core curriculum in nephrology - mcmaster university · core curriculum in nephrology...
TRANSCRIPT

Core Curriculum in Nephrology
Am J Kidne
Pathophysiology of Renal Tubular Acidosis: CoreCurriculum 2016
Manoocher Soleimani, MD,1 and Asghar Rastegar, MD 2
From the 1Department of Medicine, University of CincinnatiSchool of Medicine and Veterans Administration, Cincinnati, OH;and 2Department of Medicine, Yale University School of Medicine,New Haven, CT.Received December 25, 2015. Accepted in revised form March
29, 2016.Address correspondence to Manoocher Soleimani, MD,
Department of Medicine, University of Cincinnati School ofMedicine and Veterans Administration, 231 Albert Sabin Way,MSB G163A, Cincinnati, OH 45267-0585 (e-mail: [email protected]) or Asghar Rastegar, MD, Section ofNephrology, Department of Medicine, Yale School of Medicine,333 Cedar St, New Haven, CT 06520 (e-mail: [email protected])� 2016 by the National Kidney Foundation, Inc.0272-6386http://dx.doi.org/10.1053/j.ajkd.2016.03.422
Metabolic acidosis results from either the gain ofan acid or the loss of a base. The former is due
to exogenous or endogenous acid loads resulting inanion gap metabolic acidosis. The latter is due to theloss of a base from either the gastrointestinal orgenitourinary tract, producing nonanion gap orhyperchloremic metabolic acidosis.Renal tubular acidosis (RTA) arises from the kid-
ney’s inability to excrete enough acid or retain enoughbicarbonate (HCO3
-), resulting in a clinical syndromecharacterized by nongap metabolic acidosis, hyper-chloremia, and impaired urinary acidification. In thisCore Curriculum, we briefly summarize the role of thekidney in acid-base homeostasis and discuss clinicalpresentations, diagnoses, and treatments of RTA.
OVERVIEW OF RENAL ACID-BASE HOMEOSTASIS
Total-Body Acid-Base Homeostasis
Metabolism of food particles generates both vola-tile carbonic acid, which is excreted by the lung, andfixed acid, which is generated primarily from meta-bolism of proteins. On a Western diet containing highlevels of animal protein, an adult generates 15,000mEq of volatile acid derived from fat and carbohy-drate combustion and w1 mEq of fixed acid perkilogram of body weight from metabolism of pro-teins. This latter is initially neutralized by the bodybuffers, including HCO3
-, and then excreted by thekidney. The kidney is therefore responsible for theregeneration of lost buffers through excretion of 1mEq of hydrogen ion (H1) per kilogram of bodyweight on a daily basis. It maintains acid-base balanceby the reabsorption of all filtered HCO3
- and theregeneration of HCO3
- lost through metabolism offood particles. Both processes involve secretion ofH1, initially to reabsorb filtered HCO3
- and then togenerate new HCO3
-. The secreted H1 would eithercombine with filtered HCO3
-, resulting in HCO3-
reabsorption, or combine with urinary buffers in thetubular fluid to be excreted as titratable acids andammonium ion (NH4
1), thus leading to HCO3-
regeneration. Net acid excretion (NAE) by the kidneyequals the sum of titratable acids and ammoniumamounts minus the amount of HCO3
-:
NAE 5 titratable acids 1 NH41 2 HCO3
-
The amount of titratable acid is fixed and primarilyreflects the amount of phosphate in urine. In contrast,
y Dis. 2016;-(-):---
the amount of NH41 produced by the kidney varies
greatly, increasing dramatically in acidemic states.Because the amount of titratable acid is fixed, adecrease in NAE is primarily due to either a decreasein NH4
1 excretion or HCO3- loss (see details further
in Curriculum).
Acid-Base Regulation by the Proximal Tubule
Total filtered HCO3- per day is equal to the plasma
HCO3- concentration (24-26 mEq/L) multiplied by
glomerular filtration rate (180-200 L/d) and is esti-mated to be approximately 4,000 to 5,000 mEq/d.Approximately 85% to 90% of filtered HCO3
- isreabsorbed in the proximal tubule, with the rest beingreabsorbed by the thick ascending limb of the loop ofHenle, distal tubule, and collecting ducts. Reabsorp-tion of HCO3
- in the proximal tubule involves H1
secretion primarily by the sodium ion (Na1)/H1
exchanger isoform 3 (NHE3) and the H1-transportingadenosine triphosphatase (H1-ATPase). The secretedH1 reacts with the luminal HCO3
- to form carbonicacid, which rapidly dissociates to carbon dioxide,oxygen, and water. This latter reaction is catalyzed byluminal membrane carbonic anhydrase (CAIV). Car-bon dioxide generated by this reaction freely entersproximal tubule cells and reacts with water to formcarbonic acid, a reaction catalyzed by cytosolic car-bonic anhydrase (CAII). Carbonic acid then dissoci-ates rapidly to form HCO3
- and H1. HCO3- exits
through the basolateral membrane by the sodium bi-carbonate cotransporter (NBCE1), and H1 is againavailable to be secreted into the tubular lumen. Theschematic in Fig 1 depicts the role of apical NHE3
1

Blood
3HCO3-
NBCE1NNB E1CEE-
ATP
ADP + Pi
H+
Lumen
CO2 + H2O
T
+
O
H+
3H3
CAII
Proximal tubule cell
P + Pi
P
+
H+
NHE3NNNNHE33
Na+
Na+
HCO3- + H+
CAIVH2CO3
H2O + CO2 CV
2 H2CO3
Na+/K+-ATPase
3Na+
2K+
H+-ATPase
Figure 1. A schematic of the roles of acid-base transporters and carbonic anhydrases in acid (hydrogen ion [H1]) secretion andbicarbonate (HCO3
-) reabsorption in the kidney proximal tubule. Sodium ion (Na1)/H1 exchanger isoform 3 (NHE3) and H1-transport-ing adenosine triphosphatase (H1-ATPase) are expressed on the apical membrane, whereas the sodium bicarbonate cotransporter(NBCE1) is located on the basolateral membrane of the proximal tubule. Carbonic anhydrase IV (CAIV) and CAII are detected onthe apical membrane and in the cytoplasm, respectively. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate;CCD, cortical collecting duct; DCT, distal convoluted tubule; IMCD, inner medulla collecting duct; K1, potassium ion; OMCD, outermedulla collecting duct; PCT, proximal convoluted tubule; Pi, inorganic phosphate; TAL, thick ascending limb.
