computational study of weakly interacting complexes · computational study of weakly interacting...
TRANSCRIPT

Computational Study of Weakly Interacting Complexes
Dissertation
for the Degree of
Doktor der Naturwissenschaften (Dr. rer. nat.)
Ruhr-Universität Bochum
Elsa Sánchez García
2006

2
This work was carried out between September 2002 and March 2006 under the
supervision of Prof. Dr Luis Montero, Laboratorio de Química Computacional y Teórica,
Universidad de la Habana and Prof. Dr. Wolfram Sander, Lehrstuhl für Organische
Chemie II, Ruhr Universität Bochum.
First Referee: Prof. Dr. M. Havenith-Newen
Second Referee: Prof. Dr. L. A. Montero Cabrera
Subsidiary Subject: Prof. Dr. B. Benecke (Biochemistry)
Dissertation submitted:
Disputation: 19.07.2006

3

4
List of Publications
1. Sánchez-García, Elsa; Montero, Luis. A; Sander, Wolfram. Computational
Study of Non-Covalent Complexes between Formamide and Formic Acid. Journal of Physical Chemistry A (2006), in press.
2. Montero, Luis. A; Sánchez-García, Elsa. Similarity Analysis of Molecular Systems Formed by Amylose and Organoleptic Compounds. Revista Cubana de Física (2006), in press.
3. Sánchez-García, Elsa; Studentkowski, Marc; Montero, Luis A.; Sander,
Wolfram. Non-covalent Complexes between Dimethyl ether and Formic Acid-an
Ab initio and Matrix Isolation Study. ChemPhysChem (2005), 6(4), 618-624.
4. Sánchez-García, Elsa; George, Lisa; Montero, Luis A.; Sander, Wolfram. 1:2
Formic Acid/Acetylene Complexes: Ab initio and Matrix Isolation Studies of
Weakly Interacting Systems. Journal of Physical Chemistry A (2004), 108(52),
11846-11854.
5. George, Lisa; Sánchez-García, Elsa; Sander, Wolfram. Matrix Isolation Infrared
and ab initio Study of Formic Acid-Acetylene Interaction: Example of H…π and
C-H…O Interaction. Journal of Physical Chemistry A (2003), 107(35), 6850-
6858.
6. García, Elsa Sánchez; Montero, Luis A.; Hermida, Jose M.; Cruz, Roberto;
Gonzalez, Gerardo. Calculation of Association Energy in Acetone Clusters by the
Multiple Minima Hypersurface Approximation. Revista Cubana de Física
(2000), 17(1-2), 41-46.
7. Sánchez-García, Elsa; Mardyukov, Arthur; Studentkowski, Marc; Montero,
Luis. A; Sander, Wolfram. Furan - Formic Acid Dimers – an Ab initio and Matrix
Isolation Study, (2006) submitted.
8. Sánchez-García, Elsa; Montero, Luis. A; Sander, Wolfram. Computational Study
of Weakly Interacting Complexes between Acetylene and Oxygen Heterocycles,
(2006) in preparation.

5
Scientific Meetings (Poster = P, Oral Presentation=O)
2006 1st Workshop Forschergruppe 618, Universität Bochum, Germany. (O)
2005 International Chemical Congress of Pacific Basin Societies (Pacifichem).
Area11 - Physical and Theoretical Chemistry. Computational Quantum
Chemistry: Methodology and Application. Honolulu, Hawaii, United
States. (P)
2003 Gordon Research Conference of Physical Organic Chemistry. Plymouth,
New Hampshire, United States. (P)

6
Index
1. General Introduction ..................................................................................................... 9
1.1 The formic acid molecule......................................................................................... 10
1.2 Hydrogen bonds and weak interactions.................................................................... 11
- Some definitions of the hydrogen bond ................................................... 11
- Classification of hydrogen bonds............................................................. 12
- Some applications of the hydrogen bond................................................. 15
- Cooperativity............................................................................................ 17
- Proton transfer.......................................................................................... 18
- Methods of studying hydrogen bonds ...................................................... 19
- Spectroscopy methods.............................................................................. 20
- Diffraction methods ................................................................................. 22
- Matrix isolation ........................................................................................ 24
1.3 Quantum mechanical calculations............................................................................ 26
- The quantum-mechanical treatment of molecules. .................................. 26
- Electron correlation.................................................................................. 28
- Semiempirical methods............................................................................ 30
- Density functional theory......................................................................... 36
- Basis sets .................................................................................................. 38
- Basis set superposition error .................................................................... 41
2. The Multiple Minima Hypersurface (MMH) Approach............................................. 46
2.1 Introduction.............................................................................................................. 46
- The Multiple minima problem ................................................................. 46
- Chemical similarity searching.................................................................. 49
2.2 The Multiple Minima Hypersurface (MMH) approach ........................................... 51
3. Formic Acid Complexes with Formamide and Dimethyl ether.................................. 58
3.1 Introduction.............................................................................................................. 58
3.2 Computational methods............................................................................................ 59
3.3 Formic acid – formamide complexes. Results and discussion................................. 60
- Formic acid – formamide dimers ............................................................. 60

7
- Geometries and binding energies. Analysis of the intermolecular
interactions ............................................................................................... 60
- Comparison with other dimers ................................................................. 67
- Methods and basis set influence on the calculated geometries and binding
energies of the FMA – FA dimers............................................................ 69
- Effect of the BSSE on the calculated geometries and binding energies .. 72
- Intramolecular distances and vibrational frequencies. Calculated spectra73
- Larger systems ......................................................................................... 76
- 1:2 Formic acid – formamide complexes................................................ 76
- Analysis of the intermolecular interactions in the trimers ....................... 79
- 1:4 Formic acid – formamide complexes................................................. 86
- Comparison of FMA – FA complexes with the crystal structure ............ 88
3.4 Formic acid – dimethyl ether dimers. Results and discussion ................................. 91
- Geometries and binding energies ............................................................. 91
- Geometry optimization including BSSE.................................................. 95
- Intramolecular distances and vibrational frequencies .............................. 97
- Comparison with matrix isolation spectroscopy results........................... 99
3.5 Conclusion.............................................................................................................. 101
4. Formic Acid Complexes with π systems .................................................................. 104
4.1 Introduction .......................................................................................................... 104
4.2 Computational methods........................................................................................ 106
4.3 Formic acid – furan dimers. Results and discussion ............................................ 108
- Geometries and binding energies. .......................................................... 108
- Type (i) complexes................................................................................. 111
- Type (ii) complexes................................................................................ 117
- Other FA – furan geometries................................................................. 118
- Basis set influence on the calculated geometries of the FA – furan dimers
................................................................................................................ 122
- Effect of the BSSE on the calculated geometries and binding energies 129
- Comparison with other furan complexes ............................................... 131
- Comparison with matrix isolation spectroscopy results......................... 132

8
4.4 1:2 Formic acid – acetylene complexes. Results and discussion ......................... 136
- Geometries and binding energies ........................................................... 136
- Intramolecular distances and vibrational frequencies ............................ 141
- Comparison with matrix isolation spectroscopy results......................... 145
- Analysis of the intermolecular interactions in the trimers ..................... 148
4.5 Conclusion ............................................................................................................. 152
5. Acetylene Complexes with Oxygen Heterocycles. An Outlook............................... 155
5.1 Introduction............................................................................................................ 155
5.2 Computational methods ......................................................................................... 158
5.3 Acetylene – furan dimers. Results and discussion................................................. 159
5.4 Acetylene – THF dimers. Results and discussion.................................................. 164
5.5 Acetylene – 1,4-dioxane dimers. Results and discussion ...................................... 167
5.6 Conclusion ............................................................................................................. 168
6. General Conclusion................................................................................................... 169
7. Summary ................................................................................................................... 174
8. References................................................................................................................. 181

9
1. General Introduction
Hydrogen bonds and weak interactions play important roles in molecular recognition,
properties of condensed phases, solid state reactions, crystal engineering, and in
determining the shapes and stabilities of biomolecules.[1-3] In contrast to the conventional
strong and moderate hydrogen bonds, which have been extensively described, the nature
and characteristics of weak interactions is not an undisputed field.[3] To find out a
representative and large set of the possible molecular arrangements (minima) of hydrogen
bonded and weakly interacting complexes is quite often one of the most complicated
questions.
Therefore, the structural analysis application of the Multiple Minima Hypersurface
(MMH) approach[4-6] as a tool for localizing minima is introduced here. Randomly
arranged clusters are generated as starting points and subsequently optimized. The results
are processed with programs especially written for this purpose and the geometries are
afterward re-optimized at higher level of theory. The bases of the MMH procedure for
searching local minima are presented and discussed.
Dimers and larger aggregates of formic acid with acetylene, dimethyl ether, formamide
and furan show both strong hydrogen bonds and weak interactions and are studied using
MMH in combination with high level ab initio calculations. The comparison of the
various minima and systems allows for a detailed discussion of the individual
contributions of intermolecular interactions in formic acid complexes. An outlook to the
dimers of acetylene with furan, tetrahydrofuran and 1,4-dioxane complements the
analysis of the intermolecular interactions.
The theoretical results are compared to data from matrix isolation spectroscopy or
crystal structure analysis. The influence of the theoretical methods and the basis set
superposition errors (BSSE) on the calculated geometries and binding energies of the
complexes is also studied. The Multiple Minima Hypersurface (MMH) approach,
combined with ab initio quantum-chemical calculations is established as a very reliable
procedure for localizing weakly and moderate hydrogen bonded minima.

10
1.1 The formic acid molecule
Formic acid (HCOOH) is the smallest monocarboxylic acid and one of the simplest
molecules that forms two hydrogen bonds.[7] Therefore, its structure in the gas and
condensed phases has been much studied.[8-29] The formic acid molecule displays
rotational isomerism[8, 9] between the experimentally well characterized s-trans[10, 11] and
s-cis conformers[9, 12] (Figure 1.1). The s-trans form is 4 kcal/mol lower in energy than
the s-cis form.[7-9] Lundell et al.[13] generated the s-cis conformer by multiphoton IR
irradiation of the s-trans conformer in low-temperature matrices.[7, 13]
Figure 1.1: s-trans and s-cis conformers of the formic acid molecule
In the gas phase the monomer and the dimer of formic acid are forming an equilibrium
in which the dimer is more stable by 14 kcal/mol.[7, 14] Like acetic acid, but unlike many
others carboxylic acids which retain the dimeric structure in the crystalline state, the
crystal structure of formic acid shows an infinite polymeric chain in which each molecule
is linked to two neighbors by a hydrogen bond.[8, 15] At very low temperatures (4.5 K) the
chains of the s-trans form are found in the crystal structure, whereas chains of the s-cis
form are found at higher temperatures.[8, 15] Formic acid is a strongly hydrogen bonded
liquid[16] which probably consists of short chains similar to those observed in the solid.[8]
However, the structure of the liquid formic acid is still a subject of debate, since the
cyclic dimer, an acyclic open dimer[17], polymeric chains [7, 18] and a mixture of several of
these species[7] have been proposed as main constituents. Due to the properties of formic
acid, the hydrogen bonding with other molecules can be used as a model for many
chemical and biochemical systems which exhibit the organic acidic type of bonding, like
proteins and the base pairs in nucleic acids.[19-22]

11
1.2 Hydrogen bonds and weak interactions
Some definitions of the hydrogen bond
The evidences of hydrogen bond were observed long before it was identified and given
a name.[1] Since the beginning of the last century, scientists like Werner (1902), Hantzsch
(1910) and Pfeiffer (1914)[23, 24] used the terms “Nebenvalenz” (near valence) and “innere
Komplexalzbildung” to describe both intra- and intermolecular hydrogen bonds.[1] Moore
and Winmill[25] in 1912 used the term weak union in describing properties of amines in
aqueous solutions.
In “An Introduction to Hydrogen Bonding”, Jeffrey describes that, according to
Pauling, the concept of the hydrogen bond is attributed to M.L. Huggins and
independently to W.M Latimer and W.H Rodebush.[1] In 1922, Huggins affirmed that “a
positively charge kernel containing no electrons in its valence shell reacting with an
atom containing a lone valence pair can form a weak hydrogen bond”.[1] But two years
earlier in 1920, Latimer and Rodebush published that “The hydrogen nucleus held by two
octets constitutes a weak bond”. [1, 26] Both of them mentioned the example of the amines
in aqueous solutions described previously by Moore and Winmill.
It was Pauling who really introduced the concept of the hydrogen bond with the
statements: “Under certain conditions an atom of hydrogen is attracted by rather strong
forces to two atoms instead of only one, so it may be considered to be acting as a bond
between them. This is called a hydrogen bond” and “A hydrogen atom with only one
stable orbital cannot form more than one pure covalent bond and the attraction of the
two atoms observed in hydrogen bond formation must be due largely to ionic forces”.[27]
Therefore, Jeffrey states that hydrogen bonds are formed when the electronegativity,
according to Pauling, of A relative to H in an A-H covalent bond is such as to withdraw
electrons and leave the proton partially unshielded. To interact with this donor A-H bond,
the acceptor B must have lone-pair electrons or polarizable π electrons.[1]
The first text devoted entirely to hydrogen bonding “The Hydrogen Bond” was written
by Pimentel and McClellan in 1960.[28] They give a more general definition of hydrogen
bond: “A hydrogen bond exists between the functional group, A-H, and an atom or a

