diphtheria toxin: mode of action anddiphtheria toxin has been purified, crystal-lized, andpartially...
TRANSCRIPT

BACTrIOLOGICAL REvIwS, Mar. 1975, p. 54-85Copyright 0 1975 American Society for Microbiology
Vol. 39, No. 1Printed in U.SA.
Diphtheria Toxin: Mode of Action and StructureR. JOHN COLLIER
Department of Bacteriology and the Molecular Biology Institute, University of California, Los Angeles,California 90024
INTRODUCTION.... 54OUTLINE OF THEPROBLEM. 54INHIBITION OFPROTEIN SYNTHESIS ..56
Studies in Cell Culture ..56Studies in Cell-Free Systems ..58
ADP-RIBOSYLATION OF EF-2 ..59Identification of the Reaction ..59ADPR is Covalently Attached . ..60The Role of Toxin is Catalytic ..61Substrate Specificity ..61Reversibility and Thermodynamics ..62Factors Affecting the Rate of the Forward Reaction 62
STRUCTURE-ACTIVITY RELATIONSHIPS IN THE TOXIN 63Whole Toxin is a Proenzyme ..63Toxicity of the Various Fragments andForms. 65Properties of Fragment A ..65Variations Among Toxin Preparations ..66Nontoxic, Cross-Reacting Forms of Toxin ..67Mechanism of the ADP-Ribosylation Reaction ..68Pyridine Nucleotide Binding and ADP-Ribosylation Activity of Whole Toxin ... 70
IS THE ADP-RIBOSYLATION REACTION RESPONSIBLE FOR TOXICITY? ... 71EVENTSPRECEDING THE ADP-RIBOSYLATION REACTION IN CELLS.. 72Attachment to Specific Receptors.72Entry and Activation Processes.73Kinetics of Entry and Turnover.74
IMMUNOLOGY.75Avidity Correlates with Antibody Against Fragment B.75Immunogenicity and the Mechanism of Toxoiding.75
BIOLOGICAL FUNCTION AND ORIGIN OF DIPHTHERIATOXIN.77Biological Function.77Origin... .77
SUMMARY AND CONCLUDING REMARKS.................................. 78LITERATURE CITED............................... 80
INTRODUCTIONThe wealth of biochemical knowledge accu-
mulated in recent years has permitted fruitfulstudy of the molecular mechanisms of pathoge-nicity in a wide variety of diseases. In bacterialdiseases many aspects of the host-parasite in-teraction still remain snarled in complexity, butcertain factors have become amenable to study.Perhaps the most notable advances have oc-curred in diseases, such as diphtheria andcholera, in which a single, potent exotoxin isresponsible for the major symptoms.
In diphtheria the existence of such a toxin hasbeen known for almost a century, and its actionhas been studied in a variety of systems almostfrom the time of its discovery. Only after thebasic framework of knowledge of protein syn-thesis was established in the 1950s, however,did such studies begin to yield a consistent pic-
ture. At the present time there is good reasonto believe that the toxicity of the molecule isdue to inhibition of protein synthesis in the hu-man host, and certain aspects of the biochem-istry of its action are known in detail.
OUTLINE OF THE PROBLEMThe causative organism of diphtheria, Cor-
ynebacterium diphtheriae, is normally foundonly in the upper respiratory tract of men,cattle, and horses. Infections in man may re-main subclinical, or the bacillus may proliferateextensively upon and within the superficialepithelial layers of the pharynx, nasopharynx,or upper trachea. Such proliferation commonlyresults in the formation of a leather-likepseudomembrane, which is a characteristic di-agnostic feature of the disease. Less commonly,
54
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
infections of wounds or the skin or mucousmembranes occur at peripheral sites. However,regardless of the location of the primary infec-tions, there is little invasion of underlyingtissues, and the bacillus is rarely found insignificant numbers in the blood or internalorgans.The symptoms, in contrast to the causative
agent, are not localized (2). Death can resultfrom suffocation caused by occlusion of the airpassage by the pseudomembrane, but moreoften it is attributable to damage to internalorgans, distant from the site of infection. Insevere or fatal cases, tissue necrosis at gross ormicroscopic levels and various physiologicalchanges are observed in many organs, includingthe heart, kidneys, liver, lungs, nervous system,and others. Cardiac failure is frequently cited asthe immediate cause of death (28), but physio-logical alterations in other organs certainlycontribute and may often be the determiningfactor.
Loeffler observed in 1884 that injections ofexperimental animals with the diphtheria bacil-lus produced localized infections with wide-spread tissue damage similar to that in theclinical disease (101). He suggested that thedamage at distant sites might be caused by atoxic substance produced by the pathogen andtransported throughout the body. In 1888 Rouxand Yersin demonstrated that culture filtratesof the diphtheria bacillus caused death of exper-imental animals with a similar pattern of tissuedamage (141). Some five decades later the toxicproduct was obtained in sufficiently pure formto ascertain that it was protein in nature (49,120) and it is now clear that it is a single protein(33, 46, 47, 58, 160).That the toxin is indeed of major importance
in clinical diphtheria is shown not only by thesimilarity of the symptoms produced by puri-fied toxin in experimental animals, but also bythe fact that immunity to the toxin providesprotection against severe symptoms of the dis-ease. In recent years mass immunizationagainst diphtheria toxoid (toxin detoxified bytreatment with formaldehyde) has led to aremarkable decrease in the incidence of clinicaldiphtheria in many countries (24) and mightresult in eradication if a sufficiently high per-centage of the population were immunized.
Despite the proven role of the toxin, toxige-nicity in C. diphtheriae is not synonymous withpathogenicity (3, 5, 73). Nontoxigenic strainshave been isolated, and certain of these not onlysurvive for long periods in the upper respiratorytract, but also are able to produce mild tomoderately severe infections (5, 50). Although
the more dramatic symptoms seen in severeinfections by toxigenic strains are lacking, pseu-domembrane formation does occur. Further-more, merely the capacity to produce the toxindoes not make a strain pathogenic. The PW-8strain, which is used throughout the world toproduce high titers of toxin for preparing toxoidor for research, appears to be relatively aviru-lent (97). The pathogenicity of C. diphtheriae istherefore complex. Toxin formation can dra-matically increase the severity of an infection,but is neither necessary nor sufficient for sur-vival or pathogenicity of the bacillus.
In 1951 Freeman made the remarkable dis-covery that toxigenicity in C. diphtheriae iscorrelated with infection by certain temperatebacteriophage (53). This observation was subse-quently confirmed and extended by Barksdale,Groman, and others (4-7, 75-78). Lysogeniza-tion of a nontoxigenic strain with phage carry-ing the tox+ gene converted it to toxigenicity,and curing of phage infection yielded a nontoxi-genic strain. The implication of these resultsthat the structural gene for diphtheria toxinmight reside on a phage genome now seemsvirtually beyond doubt (63, 81, 100, 115, 158).Toxin is synthesized in a cell-free system fromEscherichia coli programmed with DNA fromcorynephage fl, carrying the tox+ gene (106),and other work with mutants of the same phagealso implies that the phage genome codes for thetoxin (158). The toxin is therefore, by definition,a phage protein.Diphtheria toxin has been purified, crystal-
lized, and partially characterized in many labo-ratories. It is an acidic, globular protein (pI14.1) with a molecular weight most recentlyestimated at 62,000 to 63,000. As far as isknown, it contains no unusual amino acids, nononprotein moieties, and no other gross featuresto distinguish it from a wide variety of othernontoxic proteins (91, 134). It is not as potent ona weight or molar basis as certain other bacte-rial toxins, such as those of the botulinum andtetanus bacilli, but its toxicity is remarkablenonetheless. About 25 ng of the toxin injectedsubcutaneously into a 250-g guinea pig is suffi-cient to cause death in 4 to 5 days (therebydefining a standard minimum lethal dose[MLD]), and less than one-thousandth of thisamount injected intradermally into the shavedback of a rabbit or guinea pig produces a visiblenecrotic reaction. Many animals, includingman, monkeys, rabbits, and various fowl, areabout as sensitive as the guinea pig on a bodyweight basis, and it may be calculated that afew micrograms is sufficient to cause death ofan unimmunized adult human. However, with
VOL. 39, 1975 55
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
rats and mice, doses about 3 orders of magni-tude greater per unit body weight are requiredfor comparable responses. Inasmuch as thetoxin is unstable at acid pH, it has no effectwhen administered orally.A guinea pig injected with a lethal dose of the
toxin remains normal in appearance and behav-ior for several hours and then gradually beginsto show signs of weakness and lethargy, whichprogress until the animal goes into shock anddies a few days later. Even when a massiveamount of the toxin is administered (severalthousand lethal doses), the animal shows nosymptoms immediately and never dies beforeabout 10 h. Thus, a period of several hours todays is required for the toxin's effects to bemanifested. Its ultimate effect becomes irrever-sible much sooner, however. With a dose of 10MLD, for example, even a large excess ofantitoxin administered 1 h later will not preventdeath. It is probable that the period of reversi-bility corresponds to the time required forabsorption of a lethal dose by the cells.The mechanism by which the toxin causes
death has been studied extensively in wholeanimals. As indicated above, gross and micro-scopic tissue damage is found in a variety ofinternal organs (the heart, suprarenals, kidneys,liver, pancreas, diaphragm, nervous tissue, andothers). The specific pattern varies somewhatfrom species to species, but in all there arewidespread morphological changes (2). Simi-larly, and not surprisingly, multiple physiologi-cal changes occur. For example, toxin-treatedanimals show decreased levels of muscle phos-phocreatine (129), reduced capacity to metabo-lize lactic acid (45) and to synthesize carbohy-drates (40), increased resistance to insulin (38),reduced cardiac capacity (164), increased levelsof potassium in the blood (164), and so forth. Itis conceivable that such diverse effects mightfollow from a specific action of the toxin on aparticular organ or cell type. However, it waslong suspected that the toxin may be relativelynonspecific.Support for this notion has come from studies
in cell culture (see reference 146 for a review).The toxin has been shown to be lethal forprimary and continuous cell cultures from avariety of animals (54, 99, 130). The firstmorphological changes in monolayer cultures ofHeLa cells, for example, may be detectedwithin several hours (3 h in one study; 106) afteraddition of high concentrations of toxin. In-creased granularity is observed, followed byrounding up of the cells and release from theglass surface (Fig. 1). Lysis and disintegration
do not seem to occur for at least some hoursafter release, however (109, 146).The relative sensitivities to toxin of cell
cultures from humans and various animalsseem to correspond approximately to those ofthe parent animals (54, 145). Thus cells fromhumans, monkeys, or chickens, for example, arekilled by relatively low concentrations of toxin,whereas those from mice and rats require con-centrations orders of magnitude higher for simi-lar effects. Cultures from various organs withina given animal species are similar in sensitivity.Despite the complications of de-differentiationand selection in culture, morphologically dis-tinct cell types, including embryonic heart,fibroblastic, and epithelial types show no obvi-ous differences in sensitivity to the toxin (19,54). This correlates with the apparent lack ofstrong tissue specificity observed in vivo andimplies that the toxin acts on a process commonto many if not all cell types.
INHIBITION OF PROTEIN SYNTHESIS
Studies in Cell CultureThe first indication of an effect of diphtheria
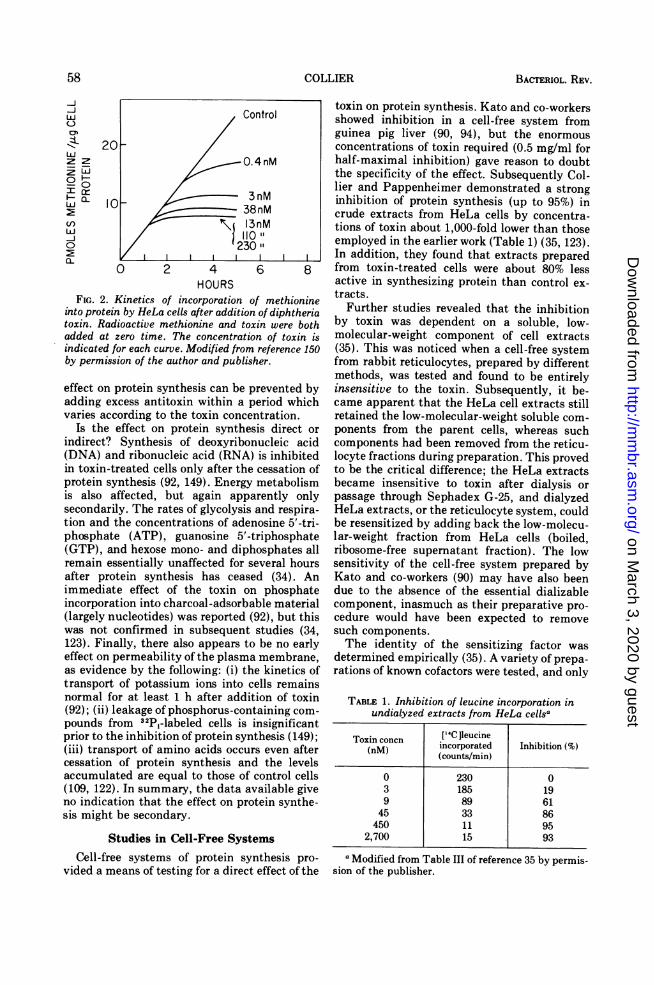
toxin on protein synthesis was observed byStrauss and Hendee in 1959 (150). While study-ing the effect of the toxin on respiration in HeLacells (no significant effect was found), they ob-served that accumulation of protein was reducedover the 12-h period of the experiment. Subse-quently, they characterized the effect of toxinon the incorporation of [35S ]methionine intoprotein in these cells. After the toxin was addedto a final concentration of about 10 nM, aminoacid incorporation proceeded normally for 1 to1.5 h and then rapidly came to a halt (Fig. 2).With lower toxin concentrations, the lag periodwas longer and the decline in the rate of proteinsynthesis more gradual, but higher concentra-tions failed to shorten the lag. Thus, about 10nM toxisn was saturating for this system. Inanother study slightly lower concentrations (3to 5 nM) were required for saturation (109).(Concentrations of toxin are frequently ex-pressed in terms of flocculating units [Lf] orMLD per unit volume. One Lf of toxin isequivalent to about 2.5 Atg of protein [8, 34 ]. Thespecific toxicity varies from preparation topreparation, but figures of 40 to 60 MLD/Lf areoften reported.)The presence of a lag seems to be invariable.
However, there is a marked variation in theminimal duration, from as little as 15 min to atleast 3 h, depending on the strain of cells andculture conditions employed (109, 123). The
56 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
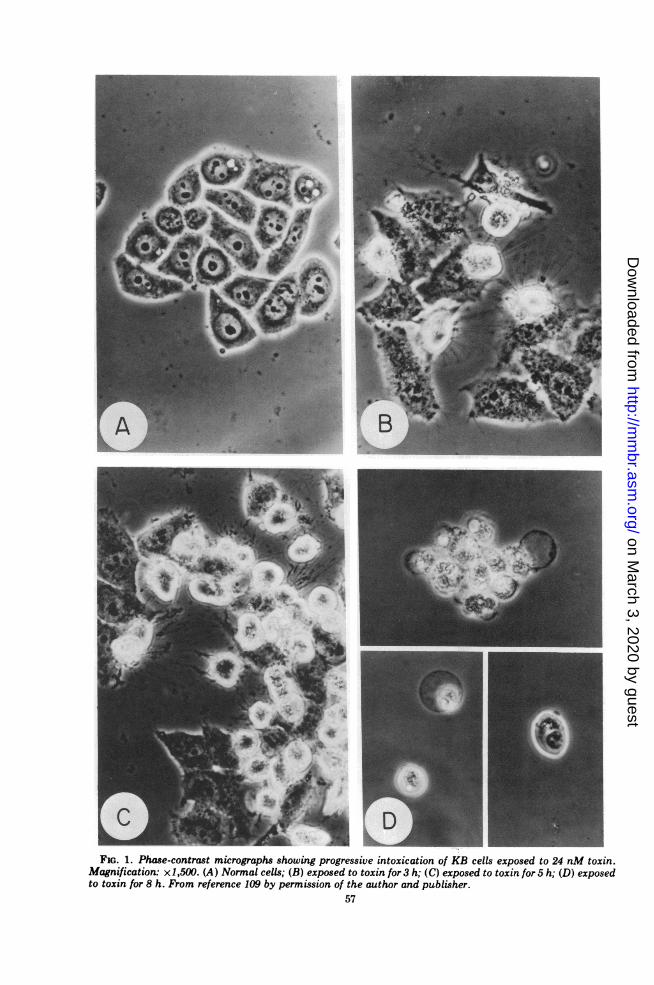
S-FIG. 1. Phase-contrast micrographs showing progressive intoxication of KB cells exposed to 24 nM toxin.
Magnification: x1,500. (A) Normal cells; (B) exposed to toxin for 3 h; (C) exposed to toxin for 5 h; (D) exposedto toxin for 8 h. From reference 109 by permission of the author and publisher.
57
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
BACTERIOL. REV.
-j
LU 1 0c3
C)~~~~~~~C20
LU ~ ~ ~ OR
00~~~~~~~~~0
01-~~~~~~~~~1
jI) I I -o 23C
0 2 ~40 2 ~HOURS
FIG. 2. Kinetics of incorporation ainto protein by HeLa cells after additiontoxin. Radioactive methionine and totadded at zero time. The concentratioindicated for each curve. Modified fromby permission of the author and publis)
effect on protein synthesis can be padding excess antitoxin within a pvaries according to the toxin conce
Is the effect on protein synthesindirect? Synthesis of deoxyribor(DNA) and ribonucleic acid (RNA)in toxin-treated cells only after theprotein synthesis (92, 149). Energyis also affected, but again appesecondarily. The rates of glycolysistion and the concentrations of aderphosphate (ATP), guanosine 5'-t(GTP), and hexose mono- and dipiremain essentially unaffected for safter protein synthesis has ceasEimmediate effect of the toxin orincorporation into charcoal-adsorba(largely nucleotides) was reported (was not confirmed in subsequent123). Finally, there also appears toeffect on permeability of the plasmaas evidence by the following: (i) thtransport of potassium ions into cnormal for at least 1 h after addit(92); (ii) leakage of phosphorus-conpounds from 32P1-labeled cells is iprior to the inhibition of protein syn(iii) transport of amino acids occurcessation of protein synthesis an(accumulated are equal to those of(109, 122). In summary, the data asno indication that the effect on prosis might be secondary.
toxin on protein synthesis. Kato and co-workersIntrol showed inhibition in a cell-free system fromguinea pig liver (90, 94), but the enormousconcentrations of toxin required (0.5 mg/ml for
4nM half-maximal inhibition) gave reason to doubtthe specificity of the effect. Subsequently Col-lier and Pappenheimer demonstrated a strong
3nM inhibition of protein synthesis (up to 95%) in3nM crude extracts from HeLa cells by concentra-fnM tions of toxin about 1,000-fold lower than those
employed in the earlier work (Table 1) (35, 123).E |ZIn addition, they found that extracts prepared6 8 from toxin-treated cells were about 80% less
active in synthesizing protein than control ex-
.fmethionine tracts.
of diphtheria Further studies revealed that the inhibitiontin were both by toxin was dependent on a soluble, low-in of toxin is molecular-weight component of cell extractsreference 150 (35). This was noticed when a cell-free system
her. from rabbit reticulocytes, prepared by differentmethods, was tested and found to be entirely
Irevented by insensitive to the toxin. Subsequently, it be-eriod which came apparent that the HeLa cell extracts stillntration. retained the low-molecular-weight soluble com-sis direct or ponents from the parent cells, whereas suchnucleic acid components had been removed from the reticu-is inhibited locyte fractions during preparation. This provedcessation of to be the critical difference; the HeLa extractsmetabolism became insensitive to toxin after dialysis orirently only passage through Sephadex G-25, and dialyzedand respira- HeLa extracts, or the reticulocyte system, couldnosine 5'-tri- be resensitized by adding back the low-molecu-triphosphate lar-weight fraction from HeLa cells (boiled,iosphates all ribosome-free supernatant fraction). The loweveral hours sensitivity of the cell-free system prepared byed (34). An Kato and co-workers (90) may have also beeni phosphate due to the absence of the essential dializableble material component, inasmuch as their preparative pro-92), but this cedure would have been expected to removestudies (34, such components.be no early The identity of the sensitizing factor was
l membrane, determined empirically (35). A variety of prepa-e kinetics of rations of known cofactors were tested, and onlyells remainstion of toxin TABLE 1. Inhibition of leucine incorporation intaining com- undialyzed extracts from HeLa cellsains gnificantthesis (149);rs even afterd the levelscontrol cellsvailable givestein synthe-
Studies in Cell-Free Systems
Toxin concn [4CCleucine(nM) incorporated Inhibition (%)(counts/min)
0 230 03 185 199 89 61
45 33 86450 11 95
2,700 15 93
Cell-free systems of protein synthesis pro- a'Modified from Table III of reference 35 by permis-vided a means of testing for a direct effect of the sion of the publisher.
