curso bioquímica 23-aminoácidos
TRANSCRIPT

Oxidación de AminoácidosAntonio E. Serrano PhD. MT.Cátedra de Bioquímica - [email protected]

Sumario• Digestión de Proteínas de
la Dieta• Degradación de
Aminoácidos• Metabolismo del
Nitrógeno• Degradacion de
Glutamina, Alanina• Aminoácidos cetogénicos
y glucogénicos• Enfermedades
Metabolicas

Degradacion de Proteínas en la Dieta
• En humanos, las proteínas ingeridas en la dieta, son degradadas a sus aminoácidos constituyentes en el tracto gastrointestinal.
Degradación en el Estómago• La entrada de proteínas en el
estómago estimula la mucosa gástrica para secretar la hormona gastrina, que estimula a su vez la secreción de HCl por las células parietales, y pepsinógeno por las glándulas gástricas (pH 1 a 2,5)
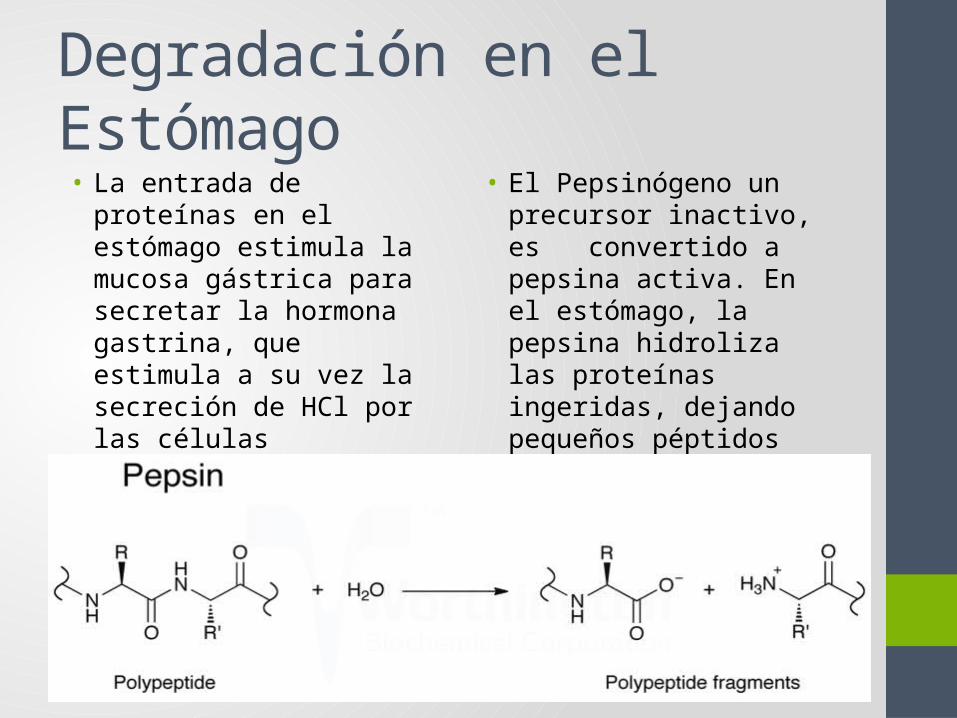
• El Pepsinógeno un precursor inactivo, es convertido a pepsina activa. En el estómago, la pepsina hidroliza las proteínas ingeridas, dejando pequeños péptidos
En el Intestino• Debido al paso de HCl hacia el
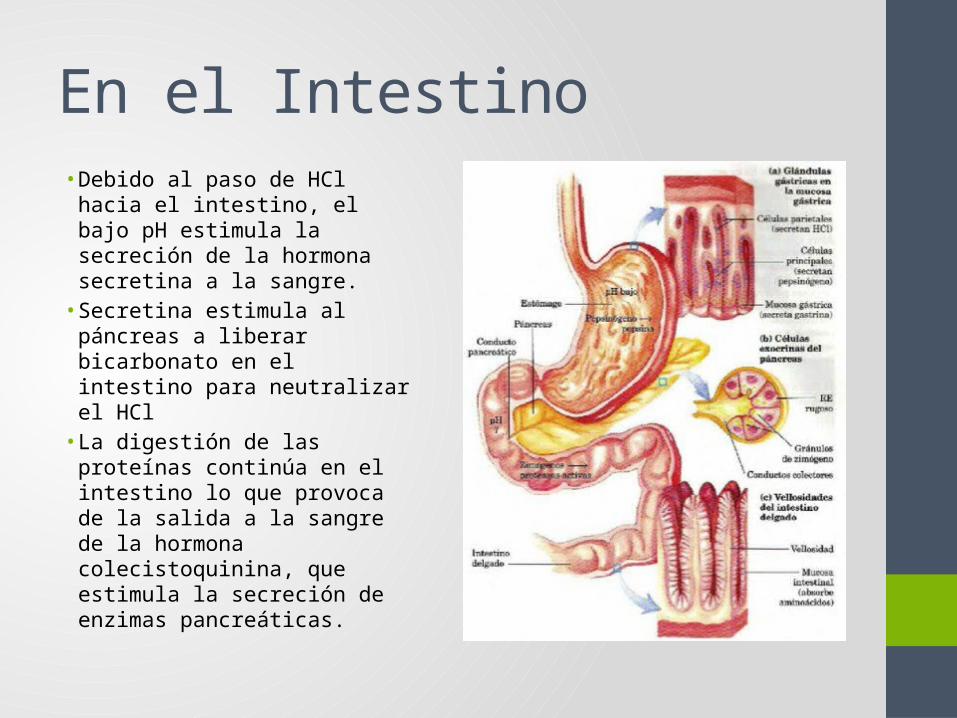
intestino, el bajo pH estimula la secreción de la hormona secretina a la sangre.
• Secretina estimula al páncreas a liberar bicarbonato en el intestino para neutralizar el HCl
• La digestión de las proteínas continúa en el intestino lo que provoca de la salida a la sangre de la hormona colecistoquinina, que estimula la secreción de enzimas pancreáticas.

