dna methylation in cancer: a gene silencing mechanism and
TRANSCRIPT

DNA Methylation and Cancer 173Tohoku J. Exp. Med., 2013, 229, 173-185
173
Received January 15, 2013; accepted January 29, 2013. Published online February 16, 2013; doi: 10.1620/tjem.229.173.Correspondence: Shinichi Fukushige, Ph.D., Department of Molecular Pathology, Tohoku University Graduate School of Medicine, 2-1
Seiryo-machi, Aoba-ku, Sendai, Miyagi 980-8575, Japan.e-mail: [email protected]. Shinichi Fukushige is a recipient of the 2011 Gold Prize, Tohoku University School of Medicine.
Invited Review
DNA Methylation in Cancer: A Gene Silencing Mechanism and the Clinical Potential of Its Biomarkers
Shinichi Fukushige1, 2 and Akira Horii1
1Department of Molecular Pathology, Tohoku University Graduate School of Medicine, Sendai, Miyagi, Japan2Center for Regulatory Epigenome and Diseases, Tohoku University Graduate School of Medicine, Sendai, Miyagi, Japan
Initiation and progression of human caner not only depends on genetic alterations but also on epigenetic changes such as DNA methylation and histone modifications. Aberrant DNA hypermethylation in the promoter regions of genes is the most well-defined epigenetic change in tumors and is associated with inappropriate gene silencing. This feature can be utilized to search for tumor-specific DNA methylation biomarkers and to examine candidate DNA biomarkers for clinical use. DNA methylation biomarker is defined as a molecular target that undergoes DNA methylation changes in carcinogenesis. Such a biomarker is useful for early detection of cancer, predicting and/or monitoring the therapeutic response, and detection of recurrent cancer. In this review, we describe the mechanism that establishes and maintains DNA methylation patterns as well as the mechanism of aberrant gene silencing in cancer, and then we introduce methods to isolate the DNA methylation biomarkers. We also summarize the current status of clinical implementation for some of the most widely studied and well-validated DNA methylation biomarkers, including tissue factor pathway inhibitor 2 (TFPI2), septin 9 (SEPT9), glutathione S-transferase pi 1 (GSTP1), and O6-methylguanine-DNA methyltransferase (MGMT), and assess the clinical potential of these biomarkers for risk assessment, early diagnosis, prognosis, treatment, and the prevention of cancer. Finally we describe the possible involvement of 5-hydroxymethylcytosine in cancer; this is a recently discovered 5-methylcytosine oxidation derivative and might have a diagnostic potential in certain cancers. Abnormal DNA methylations are leading candidates for the development of specific markers for cancer diagnosis and therapy.
Keywords: biomarker; cancer; CpG island; DNA methylation; gene silencingTohoku J. Exp. Med., 2013 Mar, 229 (3), 173-185. © 2013 Tohoku University Medical Press
IntroductionDNA methylation at position five of the cytosine ring
occurs on most CpG dinucleotides and has long been recog-nized as “the fifth base” in the mammalian genome (Fig. 1). DNA methylation is involved in a variety of biological pro-cesses, including embryonic development, X chromosome inactivation, genomic imprinting, inactivation of transpos-able elements, regulation of gene expression, and mainte-nance of epigenetic memory (Bird 2002). The enzymatic step involved utilizes S-adenosyl-methionine (SAM) as a methyl donor and is carried out by three DNA methyltrans-ferases, Dnmt1, Dnmt3a, and Dnmt3b (Okano et al. 1998; Okano et al. 1999). New DNA methylation patterns are ini-tially established by de novo DNA methyltransferases, Dnmt3a and Dnmt3b, but the DNA methylation pattern is
then maintained during DNA replication by the mainte-nance DNA methyltransferase Dnmt1. DNA methylation is essential for normal mammalian development, because tar-geted disruption of Dnmt1 or Dnmt3b results in embryonic lethality in mice, and homozygous Dnmt3a knockout mice die about 4 weeks after birth (Li et al. 1992; Okano et al. 1999). Mutations in the DNA methylation machinery have long been linked to inherited human diseases. For example, mutations in the human DNMT3B gene cause immunodefi-ciency, centromeric instability, and facial anomalies (ICF) syndrome (Okano et al. 1999; Xu et al. 1999). Aberrant DNA methylation is also linked to imprinting disorders, such as Beckwith-Wiedemann syndrome (BWS), Prader-Willi syndrome (PWS) and Angelman syndrome (AS), as well as human cancers (reviewed by Robertson 2005). The study of these diseases has provided fundamental insights
S. Fukushige and A. Horii174
that properly established and maintained DNA methylation patterns are essential for mammalian development and for the normal functioning of the adult organism.
CpG dinucleotide is under-represented in the mamma-lian genome; it occurs at only about one-fifth of the roughly 4% frequency that would be expected in human DNA, because approximately 70% of CpG dinucleotides are methylated on the cytosine base, and spontaneous hydro-lytic deamination of 5-methylcytosine (5mC) residues gives rise to T residues (Lander et al. 2001). In contrast, short stretches (an average of about 1 kb) of CpG rich regions, known as CpG islands (CGIs), which are found in the pro-moters of approximately 60% of the coding genes in the mammalian genome, are generally unmethylated in normal cells (Cooper et al. 1983; Bird et al. 1985; Gardiner-Garden et al. 1987). However, the hypermethylation of these pro-moter regions is found in virtually every type of human cancer and is associated with the inappropriate transcrip-tional silencing of genes (Fig. 2) (Jones and Laird 1999; Jones and Baylin 2002; Herman and Baylin 2003). These hypermethylated genes are composed of important catego-ries of tumor suppressor genes, including cell cycle control regulators, pro-differentiation factors, DNA repair proteins, and anti-apoptotic factors; many of these genes are known to play a role in normal development. A recent study also
showed that most methylation alterations in colon cancer occur not in promoters, but in sequences up to 2 kb distant that are called ‘CpG island shores’ and have comparatively low GC density (Irizarry et al. 2009). CGI shore methyla-tion is tightly linked with the tissue of origin (Doi et al. 2009), but the importance of those changes is not yet under-stood. In addition to promoter DNA hypermethylation, global DNA hypomethylation is another major altered methylation pattern observed in cancer (Fig. 2) (Herman and Baylin 2003; Esteller 2008). This alteration is also cor-related with different stages of cancer progression and metastasis in various tumor types, and it causes chromo-somal instability, gene mutation, and reactivation of various cancer-related genes (Goelz et al. 1985; Cui et al. 2002).
