controlling false discoveries in genetic studies
TRANSCRIPT

American Journal of Medical Genetics Part B (Neuropsychiatric Genetics) 147B:637–644 (2008)
Review ArticleControlling False Discoveries in Genetic StudiesEdwin J.C.G. van den Oord1,2*1Center for Biomarker Research and Personalized Medicine, Medical College of Virginia, Virginia Commonwealth University,Richmond, Virginia2Virginia Institute for Psychiatric and Behavioral Genetics, Richmond, Virginia
A false discovery occurs when a researcher con-cludes that a marker is involved in the etiology ofthe disease whereas in reality it is not. In geneticstudies the risk of false discoveries is very highbecause only few among the many markers thatcan be tested will have an effect on the disease. Inthis article, we argue that it may be best to usemethods for controlling false discoveries thatwould introduce the same ratio of false discov-eries divided by all rejected tests into the litera-ture regardless of systematic differences betweenstudies. After a brief discussion of traditional‘‘multiple testing’’ methods, we show that methodsthat control the false discovery rate (FDR) may bemore suitable to achieve this goal. These FDRmethods are therefore discussed in more detail.Instead of merely testing for main effects, it maybe important to search for gene–environment/covariate interactions, gene–gene interactions orgenetic variants affecting disease subtypes. Inthe second section, we point out the challengesinvolved in controlling false discoveries in suchsearches. The final section discusses the role ofreplication studies for eliminating false discov-eries and the complexities associated with thedefinition of what constitutes a replication andthe design of these studies. � 2007 Wiley-Liss, Inc.
KEY WORDS: false discoveries; genome-wide asso-ciation studies; multiple hypothesistesting; FDR; data mining; multi-stage designs
Please cite this article as follows: van den Oord EJCG.2008. Controlling False Discoveries in Genetic Studies.Am J Med Genet Part B 147B:637–644.
INTRODUCTION
A false discovery occurs when a researcher concludes that amarker is involved in the etiology of the disease whereas in
reality it is not. In genetic studies the risk of a false discovery isvery high because only few among all markers that can betested will have an effect on the disease. Indeed, it has beenspeculated that 19 out of every 20 marker-disease associationscurrently reported in the literature are false [Colhoun et al.,2003]. Phenomena such as population stratification play a rolebut failure to exclude chance is the main cause of all these falsediscoveries.
Proper methods for controlling false discoveries are impor-tant because they can prevent that a lot of time and resourcesare spend on leads that will eventually prove irrelevant andavoid a loss of confidence in research when many publicized‘‘discoveries’’ are followed by non-replication. These methodsmay become even more important considering it has recentlybecome possible to screen hundreds of thousands to a millionsingle nucleotide polymorphisms (SNPs) across the wholegenome for their association with a disease. Without propercontrol, the number of false discoveries will be proportional tothe number of markers tested and the literature would beflooded with false discoveries. The question of how to bestcontrol false discoveries is therefore appropriate and timely.
In the first section of this article we focus on significancetesting. We argue that it may be best to use a method thatwould produce the same ratio of false discoveries divided by allrejected tests regardless of systematic differences betweenstudies. This would ensure that in the long run, we obtain adesired ratio of false discoveries to all reported discoveries inthe literature. After a brief discussion of traditional ‘‘multipletesting’’ methods, we show that methods that control the falsediscovery rate (FDR) may be more suitable to achieve this goal.These FDR methods are therefore discussed in more detail.Instead of merely testing for main effects, it may be importantto search for gene–environment/covariate interactions, gene–gene interactions or genetic variants affecting disease sub-types. In the second section, we point out the challengesinvolved in controlling false discoveries in such searches. Thecontrol of false discoveries is not solely a data analysis problemand in the final section we argue that (the theory of) adaptivemultistage designs may present advantages in the search forgenetic variants affecting complex diseases.
SIGNIFICANCE TESTING
Significance testing typically starts with calculating P-values for each marker. If the calculated P-value is smallerthan a threshold P-value the null-hypothesis, assuming thatthe marker has no effect, is rejected and the test is calledsignificant. A Type I error is the error of rejecting the null-hypothesis when it is true. This results in a false discovery orfalse positive.
Controlling the Family Wise Error Rate
Traditional approaches for controlling false discoveriesattempt to maintain a desired probability that a studyproduces one or more false discoveries (see supplementalmaterial for precise definitions of error rates discussed in thisarticle.). This probability depends on the number of markers
This article contains supplementary material, which may beviewed at the American Journal of Medical Genetics websiteat http://www.interscience.wiley.com/jpages/1552-4841/suppmat/index.html.
Grant sponsor: US National Institute of Mental Health; Grantnumber: R01 MH065320.
*Correspondence to: Edwin J.C.G. van den Oord, MedicalCollege of Virginia, Virginia Commonwealth University, P.O. Box980533, Richmond, VA 23298-0533.E-mail: [email protected]
Received 19 June 2007; Accepted 18 September 2007
DOI 10.1002/ajmg.b.30650
� 2007 Wiley-Liss, Inc.

tested. For instance, if a single test is performed using athreshold P-value of 0.05, the probability of a false discovery is5% if the marker has no effect. However, if 100,000 markerswithout effects are tested using the same 0.05 threshold, theprobability of one or more false discoveries is close to one andthe study may produce about 100,000� 0.05¼ 5,000 falsediscoveries. To counteract this effect of performing multipletests, the threshold P-value needs to be adjusted. In theBonferroni correction, for example, the corrected P-valuethreshold equals the desired probability of producing one ormore false discoveries divided by the number of tests carriedout. Regardless of the number of tests, such as correction wouldensure that less then one out of every 20 studies produces oneor more false discoveries.
Technically speaking traditional methods control the so-called family wise error-rate (FWE, these methods control theerror rate for the whole set of ‘‘family’’ of tests). In the context ofgenome-wide scans this has been labeled the genome wiseerror-rate [Lander and Kruglyak, 1995; Risch and Merikan-gas, 1996]. Although the above discussed single step Bonfer-roni correction is probably the most well known procedure inthis class, it controls the FWE too conservatively at a levelsmaller than a thereby sacrificing statistical power. The Sidakcorrection [Sidak, 1967] gives exact control of the FWE whennone of the markers have an effect and are independent. Ifsome of the markers have an effect, step-wise procedures aregenerally preferable. The idea is that once one of the nullhypotheses is rejected it cannot any longer be considered true.We can therefore continue with correcting by a factor (m� 1)rather than m. Holm’s step-down procedure [Holm, 1979] wasone of the first step-wise procedures, but more powerfulvariants now exist [Hochberg and Benjamini, 1990; Dunnetand Tamhane, 1992]. In GWAS where the number of tests isvery large and the number of true effects relatively very small,the use of the Sidak correction or a step-wise procedures isunlikely to have a substantial impact on the number of teststhat are declared significant. The fact that control of the FWE issensitive to correlated tests may present to biggest challenge.To illustrate the impact of correlated tests, assume 100 per-fectly correlated tests. Whereas no correction would be neededbecause essentially only one independent test is performed, theBonferroni correction would still divide the significance levelby the number of tests carried out. Step-wise methods thataccount for such correlated tests are most powerful. In theseinstances, re-sampling [Westfall and Young, 1993] or alter-native methods [Dudbridge and Koeleman, 2004; Lin, 2004;Jung et al., 2005] for drawing repeated samples from the givendata or population suggested by the data may help to avoidmaking assumptions about the joint distribution of the teststatistics under the null hypothesis and produce more accuratecontrol of the FWE.
