differential effects of desmoglein 1 and desmoglein 3 on desmosome formation
TRANSCRIPT

ORIGINAL ARTICLESee related Commentary on page 1215
Di¡erential E¡ects of Desmoglein 1 and Desmoglein 3 onDesmosome Formation
Yasushi Hanakawa, Masayuki Amagai,n Yuji Shirakata,YokoYahata, ShoTokumaru, Kenshi Yamasaki,Mikiko Tohyama, Koji Sayama, and Koji HashimotoDepartment of Dermatology, School of Medicine, Ehime University, Ehime, Japan; nDepartment of Dermatology, School of Medicine, Keio University,Tokyo, Japan
The desmoglein plays an important part in the forma-tion of desmosomes.We constructed recombinant ade-noviruses containing desmoglein 1 and desmoglein 3derivatives partly lacking the extracellular domain (des-moglein 1DEC and desmoglein 3DEC, respectively), andfull-length desmoglein 1 and desmoglein 3 and studiedthe involvement of desmoglein 1 and desmoglein 3 indesmosome formation. During low-level expressionof desmoglein 3DEC in transduced HaCaTcells, keratininsertion at cell^cell contact sites was only partiallyinhibited and desmoplakin was partially stained atcell^cell contact sites. Low-level expression of desmo-glein 1DEC, however, resulted in complete inhibitionof keratin insertion at the cell^cell contact sites, anddesmoplakin was stained in perinuclear dots. Theseresults indicate the dominant-negative e¡ect of desmo-glein 1DEC on desmosome formation was stronger thanthat of desmoglein 3DEC. Desmoglein 1DEC coprecipi-tated plakoglobin to approximately the same extent as
desmoglein 3DEC. Therefore, we conclude that thedominant-negative e¡ect of desmoglein 1DEC is notsimply due to plakoglobin sequestration. On the otherhand, during low-level expression of full-length desmo-glein 3 and desmoglein 1, they both colocalized withdesmoplakin. During high-level expression, however,keratin insertion at cell^cell contact sites was inhibitedin desmoglein 1 but not in desmoglein 3, and desmopla-kin was stained at cell^cell contact sites in desmoglein 3but not in desmoglein 1. These data suggest desmoglein1 and desmoglein 3 expressed at low level were incorpo-rated into desmosome but at high-level expression, des-moglein 1 disrupted desmosomes but desmoglein 3 didnot. Our ¢ndings provide biologic evidence that des-moglein 1 and desmoglein 3 play a di¡erent functionalrole in cell^cell adhesion of keratinocytes. Key words:desmosomes/desmoglein 1/desmoglein 3. J Invest Dermatol119:1231 ^1236, 2002
Desmosomes are cell^cell adhesion complexes thatprovide mechanical integrity to keratinocytes bylinking to keratin intermediate ¢laments. Desmo-somes are composed of two major transmembraneproteins, desmoglein (Dsg) and desmocollin (Ama-
gai, 1996a; Kowalczyck et al, 1999; Green and Gaudry, 2000). Inhumans, three desmoglein isoforms have been identi¢ed: Dsg1,Dsg2, and Dsg3. They are encoded by individual genes and dif-ferentially distributed in tissue. Dsg2 is expressed in all desmo-some-containing tissues, including simple epithelium andmyocardium. In contrast, Dsg1 and Dsg3 are expressedin strati¢ed squamous epithelia. Dsg3 is found in the basal andsuprabasal layers of stratifying epithelia, whereas Dsg1 isdominantly expressed in the di¡erentiated upper layers ofepithelia (Arnemann et al, 1993; Shimizu et al, 1995; Amagai et al,1996b).Desmogleins play important parts in the formation and main-
tenance of desmosomes. Autoantibodies against desmogleins leadto impairment of epidermal tissue integrity. Pemphigus vulgaris
is a disease caused by autoantibodies directed against Dsg3 inwhich skin lesion biopsies exhibit suprabasilar acantholysis (Udeyand Stanley, 1999). On the other hand, pemphigus foliaceus iscaused by autoantibodies directed against Dsg1 and in this case,lesion biopsies exhibit subcorneal acantholysis. Dsg3 knockoutmice phenotypically mimicked pemphigus vulgaris patients(Koch et al, 1997) in that they displayed oral erosions and loss ofintercellular adhesion of suprabasal layers of the mucosal epithe-lium and epidermis.Several studies have suggested that both the extracellular and
cytoplasmic domains of desmogleins are critical for normal des-mosome formation. N-terminally truncated Dsg3 caused domi-nant-negative e¡ects on desmosome formation in HaCaT cells(Hanakawa et al, 2000). Expression of chimeric molecules con-taining the transmembrane domain of connexin and cytoplasmicdomain of Dsg1 disrupted desmosomes in A431 cells (Troyanovs-ky et al, 1993). Similarly, a chimeric molecule containing the ex-tracellular domain of E-cadherin and cytoplasmic domain ofDsg1 (Ecad-Dsg1) disrupted desmosomes in A431 cells (Norveland Green, 1998). In contrast, a recent report indicated that a chi-meric molecule of the extracellular domain of E-cadherin andcytoplasmic domain of Dsg3 (Ecad-Dsg3) was incorporated intodesmosomes of A431 cells (Andl and Stanley, 2001). This di¡er-ence between Ecad-Dsg1 and Ecad-Dsg3 indicates functional dif-ferences of cytoplasmic domains of Dsg1 and Dsg3. In addition,full-length Dsg1 disrupted desmosome when the expression level
Reprint requests to: Yasushi Hanakawa, MD, Ehime University Schoolof Medicine, Department of Dermatology, Shitukawa, Shigenobu, Onsen-gun, Ehime 791-0295, Japan. Email: [email protected]: Dsg, desmoglein; MOI, multiplicity of infection.