58 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
those containing oxidized nicotinamide adeninedinucleotide (NAD+) were found to sensitizedialyzed systems to the toxin. It was then shownthat the factor chromatographed with NAD+ on
Dowex-1 ion-exchange resin, and that undi-alyzed HeLa extracts became insensitive totoxin after treatment with streptococcal NAD-ase (Table 2). Goor and Pappenheimer latershowed that the oxidized but not the reducedform of the cofactor was active (71).Which component of protein synthesis is
inhibited by toxin and NAD+? Initial experi-ments showed that the transfer of labeled aminoacids into polypeptide chains from preformed[14C ]aminoacyl transfer RNA (tRNA) was
strongly inhibited, whereas the synthesis ofaminoacyl-tRNA was not (29). Thus, the toxin-sensitive component was involved in the process
of amino acid polymerization on the ribosome.Tests of the supernatant and ribosomal frac-tions then localized the effect to the former ofthese (29). Incubation of ribosomes or poly-somes with strongly inhibitory levels of toxinand NAD+ had no effect on their integrity or
activity, but the supernatant fraction was inac-tivated by a similar treatment.
At the time that these experiments wereperformed, the only components of the superna-tant fraction known to be required for aminoacid polymerization, besides GTP and certaininorganic ions, were two proteins now termedelongation factors 1 and 2 (EF-1 and EF-2;reference 27). (Earlier terms used for the same
factors were transfer factors 1 and 2, andaminoacyl transferases 1 and 2.) EF-1 is re-
quired for attachment of aminoacyl-tRNA toribosomes and EF-2 (molecular weight about100,000; reference 37) is required for the translo-cation process. The latter process involvestranslocation of peptidyl-tRNA from the accep-tor to the donor site on ribosomes and move-ment of mRNA by one nucleotide triplet aftereach round of peptide bond formation. GTP isrequired and is hydrolyzed to guanosine 5'-diphosphate (GDP) and inorganic orthophos-phate during both processes.As shown in Fig. 3, after treatment of reticu-
locyte supernatant fraction with toxin plusNAD+ and separation of the elongation factorson Sephadex G-100, EF-2 shows a markedlyreduced activity whereas EF-1 remains fullyactive (29). No effects on other components ofthe cell-free system were found, and it wasconcluded that EF-2 was probably the target ofaction of the toxin. From studies of extractsfrom toxin-treated HeLa cells, Goor and Pap-penheimer also concluded that it was one of theelongation factors which was inactivated (70).
TABLE 2. Effect of nicotinamide adenine nucleotidase(NADase) on the toxin sensitivity of undialyzed
extracts from HeLa cellSa
I14C Ileucineincorporated(counts/min) Inhibition
HeLa extractToxin (%
Control (360nM)
Untreated 767 50 93Pretreated with NADase 761 720 5Pretreated with NADase; 802 320 60250ug NAD+ added attime of initiation of incor-poration reaction
a Modified from Table IX of reference 35 by permission ofthe publisher.
bExtracts were pretreated with 900 units of NADase per mlfor 1 h at 0 C before addition to reaction mixtures.
In addition, their results suggested that thetoxin-sensitive factor was accessible only whenfree in solution. A fraction of both elongationfactors within cells is bound to ribosomes, inwhich form EF-2 is protected from inactivationby toxin and NAD+ (59, 70, 144).
ADP-RIBOSYLATION OF EF-2
Identification of the Reaction
An important clue to the mechanism ofinactivation of EF-2 came from the effect ofnicotinamide, first observed by Goor, Pappen-heimer, and Ames (72). Nicotinamide not onlyinhibited the inactivation, but at high concen-trations (on the order of 30 mM) also promotedreactivation of the inactivated factor.This finding and other data on the kinetics of
inactivation were originally interpreted in termsof a model involving formation of a ternarycomplex between the toxin, EF-2, and NAD+. Itwas supposed that the complexed EF-2 wasinactive and that nicotinamide dissociated thecomplex by competing with NAD+. The factthat a stoichiometrically equivalent amount oftoxin would be required for inactivation of EF-2detracted from the model, in view of estimatesthat only a few molecules of toxin might besufficient to kill a cell. Later Gill et al. wereunable to detect the predicted ternary complex(60). (There is evidence, however, that such acomplex may exist under certain conditions[52].)A more plausible, catalytic mechanism of
inactivation of EF-2 was demonstrated byHonjo and co-workers in 1968 (83-85). A keyexperiment involved incubation of mixtures oftoxin and purified EF-2 with each of a number
59VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

COLLIER
,, 200- Control
'EF-
I-
EF-2
z I',< 100 I0
C)
z
z
z
-lJ I l\
I _o~~~I_
-J100
L
EFFLUENT VOLUME- >FIG. 3. Fractionation of normal and toxin-treated
ribosome-free supernatant fraction from rabbit reticu-locytes on Sephadex G-100. Upper frame, controlsupernatant fraction, incubated with NAD+ in bufferbefore loading onto the column. Lower frame, super-natant fraction incubated with toxin and NAD+before chromatography. Modified from reference 29by permission of the publisher.
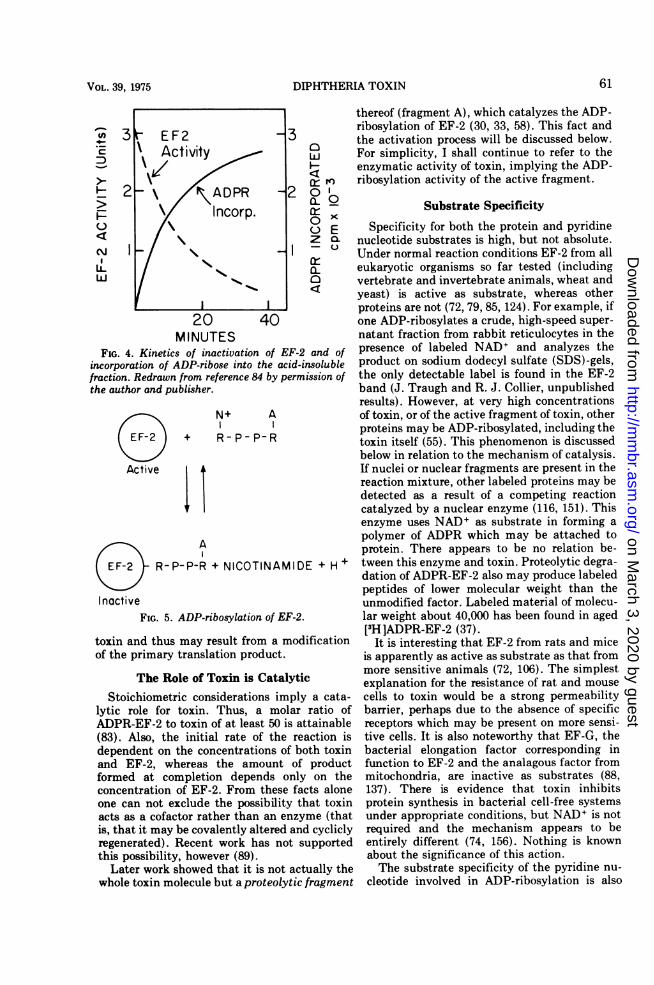
of preparations of radioactive NAD+, labeled invarious parts of the molecule. Stoichiometri-cally equivalent amounts of label were incorpo-rated into the protein fraction (trichloroaceticacid-precipitable material) from all parts ofNAD+ except the nicotinamide moiety (Table3). Label in nicotinamide was released free insolution. The protein-bound label was shown tobe stably attached to EF-2; none was foundassociated with toxin in mixtures fractionatedon hydroxylapatite. Also, the kinetics of attach-ment of label coincided with the time-course ofinactivation of EF-2 (Fig. 4).From these and other results, it was suggested
that the toxin inactivated EF-2 by promotingattachment of the adenosine diphosphate riboseportion (ADPR) of NAD+ (Fig. 5) (85):EF-2 + NAD+ = ADPR-EF-2 + nicotinamide + H+
Gill et al. independently obtained less directevidence for this reaction (60), and subsequentwork in many laboratories leaves no doubt thatthis mechanism explains the inactivation ofEF-2. The reaction also provides a simple expla-nation for the reactivation by nicotinamide.Inasmuch as nicotinamide is a reaction product,high concentrations should shift the equilib-rium position toward the left, thus reactivatingEF-2.
Formally, the reaction should be termedNAD+ :EF-2 ADPR-transfer, but for simplicity Ishall refer to it as ADP-ribosylation or ADPR-transfer. Certain important features of the reac-tion are discussed below. Other aspects will beconsidered in a later section.
ADPR is Covalently Attached
The chemical stability of the linkage betweenADPR and EF-2 and the reversibility of thereaction imply that ADPR is covalently at-tached (85). The linkage resists treatment at95 Cfor6minin0.1 NHClorO.1NNaOH, andis stable for longer periods in 5% trichloroaceticacid. Treatment with 1 N NaOH or 1 N HCl forsimilar periods causes partial hydrolysis, how-ever. The reversibility of the reaction, as dis-cussed below, implies that the ADPR-EF-2linkage conserves a significant percentage of thebond energy of the original nicotinamide-riboselinkage of NAD+.There is good evidence that EF-2 contains
only a single attachment sight for ADPR (131,140), but the nature of the linkage betweenADPR and EF-2 is not yet fully characterized.ADPR must be linked through its nicotinamidemononucleotide (NMN) ribose moiety, inas-much as ribose 5'-phosphate remains attachedafter removal of the adenosine 5'-monophos-phate (AMP) portion with snake venom phos-phodiesterase (82, 83). However, the side chainof EF-2 to which ADPR is attached remainsuncertain. The amino acid sequence of a 15-resi-due tryptic peptide surrounding the residue hasbeen determined as Phe-Asp-Val-His-Asp-Val-Thr-Leu-His-Ala-Asp-Ala-Ile-X-Arg, where Xrepresents the attachment site (138, 139). Resi-due X is weakly basic and does not correspondto any amino acid commonly occurring in pro-teins. It is present even prior to contact with
TABLE 3. Incorporation of label from NAD+ intoacid-insoluble fractiona
NAD+ employed IncorporationNAD~(pmol)
NAD+-(adenine)-14C 51.2NAD+-(adenosine)-3H 50.6NAD+-(both phosphates)-32P 50.0NAD+-(ribose in NMN)- 14C 50.0NAD+-(nicotinamide)-14C 0.3
a Reaction mixtures contained tris(hydroxy-methyl)aminomethane buffer, EF-2 from rat liver, anddiphtheria toxin, in addition to the radioactive NAD+indicated. After incubation for 10 min at 37 C, thetrichloroacetic acid-insoluble material was collectedon filters and counted. From reference 84, by permis-sion of the authors and publisher.
BACTERIOL. REV.60
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
DIPHTHERIA TOXIN
C)O
o x0) 1
a- LZ CL
0~_ Q
C]
MINUTESFIG. 4. Kinetics of inactivation of EF-2 and of
incorporation of ADP-ribose into the acid-insolublefraction. Redrawn from reference 84 by permission ofthe author and publisher.
Active
N+ A
+ R-P-P-R
A
R-P-P-R + NICOTINAMIDE + H+
InactiveFIG. 5. ADP-ribosylation of EF-2.
toxin and thus may result from a modificationof the primary translation product.
The Role of Toxin is CatalyticStoichiometric considerations imply a cata-
lytic role for toxin. Thus, a molar ratio ofADPR-EF-2 to toxin of at least 50 is attainable(83). Also, the initial rate of the reaction isdependent on the concentrations of both toxinand EF-2, whereas the amount of productformed at completion depends only on theconcentration of EF-2. From these facts aloneone can not exclude the possibility that toxinacts as a cofactor rather than an enzyme (thatis, that it may be covalently altered and cycliclyregenerated). Recent work has not supportedthis possibility, however (89).
Later work showed that it is not actually thewhole toxin molecule but a proteolytic fragment
thereof (fragment A), which catalyzes the ADP-ribosylation of EF-2 (30, 33, 58). This fact andthe activation process will be discussed below.For simplicity, I shall continue to refer to theenzymatic activity of toxin, implying the ADP-ribosylation activity of the active fragment.
Substrate Specificity
Specificity for both the protein and pyridinenucleotide substrates is high, but not absolute.Under normal reaction conditions EF-2 from alleukaryotic organisms so far tested (includingvertebrate and invertebrate animals, wheat andyeast) is active as substrate, whereas otherproteins are not (72, 79, 85, 124). For example, ifone ADP-ribosylates a crude, high-speed super-natant fraction from rabbit reticulocytes in thepresence of labeled NAD+ and analyzes theproduct on sodium dodecyl sulfate (SDS)-gels,the only detectable label is found in the EF-2band (J. Traugh and R. J. Collier, unpublishedresults). However, at very high concentrationsof toxin, or of the active fragment of toxin, otherproteins may be ADP-ribosylated, including thetoxin itself (55). This phenomenon is discussedbelow in relation to the mechanism of catalysis.If nuclei or nuclear fragments are present in thereaction mixture, other labeled proteins may bedetected as a result of a competing reactioncatalyzed by a nuclear enzyme (116, 151). Thisenzyme uses NAD+ as substrate in forming apolymer of ADPR which may be attached toprotein. There appears to be no relation be-tween this enzyme and toxin. Proteolytic degra-dation of ADPR-EF-2 also may produce labeledpeptides of lower molecular weight than theunmodified factor. Labeled material of molecu-lar weight about 40,000 has been found in aged[3H]ADPR-EF-2 (37).
It is interesting that EF-2 from rats and miceis apparently as active as substrate as that frommore sensitive animals (72, 106). The simplestexplanation for the resistance of rat and mousecells to toxin would be a strong permeabilitybarrier, perhaps due to the absence of specificreceptors which may be present on more sensi-tive cells. It is also noteworthy that EF-G, thebacterial elongation factor corresponding infunction to EF-2 and the analagous factor frommitochondria, are inactive as substrates (88,137). There is evidence that toxin inhibitsprotein synthesis in bacterial cell-free systemsunder appropriate conditions, but NAD+ is notrequired and the mechanism appears to beentirely different (74, 156). Nothing is knownabout the significance of this action.The substrate specificity of the pyridine nu-
cleotide involved in ADP-ribosylation is also
-
NI-
LLw
61VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
high, and it is probable that only NAD+ isutilized in vivo. Of other pyridine nucleotidestested only certain unnatural analogues ofNAD+ with minor structural changes can sub-stitute (71, 85, 89). Thionicotinamide-AD+ hasa Km (5 to 10 mM) similar to that of NAD+ (8mM), whereas acetylpyridine-AD+ and de-amino-NAD+ have Km values about 10-foldhigher. Oxidized NAD phosphate (NADP+),reduced NAD (NADH), reduced NADP(NADPH), and NMN are all inactive as sub-strates.
Reversibility and ThermodynamicsThe equilibrium position of the reaction lies
far to the right under normal conditions, butmay be shifted toward the left by adding highconcentrations of nicotinamide (72, 82, 85, 132),as indicated above. Honjo and co-workers dem-onstrated removal of attached ADPR and reac-tivation of EF-2 by incubation of ADPR-EF-2with nicotinamide and toxin, and in additonhave recovered an equivalent amount of au-thentic NAD+ generated by the reversal (85). Aspredicted by the reaction equation, the equilib-rium is further shifted toward the left (and thereverse reaction accelerated) by lowering the pHsomewhat. The pH optimum of the reversereaction is 5.3. Attempts to rescue toxin-treatedcells with nicotinamide have failed (72, 105).From the equilibrium position in the presence
of various concentrations of nicotinamide theequilibrium constant has been determined (85):
K (ADPR-EF-2)(nicotinamide) (H+)-63 10-4M(EF-2) (NAD+)
From this value one may calculate the standardfree energy change (AF°') as -5.2 kcal per molat pH 7 and 25 C (85). Inasmuch as the freeenergy of hydrolysis of the nicotinamide-riboselinkage of NAD+ is known to be about -9.2 kcalper mol, the value for hydrolysis of the ADPR-EF-2 linkage must be about -4.0 kcal per molat pH 7 and 25 C.The major implication of these findings with
respect to the reaction in vivo is that theinactivation of EF-2 at equilibrium should bevirtually complete under physiological condi-tions. At pH 7 and a concentration of 10,MNADI (certainly an underestimate), less than0.01% of the free EF-2 would be expected toremain in the unmodified, active form. This isconsistent with the virtually complete inhibi-tion of protein synthesis observed in tissueculture.
Factors Affecting the Rate of the ForwardReaction
Reversal of the reaction probably does notoccur to a significant extent in vivo, and thusthe rate of the forward reaction should primarilydetermine the rate of inhibition of proteinsynthesis. The forward reaction is affected by anumber of variables, but uncertainties in manyof these permit little confidence in calculationsof the rate of inactivation of EF-2 in vivo atgiven concentrations of toxin or active toxinfragment.The transfer of ADPR to EF-2 has a pH
optimum of 8.2 to 8.5, but is rapid even atpH 7 (33, 69, 73, 84). In our experience thereare no requirements for specific ions (33). Aslight stimulation by Mg2+ has been reported(69), but we have found no inhibition byethylenediaminetetraacetic acid (EDTA), ex-cept for that due to its contribution to ionicstrength. The reaction is sensitive to ionicstrength, being inhibited 25 to 40% by 30 mMNaCl, KCl, or NH.Cl (33). MgCl2 and magne-sium acetate are each about 10-fold more inhib-itory on a molar basis.The finding that thiols stimulate the reaction
(30, 36) appears to be due solely to theircapacity to promote reductive activation of thetoxin (33, 60) as described below. Thiols are alsorequired for protection of EF-2 activity in pro-tein synthesis (111), but N-ethyl-maleimidetreatment does not block its activity as sub-strate for ADP-ribosylation (60). However, an-other sulfhydryl reagent, p-hydroxymercuriben-zoate, does inactivate EF-2 as substrate (140),suggesting that certain critical sulfhydryls maybe reactive with this but not the former reagent.