12
group of atoms B, in the same or different molecules when (a) there is evidence of bond
formation (association or chelation), (b) there is evidence that this new bond linking A-H
and B specifically involves a hydrogen atom already bonded to A”.[28] It is important to
realize that the Pimentel and McClellan definition makes no assumptions about the nature
of the A and B atoms, and that it enables an evaluation of the hydrogen bonding potential
of groups like C-H and π acceptors.[29]
As Jeffrey points out, a lot has been written about hydrogen bonds, and some concepts
are been continuously rediscovered.[1] Thus, the C-H hydrogen bonds are currently a
point of interest of the scientific community, but they were reviewed more than 50 years
ago by Hunter.[30] However, nowadays the definition of hydrogen bond by Pimentel and
McClellan is the most accepted due to its practical applications and suitability for both
experimental and theoretical investigators.[1]
Classification of hydrogen bonds
The hydrogen bonds are classified by Jeffrey in three categories according to their
energies and the nature of the A-H…B interactions.[1] The strong hydrogen bonds have
bond energies between 15 – 40 kcal/mol and the A-H…B interaction is mostly covalent
with H…B bond lengths from 1.2 to 1.5 Å and bond angles of 175 – 180°. They have an
electron density deficient donor group or an acceptor group with an excess of electron
density.[1]
Moderate hydrogen bonds are those which have bond energies between 4 – 15 kcal/mol
and the A-H…B interactions are mostly electrostatics with H-B bond lengths of 1.5 – 2.2
Å and bond angles of 130 – 180°.[1] They are mostly formed by neutral donor and
acceptor groups in which the donor A atoms are more electronegative than the hydrogen
and the acceptor B atoms have lone-pair unshared electrons. According to Jeffrey, these
are the most common hydrogen bonds in chemistry and nature and essential components
of the structure and function of biological molecules.[1]
Weak hydrogen bonds have bond energies between 1 – 4 kcal/mol and the A-H…B
interactions are basically electrostatic although probably also involving electron
correlation, with H-B bond lengths of 2.2 – 3.2 Å and bond angles of 90 – 150°. They are

13
formed when the hydrogen atom is covalently bonded to a slightly more electroneutral
atom relative to hydrogen, as in C-H or when the acceptor group has π electrons, such as
C≡C or an aromatic ring. These interactions have similar energies and geometries than
van der Waals complexes, however, differ from the latter by a directional involvement of
the A-H bond.[1]
As can be seen in Figure 1.2,[31] Desiraju classifies the hydrogen bonds in a very
similar way to Jeffrey, but he names the strong hydrogen bonds “very strong”, and the
moderate “strong”. This distinction comes from supramolecular considerations, since
Desiraju means by “strong” bonds those that are able to control crystal and
supramolecular structure effectively. By weak, Desiraju and Steiner mean hydrogen
bonds whose influence on crystal structure and packing is variable.[29] In this sense, a
“strong” hydrogen bond is one which is much stronger than a van der Waals interaction
while a weak hydrogen bond is one which is not. In order to be consistent, the
classification made by Jeffrey is used here and Desiraju’s classification is referred in
“italics” if necessary. Thus, according to Desiraju`s classification, the O-H…O=C and N-
H…O=C interactions are “strong” (moderate for Jeffrey) hydrogen bonds. The C-H…O,
C-H…N, N-H…π, O-H…π and C-H…π interactions are weak hydrogen bonds.
Desiraju and Steiner also classify the hydrogen bonds as “conventional” and “non-
conventional”,[29] based on the “conventionality” of the donor and acceptor groups,
where the categories “strong” and “conventional” have many points in common.
However, there are “strong” non-conventional hydrogen bonds as well as there are weak
conventional hydrogen bond types. The classification of hydrogen bonds still is a
controversial field. For instance, methyl donors are borderline cases between weak
hydrogen bonds and van der Waals interactions because of their large dispersion
contribution.

14
Figure 1.2: Desiraju’s classification of hydrogen bonds. This figure has been taken
from “Hydrogen Bridges in Crystal Engineering: Interactions without Borders” by G. R.
Desiraju[31]
The C-H…π interaction is another borderline case, since it is considered by some
authors like Nishio[3] as the weakest hydrogen bond occurring between a soft acid (CH)
and a soft base (π electrons), whereas others will not name it as a hydrogen bond at all.
Nevertheless, it has gradually become accepted that the C-H…π interaction plays a role
in a variety of chemical and biochemical phenomena like the stabilization of proteins
structures, the conformation of coordination compounds and the selectivity in organic
reactions.
Desiraju and Steiner point out some differences between “strong” and weak hydrogen
bonds:[29]
• The van der Waals cut-off criterion in the H…B distance for the assignment of
hydrogen bond character is inappropriate for weak hydrogen bonds. This

15
criterion does not stand on experimental or theoretical ground, but has only
been established for reasons of apparent convenience. This criterion works
reasonably well for “strong” hydrogen bonds which are almost always short
enough to fulfill it. But even for these, due to sterical reasons, bonds like N-
H…O can be elongated beyond the van der Waals separation. Weak hydrogen
bonds, especially A-H…π interactions are even longer.[29]
• The results of crystallographic and spectroscopic investigations do not
necessarily agree to each other as well as they do for “strong” hydrogen bonds.
Unlike the “strong” hydrogen bonds, large distortions are possible with very
little changes of the energies in weakly bonded systems, due to their shallow
potential energy surfaces. Consequently, the correlation between
crystallographic and spectroscopic properties is very variable.[29]
• The hydrogen bond in general is considered as the initial state of a proton
transfer process, but only for “strong” hydrogen bonds do such proton transfer
processes occur with significant rates.[29]
Some applications of the hydrogen bond
As Nishio explains, noncovalent forces play an important role in chemical reactions,
molecular recognition, and in many biochemical and chemical processes. While strong
covalent bonds bind the atoms together in a molecule, the noncovalent and weak
interactions determine the shape and the conformation of the molecule.[3]
Therefore, hydrogen bonding is very relevant to supramolecular chemistry. For
instance, the role of hydrogen bonding in determining the packing motifs of molecules in
crystals requires the recognition and understanding of the cooperative systems of
hydrogen bonding. As with molecules, any description of supramolecular structure
requires the knowledge of connectivity and geometry. For Jeffrey the connectivity is the
hydrogen-bonding pattern.[1] A knowledge of commonly occurring hydrogen-bond
patterns associated with particular donor and acceptor function groups can be used to
synthesize new supramolecular complexes.[1, 32]

16
Intermolecular hydrogen bonding also plays an important role in the way molecules
assemble in liquid crystals.[1] According to the name, liquid crystals are supramolecular
assemblies in one and two dimensions which constitute a state of matter between crystals
and liquids. They are also known as ordered liquids.[1] Ferroelectric and others liquid
crystals have been made through hydrogen bonding, and in some liquid crystals the
function of the hydrogen bonding is to increase the length of the rods.[1]
Molecular inclusion is another large and growing field of supramolecular chemistry. In
inclusion compounds like the hydrates and the cyclodextrines, hydrogen bonding is an
essential component of the host structure.[1]
The hydrogen-bonded helical and sheet structures proposed for proteins by Pauling,
Corey and Branson and the hydrogen bonded base-pair in the structure of DNA by
Watson and Crick are evidences of the importance of hydrogen bonding in the structure
and function of biological macromolecules.[1] Jeffrey explains that strong hydrogen bonds
are rare in biological structures since they are too rigid and not easily broken. For
example, the salt bridges in proteins and the P-OH…O=P bonds in nucleic acids are
hydrogen bonds. These bonds are generally interrupted by water molecules, which do not
form very strong hydrogen bonds, either as donors or acceptors. The weak interactions
like the C-H…O hydrogen bonds play also a role in biological structures.[1]
Hydrogen bonding is the major factor in determining the structure of the nucleic acids.
Inter- and intramolecular hydrogen bonding schemes have been also proposed for
polysaccharides. In protein structures, the peptide N-H and C=O groups form
intramolecular N-H…O=C hydrogen bonds which determine the conformation of the
peptide main chain, being responsible for the formation of helical or sheet structures. The
formation of these hydrogen bonds results in additional π character of the peptide C-N
bond, which results in a more rigid planar conformation. The side groups contain
hydrogen bond donor and acceptor groups which form the hydrogen bonds between the
polypeptide chains. [1]

17
Cooperativity
Hydrogen bonds show cooperative effects. Accordingly, the energy of an array of n
interlinked hydrogen bonds is larger than the sum of n-isolated hydrogen bonds, as
described by Desiraju.[29] Or, according to Jeffrey’s definition, cooperativity or non-
additivity represents the difference between calculating energies using atom-pair
potentials and many-atom potentials.[1] This non-additive property can be applied in
general to all intermolecular interactions. Cooperativity occurs because of the ability of
donor and acceptor groups to form hydrogen bonds is further increased by an increase in
polarity when the hydrogen bonds are part of a collective ensemble. Two different
mechanisms to producing this effect are described:[29]
Functional groups acting simultaneously as hydrogen bond donors and acceptors form
extended chains or rings in which the individual hydrogen bonds enhance each other’s
strength by mutual polarization.[29] This occurs mainly with hydroxyl groups and was
recognized in the crystal structure of small carbohydrates by Jeffrey.[1] From the crystal
structure of the cyclodextrins this effect was identified by Saenger.[33, 34] Since there are
no multiple bonds involved, it has been described by Jeffrey and Saenger as σ-bond
cooperativity.[1]
Charge flow in suitably polarizable π-bond systems increases donor and acceptor
strengths. This cooperativity involves hydrogen bonding between molecules with
conjugated multiple π systems and is also described as Resonance-Assisted Hydrogen
Bonding (RAHB).[1, 29, 35] This descriptor was first applied to hydrogen bonding in β-
diketone moieties and has since been extended and made more general by Gilli, Bertolasi
and Ferreti.[35] In some biological structures it is called π-cooperativity by Jeffrey and
Saenger.[1]
Cooperativity is particularly important in hydrogen bonding because of the diffuse
nature and high polarizability of the hydrogen and lone-pair electron densities; and
according to Jeffrey,[1] the most evident structural manifestation of σ-bond cooperativity
is the predominance of linear chains of …O-H…O-H…O-H… bonds in the crystal
structures of the monosaccharides and the cyclic hydrogen bond structures of

18
oligosaccharides and cyclodextrins. In carbohydrate hydrates, the water molecules use
their double-donor double-acceptor properties to link the chains of hydroxyl bonds into
three-dimensional nets. [1]
As Jeffrey explains, RAHB is important in many biological structures and has been
observed in the crystal structures of purines, pyrimidines and their complexes. In these
crystal structures, the hydrogen bonding extends beyond the base pairs to other
molecules, and infinite hydrogen bond – π bond networks link the molecules throughout
the crystal structures. In the nucleic acids, base-pairing between purines and pyrimidines
involves the hydrogen bonds which link two conjugated ring systems. Thus, RAHB plays
an important role in increasing the delocalization energy of the molecules involved and in
strengthening the hydrogen bonding. In proteins, the main chain is not an extended
conjugated system, since two peptide units are separated by a single C-C bond. However,
there is RAHB in the pleated-sheet hydrogen bond structures running laterally across the
main chains.[1]
Jeffrey describes that the first evidence of the Polarization Enhanced Hydrogen
Bonding came from the ab initio calculations of del Bene and Pople on cyclic and chain
water polymers.[36] The calculations of the cyclic sequential (H2O)n shows a increase of
hydrogen bond energy per bond from 5.6 kcal/mol for n = 3 to 10.6 kcal/mol for n = 5
and 10.8 kcal/mol to n = 6, simultaneously there was a corresponding shortening of the
calculated H…O bond lengths from 1.57 to 1.45 Å. An example of polarization via a
combination of σ and π bonds are the (HCN)n chains. Desiraju also explains that the
ethynyl group is of particular relevance to the phenomenon of cooperativity because it
can simultaneously form C-H…B and A-H…π hydrogen bonds.[29] Another case of
cooperativity is the steroid danazole where the hydrogen bonds form a cooperative
pattern with infinite chains of C-H…O and O-H…π interactions.[29]
Proton transfer
Hydrogen bonding can ultimately lead to proton transfer, but it is important to stress
the differences between proton transfer and hydrogen bonding.[1] The pyridine –
hydrogen fluoride complexes are one example for the transition of hydrogen bonding to