The increasing recognition of the importance of epi-genetic changes in cancer pathogenesis has led to a shift in the approaches that are used to identify genes affected by this process. Identification of genes that are specifically hypermethylated or hypomethylated might lead to the dis-covery of new factors that are important for tumor initiation and progression. Genome methylation patterns are also being developed as biomarkers for tumor types, risk assess-ment, early detection, monitoring of prognosis, and as indi-cators of susceptibility or response to therapy. Molecular alterations of the epigenome, especially DNA methylation,
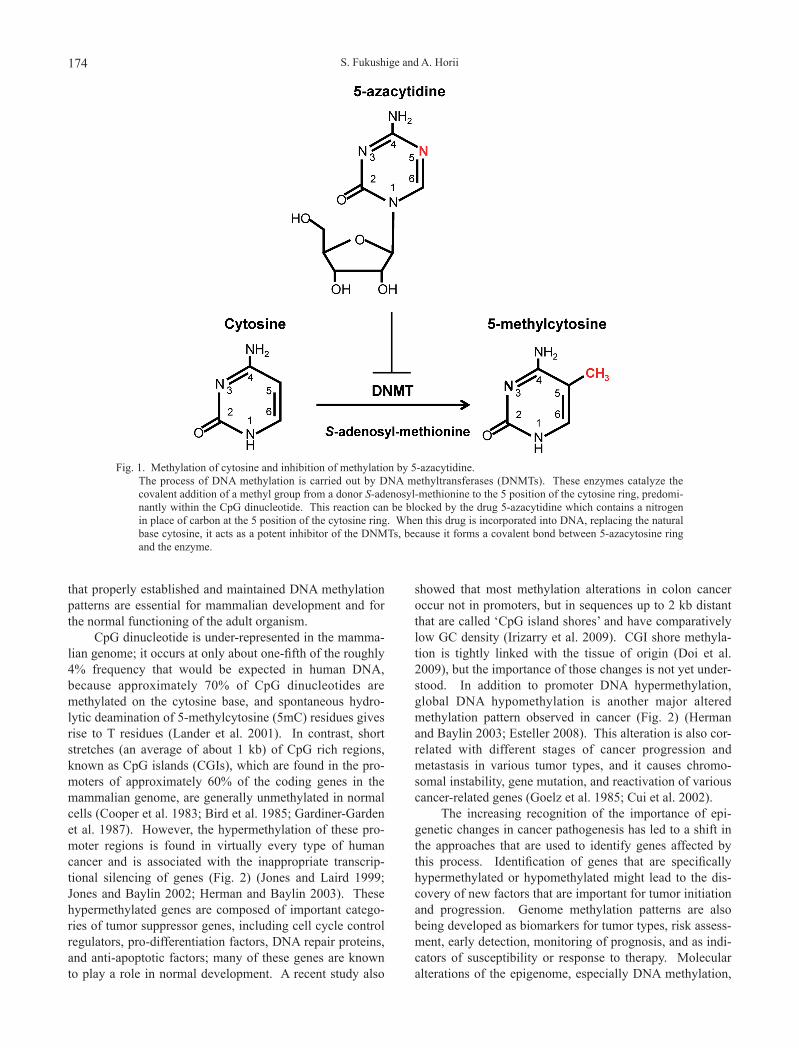
Fig. 1. Methylation of cytosine and inhibition of methylation by 5-azacytidine. The process of DNA methylation is carried out by DNA methyltransferases (DNMTs). These enzymes catalyze the
covalent addition of a methyl group from a donor S-adenosyl-methionine to the 5 position of the cytosine ring, predomi-nantly within the CpG dinucleotide. This reaction can be blocked by the drug 5-azacytidine which contains a nitrogen in place of carbon at the 5 position of the cytosine ring. When this drug is incorporated into DNA, replacing the natural base cytosine, it acts as a potent inhibitor of the DNMTs, because it forms a covalent bond between 5-azacytosine ring and the enzyme.

DNA Methylation and Cancer 175
have emerged as alternative targets of biomarker research and display potentially great clinical value. Therefore, we will focus on DNA methylation based biomarkers in this review.
Mechanisms to Establish and Maintain DNA Methylation Status
Although DNA methylation at CGIs is thought to be important for cancer initiation and progression, mechanisms that establish and maintain DNA methylation patterns dur-ing tumorigenesis or normal development remain poorly understood. In general, there are at least three possibilities for the mechanisms. First, an initial random methylation event provides a growth advantage to cells resulting in their clonal selection and proliferation (Jones and Baylin 2007). Second, cis-acting factors such as transcription factor PU.1 (Suzuki et al. 2006) and estrogen receptor ERα (Metivier et al. 2008) or histone methyltransferases such as G9a (Feldman et al. 2006; Tachibana et al. 2008) and enhancer of zeste homolog 2 (EZH2) (Viré et al. 2006) recruit DNA methyltransferases to methylation target sites. Third, loss of certain transcription factors leads to the spreading of DNA methylation into affected CGIs (Turker 2002).
In early work using transgenic mice, two groups showed that a small number of transcription factor binding sites at a promoter region, specifically those for Sp1, are necessary to protect the adenine phosphoribosyltransferase (Aprt) gene from de novo methylation (Brandeis et al. 1994; Macleod et al. 1994). Furthermore, Gebhard et al. (2010) examined global methylation-prone and methylation-resis-tant CGIs of human genome in acute leukemia cell lines as well as in normal blood monocytes and found that general transcription factor binding at CGIs correlates with resis-tance to de novo DNA methylation. Recently, Lienert et al.
(2011) used an elegant experimental system and proposed the concept of MDR (methylation-determining region) for the first time (Fig. 3). They showed that promoter sequences of approximately 1 kb named MDRs are gener-ally sufficient to precisely recapitulate DNA methylation patterns in embryonic stem (ES) cells and to replicate the changes that occur during differentiation. On the basis of truncation and mutagenesis analyses, they also demon-strated that DNA binding motifs within MDRs are critical for generating a hypomethylated state. By analogy with these findings, it is also possible to consider that the loss of MDR protective activity caused by decreased expression of transcription factors and/or mutations in transcription factor binding sites defines aberrant targets of de novo methyla-tion in cancer. In this context, it is important to understand the mechanism of formation of the CpG island methylator phenotype (CIMP), coordinated methylation of a subset of CGIs in tumors, because many CIMP loci are known to be polycomb targets (Weisenberger et al. 2006; Widschwendter et al. 2007). The effort to find MDRs and their associated transcription factors is especially important in cancer research, because these factors may become excellent can-didates with diagnostic and therapeutic implications in the future.
Relationship between Hypermethylated Genes and Tumorigenesis
Recent advances in epigenome analysis which allowed precise mapping of DNA methylation across normal and cancer genomes have provided evidence that almost all can-cers carry hundreds of genes with abnormal hypermethyl-ation (Baylin and Jones 2011). Among these, some genes show age-related DNA methylation (Maegawa et al. 2010). However, similarly to DNA mutations, a number of tumor
Fig. 2. DNA methylation of a typical tumor suppressor gene in normal and cancer cells. Boxes with number indicate exons and lines indicate introns as well as the regions outside the genes. Short stretches of
CpG rich sequences, usually around 1 kb in size, are termed CpG islands (CGIs). In normal cells, most CpG sites out-side of CGIs, including gene bodies, repeat elements (indicated by boxes with arrows), and pericentromeric regions, are methylated, whereas most CGIs are unmethylated. The unmethylated state of the CGIs in a gene promoter permits active gene expression (arrow in upper panel). In cancer cells, on the other hand, many CpG sites outside of CGIs are unmethylated, but most CGIs are methylated. Aberrant promoter methylation serves to turn off the gene (lower panel). Filled and open circles represent methylated and unmethylated CpG sites, respectively.

S. Fukushige and A. Horii176
suppressor and candidate tumor suppressor genes have been found to be hypermethylated in multiple tumor types from cancer cell lines and primary tumor samples. One of inter-esting features of these genes is the high frequency of tar-geting of genes involved in normal development. For example, these genes include cyclin-dependent kinase inhibitor 2A (CDKN2A), which regulates stem cell number and cell cycle functions (Park et al. 2003), GATA binding protein 4 and 5 (GATA-4 and -5), which are crucial for proper epithelial differentiation (Akiyama et al. 2003), E-cadherin (CDH1), which controls cell-cell adhesion (Hirohashi 1998), and death-associated protein kinase (DAPK), which functions as anti-apoptotic gene (Michie et al. 2010). These observations support the idea of a stem or precursor cell origin for cancer and explain the targeting of at least some genes for DNA methylation and gene silenc-ing during tumor initiation and progression. Furthermore, like the majority of gene mutations, the roles of the hun-dreds of DNA hypermethylated genes other than genes functioning as tumor suppressors may be their aggregation in the same signaling pathway, thus helping derive the can-cer phenotype (Wood et al. 2007).
General Mechanisms of Gene Silencing Associated with Promoter Methylation
There are two general mechanisms by which DNA methylation inhibits gene expression (Fig. 4). First of all, methylation of cytosine bases can directly inhibit the asso-ciation of some transcription factors with their cognate DNA recognition sequences (Watt and Molloy 1988). The study of CCCTC-binding factor (CTCF) in genomic imprinting at the H19 (H19, imprinted maternally expressed transcript)/Igf2 (insulin-like growth factor 2) locus has pre-sented strong evidence for involvement of this mechanism (Bell and Felsenfeld 2000; Hark et al. 2000; Holmgren et al. 2001). CTCF, an 82 kDa protein with 11 zinc fingers, is associated with chromatin boundaries (Bell et al. 1999) and
can insulate a promoter from the influence of remote enhancers. At the maternal allele, the Igf2 gene is silent, because CTCF is bound between its promoter and a down-stream enhancer. On the other hand, at the paternal allele, these CpG-rich binding sites are methylated, and the bind-ing of CTCF is prevented, resulting in the loss of enhancer blocking and thereby allowing the downstream enhancer to activate Igf2 expression.
