricular myocardium [149,170].
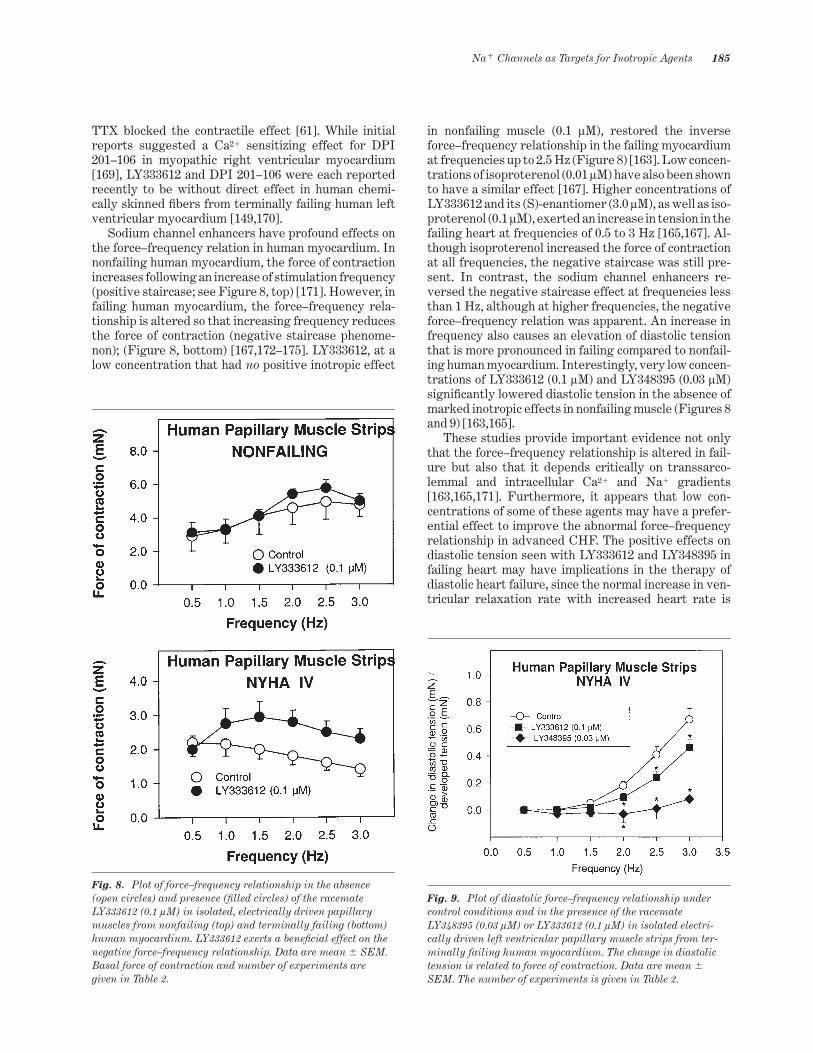
Sodium channel enhancers have profound effects onthe force–frequency relation in human myocardium. Innonfailing human myocardium, the force of contractionincreases following an increase of stimulation frequency(positive staircase; see Figure 8, top) [171]. However, infailing human myocardium, the force–frequency rela-tionship is altered so that increasing frequency reducesthe force of contraction (negative staircase phenome-non); (Figure 8, bottom) [167,172–175]. LY333612, at alow concentration that had no positive inotropic effect
in nonfailing muscle (0.1 lM), restored the inverseforce–frequency relationship in the failing myocardiumat frequencies up to 2.5 Hz (Figure 8) [163]. Low concen-trations of isoproterenol (0.01 lM) have also been shownto have a similar effect [167]. Higher concentrations ofLY333612 and its (S)-enantiomer (3.0 lM), as well as iso-proterenol (0.1 lM), exerted an increase in tension in thefailing heart at frequencies of 0.5 to 3 Hz [165,167]. Al-though isoproterenol increased the force of contractionat all frequencies, the negative staircase was still pre-sent. In contrast, the sodium channel enhancers re-versed the negative staircase effect at frequencies lessthan 1 Hz, although at higher frequencies, the negativeforce–frequency relation was apparent. An increase infrequency also causes an elevation of diastolic tensionthat is more pronounced in failing compared to nonfail-ing human myocardium. Interestingly, very low concen-trations of LY333612 (0.1 lM) and LY348395 (0.03 lM)signi~cantly lowered diastolic tension in the absence ofmarked inotropic effects in nonfailing muscle (Figures 8and 9) [163,165].
These studies provide important evidence not onlythat the force–frequency relationship is altered in fail-ure but also that it depends critically on transsarco-lemmal and intracellular Ca21 and Na1 gradients[163,165,171]. Furthermore, it appears that low con-centrations of some of these agents may have a prefer-ential effect to improve the abnormal force–frequencyrelationship in advanced CHF. The positive effects ondiastolic tension seen with LY333612 and LY348395 infailing heart may have implications in the therapy ofdiastolic heart failure, since the normal increase in ven-tricular relaxation rate with increased heart rate is
Fig. 8. Plot of force–frequency relationship in the absence(open circles) and presence (~lled circles) of the racemateLY333612 (0.1 lM) in isolated, electrically driven papillarymuscles from nonfailing (top) and terminally failing (bottom)human myocardium. LY333612 exerts a bene~cial effect on thenegative force–frequency relationship. Data are mean 6 SEM.Basal force of contraction and number of experiments aregiven in Table 2.
Fig. 9. Plot of diastolic force–frequency relationship undercontrol conditions and in the presence of the racemateLY348395 (0.03 lM) or LY333612 (0.1 lM) in isolated electri-cally driven left ventricular papillary muscle strips from ter-minally failing human myocardium. The change in diastolictension is related to force of contraction. Data are mean 6
SEM. The number of experiments is given in Table 2.
Na1 Channels as Targets for Inotropic Agents 185

compromised by even small increases in heart rate[176]. Nevertheless, because other in vitro evidencedemonstrates that in some circumstances Na1 channelenhancers may increase twitch duration and diastolictension in failing and nonfailing human myocardium[61,137,165], caution must be exercised in extrapolat-ing these ~ndings to clinical use.
Preclinical pharmacology in vivo
Inotropic effects. In general, the inotropic effectsand enantiomeric selectivity of Na1 channel enhancersin vivo parallel the effects described in vitro. LY333612and its (S)-enantiomer increased cardiac contractilityin rats, rabbits, and dogs when administered intrave-nously [130,177,178]. LY333612 was more active thanDPI 201-106 in increasing contractility in anesthetizeddogs, commensurate with its greater in vitro potency(Figure 10). Also consistent with in vitro data, thepropylamino-analogue, LY348395, was 10 times morepotent than LY333612 when administered i.v. to rats
and rabbits [130]. DPI 201-106 and its weaker conge-ner, SDZ 218-135 [147], also increased contractility invivo in rats. In conscious dogs, intravenous DPI 201-106 increased contractility to the same extent as dobu-tamine, but unlike dobutamine did not affect heart rate[179]. As anticipated, the (S)-enantiomer of DPI 201-106 accounts for all the positive inotropic activity of theracemate in conscious dogs [180].