19
proton transfer. For the 1:1 pyridine – HF complex there is an F-H…N hydrogen bond
and no proton transfer although the F-H distance is long, 1.13 Å, and the H...N distance is
short, 1.32 Å. In the 1:2 and 1:3 complexes, there is proton transfer forming the
pyridinium cation.[1]
The fact that hydrogen bonding facilitates, or restricts, proton transfer is considered as
the most important chemical property of the hydrogen bond by authors like Jeffrey.[1] The
facility of hydrogen bonds to transmit H+(or H3O+) and OH- ions in water or an aqueous
media provides a catalysis mechanism for many reactions. In the field of molecular
biology proton transfer has been recognized as a significant component of enzyme
catalysis and the transmission of ions through membranes.[1]
Methods of studying hydrogen bonds
The following methods are frequently used to study hydrogen bonded systems,
according to Jeffrey:[1]
• Spectroscopy methods: They depend on exciting the vibrational or rotational
energy levels of molecules, resulting in the absorption, or emission of the incident
radiation at specific frequencies. This radiation can be electromagnetic or
neutrons. Spectroscopy methods include infrared and Raman, microwave and
NMR, among others. They provide information relating to structure and processes
on a picosecond time scale (10-10 – 10-15 sec). NMR spectroscopy provides
information at 10 – 10-4 sec.
• Diffraction methods: They depend on the three-dimensional periodicity of the
atoms in crystals to provide a diffraction grating for X-rays or neutrons of wave
lengths comparable to the interatomic distances. Diffraction by liquids gives
much less information, even when X-ray and neutron diffraction results are
combined for simple liquid such as water. As already mentioned, diffraction
methods include X-ray and neutron diffraction. They provide information on a 10
– 103 sec time scale.

20
• Thermochemical methods: Thermodynamic methods involve either direct
calorimetry or using the effect of hydrogen bonding on physical properties at
different concentrations or temperatures to determine the equilibrium constants
for the formation of the hydrogen bond. Thermochemical methods include
calorimetry of heats of mixing or dilution and the determination of enthalpies
directly or through the measurements of equilibrium constants. Like diffraction
methods, thermodynamic methods provide information on a 10 – 103 sec time
scale.
• Theoretical methods: They include ab-initio, density functional, semi-empirical,
and empirical methods.
Here we are presenting some aspects related to spectroscopic and diffraction methods,
according to Jeffrey’s point of view.[1]
Spectroscopy methods
Spectroscopic methods are more general and sensitive than diffraction methods. They
are used to identify hydrogen bonding in all states of matter. For example, the weak C-
H…O hydrogen bonds were identified by spectroscopists long before they were
recognized by the crystallographers.[1]
Infrared spectroscopy is the most used tool for identifying hydrogen bonding. Near
infrared spectroscopy uses the electromagnetic frequency ranges of 10 000 – 4000 cm-1,
middle, 4000–200 cm-1, and far 200–10 cm-1. Raman spectra are recorded in the range of
4000 – 10 cm-1. Most infrared studies of hydrogen bonding are in the mid IR range. With
these methods the hydrogen bonding is investigated by observing the transitions between
the vibrational levels of the molecules involved in hydrogen bonding.
Jeffrey points out some general IR criteria for hydrogen bonding:[1]
• The A – H stretching frequency νs, is shifted to lower frequencies (red shift).
This is accompanied with an increase in intensity and band width compared to
the isolated monomers.

21
• The A – H bending frequencies νb, move to higher frequencies.
• Upon cooling, νs shifts to high frequencies with increase in intensity and decrease
in band width, νb moves to lower frequencies with decrease in band width.
• Isotopic substitution of H by D lowers νs frequencies by a factor of around 0.75.[1]
With the introduction of interferometers in place of dispersive elements in IR
spectroscopy it is possible to record data of all frequencies at the same time. This method
is known as Fourier transform infrared spectroscopy and delivers in addition more
radiation with greater stability.[37]
The correlations between stretching frequencies and hydrogen-bond geometries have
been studied. A linear relationship has been found between νA-H and the A----B bond
distances for the strong O-H…O hydrogen bonds with νs 2700 cm-1 to 750 cm-1 and O----
O from 2.60 to 2.45 Å. For weaker bonds with O----O > 2.6 Å, the relationship is getting
curved, the agreement deteriorated, and the frequency shifts become increasingly
insensitive to changes in O----O distances.
The microwave rotational spectroscopy uses electromagnetic radiation in the frequency
region 109 – 1011 Hz to record the vibrational and rotational spectra of hydrogen-bonded
dimers and 1:1 adducts in gas phase. The analysis of these spectra provides information
about rotational constants, centrifugal distortion constants, nuclear quadrupole and
nuclear spin-nuclear spin coupling constants, and the Stark and Zeeman effects.
Molecular geometries, bond energies, force constants, electric dipole moments, electric
charges distributions, and electric quadrupole moments are derived from these
measurements. This method is sensitive enough to give information about very weak
hydrogen bonds and provides hydrogen bonding information that it is not compromised
by solvent effects or crystal field effects.[1]
There are two experimental methods in gas-phase microwave rotational spectroscopy:
One uses Stark modulated microwave spectroscopy with binary gas mixtures at
temperatures ≥ 175ºC and pressures of 50 mTorr and the second uses Fourier transform
microwave spectroscopy of a pulse of gas mixture diluted in argon and expanded
supersonically into an evacuated Fabry-Perot cavity.[1] Since gas-phase microwave

22
rotational spectroscopy measures the distances between centers of mass of the donor and
acceptors molecules, and hydrogen atoms make only a very small contribution to the
molecular mass, there are ambiguities in the measurements of geometries for weakly
bonded dimers where the A-H…B interaction is not linear.[1]
NMR spectroscopy is another very sensitive method for identifying hydrogen bonding.
It is less widely applied than infrared spectroscopy, because of the complexity of
hydrogen bonding in solution due to the uncertainty in identifying the particular bonds
and the number of molecules involved. NMR spectroscopy measures the degree to which
the proton is shielded by its electronic environment in terms of chemical shifts. These
shifts provide evidence of hydrogen bonding in liquids and solution and their magnitude
is quantitatively proportional to the strength of the hydrogen bond. As with infrared
spectroscopy, the change in chemical shift with concentration or temperature can give
equilibrium constants and therefore thermodynamic data. The sensitivity of 1H NMR to
changes in the electronic environment makes it a useful probe for detecting hydrogen
bonding from weak donors, such as C-H, and weak acceptors, such as multiple bonds and
aromatic rings.[1] With the development of multi-dimensional methods, NMR
spectroscopy has become a powerful tool for elucidating molecular structure in solution.
However, solution NMR spectroscopy has only relative little impact on the study of
hydrogen bonds, because of the complexity of the liquid state. The results of the solid-
state NMR spectroscopy can be correlated with those of crystal structure analysis and
therefore, solid-state NMR spectroscopy has become a tool for studying hydrogen
bonding.[1]
Diffraction methods
Location of the hydrogen atoms is essential to understand the nature of the hydrogen
bond, and crystal structure analysis by means of neutron diffraction is the most definitive
method for locating hydrogen atoms in hydrogen bonds. Together with infrared
spectroscopy, neutron diffraction provides a basis for distinguishing between strong,
moderate, and weak bonds. It also gives information to differentiate between two-, three-,
and four-center bonds. In strong hydrogen bonds the A-H…B bonds are almost collinear
and the covalent A-H bond length elongates to become almost equal to that of the

23
hydrogen bond. In moderate and weak hydrogen bonds the extension of the covalent A-H
bond is small and is marginally observable, but the A-H…B angles may deviate
significantly from 180º.[1]
Some advantages and disadvantages of X-ray vs. neutron diffraction single crystal
analysis are presented by Jeffrey:[1]
• An important aspect to consider is the availability of each method: While X-rays
are available on demand from laboratory instruments; neutron diffraction
equipments are only available in national or international specialized centers.
• For X-ray diffraction the time required for collecting the data is one day or less
for routine work and small crystals (~0.01 mm3, ~0.01 mg) can be used. Neutron
diffraction requires large crystals (~1 mm3, 1 – 2 mg) and a few weeks of data
collection time.
• With the X-ray diffraction method the hydrogen atoms are poorly located,
especially O-H with an accuracy of approximately 0.1 Å. Neutron diffraction
hydrogen positional parameters are comparable in accuracy to C, N, and O
(~0.001 Å)
• In X-ray diffraction experiments it can be difficult to distinguish between thermal
motion and disorder even for nonhydrogen atoms because of a fall-off in intensity
with scattering angle. In neutron diffraction fall-off in intensity with scattering
angle is only due to its thermal motion. Therefore it is easier to distinguish from
disorder.
• For X-ray diffraction, careful absorption corrections are necessary for other than
first-row atoms. In neutron diffraction the absorption is negligible, except for
crystals containing B, Cd, Sm, Li.[1]
One important advantage of both X-ray and neutron diffraction crystal structure analyses
over every other method of structure analysis is that both methods are over-determined.

24
Except for macromolecules such as proteins, the number of observations exceeds the
number of variable parameters, generally by a factor between five and ten.[1]
Matrix isolation
The matrix isolation technique was first introduced in 1954 by Pimentel and co-
workers,[38] who used the technique for systematic studies of free radicals and other
unstable or transient species. Matrix isolation was developed independently by Norman
and Porter.[39] The matrix isolation technique is used for trapping and producing chemical
species and preserving them in solidified inert (or occasionally reactive) gases at low
temperatures between 10 – 40 K. The matrices are formed, most of the time, by a non-
reactive substance like rare gases or solid nitrogen. The low temperatures required are
achieved by cryostats with closed helium-cycles.
Since solidified inert gases are used as matrix, interactions between the reaction
medium and the molecules to be studied are weak. To avoid reactions between isolated
molecules, the samples are highly diluted (1000 : 1) during the preparation of the matrix;
the molecules are thus spatially separated while embedded into the matrix lattice. In most
cases, rearrangements of trapped molecules are ruled out at 10 K by sufficiently high
energy barriers.[40]
For preparing the matrices, an excess of inert gas is condensed simultaneously with the
substance to be examined or a suitable precursor onto a cooled spectroscopic window,
usually CsI for IR spectroscopy and quartz or sapphire windows for UV/Vis
spectroscopy. Solids and liquids should have a sufficient vapor pressure (about 10-6
mbar) at temperatures which will not lead to decomposition. Gases can be mixed with
argon in an appropriate ratio before the deposition is performed.[40]
By codeposition of more than one substance, controlled reactions under matrix
conditions can be achieved. The matrix should have a temperature that is about 30 % of
the melting point of the noble gas (e. g. 30 K for Argon). Under these conditions, smaller
molecules like ozone, carbon monoxide or oxygen are able to diffuse through lattice gaps
to encounter a reaction partner. Another kind of reactions of matrix-isolated molecules

25
are photochemically induced processes by irradiation at a suitable wavelength using
mercury high pressure lamps or lasers.[40]
In order to characterize matrix isolated species, infrared, UV/VIS and EPR
spectroscopy are frequently used. Matrix isolation experiments allow, among other
applications to study unstable molecules generated by photolysis or gas phase
thermolysis, to observe directly reaction intermediates, to generate and to study novel
reactive species, to determine the structures of reactive species and to freeze out and to
study particular molecular conformations. An example for the latter is cyclohexane in its
chair and twist conformations.[41]
Another important application of the matrix isolation technique is the study of weakly
bound systems like H-bonded, charge transfer, and van der Waals complexes that can be
isolated in low temperature matrices, despite they dissociate under normal temperature
conditions due to the weak intermolecular forces. The IR bands of the components of
these complexes are significantly perturbed which provides an insight into the
intermolecular interactions. Therefore, matrix isolation, combined with spectroscopic
methods such as infrared spectroscopy, is a very important tool for the study of hydrogen
bonding.[40, 42]

26
1.3 Quantum mechanical calculations
Computational chemistry has become an important method for understanding hydrogen
bonding. In addition to the global minimum, computational methods can locate secondary
minima and stationary points of higher order. It is also possible to study the
interconversion pathways from one minimum to another and the magnitudes and shapes
of energy barriers along these paths. In addition, quantum chemical methods greatly
improve the understanding of the perturbations in vibrational spectra that accompany the
formation of a hydrogen bond. A frequent problem of experimental studies of hydrogen
bonded complexes is separating the intrinsic properties of the complex from the
perturbations due to interactions with the solvent. In this respect, an advantage of
computational methods is that they are free of complicating solvent effects.[43]
The quantum-mechanical treatment of molecules.
Quantum chemical methods are based on the time-independent Schrödinger equation:
H r R E r RΨ Ψ( , ) ( , )= (2.1)
where Ψ(r,R) is the wave function that represents the “trajectories” of the particles and
should be single-valued, quadratically integrable and continuous. H is the Hamiltonian
operator that returns the system energy, E, as an eigenvalue. For a molecule with n-
electrons and N nuclei the Hamiltonian operator is:
H(r,R)= Tel+ T nucl + V nucl,el + V el,el+ V nucl,nucl (2.2)
where:
Tel operator of the kinetic energy of the electrons.
Tnucl operator of the kinetic energy of the nuclei.
Vnucl,el attraction potential between the electrons and the nuclei.
Vel,el repulsion potential between the electrons.
Vnucl,nucl repulsion potential between the nuclei.
r set of coordinates of the n electrons.