Certain compounds related to NAD+ inhibitthe reaction by competing for the NAD+ bind-ing site on the active toxin fragment. The mostpotent inhibitor of this type is adenine (74, 89),but its Ki (40,uM) is still well above the Km ofNAD+ (8 ,M). Adenosine is less effective thanadenine by at least an order of magnitude, andAMP, ADP, ATP, ADPR, and NADP+ are lessinhibitory by two orders of magnitude (69, 89).Nicotinamide (K, 0.2 mM) also inhibits thereaction weakly.NADH has been found to inhibit weakly (Ki
30,uM) if at all in assays with EF-2 from rabbitreticulocytes (89), although an early reportsuggested a strong inhibition (Ki 0.2 AM) withrat liver EF-2 (69). The results in the reticulo-cyte system are supported by the finding thatNADH binds only weakly to the active fragmentof toxin (89). Inasmuch as the ratio of the
62 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
DIPHTHERIA TOXIN
intracellular concentration of NAD+ to its Km islikely to be >1, whereas the ratio for all theinhibitors listed is probably < 1, such inhibitorsprobably affect the reaction very little in vivo.Other, unnatural analogues of NAD+ inhibitthe reaction (89), but can be of no physiologicalsignificance.The rate of ADP-ribosylation is also affected
by interactions of EF-2. EF-2 binds to ribo-somes (to the 60S subunit [154]) in the course ofthe translocation process and is apparentlyinaccessible to ADP-ribosylation while bound(59, 70, 96, 133). Gill and Dinius have estimatedthat over 75% of EF-2 is ribosome bound inextracts from rat liver (57). The rate of releaseof EF-2 during protein synthesis may be thelimiting factor in the inactivation process undercertain circumstances and may at least in partdetermine the duration of the lag period beforeprotein synthesis is affected in toxin-treatedcells (60).EF-2 can also bind GTP, GDP, and various
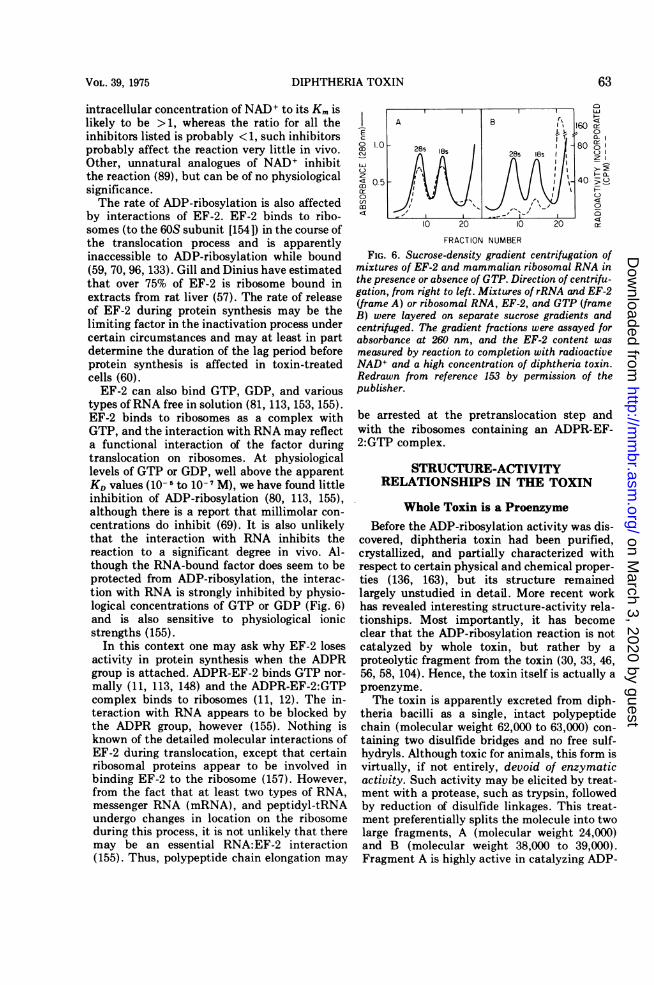
types ofRNA free in solution (81, 113, 153, 155).EF-2 binds to ribosomes as a complex withGTP, and the interaction with RNA may reflecta functional interaction of the factor duringtranslocation on ribosomes. At physiologicallevels of GTP or GDP, well above the apparentKD values (10-5 to 10-7 M), we have found littleinhibition of ADP-ribosylation (80, 113, 155),although there is a report that millimolar con-centrations do inhibit (69). It is also unlikelythat the interaction with RNA inhibits thereaction to a significant degree in vivo. Al-though the RNA-bound factor does seem to beprotected from ADP-ribosylation, the interac-tion with RNA is strongly inhibited by physio-logical concentrations of GTP or GDP (Fig. 6)and is also sensitive to physiological ionicstrengths (155).
In this context one may ask why EF-2 losesactivity in protein synthesis when the ADPRgroup is attached. ADPR-EF-2 binds GTP nor-mally (11, 113, 148) and the ADPR-EF-2:GTPcomplex binds to ribosomes (11, 12). The in-teraction with RNA appears to be blocked bythe ADPR group, however (155). Nothing isknown of the detailed molecular interactions ofEF-2 during translocation, except that certainribosomal proteins appear to be involved inbinding EF-2 to the ribosome (157). However,from the fact that at least two types of RNA,messenger RNA (mRNA), and peptidyl-tRNAundergo changes in location on the ribosomeduring this process, it is not unlikely that theremay be an essential RNA:EF-2 interaction(155). Thus, polypeptide chain elongation may
E
o 1.0
z
atcr0
A B r ~.1 -3- 0
28sl-28Oo'I
z
--I I ~ ~ ~ ~ ~ ~~I--
0 20 0 ~~~~20 a
FRACTION NUMBER
FIG. 6. Sucrose-density gradient centrifugation ofmixtures of EF-2 and mammalian ribosomal RNA inthe presence or absence of GTP. Direction of centrifu-gation, from right to left. Mixtures of rRNA and EF-2(frame A) or ribosomal RNA, EF-2, and GTP (frameB) were layered on separate sucrose gradients andcentrifuged. The gradient fractions were assayed forabsorbance at 260 nm, and the EF-2 content wasmeasured by reaction to completion with radioactiveNAD+ and a high concentration of diphtheria toxin.Redrawn from reference 153 by permission of thepublisher.
be arrested at the pretranslocation step andwith the ribosomes containing an ADPR-EF-2:GTP complex.
STRUCTURE-ACTIVITYRELATIONSHIPS IN THE TOXIN
Whole Toxin is a ProenzymeBefore the ADP-ribosylation activity was dis-
covered, diphtheria toxin had been purified,crystallized, and partially characterized withrespect to certain physical and chemical proper-ties (136, 163), but its structure remainedlargely unstudied in detail. More recent workhas revealed interesting structure-activity rela-tionships. Most importantly, it has becomeclear that the ADP-ribosylation reaction is notcatalyzed by whole toxin, but rather by aproteolytic fragment from the toxin (30, 33, 46,56, 58, 104). Hence, the toxin itself is actually aproenzyme.The toxin is apparently excreted from diph-
theria bacilli as a single, intact polypeptidechain (molecular weight 62,000 to 63,000) con-taining two disulfide bridges and no free sulf-hydryls. Although toxic for animals, this form isvirtually, if not entirely, devoid of enzymaticactivity. Such activity may be elicited by treat-ment with a protease, such as trypsin, followedby reduction of disulfide linkages. This treat-ment preferentially splits the molecule into twolarge fragments, A (molecular weight 24,000)and B (molecular weight 38,000 to 39,000).Fragment A is highly active in catalyzing ADP-
63VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
COLLIER
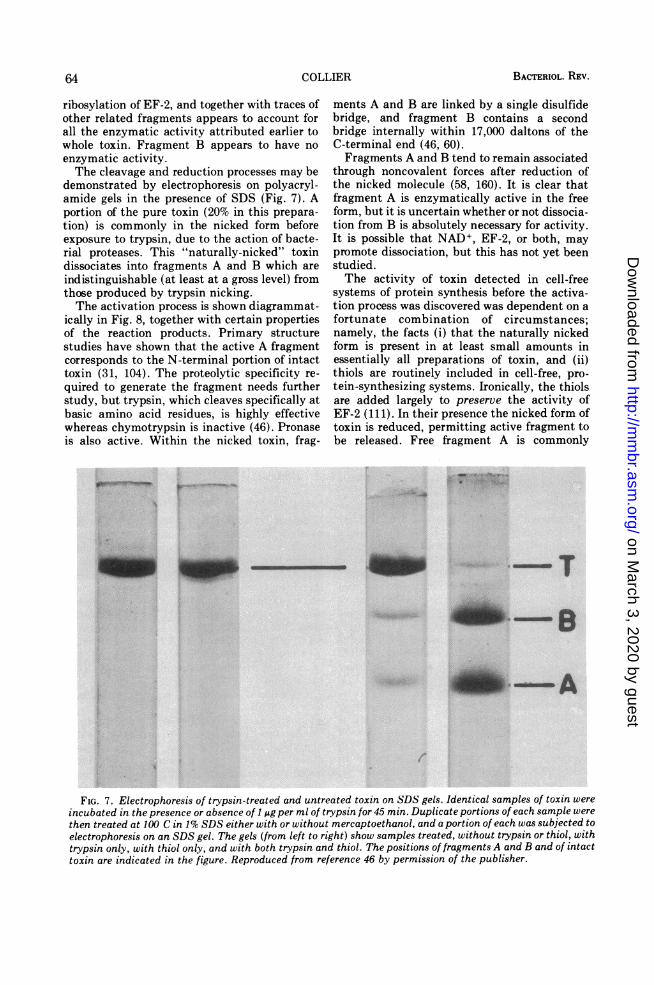
ribosylation of EF-2, and together with traces ofother related fragments appears to account forall the enzymatic activity attributed earlier towhole toxin. Fragment B appears to have noenzymatic activity.The cleavage and reduction processes may be
demonstrated by electrophoresis on polyacryl-amide gels in the presence of SDS (Fig. 7). Aportion of the pure toxin (20% in this prepara-tion) is commonly in the nicked form beforeexposure to trypsin, due to the action of bacte-rial proteases. This "naturally-nicked" toxindissociates into fragments A and B which areindistinguishable (at least at a gross level) fromthose produced by trypsin nicking.The activation process is shown diagrammat-
ically in Fig. 8, together with certain propertiesof the reaction products. Primary structurestudies have shown that the active A fragmentcorresponds to the N-terminal portion of intacttoxin (31, 104). The proteolytic specificity re-quired to generate the fragment needs furtherstudy, but trypsin, which cleaves specifically atbasic amino acid residues, is highly effectivewhereas chymotrypsin is inactive (46). Pronaseis also active. Within the nicked toxin, frag-
ments A and B are linked by a single disulfidebridge, and fragment B contains a secondbridge internally within 17,000 daltons of theC-terminal end (46, 60).Fragments A and B tend to remain associated
through noncovalent forces after reduction ofthe nicked molecule (58, 160). It is clear thatfragment A is enzymatically active in the freeform, but it is uncertain whether or not dissocia-tion from B is absolutely necessary for activity.It is possible that NAD+, EF-2, or both, maypromote dissociation, but this has not yet beenstudied.The activity of toxin detected in cell-free
systems of protein synthesis before the activa-tion process was discovered was dependent on afortunate combination of circumstances;namely, the facts (i) that the naturally nickedform is present in at least small amounts inessentially all preparations of toxin, and (ii)thiols are routinely included in cell-free, pro-tein-synthesizing systems. Ironically, the thiolsare added largely to preserve the activity ofEF-2 (111). In their presence the nicked form oftoxin is reduced, permitting active fragment tobe released. Free fragment A is commonly
FIG. 7. Electrophoresis of trypsin-treated and untreated toxin on SDS gels. Identical samples of toxin wereincubated in the presence or absence of 1 A.g per ml of trypsin for 45 min Duplicate portions of each sample werethen treated at 100 C in 1% SDS either with or without mercaptoethanol, and a portion of each was subjected toelectrophoresis on an SDS gel. The gels (from left to right) show samples treated, without trypsin or thiol, withtrypsin only, with thiol only, and with both trypsin and thiol. The posittions of fragments A and B and of intacttoxin are indicated in the figure. Reproduced from reference 46 by permission of the publisher.
BACTERIOL. REV.642
, . !- .1-.
I
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
S-S S-SN C
ftttSmall*\peptil~es Trypsin
S-S S-SN _ __CIs-ss-s
RSH
SHNC +
SH SH SHNC
INTACT TOXINm wt 63, 000enzymically inactivetoxic
NICKED TOXINm wt 63, 000enzymically inactivetoxic
FRAGMENT A FRAGMENT Bm wt 24, 000 m wt 39, 000enzymrically active enzymically inactivenon-toxic non-toxic
FIG. 8. Sequence of events in the expression ofenzymatic activity (ADP-ribosylation of EF-2) indiphtheria toxin.
present in impure toxin preparations and mayhave been another source of activity in someexperiments (30).
Toxicity of the Various Fragments andForms
Preparations of toxin entirely free of thenicked form are not available, but samplesoriginally containing 80% intact toxin appear tohave the same toxicity for guinea pigs eitherbefore or after nicking with trypsin (46, 58).(Lethality tests used for these estimates areusually not more accurate than 20%.) Simi-larly, measurements of the kinetics of inhibitionof protein synthesis in HeLa cells reveal onlysmall differences between nicked and 80% in-tact samples. With the fully nicked material thelag period is reduced slightly, but the kineticsare otherwise similar (46). This suggests thatHeLa cells contain a mechanism for nickingintact toxin, and that this step is not ratelimiting.Fragment A, despite its enzymatic activity, is
not toxic for guinea pigs in doses of 8 nmol perkg body weight (46, 58). In contrast, whole toxinis lethal at molar concentrations 2,000 timeslower (4 pmol per kg body weight). It has alsobeen shown that fragment A does not inhibitprotein synthesis in Hela cells at concentrationsup to 4 AM. These somewhat surprising resultssuggest that fragment B is also required fortoxicity. Work with nontoxic, immunologicallycross-reacting forms of toxin has confirmed thisnotion, and there is now good reason to believethat fragment B is needed for attachment oftoxin to cells. Thus, free fragment A is inactiveon whole cells probably because it cannottraverse the plasma membrane to a significantextent.
Tests of purified fragment B have revealed no
toxicity or ADP-ribosylation activity. Althoughthe instability of the fragment (see below)makes such measurements suspect, the absenceof these activities is supported by studies withcertain nontoxic, mutant forms of toxin, de-scribed below.
Properties of Fragment AA prominent feature of fragment A is its
stability. It may be heated at 100 C at neutralpH or exposed to pH values of 2 or 12 at roomtemperature for brief periods with little loss ofactivity (46, 58). Also, it is relatively resistant totrypsin or chymotrypsin, especially in the pres-ence of NAD+ (31, 32, 89). On the other hand,fragment B denatures and precipitates rapidlyafter dissociation of nicked toxin and can onlybe maintained in solution with high concentra-tions of urea or guanidine, or with detergents.For these reasons, and because of its enzymaticactivity, fragment A has been studied moreextensively than B.
Figure 9 shows a tentative, partial amino acidsequence of fragment A based on work nearingcompletion at the University of California atLos Angeles (R. J. DeLange, R. Drazin, and R.J. Collier, unpublished data). The N-terminalsequence is identical with that reported earlierby Michel et al. for fragment A and whole toxin(104).The most noteworthy aspect of the sequence
as it stands is the heterogeneity at the C-ter-minus. From digests with CNBr it is possible toisolate three C-terminal peptides, which termi-nate in the sequences ---Asn-Arg-COOH,---Asn-Arg-Val-Arg-COOH, and ---Asn-Arg-Val-Arg-Arg-COOH. Trypsin apparently attacks atany of three, closely spaced arginines, produc-ing three major forms of fragment A. Eachadditional arginine adds an extra positivecharge at neutral pH, which probably explainsthe electrophoretic heterogeneity of the frag-ment. Under nondenaturing conditions, thefragment forms three major bands on polyacryl-amide gels, all of which are active (see Fig. 11)(89). It also separates into three bands onisoelectric focusing (pI values of 4.56, 4.72, and4.85).A second point to note in the sequence is that
the single half-cystine residue is near the C-ter-minus (the fourth residue from the proximalarginine) (31). This fact, together with otherevidence placing the corresponding half-cystineof fragment B near its N-terminus, indicatesthat the trypsin-sensitive region of the toxin isenclosed within a disulfide loop of limited size(it is no greater than 40 residues, but may bemuch smaller).
VOL. 39, 1975 65
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

CNBr-I1 10 14 1 20
H2N-Gly-Ala-Asp-Asp-Val-Val-Asp-Ser-Ser-Lys-Ser-Phe-Val-Met-Glu-Asx-Phe-Ser-Ser-Tyr-His-Gly-Thr-30 40
(Lys,Pro)-Gly-Tyr-Val-Asp-Ser-Ile-Gln-Lys-Gly-Ile-Gln-Lys-Pro-Lys-Ser-Gly-Thr-Glx-Gly-Tyr-Asx-Asx-Asx-50 60 70
Asx-Lys-Gly-Phe-Tyr-Ser-Thr-Asp-Asn-Lys-Tyr-Asp-Ala-Ala-Gly-Tyr-Ser-Val-Asp-Asn-Asx-Pro-Glu-Leu-80 90
Ser-Gly-Lys-Ala-Gly-Gly-Val-Val-Lys-Val-Thr-Tyr-Pro-Gly-Leu-Thr-Lys-Val-Leu-Ala-Leu-Lys-Val-Asp-Asn-Ala-Glu-CNBr-II
100 110 1141 120Thr-Ile-Lys-Lys-Glu-Leu-Gly-Leu-Ser-Leu-Thr-Glu-Pro-Leu-Met-Glu-Val-Gly-Thr-Gln-Glu-Glu-Phe-Ile-
T-11 130 140
Lys-Arg-Val-Val-Leu-Ser-Leu-Pro-Phe-Ala-Glu-Gly-Ser-Ser-Val-Glu-Sgr-Tyr-Ile-Asn-Asn-Trp-Glu-Gln-Ala-Lys-T-2 T-3 T-4
150 160 1621 1170 1Ala-Leu-Ser-Val-(Glu,Glu,Leu)-Ile-Asn-Phe-Glu-Thr-Arg-Phe-Gly-Asp-Gly-Ala-Ser-Arg-Gly-Lys-Arg-Gly-
CNBr-III CNBr-IV CNBr-V1 180 1 190 192
Gln-Asp-Ala-Met-Tyr-Glu-Tyr-Met-Ala-Gln-Ala-Cys-Ala-Gly-Asn-Arg-Val-Arg-Arg-COOHT I
T-5
Other C-termini found ----- Val-Arg-COOHT-6
FIG. 9. Tentative, partial sequence of fragment A from diphtheria toxin. The residue numbers have beenadded for convenience only, inasmuch as two of the three tryptophans have not been placed in the sequence.Further studies may necessitate changes in the sequence.
Other points of interest in the sequence arethe concentration of all arginines in the last 68residues and the clustering of two to four Asxresidues at various places in the sequence. Theonly histidine in fragment A is near the N-ter-minus.The longest form of fragment A may be
attached directly to the N-terminus of B withinintact toxin, but it remains possible that one ormore small linking peptides (estimated toamount to no more than 10 residues) may besplit out by trypsin (104). The heterogeneity atthe N-terminus of B reported by Michel et al.(equivalent amounts of glycine and serine andlesser amounts of valine and lysine) is consist-ent with this (104). We have found predomi-nantly glycine at the N-terminus and serine atthe C-terminus of B. Michel et al. have reportedseveral amino acids, not including serine, at theC-terminus (104).The active-site residues of fragment A have
not been studied, but it is known that destruc-tion of a single tryptophan (J. Kandel and R. J.Collier, unpublished data), or a single tyrosine(14) by chemical modification brings about acomplete or almost complete loss of enzymaticactivity. The half cystine is not important forcatalysis, inasmuch as reaction with any of anumber of sulfhydryl reagents does not affectthe activity (46, 58).The sedimentation coefficient of fragment A
has been determined accurately as 2.19 S(89) bythe method of differential sedimentation, usingribonuclease as a standard (1.84S). No changewas observed in the presence of NAD+.
