diagnosis management wegener's granulomatosis...
TRANSCRIPT

Britishgournalof phthalmology, 1991,75,201-207
Diagnosis and management of systemic Wegener'sgranulomatosis presenting with anterior ocularinflammatory disease
Stephen J Charles, Paul A R Meyer, Peter G Watson
AbstractThe ocular and systemic features of 10 patientswhose Wegener's granulomatosis presentedwith corneoscleral inflammatory disease aredescribed. Marginal corneal infiltrates wereseen in all patients with anterior scleritis andwere a valuable sign of disease activity. Nineout of 10 patients had symptoms of systemicvasculitis on presentation; seven had renalimpairment; three had chest x-ray abnor-malities. Autoantibodies against neutrophilcytoplasmic determinants (ANCA) werepresent in all cases. In seven patients thescleritis responded well to pulsed immuno-suppressive therapy followed by long term oralsteroids and cyclophosphamide. Oral steroidtherapy alone failed to control severe disease.Corneoscleral disease was not a cause ofvisualloss. It is important to realise that inflammatorycorneoscleral disease may be the presentingfeature ofa severe systemic vasculitis.
Wegener's granulomatosis (WG) is a multifocalnecrotising vasculitis which most commonlyaffects the lungs, upper respiratory tract, andkidneys.' 2 In consequence patients may presentwith haemoptysis from cavitating areas of pul-monary consolidation, sinusitis and serous otitismedia, or renal failure resulting from chronicglomerulonephritis. Untreated, the disease usedto be rapidly fatal, usually as a result of renalfailure.3The eye is involved in between 28%4 and 58%1
of cases, and patients often present with theocular manifestations. There may be orbital
Department ofOphthalmology,Addenbrooke's Hospital,Cambridge CB2 2QQS J CharlesPA R MeyerP G WatsonCorrespondence to:Mr S J Charles, FRCS.Accepted for publication26 October 1990
invasion by paranasal sinus granulomata,5producing painful proptosis with ophthal-moplegia and optic nerve compression, or a focalvasculitis6 causing scleritis and peripheralcorneal disease. In the past ocular involvementcaused severe visual loss regardless oftreatment.8The affected tissues show a necrotising
vasculitis with granuloma formation,' andleucocyte fragments have been demonstrated inthe lumina of the inflamed vessels by electronmicroscopy.9 The characterisation of the diseasehas recently been facilitated by the detection ofautoantibodies against neutrophil cytoplasmicdeterminants (ANCA) in patients with systemicvasculitis, especially WG and microvascularpolyarteritis (MPA)."''2 Two types of autoanti-body have recently been identified: c-ANCAactive against a 29 kD protease'3 and p-ANCAdirected against myeloperoxidase.14 The formerappears to be specific for WG, while p-ANCAhas been found in other forms of vasculitis suchas MPA."5The treatment of choice in WG has been a
combination of systemic corticosteroids and alymphocytotoxic agent, of which cyclo-phosphamide is the most effective.216 Pulsedintravenous corticosteroids and cyclo-phosphamide,'7 and plasma exchange,'8 havealso been used to control the acute phase of thedisease.
In this study we describe the ocular andsystemic features of a series of patients whoseWG presented with anterior ocular disease. Wealso consider the consequences for ocularmorbidity when treatment is directed against theunderlying systemic disease.
Table I Results ofinvestigations at presentation with eye disease
Scleritis without necrosis Scleritis with necrosis
withoutgutter with gutter
Patient: 1 2 3 4 5 6 7 8 9 10Investigation
CXR changes - - - + - - + + - -Sinus XR changes + - + - + + + + + -Hb(gdl) 13-8 12-1 10-3 11-6 13-2 12-4 9 4 11-3 8-0 15-3WBC(x101/l) 5 9 10-8 13-5 14-8 7-9 12-0 11-8 11-2 10-8 8-9Platelets (x 109/1) 432 534 847 495 263 386 832 814 651 243ESR (mm/h) 19* 123 129 83 32* 114 121 89 >140 35CRP (mg/i) ND 123 192 109 9* 97 138 57 284 16Immune complexes
(normal <30"gfl) <30 10 28 4 ND 40 72 33 ND NDComplement N Raised N N N Raised Raised N N ND
C3C4 C4 C3C4Raised immunoglobulins N IgA IgG N N IgG IgG IgA ND N
IgGRaised ANCA titre t + + + 4 + + + + tCreatinine clearance ml/min ND 84-5 62-5 98-8 71-3 53-1 41-9 20-5 3-84 126Biopsy: Renal + +
Nasal + + + + + + +Other +
*On steroid therapy. ND=not done. N-normal. tANCA testing not yet available when patient I initially presented,but ANCA was elevated when tested later. tBecame raised.