In anesthetized dogs subjected to coronary arteryocclusion 5–7 days before study, intravenous DPI 201-106 and dobutamine increased contractility to the sameextent, but only dobutamine increased heart rate andcardiac output [181]. In anesthetized dogs, LY333612 (3mg/kg, i.v.) increased myocardial contractility to aboutthe same extent in the nonischemic setting as in thepresence of acute LAD occlusion [177]. In anesthetizeddogs with chronic heart failure induced by intermittentintracoronary microembolization, LY366634 (3, 5, and10 lg/kg/min) increased the rate of left ventricularpressure development and fractional area of shorten-ing and did not change or only slightly decreased heartrate [182].
Electrophysiology. The prolongation of the cardiacAPD and the QT interval due to interference withinactivation mechanisms could serve as a substrate forafterdepolarizations and malignant arrhythmias [61].Indeed, as mentioned earlier, the LTQ3-associated mu-tation in the inactivation region of the channel is asso-ciated with torsade des pointes. Since in many studiescited above, LY333612 caused less prolongation ofAPD than DPI 201-106, the QT interval might likewisebe expected to be affected less. Accordingly, despitehigher ef~cacy in increasing contractility in anesthe-tized dogs, LY333612 caused signi~cantly less prolon-gation of QTc [183] (Figure 10, bottom). In dogs withischemic heart failure, the QTc interval increased byonly 28 msec at the highest administered dose (10l/kg/min) [182]. The basis for the increase in QT withthese agents remains controversial. In conscious dogs,the increase in QT interval by DPI 201-106 was en-hanced after autonomic blockade [179], but anotherreport suggested that the small increase in QT intervalcaused by DPI 201-106 in conscious dogs might be neu-ral in origin (possibly related to bradycardia) and wasprevented by autonomic blockade.
To evaluate any potential arrhythmogenic effectsrelated to QT prolongation, LY333612, its (S)-enan-tiomer (LY366634), and DPI 201-106 were studied inautonomic-blocked, anesthetized dogs before and afteracute coronary stenosis [177]. At doses eliciting a pro-found inotropic response, none of these Na1 channelmodulators signi~cantly changed myocardial excitabil-ity, conduction time, ventricular refractoriness, or in-duced arrhythmias. In contrast, equieffective doses ofnorepinephrine and ouabain caused ventricular ar-rhythmias and modi~ed excitability. An absence ofproarrhythmic effects of LY333612, LY366634, andLY348395 was observed in aconitine-induced arrhyth-
Fig. 10. Effect of vehicle (n=6), LY333612 (n=8), and DPI 201-106 (n=8) on myocardial contractility (dP/dtmax) and QTc inter-val in anesthetized dogs. For a given increase in contractility,LY333612 causes less change in QTc interval compared to DPI201-106. Data are mean 6 SEM. Asterisks indicate resultssigni~cantly different from vehicle and DPI 201-106.
186 Steinberg et al.

mia in guinea pig auricle, in the reperfused guinea pigLangendorff heart, and in rats with regional ischemia[184]. Indeed, DPI 201-106 prevented, while dobu-tamine worsened, reentrant ventricular arrhythmiasin dogs subjected to programmed electrical stimulationin the presence of subacute ischemia [181]. DPI 201-106and SDZ 218-135 (the less potent analogue of DPI 201-106) also had antiarrhythmic activity against coronaryartery reperfusion and aconitine-induced arrhythmiasin rats [119,147]. The antiarrhythmic effects of DPImight derive from the class III effect due to either theprolonged APD accompanying the enhanced Na1 cur-rent [185] or direct local anesthetic effects [119] leadingto slowed intramyocardial conduction [181]. Most stud-ies therefore suggest that the proarrhythmic potentialof this class of drugs should be small and that theremay be differences in effects on APD and refractori-ness among various compounds. Nevertheless, carefulclinical evaluations will be required to evaluate poten-tial bene~cial or adverse cardiac electrophysiologicalactions in heart failure patients (vide infra).
Potential interactions with cardiac glycosides.
Although the Na1 channel enhancers do not directlyinhibit Na1–K1-ATPase, they increase intracellularNa1 and thereby could potentiate inotropic and elec-trophysiologic effects of digitalis. For instance, DPI201-106 (or its (S)-enantiomer) potentiated the contrac-tile effects of ouabain in isolated rabbit papillary mus-cles and in anesthetized dogs [154]. After pretreatmentwith low concentrations of LY333612 (0.03 lM), guineapig left atria were slightly more sensitive to the inot-ropic effect of ouabain [135]. Increased sensitivity toouabain was also reported by Herzig et al. [158] whofound that the threshold dose of ouabain necessary toinduce arrhythmia in guinea pig left atria was loweredby a factor of 2–3 in the presence of LY333612 or DPI201-106. The toxic effects of ouabain were also en-hanced in human myocardium in the presence of 0.1 lMor 1.0 lM LY333612 [164]. However, since the contrac-tile effects of digitalis are also potentiated, it is unclearwhether the therapeutic ratio of digitalis would be ad-versely affected [158,179]. There have been as yet noreports that coadministration in vivo of Na1 channelenhancers and cardiac glycosides are arrhythmogenic.DPI 201-106 did not in_uence the arrhythmogenic ef-fects of ouabain in anesthetized guinea pigs [119] andactually protected against its arrhythmogenic effectsin anesthetized dogs [154].
Clinical Pharmacology
The only clinical data available on Na1 channel modula-tors relate to the racemate DPI 201-106. In heart failurepatients with ischemic [186] or dilated cardiomyopathy[187], short-term intravenous administration of DPI201-106 increased all indices of contractility without af-fecting heart rate or pressure. Moreover, in both catego-
ries of patients, positive inotropic activity was accompa-nied by signi~cant decreases in left ventricular end-diastolic pressures and other indices of preload. Usingindirect measures of MVO2 and catheter-based pres-sure–volume loops Thormann et al. [187] concluded thatin comparison to dobutamine, DPI 201-106 appearedmore ef~cacious in shifting the ventricular functioncurve (end-diastolic pressure vs. stroke work index) up-wards and to the left and in reducing ventricular vol-umes, preload, and MVO2. The main difference betweenthe drugs was that dobutamine alone increased heartrate. One patient developed an episode of atrial ~brilla-tion [186], but otherwise no arrhythmogenic or adverseQTc effects were noted in these short-term studies. Instudies using programmed electrical stimulation, atrialrefractoriness and AV nodal conduction were de-creased, but in 5 of 6 patients, premature atrial stimula-tion induced atrial ~brillation [188]. No ventricular ar-rhythmias were induced, although the ventricularrefractory period and QTc interval were increased. Thebasis of the effect on atrial refractoriness and inducibil-ity is not known, but increases in re_ex vagal tone (per-haps due to an increase in systolic pressure associatedwith the rather high plasma levels of drug achieved)were suggested [188].