Variations Among Toxin PreparationsAll preparations of toxin examined contain at
least a trace of the nicked form, and some arealmost entirely nicked (33, 60). Variation in theproportions of the nicked and intact formsdepends on the action of bacterial proteases inthe culture or during isolation and purificationof the toxin (60). Traces of such proteases oftenremain even after several steps of purification,and may increase the degree of nicking duringhandling. Phenylmethylsulfonyl fluoride inhib-its such proteolytic activity (60).
Reduction and dissociation of most prepara-tions yield predominantly fragments A and Bbut other fragments are sometimes seen (Fig.10). Gill and Dinius have found certain prepara-tions which contain a species of nicked toxinwhich yields fragments E and F (molecularweight 34,000 and 28,000, respectively) (60).Fragments E and F each contain an intactdisulfide bridge and are apparently normallybound together only by noncovalent forces.Fragment F is enzymatically active and has thestability properties of fragment A, which itcontains. How these unusual fragments aregenerated has not been studied but it may
66 COLLIER BACTERIOL. REV.
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
involve a protease of different specificity fromthat producing A-B nicked toxin.
Although not consistently observed in otherlaboratories, it has been reported that the site ofpreferential cleavage by trypsin is altered de-pending on the presence or absence of thiols(60). Highly specific cleavage at the A-B junc-tion was observed ohly in the presence of a thiol.In its absence, preferential cleavage occurred ata point 7,000 daltons from the N-terminus,producing fragment C (molecular weight55,000) plus the N-terminal fragment, whichwas not recovered (Fig. 10). Fragment C wassusceptible to further attack by trypsin at theA-B junction, producing fragments D (molecu-lar weight 17,000) and B in disulfide linkage.Free fragment D obtained after reduction is notenzymatically active, which has led to specula-tion that A may be the smallest enzymaticallyactive fragment. The fact that the variation inspecificity of the site of cleavage by trypsin withthiols is not uniformly observed may be due toundefined differences among various toxinpreparations.A dimeric form of whole toxin (6.6 to 6.8S)
has been found and constitutes a sizable frac-tion (sometimes 100%) of certain toxin prepara-tions (67, 122). SDS causes dissociation in theabsence of thiols (56). (However, Goor hasreported that 0.33 M dithiothreitol in the pres-ence of 0.08 M EDTA dissociates the dimer[67].) The dimeric form appears to have similarbut perhaps slightly reduced toxicity comparedto the monomer (67, 135). There is evidencethat it may be formed during ammonium sul-fate precipitation of the toxin (135).There are numerous suggestions in the litera-
FRAGMENT F FRAGMENT Emwt 28,000 S-S s-s / mwt34,000enzymically
active AI Unknown protease
INTACT TOXIN
ITrypsins-s s-S
fttt --FRAGMENT C
/ Trypsin + RSH m wt 57,000Unrecovered SHpeptide(s) + FRAG. B
FRAGMENT Dm wt 17,000enzymically inactive
FIG. 10. Alternative modes of cleavage of intacttoxin.
ture that other variations may exist in wholetoxin, besides those which have been described.This is evidenced, for example, by fractionalimmunological reactivity with antifragment A(127), variations in pyridine nucleotide binding(103), and heterogeneous electrophoretic pat-terns (J. Kandel and R. J. Collier, unpublisheddata). Studies are under way to analyze andcorrelate some of these variations.
Free fragment A is often found in toxinpreparations, and, in fact, discovery of thispeptide in crude toxin was the first indication ofan interesting structure-activity relationship inthe toxin (30). The free fragment probablyarises by reduction or disulfide interchange ofnaturally nicked toxin, promoted by low levelsof thiols in the culture medium. The fact thatthe contaminating fragment does not readilyform disulfide-linked dimers implies that itssulfhydryl group may have been oxidized or is inthe form of a mixed disulfide, perhaps with ahalf-cystine from cystine in the culture me-dium. Free fragment B is not found in crudetoxin, presumably because of its insolubilityand susceptibility to proteolysis.
Nontoxic, Cross-Reacting Forms of ToxinVarious nontoxic or partially toxic, immuno-
logically cross-reacting forms of toxin (CRMs)have been isolated recently (125, 158-162).Mutants of phage have been selected whichinduce the formation of such CRMs when theylysogenize nontoxigenic strains of C. diph-theriae. Analysis of the properties of theseproteins has demonstrated the feasibility of thegenetic approach to the study of the toxin andhas yielded considerable information about itsstructure and activity.
Certain of the CRMs are the same size asintact toxin and presumably represent missensemutations (CRMs 197, 228, and 176), whereastwo others (CRMs 30 and 45) are N-terminalfragments, perhaps resulting from chain-termi-nation mutations (Table 4). In all five CRMs,the A-B junction remains susceptible to trypticattack, thus permitting analysis of the peptideproducts on SDS gels. CRMs 30 and 45 are bothenzymatically active and yield fragment A withthe normal level of activity. Their lack oftoxicity is due to the absence of large C-termi-nal sections of fragment B, thus underscoringthe necessity of this portion for toxicity. CRM45 contains only two half-cystine residues,which localizes the second disulfide within themissing 17,000- to 18,000-dalton, C-terminalpeptide.The fragment A portions of the other three
CRMs all have altered activities. Fragment A
Ehmimm
67VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
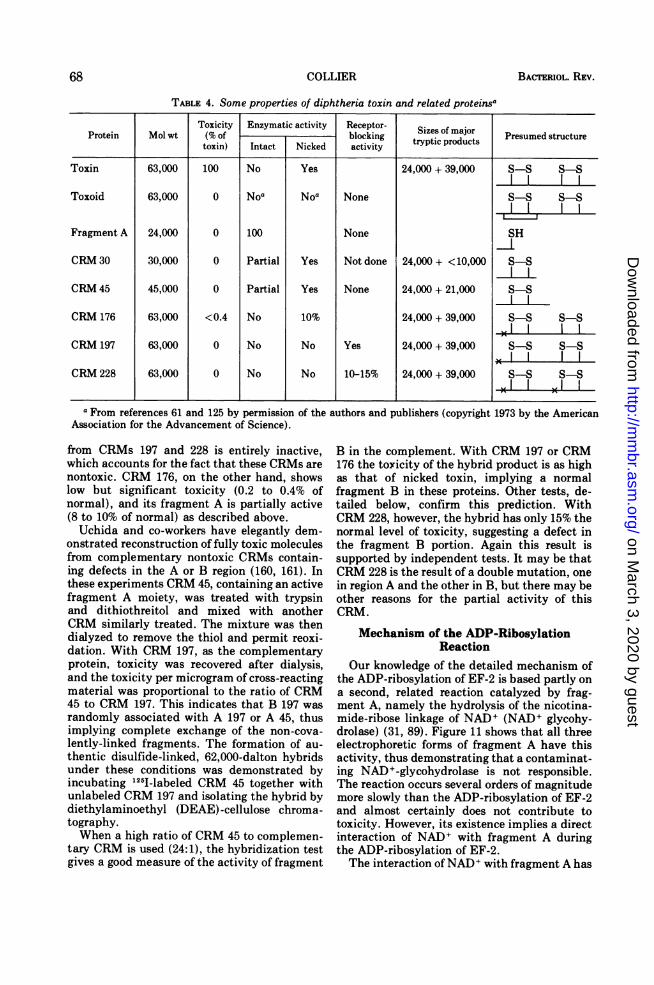
TABLE 4. Some properties of diphtheria toxin and related proteinsa
Toxicity Enzymatic activity Receptor- Sizes of majorProtein Mol wt (% of blocking tryptic products Presumedtoxin) Intact Nicked activity
Toxin 63,000 100 No Yes 24,000 + 39,000 S-S S-S
Toxoid 63,000 0 Noa NOa None S-S S-S
Fragment A 24,000 0 100 None SH
CRM 30 30,000 0 Partial Yes Not done 24,000 + <10,000 S-S
CRM 45 45,000 0 Partial Yes None 24,000 + 21,000 S-S
CRM 176 63,000 <0.4 No 10% 24,000 + 39,000 S-S S-S
CRM 197 63,000 0 No No Yes 24,000 + 39,000 5-5 S-S
CRM 228 63,000 0 No No 10-15% 24,000 + 39,000 S-S S-S
aFrom references 61 and 125 by permission of the authors and publishers (copyright 1973 by the AmericanAssociation for the Advancement of Science).
from CRMs 197 and 228 is entirely inactive,which accounts for the fact that these CRMs arenontoxic. CRM 176, on the other hand, showslow but significant toxicity (0.2 to 0.4% ofnormal), and its fragment A is partially active(8 to 10% of normal) as described above.Uchida and co-workers have elegantly dem-
onstrated reconstruction of fully toxic moleculesfrom complementary nontoxic CRMs contain-ing defects in the A or B region (160, 161). Inthese experiments CRM 45, containing an activefragment A moiety, was treated with trypsinand dithiothreitol and mixed with anotherCRM similarly treated. The mixture was thendialyzed to remove the thiol and permit reoxi-dation. With CRM 197, as the complementaryprotein, toxicity was recovered after dialysis,and the toxicity per microgram of cross-reactingmaterial was proportional to the ratio of CRM45 to CRM 197. This indicates that B 197 wasrandomly associated with A 197 or A 45, thusimplying complete exchange of the non-cova-lently-linked fragments. The formation of au-thentic disulfide-linked, 62,000-dalton hybridsunder these conditions was demonstrated byincubating 125I-labeled CRM 45 together withunlabeled CRM 197 and isolating the hybrid bydiethylaminoethyl (DEAE)-cellulose chroma-tography.When a high ratio of CRM 45 to complemen-
tary CRM is used (24:1), the hybridization testgives a good measure of the activity of fragment
B in the complement. With CRM 197 or CRM176 the toxicity of the hybrid product is as highas that of nicked toxin, implying a normalfragment B in these proteins. Other tests, de-tailed below, confirm this prediction. WithCRM 228, however, the hybrid has only 15% thenormal level of toxicity, suggesting a defect inthe fragment B portion. Again this result issupported by independent tests. It may be thatCRM 228 is the result of a double mutation, onein region A and the other in B, but there may beother reasons for the partial activity of thisCRM.
Mechanism of the ADP-RibosylationReaction
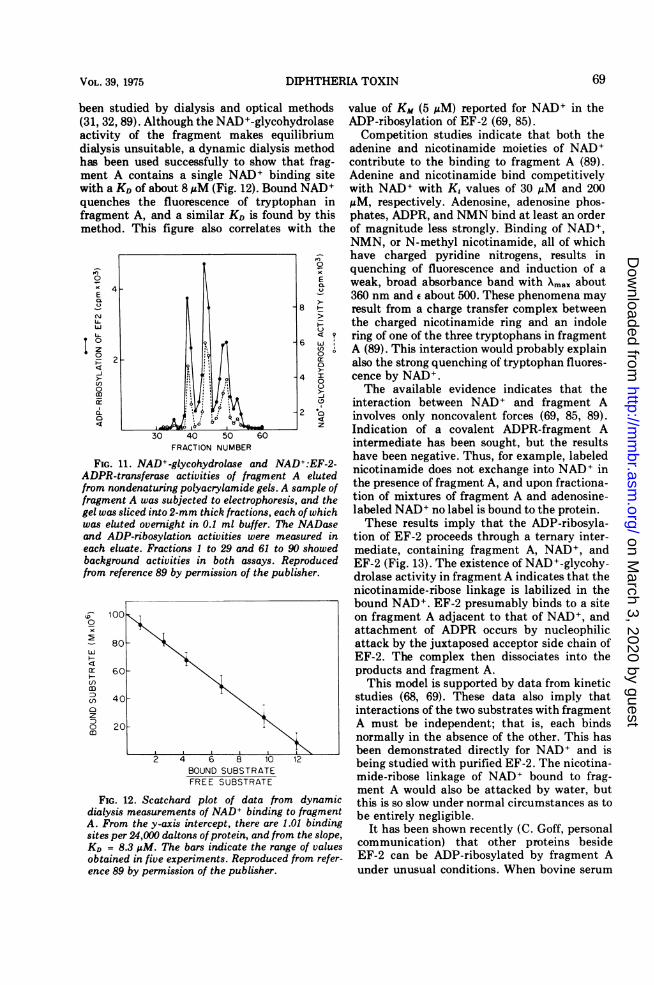
Our knowledge of the detailed mechanism ofthe ADP-ribosylation of EF-2 is based partly ona second, related reaction catalyzed by frag-ment A, namely the hydrolysis of the nicotina-mide-ribose linkage of NAD+ (NAD+ glycohy-drolase) (31, 89). Figure 11 shows that all threeelectrophoretic forms of fragment A have thisactivity, thus demonstrating that a contaminat-ing NAD+-glycohydrolase is not responsible.The reaction occurs several orders of magnitudemore slowly than the ADP-ribosylation of EF-2and almost certainly does not contribute totoxicity. However, its existence implies a directinteraction of NAD+ with fragment A duringthe ADP-ribosylation of EF-2.The interaction ofNAD+ with fragment A has
68 COLLIER BACTERIOL. REV.
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
DIPHTHERIA TOXIN
been studied by dialysis and optical methods(31, 32, 89). Although the NAD+-glycohydrolaseactivity of the fragment makes equilibriumdialysis unsuitable, a dynamic dialysis methodhas been used successfully to show that frag-ment A contains a single NAD+ binding sitewith a KD of about 8 MM (Fig. 12). Bound NAD+quenches the fluorescence of tryptophan infragment A, and a similar KD is found by thismethod. This figure also correlates with the
0
o E
E
CU
0 ~~ ~~96 U:z U~~~~~~~~~~~~~~~~~~~~~~~~~~()
(1 0
_J~~~~~~~~~~~~~~~~~~~~-
0~~~~~~~~~~~~~~~o1,0 z
30 40 50 60FRACTION NUMBER
FIG. 11. NAD+-glycohydrolase and NAD+:EF-2-ADPR-transferase activities of fragment A elutedfrom nondenaturing polyacrylamide gels. A sample offragment A was subjected to electrophoresis, and thegel was sliced into 2-mm thick fractions, each of whichwas eluted overnight in 0.1 ml buffer. The NADaseand ADP-ribosylation activities were measured ineach eluate. Fractions 1 to 29 and 61 to 90 showedbackground activities in both assays. Reproducedfrom reference 89 by permission of the publisher.
0
8001
r 60-
D
2 40 _
o 20M
BOUND SUBSTRATEFREE SUBSTRATE
FIG. 12. Scatchard plot of data from dynamicdialysis measurements of NAD+ binding to fragmentA. From the y-axis intercept, there are 1.01 bindingsites per 24,000 daltons ofprotein, and from the slope,KD = 8.3 AM. The bars indicate the range of valuesobtained in five experiments. Reproduced from refer-ence 89 by permission of the publisher.
value of Km (5 MM) reported for NAD+ in theADP-ribosylation of EF-2 (69, 85).
Competition studies indicate that both theadenine and nicotinamide moieties of NAD+contribute to the binding to fragment A (89).Adenine and nicotinamide bind competitivelywith NAD+ with Ki values of 30 MM and 200MM, respectively. Adenosine, adenosine phos-phates, ADPR, and NMN bind at least an orderof magnitude less strongly. Binding of NAD+,NMN, or N-methyl nicotinamide, all of whichhave charged pyridine nitrogens, results inquenching of fluorescence and induction of aweak, broad absorbance band with Xmax about360 nm and e about 500. These phenomena mayresult from a charge transfer complex betweenthe charged nicotinamide ring and an indolering of one of the three tryptophans in fragmentA (89). This interaction would probably explainalso the strong quenching of tryptophan fluores-cence by NAD+.The available evidence indicates that the
interaction between NAD+ and fragment Ainvolves only noncovalent forces (69, 85, 89).Indication of a covalent ADPR-fragment Aintermediate has been sought, but the resultshave been negative. Thus, for example, labelednicotinamide does not exchange into NAD+ inthe presence of fragment A, and upon fractiona-tion of mixtures of fragment A and adenosine-labeled NAD+ no label is bound to the protein.These results imply that the ADP-ribosyla-
tion of EF-2 proceeds through a ternary inter-mediate, containing fragment A, NAD+, andEF-2 (Fig. 13). The existence of NAD+-glycohy-drolase activity in fragment A indicates that thenicotinamide-ribose linkage is labilized in thebound NAD+. EF-2 presumably binds to a siteon fragment A adjacent to that of NAD+, andattachment of ADPR occurs by nucleophilicattack by the juxtaposed acceptor side chain ofEF-2. The complex then dissociates into theproducts and fragment A.This model is supported by data from kinetic
studies (68, 69). These data also imply thatinteractions of the two substrates with fragmentA must be independent; that is, each bindsnormally in the absence of the other. This hasbeen demonstrated directly for NAD+ and isbeing studied with purified EF-2. The nicotina-mide-ribose linkage of NAD+ bound to frag-ment A would also be attacked by water, butthis is so slow under normal circumstances as tobe entirely negligible.
It has been shown recently (C. Goff, personalcommunication) that other proteins besideEF-2 can be ADP-ribosylated by fragment Aunder unusual conditions. When bovine serum
69VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
HS,
A N
R-P-P-R(NAD S)
KD- 8IMM
/FRAG.AX
A Ng
R-P-P-R
NA&6FGLYCOHYDROLASE H20 EF2
REACTION
FRAG. A
ADPR+/
NICOTINAMIDE +HHSh EF-2
FRAG.AP \
R-P-P-R'-- :X
HSK
EF-2
E EF-2 a
NADIR
A X/R-P-P-R
+ NICOTINAMIDE +H
FIG. 13. Proposed mechanism for the ADP-ribosy-lation of EF-2 and NAD+-glycohydrolase activities offragment A.
albumin or RNA polymerase core enzyme wasincubated with adenine-labeled NAD+ and anextraordinarily high concentration of fragmentA (20 mM, which is about 106 times greater thanthat normally employed in our assays), labelwas incorporated into the serum albumin, the yand fl' subunits of RNA polymerase, and frag-ment A itself. The a subunit incorporated nolabel, and in those chains which were labeledthe efficiency of labeling was low, with onlyabout one ADPR incorporated per thousandmolecules of protein. These results suggest thata variety of proteins may be reactive if thereaction is "forced," but EF-2 is probably themost efficient substrate by several orders ofmagnitude.
Pyridine Nucleotide Binding andADP-Ribosylation Activity by
Whole Toxin
When fragment A is linked to B within intactor nicked toxin it seems to be virtually if notcompletely inactive in ADP-ribosylating EF-2.Whether this lack of activity is due to interfer-
ence with the binding of either substrate, orboth, or to other alterations of fragment A is notknown. However, there is some pertinent infor-mation available.