Soleimani and Rastegar
and H1-ATPase, basolateral NBCE1, and membraneand cytoplasmic carbonic anhydrases in acid (H1)secretion and HCO3
- reabsorption in the kidneyproximal tubule.
Acid-Base Regulation in the Distal Nephron(Collecting Duct)
The collecting duct has a main role in systemicacid-base homeostasis by fine-tuning acid and baseexcretion. The cortical collecting duct expresses 3distinct cell types: A-intercalated cells, which secreteH1 (acid); B-intercalated cells, which secrete HCO3
-
(base); and principal cells, which reabsorb Na1 andwater and secrete potassium ion (K1). A-intercalatedcells secrete acid (H1) primarily by apicalH1-ATPases (and H1/K1-ATPases), generating newHCO3
- under the control of cytosolic CAII. Thegenerated HCO3
- is transported to the blood in ex-change for chloride ions (Cl-) by the basolateral Cl-/HCO3
- exchangers, including the kidney anionexchanger 1 (AE1). Contrary to A-intercalated cells,which are detected along the length of the cortical andmedullary collecting ducts, B-intercalated cells arepredominantly detected in the cortical collecting ductand are almost absent in the medullary collectingduct. B-intercalated cells secrete HCO3
- into thecortical collecting duct lumen in exchange for the
2
luminal Cl-, primarily by the apical Cl-/HCO3-
exchanger pendrin. The intracellular acid generatedsubsequent to HCO3
- secretion in B-intercalated cellsis transported into blood membrane by the basolateralH1-ATPase.Secreted H1 by A-intercalated cells is buffered by
titratable acids (mainly phosphate) and ammonia(NH3; to generate NH4
1), matching daily acid pro-duction. Although the amount of urinary phosphate isfixed, urinary NH4
1 level varies because it is stimu-lated by systemic acidemia (and hypokalemia). H1
secretion by H1-ATPase is modulated by the activityof the epithelial sodium channel (ENaC) in principalcells and by angiotensin II, aldosterone, and thecalcium sensing receptor. Aldosterone plays a keyrole in H1 secretion by stimulating ENaC andH1-ATPase. Aldosterone deficiency is characterizedby sodium wasting, hyperkalemia, and RTA (seeHyperkalemic RTA section). The schematic in Fig 2shows the localization of acid-base transporters andCAII in the cortical collecting duct and their role inacid-base transport.
Physiology of Ammoniagenesis and HCO3-
Regeneration
NH3generates in the proximal tubule frommetabolismof glutamine through the process of ammoniagenesis.
Am J Kidney Dis. 2016;-(-):---
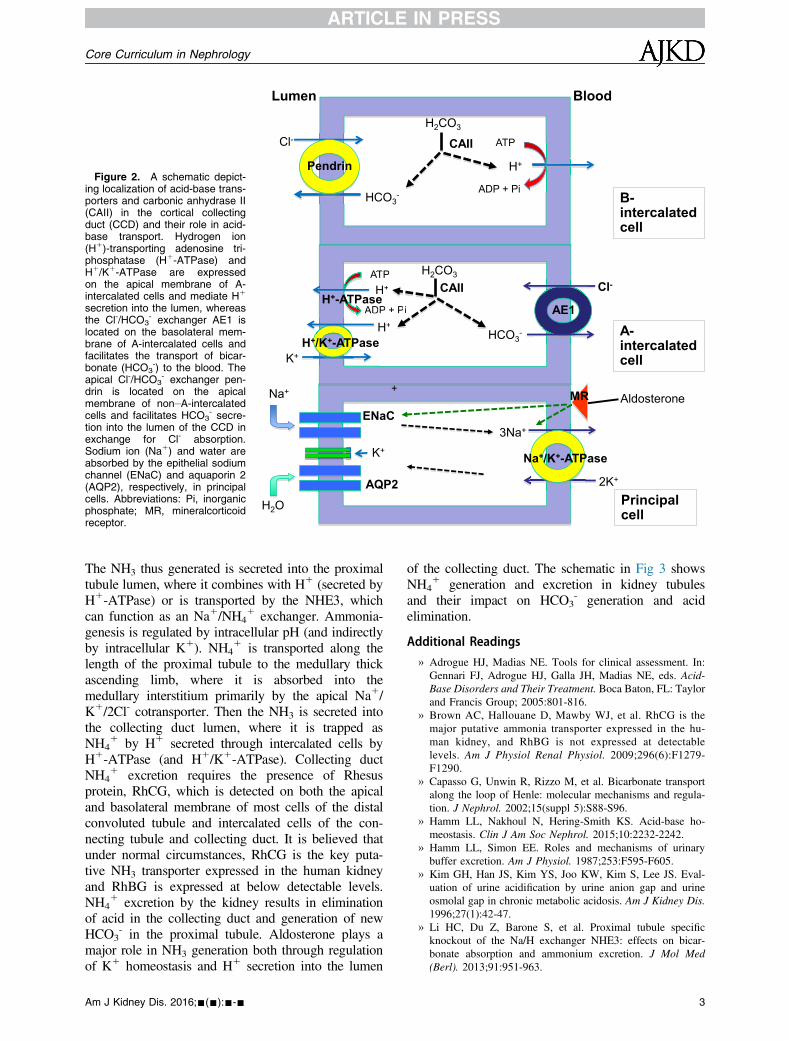
Figure 2. A schematic depict-ing localization of acid-base trans-porters and carbonic anhydrase II(CAII) in the cortical collectingduct (CCD) and their role in acid-base transport. Hydrogen ion(H1)-transporting adenosine tri-phosphatase (H1-ATPase) andH1/K1-ATPase are expressedon the apical membrane of A-intercalated cells and mediate H1
secretion into the lumen, whereasthe Cl-/HCO3
- exchanger AE1 islocated on the basolateral mem-brane of A-intercalated cells andfacilitates the transport of bicar-bonate (HCO3
-) to the blood. Theapical Cl-/HCO3
- exchanger pen-drin is located on the apicalmembrane of non–A-intercalatedcells and facilitates HCO3
- secre-tion into the lumen of the CCD inexchange for Cl- absorption.Sodium ion (Na1) and water areabsorbed by the epithelial sodiumchannel (ENaC) and aquaporin 2(AQP2), respectively, in principalcells. Abbreviations: Pi, inorganicphosphate; MR, mineralcorticoidreceptor.
