development of an expression system for a dehydrogenase
TRANSCRIPT

FinalThesis
Developmentofanexpressionsystemforadehydrogenase
AxelVeibäckLITH‐IFM‐A‐EX‐‐10/2235‐‐SE
DepartmentofPhysics,ChemistryandBiologyLinköpingUniversitySE‐58183Linköping

i
Abstract Inrecentyears,biocatalyticalstepsinchemicalsynthesisarebecomingincreasinglyimportantforeconomicalandenvironmental‐friendlyproduction.InordertoevaluatetheuseofenzymesinaprocessatCambrexKarlskogaAB,anexpressionsystemwasdevelopedforadehydrogenase.AsyntheticgenewasclonedintoEscherichiacoliDH5αcells,usingthepTZ19Rexpressionvector,aspreviouslydescribedintheliterature.Proteinexpressionwascarriedoutat25°C,30°Cand37°CandresultsweremeasuredusingSDS‐PAGEandactivityassays.Toimproveexpression,thegenewasmodifiedinthreewaysusingPCR,yieldingeightclones:ItwasinsertedintothepSE420expressionvector,shortenedtoavoidinclusionbodyformationandamissingnucleotidewasinsertedintothesequence.Aprotocolforinclusionbodyscreeningwasalsodeveloped.Finally,anassayfordeterminingthekineticconstantsofdehydrogenasewasdesigned.Itisconcludedthatfurtherexperimentsmustbedonetoobtainexpressionofthedehydrogenaseandrecommendationsforadditionalworkaregiven.
Keywords:biocatalysis,proteinexpression,geneexpression,inclusionbodies,SDS‐PAGE,polymerasechainreaction,pTZ19R,pSE420

ii
Sammanfattning Biokatalytiskaprocessteghardesenasteårenblivitettalltviktigareinslagikemisksyntesförattåstadkommaekonomiskochmiljövänligproduktion.FörattutvärderaanvändandetavenzymerienprocesshosCambrexKarlskogaAButveckladesettexpressionssystemförettdehydrogenas.EnsyntetiskgenklonadesiniEscherichiacoliDH5αochuttrycktesmedhjälpavexpressionsvektornpTZ19R,somtidigarefinnsbeskrivetilitteraturen.Proteinuttrycketutfördesvid25°C,30°Coch37°CochresultatetmättesmedhjälpavSDS‐PAGEochaktivitetsmätningar.Genenfördehydrogenasetmodifieradespåtresätt,vilketgavupphovtillåttavarianter.GenenfördesövertillexpressionsvektornpSE420,kortadesförattundvikabildningavinklusionskropparochennukleotidsomfattadesfrångensekvensenåterinfördes.Ettprotokollutarbetadesävenförundersökningavinklusionskroppar.Tillsistsammanställdesenmetodförattundersökadekinetiskakonstanternahosdehydrogenaset.Slutsatsenavarbetetärattfortsattastudiermåsteutförasföratterhållauttryckavdehydrogenasetochrekommendationergesförframtidaundersökningar.

iii
A note on secrecy Thisthesisispublicwork.However,forthistobepossibleandnotpresentadisadvantageforCambrexKarlskogaAB,partsofthisworkthatcouldbeusedtoidentifytheexactprocesshavebeenomitted.Thisincludesthenameofthesubstrateusedandproductformedandthenameandmolecularweightoftheenzymecentraltothisreport,aswellasliteraturereferencesdealingwiththedehydrogenase.However,theprocessandmethodsofdevelopingtheexpressionsystem,togetherwithresultsanddiscussionthereofhavenotbeenleftout.
Theenzymeusedwillbereferredtoasdehydrogenasethroughoutthisreport.
Thesubstrateandproductwillbereferredtoassubstrateandproduct.

iv
Abbreviations ADH Alcoholdehydrogenase
dH2O Deionisedwater(MilliQwater)
DNA Deoxyribonucleidacid
EDTA Ethylenediaminetetraaceticacid
HPLC Highperformanceliquidchromatography
IPTG Isopropylβ‐D‐1‐thiogalactopyranoside
LB Lysogenybroth
LB/Amp Lysogenybrothwithampicillin
NADP Nicotinamideadeninedinucleotidephosphate(oxidizedform)
NADPH Nicotinamideadeninedinucleotidephosphate(reducedform)
PCR Polymerasechainreaction
PMMA Polymethylmethacrylate
PMSF Phenylmethylsulfonylflouride
SDS‐PAGE Sodiumdocecylsulfatepolyacrylamidegelelectrophoresis
SOB Superoptimalbroth
SOC Superoptimalbrothwithcataboliterepression

v
Table of contents Abstract iSammanfattning iiAnoteonsecrecy iiiAbbreviations ivTableofcontents v1 Introduction 12 Theoreticalbackground 22.1 Biocatalysis 22.2 Dehydrogenases 22.3 Thesoughtreaction 32.4 Coenzymeregeneration 32.5 Reactionmedium 42.6 Geneexpression 42.6.1 Recombinantgeneexpression 42.6.2 Controlofgeneexpression 42.6.3 T7Expressionsystem 52.6.4 Expressionvectors 52.6.5 ComplicationswithE.coli 62.6.6 Inclusionbodies 72.6.7 Proteolysis 7
2.7 Experimentalmethods 72.7.1 Restrictionendonucleases 72.7.2 Ligation 82.7.3 Polymerasechainreaction 82.7.4 Geneticengineering 9
2.8 Kineticcharacterization 103 Experimentaldetails 113.1 Cloning 113.1.1 Restrictionendonucleasecleaving 113.1.2 DNAseparation 113.1.3 Concentrationmeasurements 123.1.4 Ligation 123.1.5 Transformation 12
3.2 Analyticalmethods 123.2.1 Plasmidpreparation 123.2.2 Sequencing 123.2.3 Cellcultivation 133.2.4 Celllysis 133.2.5 Proteinseparation 133.2.6 Enzymeactivityassays 133.2.7 Inclusionbodyscreening 14
3.3 DNAmodifications 143.3.1 Implementedmodifications 143.3.2 Polymerasechainreaction 153.3.3 CloningofmodifiedDNA 15
3.4 Kineticcharacterization 16

vi
4 Resultsanddiscussion 174.1 Proteinexpression 174.2 Kineticcharacterization 21
5 Conclusions 246 Recommendations 257 Acknowledgements 268 References 27Appendix:Growthmedium 29

1
1 Introduction Theuseofbiocatalyticalmethodsfortheproductionofchemicalsandpharmaceuticalsarebecomingincreasinglypopular.CambrexKarlskogaABhasahistoryofmanufacturingchemicalsthatdatesbacktothelaboratoriesofAlfredNobelinthelate19thcentury.Thecompanysupportsthepharmaceuticalindustrybymanufacturingactivepharmaceuticalingredients,andprovidesresearchanddevelopmentfortheirclientsaswellasin‐houseproducts.Duringthelastdecadesinitiativeshavebeentakentofurtherinvestigatetheuseofbiocatalyticmethodsinthefabricationofchemicalproducts.ThebiocatalystdepartmentatCambrexwasmovedtoKarlskogain2008.
ForanewproductatCambrex,biocatalyticpathwaysarebeingconsideredformanufacturing.Aninterestingenzyme,previouslyreportedintheliterature,wastargetedandinvestigated.Asyntheticgenewasordered,basedonsequenceinformationfromtheliterature.TheenzymehaspreviouslybeenmanufacturedinEscherichiacoliDH5αcells,usingpTZ19Rexpressionvectors,butproductiondetailsarenotavailable.Inordertocompleteandevaluateaproductionprocess,anexpressionsystemfortheenzymemustbesetup.
Theaimofthisprojectistodevelopandoptimizeanexpressionsystemforaspecificdehydrogenaseenzyme.Thisincludescloningthegeneintoasuitablehost,optimizingexpressionconditionsandmeasuringenzymeactivity.

2
2 Theoretical background
2.1 Biocatalysis Inrecentyears,biocatalyticproductionofchemicalshasdevelopedintoawidelyusedmanufacturingmethod.Itcanhaveseveraladvantagesovertheconventionalchemicalapproachforproduction,mostsignificantlytheselectivitiesoftheenzymesusedandthemildoperationalconditionsofthereactions[Panke,2004].Protectiveblockinganddeblockingstepsofotherlabilefunctionalgroupsonthesubstratecanbeeliminatedthankstothesite‐specificenzymes,aswellasreactionsdemandinghighpressureandhightemperatures.Thismeansthatbiocatalysisinmostcasesisconsideredamoreeconomicalandenvironmental‐friendlymethodthanthechemicalroute[Ishige,2005].Thisiswidelyseenasapromisingresearchfieldandmanyinitialproblemstothetechniquesuchastheavailabilityofbiocatalystsandtheiroperationalstabilityhavebeenhelpedthroughmolecularbiologymethods,suchasdirectedevolution[Schoemaker,2003].Somedrawbacksofbiocatalysisarethatsinceeachenzymeisrelativelyspecific,amatchingenzymethatproducestheproductinasatisfyingyieldmustbefound,whichcanprovetediouswork.Theenantiospecificnatureofenzymesisnotalwayspositive,andacompletelydifferentenzymeisusuallyneededtocoverbothformsofasubstrate.Enzymesworkbestinwater,whereaswaterisusuallynotsuitablefororganicreactions.Enzymaticreactionsinothersolventswillusuallywork,butwithsomelossofactivity[Faber,2000].Advancedchemicalroutesarelikelytoremainimportantinthefuture,butacombinationofchemicalroutesandbiocatalysiswheresuitablewillprobablybemorecommon[Schoemaker,2003].
Biocatalysiscanbecarriedoutwithisolatedenzymesorwholecellsystems.Usingwholecellcatalysiscanbebotheasierandmoreinexpensive.Enzymesarekeptintheirnaturalsurroundingsandaregenerallymorelong‐termstablethanfreeenzymes[Ishige,2005].Whetherwholecellsystemscouldbeuseddependsonmanyfactors,themostimportantbeingtypeofreaction,cofactorrecyclingneedsandscaleofbiocatalysis[Faber,2000].However,reactionsaremoreunpredictableduetosidereactionsandthemorecomplexcomposition.Usingwholecellsystemsinanindustrialscalecouldprovedifficult[Ischige,2005].
2.2 Dehydrogenases Theenzymesinvolvedinoxidationandreductionofbiologicalcompoundsarecalledoxidoreductases[Botham,2009].Oxidoreductasesaccountforaboutonefourthofallknownenzymes[Liu,2007].Theyareclassifiedintofourgroups:oxidases,dehydrogenases,hydroperoxidasesandoxygenases.Thisstudyfocusesonenzymesfromthesecondgroup,thedehydrogenases.Dehydrogenaseshelptransferhydrogenbetweenasubstrateandacoenzymeorhydrogencarriers.Thesearetypicallynicotinamidecoenzymesorriboflavin[Botham,2009].Nicotinamideadeninedinucleotide(NAD)andNicotinamideadeninedinucleotidephosphate(NADP)arethetwomostcommonlyencounteredcoenzymes.Theenzymegroupiscapableoftransformingprimaryandsecondaryalcoholstoaldehydesandketonesandprimaryaminestoprimaryimines.Dehydrogenasesusingflavinasacofactorarealsocapableofintroducingadoublebondbetweentwocarbonatoms[Bugg,2004].Dehydrogenaseshavefoundwidespreaduseinclinicalandfoodanalysis[Hummel,1989]andinprocessessuchassynthesisofchiralcompounds,preparationandmodificationsofpolymers,biosensorsanddegradationoforganicpollutants[Liu,2007].