As a second example, one family of proteins that rec-ognize methyl-CpG, known as methyl-CpG binding pro-teins (MBPs), can elicit the repressive potential of methyl-ated DNA (Bird and Wolffe 1999). MBPs fall into two families: first, the methyl-CpG binding domain (MBD) pro-teins, MeCP2, MBD1, MBD2, MBD3, and MBD4, which share an approximately 80 amino acid MBD and repress transcription through a transcriptional repression domain (TRD) (Hendrich and Bird 1998), and second, the Kaiso-like proteins, Kaiso, ZBTB4 (zinc finger and BTB domain containing 4), and ZBTB38, which lack the MBD, but rec-ognize DNA sequences containing methyl-CpG sequences through a zinc-finger domain and repress transcription through a POZ/BTB domain (Prokhortchouk et al. 2001; Filion et al. 2006). It has long been thought that all but MBD4 are associated with methylation-dependent tran-scriptional repression and that MBD4 plays an important role in G-T mismatch repair, because only MBD4 contains a C-terminal DNA glycosylase catalytic domain (Hendrich et al. 1999). Surprisingly, we also found transcriptional repression activity in MBD4 as well as other MBPs (Kondo et al. 2005). Therefore, all known MBPs can mediate silencing of gene expression through methyl-CpG sequences. MBPs directly recognize methylated DNA and recruit transcriptional corepressor molecules to silence tran-scription and to modify surrounding chromatin (Fig. 4). The methyl-CpG binding protein 1 (MeCP1) complex was first described to have methyl-CpG binding activity and repress transcription (Bird and Wolffe 1999). MeCP1 con-
Fig. 3. Model of MDR function in mediating DNA hypomethylation. In a normal cell, exclusion of DNA methylation from a domain (gray oval) is established and maintained in tumor sup-
pressor genes (TSGs) by the binding of transcription factors (TFs, green and orange circles) to discrete sites within it (left panel). DNA methyltransferases (DNMTs, blue circles) are prevented from accessing the protected domain, but they can access to flanking DNA with methyl groups (red circles). In a cancer cell, loss of MDR protecting activity, for example, through loss of transcription factors and/or through mutations in transcription factor binding sites, may result in aberrant hypermethylation in cancer-related genes (right panel).

DNA Methylation and Cancer 177
sists of the nucleosome remodeling and histone deacety-lation (NuRD) corepressor complex and MBD2, and MBD3 is a component of NuRD (Feng and Zhang 2001). MeCP2 and MBD4 associate with a corepressor complex contain-ing the transcriptional repressor Sin3A and histone deacety-lases (Nan et al. 1998; Kondo et al. 2005). MBD1 interacts with the histone H3 lysine 9 (H3K9) methyltransferase SETDB1 (SET domain, bifurcated 1) and modifies the sur-rounding chromatin by histone methylation (Sarraf and Stancheva 2004). Proteins that interact with MBPs and enhance transcriptional repression activities have also been studied (Jiang et al. 2004; Fukushige et al. 2006). The pres-ence of multiple MBPs with repressive properties supports the idea that these may be important mediators of the meth-ylation signal. However, the question of whether each MBP has its own property remains to be elucidated.
Methods for Identifying Genes with Cancer-Specific DNA Methylation
A number of techniques have been used to identify genes which are specifically methylated in cancer. The gold standard and the most direct method used to search for methylated genes is bisulfite genomic sequencing, which maps 5mC residues at single-base resolution (Frommer et al. 1992). This method utilizes bisulfite-induced modifica-tion of genomic DNA, under conditions whereby cytosine is converted to uracil, but 5mC remains nonreactive (Fig. 5). Although large-scale bisulfite sequencing of a human genome has been successfully initiated (Eckhardt et al. 2006), more indirect methods have been utilized preferen-tially so far, because whole-genome bisulfite sequencing is a time- and resource-intensive task (Suzuki and Bird 2008).
Fig. 4. Mechanisms of transcriptional repression by DNA methylation. A stretch of nucleosome is shown with methylated CpGs (red circles). Below the diagram is a transcription factor (green
circle) that is unable to bind its recognition site when DNA is methylated. Above the diagram is a protein complex that can be attracted by DNA methylation, including the methyl-CpG binding protein (MBP, orange oval) and the corepres-sor complex (gray circles).
Fig. 5. Bisulfite conversion. DNA is denatured and then treated with sodium bisulfite
to convert unmethylated cytosine to uracil, which is con-verted to thymine by PCR. Following bisulfite conver-sion, the DNA strands are no longer complementary, and primers are designed to assay the methylation status of a specific strand by bisulfite sequencing or methylation-specific PCR (MSP).

S. Fukushige and A. Horii178
As alternatives, methylated DNA fragments are first puri-fied and then coupled with a genomic microarray to map them at specific genomic loci or sequence them extensively. Two approaches have been used to purify methylated DNA fragments (Fig. 6). One approach is based on the sensitiv-ity of methyl-specific restriction enzymes to CpG methyla-tion within their cleavage recognition sites (Schumacher et al. 2006). Another approach uses protein affinity to enrich methylated DNA sequences with either an anti-5mC anti-body (methylated DNA immunoprecipitation; MeDIP) (Weber et al. 2005) or the MBD of MBPs (methyl-binding domain affinity purification; MAP) (Cross et al. 1994). All of these methods have proven useful for identifying hyper-methylated CGIs; however, because it is difficult to know which methylated CGIs are involved in transcriptional repression, we need additional experiments to obtain this information. Therefore, in order to identify more genes silenced by DNA methylation, global changes in the gene expression profiles of cell lines have been analyzed after treatment with DNA demethylating agents such as 5-aza-2′-deoxycytidine (5-Aza-CdR; decitabine) or 5-azacytidine (5-Aza-CR; azacitidine; see Fig. 1) (Suzuki et al. 2002). Candidate genes targeted for aberrant DNA methylation in cancer cells are identified, and then a number of clinical samples are examined for DNA methylation and gene expression. Although this strategy is useful for identifying a substantial number of candidate genes, it also identifies genes whose promoters seem to be unmethylated (Soengas et al. 2001; Suzuki et al. 2002), thus leading to the develop-ment of more specific and sensitive methods.
As described above, MBPs directly bind to hypermeth-ylated promoters of human genes and are associated with transcriptional silencing. Therefore, if the genes governed by MBPs are specifically reactivated, it should be possible to uncover them. We developed a method termed “methyl-CpG targeted transcriptional activation (MeTA)” (Fig. 7) that employs a fusion gene comprised of the MBD from MBD2 and the NFκB transcriptional activation domain (TAD) (Fukushige et al. 2008). We found that MeTA recruits p300/CREB-binding protein (CBP) in the hyper-methylated promoter regions and reactivates epigenetically silenced tumor suppressor genes in human cancer cells (Fukushige et al. 2009). Microarray coupled with MeTA (MeTA-array) provides not only the information about methylated genes but also about transcriptional repression in a single experiment (Sato et al. 2011). Therefore, it is more advantageous to use this technique to identify methyl-ated genes involved in transcriptional repression than to use strategies such as MeDIP, MAP, or DNA demethylating agents coupled with microarray. In fact, we applied the MeTA-array to pancreatic cancer and successfully identi-fied tumor-specific methylated genes (Shimizu et al. 2011).
Advantages of DNA Methylation Over Genetic Mutations as Biomarkers
DNA methylation biomarkers, which undergo DNA methylation changes in carcinogenesis, provide a range of opportunities for early detection, diagnosis, prognosis, ther-apeutic stratification and post-therapeutic monitoring of cancer. DNA methylation biomarkers can offer several
Fig. 6. Two approaches to purifying methylated DNA fragments. (A) Detection of methylated DNA fragments with methylation-sensitive restriction enzymes. A methylated (m) region
of genomic DNA is digested with HpaII (left) or MspI (right). Smaller fragments are discarded (red X), enriching for methylated DNA in the HpaII-treated sample, when compared to the MspI-treated one. (B) Affinity enrichment of methylated DNA. Genomic DNA is denatured and then affinity purified with either an anti-5mC antibody (green, meth-ylated DNA immunoprecipitation; MeDIP) or a methyl-CpG binding domain (orange, methyl-binding domain affinity purification; MAP) that can be attached to a column.