201
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from

Charles, Meyer, Watson
Patients and methodsBetween 1986 and 1989 ten patients (six femalesand four males) were seen in Addenbrooke'sHospital with scleritis complicating WG. Allpresented with scleritis, with or without cornealguttering, and had features compatible with WGin nasal mucosal or renal biopsies, together withincreased ANCA.Each patient underwent full medical examin-
ation, and their investigations are listed in Table1. The ANCA were measured by a solid phaseradioimmunoassay (RIA); test sera werequantified as a percentage of the binding of areference strong positive serum (normal range<16%). Indirect immunofluorescence (IIF)was performed to distinguish granular cyto-plasmic staining (c-ANCA) from a perinuclearstaining pattern (p-ANCA).
Clinical findingsThe patients were divided into two groupsaccording to the absence of presence of scleralnecrosis. The age at onset of disease ranged from38-65 years (mean 56-6 years).
SCLERITIS WITHOUT SCLERAL NECROSIS
Systemic diseaseThe first patient in this group appeared to sufferfrom scleritis alone, but developed renal failurefour years later. Two other patients sufferedfrom arthralgia at the time of their presentation;one also complained of epistaxis. Both werefound to have renal disease. None had' anyevidence of lung disease.
Renal biopsies in two patients demonstratedrespectively mesangioproliferative glomerulo-nephritis and acute glomerular necrosis. In thethird patient nasal mucosal biopsy showednecrotising vasculitis affecting small arteries andvenules, with granuloma formation. The ANCAtitre (RIA) was raised in all three cases, and IIFconfirmed granular cytoplasmic staining in twocases.
Ocular diseaseAll patients had anterior scleritis (Fig 1), inwhich different sectors became involved duringthe course of the disease; a mild anterior uveitiswas also present in one.
Limbal subepithelial infiltrates were seen intwo cases, but thinning was not present. In onecase areas of corneal epithelial disturbance wereseen.
Case history: patient 2: scleritis, limbal ischaemiaand proteinuriaA 5 1-year-old woman with a two-year history ofrecurrent episcleritis presented with rightanterior scleritis and marginal corneal sub-epithelial infiltrates. She also complained of apainful swollen foot. The erythrocyte sedi-mentation rate (ESR) was 123 mm/h and despitenormal urea and electrolytes there was heavyproteinuria and reduced creatinine clearance.Chest and sinus x-rays were normal. The ANCA
Figure I Anterior scleritis without scleral necrosis (patient 3)may be the presentingfeature ofWG. Scleral oedema isaccompanied by episcleral venous congestion.
titre was not raised and the eyes settled withouttreatment.Four weeks later the ANCA titre rose (RIA=
44%), and renal biopsy showed mesangialproliferation compatible with, but not diagnosticof, a systemic vasculitis such asWG or MPA. Notreatment was instituted.Four months later she developed bilateral
multifocal marginal corneal epithelial defects(Fig 2) and granular subepithelial deposits.Herpetic infection was suspected, but no viruscould be isolated, and there was no response totopical antiviral agents. Haemoglobin videoimaging showed severe limbal hypoperfusion,and the epithelial defects were thereforeattributed to limbal ischaemia. Her systemicvasculitis was controlled by cyclophosphamideand prednisolone, and, after two weeks, per-fusion of the limbal arcades had returned tonormal and the epithelial defects had dis-appeared. Leucopenia and alopecia promptedreduction of the cyclophosphamide dose. Shehas had episodes of arthralgia but is currentlycontrolled on cyclophosphamide (1 5 mg/kg)and prednisolone 10 mg daily.
SCLERITIS WITH SCLERAL NECROSIS
Systemic diseaseSeven patients (four males, three females)presented with necrotising scleritis. All hadsymptoms of systemic disease; arthralgia (3),cough and breathlessness (2), pain over the
Figure 2 Unusual marginal corneal epithelial defectdemonstrated by rose-Bengal stain (patient 2). Haemoglobinvideoimaging showed hypoperfusion ofthe adjacent limbalarcades. The lesion was not affected by antiviral therapy butresolved after immunosuppression.
