computer modeling dr. guanhua chen department of chemistry university of hong kong
Post on 19-Dec-2015
219 views
TRANSCRIPT

Computer Modeling
Dr. GuanHua CHEN
Department of Chemistry
University of Hong Konghttp://yangtze.hku.hk/lecture/comput06-07.ppt

Computational Chemistry
• Quantum Chemistry
SchrÖdinger Equation
H = E• Molecular Mechanics
F = Ma
F : Force Field
• Bioinformatics

Computational Chemistry Industry
Company Software
Gaussian Inc. Gaussian 94, Gaussian 98Schrödinger Inc. Jaguar Wavefunction SpartanQ-Chem Q-ChemAccelrys InsightII, Cerius2
HyperCube HyperChemInformatixCelera Genomics
Applications: material discovery, drug design & research
R&D in Chemical & Pharmaceutical industries in 2000: US$ 80 billionBioinformatics: Total Sales in 2001 US$ 225 million
Project Sales in 2006 US$ 1.7 billion

Vitamin CC60
Cytochrome c
heme
OH + D2 --> HOD + D
energy

Quantum Chemistry Methods
• Ab initio Molecular Orbital Methods
Hartree-Fock, Configurationa Interaction (CI)
MP Perturbation, Coupled-Cluster, CASSCF
• Density Functional Theory
• Semiempirical Molecular Orbital Methods Huckel, PPP, CNDO, INDO, MNDO, AM1
PM3, CNDO/S, INDO/S

H E
SchrÖdinger Equation
HamiltonianH = (h2/2m
h2/2me)ii2
i e2/ri+ ZZer
ije2/rij
Wavefunction
Energy
One-electron terms: (h2/2m
h2/2me)ii2i e2/ri
Two-electron term:
ije2/rij

1. Hartree-Fock EquationF i = i i
F Fock operator
i the i-th Hartree-Fock orbital
i the energy of the i-th Hartree-Fock orbital
Hartree-Fock MethodOrbitals

2. Roothaan Method (introduction of Basis functions)i = k cki k LCAO-MO
{k } is a set of atomic orbitals (or basis functions)
3. Hartree-Fock-Roothaan equation j ( Fij - i Sij ) cji = 0
Fij iF j Sij ij
4. Solve the Hartree-Fock-Roothaan equation self-consistently (HFSCF)

Graphic Representation of Hartree-Fock Solution
0 eV
IonizationEnergy
ElectronAffinity

Basis Set i = p cip p
{k } is a set of atomic orbitals (or basis functions)
STO-3G, 3-21G, 4-31G, 6-31G, 6-31G*, 6-31G**------------------------------------------------------------------------------------- complexity & accuracy
# HF/6-31G(d) Route section water energy Title
0 1 Molecule Specification O -0.464 0.177 0.0 (in Cartesian coordinatesH -0.464 1.137 0.0H 0.441 -0.143 0.0
A Gaussian Input File for H2O

Gaussian type functionsgijk = N xi yj zk exp(-r2)
(primitive Gaussian function)p = u dup gu
(contracted Gaussian-type function, CGTF)u = {ijk} p = {nlm}

Electron Correlation: avoiding each other
The reason of the instantaneous correlation:Coulomb repulsion (not included in the HF)
Beyond the Hartree-FockConfiguration Interaction (CI)Perturbation theoryCoupled Cluster MethodDensity functional theory

Configuration Interaction (CI)
+
+ …

Single Electron Excitation or Singly Excited

Double Electrons Excitation or Doubly Excited

Singly Excited Configuration Interaction (CIS): Changes only the excited states
+

Doubly Excited CI (CID):Changes ground & excited states
+
Singly & Doubly Excited CI (CISD):Most Used CI Method

Full CI (FCI):Changes ground & excited states
++
+ ...