The hemodynamic effects of single oral doses of DPI201-106 have been reported. Using either systolic timeintervals in normal volunteers [189], or invasive cathe-ter techniques in patients with class III failure [190],DPI 201-106 increased indices of contractility and de-creased preload. In normal volunteers, a 30-mg doseincreased preejection period without changing QTc in-terval. Doubling the dose signi~cantly increased theQTc interval by about 22 msec and caused minor T-wave abnormalities. Higher doses (80 to 100 mg) ad-ministered to heart failure patients increased systolicfunction (cardiac index and stroke volume) and tendedto decrease ~lling pressures; these changes were ac-companied by a 7% increase in QTc interval [190]. Noarrhythmogenic effects were noted in these single-dose studies, although at high plasma concentrations(.80 ng/mL) there was a tendency for increased ven-tricular ectopy [190].
Summary and Future Directions
Despite their inherent de~ciencies, inotropic catecho-lamines still represent the major short-term pharma-cologic means to support the failing heart [14]. Theexperimental and early clinical data summarized in thisreview clearly demonstrate that Na1 channel en-hancers are powerful inotropic drugs in the setting ofadvanced heart failure. In short-term use, they willlikely improve the signs and symptoms of CHF as wellas the quality of life in end-stage disease. Whetherthese agents will be used in longer-term situations de-pends on additional studies designed to address anumber of important issues.
Na1 Channels as Targets for Inotropic Agents 187

Whether increasing contractility in the failing heartby cAMP- independent mechanisms will provide a fa-vorable effect on myocardial oxygen demand ratios andimprove long-term prognosis is unknown. It remainsunclear whether agents that increase Ca21 availabilityto the contractile apparatus will obviate the potentiallydetrimental effects of cAMP on cardiac energetics. Thenegative prognostic implications of increasing energydemands on an already “energy-starved” heart arewell-known [191]. What is not known and must be as-certained by additional clinical trials in heart failurepatients is whether the balance between energy utili-zation and expenditure can be improved by agents thatmodify Ca21-transport abnormalities in the absence ofaltering cAMP levels. Preliminary short-term resultswith DPI 201-106 in this regard were encouraging[187]. Although the Na1 channel enhancers increasecontractility in the failing heart without increasingheart rate, it is unknown whether these agents mightalso possess the bene~cial coupling between cardiacand peripheral hemodynamics that appears to be soimportant in the therapeutic spectrum of dobutamine[14]. Moreover, since the experimental data reviewedabove suggest that drugs in this class can differ dra-matically in their effect on the QTc interval, carefulclinical trials in heart failure patients are required toshow improved cardiac function without adverse ef-fects on cardiac electrophysiology (see [59]).
Finally, one of the most intriguing properties of somecompounds in this class relates to their bene~cial effectson the impaired systolic and diastolic force–frequencyrelationship in isolated cardiac muscles from end-stageCHF patients (Figures 8 and 9). It is appealing to con-sider the potential therapeutic utility of a compoundthat in appropriate doses might only minimally affectbasal contractility (Figure 7) yet might permit an in-crease in ejection fraction and diastolic function with in-creased physical activity. Such an agent would make animportant short-term improvement in symptoms andquality of life for patients with end-stage systolic or dia-stolic dysfunction, even if a reversal of ventricular re-modeling could not be demonstrated.
Acknowledgements
The authors wish to thank Dr. R.W. Schwinger and colleagues fortheir permission to use portions of original work, reproduced infull or in part and Dr. W. J. Crumb for permission to reproduceportions of unpublished work on human atrial myocytes. Thanksalso to Dr. S. A. Wiest for carefully reviewing the manuscript.
References
1. The SOLVD investigators. Effect of enalapril on mortalityand the development of heart failure in asymptomatic pa-tients with reduced left ventricular ejection fractions. NEngl J Med 1992;327:685–691.
2. Kannel WB, Belanger AJ. Epidemiology in heart failure.Am Heart J 1991;121:951–957.
3. Miller L. Candidate selection for heart transplantation.Cardiol Clin 1995;13:93–100.
4. American Heart Association. Heart and Stroke Facts: Sta-tistical Supplement. 1997
5. Ramahi TM, Lee FA. Medical therapy and prognosis inchronic heart failure. Lessons from clinical trials. CardiolClin 1995;13:5–26.
6. National Heart Lung and Blood Institute Data Fact Sheet.Congestive Heart Failure Update. 1996
7. O’Connell JB, Bristow MR. Economic impact of heart failurein the United States: time for a different approach. J HeartLung Transplant 1994;13 (Suppl):S107–S112.
8. Winker M, Glass R. The aging global population, (editorial).JAMA 1996;276:1758–1759.
9. Braunwald E. ACE inhibitors—a cornerstone of the treat-ment of heart failure. N Engl J Med 1991;325:351–353.
10. Pfeffer M, Stevenson L. b-Adrenergic blockers and survivalin heart failure. N Engl J Med 1996;334:1396–1397.
11. Higgins R, Silverman N. Status of permanent cardiac re-placement for end-stage congestive heart failure. HeartFailure Rev 1995;1:39–51.
12. McCarthy PM. HeartMate implantable left ventricular as-sist device: bridge to transplantation and future applica-tions. Ann Thorac Surg 1995;59:S46–S51.
13. Shipley J, Hess M. Inotropic therapy for the failing myocar-dium. Clin Cardiol 1995;18:615–619.
14. Leier C. Positive inotropic therapy: an update and newagents. Curr Prob Cardiol 1996;21:523–581.
15. Afridi I, Quinones M, Zoghbi W, Cheirif J. Dobutaminestress echocardiography: sensitivity, speci~city, and predic-tive value for future cardiac events. Am Heart J 1994;127:1510–1515.
16. Miller L, Merkle E, Jennison S. Outpatient use of dobu-tamine to support patients awaiting heart transplantation. JHeart Lung Transplant 1994;13:S126–S129.
17. Farah A. Positive inotropic agents. Annu Rev PharmacolToxicol 1984;24:275–328.
18. Sasayama S. Inotropic agents in the treatment of heart fail-ure: despair or hope. Cardiovasc Drugs Ther 1996;10:703–709.
19. Packer M. A novel approach to the development of positiveinotropic agents for chronic heart failure. J CardiovascPharmacol 1995;26:S52–S56.
20. Eichhorn E, Bristow M. Medical therapy can improve thebiologic properties of the chronically failing heart: a new erain the treatment of heart failure. Circulation 1996;94:2285–2296.
21. Varró A, Papp J. Classi~cation of positive inotropic actionsbased on electrophysiologic characteristics: where shouldcalcium sensitizers be placed? J Cardiovasc Pharmacol1995;26:S32–S44.
22. The Digitalis Investigation Group. The effect of digoxin onmortality and morbidity in patients with heart failure. NEngl J Med 1997;336:525–533.
23. Jones L. Sarcolemmal enzymes mediating b-adrenergic ef-fects on the heart. In Bronner F, Shamoo A, eds. CurrentTopics in Membranes and Transport: Regulation of Cal-cium Transport Across Muscle Membranes. New York:Academic Press, 1985:11–41.
24. Remme W. Inotropic agents for heart failure: what if digoxinincreases mortality? Br Heart J 1994;72:S92–S99.
25. Hassenfuss G, Holubarsch C, Heiss H, et al. Myocardial
188 Steinberg et al.

energetics in patients with dilated cardiomyopathy.In_uence of nitroprusside and enoximone. Circulation 1989;80:51–64.