A-intercalated cell
Lumen Blood
Principal cell
ATP
ADP + Pi
H+
H2O
AQP2
Cl- ATAA P
ADP +
H
HCO3-
AE1
ENaC
K+
+
Cl-
HCO3- B-
intercalated cell
Pendrin H+
ATP
ADP + Pi
K+
H2CO3
H+
H2CO3
-
CAII
H+-ATPase CAII
H
+ Pi+
+
H+/K+-ATPase
Na+
Na+/K+-ATPase
3Na+
2K+
Aldosterone MR
Core Curriculum in Nephrology
The NH3 thus generated is secreted into the proximaltubule lumen, where it combines with H1 (secreted byH1-ATPase) or is transported by the NHE3, whichcan function as an Na1/NH4
1 exchanger. Ammonia-genesis is regulated by intracellular pH (and indirectlyby intracellular K1). NH4
1 is transported along thelength of the proximal tubule to the medullary thickascending limb, where it is absorbed into themedullary interstitium primarily by the apical Na1/K1/2Cl- cotransporter. Then the NH3 is secreted intothe collecting duct lumen, where it is trapped asNH4
1 by H1 secreted through intercalated cells byH1-ATPase (and H1/K1-ATPase). Collecting ductNH4
1 excretion requires the presence of Rhesusprotein, RhCG, which is detected on both the apicaland basolateral membrane of most cells of the distalconvoluted tubule and intercalated cells of the con-necting tubule and collecting duct. It is believed thatunder normal circumstances, RhCG is the key puta-tive NH3 transporter expressed in the human kidneyand RhBG is expressed at below detectable levels.NH4
1 excretion by the kidney results in eliminationof acid in the collecting duct and generation of newHCO3
- in the proximal tubule. Aldosterone plays amajor role in NH3 generation both through regulationof K1 homeostasis and H1 secretion into the lumen
Am J Kidney Dis. 2016;-(-):---
of the collecting duct. The schematic in Fig 3 showsNH4
1 generation and excretion in kidney tubulesand their impact on HCO3
- generation and acidelimination.
Additional Readings
» Adrogue HJ, Madias NE. Tools for clinical assessment. In:Gennari FJ, Adrogue HJ, Galla JH, Madias NE, eds. Acid-Base Disorders and Their Treatment. Boca Baton, FL: Taylorand Francis Group; 2005:801-816.
» Brown AC, Hallouane D, Mawby WJ, et al. RhCG is themajor putative ammonia transporter expressed in the hu-man kidney, and RhBG is not expressed at detectablelevels. Am J Physiol Renal Physiol. 2009;296(6):F1279-F1290.
» Capasso G, Unwin R, Rizzo M, et al. Bicarbonate transportalong the loop of Henle: molecular mechanisms and regula-tion. J Nephrol. 2002;15(suppl 5):S88-S96.
» Hamm LL, Nakhoul N, Hering-Smith KS. Acid-base ho-meostasis. Clin J Am Soc Nephrol. 2015;10:2232-2242.
» Hamm LL, Simon EE. Roles and mechanisms of urinarybuffer excretion. Am J Physiol. 1987;253:F595-F605.
» Kim GH, Han JS, Kim YS, Joo KW, Kim S, Lee JS. Eval-uation of urine acidification by urine anion gap and urineosmolal gap in chronic metabolic acidosis. Am J Kidney Dis.1996;27(1):42-47.
» Li HC, Du Z, Barone S, et al. Proximal tubule specificknockout of the Na/H exchanger NHE3: effects on bicar-bonate absorption and ammonium excretion. J Mol Med(Berl). 2013;91:951-963.
3

G
PCT
TAL
DCT CCD
OMCD
IMCD
Blood
3HCO3-
NBCE1NBCNN EEEE1
3------
ATP
ADP + Pi
H+
Lumen
(H+)
ATPAA
NHE3Na+
Na+
H+-ATPase
Gln
Glu
α-ketoglutarate
G
u
ketogluta
HCO3-
3H
NH3
+ Pi
+)NH4
+
NH3
NH4+
H++
NHH
NH4+
NH4+
NH4++
A-intercalated cell
H+
H+
NH33
NH3
NH3
NH4+NH4+
NNH3
H+
Proximal tubule cell
ATP
ADP + Pi
Figure 3. Generation and excretion of ammonium ion (NH41) in kidney tubules and their impact on bicarbonate (HCO3
-) generationand acid elimination. NH4
1 is primarily generated in the proximal tubule consequent to the metabolism of glutamine. NH41 transport
along the length of the nephron, its absorption in the thick ascending limb, and its secretion into the collecting duct are shown. Abbre-viations: CCD, cortical collecting duct; DCT, distal convoluted tubule; G, glomerulus; Gln, glutamine; Glu, glutamate; IMCD, inner me-dulla collecting duct; NBCE1, sodium bicarbonate cotransporter; OMCD, outer medulla collecting duct; TAL, thick ascending limb.
Soleimani and Rastegar
» Purkerson JM, Schwartz GJ. The role of carbonic anhydrasesin renal physiology. Kidney Int. 2007;71:103-115.
» Sastrasinh S, Tannen RL. Effect of potassium on renal NH3production. Am J Physiol. 1983;244:F383-F391.
» Tannen RL. Relationship of renal ammonia production andpotassium homeostasis. Kidney Int. 1977;11:453-465.
» Wagner CA, Devuyst O, Bourgeois S, Mohebbi N. Regulatedacid-base transport in the collecting duct. Pflugers Arch.2009;458:137-156.
» Weiner ID, Hamm LL. Molecular mechanisms of renalammonia transport. Annu Rev Physiol. 2007;69:317-340.