Manuscript received January 10, 2002; revised June 24, 2002; accepted forpublication August 29, 2002
0022-202X/02/$15.00 � Copyrightr 2002 by The Society for Investigative Dermatology, Inc.
1231

was high; however, full-length Dsg1 was incorporated into des-mosome when the expression level was low in A431 cells (Norveland Green, 1998). These data suggest that Dsg1 and Dsg3 expres-sion have di¡erent e¡ects on desmosome formation and integrity.To characterize further and de¢ne these di¡erences, we con-structed full-length and N-terminally truncated mutants ofDsg1 and Dsg3, and introduced them into cultured keratinocytesusing an adenovirus vector. Taking advantage of the adenovirusvector to control expression level by changing multiplicity ofinfection (MOI), we evaluated the e¡ects of expression levels ofthese isoforms on the integrity of a desmosome^keratin struc-tured complex.
MATERIALS AND METHODS
Cell culture The human embryonic kidney cell line 293 was obtainedfrom American Type Tissue Culture (ATCC; Rockville, MD). The humannaturally immortalized keratinocyte HaCaT cell line was a kind gift fromDr Norbert Fusenig (German Cancer Research Center, Heidelberg,Germany). These cells were cultured in Dulbecco modi¢ed Eagle’smedium supplemented with 10% fetal bovine serum.
Plasmid construction The full-length cDNA encoding human Dsg1was a kind gift from Dr Kathleen Green (North-western University,Chicago, IL). The construction of the Dsg3 mutant, in which a large partof the extracellular domain was deleted and a seven c-myc tag was inserted£anking the C-terminal end, has been described previously (Hanakawaet al, 2000). A mutant of Dsg1 (Dsg1DEC), with a large part of theextracellular domain deleted and with a seven c-myc tag £anking theC-terminal end, was also constructed. Dsg1 DNA fragments (nucleotides13^402 and 1726^3360) were generated by polymerase chain reactionusing primers DG1F150 (50 -TTTTAGGGTGGGGATCCAGAC-30), DG1F130(50 -GGGGTACCGTCTTCACCTTCACGACAGGC-30, KpnI site at the 30end), DG1F250 (50 -GGGGTACCGGCAGACAAGAAAGTACT-30, KpnIsite at the 50 end), and DG1F230 (50 -CGGAATTCCTTGCTATAT-TGCACGGT-30, EcoRI site at the 30 end). The polymerase chain reactionproducts were subcloned into pcDNA1^7myc/Amp to generatepcDsg1DEC. pcDNA1^7myc/Amp was a kind gift from Dr KathleenGreen (North-western University; Roth et al, 1991).
Adenovirus vector construction and infection The cosmid cassettepAxCAw, pAxCALNLw (Kanegae et al, 1996), the loxP-NeoR-loxP unitunder a CA promoter consisting of a cytomegalovirus enhancer andchicken b-actin promoter (Niwa et al, 1991), the nuclear localizing signal-tagged Cre recombinase-expressing adenovirus (AxCANCre), the controladenovirus Ax1w, and the parent virus Ad5-dLX (Miyake et al, 1996)were all kind gifts from Dr Izumu Saito (Tokyo University, Japan).Fragments of Dsg1DEC were subcloned into the adenovirus cosmidcassette pAxCALNLw. Adenovirus containing CALNL and Dsg1DEC(AxCALNLDsg1DEC) were generated by the COS-TPC method(Miyake et al, 1996). The cosmid DNA was mixed with the EcoT22I-digested DNA-terminal protein complex of Ad5-dLX, and used tocotransfect 293 cells. Recombinant viruses were generated throughhomologous recombination in 293 cells. Virus stocks were prepared by astandard procedure (Miyake et al, 1996). Concentrated, puri¢ed virusstocks were prepared by the CsCl gradient method and the virus titer waschecked using the plaque formation assay. HaCaT cells were doublyinfected with AxCANCre and Ax1w, AxCALNLDsg1DEC, orAxCALNLDsg3DEC. HaCaT cells were infected with AxCANCre at aMOI of 5 and Ax1w, AxCALNLDsg1DEC, or AxCALNLDsg3DEC at aMOI of 5 (low expression) or were infected with AxCANCre at a MOIof 15 and Ax1w, AxCALNLDsg1DEC, or AxCALNLDsg3DEC at a MOIof 15 (high expression).Adenovirus expressing mouse Dsg1-FLAG and mouse Dsg3-FLAG have
already been shown elsewhere (Amagai et al, 2002). Brie£y, cDNAencoding mouse Dsg1-FLAG and mouse Dsg3-FLAG were subclonedinto the adenovirus cosmid cassette pAxCAw. Adenovirus containing CApromoter and cDNA encoding mouse Dsg1-FLAG and mouse Dsg3-FLAG (AxmDsg1F and AxmDsg3F) were generated by the COS-TPCmethod (terminal protein complex). Puri¢ed, concentrated, and titer-checked viruses were infected to HaCaT cells at a MOI of 10 and 30.