27
R set of coordinates of the N nuclei
The Born-Openheimer approximation is used to simplify the solution of the
Schrödinger equation. Under typical physical conditions, the nuclei of molecular systems
are moving much more slowly than the electrons since the mass of a typical nucleus is
thousands of times greater than that of an electron. Consequently, according to the Born-
Openheimer approximation, the electronic energies are computed for fixed nuclear
positions. Therefore, the nuclear kinetic energy term is taken to be independent of the
electrons, correlation in the attractive electron-nuclear potential energy term is
eliminated, and the repulsive nuclear-nuclear potential energy term becomes a simply
evaluated constant for a given geometry.[44]
Due to the electron-electron repulsion term in the Hamiltonian, an exact solution to the
Schrödinger equation is not possible for systems with more that one electron. However, a
number of simplifying assumptions and procedures do make an approximate solution
possible for a large range of molecules. As Scheiner explains in “Hydrogen bonding: A
Theoretical Perspective”,[43] the usual method is the Hartree-Fock (HF) approximation
where electron 1 is considered to move in the field of the electron cloud associated with
the probability distribution of all other electrons. The same idea is applied to electron 2
which moves in the time-averaged field of electron 1 plus all the others, and so on.
Solution of the 1-electron Hartree-Fock equation for each electron changes its probability
density, thereby altering the field it sets up for the other electrons. Consequently, the
equations are solved iteratively, until the 1-particle wave function and the fields
generated there from no longer change appreciably from one cycle to the next. Because
of this, sometimes the SCF abbreviation of self consistent field is used synonymously
with HF.[43]
The Hartree-Fock approximation neglects the electron correlation. Since the electrons
are constantly aware of each others’ presence via their electrostatic repulsion, they tend to
correlated their motions to avoid one another. The electron correlation lowers the energy
of the system and affects the overall electron density of the system.[43]

28
Electron correlation
There are different approaches to the electron correlation problem. The conceptually
simplest is configuration interaction (CI)[45] which takes the Hartree-Fock solution as a
starting point, or reference configuration.[43] Other configurations are generated by
permitting the excitation of one electron from the subset of occupied molecular orbitals to
the subset of unoccupied or “virtual” MOs. The complete list of single excited
configurations is generated by considering all possible excitations with the same spin
state as the ground state under study. The list is then extended to double excitations,
accounting for all possible combinations of excitations of two electrons from the
occupied to the virtual MOs. A full-CI list is generated by progressing to include triple,
quadruple, and higher excitations, until all n electrons have been excited. The correlated
wave function is then expressed as a linear combination of the reference, Hartree-Fock
configuration, plus small amounts of all the possible excitations. Variational treatment of
this trial wave function leads to the correlation energy by adjusting the relative amount
that each particular configuration contributes to the final correlated wave function.[43]
One problem is that even for small systems, the number of configurations generated by
all possible excitations is out the reach of any computer. For this reason, one of the
common points of termination of the list is after the inclusion of all single and double
excitations (CISD). One problem with termination of the full CI expansion is the size-
consistency problem.[43] This means that the same treatment of a complex is
fundamentally different than that of the subunits of which it is composed. For instance,
for a dimer, the CISD treatment would permit double excitations for each monomer but,
instead of permitting quadruple excitations within the dimer, taking into account
simultaneous double excitations of each of the monomers, CISD terminates the excitation
list at doubles in the complex. Therefore, truncated CI treatments handle poorly with
molecular interactions like hydrogen bonds. Another means of introducing size
consistency is by quadratic approximation, QCISD.[46]The approach achieves this size
consistency by giving up its variational character.[43] It must be pointed out that single
excitations only improve the quality of the total wave function, mostly regarding the so

29
called “empty” Hartree-Fock levels, but, according to the Brillouin theorem, do not affect
the total energy of the system.
Other procedures, like the Coupled pair theories[47, 48] are size consistent but are not
variational. This means that in principle, it is possible to obtain a value of energy lower
than the true energy of the system. In the independent electron-pair approximation
(IEPA), the total correlation energy is partitioned into a sum of contributions from each
occupied pair of spin orbitals. A different correlation wave function is constructed for
each pair, letting their electrons be excited into the virtual MOs of the reference
configuration. The total correlation energy then corresponds to the sum of all pair
energies.
When the IEPA approach is extended to incorporate coupling between different pairs,
becomes a coupled-pair theory. In terms of excitations from the original Hartree-Fock
determinant, the correlation energy depends directly upon the double excitations, but
their contributions involve quadruple excitations in an indirect way, and the latter are
linked to hextuple excitations, and so on. The coupled-cluster approximation expresses
this relationship in a closed set of equations. [49] When the applications of coupled-cluster
theory include only double excitations it is identified as CCD.[50] More general versions
of the theory that include also single and higher excitations are abbreviated as CCSD.[51]
Various approximations have been suggested to coupled-cluster since it is highly
demanding of computer resources. One is the linear coupled-cluster approximation (L-
CCA) which sets certain products equal to zero, and is equivalent to doubly-excited many
body perturbation theory. If instead of ignoring all the product terms set equal to zero in
L-CCA, some of them are retained, ones arrives to the coupled electron pair
approximation (CEPA).[43]
The correlation method mostly used to calculate hydrogen bonded systems is the
Møller−Plesset perturbation theory.[52, 53] This approach considers the true Hamiltonian
as a sum of its Hartree-Fock part plus an operator corresponding to electron correlation.
In other words, the unperturbed Hamiltonian consists of the interaction of the electrons
with the nuclei, plus their kinetic energy, to which is added the Hartree-Fock potential:
the interaction of each electron with the “time-averaged” field generated by the others.

30
The perturbation or “correction” operator therefore becomes the difference between the
expected exact interelectronic repulsion operator, with its instantaneous correlation
between electrons, and the latter Hartree-Fock potential.
The first correction to the Hartree-Fock energy appears as the second-order
perturbation energy. The energy including this correction is known as MP2. The MP3
level involves additional terms, but remains restricted to double substitutions from the
reference configuration. At fourth order, there are contributions from single, triple, and
quadruple excitations, as well as doubles. One strong advantage of the MP theory is that,
in addition to its computational efficiency, it is size consistent, so it is a good choice for
different types of molecular interactions. According to Schneiner,[43] several calculations
indicate that MP2 provides results in excellent agreement with the much more
computationally demanding MP4. Therefore, the literature of correlated calculations of
hydrogen bonds is dominated by Møller−Plesset theory.[43]
In certain cases, a single determinant does not offer an adequate representation of the
electronic structure. In such cases, it is useful to perform a Multi-Configurational SCF
calculation (MCSCF) in which a number of different electron configurations among the
Hartree – Fock orbitals are chosen as important and their adjustable parameters like
orbital coefficients are variationally optimized.[54] This procedure is arbitrary in the
choice of which configurations are considered being important. The calculation can be
more objective by including all excitations between a subset of occupied MOs and a
subset of vacant orbitals. These excitations have some restrictions like multiplicity or
order of excitation. The orbitals selected for the excitations are the active space and the
method is called Complete Active Space Self Consistent Field (CASSCF).[55]
Semiempirical methods
Using semiempirical methods allows to calculate large systems when the ab initio
treatment is too demanding computationally. Semiempirical approaches are developed
under the same formalism than ab initio methods, but the semiempirical methods neglect
many smaller integrals.[56] To compensate for these approximations, empirical
parameters are introduced into the remaining integrals and their values are assigned on
the basis of calculations or experimental data.[56] According to Jensen, the various

31
semiempirical methods are defined by how many integrals are neglected, and how the
parameterization is done.[56]
In “Semiempirical Methods”,[57] Thiel points out that, among other applications,
semiempirical methods are useful as a previous approach to a computational problem
before proceeding with higher-level of theory. Compared with ab initio or density
functional methods, semiempirical calculations are much faster and therefore can be used
for calculating larger systems. But semiempirical methods have the disadvantage of being
less accurate and the errors are less systematic.[57]
According to Thiel, the quantum-chemical semiempirical treatments can be defined
depending on:[57]
• The basic theoretical approach: Most semiempirical methods are based on MO
theory and use a minimal basis set for the valence electrons. Electron
correlation is treated explicitly only when necessary for an appropriate zero-
order description.
• The integral approximation and the types of interactions included: According to
that, there are three levels of integral approximation:[57] CNDO (complete
neglect of differential overlap), INDO (intermediate neglect of differential
overlap), and NDDO (neglect of diatomic differential overlap). Unlike CNDO
and INDO which truncate after the monopole, NDDO keeps the higher
multipoles of charge distributions in the two-center interactions.[57]
• The evaluation of integrals: The integrals can be estimated directly from
experimental data, calculated from analytical formulas or from appropriate
parametric expressions. One-center integrals can be calculated from atomic
spectroscopic data. The selection between analytical formulas or parametric
expressions depends mostly on the consideration of how to model the
interactions.
• The parameterization: the semiempirical MO methods are parameterized to
reproduce experimental reference data (or, possibly, accurate high-level
theoretical predictions as substitutes for experimental data). The reference

32
properties are chosen to be representative for the intended applications. The
quality of semiempirical results depends very much of the parameterization.[57]
As Thiel states, the most popular semiempirical methods for studying ground-state
potential surfaces are based on the MNDO model. MNDO is a valence-electron self-
consistent-field (SCF) MO treatment which uses a minimal basis of atomic orbitals and
the NDDO integral approximation.[57] The total energy of a molecule is the sum of its
electronic energy and the core-core repulsion energies.
The MNDO model includes only one-center and two-center terms, so it is
computationally more efficient. The one-center terms are taken from atomic
spectroscopic data, and slight adjustments are permitted in the optimization to take into
account the differences between free atoms and atoms in a molecule.[57]
In “Hydrogen Bonding by Semiempirical MO Methods”,[58] Hadzi and Koller state that
the MNDO approximation includes the terms of: one-centre one-electron energies, which
parameters are taken from atomic spectroscopic data and are allowed in the optimization
to account for differences between atoms in molecules and free atoms; the one-centre,
two-electron repulsion integrals Coulomb and exchange integrals, which are derived from
spectroscopic data with some adjustments and are smaller than the analytically calculated
to partially consider the electron correlation ; the two-centre one-electron resonance
integrals that represent the electronic kinetic energy and electrostatic core-electron
energies; the two-centre, one-electron integrals representing the core-electron attractions;
the two-centre two electron repulsion integrals which are evaluated by semiempirical
parametric formulas that simulate multipole-multipole interactions; and the two-centre,
core-core repulsion terms composed by an electrostatic and an additional effective part,
the effective term represents the Pauli repulsion and compensate the errors of the
model.[58]
MNDO, AM1 and PM3 methods are standard implementations of the MNDO model
that have been parameterized mainly with respect to ground-state properties, with special
attention on the energies and geometries of organic molecules. AM1 and PM3 give some
improvement in accuracy over the original MNDO method, but the mean absolute errors
remain of the same order of magnitude.[57]