The False Discovery Rate
Rather than controlling the probability that a studyproduces one or more false discoveries, it can be argued thatit may be better to use a method that would produce the sameratio of false discoveries divided by all rejected tests regardlessof systematic differences between studies. This would ensurethat in the long run, we obtain a desired ratio of falsediscoveries to all reported discoveries in the literature. Thisratio is called the marginal FDR [Tsai et al., 2003]. Themarginal FDR can also be interpreted as the probability that arandomly selected discovery from the literature is false. Inthese cases, it is labeled as the false positive report probability[Thomas and Clayton, 2004; Wacholder et al., 2004] or,following Morton [1955], the proportion of false positives[Fernando et al., 2004]. Although in this article, I confinemyself to a more frequentist approach to the false discovery
issue, it should also be noted that to a certain extent FDR alsoallows you to be a frequentist and Bayesian at the same time[Efron and Tibshirani, 2002].
The marginal FDR is closely related to indices such asBenjamini and Hochberg’s [1995] FDR and Storey’s [2002]positive false discovery rate (pFDR). Loosely speaking, themarginal FDR is a theoretical construct that represents anideal goal. The FDR or pFDR can be viewed as tools thatresearchers can use in practice to make decisions about whichtests to call significant to achieve that goal. For sake ofsimplicity, however, we will use the term (marginal) FDR fornow and explain some of the differences between thesemeasures below.
The FDR is not merely another statistical technique anddiffers in fundamental ways from the FWE. First, the FWEfocuses exclusively on the risk of false discoveries. Because thisrisk is high in a genome-wide association study (GWAS) withsay 500k markers, large studies will be heavily penalized viavery small threshold P-values. However, large studies will notonly produce more false discoveries, they are also likely todiscover more true positives. The FDR ‘‘rewards’’ large studiesfor finding more true discoveries by focusing on the proportionof false discoveries divided by all rejected tests (including falsebut also true discoveries). Considering true positives maymake sense in the context of finding genetic variants forcomplex diseases. That is, due to small effect sizes, the power todetect genes is already modest. Instead of further sacrificingpower, it may be better to allow an occasional false discovery toimprove the chances of finding effects. Furthermore, becausethere will be multiple genes with small effects the consequen-ces of a false discovery are not that severe. This would, forexample, be different for single gene disorders where adiscovery implies the strong claim that one has found thecause, which has important scientific and clinical implications.
A second difference is that in contrast to the FWE, thenumber of tests that are performed are not important for thecontrol of the (marginal) FDR. Instead, an important param-eter is p0, which can either be interpreted as the proportion ofmarkers without effect on the disease or as the probability thata randomly selected marker has no effect. The higher theproportion of markers without effect, the more likely it is that asignificant result is a false discovery. This makes intuitivesense: if p0 is one all discoveries are false, whereas if p0 is zeronone of the discoveries are false. A higher p0 therefore implies alower threshold P-value for obtaining the same FDR.
From a theoretical perspective, it seems more sensible tobase the control of false discoveries on p0 rather than thenumber of tests carried out. For example, assume that 100,000researchers each test a single marker. Because each researcherperforms only one test, from his or her perspective nocorrection for multiple testing is necessary. However, if allsignificant results were published the researchers togetherwould introduce 100,000� 0.05¼ 5,000 false discoveries intothe literature. Furthermore, assume that one of the research-ers would have had the budget to type all 100,000 markers. Inthis case, s/he would then have applied a correction for multipletesting and instead of 500 there would probably not be a singlefalse discovery. The basic problem is that the number of tests isarbitrary depending on factors such as budget, publicationstrategy, and genotyping capacity. It can therefore not be usedto control the accumulation of false discoveries in the literatureat a desired level. In contrast, parameter p0 is not arbitrary andprovides a better basis for applying similar standards todifferent studies which is needed for controlling this accumu-lation. Thus, it is the fact that p0 is close to one in geneticstudies rather than the number of tests that creates the highrisk for false discoveries.
A numerical example. In Figure 1 we demonstrate therelation between the FDR and FWE numerically assuming
638 van den Oord
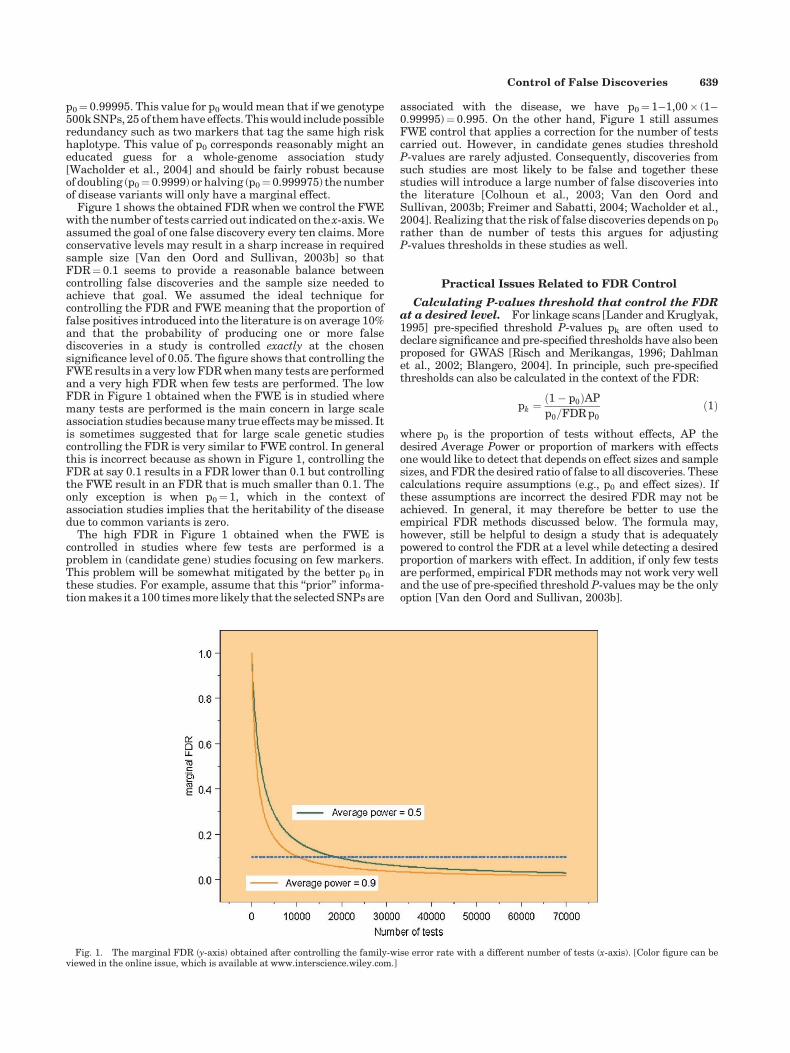
p0¼ 0.99995. This value for p0 would mean that if we genotype500k SNPs, 25 of them have effects. This would include possibleredundancy such as two markers that tag the same high riskhaplotype. This value of p0 corresponds reasonably might aneducated guess for a whole-genome association study[Wacholder et al., 2004] and should be fairly robust becauseof doubling (p0¼ 0.9999) or halving (p0¼ 0.999975) the numberof disease variants will only have a marginal effect.