Antibodies The following monoclonal antibodies were used: AE1 andAE3 (murine anti-keratin intermediate ¢lament; PROGEN, Heidelberg,Germany); 9E10.2 (murine anti-myc; American Type Tissue Culture); anti-b-catenin (murine; Transduction Laboratories, Lexington, KY); DPI/II
(murine anti-desmoplakin; PROGEN); PG5.1 (murine anti-plakoglobin;PROGEN); PP1-5C2 (murine anti-plakophilin 1; PROGEN); anti-plakophilin 2 (murine; PROGEN); anti-involucrin (Abcam, Cambridge,U.K.); and 6H6 (murine anti-Dsg3). The following rabbit anti-sera wereused: Z622 (anti-pan-keratin; DAKO, Copenhagen, Denmark); anti-myc(a kind gift from Dr John Stanley, University of Pennsylvania, PA); anti-keratin 1 (CONVENCE, Richmond, CA); anti-loricrin (CONVENCE);and anti-FLAG (Abcam). Fluorescein isothiocyanate-conjugated goat anti-mouse or goat anti-rabbit antibodies (Kirkegaard and Perry Laboratories,Gaithersburg, MD) and rhodamine-conjugated goat anti-rabbit antibody(BioSource, Camarillo, CA) were used.
Immunohistochemistry Cells grown on Laboratory-Tech 4-wellculture slides (Nalge-Nunc, Napierville, IL) were ¢xed with methanol at^201C for 20 min and permeabilized with 0.05% Triton X-100 in Tris-bu¡ered saline containing 1 mM CaCl2 (TBS-Ca
2þ ) at room temperaturefor 5 min. After incubation with 1% bovine serum albumin inTBS-Ca2þ
for 20 min at room temperature, the cells were incubated with variousantibodies at the appropriate dilution in 1% bovine serum albumin inTBS-Ca2þ for 1 h at room temperature. The samples were furtherincubated with £uorescein isothiocyanate- or rhodamine-conjugatedsecondary antibodies at the appropriate dilution in 1% bovine serumalbumin in TBS-Ca2þ for 1 h at room temperature. Stained cells wereexamined using a confocal laser microscope (Zeiss, Oberkachen,Germany).
Immunoblotting and immunoprecipitation For immunoblotting,cells were lyzed in sodium dodecyl sulfate sample bu¡er (62.5 mM Tris^HCl, pH 7.5, 1% sodium dodecyl sulfate, 0.0025% bromophenol blue,10% glycerol, 2.5% 2-mercaptoethanol) and subjected to immunoblottingusing enhanced chemi£uorescence according to the manufacturer’sprotocol (Amersham Pharmacia Biotech, Uppsala, Sweden). For solubleand insoluble fractionation, the cells were scraped in 1% Nonidet P-40and 1% Trion-X-100 in TBS-Ca2þ on ice. Following centrifugation at15,000 r.p.m. (30,000g) for 10 min at 41C, the supernatant was collected asthe soluble fraction. The pellet was dissolved in sodium dodecyl sulfatesample bu¡er as the insoluble fraction. Immunoprecipitation was carriedout on adenovirus-infected cells extracted with 1% Nonidet P-40, 1%Triton X-100, 2 mM CaCl2, and 150 mM NaCl in 10 mM Tris^HCl, pH7.4, in the presence of protease inhibitors (2 mg aprotinin per ml, 2 mgleupeptin per ml, phenylmethylsulfonyl £uoride 1 mM). Aliquots of celllysates were preadsorbed with normal mouse IgG or normal rabbit IgGand protein G-Sepharose (Amersham Pharmacia Biotech), thenprecipitated with various antibodies and protein G-Sepharose.Immunoprecipitates were eluted from protein G-Sepharose with sodiumdodecyl sulfate sample bu¡er and subjected to immunoblotting. Theimmunoblots were analyzed by using ImageQuantTM (AmershamPharmacia Biotech) software to determine the relative ratio in theimmunoprecipitated complex.