Tripsina y Quimiotripsina• Hidrolizan los péptidos que
fueron producidos por la pepsina en el estómago.
• La mezcla de aminoácidos libres es transportada a enterocitos y entran a la sangre y viajan al hígado
• La degradación de pequeños péptidos en el intestino delgado es completada por otras peptidasas intestinales.• Carboxipeptidasas A y B:
Remueven residuos COO- • Aminopeptidasa que hidroliza
los residuos NH3-terminales.

Resumen• Las células parietales y la
células de las glándulas gástricas secretan sus productos en respuesta a la hormona gastrina
• La pepsina comienza el proceso de degradación de proteínas en el estómago
• Los aminoácidos son absorbidos a través de las células epiteliales (mucosa intestinal)

Degradación de Aminoácidos• DESTINOS DE AMINOACIDOS:1. Biosíntesis • Proteínas • Hormonas2. Obtención de Energía• A partir de esqueleto carbonado3. Destino de grupo amino :• Formación de urea (en mamíferos)• Biosíntesis de aminoácidos y de nucleótidos

Degradación Oxidativa• En animales, ocurre en 3 circunstancias:1. Durante la síntesis normal y degradación de proteínas celulares,• Algunos aminoácidos que son liberados desde la hidrólisis de
proteínas y que no son necesarios para una nueva síntesis de proteínas bajo degradación oxidativa.
2. Cuando una dieta es rica en proteínas• La ingesta de aminoácidos excede las necesidades del
organismo para la síntesis de proteínas.3. Durante el almacenaje o la Diabetes Mellitus incontrolada.• Cuando los hidratos de carbono no son utilizados
apropiadamente, las proteínas son usadas como combustible.

Degradación de aminoácidos• Los a.a. pierden sus
grupos amino para formar a-cetoácidos, el “esqueleto carbonado” de los aminoácidos.
• Los a-cetoácidos bajo oxidación a CO2 y H2O, proveen 3 y 4 carbonos que pueden ser convertidos en glucosa Gluconeogénesis (Músculo y Cerebro principalmente)

Metabolismo de Aminoácidos
• La mayoría de los aminoácidos son metabolizados en el hígado.• El esqueleto carbonado se transforma en acetil-Co A y en
intermediarios del Ciclo del ácido cítrico.• El grupo amino es reciclado y utilizado en diversas rutas
biosintéticas y el exceso entra al ciclo de la urea para ser excretado.