Figure 1 shows the obtained FDR when we control the FWEwith the number of tests carried out indicated on the x-axis. Weassumed the goal of one false discovery every ten claims. Moreconservative levels may result in a sharp increase in requiredsample size [Van den Oord and Sullivan, 2003b] so thatFDR¼ 0.1 seems to provide a reasonable balance betweencontrolling false discoveries and the sample size needed toachieve that goal. We assumed the ideal technique forcontrolling the FDR and FWE meaning that the proportion offalse positives introduced into the literature is on average 10%and that the probability of producing one or more falsediscoveries in a study is controlled exactly at the chosensignificance level of 0.05. The figure shows that controlling theFWE results in a very low FDR when many tests are performedand a very high FDR when few tests are performed. The lowFDR in Figure 1 obtained when the FWE is in studied wheremany tests are performed is the main concern in large scaleassociation studies because many true effects may be missed. Itis sometimes suggested that for large scale genetic studiescontrolling the FDR is very similar to FWE control. In generalthis is incorrect because as shown in Figure 1, controlling theFDR at say 0.1 results in a FDR lower than 0.1 but controllingthe FWE result in an FDR that is much smaller than 0.1. Theonly exception is when p0¼ 1, which in the context ofassociation studies implies that the heritability of the diseasedue to common variants is zero.
The high FDR in Figure 1 obtained when the FWE iscontrolled in studies where few tests are performed is aproblem in (candidate gene) studies focusing on few markers.This problem will be somewhat mitigated by the better p0 inthese studies. For example, assume that this ‘‘prior’’ informa-tion makes it a 100 times more likely that the selected SNPs are
associated with the disease, we have p0¼ 1–1,00� (1–0.99995)¼ 0.995. On the other hand, Figure 1 still assumesFWE control that applies a correction for the number of testscarried out. However, in candidate genes studies thresholdP-values are rarely adjusted. Consequently, discoveries fromsuch studies are most likely to be false and together thesestudies will introduce a large number of false discoveries intothe literature [Colhoun et al., 2003; Van den Oord andSullivan, 2003b; Freimer and Sabatti, 2004; Wacholder et al.,2004]. Realizing that the risk of false discoveries depends on p0
rather than de number of tests this argues for adjustingP-values thresholds in these studies as well.
Practical Issues Related to FDR Control
Calculating P-values threshold that control the FDRat a desired level. For linkage scans [Lander and Kruglyak,1995] pre-specified threshold P-values pk are often used todeclare significance and pre-specified thresholds have also beenproposed for GWAS [Risch and Merikangas, 1996; Dahlmanet al., 2002; Blangero, 2004]. In principle, such pre-specifiedthresholds can also be calculated in the context of the FDR:
pk ¼ ð1 � p0ÞAP
p0=FDR p0
ð1Þ
where p0 is the proportion of tests without effects, AP thedesired Average Power or proportion of markers with effectsone would like to detect that depends on effect sizes and samplesizes, and FDR the desired ratio of false to all discoveries. Thesecalculations require assumptions (e.g., p0 and effect sizes). Ifthese assumptions are incorrect the desired FDR may not beachieved. In general, it may therefore be better to use theempirical FDR methods discussed below. The formula may,however, still be helpful to design a study that is adequatelypowered to control the FDR at a level while detecting a desiredproportion of markers with effect. In addition, if only few testsare performed, empirical FDR methods may not work very welland the use of pre-specified threshold P-values may be the onlyoption [Van den Oord and Sullivan, 2003b].
Fig. 1. The marginal FDR (y-axis) obtained after controlling the family-wise error rate with a different number of tests (x-axis). [Color figure can beviewed in the online issue, which is available at www.interscience.wiley.com.]
Control of False Discoveries 639

Table I reports threshold P-values that control the marginalFDR at a level of 0.1 for scenarios that might be of practicalinterest. Thus, threshold P-values of �5� 10�6 would beneeded in a GWAS (p0¼ 0.99995) that would have good powerto detect effects with that threshold. This would generally bemore liberal compared to controlling the FWE in a GWAS with500k to 1 million SNPs that would require P-value thresholdsin the range of 10�7 to 10�8. Furthermore, the table showsthat in a very good candidate gene study assuming that theprior probability of selecting a marker with effect increased100 times, p0¼ 0.995¼ 1–100� (1–0.99995), assuming, thre-shold P-values of �5� 10�4 (e.g., 0.0005) may be needed tocontrol the FDR at an acceptable level. This threshold is, forinstance, considerably lower than the 0.05 commonly used inpractice to declare significance in candidate gene studies.
Q-values and sequential P-value methods. FDRs canbe estimated in multiple ways and many standard computerpackages (e.g., R, SAS) have such estimation proceduresimplemented. The first approach is to estimate the FDR for achosen threshold P-value t. If the m P-values are denoted pi,i¼ 1. . .m, this can be done using the formula:
dFDRðtÞ ¼ mt
#fpi � tg ð2Þ
Thus, the FDR is estimated by dividing the estimatednumber of false discoveries (is number of tests times theprobability t of rejecting a marker without effect) by the totalnumber of significant markers (i.e., total number of P-valuessmaller than t) that includes the false and true positives. Toavoid arbitrary choices, each of the observed P-values can beused as a threshold P-value t. The resulting FDR statistics arethen called q-values [Storey, 2003; Storey and Tibshirani,2003].