202
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from

Diagnosis and management ofsystemic Wegener's granulomatosis presenting with anterior ocular inflammatory disease
Figure 3 Anterior scleral necrosis with conjunctival cover(Patient 4). Noteyellow colour ofnecrotic sclera and theadjacent marginal corneal infiltrates, which are multifocal,grey and have diffuse edges.
maxillary sinuses (6), epistaxis (6), and deafness(3).
Sinus x-rays revealed mucosal thickening orsinus pacification in five patients. Three caseshad chest x-ray evidence of pulmonary con-solidation, two with cavitation (Fig 6). Creatinineclearance indicated renal impairment in fivepatients.
Nasal mucosal biopsy provided histologicalconfirmation of vasculitis in six patients andshowed granuloma formation in five. Conjunc-tival biopsy (prior to referral) had shownvasculitis in a further case. The ANCA titre wasalso raised in all these patients. However, onehad a normal titre at presentation, which wasraised 18 months later. IIF could be demon-
rtgure DA Corneal gutterng central to areas of scleralnecrosis with loss ofconjunctival cover (patient 7). Notecollagen plug in corneal gutter (arrowed) and necrotic tissueemergingfrom conjunctival defects.
Figure 5B Same patient one week later, after two pulses ofintravenous cyclophosphamide and methylprednisolone. Thecorneal plugfrom the nasal gutter has been lost, revealing alimbal perforation.
Figure 4 Anterior scleral necrosis with loss ofconjunctivalcover. The scleral necrosis is confluent with a circumferentialcorneal melt (patient 8).
strated in six cases, and in all of these c-ANCAwas confirmed.
Ocular diseaseScleritis. Scleral necrosis appeared to arise at asingle focus, which evolved either by enlarge-ment or by the development of satellite lesions(Fig SA). Superior sclera was involved in foursubjects, inferior in one and nasal in three.Ultrasound demonstrated spread to post-equatorial sclera in two patients. Necrotic scleraappeared yellow when viewed with low intensity,diffuse light (Fig 3). The overlying conjunctivaremained intact in two patients (Fig 3) but hadbecome lost in five by the time of presentation(Figs 4, 5A, 7A).
Subconjunctival haemorrhage was present infive patients.Limbal changes. The scleral necrosis trans-
gressed the limbus in three patients (Fig 4) andevidence of limbal vascular remodelling wasfound in the remainder.
Corneal infiltrates. Multifocal stromal infil-trates below Bowman's membrane extendedcentrally from the limbus in every patient (Fig3). Disease activity was accompanied by enlarge-ment of the infiltrates and loss of definition oftheir borders; Conversely, during periods ofremission they became smaller and more discrete(Fig 7d).
Corneal guttering was present in four patients.Gutters arose only central to areas of scleralnecrosis (Fig 5A). Circumferential progressionwas characterised by stromal oedema at the
Figure SC Lamellar corneoscleral graft perfonned aftermedical control ofdisease activity.
203
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from
Charles, Meyer, Watson
leading edge. The central limit of the gutter wasshelving in three cases and steep in one. Limbalperforation occurred in one patient (Fig 5B) butanterior chamber depthwas maintained, allowingcorrective corneoscleral lamellar patch graftingto be delayed until disease activity had been fullycontrolled (Fig 5C).
Uveitis. A few cells and minimal flare werepresent in four cases.
Case history: patient four: nodular scleritis withcavitating lung lesionsA 39-year-old woman, with a past history ofpolyarthralgia, developed left anterior scleritis.This initially responded to high dose oralsteroids, but then relapsed accompanied by acough, a sore throat and malaise. She firstattended our unit with visual acuities of 6/5 ODand 6/36 OS, left nodular scleritis with dis-ruption of the adjacent limbal arcade, and super-ficial marginal stromal corneal infiltrates (Fig 3).
