die oxydation organischer verbindungen mit luft-sauerstoff
TRANSCRIPT

Rieche: Die O x y d a t i o n organischer V e r b i n d u n g e n m i t L u f t - S a u e r s t o f f
Die Oxydation organischer Verbindungen mit Luft-Sauerstoff V o n Dr . A L F R E D R I E C H E
D o z e n t an. der U n i v e r s i t d t L e i p z i g
Binpq. 28. Mai 1937
I n h a l t : 1 . Oxyydalwn von AIdehyden. - 2. Oxg&wn von Ketunen. - 3. Oxydafwn von Olefinen. - 4. Oxyydatwn geaattigter Verbindungen, Fettsiiuren und Kofinwaseer- stoffe. - 5. Oxydution von Athern.
ei der Oxydation organischer Verbindungen mit Luft- B Sauerstoff treten als erste Reaktionsstufen organische Peroxyde auf. Der molekulare Sauerstoff ist als eine ungesattigte Verbindung zu betrachten, die Additions- reaktionen eingeht. Wasserstoff wird zu Hydroperoxyd, Metalle werden zu Metallperoxyden und organische Ver- bindungen zu organischen Peroxyden angelagert .
Die Peroxydgruppe hat das Bestreben, die 4 0 - B i n - dung durch Wasserstoff oder organische Reste abzusattigen, erkennbar an den Oxydationswirkungen.
Wenn Sauerstoff auf organische Stoffe einwirkt, so sind nicht immer Peroxyde als Zwischenstufen faBbar. Die Herstellung einer groBen Zahl der verschiedensten Typen organischer Peroxyde und das Studium ihrer Reaktionen und ihres Zerfalls hat ergeben, daB sie sehr reaktionsfahig sind und unter den Bedingungen einer energischen Sauerstoff- oxydation meist nicht bestandig sein konnen.
Die Peroxyde konnen sich selbst zersetzen, wirken dehydrierend oder losen bestimmte Folgereaktionen aus, wie z. B. Polymerisationen ungesttigter Verbindungen. Auch im biologischen Geschehen treten bei Oxydations- vorgangen Peroxyde als Zwischenstufen auf. Wenn diese auch nur in wenigen Fallen nachgewiesen werden konnten, so beweist doch die weite Verbreitung Sauerstoff und Per- oxyde umsetzender Exbyme - Oxydasen, Peroxydasen und Katalasen -in fast allen lebenden Zellen ihr Vorhandensein.
Die organischen Peroxyde besitzen also Bedeutung vor allem fur zwei Gebiete: 1. Oxydationsvorgange, bei denen Peroxyde Zwischen- oder
2. Peroxydisch angeregte oder durch Bildung und Zerfall
Hier soll nur auf das erste Teilgebiet eingegangen werden: Die Oxyda t ion organischer S tof fe , soweit diese von Bedeu tung f u r d ie Technik ist').
Ganz allgemein beschleunigen bestimmte Faktoren, wie Metallsalze, als Ubertrager wirkende organische Verbindun- gen, Licht und bei Belichtung als Sensibilisatoren wirkende Stoffe, die Oxydation. Die Oxydat ions theor ien von Manchot, WieEand, Haber, Frank und Wilbtiit@r konnen hier nur eben gestreift werdenl). Ihnen liegt vor allem die Er- kenntnis zugrunde, da13 Fe" wirksamer als Fe"' ist. Manchot nimmt Peroxyde des Eisens als Zwischenstufen an. Nach Wiehnd bildet Fe" mit dem zu oxydierenden Substrat einea Komplex, in dem Wasserstoff aktiviert ist, so daB er an 0, zu H,O, angelagert werden kann. Auf die K e t t e n - theor ie wird am Beispiel der Aldehydoxydation noch ein- gegangen .
Endstufen der Reaktion sind.
von Peroxyden fortgefuhrte Reaktionen.
1. Oxydation von Aldehyden. Alkohole konnen mit chemischen Mitteln leicht zu
Aldehyden oxydiert werden. Die Weiteroxydation zu Sauren mit denselben Mitteln geht schlecht . Leicht verlauft die Oxydation von Aldehyden zu Sauren mit Luftsauerstoff, besonders bei Gegenwart von etwas Eisen-, Kobalt- oder Mangansalz. Als Zwischenstufe tritt Persaure auf, die sich - z. B. beim Acetaldehyd kristallin abscheiden kann. Mit Essigdureanhydrid kann die Persiiure unter Bildung von
Diacylperoxyd abgefangen werden. Aus der iiblichen Vor- stellung heraus, daB die C=O-Doppelbindung des Aldehyds Sauerstoff ,,addiert", nahmen Engkr und Weipberg ein Zwischenprodukt an, das sich zu Persaure umlagert :
0 R - C H O + 0, -+ R-C/O)O -+ R-C<,,,
H / \o
Das Auftreten einer Sauerstoffaddition an die C=O- Doppelbindung erscheint schon deshalb als unwahrschein- lich, weil auch Aldehydhydrat mit O2 Persaure bildet. Aus diesen und zahlreichen noch spater bei der Oxydation anderer Qngesattigter Verbindungen zu machenden Beobachtungen mochte ich bei den Aldehyden annehmen, daI3 sich der Sauerstoff n i ch t a n d ie Doppelb indung a d d i e r t , sonde rn zwischen C u n d H e inschieb t , da13 sich also
die hypothetischen Radikale R-C( und H- an 0, addieren.