3
2.3 The sought reaction Thesoughtreactionistooxidizeasecondaryalcoholtoaketoneusingthedehydrogenase.TheenzymeusedrequiresNADPasacoenzyme(Figure1).
Thesubstrateusedisreadilyavailable,butcontainslabilefunctionalgroups.Bybeingabletoperformthisreaction,aloneorincombinationwithchemicalmethodsthataddressesotherfeaturesinthesubstrate,theeconomicalvaluecanbegreatlyincreased.Usinganenzymeforthereactionwilladdressaspecifichydroxylgrouponthesubstrate,leavingotherfunctionalgroupsuntouched.
Generally,thereversereactionwhereketonesarereducedismorepopular.Astudyinvestigating205selectedpatentsintheareaofbiocatalysisandbiotransformationbetween2000and2004revealed35patents(or17%)focusingonketonereductionwhereasthereversereactionwasrare[Panke,2004].
Figure1:Thereactionoftheexamineddehydrogenase:AsecondaryalcoholisoxidizedtoaketonewiththehelpofthecoenzymeNADP.
2.4 Coenzyme regeneration Adrawbackwithbiocatalysisisthatcofactor‐dependentenzymesarealmostexclusivelytiedtotheirnaturalcofactors,inthiscaseNADP[I].Thesecofactorsaregenerallyexpensiveandrelativelyunstableandcannotbereplacedbymoreeconomicalman‐madesubstitutes.Thishasledtothatinsyntheticreactionsdemandingcofactors,asubstoichiometricamountisoftenusedandinstead,acoupledreactiondemandingthecorrespondingoxidizingorreducingagentisusedtoregeneratetheoriginalcofactor.Thisrecyclingisnotatrivialtask,despiteprogressesmadeinthefield[Faber,2000].Traditionally,reactionshavebeenperformedusingwholecellsystems,wheresomerecyclingisperformednaturally,butwiththeincreasinginterestofimmobilizedenzymaticreactionssincethelate1990’s,newwaysofcoupledreactionsarebeinginvestigated[Liu,2007].
[I]Nicotinamideadeninedinucleotidephosphate(NADP)

4
2.5 Reaction medium Thereareseveralfactorsthatneedtobeconsideredwhilesettinguptheenzymaticreaction:temperature,reactantconcentrations,theformoftheenzyme,suitablesolvent,levelofresidualwaterandacid‐baseconditions[Halling,2002].
Generally,enzymesarethemostefficientinaqueoussolutions,butthisisnotalwaysthebestchoice.Sincebiotransformationsoftenincludemoleculeswithalowsolubilityinwater,reactionscouldworkbetterinsomeorganicmediaorasolventwithalowamountofwater[Schoemaker,2003].Itisthereforeoftenpreferredtocarryoutthereactioninanon‐aqueousmedium.Reasonsbehindthisisthatreactantsmightnotbesolubleinwater,equilibriaandkineticsofthereactioncouldbechangedduetothesolvent,differentmediacouldbeusedtotunespecificity,side‐reactionsandmicrobialgrowthinaqueousmediacanbeavoidedandtheprocessisbetterintegratedtothechemicalstepsofthereaction[Halling,2002].
Enzymesinaqueousmediaareusuallyintheirdissolvedform,whileadifferentsolventoftenrequiressomekindofimmobilization,throughasolidsupportorcrossed‐linkedproteincrystals.Thismakestheprocessmoreadvanced.Itisalsoimportanttoinvestigatetheeffectonwatercontentonenzymebehavior.Toolowamountofwatercommonlyhindersthecatalyticactivity,whereasstabilityisincreased[Halling,2002].
2.6 Gene expression
2.6.1 Recombinant gene expression Toexpressaspecificgene,thegeneisinsertedintoanexpressionvectorandtransformedintoahost.Prokaryoticsystemsareverycommon,butforspecificpurposesandrequirementsfromtheproteinexpressed,eukaryoticsystemssuchasyeast,insectormammaliancellsareavailableaswell.Ofthebacterialexpressionsystems,E.coliisbyfarthemostcommonlyusedforhigh‐levelproteinexpression.Itpresentsanumberofadvantages,includingeasycultivationandmanipulationusingsimpleequipment,availabilityofstrainsandvectorsformaximizedexpression,greatknowledgeaboutthegeneticsandphysiologyofE.coliand,inoptimalcases,rapidproteinproduction[Appelbaum,2003].Although,italsopresentsafewlimitationsforproduction.Theproductcannotbemodifiedaftertranslation,thushinderingprocesseslikeglycosylation,acetylation,amidationandphosporylation[Ling,2001].Morecomplicationswillbefurtherdiscussedindepthlater.
Eachspecificgeneexpressionpresentsitsownchallenges,andageneral,all‐purposeprotocolforgeneexpressioncannotbemade.Staderrecommendsahigh‐copy‐numberexpressionvector,togethigheryields,andatightgeneregulation,becausemanyproteinsaretoxictothecellwhenproducedinhighnumbers[Stader,1995].
2.6.2 Control of gene expression TherelativeamountsofdifferentproteinsinE.colivarybetweenlessthan0.01%uptoabout2%ofthetotal.Thismakesitevidentthatgenetranscriptionisregulatedbythecell.Theseregulationsystemscanalsobeusedforsyntheticproduction.Regulationineukaryotesiscontrolledbyanumberofregulatorysignals,whereasinprokaryotesthemainmethodsaretranscriptionalcontrolandposttranscriptionalcontrol,thelatterconsistingofthedestructionofsynthesizedmRNAandproteinsynthesisrateregulation.Themoreimportantofthetwoistranscriptionalcontrol,consistingofrepressors,operatorsandpromoters[Watson,2007].

5
Oneofthemostextensivelystudiedandwell‐usedregulatorsisthelacoperon.Itconsistsofaregulatorygenecodingforarepressorprotein,acontrolsiteconsistingofthepromoterandoperatorandlastlythestructuralgenes,codingforβ‐galactosidase,lactosepermeaseandthiogalactosidasetransacetylase,proteinsthathelpthecellmetabolizelactose.Withoutlactoseavailable,therepressorproteinbindstotheoperatorsite,therebyhinderingtranscriptionofthegene.Whenlactoseisaddedtothecells,itbindstotherepressorprotein,allowingDNApolymerasetobindtothepromoterandtranscribethegenes(Figure2).Innature,thiswouldleadtoamoreefficientlactosemetabolism,butbysubstitutingthetranscriptionalgeneswithageneofinterest,thesamesystemisusedforrecombinantgeneproduction.Lactoseisoftensubstitutedwiththeanalogueisopropylβ‐D‐1‐thiogalactopyranoside(IPTG)[Watson,2007].
Figure2:Theprincipleofthelacoperon,initsrepressedstate.Theregulatorygene(I)producesarepressorprotein(1),whichbinds(2)totheoperator(O),therebyhinderingtranscriptioninitiationatthepromoter(P).Withlactosepresent,step2isobstructedandtherepressorproteindoesnotbindtotheoperator.DNApolymerasethenbindstotheoperatorandisfreetotranscribethestructuralgenes(Z,YandA).
2.6.3 T7 Expression system TheT7expressionsystemisasystemforstrong,regulatableexpressionofaspecificrecombinantgene.ItisbasedontheT7RNApolymerase,isolatedfrombacteriophageT7,capableofelongatingchainsaboutfivetimesfasterthanRNApolymerasefromE.coli.ThebacterialstrainincludesalacUV5promoter,governingtheproductionofT7RNApolymerase.TheadditionofIPTGtothecultureinducesthepromoter,leadingtoproductionofT7RNApolymerase,whichinturnusestheT7promoterintheplasmidtotranscribethetargetDNA[Studier,1990].Thissystemdoesnoteliminatetheneedforrepressionusingarepressorprotein,butchangestherepressingneedsfromtheactualgenetotheT7RNApolymeraseexpressionsite[Stader,1995].
2.6.4 Expression vectors Efficientexpressionvectorscontainastrongpromoterthatcanberegulated,aribosomebindingsequencespecificforE.coliandastrongterminationsequence.Directlyfollowingthetranscriptionstart,amultiplecloningsitecanusuallybefound.Thisisusedtoinsertthegeneofinterest.Becauseofthegapbetweenthetranscriptionstartandthestartoftheactualgene,asmallpeptidefromtheexpressionvectorisfusedtogetherwiththerequestedprotein.Thiscanhaveseveraladvantages.Firstly,thetranslationofmRNAisaffectedbytheribosomebindingsite,butalsothestartofthecodingregionhasbeenshowntointerfere.IntroducingthesequencewithnaturalE.colisequencesisawayofavoidingdifficultieswhenribosomebindingtakesplace.Secondly,thebacterialpeptidemaypreventthepeptideofbeingdegradedbythehostcell,asthecellrecognizesthesequence.Foreignproteinsareoftendestroyedbythehost.Thirdly,theextrasequencecouldbeusedtotagtheproteinwithasignalpeptidethatdirectstheproteintodifferentpositionswithinthecell,intheperiplasmicspacebetweentheinnerandoutermembraneorintotheculturemedium.Theextrasequencecouldalsobe