DNA Methylation and Cancer 179
advantages over genetic and serum markers (Laird 2003; Miyamoto and Ushijima 2005; Schuebel et al. 2007). First, the incidences of aberrant DNA methylation of specific CGIs are higher than those of genetic defects. For example, Sjöblom et al. (2006) sequenced 13,023 genes in colorectal and breast cancer, and estimated an average of only 14 sig-nificant mutations per tumor, suggesting that a relatively small number of genetic events may be sufficient to drive tumorigenesis. On the other hand, by screening cell lines and validating tumor-specific hypermethylation in a panel of primary human colorectal cancer samples, Schuebel et al. (2007) estimated that nearly 5% or more of all known genes may be promoter methylated in an individual colorec-tal cancer. Second, the aberrant DNA methylation seen in cancer cells can be sensitively detected, even when it is embedded in substantial amounts of contaminating normal DNA. Third, detection of aberrant DNA methylation is technically simple; it can be detected using methylation-specific PCR (MSP). DNA hypermethylation only occurs at rather small promoter regions called CGIs, whereas genetic mutations occur throughout the length of the gene. Fourth, aberrant DNA methylation seems to occur in early-stage tumors, causing loss- and/or gain-of-function of key processes and signaling properties. Therefore, detection of aberrant DNA methylation is potentially a good early indi-
cator of existing cancer and even of risk assessment for the future development of cancer. Although, as described above, DNA methylation biomarkers have several advan-tages over genetic markers it has been clearly shown that a combination of both types could exploit the advantages of both techniques. For example, one combination of both techniques in a stool DNA test allows the detection of cur-able stage colorectal cancer and large adenomas with high levels of accuracy (Ahlquist et al. 2012).
DNA Methylation as a BiomarkerDNA methylation has great potential to be used as a
nucleic acid-based biomarker for assessing cancer risk, early detection, prognosis, and predicting therapeutic responses (Table 1) (Baylin and Jones 2011; Heyn and Esteller 2012; Fukushige and Horii in press). The number of potential DNA methylation marker genes and knowledge of their roles in cancer are rapidly increasing owing to the development of recent genome-wide techniques for their identification and subsequent functional analyses (Carmona and Esteller 2011; Heyn and Esteller 2012). In addition, the development of more sensitive methods, including nan-otechnology assays, facilitates methylation detection in patient samples (Bailey et al. 2009; Li et al. 2009). One of the most important criteria for clinically useful biomarkers
Fig. 7. Outline of MeTA procedure. (A) Each member of the methyl-CpG binding domain (MBD) protein family is composed of two domains, MBD and
the transcriptional repression domain (TRD), and it represses transcription by recruiting corepressor complexes. Filled circles represent methylated CpG sites. (B) In MeTA, TRD is replaced by transcriptional activation domain (TAD) which recruits p300/CREB-binding protein (CBP) coactivators. As a result, the genes governed by MBD are reactivat-ed. (C) An example of MeTA. Transfection of the MeTA-inducing DNA construct into 293T cells reactivates the silenced MLH1 gene, as indicated by Reverse transcription (RT)-PCR analysis.
Table 1. A selected list of potential DNA-based biomarkers in cancer.
Gene name Gene function Application Cancer type Tissue examined
SEPT9 Cell cycle control Early detection Colon BloodTFPI2 Extracellular
matrix proteinEarly detection Colon Stool
CDKN2A & CDH13 Cell cycle control & Cell-cell adhesion
Prognosis NSCLC Tumor, Mediastinal lymph node biopsy
MGMT DNA repair Predicting drug sensitivity
Glioma, Colon, Lung, Lymphoma
Tumor
GSTP1 Detoxification Predicting drug sensitivity
Prostate, Breast, Kidney
Tumor, Urine

S. Fukushige and A. Horii180
is whether they are applicable to surrogate tissues such as blood or other body fluids that can be obtained through minimally invasive procedures. The sensitive and specific detection of tumor-specific DNA methylation patterns at distal sites makes DNA methylation a biomarker of choice for early detection of cancer and the clinical management of cancer patients. In this context, DNA methylation of tis-sue factor pathway inhibitor 2 (TFPI2) in the stool is a highly sensitive and specific marker and a useful adjunct to the noninvasive strategies for screening of colorectal can-cers (Glöckner et al. 2009). TFPI2 methylation was detected in stool DNA from stage I to III colorectal cancer patients with a sensitivity of 76% to 89% and a specificity of 79% to 93%. In addition, a sieving strategy for success-ful identification and ranking of blood-based DNA methyla-tion biomarker candidates has been developed for colorectal cancer (Lofton-Day et al. 2008). In particular, hypermeth-ylation of septin 9 (SEPT9) is reported to detect colorectal cancer in blood-based samples from patients with a sensi-tivity of 90% and a specificity of 88% (Warren et al. 2011). Glutathione S-transferase pi 1 (GSTP1) is involved in cellu-lar detoxification of xenobiotics and carcinogens; it was first identified as a hypermethylated gene in prostate cancer in 1994 (Lee et al. 1994). DNA methylation of GSTP1 has been consistently validated in more than 30 independent, peer-reviewed studies, and the meta-analysis pooling these almost 3,500 subjects has established GSTP1 as a promis-ing biomarker of prostate cancer with a sensitivity of 82% and a specificity of 95% (Van Neste et al. 2012). GSTP1 hypermethylation in tumor biopsy and urine samples has been extensively analyzed and provides value as an alterna-tive to conventional methods of prostate cancer detection (Cairns et al. 2001; Hoque et al. 2005; Rosenbaum et al. 2005). In particular, the combination of GSTP1 with the serum-based biomarker, prostate-specific antigen (PSA) or additional DNA methylation biomarkers such as adenoma-tous polyposis coli (APC) and endothelin receptor type B (EDNRB) should improve the sensitivity of diagnosis (Sunami et al. 2009; Rogers et al. 2006). Mapping the pat-terns of DNA methylation has also recently been proposed to help in the identification of cancers of unknown primary site (Fernandez et al. 2012).
DNA methylation can be used as a molecular progno-sis biomarker of potentially curable, stage I non-small-cell lung cancer (NSCLC) (Brock et al. 2008). Methylation of the promoter region of four genes, p16 (CDKN2A), H-cadherin (CDH13), Ras association domain family 1A (RASSF1A), and APC, in primary tumor and mediastinal lymph node biopsy samples strongly correlates with early recurrence and short survival (Brock et al. 2008). The cur-rent method for risk assessment of recurrence in patients with stage I NSCLC is imprecise; one third of such tumors recur after curative surgery. The validation of these find-ings may allow re-staging of NSCLC at the molecular level and may thus identify high risk patients who require special adjuvant therapies.
DNA methylation biomarkers can also predict responses to chemotherapy; the best example is promoter hypermethylation at O6-methylguanine-DNA methyltrans-ferase (MGMT), a DNA repair enzyme, in glioma. MGMT hypermethylation was detected in 40% of glioma and colorectal cancer in a study of more than 500 primary human tumors (Esteller et al. 1999). MGMT protects cells against mutations by removing alkyl groups from guanine bases, which are introduced by alkylating agents such as nitrosamides. Therefore, the promoter hypermethylation status of MGMT is associated with glioma sensitivity to alkylating agents such as carmustine and temozolomide and to radiotherapy in patients (Esteller et al. 2000; Hegi et al. 2005). A better response to treatment with alkylating agents in part depends on unrepaired alkylated guanine residues initiating cycles of futile mismatch repair (MMR), which can lead to cell death (Kaina et al. 2007). Therefore, treat-ment of MGMT-deficient glioma with alkylating therapy introduces a strong selective pressure for losing MMR function (Casorelli et al. 2008). In fact, recent literature has shown that the MMR genes are mutated with characteristic CG to AT transitions at non-CpG sites resulting from unre-paired alkylated guanine residues. In spite of its usefulness, we always have to bear in mind that there is the possibility that patients who initially respond to the alkylating therapy may evolve not only into treatment resistance, but also into a MMR defective hypermutator phenotype (TCGA Research Network 2008).