H = H0 + H’H0n
(0) = En(0)n
(0)
n(0) is an eigenstate for unperturbed system
H’ is small compared with H0
Perturbation Theory

Moller-Plesset (MP) Perturbation Theory
The MP unperturbed Hamiltonian H0
H0 = m F(m)
where F(m) is the Fock operator for electron m.And thus, the perturbation H’
H’ = H - H0
Therefore, the unperturbed wave function is simply the Hartree-Fock wave function . Ab initio methods: MP2, MP3, MP4

= eT(0)
(0): Hartree-Fock ground state wave function: Ground state wave functionT = T1 + T2 + T3 + T4 + T5 + …Tn : n electron excitation operator
Coupled-Cluster Method
=T1

CCD = eT2(0)
(0): Hartree-Fock ground state wave functionCCD: Ground state wave functionT2 : two electron excitation operator
Coupled-Cluster Doubles (CCD) Method
=T2

Complete Active Space SCF (CASSCF)
Active space
All possible configurations

Density-Functional Theory (DFT)Hohenberg-Kohn Theorem: Phys. Rev. 136, B864 (1964)
The ground state electronic density (r) determines uniquely all possible properties of an electronic system
(r) Properties P (e.g. conductance), i.e. P P[(r)]
Density-Functional Theory (DFT)E0 = h2/2me)i <i |i
2 |i > dr e2(r) /
r1 dr1 dr2 e2/r12 + Exc[(r)]
Kohn-Sham Equation Ground State: Phys. Rev. 140, A1133 (1965)
FKS i = i i
FKS h2/2me)ii2 e2 / r1jJj + Vxc
Vxc Exc[(r)] / (r)
A popular exchange-correlation functional Exc[(r)]: B3LYP

B3LYP/6-311+G(d,p) B3LYP/6-311+G(3df,2p)
RMS=21.4 kcal/mol RMS=12.0 kcal/mol
RMS=3.1 kcal/mol RMS=3.3 kcal/mol
B3LYP/6-311+G(d,p)-NEURON & B3LYP/6-311+G(d,p)-NEURON: same accuracy
Hu, Wang, Wong & Chen, J. Chem. Phys. (Comm) (2003)

Time-Dependent Density-Functional Theory (TDDFT)
Runge-Gross Extension: Phys. Rev. Lett. 52, 997 (1984)
Time-dependent system (r,t) Properties P (e.g. absorption)
TDDFT equation: exact for excited states
Isolated system
Open system
Density-Functional Theory for Open System ???
Further Extension: X. Zheng, F. Wang & G.H. Chen (2005)
Generalized TDDFT equation: exact for open systems
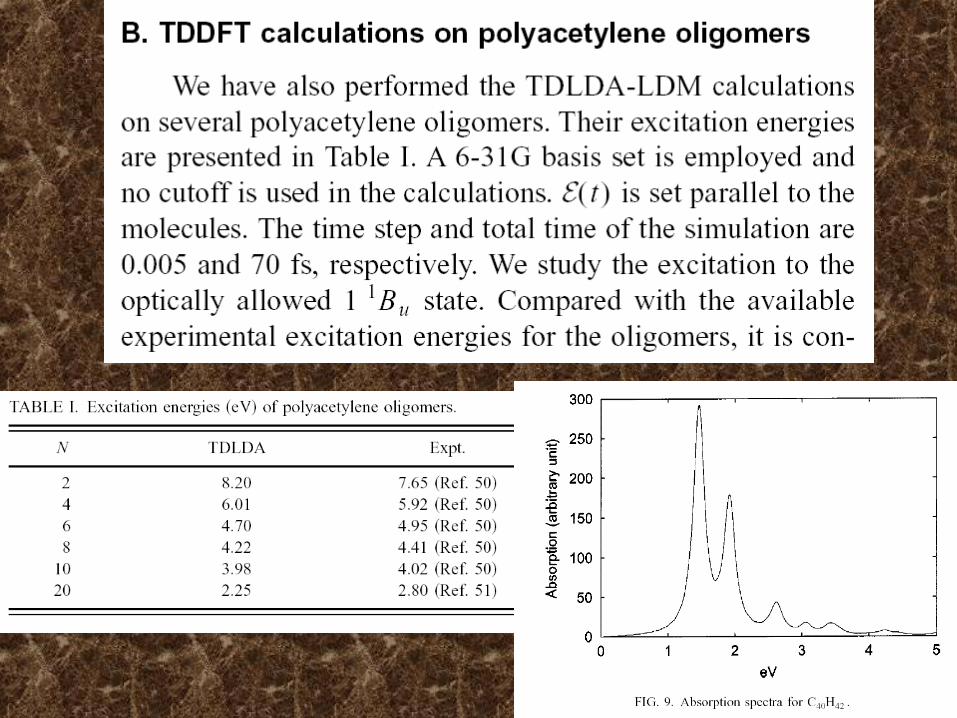
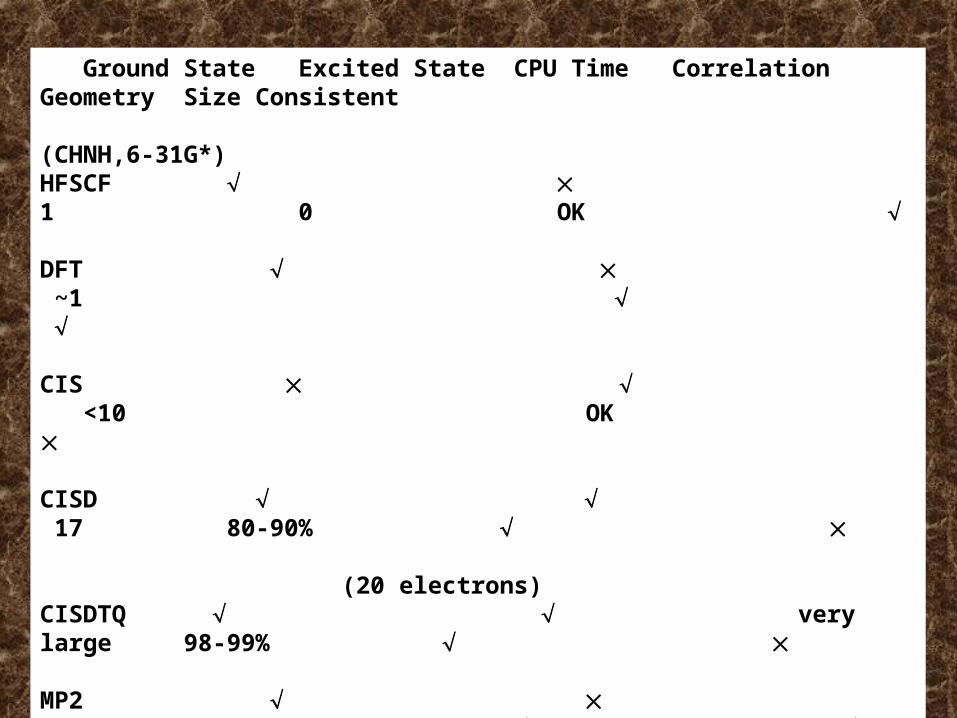
Ground State Excited State CPU Time Correlation Geometry Size Consistent (CHNH,6-31G*)HFSCF 1 0 OK
DFT ~1
CIS <10 OK
CISD 17 80-90% (20 electrons)CISDTQ very large 98-99%
MP2 1.5 85-95% (DZ+P)MP4 5.8 >90% CCD large >90%
CCSDT very large ~100%