Dieser Vorstellung wird auch die Rad ika l -Ke t t en - t heo r i e von Frank, Haber und Willskittefl), wie sie die letzteren beiden Autoren auf die Aldehydoxydation an- wenden, durchaus gerecht :
0
CH,-CO + 0, --+ CH,---C=O
(1) I 0 0 4
4
CH,--C=O + Ha0 + CHS-CHO -+ 2 C H S 4 O O H + OH
+ (2) 0 0 4
(3) CH,CHO + OH + C H , - C O + H,O
4 4 (Die kleinen Pfeile bedeuten, daO es sich jeweils urn ein Radikal
Persaure liefert mit Aldehyd ein zersetzliches Peroxyd als Zwischenstufe, das gemaB Formel 2 eine intermolekulare Disproportionierung erleidet :
handelt .)
0 I1 H
0 R 4 - Q 0-C-R -+ 2 RCOOH.
H
Die Disproportionierung entspricht in ihrem Verlauf durchaus der des OxymethylathylperoxydsS) :
H
0 H
C,H,-0 0-CH + C,H,OH + HCOOH.
2. Oxydation von Ketonen. tfber die Oxydation von Ketonen mit Sauerstoff ist in
der wissenschaftlichen Literatur fast nichts berichtet worden. In der Patentliteratur sind Verfahren bekannt, wonach Ketone durch Sauerstoff unter verhaltnismiil3ig milden Bedingungen glatt in Sauren iibergefiihrt werden
~~
1) Vgl. dam die znsammenfaesende Datstellung: A. Rieche: Die Bedeutung der organischen Peroxyde fiir die chemische Wissen- schaft und Technik, Enke 1936.
*) P. Haber, Naturwiss. 19, 450 [1931]; diese Ztschr. 46, 51 [1933], Haber u. WiUstiifter, Ber. dtsch. &em. ,Ges. M, 2844 [1931].
5 ) R k h e u. Meieter, ebenda 68, 2642 [1930].
A n g c w a n d l c C h c m i e

Rieche: D i e Oxydat ion organiacher Verbindungen mi t Luf t -Sauers to f f
koimen4). Aus Methylathylketon entsteht Essigsaure, Aceton liefert Ameisensaure und Essigsaure, wogegen Aceto- phenon Benzoesaure und Ameisensaure liefert . Auch hier is% unter Beriicksichtigung der spater noch zu erorternden Beobachtungen bei Olefinen und Athern ebensowenig wie bei den Aldehyden eine Anlagerung von 0, an die C=O- Bindung anzunehmen. Unter Anwendung der von mir seit langerer Zeit vcrtretenen Anschauung des 'Zwischenschiebens von 0, zwischen R und H stellt sich die Ketonoxydation wie folgt dar:
H H
H I1 0 II CH,-C--C-CH, + 0*+ CH+2--C-CH8 -+ CH,--COOH + CH,CHO
0 0 0 H
H H
H I1 0 II HC--C-CH, + 0, -+ HC-C-CH, + HCOOH + CH,--CkO.
ij 0 0 H
Als Zwischenprodukte sind also Hydrope roxyke tone anzunehmen, die unter dem EinfluB von Metallsalzen Dis- proportionierung in S a u r e und Aldehyd erleiden konnen. Der Aldehyd wird dann durch Luftsauerstoff in der erorterten Weise weiter zu Saure oxydiert. Es ist aber auch moglich, daB Hydroperoxyketone sich durch Platzwechsel von Acyl- rest und einem H-Atom zu Pe r sau rees t e rn umlagern, die sich dann zu Saure und Aldehyd disproportionieren:
H H C H S 4 4 4 H S -+ CHZ-C H + CHs--CHO + CH,-COOH
0 I I 0 0 0 0 H ( I
O=C--CH,
3. Oxydation von Olefinen. Wahrend Aldehyde allgemein zur Sauerstoffaufnahme
neigen, reagieren Olefine nur bei Vorliegen bestimmter giinstiger Konfigurationen glatt mit Sauerstoff. Vorgange von groBter praktischer Bedeutung, wie die Polymerisation ungesattigter Verbindungen, das Trocknen der ole in der Anstrichtechnik, das Verharzen von Schmierolen und Treib- stoffen, das Ranzigwerden von Speiseolen und Fetten, sie alle gehoren in dieses Gebiet und sind zum groI3en Teil als Folgereaktion der Peroxydbildung anzusehen. Hier sollen aber nur solche verhaltnismiiI3ig einfachen Vorgknge be- sprochen werden, bei denen Peroxyde gefaBt werden konnten.
Bei der Einwirkung von Sauerstoff auf organische Stoffe miissen wir zwei T y p e n von Reak t ionen unter- scheiden : 1.
2.