5-Hydroxymethylcytosine and its Potential as a Biomarker
Ten-eleven translocation (TET) proteins have recently been demonstrated to have the capacity to convert 5mC to 5-hydroxymethylcytosine (5hmC), known as the sixth base, raising the possibility that 5mC distribution can be dynami-cally regulated by the TET family of DNA hydroxylases (Tahiliani et al. 2009; Ito et al. 2010). Furthermore, the presence of novel 5mC oxidation derivatives such as 5hmC, 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) in genomic DNA may provide an additional layer of epigene-tic information or represent intermediates in the process of DNA demethylation (Fig. 8) (Ito et al. 2011). As Nestor et al. (2010) clearly demonstrated, it is important to notice that 5mC and 5hmC are experimentally indistinguishable by established 5mC mapping techniques using methyl-sensi-tive enzymes and the gold-standard bisulfite sequencing. Therefore, it is necessary to re-evaluate existing 5mC data-sets carefully in the context of the possible presence of 5hmC. Recently, an oxidative bisulfite sequencing (oxBS-Seq) method has been developed for quantitative mapping of 5hmC in genomic DNA at single nucleotide resolution (Booth et al. 2012). High levels of 5hmC were detected in nearly all mouse embryonic tissues, but were restricted to brain and bone marrow in adult human and mouse tissues (Globisch et al. 2010; Ruzov et al. 2011). In addition, high levels of 5hmC are not only present in human and mouse

DNA Methylation and Cancer 181
embryonic stem (ES) cells and lost during differentiation, but they also reappear during the generation of induced plu-ripotent stem (iPS) cells, suggesting a biological role in pluripotency for this epigenetic marker (Ruzov et al. 2011). Although information about 5hmC in cancer is still limited, frequent TET2 mutations have been reported to be associ-ated with decreased 5hmC levels in various myeloid leuke-mias (Delhommeau et al. 2009; Langemeijer et al. 2009). Furthermore, Lian et al. (2012) recently reported that “loss of 5hmC” is an epigenetic hallmark of melanoma and that genome-wide mapping of 5hmC reveals loss of the 5hmC landscape in the melanoma epigenome. The TET family is directly responsible for the generation of 5hmC, and the catalytic reaction requires cofactor α-ketoglutarate (α-KG) (Tahiliani et al. 2009; Ito et al. 2010), which is mainly con-trolled by isocitrate dehydrogenases (IDHs) (Xu et al. 2011). Frequent mutations of IDH1 or IDH2 are also found in glioma, myeloid leukemia, and melanomas (Dang et al. 2010; Krell et al. 2011; Shibata et al. 2011). These studies suggest the important role of 5hmC, TET, and IDH in can-cer; however, it is still uncertain how this epigenetic mark and these related enzymes are associated with cancer initia-tion and progression and whether they are suitable for use as biomarkers.
ConclusionAlthough DNA methylation have rich potential as bio-
markers in clinical applications and although an increasing number of promising candidates have been identified in research laboratories throughout the world, their use in clin-ics is still very limited. This is due in part to the fact that they are insufficiently sensitive and specific for a diagnostic test, perhaps because of our yet insufficient knowledge of disease genomes and DNA methylomes. Therefore, we have to determine which profiles predict disease outcome in terms of patient prognosis and treatment response by com-bining and integrating enormous data sets obtained from various approaches, including genomics, transcriptomics, and epigenomics. These efforts will produce a group of effective, cancer specific biomarkers that will more accu-rately diagnose and predict disease prognosis. Probably, some combination of the various biomarkers, including genetic, epigenetic, and serum ones, will facilitate more reliable diagnoses, and DNA methylation biomarkers will be central to this development.
AcknowledgmentsWe are grateful to Dr. B.L.S. Pierce (University of Mary-
Fig. 8. Proposed models of DNA demethylation pathways. DNA methylation (5mC) is established and maintained by DNA methyltransferases (DNMTs). TET family proteins cat-
alyze 5mC oxidation to 5hmC. 5hmC can be further oxidized by TET proteins to produce 5fC and 5caC, and the latter may be recognized and excised by thymine DNA glycosylase (TDG) to generate cytosine. Alternatively, 5hmC may be further deaminated to become 5hmU by activation-induced cytidine deaminase (AID). 5hmU in turn can be converted to cytosine following base-excision repair (BER) pathway-mediated demethylation. It remains unknown whether there are decarboxylases or deformylases that can remove the modification directly. 5mC and its oxidative derivatives may undergo passive demethylation during DNA replication.

S. Fukushige and A. Horii182
land University College) for editorial work in the preparation of this manuscript. This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS).
Conflict of InterestWe declare no conflict of interest.
ReferencesAhlquist, D.A., Zou, H., Domanico, M., Mahoney, D.W., Yab, T.C.,
Taylor, W.R., Butz, M.L., Thibodeau, S.N., Rabeneck, L., Paszat, L.F., Kinzler, K.W., Vogelstein, B., Bjerregaard, N.C., Laurberg, S., Sørensen, H.T., Berger, B.M. & Lidgard, G.P. (2012) Next-generation stool DNA test accurately detects colorectal cancer and large adenomas. Gastroenterology, 142, 248-256.
Akiyama, Y., Watkins, N., Suzuki, H., Jair, K.W., van Engeland, M., Esteller, M., Sakai, H., Ren, C.Y., Yuasa, Y., Herman, J.G. & Baylin, S.B. (2003) GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol. Cell. Biol., 23, 8429-8439.
Bailey, V.J., Easwaran, H., Zhang, Y., Griffiths, E., Belinsky, S.A., Herman, J.G., Baylin, S.B., Carraway, H.E. & Wang, T.H. (2009) MS-qFRET: a quantum dot-based method for analysis of DNA methylation. Genome Res., 19, 1455-1461.
Baylin, S.B. & Jones, P.A. (2011) A decade of exploring the cancer epigenome-Biological and translational implications. Nat. Rev. Cancer, 11, 726-734.
Bell, A.C. & Felsenfeld, G. (2000) Methylation of a CTCF-depen-dent boundary controls imprinted expression of the Igf2 gene. Nature, 405, 482-485.
Bell, A.C., West, A.G. & Felsenfeld, G. (1999) The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell, 98, 387-396.
Bird, A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev., 16, 6-21.
Bird, A., Taggart, M., Frommer, M., Miller, O.J. & Macleod, D. (1985) A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell, 40, 91-99.
Bird, A. & Wolffe, A.P. (1999) Methylation-induced repression-Belts, braces and chromatin. Cell, 99, 451-454.
Booth, M.J., Branco, M.R., Ficz, G., Oxley, D., Krueger, F., Reik, W. & Balasubramanian, S. (2012) Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science, 336, 934-937.
Brandeis, M., Frank, D., Keshet, I., Siegfried, Z., Mendelsohn, M., Nemes, A., Temper, V., Razin, A. & Cedar, H. (1994) Sp1 elements protect a CpG island from de novo methylation. Nature, 371, 435-438.
Brock, M.V., Hooker, C.M., Ota-Machida, E., Han, Y., Guo, M., Ames, S., Glöckner, S., Piantadosi, S., Gabrielson, E., Pridham, G., Pelosky, K., Belinsky, S.A., Yang, S.C., Baylin, S.B. & Herman, J.G. (2008) DNA methylation markers and early recurrence in stage I lung cancer. N. Engl. J. Med., 358, 1118-1128.
Cairns, P., Esteller, M., Herman, J.G., Schoenberg, M., Jeronimo, C., Sanchez-Cespedes, M., Chow, N.H., Grasso, M., Wu, L., Westra, W.B. & Sidransky, D. (2001) Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin. Cancer Res., 7, 2727-2730.
Carmona, F.J. & Esteller, M. (2011) DNA methylation in early neoplasia. Cancer Biomark., 9, 101-111.
Casorelli, I., Russo, M.T. & Bignami, M. (2008) Role of mismatch repair and MGMT in response to anticancer therapies. Anti-cancer Agents Med. Chem., 8, 368-380.
Cooper, D.N., Taggart, M.H. & Bird, A.P. (1983) Unmethylated
domains in vertebrate DNA. Nucleic Acids Res., 11, 647-658.Cross, S.H., Chariton, J.A., Nan, X. & Bird, A.P. (1994) Purifica-
tion of CpG islands using a methylated DNA binding column. Nat. Genet., 6, 236-244.