Reactant
Product
Transition State: one negative frequency
Reaction Coordinate
Search for Transition State
G
k e-G/RT

#b3lyp/6-31G opt=qst2 test
the first is the reactant internal coordinate
0 1OH 1 oh1 H 1 oh1 2 ohh1
oh1 0.90ohh1 104.5
The second is the product internal coordinate
0 1OH 1 oh2H 1 oh3 2 ohh2
oh2 0.9oh3 10.0ohh2 160.0
Gaussian Input File for Transition State Calculation

Semiempirical Molecular Orbital Calculation
Extended Huckel MO Method (Wolfsberg, Helmholz, Hoffman)
Independent electron approximation
Schrodinger equation for electron i
Hval = i Heff(i)
Heff(i) = -(h2/2m) i2 + Veff(i)
Heff(i) i = i i

LCAO-MO: i = r cri r
s ( Heff
rs - i Srs ) csi = 0
Heffrs rHeff s Srs
rs Parametrization: Heff
rr rHeff r minus the valence-state ionization potential (VISP)

Atomic Orbital Energy VISP--------------- e5 -e5
--------------- e4 -e4
--------------- e3 -e3
--------------- e2 -e2
--------------- e1 -e1
Heff
rs = ½ K (Heffrr + Heff
ss) Srs K:
13

CNDO, INDO, NDDO(Pople and co-workers)
Hamiltonian with effective potentialsHval = i [ -(h
2/2m) i2 + Veff(i) ] + ij>i e
2 / rij
two-electron integral:(rs|tu) = <r(1) t(2)| 1/r12 | s(1) u(2)>
CNDO: complete neglect of differential overlap (rs|tu) = rs tu (rr|tt) rs tu rt

INDO: intermediate neglect of differential overlap(rs|tu) = 0 when r, s, t and u are not on the same atom.
NDDO: neglect of diatomic differential overlap(rs|tu) = 0 if r and s (or t and u) are not on the same atom.
CNDO, INDO are parametrized so that the overallresults fit well with the results of minimal basis abinitio Hartree-Fock calculation.
CNDO/S, INDO/S are parametrized to predict optical spectra.