26. Bristow M, Ginsburg R, Minobe W, et al. Decreased catecho-lamine sensitivity and b-adrenergic-receptor density in fail-ing human hearts. N Engl J Med 1982;307:205–211.
27. Böhm M, Diet F, Feiler G, Kemkes B, Kreuzer E, WeinholdC, Erdmann E. Subsensitivity of the failing human heart toisoprenaline and milrinone is related to b-adrenoceptordownregulation. J Cardiovasc Pharmacol 1988;12:726–732.
28. Haft J. Cardiovascular injury induced by sympatheticcatecholamines. Prog Cardiovasc Dis 1974;17:73–85.
29. Lubbe WF, Podzuweit T, Opie LH. Potential arrhythmo-genic role of cyclic adenosine monophosphate (cAMP) andcytosolic calcium overload: implications for prophylactic ef-fects of beta-blockers in myocardial infarction and proar-rhythmic effects of phosphodiesterase inhibitors. J Am CollCardiol 1992;19:1622–1633.
30. Mann DL, Kent RL, Parsons B, Cooper G. Adrenergic ef-fects on the biology of the adult mammalian cardiocyte. Cir-culation 1992;85:790–804.
31. Ebbesen P. Myocardial degeneration in mice treated withdibutyryl cyclic AMP and/or theophylline. Virchows ArchPathol Anat Histol 1976;372:89–95.
32. Lee J, Downing S. Cyclic AMP and the pathogenesis ofmyocardial injury. Res Commun Chem Pathol Pharmacol1971;23:200–204.
33. Dhalla K, Rupp H, Beamish R, Dhalla N. Mechanisms ofalterations in cardiac membrane Ca21 transport due to ex-cess catecholamines. Cardiovasc Drug Ther 1996;10:231–238.
34. Feldman MD, Copelas L, Gwathmey JK, et al. De~cientproduction of cyclic AMP: pharmacologic evidence of an im-portant cause of contractile dysfunction in patients withend-stage heart failure. Circulation 1987;75:331–339.
35. Packer M. The development of positive inotropic agents forchronic heart failure: How have we gone astray? J Am CollCardiol 1993;22:119A–126A.
36. Rahimtoola S. Digitalis and William Withering: the clinicalinvestigator (editorial). Circulation 1975;52:969–971.
37. Rahimtoola S, Tak T. The use of digitalis in heart failure.Curr Probl Cardiol 1996;21:787–853.
38. Uretsky B, Young J, Shahidi F, Yellen L, Harrison M, JollyM. Randomized study assessing the effect of digoxin with-drawal in patients with mild to moderate chronic congestiveheart failure: results of the PROVED study. J Am CollCardiol 1993;22:955–962.
39. Packer M, Gheorghiade M, Young J, et al. Withdrawal ofdigoxin from patients with chronic heart failure treated withangiotensin-converting-enzyme inhibitors. N Engl J Med1993;329:1–7.
40. Repke K. Toward the discovery of digitalis derivatives withinotropic selectivity. Drug Dis Today 1997;2:110–116.
41. Kokubun S, Prod’hom C, Becker H, Porzig H, Reuter H.Studies on Ca12 channels in intact cardiac cells: Voltage-de-pendent effects and cooperative interactions of dihy-dropyridine enantiomers. Mol Pharmacol 1986;30:571–584.
42. Uechi M, Sato N, Asai K, Kudej R, Vatner S. A novel calciumpromoter improves inotropic state and mechanical ef~ciencyin conscious dogs with heart failure (abstract). J Am CollCardiol 1997;29:323A.
43. Gross R, Bechem M, Dayser M, Schramm M, Tariel R,Thomas G. Cardiovascular effects of dihydropyridine-typecalcium antagonist and agonists. In Bayer Symposium IX.
Effects of the Calcium Agonistic Dihydropyridine BAY K8644 on the Heart. Berlin: Springer Verlag, 1985:218–312.
44. Fujino K, Sperelakis N, Solaro R. Sensitization of dog andguinea pig heart myo~laments to Ca21 activation and theinotropic effect of pimobendan: comparison with milrinone.Circ Res 1988;63:911–922.
45. Végh A, Papp J, Udvary É, Kaszala K. Hemodynamic ef-fects of calcium-sensitizing agents. J Cardiovasc Pharmacol1995;26:S20–S31.
46. Haikala H, Lindén I-B. Mechanism of action of calcium-sen-sitizing drugs. J Cardiovasc Pharmacol 1995;26:S10–S19.
47. Lilleberg J, Sundberg S, Nieminen M. Dose-range study ofa new calcium sensitizer, levosimendan, in patients with leftventricular dysfunction. J Cardiovasc Pharmacol 1995;26:S63–S69.
48. Toivonen L, Vitasalo M, Lehtonen L, Sundberg S, NieminenM. Electrophysiological effects of a calcium sensitizer inot-rope levosimendan in patients with normal heart functionabstract. J Am Coll Cardiol 1997;29:41A.
49. Neumann J, Boknik P, Schmitz W, Scholz H, ZimmermannN. Comparison of the stereoselective effects of a thiadiazi-none derivative on contractile parameters and protein phos-phorylation in the mammalian ventricle. J Cardiovasc Phar-macol 1995;25:789–793.
50. Neumann J, Eschenhagen T, Grupp I, Haverich A, Herzig J.Positive inotropic effects of the calcium sensitizer CGP48506 in failing human myocardium. J Pharmacol Exp Ther1996;277:1579–1585.
51. Feldman AM, Bristow MR, Parmley WW, et al. Effects ofvesnarinone on morbidity and mortality in patients withheart failure. N Engl J Med 1993;329:149–155.
52. Iljima T, Taira N. Membrane current changes responsiblefor the positive inotropic effect of OPC-8212, a new positiveinotropic agent, in single ventricular cells of the guinea pigheart. J Pharmacol Exp Ther 1987;240:657–662.
53. Focaccio A, Peeters G, Movsesian M, Roden R, Eki Y, KrallJ, Bristow M. Mechanism of action of OPC-8490 in humanventricular myocardium. Circulation 1996;93:817–825.
54. Lathrop D, Nanasi P, Schwartz A, Varro A. Ionic basis forOPC-8212-induced increase in action potential duration inisolated rabbit, guinea pig and human ventricular myocytes.Eur J Pharmacol 1993;240:127–137.
55. Hosokawa T, Mori T, Fujiki H, et al. Cardiovascular actionsof OPC-18790: a novel positive inotropic agent with littlechronotropic action. Heart Vessels 1992;7:66–75.
56. Scholtysik G, Salzmann R, Berthold R, Herzig U, Quast U,Markstein R. DPI 201-106, a novel cardiotonic agent. Com-bination of cAMP-independent positive inotropic, negativechronotropic, action potential prolonging and coronary dila-tory properties. Naunyn Schmiedebergs Arch Pharmacol1985;329:316–325.
57. Buggisch D, Isenberg G, Ravens U, Scholtysik G. The roleof sodium channels in the effects of the cardiotonic com-pound DPI 201-106 on contractility and membrane poten-tials in isolated heart preparations. Eur J Pharmacol1985;118:303–311.
58. Doggrell S, Brown L. Ion channel modulators in the treat-ment of congestive heart failure. Exp Opin Invest Drugs1996;5:495–512.