33
As a result of too repulsive interactions in the core-core potential, MNDO
overestimates the repulsion between two atoms 2 - 3 Å apart.[56] As a solution to this, in
the Austin Model 1 (AM1) by Dewar,[59] the core-core function was modified by adding
Gaussian functions, and the whole model was parameterized again. The Gaussian
functions were added somehow as patches onto the basic parameters, which explains why
different number of Gaussians are used for each atom.[56] According to Jensen in
“Introduction to Computational Chemistry”, some improvements and limitations of the
AM1 model are:[56]
• AM1 predicts the strength of hydrogen bonds more or less correctly, but the
geometry is frequently wrong.
• The activation energies are much better than with MNDO.
• Hypervalent molecules are improved compared to MNDO, but still there are
significant errors.
• Alkyl groups are systematically too stable by around 2 kcal/mol per CH2 group.
Nitro compounds are systematically too unstable. The gauche conformation of
ethanol is predicted to be more stable than the trans.
• Peroxide bonds are ~0.17 Å too short.
• When atoms are around 3 Å apart, phosphor compounds show incorrect
geometries.
The Modified Neglect of Diatomic Overlap, Parametric Method Number 3 (MNDO-
PM3)[60] is a reparameterization of the AM1 with all the parameters automatic fully
optimized. The AM1 expression for the core-core repulsion was kept, except that only
two Gaussians were assigned to each atom. These Gaussian parameters are included as an
integral part of the model, and allowed to vary freely.[56]
The PM3 method has been parameterized using the standard heats of formation of a
large set of typical reference molecules. It has been designed to reproduce standard heats
of formation from total energies (after the inclusion of accurate experimental atomization
heats) in the case of molecular geometries corresponding to the minimal SCF value of

34
trial molecules. PM3 is considered to have the best set of parameters for the given set of
experimental data.[56]
Jensen points out some limitations of the PM3 model:[56]
• Almost all sp3-nitrogens are predicted to be pyramidal, contrary to experimental
observation. The charge in nitrogen atoms is frequently of wrong sign and
magnitude.
• Hydrogen bonds are ~0.1 Å too short. Bonds between Si and Cl, Br and I are
also underestimated.
• The gauche conformation of ethanol is predicted to be more stable than the
trans. H2NNH2 is predicted to have a C2h structure, while the experimental is
C2. ClF3 is predicted D3h, while the experimental structure is C2v.[56]
Another point to take into account about the PM3 calculations is that, as described by
Csonka et al.,[61, 62] the PM3 Hamiltonian has a tendency to create wrong geometries with
H-H interactions between 1.8 and 2.0 Å due to parameterizations errors.
Jensen mentions other limitations which are common to MNDO, AM1 and PM3:[56]
• The rotational barriers for bonds with partial double bond character are
significantly too low.
• The bond length to nitrosyl groups is underestimated.
• The parameters for metals which are included are based on only a few
experimental data.
• For weak interactions, like van der Waals complexes or hydrogen bonds, the
minimum geometry is wrong or the interaction is too weak.
However, there are some distinctions about the use of the semiempirical methods in the
calculations of hydrogen bonding and weak interactions. For instance, in “An
Introduction to Hydrogen Bonding”, Jeffrey[1] states that the semi-empirical methods
such as MNDO and AM1 are considered to be inappropriate for simulating moderate or
weak hydrogen bonding due to an overestimation of the exchange repulsion at hydrogen

35
bond distances and PM3 is said to give better results for systems like the water dimer.[1]
The Semi-ab initio Method 1 (SAM1) was developed by Dewar, Jie and Yu[63] and it is
said to correct this deficiency.[1] SAM1 is based on the NDDO approximation, but instead
of replacing all integrals by parameters, the one- and two-centre electron integrals are
calculated directly from the atomic orbitals.[56] However, for systems like the ammonia
dimer the SAM1 calculations do not lead to correct results.[58] For the formic acid dimer
the AM1, PM3 and SAM1 overestimate the association enthalpy and AM1 overestimates
the O…O distances while PM3 gives better values.[58] Turi and Dannenberg[64] found
similar results for the acetic acid dimer, and they found that the calculated semiempirical
vibrational frequencies were in good agreement with the MP2 results.[58]
Other authors like Zheng and Merz[20] in their studies of hydrogen bonding interactions
relevant to biomolecular structures state that the AM1 geometries were in poor agreement
with ab initio structural results and the PM3 method gives geometries similar to the ab
initio ones. On the other hand, Turi and Dannenberg[65] in their molecular orbital studies
of C-H…O bonded complexes found a good agreement between the energies and
structures at the AM1 and ab initio levels, while the PM3 results were erratic. All of that
shows that, so far, there is no conclusive criterion concerning the selection of an AM1 or
PM3 hamiltonian for the semiempirical calculations of a given system.
According to Thiel,[57] although semiempirical methods can be frequently used with
useful accuracy and at very low computational costs, some general limitations should be
taken into account. One of these is the fact that the errors in semiempirical calculations
are less systematic and harder to correct compared to ab initio or DFT methods. Another
point is that the accuracy of the semiempirical results varies with the classes of
compounds and these variations are more pronounced than in high-level ab initio and
DFT calculations. An additional aspect to consider is that, unlike ab initio and DFT
methods, semiempirical methods require reliable experimental or theoretical reference
data for the parameterizations and they can be used only in molecules which elements
have been parameterized.[57]

36
Density functional theory
The Density Functional Theory (DFT) is based in a one-to-one correspondence
between the electron density of a system and the energy.[56] That means that the ground-
state electronic energy is determined completely by the electronic density.[56, 66] The
advantage of this approach is that the electron density is independent of the number of
electrons. That means that, while the complexity of a wave function increases with the
number of electrons, the electron density is independent of the system size. The problem
is to find the appropriate density functional. Therefore, the goal of the research in DFT is
the design of functionals connecting the electron density with the energy.[56]
The use of DFT methods is based on the introduction of the Kohn and Sham (KS)
formalism[67] which splits the kinetic energy functional in two parts, one that can be
calculated exactly and a small correction term. The kinetic energy is calculated assuming
non-interacting electrons and the remaining kinetic energy is included into an exchange-
correlation term.
Therefore, and accordingly to Kohn and Sham, the functionals used by DFT methods
part the electronic energy into several terms:[68]
E = ET + EV + EJ + EXC (2.3)
Where ET is the kinetic energy term, EV includes terms of nuclear-electron attraction
and nuclear-nuclear repulsion. EJ is the electronic repulsion term and EXC is the
exchange-correlation term that includes the remaining part of the electron-electron
interactions.[68] Since the functional form of the exchange-correlation energy is still
unknown, the main problem of DFT is to find the adequate formulas for this exchange-
correlation term. Therefore, the difference between DFT methods is the choice of the
functional form of the exchange-correlation energy.
The Local Density approximation (LDA) assumes that the local density can de treated
as a uniform electron gas and that means that the density is a slowly varying function.[56]
The exchange-correlation energy is very frequently separated into exchange and
correlation parts. In the LDA approximation, the correlation energy of a uniform gas has
been determined using Monte Carlo methods for different densities and an analytic

37
interpolation formula was developed by Vosko, Wilk and Nusair (VMN)[69] to use these
results in DFT calculations.
According to Guo, Sirois et al. in “Density Functional Theory and its Aplications to
Hydrogen-bonded Systems”,[70] the most used LDA functionals use the Slater functional
for exchange and the VMN formula for the correlation energy. LDA provides reliable
results for molecular properties as structures, vibrations and ionization potentials, but it is
not able to provide an adequate description of hydrogen bonding interactions.[70]
The Gradient Corrected or Generalized Gradient Approximation (GGA) methods are
improvements of the LDA approach that consider a non-uniform electron gas. In the
GGA methods the exchange and correlation energies depend not only on the electron
density, but also on derivatives of the density.[56] Very popular GGA exchange
functionals are those of Becke (B)[71], Perdew and Wang (P),[72] among others.
Commonly used GGA correlation functionals are the Perdew (P86), and Lee-Yang-Parr
(LYP)[73] functionals.
Jensen states that the models that include exact exchange are called hybrid methods,[56]
and Guo, Sirois et al.[70] affirm that hybrid functionals are those that include a component
of Hartree-Fock exchange. One example of this is the very accepted GGA exchange
functionals Becke 3 parameter (B3)[74] hybrid functional.
As already mentioned, the advantage of DFT is that only the total density is considered.
In addition, DFT has a computational cost which is similar to HF theory with the
possibility of providing more accurate results. Some DFT methods are very successful in
studying properties of molecules like structural parameters, vibrational frequencies and
electrostatic potentials, among others. For instance, an study of the hydrogen-bonded
formic acid dimer shows that the energies and the barriers for the symmetrical double
proton transfer are well described by the DFT approach with BLYP functionals.[70]
According to Guo, Sirois et al., the GGA and hybrid functionals are very good for the
study of hydrogen-bonded complexes, since they provide reasonably accurate binding
energies, hydrogen bond geometries and thermodynamic properties for small neutral
complexes.[70] DFT methods predict cooperative effects that agree with MP2 calculations

38
for water polymers and neutral complexes containing a peptide linkage. They also give
good dipole moments and polarizabilities for hydrogen-bonded systems like the water
dimer.[70] However, as Jensen states, unlike the mainly electrostatic interactions, the
dispersive weak interactions are poorly described by the current functionals. In addition,
DFT methods are inappropriate for excited states of the same symmetry as the ground
state.[56]
On the other hand, the developing of DFT functionals is a growing field. Recently,
Truhlar and coworkers have developed new DFT methods for the calculation of π
hydrogen bonding systems.[75, 76] They calculated systems like the dimers of benzene with
water and ammonia, among others. They found that their MPW1B95, MPWB1K,
PW6B95, and PWB6K methods predict accurately the energies and geometries of π
hydrogen bonded systems, in cases where the B3LYP functional fails and the PW91 is
less accurate. They also emphasize the application of their PWB6K functional for
calculating large π hydrogen bonded systems and stacking interactions in the DNA base
pairs and amino acid pairs.[75, 76]
Basis sets
Most quantum mechanical treatments describe each molecular orbital as a linear
combination of atomic orbitals (LCAO approximation).[43, 77, 78] In this approximation,
each atom has assigned to it certain functions that resemble the standard s, p, d and so
atomic orbitals that are centered at the nucleus. Whereas the hydrogen-like orbitals die
off as exp(-ζr), where r is the distance from the nucleus and ζ a constant, the integrals
using this form of the orbital are difficult to evaluate. These Slater-type orbitals (STOs)
are usually replaced by a small number of Gaussian functions, where exp(-ζr) is replaced
by exp(-αr2). The quadratic dependence of r in the exponent greatly simplifies the form
of the integrals, particularly those that involve several atomic centers simultaneously. In
fact, it is computationally more efficient to evaluate a large number of integrals involving
Gaussians than a much smaller number of STO integrals. In addition, a series of
Gaussians with progressively larger values of orbital exponent α can fairly closely
reproduce a Slater-type function. Consequently, most modern quantum chemical

39
calculations are performed using basis sets composed exclusively of Gaussian
functions.[43]
The collections of orbitals that are applied to calculations are called basis sets. The
smallest basis sets uses one orbital to represent each of the orbitals of each shell that is
full or partially filled. The STO-3G[79] is one minimal basis set where each Slater-type
orbital is replaced by a contraction of three primitive Gaussian functions.[43]
Minimal basis sets are improved by doubling the number of functions to provide more
flexibility. A “double-ζ” basis set is similar to minimal, except that each atomic orbital is
split into two. The flexibility of a “DZ” basis permits each orbital to expand or contract in
size to conform to the environment in which the atom finds itself. Triple-ζ or TZ basis set
provides even more flexibility. Splitting the valence shell is worthwhile because the inner
shell electrons are little affected by changes in the bonding environment around the atom.
Therefore, there are split valence basis sets like the 6-31G[80] or the 6-311G.[81] In the 6-
31G basis set the 6 refers to the number of Gaussian primitives used to describe the inner
shell, 1s orbital. The 3 and 1 indicate 3 primitives for the inner and 1 primitive for the
outer valence orbitals. The basis set 6-311G is similar except that a third set of functions
are added, by a single Gaussian, to split the valence shell three ways.[43]
The inner and outer s orbitals of an atom in a double-ζ basis set are both spherical. So,
while the presence of two of them permits the orbital to expand, it can do it isotropically
only, with no stretching in any one direction over another. This “polarization” in a given
direction is necessary in many situations. Therefore, the flexibility is added in the form of
basis functions corresponding to one quantum number of higher angular momentum than
the valence orbitals.[44] So, p-orbitals added to hydrogen are called polarization
functions. Analogously, the p-orbitals of C or O can be polarized by a small amount of a
d-orbital of appropriate symmetry.[43]
To indicate when polarization functions have been added to the basis set, various
conventions are used. The addition of the P in the DZP indicates a polarized double-ζ
basis set but does not clarify whether all atoms have had polarization functions added, or
only some of them. In most cases, the P indicates polarization functions on all atoms.