MINDO, MNDO, AM1, PM3(Dewar and co-workers, University of Texas, Austin) MINDO: modified INDOMNDO: modified neglect of diatomic overlap AM1: Austin Model 1PM3: MNDO parametric method 3 *based on INDO & NDDO *reproduce the binding energy

Relativistic Effects
Speed of 1s electron: Zc / 137
Heavy elements have large Z, thus relativistic effects areimportant.
Dirac Equation:Relativistic Hartree-Fock w/ Dirac-Fock operator; orRelativistic Kohn-Sham calculation; orRelativistic effective core potential (ECP).

(1) Neglect or incomplete treatment of electron correlation
(2) Incompleteness of the Basis set
(3) Relativistic effects
(4) Deviation from the Born-Oppenheimer approximation
Four Sources of error in ab initio Calculation

Quantum Chemistry for Complex Systems

Quantum Mechanics / Molecular Mechanics (QM/MM) Method
Combining quantum mechanics and molecular mechanics methods:
QM
MM

Hamiltonian of entire system:H = HQM +HMM +HQM/MM
Energy of entire system:E = EQM(QM) + EMM(MM) + EQM/MM(QM/MM)EQM/MM(QM/MM) = Eelec(QM/MM) + Evdw(MM) + EMM-bond(MM)
EQM(QM) + Eelec(QM/MM) = <| Heff |>
Heff = -1/2 ii2 + ij 1/rij - i Z/ri - i q/ri
+ i Vv-b(ri) + ZZ/r + Zq/r
QM
MM

Quantum Chemist’s Solution
Linear-Scaling Method: O(N)
Computational time scales linearly with system size
Time
Size

Linear Scaling Calculation for Ground State
W. Yang, Phys. Rev. Lett. 1991
Divide-and-Conqure (DAC)

Superoxide Dismutase (4380 atoms)
York, Lee & Yang, JACS, 1996
Strain, Scuseria & Frisch, Science (1996):LSDA / 3-21G DFT calculation on 1026 atom RNA Fragment

Liang, Yokojima & Chen, JPC, 2000
Linear Scaling Calculation for Excited State

LDM-TDDFT: CnH2n+2
Fast Multiple Method

LODESTAR: Software Package for Complex Systems
Characteristics :O(N) Divide-and-ConquerO(N) TDHF (ab initio & semiemptical)
O(N) TDDFT
CNDO/S-, PM3-, AM1-, INDO/S-, & TDDFT-LDM
Light Harvesting SystemNonlinear Optical

Photo-excitations in Light Harvesting System II
generated by VMD
strong absorption: ~800 nm
generated by VMD


Carbon Nanotube

Quantum mechanical investigation of the field emission from Quantum mechanical investigation of the field emission from the tips of carbon nanotubesthe tips of carbon nanotubes
Zettl, PRL 2001Zheng, Chen, Li, Deng & Xu, Phys. Rev. Lett. 2004

Molecular Mechanics Force Field
• Bond Stretching Term
• Bond Angle Term
• Torsional Term
• Electrostatic Term
• van der Waals interaction
Molecular Mechanics
F = Ma
F : Force Field

Bond Stretching PotentialEb = 1/2 kb (l)2
where, kb : stretch force constantl : difference between equilibrium & actual bond length
Two-body interaction

Bond Angle Deformation PotentialEa = 1/2 ka ()2
where, ka : angle force constant
: difference between equilibrium & actual bond angle
Three-body interaction

Periodic Torsional Barrier PotentialEt = (V/2) (1+ cosn )where, V : rotational barrier
: torsion angle n : rotational degeneracy
Four-body interaction

Non-bonding interaction
van der Waals interactionfor pairs of non-bonded atoms
Coulomb potential
for all pairs of charged atoms

Force Field Types
• MM2 Molecules
• AMBER Polymers
• CHAMM Polymers
• BIO Polymers
• OPLS Solvent Effects

Algorithms for Molecular Dynamics
Runge-Kutta methods:
x(t+t) = x(t) + (dx/dt) t
Fourth-order Runge-Kutta
x(t+t) = x(t) + (1/6) (s1+2s2+2s3+s4) t +O(t5) s1 = dx/dt s2 = dx/dt [w/ t=t+t/2, x = x(t)+s1t/2] s3 = dx/dt [w/ t=t+t/2, x = x(t)+s2t/2] s4 = dx/dt [w/ t=t+t, x = x(t)+s3 t]
Very accurate but slow!