59. Ravens U, Amos G, Ehring T, Heusch G. BDF 9148—asodium channel modulator with positive inotropic action.Cardiovasc Drug Rev 1995;13:260–274.
60. Kihara Y, Gwathmey JK, Grossman W, Morgan JP. Mecha-nisms of positive inotropic effects and delayed relaxation
Na1 Channels as Targets for Inotropic Agents 189

produced by DPI 201-106 in mammalian working myocar-dium: effects on intracellular calcium handling. Br J Phar-macol 1989;96:927–939.
61. Gwathmey J, Slawsky M, Briggs G, Morgan J. Role of intra-cellular sodium in the regulation of intracellular calcium andcontractility. Effects of DPI 201-106 on excitation—contrac-tion coupling in human ventricular myocardium. J Clin In-vest 1988;82:1592–1605.
62. Hodgkin AL. The Conduction of the Nervous Impulse. Liv-erpool: Liverpool University Press, 1967.
63. Hille B. Ionic Channels of Excitable Membranes. Sunder-land: Sinauer Associates, Inc., 1992.
64. Tamkun MM, Knittle TJ, Deal KK, et al. Molecular physiol-ogy of voltage-gated potassium and sodium channels: ionchannel diversity within the cardiovascular system. InSpooner PM, Brown AM, Catterall WA, Kaczorowski GJ,Strauss HC, eds. Ion Channels in the Cardiovascular Sys-tem. New York: Futura, 1994:287–316.
65. Gellens ME, George AL, Chen L, Chahine M, Horn R, Bar-chi R, Kallen RG. Primary structure and functional expres-sion of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci USA1992;89:554–558.
66. Cohen S, Barchi R. Cardiac sodium channel structure andfunction. Trends Cardiovasc Med 1992;2:133–140.
67. Isom L, deGongh K, Patton D, et al. Primary structure andfunctional expression of the b1 subunit of the rat brain so-dium channel. Science 1992;256:839–842.
68. Catterall WA, Scheuer T, West JW, Numann R, Patton ED,Duff HJ, Goldin AG. Structure and modulation of voltage-gated sodium channels. In Spooner P, Brown A, Catterall W,Kaczorowski G, Strauss H, eds. Ion Channels in the Cardio-vascular System. New York: Futura, 1994:317–340.
69. Patton DE, Issom LL, Catterall WA, Goldin AL. The adultrat brain b1 subunit modi~es activation and inactivation gat-ing of multiple sodium channel a subunits. J Biol Chem1994;269:17649–17655.
70. McAllister RE, Noble D, Tsien RW. Reconstruction of theelectrical activity of cardiac Purkinje ~bres. J Physiol(Lond) 1975;251:1–59.
71. Corabeouf E. Aspects cellulares de l’electrogenesecardiaque chez les vertebres. J Physiol (Paris) 1960;52:323–417.
72. Hoffman BF, Crane~eld PF. Electrophysiology of the Heart.New York: McGraw-Hill, 1960.
73. Fozzard HA. Conduction of the action potential. In Berne R,Sperelakis N, Geiger S, eds. Handbook of Physiology. Vol-ume 1, The Heart; Section 2: The Cardiovascular System. Be-thesda, MD: American Physiological Society, 1979:335–356.
74. Catterall WA. Structure and function of voltage-gated ionchannels. Annu Rev Biochem 1995;64:493–531.
75. Brown AM, Lee KS, Powell T. Voltage clamp and internalperfusion of single heart cells. J Physiol (Lond) 1981;318:455–477.
76. Fozzard HA. Cardiac electrogenesis and the sodium chan-nel. In Spooner P, Brown A, Catterall W, Kaczorowski G,Strauss H, eds. Ion Channels in the Cardiovascular Sys-tem. New York: Futura, 1994:81–100.
77. Saint DA, Ju YK, Gage PW. A persistent sodium current inrat ventricular myocytes. J Physiol (Lond) 1992;453:219–231.
78. Bkaily G, Jasmin G, Tautu C, et al. A tetrodotoxin andMn21-insensitive Na1 current in Duchenne muscular dystro-phy. Muscle Nerve 1990;13:939–948.
79. Jacques D, Bkaily G. Presence of early embryonic slow Na1
channels in cardiomyopathic hamster (abstract). Biophys J1991;59:259a.
80. Armstrong CM. Sodium channels and gating currents.Physiol Rev 1981;61:644–683.
81. Strichartz G, Rando T, Wang G. An integrated view of themolecular toxinology of sodium channel gating in excitablecells. Annu Rev Neurosci 1987;10:237–267.
82. Grant A, Starmer C, Strauss H. Antiarrhythmic drug ac-tion. Blockade of the inward sodium current. Circ Res1984;55:427–439.
83. Noda M, Suzuki H, Numa S, Stühmer W. A single pointmutation confers tetrodotoxin and saxitoxin insensitivity onthe sodium channel-II. FEBS Lett 1989;259:213–216.
84. Terlau H, Heinemann SH, Stühmer W, Pusch W, Conti F,Imoto K, Numa S. Mapping the site of block by tetrodotoxinand saxitoxin of sodium channel II. FEBS Lett1991;293:93–96.
85. Satin J, Kyle JW, Chen M, Rogart RB, Fozzard HA. Thecloned cardiac Na channel alpha-subunit expressed inXenopus oocytes shows gating and blocking properties ofnative channels. J Membr Biol 1992;130:11–22.
86. Backx PH, Yue DT, Lawrence JH, Marban E, Tomaselli GF.Molecular localization of an ion-binding site within the poreof mammalian sodium channels. Science 1992;257:248–251.
87. Heinemann SH, Terlau H, Imoto K. Molecular basis forpharmacological differences between brain and cardiac so-dium channels. P_ügers Arch Eur J Physiol 1992;422:90–92.
88. Dudley SC, Fozzard HA. Current understanding of thestructure of the voltage-gated sodium channel. In Morad M,Ebashi S, Trautwein W, Kurachi Y, eds. Molecular Physiol-ogy and Pharmacology of Cardiac Ion Channels and Trans-porters. Dordrecht: Kluwer Academic, 1996:39–52.
89. Lipkind GM, Fozzard HA. A structural model of the tetro-dotoxin and saxitoxin binding site of the Na1 channel. Bio-phys 1994;66:1–13.
90. Stühmer W, Conti F, Suzuki H, Wang X, Noda M. Structuralparts involved in activation and inactivation of the sodiumchannel. Nature 1989;339:597–603.
91. Vassilev PM, Scheuer T, Catterall WA. Identi~cation of anintracellular peptide segment involved in sodium channelinactivation. Science 1988;241:1658–1661.
92. Vassilev PM, Scheuer T, Catterall WA. Inhibition of inacti-vation of single sodium channels by a site-directed antibody.Proc Natl Acad Sci USA 1989;86:8147–8151.
93. McPhee J, Ragsdale D, Scheuer T, Catterall W. A mutationin segment IVS6 disrupts fast inactivation of sodium chan-nels. Proc Natl Acad Sci USA 1994;91:12346–12350.
94. McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A criti-cal role for the transmembrane segment IVS6 of the sodiumchannel a subunit in fast inactivation. J Biol Chem1995;270:12025–12034.