Die C=C-Doppelbindung reagiert unter Addition von O,? es bilden sich polymere Peroxyde. Diese Reaktion kommt naturlich nur bei Olefinen in Frage. Organische Stoffe lagern sich unter Sprengung einer R-R- oder R-H-Bindung an Sauerstoff an:
a) R-R + 0, -+ R-00-R b) R-H + 0, + R 4 O - H C) 2R-H + 20, + R 4 0 - R 3. H,O,.
Mit der Bildung von R-00-R ist wohl-nur dann zu rechnen, wenn der Sauerstoff unter Bedingungen einwirkt, wo Radikalbildung R- miiglich ist, z. B. bei Zimmer- temperatur nur bei den dissoziierenden Athanen. Bei hohen Temperaturen jedoch kommt Radikaldissoziation
9 D. R. P. 583704, 590365, 597973 ( I . G. Farbeninduatrie)' Erfinder Dr. Flcmming u. Dr. Spew. Henn Dr. Flemming verdanke ich den freundlichen Hinweis auf die Analogie des von uns unter- suchten Mechanismus der dtherautoxydation und der Oxydation der Ketone.
und'Addition in dem obigen Sinne wohl bei fast allen organi- schen Stoffen in Frage.' Das Zwischenschieben von 0, zwischen C-H unter milden Reaktionsbedingungen kommt jedoch bei allen den Stoffen in Betracht, bei denen eine Akt iv ie rung der C-H-Bindung aus Griinden, die im Molekiilbau liegen, eingetreten ist.
Die olefinische Doppelbindung lagert 0, an, Z. B. bei den Fu lve n en , beim asymmetrischen Dip h en yla t h yl e n (Staudinger)5), Linolensaure (Golds~hrnidt)~) und anderen. Es bilden sich stets po lymere khylenperoxyde:
)c=c<+ 0, +>-c<+ 0 0
polymerisiert sich zu:
I I I I I I I I
oe-c--C-oo--C--C usw.
Lange bekannt ist auch die Peroxydbi ldung be i Terpenen . Auch hier erhdt man polymere Peroxyde. Te rpen t ino l und die reinen Terpene, Terp inen und Phe l l andren , liefern Peroxyde, wobei von den letzteren 1 Mol mit 1 0, reagiert. Bei der Einwirkung von Sauerstoff auf organische Stoffe kommt die Reaktion erst nach Ver- streichen einer gewissen , , Indukt ionszei t" in Gang. Diese Induktionsperiode wird durch Zusatz einer kleinen Menge des in Autoxydation befindlichen Stoffes zu dem noch nicht angegriffenen, in der Induktionsperiode befindlichen sofort iiberwunden. Diese ,,Ansteckung", wie man diese tiber- tragung fast nennen konnte, die z. B. sehr deutlich beim k h e r zu beobachten ist, diirfte wohl allgemein in Frage kommen. Bode&?) zeigte, daB bei den Terpenen die Induktionszeit abgeliiirzt wird durch , ,Animpfen" mit den in Oxydation befindlichen Produkten, nicht jedoch den fertigen Peroxyden. Dabei ergab sich eine gewisse Spezifitat, indem in Oxydation befindliches Terpinen auch nur die Terpinenautoxydation induzierte. Das alles deutet darauf hin, dal3 labile Anlagerungsverb indungen des L u f t - sauers tof fs an die Olef ine als Induktoren wirken. Diese Spezifitat wird sich aber nur auf bestimmte Stoffe be- schranken und wird keine Allgemeingiiltigkeit besitzen. Ein wichtiger Beitrag zur Klarung dieser Fragen wurde von Ziegkr geliefert8). Dissoziierende Athane werden durch 0, in Losung schneller oxydiert, als sie dissoziieren. Es findet also die Oxydation des A t h a n s statt. Die Reaktion wird eingeleitet durch ein Anlagerungsprodukt des Radikals
R&. . . an 0,.
Dieses primare Anlagerungsprodukt ist der Trager einer Kettenreaktion und bildet mit dem Athan das Peroxyd:
R
R/
R \ \-00- + R L c 4 +
R R-C.. . . + 0, 3 R 7 R / R R ' \R
R
Es ist anzunehmen, daI3 die Oxydation auch bei anderen organischen Verbindungen ahnlich verlauft . Zwar kann a d e r bei den dissoziierenden Athanen sonst nur bei hohen Temperaturen eine wirkliche Radikalbildung angenommen werden. Bei tieferen Temperaturen dagegen lagert sich der
6, Staudinger, Ber. dtsch. &em. Ges. 68, 1075 [1925].
') Bodendorf, Arch. Pharmaz. Ber. dtsch. pharmaz. Ces. 271,
8 ) Zicglm u. E d d , Liebigs Ann. Chem. m, 277 [1930], 804,
Uoldachmidt u. Freudenberg. ebenda 67, 1991 [1934].
1 [1931].