RESULTS
Desmosome disruption is greater with Dsg1DEC than withDsg3DEC A Dsg1 mutant was constructed by deleting a largepart of the extracellular domain and adding a seven c-myc tag(Dsg1DEC; Fig 1). The corresponding Dsg3 mutant has beenpreviously described (Hanakawa et al, 2000; Fig 1). To preventthe expression of toxic, truncated products that interfered withthe production of recombinant virus, the loxP gene sequence,disrupted by a neomycin-resistance cassette, was interposedbetween the CAG promoters and the coding regions in themutants (Kanegae et al, 1996). Co-infection of keratinocytes(HaCaT cells) with adenovirus-expressing Cre recombinaseremoved the stu¡er sequence and activated the expression of themutant desmogleins. The expression of mutant proteins wasdetected at 6 h and reached a plateau between 24 and 36 h afterinfection (data not shown). Almost all cells expressed the mutantproteins when HaCaT cells were coinfected with adenoviruscarrying mutant desmogleins and Cre recombinase, £ankedwith the nuclear localization signal (NCre) at a MOI of 30(mutant: Cre¼1 : 1) or a MOI of 10. The relative expressionlevels of both mutants were essentially the same when HaCaTcells were infected at the same MOI, with a MOI of 30 causinggreater expression than MOI of 10 (Fig 2). No toxicity wasapparent in cells infected with control adenovirus (Ax1w) or
1232 HANAKAWA ETAL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
NCre adenovirus at MOI of 10 or 30 in phase microscope. Weexamined the e¡ects of these di¡erent expression levels ofmutant desmogleins on HaCaT cells, at 36 h after infection.There were no apparent morphologic changes in cells infectedwith Dsg1DEC adenovirus or Dsg3DEC adenovirus whencompared with cells infected with the control adenovirus, usingphase microscopy (data not shown). The e¡ects of mutantdesmogleins on the formation of adherens junctions wereexamined by immuno£uorescence staining using doublestaining for b-catenin (a marker of adherens junctions) and myc.No di¡erences were observed in b-catenin staining patterns 36 h
after infection with control adenovirus, Dsg1DEC adenovirus, orDsg3DEC adenovirus at a MOI of 30 (data not shown).We next studied desmosomal changes in HaCaT cells induced
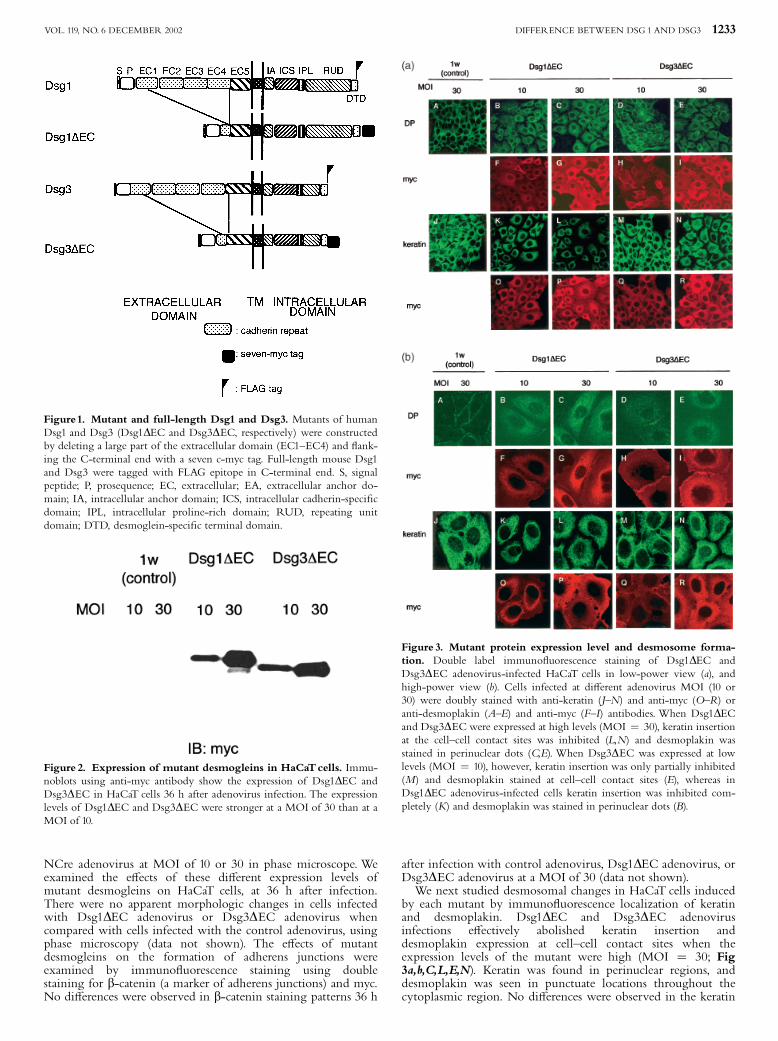
by each mutant by immuno£uorescence localization of keratinand desmoplakin. Dsg1DEC and Dsg3DEC adenovirusinfections e¡ectively abolished keratin insertion anddesmoplakin expression at cell^cell contact sites when theexpression levels of the mutant were high (MOI ¼ 30; Fig3a,b,C,L,E,N). Keratin was found in perinuclear regions, anddesmoplakin was seen in punctuate locations throughout thecytoplasmic region. No di¡erences were observed in the keratin
Figure1. Mutant and full-length Dsg1 and Dsg3. Mutants of humanDsg1 and Dsg3 (Dsg1DEC and Dsg3DEC, respectively) were constructedby deleting a large part of the extracellular domain (EC1^EC4) and £ank-ing the C-terminal end with a seven c-myc tag. Full-length mouse Dsg1and Dsg3 were tagged with FLAG epitope in C-terminal end. S, signalpeptide; P, prosequence; EC, extracellular; EA, extracellular anchor do-main; IA, intracellular anchor domain; ICS, intracellular cadherin-speci¢cdomain; IPL, intracellular proline-rich domain; RUD, repeating unitdomain; DTD, desmoglein-speci¢c terminal domain.
Figure 2. Expression of mutant desmogleins in HaCaTcells. Immu-noblots using anti-myc antibody show the expression of Dsg1DEC andDsg3DEC in HaCaT cells 36 h after adenovirus infection. The expressionlevels of Dsg1DEC and Dsg3DEC were stronger at a MOI of 30 than at aMOI of 10.