95. Armstrong CM, Benzanilla F. Inactivation of the sodiumchannel, II: gating current experiments. J Gen Physiol1977;70:567–590.
96. Patton DE, West JW, Catterall WA, Goldin AL. Amino-acidresidues required for fast Na1-channel inactivation: chargeneutralizations and deletions in the III–IV linker. Proc NatlAcad Sci USA 1992;89:10905–10909.
97. West J, Patton D, Scheuer T, Wang Y, Goldin A, Catterall W.A cluster of hydrophobic amino acid residues required forfast Na1 channel inactivation. Proc Natl Acad Sci USA1992;89:10910–10914.
98. Hartmann HA, Tiedeman AA, Chen S-F, Brown AM, Kirsch
190 Steinberg et al.

GE. Effects of III–IV linker mutations on human heart Na1
channel inactivation gating. Circ Res 1994;75:114–122.99. Valenzuela C, Bennett PB. Gating of cardiac Na1 channels in
excised membrane patches after modi~cation by alpha-chy-motrypsin. Biophys J 1994;67:161–171.
100. Zhang J, Ellinor P, Aldrich R, Tsien R. Molecular determi-nants of voltage-dependent inactivation in calcium channels.Nature 1994;372:97–100.
101. Wang S, Wang G. A mutation in segment I-S6 alters slowinactivation of sodium channels. Biophys J 1997;72:1633–1640.
102. Hoshi T, Zagotta W, Aldrich R. Biophysical and molecularmechanisms of Shaker potassium channel inactivation. Sci-ence 1991;250:533–568.
103. Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F.Paramyotonia congenita: the R1448P Na1 channel mutationin adult human skeletal muscle. Ann Neurol 1996;39:599–608.
104. Mitrovic H, George AL, Heine R, et al. K(1)-aggravatedmyotonia: destabilisation of the inactivated state of the hu-man muscle Na1 channel by the V1589M mutation. JPhysiol (Lond) 1994;478:395–402.
105. Bennett PB, Yazawa K, Makita N, George AL. Molecularmechanism for an inherited cardiac arrhythmia. Nature1995;376:683–685.
106. Wang G, Dugas M, Armah B, Honerjäger P. Interactionbetween DPI 201-106 enantiomers at the cardiac sodiumchannel. Mol Pharmacol 1995;37:17–24.
107. Dumaine R, Wang Q, Keating MT, Hartmann HA, SchwartzPJ, Brown AM, Kirsch GE. Multiple mechanisms of Na1-channel linked Long-QT syndrome. Circ Res 1996;78:916–924.
108. Keating M. Genetic approaches to cardiovascular disease.Supravalvular aortic stenosis, Williams syndrome, and LongQT syndrome. Circulation 1995;92:142–147.
109. Keating M, Sanguinetti M. Molecular genetic insights intocardiovascular disease. Science 1996;272:681–685.
110. Priori SG, Napolitano C, Cantu F, Brown AM, Schwartz PJ.Differential response to Na1 channel blockade, b-adrenergicstimulation, and rapid pacing in a cellular model mimickingthe SCN5A and HERG defects present in Long-QT syn-drome. Circ Res 1996;78:1009–1015.
111. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syn-drome patients with mutations of the SCN5A and HERGgenes have differential responses to Na1 channel blockadeand to increases in heart rate. Implications for a gene-speci~c therapy. Circulation 1995;92:3381–3386.
112. Honerjäger P. Cardioactive substances that prolong theopen state of sodium channels. Rev Physiol Biochem Phar-macol 1982;92:1–74.
113. Fujiwara M, Muramatsu I, Hidaka H, Ikushima S, Ashida K.Effects of goniopora toxin, a polypeptide isolated from coral,on electromechanical properties of rabbit myocardium. JPharmacol Exp Ther 1979;210:153–157.
114. Hoey A, Amos G, Wettwer E, Ravens U. Differential effectsof BDF 9148 and DPI 201-106 on action potential and con-tractility in rat and guinea-pig mycardium. J CardiovascPharmacol 1994;23:907–915.
115. Romey G, Quast U, Pauron D, Frelin C, Renaud J, Lazdun-ski M. Na1 channels as sites of action of the cardiotonic agentDPI 201-106 with agonist and antagonist enantiomers. ProcNatl Acad Sci USA 1987;84:896–900.
116. Ravens U, Wettwer E, Pfeifer T, Himmel H, Armah B.Characterization of the effects of the new inotropic agent
BDF 9148 in isolated papillary muscles and myocytes of theguinea-pig heart. Br J Pharmacol 1991;104:1019–1023.
117. Honerjäger P, Dugas M, Wang G. Effects of a new indol-2-carbonitrile, BDF 9148, and its two enantiomers on sodiumcurrent in rat heart cells (abstract). Naunyn SchmiedebergsArch Pharmacol 1990;341:R51.
118. Scholtysik G. Cardiac Na1-channel activation as a positiveinotropic principle. J Cardiovasc Pharmacol 1989;14:S24–S29.
119. Scholtysik G, Williams F. Antiarrhythmic effects of DPI201-106. Br J Pharmacol 1986;89:287–292.
120. Krafte D, Davison K, Dugrenier N. Pharmacological modu-lation of human cardiac Na1-channels. Eur J Pharmacol1994;266:245–254.
121. Nilius B, Benndorf K, Markwardt F, Franke T. Modulationof single cardiac sodium channels by DPI 201-106. GenPhysiol Biophys 1987;6:409–424.
122. Kohlhardt M, Fröbe U, Herzig J. Modi~cation of single car-diac Na1-channels by DPI 201-106. J Membrane Biol1986;89:163–172.
123. Kohlhardt M, Fröbe U, Herzig J. Removal of inactivationand blockade of cardiac Na1 channels by DPI 201-106: differ-ent voltage dependencies of drug actions. NaunynSchmiedebergs Arch Pharmacol 1987;335:183–188.
124. Langer G, Peskoff A, Post J. How does the Na1/Ca21-ex-changer work in the intact cardiac cell? J Mol Cell Cardiol1993;25:637–639.
125. Glossmann H, Zech C, Striessnig J, Staudinger R, Hall L,Greenberg R, Armah B. Very high af~nity interaction ofDPI 201-106 and BDF 8784 enantiomers with the phenylal-kylamine-sensitive Ca2(1)-channel in Drosophila head mem-branes. Br J Pharmacol 1991;102:446–452.
126. Siegl PK, Garcia ML, King VF, Scott AL, Morgan G,Kaczorowski GJ. Interactions of DPI 201-106, a novel cardio-tonic agent, with cardiac calcium channels. NaunynSchmiedebergs Arch Pharmacol 1988;338:684–691.
127. Holck M, Osterrieder W. Interaction of the cardiotonic agentDPI 201-106 with cardiac Ca21-channels. J CardiovascPharmacol 1988;11:478–482.
128. Hof R, Hof A. Mechanism of the vasodilator effects of thecardiotonic agent DPI 201-106. J Cardiovasc Pharmacol1985;7:1188–1192.