162 [1933]; Ziqler u. Orth, Ber. dtsch. chem. a. a, 628 [1932]. d nne tctlndtc Chcnr ie SLlahe 19.17 Nr. 2.8 52x

R i e c h e . Die O x y d a t i o n o r g a n i s c h e r V e r b i n d u n g e n mi l L u f t - S a u e r s t o f f
Sauerstoff an das organische Molckiil an und schiebt sich mag diese zu spalten, etwa wie Lavul insaureperoxyd in zwischen eine aktivierte CH-Bindung ein. Addition der Berns te insaurees te r iibergeht (Pummerer) ll). Radikale oder ,,Einschieben" des 0, unterscheiden sich in Von besonderem Interesse ist noch der Ablauf der ihrem Endeffekt nicht voneinander. Gleichzeitig ergibt sich Oxydat ion des Athylens selbst. D~ Athylen mit aus den Versuchen von ziegkr eine Deutungsmoglichkeit Sauerstoff erst merklich bei hoheren Temperaturen reagiert fur die vereinzelt beobachtete ,,Spzifitat" der Induktoren. und unter diesen Bdingungen etwa auftretende Peroxyde R-00- kann wohl in einzclncn Pallen nur mit identischem zersetzt werden, so konnte man peroxyde nicht fassen. Die R-R reagieren Zu R-4O-R. Bei Gegenwart cines andexen Natur der entstehenden Oxydationsprodukte zeigt aber ein- Stoffes, etwa Rl-Rl, lauft die Reaktion nicht weiter. deutig, die erste Stufe der Reaktion ein Peroxyd ist.
Die Vorstellupg erscheint berechtigt, da13 auch stabile der Beobachtung ~h~~~~ und Himhiwdia) , Kohlenwasserstoffe bei hoherer Temperatur mit 0, ahnlich daO die Geschwindigk& der Oxydation mehr vom padial- reagieren wie etwa Triphenylmethyl bei ZimmertemPeratur. druck des Athylens als von dem des Sauerstoffs abhangt, ist Bei der Explos ion von Brenns tof f -Luf t -Gemischen auf das Vorzegen einer K e t t e n r e a k t i o n zu schlieBen, bei im Verbrennungsmotor liegen sokhe BdinWngen VOr. der ein Anlagerungsprodukt des Sauerstoffs an Athylen mit Leicht dissoziierendc Brennstoffe spalten R- oder H- ab, Athylen reagiert. Die Natur der Oxydationsprodukte wurde die mit 0, zu Peroxyden reagieren. Die Peroxyde wirken eingehend untersucht. Nach Lenher13) entstehen Athylen- als Reaktionszentren und fuhren zu einer vorzeitigen Ver- oxyd, Glykol, Fomaldehyd, ~ ~ ~ i ~ ~ ~ ~ g ~ ~ , Wasser, puffung. Es tritt ,,Klopfen" ein. s o kann man denn such, Kohlenoxyd, Kohlendioxyd und Wasserstoff, ferner Dioxy- wie A . w. Schmidt') zeigte, organische PeroxYde mit Wtem methylperoxyd, j edoch kein Acetaldehyd (im Gegensatz zu Erfolg als Proklopfmittel zur Steigerung der Ziindwilligkeit dem Befund .van hter, hi^ und wkhr)i4), Unter Ab- von Dieseltreibolen verwenden. Aromatische Kohlen- wei&ung in einigen punkten henher mijchte ich den
halb geringere Neigung zum Klopfen, weil sie stabil sind A ~ s erste Stufe tritt Athylenperoxyd auf, das schon als und weniger zur Bildung von Radikalen neigen. monomere Stufe oder auch in teilweise polymerisiertem Zu-
Der Befund, daB in einzelnen Fiillen von Olefinoxyda- stande sich weiter umsetzt: tion der Sauerstoff zweifellos an der Doppelbindung angreift,
stoff bei Olcfinen etwa ahnlich wie Ozon stets mit der
der Sauerstoff sich in den meisten Fallen in seiner Reaktions- weise grundsatzlich vom Ozon unterscheidet, und daB er Die Formaldehydbildung wird analog der von vielfach n i ch t a n de r Doppelb indung angreif t , Staudinger untersuchten Spaltung vom asymmetrischen Di- so n der n s ic h , wie bei Aldehyden, Athern und Ketonen, phenylathylenperoxyd zu Benzophenon und Formaldehyd zwischen e ine der Dbppe lb indung oder dem Saue r - verlaufen. Athylenperoxyd kann aber auch mit einem s t off b en a c h b a r t e C-H - B i n du n g ei n s c h i e b t . Die weiteren Athylenmolekul reagieren unter Bildung von Doppelbindung begiinstigt also 'die Sauerstoffaufnahme, B thy lenoxy d , ahnlich wie Renzopersaure mit Olefinen indem sie die C-H-Bindung aktiviert. Wahrend Alkyl- Oxyde gibt:
Reaktion durch die Nachbarschaft eines aromatischen Restes (siehe z. B. Toluol) oder eines 0-Atoms begiinstigt (Alkohole, Ather). Dasselbe gilt auch fur die Nachbarschaft einer Athylengruppc. Te t r a l in und Cyclohexen oxydieren sich bekanctlicll zu I'c- oxyden folgender Konstitution:
wasserstoffe und ganz besonders Isooktan zeigen wohl des- Vorgang der Athylenoxydation folgendermafien erklaren :
fuhrte zu der allgemein verbreitcten Ansicht, dafl der Sauer-
Doppelbindung reagiert. Es sol1 nun gezeigt werden, daI3
CH, = CHa 4- 0, -+ -cHz-cHa-oo- -+ PC"Yrnerisiert sich +
--cH, CH,-O O-CH, CH,-O 0 - c ~ ~ - CH*-O 0- usw. -+
-+ liefert Fonnaldehyd.