» Weiner ID, Verlander JW. Renal acidification mechanisms.In: Brenner B, ed. Brenner and Rector’s The Kidney. 9th ed.Philadelphia, PA: W.B. Saunders; 2011:293-325.
RENAL TUBULAR ACIDOSIS
Based on clinical presentation and pathophysiologicmechanism, RTA is classified into proximal (pRTAor type II), distal (dRTA or type I), and hyperkalemic(or type IV) RTA (RTA type III [ie, combined pRTAand dRTA] is not detailed in this Curriculum becauseof its rarity).
Proximal RTA
pRTA is caused by defective HCO3- reabsorption
in the proximal tubule (Figs 1 and 3). This couldbe due to defects in H1 secretion by NHE3or H1-ATPase, impairment of luminal or cytosolic
4
carbonic anhydrase, or defects in the exit of HCO3- by
the Na1/3HCO3- cotransporter NBCE1. pRTA could
also present as a component of Fanconi syndrome dueto generalized impairment of solute reabsorption inthe proximal tubule, manifesting as renal loss ofglucose, amino acids, phosphate, uric acid, and otherorganic anions.
Classification of pRTA
pRTA can present in hereditary or acquired forms.Hereditary pRTA. Hereditary pRTA may manifest
as autosomal recessive, autosomal dominant, or spo-radic forms. Patients with autosomal recessive pRTApresent with short stature, cataracts, and central ner-vous system calcification, and the condition is linkedto mutations in the Na1/3HCO3
- cotransporterNBCE1 (Fig 1). In severe forms of pRTA, serumHCO3
- concentrations , 10 mEq/L and bloodpH , 7.1 have been reported. Although inactivationof NHE3 could result in HCO3
- wasting and pRTA inanimal models, mutations in NHE3 causing pRTA inhumans have not been reported. Mutations in CAIIlead to recessive mixed pRTA and dRTA, or type IIIRTA, with a predominance of distal acidificationdefect. Patients with CAII mutations present withHCO3
- wasting, inability to lower urine pH to ,5.5,
Am J Kidney Dis. 2016;-(-):---

Core Curriculum in Nephrology
and decreased NH41 excretion. The gene(s) involved
in autosomal dominant pRTA has (have) not yetbeen identified. The most common forms of familialFanconi syndrome are due to cystinosis and Wilsondisease. Other inherited causes of pRTA include:� Lowe syndrome (oculocerebrorenal syndrome
[OCRL]), an X-linked disorder characterizedby cataract, mental retardation, and Fanconi-likepRTA, results from mutations in the OCRL gene,encoding a-phosphatidylinositol (4,5)-biphosphatephosphatase (PIP2P);
� Fanconi-Bickel syndrome, an autosomal recessivedisorder characterized by pRTA and impaired uti-lization of glucose and galactose, is due to defectsin monosaccharide transport across the tubularmembranes; and
� Dent disease, an X-linked recessive disorder char-acterized by low-molecular-weight proteinuria,hypercalciuria, nephrocalcinosis, and neph-rolithiasis, results from mutations in either thechloride channel gene CLCN5 or the OCRL gene.
Acquired pRTA. Most common causes includetubular injury by light chains, amyloidosis, multiplemyeloma, autoimmune disorders, toxins such ascadmium or lead, and drugs, including ifosfamide,valproic acid, carbonic anhydrase inhibitors, andvarious antiretrovirals such as tenofovir (especiallywhen administered to patients with human immuno-deficiency virus [HIV] infection who are concomi-tantly receiving protease inhibitors such as ritonaviror reverse transcriptase inhibitors such as didanosine).Acquired pRTA may present as Fanconi syndrome,characterized by glycosuria, aminoaciduria, phos-phaturia, and uricosuria.Clinical Presentation and Diagnosis of pRTA.
Patients with pRTA can be asymptomatic or couldpresent with weakness/paralysis due to severe hypo-kalemia and, in rare cases, with bone pain/fracture dueto osteomalacia. Hyperchloremic metabolic acidosis inpRTA tends to be milder because distal HCO3
- recla-mation is intact and bicarbonaturia disappears whenHCO3
- load falls below the HCO3- tubular maximum
(often at serum HCO3- level of 14-18 mEq/L).
The key laboratory finding is the presence ofhyperchloremic metabolic acidosis with hypokalemiaand variable urinary pH,with alkaline urine pH if serumHCO3
- concentration is above the tubular maximumand pH, 5.3when it is below the tubularmaximum. Ifthe diagnosis of pRTA is not clear, urinary pH shouldbe measured after an acid load with ammonium chlo-ride (0.1 g per kilogram of body weight). In patientswith pRTA, but not dRTA, urine pH decreases to,5.3.Because furosemide also decreases urine pH and iseasier to use than ammonium chloride, measuring urinepH after the use of furosemide has been suggested in
Am J Kidney Dis. 2016;-(-):---
these patients.However, this test has unacceptable ratesof false-positive and -negative results. Anotherapproach is to treat patients with HCO3
- at 1 to 2mEq/kg/d and check serum HCO3
- concentration after2 to 3 weeks. In dRTA, serum HCO3
- concentrationapproaches the reference range, whereas it will remainsignificantly below normal in pRTA and urine will be(or become) markedly alkaline due to HCO3
- wasting.
Treatment of pRTA
The goal is to increase serum HCO3- concentration
as close to normal as possible. However, this is oftenvery difficult due to the decrease in HCO3
- tubularmaximum. Oral HCO3
- at doses of 10 to 15 mEq/kg/dwith potassium supplementation is often used. Hy-drochlorothiazide may be helpful by increasing theHCO3
- tubular maximum; however, hypokalemiamust be prevented with supplemental potassium and/or treated. In patients with Fanconi syndrome, phos-phate supplementation is often required.
Distal RTA
dRTA reflects a failure to reabsorb HCO3- by
intercalated cells in the collecting duct, resulting inpersistent alkaline urine. It is characterized byimpaired acid excretion and the inability to reduceurinary pH to ,5.3 when confronted with sponta-neous acidemia or acid loading. The defect in H1
secretion in the collecting duct leads to reduced NAEsubsequent to decreased NH4
1, titratable acid excre-tion, and HCO3
- wasting. This results in a decrease inserum HCO3
- concentration and generation ofhyperchloremic metabolic acidosis. The abnormalitiesin H1 secretion in the collecting duct are secondary todefects in H1-ATPase, cytosolic CAII, or kidneyAE1. NH4
1 excretion is reduced in dRTA due toimpaired trapping of luminal NH3 in the collectingduct subsequent to defects in H1 secretion. Oneunique feature of dRTA is very low urinary citratelevels. This is due to the increase in reabsorption ofcitrate by the proximal tubule in response to intra-cellular acidosis.