Algorithms for Molecular Dynamics
Verlet Algorithm:
x(t+t) = x(t) + (dx/dt) t + (1/2) d2x/dt2 t2 + ... x(t -t) = x(t) - (dx/dt) t + (1/2) d2x/dt2 t2 - ...
x(t+t) = 2x(t) - x(t -t) + d2x/dt2 t2 + O(t4)
Efficient & Commonly Used!
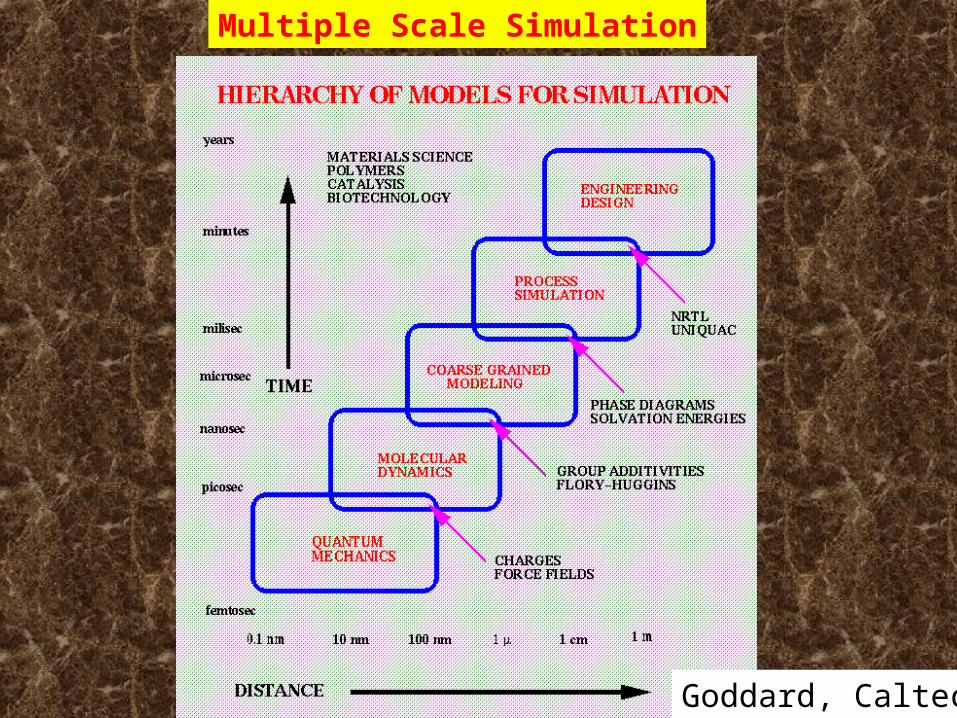
Goddard, CaltechGoddard, Caltech
Multiple Scale Simulation

Large Gear Drives Small Gear
G. Hong et. al., 1999

Nano-oscillators
Zhao, Ma, Chen & Jiang, Phys. Rev. Lett. 2003
Nanoscopic Electromechanical Device (NEMS)