Metabolismo de Aminoácidos

Metabolismo del Nitrógeno• Glutamato,
glutamina y alanina juegan un importante rol en el metabolismo de nitrógeno (grupo amino de aminoácidos)

Catabolismo de Aminoácidos• Remoción de los grupos α-
amino, promovidos por enzimas aminotransferasas o transaminasas
• En estas reacciones de transaminación, el grupo α-amino es transferido al carbono α de acetoglutarato, generando un análogo al α-cetoácido del aminoácido.
aminotransferasa

Deaminación de Glutamato por la Glutamato deshidrogenasa
• Pérdida del grupo amino desde el glutamato.• Cataliza la deaminación
oxidativa del glutamato a acetoglutarato.• Es la única enzima que
puede utilizar tanto NAD+ como NADP+ como aceptor de equivalentes de reducción.
Amonio

Degradación de Glutamina• El amonio es tóxico para los
tejidos animales.• Se debe transformar en un
metabolito no tóxico para ser transportado por la sangre hasta el hígado.
• La glutamina es una forma eficiente de transportar el exceso de amonio.
• La glutamina se encuentra en la sangre en concentraciones mayores que el resto de las aminoácidos
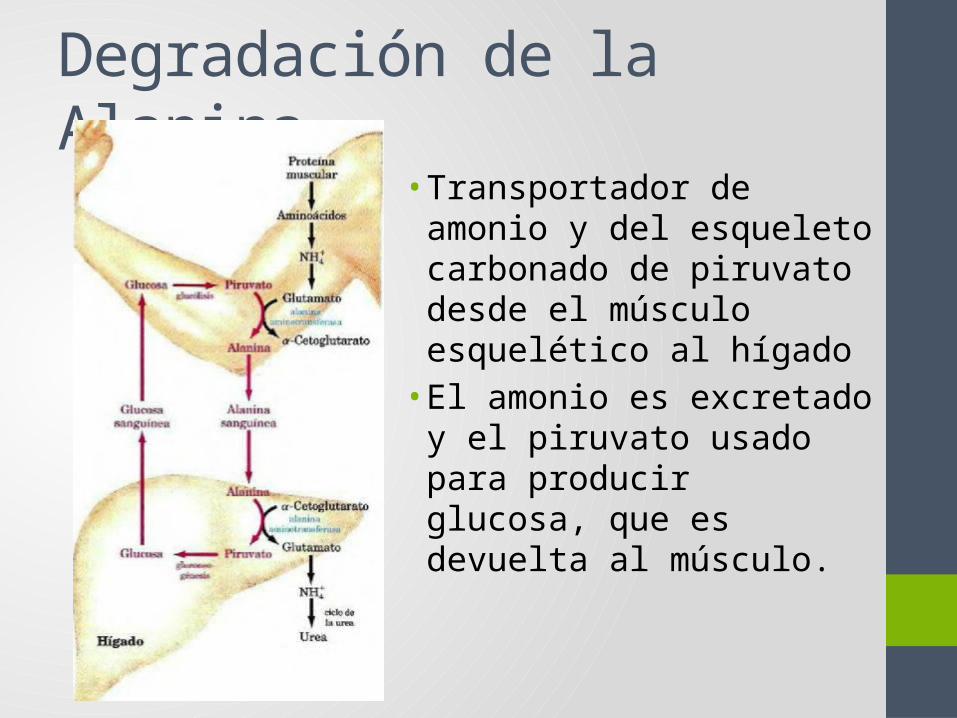
Degradación de la Alanina• Transportador de amonio y
del esqueleto carbonado de piruvato desde el músculo esquelético al hígado• El amonio es excretado y el
piruvato usado para producir glucosa, que es devuelta al músculo.