Classification of dRTA
dRTA can be hereditary (primary) or acquired(secondary).Hereditary dRTA. The vast majority of inherited
forms of dRTA are due to defects in AE1 orH1-ATPase. In addition, cytosolic CAII gene muta-tions are associated with a mixed picture of pRTA anddRTA. Mutations in the basolateral Cl-/HCO3
-
exchanger AE1 lead to the impairment of HCO3- ab-
sorption in A-intercalated cells in the collecting duct.Both autosomal recessive and autosomal dominantmutations have been reported. Mutations in apicalH1-ATPase in A-intercalated cells impair H1 sec-retion into the lumen of the collecting duct. Some
5

Box 2. Measuring Urinary Ammonium Excretion
� Because urinary ammonia is not directly measured by the
clinical laboratory, it is estimated by measuring UAG or
urine osmolar gap
� Both calculations only give a qualitative and not a
quantitative measure of urinary ammonia
� UAG
� Urinary cations [Na1 1 K1] – urinary anions [Cl-]
� In patients with hyperchloremic metabolic acidosis
and normal kidneys, UAG would be , 0
� A positive UAG indicates low urinary NH41
� UAG cannot be used if:
- Urine pH. 7, as this would indicate the presence
of bicarbonate in urine
- Other organic anion(s) is (are) present in urine
Soleimani and Rastegar
patients with H1-ATPase mutations present withsensorineural deafness. The mutations in H1-ATPaseare predominantly of the autosomal recessive form.Mutations in CAII impair HCO3
- reabsorption in theproximal tubule and collecting duct (mixed dRTA andpRTA). It also affects bone osteoclasts resulting inosteopetrosis.Acquired dRTA. The most common causes are
autoimmune diseases such as Sjögren syndrome, sys-temic lupus erythematosus, rheumatoid arthritis, andhypergammaglobulinemia; kidney transplantation;sickle cell disease; and drugs including ifosfamide(more commonly causing pRTA than dRTA),amphotericin B, lithium carbonate, and intravenouslyadministered bisphosphonates such as zoledronate. InSjögren syndrome, 3% to 6.5% of patients havecomplete dRTA, whereas up to 33% have incompletedRTA. A small number have combined dRTA andpRTA, including Fanconi syndrome. A variety of an-tibodies against H1-ATPase, kidney AE1 transporter,and CAII have been reported in these patients. Box 1summarizes the causes of classical dRTA (type 1).
Clinical Presentation and Diagnosis
Patients with dRTA often present with signs andsymptoms related to severe hypokalemia, includingproximal muscle weakness, polydipsia, and polyuria.In milder cases, symptoms related to renal calculi maybe the first sign of an abnormality in acid secretion.Laboratory studies show hyperchloremic metabolicacidosis with persistent urine pH . 5.3 often associ-ated with hypokalemia. Patients with dRTA andhyperkalemic RTAs have low urinary NH4
1 (positiveurinary anion gap and low urine osmolal gap),whereas patients with hyperchloremic metabolicacidosis due to gastrointestinal HCO3
- loss (diarrhea)usually have elevated urinary anions (negative urinaryanion gap and high urine osmolal gap of more than300-400 mEq/L). Box 2 shows a summary of urinaryNH4
1 estimation by measuring urinary anion gap andurinary osmolal gap. Figure 4 is a schematic depictingthe differential diagnosis of hyperchloremic acidosis
Box 1. Cause of Classical Distal Renal Tubular Acidosis (Type I)
1. Primary
a. Familial
b. Sporadic
2. Secondary
a. Systemic diseases: Sjogren syndrome, PBS, systemic
lupus erythematosus
b. Nephrocalcinosis: hyperparathyroidism, milk/alkali,
vitamin D
c. Drugs: amphotericin B
d. Tubulointerstitial disease: Obstructive uropathy
e. Miscellaneous
Abbreviation: PBS, primary biliary cirrhosis.
6
(hyperchloremic metabolic acidosis) with respect tothe source of HCO3
- loss (gastrointestinal vs kidney)and the delineating features of serum K1 concentra-tion, urinary NH4
1 excretion, and urine pH in dis-tinguishing various types of hyperchloremicmetabolic acidosis. The pathophysiology of hyper-kalemic RTA (type IV) is discussed in more detail inthe following sections.
Treatment of dRTA
dRTA, in contrast to pRTA, has a relatively smalldaily loss of HCO3
-, often in the range of 1 to 2 mEq/kg/d. The amount of supplemental sodium or potas-sium bicarbonate is therefore 1 to 2 mEq/kg/d. Inchildren, normal growth is dependent on an adequatesupply of HCO3
- and maintenance of normal serumHCO3
- concentration. This often requires an HCO3-
dose of 4 to 8 mEq/kg/d.
Incomplete dRTA
Incomplete dRTA is defined as having normalserum HCO3
- concentration while lacking the abilityto acidify urine when challenged with an acid-loadingtest. This should be considered in patients with idio-pathic renal calculi with alkaline urine, as wellas patients with Sjögren syndrome, children withposterior urethral valve disease, and patientswith amphotericin toxicity. Kidney stones are due
� Urinary osmolal gap
� Measured urinary osmolality minus (2) calculated
urinary osmolality, which can be written as [(urinary
Na1 1 urinary K1) 3 2 1 urinary urea nitrogen/2.8 1urinary glucose/18]
� Normal osmolal gap is usually 80-150 mEq/L
� An elevated urine osmolal gap indicates high urine
NH41
� Urinary osmolal gap cannot be used if urine contains
another neutral compound such as mannitol
� Urinary osmolal gap is not affected by the presence of
other anions
Note: Estimation of urinary NH41 excretion can be used as a
tool to differentiate various types of hyperchloremic metabolic
acidosis.