Computer-Aided Drug Design
GENOMICS
Human Genome Project

Computer-aided drug design
Chemical Synthesis
Screening using in vitro assay
Animal Tests
Clinical Trials
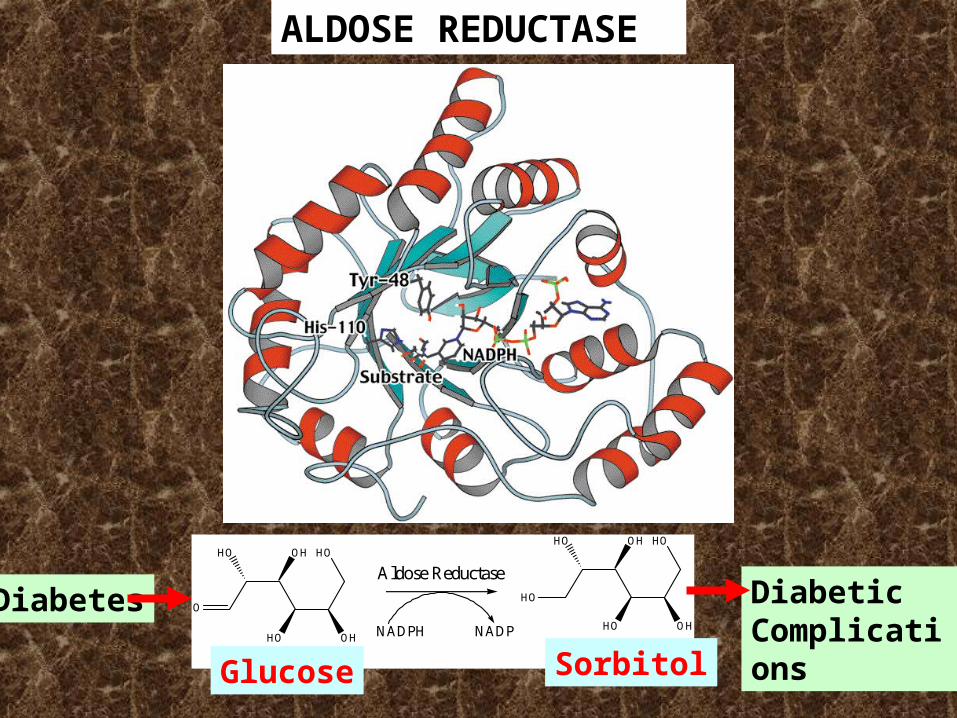
ALDOSE REDUCTASE
O
HO OH
HO OH
HO
glucose
HO
HO OH
HO OH
HO
sorbitol
Aldose Reductase
NADPH NADP
Diabetes DiabeticComplications
Glucose Sorbitol

Design of Aldose Reductase Inhibitors
Aldose Reductase
Inhibitor

Database for Functional GroupsDescriptors: Electron negativityVolume

2.5 3.0 3.5 4.0 4.5
2.5
3.0
3.5
4.0
4.5
5.0
Exp
erim
enta
l val
ues
Fig 3 QSAR OF INHIBITOR CONCENTRATION OF INHIBITING AR Log(IC50
)
NH
NMe
NH
HN
O
O
O
5'
6'
7'8'
X
-0.1 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Exp
erim
etal
val
ues
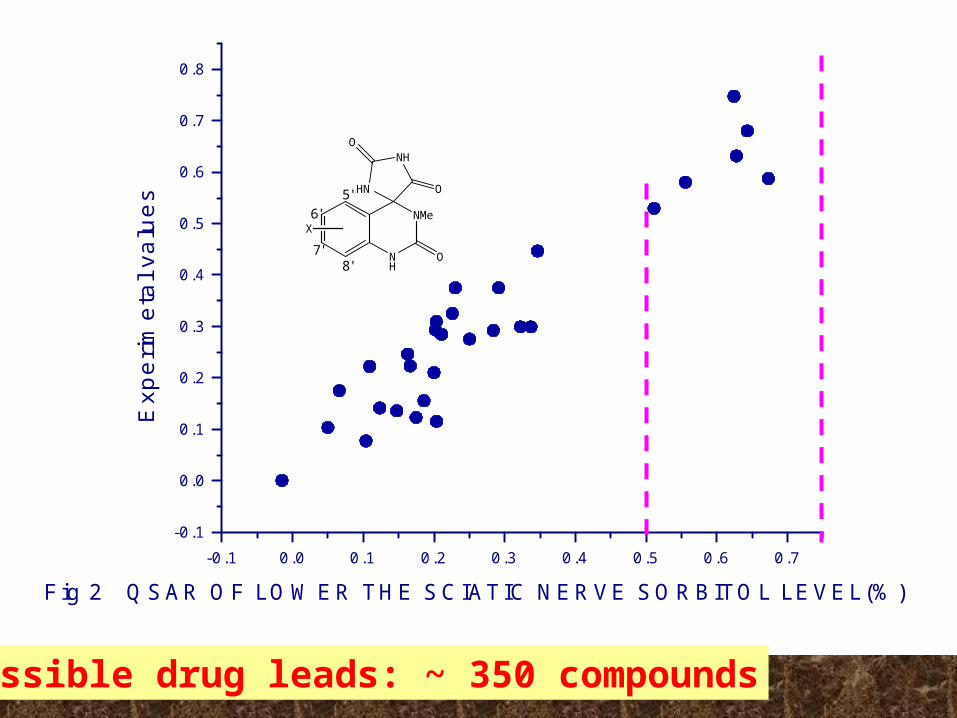
Fig 2 QSAR OF LOWER THE SCIATIC NERVE SORBITOL LEVEL(%)
NH
NMe
NH
HN
O
O
O
5'
6'
7'8'
X
Possible drug leads: ~ 350 compounds