Curiously, despite the fact that whole toxincannot ADP-ribosylate EF-2, it seems to ADP-ribosylate itself if incubated long enough withNAD+ (55, 62, 64). The reaction requires hoursto days to reach completion, and the maximalextent of labeling corresponds to only about 0.6ADPR per toxin molecule. The intact form oftoxin appears to be modified at about the samerate as the nicked form. Reduction is notrequired. There seems to be more than one sightwithin toxin which may accept ADPR as evi-dence by the fact that incorporated label isfound in both A and B fragments separated ongels, and also by the fact that the release ofADPR in 0.1 N NaOH does not follow first-orderkinetics.The available data suggest but do not prove
that the ADP-ribosylation of whole toxin mayrepresent an intramolecular modification; thatis, one in which a given toxin molecule catalyzesattachment of ADPR to itself and no othermolecules. Studies of the kinetics of the reac-tion as a function of the toxin concentration willbe useful in deciding if this is indeed the case.The existence of this reaction provides strong
evidence that at least a fraction of the popula-tion of whole toxin molecules is capable ofbinding NAD+. The nature of the reactionsuggests that the binding would be to the site onthe fragment A moiety, and this is supported bythe finding that CRM 197 lacks the self-modifi-cation reaction, as well as ADP-ribosylation andNAD+-glycohydrolase activities.The binding of NAD+ and other pyridine
nucleotides by whole toxin has been studied,but the problem needs still more work (103, 112,147, 148). Measurements of quenching of toxinfluorescence by NAD+ (103) seem to indicatethat the nucleotide binds to nicked with anaffinity of the same order (KD 9.7 to 13.6 ,M) asthat seen with fragment A, and to intact toxinwith a slightly lower affinity (KD 28 uM).However, the number of sites remains in doubtbecause of the relatively low quenching ob-served (which limits the accuracy of the data)and also because the calculated dissociationconstants are about an order of magnitudegreater than the concentration of toxin used.Michel and Dirkx (103) have calculated thatintact toxin binds two molecules of NAD+whereas the nicked form binds only one. Incontrast, preliminary experiments in my labo-ratory with dialysis techniques suggest thatwhole toxin binds far less than a molar equiva-lent of NAD+ regardless of whether it is nicked
70 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
or largely intact (J. Kandel and R. J. Collier,unpublished data). Unknown sources of varia-tion among different toxin preparations may beresponsible for these discrepancies. GTP hasbeen reported to inhibit competitively the toxinNAD+ interaction (148).Two laboratories have reported that whole
toxin binds NADH more strongly (KD 0.5-0.7AiM) than NAD+ (102, 103, 112). One batch oftoxin which had been nicked by bacterial pro-teases seemed to contain two NADH bindingsites, whereas another manifested only one site,either before or after nicking with trypsin (103).NADP and NADPH bound with affinities simi-lar to NAD+ and NADH, respectively, but therewas not an exact correlation in the calculatednumber of binding sites. Although it is temptingto construct models on the basis of fluorescencedata alone, it would perhaps be safe to awaitconfirmation by results from dialysis experi-ments.The question of whether whole toxin can bind
EF-2 has not yet been studied carefully. Thereis a report that toxin forms a stable, ternarycomplex with NAD+ and EF-2 (52), but anotherlaboratory has sought such a complex withoutsuccess, using different techniques (60).
IS THE ADP-RIBOSYLATIONREACTION RESPONSIBLE FOR
TOXICITY?
In cell-free systems there is little doubt thatthe ADP-ribosylation of EF-2 is responsible forthe observed inhibition of protein synthesis, butone must question whether this reaction ac-tually occurs in vivo and whether it is responsi-ble for toxicity. Although there is still room fordoubt, a strong case can be made for anaffirmative answer.
(i) Persuasive evidence comes from a consid-eration of the properties of CRMs 176 and 197(159). Each of these apparently contains asingle amino acid substitution which alters theenzymatic activity of the molecules, and mostimportantly, it is found that toxicity is affectedsimilarly. Thus, CRM 197 is devoid of en-zymatic activity and is entirely nontoxic,whereas CRM 176 has a low specific activity inthe ADP-ribosylation reaction and is partiallytoxic. The correlation is also valid for theinhibition of protein synthesis in tissue culture;CRM 197 is totally inactive and CRM 176 ispartially active. The toxicity ofCRM 176 (0.2 to0.4% of normal) is not precisely the same as itsspecific activity (8 to 10% of normal), but thismay be due to an altered stability or otherfactors operative in vivo.
Although one would like to have a largernumber of mutants with specific alterations inADP-ribosylation activity, the evidence pro-vided by these two is strong. Unless toxin has asecond, more important activity which is simi-larly affected by these mutations, toxicity mustinvolve ADP-ribosylation. The two amino acidsubstitutions in question lie within the enzy-matically active region of the peptide chain anddo not significantly affect the function of theremainder of the toxin molecule.
(ii) There is good, although indirect, evidencethat the ADP-ribosylation of EF-2 actuallyoccurs in cultured cells treated with toxin. Thedefinitive reaction product, ADPR-EF-2, hasnot been isolated from toxin-treated cells, butits existence is implied by the fact that proteinsynthetic activity of extracts from such cells isrestored by nicotinamide (60, 105). (Additionaltoxin may be required to accelerate reversal ofthe ADP-ribosylation reaction [60].) Also, thetotal elongation factor activity (EF-1 and EF-2combined) declines by 90% within the lag periodof toxin-treated HeLa cells (60).
(iii) For reasons of complexity, the leastcompelling evidence comes from studies inwhole animals. However, it appears that thedata are generally consistent with the resultsfrom simpler systems.
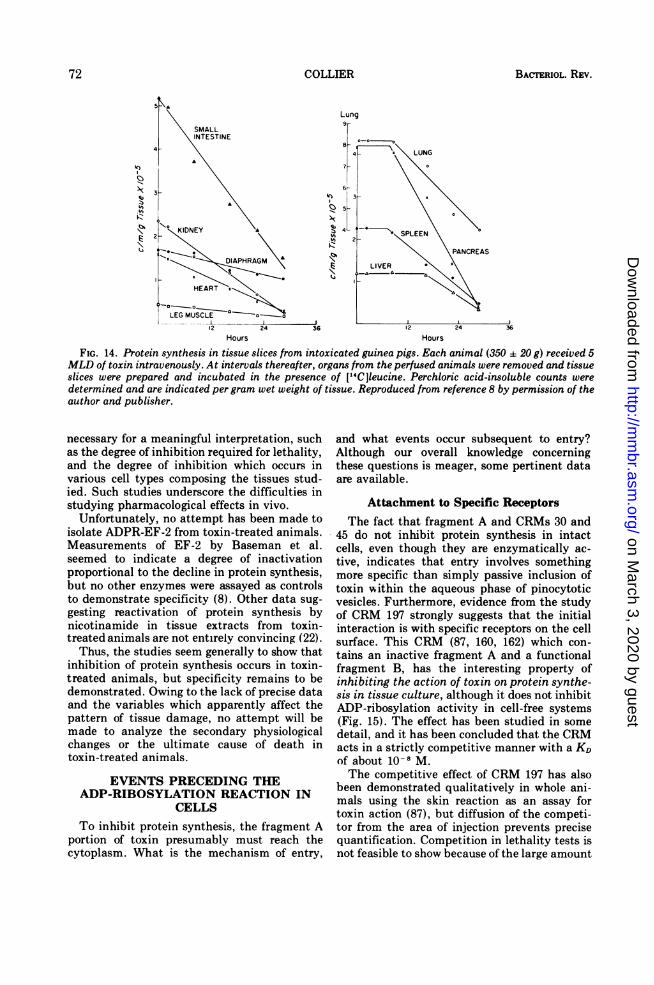
Effects of toxin on protein synthesis in ani-mals have been documented by two groups, butthe pattern of inhibition seems to vary accord-ing to the site of injection, the dose, and perhapsother factors. Baseman et al. injected guineapigs with 5 MLD toxin intravenously and mea-sured protein synthesis in tissue slices preparedfrom animals sacrificed at intervals up to thetime of death (30 h) (8). A decline was found inall organs tested, although in some the declinewas preceded by a lag of several hours (Fig.14). Bonventre and co-workers noted similar,widespread effects under certain conditions(20), but found preferential effects on muscula-ture (cardiac, skeletal, diaphragmatic) underothers (18, 20-22, 142). It may be that theseresults can be explained by a greater inherentsensitivity of musculature to toxin, which ismanifested only at low toxin concentrations.
In another study, Bonventre reported little orno inhibition of protein synthesis in most tissuesof guinea pigs injected with 1.4 MLD of toxin, ifthis total was divided into four equal doseswhich were administered at daily intervals (17).However, inhibitions ranging up to about 50%were observed in two tissues, pancreas andskeletal muscle. Such results may be variouslyinterpreted for or against the assumption thatthe inhibition of protein synthesis explainstoxicity. Unfortunately, we lack certain data
71VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
BACTERIOL. REV.
14)
6NKq)Z3V$.V.
1.11.(i
Lung9r
Hours Hours
FIG. 14. Protein synthesis in tissue slices from intoxicated guinea pigs. Each animal (350 a 20 g) received 5MLD of toxin intravenously. At intervals thereafter, organs from the perfused animals were removed and tissueslices were prepared and incubated in the presence of [14C]leucine. Perchloric acid-insoluble counts weredetermined and are indicated per gram wet weight of tissue. Reproduced from reference 8 by permission of theauthor and publisher.
necessary for a meaningful interpretation, suchas the degree of inhibition required for lethality,and the degree of inhibition which occurs invarious cell types composing the tissues stud-ied. Such studies underscore the difficulties instudying pharmacological effects in vivo.
Unfortunately, no attempt has been made toisolate ADPR-EF-2 from toxin-treated animals.Measurements of EF-2 by Baseman et al.seemed to indicate a degree of inactivationproportional to the decline in protein synthesis,but no other enzymes were assayed as controlsto demonstrate specificity (8). Other data sug-gesting reactivation of protein synthesis bynicotinamide in tissue extracts from toxin-treated animals are not entirely convincing (22).
Thus, the studies seem generally to show thatinhibition of protein synthesis occurs in toxin-treated animals, but specificity remains to bedemonstrated. Owing to the lack of precise dataand the variables which apparently affect thepattern of tissue damage, no attempt will bemade to analyze the secondary physiologicalchanges or the ultimate cause of death intoxin-treated animals.
EVENTS PRECEDING THEADP-RIBOSYLATION REACTION IN
CELLSTo inhibit protein synthesis, the fragment A
portion of toxin presumably must reach thecytoplasm. What is the mechanism of entry,
and what events occur subsequent to entry?Although our overall knowledge concerningthese questions is meager, some pertinent dataare available.
Attachment to Specific ReceptorsThe fact that fragment A and CRMs 30 and
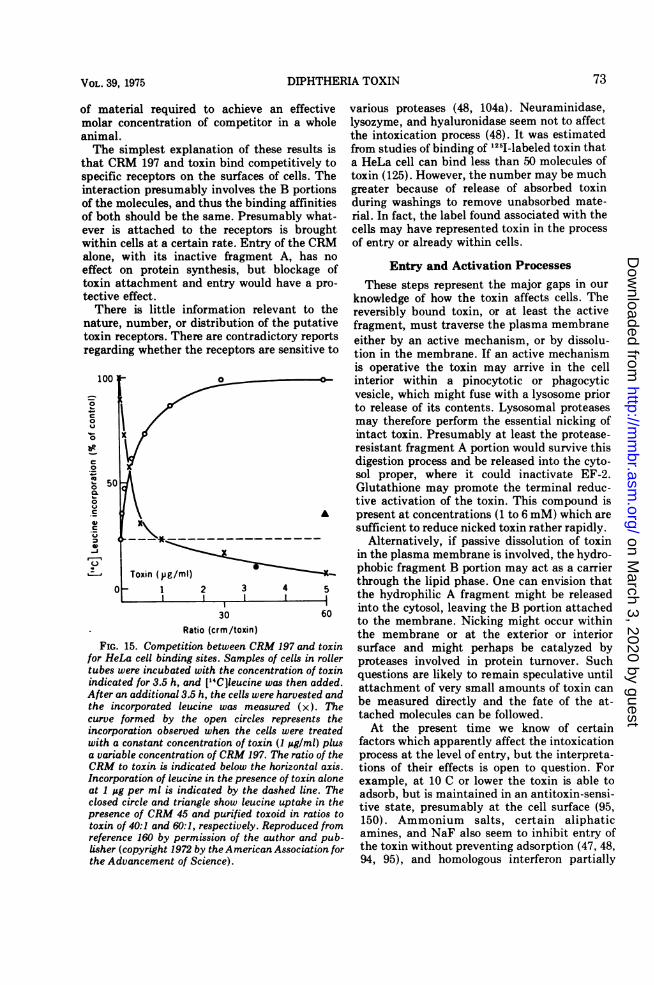
45 do not inhibit protein synthesis in intactcells, even though they are enzymatically ac-tive, indicates that entry involves somethingmore specific than simply passive inclusion oftoxin within the aqueous phase of pinocytoticvesicles. Furthermore, evidence from the studyof CRM 197 strongly suggests that the initialinteraction is with specific receptors on the cellsurface. This CRM (87, 160, 162) which con-tains an inactive fragment A and a functionalfragment B, has the interesting property ofinhibiting the action of toxin on protein synthe-sis in tissue culture, although it does not inhibitADP-ribosylation activity in cell-free systems(Fig. 15). The effect has been studied in somedetail, and it has been concluded that the CRMacts in a strictly competitive manner with a KDof about 10-8 M.The competitive effect of CRM 197 has also
been demonstrated qualitatively in whole ani-mals using the skin reaction as an assay fortoxin action (87), but diffusion of the competi-tor from the area of injection prevents precisequantification. Competition in lethality tests isnot feasible to show because of the large amount
72 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
DIPHTHERIA TOXIN
of material required to achieve an effectivemolar concentration of competitor in a wholeanimal.The simplest explanation of these results is
that CRM 197 and toxin bind competitively tospecific receptors on the surfaces of cells. Theinteraction presumably involves the B portionsof the molecules, and thus the binding affinitiesof both should be the same. Presumably what-ever is attached to the receptors is broughtwithin cells at a certain rate. Entry of the CRMalone, with its inactive fragment A, has noeffect on protein synthesis, but blockage oftoxin altective
Therenature,toxin reregardii
100I
0
C
0
QI*-
0
0
6.
500
CLb-0
.uA_r-J-
0o
FIG.
for Heltubes u
indicat
After at
the inccurve Yincorpowith a
a varialCRM toIncorpcat 1 ggclosed i
presenctoxin o0referentUisher (athe Ad
various proteases (48, 104a). Neuraminidase,lysozyme, and hyaluronidase seem not to affectthe intoxication process (48). It was estimatedfrom studies of binding of l25l-labeled toxin thata HeLa cell can bind less than 50 molecules oftoxin (125). However, the number may be muchgreater because of release of absorbed toxinduring washings to remove unabsorbed mate-rial. In fact, the label found associated with thecells may have represented toxin in the processof entry or already within cells.
Entry and Activation Processesttachment and entry would have a pro- These steps represent the major gaps in oureffect. knowledge of how the toxin affects cells. Thee is little information relevant to the reversibly bound toxin, or at least the activenumber, or distribution of the putative fragment, must traverse the plasma membraneDceptors. There are contradictory reports either by an active mechanism, or by dissolu-ng whether the receptors are sensitive to tion in the membrane. If an active mechanism
is operative the toxin may arrive in the cell0 interior within a pinocytotic or phagocytic
vesicle, which might fuse with a lysosome priorto release of its contents. Lysosomal proteasesmay therefore perform the essential nicking ofintact toxin. Presumably at least the protease-resistant fragment A portion would survive thisdigestion process and be released into the cyto-sol proper, where it could inactivate EF-2.Glutathione may promote the terminal reduc-tive activation of the toxin. This compound is
A present at concentrations (1 to 6 mM) which aresufficient to reduce nicked toxin rather rapidly.
I---*---~-~--~-.--- Alternatively, if passive dissolution of toxinin the plasma membrane is involved, the hydro-
Toxin (pg/ml) phobic fragment B portion may act as a carrier-l 2 3 4 through the lipid phase. One can envision that
I2
I I I the hydrophilic A fragment might be released
30 60 into the cytosol, leaving the B portion attachedto the membrane. Nicking might occur withinRatio (crm/toxin) the membrane or at the exterior or interior
15. Competition between CRM 197 and toxin surface and might perhaps be catalyzed byLa cell binding sites. Samples of cells in roller proteases involved in protein turnover. Suchvere incubated with the concentration of toxin questions are likely to remain speculative untiled for 3.5 h, and [CCl]eucine was then added. attachment of very small amounts of toxin cann additional 3.5 h, the cells were harvested andcorporated leucine was measured (x). The be measured directly and the fate of the at-formed by the open circles represents the tached molecules can be followed.ration observed when the cells were treated At the present time we know of certainconstant concentration of toxin (1 ;Lg/ml) plus factors which apparently affect the intoxicationble concentration of CRM 197. The ratio of the process at the level of entry, but the interpreta-o toxin is indicated below the horizontal axis. tions of their effects is open to question. For)ration of leucine in the presence of toxin alone example, at 10 C or lower the toxin is able toper ml is indicated by the dashed line. The adsorb, but is maintained in an antitoxin-sensi-
circle and triangle show leucine uptake in the tive state, presumably at the cell surface (95,'e of CRM 45 and purified toxoid in ratios to 150). Ammonium salts, certain aliphaticf 40:1 and 60:1, respectively. Reproduced from a50)- A nd salso ertain entry ofce 160 by permission of the author and pub- amines, and NaF also seem to inhibit entry ofcopyright 1972 by the American Association for the toxin without preventing adsorption (47, 48,vancement of Science). 94, 95), and homologous interferon partially
73VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
protects against the toxin, perhaps by an effectat the same level (110). In contrast, the sensitiv-ity of toxin-resistant mouse cell lines to thetoxin may be enhanced by polycations, such aspolyornithine or poly-DEAE-dextran (106). Theeffects of these factors may perhaps be ex-plained by their stimulation or inhibition ofendocytotic activity, but further studies areneeded to confirm this. If this is the case, thetoxin would presumably enter the cell attachedto membrane receptor sites on the interiorsurfaces of endocytotic vesicles.
Cell lines which are resistant to the toxin willcertainly be useful in studying the adsorptionand entry processes. A block at this level hasbeen inferred not only for cell lines from toxin-resistant animals, but also for toxin-resistantvariants of normally sensitive cell lines. Moehr-ing and Moehring have selected for clones ofhuman KB cells which are resistant to moderatelevels of toxin (107). The change to toxinresistance seems to be stable, in that resistanceis maintained when the cells are cultivated inthe absence of toxin. Protein synthesis wasstudied in extracts of one of the resistant strainsand was found to remain highly sensitive to thetoxin, thus indicating that resistance was notdue to an alteration of EF-2. Other evidencesuggested that changes had occurred in the cellsurface. Thus, resistant clones formed colonieswith different morphology and were resistant todispersion by trypsin. In addition, toxin resist-ance seemed to be accompanied by an increasedresistance to certain RNA viruses, includingpoliovirus, Mengo virus, vesicular stomatitisvirus, and Newcastle disease virus (108). Ad-sorption of the viruses occurred, but uncoatingand release of viral RNA seemed to be inhibitedin certain resistant cells. In two strains therewas also a reduced production of viral mRNA. Ithas been suggested that an alteration may haveoccurred in a proteolytic activity required bothfor nicking of toxin and uncoating of certainviruses (108), but the relationship of virusresistance to toxin resistance remains uncer-tain.