6
optimizedforaffinitychromatographyforeasyrecoveryinthepurificationsteps[Brown,2006].
Inthisstudy,twoexpressionvectorsareexamined(Figure3).pTZ19Risinitiallychosen.Thisvectorhasbeenusedearlierwiththespecificgene.pSE420isusedasanalternativeexpressionvectorandisintroducedinlaterexperiments.
pTZ19Rcontainsageneforβ‐lactamase,givingthehostampicillinresistance,foreasyselectionofcoloniescontainingtheplasmidgrownonplateswithampicillin.Itincludesalacpromoter,andagenecodingfortheN‐terminalfragmentofβ‐galactosidase.Themultiplecloningsiteispositionedbetweenthepromoterandthefragment,allowingforblue/whitescreening.ThevectoralsocontainsaT7promoter,allowingforhighproductionratesofmRNA[Fermentas(2)].
pSE420alsocontainsaβ‐lactamasegeneforampicillinresistance.Itusesthetrcpromoterforhigh‐leveltranscription,thelacOoperonfortranscriptionalregulationusingIPTGandthelacIrepressortoensurethatnoprematureexpressionoccurs[Biocompare].
Figure3:Theexpressionvectorsused:pTZ19RandpSE420withmarkedfeaturesandusedrestrictionsitestogetherwiththeirpositionsinthegene.
2.6.5 Complications with E. coli TherearesomecomplicationsintheexpressionofaforeigngeneinE.coli.Generallyspeaking,theinsertedgenemustnotcontainsequencesthatcanbeinterpretedasterminationsignalsformRNAproduction,thegenemustnotcontainintronsandthecodonbiasofthegenemustbecompatiblewiththatofthehost[Brown,2006].Forexample,only4%oftheleucinsinE.coliareencodedbytheCUAcodonand50%bytheCUGcodon[KazusaDNAResearchInstitute].Ifahighnumberofaminoacidsareencodedbyalesser‐usedcodon,transcriptionmaybehampered.However,theseproblemspredominantlyarisewhileworkingwitheukaryoticgenes,andinthecasethattheyoccurcanbeworkedoutbymodifyingthegene.Otherproblems,withtheirbasisinE.coliitself,arethatposttranslationalmodificationscannotbeperformed;meaning

7
proteinsrequiringsuchalterationsneedtobeproducedinanothercelltype.Therecombinantproteinmightnotbefoldedcorrectly,anddisulfidebondsaregenerallynotsynthesized.Errorsinfoldingleadstotheproductionofinclusionbodies,clustersofaggregatedprotein.Lastly,thecellmightdegradetheprotein.Themechanismsbehindthisselectionarelargelyunknown[Brown,2006].
2.6.6 Inclusion bodies Athighexpressionratesofplasmid‐encodedgenes,manypolypeptidesfailtoundergocorrectfoldingandinclusionbodiesconsistingofanaggregatedproteincomplexareformed,oraredegradedbythecell.Toavoidinclusionbodies,differentgrowthparameterscanbetested.Itmightbenecessarytolowertheyieldofproteininordertogetafunctioningprotein[MarCarrió,2001;Georgiou,1996].
Somecommonwaysofloweringtheyieldaretouseaweakpromoterorastrongpromoterunderpartialinductionconditions.Loweringthetemperaturealsodecreasesthedrivingforceforproteinself‐associationandtherateofproteinsynthesis,leadingtolessinclusionbodyformation.Atleastonestudyshowsthatrichmediumcellcultureshelptheproductionofprotein[Georgiou,1996].SomeotherfactorsthatcouldaffectinclusionbodyformationsinvolvesextracellularpH,usedcarbonsource[Strandberg,1991],aminoacidcompositionofthegene,thepresenceofchaperonesandfoldasesandtheexpressionoftheproteinasafusionwithahighlysolublepolypeptide[Georgiou,1996].
Insomecases,inclusionbodiescanprovebeneficial,andbecauseoftheirgeneralstabilitytheycaneasilybeisolatedfromthehost.Itisthenneededtodenaturetheaggregatedproteinandcarefullyallowtherefoldingprocesstotakeplace.However,notallproteinslendthemselvestothiskindofextraction.Also,inlargescalethisisafairlyexpensivetask[Georgiou,1996].
AsolutionofTris,EDTA,lysozymeandTritonX‐100canbeusedtosolubilizethecellenvelopeandreleaseDNA.SonicationreducestheviscositycausedbythedischargedDNAand8Mureaiscommonlyusedforthesolubilizationofinclusionbodies[Stader,1995].
2.6.7 Proteolysis Inanormalcell,theproteolysismachinerymakessurethatmisfoldedproteinsaredegradedanddonotaccumulatewithinthecell.Apartfromcleaningupnon‐functionalpolypeptides,thisallowsforaminoacidrecycling.Targetsforproteindegradationincludeproteinsthathavefoldedincorrectlyandprematurelyterminatedpolypeptides.InE.colicytoplasm,thedegradationisinitiatedbyfiveheatshockproteases,Lon,ClpYQ/HslUV,ClpAP,ClpXpandFtsH.Peptidases,capableofdegradingpeptides2‐5residuesinlengthcontinuethereaction[Baneyx,2004].
2.7 Experimental methods
2.7.1 Restriction endonucleases RestrictionendonucleasesareusedingenecloningtocleaveDNAmoleculesatspecificsites.Theirdiscoveryinthe1960srevolutionizedtheuseofrecombinantDNAtechnology.TherestrictionendonucleasetypeIIrecognizesspecificDNAsequences,frequentlypalindromicsequences,andcleaveatorclosetotherecognitionsite[Mani,2007](Figure4).Itisoftenfavorabletocleavewiththetypeofenzymesthat

8
produceasomewhatstaggeredcleavage,resultinginshortoverhangsattheendscalledstickyends[Brown,2006].ThismakesitpossibletoeasierfitdifferentDNAfragmentstogether,inthiscasethegeneexpressingthedehydrogenaseandtheexpressionvector.
Figure4:DNAcleavedbyarestrictionenzyme.XbaIrecognizesthesequenceTCTAGAandleavesthecleavedDNAwithstickyends.
2.7.2 Ligation DNAfragmentsareassembledusingligation.T4DNAligasecatalysestheformationofthebondbetweenthephosphategrouplocatedonthe5’strandandthehydroxylgrouponthe3’strand.Sinceonlyoneofthestrandsneedstobephosphorylatedduringligation,itisusefultotreatthevectorwithadephosphorylasetopreventself‐ligationofvectorsthatarenotfullycleaved.ThedephosporylatedDNAstillcontainsa3’hydroxylgroup,leavingthevectorcapableofligatingwithanon‐treatedDNAfragment[Quail,2005;Ukai,2002].
2.7.3 Polymerase chain reaction Thepolymerasechainreaction(PCR)isanimportanttoolinmolecularbiologytoamplifyDNA.Itisalsoausefulmeantomakemodificationstoplasmids,suchasalteringbases,insertionsanddeletions[Chamberlain,2004].AbriefsummaryoftheprinciplebehindPCRisshowninFigure5.
ThePCRcycleconsistsofthreetemperaturesteps:denaturation,annealingandelongation(Table1).ThedenaturationstepservestoseparatetheDNAstrands,theannealingstepprovidesprimerstickingtothesinglestrandedDNAandduringtheelongationstep,thepolymerasesynthesizesanewcorrespondingstrand.Beforethefirstcycle,thereisalongerdenaturationstepmakingsuretheDNAtemplateiscompletelydenatured,andafterthelastcyclethereisalongerelongationstepmakingsuretheprotrudingendsofthesynthesizedproductiscompletelyfilled.AtypicalPCRconsistsof25‐35cycles[Metzker,2009;Chamberlain,2004].
Table1:RecommendedthermalcyclingconditionsforPfuDNApolymerase[Fermentas(1)]
Step Temperature(°C) Time(min)Initialdenaturation 95 1‐3Denaturation 95 0.5‐2Annealing 37‐70(primerdependent) 0.5‐2Extension 70‐75 2/kbpFinalextension 70‐75 5

9
Figure5:TheprincipleofPCR.TemplateDNA(1)isheatedsothestrandsseparatefromeachother(2).Loweringthetemperaturecausesoligonucleotideprimerstoadheretotheloosestrands(3).AheatstabileDNApolymeraseaddstotheprimersequencesandconstructsanewstrandfromthetemplate(4),whichcanbeusedinanewreactioncycle.Figureadaptedfrom[Metzker,2009].
2.7.4 Genetic engineering Geneticengineeringcanbeusedtoalterenzymecharacteristicsor,asinthiscase,correcterrorsinDNAsequencesandmakemodificationstowardsbetterproteinyields.InordertoinsertanucleicacidinanexistingDNAsequence,site‐specificmutagenesisisused.OnemethodtoobtainthisresultisbyusingPCRwithapairofmutagenicprimersthatdoesnotfullymatchthetemplatesequence.Theprimersbindtothetemplateandformthestartingmaterialfornewstrands[Brown,2006].
ThroughPCRandcleverprimerdesignitispossibletoaddarestrictionsitetoanamplifiedgenesequence.Thisisdonebyaddingmismatchingbasesinthe5’endoftheprimer(Figure6)Theremainingbasesoftheprimerbindtothetemplate,andnewlysynthesizedDNAwillcontaintherestrictionsiteadded.ThisisusefulwhenthePCRproductwillbecleavedandputintoanewplasmid.

10
Figure6:AddingaEcoRIrestrictionsitetoanewlysynthesizedPCRproductbyaddingthenecessarybasesinthe3’endoftheprimer.Additionalbasescanalsobeadded.
Asequenceofaminoacidscanberemovedfromtheedgesofagenebyintroducingaprimerwithamismatching5’endcontainingarestrictionsiteandastartcodon.Withthestartcodonplacedinsidethegene,theprecedingaminoacidswillnotbeamplifiedandtherestrictionsitecanbeusedtoacquirestickyendsforplasmidinsertion[Chamberlain,2004].
2.8 Kinetic characterization Thekineticpropertiesofanenzymecanbeanalyzedbymeasuringtheinitialvelocityofthereactionandfittingtheresulttoamathematicmodel.ByfarthemostcommonmodelistheMichaelis‐Mentenmodel:
€
V0 =Vmax[S]
[S]+ KM
(1)
Thismodeldescribesthesimplesttypeofenzymaticreactions,andanumberofsimplificationsmustbemadeinordertofitthedatatothemodel.Usinghighstoichiometricamountsofco‐enzymeinrelationtosubstrateconcentrationscanbedonetoobtainapseudo‐first‐orderreaction.Initialvelocityismeasuredasclosetothereactioninitiationaspossibleinordertomakemeasurementsbeforehighconcentrationsofproductareformed[Cornish‐Bowden,2004;Berg,2002].
Graphicalrepresentationscanbeusedtoestimatethekineticconstants.Themostpopularmethodreliesonthedoublereciprocalplot,orLineweaver‐Burkplot(Equation2).Itwasfirstusedinthe1930’s,andeventhoughitcontainsflawssuchasmisleadingimpressionsofexperimentalerroritcontinuepopular.Instead,forshowinghowwellthedataagreestothemathematicalinterpretation,theHanes‐Woolfplotispreferred(Equation3)[Cornish‐Bowden,2004].
€
1V0
=KM
Vmax1[S]
+1
Vmax (2)
€
[S]V0
=1
Vmax[S]+ KM
Vmax (3)