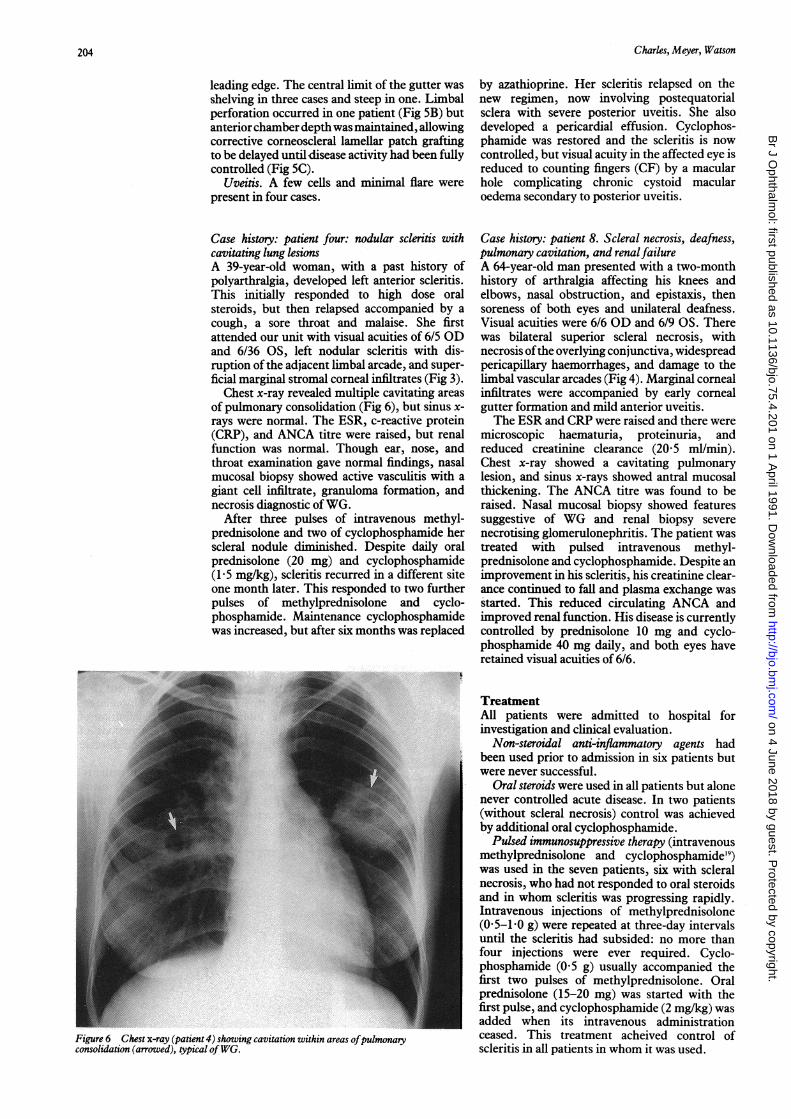
Chest x-ray revealed multiple cavitating areasof pulmonary consolidation (Fig 6), but sinus x-rays were normal. The ESR, c-reactive protein(CRP), and ANCA titre were raised, but renalfunction was normal. Though ear, nose, andthroat examination gave normal findings, nasalmucosal biopsy showed active vasculitis with agiant cell infiltrate, granuloma formation, andnecrosis diagnostic ofWG.
After three pulses of intravenous methyl-prednisolone and two of cyclophosphamide herscleral nodule diminished. Despite daily oralprednisolone (20 mg) and cyclophosphamide(15 mg/kg), scleritis recurred in a different siteone month later. This responded to two furtherpulses of methylprednisolone and cyclo-phosphamide. Maintenance cyclophosphamidewas increased, but after six months was replaced
Figure 6 Chest x-ray (patient 4) showing cavitation within areas ofpulmonaryconsoltdatwon (arrowed), typical ofWG.
by azathioprine. Her scleritis relapsed on thenew regimen, now involving postequatorialsclera with severe posterior uveitis. She alsodeveloped a pericardial effusion. Cyclophos-phamide was restored and the scleritis is nowcontrolled, but visual acuity in the affected eye isreduced to counting fingers (CF) by a macularhole complicating chronic cystoid macularoedema secondary to posterior uveitis.