For other methods a researcher needs to choose the level atwhich to control the FDR statistic. The method then estimatesthe threshold P-value. For example, the so-called sequentialP-value method proposed by Benjamini and Hochberg [1995]first sorts the P-values and then applies a simple rule to decidewhich tests are significant [Benjamini and Hochberg, 1995]. Itmay not be immediately transparent why this controls theFDR, and we therefore provide supplementary materialshowing that these sequential P-value methods perform thesame calculation as in (1). In addition to being very similarform a theoretical perspective [Black, 2004; Storey et al., 2004],q-value methods will probably also operate similarly inpractice because researchers will only report markers withq-values below a certain FDR cut-off as discoveries.
FDR, pFDR, and local FDR. Controlling Benjamini andHochberg’s [1995] FDR at level q in studies where few tests areperformed may result in a proportion of false to all discoveriesin the literature that is much higher than q [Zaykin et al.,2000]. The positive FDR (pFDR) attempts to correct for this[Storey, 2002]. The disadvantage of the pFDR is that it requiresadditional information to be estimated from the data. This mayin some situations offset its clearer interpretation andtheoretical appeal. As the number of tests carried outincreases, the different FDR indices will become equivalentto each other and the marginal FDR [Storey, 2003; Tsai et al.,
2003]. How fast this happens depends on p0, average power AP,and the level at which the FDR is controlled. In general, foradequately powered GWAS involving hundreds of thousands ofmarkers there should be little difference. For studies testing amarker set with a better p0 (e.g., candidate genes, replicationstudies) 100–200 makers could suffice.
For a proper interpretation it is important to note that theabove FDRs averages the probabilities of being a falsediscovery across all significant markers [Finner and Roters,2001; Glonek and Soloman, 2003]. For instance, a marker mayhave a 90% probability of being a false discovery but still besignificant at an FDR level of 0.1 because it was testedsimultaneously with unrelated markers having very lowprobabilities. This also applies to q-values that at first glancemay seem to provide marker specific evidence. One conse-quence is that it is not possible to combine the FDRs fromdifferent markers. For example, to examine whether a certainbiological pathway is involved, a researcher may want tocombine the evidence from all the genes in that pathway[Aubert et al., 2004]. To quantify the probability that a specificmarker is a false discovery, we need to estimate so-called localFDRs. These local FDRs, however, generally require data froma large number of tests to be estimated reliably [Liao et al.,2004].
Estimating p0 and the effect size. Parameter p0 isunknown and it is not uncommon to assume p0¼ 1 in empiricalresearch. This will control the FDR conservatively as highervalues of p0 will result in smaller values of threshold P-value pk
(see Eq. 1). To avoid this conservative bias, p0 can also beestimated from the data [Schweder and Spjøtvoll, 1982;Benjamini and Hochberg, 2000; Mosig et al., 2001; Turkheimeret al., 2001; Allison et al., 2002; Storey, 2002; Hsueh et al., 2003;Pounds and Morris, 2003; Pounds and Cheng, 2004; Dalmassoet al., 2005; Meinshausen and Rice, 2006; Efron et al., 2001].For genetic studies the best estimators seem to be those thattake advantage of the knowledge that p0 has to be close to one[Meinshausen and Rice, 2006; Kuo et al., 2007]. However, formost standard situations where the FDR is controlled at a low(say 0.1) level, the use of an estimate should no make too muchof a difference because an accurate estimate will be close to one.
In genetic studies there will typically be a large range ofeffect sizes. In most cases such as GWAS we are (necessarily)focusing on markers with effect above a certain thresholdrather than the substantial number of markers showing effectsthat are real but too small to be reliably detected. It has evenbeen argued that these very small effects should perhaps betterbe viewed as ‘‘null-markers’’ and that the FDR should becontrolled for effects above a certain threshold. For example,FDR can be low because some of the markers out of thepotentially large pool of markers with very small effects aresignificant. In an independent replication study these markersare, however, unlikely to replicate due to low power. When theFDR is controlled at say the 0.1 level, it may therefore be betterthat this ratio pertains to the markers with effects above acertain threshold only. In the context of expression arrays,Efron [2004a] proposed a re-sampling technique for thispurpose that is drawing repeated samples from the data todetermine an ‘‘empirical’’ null distribution comprising bothtrue null markers plus the markers with very small effects.However, in the context of genetic association studies where weoften have very good approximations to the tests statisticdistribution, a potentially more precise parametric approach tocontrol the FDR for effect above a certain threshold is alsoconceivable [Bukszar and Van den Oord, 2007a,b].
Correlated tests due to linkage disequilibrium. Ingenetics correlated tests can be expected because of linkagedisequilibrium between markers. Compared to methods tocontrol the FWE, FDR methods appear relatively robustagainst the effects of correlated tests. This has been shown
TABLE I. Threshold P-Values Required for Controlling the FDRat the 0.1 Level
Average power with threshold P-value
p0 0.5 0.8 0.90.99995 2.78� 10�6 4.44�10�6 5.00�10�6
0.995 2.79� 10�4 4.47�10�4 5.03�10�4
640 van den Oord

theoretically for certain forms of dependence [Benjamini andHochberg, 1995; Storey, 2003; Tsai et al., 2003; Van den Oordand Sullivan, 2003a; Fernando et al., 2004] and throughsimulations [Brown and Russell, 1997; Korn et al., 2004]. Asthe nature and size of the correlations play a role [Efron, 2006],it is important to note that this robustness seems to generalizeto the context of genetic studies [Sabatti et al., 2003; Van denOord and Sullivan, 2003a; Van den Oord, 2005]. An intuitiveexplanation for this robustness of FDR methods is that FDRmethods estimate of the ratio of false to total discoveries in astudy. Correlated tests mainly increase the variance of theseestimates. However, the FDR indices themselves that are themeans of these estimates tend to be robust.
EXPLORATORY ANALYSES, DATA MINING,AND MODEL DISCOVERY
Instead of merely testing for main effects, it may beimportant to search for gene–environment/covariate interac-tions [Collins, 2004], gene–gene interactions [Carlborg andHaley, 2004] or genetic variants affecting disease subtypes[Kennedy et al., 2003]. As long as these searches are (1)performed systematically for a certain model and (2) testresults can be summarized by a P-value, the above methods canbe used to control false discoveries.