gruppen sonst ziemlich schwer zu oxydieren sind, wird die -CHz4H2-OO- + CH, = CH, + 2 CH,--CH,
\O/
Die Bildung von H,O, kann in ahnlicher Weise er-
-cH, CH, 00- + H O H + H ~ - C H , cH, OOH + cH, C q , + H,o, folgen:
(11) (1)
Fruhcr wurde das C y cl o hexenp er o x y d als A t h y len - peroxyd (I) formuliert. Criegee fand aberlO), da@ es ein Hydrope roxyd (11) ist. Es wird sich in Zukunft wohl noch ofter erweisen, da13 die Oxydationsprodukte einfacher , ungesattigter Kohlenwasserstoffe die Konstitution von Hydroperoxyden besitzen.
Ahnlich liegen die Verhaltnisse auch bei ungesattig- ten F e t t e n und.Olen. Da ungesattigte Fette U. a. be- sonders leicht zum Ranzigwerden neigen, so konnte man zunachst auch einen ausschliefllichen Angriff auf die Doppelbindungen annehmen. Ungesattigte Kohlenwasser- stoffe werden jedoch bekanntlich leicht zu a .p -un- g e s a t t i g t e n K e t o n e n oxydiert. Bei der Linol - s a u r e ist es bekannt, daB der oxydative Angriff an der zwischen den olefinischen Gruppen liegenden CH,-Gruppe erfolgt (Buuer). Auch hier ist ein Alkylhydroperoxyd als Zwischenstufe anzunehmen. Der peroxydische Rest greift dann auf die benachbarten C-C-Bindungen uber und ver-
*) A. W . 8 d d d t . Braunkohle 86, 535 [1936]. lo) UiicgCe, ulr#gs Ann. &em. 6M, 84 [1936].
\C/ Y + CH, = CH, -+ CH, CH, + CHaOH CI-
\O/
H,O, -t 2CH,O gibt in bekannter Weise Dioxy- me thy lpe roxyd CH,OH-OO--CH,OH, das in H, + ZHCOOH zerfallt .
Glyoxal konnte aus Formaldehyd entstehen oder durch intramolekulare Disproportionierung des polymeren Athylenperoxyds : .
CH, CH, OO-HCH-HCH-OO-CHl- CH,-00-HCH-HCH - A A A
--CH+X*OH + OW-HCO + HOCH2--CH10H + OHC-CH(
Hierbei wiirden also gleich vie1 Molekiile Glykol und Glyoxal entstehen. Demnach erscheint die Entstehung der erwahnten Oxydationsprodukte als durchaus erklarlich.
ES bleibt nun noch die von Lenher gefundene Bildung von Propylen zu erklaren. Lenher nimmt die intermediare Bildung von ,,Methylen" an, das sich an Athylen addiert:
= CH, + )CH, -P CH, = CH--CH,
11) P u m w e f , &dkch, E b e m y e t . Diss. Ueflach, Erlangen 1929,
1,) Proc. Roy. Soc., London, Ser. A 136, 277 [1929]. li) J. Amer. chem. Sot. 68, 3737-3765 r19311.
Ber. dtsch. chem. Ges. 64, 807 [1931].
la) j. Soc. &em. Ind., Chem & Ind. 41,- 303-T [1922]; 43, 415 T [1923].

R i e c h,e : Il i e O x y d a 1 i o n o T Q an i R c h e r 1' e T h i n dir, ng c n m i I I, uf t -So 71. e r s t o f f
, ,Methylen" konnte aus Athylenperoxyd entstehen : Der zunachst nur hypothetische Hydroperoxydiathyl- ather konnte synthetisch aus Oxyathylhydroperoxyd und Athanol erhaltcn werden : -CHa-CH2--O0- + HCOOH -t / \CH,.
4. Oxydation ge&ttigter H O H H
CH, CH0 +H,O, -+CH, C/ + €IOC,H, + CH,-C-- 0---C,H, + H,O Verbindungen, FettGuren und Kohlenwasserstoffe. \OOH I
0 0 Der oxydative Abbau der Fettsauren durch chemische
Mittel ist noch VerhaltnisrniiDig wenig untersucht. Im H
Mittelpunkt des Interesses standen fur den Chemiker die Ole der Ansttichtechnik. Die Leinolsaure wird durch Sauer- stoff bis herunter zu Pelargonsaure, Capronsaure, C,-Saure abgebaut. Bei der OxYdation gesa t t i g t e r Sauren wurde man vom chemischen Standpunkt aus die Bildung von a-Ketosauren envarten, die unter Decarboxylierung Aldehyde @ben maten. HieT liegen Wert~~olle unter- suchungen von physiologischer Seite vor16). Der Abbau von Sauren mit gerader Kohlenstoffzahl geht im tierischen ather, Organismus iiber p-Ketosauren vor sich. Ahnlicli verlauft die Oxydation von Sauren mit Luftsauerstoff. Die , ,Parfum- ranzigkeit" an' der Zuft oxydierter Fette beruht auf der Bildung von Methylketonen, die durch 'Decarboxylierung der p-Ketosauren entstehen : R-CH,-CH,--COOH -> R--c<H, --COOH +K--c.-.cH, +. ~ 0 , .