Cui, H., Onyango, P., Brandenburg, S., Wu, Y., Hsieh, C.L. & Fein-berg, A.P. (2002) Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res., 62, 6442-6446.
Dang, L., Jin, S. & Su, S.M. (2010) IDH mutations in glioma and acute myeloid leukemia. Trends Mol. Med., 16, 387-397.
Delhommeau, F., Dupont, S., Della Valle, V., James, C., Trannoy, S., Massé, A., Kosmider, O., Le Couedic, J.P., Robert, F., Alberdi, A., Lécluse, Y., Plo, I., Dreyfus, F.J., Marzac, C., Casadevall, N., Lacinbe, C., Romana, S.P., Dessen, P., Soulier, J., Viguié, F., Fontenay, M., Vainchenker, W. & Bernard, O.A. (2009) Mutation in TET2 in myeloid cancers. N. Engl. J. Med., 360, 2289-2301.
Doi, A., Park, I.H., Wen, B., Murakami, P., Aryee, M.J., Irizarry, R., Herb, B., Ladd-Acosta, C., Rho, J., Loewer, S., Miller, J., Schlaeger, T., Daley, G.Q. & Feinberg, A.P. (2009) Differen-tial methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet., 41, 1350-1353.
Eckhardt, F., Lewin, J., Cortese, R., Rakyan, V.K., Attwood, J., Burger, M., Burton, J., Cox, T.V., Davies, R., Down, T.A., Haefliger, C., Horton, R., Howe, K., Jackson, D.K., Kunde, J., Koenig, C., Liddle, J., Niblett, D., Otto, T., Pettett, R., Seemann, S., Thompson, C., West, T., Rogers, J., Olek, A., Berlin, K. & Beck, S. (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet., 38, 1378-1385.
Esteller, M. (2008) Epigenetics in cancer. N. Engl. J. Med., 358, 1148-1159.
Esteller, M., Garcia-Foncillas, J., Andion, E., Goodman, S.N., Hidalgo, O.F., Vanaclocha, V., Baylin, S.B. & Herman, J.G. (2000) Inactivation of the DNA-repair gene MGMT and the clinical response of glioma to alkylating agents. N. Engl. J. Med., 343, 1350-1354.
Esteller, M., Hamilton, S.R., Burger, P.C., Baylin, S.B. & Herman, J.G. (1999) Inactivation of the DNA repair gene O6-methyl-guanine-DNA methyltransferase by promoter hypermethyl-ation is a common event in primary human neoplasia. Cancer Res., 59, 793-797.
Feldman, N., Gerson, A., Fang, J., Li, E., Zhang, Y., Shinkai, Y., Cedar, H. & Bergman, Y. (2006) G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat. Cell Biol., 8, 188-194.
Feng, Q. & Zhang, Y. (2001) The MeCP1 complex represses tran-scription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev., 15, 827-832.
Fernandez, A.F., Assenov, Y., Martin-Subero, J.I., Balint, B., Siebert, R., Taniguchi, H., Yamamoto, H., Hidalgo, M., Tan, A.C., Galm, O., Ferrer, I., Sanchez-Cespedes, M., Villanueva, A., Carmona, J., Sanchez-Mut, J.V., Berdasco, M., Moreno, V., Capella, G., Monk, D., Ballestar, E., Ropero, S., Martinez, R., Sanchez-Carbayo, M., Prosper, F., Agirre, X., Fraga, M.F., Graña, O., Perez-Jurado, L., Mora, J., Puig, S., Prat, J., Badimon, L., Puca, A.A., Meltzer, S.J., Lengauer, T., Bridgewater, J., Bock, C. & Esteller, M. (2012) A DNA meth-ylation fingerprint of 1628 human samples. Genome Res., 22, 407-419.
Filion, G.J.P., Zhenilo, S., Salozhin, S., Yamada, D., Prokhortchouk, E. & Defossez, P.A. (2006) A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol., 26, 169-181.
Frommer, M., McDonald, L.E., Millar, D.S., Collins, C.M., Watt, F., Grigg, G.W., Molloy, P.L. & Paul, C.L. (1992) A genomic

DNA Methylation and Cancer 183
sequencing protocol that yields a positive display of 5-methyl-cytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA, 89, 1827-1831.
Fukushige, S. & Horii, A. Road to early detection of pancreatic cancer: Attempts to utilize epigenetic biomarkers. Cancer Lett., in press [Epub ahead of print 23 Mar 2012] (doi: 10.1016/j.canlet.2012.03.022)
Fukushige, S., Kondo, E., Gu, Z., Suzuki, H. & Horii, A. (2006) RET finger protein enhances MBD2- and MBD4-dependent transcriptional repression. Biochem. Biophys. Res. Commun., 351, 85-92.
Fukushige, S., Kondo, E. & Horii, A. (2008) Methyl-CpG targeted transcriptional activation allows re-expression of tumor suppressor genes in human cancer cells. Biochem. Biophys. Res. Commun., 377, 600-605.
Fukushige, S., Kondo, E. & Horii, A. (2009) Methyl-CpG targeted recruitment of p300 reactivates tumor suppressor genes in human cancer cells. Biochem. Biophys. Res. Commun., 379, 1021-1026.
Gardiner-Garden, M. & Frommer, M. (1987) CpG islands in verte-brate genomes. J. Mol. Biol., 196, 261-282.
Gebhard, C., Benner, C., Ehrich, M., Schwarzfischer, L., Schilling, E., Klug, M., Dietmaier, W., Thiede, C., Holler, E., Andreesen, R. & Rehi, M. (2010) General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res., 70, 1398-1407.
Globisch, D., Munzel, M., Muller, M., Michalakis, S., Wagner, M., Koch, S., Bruckl, T., Biel, M. & Carell, T. (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One, 5, e15367.
Glöckner, S.C., Dhir, M., Yi, J.M., McGarvey, K.E., Van Neste, L., Louwagie, J., Chan, T.A., Kleeberger, W., de Bruïne, A.P., Smits, K.M., Khalid-de Bakker, C.A., Jonkers, D.M., Stockbrügger, R.W., Meijer, G.A., Oort, F.A., Iacobuzio-Donahue, C., Bierau, K., Herman, J.G., Baylin, S.B., Van Engeland, M., Schuebel, K.E. & Ahuja, N. (2009) Methyla-tion of TFPI2 in stool DNA: a potential novel biomarker for the detection of colorectal cancer. Cancer Res., 69, 4691-4699.
Goelz, S.E., Vogelstein, B., Hamilton, S.R. & Feinberg, A.P. (1985) Hypomethylation of DNA from benign and malignant human colon neoplasms. Science, 228, 187-190.
Hark, A.T., Schoenherr, C.J., Katz, D.J., Ingram, R.S., Leborse, J.M. & Tilghman, S.M. (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature, 405, 486-489.
Hegi, M.E., Diserens, A.C., Gorlia, T., Hamou, M.F., de Tribolet, N., Weller, M., Kros, J.M., Hainfellner, J.A., Mason, W., Mariani, L., Bromberg, J.E., Hau, P., Mirimanoff, R.O., Cairncross, J.G., Janzer, R.C. & Stupp, R. (2005) MGMT gene silencing and benefit from temozolomide in glioblas-toma. N. Engl. J. Med., 352, 997-1003.
Hendrich, B. & Bird, A. (1998) Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol., 18, 6538-6547.
Hendrich, B., Hardeland, U., Ng, H.H., Jiricny, J. & Bird, A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301-304.
Herman, J.G. & Baylin, S.B. (2003) Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med., 349, 2042-2054.
Heyn, H. & Esteller, M. (2012) DNA methylation profiling in the clinic: applications and challenges. Nat. Rev. Genet., 13, 679-692.
Hirohashi, S. (1998) Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am. J. Pathol., 153, 333-339.
Holmgren, C., Kanduri, C., Dell, G., Ward, A., Mukhopadhya, R., Kanduri, M., Lobanenkov, V. & Ohlsson, R. (2001) CpG methylation regulates the Igf2/H19 insulator. Curr. Biol., 11, 1128-1130.
Hoque, M.O., Topaloglu, O., Begum, S., Henrique, R., Rosenbaum, E., Van Criekinge, W., Westra, W.H. & Sidransky, D. (2005) Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J. Clin. Oncol., 23, 6569-6575.