Kinetics of Entry and TurnoverBy whatever mechanism toxin enters, it be-
gins to inactivate EF-2 within minutes, longbefore the end of the lag period. Gill et al. haveshown that the total elongation factor activityin the supernatant fraction of toxin-treated cellsdrops markedly within 20 min although proteinsynthesis is not affected until 1 h (60). Thediscrepancy here apparently results from thefact that EF-2 is not normally the limiting
factor in protein synthesis. Gill and Dinius haveshown that EF-2 is present in a molar ratio of1.2 molecules per ribosome in a variety oftissues (57). Apparently only after this ratiodrops as a result of ADP-ribosylation to someas-yet-undetermined low value does EF-2 beginto limit the rate of protein synthesis.Entry of toxin continues well beyond the end
of the lag period, but the exact duration is notknown. Protracted entry is well demonstratedwith CRM 176, which has a fragment A with 8to 10% the normal specific activity, and whichinhibits protein synthesis in HeLa cells to alesser extent than similar levels of toxin (162). Ifcells are treated with CRM 176 for 1.5 or even 3h, and then washed, the decline in the rate ofprotein synthesis halts, and the cells recover.Inasmuch as the decline continues in cellsfurther treated with the CRM, entry must beoccurring at these times. In cells treated withtoxin for 1.5 h and resuspended in toxin-freemedium (containing CRM 197 to prevent fur-ther entry of toxin), the decline in the rate ofprotein synthesis was only marginally affectedby the removal of toxin, and the cells did notrecover.The recovery of CRM 176-treated cells pre-
sumably depends on degradation of the A176present in the cytoplasm, and it is reasonablethat any protein brought within cells should besubject to the protein turnover system (66). Thegreater stability and/or specific activity of nor-mal fragment A presumably accounts for thelack of recovery of cells treated similarly withtoxin.
After the lag period the decline in the rate ofprotein synthesis in toxin-treated cells appearsto follow first-order kinetics (162). It has beensuggested that this implies the presence of avirtually constant concentration of fragment Awithin cells, presumably a steady state betweenentry and degradation. This is plausible, butother explanations for such kinetics must beconsidered in view of the number of unknownsaffecting the intracellular reaction.
Calculations of the specific activity of frag-ment A in the intracellular milieu cannot yet beperformed with confidence because of the largenumber of uncertainties in variables affectingenzymatic activity and the substrate activityand availability of EF-2. Therefore, it is notpossible to predict accurately on this basis theconcentration of intracellular fragment Aneeded to inhibit protein synthesis at a givenrate. Unfortunately this concentration is too lowto measure directly.There has been considerable speculation
74 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
about the minimum number of toxin moleculesrequired to kill a cell (48, 61, 122, 123). Withcertain cell lines it is clear that concentrationsof toxin equivalent to several hundred mole-cules per cell are sufficient to cause deathwithin 3 to 4 days (54, 130). The actual numbermay be much lower, however, because a signifi-cant percentage of the toxin may not have beentaken up by cells. Studies of the uptake of1251-labeled toxin by HeLa cells suggest that 50molecules of toxin or less may be sufficient tocause cell death (122), and from the fact thatthe kinetics of adsorption of a lethal dose oftoxin by HeLa cells are first order, it has beensuggested that a single molecule of toxin may belethal (48). In view of the complexities andunknowns in the intoxication process, onewould like to have independent confirmation ofthe last figure.
IMMUNOLOGY
Avidity Correlates with Antibody AgainstFragment B
Only recent studies pertaining directly to thestructure and activity of the toxin will beconsidered here; earlier studies which contrib-uted so much to the understanding of antigen-antibody interactions (121) will be ignored.Pappenheimer, Uchida, and Harper have re-
ported an immunological study of the toxinmolecule making use of the fragmentation tech-niques and CRMs available (127). As studied byimmunodiffusion in the presence of 0.5 M urea,fragments A and B are immunologically dis-tinct, and both formed lines of partial identitywith toxin when tested against antitoxin (Fig.16) (9, 127). CRM 197 is immunologically indis-tinguishable from toxin, whereas CRM 45 showspartial identity. Both fragments A and B formlines of partial identity with CRM 45.Other findings on antisera or absorbed sera
directed against specific portions of the toxinmolecule were less predictable. By adsorption ofantitoxin with CRM 45, a serum was obtainedwhich was specific for the C-terminal 17,000daltons of the toxin. Interestingly, the avidity ofthis serum was greater than the unadsorbedsample; that is, it had an increased capacity perunit of antibody protein, for neutralization oftoxicity. This finding suggested that antibodiesagainst the C-terminal region of the toxin havea greater capacity for neutralization than thoseagainst the remaining portion.
Other results neatly confirmed this predic-tion. Thus antisera against purified fragment A,and which quantitatively precipitated the frag-
ment, were found to contain almost no capacityto neutralize toxicity. Furthermore, among anumber of antisera tested, the avidity wasinversely related to the proportion of anti-frag-ment A, expressed as percent of total toxin-precipitable antibody. Thus, there appears tobe a basic difference in neutralizing capacity ofantibodies directed against the N- and C-termi-nal regions of the molecule.That antibodies against a region necessary for
attachment to cells should block toxic activityis not surprising, but the lack of neutralizingcapacity of those against the fragment A regionrequires explanation.The low avidity of anti-fragment A antibodies
could be due to a low affinity constant, permit-ting dissociation of toxin-antitoxin complexes invivo. Alternatively, an antibody of high affinitymight become detached during or after theadsorption and entry of toxin into cells. Forexample, this might occur as a result of analtered conformation of the receptor-boundtoxin, or might take place after traversal of theplasma membrane, perhaps following cleavageor reduction events. Complexes of antitoxinwith fragment A or nicked toxin are dissociatedin the presence of thiols, yielding active frag-ment A (127). (This finding probably explainsthe early observation that washed toxin-antitoxin floccules are slightly active in inhibit-ing protein synthesis in cell-free systems (35,123]).A more complex situation is suggested by the
fact that anti-fragment A antibody precipitatesonly a fraction of 125I-labeled toxin (127). Fur-thermore, in antibody excess, unprecipitatedtoxin did not appear to be complexed withimmunoglobulin, as judged by Sephadex chro-matography. This suggests that the fragment Adeterminants may be obscured in a fraction ofthe population of toxin molecules. This wouldpartially explain the lack of neutralizing capac-ity of anti-fragment A antibodies.
Immunogenicity and the Mechanism ofToxoiding
Pappenheimer et al. reported other interest-ing results relevant to the immunogenicty ofdiphtheria toxin (127). Antitoxins elicitedagainst formaldehyde-treated toxin, or toxoid,in rabbits or horses varied in content of anti-fragment A antibodies and hence in avidity. Intwo sera of moderate to high avidity, about 30%of the toxin-precipitable antibodies wereagainst fragment A, whereas another horse-antitoxic globulin preparation (number 5353) ofparticularly high avidity was found to contain
75VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from
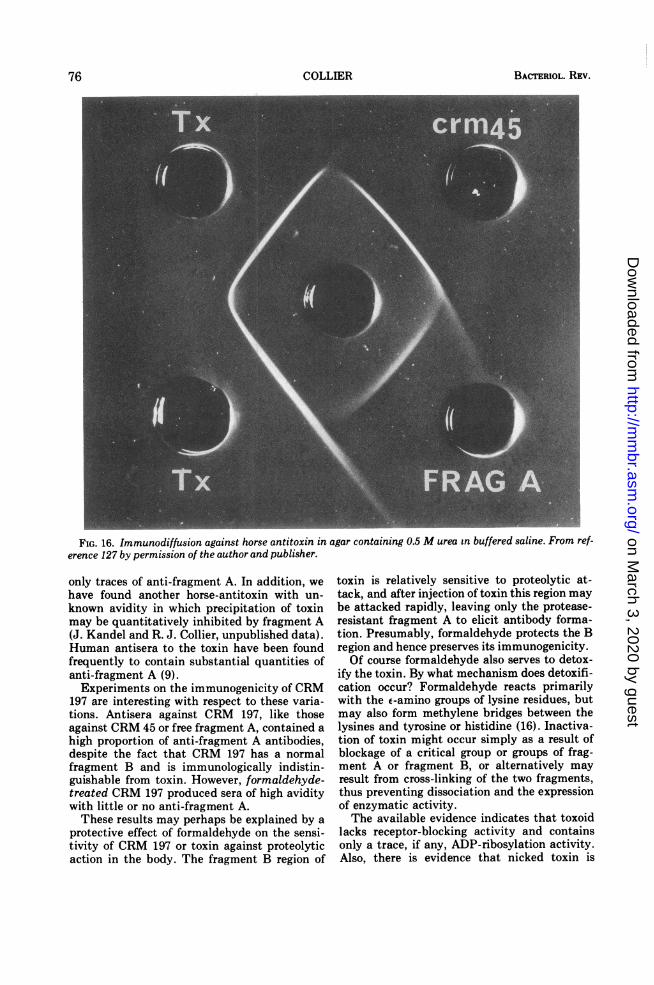
BACTERIOL. REV.
FIG. 16. Immunodiffusion against horse antitoxin in agar containing 0.5 M urea in buffered saline. From ref-erence 127 by permission of the author and publisher.
only traces of anti-fragment A. In addition, we
have found another horse-antitoxin with un-
known avidity in which precipitation of toxinmay be quantitatively inhibited by fragment A(J. Kandel and R. J. Collier, unpublished data).Human antisera to the toxin have been foundfrequently to contain substantial quantities ofanti-fragment A (9).Experiments on the immunogenicity of CRM
197 are interesting with respect to these varia-tions. Antisera against CRM 197, like thoseagainst CRM 45 or free fragment A, contained a
high proportion of anti-fragment A antibodies,despite the fact that CRM 197 has a normalfragment B and is immunologically indistin-guishable from toxin. However, formaldehyde-treated CRM 197 produced sera of high aviditywith little or no anti-fragment A.These results may perhaps be explained by a
protective effect of formaldehyde on the sensi-tivity of CRM 197 or toxin against proteolyticaction in the body. The fragment B region of
toxin is relatively sensitive to proteolytic at-tack, and after injection of toxin this region maybe attacked rapidly, leaving only the protease-resistant fragment A to elicit antibody forma-tion. Presumably, formaldehyde protects the Bregion and hence preserves its immunogenicity.Of course formaldehyde also serves to detox-
ify the toxin. By what mechanism does detoxifi-cation occur? Formaldehyde reacts primarilywith the E-amino groups of lysine residues, butmay also form methylene bridges between thelysines and tyrosine or histidine (16). Inactiva-tion of toxin might occur simply as a result ofblockage of a critical group or groups of frag-ment A or fragment B, or alternatively mayresult from cross-linking of the two fragments,thus preventing dissociation and the expressionof enzymatic activity.The available evidence indicates that toxoid
lacks receptor-blocking activity and containsonly a trace, if any, ADP-ribosylation activity.Also, there is evidence that nicked toxin is
76 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
resistant to dissociation by thiols and SDS afterformaldehyde treatment (9, 100, 104). It isclear, then, that the fragment B region isinactivated, but it is not yet known whether thelack of activity of fragment A within toxoid isdue to modification of essential groups or to theinability to be dissociated from B. Beugnier andZanen have reported evidence favoring theformer possibility (13). All of the mechanismsmentioned may in fact occur and contribute tovarious degrees to detoxification.
BIOLOGICAL FUNCTION AND ORIGINOF DIPHTHERIA TOXIN
With some solid information on the structureand biochemical activity of diphtheria toxin inhand, one must consider whether we are anycloser to understanding the origin and raisond'itre of the toxin. How did the toxin evolve,and what selective advantage accrues to phagecarrying the tox+ gene and/or to their hostbacteria?
Biological FunctionIn essence, we must examine the possible
role(s) of the toxin in the lysogen-human hostinteraction, and that between the phage and itsbacterial host.
(i) The lysogen-human host interaction.Our tendency in thinking about a selectiveadvantage or biological function of toxin pro-duction is to focus on the dramatic, highlymalignant infections of C. diphtheriae, but in sodoing we are probably misled. In early surveysof naturally acquired immunity to the toxin inchildren, it was found that most infectionssufficient to cause such immunity do not resultin serious disease (25). Thus, as with manyother pathogens, the number of subclinicalinfections appears to outnumber greatly theclinical ones. In addition, it seems quite certainthat death of the human host does not benefiteither the lysogen or the phage, inasmuch as adeceased host is unable to transmit either. Bothof these considerations lead us to conclude thatthe selective advantage of toxicity, if any, isprobably exerted in mild or symptom-free upperrespiratory infections, rather than the severeforms of the disease. Thus, highly malignantdiphtheria may perhaps best be viewed as abiological accident, of little significance to thelong-term survival of toxigenic strains.We certainly do not know the true function
of the toxin, but one can conceive that irritationof the epithelial tissues of the upper respiratorytract by the toxin might facilitate growth, orperhaps promote dissemination by inducing
coughing or sneezing. Alternatively, the toxinmight provide a local defense against phagocy-tosis. Whatever the effect, it is somewhat anti-climactic in comparison with the more malignmanifestations of toxicity.
(ii) The phage-bacterium interaction. Thisproblem is at least amenable to experimentalstudy. Given that the structural gene for toxinresides within a phage genome, one must firstask whether the toxin plays a direct role inphage replication. It has been argued that thetoxin cannot be essential for phage replicationinasmuch as the mutations producing CRMs 30,45, 176, 197, and 228 do not affect the one-stepgrowth curves after ultraviolet induction oflysogens carrying these mutants (158, 159).However, mutations which alter the toxicity ofthe toxin would not necessarily block a differentfunction in phage replication. Moreover, itshould be noted that the isolation procedureemployed selected for viable phage mutants inthe first place, thereby inserting a bias into theresults.
Iglewski and co-workers have published evi-dence suggesting that fragment B from thetoxin may form part of the phage B virion (51).
Virions of a virulent mutant of is, purified bydifferential centrifugation and isopycnic band-ing in CsCl gradients, show a major proteinband in SDS gels which comigrates with frag-ment B. Also, it was reported that such purifiedphage exhibited blocking activity in protectingKB cells from diphtheria toxin, implying thepresence of functionally active fragment B. Amajor weakness in these results is the possibilityof copurification or adsorption of fragment Bwhich is not an integral part of the phage. Also,one would like more positive evidence (e.g.,amino acid sequence data) confirming that theprotein observed on gels is in fact fragment B.At present the data seem insufficient to
decide definitely for or against an essentialfunction for the toxin in phage replication. Evenif no essential role can be found, it is stillpossible that the toxin could perform a directbut nonessential function in replication. Fi-nally, in the absence of the latter, the phagemight benefit indirectly by an unknown mecha-nism by virtue of the effects of toxin on thehuman host.
OriginGiven the location of the structural gene for
diphtheria toxin, it is simplest to suppose thattox+ originated from a phage gene, even if itnow serves neither an essential nor a facultativefunction in phage replication. This notion has
VOL. 39, 1975 77
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

COLLIER
recently received indirect support from a find-ing by Goff in E. coli infected with phage T4 (64,65). In such cells the a subunit ofRNA polymer-ase was found to be modified by covalentattachment of the ADPR moiety of NAD+.The facts that this modification (i) involves
the same group-transfer from the same pyridinenucleotide substrate, (ii) is catalyzed by a
phage-encoded enzyme, and (iii) is the firstexample in prokaryotes of ADP-ribosylation of aprotein, invite speculation that it may be re-
lated to the origin of diphtheria toxin. One can
conceive that the ADP-ribosylation activity ofthe fragment A portion of the toxin may haveoriginated from a phage enzyme which cata-lyzed a similar group-transfer to a subunit ofRNA polymerase or some other protein involvedin phage replication. The gene for this enzymemight have been joined by gene fusion with thatof another protein (the phage coat protein?)corresponding to the B portion of the toxin, withgene duplication accounting for essential genefunctions destroyed in the fusion process.Whether this hypothesis deserves much cre-
dence will depend in part upon whether ADP-ribosylation of proteins is found in other phageinfections, including those by corynephages. Nosignificant incorporation of ADPR from NAD+has been detected in extracts from the PW-8strain of C. diphtheriae incubated either in thepresence or absence of fragment A, suggestingthat such cells contain no protein acceptor forADPR. However, such an acceptor might easilyhave been overlooked if it were labile or presentin low concentrations. Goff has found (personalcommunication) that fragment A does not cata-lyze ADP-ribosylation of the a subunit of E. coliRNA polymerase, but the corresponding en-zyme from diphtheria bacilli has not beentested.
Alternatively one can conceive that the toxinmay have originated from NAD+-glycohydro-lase or a NAD+-linked dehydrogenase. Thetransition to an enzyme capable of transferringthe ADPR moiety to a specific protein mightwell be relatively minor in either case. Presum-ably the substrate activity of EF-2 could only beaccounted for by a chance affinity for theenzyme.
Other authors have suggested that the genefor diphtheria toxin may have originated in thegenome of a eukaryotic host (158). The appar-ent rationale for this is that the only knownefficient protein substrate for the toxin is a
eukaryotic intracellular protein. It is supposedthat the ancestral gene for the toxin might haveencoded a regulatory protein which modified
the activity of EF-2 or a similar protein, andthat the phage picked up this gene through itsclose association with a eukaryotic host. Thereis, however, no evidence for such a regulatoryprotein at the present time.None of the above theories offers an explana-
tion for the origin of the binding site on toxin forspecific cell surface receptors. Speculation onthis point is largely futile until we know moreabout the chemistry of the receptors.
SUMMARY AND CONCLUDINGREMARKS
The following is suggested as a possible se-quence of events in the action of diphtheriatoxin in clinical diphtheria.
(i) The toxin is excreted by C. diphtheriae inthe form of a single polypeptide chain, molecu-lar weight 63,000, and gains entry into the bodythrough lesions in the epithelium at the site ofthe infection.
(ii) It is transported throughout the body bythe blood and lymphatic systems and attacksmany or most types of cells to which it isexposed.
(iii) The toxin attaches with a KD of about 10nM to specific receptors on the cell surface, thenature and number of which are unknown.
(iv) While attached to the surface mem-brane, it is transported within the cell byendocytosis. The toxin may have been partiallynicked before it arrives at the cell surface, butnicking may also occur intracellularly, perhapswithin the endocytotic vesicle through the ac-tion of lysozomal or other proteases.
(v) A certain percentage of the toxin mole-cules, or at least the fragment A portion,survives and is released into the cytosol, wherereduction and release of fragment A may bepromoted by glutathione.
(vi) Fragment A inactivates the free form ofEF-2 by catalyzing attachment of the ADPRmoiety of intracellular NAD+. The modifiedfactor can still attach to ribosomes and bindGTP, but is inactive in promoting transloca-tion.
(vii) EF-2 apparently does not normally limitthe rate of protein synthesis, and only after asizable fraction has been inactivated does therate of protein synthesis decline. After thisinhibition, other cellular processes are affected,producing necrosis, gross physiological changes,and sometimes death.
Certain aspects of this sequence are wellsupported by data, in particular, the enzymol-ogy of the toxin and its effect on proteinsynthesis. On the other hand, we are badly in
78 BACTERIOL. REV.
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
need of more evidence regarding the attachmentand entry phenomena. In view of the rapidadvance of techniques for studying biologicalmembranes, one can hope that such data will beforthcoming soon.
In conclusion one should perhaps considerpossible implications of the studies reviewedhere. First, what generalizations are possible?With regard to the ADP-ribosylation reac-
tion, there is no evidence to suggest it may be inany way general among toxins (163). Althoughthe precise actions of most bacterial toxins arenot known, their gross activities give no sugges-tion that they may act like diphtheria toxin.On the other hand, the structure-activity
relationships evidenced in diphtheria toxin mayrepresent a more general phenomenon. In cer-tain other bacterial toxins and in two toxinsfrom plants there is evidence that the functionsof attachment to cells and of toxic modificationof cells (the effector function) may reside indifferent polypeptide chains. The data are clearin the case of the plant toxins, ricin and abrin,both of which inhibit protein synthesis bymechanisms unrelated to ADP-ribosylation(117-119). Each of these contains two chains indisulfide linkage. One of the chains (the Bchain) is active in attachment to cells, and theother (A chain) inhibits protein synthesis. Theactivity of the A chain is apparently expressedonly after reduction and dissociation from the Bchain. Both toxins attach to galactose-contain-ing receptors on the cell surface.Another example is cholera toxin. The toxin
appears to be dissociable under nonreducingconditions into a subunit (molecular weight66,000) which is devoid of biological activity butwhich competitively binds to the same mem-brane site as the toxin, and another (molecularweight 36,000) which presumably has the effec-tor activity of the toxin (43). The toxin appearsto act by stimulating adenyl-cyclase in cells byan undefined mechanism. The binding sites onthe cell surface have been shown to be ganglio-sides (41, 42).