TYR48 LYS77
HIS110
TRP111
PHE122
TYP219
TRP20
CYS298LEU300
NADPH
TRP79
VAL47
Aldose Reductase Active Site Structure
Cerius2 LigandFit

To further confirm the AR-ARI binding,We perform QM/MM calculations on drug leads.
CHARMM
5'-OH, 6'-F, 7'-OH
NH
NMe
NH
HN
O
O
O
5'
6'
7'8'
X
Binding energy is found to be –45 kcal / mol

Docking of aldose reductase inhibitor
Cerius2 LigandFit
Aldose reducatse
(4R)-6’-fluoro-7’-hydroxyl-8’-bromo-3’-methylspiro-[imidazoli-dine-4,4’(1’H)-quinazoline]-2,2’,5(3’H)-trione
Inhibitor
Hu & Chen, 2003

Interaction energy between ligand and protein
Quantum Mechanics/Molecular Mechanics (QM / MM)
Hu & Chen, 2003

a:Inhibitor concentration of inhibit Aldose Reductase;b: the percents of lower sciatic nerve sorbitol levelsc: interaction with AR in Fig. 4
NH
NMe
NH
HN
O
O
O
5'
6'
7'8'
X
Our Design Strategy
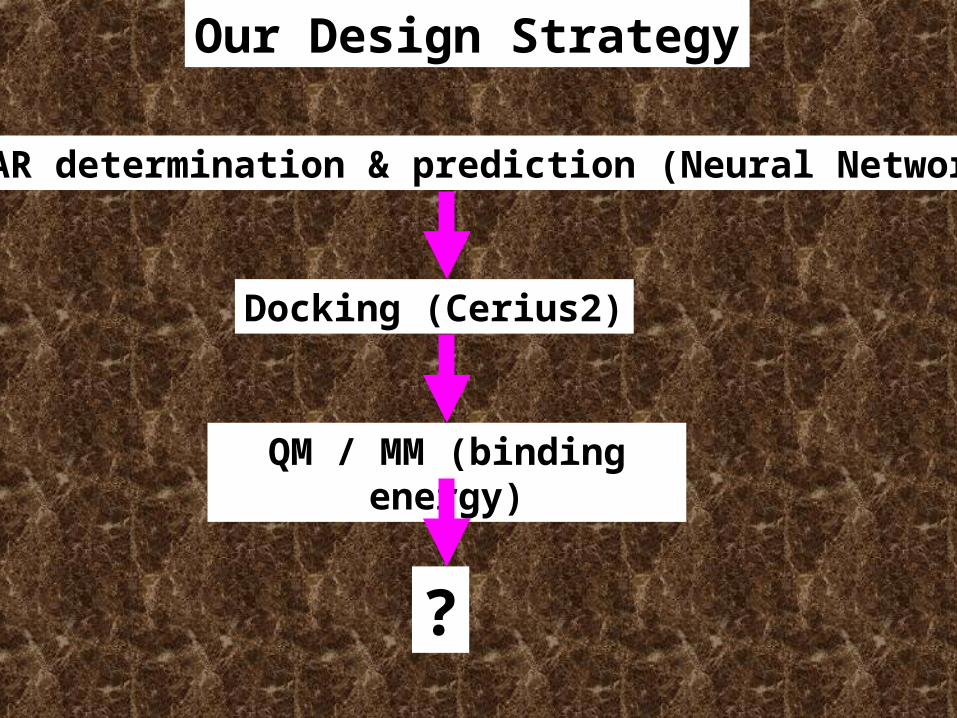
QSAR determination & prediction (Neural Network)
Docking (Cerius2)
QM / MM (binding energy)
?