11
3 Experimental details
3.1 Cloning
3.1.1 Restriction endonuclease cleaving ThesyntheticgenefordehydrogenasewasdeliveredfromDNA2.0(MenloPark,USA)inacloningvectorwithcleavingsitesandkanamycinresistancegene.Approximately200‐500ngofDNAwasmixedwith1µlofeachrestrictionenzyme,XbaIandSacI(Table2),2µlof10xbufferanddH2Oinatotalreactionvolumeof20µl.Themixturewasincubatedat37°Cfor15minutes.CleavingwasperformedusingFermentasbrandFastDigestenzymestogetherwithbufferprovided.Whencleavedproductwastobeanalyzedonagarosegel,greenbufferfromthesamemanufacturerwasusedinstead,eliminatingtheneedofloadingdye.
Aftercleaving,expressionvectorswerefurtherdephosphorylatedthroughadding1µlofFastAPand2.2µlofFastAP10xbufferandincubatedfor15minutesat37°C.
Table2:Restrictionenzymesused,theirrecognitionsequences,locationsanduses.BoldAmeansthatitrecognizesthemethylatedvariant.
Restrictionenzyme
Recognitionsequence
Locationanduse
Xbal TCTAGA Locatedupstreamfromthegene.UsedwithpTZ19R.SacI GAGCTC Locateddownstream.UsedwithpTZ19RandpSE420.EcoRI GAATTC Locatedupstream.UsedwithpSE420.DpnI GATC About20locations.UsedtodigestmethylatedDNA
3.1.2 DNA separation Agarosegelwasmadebydissolving2gofagarosein200mlofTBEelectrophoresisbufferbyheatinginamicrowaveoven.Aftercoolingforafewminutes,20µlofgelredwasaddedforcolormarkup,gentlymixedandthegelwasmouldedwithasmallorlargecomb,givingthewellsdifferentsizes.Smallwellsholdabout15µlofsampleandlargeholdabout35µl,dependingonhowthegelswerecast.Smallwellsarefirstusedfortestingwithminimalsamplewasteandlargewellsareusedforfurtherpurification.
Productfromrestrictionendonucleasecleavingcanbeloadeddirectly.Smallgelswereloadedbymixing2µlof6xLoadingDyewith10µlofsample,andlargewellswereloadedwith5µlloadingdyeto25µlsample,orproportionalamounts.
AhighandalowMassRulerDNAladderwereloadedoneachgel,andthesampleswereseparatedbyapplying110Vcurrentforabout90minutes.
FinishedgelswerephotographedinUVlightorusedforgelextraction.
AppropriatebandsfromlargewellswereexaminedquicklyinUVlight,cutoutandweighed.GelextractionwasperformedusingQIAquickGelExtractionKitaccordingtomanufacturer’sinstructions,withtheexceptionofelutingin25µlofdH2O.

12
3.1.3 Concentration measurements AllconcentrationmeasurementswerecarriedoutonaCary100BioSpectrophotometer(Varian,PaloAlto,USA),usingquartzcuvettes.Sampleswerediluted1:100indH2O,andabsorbancewasmeasuredat260nmand280nm.ConcentrationwascalculatedusingtheLambert–BeerLaw(
€
A = εlc ),with
€
εdsDNA = 0.020(ng /µl)−1cm−1fordoublestranded
DNA.PurityoftheDNAwithrespecttopolypeptidecontaminationwasmeasuredbyexaminingtheratioA260/A280.
Measuringabsorbancelowerthan0.1isgenerallyconsideredinaccuratebecauseofthehighimpactofnoiseandfluctuations[Sambrook,2001].However,itisconvenienttodoaquicktestwiththesamedilutionfactorforallsamples,bearinginmindtheresultcouldbeimprecise.Onlyasmallamountofsampleisneeded,andtheresultisgenerallytrustable.Fortheseapplicationsitisconsideredsufficient.
3.1.4 Ligation Ligationwasperformedusingdifferentweightratiosofvectorandgenefragment.Theratiosexaminedwere1:1,1:3and3:1.DNAfromvectorandgenefragmentweremixedwith2µlofligasebuffer,1µlofT4DNAligaseand1µlofATP.Thereactionvolumewasadjustedto20µlwithdH2O.Thereactionwasleftat37°Cfortwohours.
3.1.5 Transformation TransformationwasperformedusingfrozencompetentDH5αE.colicellspreparedatthelaboratoryandstoredat‐80°C.Tubeswith100µlDH5αcellswerethawedonice.TheDNAfromligationreactionwasdiluted5‐foldin10mMTris‐HCl(pH7.5)1mMEDTA.PlasmidDNAwasdiluted50‐fold.1µlofDNAwasaddedtothecellsfollowedbyincubationfor30minutesonice.Toperformthetransformation,thecellswereheat‐shockedfor45secondsin42°Cwaterbathbeforebeingputbackonicefortwominutes.900µlofSOCmedium(SeeAppendix)wasaddedtoeachtubeandthecontentswastransferredto15mlplastictubes.Thetubesareincubatedat37°C,180rpmfor1hour.400µlofthecellswerespreadonpreheatedLB/Ampplates(SeeAppendix)andincubatedat37°Covernight.GrownsinglecolonieswerepickedfromtheplateandtransferredontoanewLB/Ampplateandwereincubatedat37°Covernight.Transformedcellswithoriginalgeneconstructwereinsteadgrownonplateswithkanamycin.
3.2 Analytical methods
3.2.1 Plasmid preparation Plasmidpreparationwasusedtoobtainsmallamountsofplasmidsforagarosegelanalysisorsequencing.Abacterialculturewaspickedwithaninoculatorandputin5mlofmedium.LB/Ampmedium(SeeAppendix)wasusedforallapplicationsexcepttheoriginalgeneconstructforwhichLBwithkanamycinwasused.Samplesweregrownovernightat37°C,180rpm.Thesampleswerethenspundownat4000rpmfor15minutesandthesupernatantwasdiscarded.PurificationoftheharvestedcellswascarriedoutusingFermentasGeneJETPlasmidMiniprepKitaccordingtoinstructions,butelutedin30µlofdH2O.Resultingproductwasuseddirectlyorstoredin‐20°C.
3.2.2 Sequencing SequencingwasperformedbyEurofinsMWGOperon(Ebersberg,Germany).Samplesweremadethroughplasmidpreparation,concentrationdeterminedanddilutedindH2Otoholdafinalconcentrationofaround80ng/µlandavolumeof20µl.

13
3.2.3 Cell cultivation Cellcultivationwascarriedoutinautoclavedwide‐neckedErlenmeyerflasks(E‐flasks),withacapofaluminiumfoil.Allhandlingofcellculturestookplaceonasterilebench.
CellsfromafreshplatewasinoculatedintoanE‐flaskwithLB/Ampmediumandgrownovernightunderdifferenttemperatures:25°C,30°Cand37°C.TheculturewasthenscaledupbytransferringintolargerE‐flasksandaddingadditionalLBmedium.ForexactamountsandE‐flasksizes,seeTable3.Afterthreehours,proteinexpressionwasinducedbyaddingIPTGto1µM,oraspecifiedconcentration.After2or4hours,expressionwashaltedbyputtingtheflasksonice.Cellculturewasharvestedbytransferringinto50mlplastictubesandcentrifugedfor15minutes,4000rpm,4°C.Thesupernatantwasdiscarded.
Table3:DifferentculturesizesaswellasEflasksandmediumvolumesused.
Culturesize
Initialmediumvolume
InitialEflasksize
Scaleupmediumvolume
ScaleupEflasksize
Small 5ml 50ml 20ml 100mlMedium 20ml 100ml 80ml 500mlLarge 40ml 500ml 160ml 1000ml
3.2.4 Cell lysis Asmallamountoftheharvestedcellpelletwascollectedusingthetipofapipetteandresuspendedin300µloflysisbuffer(1mg/mllysozymein100mMTrisHCl,pH6.8).Itwasincubatedonheat‐plate,30°C,350rpmfor1hour.Thesamplewasthencentrifugedfor10minutes,13200rpm,andthesupernatantwasrecoveredintofreshtubes.
3.2.5 Protein separation Toanalyzesamplesforproteins,sodiumdocecylsulfatepolyacrylamidegelelectrophoresis(SDS‐PAGE)wasperformed.13µllysedcellsamplewasmixedwith5µlofSDSloadingdyeand2µlofdithiothreiolreducingagent.Themixturewasheatedonheatingplate,70°Cor90°Cfor10minutes.Sampleswereloadedonaprecast10%TEO‐ClSDSgel(CBSScientific,SolanaBeach,USA)togetherwithaproteinstandard.SeeBluePlus2PrestainedStandardwasusedinearlyexperimentsandPagerulerPrestainedProteinLadderwasusedinlaterexperiments.Thegelwasrunat200Vforapproximately45minutes,thensoakedinCoomassieBrilliantBluesolution(2.4%CoomassieBrilliantBlue,60%methanol,12%aceticacidindH2O)foratleast1hour.Gelwasdestainedbysoakingindestainingfluid(10%methanol,10%aceticacidindH2O).
3.2.6 Enzyme activity assays Enzymeactivitywasmeasuredbylettingenzymefromalargeculturereactin50mMglycinebuffer,pH9.5,containing3mMsubstrateand3mMNADP.Thetotalvolumewas10mlinastirredvialthatwaskeptat30°C.
ReductionofNADPwasmeasuredafter24hoursat340nminaspectrophotometer,asdescribedintheliterature[Dell,2009].Measurementsweredoneindisposablepolymethylmethacrylate(PMMA)cuvettes.Samplesfromthereactionwerediluted1:20togiveamaximalabsorbanceof1ifNADPHwasformedto100%,calculatedwithLambert‐BeerLawusingtheextinctioncoefficient
€
εNADP = 6.220mM−1cm−1[Dawson1985].