Abbreviations: NH41, ammonium ion; UAG, urinary anion gap.
Am J Kidney Dis. 2016;-(-):---

Figure 4. The differential diagnosis of hyper-chloremic acidosis (hyperchloremic metabolicacidosis). The pathogenesis of renal tubularacidosis (RTA) with respect to the source of bi-carbonate (HCO3
-) loss (gastrointestinal [GI] vskidney) and the delineating features of serum po-tassium ion (K1) and urinary ammonium ion(NH4
1) excretion (UNH41) and urine pH (UpH).
Caveat: Urinary NH41 is variable in proximal
RTA.
Hyperchloremic acidosis
GI loss of bicarbonate
Defect in bicarbonate regeneration (type IV)
Renal loss of bicarbonate
UNH4+
K+
Proximal RTA (RTA type II)
UpH > 5.3 UpH = variable
UpH < 5.3
Distal RTA (RTA type I)
UNH4+
K+
Core Curriculum in Nephrology
primarily to low urinary citrate levels combined witha high urine pH (Fig 5).
Kidney Stones and Nephrocalcinosis in RTA
Kidney stones and nephrocalcinosis are primarilyseen in dRTA. The pathogenesis includes low uri-nary citrate, high urinary calcium, and high urinarypH favoring calcium phosphate precipitation. Theuse of carbonic anhydrase inhibitors (eg, acetazol-amide, topiramate, and zoniramate) results in asimilar scenario and is associated with an increasedincidence of kidney stones. Patients with pRTA havenormal urinary citrate levels and their urinary pH iscommonly in an acidic range, protecting againststone formation.
Mechanism of Hypokalemia in pRTA and dRTA
Hypokalemia is a common feature of both pRTAand dRTA. The degree of hypokalemia varies and isoften more severe in patients with dRTA. In heredi-tary dRTA, hypokalemia is a cardinal clinical feature,independent of the genetic mutation, although it ismore severe in autosomal recessive forms due to amutation in subunits of the H1-ATPase pump. Themechanism of hypokalemia is only partially
Figure 5. Clinical and laboratory features,pathogenesis, and major complications of incom-plete distal renal tubular scidosis (idRTA). Abbrevi-ation: NH3, ammonia.
Clinicalfe
• Normal electto lower urine• Initial phase:low urine citra• Late phase: low urine citra• Complicationnephrocalcinokalemia• Etiology: AmSjögren syndridiopathic ston
Am J Kidney Dis. 2016;-(-):---
understood and involves an increase in aldosteronelevels due to sodium wasting, as well as metabolicacidosis. In amphotericin B–induced dRTA, both theimpairment in H1 secretion and K1 loss are due to adefect in membrane permeability. However, thisdefect is unique to amphotericin B toxicity and hasnot been shown in other forms of dRTA. In pRTA,treatment with HCO3
- supplement will result inmarked bicarbonaturia. An increase in urinary HCO3
-
excretion may obligate an increase in K1 secretion,thereby worsening the hypokalemia. Please refer toFig 4 for a summary of features of various typesof hyperchloremic metabolic acidosis with regardto serum K1 level, urine pH, and urine NH4
1
excretion.
Additional Readings
» Batlle D, Moorthi KM, Schlueter W, Kurtzman N. Distalrenal tubular acidosis and the potassium enigma. SeminNephrol. 2006;26:471-478.
» Batlle DC, Hizon M, Cohen E, Gutterman C, Gupta R. Theuse of the urinary anion gap in the diagnosis of hyper-chloremic metabolic acidosis. N Engl J Med. 1988;318:594-599.
» Batlle DC, Mozes MF, Manaligod J, et al. The pathogenesisof hyperchloremic metabolic acidosis associated with kidneytransplantation. Am J Med. 1981;70:786-796.
& laboratory aturesrolyte with inability pH < 5.5 high urine NH3 with teLow urine NH3 and te: Nephrolithiasis/ sis and hypo-
photericin B, ome, endemic RTA, e formers
Pathogenesis
Decrease urine citrate
Nephrocalcinosis/nephrolithiasis
Interstitial damage
Decrease urine NH3
Complete RTA
Intracellular acidosis
7

Soleimani and Rastegar
» Borthwick KJ, Karet FE. Inherited disorders of theH1-ATPase. Curr Opin Nephrol Hypertens. 2002;11:563-568.
» Both T, Hoorn EJ, Zietse R, et al. Prevalence of distal renaltubular acidosis in primary Sjogren’s syndrome. Rheuma-tology. 2015;54:933-939.
» Brenner RJ, Spring DB, Sebastian A, et al. Incidence ofradiographically evident bone disease, nephrocalcinosis, andnephrolithiasis in various types of renal tubular acidosis. NEngl J Med. 1982;307:217.
» Defranco PE, Haragsim L, Schmitz PG, et al. Absence ofvacuolar H-ATPase pump in the collecting duct of a patientwith hypokalemic distal renal tubular acidosis and Sjogren’ssyndrome. J Am Soc Nephrol. 1995;6:295-301.
» Goldfarb DS. A woman with recurrent calcium phosphatekidney stones. Clin J Am Soc Nephrol. 2012:7:1172-1178.
» Hamm LL. Renal handling of citrate. Kidney Int.1990;38:728.
» Han JS, Kim GH, Kim J, et al. Secretory-defect distal renaltubular acidosis is associated with transporter defect in H(1)-ATPase and anion exchanger-1. J Am Soc Nephrol.2002;13:1425-1432.
» Haque SK, Ariceta G, Batlle D. Proximal renal tubularacidosis: a not so rare disorder of multiple etiologies. NephrolDial Transplant. 2012;27:4273-4287.
» Igarashi T, Sekine T, Inatomi J, Seki G. Unraveling themolecular pathogenesis of isolated proximal renal tubularacidosis. J Am Soc Nephrol. 2002;13:2171-2177.
» Karet FE. Inherited distal renal tubular acidosis. J Am SocNephrol. 2002;13:2178-2184.
» Karet FE, Gainza FJ, Gyory AZ, et al. Mutations in thechloride bicarbonate exchanger gene AE1 cause auto-somal dominant but not autosomal-recessive distal renaltubular acidosis. Proc Natl Acad Sci U S A. 1998;95:6337-6342.