Software in Department
1. Gaussian2. Insight II CHARMm: molecular dynamics simulation, QM/MM Profiles-3D: Predicting protein structure from sequences SeqFold: Functional Genomics, functional identification
of protein w/ sequence and structure comparison NMR Refine: Structure determination w/ NMR data 3. Games4. HyperChem5. AutoDock (docking)6. MacroModel6. In-House Developed Software
LODERSTARNeural Network for QSARMonte Carlo & Molecular Dynamics

Lecture Notes for Physical Chemistry
Year 2 1.Intermediate Physical Chemistry (CHEM2503) (Powerpoint format .ppt)
Year 3 1.Advanced Physical Chemistry (Powerpoint format .ppt) 2.Electronic Spectroscopy (Powerpoint format .ppt) 3.Electronic Spectroscopy (assignment) (rar file)
Postgraduate Course 1.Research Techniques in Chemistry (Powerpoint format .ppt) Course Work Download Molecule
M.Sc Course 1.Computational Modeling of Macromolecular Systems (Powerpoint format .ppt) Download Molecule

Step 1: Build up the structure of the formaldehyde.1. Run HYPERCHEM software in the start menu.2. Double click the drawing tool to open the elements table dialogue box and select carbon atom.
Close the element table. (Drawing tool)3. L-click the cursor on the workspace. A carbon atom will appear.
(Make sure drawing tool is selected. R-click on the atom if you want to delete it)4. Repeat (2) and choose oxygen instead of carbon. Move the cursor to the carbon centre and drag the mouse from the carbon centre to an empty workspace. (A single bond is created between carbon atom and oxygen atom.)5. L-click the bond between carbon and oxygen to create a double bond.6. L-click on Build in the menu bar and switch on ‘add H & model build’ (i.e. make sure a tick
appeared on the left of this function.).
Step 2: Optimize the structure using RHF and 6-31G* basis set.7. L-Click on Setup in the menu bar and L-click ab Initio;
L-Click on 6-31G*; then, L-Click on Options button; Select RHF, set Charge to 0 and Multiplicity to 1 (default for charge 0);L-Click OK buttons after modifications were done.
8. L-Click on Compute in the menu bar and select Geometry Optimization;Select Polak-Ribiere and set RMS gradient to 0.05 and max cycles to 60;
L-Click OK button (The calculation will be started. Repeat the step till “Conv=YES” appears in the status line.).Record the energy appeared in the status line9. L-Click on Compute in the menu bar and select Orbitals.Record energy levels and point groups of required molecular orbitals (MO)(Optional: You can draw the contour plot of the selected orbital and visualize the shape of the orbital.)10. L-Click on Compute in the menu bar and select Vibrations.11. L-Click on Compute in the menu bar and select Vibrational Spectrum.Record the frequencies of different vibrational modes and their corresponding oscillator strengths.(Optional: You can turn on animate vibrations, select any vibrational modes, and L-Click on OK button. The molecule begins to vibrate. To suspend the animation, L-Click on Cancel button.)
H
C
O
H
Formaldehyde
HYPERCHEM ExercisePart A: Study the electronic structure and vibrational spectrum of formaldehyde
Procedures:

Part B: Molecular Dynamics of Tetrapeptide1. L-click Databases on the menu bar. Choose Amino Acids.2. Select Beta sheet.3. L-click Ala, Tyr, Asp and Gly to create tetrapeptide Ala-Tyr-Asp-Gly.4. L-click on rotate-out-of-plane tool and use it to rotate the molecule to a proper angle for observation and measurements.(Rotate-out-of-plane tool) 5. L-Click on Setup in the menu bar and L-click Molecular Mechanics;
L-Click on MM+;L-Click OK buttons after modifications were done.
6. L-Click on Compute in the menu bar and select Geometry Optimization;7. Record the total energies.8. L-Click on Compute in the menu bar and L-click Molecular Dynamics;
Run molecular dynamics at 0K and 300K with constant temperature.Simulation Time: 1ps
9. Record the total energies.
Part C: Molecular Dynamics of Ribosomal ProteinProcedures:10. Use a web-browser and Go to http://yangtze.hku.hk/lecture_notes.htm.11. R-click the title labeled “Download molecule” and save it in a folder in your local disk (C:\).12. L-click on File in the menu bar and select open to load in the molecule.
(You should notify that this file has extension filename .ENT and is in PDB format.)13. L-click on rotate-out-of-plane tool and use it to rotate the molecule to a proper angle for
observation and measurements.(Rotate-out-of-plane tool) 14. L-Click on Setup in the menu bar and L-click Molecular Mechanics;
L-Click on MM+;L-Click OK buttons after modifications were done.
15. L-Click on Compute in the menu bar and L-click Molecular Dynamics;Run molecular dynamics at 300K with constant temperature.Simulation Time: 1ps
16. Record the total energy.