Figure 3. Mutant protein expression level and desmosome forma-tion. Double label immuno£uorescence staining of Dsg1DEC andDsg3DEC adenovirus-infected HaCaT cells in low-power view (a), andhigh-power view (b). Cells infected at di¡erent adenovirus MOI (10 or30) were doubly stained with anti-keratin (J^N) and anti-myc (O^R) oranti-desmoplakin (A^E) and anti-myc (F^I) antibodies. When Dsg1DECand Dsg3DECwere expressed at high levels (MOI ¼ 30), keratin insertionat the cell^cell contact sites was inhibited (L,N) and desmoplakin wasstained in perinuclear dots (C,E). When Dsg3DEC was expressed at lowlevels (MOI ¼ 10), however, keratin insertion was only partially inhibited(M) and desmoplakin stained at cell^cell contact sites (E), whereas inDsg1DEC adenovirus-infected cells keratin insertion was inhibited com-pletely (K) and desmoplakin was stained in perinuclear dots (B).
DIFFERENCE BETWEEN DSG 1 AND DSG3 1233VOL. 119, NO. 6 DECEMBER 2002

or desmoplakin staining patterns 36 h after infection with controladenovirus (Fig 3a,b,A,J). In contrast, when the expression levelwas low (MOI¼10), Dsg3DEC adenovirus partially a¡ected onkeratin insertion and desmoplakin expression (Fig 3a,b,D,M),whereas Dsg1DEC adenovirus completely inhibited keratininsertion and desmoplakin expression at the cell^cell contact site(Fig 3a,b,B,K). In low-level expression, the Dsg3DEC mutantproteins were detected with anti-myc antibodies at cell^cellboundaries and in the cytoplasm (Fig 3b,H,Q); however, theDsg1DEC mutant proteins were detected mainly in thecytoplasm but not at cell^cell contact sites (Fig 3b,F,O). Inhigh-level expression, both the Dsg3DEC and Dsg1DEC mutantproteins were detected at cell^cell boundaries and in thecytoplasm (Fig 3b,G,I,P,R).
Plakoglobin binds equally to Dsg1DEC and Dsg3DEC Wecombined immunoprecipitation with immunoblotting in orderto detect plakoglobin bound to either Dsg1DEC or Dsg3DEC(Fig 4). Cell extracts from HaCaT cells expressing mutantdesmogleins were immunoprecipitated with an anti-mycantibody, then subsequently immunoblotted with anti-myc andanti-plakoglobin antibodies. Plakoglobin coprecipitated with theDsg1DEC and Dsg3DEC proteins expressed by the two mutantswith similar e⁄ciencies (Fig 4A).When the order in which theantibodies were used was reversed, i.e., immunoprecipitationwith anti-plakoglobin followed by immunoblotting with anti-myc antibody, plakoglobin was again coprecipitated withDsg1DEC and Dsg3DEC at similar levels (Fig 4B). We couldnot detect any association of plakophilin 1 and plakophilin 2with Dsg1DEC or Dsg3DEC (data not shown).Several published data indicate that disruption of cell adhesion
leads to altered di¡erentiation in keratinocytes (Allen et al, 1996;Zhu and Watt, 1996). To test whether Dsg1DEC and Dsg3DECmight a¡ect di¡erentiation in HaCaT cells, we analyzed thedi¡erentiation marker of keratinocytes after transduction withDsg1DEC and Dsg3DEC. Western blotting of Dsg1DEC andDsg3DEC expressing HaCaT cell extracts stained with anti-keratin 1, anti-involucrin, and anti-loricrin antibody antibodiesshowed no change (data not shown), suggesting dominant-negative e¡ects on desmosomes did not a¡ect di¡erentiation inour system.
Full-length Dsg1 and Dsg3 are incorporated intodesmosome in low-level expression We also tried to ¢nd thefunctional di¡erence in full-length Dsg1 and Dsg3 in desmosomeformation. We constructed adenovirus containing full-lengthDsg1 and Dsg3 with FLAG epitope tag on the C-termini andtransduced HaCaT cells. Western blots stained with anti-FLAG
antibody showed the relative expression levels of Dsg1 and Dsg3were essentially the same when HaCaT cells were infected at thesame MOI (Fig 5). Double immuno£uorescence staining of anti-FLAG and anti-desmoplakin showed colocalization of Dsg1 andDsg3 with desmoplakin at cell^cell contact site when theexpression level was low, although in the merged picture redstaining of anti-FLAG was stronger compared with desmoplakinstaining (Fig 6). These results show the incorporation of Dsg1and Dsg3 into desmosomes when their expression levels werelow.
In high-level expression, full-length Dsg1 disruptsdesmosome formation but Dsg3 does not On the otherhand, when the expression levels were high, Dsg1 and Dsg3showed di¡erent e¡ects on desmosome formation. In high levelfull-length expressing HaCaT cells, double immuno£uorescencestaining of anti-FLAG and anti-keratin showed insertion ofkeratin to cell^cell contact site in Dsg3 (Fig 7A,B). Disruptionof insertion to cell^cell contact site and shrinking aroundnucleus of keratin, however, was observed in Dsg1. Doublestaining of FLAG and desmoplakin showed dot staining ofdesmoplakin at cell^cell contact site in Dsg3, suggesting Dsg3 isstill incorporated into desmosomes at high-level expression. Incontrast, high-level expression of Dsg1 changed staining ofdesmoplakin to a di¡use cytoplasmic pattern. These resultssuggest that when full-length protein expression levels werehigh, Dsg1 disrupts desmosome formation but Dsg3 is stillincorporated into desmosomes. In high-level expression,full-length Dsg3 localized mainly at cell^cell contact site(Fig 7A,C); however, Dsg1 localized both cell^cell contact site
Figure 4. Coprecipitation of plakoglobin with Dsg1DEC andDsg3DEC. Extracts from HaCaT cells infected with the desmoglein mu-tants were immunoprecipitated with polyclonal anti-myc (A) or anti-pla-koglobin (B) antibodies, and immunoblotted with either monoclonalanti-myc or anti-plakoglobin antibodies. Plakoglobin coprecipitated withboth Dsg1DEC and Dsg3DEC to the same extent and this was also truewhen the order of addition of the antibodies was reversed.