» Quigley R. Proximal renal tubular acidosis. J Nephrol.2006;19(suppl 9):S41-S45.
» Ren H, Wang WM, Chen XN, et al. Renal involvement andfollowup of 130 patients with primary Sjögren’s syndrome. JRheumatol. 2008;35:278-284.
» Rodríguez-Soriano J, Vallo A. Renal tubular acidosis.Pediatr Nephrol. 1990;4:268-275.
» Sebastian A, McSherry E, Morris RC Jr. Renal potassiumwasting in renal tubular acidosis (RTA). J Clin Invest.1971;50:667.
» Sly WS, Whyte MP, Sundaram V, et al. Carbonic anhydraseII deficiency in 12 families with the autosomal recessivesyndrome of osteopetrosis with renal tubular acidosisand cerebral calcification. N Engl J Med. 1985;313:139-145.
» Soleimani M. SLC26 Cl-/HCO3 exchangers in the kidney:roles in health and disease. Kidney Int. 2013;84:657-666.
» Takemoto F, Hoshino J, Sawa N. Autoantibodies againstcarbonic anhydrase II are increased in renal tubular acidosisassociated with Sjögren syndrome. Am J Med. 2005;118:181-184.
» Tannen RL, Falls WF, Brackett NC. Incomplete renal tubularacidosis: some clinical and physiological features. Nephron.1975;15:111-123.
» Walsh SB, Shirley DG, Wrong OM, Unwin RJ. Urinaryacidification assessed by simultaneous furosemide and flu-drocortisone treatment: an alternative to ammonium chloride.Kidney Int. 2007;71:1310-1316.
» Welch BJ, Graybeal D, Moe OW, Maalouf NM, Sakhaee K.Biochemical and stone-risk profiles with topiramate treat-ment. Am J Kidney Dis. 2006:48:555-563.
» Wrong O, Davies HE. The excretion of acid in renal disease.Q J Med. 1959;28:259-313.
8
Hyperkalemic RTA
Hyperkalemic RTA (type IV) is due to a defect inregeneration of HCO3
- secondary to lack of adequateurinary NH4
1. The most common cause of lowurinary NH4
1 in hyperkalemic RTA is hypo-aldosteronism in conjunction with hyperkalemia.Hypoaldosteronism should be considered in allpatients with persistent hyperkalemia for whomthere is no obvious cause (eg, kidney failure, useof potassium supplements, or potassium-sparingdiuretics). A hypoaldosterone state could be due tohyporeninism seen in patients with kidney disease orfrom a defect in the renin-angiotensin-aldosteronepathway. In hyperkalemic RTA with normal orhigh aldosterone levels, the defect is in responseto aldosterone; for example, patients on ENaCblockers. Hyperkalemia can impair NH4
1 excretionby 2 major mechanisms: intracellular alkalosisdue to the entry of K1 into proximal tubule cellsin exchange for Na1 and H1, inhibiting ammo-niagenesis, or a decrease in medullary NH4
1
absorption through inhibition of its reabsorptionin the thick ascending limb of loop of Henleby the Na1/K1/2Cl- cotransporter, where K1
and NH41 compete for the same site on this
transporter.
Classification of Hyperkalemic RTA
Hyperkalemic RTA can be inherited or acquired.Hereditary hyperkalemic RTA. Inherited hypo-
aldosteronism is due to a decrease in aldosteronesynthesis, such as in congenital isolated hypo-aldosteronism or pseudohypoaldosteronism type 2(Gordon syndrome), due to an increase in sodiumchloride absorption in the distal convoluted tubuleresulting in secondary hypoaldosteronism. It couldalso be due to resistance to aldosterone action as seen intype 1 pseudohypoaldosteronism.Acquired hyperkalemic RTA. Acquired hypo-
aldosteronism may be secondary to hyporeninism,which is most commonly seen in patients with mildto moderate chronic kidney disease due to diabeticnephropathy or chronic interstitial nephritis. Primaryadrenal insufficiency can result from autoimmuneadrenalitis, infectious adrenalitis (eg, HIV), and otherdisorders. Hypoaldosteronism may also present inseverely ill patients due to unknown mechanisms.Obstructive uropathy may present with hyperkalemicRTA due to impaired H1 and K1 secretion in thecollecting duct. Urinary pH in these patients maybe .5.5 despite the presence of acidemia. Drugsare a major cause of acquired hyperkalemic RTA(Fig 6). The major drugs and mechanisms resultingin acquired hyperkalemic RTA are summarized asfollows:
Am J Kidney Dis. 2016;-(-):---

Core Curriculum in Nephrology
� Potassium-sparing diuretics: these drugs causehyperkalemic RTA by either blocking the action ofaldosterone on the collecting tubule cells throughcompeting for the aldosterone receptor (spi-ronolactone or eplerenone) or inhibiting the ENaC(amiloride)
� Antibiotics: 2 antibiotics, trimethoprim and pent-amidine, can cause hyperkalemia by inhibitingENaC
� Nonsteroidal anti-inflammatory drugs: nonsteroidalanti-inflammatory drugs interfere with the secretionof renin and also impair angiotensin II–inducedrelease of aldosterone
� Calcineurin inhibitors: both cyclosporine andtacrolimus can cause hyperkalemia by inducingaldosterone resistance. The effect of tacrolimus isthrough activation of the thiazide-sensitive Na1/Cl-
cotransporter and inhibition of the renal outermedullary K1 channel (ROMK) in the distalnephron
� Angiotensin inhibitors: angiotensin-convertingenzyme inhibitors, angiotensin II receptor blockers,
Figure 6. Pathogenesis of hyperkalemia. Medications, diseasespotassium ion (K1) homeostasis. Abbreviations: Na1, sodium ion;Palmer (“A Physiologic-Based Approach to the Evaluation of a Patieof the National Kidney Foundation.