Ciclo de la alanina
• 1. En músculo: Transaminación de piruvato para producir alanina, que viaja al hígado por el torrente sanguíneo.
• 2. En el hígado: Transaminación de alanina a piruvato que pasa a la gluconeogénesis.
• 3. La Glucosa producida se libera al torrente sanguíneo.• Este proceso ayuda a mantener el balance de nitrógeno (se
transporta NH4+ al hígado)

Degradación de los aminoácidos cetogénicos y glucogénico
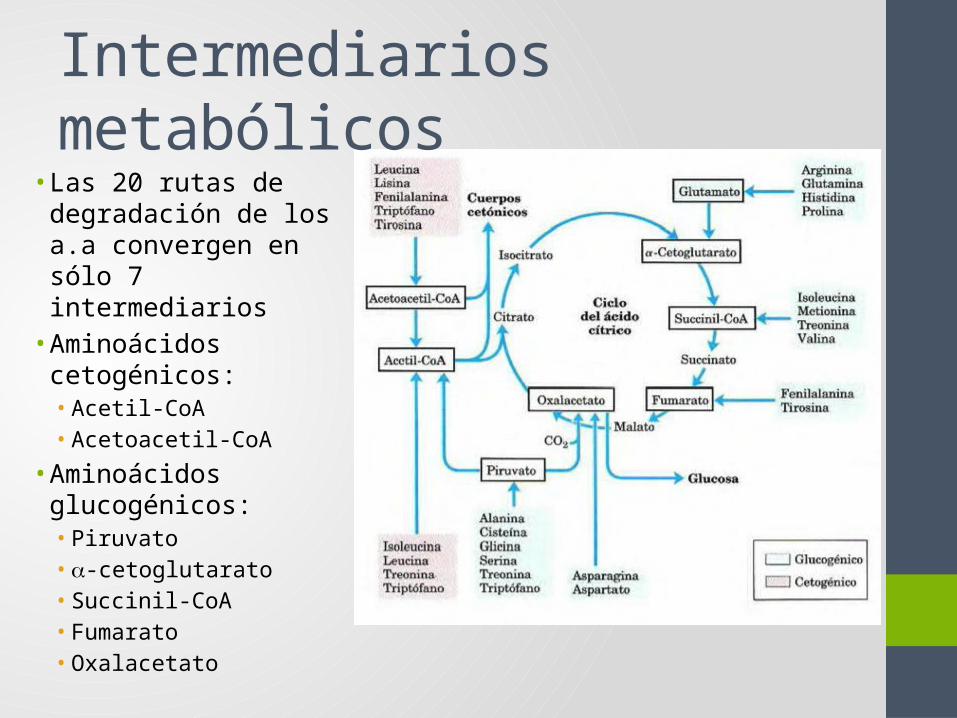
Intermediarios metabólicos• Las 20 rutas de
degradación de los a.a convergen en sólo 7 intermediarios
• Aminoácidos cetogénicos:• Acetil-CoA• Acetoacetil-CoA
• Aminoácidos glucogénicos:• Piruvato• a-cetoglutarato• Succinil-CoA• Fumarato• Oxalacetato

Aminoácidos Esenciales

Enfermedades Metabólicas Asociadas• Ausencia de enzimas
cada uno de los aminoácidos de lo contrario se presentan enfermedades asociadas al metabolismo.
• Las personas con algún defecto en las enzimas que participan por ej. en el ciclo de la urea no pueden tolerar una dieta rica en proteínas y deben ser tratadas

Enfermedades Genéticas

Fenilcetonuria (PKU)• No se puede metabolizar la
fenilalanina en el hígado.• Carencia de la enzima:
fenilalanina hidroxilasa. La condición debe ser identificada inmediatamente luego del nacimiento.
• Tratamiento consiste de una dieta baja en Fen y el monitoreo es hasta los 10 años
• Es un defecto genético. Acumulación de fenilalanina o sus metabolitos (fenilpiruvato, fenilactato y fenilacetato) producen retardo mental severo

Albinismo• Fenilalanina se convierte en tirosina.• Tirosina es precursor de la síntesis de melanina, el pigmento oscuro de
la piel y el pelo presente en los melanocitos.• Una deficiencia en la enzima tirosinasa produce inhibición de la
síntesis de melanina y por lo tanto falta de pigmentación.