Figure 5. Expression of full-length desmogleins in HaCaTcells. Im-munoblots using anti-FLAG antibody show the expression of full-lengthDsg1 and Dsg3 in HaCaT cells 36 h after adenovirus infection. The expres-sion levels of Dsg1 and Dsg3 were stronger at a MOI of 30 than at a MOIof 10.
Figure 6. Full-length Dsg1 and Dsg3 were incorporated into des-mosome at low-level expression. HaCaT cells were transduced withadenovirus containing full-length Dsg1 and Dsg3 at a MOI of 10. Doubleimmuno£uorescence staining of anti-FLAG (A,D) and anti-desmoplakin(B,E) showed colocalization of Dsg1 and Dsg3 with desmoplakin at cell^cell contact site. C Shows a merged picture of A and B, and F shows amerged picture of D and E.
1234 HANAKAWA ETAL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY

and cytoplasmic region (Fig 7E,G). Although full-length Dsg1in cytoplasm showed reticular pattern, we cannot explain thereason for such a staining pattern.
DISCUSSION
We investigated the roles of Dsg1 and Dsg3 in desmosome for-mation in HaCaT keratinocytes with adenovirus vectors contain-ing dominant-negative mutants and full-length forms of Dsg1and Dsg3. When the mutant desmoglein expression levels werehigh, both Dsg1DEC and Dsg3DEC inhibited the formation ofdesmosomes. When expression levels were low, however,Dsg3DEC only partially inhibited the formation of desmosomes,whereas Dsg1DEC completely inhibited the formation of desmo-somes. These ¢ndings indicate that the dominant-negative e¡ecton desmosome formation of the mutated Dsg1was stronger thanthat of mutated Dsg3. On the other hand, when the full-lengthdesmoglein expression levels were low, both Dsg1 and Dsg3 in-corporated into desmosomes. In contrast, when expression levelswere high, Dsg1 inhibited the formation of desmosomes, whereasDsg3 did not. These ¢ndings indicate that the functional di¡er-ence between the full-length form of Dsg1 and Dsg3 at least inless di¡erentiated cells in proliferating in culture.Why is the e¡ect of the Dsg1DEC mutant stronger than that of
Dsg3DECD One possibility is that mutated desmoglein interactswith desmosome components. By combining immunoprecipita-tion with subsequent immunoblotting, we found that the cyto-plasmic domains of Dsg1DEC and Dsg3DEC interacted withplakoglobin to the same extent. Recently it has been reportedthat the binding ratio of plakoglobin/full-length Dsg1 exhibitedless than 2 : 1 but was still higher compared with Dsg3 (Bannonet al, 2001). Sequestration of plakoglobin by the mutant desmo-glein proteins would make it inaccessible to native desmogleinand might partly explain the dominant-negative e¡ect. The ideaof di¡erential sequestering of plakoglobin, however, is contrain-dicated by the ¢nding that there were no di¡erences in plakoglo-bin-binding capacity between Dsg1DEC and Dsg3DEC. It seemslikely that molecules other than plakoglobin contribute to theformation of desmosomes. Another armadillo gene family mem-ber of plakophilin (Hatzfeld et al, 1994; Heid et al, 1994; Mertenset al, 1996; Bornslaeger et al, 2001) is now thought to be a majordesmosomal plaque protein; however, we could not detect bind-ing of plakophilin 1 or plakophilin 2 to Dsg1DEC or Dsg3DEC(data not shown).We cannot rule out the possibility that indirect
interactions occur between the mutant cadherins and plakophilin1 and plakophilin 2.The other possibility for the disruption of desmosome is in-
stability of desmosomes at cell^cell contact sites. Exfoliative toxinfrom Staphylococcus aureus cleaves the extracellular domain ofDsg1 and causes dysfunction of Dsg1 (Amagai et al, 2001, 2002).Keratinocytes in neonatal mice injected with exfoliative toxin Aor B showed internalization of Dsg1 and resultant blister forma-tion in the upper layers of the epidermis. We speculate thatcleaved Dsg1might be unstable in the desmosome because it doesnot bind its partner, and such unstabilized desmosomes could beeasily internalized; however, these observed internalizations onlyhappened in living epidermis but were not seen in the cryosec-tion of epidermis (Amagai et al, 2002). It is probable that the dy-namics of a living cell are a prerequisite for desmosomeinternalization after the dysfunction of desmogleins. These dataare consistent with our ¢ndings of a retracted, perinuclear