Irizarry, R.A., Ladd-Acosta, C., Wen, B., Wu, Z., Montano, C., Onyango, P., Cui, H., Gabo, K., Rongione, M., Webster, M., Ji, H., Potash, J.B., Sabunciyan, S. & Feinberg, A.P. (2009) The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet., 41, 178-186.
Ito, S., D’Alessio, A.C., Taranova, O.V., Hong, K., Sowers, L.C. & Zhang, Y. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specifica-tion. Nature, 466, 1129-1133.
Ito, S., Shen, L., Dai, Q., Wu, S.C., Collins, L.B., Swenberg, J.A., He, C. & Zhang, Y. (2011) Tet proteins can convert 5-methyl-cytosine to 5-formylcytosine and 5-carboxycytosine. Science, 333, 1300-1303.
Jiang, C.L., Jin, S.G. & Pfeifer, G.P. (2004) MBD3L1 is a tran-scriptional repressor that interacts with methyl-CpG-binding protein 2 (MBD2) and components of the NuRD complex. J. Biol. Chem., 279, 52456-52464.
Jones, P.A. & Baylin, S.B. (2002) The fundamental role of epigen-etic events in cancer. Nat. Rev. Genet., 3, 415-428.
Jones, P.A. & Baylin, S.B. (2007) The epigenomics of cancer. Cell, 128, 683-692.
Jones, P.A. & Laird, P.W. (1999) Cancer epigenetics comes of age. Nat. Genet., 21, 163-167.
Kaina, B., Christmann, M., Naumann, S. & Roos, W.P. (2007) MGMT: Key node in the battle against genotoxicity, carcino-genicity and apoptosis induced by alkylating agents. DNA Repair (Amst), 6, 1079-1099.
Kondo, E., Gu, Z., Horii, A. & Fukushige, S. (2005) The thymine DNA glycosylase MBD4 represses transcription and is associ-ated with methylated p16INK4a and hMLH1 genes. Mol. Cell. Biol., 25, 4388-4396.
Krell, D., Assoku, M., Galloway, M., Mulholland, P., Tomlinson, I. & Bardella, C. (2011) Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH mutations in glioblastoma. PLoS One, 6, e19868.
Laird, P.W. (2003) The power and the promise of DNA methyla-tion markers. Nat. Rev. Cancer, 3, 253-266.
Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860-921.
Langemeijer, S.M., Kuiper, R.P., Berends, M., Knops, R., Aslanyan, M.G., Massop, M., Stevens-Linders, E., van Hoogen, P., van Kessel, A.G., Raymakers, R.A., Kammping, E.J . , Verhoef , G.E. , Verburgh, E. , Hagemeijer, A. , Vandenberghe, P., de Witte, T., van der Reijden, B.A. & Jansen, J.H. (2009) Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet., 41, 838-842.
Lee, W.H., Morton, R.A., Epstein, J.I., Brooks, J.D., Campbell, P.A., Bova, G.S., Hsieh, W.S., Isaacs, W.B. & Nelson, W.G. (1994) Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. USA, 91, 11733-11737.
Li, E., Bestor, T.H. & Jaenisch, R. (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell, 69, 915-926.

S. Fukushige and A. Horii184
Li, M., Chen, W.D., Papadopoulos, N., Goodman, S.N., Bjerregaard, N.C., Laurberg, S., Levin, B., Juhl, H., Arber, N., Moinova, H., Durkee, K., Schmidt, K., He, Y., Diehl, F., Velculescu, V.E., Zhou, S., Diaz, L.A. Jr., Kinzler, K.W., Markowitz, S.D. & Vogelstein, B. (2009) Sensitive digital quantification of DNA methylation in clinical samples. Nat. Biotechnol., 27, 858-863.
Lian, C.G., Xu, Y., Ceol, C., Wu, F., Larson, A., Dresser, K., Xu, W., Tan, L., Hu, Y., Zhan, Q., Lee, C.W., Hu, D., Lian, B.Q., Kleffel, S., Yang, Y., Neiswender, J., Khorasani, A.J., Fang, R., Lezcano, C., Duncan, L.M., Scolyer, R.A., Thompson, J.F., Kakavand, H., Houvras, Y., Zon, L.I., Mihm, M.C.Jr., Kaiser, U.B., Schatton, T., Woda, B.A., Murphy, G.F. & Shi, Y.G. (2012) Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell, 150, 1135-1146.
Lienert, F., Wirbelauer, C., Som, I., Dean, A., Mohn, F. & Schübeler, D. (2011) Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet., 43, 1091-1097.
Lofton-Day, C., Model, F., Devos, T., Tetzner, R., Distler, J., Schuster, M., Song, X., Lesche, R., Liebenberg, V., Ebert, M., Molnar, B., Grützmann, R., Pilarsky, C. & Sledziewski, A. (2008) DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem., 54, 414-423.
Macleod, D., Charlton, J., Mullins, J. & Bird, A.P. (1994) Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev., 8, 2282-2292.
Maegawa, S., Hinkal, G., Kim, H.S., Shen, L., Zhang, L., Zhang, J., Zhang, N., Liang, S., Donehower, L.A. & Issa, J.P. (2010) Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res., 20, 332-340.
Metivier, R., Gallais, R., Tiffoche, C., Le Péron, C., Jurkowska, R.Z., Carmouche, R.P., Ibberson, D., Barath, P., Demay, F., Reid, G., Benes, V., Jeltsch, A., Gannon, F. & Salbert, G. (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature, 452, 45-50.
Michie, A.M., McCaig, A.M., Nakagawa, R. & Vukovic, M. (2010) Death-associated protein kinase (DAPK) and signal transduc-tion: regulation in cancer. FEBS J., 277, 74-80.
Miyamoto, K. & Ushijima, T. (2005) Diagnostic and therapeutic applications of epigenetics. Jpn. J. Clin. Oncol., 35, 293-301.
Nan, X., Ng, H.H., Johnson, C.A., Laherty, C.D., Turner, B.M., Eisenman, R.N. & Bird, A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393, 386-389.
Nestor, C., Ruzov, A., Meehan, R. & Dunican, D. (2010) Enzy-matic approaches and bisulphate sequencing cannot distin-guish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques, 48, 317-319.
Okano, M., Bell, D.W., Haber, D.A. & Li, E. (1999) DNA methyl-transferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99, 247-257.
Okano, M., Xie, S. & Li, E. (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyl-transferases. Nat. Genet., 19, 219-220.
Park, I.K., Qian, D., Kiel, M., Becker, M.W., Pihalja, M., Weissman, I.L., Morrison, S.J. & Clarke, M.F. (2003) Bmi-1 is required for maintenance of adult self-renewing haemato-poietic stem cell. Nature, 423, 302-305.
Prokhortchouk, A., Hendrich, B., Jorgensen, H., Ruzov, A., Wilm, M., Georgiev, G., Bird, A. & Prokhortchouk, E. (2001) The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev., 15, 1613-1618.
Robertson, K.D. (2005) DNA methylation and human disease. Nat. Rev. Genet., 6, 597-610.
Rogers, C.G., Gonzalgo, M.L., Yan, G., Bastian, P.J., Chan, D.Y., Nelson, W.G. & Pavlovich, C.P. (2006) High concordance of gene methylation in post-digital rectal examination and post-biopsy urine samples for prostate cancer detection. J. Urol.,
176, 2280-2284.Rosenbaum, E., Hoque, M.O., Cohen, Y., Zahurak, M.,
Eisenberger, M.A., Epstein, J.I., Partin, A.W. & Sidransky, D. (2005) Promoter hypermethylation as an independent prog-nostic factor for relapse in patients with prostate cancer following radical prostatectomy. Clin. Cancer Res., 11, 8321-8325.
Ruzov, A., Tsenkina, Y., Serio, A., Dudnakova, T., Fletcher, J., Bai, Y., Chebotareva, T., Pells, S., Hannoun, Z., Sullivan, G., Chandran, S., Hay, D.C., Bradley, M., Wilmut, I. & De Sousa, P. (2011) Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian develop-ment. Cell Res., 21, 1332-1342.
Sarraf, S.A. & Stancheva, I. (2004) Methyl-CpG binding protein MBD1 couples histone methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell, 15, 595-605.