Case history: patient 8. Scleral necrosis, deafness,pulmonary cavitation, and renalfailureA 64-year-old man presented with a two-monthhistory of arthralgia affecting his knees andelbows, nasal obstruction, and epistaxis, thensoreness of both eyes and unilateral deafness.Visual acuities were 6/6 OD and 6/9 OS. Therewas bilateral superior scleral necrosis, withnecrosis ofthe overlying conjunctiva, widespreadpericapillary haemorrhages, and damage to thelimbal vascular arcades (Fig 4). Marginal cornealinfiltrates were accompanied by early cornealgutter formation and mild anterior uveitis.The ESR and CRP were raised and there were
microscopic haematuria, proteinuria, andreduced creatinine clearance (20-5 ml/min).Chest x-ray showed a cavitating pulmonarylesion, and sinus x-rays showed antral mucosalthickening. The ANCA titre was found to beraised. Nasal mucosal biopsy showed featuressuggestive of WG and renal biopsy severenecrotising glomerulonephritis. The patient wastreated with pulsed intravenous methyl-prednisolone and cyclophosphamide. Despite animprovement in his scleritis, his creatinine clear-ance continued to fall and plasma exchange wasstarted. This reduced circulating ANCA andimproved renal function. His disease is currentlycontrolled by prednisolone 10 mg and cyclo-phosphamide 40 mg daily, and both eyes haveretained visual acuities of 6/6.
TreatmentAll patients were admitted to hospital forinvestigation and clinical evaluation.
Non-steroidal anti-inflammatory agents hadbeen used prior to admission in six patients butwere never successful.
Oral steroids were used in all patients but alonenever controlled acute disease. In two patients(without scleral necrosis) control was achievedby additional oral cyclophosphamide.
Pulsed immunosuppressive therapy (intravenousmethylprednisolone and cyclophosphamide'9)was used in the seven patients, six with scleralnecrosis, who had not responded to oral steroidsand in whom scleritis was progressing rapidly.Intravenous injections of methylprednisolone(05-1 -0 g) were repeated at three-day intervalsuntil the scleritis had subsided: no more thanfour injections were ever required. Cyclo-phosphamide (0 5 g) usually accompanied thefirst two pulses of methylprednisolone. Oralprednisolone (15-20 mg) was started with thefirst pulse, and cyclophosphamide (2 mg/kg) wasadded when its intravenous administrationceased. This treatment acheived control ofscleritis in all patients in whom it was used.
204
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from

Diagnosis and management ofsystemic Wegener's granulomatosis presenting with anterior ocular inflammatory disease
Figure 7A Necrotising scleral granuloma with loss of Figure 7C After involution ofthe granulation tissue, theconjunctival cover (patient 5). exposed sclera became covered by afibrovascular
membrane. This contracted, distorting the conjunctival_______vascular pattern.
Figure 7B The same patientfive weeks later, showingexcessive granulation tissue which developed duringrecovery. It was centred on a perforating deep scleralartery.
Plasma exchange was used in two patients withsevere progressive renal disease: one had receivedpulsed immunosuppressive therapy, which hadappeared to control the scleritis but not the renaldisease; the other presented in severe renalfailure and was also treated with oral cyclo-phosphamide and prednisolone.
Azathioprine was substituted for cyclo-phosphamide in one patient in remission, andprompted an immediate relapse.
Surgery was required in one patient. A corneo-scleral patch graft was performed for a limbalperforation after the disease had been controlledmedically (Fig 5C).
CLINICAL PROGRESSIn areas of conjunctival necrosis exposed sclerabecame covered by fibrovascular tissue fromadjacent conjunctiva. These neovascular mem-branes contracted, distorting the surroundingconjunctival vascular pattern (Figs 7A, B, C).Granulation tissue was pronounced in one patient(Fig 7B). It arose in a zone of scleral necrosis, wascentred on a perforating deep scleral artery, andlater became infarcted.During remission marginal corneal infiltrates
became smaller, better defined, and morerefractile in nature but did not disappearcompletely (Fig 7D).
FOLLOW-UPSix out of 10 patients required no further pulsedimmunosuppressive therapy to control theirscleritis (follow-up 11 to 72 months, mean 25
Figure 7D Corneal stromal infiltrates during remission,demonstrated by retroillumination in the same eye.
months). One (two cases) or two (two cases)further pulsed treatments were necessary in fourpatients.