14
Theenzymeassaywasevaluatedwithastockenzymesolutionofalcoholdehydrogenase(ADH).TwotypesofADHwereusedtogetherwithtwotypesofalcoholsubstrates.TheenzymesusedwereADHTfromThermoanaerobacterandADHTSfromamicrobialsource,bothfromJülichFineChemicals(nowCodexis,RedwoodCity,USA).Substratesusedwere1‐pentanol[II]andα‐phenylethanol[III].UsingLambert‐BeerLawasdescribedbefore,thesampleanalyzedneededtocontain0.16mMNADPinitiallytogiveamaximumabsorbanceof1at100%yield.Amountswereenlarged100timesforthereactionanddilutedformeasurements;thereactionwasthereforerunwithaNADPconcentrationof16mM,using16mM1‐pentanoland32mMα‐phenylethanol.Sincetheenzymesusedarestereospecificandα‐phenylethanolisaracemicsolution,doubleamountswereused.Reactionswereperformedinstirredvialskeptat30°Cwithatotalvolumeof5mlin50mMglycinebuffer,pH9.5.Measurementsweremadeafter24hours.
[II]1pentanol [III]αphenylethanol
3.2.7 Inclusion body screening SampleswerescreenedforinclusionbodiesbyadaptingaprotocolfromVanderbiltUniversityforinclusionbodyproteinpurification[PurifyingProteinfromInclusionBodies].
Theharvestedcellpelletfromamediumsizecellculturewasweighedandresuspendedin3volumesoflysisbuffer(50mMTris‐HCl,pH8.0,5mMEDTA,10mMNaCl).PMSF(100mMin99.5%ethanol)wasaddedtogiveafinalconcentrationof1mM,and16µloflysozyme(50mg/mlin100mMTris‐HCl,pH6.8)wasaddedforeachgramofcellpellet.Thetubewasmixedandputat37°Cinawaterbathfor20minutes.Theresultingviscoussolutionwastransferredto25mlbeakersandsonicatedat20%amplificationfor30secondswithaVibracellVCX750sonicator(Sonics&Materials,Newtown,USA)usingaCV33probe.Thesolutionwasthendividedintosmallertubesandcentrifugedfor30minutesat13200rpm.Thesupernatantwasremoved,butsavedforfurtheranalysis.Thecellpelletisresuspendedinthesameamountofbuffer(20mMNa2HPO4,pH7.2,20mMNaCl,5mMEDTA,25%(w/v)sucrose),PMSFwasaddedtogiveafinalconcentrationof1mMand10µlofTritonX‐100wasaddedpermlsolution.Thesolutionwascentrifugedfor30minutesat13200rpm.Supernatantwasremoved,butsavedforfurtheranalysis.Thecellpelletwasresuspendedin8.0Mureasolutionina50°Cwaterbath.
3.3 DNA modifications
3.3.1 Implemented modifications ThreeDNAmodificationswereperformed,resultingineightclones.ThepTZ19RexpressionvectorwaschangedtopSE420toperformsimultaneousexperimentswithanalternativevector.Toavoidinclusionbodyformationpossiblycausedbyextraaminoacidsaddedtotheenzymebyexpressionvectordesign,thegenewasshortenedwith12
15
or13aminoacidsforthepTZ19RvectorandpSE420vector,respectively.Finally,sequencingofthecodinggenerevealedanerrorconsistingofamissingguanidine,whichwasadded.PCR‐basedmethodswereusedtomodifytheDNA.Thefirsttwoalterationswereperformedinparallelbyamplifyingfragments,andthelastbyamplifyingthewholeplasmid.
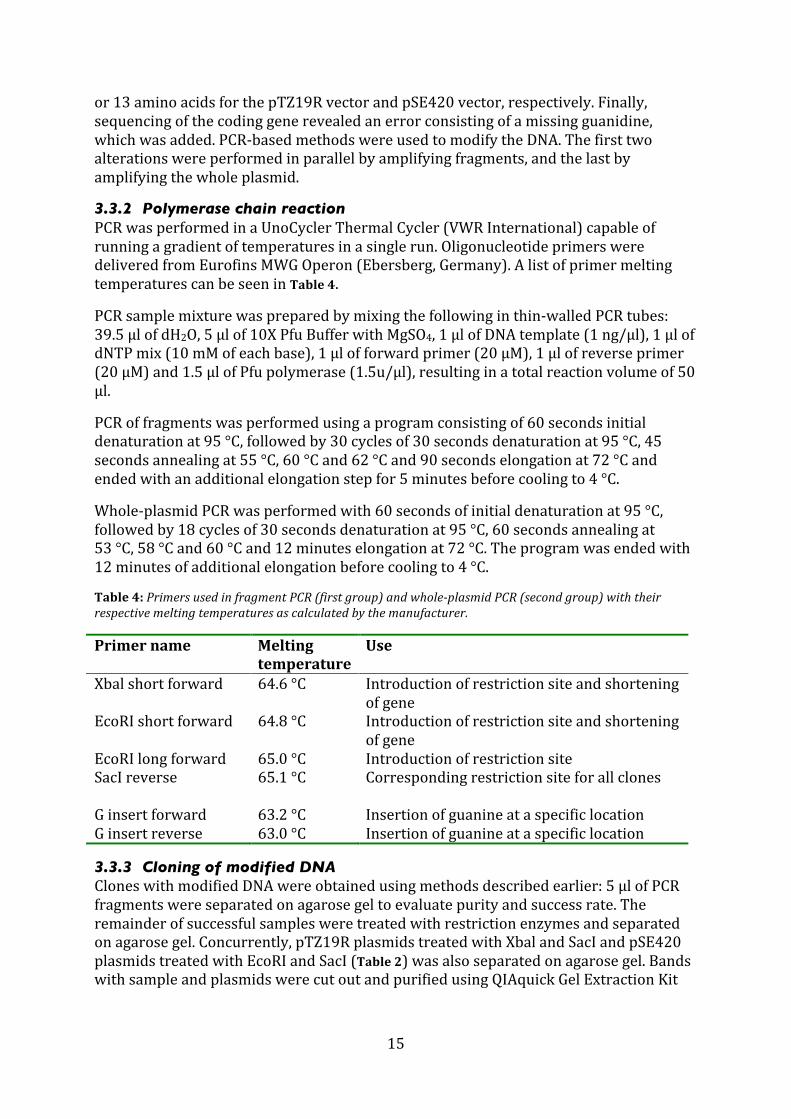
3.3.2 Polymerase chain reaction PCRwasperformedinaUnoCyclerThermalCycler(VWRInternational)capableofrunningagradientoftemperaturesinasinglerun.OligonucleotideprimersweredeliveredfromEurofinsMWGOperon(Ebersberg,Germany).AlistofprimermeltingtemperaturescanbeseeninTable4.
PCRsamplemixturewaspreparedbymixingthefollowinginthin‐walledPCRtubes:39.5µlofdH2O,5µlof10XPfuBufferwithMgSO4,1µlofDNAtemplate(1ng/µl),1µlofdNTPmix(10mMofeachbase),1µlofforwardprimer(20µM),1µlofreverseprimer(20µM)and1.5µlofPfupolymerase(1.5u/µl),resultinginatotalreactionvolumeof50µl.
PCRoffragmentswasperformedusingaprogramconsistingof60secondsinitialdenaturationat95°C,followedby30cyclesof30secondsdenaturationat95°C,45secondsannealingat55°C,60°Cand62°Cand90secondselongationat72°Candendedwithanadditionalelongationstepfor5minutesbeforecoolingto4°C.
Whole‐plasmidPCRwasperformedwith60secondsofinitialdenaturationat95°C,followedby18cyclesof30secondsdenaturationat95°C,60secondsannealingat53°C,58°Cand60°Cand12minuteselongationat72°C.Theprogramwasendedwith12minutesofadditionalelongationbeforecoolingto4°C.
Table4:PrimersusedinfragmentPCR(firstgroup)andwholeplasmidPCR(secondgroup)withtheirrespectivemeltingtemperaturesascalculatedbythemanufacturer.
Primername Meltingtemperature
Use
Xbalshortforward 64.6°C Introductionofrestrictionsiteandshorteningofgene
EcoRIshortforward 64.8°C Introductionofrestrictionsiteandshorteningofgene
EcoRIlongforward 65.0°C IntroductionofrestrictionsiteSacIreverse 65.1°C Correspondingrestrictionsiteforallclones Ginsertforward 63.2°C InsertionofguanineataspecificlocationGinsertreverse 63.0°C Insertionofguanineataspecificlocation
3.3.3 Cloning of modified DNA CloneswithmodifiedDNAwereobtainedusingmethodsdescribedearlier:5µlofPCRfragmentswereseparatedonagarosegeltoevaluatepurityandsuccessrate.Theremainderofsuccessfulsamplesweretreatedwithrestrictionenzymesandseparatedonagarosegel.Concurrently,pTZ19RplasmidstreatedwithXbalandSacIandpSE420plasmidstreatedwithEcoRIandSacI(Table2)wasalsoseparatedonagarosegel.BandswithsampleandplasmidswerecutoutandpurifiedusingQIAquickGelExtractionKit

16
andconcentrationwasmeasured.Plasmidsandfragmentswereligatedovernightat37°C.Ligationproductswereusedfortransformation.
Whole‐plasmidPCRproductswereevaluatedbyseparating5µlonagarosegel,andsuccessfulsamplesweretreatedwithDpnIrestrictionenzymetodegradetheoriginaltemplate.TheproductwaspurifiedandconcentratedusingQIAquickPCRPurificationKit,poolingthesamplesofthesamekindandelutingin10µlofdH2O.Theresultingproductwasuseddirectlyfortransformationwithoutfurtherdilution.
3.4 Kinetic characterization AnassayforkineticcharacterizationofdehydrogenasewasmadeandtestedwithADHTfromThermoanaerobacter,usingthesameprinciplesasfortheactivitytests.ReactionswerecarriedoutinPMMAcuvettesandmeasuredspectophotometrically.Reactioncomponentswereaddedtoobtainafinalvolumeof1ml.Astheenzymewasadded,thecuvettewasquicklymixedbyinvertingandinitialreactionvelocitywasmeasuredduringthefirst10secondsofthereaction.Enzymeamountwaskeptconstantat0.15UandNADPat5mM.Thesubstrateconcentrationwerevariedbetween0.005and1.0mMineightmeasurementpoints(0.005,0.01,0.025,0.05,0.1,0.25,0.5,1.0mM).Reactionswerecarriedoutinglycinebuffer(50mMglycine,pH9.5)atroomtemperature.