40
Another designation is an asterisk *. In 6-31G*, the single asterisk indicates polarization
functions of d-type added to the non-hydrogen atoms. A second asterisk would inform of
p-functions on hydrogen, too. One more designation is to indicate the numbers of
polarization functions in parenthesis. Thus, the 6-31G** could be also described as 6-
31G(d,p). Doubling the d-functions, but leaving a single set the p-functions of hydrogen,
would be indicated as 6-31G(2d,p). This notation makes possible also the representation
of orbitals with higher angular momentum, as for example 6-311G(3df,2pd).[43]
According to Cramer, an alternative way to introduce polarization is to allow basis
functions to be centered away from atoms. They are called floating Gaussian orbitals
(FLOGOs) and they are rarely employed in modern calculations, since the process of
geometry optimization is considerably more complicated.[44]
Another way to provide flexibility is the use of functions of, for example, s or p
symmetry, with small orbital component. They are called diffuse functions and are
especially useful for describing systems like anions since they permit the overload of
electrons to better avoid one another since they take advantage of the large expansion of
space over which this orbital extends. The + symbol is used to indicate the presence of
such functions. For example, the 6-31+G* includes a diffuse sp-shell on non-hydrogen
atoms; a second + indicates diffuse functions on H as well.[44]
Correlation consistent (cc) basis sets have been designed[82] specifically to be suitable
to calculations involving electron correlation which has been taken into account at the
basis atomic level, because they have been constructed from the results of correlated
atomic calculations.[56] The smallest is the cc-pVDZ (correlation-consisted, polarized
valence double zeta). These basis sets can be augmented (prefix aug) by additional
functions optimized for atomic anions to describe diffuse electronic distributions. This
augmentation consists of adding one extra function with a smaller exponent for each
angular momentum. For example, the aug-cc-pVDZ has additionally 1s-, 1p- and 1d-
functions.

41
Basis set superposition error
If a bimolecular interaction is considered, the AB interaction energy at the same level
of theory for the monomers and the dimer, can be defined as:[44]
ΔEcomplexation = E(A…B)*ab – [E(A)a + E(B)b] (2.4)
The asterisk (*) denotes the geometry of the complex. The basis functions a and b are
associated with the monomers A and B, respectively, and they are both (ab) used in the
calculation of the complex. Consequently, there are more basis functions employed in the
calculation of the complex than in either of the monomers. The greater flexibility of the
basis set for the complex can provide an artificial lowering of the energy when one of the
monomers “borrows” basis functions of the other to improve its own wave function.[44]
This artificial stabilization of the complex, due only to its larger basis set, in comparison
to the smaller sets of the monomers, is called basis set superposition error (BSSE).
One approximate way to correct this phenomenon is the counterpoise (CP) correction
proposed by Boys and Bernardi.[83] To estimate how much of this complexation energy is
due to BSSE, four additional energy calculations are needed. Using basis set a for A and
basis set b for B, the energies of each of the two fragments are calculated with the
geometry they have in the complex. Two additional energy calculations of the fragments
at the complex geometry are then carried out with the full ab basis set. For example, the
energy of A is calculated in the presence of both the normal a basis functions and with
the b basis functions of fragment B located at the corresponding nuclear positions, but
without the B nuclei present. Such basis functions located at fixed points in space are
called ghost orbitals. The fragment energy for A will be lowered owing to these ghost
functions, since the basis becomes more complete. The CP correction is defined as:
ΔECP = E(A)*ab + E(B)*
ab – E(A)*a – E(B)*
b (2.5)
The counterpoise corrected complexation energy is given as:
ΔEcomplexation CP corrected = ΔEcomplexation - ΔECP (2.6)
ΔECP is an approximate correction that gives an estimate of the BSSE effect, but it does
not provide either an upper or lower limit. Another point to take into account is that this

42
is a single point correction. If the potential energy surface where the minima were found
was “BSSE contaminated”, the results will only tell us how bad the reached minimum is,
and the interatomic distances in the dimer will be wrong to an unknown extent.
Algorithms to perform this correction at each step of the optimization procedure are
available but they are computationally demanding.[84-89] Recently, Crespo, Montero et
al.[90] discussed the influence of the BSSE in the geometries and energies of methane –
nitric oxide dimers at the MP2 and DFT levels of theory with various basis sets. They
state that weak interactions determined by dispersive forces cannot be predicted with
standard non BSSE corrected PES and that DFT results are erratic, even within BSSE
corrected PES.[90]
Hobza and Havlas[91] studied the PES of hydrogen bonded-systems such as the water
dimer, hydrogen fluoride dimer, formamide dimer and formic acid dimer. They conclude
that the CP-corrected and standard PES of these complexes differ, depending on the level
of theory and that the optimization on the standard PES using medium basis sets
sometimes leads to completely wrong geometries, whereas CP-corrected PES yields the
correct structure.
Simon et al.[84] studied the effect of the counterpoise correction on the geometries and
vibrational frequencies of 15 hydrogen bonded systems at the DFT and MP2 levels of
theory, using the 6-31++G(d,p) basis set. They found larger changes at the MP2 level of
theory and that, in general, the CP correction increases the hydrogen bond distance,
decreases the intermolecular stretching frequency and decreases the red shifts of the
donor A-H stretching vibrational frequency. In addition, they observed an interesting
relationship between the percentage of BSSE, the relative changes of the hydrogen bond
distances and intermolecular stretching frequencies. This relationship allows for the
estimation of these properties on the CP-corrected PES, from one single point
counterpoise calculation on the standard PES.[84]
As shown, the calculation of BSSE-corrected potential energy surfaces is a rising field
with many perspectives and open questions. However, the main disadvantage of the
current implementations of the algorithms to perform this correction at each step of the
optimization procedure is the high demand of computational time, which makes this type

43
of calculations prohibitive in many cases such as for bigger systems or when using larger
basis sets.
According to Scheiner in “Hydrogen Bonding: A Theoretical Perspective”, there are
other important facts to consider about the BSSE:[43]
• In general, BSSE is reduced as the basis set becomes larger and more flexible.
However, there is no strict correspondence, and the BSSE in some cases can
become larger with the increasing of the size of the basis set.
• The BSSE rises rapidly as the two molecules approach one another. Whereas
SCF BSSE can be reduced to negligible proportions with large basis sets, the
superposition error at correlated levels goes down much more slowly, persisting
at large values, even with very flexible bases.
An alternative to the CP method is the Chemical Hamiltonian Approach (CHA)[92] that
attempts to isolate the superposition error directly in the Hamiltonian operator. That
means that the CHA is based on omitting from the Hamiltonian the intermolecular
contributions that cause the BSSE, while keeping all the physically true interaction terms.
Omitting these terms, a Hamiltonian which leads to the BSSE-free wave functions is
achieved. Thus, the CHA ensures that the description of each monomer or fragment
isolated and within the complex is consistent.[43, 93, 94]
According to Meyer et al.,[95] the CHA method performs a detailed analysis of different
terms in Hamiltonians and distinguishes three different types:
• The terms corresponding to true intramolecular interactions of the monomers
treated in their own molecular basis.
• The terms describing the true intermolecular interactions.
• Some finite basis correction terms responsible of BSSE.
As Bende[93] points out, several different approaches have been developed using the
CHA scheme both at the HF level and using second order perturbation theory.[96-98] Many
calculations have been performed in the last decade by applying this CHA to study the

44
structures and interaction energies for different van der Waals and hydrogen bonded
systems.[86, 93, 94] Different systems were investigated, from the small “bimolecular
complexes” like formamide dimers by Bende et al.[93] to large DNA basis pairs,[99] by
using a variety of different basis sets. It has been concluded that in all cases a remarkable
agreement has been found with the results given by the CP method, despite of the fact
that both schemes are conceptually very different. However, Scheiner states that the CHA
approach has been criticized on the grounds of its inconsistency with the results of
symmetry adapted perturbation theory and because, since the BSSE is a non-physical
phenomenon, this Hamiltonian is non-Hermitian.[43]
According to Salvador et al., the removal of the BSSE in molecular complexes
composed by more than two fragments is not extensively discussed in the literature.[87] A
few years ago, Turi and Dannenberg[100] pointed out the ambiguity of the counterpoise
correction when studying growing chains of hydrogen fluoride. They showed that the
BSSE computed for the addition of a new HF monomer to the (HF)n aggregate depends
upon whether the incoming monomer is added to the H or to the F end of the aggregate.
Therefore, one can obtain different interaction energies for the same chemical process,
which is obviously wrong. They proposed the use of the counterpoise method by defining
as many fragments as there are monomer subunits in the complex, with the BSSE defined
as the difference between the energy of each monomer in its own basis set and that of the
whole aggregate. This method solves the problem of the ambiguity of the CP correction
but is unable to explain all the effects of the incoming monomer on the interaction and
BSSE already present in the molecular aggregate.
As Salvador explains, another way to take into account the high-order BSSE effects
within the counterpoise framework is based on a hierarchical counterpoise scheme[101] for
N-body clusters that treats the basis set extension effects of all the monomers, dimers,
trimers and so on, present in an aggregate. Another counterpoise scheme is the pairwise
additive function counterpoise (PAFC)[102] where the counterpoise correction is carried
out over pairs of fragments.
In short, three counterpoise schemes for trimers are mentioned: the site-site[102] function
counterpoise which includes only the basis set extensions of the monomers in the whole

45
basis set, the Valiron-Mayer hierarchical function counterpoise which includes the
differences, for each dimer in the aggregate, between the dimer interaction energy
computed within the dimer-centered basis set (DCBS) and the basis set of the whole
trimer (TCBS); and the pairwise additive function counterpoise in which the three-body
interaction terms are not corrected according to the counterpoise scheme and instead,
only the two-body interaction energies are corrected by using DCBS.
The N-body cluster generalizations for these three function counterpoise schemes, the
site-site (SSFC), pairwise additive(PACF) and Valiron-Mayer (hierarchical) (VMFC) are
as follows:[87]
Figure 1.3: The N-body cluster generalization for the site-site (SSFC), pairwise
additive(PACF) and Valiron-Mayer (hierarchical) (VMFC) counterpoise schemes.
Figure taken from “Counterpoise-corrected geometries and harmonic frequencies of N-
body clusters: Application to (HF)n(n=3,4)” by Salvador and Szczesniak.[87]

46
2. The Multiple Minima Hypersurface (MMH) Approach
2.1 Introduction
The multiple minima problem
According to Cramer in “Essentials of Computational Chemistry, Theories and
Models”,[44] the first step for making the theory closer to the experiment is to consider not
just one structure for a given chemical formula, but all possible structures. This involves
the full characterization of the Potential Energy Surface (PES) for a given chemical
formula. The PES is a hypersurface defined by the potential energy of a collection of
atoms over all possible arrangements and has 3N - 6 coordinate dimensions (for linear
molecules 3N - 5). Therefore, each structure, which is a point on the PES, can be defined
by a vector X where[44]
X ≡ ( x1, y1 , z1 , x2 , y2 , z2 ,… , xN , yN , zN ) (3.1)
and xi, yi, and zi are the Cartesian coordinates of atom i.
Of particular interest on PES are the local minima, which correspond to optimal
molecular structures and saddle points which are lowest energy barriers on paths
connecting minima. The saddle points are related to the chemical concept of the transition
state. Therefore, a complete PES provides, for a given collection of atoms, complete
information about all possible chemical structures and all isomerization pathways
interconnecting them.[44]
However, the problem of locating the global minimum or an important and
representative set of local minima in polyatomic systems is a very difficult computational
task, taking into account that the number of minima increases very much with the number
of variables. This problem is frequently called the Multiple Minima Problem in the
literature.[56]
In “Introduction to Computational Chemistry” Jensen describes some methods which
are commonly used for conformational sampling in larger systems:[56]
• Stochastic and Monte Carlo methods
• Molecular dynamics