Finally, in some clostridial toxins there isevidence for a form containing two disulfide-linked chains. In certain botulinal toxins thereis an activated form created by proteolyticnicking, and reduction produces a loss of toxic-ity (10, 44, 152). A similar situation may existwith tetanus toxin (39). The specific functionsof the chains are not well defined in either case.
It will be interesting to know whether thereceptor-attachment and effector functions ofprotein toxins generally reside on separate pep-tide chains. The various toxins almost certainly
do not have a common origin, but evolutionaryconstraints may exist which dictate such astructure. Different locations of the receptor-binding and effector activities might be tavoredif the receptor-attachment region remains per-manently fixed to the membrane; release of theeffector region into the cytosol might be facili-tated if it were on a separate chain.
Diphtheria toxin has also proven to be auseful tool in studying certain aspects of proteinsynthesis. In addition to inactivating EF-2 andblocking translocation specifically, the toxinoffers the advantage of attaching a specificradioactive label to EF-2. This has provenuseful in quantifying EF-2 in crude tissue ex-tracts and in studying the mechanism of trans-location in detail (11, 12, 36, 57, 59, 153-155).An example of the latter application is recentwork using ADPR-EF-2 to identify by chemicalcross-linking the proteins to which EF-2 bindson the ribosome (157).What about possible medical applications of
the research? It was hoped that a means oftherapy useful in clinical diphtheria beyondrescue with antitoxin would have been revealed.Knowledge of the intracellular events mighthave provided a means of reversing or at leastlimiting the lethal action. Unfortunately, thishas not occurred. To reverse the reaction withnicotinamide, one would need intracellular lev-els of this compound which themselves would betoxic even if they were attainable. Althoughcertain of the CRMs block the toxin's activityon whole cells, they are no more useful thanantitoxin because they do not affect toxin whichhas already entered the cell.With regard to prophylaxis against diph-
theria, there is really no need to improve uponthe efficacy of toxoid for immunization. How-ever, use of one of the nontoxic CRMs for thepreparation of toxoid should eliminate the occa-sional problem of partial reversion of toxoid tothe toxic state (1).
Finally, there appears to be a remote possibil-ity that our knowledge about diphtheria toxinmay have practical applications outside therealm of bacteriology. It has been reportedrecently that certain types of malignant cellsfrom mice or humans may be inherently moresensitive to diphtheria toxin than normal cellsfrom the corresponding organism (26, 86). Inaddition, there have been other reports suggest-ing that the toxin may be converted into a morespecific cytotoxic agent by covalently conjugat-ing it to antibodies directed against specificcellular antigens (114, 128, 143). It is difficult topredict if such studies may ever reach the stage
79VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
of practical therapy, but this possibility shouldnot be neglected.
ACKNOWLEDGMENTSI wish to thank A. M. Pappenheimer, Jr., for
introducing me to the study of the action of diphtheriatoxin, and Lane Barksdale for his continued, stimu-lating support. Valuable criticism and help with themanuscript were provided by Drs. Barksdale andSamuel Raeburn.The work in my laboratory has been supported by
Public Health Service Grant AI-07877 from the Na-tional Institute of Allergy and Infectious Diseases andby National Science Foundation Grant GB-32406,and could not have been done without the collabora-tive efforts of Dominic Chung, R. J. DeLange, RaeDrazin, Gerald Hodge, Judith Kandel, Meryl Maler,and Jolinda Traugh.
This review was developed from the Eli Lilly andCo. Award Lecture given at the 1972 ASM AnnualMeeting. It was prepared in part while I was on sab-batical leave in the laboratory of Fran~ois Gros, In-stitut Pasteur, Paris, where I was supported by afellowship from the John Simon Guggenheim Foun-dation.
LITERATURE CITED1. Akama, K., S. Kameyama, S. Otani, S. Sada-
hiro, and R. Murata. 1971. Reversion of tox-icity of diphtheria toxoid. Jpn. J. Med. Sci.Biol. 24:183-187.
2. Andrewes, F. W., W. Bulloch, S. R. Douglas, G.Dreyer, A. D. Gardner, P. Fildes, J. C. G.Ledingham, and C. G. L. Wolf. 1923. Diph-theria, its bacteriology, pathology, and immu-nology. Medical Research Council, London.
3. Barksdale, L. 1970. Corynebacterium diph-theriae and its relatives. Bacteriol. Rev.34:378-422.
4. Barksdale, L., and S. B. Arden. 1974. Persistingbacteriophage infections, lysogeny, and phageconversions. Annu. Rev. Microbiol.28:265-298.
5. Barksdale, L., L. Garmise, and K. Horibata.1960. Virulence, toxinogeny, and lysogeny inCorynebacterium diphtheriae. Ann. N.Y.Acad. Sci. 88:1093-1108.
6. Barksdale, W. L., L. Garmise, and R. Rivera.1961. Toxinogeny in Coiynebacterium diph-theriae. J. Bacteriol. 81:527-540.
7. Barksdale, W. L., and A. M. Pappenheimer, Jr.1954. Phage-host relationships in nontoxigenicand toxigenic diphtheria bacilli. J. Bacteriol.67:220-232.
8. Baseman, J. B., A. M. Pappenheimer, Jr., D. M.Gill, and A. A. Harper. 1970. Action of diph-theria toxin in the guinea pig. J. Exp. Med.132:1138-1152.
9. Bazaral, M., P. J. Goseienski, and R. N. Ham-burger. 1973. Characteristics of human anti-body to diphtheria toxin. Infect. Immun.7:130-136.
10. Beers, W. H., and E. Reich. 1969. Isolation andcharacterization of Clostridium botulinum
type B toxin. J. Biol. Chem. 244:4473-4479.11. Bermek, E. 1972. Formation of a complex involv-
ing ADP-ribosylated human translocation fac-tor, guanosine nucleotide, and ribosomes.FEBS Lett. 23:95-99.
12. Bermek, E., H. Tsai, J. Tsai, and U. Ueer. 1974.Studies of ADP-ribosylated human transloca-tion factor, p. 325-332. In M. Harris (ed.),Poly (ADP-ribose), an international sympo-sium. U.S. Govt. Printing Office. Washing-ton, D.C.
13. Beugnier, N., and J. Zanen. 1972. Etude de ladetoxification de la toxine diphterique. Arch.Int. Physiol. Biochim. 80:389-390.
14. Beugnier. N., and J. Zanen. 1973. Mise enevidence d'une tyrosine dans le site en-zymatique de la toxine diphterique. Arch. Int.Physiol. Biochim. 81:581.
15. Bizzini, B., R. 0. Prudhomme, A. Turpin, andM. Raynaud. 1963. Essai de mise en evidencede liaisons disulfure dans la toxine tetaniqueet la toxine diphterique. Bull. Soc. Chim. Biol.45:925-932.
16. Blass, J., M. Bizzini, and M. Raynaud. 1967.Etudes sur le mechenisme de la de'toxificationdes toxines proteiques par le formol. Bull. Soc.Chim. Fr. 10:3957-3965.
17. Bonventre, P. F., and J. G. Imhoff. 1966. Studieson the mode of action of diphtheria toxin. V.Protein metabolism in a guinea pig modelsimulating chronic diphtheritic toxemia. In-fect. Immun. 7:556-560.
18. Bonventre, P. F., and J. G. Imhoff. 1966. Studieson the mode of action of diphtheria toxin. I.Protein synthesis in guinea pig tissues. J. Exp.Med. 124:1107-1122.
19. Bonventre, P. F., and J. M. Imhoff. 1967. Stud-ies on the mode of action of diphtheria toxin.II. Protein synthesis in primary heart cultures.J. Exp. Med. 126:1079-1086.
20. Bonventre, P. F., and C. B. Saelinger. 1972.Inhibition of protein synthesis after intrave-nous or intramuscular challenge with diph-theria toxin. Infect. Immun. 6:418-421.
21. Bowman, C. G., and P. F. Bonventre. 1970.Studies on the mode of action of diphtheriatoxin. III. Effect on subcellular components ofprotein synthesis from the tissues of intox-icated guinea pigs and rats. J. Exp. Med.131:659-674.
22. Bowman, C. G., and P. F. Bonventre. 1970.Specific reversal of diphtheria toxin mediatedinhibition of protein synthesis in guinea pigtissues. Biochem. Biophys. Res. Commun.41:1148-1154.
23. Bowman, C. G., J. G. Imhoff, and P. F. Bonven-tre. 1970. Specificity of diphtheria toxin actionon heart and muscle tissues of guinea pigs.Infect. Immun. 2:686-688.
24. Brooks, G. F., J. V. Bennett, and R. A. Feldman.1974. Diphtheria in the United States,1959-1970. J. Infect. Dis. 129:172-178.
25. Burnet, F. M., and D. O. White. 1972. Naturalhistory of infectious disease, 4th Ed. Cam-
80 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
bridge Univ. Press, London.26. Buzzi, S., and I. Maistrello. 1973. Inhibition of
growth of Ehrlich tumors in Swiss mice bydiphtheria toxin. Cancer Res. 33:2349-2353.
27. Caskey, T., P. Leder, K. Moldave, and D.Schlessinger. 1972. Translation: its mecha-nism and control. Science 176:195-197.
28. Chin, K. Y., and C. H. Huang. 1941. Myocardialnecrosis in diphtheria. Am. Heart. J.22:690-701.
29. Collier, R. J. 1967. Effect of diphtheria toxin on
protein synthesis: inactivation of one of thetransfer factors. J. Mol. Biol. 25:83-98.
30. Collier, R. J., and H. A. Cole. 1969. Diphtheriatoxin subunit active in vitro. Science164:1179-1182.
31. Collier, R. J., R. J. DeLange, R. Drazin, J.Kandel, and D. Chung. 1974. Enzymology ofdiphtheria toxin, p. 287-304. In M. Harris(ed.), Poly (ADP-ribose), an internationalsymposium. U.S. Govt. Printing Office,Washington, D.C.
32. Collier, R. J., R. Drazin, J. Kandel, R. J.DeLange, and D. W. Chung. 1974. Covalentalterations of protein structure in the inhibi-tion of protein synthesis by diphtheria toxin,p. 245-256. In E. Fischer, E. Krebs, H. Neu-rath, and E. Stadtman (ed.), Metabolic inter-conversion of enzymes. Springer-Verlag, Ber-lin.
33. Collier, R. J., and J. Kandel. 1971. Structure andactivity of diphtheria toxin. I. Thiol-depend-ent dissociation of a fraction of toxin intoenzymically active and inactive fragments. J.Biol. Chem. 246:1496-1503.
34. Collier, R. J., and A. M. Pappenheimer, Jr. 1964.Studies on the mode of action of diphtheriatoxin. I. Phosphorylated intermediates in nor-
mal and intoxicated HeLa cells. J. Exp. Med.120:1007-1018.
35. Collier, R. J., and A. M. Pappenheimer, Jr. 1964.Studies on the mode of action of diphtheriatoxin. II. Effect of toxin on amino acid incor-poration in cell-free systems. J. Exp. Med.120:1019-1039.
36. Collier, R. J., and J. Traugh. 1969. Inactiva-tion of aminoacyl transferase II by diphtheriatoxin. Cold Spring Harbor Symp. Quant. Biol.34:589-594.
37. Collins, J. F., S. Raeburn, and E. S. Maxwell.1971. Aminoacyl-transferase II from rat liver.II. Some physical and chemical properties ofthe purified enzyme and its adenosine diphos-phate ribose derivative. J. Biol. Chem.246:1049-1054.
38. Corkill, B. 1932. The influence of toxaemia on
carbohydrate metabolism. J. Physiol. 75:381-404.
39. Craven, C. J., and D. J. Dawson. 1973. The chaincomposition of tetanus toxin. Biochim. Bio-phys. Res. Commun. 317:277-285.
40. Cross, M. C. A., and E. Holmes. 1937. The effectof toxaemia and carbohydrate synthesis. Br. J.Exp. Pathol. 18:370.
41. Cuatrecasas, P. 1973. Gangliosides and mem-brane receptors for cholera toxin. Biochemis-try 12:3558-3566.
42. Cuatrecasas, P. 1973. Interaction of Vibrio chole-rae enterotoxin with cell membranes. Bio-chemistry 12:3547-3558.
43. Cuatrecasas, P., I. Parikh, and M. D. Hollen-berg. 1973. Affinity chromatography and struc-tural analysis of Vibrio cholerae enterotoxin-ganglioside agarose and the biological effectsof ganglioside containing soluble polymers.Biochemistry 12:4253-4264.
44. DasGupta, B. R., and H. Sugiyama. 1972. Acommon subunit structure in Clostridiumbotulinum type A, B, and E toxins. Biochem.Biophys. Res. Commun. 48:108-112.
45. Dawson, C. R., and E. Holmes. 1939. Themetabolism of lactic acid in diphtheritic tox-aemia. Br. J. Exp. Pathol. 20:357-370.
46. Drazin, R., J. Kandel, and R. J. Collier. 1971.Structure and activity of diphtheria toxin. II.Attack by trypsin at a specific site within theintact toxin molecule. J. Biol. Chem.246:1504-1510.
47. Duncan, J. L., and N. B. Groman. 1967. Studieson the activity of diphtheria toxin. I. Poliovirus replication in intoxicated HeLa cells. J.Exp. Med. 125:489-500.
48. Duncan, J. L., and N. B. Groman. 1969. Activityof diphtheria toxin. II. Early events in theintoxication of HeLa cells. J. Bacteriol.98:963-969.
49. Eaton, M. D. 1936. The purification and concen-tration of diphtheria toxin. J. Bacteriol.31:347-383.
50. Edward, D. G., and V. D. Allison. 1951. Diph-theria in immunized persons, with observa-tions on a diphtheria-like disease associatedwith non-toxigenic C. diphtheriae. J. Hyg.49:205-219.
51. Elwell, L. P., and B. H. Iglewski. 1972. Diph-theria toxin: evidence for presence of fragmentB in corynebacteriophage. Biochem. Biophys.Res. Commun. 49:609-614.
52. Everse, J., D. A. Gardner, N. 0. Kaplan, W.Galisinski, and K. Moldave. 1970. The forma-tion of a ternary complex between diphtheriatoxin, aminoacyltransferase II, and diphos-phopyridine nucleotide. J. Biol. Chem.245:899-901.
53. Freeman, V. J. 1951. Studies on the virulence ofbacteriophage-infected strains of Corynebac-terium diphtheriae. J. Bacteriol. 61:675-688.
54. Gabliks, J., and M. Solotorovsky. 1962. Cellculture reactivity to diphtheria staphylococ-cus, tetanus, and Escherichia coli toxins. J.Immunol. 88:505-512.
55. Gill, D. M. 1974. Self-modification of diphtheriatoxin, p. 333-337. In M. Harris (ed.), Poly(ADP-ribose), an international symposium.U.S. Govt. Printing Office, Washington, D.C.
56. Gill, D. M., and L. L. Dinius. 1971. Observationson the structure of diphtheria toxin. J. Biol.Chem. 246:1485-1491.
81VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
57. Gill, D. M., and L. L. Dinius. 1973. The elonga-tion factor 2 content of mammalian cells.Assay method and relation to ribosome num-
ber. J. Biol. Chem. 248:654-658.58. Gill, D. M., and A. M. Pappenheimer, Jr. 1971.
Structure-activity relationships in diphtheriatoxin. J. Biol. Chem. 246:1492-1495.
59. Gill, D. M., A. M. Pappenheimer, Jr., and J. B.Baseman. 1969. Studies on transferase II usingdiphtheria toxin. Cold Spring Harbor Symp.Quant. Biol. 34:595-602.
60. Gill, D. M., A. M. Pappenheimer, Jr., R. Brown,and J. J. Kurnick. 1969. Studies on the modeof action of diphtheria toxin. VII. Toxin-stimulated hydrolysis of nicotinamide adeninedinucleotide in mammalian cell extracts. J.Exp. Med. 129:1-21.
61. Gill, D. M., A. M. Pappenheimer, Jr., and T.Uchida. 1973. Diphtheria toxin, protein syn-
thesis, and the cell. Fed. Proc. 32:1508-1515.62. Gill, D. M., and D. M. Steinhaus. 1974. Modifi-
cation of diphtheria toxin by NAD+. J. Hyg.Epidemiol. Microbiol. Immunol. 18:316-323.
63. Gill, D. M., T. Uchida, and R. A. Singer. 1972.Expression of diphtheria toxin genes carriedby integrated and nonintegrated phage beta.Virology 50:664-668.
64. Goff, C. G. 1974. Chemical structure of a modifi-cation of the Escherichia coli ribonucleic acidpolymerase and polypeptides induced by bac-teriophage T4 infection. J. Biol. Chem.249:6181-6190.
65. Goff, C. G. 1974. T4-induced modification ofEscherichia coli RNA polymerase in vitro, p.235-244. In E. Fischer, E. Krebs, H. Neurath,and E. Stadtman (ed.), Metabolic conversionof enzymes. Springer-Verlag, Berlin.
66. Goldberg, A. L., and J. F. Dice. 1974. Intracellu-lar protein degradation in mammalian andbacterial cells. Annu. Rev. Biochem.43:835-869.
67. Goor, R. S. 1968. New form of diphtheria toxin.Nature (London) 217:1051-1053.
68. Goor, R. S., and E. S. Maxwell. 1969. A proposedmechanism for ADP-ribosylation of aminoacyltransferase II by diphtheria toxin. Cold SpringHarbor Symp. Quant. Biol. 34:609-610.
69. Goor, R. S., and E. S. Maxwell. 1970. Thediphtheria toxin-dependent adenosine diphos-phate ribosylation of rat liver aminoacyltransferase II. J. Biol. Chem. 245:616-623.
70. Goor, R. S., and A. M. Pappenheimer, Jr. 1967.Studies on the mode of action of diphtheriatoxin. III. Site of toxin action in cell-freeextracts. J. Exp. Med. 126:899-912.
71. Goor, R. S., and A. M. Pappenheimer, Jr. 1967.Studies on the mode of action of diphtheriatoxin. IV. Specificity of the cofactor (NAD)requirement for toxin action in cell-free sys-
tems. J. Exp. Med. 126:913-921.72. Goor, R. S., A. M. Pappenheimer, Jr., and E.
Ames. 1967. Studies on the mode of action ofdiphtheria toxin. V. Inhibition of peptidebond formation by toxin and NAD in cell-free
systems and its reversal by nicotinamide. J.Exp. Med. 126:923-939.
73. Gordon, H., and D. G. Fleck. 1971. An epidemicof diphtheria carriers. Public Health85:228-232.
74. Goto, N., I. Kato, and H. Sato. 1968. Theinhibitory effect-of diphtheria toxin on amino
acid incorporation by a bacterial cell-freesystem. Jpn. J. Exp. Med. 38:185-192.
75. Groman, N. B. 1953. Evidence for the inducednature of the change from nontoxigenicity totoxigenicity in Corynebacterium diphtheriae,as a result of exposure to specific bacterio-phage. J. Bacteriol. 66:184-191.
76. Groman, N. B. 1955. Evidence for the active roleof bacteriophage in the conversion of nontoxi-genic Corynebacterium diphtheriae to toxinproduction. J. Bacteriol. 69:9-15.