129. Hoey A, Lehmkuhl A, Sadony V, Ravens U. Inotropic ac-tions of BDF 9148 and DPI 201-106 and their enantiomers inguinea-pig, rat and human atria. Eur J Pharmacol1993;231:477–480.
130. Raap A, Mest H-J, Armah B, Grott W, Stenzel W, BlechaczW, Thomsen P. BDF 9198, a new Na1-channel activatinginotropic agent with higher in vitro and in vivo potency asBDF 9148 and DPI 201-106 (Abstract). J Mol Cell Cardiol1995;27:A124.
131. Press J, Falotico R, Hajos Z, et al. Synthesis and SAR of6-substituted purine derivatives as novel selective positiveinotropes. J Med Chem 1992;35:4509–4515.
132. Miltz W, Zierhut W, Buccheri E, Schultz R, Stirnimann R.Determination of the active conformation of 6-amino-a-[(4-diphenylmethyl-1-piperazinyl)methyl-9H-purine]-9-ethanol: a positive inotropic agent. Bioorg Med Chem Lett1993;3:1233–1239.
133. Estep K, Josef K, Bacon E, et al. Synthesis and struc-ture–activity relationships of 6-heterocyclic-substitutedpurines as inactivation modi~ers of cardiac sodium channels.J Med Chem 1995;38:2582–2595.
134. Armah B, Pfeifer T, Ravens U. Reversal of the cardiotonic
Na1 Channels as Targets for Inotropic Agents 191

and action-potential prolonging effects of DPI 201-106 byBDF 8784, a methyl-indole derivative. Br J Pharmacol1989;96:807–816.
135. Brasch H, Iven H. Inotropic and electrophysiological effectsof BDF 9148, a congener of DPI 201-106, in guinea-pig atriaand papillary muscles. Br J Pharmacol 1991;103:1939–1945.
136. Amos GJ, Ravens U. The inotropic agents DPI 201-106 andBDF 9148 differentially affect potassium currents of guinea-pig ventricular myocyates. Naunyn Schmiedebergs ArchPharmacol 1994;350:426–433.
137. Hoey A, Amos G, Ravens U. Comparison of the action po-tential prolonging and positive inotropic activity of DPI 201-106 and BDF 9148 in human ventricular myocardium. J MolCell Cardiol 1994;26:985–994.
138. Kawamura A, Wahler G, Solaro R. RWJ-24517, a positiveinotropic agent, has novel effects on action potentials inguinea pig myocardium. J Cardiovasc Pharmacol 1993;22:519–525.
139. Raap A, Armah B, Mest H-J, Stenzel W, Schloos J, BlechaczW. Investigations of the mechanism of the positive inotropicaction of BDF 9148; comparison with DPI 201-106 and theenantiomers. J Cardiovasc Pharmacol 1997;29:164–173.
140. Beck W, Benz I, Bessler W, Jung G, Kohlhardt M. Respon-siveness of cardiac Na1 channels to a site-directed antis-erum against the cytosolic linker between domains III andIV and their sensitivity to other modifying agents. J Mem-brane Biol 1993;134:231–239.
141. Grant AO. The electrophyisology of the cardiac sodiumchannel. Trends Cardiovasc Med 1991;1:321–330.
142. Quant F, Narahashi T. Modi~cation of single Na1 channelsby batrachotoxin. Proc Natl Acad Sci USA 1982;79:6732–6736.
143. Wang G, Dugas M, Armah B, Honerjäger P. Sodium channelcomodi~cation with full activator reveals veratridine reac-tion dynamics. Mol Pharmacol 1990;37:144–148.
144. Nilius B, Vereecke J, Carmeliet E. Properties of the burst-ing sodium channel in guinea pig ventricular myocytes.P_ügers Arch 1989;413:234–241.
145. Schüttler K, Wettwer E, Ravens U. The S-enantiomer ofBDF 9148 affects sodium current deactivation in guinea-pigventricular myocytes (Abstract). P_ügers Arch 1991;418:R86.
146. Studer R, Reinecke H, Bilger J, et al. Gene expression of theNa1/Ca212 exchanger in endstage human heart failure. CircRes 1994;75:443–453.
147. Zierhut W, Salzmann R, Gormann G, Rüegg U, Hof R. Phar-macological actions of SDZ 218-135, a novel positive inot-ropic agent. Cardiovasc Drug Ther 1994;8:235–244.
148. Armah Bl, Brückner R, Stenzel W. BDF 9148: A novel inot-ropic agent that transiently prolongs cardiac action poten-tial duration (Abstract). Naunyn Schmiedebergs ArchPharmacol 1990;341:R50.
149. Müller-Ehmsen J, Frank K, Brixius K, Schwinger R. In-crease in force of contraction by activation of the Na1 /Ca21-exchanger in human myocardium. Br J Clin Pharmacol1997;43:399–405.
150. Kirby M, McCall E, Orchard C, Boyett M. The role ofNa1–Ca21 exchange in paired pulse potentiation of ferretventricular muscle. J Physiol (Lond) 1993;472:415–442.
151. Barcenas-Ruiz L, Beuckelmann DJ, Wier WG. Sodium–cal-cium exchange in heart: membrane currents and changes in[Ca21]i. Science 1987;238:1720–1722.
152. Carafoli E. Intracellular calcium homeostasis. Ann Rev Bio-chem 1987;56:395–433.
153. Beuckelmann D, Wier WG. Sodium–calcium exchange inguinea-pig cardiac cells: exchange current and changes inintracellular Ca21. J Physiol (Lond) 1989;414:499–520.
154. Scholtysik G, Salzmann R, Gerber W. Interaction of DPI201-106 with cardiac glycosides. J Cardiovasc Pharmacol1989;13:342–347.
155. Brasch H. Positive inotropic but no chronotropic effect ofBDF 9148 in guinea-pig atria (Abstract). NaunynSchmiedebergs Arch Pharmacol 1990;341:R 51.
156. Raap A, Armah B, Grott W, Steinberg M, Mest H-J. Theeffect of BDF 9148, its enantiomers and DPI 201-106 on thecardiac and hemodynamic pro~le in anesthetized dogs (un-published). 1997.
157. Doggrell S, Bishop B, Brosch S. The effects of veratridineand BDF 9148 on the action potentials and contractility ofthe rat right ventricle. Gen Pharmacol 1995;26:593–601.
158. Herzig S, Lilienthal E, Mohr K. The positive inotropic drugsDPI 201-106, BDF 9148, and veratridine increase ouabaintoxicity and [3H]ouabain binding in guinea-pig heart. JCardiovasc Pharmacol 1991;18:182–189.
159. McCune S, Baker P, Stills H. SHHF/Mcc-cp rat; model ofobesity, non-insulin dependent diabetes, and congestiveheart failure. ILAR News 1990;32:23–27.
160. Bers D. Excitation–Contraction Coupling and CardiacContractile Force. Dordrecht: Kluwer Academic, 1991.
161. McCall E, Steinberg M. The effects of LY 368052 on contrac-tion in isolated cardiac myocytes (Abstract). Biophys J1997;72:A166.
162. Schwinger R, Böhm M, Mittmann C, La Rosée K, ErdmannE. Evidence for a sustained effectiveness of sodium channelactivators in failing human myocardium. J Mol Cell Cardiol1991;22:461–471.