47
• Simulated annealing
• Genetic Algorithms
• Diffusion methods
• Distance Geometry methods
He emphasizes that none of them guaranty to find the global minimum, but in many
cases they provide a local minimum which is close in energy to the global one, but not
necessary close in terms of structure.
The Stochastical and Monte Carlo (MC) methods start from a given geometry, usually
a local minimum, to generate new configurations by adding a random perturbation to one
of more atoms.[56] In Monte Carlo methods, if the energy of the new geometry is lower
than the present, this new geometry is taken as starting point for the next step. If not, the
Boltzmann factor e -ΔE/KbT is calculated and compared to a random number between 0 and
1. If the Boltzmann factor is less than this number the new geometry is accepted,
otherwise the next step is taken from the old geometry. In this way, a sequence of
configurations is generated from which geometries may be selected for following
minimization. To have a fair acceptance radio, the step size must be reasonably small.[56]
In Stochastical methods the random perturbation is usually larger compared to Monte
Carlo and a standard minimization is performed starting at the perturbed geometry. Then,
a new perturbed geometry is generated and minimized, and so on.[56] It is important to
consider the length of the perturbing step, because small perturbations basically return the
geometry to the starting minimum and large perturbations may lead to high energy
structures that minimize to high energy local minima. Another point to consider is that
when the perturbing step is done in Cartesian coordinates, many of the perturbed
geometries are high in energy, since two o more atoms are moved close together by the
perturbation.[56] The perturbation can be done in a selected set of internal coordinates,
too. The perturbation step may be taken from the last minimum found or from all the
previous found minima, weighted by a probability factor. Therefore low energy minima
are more frequently used than the higher energy ones.
Molecular Dynamics (MD) methods solve the Newton equation of motion for atoms on
an energy surface.[56] The energy of the molecule is distributed in potential and kinetic
energy, and therefore, molecules are able to rise above barriers separating minima if the

48
barrier height is less than the total energy minus the potential energy. The energy is
related to the simulation temperature and with energy high enough, the dynamics would
sample the entire surface. However, this will not be practical by means of computational
time. Very small time steps must be used for integrating Newton’s equation, therefore the
simulation time is short. That, combined with the use of temperatures of few hundreds or
thousands of degrees, means that only the local area around the starting point is sampled
and only relatively small barriers of few kcal/mol can be overcome. Different local
minima can be generated by selecting configurations at appropriate intervals during the
simulation and subsequently minimizing these structures.[56]
According to Jensen,[56] the Simulated Annealing (SA) techniques choose high initial
temperatures 2000 – 3000 K to initiate an MD or MC run, during which the temperature
is slowly reduced. The molecule is initially allowed to move over a large area, but with
the decreasing of the temperature it becomes trapped in a minimum. That means that, in
principle, if the cooling is done infinitely slowly at infinite run time, the resulting
minimum would be the global minimum. In practice, the MD or MC run is so short that
only the local area is sampled.[56]
Other methods are the Genetic Algorithms (GA) which are based on biological concepts
and terminology.[56] Accordingly, there is a “population” of structures, each characterized
by a set of “genes”. The parent structures generate the new structures which have a
mixture of the parent genes, and “mutations” are allowed to occur in the process. Their
energies are minimized and the best new and parent structures are selected and used for
the next generation and so on. The population is allowed to develop for hundreds of
generations. The criterion for selecting the best structures in each generation may vary
and can be, for instance, that the good structures are those with lower energy. An
example of mutation can be the random variation of geometrical parameters to produce
conformations outside the restricted range of the current population. The size of the
population, the mutation rate and the amount of structures selected in each generation,
can be adjusted, among other parameters. Due to their reliability and facilities for
implementation, the use of genetic algorithms is becoming very popular.[56]
Diffusion methods are those where the energy function is changed to contain only one
minimum.[56] One change may be the addition of a contribution proportional to the
second derivative of the function (local curvature). Consequently, the minima are
increased in energy and maxima and saddle points are reduced in energy, and finally only

49
one minimum remains. Using the single minimum geometry of the modified potential,
the process can be reversed and a minimum on the original surface is obtained. This
minimum is frequently, but not necessarily the global minimum.[56]
In Distance Geometry methods, test geometries are produced from a set of lower and
upper limits on distances between all pairs of atoms, and therefore many different trial
sets of distances can be generated by assigning random numbers between these limits.
The random distance matrix can be translated into a three-dimensional structure and trial
conformations are generated, which can be optimized using conventional methods.[56]
Chemical similarity searching
As Willett, Barnard and Downs explain in “Chemical Similarity Searching”, the
chemical similarity searching is an alternative and complementary method to the
substructure searching.[103] The substructure searching is a mechanism that involves the
recovery of all those molecules in a database that contain a query substructure defined by
the user, irrespective of the environment in which the query substructure occurs.[103] One
limitation of the substructure searching is that the database structure must include the
entire query substructure. In the similarity searching, a query involves the specification
of an entire molecule or structure as reference which is characterized by one or more
structural descriptors, and this set is compared to the corresponding set of descriptors for
each of the molecules in the database. This comparison allows to measure of similarity
between the reference structure and each of the database structures. Therefore, with the
increasing importance of databases of two-dimensional and three-dimensional molecular
structures in chemical research, the chemical similarity searching is becoming a powerful
source of useful and interesting information for chemists, especially in the areas of
molecular design and computer-aided synthetic planning.[103, 104]
According to Willet et al.,[103] the similarity coefficients provide a quantitative measure
of the degree of structural relatedness between a pair of structural representations. Some
coefficients are measures of the distance or dissimilarity between objects and have value
of 0 for identical objects, whereas others measure similarity and have their maximum
value for identical objects. In most cases the values taken for a coefficient are in the
range from 0 to 1.[103] Since the idea of determining a numerical measure of the similarity
between two atoms is common to different sciences apparently not related to each other,
the most used similarity coefficients have been sometimes reinvented, as shown in Figure
2.1.[103]

50
Figure 2.1: Description of two similarity coefficients commonly used in chemical information. Figure taken from “Chemical Similarity
Searching” by Willett, Barnard and Downs.[103]

51
There are several similarity coefficients that can be used for similarity searching.
However, the Tanimoto index has the advantage of the accessibility of its formula for
dichotomous variables for implementation in different subjects (Figure 2.1). When
coefficients are monotonic to each other, it can be shown analytically that they will
always produce identical similarity rankings of objects against a specified reference, even
though the actual coefficient values are different.[103] The Tanimoto index is monotonic
with the Dice coefficient. The use of the Tanimoto coefficient for similarity searching is
described in several studies in the scientific literature.[103-105]
2.2 The Multiple Minima Hypersurface (MMH) approach
The Multiple Minima Hypersurface (MMH) approach combines the quantum chemical
procedures for calculating the cluster energies in local minima of supermolecules and the
statistical thermodynamics approach for the evaluation of macroscopic properties. It was
first introduced in 1998 with the study of the pH-dependent equilibria of 2,4-diamino-5-
phenylthiazole tautomer molecules in water by Montero, Esteva et al.[5]
This approach was developed by Montero and coworkers with the original purpose of
evaluating the thermodynamic association functions of various molecular clusters using
the partition function.[4-6, 106] Therefore, it is advisable to explore the multiple minima
hypersurface (MMH) of the supermolecular systems. This exploration is made by
generating several sets with initial random geometries and following a gradient pathway
to search the local minimum that could be statistically significant for the partition
function. Therefore, it is of key importance to find the appropriate collection of
supermolecules with their respective energies and geometries that represent a set of the
most important states which are significant to the thermodynamic properties of the whole
system. Their energies can be added to the partition function, and by using statistical
thermodynamics the thermodynamic association functions can be calculated through the
partition function.
Here, the MMH approach is arbitrarily divided in two parts: The statistical
thermodynamic analysis and the structural analysis. The statistical thermodynamic
component of this method is not discussed here, since this work is only based on the
structural analysis. The study of the various possible geometrical arrangements of a given
system and the molecular interactions in the system is called structural analysis. The
generation and calculation of different geometries was originally conceived as a

52
necessary step for the thermodynamical analysis. Here, the structural analysis is further
developed as an independent tool for exploring the potential energy surfaces of a variety
of systems.
Therefore, the MMH approach as a very useful and reliable tool for localizing the
minima of hydrogen-bonded and weakly interacting systems is introduced here. The
MMH procedure provides a way to study the multiple possible molecular arrangements
for a given system via the exploration of large numbers of automatically generated
starting geometries. The MMH approach has the advantage that each structure is treated
and optimized independently, which is especially important in the treatment of very flat
potential energies surfaces without a well defined “global” minimum. The number of
minima increases very much with the number of variables; therefore the “global
optimization” is an extremely demanding task. Many times a “global minimum” is just a
local minimum with low energy.
In many instances the finding of the “global minimum” is not sufficient to describe the
properties of the system. The statistical thermodynamic approach of MMH, which is not
discussed here, shows the importance of considering local minima.[4-6, 106] The structural
analysis shows that many interactions which contribute significantly to the stabilization
of the system appear in various “local” minima. Additionally, sometimes the global
minimum is not clearly defined since it depends on the level of theory or the accuracy of
the calculation. Only by a careful and systematic exploration of the PES of the system it
is possible to determine the set of geometries representative for the interactions that
stabilize the system and provide an adequate description of the association process.
Frequently, the starting geometry for the optimization of the “global” minimum of a
system depends on “chemical intuition”, ignoring other alternatives. This may be less
severe in very simple systems, but for larger and more complicated systems, the use of a
systematic and reliable procedure for searching the minima, like MMH, becomes a
necessity. Even for some small systems, MMH can provide surprising structures that
otherwise might be excluded.
There are several steps in the MMH procedure:
1. Generation of starting geometries
The program for generating random sets was especially written for this purpose and is
called GRANADA. The GRANADA program[4] inputs molecular set data, such as the

53
radius of distribution around the coordinate origin in one monomer X (or more general,
subunit X) and the number of molecules of the other subunit or monomer Y to be taken
into account. As can be seen, the generation of geometries is not only restricted to dimers,
since the amount of molecules of monomer Y can be more than one. The definition of
which monomer is X and which one is Y is arbitrary and depends on the user. The user
also determines the number of randomly generated geometries. In most cases, here,
around 1000 randomly arranged clusters or complexes are generated as starting points.
(Figure 2.2)
Figure 2.2: Selection of random geometries of the FA – FMA dimer, produced with
the GRANADA program
Then, the GRANADA program outputs the desired series of different arrangements of
X and Y molecules as input files for MOPAC. Each Y molecule is situated in a randomly
selected new center of coordinates with respect to the X molecule and is rotated, also
randomly, around the three coordinate axes. All cases where the newly generated
molecules overlap the van der Waals volume of any existing molecule, are discarded.
Seeds for the random number generating routines are taken for each new molecular set
from the product of the seconds and the hundredths of seconds of the computer clock
when the program begins each calculation, to avoid any equivalent random number
series. Randomness has been carefully tested.

54
2. Preliminary calculation of the energy
To calculate the preliminary energies of all the generated molecular arrangements,
semiempirical Hamiltonians are used. Since hundreds of thousands of SCF cycles, with
their respective supermolecular geometries, are computed during the process, the amount
of calculations involved at this level of refinement makes the use of ab initio methods not
possible. Despite of controversial opinions, semiempirical methods have been confirmed
as a good choice for this previous discrimination of geometries. In addition, such
calculations have important advantages: computations are fast which allows for the
exploration of large amounts of possible starting geometries and correlation effects are
implicitly considered during the parameterization procedures with respect to
experimental values. Therefore, the PM3 method which is, for instance, able to reproduce
the hydrogen-bonding patterns between water molecules,[107] is selected. Since the choice
of an adequate semiempirical Hamiltonian is still a controversial field, the geometries are
in addition optimized with the AM1 Hamiltonian and both results are compared.
All the semiempirical calculations are performed with the MOPAC program.[108-110] The
minimization method selected is the Eigenvector following (EF),[111, 112] which is
designed to search for critical final structures, as transition states. The EF is supposed to
avoid unexpected molecular rearrangements in the final steps of the minimization path
due to any possible overestimation of geometry changes when the molecules are too close
together. In fact, this very common problem of MOPAC optimization of nonstandard
structures, with the keyword precise, is avoided in all of the calculations performed.
Therefore, the procedure involves a large number of molecular arrangements during the
gradient-driven path to explore the multiple minima hypersurface (MMH) of such
systems. The followed path leads to local minima of potential energy of the molecular
cluster.
3. First discrimination and similarity analysis
Once the geometries of all the starting structures are optimized at a semiempirical level,
the next step is the geometrical and energetic discrimination of the resulting complexes.
The program for processing the MOPAC output data is called Q3.[4] This program
performs the statistical thermodynamic analysis and discards all optimized structures
which are degenerated. In a first approach, all structures with the same energy compared
to a previous one, are discarded. Later, it is necessary to consider that there are two types
of degeneracy in this kind of potential energy surface minima. The first consists of