17
4 Results and discussion
4.1 Protein expression ThegenefordehydrogenasewasclonedfromthevectoritwasdeliveredinandinsertedintothepTZ19Rexpressionvector.TheresultingplasmidwastransformedintoDH5αE.coliandgrownonLB/Ampplates.Transformedcellsgrewwell.Theresultwasanalyzedonagarosegel,asseeninFigure7.BoththepTZ19Rvectorandtheoriginalsyntheticgenevectorhaveapproximatelythesamemolecularweight(b).Thegenefragment(a)ispresentinallsamplesbuttheemptypTZ19Rvector(well2),asexpected.Thismeansthatthecloningstepshavesucceededandgeneexpressioncanbestarted.
Figure7:Agarosegelseparationofgeneandvector.1:Originalgeneconstruct,2:EmptypTZ19Rvector,3,4,5:DehydrogenasegeneinsertinpTZ19R,a:Genefragment,b:Vector
Todetermineanidealgrowthandexpressiontemperature,cultureswiththedehydrogenasegeneweregrownat37°C,30°Cand25°C.Acombinedtestwheregrowthtookplaceat37°Candexpressionat30°Cwasalsoperformed.Thecellswereinducedwithafinalconcentrationof1.0mMIPTGandharvestedaftertwohoursofexpression.AculturewithemptypTZ19Rvectorswasusedasnegativecontrol.SamplesweretakenforSDS‐PAGEseparation(Figure8).Thegelseparationdidnotrevealanyclearbandspresentinanyoftheculturesgrowndifferingfromthenegativecontrol,nordidthedifferenceindenaturingtemperatureseemtohaveaneffect.However,thegelrevealsdifferencesinintensityofthebands,wherehighercultivationtemperaturesappeartogiverisetoastrongerbackgroundofcellproteins.
Activitytestingwasinitiatedtoexamineiftherewouldbeanysignsofactiveenzymepresentinthesamples.Twolargebatchesofenzymewerepreparedfortwotemperatureconditions:25°Cgrowthandexpressionand37°Cgrowth,30°Cexpression.Allavailableenzymefromlysedcellswasusedintheassay.However,noenzymaticactivitycouldbedetected.HPLCmeasurementswereperformedtoconfirmthatthesubstratehadnotspontaneouslydecomposed.

18
Figure8:InitialSDSPAGEseparationofcellculturesgrownandexpressedat25°C,30°C,37°Candasamplegrownat37°Candexpressedat30°C.Twodifferentdenaturingtemperatures(inparenthesis)areused.1:37/30°Csample(70°C),2:37°Csample(70°C),3:Negativecontrol(70°C),4:30°Csample(70°C),5:25°Csample(70°C),6:37/30°Csample(90°C),7:37°Csample(90°C),8:Negativecontrol(90°C),9:30°Csample(90°C),10:25°Csample(90°C).
ToconfirmthattheenzymaticassaywasfunctionalandthatNADPwasactive,anenzymeassayusingstockalcoholdehydrogenaseandalcoholswasprepared.Amountswerechosenastocorrespondto0%yieldatabsorbance0at340nm,and100%yieldatabsorbance1,givinganapproximationoftheactivity.Table5showsthatADHTexhibits16%yieldandADHTS52%yieldwith1‐pentanol,whilebothenzymesshow100%yieldwhenα‐phenylethanolisusedasasubstrate.Theseresultsalsosuggestthatmeasurementsusingthisassayweresomewhatinaccuratesincethemeasuredabsorbancesurpasses1.0.
Table5:Absorbancemeasuredinactivitytestingoftwotypesofalcoholdehydrogenaseandtwotypesofsubstrates.Negativecontroldoesnotcontainanyenzyme.
Substrate Enzyme Absorbance340nm1‐pentanol ‐ 0.01521‐pentanol ADHT 0.15791‐pentanol ADHTS 0.5232α‐phenylethanol ‐ 0.0212α‐phenylethanol ADHT 1.0532α‐phenylethanol ADHTS 1.0642
ToinvestigatetheeffectofdifferentIPTGconcentrations,cellsweregrownat30°CandinducedwithsixdifferentIPTGconcentrations:0.01mM,0.1mM,0.25mM,0.5mM,0.75mMand1.0mM.SampleswereanalyzedonSDS‐PAGE.Nosignificantvariationinexpressioncouldbedetected.
Inconclusion,activeenzymecouldbefoundneitheratSDS‐PAGEanalysis,noratenzymeactivityassays.Thereasonbehindthismightbeformationofinclusionbodiesduringexpression.Apossibleexplanationforinclusionbodyformation,evenatlowertemperaturesandIPTGconcentrations,mightbethetwelveextraaminoacidsthatwereattachedtotheproteinfromtheexpressionvector.Thissequencecouldcausetheproteintofoldincorrectly,thusrenderinginutileprotein.Itwasalsoconcludedthatusingabetter‐knownexpressionvectorcouldhelpunderstandingtheproteinexpression.Threenewclonesweredesigned,onewithashortergeneinthepTZ19R

19
vector,andtwousingthenormalgeneandashortervariantinthepSE420vector.ThecloneswerepreparedbyfragmentPCRusingappropriateprimersandthreedifferentannealingtemperatures,seeFigure9.Allsamplesexceptsample7wereconsideredfunctionalforfurtherligationandtransformation.
Figure9:AgarosegelseparationofPCRfragmentsfortheconstructionofcloneswithshortergeneandpSE420expressionvector.Wellcontents:SamplestreatedwithXbalshortforwardprimerandannealingtemperaturesetto55°C(1),60°C(2)and62°C(3).SamplestreatedwithEcoRIshortforwardprimerandannealingtemperaturesettoto55°C(4),60°C(5)and62°C(6).SamplestreatedwithEcoRIlongforwardprimerandannealingtemperaturesetto55°C(7),60°C(8)and62°C(9).
Uponcompletion,transformedsamplesweresentforsequencingandexpressionwasstudiedonthenewclonesusingthesamemethodasbefore.Testswerecarriedoutin37°C(Figure10)and30°Cusing1mMIPTGand2hourexpressiontime.Theresultswerenegative;nogeneexpressioncouldbeidentifiedonthegels.Furthertestingwasabortedduetotheresultsofthesequencing.
Figure10:SDSPAGEseparationofcellculturesgrownat37°C.1:Negativecontrol,2:OriginalgeneinpTZ19R,3:ShortenedgeneinpTZ19R,4:OriginalgeneinpSE420,5:ShortenedgeneinpSE420.
SequencingoftheclonesshowedthattheDNAmodificationshadbeenexecutedcorrectly.However,adeletionofasingleguaninenucleotidewasfoundneartheendofthegenesequence,confirmedinallfivesequencesthatcoveredthisregion.Thisindicatedthattherewasanerrorinthegenesequencepresentinalloftheclones,thusprobablyoriginatingfromanerrorintheoriginalgene.Thisframeshiftofthetranslationofthefollowingnucleotidesleadstomisreadingofthestopcodonandtheproteinexpressioncontinuesaftertheproteinshouldhaveended.Thiscouldbethereasonbehindthelackofgeneproductintheanalysis.
Therefore,inordertocorrectthemissingnucleotide,theexpressionvectorswerecopiedusingPCRwithprimersdesignedwiththemissingbaseinserted.Theoriginal

20
templatewasdigestedwithDpnIendonucleasestoeliminateplasmidswithouttheinsert.AscanbeseeninFigure11,copynumberswerelow.
Figure11:AgarosegelseparationofwholeplasmidPCRproductswithinsertedguanine.Wellcontents:pTZ19RGinsertwithannealingtemperatureof53°C(1),58°C(2)and60°C(3).pTZ19RshortGinsertwithannealingtemperatureof53°C(4),58°C(5)and60°C(6).pSE420Ginsertwithannealingtemperatureof53°C(7),58°C(8)and60°C(9).pSE420shortGinsertwithannealingtemperatureof53°C(10),58°C(11)and60°C(12).
Simultaneously,amethodforexaminingthecellpelletforinclusionbodieswasinvestigated.SamplesweretakenfromdiscardedsupernatantsduringtheprocessandseparatedonSDS‐PAGEgels.Expressionwasmeasuredonthenewcloneswithinsertedguanineusingtheinclusionbodyscreeningprotocolinordertoexamineboththesolubleandinsolubleproteincontent.Expressiontimewasincreasedtofourhourstoinvestigateifalongerexpressiontimecouldhelpaugmenttheyield.SDS‐PAGEgelsshowednonewbandsineitheranalysis.
Figure12:SDSPAGEseparationofinclusionbodyscreeningsamplesgrownat30°Ccontaining:firstcentrifugationsupernatantofnegativecontrol(1)andexpressionsample(2),secondcentrifugationsupernatantofnegativecontrol(3)andexpressionsample(4),8MUreasuspensionofnegativecontrol(5)andexpressionsample(6).
21
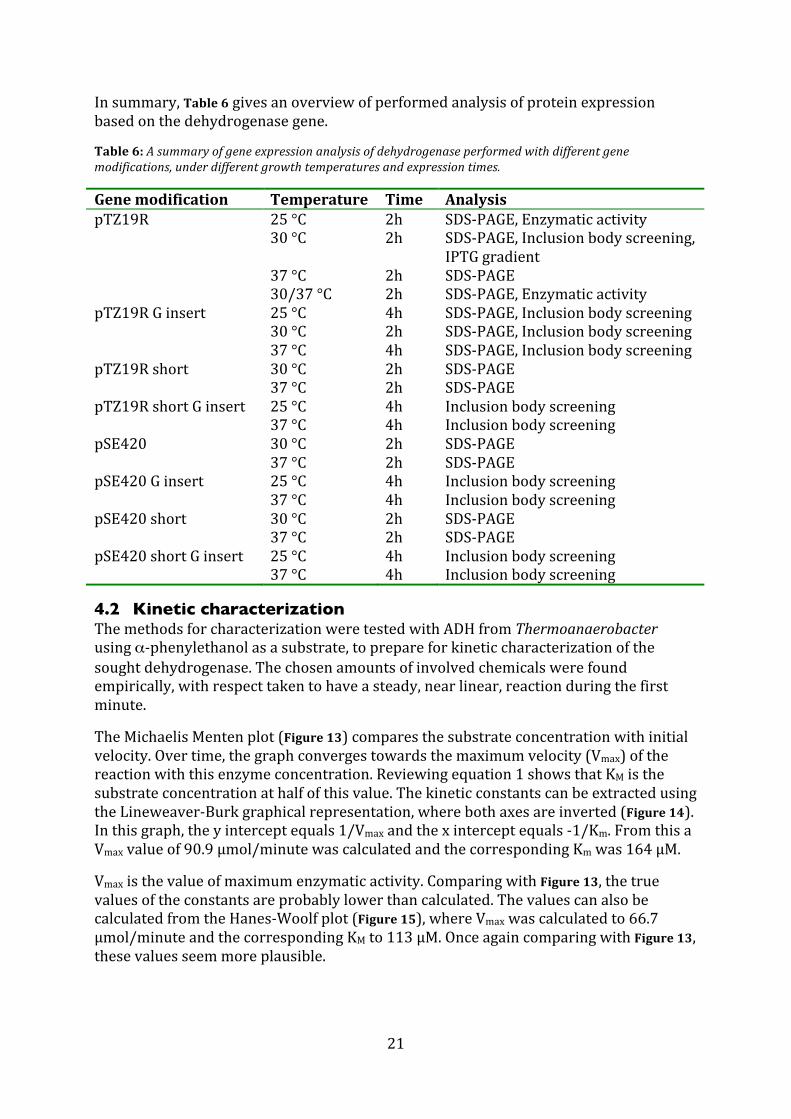
Insummary,Table6givesanoverviewofperformedanalysisofproteinexpressionbasedonthedehydrogenasegene.
Table6:Asummaryofgeneexpressionanalysisofdehydrogenaseperformedwithdifferentgenemodifications,underdifferentgrowthtemperaturesandexpressiontimes.
Genemodification Temperature Time AnalysispTZ19R 25°C 2h SDS‐PAGE,Enzymaticactivity 30°C 2h SDS‐PAGE,Inclusionbodyscreening,
IPTGgradient 37°C 2h SDS‐PAGE 30/37°C 2h SDS‐PAGE,EnzymaticactivitypTZ19RGinsert 25°C 4h SDS‐PAGE,Inclusionbodyscreening 30°C 2h SDS‐PAGE,Inclusionbodyscreening 37°C 4h SDS‐PAGE,InclusionbodyscreeningpTZ19Rshort 30°C 2h SDS‐PAGE 37°C 2h SDS‐PAGEpTZ19RshortGinsert 25°C 4h Inclusionbodyscreening 37°C 4h InclusionbodyscreeningpSE420 30°C 2h SDS‐PAGE 37°C 2h SDS‐PAGEpSE420Ginsert 25°C 4h Inclusionbodyscreening 37°C 4h InclusionbodyscreeningpSE420short 30°C 2h SDS‐PAGE 37°C 2h SDS‐PAGEpSE420shortGinsert 25°C 4h Inclusionbodyscreening 37°C 4h Inclusionbodyscreening
4.2 Kinetic characterization ThemethodsforcharacterizationweretestedwithADHfromThermoanaerobacterusingα‐phenylethanolasasubstrate,toprepareforkineticcharacterizationofthesoughtdehydrogenase.Thechosenamountsofinvolvedchemicalswerefoundempirically,withrespecttakentohaveasteady,nearlinear,reactionduringthefirstminute.
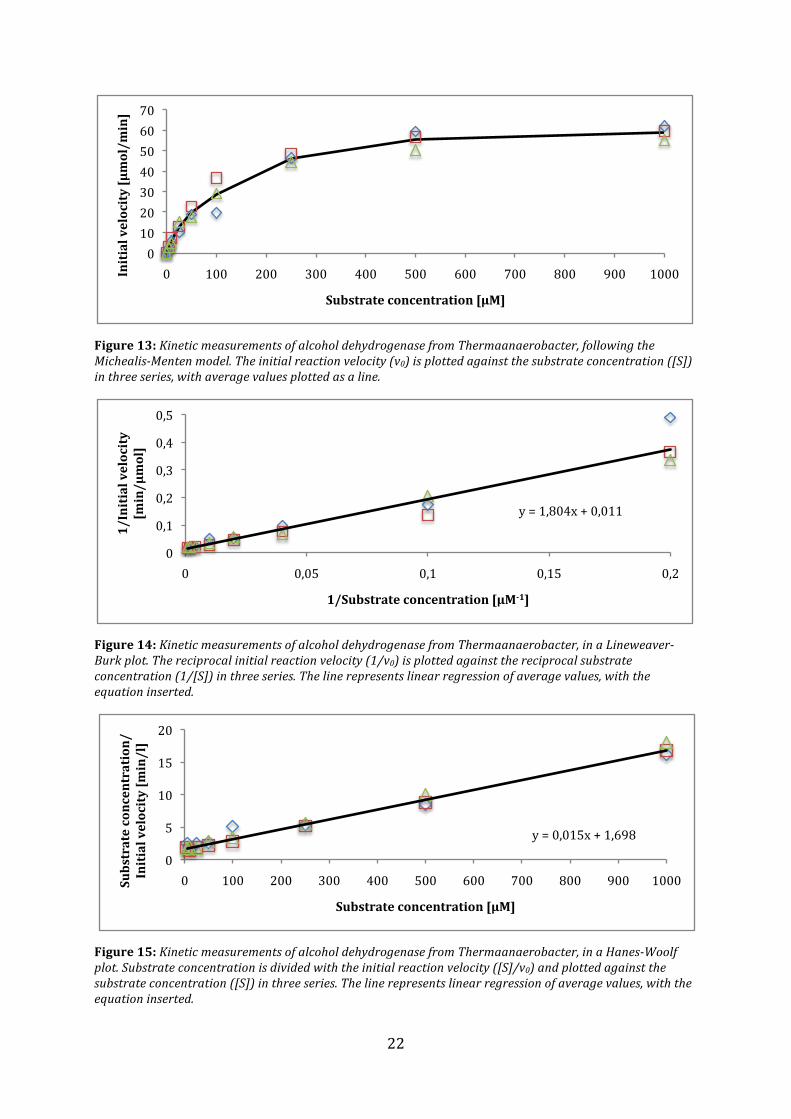
TheMichaelisMentenplot(Figure13)comparesthesubstrateconcentrationwithinitialvelocity.Overtime,thegraphconvergestowardsthemaximumvelocity(Vmax)ofthereactionwiththisenzymeconcentration.Reviewingequation1showsthatKMisthesubstrateconcentrationathalfofthisvalue.ThekineticconstantscanbeextractedusingtheLineweaver‐Burkgraphicalrepresentation,wherebothaxesareinverted(Figure14).Inthisgraph,theyinterceptequals1/Vmaxandthexinterceptequals‐1/Km.FromthisaVmaxvalueof90.9µmol/minutewascalculatedandthecorrespondingKmwas164µM.
Vmaxisthevalueofmaximumenzymaticactivity.ComparingwithFigure13,thetruevaluesoftheconstantsareprobablylowerthancalculated.ThevaluescanalsobecalculatedfromtheHanes‐Woolfplot(Figure15),whereVmaxwascalculatedto66.7µmol/minuteandthecorrespondingKMto113µM.OnceagaincomparingwithFigure13,thesevaluesseemmoreplausible.
22
Figure13:KineticmeasurementsofalcoholdehydrogenasefromThermaanaerobacter,followingtheMichealisMentenmodel.Theinitialreactionvelocity(v0)isplottedagainstthesubstrateconcentration([S])inthreeseries,withaveragevaluesplottedasaline.
Figure14:KineticmeasurementsofalcoholdehydrogenasefromThermaanaerobacter,inaLineweaverBurkplot.Thereciprocalinitialreactionvelocity(1/v0)isplottedagainstthereciprocalsubstrateconcentration(1/[S])inthreeseries.Thelinerepresentslinearregressionofaveragevalues,withtheequationinserted.
Figure15:KineticmeasurementsofalcoholdehydrogenasefromThermaanaerobacter,inaHanesWoolfplot.Substrateconcentrationisdividedwiththeinitialreactionvelocity([S]/v0)andplottedagainstthesubstrateconcentration([S])inthreeseries.Thelinerepresentslinearregressionofaveragevalues,withtheequationinserted.
010203040506070
0 100 200 300 400 500 600 700 800 900 1000Initialvelocity[µmol/m
in]
Substrateconcentration[µM]
y=1,804x+0,011
0
0,1
0,2
0,3
0,4
0,5
0 0,05 0,1 0,15 0,2
1/Initialvelocity
[min/µmol]
1/Substrateconcentration[µM1]
y=0,015x+1,698
0
5
10
15
20
0 100 200 300 400 500 600 700 800 900 1000Substrateconcentration/
Initialvelocity[min/l]
Substrateconcentration[µM]

23
Inordertocomparethisenzymetoothers,theamountofenzymeusedisanimportantfactortoconsider.Sinceenzymeconcentrationisunknowninthestocksolution,absorbanceat280nmismeasuredtoestimatethisvalue.Theconcentrationismeasuredata1:100dilution,andgaveanabsorbanceof0.4274.
Usingtheestimatedextinctioncoefficient
€
εADH = 30.940mM−1cm−1[SwissInstituteof
Bioinformatics]andassuming100%purity,thestockenzymeconcentrationisestimatedto1.4mM,or52grams/liter.Furthermore,thissignifiesthateachreactioncontained2.6µgenzyme.Thus,thecalculatedspecificactivityofthisenzymeis26000µmolmin‐1mg‐1forreactionswithα‐phenylethanolasasubstrate.Thisvalueisanestimate;bearinginmindthatreactionconditionssuchastemperatureisnotoptimalandtheconcentrationcalculatedisapproximate.

24
5 Conclusions ThedehydrogenasegenewassuccessfullyclonedintoDH5αE.colicellsusingthepTZ19Rexpressionvectoraspreviouslydescribedintheliterature.However,initialanalysisofproteinexpressionindicatedthattheenzymewasnotexpressedcorrectly.Themostprobableexplanationwasformationofinclusionbodiesandtodealwiththis,threenewclonesweresetuptobeanalyzedsimultaneously.Toinvestigatetheimpactoftheexpressionvector,thegenewasclonedintothepSE420vectorasanalternative.
Sincethecombinationofrestrictionsitesandexpressionvectorscaused12extraaminoacidstobeaddedtothebeginningofthegene,thiswasthoughtasapossiblecauseforinclusionbodyformation.Therefore,12aminoacidswereremovedfromthebeginningofthegene,substitutedwiththeaminoacidscodedbytheexpressionvector.TheusageofarestrictionsitecontainingtheATGstartsequence,suchasNcoI(CCATGG)orNdeI(CATATG)wouldhaveminimizedthislossifavailableandcorrectlysituatedinthereadingframe.However,availablerestrictionsitesintheexpressionvectorswerealsopresentinthegene,therebyblockingtheirusage.
SequencingofthefinishedDNAmaterialrevealedanerrorinthegene,asinglenucleotidedeletion,presentinallfourclones.Sincethiscausesaframeshiftofthetranslationofthegene,renderingallfollowingaminoacidschanged,thegeneticcodewascorrectedinallclones.Followingtestingofgeneexpressionstillindicatedthattheproteinwasnotexpressedcorrectly.Analysisofinclusionbodycontentinexpressedcellsalsoshowedlittleornoexpressionoftheprotein.
Additionally,theoriginalgenesequenceusedwasfoundtocontainerrorscausedbyusinganobsoleteversionofthesequencereleasedbytheauthors.Theerrorconsistsofasingleaminoacidchangeinonelocationandasequenceofsixaminoacidschangedinanotherlocation.Thiserrorcouldrenderanon‐functionalprotein,butshouldhowevernotimpactproteinexpression.
Thereareafewpossibleexplanationsastowhytheproteinisnotexpressedcorrectly.SincetheproteinisnotnativetoE.coli,thecodonusageintheproteinmightbesuboptimalforexpressionoutsideitsnativehost,therebyhamperingproteinexpression.Anotherpossibleexplanationisthattheproteinisdegradedbythecell.TheDH5αstrainofE.colicontainsgenesforLonprotease,naturallyusedbythecelltodegrademisfoldedorunfoldedproteins.Iftheproteinisfoldedincorrectly,itmightbedegradedbytheseproteases,therebynotshowingupinanalysis.

25
6 Recommendations Correctingtheerrorsremaininginthegeneticsequencemightberequiredfortheexpressionofactiveprotein.Thereisachancethattheintroducederrorsdonotaffectproteinfolding,buttherewilllikelybeapositiveeffectinthefinishedproductiftheseaminoacidsarecorrect.ItispossibletousemultiplePCR:stocorrecttheseinaccuracies.
Thecodonusageinthegeneshouldbeoptimized,usingthemostcommoncodonsinthehostforanefficienttranslationprocess.However,doingthisusingPCRwillproveatediousprocesswhereonlyafewbasescanbeaffectedforeachcyclerun.Acompletelynewgene,usingtheupdatedsequenceandoptimizedcodonsmightbeabetteralternative,albeitcostly.
Toimpedeproteolyticactivity,abacterialstrainknockedoutforproteasesshouldbeused.TheBL21strainofE.coli,whichcanbeusedlicence‐free,fallsunderthiscategory.BL21(DE3),cellslysogenizedwiththeDE3phage,arealsocapableofusingtheT7expressionsystemavailableonthepTZ19Rexpressionvector.TheT7systemisoftenusedforhighexpressionratesandmightproveanadvantage.
Afterproteinexpressionisobtained,itshouldbeoptimizedwithrespecttotemperature,IPTGconcentration,thetimeofinductionandexpressionduration.Growthconditions,suchasmediumusedtogetherwiththesubstrate,shouldbedetermined.Theactivitytestproposedinthisreportcanbeusedtoassesstheefficiencyoftheenzyme.

26
7 Acknowledgements IwouldliketothankCambrexKarlskogaABgivingmetheopportunitytodothisthesisandthestaffforwelcomingmeintotheworkplace.SpecialthanksgoestomysupervisorCeciliaBrannebyforinvaluablehelpinthelaboratory,discussionsandwhilewritingthisthesis,LinaLindbergforhelpingmeinthelaboratoryandforendlessdiscussions,ThomasNorström,forHPLCanalysisandPärHolmbergfordiscussionsaroundthesubstrate.
IwouldalsoliketothankmyexaminerUnoCarlssonandLars‐GöranMårtenssonatLinköpingUniversityforinterestinginputonmyresults,andmyopponentAnnelieBorgström.
Finally,IwouldliketothankChristinaandAndersRubensonforwelcomingmetoKarlskogaandkindlyofferinghousingduringmystay,familyandfriendsforyourencouragement.

27
8 References AppelbaumER,ShatzmanAR:Procaryoticinvivoexpressionsystems,InSJHiggins,BDHames(Eds):ProteinExpression:aPracticalApproach,Oxford:OxfordUniversityPress,2003
BaneyxF,MujacicM:RecombinantproteinfoldingandmisfoldinginEscherichiacoli,NatureBiotechnology,2004;22(11):1399‐1408
BergJM,TymoczkoJL,StryerL:Biochemistry,5thed.,NewYork:W.H.FreemanandCompany,2002
Biocompare:pSE420ExpressionvectorfromSigmaAldrich,Availableonlineat:<http://www.biocompare.com/ProductDetails/349908/pSE420‐Expression‐vector.html>(Downloaded2010‐02‐15)
BothamKM,MayesPA:BiologicOxidation,InMurrayRK,BenderDA,BothamKM,KennelyPJ,RodwellVW,WeilPA(Eds):Harper'sIllustratedBiochemistry,28thed.,NewYork:McGraw‐Hill,2009
BrownTA:GeneCloningandDNAanalysis,5thed.,Oxford:BlackwellPublishing,2006
BuggTDH:IntroductiontoEnzymeandCoenzymeChemistry,2nded.Oxford:BlackwellPublishing,2004
CarrióMM,VillaverdeA:Proteinaggregationasbacterialinclusionbodiesisreversible,FEBSLetters,2001;489(1):29‐33
ChamberlainJ:PCRmediatedmutagenesis,InEncyclopediaofLifeSciences,Chichester:JohnWiley&Sons,<http://www.els.net>,2004
Cornish‐BowdenA:FundamentalsofEnzymeKinetics,3rded.,London:PortlandPress,2004
DawsonRB:Dataforbiochemicalresearch,3rded.,Oxford:ClarendonPress,1985
DellEJ,GanskeF:DetectionofNADHandNADPHwiththeOmega’sHighSpeed,FullUV/VisAbsorbanceSpectrometer,BMGLabtechInc.Availableonlineat:<http://www.analytica‐world.com/articles/e/86152>(Downloaded2009‐12‐03)
FaberK:BiotransformationsinOrganicChemistry:ATextbook,4thed.,Heidelberg:SpringerVerlag,2000
Fermentas:(1)ProtocolforPCRwithPfuDNAPolymerase,productinformationforPfuDNAPolymerase
Fermentas:(2)pTZ19RDNA.Availableonlineat:<http://fermentas.com/en/products/all/molecular‐cloning/vectors‐phage/sd014‐ptz19r‐dna>(Downloaded2010‐02‐15)
GeorgiouG,ValaxP:ExpressionofcorrectlyfoldedproteinsinEscherichiacoli,CurrentOpinioninBiotechnology,1996;7(2):190‐197
HallingP:EnzymicConversionsinOrganicandOtherLowWaterMedia,InDrauzK,WaldmannH(Eds.):EnzymeCatalysisinOrganicSynthesis,Weinheim:Wiley‐VCH2002

28
HummelW,KulaMR:Dehydrogenasesforthesynthesisofchiralcompounds,EuropeanJournalofBiochemistry,1989;184(1):1‐13
IshigeT,HondaK,ShimizuS:Wholeorganismbiocatalysis,CurrentOpinioninChemicalBiology,2005;9(2):174‐180.
KazusaDNAResearchInstitute:CodonUsageDatabase,Entry:EscherichiacoliW3110.Availableonlineat:<http://www.kazusa.or.jp/codon/cgi‐bin/showcodon.cgi?species=316407>(Downloaded2010‐03‐31)
LingMM:GeneExpressioninEscherichiacoli,InEncyclopediaofLifeSciences,Chichester:JohnWiley&Sons,<http://www.els.net>,2001
LiuW,WangP:Cofactorregenerationforsustainableenzymaticbiosynthesis,BiotechnologyAdvances,2007;25(4):369‐384
ManiM,KandavelouK,ChandrasegaranS:RestrictionEnzymes,InEncyclopediaofLifeSciences,Chichester:JohnWiley&Sons,<http://www.els.net>,2007
MetzkerML,Caskey,CT:PolymeraseChainReaction(PCR),InEncyclopediaofLifeSciences,Chichester:JohnWiley&Sons,<http://www.els.net>,2009
PankeS,HeldM,WubboltsM:Trendsandinnovationsinindustrialbiocatalysisfortheproductionoffinechemicals,CurrentOpinioninBiotechnology,2004;15(4):272‐279
PurifyingProteinfromInclusionBodies,Availableonlineat<http://structbio.vanderbilt.edu/chazin/wisdom/labpro/inclusion.html>(Downloaded2009‐12‐12)
QuailMA:DNACloning,InEncyclopediaofLifeSciences,Chichester:JohnWiley&Sons,<http://www.els.net>,2005
SambrookJ,RusselDW:Molecularcloning,alaboratorymanual,3rded.ColdSpringHarbor:ColdSpringHarborLaboratoryPress,2001
SchoemakerHE,MinkD,WubboltsMG:DispellingtheMyths–BiocatalysisinIndustrialSynthesis,Science,2003;299(5613):1694‐1697
StaderJ:GeneExpressioninRecombinantEscherichiacoli,InSmithA(Ed):GeneExpressioninRecombinantMicroorganisms,NewYork:MarcelDekker,1995
StudierFW,RosenbergAH,DunnJJ,DubendorffJW:UseofT7RNAPolymerasetoDirectExpressionofClonedGenes,MethodsinEnzymology,1990;185:60‐89
SwissInstituteofBioinformatics:ExpasyProtParam,Entry:SwissProtaccessionnumberP14941,Avalableonlineat<http://expasy.org/tools/protparam.html>
UkaiH,Ukai‐TadenumaM,OgiuT,TsujiH:Anewtechniquetopreventself‐ligationofDNA,JournalofBiotechnology,2002;97(3):233‐242
WatsonJD,CaudyAA,MyersRM,WitkowskiJA:RecombinantDNA,3rded.,NewYork:W.H.FreemanandCompany,2007

29
Appendix: Growth medium Thegrowthmediumusedwasastandardlysogenybroth(LB)consistingof:
• 10goftryptone• 5gofyeastextract• 5gofNaCl• dH2Otoatotalvolumeof1literandautoclavedat121°Cfor20minutes.
Toobtainaselectivemedium,ampicillinwasaddedtogiveafinalconcentrationof100µg/ml.
Agarplatesweremadeinthesamefashionwiththeadditionof15gofagarose.
KanamycinplateswerepreparedbyusingLBplatesandadding30µlofkanamycinin100µlofdH2O.Thismethodwasfunctionalandeliminatedtheneedofmakingkanamycin‐specificplatesastheywereusedinlownumbers.
Intransformation,anextrarichmediumwasused.TheSuperOptimalBroth(SOB)basewasmadebeforehandandtheunstablefinishedcataboliterepressionmedium(SOC)waspreparedrightbeforeuse.
SOB:(1liter)
• 20goftryptone• 5gofyeastextract• 0.58gofNaCl• 0.19gofKCl• 4mlofNaOH,1M• dH2Otoatotalvolumeof1literandautoclavedat121°Cfor20minutes.
SOC:(1ml)
• 960µlofSOB• 10µlofMgCl2• 10µlofMgSO4• 20µlofglucose,1M,sterilized