77. Groman, N. B., and M. Eaton. 1955. Geneticfactors in Corynebacterium diphtheriae con-
version. J. Bacteriol. 70:637-640.78. Groman, N. B., M. Eaton, and Z. K. Booher.
1958. Studies of mono and polylysogenic Co-rynebacterium diphtheriae. J. Bacteriol. 75:320-325.
79. Hameister, H., and D. Richter. 1970. Inhibitionof peptide chain elongation in a cell-freesystem from yeast by preincubation of theyeast peptidyl-translocase with diphtheriatoxin and NAD. J. Physiol. Chem.351:532-536.
80. Henriksen, O., and E. S. Maxwell. 1973. Interac-tion of guanosine nucleotides with elongationfactor 2. Fed. Proc. 32:494.
81. Holmes, R. K., and L. Barksdale. 1969. Geneticanalysis of tox+ and tox- bacteriophages ofCorynebacterium diphtheriae. J. Virol.3:586-598.
82. Honjo, T., and 0. Hayaishi. 1973. EnzymaticADP-ribosylation of proteins and regulation ofcellular activity. Curr. Topics Cell. Regul.7:87-127.
83. Honjo, T., Y. Nishizuka, 0. Hayaishi, and I.
Kato. 1968. Diphtheria toxin-dependent aden-osine diphosphate ribosylation of aminoacyltransferase II and inhibition of protein synthe-sis. J. Biol. Chem. 243:3553-3555.
84. Honjo, T., Y. Nishizuka, and 0. Hayaishi. 1969.Adenosine diphosphoribosylation of amino-acyl transferase II by diphtheria toxin. ColdSpring Harbor Symp. Quant. Biol. 34:603-608.
85. Honjo, T., Y. Nishizuka, I. Kato, and 0. Hayai-shi. 1971. Adenosine diphosphate ribosylationof aminoacyl transferase II and inhibition ofprotein synthesis by diphtheria toxin. J. Biol.Chem. 246:4251-4260.
86. Iglewski, B. H., and M. B. Rittenberg. 1974.Selective toxicity of diphtheria toxin for ma-
lignant cells. Proc. Nat. Acad. Sci. U.S.A.71:2707-2710.
87. Ittelson, T. R., and D. M. Gill. 1973. Diphtheriatoxin: specific competition for cell receptors.Nature (London) 242:330-332.
82 COLLIER
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
88. Johnson, W., R. J. Kuchler, and M. Soloto-rovsky. 1968. Site in cell-free protein synthesissensitive to diphtheria toxin. J. Bacteriol.96:1089-1098.
89. Kandel, J., R. J. Collier, and D. W. Chung. 1974.Interaction of fragment A from diphtheriatoxin with nicotinamide adenine dinucleotide.J. Biol. Chem. 249:2088-2097.
90. Kato, I. 1962. Mode of action of diphtheria toxinon protein synthesis. I. Effect of diphtheriatoxin on C 4 amino acid incorporation intomicrosomes and mitochondria in vitro. Jpn. J.Exp. Med. 32:335-343.
91. Kato, I., H. Nakamura, I. Uchida, J. Koyama,and T. Katsura. 1960. Purification of diph-theria toxin. II. The isolation of crystallinetoxin-protein and some of its properties. Jpn.J. Exp. Med. 30:129-145.
92. Kato, I., and A. M. Pappenheimer, Jr. 1960. Anearly effect of diphtheria toxin on the metabo-lism of mammalian cells growing in culture. J.Exp. Med. 112:329-349.
93. Kato, I., and H. Sato. 1962. Mode of action ofdiphtheria toxin on protein synthesis. II. Ef-fect of diphtheria toxin on biosynthesis ofserum albumin in cell-free systems. Jpn. J.Exp. Med. 32:495-504.
94. Kim, K., and N. B. Groman. 1965. Mode ofinhibition of diphtheria toxin ammoniumchloride. J. Bacteriol. 90:1557-1562.
95. Kim, K., and N. B. Groman. 1965. In vitroinhibition of diphtheria toxin action by am-monium salts and amines. J. Bacteriol.90:1552-1556.
96. Kloppstech, K. 1969. Characteristics of the reac-tion between diphtheria toxin, pyridine coen-zymes, and the GTP-splitting transfer factorFl. Hoppe-Seylers Z. Physiol. Chem.350:1377-1384.
97. Lampidis, T., and L. Barksdale. 1971. Park-Wil-liams number 8 strain of Corynebacteriumdiphtheriae. J. Bacteriol. 105:77-85.
98. Lecroisey, A., B. Bizzini, J. Blass, and M.Raynaud. 1972. Relations entre la structure dela toxine et de l'anatoxine diphteriques. C. R.Acad. Sci. Paris 274:2395-2397.
99. Lennox, E. S., and A. S. Kaplan. 1957. Action ofdiphtheria toxin on cells cultivated in vitro.Proc. Soc. Exp. Biol. Med. 95:700-702.
100. Lightfoot, H. N., and B. H. Iglewski. 1974.Synthesis of diphtheria toxin in E. coli cell-free extract. Biochem. Biophys. Res. Com-mun. 56:351-357.
101. Loeffler, F. 1884. Untersuchungen uber die be-deutung der mikroorganismen fur die en-stehung der diphtherie beim menschen, beider traube und beim kalbe. Mitt. Klin. Ge-sundh. Berlin 2:421-499.
102. Michel, A., and J. Dirkx. 1973. Liaison denucleotides a la toxine diphterique et sonfragment A. Arch. Int. Physiol. Biochim.81:591.
103. Michel, A., and J. Dirkx. 1974. Fluorescencestudies of nucleotides binding to diphtheria
toxin and its fragment A. Biochim. Biophys.Acta 365:15-24.
104. Michel, A., J. Zanen, C. Monier, C. Crispeels,and J. Dirkx. 1972. Partial characterization ofdiphtheria toxin and its subunits. Biochim.Biophys. Acta 257:249-256.
104a. Moehring, T. J., and J. P. Crispell. 1974.Enzyme treatment of KB cells-altered effectof diphtheria toxin. Biochem. Biophys. Res.Commun. 60:1446-1452.
105. Moehring, T. J., and J. M. Moehring. 1968.Response of culture of mammalian cells todiphtheria toxin. III. Inhibition of proteinsynthesis studies at the subcellular level. J.Bacteriol. 96:61-69.
106. Moehring, J. M., and T. J. Moehring. 1968. Theresponse of cultures of mammalian cells todiphtheria toxin. II. The resistant cell: en-hancement of toxin action by poly-L-orni-thine. J. Exp. Med. 127:541-553.
107. Moehring, T. J., and J. M. Moehring. 1972.Response of cultured mammalian cells todiphtheria toxin. IV. Isolation of KB cellsresistant to diphtheria toxin. Infect. Immun.6:487-492.
108. Moehring, T. J., and J. M. Moehring. 1972.Response of cultured mammalian cells todiphtheria toxin. V. Concurrent resistance toribonucleic acid viruses in diphtheria toxinresistant KB cell strains. Infect. Immun.6:493-500.
109. Moehring, T. J., J. M. Moehring, R. J. Kuchler,and M. Solotorovsky. 1967. The response ofcultured mammalian cells to diphtheria toxin.I. Amino acid transport, accumulation, andincorporation in normal and intoxicated sensi-tive cells. J. Exp. Med. 126:407-422.
110. Moehring, T. J., J. M. Moehring, and W. R.Stinebring. 1971. Response of interferontreated cells to diphtheria toxin. Infect. Im-mun. 4:747-752.
111. Moldave, K., W. Galisinski, P. Rao, and J. Siler.1969. Studies on the peptidyl-tRNA translo-case from rat liver. Cold Spring Harbor Symp.Quant. Biol. 34:347-356.
112. Montanaro, L., and S. Sperti. 1967. Binding ofnicotinamide adenine dinucleotides to diph-theria toxin. Biochem. J. 105:635-640.
113. Montanaro, L., S. Sperti, and A. Mattioli. 1971.Interaction of ADP-ribosylated aminoacyl-transferase II with GTP and with ribosomes.Biochim. Biophys. Acta 238:493-497.
114. Moolten, F. L., and S. R. Cooperband. 1970.Selective destruction of target cells by diph-theria toxin conjugated to antibody directedagainst antigens on the cells. Science169:68-70.
115. Murphy, J. R., A. M. Pappenheimer, Jr., and S.T. deBorms. 1974. Synthesis of diphtheriatox-gene products in Escherichia coli extracts.Proc. Nat. Acad. Sci. U.S.A. 71:11-15.
116. Nishizuka, Y., K. Veda, K. Nakazawa, and 0.Hayaishi. 1967. Studies on the polymer ofadenosine diphosphate ribose. I. Enzymic for-
83VOL. 39, 1975
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

BACTERIOL. REV.
mation from nicotinamide adenine dinucleo-tide in mammalian nuclei. J. Biol. Chem.242:3164-3171.
117. Olsnes, S. 1972. Toxic proteins inhibiting proteinsynthesis. Naturwissenschaft. 59:497-502.
118. Olsnes, S., and A. Pihl. 1973. Different biologicalproperties of the two constituent peptidechains of ricin, a toxic protein inhibitingprotein synthesis. Biochemistry 12:3121-3126.
119. Olsnes, S., K. Refsnes, and A. Pihl. 1974. Mech-anism of action of the toxic lectins abrin andricin. Nature (London) 249:627-631.
120. Pappenheimer, A. M., Jr. 1937. Diphtheriatoxin. I. Isolation and characterization of atoxic protein from C. diphtheriae filtrates. J.Biol. Chem. 120:543-553.
121. Pappenheimer, A. M., Jr. 1942. Studies ondiptheria toxin and its reaction with anti-toxin. J. Bacteriol. 43:273-289.
122. Pappenheimer, A. M., Jr., and R. Brown. 1968.Studies on the mode of action of diphtheriatoxin. VI. Site of action of toxin in living cells.J. Exp. Med. 127:1073-1086.
123. Pappenheimer, A. M., Jr., R. J. Collier, and P.A. Miller. 1963. Effect of diphtheria toxin onthe metabolism of mammalian cells in tissueculture, p. 21-35. In M. Solotorovsky (ed.),Cell culture in the study of bacterial disease.Rutgers University Press, New Brunswick,New Jersey.
124. Pappenheimer, A. M., Jr., and D. M. Gill. 1972.Inhibition of protein synthesis by activateddiphtheria toxin, p. 134-149. In E. Munoz, F.Garcia-Ferrandiz, and D. Vasquez (ed.), Mo-lecular mechanisms of antibiotic action onprotein synthesis. Elsevier, Amsterdam.
125. Pappenheimer, A. M., Jr., and D. M. Gill. 1973.Diphtheria. Science 182:353-358.
126. Pappenheimer, A. M., Jr., H. P. Lundgren, andJ. W. Williams. 1940. Studies on the molecu-lar weight of diphtheria toxin, antitoxin, andtheir reaction products. J. Exp. Med.71:247-262.
127. Pappenheimer, A. M., Jr., T. Uchida, and A. A.Harper. 1972. An immunological study of thediphtheria toxin molecule. Immunochemistry9:891-906.
128. Philpott, G. W., R. J. Bower, and C. W. Parker.1973. Improved selective cytotoxicity with anantibody-diphtheria toxin conjugate. Surgery73:928-935.
129. Pinchot, G. B., and W. L. Bloom. 1950. Altera-tions in the level of muscle phosphocreatine ofguinea pigs produced by the injection of diph-theria toxin. J. Biol. Chem. 184:9-16.
130. Placido-Sousa, C., and D. G. Evans. 1957. Theaction of diphtheria toxin on tissue culturesand its neutralization by antitoxin. Br. J. Exp.Pathol. 38:644-649.
131. Raeburn, S., J. F. Collins, H. M. Moon, and E.S. Maxwell. 1971. Aminoacyl-transferase IIfrom rat liver. I. Purification and enzymaticproperties. J. Biol. Chem. 246:1041-1048.
132. Raeburn, S., R. S. Goor, J. F. Collins, and E. S.
Maxwell. 1970. Alteration in the ionic proper-ties of aminoacyl transferase II from rat liverby NAD+ and diphtheria toxin. Biochim.Biophys. Acta 199:294-297.
133. Raeburn, S., R. S. Goor, J. A. Schneider, and E.S. Maxwell. 1968. Interaction of aminoacyl-transferase II and guanosine triphosphate:inhibition by diphtheria toxin and nicotina-mide adenine dinucleotide. Proc. Nat. Acad.Sci. U.S.A. 61:1428-1434.
134. Raynaud, M., B. Bizzini, and E. H. Relyveld.1965. Composition en amino-acides de la tox-ine diphterique. Bull. Soc. Chim. Biol.47:261-266.
135. Relyveld, E. H. 1970. Formation de la toxinediphterique lourde. C. R. Acad. Sci. Paris270:410-413.
136. Relyveld, E. H., and S. B. Efraim. 1959. Latoxine diphterique. Ann. Inst. Pasteur97:697-717.
137. Richter, D., and F. Lipmann. 1970. Separationof mitochondrial and cytoplasmic peptidechain elongation factors from yeast. Biochem-istry 9:5065-5070.
138. Robinson, E. A., 0. Henriksen, and E. S. Max-well. 1974. Elongation factor 2. Amino acidsequence at the site of adenosine diphosphateribosylation. J. Biol. Chem. 249:5088-5093.
139. Robinson, E. A., 0. Henriksen, and E. S. Max-well. 1974. The site of ADP-ribosylation ofelongation factor 2, p. 315-320. In Poly (ADP-ribose): an international symposium. U.S.Govt. Printing Office, Washington, D.C.
140. Robinson, E. A., and E. S. Maxwell. 1972.Chemical properties of elongation factor 2.Amino acid composition, NH2 terminal resi-due, and sulfhydryl reactivity. J. Biol. Chem.247:7023-7028.
141. Roux, E., and A. Yersin. 1888. Contribution al'etude de la diphterie. Ann. Inst. Pasteur2:629-661.
142. Saelinger, C. B., J. G. Imhoff, and P. F. Bonven-tre. 1973. Studies on the mode of action ofdiphtheria toxin. VI. Inhibition of proteinsynthesis induced by local infection with tox-inogenic Corynebacterium diphtheriae. Proc.Soc. Exp. Biol. Med. 142:41-45.
143. Samagh, B. S., and K. F. Gregory. 1972. Anti-body to lactic dehydrogenase. V. Use of as acarrier for introducing diphtheria toxin intomouse tumor cells. Biochim. Biophys. Acta273:188-198.
144. Simulson, M. E., C. Rideau, and S. Raeburn.1970. Diphtheria toxins requirement for activeprotein synthesis for inactivation of aminoacyltransferase II in the intact mammalian cell.Biochim. Biophys. Acta 224:269-271.
145. Solotorovsky, M., and J. Gabliks. 1965. Thebiological action of diphtheria toxin in cell andtissue culture, p. 5-15. In M. Solotorovsky(ed.), Cell culture in the study of bacterialdisease. Rutgers University Press, New Bruns-wick, New Jersey.
146. Solotorovsky, M., and W. Johnson. 1970. Tissue
COLLIER84-
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from

DIPHTHERIA TOXIN
culture and bacterial protein toxins, p.277-327. In S. J. Ajl, S. Kadis, and T. C.Montie (ed.), Microbial toxins, vol. 1. Aca-demic Press Inc., New York.
147. Sperti, S., and L. Montanaro. 1968. Competitivebinding of adenine and nicotinamide adeninedinucleotide to diphtheria toxin. Biochem. J.107:730-732.
148. Sperti, S., L. Montanaro, and A. Mattioli. 1971.Studies of diphtheria toxin. The effect of GTPon the toxin-dependent adenosine diphos-phate ribosylation of rat liver amino acyltransferase II. Chem. Biol. Interactions3:141-148.
149. Strauss, N. 1960. The effect of diphtheria toxinon the metabolism of HeLa cells. II. Effect onnucleic acid metabolism. J. Exp. Med.112:351-359.
150. Strauss, N., and E. D. Hendee. 1959. The effectof diphtheria toxin on the metabolism of HeLacells. J. Exp. Med. 109:144-163.
151. Sugimura, T. 1973. Poly (adenosine phosphateribose), p. 127-151. In W. E. Cohn (ed.),Progress in nucleic acid research and molecu-lar biology, vol. 13. Academic Press Inc., NewYork.
152. Sugiyama, H., B. R. DasGupta, and K. H. Yang.1973. Disulfide-toxicity relationship of botuli-nal toxin types A, E, and F. Proc. Soc. Exp.Biol. Med. 143:589-591.
153. Traugh, J., and R. J. Collier. 1970. Binding oftransfer factor II to ribosomal RNA and inhi-bition of the binding by guanosine nucleo-tides. Biochem. Biophys. Res. Commun.40:1437-1444.
154. Traugh, J., and R. J. Collier. 1971. Interaction oftransferase II with the 60s ribosomal subunit.FEBS Lett. 14:285-288.
155. Traugh, J., and R. J. Collier. 1971. Interaction oftransferase II with polynucleotides and inhibi-tion of the interaction by guanosine nucleo-tides. Biochemistry 10:2357-2365.
156. Tsugawa, A., Y. Ohsumi, and I. Kato. 1970.Inhibitory effect of diphtheria toxin on aminoacid incorporation in Escherichia coli cell-freesystem. J. Bacteriol. 104:152-157.
157. Ucer, U., and E. Bermek. 1974. Cross-linking ofADP-ribosylated human translocation factorto ribosomes. FEBS Lett. 38:161-165.
158. Uchida, T., D. M. Gill, and A. M. Pappen-heimer, Jr. 1971. Mutation in the structuralgene for diphtheria toxin carried by temperatephage B. Nature N. Biol. 233:8-11.
159. Uchida, T., A. M. Pappenheimer, Jr., and R.Gregory. 1973. Diphtheria toxin and relatedproteins. I. Isolation and properties of mutantproteins serologically related to diphtheriatoxin. J. Biol. Chem. 248:3838-3844.
160. Uchida, T., A. M. Pappenheimer, Jr., and A. A.Harper. 1972. Reconstitution of diphtheriatoxin from two nontoxic cross-reacting mutanttoxins. Science 175:901-903.
161. Uchida, T., A. M. Pappenheimer, Jr., and A. A.Harper. 1973. Diphtheria toxin and relatedproteins. Iml. Reconstitution of hybrid "diph-theria toxin" from nontoxic mutant proteins.J. Biol. Chem. 248:3851-3854.
162. Uchida, T., A. M. Pappenheimer, Jr., and A. A.Harper. 1973. Diphtheria toxin and relatedproteins. II. Kinetic studies on intoxication ofHeLa cells by diphtheria toxin and relatedproteins. J. Biol. Chem. 248:3845-3850.
163. Van Heyningen, W. E., and S. N. Arseculeratne.1964. Exotoxins. Annu. Rev. Microbiol.18:195-216.
164. Wiester, M. J., P. F. Bonventre, and G. Grupp.1973. Estimate of myocardial damage inducedby diphtheria toxin. J. Lab. Clin. Med.81:354-364.
165. Yoneda, M. 1957. A new culture method de-signed for kinetic studies on diphtheria toxinproduction. Br. J. Exp. Pathol. 38:190-193.
166. Yoneda, M., and A. M. Pappenheimer, Jr. 1957.Some effects of iron deficiency on the extracel-lular products released by toxigenic and non-toxigenic strains of Corynebacterium diph-theriae. J. Bacteriol. 74:256-264.
167. Zanen, J., F. Depuydt, and J. Dirkx. 1968.Action d-agents denaturants sur la toxinediphterique. Arch. Int. Physiol. Biochim.76:601-602.
VOL. 39, 1975 85
on March 3, 2020 by guest
http://mm
br.asm.org/
Dow
nloaded from