Die Methylketone werden vermutlich, wie bereits bei der Oxydation einfacher aliphatischer Ketone beschrieben, zu einer hochmolekularen Fettsaure und zu Ameisensaure weiter oxydiert.
Der Mechanismus des Ablaufs der Fettsaureoxydation
durchgefiihrte Verfahrerf der ?erstdung von Fettsauren durch Paraffinoxydation von Bedeutung, denn neben dem
Die Verbindung liefert alle die im autoxydierten Ather zu findenden produkte, die in dem Schema aufgefiihh sind, inshesondere such beim Emarmen das hochexplosive Athylidenperoxyd, das ,&herperoxyd'. Weniger zur Oxy- dation als Athylather nd@ Dimethylather, wesentfich starker oxydabel ist Dibenzylather, nurch die Nachbar- stellung der Phenylgruppe ist die CH,-Gruppe besonders aufgelockert. Isopropy~~ther verhalt sich etwa wie Athyl-
Zusammenfassung . Die geschilderten Oxydationsvorgange zeigen, daB der
Sauerstoff im Gegensatz zum Ozon in vielen Fallen n i ch t mi t der Doppelb indung r eag ie r t , sondern s ich
0 0 zwischen eine C--H-Bindung einschiebt . Wir miissen uns mit dem Gedanken vertraut machen, da13 diese Keak- +.ionsweise die hgufigere ist. Diese neue Erkenntnis zeigt den Ablauf verschiedener Oxydationsvorgange in einem anderen Licht. Ein schoner Beweis fur die Richtigkeit dieser schon lange von uns geaderten Anschauungen wurde
criegee geliefert17). D~~ Tetrafin vermag sich statt an durch Luftsauerstoff ist im Hinblick auf das .techdsch Sauerstoff such an chinon anzdagern und bildet die dem
TetraEnperoxyd analog gebaute verbindung.
/\I\ ~ I1
\/\/ /\
Ausgangsparaffin wird auch ein Teil schon gebildeter Fett-
eines Paraffinkohlenwasserstoffs in Frage kommenden Reak-
scheinlichkeit : \..- .
/\A
\/\/ saure oxydiert. Unter den fur den Ablauf der Oxydation I . H
tionen besitzt folgende als Teilreaktion eine gewisse Wahr- /'C)-(--) - O H I1 OOH
H H
H 0 bindungen stellt sich nun folgendermafien dar : K--CH,--C---CH, .K + 0, + R-- CHa-CC---CH2---R -+ Die Reaktion des Sauerstoffs mit organischen Ver-
0 €1
K-CH, CH- 0 CH,---R + K- CH2---CH0 f HOCHz---R I 'l*O*
0 H
Y K- - CH,-COOH
Wenn eine Doppelbindung im Molekul ist, etwa durch Spaltung entstanden, so wird die Oxydation neben der Doppelbindung erfolgen.
5. Oxydation von khern. Der Ablauf der Atheroxydation wurde schon vor einiger
Zeit von uns geklartle). Es konnte gezeigt werden, daD hier ein Zwischenschieben von 0, zwischen C-H eintritt. Aus Ather bildet sich H y d r op e r o x y d i a t h y 1 a t h e r :
H H
H 91 0 diathylather El 0
H
Hydroperoxy- CZHb-O-C--CH, + 0, -+ C,H,-O-C-CH,
H
(CH*--C--OO-), CH,---CHO + Ha02 Kthylidenperoxyd (polymer)
Knoop: Orydationen h Tierkorper, Enke 1931. I') Riadrsn.Ildsirttr, diese Ztschr. 44,896 [1931]; 49, 101 [19361;
R. Neu, ebenda 46, 519 [1932].
1. K-K + 0, + R-00---R
H H H H
0 0
2. R--C=C-R + 0, --f R-C-C-R --f polym. -4tllylenperoxyd
3. R- -H + 0, + KOOH
0
(00, 3a. R-CHO + 0, + R--c/
H H 3b. R--C--C-R+02 -+ R 4 - C - R
H I I 0 I1 0 0 H
H H
0
3 ~ . K-C-OH + 0, + R--C--OH H 0
0 H
H H 3d. R-C-0-R -+ RX-0-R
H 0 0 H
Bei organischen Stoffen konnen sich also die Kohlen- wasserstoffreste an Sauerstoff anlagern (l), wie beim Tri-
1') Criegee, diese Ztschr. 49, 323 [19361.