163. Flesh M, Schwinger R, Schiffer F, et al. Evidence for func-tional relevance of an enhanced expression of the Na1–Ca21
exchanger in the failing human myocardium. Circulation1996;94:992–1002.
164. Schwinger R, Böhm M, LaRosee K, Schmidt U, Schulz C,Erdmann E. Na1-channel activators increase cardiac gly-coside sensitivity in failing human myocardium. JCardiovasc Pharmacol 1992;19:554–561.
165. Schwinger RHG, Müller-Ehmsen J, Böhm M, Erdmann E.Enantioselective inotropic actions of the Na1-channel acti-vators BDF 9148, BDF 9196 and BDF 9167 in human failingand nonfailing myocardium. J Pharmacol Exp Ther1996;276:1180–1188.
166. Schwinger R, Müller-Ehmsen J, Frank K, Koch K, ErdmannE. Enhanced sensitivity of the failing human myocardium tocardiac glycosides and Na1-channel activators. Am Heart J1996;131:988–993.
167. Schwinger R, Böhm M, Müller-Ehmsen J, et al. Effect ofinotropic stimulation on the negative force–frequency rela-tionship in the failing human heart. Circulation 1993;88:2267–2276.
168. Modersohn D, Rüffert B, Sutowicz M, Piorkowski C, Bau-mann G. In_uence of three Na-channel activators BDF 9148,BDF 9196 and BDF 9198 on isometric, isotonic and auxo-tonic contractions of isolated human failing myocardium (ab-stract). P_ügers Arch 1996;431:R132.
169. Hajjar R, Gwathmey J, Briggs G, Morgan J. Differentialeffect of DPI 201-106 on the sensitivity of the myo~lamentsto Ca21 in intact and skinned trabeculae from control andmyopathic human hearts. J Clin Invest 1988;82:1578–1584.
170. Brixius K, Zobel C, Müller-Ehmsen J, Reuter H, Pietsch M,Frank K, Schwinger R. The Na1-channel modulators BDF
192 Steinberg et al.

9148 and DPI 201-106 exert positive inotropic effect withoutdirect interaction with the contractile proteins on humanmyocardium (Abstract). J Am Coll Cardiol 1997;29:403A.
171. Bers D. Excitation–Contraction Coupling and CardiacContractile Force. Dordrecht: Kluwer Academic, 1991.
172. Mulieri L, Hasenfuss G, Leavit TB, Allen P, Alpert N. Al-tered myocardial force–frequency relation in human heartfailure. Circulation 1992;85:1743–1750.
173. Buckley N, Penefsky Z, Litwak R. Comparative force–fre-quency-relationships in human and other mammalian ven-tricular myocardium. P_ügers Arch 1972;332:259–270.
174. Gwathmey J, Slawsky M, Hajjar R, Briggs G, Morgan J.Role of intracellular calcium handling in force-interval rela-tionships of human ventricular myocardium. J Clin Invest1990;85:1599–1613.
175. Böhm M, LaRosee K, Schmidt U, Schulz C, Schwinger R,Erdmann E. Force-frequency relationship and inotropicstimulation in the nonfailing and failing human myocardium:implications for the medical treatment of heart failure. ClinInvest 1992;70:421–425.
176. Gaasch W. Diagnosis and treatment of heart failure based onleft ventricular systolic or diastolic dysfunction. JAMA1994;271:1276–1280.
177. Baumgart D, Ehring T, Krajcar M, Skyschally A, Heusch G.Characterization of the inotropic and arrhythmogenic actionof the sodium channel activator BDF 9148: a comparison toits S-enantiomer BDF 9196, to its congener DPI 201-106, tonorepinephrine, and to ouabain. Basic Res Cardiol 1994;89:61–79.
178. Raap A, Grott W, Mest H-J, Blechacz W. Cardiovasculareffects of the inotropic agent, BDF 9148, and its enantiomersin dogs (abstract). Can J Physiol Pharmacol 1994;72:100.
179. Gérard J-L, Berdaux A, Giudicelli J-F. Cardiac and hemody-namic pro~le of the new cardiotonic agent, DPI 201-106, inthe conscious dog. Eur J Pharmacol 1989;165:39–49.
180. Gérard J-L, Berdeaux A, Giudicelli J-F. Cardiac andhemodynamic effects of S(-)- and R(+)-DPI 201-106 and ofracemic DPI 201-106 in conscious dogs. Eur J Pharmacol1990;184:321–323.
181. Ozaki T, Uematsu T, Nagashima S, Nishimoto M, NakashimaM. Effects of DPI 201-106, a novel cardiotonic agent, onhemodynamics, cardiac electrophysiology and arrhythmiasinduced by programmed ventricular stimulation in dogs
with subacute myocardial infarction: a comparative studywith dobutamine. Naunyn Schmiedebergs Arch Pharmacol1991;344:478–487.
182. Tanimura M, Shevlyagin S, Shimoyama H, Steinberg M,Borzak S, Goldstein S, Sabbah H. LY366634, a novel sodiumchannel activator, improves left ventricular function in dogswith chronic heart failure (abstract). J Am Coll Cardiol1997;29:403A.
183. Raap A, Armah B, Hofferber E, Stenzel W, Blechacz W.BDF 9148: Comparison of action potential duration in vitroand prolongation of the Q-T interval in anesthetized dogs(time course study) (abstract). Naunyn SchmiedebergsArch Pharmacol 1991;344:R85.
184. Wilkens B. Ein_uss von Natrium-Kanal-Modulatoren aufexperimentelle Herzarrhythmien. Tierärztliche Hochschule,Hannover, Germany: 1995.
185. Novosel D, Hof A, Zierhut W, Hof R. Proarrhythmic effectsof DPI 201-106 elicited with programmed electrical stimula-tion: a comparison with sotalol. J Cardiovasc Pharmacol1993;21:967–972.
186. Hogan J, Greenbaum R, Lunnon M, Hilson A, Evans T.Haemodynamic effects of DPI 201-106, following single in-travenous dose administration to patients with moderatecardiac failure. Eur Heart J 1988;9:498–502.
187. Thormann J, Kramer W, Kindler M, Kremer F, Schlepper M.Comparative ef~cacy of the new cardiotonic agent DPI 201-106 versus dobutamine in dilated cardiomyopathy: analysisby serial pressure/volume relations and “on line” MVO2 as-sessment. J Cardiovasc Pharmacol 1986;8:749–757.
188. Butrous G, Debbas N, Erwin J, et al. Clinical cardiac elec-trophysiologic evaluation of the positive inotropic agent,DPI 201-106. Eur Heart J 1988;9:489–497.
189. Rüegg P, Nüesch E. The effect of a new inotropic agent, DPI201-106, on systolic time intervals and the electrocardio-gram in healthy subjects. Br J Clin Pharmacol 1987;24:453–458.
190. Kostis J, Lacy C, Raia J, Dworkin J, Warner R, Casazza L.DPI 201-106 for severe congestive heart failure. Am JCardiol 1987;60:1334–1339.
191. Katz A. The myocardium in congestive heart failure. Am JCardiol 1989;63:12A–16A.
Na1 Channels as Targets for Inotropic Agents 193