stain-ing of keratin ¢lament network in HaCaT cells expressing highamounts of mutant desmogleins. The reason for the disruptionof the desmosome with full-length Dsg1 might be similar. Wespeculate that Dsg1 might associate with more desmosomal mo-lecules than Dsg3, based on our results with HaCaT cells withregard to basal layer keratinocytes in the epidermis.When Dsg1cannot ¢nd enough amount partners to stabilize on the cell sur-face, Dsg1 might be easily internalized with already interacteddesmosomal proteins. A recent report that Dsg2 e⁄ciently incor-porated into desmosomes but Dsg1 did not in MDCK and A431cells (Ishii et al, 2001) support the idea that cell and di¡erentiationspeci¢c capacity for isotype dependent incorporation of desmo-gleins into desmosome.This study has shown a functional di¡erence between Dsg1
and Dsg3 on desmosome formation in keratinocytes; however,why does the human body need di¡erent isoforms of Dsg1 andDsg3 in strati¢ed epithelia? Dsg3 knockout mice showed erosionsin the oral mucosa and in areas subject to mechanical irritation(Koch et al, 1997), although the epidermis was intact in nonme-chanically stressed areas. This result suggests that Dsg1 may com-pensate functionally for a de¢ciency in Dsg3 (Mahoney et al,1999;Wu et al, 2000). If this hypothesis is correct, small amountsof Dsg1 in the lower part of the epidermis are enough to producedesmosomes, although normal desmosomal function may becompromised in such situations. Although expression levels ofDsg1 are lower than Dsg3, Dsg1 may play an important part indesmosome formation in basal layer keratinocytes in epidermisand in certain circumstances, compensate for the lack of Dsg3function. The distribution of Dsg1 and Dsg3 is di¡erent betweenthe epidermis and mucous membrane (Shirakata et al, 1998). Inmucous membranes, Dsg1 is less expressed in upper layers com-pared with epidermis. Recently, involucrin promoter driventransgenic expression of Dsg3 in mouse showed altered stratumcorneum and increased transepidermal water loss in epidermis(Elias et al, 2001). These data suggest that Dsg1 and Dsg3 contri-bute not only to the formation of desmosomes but also tostrati¢cation. Strictly organized expression of Dsg1 and Dsg3might be needed to architect the di¡erence in epidermis and mu-cous membrane, which have di¡erent functions, such as waterloss protection in epidermis and water absorption in mucousmembrane.In summary, the discovery of the di¡erence in e¡ects on des-
mosome by Dsg1DEC and Dsg3DEC mutants and by full-lengthforms of Dsg1 and Dsg3 support the hypothesis of functionaldi¡erences in desmoglein activities during desmosome formationin keratinocytes. Further investigations are needed in order toclarify the precise roles of di¡erent desmoglein isotopes inhuman tissue.
We especially thank Dr John Stanley for insightful discussion and critical reading ofthis manuscript.We thank MsTerukoTsuda and Mrs Akiko Kon for expert technicalassistance. We also thank Dr Izumu Saitou for the adenovirus expression systemand adenovirus Cre-loxP system, Dr June-ichi Miyazaki for the CAG promoter,
Figure 7. Full-length Dsg1 disrupted desmosome but Dsg3 did notat high-level expression. HaCaT cells were transduced with adenoviruscontaining full-length Dsg1 and Dsg3 at a MOI of 30. Double immuno-£uorescence staining of anti-FLAG and anti-keratin showed insertion ofkeratin to cell^cell contact site in Dsg3 (A,B). Keratin insertion to cell^cellcontact site was inhibited in Dsg1 (E,F). Double staining of FLAG and des-moplakin showed dot staining of desmoplakin at cell^cell contact site andlinear cell^cell staining of FLAG in Dsg3 (C,D). In contrast, high-level ex-pression of Dsg1 changed staining of desmoplakin to di¡use cytoplasmicpattern (G,H).
DIFFERENCE BETWEEN DSG 1 AND DSG3 1235VOL. 119, NO. 6 DECEMBER 2002

Dr Norbert Fusenig for the HaCaTcells, Dr Kathleen Green for the pcDNA1^7myc/Amp and cDNA encoding human Dsg1, and Dr John Stanley for the polyclonal anti-myc antibody.This work was supported by a Health Sciences Research Grant for Re-search on Speci¢c Diseases from the Ministry of Health, labor andWelfare of Japanand a Grant-in-Aid of Scienti¢c Research from the Ministry of Education, Cluture,Sports, Science andTechnology of Japan.