Sato, Y., Horii, A. & Fukushige, S. (2011) Microarray coupled with methyl-CpG targeted transcriptional activation (MeTA-array) identifies hypermethylated genes containing the strin-gent criteria of CpG islands at high frequency. Epigenetics, 6, 752-759.
Schuebel, K.E., Chen, W., Cope, L., Glockner, S.C., Suzuki, H., Yi, J.M., Chan, T.A., Neste, L.V., Crikinge, W.V., van den Bosch, S., van Engeland, M., Ting, A.H., Jair, K., Yu, W., Toyota, M., Imai, K., Ahuja, N., Herman, J.G. & Baylin, S.B. (2007) Comparing the DNA hypermethylome with gene mutations in human colorectal cancer. PLoS Genet., 3, 1709-1723.
Schumacher, A., Kapranov, P., Kaminsky, Z., Flanagan, J., Assadzadeh, A., Yau, P., Virtanen, C., Winegarden, N., Cheng, J., Gingeras, T. & Petronis, A. (2006) Microarray-based DNA methylation profiling: Technology and applications. Nucleic Acids Res., 34, 528-542.
Shibata, T., Kokubu, A., Miyamoto, M., Sasajima, Y. & Yamazaki, N. (2011) Mutant IDH1 confers an in vivo growth in a mela-noma cell line with BRAF mutation. Am. J. Pathol., 178, 1395-1402.
Shimizu, H., Horii, A., Sunamura, M., Motoi, F., Egawa, S., Unno, M. & Fukushige, S. (2011) Identification of epigenetically silenced genes in human pancreatic cancer by a novel method “microarray coupled with methyl-CpG targeted transcriptional activation” (MeTA-array). Biochem. Biophys. Res. Commun., 411, 162-167.
Sjöblom, T., Jones, S., Wood, L.D., Parsons, D.W., Lin, J., Barber, T.D., Mandelker, D., Leary, R.J., Ptak, J., Silliman, N., Szabo, S., Buckhaults, P., Farrell, C., Meeh, P., Markowitz, S.D., Willis, J., Dawson, D., Willson, J.K.V., Gazdar, A.F., Hartigan, J., Wu, L., Liu, C., Parmigiani, G., Park, B.H., Bachman, K.E., Papadopoulos, N., Vogelstein, B., Kinzler, K.W. & Velculescu, V.E. (2006) The consensus coding sequences of human breast and colorectal cancers. Science, 314, 268-274.
Soengas, M.S., Capodieci, P., Polsky, D., Mora, J., Esteller, M., Opitz-Araya, X., McCombie, R., Herman, J.G., Gerald, W.L., Lazebnik, Y.A., Cordón-Cardó, C. & Lowe, S.W. (2001) Inac-tivation of the apoptosis effector Apaf-1 in malignant mela-noma. Nature, 409, 207-211.
Sunami, E., Shinozaki, M., Higano, C.S., Wollman, R., Dorff, T.B., Tucker, S.J., Martinez, S.R., Mizuno, R., Singer, F.R. & Hoon, D.S. (2009) Multimarker circulating DNA assay for assessing blood of prostate cancer patients. Clin. Chem., 55, 559-567.
Suzuki, H., Gabrielson, E., Chen, W., Anbazhagan, R., van Engeland, M., Weijenberg, M.P., Herman, J.G. & Baylin, S.B. (2002) A genomic screen for genes upregulated by demethyl-ation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet., 31, 141-149.
Suzuki, M., Yamada, T., Kihara-Negishi, F., Sakurai, T., Hara, E., Tenen, D.G., Hozumi, N. & Oikawa, T. (2006) Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene, 25, 2477-2488.

DNA Methylation and Cancer 185
Suzuki, M.M. & Bird, A. (2008) DNA methylation landscape: provocative insights from epigenomics. Nat. Rev. Genet., 9, 465-476.
Tachibana, M., Matsumura, Y., Fukuda, M., Kimura, H. & Shinkai, Y. (2008) G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J., 27, 2681-2690.
Tahiliani, M., Koh, K.P., Shen, Y., Pastor, W.A., Bandukwala, H., Brudno, Y., Agarwal, S., Iyer, L.M., Liu, D.R., Aravind, L. & Rao, A. (2009) Conversion of 5-methylcytosine to 5-hydroxy-methylcytosine in mammalian DNA by MLL partner TET1. Science, 324, 930-935.
The Cancer Genome Atlas Research Network (2008) Comprehen-sive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455, 1061-1068.
Turker, M.S. (2002) Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene, 21, 5388-5393.
Van Neste, L., Herman, J.G., Otto, G., Bigley, J.W., Epstein, J.I. & Van Criekinge, W. (2012) The epigenetic promise for prostate cancer diagnosis. Prostate, 72, 1248-1261.
Viré, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., Morey, L., Van Eynde, A., Berrnard, D., Vanderwinden, J.M., Bollen, M., Esteller, M., Di Croce, L., de Launoit, Y. & Fuks, F. (2006) The Polycomb group protein EZH2 directly controls DNA methylation. Nature, 439, 871-874.
Warren, J.D., Xiong, W., Bunker, A.M., Vaughn, C.P., Furtado, L.V., Roberts, W.L., Fang, J.C., Samowitz, W.S. & Heichman, K.A. (2011) Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med., 9, 133.
Watt, F. & Molloy, P.L. (1988) Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev., 2, 1136-1143.
Weber, M., Davies, J.J., Wittig, D., Oakeley, E.J., Haase, M., Lam, W.L. & Schübeler, D. (2005) Chromosome-wide and promoter-specific analyses identify sites of differential DNA
methylation in normal and transformed human cells. Nat. Genet., 37, 853-862.
Weisenberger, D.J., Siegmund, K.D., Campan, M., Young, J., Long, T.I., Faasse, M.A., Kang, G.H., Widschwendter, M., Weener, D., Buchanan, D., Koh, H., Simms, L., Barker, M., Leggett, B., Levine, J., Kim, M., French, A.J., Thibodeau, S.N., Jass, J., Haile, R. & Laird, P.W. (2006) CpG island methylator phenotype underlies sporadic microsatellite insta-bility and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet., 38, 787-793.
Widschwendter, M., Fiegl, H., Egle, D., Mueller-Holzner, E., Spizzo, G., Marth, C., Weisenberger, D.J., Campan, M., Young, J., Jacobs, I. & Laird, P.W. (2007) Epigenetic stem cell signature in cancer. Nat. Genet., 39, 157-158.
Wood, L.D., Parsons, D.W., Jones, S., Lin, J., Sjöblom, T., Leary, R.J., Shen, D., Boca, S.M., Barber, T., Ptak, J., Silliman, N., Szabo, S., Dezso, Z., Ustyanksky, V., Nikolskaya, T., Nikolsky, Y., Karchin, R., Wilson, P.A., Kaminker, J.S., Zhang, Z., Croshaw, R., Willis, J., Dawson, D., Shipitsin, M., Willson, J.K., Sukumar, S., Polyak, K., Park, B.H., Pethiyagoda, C.L., Pant, P.V., Ballinger, D.G., Sparks, A.B., Hartigan, J., Smith, D.R., Suh, E., Papadopoulos, N., Buckhaults, P., Markowitz, S.D., Parmigiani, G., Kinzler, K.W., Velculescu, V.E. & Vogelstein, B. (2007) The genomic landscapes of human breast and colorectal cancers. Science, 318, 1108-1113.
Xu, G.L., Bestor, T.H., Bourc’his, D., Hsieh, C.L., Tommerup, N., Bugge, M., Hulten, M., Qu, X., Russo, J.J. & Viegas-Pequignot, E. (1999) Chromosome instability and immunode-ficiency syndrome caused by mutations in a DNA methyltrans-ferase gene. Nature, 402, 187-191.
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S.H., Ito, S., Yang, C., Wang, P., Xiao, M.T., Liu, L.X., Jiang, W.Q., Liu, J., Zhang, J.Y., Wang, B., Frye, S., Zhang, Y., Xu, Y.H., Lei, Q.Y., Guan, K.L., Zhao, S.M. & Xiong, Y. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-keto gluta-rate-dependent dioxygenases. Cancer Cell, 19, 17-30.