However, very often such searches are not done systemati-cally. For example, after failing to find main effects or replicatea previously reported association, researchers may startexploring interactions between genes and environmentalfactors, test for effects in subgroups of patients, performmultimarker and haplotype analyses, test for effects in subsetsof the whole sample, etc. The more extensive these searches,the more likely that a ‘‘significant’’ finding will eventually beobtained. The problem is that these models may fit or haveindividual components (e.g., an interaction term) that seemsignificant because they capitalize on random fluctuations inthe data. The ‘‘significant’’ findings are, however, deceptivebecause they may be unlikely to replicate in independent datasets. In this context, one could wonder how much of the patternof all the different ‘‘significant’’ findings for genes such asDysbindin are the result of the exploratory nature of some ofthe replication studies. To properly assess whether results ofsuch exploratory searches will ‘‘replicate’’ in independent datasets, we need to correct for the complexity of the search process[Shen et al., 2004]. In addition, the complexity of the modelplays a role and needs to be taken into account as well. Forexample, (regression) models that have many parameters bycomparison to the amount of data available may still explain asubstantial proportion of explained variance in the outcome(i.e., show a good fit). Finally, the form of the model is relevantbecause, even if the number of parameters is the same, modelsdiffer in their ability to fit random data [Rissanen, 1978]. Thecontrol of false discoveries after such (model) searches is anactive area of statistical research and in many instancesreplication may currently the safest approach to validateresults obtained from such searches.
Rather than doing such searches manually, the options tocontrol false discoveries increase by using computer algo-rithms. However, even with computers, exhaustive searchesmay not be possible. For example, for a two-locus, two-allele,fully penetrant models with disease simply classified as absentversus present, even with strictest definition of what isessentially the same pattern there are already 58 non-redundant two-locus models [Li and Reich, 2000]. When eitherthe covariate or outcome variable are continuous the number ofmodels will increase dramatically because of possible non-linear relations. One approach is to confine the analysis to aspecific model and then test this model systematically for allmarkers. However, this may miss effects that involve all
the non-tested models. Alternatively, machine learningapproaches (e.g., data mining) can be used that attempt tofind other models. These methods typically perform searches inan intelligent way and avoid considering all possible alter-native models [Hastie et al., 2001; Hahn et al., 2003].
When searches are performed by computers, covariancepenalties can be used to account for the complexity of the modeland search process. Covariance penalties are related to thedegrees of freedom (of a test). Loosely speaking, covariancepenalties reflect the extent to which a model can fit randomdata or, alternatively, the extent to which model fit depends(i.e., covaries) on the random features of the data used to derivethe model. A fit index that is corrected by a covariance penaltyessentially estimates the fit of that model in an independentreplication data set. The most simple covariance penaltiesmerely penalize models for the number of parameters theyestimate. However, covariance penalties exist that also try tocapture the complexity of the form of the model and the searchprocess [Ye, 1998; Shen and Ye, 2002; Efron, 2004b]. Forexample, Owen for example suggested using three degrees offreedom for testing a single predictor term in a specific kind ofnon-parametric regression model. More popular are the useof cross-validation [Stone, 1974] and related techniques can beviewed as non-parametric (i.e., based on few or weak statisticalassumptions) estimates of covariance penalties [Efron, 2004b].However, it should be noted that these non-parametricapproaches can result in imprecise corrections of fit indices[Efron, 2004b].
Searching for Models Using Biological Knowledge
Off-the-shelf data-mining and machine-learning techniquesmay help to search for models but can produce artificial modelsdifficult to interpret from a biological perspective. So, it isimportant to use available knowledge to constrain searches tothose models that are biologically meaningful. Althoughknowledge is too limited to pre-specify explicit models linkinggenotypes to phenotypes, we often do have partial information[Strohman, 2002; Alon, 2003; Vazquez et al., 2004]. Forexample, transcription networks comprise smaller substruc-tures called motifs [Lee et al., 2002; Shen-Orr et al., 2002],metabolic networks are subject to well-established organiza-tion principles [Kell, 2004], and genetic effects can be assumedto be mediated by more or less coherent biological or pathogenicprocesses that can be represented in models by latent variables[Bollen, 2002; Van den Oord and Snieder, 2002; Van den Oordet al., 2004]. Using specific machine learning techniques it ispossible to search through complex data sets efficiently whileincorporating such biological knowledge [Kell, 2002; Goodacre,2005] thereby reducing the probability of false positive andartificial findings.
REPLICATION STUDIES
Replication is generally perceived as a key step to rule outfalse discoveries. The answer to the questions what constitutes areplication and how best can it be achieved is, however, notstraightforward [Chanock et al., 2007]. An important issue iswhether it is necessary to require precise replication (the samephenotype, genetic marker, genotype, statistical test, anddirection ofassociation) of if less precise definitionsof replicationsuffice (e.g., any significant marker in the same gene) [Sullivan,2007]. The problem with less precise definitions is that the‘‘replication’’ study partly becomes an exploratory analysis andsubject to the above described phenomenon that the moreextensive the search process the more likely it is that results will‘‘replicate.’’ Researchers sometimes justify less precise defini-tions based on the complexity of the genetics effects on thepsychiatric disorder (e.g., locus heterogeneity, different family
Control of False Discoveries 641

history, disease subtypes, or differences by genetic ancestry).While such mechanisms might sometimes be true, theseexplanations tendtominimize the possibility that aconsiderablymore parsimonious explanation is responsible for the results(i.e., a false positive association) [Sullivan, 2007].
Even when a precise definition of replication is used,declaring significance is not unambiguous. Rules such asP-values smaller than 0.05 suggest replication are arbitrary.That is, depending on factors such as effect sizes, sample sizes,and the prior probability, this rule may result in significantfindings that have very different probabilities of being a truereplication. More meaningful decision rules will be neededsuch as the use of threshold P-values ensuring that a desiredratio of false discoveries to all reported discoveries in theliterature is achieved. A very simple approach of is to calculatethe local FDR (fdr) discussed in the previous section. That is,each marker has potentially two states (1) it is related to thedisease and (2) it is unrelated to the disease. Given the testresult in the replication study we can now estimate the(posterior) probability that it is unrelated to the disease.
fdrðiÞ ¼ PrðH0i¼ truejT ¼ tiÞ ¼ p0ðiÞf0ðtiÞ
p0if0ðtiÞ þ ð1 þ p0i
ÞfciðtiÞð3Þ
where H0istates the null hypothesis that marker i is unrelated
to the disease, ti is the value of test statistic T for marker i in thereplication study, f0 the density function under the nulldistribution and fci
the density function under the alternativedistribution where ci is the (effect size) parameter for marker ithat affects the test statistic distribution under the alternative.To estimate fdr(i) we can use the effect size we observe in thereplication study as the effects in the initial study is typicallybiased upward [Goring et al., 2001; Ioannidis et al., 2001].Although p0i
can be estimated, a simple approach would be toassume a range of values and examine for what value fdr(i) would be sufficiently small (say 0.1). Although moresophisticated method are conceivable, this simple methodwould at least provide a better interpretation of what we call areplication and makes an attempt to start standardizing thecriterion across studies for declaring significance.