A b # ~ w a m d l e Chemic L a -sn 5.2 3
K I e m in : N e u e r e P r o b 1 e me d er a n o r g a n iac h en C he mi e
phenylmethyl, dann bekommen wir Dialkylperoxyde. Aus bestimmten Athylenverbindungen erhalt man durch Addition an die Doppelbindung polymere Athy len - peroxyde (2). Lagern sich der Kohlenstoffrest und das losgeloste H-Atom an, so erhalten wir Alkylhydro- p e r o x y d e (3 a 4 ) . Wir finden diesen haufigsten der Falle bei Kohlenwasserstoffen, Alkoholen, Athern, Aldehyden, Ketonen. Und schliefllich konnen sich Wasserstoffatome
Neuere Probleme der anorganischen Chemie V o n Prof. Dr. W I L H E L M K L E M M , T . H . D a n z i g - L a n g f u h r
Z u s a m m e n f a s s e n d e r V o r t r a g anl l iPl ich d e s R e i c h s t r e f f e n s der D e u t s c h r n C h e m i k t i i n F r a n k f u r t ( M a i n ) a m 7 . J u l i 1 9 3 7
Eingeg. 8. Juni I937
allein an den Sauerstoff anlagern, (z. B. bei biologischen Dehydrierungen); dann erhalten wir Wasserstoffperoxyd. Und damit kehren wir zum Ausgangspunkt zuruck. Es besteht zwischen Wasserstoff, Alkali- und Erdalkalimetallen einerseits und organischen Stoffen andererseits kein grund- satzlicher Unterschied, wenn sie mit Sauerstoff reagieren : Sie addieren sich zunachst an den Sauerstoff unter Bildung von Peroxyden. [A. 72.1
n Geburtstagen, die ein Lebensjahrzehnt beschliefien, A blickt man gern zuruck, um den Lauf der Entwicklung in denvergangenen Jahrzehnten noch einmal in seinen wesent- lichen Ziigen zu verfolgen. Man versucht aber auch, etwas den Schleier zu heben, der die Zukunft verhdlt, und spricht Hoffnungen und Wunsche aus. So sol1 auch anlal3lich des diesjahrigen Jubilaumstreffens der deutschen Chemiker fur eines der Fachgcbiete, die der Verein in seiner Fachgruppen- arbeit besonders berucksichtigt hat, ein solcher Ruck- und Ausblick angestellt werden. Dies scheint um so notwendiger, als weiten Kreisen von Chemikern noch nicht so recht zum RewuBtsein gekommen ist, daq die anorganische Chemic etwa seit der Jahrhundertwende eine Wiederbelebung er- fahren hat, an der nicht zuletzt deutsche Forscher - und zwar sowohl Chemiker als auch Physiker - entscheidenden Anteil haben.
Selbstverstandlich %t es unmoglich, eine solche Be- trachtung a d al le Gebiete der anorganischen Chemie auszudehnen ; vielmehr sollen sich die Ausfuhrungen auf die Frage beschranken, die letzten Endes das Kern - problem der gesamten Chemie ist: Die E n t s t e h u n g von Verbindungen a u s den Elementen . Welches sind die Gesetzmaoigkeiten, die man in bezug auf die Zu- sammensetzung, die Bestandigkeit und die Eigenschaften der chemischen Verbindungen kennt, und inwieweit kann man diese in leicht iibersichtlicher Weise deuten?
Dabei sei beziiglich des Stoffmaterials nicht allzu eng- herzig verfahren; wenn sich die Betrachtungen auch in der Hauptsache auf die wirklich im Gleichgewicht bestandigen Verbindungen beschraken, so werden doch auch gelegentlich solche Stoffe beriicksichtigt, die, wie z. B. KC10,. instabil sind und iiberhaupt nur dargestellt werden konnen, weil ihre Zersetzungsgeschwindigkeit bei nicht zu hohen Temperaturen iiuiU8erst klein ist.
I. Vor einem halben Jahrhundert glaubte man eine Reihe
von Erkenntnissen gewonnen zu haben, durch die die Probleme der chemischen Bindung weitgehend gelost schienen. In den Lehrbuchern findet sich als eines dieser ,, Grundgesetze" der Chemie das Gesetz der kon- s t a n t e n u n d mul t ip len Propor t ionen . Ferner hatte sich aus den Kampfen um das Wesen der chernischen Bindung, aus denen die Namen Berzeliua, Dumas und KekulC hervorragen, der Begriff der ,,Wertigkeit" her- ausgeschalt, die die Zusammensetzung der chemischen Ver- bindungen regelt. Wir wissen heute, daI3 diese beiden Be- griffe sich auf dem Gebiete der organischen Chemie aus- gezeichnet bewahrt haben. Fiir das Gesamtgebiet der Chemie 'dagegen besitzen sie durchaus nicht die allgemeine Bedeutung, die man ihnen zugeschrieben hatte. Da13 der
I n h a l t : AUgemeinee (I.-I V . ) . A . Verhindungen z w k h e n unedlen Metallen und NichthlmataUen (Ionenbindung). B . Verbindungen aw Nichtmetallen (Atombindung). C . Intermetallkche Verbindungen. D . Verbindungen der ttbergangeebmente (Maraganiden). V .