REFERENCES
Allen E, Yu QC, Fuchs E: Mice expressing a mutant desmosomal cadherin exhibitabnormalities in desmosomes, proliferation, and epidermal di¡erentiation.J Cell Biol 133:1367^1382, 1996
Amagai M: Pemphigus: autoimmunity to epidermal cell adhesion molecules. AdvDermatol 11:319^352, 1996a
Amagai M, Koch PJ, NishikawaT, Stanley JR: Pemphigus vulgaris antigen (desmo-glein 3) is localized in the lower epidermis, the site of blister formation in pa-tients. J Invest Dermatol 106:351^355, 1996b
Amagai M, Matsuyoshi N,Wang ZH, Andl C, Stanley JR: Toxin in bullous impet-igo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat Med6:1275^1277, 2001
Amagai M,Yamaguchi T, HanakawaY, Nishifuji K, Sugai M, Stanley JR: Staphylo-coccal exfoliative toxin B speci¢cally cleaves desmoglein 1. J Invest Dermatol118:845^850, 2002
Andl CD, Stanley JR: Central role of the plakoglobin-binding domain for desmo-glein 3 incorporation into desmosomes. J Invest Dermatol 117:1068^1074, 2001
Arnemann J, Sullivan KH, Magee AI, King IA, Buxton RS: Strati¢cation-relatedexpression of isoforms of the desmosomal cadherins in human epidermis.J Cell Sci 104:741^750, 1993
Bannon LJ, Cabrera BL, Stack MS, Green KJ: Isoform-speci¢c di¡erences in the sizeof desmosomal cadherin/catenin complexes. J Invest Dermatol 117:1302^1306,2001
Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP,Green KJ: Plakophilin 1 interferes with plakoglobin binding to desmoplakin,yet together with plakoglobin promotes clustering of desmosomal plaquecomplexes at cell^cell borders. J Cell Sci 114:727^738, 2001
Elias PM, Matsuyoshi N,Wu H, Lin C,Wang ZH, Brown BE, Stanley JR: Desmo-glein isoform distribution a¡ects stratum corneum structure and function.J Cell Biol 153:243^249, 2001
Green KJ, Gaudry CA: Are desmosomes more than tethers for intermediate ¢la-ments? Nat Rev Mol Cell Biol 1:208^216, 2000
HanakawaY, Amagai M, Shirakata Y, Sayama K, Hashimoto K: Di¡erent e¡ects ofdominant negative mutants of desmocollin and desmoglein on the cell^celladhesion of keratinocytes. J Cell Sci 113:1803^1811, 2000
Hatzfeld M, Kristjansson GI, Plessmann U,Weber K: Band 6 protein, a major con-stituent of desmosomes from strati¢ed epithelia, is a novel member of the ar-madillo multigene family. J Cell Sci 107:2259^2270, 1994
Heid HW, Schmidt A, Zimbelmann R, et al: Cell type-speci¢c desmosomal plaqueproteins of the plakoglobin family: plakophilin 1 (band 6 protein). Di¡erentia-tion 58:113^131, 1994
Ishii K, Norvell SM, Bannon LJ, Amargo EV, Pascoe LT, Green KJ: Assembly ofdesmosomal cadherins into desmosomes is isoform dependent. J Invest Dermatol117:26^35, 2001
Kanegae Y, Takamori K, Sato Y, Lee G, Nakai M, Saito I: E⁄cient gene activationsystem on mammalian cell chromosomes using recombinant adenovirusproducing Cre recombinase. Gene 181:207^212, 1996
Koch PJ, Mahoney MG, Ishikawa H, et al: Targeted disruption of the pemphigusvulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte celladhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol137:1091^1102, 1997
Kowalczyck AP, Bornslaeger EA, Norvell SM, Palka HL, Green KJ: Desmosomes.intercellular adhesive junctions specialized for attachment of intermediate¢laments. Int Rev Cytol 185:237^302, 1999
Mahoney MG,Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR: Expla-nations for the clinical and microscopic localization of lesions in pemphigusfoliaceus and vulgaris. J Clin Invest 103:461^468, 1999
Mertens C, Kuhn C, Franke WW: Plakophilins 2a and 2b. constitutive proteins ofdual location in the karyoplasm and the desmosomal plaque. J Cell Biol135:1009^1025, 1996
Miyake S, Makimura M, Kanegae Y, et al: E⁄cient generation of recombinant ade-noviruses using adenovirus DNA-terminal protein complex and a cosmidbearing the full-length virus genome. Proc Natl Acad Sci USA 93:1320^1324,1996
Niwa H, Yamamura K, Miyazaki J: E⁄cient selection for high-expression transfec-tants with a novel eukaryotic vector. Gene 108:193^199, 1991
Norvell SM, Green KJ: Contributions of extracellular and intracellular domains offull length and chimeric cadherin molecules to junction assembly in epithelialcells. J Cell Sci 111:1305^1318, 1998
Roth MB, Zahlerm AM, Stolkm JA: A conserved family of nuclear phosphoproteinslocalized to sites of polymerase II transcription. J Cell Biol 115:587^596, 1991
Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, NishikawaT: Pemphi-gus vulgaris and pemphigus foliaceus sera show an inversely graded bindingpattern to extracellular regions of desmosomes in di¡erent layers of humanepidermis. J Invest Dermatol 105:153^159, 1995
Shirakata Y, Amagai M, HanakawaY, Nishikawa T, Hashimoto K: Lack of mucosalinvolvement in pemphigus foliaceus may be due to low expression of desmo-glein 1. J Invest Dermatol 110:76^78, 1998
Troyanovsky SM, Eshkind LG, Troyanovsky RB, Leube RE, Franke WW: Contri-butions of cytoplasmic domains of desmosomal cadherins to desmosomeassembly and intermediate ¢lament anchorage. Cell 72:561^574, 1993
Udey MC, Stanley JR: PemphigusQdiseases of antidesmosomal autoimmunity.JAMA 282:572^576, 1999
Wu H,Wang ZH,Yan A, et al: Protection against pemphigus foliaceus by desmoglein3 in neonates. N Engl J Med 343:31^35, 2000
Zhu AJ, Watt FM: Expression of a dominant negative cadherin mutant inhibitsproliferation and stimulates terminal di¡erentiation of human epidermalkeratinocytes. J Cell Sci 109:3013^3023, 1996
1236 HANAKAWA ETAL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY