darwin: a hardware-acceleration framework for … talks/yatish_turakhia.pdfpolymorphism (snp)...
TRANSCRIPT

Darwin: A Hardware-acceleration Framework for Genomic Sequence Alignment
Prof. Bill Dally (Electrical Engineering and Computer Science)Prof. Gill Bejerano (Computer Science, Developmental Biology and Pediatrics)
Yatish Turakhia EE PhD candidate Stanford University

Overview
2
1. DNA sequencing and long read assembly
2. Darwin: A Hardware-acceleration Framework for Genomic Sequence Alignment
3. Ongoing Work

3
PART I: DNA Sequencing and Long Reads

Human Genome Project
4
} Complete human genome draft published in 2003 through “DNA sequencing”
} $3billion and 13 years
HGC Celera
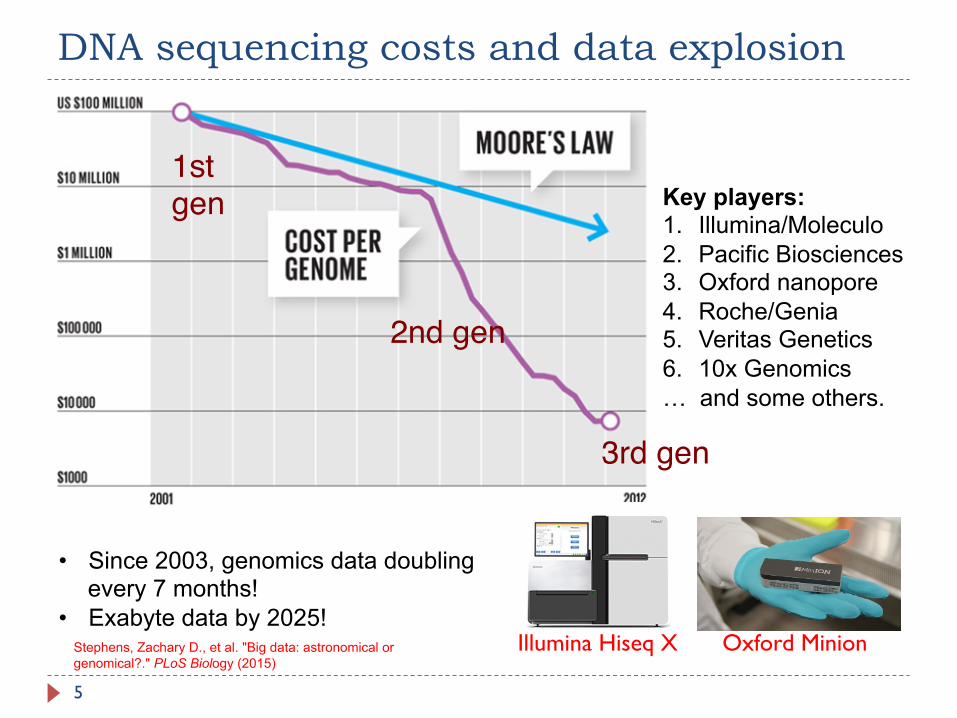
DNA sequencing costs and data explosion
5
• Since 2003, genomics data doubling every 7 months!
• Exabyte data by 2025!
1st gen
2nd gen
3rd gen
Key players: 1. Illumina/Moleculo 2. Pacific Biosciences 3. Oxford nanopore 4. Roche/Genia 5. Veritas Genetics 6. 10x Genomics … and some others.
Illumina Hiseq X Oxford Minion Stephens, Zachary D., et al. "Big data: astronomical or genomical?." PLoS Biology (2015)

DNA sequencing costs and data explosion
6
[nasaspaceflight.com]
Oxford Minion

Some questions we can now ask and budding industry ecosystem around it
7
What genomic mutations predispose us to disease? Genentech/Roche, Myriad, Counsyl, Rosetta, Veritas,23andMe, HLI, Calico, etc.
Where did we come from? How are we different from each other? Ancestry.com, 23andMe etc.
What in our genomes make us different from other species? J. Craig Venter Institute
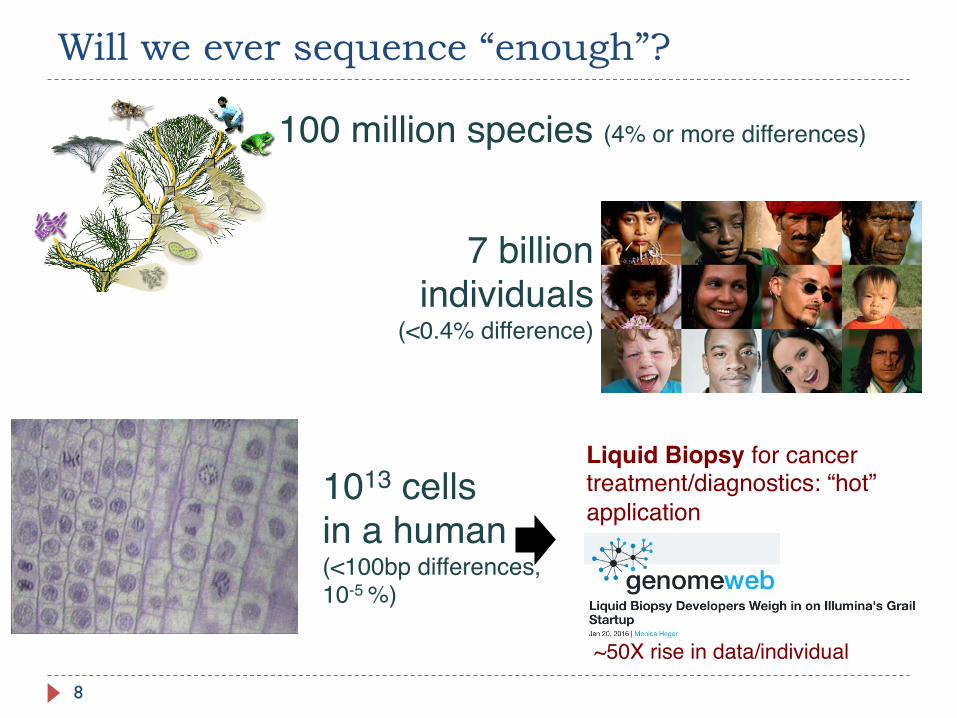
Will we ever sequence “enough”?
8
100 million species (4% or more differences)
7 billion individuals
(<0.4% difference)
1013 cells in a human(<100bp differences, 10-5 %)
Liquid Biopsy for cancer treatment/diagnostics: “hot” application
~50X rise in data/individual

Genome assembly
9
} No sequencing technology as of today can read whole genome/chromosome string from start to end:} Illumina: larger length => larger (phase) error } PacBio/Nanopore: hard to keep long DNA fragments intact
} Replicate -> fragment & read -> assemble (software)} Fragments called “reads”} Number of copies is called coverage} Assembly is the process of reconstructing the original sequence
from fragments} Higher coverage reduces sequencing gaps
C
Sequencing gap

Types of Genome assembly
10
1. De novo / ab initio assembly:
2. Resequencing / Reference-guided / Comparative assembly:
Reference Genome
Reads
???
Contigs
Overlap reads and extend

Types of Genome assembly
11
1. De novo assembly:} No reference-bias} Works better than reference-guided when difference wrt reference is large} More fragmented assembly} Computationally intensive ~ O(n2) for n reads
2. Reference-guided / Resequencing / Comparative assembly:} Good for finding small differences (eg. SNPs)
} Less fragmented assembly with high-quality reference} Fast ~O(n) for n reads} Reference bias} Cannot capture large differences (reads don’t map to reference)
Single Nucleotide Polymorphism (SNP)
TGCTGAGA TGCCGAGA

Sequencing Technologies
12
Illumina Hiseq X PacBio Sequel Oxford Nanopore PromethIon1
Mean read length 35-200bp 10,000-15,000bp 10,000 (up to 300Kbp)
Error rates 0.1% to 2.0% ~14% ~25% to 40% 2
Error profile Systematic*, more substitutions
Stochastic, more indels**
Stochastic, more indels**
Throughput (genomes/yr)
18,000 human genomes/yr (30X)
90-120 human genomes/yr (30X)
~17,000 human genomes/yr (30X)
Consensus accuracy >99.99% (30X) >99.98% (30X) >98.0% (30X)
De novo assemblyAlgorithms
~3,000 CPU hoursABySS, Celera,
Velvet etc
>15,000 CPU hoursFALCON,
DALIGNER
>100,000 CPU hoursCanu (Celera)
Reference guidedAlgorithms
20-30 CPU hoursBWA
~1,300 CPU hoursBWA-MEM
~20,000 CPU hoursGraphMap (30X)
* Biased against GC content** indels = insertions + deletions
Bina/Roche (Cloud), Edico Genome (Hardware)
DNAnexus (Cloud)1 48 flow cells2 R7.3 (superseded by R9)

Long Reads: Holy grail of sequencing
13
} Fewer and larger contigs in de novo assembly
} Better at handling structural variants (variants >1kb)
[Source: pacbio.com]
Inversion
Mobile Element orPseudogene Insertion
Tandem Duplication
short
long
short
long
short
long
30X

14
PART II: Darwin FrameworkPreprint: http://biorxiv.org/content/early/2017/01/24/092171

Smith-Waterman Alignment
15
} Given: Reference (R) and size m and Query (Q) of size n } Two step procedure
1. Compute scores H of every cell in the dynamic programming (DP) matrix using Smith-Waterman equations and scoring matrix W
2. Traceback from high-scoring cells to obtain final alignments
Smith-Waterman equations
Scoring Matrix WA C G T
A 2 -1 -1 -1C -1 2 -1 -1G -1 -1 2 -1T -1 -1 -1 2
gap = 1
* G G C G A C T T T* 0 0 0 0 0 0 0 0 0 0
G 0 2 2 1 2 1 0 0 0 0
G 0 2 4 3 3 2 1 0 0 0
T 0 1 3 3 2 2 1 3 2 2
C 0 0 2 5 4 3 4 3 2 1
G 0 2 2 4 7 6 5 4 3 0
T 0 1 1 3 6 6 5 7 6 5
T 0 0 0 2 5 5 5 7 9 8
T 0 0 0 1 4 4 4 7 9 11
Que
ry (Q
)
Reference (R)
G G - C G A C T T T| | | | | | | G G T C G - - T T T

Strategies for long sequence alignment
16
Algorithm Time Space(compute-
intensive step)
Optimal
Smith-Waterman O(mn) O(mn) YesHirschberg O(mn) O(m+n) Yes
Banded Smith-Waterman
O(n) O(n) No
X-drop O(n) O(n) NoGACT O(n) O(1) No
Profound hardware design implications
Prior hardware-accelerators have either:1. Assumed a small upper bound on sequence length n2. Left trace-back of alignment to software – slow!
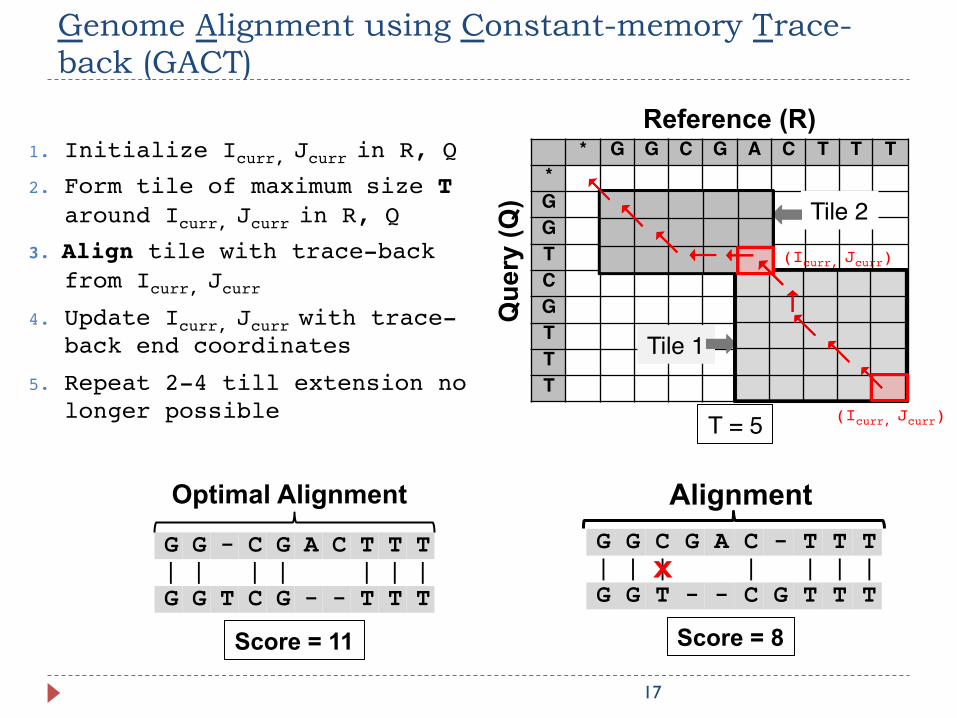
Genome Alignment using Constant-memory Trace-back (GACT)
17
* G G C G A C T T T*GGTCGTTT
Reference (R)
Que
ry (Q
)
Tile 1
Tile 2
1. Initialize Icurr, Jcurr in R, Q
2. Form tile of maximum size T around Icurr, Jcurr in R, Q
3. Align tile with trace-back from Icurr, Jcurr
4. Update Icurr, Jcurr with trace-back end coordinates
5. Repeat 2-4 till extension no longer possible
C - T T T| | | | C G T T T
Alignment G G C G A| | | G G T - -
X
Score = 8
G G - C G A C T T T| | | | | | | G G T C G - - T T T
Optimal Alignment
Score = 11
(Icurr, Jcurr)
(Icurr, Jcurr)
T = 5

Banded Smith-Waterman And GACT
Banded Smith-Waterman (25KB trace-back memory)
GACT (25KB trace-back memory)
18
• Insertion rate (9.02%) > Deletion rate (4.49%)• Alignment drifts from main diagonal – banding difficult!• GACT enforces adaptive banding with successive tiles
Score = 0.47 * OPT Score = OPT
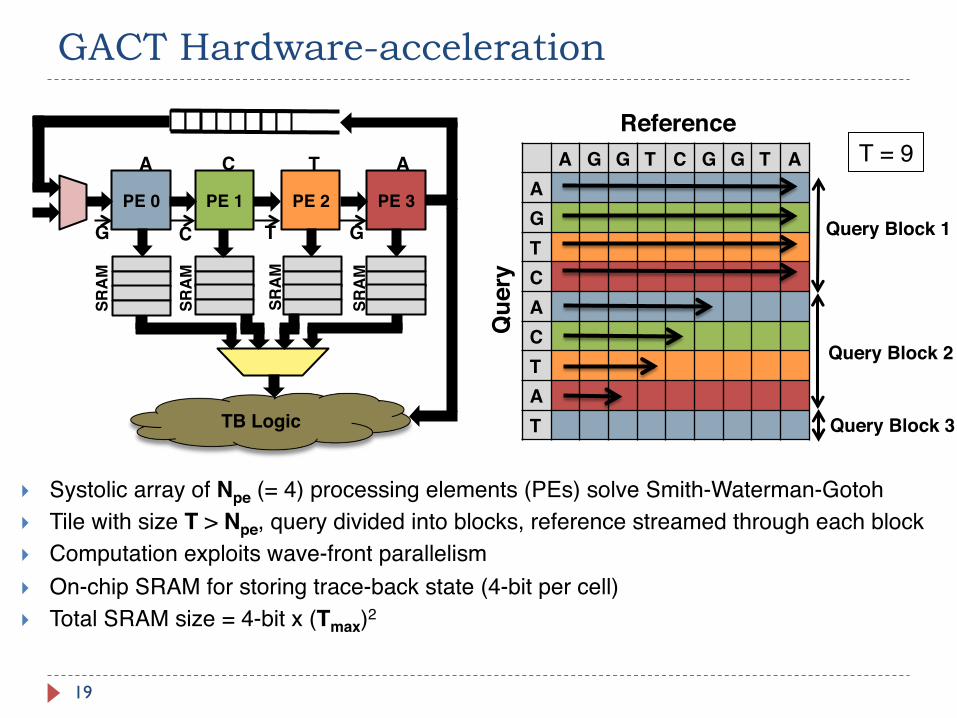
GACT Hardware-acceleration
19
A G G T C G G T AAGTCACTAT
Query Block 1
Query Block 2
Query Block 3
Reference
Que
ry
PE 0 PE 1 PE 2 PE 3
TB Logic
A C T A
G C T G
SRA
M
SRA
M
SRA
M
SRA
M
} Systolic array of Npe (= 4) processing elements (PEs) solve Smith-Waterman-Gotoh} Tile with size T > Npe, query divided into blocks, reference streamed through each block} Computation exploits wave-front parallelism} On-chip SRAM for storing trace-back state (4-bit per cell) } Total SRAM size = 4-bit x (Tmax)2
T = 9

GACT ASIC Design
20
Processing Element (PE)} Scoring using 10-bit signed integers} 3-bit internal representation for
alphabet = {A, C, G, T, N}} Area: 0.003mm2
Array (64 PEs, Tmax = 512)} TB memory: 128KB (2KB/PE)} Area: 1.37mm2
} Power: 137mW} Frequency: 847MHz
} FPGA prototype: 250MHz
Layout: Processing Element (PE)
TSMC 40nm

GACT Performance
21
23562
4712
0
5000
10000
15000
20000
25000
1000 2000 3000 4000 5000 6000 7000 8000
GA
CT
Thro
ughp
ut(A
lignm
ents
/Sec
ond)
Sequence length (in bp)
• Throughput: ~70,000 GACT tiles/second (T=300) • 762X faster than software (SeqAn Banded SW)• 39,000X more energy-efficient• 65% software overhead (append/prepend TB pointers, extract sequences etc)• With oracle mapper, 54X reads can be aligned in 1.6 hours with only 1 GACT
array (<2mm2, 137mW)
Communication overhead ignored
2350
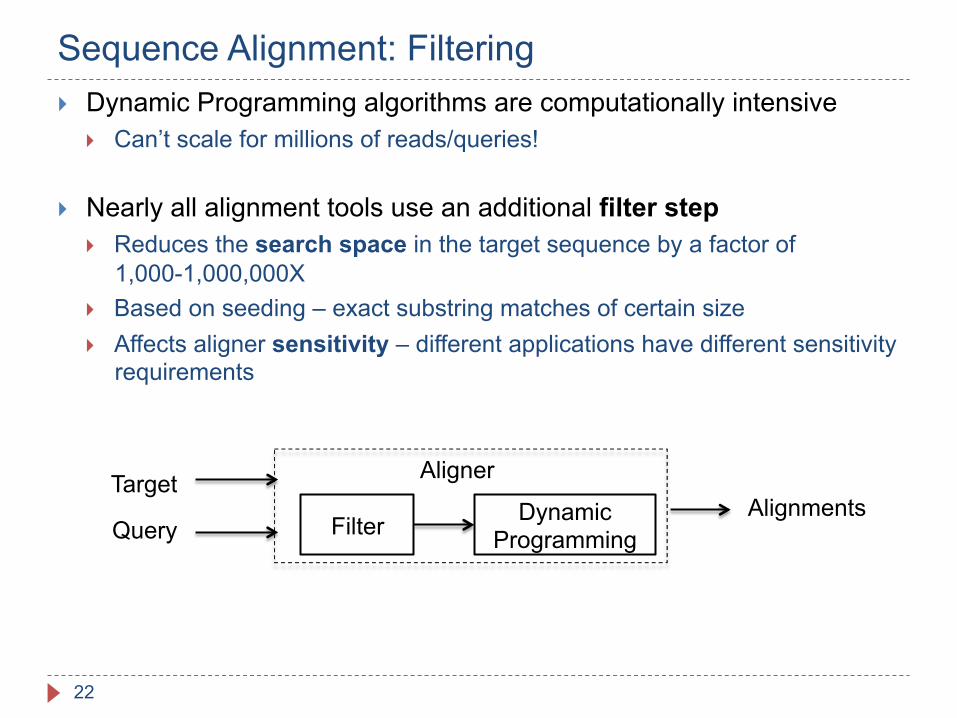
Sequence Alignment: Filtering
22
} Dynamic Programming algorithms are computationally intensive } Can’t scale for millions of reads/queries!
} Nearly all alignment tools use an additional filter step } Reduces the search space in the target sequence by a factor of
1,000-1,000,000X } Based on seeding – exact substring matches of certain size } Affects aligner sensitivity – different applications have different sensitivity
requirements
Target
Query Filter Dynamic Programming
Alignments Aligner

Seed Position table based exact matching
23
R = AGCTATACTA
AA
AC 6
AG 0
AT 4
CA
CC
CG
CT 2 7
GA
GC 1
GG
GT
TA 3 5 8
TC
TG
TT
Seed Positions Q = GCTA
GC:1 CT: 2, 7 TA: 3, 5, 8
Slope=1
R
Q
For human genome, position table size > 12GB (4B x 3 x 109) Algorithms like BWA use compressed tables at cost of performance (larger number of memory accesses)
3210
1 2 3 4 5 6 7 8

Diagonal-band Seed Overlapping based Filtration Technique (D-SOFT)
} Divide R into NB bins (diagonal bands)} Use N seeds of size k bp from different offsets in Q} Lookup positions of seeds in R and assign each seed hit to
corresponding bin (diagonal band)} Count non-overlapping Q base-pairs covered by seed hits for
each bin and filter based on threshold h (same as DALIGNER)
Bin 1 Bin 2 Bin 3 Bin 4 Bin 5 Bin 6
Reference (R)
Que
ry (Q
)
123456789
106 5 9 4 0 5
NB = 6N = 10 k = 4h = 7
24

D-SOFT: Overlap detection
25
1. Concatenated reads as reference (R)
2. Construct seed position table for R
3. Use forward and reverse-complement of every read as query (Q)
4. When number of reads too large, divide reads into blocks and repeat 1-3 for every block
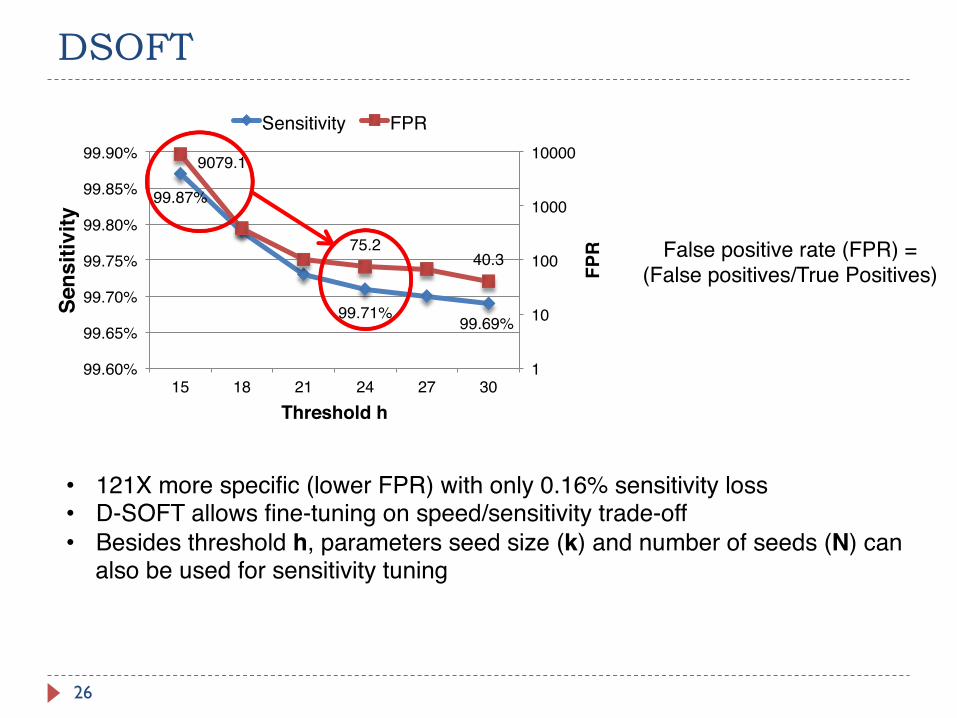
DSOFT
26
99.87%
99.71% 99.69%
9079.1
75.240.3
1
10
100
1000
10000
99.60%
99.65%
99.70%
99.75%
99.80%
99.85%
99.90%
15 18 21 24 27 30
FPR
Sens
itivi
ty
Threshold h
Sensitivity FPR
False positive rate (FPR) = (False positives/True Positives)
• 121X more specific (lower FPR) with only 0.16% sensitivity loss• D-SOFT allows fine-tuning on speed/sensitivity trade-off• Besides threshold h, parameters seed size (k) and number of seeds (N) can
also be used for sensitivity tuning

Darwin: System Overview
27
} Darwin is an alignment framework combining DSOFT filtering with hardware-accelerated GACT
} Darwin uses dynamic-programming based GACT to filter DSOFT false positives
} Absolute threshold htile on first GACT tile score
} Near-zero (<0.05%) additional loss of sensitivity
} DSOFT settings alone determine Darwin sensitivity – useful feature!
htile = 70

Experimental Setup
28
Read Type Substitution Insertion Deletion Total ErrorPacBio 1.50% 9.02% 4.49% 15.01%ONT 2D1 16.50% 5.10% 8.40% 30.0%ONT 1D1 20.39% 4.39% 15.20% 39.98%1R7.3 chemistry used here superseded by the new R9 chemistry
PBSIM simulator to generate reads from human reference assembly (GRCh38)
Software Baseline• Intel Xeon E5-26200 CPU @ 2.0GHz, single thread executionDarwin• D-SOFT on software (single thread) with above processor configuration• GACT single array ASIC performance extracted from FPGA prototype
Reference-guided:• PacBio: BWA-MEM• Oxford nanopore (ONT): GraphMapDe Novo:• PacBio: DALIGNER

Results
29
Reference-guided
De novo
Darwin’s bottleneck: DSOFT (80% runtime)

GACT Consensus
30
} GACT deviations from optimal alignment can be statistically corrected} Easier to correct GACT alignment errors than sequencing errors – 0.7%
sequencing errors persist even at 15X coverage (PacBio)

31
PART III: Ongoing Work

D-SOFT hardware-acceleration
32
} D-SOFT runtime spent mostly on random accesses in updating bin counts (60-120MB memory)
} Seed lookup table (16GB) has sequential access – fast!
} D-SOFT accelerator can result in 10-100X faster filtration
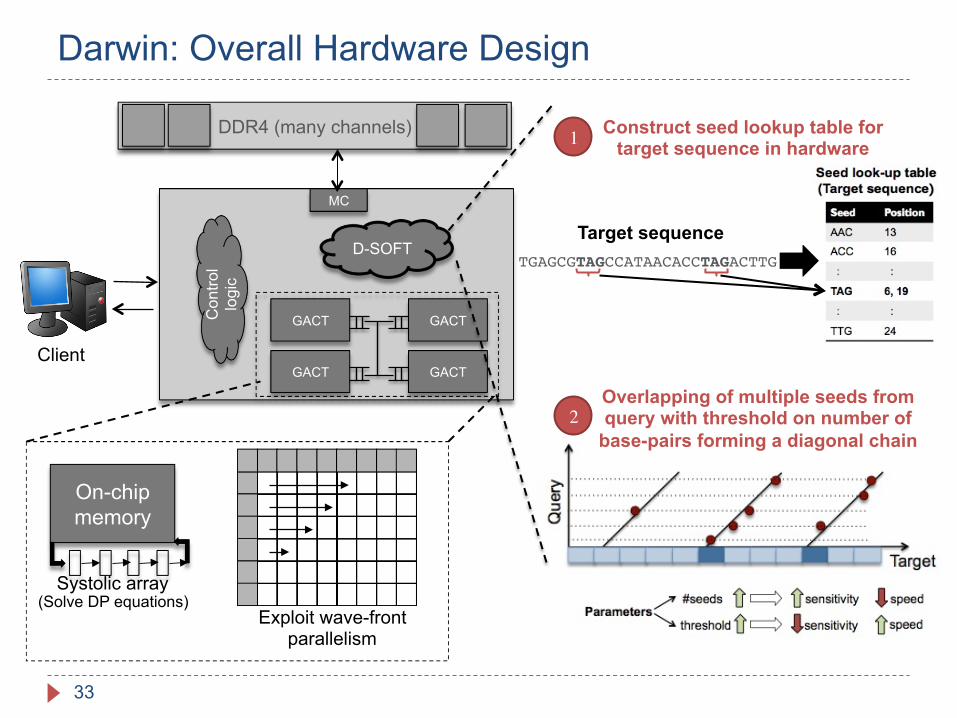
Darwin: Overall Hardware Design
33
TGAGCGTAGCCATAACACCTAGACTTG
Construct seed lookup table for target sequence in hardware
Target sequence
1
2Overlapping of multiple seeds from query with threshold on number of
base-pairs forming a diagonal chain
D-SOFT
MC
GACT GACT
GACT GACT
Con
trol
logi
c
DDR4 (many channels)
Client
On-chip memory
Systolic array
Exploit wave-front parallelism
(Solve DP equations)

Darwin: Projected Speedup And Cost (approx)
} D-SOFT: 128MB on-chip memory (4-8 banks) + Crossbar + Logic } <180mm2
} <5W} 4 x 8GB DDR-4
} 20 GACT arrays} <40mm2
} <3W } Up to 1.4M tiles/sec (~1GBps) with T=300
} >1,000X speedup over software (15mm x 15mm, 8W chip)} Maintaining high utilization might be
challenging
} 1000 CPU hours - > minutes!
#Seed positions
Effective Bandwidth
(GBps)64 1.70
256 3.981024 5.81
Volume Cost (Mask + Design/Verif + Wafer+ Test +
DRAM)1K $10K
10K $1.2K
100K $350
DDR-4 Bandwidth/Channel (1333MHz)
Approx ASIC cost (40nm)
34

Darwin: Two API styles
35
D-SOFTH/W
GACTH/W
GACT APID-SOFT API
S/W
Darwin
D-SOFTH/W
GACTH/W
GACT API
S/W
Darwin
FlexibleLess efficient
InflexibleEfficient
D-SOFT API

Thank you!
Questions or feedback?
36