Wer t ig kei t s b egr if f bei den intermetallischen Ver- bindungen und halbmetallischen Stoffen versagt, braucht heute nicht mehr besonders betont zu werden. Aber auch das Gesetz der kons t an ten und mul t ip len P r o p o r - t ionen ist - worauf zuerst wohl mit Nachdruck G. P. Hiittiql) hingewiesen hat - ein Grenzgesetz , das nur fur einen Teil der chemischen Verbindungen gilt, und zwar in erster Linie fur die Verbindungen der Nichtmetalle unter- einander sowie fur die der Metalle mit den Halogenen und den Sauerstoffsauren.
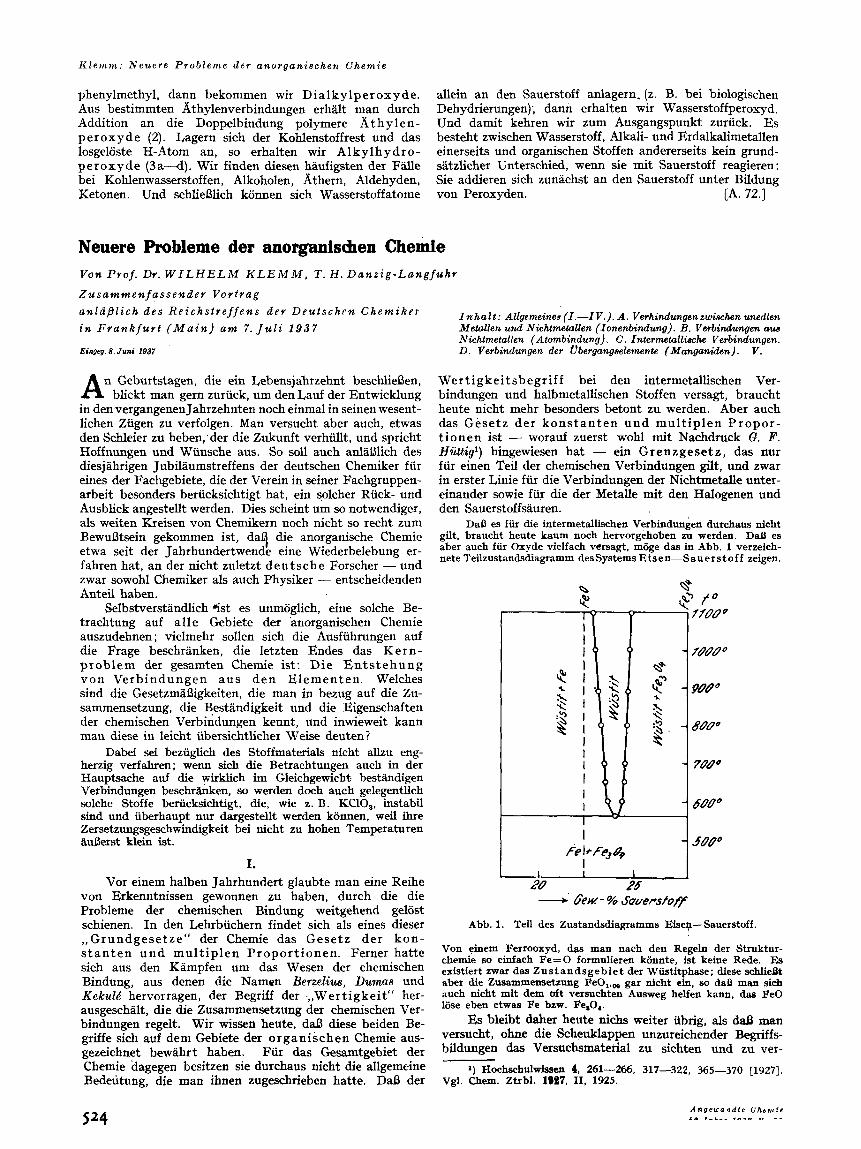
DaD es fiir die intermetallischen Verbindungen durchaus nicht gilt, braucht heute kaum noch hervorgehoben zu werden. DaB es aber auch fiir Oxyde vielfach versagt, mBge das in Abb. 1 verzeich- nete Teilzustandsdiagramm des Systems €?is e n-S auer s t o f f zeigen.
L I I I I I I I I t I I I I 7
-1-
4' GeHc - % Suuersto f f 20 25
Abb. 1. Teil des Zustandsdiagramms Eisep-Sauerstoff.
Von einem Ferrooxyd, d- man nach den Regeln der Struktur- chemie so einfach Fe=O formulieren kBnnte, ist keine Rede. Es existiert zwar das Zustandsgebiet der Wiistitphase; diese schlidt aber die Zusammensetzung FeO,,, gar dcht ein, 90 daB man sieh auch nicht mit dem bft versuchten Ausweg helfen kann, das FeO lijse eben etwas Fe bzw. Peso,.
Es bleibt daher heute nichs weiter iibrig, als dd3 man versucht, ohne die Scheuklappen unzureichender Begiffs- bildungen das Versuchsmaterial zu sichten und zu ver-
1) HochschdwkSa 4, 261-266, 317-322, 365-370 [1927]. Vgl. Chem. Ztrbl. lSS7, 11, 1925.
524 A n g c t r a a d l r C h e a i i e <" ,_.__ ..."" .. ^ ^