Five patients had relapses of their systemicWG giving rise to acute renal failure (1), pro-gressive renal failure (2), pericardial effusion (1),rashes (1), and arthralgia (3). One patient nowrequires haemodialysis.Two out of 10 patients lost visual acuity in one
eye after presentation: from 6/36 to countingfingers due to a macular hole secondary tocystoid macular oedema and uveitis in one; andfrom 6/24 to 5/60 due to retinal vascular occlusioncomplicating thrombocytosis in the other.Anterior segment changes such as guttering andperforation did not produce visual loss.
Intravenous methylprednisolone and cyclo-phosphamide were well tolerated apart fromtransient hyperglycaemia and electrolytedisturbances. Four patients became mildlycushingoid (with weight gain and facial changes),and one developed osteoporosis on prolongedsystemic steroids. Alopecia was symptomatic intwo patients. Pneumonia associated withcyclophosphamide-induced leucopenia andrequiring admission to hospital arose in twopatients. One patient suffered from herpessimplex virus pharyngitis. Later, when his WGwas inactive, he died of a viral myocarditis.Although he was not severely leucopenic (leuco-cytes 3-3 x 109/dl), this may have been related tohis immunosuppression.
DiscussionThis paper considers the manner in which WG
205
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from

Charles, Meyer, Watson
may present in an eye clinic, though our casesreflect a bias in this centre towards corneoscleralinflammatory disease. The mildest scleritis wasaccompanied by critical involvement of othersystems, and even when no symptoms werevolunteered they could be revealed by carefulquestioning. The discovery of systemic diseasewas vitally important to all our patients, andtreatment of this, rather than the eye in isolation,reduced ocular morbidity.The scleral changes with which these patients
presented included episcleral vasodilatation andoedema, scleral oedema, and scleral necrosis,which was often accompanied by destruction ofthe overlying conjunctiva. Although conjunctivalnecrosis is a dramatic physical sign, the presenceof scleral necrosis correlated best with the extentof systemic involvement.
It has been proposed that corneoscleral des-tructive disease straddling the limbus charac-terises systemic vasculitis.2021 Every patient withscleral necrosis did have multifocal, peripheralcorneal infiltrates beneath Bowman's membrane,showing the vulnerability ofthe limbal arcades inthis condition. This is to be the subject of afurther paper. Corneal guttering arose less fre-quently, and always adjacent to necrotic sclera.
Although scleritis was the presenting symptomin all our patients, it was not responsible forvisual loss. When this occurred, it resulted fromcystoid macular oedema complicating uveitis inone case and retinal vascular occlusion in another.The latter was associated with reactive thrombo-cytosis, highlighting the susceptibility ofdamaged vascular beds to alterations in bloodviscosity.
Despite their ophthalmological presentation,our patients gave histories revealing systemicdisease, ANCA levels were raised, and investi-gations confirmed involvement of respiratorytract and kidneys in all but two. Our inclusion oftwo subjects (2, 10) with scleritis and raisedANCA titres, but without the full clinical criteriaof Fauci et al, I reflects our view that thisinvestigation may enable diagnosis earlier in thenatural history of the disease. Both patientsalready had widespread vasculitis requiringsystemic treatment.
CLINICAL ASSESSMENTAlthough a careful history, and systemic andotolaryngological examination, exposed systemicinflammatory disease in our patients, furtherinvestigations were helpful in confirming thediagnosis and systems affected, and monitoringresponse to treatment.Even in the absence ofsymptomswe frequently
found radiological evidence of sinusitis orpulmonary cavitation. Renal impairment, whichwas found at presentation in most ofour patients,was also always asymptomatic. Half the patientshad significant proteinuria, and creatinineclearance was reduced in seven out of 10.Furthermore, renal disease was seen in patientswith and without scleral necrosis: hence mildscleritis may be the presenting feature ofincipientrenal failure.The activity of WG is reported to be better
estimated by CRP than ESR,"1 but both investi-
gations proved equally sensitive during theassessment and management of our patients.Seven subjects presented with an ESR of over80 mm/h, and two of the remainder were alreadyon steroid therapy. In addition there was usuallyanaemia, leucocytosis, and thrombocytosis.Immune complexes, complement, and serum
immunoglobulins were raised in some patients,but, as in other studies, these were not consistentfindings.23 One patient had a positive test forrheumatoid factor, but with the exception ofANCA other autoantibodies were not found inour patients.The initiation of long term immuno-
suppressive therapy demands confident diag-nosis of the underlying systemic disease. In thisstudy the only diagnostic histology was obtainedfrom nasal mucosal biopsy in one case afternormal findings on otolaryngological examin-ation. We consider this to be the most appropriatebiopsy site in patients presenting with anteriorocular disease, even in the absence of upperrespiratory symptoms. However, precise histo-logical confirmation may still be difficult in bothsevere'8 and early disease,24 and inconclusivehistology should never delay treatment.As in previous reports8 18 the multifocal nature
of this disease precluded the use of histology as asole criterion for inclusion in our study. TheANCA titre (RIA) was increased in every case,usually at presentation but later in the course ofthe disease in two patients. Whenever it could becharacterised by IIF, granular cytoplasmicstaining was found, supporting the diagnosis ofWG as opposed to other forms of vasculitis. Thequantification of ANCA by RIA also provided auseful indicator of disease activity with which tomonitor treatment.