Am J Kidney Dis. 2016;-(-):---
and renin inhibitors can cause hyperkalemia byreducing aldosterone synthesis
� Heparin and low-molecular-weight heparin:can cause hyperkalemia by inhibiting adrenalgland release of aldosterone from the zonaglomerulosa
Diagnosis of Hyperkalemic RTA
Diagnosis of hyperkalemic RTA is usuallystraightforward and based on the initial clinical andlaboratory presentation. However, in patients withmore complex cases, further studies are done todocument low NH4
1 and K1 excretion. This includesestimating urinary NH4
1 excretion by measuring uri-nary anion and/or urinary osmolal gaps (Box 2). UrinepH should be appropriately acidic (pH , 5.5),although pH. 5.5 is seen in obstructive uropathy.Low urinary K1 excretion can be assessed by atranstubular K1 gradient or urinary potassium-creatinine ratio. To establish the cause of hyper-kalemic RTA, specific causes including drugs shouldbe sought. When indicated, random serum renin and
, and the renin-angiotensin II-aldosterone axis and their role inNSAID, nonsteroidal anti-inflammatory drug. Reproduced fromnt With Hyperkalemia” AJKD 2010;56:387-393) with permission
9

Proximal RTA (II) Distal RTA (I) Type IV RTA
Urine pH: <5.2 >7.0 <5.2 >5.5 >5.5 <5.2
Early stage Late stage Early stage Late stage
[K+]: Osteomalacia:
Nephrocalcinosis: Urine citrate: Urine NH4
+:
Normal Absent Absent Normal Normal
Low Present Absent Normal
Low/?Normal
Low Present Present
Low Low
Normal/high Absent Absent Normal
Low
Normal
Figure 7. Pathogenesis, classification, and clinical presentation of renal tubular acidosis (RTA). The pathogenesis and contrastingfeatures of proximal RTA (type II), distal RTA (type I), and hyperkalemic RTA (type IV) are depicted.
Box 3. Long-term Treatment of Hyperkalemia in Patients With
Hyperkalemic Renal Tubular Acidosis
1. Discontinue all drugs affecting potassium
2. Restrict dietary potassium
3. Control hyperglycemia
4. Treat metabolic acidosis
5. Treat volume depletion
6. Use loop diuretics
7. Mineralocorticoids
8. Kayexalate
Soleimani and Rastegar
aldosterone levels are helpful in establishing thediagnosis of hypoaldosteronism, with or withouthyporeninism. Urinary obstruction should always beconsidered when other more common causes havebeen ruled out. Figure 7 summarizes the distinctivefeatures of pRTA (type II), dRTA (type I), andhyperkalemic RTA (type IV) with respect to theirpathogenesis and emphasis on the contrasting pres-ence of nephrocalcinosis and generation of kidneystones, urine citrate, and NH4
1 excretion.
Treatment of Hyperkalemic RTA
The goal of the treatment initially is to normalizeserum K1 concentration because this may improvemetabolic acidosis by increasing urinary NH3
responsible for buffering secreted H1, as well as byenhancing HCO3
- generation in the proximal tubuleby enhanced glutamine metabolism (Fig 3). If this isinadequate, attempts should be made to improvemetabolic acidosis. Box 3 discusses long-termtreatment of hyperkalemia in patients with hyper-kalemic RTA.
Additional Readings
» Arruda JA, Batlle DC, Sehy JT, Roseman MK,Baronowski RL, Kurtzman NA. Hyperkalemia and renalinsufficiency: role of selective aldosterone deficiency andtubular unresponsiveness to aldosterone. Am J Nephrol.1981;1:160-167.
» Batlle DC. Hyperkalemic hyperchloremic metabolicacidosis associated with selective aldosterone deficiencyand distal renal tubular acidosis. Semin Nephrol. 1981;1:260-273.
10
» Batlle DC, Arruda JA, Kurtzman NA. Hyperkalemic distalrenal tubular acidosis associated with obstructive uropathy. NEngl J Med. 1981;304:373-380.
» Batlle DC, Itsarayoungyen K, Arruda JA. Hyperkalemichyperchloremic metabolic acidosis in sickle cell hemoglo-binopathies. Am J Med. 1982;72:188-192.
» DuBose TD Jr. Molecular and pathophysiologic mechanismsof hyperkalemic metabolic acidosis. Trans Am Clin ClimatolAssoc. 2000;111:122-133, discussion 133-124.
» Furgeson SB, Linas S. Mechanisms of type I and type IIpseudohypoaldosteronism. J Am Soc Nephrol. 2010;21:1842-1845.
» Karet FE. Mechanisms in hyperkalemic renal tubularacidosis. J Am Soc Nephrol. 2009;20:251-254.
» Kutyrina IM, Nikishova TA, Tareyeva IE. Effects of heparin-induced aldosterone deficiency on renal function in patientswith chronic glomerulonephritis. Nephrol Dial Transplant.1087;2:219-223.
» Perazella MA, Rastegar A. Disorders of potassium andacid-base metabolism in association with renal disease.In: Schrier RW, Coffman TM, Falk RJ, Molitoris BA, Niel-son EJ, eds. Schrier Diseases of the Kidney.Philadelphia, PA: Lippincott Williams & Wilkens &Wolters Kluwer; 2013:2082-2116.
» Sastrasinh S, Tannen RL. Effect of potassium on renal NH3
production. Am J Physiol. 1983;244:F383-F391.
Am J Kidney Dis. 2016;-(-):---

Core Curriculum in Nephrology
» Schambelan M, Sebastian A, Biglieri EG. Prevalence, path-ogenesis, and functional significance of aldosterone defi-ciency in hyperkalemic patients with chronic renalinsufficiency. Kidney Int. 1980;17:89-101.
» Sebastian A, Schambelan M, Lindenfeld S. Amelioration ofmetabolic acidosis with fludrocortisone therapy in hypo-reninemic hypoaldosteronism. N Engl J Med. 1977;197:576-583.
» Tannen RL. Relationship of renal ammonia productionand potassium homeostasis. Kidney Int. 1977;11:453-465.
Am J Kidney Dis. 2016;-(-):---
» Velazquez H, Perazella MA, Wright FS, Ellison DH. Renalmechanism of trimethoprim induced hyperkalemia. AnnIntern Med. 1993;119:296-301.
ACKNOWLEDGEMENTSSupport: None.Financial Disclosure: The authors declare that they have no
relevant financial interests.Peer Review: Evaluated by 3 external reviewers, the Education
Editor, and the Editor-in-Chief.
11