Replication studies are often designed in an opportunisticfashion (e.g., dictated by available sample). However, using the(theory of) multistage designs, replication studies can bedesigned in a cost-effective manner. In the multistage designsas intended here, all the markers are only genotyped and testedin Stage 1. The most promising markers are then genotyped inStage 2 using other/new samples in the second/replicationstage [Saito and Kamatani, 2002; Satagopan et al., 2002;Aplenc et al., 2003; Satagopan and Elston, 2003; Van den Oordand Sullivan, 2003a,b; Lowe et al., 2004]. The theory ofmultistage designs allow you to calculate optimal simple sizesfor the replication part taking the sample size of Stage 1 intoaccount if that is not under the control of the investigator. Inaddition, multistage designs offer the possibility to useinformation collected at the first stage(s) to design the secondreplication stage. In contrast, the design of single-stage largescale association study is completely based on assumptionsabout effect sizes, proportion of markers with effect etc. Theproblem is that if these assumptions are incorrect the goalsmay not be achieved or could have been achieved at much lowercost. This idea of adaptive [Bauer and Brannath, 2004b] or self-designing studies [Fisher, 1998] where information fromearlier stages is used to improve the design of later stages isfor instance used in clinical trials. A simple example in thepresent context would be to test for population stratification/ascertainment bias in a Stage 1 case control sample and thenperform a family-based follow up study rather than anothercase-control study if needed. Another example involves the useof statistical procedures to ensure adequately powered follow
up studies or to determine the P-value threshold ensuringthat a sufficiently large proportion of markers with effectare selected for the next stage. The latter is important becausewhereas false discoveries can always be eliminated in futurestudies, markers with effects that have been eliminated cannever be recovered. A final example involves the carefulintegration of all the findings with those already out in theliterature to maximize the probability that the relevantmarkers are selected for the next stage. Particularly for studiesas expensive as GWAS, it may be better to perform interimanalyses and adjust the study design if that turns out to benecessary to achieve the goals or save costs.
Rather than analyzing replication data separately, a jointanalysis of the initial study and replication stage will give amore powerful test. Standard tests cannot be used for suchcombined data because only the markers that are significant inthe first stage are selected for the second replication stage andthe test statistics at both stages are dependent as a result of thepartly overlapping data [Satagopan et al., 2002; Van den Oordand Sullivan, 2003a]. Simulations could be used instead [Loweet al., 2004]. However, testing in both stages at a significancelevel of say a¼ .001 implies that out of every million simulatedsamples only one sample (1 million� 0.001� 0.001) will berejected at Stage 2. Thus, if one would like 1,000 rejectedsamples at Stage 2 to estimate the critical values needed forsignificance testing, a billion samples need to be simulated. Ifavailable, theoretical approximations may be preferred such asthe use of a general approximation assuming (bi-variate)normality of the test statistics at both stages [Satagopan et al.,2002], when the test statistic is the difference between theallele frequencies in cases and controls [Skol et al., 2006], andwhen Pearson’s Chi-square statistic is used to analyze acontingency table [Bukszar and Van den Oord, 2006]. How-ever, the correct distribution of the test statistic may not beknown and combining the raw data may not always be possible(e.g., if the first stage was done by other group or if the samplesin the two stages are different such as family based versus case-control studies). In these instances, researchers could resort tocombing the P-values across stages. Many techniques areavailable in the meta-analysis literature for this purpose[Sutton et al., 2000; Bauer and Brannath, 2004a] althoughmost of these techniques may need to be slightly modified toaccount for the fact that in multistage designs there is aselection of the most significant P-values in Stage 1.
CONCLUSIONS
Proper methods for controlling false discoveries are impor-tant to prevent that time and resources are spend on leads thatwill eventually prove irrelevant. In the context of findingcommon variants for complex diseases, an ideal goal may beideal to try to achieve the same ratio of false discoveries dividedby all rejected tests regardless of systematic differencesbetween studies. This ideal can never be achieved using(traditional) methods where corrections for ‘‘multiple testing’’are based on the number of tests carried out. This is because thenumber of tests is arbitrary depending on factors such asbudget, publication strategy, and genotyping capacity. Insteadmethods that control the FDR may be more suitable.
If the aim is to control the FDR control at a low level (say 0.1),FDR control in GWAS will be relatively straightforward. Thatis, a very simple method can be used that is likely to give resultvery similar results to the more complex FDR variants (e.g.,pFDR, include p0 estimates). Controlling the FDR in studieswhere very few markers are tested (e.g., candidate genestudies) is difficult. Rather then using an empirical method,the use predetermined P-value threshold such as �5� 10�4
may sometimes be the best option.
642 van den Oord

Exploratory analyses are potentially an important source offalse discoveries. The problem is to properly account for thephenomenon that the more extensive the search process, themore likely it is that a ‘‘significant’’ finding will be obtained.Rather than performing such searches manually, the possi-bility to control false discoveries increases by using computeralgorithms. However, as statistical techniques are still devel-oping to account for the complexities involved in controllingfalse positives in exploratory searches, independent replica-tion/validation may eventually be needed.
The answer to the questions what constitutes a replicationand how best can it be achieved is not straightforward. A strictdefinition what constitutes a replication may be preferred toavoid that the replication study becomes an exploratory studyand subject to the above described phenomenon that the moreextensive the search process the more likely it is that resultswill ‘‘replicate.’’ Depending on factors such as effect sizes andsample sizes, rules such as P-values smaller than 0.05 suggestreplication may result in significant findings that have verydifferent probabilities of being a true replication. Lessarbitrary decision rules, such as rules based on the posteriorprobability that the replication is a false positive, will beneeded to help interpret replication findings and start stand-ardizing the criterion across replication studies. Rather thenbeing deigned in an opportunistic fashion (e.g., dictated byavailable sample) replication studies can be designed in a cost-effective manner using the (theory of adaptive) multistagedesigns.
ONLINE LINKS
Supplementary information is available in Edwin van denOord’s web site: http://www.vipbg.vcu.edu/�edwin/.
ACKNOWLEDGMENTS
I would like to thank Joseph McClay for his comments on anearlier draft of this article and Rebecca Ortiz for her help withpreparing the article.
REFERENCES
Allison DB, Gadbury G, Heo M, Fernandez J, Lee C-K, Prolla TA, et al. 2002.A mixture model approach for the analysis of microarray geneexpression data. Comput Stat Data Anal 39:1–20.
Alon U. 2003. Biological networks: The tinkerer as an engineer. Science301:1866–1867.