TREATMENTOral steroids alone were insufficient to abort theprogression of scleral necrosis. However, pulsedimmunosuppressive therapy with intravenousmethylprednisolone and cyclophosphamidesuccessfully controlled scleritis whenever it wasused. In the acute phase co-existing respiratoryand renal disease had to be closely monitored,and two ofour patients required plasma exchangebecause of progressive renal disease. Exacer-bations were common, particularly followingchanges in treatment, but responded well tofurther pulsed immunosuppressive therapy.
Pulsed methylprednisolone and cyclo-phosphamide were well tolerated by all thesepatients. However, methylprednisolone therapymay be hazardous, and deaths have beenreported."3 The complications of prolonged oralprednisolone therapy are well known, and cyclo-phosphamide also has many side effects,including leucopenia,'6 haemorrhagic cystitis,"alopecia,'8 teratogenesis,1 0 and malignancy.3'Two of our patients developed profound cyclo-phosphamide-induced leucopenia, and in bothcases this was followed by pneumonia.We routinely check blood counts twice weekly
during the inception of cyclophosphamidetherapy, and monthly thereafter. However, thecontrol of WG with immunosuppressive drugsmay itself put a patient's life at risk, and many
206
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from

Diagnosis and management ofsystemic Wegener's granulomatosis presenting with anterior ocular inflammatory disease
ophthalmologists will wish to collaborate withphysicians experienced in lymphocytotoxictherapy.
Ophthalmologists must be able to recognisethe patient whose eye disease is the presentingfeature of a life-threatening systemic vasculitis.Previous studies have described inexorableprogression ofthe ocular disease in WG, but withprompt diagnosis and treatment of the under-lying systemic disease this need not occur.
The authors thank DrCM Lockwood for his advice and assistancewith ANCA analysis.
Paul Meyer is in receipt of a research fellowship from theWellcome Trust.
1 Godman GC, Churg J. Wegener's Granulomatosis. Pathologyand review of the literature. Arch Pathol 1954; 58: 533-53.
2 Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener's granu-lomatosis: prospective clinical and therapeutic experiencewith 85 patients for 21 years. Ann Intern Med 1983; 98: 76-85.
3 Walton EW. Giant cell granuloma of the respiratory tract(Wegener's granulomatosis). BrMedJ 1958; ii: 265-70.
4 Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA.Ocular complications of Wegener's granulomatosis.Ophthalmology 1983; 90: 279-90.
5 Straatsma BR. Ocular manifestations of Wegener'sgranulomatosis. AmJ7 Ophthalmol 1957; 44: 789-99.
6 Brady HR, Israel MR, Lewin WH. Wegener's granulomatosisand corneoscleral ulcer.JAMA 1%5: 193: 248-9.
7 Austin P, Green WR, Sallyer DC, et al. Peripheral cornealdegeneration and occlusive vasculitis in Wegener's granulo-matosis. AmJ Ophthalmol 1978; 85: 311-7.
8 Spalton DJ, Graham EM, Page NGR, Sanders MD. Ocularchanges in limited forms ofWegener's granulomatosis. BrJOphthalmol 1981; 65: 553-63.
9 Donald KJ, Edwards RL, McEvoy JDS. An ultrastructuralstudy of the pathogenesis of tissue injury in limitedWegener's granulomatosis. Pathology 1976; 8: 161-9.
10 Van der Woude FJ, Rasmussen N, Lobatto S, et al. Auto-antibodies against neutrophils and monocytes; tool fordiagnosis and marker for disease activity in Wegener'sgranulomatosis. Lancet 1985; i: 425-9.
11 Gross WL, Ludeman G, Kiefer G, Lehmann H. Anti-cytoplasmic antibodies in Wegener's granulomatosis. Lancet1986; i: 806.
12 Savage COS, Jones S, Winearls CG, Marshall PD, LockwoodCM. Prospective study of radioimmunoassay for antibodiesagainst neutrophil cytoplasm in the diagnosis of systemicvasculitis. Lancet 1987; i: 1389-93.
13 GoldschmedingR,Huinink DTB, Faber N, et al. Identificationof the ANCA antigen as a novel myeloid lysosomal serineprotease. APMIS 1989; 97 (suppl 6): 46.
14 Falk RJ, Jennett JC. Anti-neutrophil cytoplasmic autoanti-bodies with specificity for myeloperoxidase in patients withsystemic vasculitis and idiopathic necrotizing and crescenticglomerulonephritis. N EnglJ Med 1988; 318: 1651-7.
15 Cohen Tervaert JW, Goldschmeding R, Elema JD, et al.Autoantibodies against myeloid lysosomal enzymes increscentic glomerulonephritis. Kidney Int 1990; 37: 799-806.
16 Hollander D, Manning RT. The use of alkylating agents in thetreatment of Wegener's granulomatosis. Ann Intern Med1%7; 67: 393-8.
17 Harrison HL, Linshaw MA, Lindsley CB, Cuppage FE. Boluscorticosteroids for initial treatment of Wegener's granulo-matosis.JAMA 1980; 244: 1599-600.
18 Pinching AJ, Lockwood CM, Pussell BA, et al. Wegener'sgranulomatosis: observations on 18 patients with severerenal disease. QJ Med 1983; 52: 435-60.
19 Meyer PAR, Watson PG, Franks W, Dubord P. 'Pulsed'immunosuppressive therapy in the treatment of immuno-logically induced corneal and scleral disease. Eye 1987; 1:487-95.
20 Watson PG, Booth Mason S. Fluorescein angiography in thedifferential diagnosis of sclerokeratitis. Br Ophthalmol1987; 71: 145-51.
21 Watson PG. Vascular changes in peripheral corneal destruc-tive disease. Eye 1990; 4: 65-73.
22 Hind CRK, Winearls CG, Lockwood CM, Rees AJ, PepysMB. Objective monitoring of activity in Wegener'sgranulomatosis by measurement of serum C-reactive proteinconcentration. Clin Nephrol 1984; 21: 341-5.
23 Shillitoe EJ, Lehner T, Lessoff MH, Harrison DFN.Immunological features of Wegener's granulomatosis.Lancet 1974; i: 281-4.
24 McCluskey RT, Fienberg R. Vasculitis in primary vasculitides,granulomatoses and connective tissue diseases. Hum Pathol1983; 14: 305-15.
25 Thompson JF, Chalmers DH, Wood RF, Kirkham SR,Morris PJ. Sudden death following high-dose intravenousmethylprednisolone. Transplantation 1983; 36; 594-9.
26 Fauci AS, WolffSM, Johnson JS. Effect of cyclophosphamideon the immune response in Wegener's granulomatosis.N EnglJ Med 1971; 285; 1493-6.
27 Philips FS, Sterberg SS, Cronin AP, Vidol PM. Cvclo-phosphamide and urinary bladder toxicity. Cancer Res 1961;21: 1577-89.
28 Anonymous. Drug causes ofhair loss. Drug TherBull 1978; 16:77.
29 Toledo TM, Harper RC, Moser RH. Fetal effects duringcyclophosphamide and irradiation therapy. Ann Intern Med1971; 74: 87-91.
30 Greenberg LH, Verdes P. Tanaka KR. Congenital anomaliesprobably induced by cyclophosphamide. JAMA 1964; 188:423-6.
31 Marshall V. Premalignant and malignant skin tumours inimmunosuppressed patients. Transplantation 1974; 17:272-5.
207
on 4 June 2018 by guest. Protected by copyright.
http://bjo.bmj.com
/B
r J Ophthalm
ol: first published as 10.1136/bjo.75.4.201 on 1 April 1991. D
ownloaded from