Aplenc R, Zhao H, Rebbeck TR, Propert KJ. 2003. Group sequential methodsand sample size savings in biomarker-disease association studies.Genetics 163:1215–1219.
Aubert J, Bar-Hen A, Daudin JJ, Robin S. 2004. Determination of thedifferentially expressed genes in microarray experiments using localFDR. BMC Bioinformatics 5:125.
Bauer P, Brannath W. 2004a. The advantages and disadvantages ofadaptive designs for clinical trials. Drug Discov Today 9:351–357.
Bauer P, Brannath W. 2004b. The advantages and disadvantages ofadaptive designs for clinical trials. Drug Discov Today 9:351–357.
Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: Apractical and powerful approach to multiple testing. J R Stat Soc B57:289–300.
Benjamini Y, Hochberg Y. 2000. On adaptive control of the false discoveryrate in multiple testing with independent statistics. J Educ Behav Stat25:60–83.
Black MA. 2004. A note on the adaptive control of false discovery rates. J RStat Soc B 66:297–304.
Blangero J. 2004. Localization and identification of human quantitativetrait loci: King harvest has surely come. Curr Opin Genet Dev 14:233–240.
Bollen KA. 2002. Latent variables in psychology and the social sciences.Annu Rev Psychol 53:605–634.
Brown BW, Russell K. 1997. Methods of correcting for multiple testing:Operating characteristics. Stat Med 16:2511–2528.
Bukszar J, Van den Oord EJCG. 2006. Optimization of two-stagegenetic designs where data are combined using an accurate andefficient approximation for Pearson’s statistic. Biometrics 62:1132–1137.
Bukszar J, Van den Oord EJCG. 2007a. Estimating effect sizes in large scalegenetic association studies. Submitted for publication.
Bukszar J, Van den Oord EJCG. 2007b. Estimating the proportion ofmarkers without effect and average effect size in large scale geneticassociation studies. Submitted for publication.
Carlborg O, Haley CS. 2004. Epistasis: Too often neglected in complex traitstudies? Nat Rev Genet 5:618–625.
Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G,et al. 2007. Replicating genotype-phenotype associations. Nature447:655–660.
Colhoun HM, McKeigue PM, Davey SG. 2003. Problems of reporting geneticassociations with complex outcomes. Lancet 361:865–872.
Collins FS. 2004. The case for a US prospective cohort study of genes andenvironment. Nature 429:475–477.
Dahlman I, Eaves IA, Kosoy R, Morrison VA, Heward J, Gough SC, et al.2002. Parameters for reliable results in genetic association studies incommon disease. Nat Genet 30:149–150.
Dalmasso C, Broet P, Moreau T. 2005. A simple procedure for estimating thefalse discovery rate. Bioinformatics 21:660–668.
Dudbridge F, Koeleman BP. 2004. Efficient computation of significancelevels for multiple associations in large studies of correlated data,including genomewide association studies. Am J Hum Genet 75:424–435.
Dunnet CW, Tamhane AC. 1992. A step up multiple test procedure. J AmStat Assoc 87:162–170.
Efron B. 2004a. Large-scale simultaneous hypothesis testing: The choice of anull hypothesis. J Am Stat Assoc 99:96–104.
Efron B. 2004b. The estimation of prediction error: Covariance penalties andcross-validation. J Am Stat Assoc 99:619–632.
Efron. 2006. Correlation and Large-Scale Simultaneous Significance Test-ing. Stanford Technical Report.
Efron B, Tibshirani R. 2002. Empirical bayes methods and false discoveryrates for microarrays. Genet Epidemiol 23:70–86.
Efron B, Tibshirani R, Storey JD, Tusher VG. 2001. Empirical Bayesanalysis of a microarray experiment. J Am Stat Assoc 96:1151–1160.
Fernando RL, Nettleton D, Southey BR, Dekkers JC, Rothschild MF, SollerM. 2004. Controlling the proportion of false positives in multipledependent tests. Genetics 166:611–619.
Finner H, Roters M. 2001. On the false discovery rate and expected Type Ierrors. Biometrical J 8:985–1005.
Fisher LD. 1998. Self-designing clinical trials. Stat Med 17:1551–1562.
Freimer N, Sabatti C. 2004. The use of pedigree, sib-pair and associationstudies of common diseases for genetic mapping and epidemiology. NatGenet 36:1045–1051.
Glonek G, Soloman P. 2003. Discussion of resampling-based multipletesting for microarray data analysis by Ge, Dudoit and Speed. Test 12:1–77.
Goodacre R. 2005. Making sense of the metabolome using evolutionarycomputation: Seeing the wood with the trees. J Exp Bot 56:245–254.
Goring HH, Terwilliger JD, Blangero J. 2001. Large upward bias inestimation of locus-specific effects from genomewide scans. Am J HumGenet 69:1357–1369.
Hahn LW, Ritchie MD, Moore JH. 2003. Multifactor dimensionalityreduction software for detecting gene-gene and gene-environmentinteractions. Bioinformatics 19:376–382.
Hastie T, Tibshirani R, Friedman J. 2001. The elements of statisticallearning: Data mining, inference, and prediction. New York: SpringerVerlag.
Hochberg Y, Benjamini Y. 1990. More powerful procedures for multiplesignificance testing. Stat Med 9:811–818.
Holm S. 1979. A simple sequentially rejective multiple test procedure. ScandJ Stat 6:65–70.
Hsueh H, Chen J, Kodell R. 2003. Comparison of methods for estimating thenumber of true null hypotheses in multiplicity testing. J Biopharm Stat13:675–689.
Control of False Discoveries 643

Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. 2001.Replication validity of genetic association studies. Nat Genet 29:306–309.
Jung SH, Bang H, Young S. 2005. Sample size calculation for multipletesting in microarray data analysis. Biostatistics 6:157–169.
Kell DB. 2002. Genotype-phenotype mapping: Genes as computer programs.Trends Genet 18:555–559.
Kell DB. 2004. Metabolomics and systems biology: Making sense of the soup.Curr Opin Microbiol 7:296–307.
Kennedy JL, Farrer LA, Andreasen NC, Mayeux R, George-Hyslop P. 2003.The genetics of adult-onset neuropsychiatric disease: Complexities andconundra? Science 302:822–826.
Korn EL, Troendle J, McShane L, Simon R. 2004. Controlling the number offalse discoveries: Application to high-dimensional genomic data. J StatPlann Inference 124:379–398.
Kuo P, Bukszar J, Van den Oord EJCG. 2007. Estimating the number andsize of the main effects in genome-wide case-control association studies.BMC Proc.
Lander E, Kruglyak L. 1995. Genetic dissection of complex traits: Guidelinesfor interpreting and reporting linkage results. Nat Genet 11:241–247.
Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, et al.2002. Transcriptional regulatory networks in Saccharomyces cerevisiae.Science 298:799–804.
Li W, Reich J. 2000. A complete enumeration and classification of two-locusdisease models. Hum Hered 50:334–349.
Liao JG, Lin Y, Selvanayagam ZE, Shih WJ. 2004. A mixture model forestimating the local false discovery rate in DNA microarray analysis.Bioinformatics 20:2694–2701.
Lin DY. 2004. An efficient Monte Carlo approach to assessing statisticalsignificance in genomic studies. Bioinformatics 21:781–787.
Lowe CE, Cooper JD, Chapman JM, Barratt BJ, Twells RC, Green EA, et al.2004. Cost-effective analysis of candidate genes using htSNPs: A stagedapproach. Genes Immun 5:301–305.
Meinshausen N, Rice J. 2006. Estimating the proportion of false nullhypotheses among a large number of independently tested hypotheses.Ann Stat 34:373–393.
Morton NE. 1955. Sequential tests for the detection of linkage. Am J HumGenet 7:277–318.
Mosig MO, Lipkin E, Khutoreskaya G, Tchourzyna E, Soller M, FriedmannA. 2001. A whole genome scan for quantitative trait loci affecting milkprotein percentage in Israeli-Holstein cattle, by means of selective milkDNA pooling in a daughter design, using an adjusted false discovery ratecriterion. Genetics 157:1683–1698.
Pounds S, Cheng C. 2004. Improving false discovery rate estimation.Bioinformatics 20:1737–1745.
Pounds S, Morris SW. 2003. Estimating the occurrence of false positives andfalse negatives in microarray studies by approximating and partitioningthe empirical distribution of P-values. Bioinformatics 19:1236–1242.
Risch N, Merikangas K. 1996. The future of genetic studies of complexhuman diseases. Science 273:1516–1517.
Rissanen J. 1978. Modeling by shortest data description. Automatica14:465–471.
Sabatti C, Service S, Freimer N. 2003. False discovery rate in linkage andassociation genome screens for complex disorders. Genetics 164:829–833.
Saito A, Kamatani N. 2002. Strategies for genome-wide association studies:Optimization of study designs by the stepwise focusing method. J HumGenet 47:360–365.
Satagopan JM, Elston RC. 2003. Optimal two-stage genotyping inpopulation-based association studies. Genet Epidemiol 25:149–157.
Satagopan JM, Verbel DA, Venkatraman ES, Offit KE, Begg CB. 2002. Two-stage designs for gene-disease association studies. Biometrics 58:163–170.
Schweder T, Spjøtvoll E. 1982. Plots of P-values to evaluate many testssimultaneously. Biometrika 69:493–502.
Shen X, Ye J. 2002. Adaptive model selection. J Am Stat Assoc 97:210–221.
Shen X, Huang H, Ye J. 2004. The estimation of prediction error: Covariancepenalties and cross-validation: Comment. J Am Stat Assoc 99:634–637.
Shen-Orr SS, Milo R, Mangan S, Alon U. 2002. Network motifs in thetranscriptional regulation network of Escherichia coli. Nat Genet31:64–68.
Sidak Z. 1967. Rectangular confidence regions for the means of multivariatedistributions. J Am Stat Assoc 62:626–633.
Skol AD, Scott LJ, Abecasis GR, Boehnke M. 2006. Joint analysis is moreefficient than replication-based analysis for two-stage genome-wideassociation studies. Nat Genet 38:209–213.
Stone M. 1974. Cross-validatory choice and assessment of statisticalpredictions. J R Stat Soc B 36:111–147.
Storey J. 2002. A direct approach to false discovery rates. J R Stat Soc B64:479–498.
Storey J. 2003. The positive false discovery rate: A Bayesian interpretationand the q-value. Ann Stat 31:2013–2035.
Storey J, Tibshirani R. 2003. Statistical significance for genome-widestudies. Proc Natl Acad Sci 100:9440–9445.
Storey J, Taylor JE, Siegmund D. 2004. Strong control, conservative pointestimation and simultaneous conservative consistency of false discoveryrates: A unified approach. J R Stat Soc B 66:187–205.
Strohman R. 2002. Maneuvering in the complex path from genotype tophenotype. Science 296:701–703.
Sullivan PF. 2007. Spurious genetic associations. Biol Psychiatry 61:1121–1126.
Sutton A, Abrams K, Jones D, Sheldon T, Song F. 2000. Methods for meta-analysis in medical research.Chichester. UK: Wiley.
Thomas DC, Clayton DG. 2004. Betting odds and genetic associations. J NatlCancer Inst 96:421–423.
Tsai CA, Hsueh HM, Chen JJ. 2003. Estimation of false discovery rates inmultiple testing: Application to gene microarray data. Biometrics59:1071–1081.
Turkheimer FE, Smith CB, Schmidt K. 2001. Estimation of the number of‘‘true’’ null hypotheses in multivariate analysis of neuroimaging data.Neuroimage 13:920–930.
Van den Oord EJCG. 2005. Controlling false discoveries in candidate genestudies. Mol Psychiatry 10:230–231.
Van den Oord EJCG, Snieder H. 2002. Including measured genotypes instatistical models to study the interplay of multiple factors affectingcomplex traits. Behav Genet 32:1–22.
Van den Oord EJCG, Sullivan PF. 2003a. A framework for controlling falsediscovery rates and minimizing the amount of genotyping in the searchfor disease mutations. Human Heredity 56:188–199.
Van den Oord EJCG, Sullivan PF. 2003b. False discoveries and models forgene discovery. Trends Genet 19:537–542.
Van den Oord EJCG, MacGregor AJ, Snieder H, Spector TD. 2004. Modelingwith measured genotypes: Effects of the vitamin D receptor gene, age,and latent genetic and environmental factors on measures ofbone mineral density. Behav Genet 34:197–206.
Vazquez A, Dobrin R, Sergi D, Eckmann JP, Oltvai ZN, Barabasi AL. 2004.The topological relationship between the large-scale attributes and localinteraction patterns of complex networks. Proc Natl Acad Sci USA101:17940–17945.
Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. 2004.Assessing the probability that a positive report is false: An approach formolecular epidemiology studies. J Natl Cancer Inst 96:434–442.
Westfall P, Young SS. 1993. Resampling-based multiple testing. New York:Wiley.
Ye J. 1998. On measuring and correcting the effects of data mining andmodel selection. J Am Stat Assoc 93:120–131.
Zaykin D, Young S, Westfall P. 2000. Using the false discovery rate in thegenetic dissection of complex traits. Genetics 154:1917–1918.
644 van den Oord