55
clusters which are identical, which means that they have both the same energy and
molecular geometry, called similarity degeneracy (SD). The second consists of clusters
with different molecular geometry but the same energy, called valid degeneracy (VD). In
the latter, to follow only an energy criterion for the discrimination between structures
might lead to neglect important contributions.
Therefore, a subroutine called Tanimoto is introduced in the program to analyze the
similarity among molecular arrangements, to discard the SD and keep the VD and
preserving, in general, those degenerate clusters when symmetry provides the same
molecular configuration but molecules themselves are exchanged. The Tanimoto
procedure uses the Tanimoto similarity index to calculate the similarity between
structures pair by pair. For this purpose it first converts internal coordinates to Cartesians
for all atoms in a given structure. Then, it obtains the matrix of the position vector
modules with respect to an origin fixed at atom 1, called [D]:
[ ] [ ]NrrrrD ,...,,, 321≡ (3.2)
where N is the total number of atoms.
The Tanimoto similarity index T corresponding to a comparison of cluster A with B is
determined by the expression:
( )MBAMT −+= (3.3)
where A, B, M are calculated using the following expressions:
Bi
N
i
Ai rrM ∑
=
=1
(3.4)
∑=
=N
i
Ai
Ai rrA
1 (3.5)
∑=
=N
i
Bi
Bi rrB
1
(3.6)
where Air is an element of [DA] and B
ir of [DB] that belongs to A and B structures,
respectively.
The Q3 program compares all clusters with an energy difference of less than 0.096
kJ/mol compared to any previous one. This is an arbitrary limit of 10-3 ev to consider that

56
these clusters have the same energy, by far below the accuracy of semiempirical methods.
Then, a limit value of discrimination to consider the clusters equal or not from a
geometrical point of view is defined. For example, if the calculated value of T is equal or
larger than T' = 0.85 (highest limit value of the Tanimoto index), these molecular
arrangements are equivalent and geometrically SD degenerated clusters. If T is less than
0.85 the clusters are different, even when they have the same energy. The latter case
corresponds to VD. As would be expected, the selection of the most adequate highest
limit value of the Tanimoto index T’ depends on the complexity of the system
investigated. Several values were tested, and in general, for bimolecular and simple
trimolecular complexes, the T' = 0.85 value provides a good reference for the geometric
discrimination.
It is important to include structures with VD to validate the partition function in the
statistical thermodynamic analysis. Given that the 10-3 ev energies degeneracy limit is by
far below the accuracy of semiempirical Hamiltonians, it is expected that keeping the VD
structures would not play a key role for the structural analysis. However, since the
geometries of all these structures are subsequently refined at higher level of theory, there
is a possibility, especially when the PES is very flat, that some of these VD structures
might lead to different minima at other level of theory. In addition, the inclusion of the
similarity analysis in the search of supermolecular geometries is a conceptual advance in
the theoretical development of the MMH approach.
4. Refinement of the geometries
This step is not included in the statistical thermodynamic original MMH approach, in
which the thermodynamic analysis is performed in the third step, using the Q3 program,
as already mentioned. But for the purposes of the structural analysis, the semiempirical
results provide just a preliminary overview of the interactions in the complex. For this
reason, the set of relevant semiempirical local minima are refined using DFT and MP2
methods.
The selection of the next level of theory depends very much on the characteristics of
the system to study. For example, for very weak interacting systems the use of the
currently available popular DFT methods is not recommended. However for moderate-
“strong” hydrogen bonded systems like the formic acid – formamide dimer, DFT
provides results in good agreement with the ab initio calculations. Here, for an initial
discrimination of the semiempirical structures, the MP2 method with a small – medium

57
basis set like the 6-31G(d,p) is used, and the size of the basis set is gradually increased,
including correlation-consistent basis sets. During all the process, including the analysis
of the initial semiempirical geometries, each structure is individually analyzed and
calculated. This point gains special relevance in very weakly interacting systems.
The tight criterion is used in the optimization of geometries and force constants are
calculated when necessary. In weak interacting systems the distortions of the geometries
from the imaginary frequency in transition states are carefully followed. Many times this
leads to new local minima, as expected in very flat potential energy surfaces. The BSSE-
CP corrections during the optimizations of the geometry are included only with the
smaller basis set since these corrections are very demanding when larger basis sets are
used. The utilization of different basis sets, the calculation of vibrational spectra, the
analysis of the individual contributions that stabilize each structure, together with the
comparison of the calculated minima or their properties with experimental results and
similar complexes from the literature, allow for a better characterization of the systems
under study.

58
3. Formic Acid Complexes with Formamide and Dimethyl ether
3.1. Introduction
Hydrogen-bonded complexes have been subject to a large number of studies in
chemistry and life sciences. Oxygen atoms as hydrogen bond acceptors are of particular
interest since they play a key role in biological processes.
Formic acid (FA) and its interactions with other molecules provide a very good model
to understand a large variety of hydrogen bond interactions, from classical and strong to
weaker and non-classical bonds.[20, 113-118] The proton transfer mechanism of the FA
dimer has been extensively described,[119-127] and studies of the complexes of FA with
other molecules like water were carried out in our group using matrix isolation
spectroscopy in combination with ab initio methods.[117]
The formamide (FMA) molecule is the simplest molecule that contains a peptide
linkage. Therefore, it can be used as a simple model of hydrogen bond interactions
involving carboxylic acids and amino groups in biological systems, like protein-protein
and protein- substrate interactions.[21, 115] Complexes involving the interactions of ethers
with molecules of biological interest are also described in literature,[128-130] including
matrix isolation and ab initio studies of dimethyl ether (DME) complexes with water,[131]
methanol,[132, 133] hydrogen peroxide[134] and hydracids.[135]
FA – FMA complexes are very interesting hydrogen bonded systems. In addition to its
biological interest, they provide good models for studying the competition between non-
covalent interactions involving nitrogen and oxygen atoms in the same molecule. The
FMA homodimers, as well as its complexes with other molecules like water and
methanol have been intensively studied.[136-138] However, only few reports are found
about FA – FMA heterodimers. The computational study of the electron-density
dependent properties of FA, FMA and their homo and heterodimers made by Gálvez and
Gómez,[115] the crystallographic structure by Nahringbauer in 1968[139] and the ab initio
calculations of Neuheuser[140] are of special interest.
Here, several minima of the FA – FMA potential energy surface are described and their
geometries, binding energies and vibrational spectra are discussed. In addition, the 1:2
and 1:4 FA – FMA complexes are investigated. The FA – FMA trimers allow for the

59
analysis of the influence of a third molecule on the dimer properties, e. g. an additional
FMA molecule on the FA – FMA dimer or a formic acid molecule on the FMA dimer.
The structures of the FA – FMA dimers and trimers are compared to the FMA – water
and FMA – methanol dimers from literature data and with the reported FA – FMA crystal
structure.
The calculated structures, binding energies and vibrational properties of FA – DME
complexes are also discussed using ab initio calculations. The FA – DME system exhibits
classical as well as weak OH…O, C=O…H, C-O…H and CH…O hydrogen-bonding
interactions, making these complexes very interesting for theoretical and experimental
research. The results obtained with various computational methods and basis sets are
discussed, as well as the influence of the basis set superposition error (BSSE) on the
calculated energies and geometries of the different complexes. The calculated vibrational
spectra of the dimers are compared to experimental matrix isolation spectra.
3.2. Computational methods
The Multiple Minima Hypersurface (MMH) approach is used for searching
configurational minima in the FA – FMA and FA – DME systems. For each case around
one thousand randomly arranged clusters were generated as starting points, and the
resulting geometries were optimized and analyzed using PM3 and AM1 semiempirical
quantum mechanical Hamiltonians. These semiempirical results provided a preliminary
overview of the interactions in the complexes, and the relevant configurations were
further refined using DFT and ab initio methods. The AM1 Hamiltonian did not
contribute with new minima to the PM3 structures which were further refined using ab
initio methods.
In the case of the 1:2 and 1:4 FA – FMA complexes, 250 and 198 random geometries,
respectively, were taken as starting point for the PM3 geometry optimizations. A
selection of the resulting geometries was optimized using the B3LYP density functional
and Dunning’s correlation consistent triple ζ basis set.
The ab initio and DFT computations were performed using the Gaussian 98,[141]
Gaussian 03,[141] and MOLPRO[142] programs. In all cases the equilibrium geometries
and vibrational frequencies were calculated using second order Møller−Plesset
perturbation theory (MP2). Pople’s 6-31G(d,p) and 6-311++G(d,p) basis set as well of

60
augmented and non augmented Dunning’s correlation consistent double and triple ζ basis
sets (cc-pVDZ, aug-cc-pVDZ, cc-pVTZ and aug-cc-pVTZ) were used.
For the FA – FMA complexes the geometries and frequencies were calculated also
using the density functional theory (DFT) with the B3LYP hybrid functional[73, 74] and
single point calculations were done with coupled clusters of single and double
substitutions (with non iterative triples) CCSD(T)/aug-cc-pVTZ.
The stabilization energies were calculated by subtracting the energies of the monomers
from those of the complexes and corrected for the basis set superposition errors (BSSE)
using the counterpoise (CP) scheme of Boys and Bernardi. ZPE corrections were also
included.
To investigate the influence of the basis set superposition errors (BSSE) on the
geometries of the complexes, the most stable FA – FMA and FA – DME dimers were
optimized at the MP2/6-31G(d,p) level of theory using the CP scheme during the
optimization process. In addition, the geometries were optimized without BSSE at the
same level of theory to compare the influence of the BSSE on the binding energies as
well as on the geometries. The small 6-31G(d,p) basis set was selected for this purpose
since the BSSE is expected to be more pronounced with small basis sets, and in addition
the computations are less demanding.
3.3. Formic acid – formamide complexes. Results and discussion
Formic acid – formamide dimers
Geometries and binding energies. Analysis of the intermolecular interactions
Nine FA – FMA complexes A – I were localized after MMH search and refined with
both DFT and MP2 calculations (Figure 3.1). The use of Dunning´s cc-pVDZ, aug-cc-
pVDZ, cc-pVTZ, and aug-cc-pVTZ basis sets (Table 3.1) revealed that the geometries of
the complexes are almost independent of the basis sets used. Therefore, the hydrogen
bond distances and angles are discussed here at the MP2/aug-cc-pVTZ level of theory,
only.

61
Seven basic types of interactions (1) – (7) can be differentiated in the FA – FMA
complexes:
(1) NHFMA…O=CFA interaction between the amide hydrogen atom of FMA and the
carbonyl oxygen atom of FA.
(2) C=OFMA…HOFA interaction between the carbonyl oxygen atom of FMA and the
hydroxyl hydrogen atom of FA.
(3) (O)CHFMA…O=CFA interaction between the aldehyde hydrogen atom of FMA and
the carbonyl oxygen atom of FA.
(4) NHFMA…(H)OCFA interaction between the amide hydrogen atom of FMA and the
hydroxyl oxygen atom of FA.
(5) C=OFMA…HC(O)FA interaction between the carbonyl oxygen atom of FMA and
the aldehyde hydrogen atom of FA.
(6) HN(H)FMA…HOFA interaction between the nitrogen atom of FMA and the
hydroxyl hydrogen atom of FA.
(7) (O)CHFMA…(H)OCFA interaction between the aldehyde hydrogen atom of FMA
and the hydroxyl oxygen atom of FA.

62
TABLE 3.1: Calculated binding energies and ZPE and BSSEa corrected binding
energies (in kcal/mol) of the FA – FMA dimers A – I
MP2 cc-pVDZ aug-cc-pVDZ cc-pVTZ ΔE ΔE (ZPE) ΔE (BSSE) ΔE ΔE (ZPE) ΔE (BSSE) ΔE A -18.37 -15.92 -11.60 -16.53 -14.30 -14.21 -16.90 B -14.56 -12.57 -8.67 -12.87 -11.09 -11.03 -13.21 C -11.38 -9.72 -6.69 -9.93 -8.36 -8.23 -10.16 D -10.06 -8.29 -5.34 -8.97 -7.37 -7.41 -8.89 E -10.21 -8.03 -5.19 -7.54 -5.85 -5.81 -8.04 F -7.58 -6.33 -3.67 -6.38 -5.16 -4.97 -6.12 G -6.57 -4.95 -3.15 -5.42 -4.34 -4.29 -5.58 H -6.26 -5.21 -2.47 -5.50 -4.56 -4.31 -5.34 I -5.37 -4.50 -2.15 -4.64 -3.82 -3.52 -4.37 MP2 B3LYP aug-cc-pVTZ cc-pVTZ
CCSD(T)/cc-pVTZ // MP2/aug-cc-pVTZ
ΔE ΔE (ZPE) b ΔE (BSSE) ΔE ΔE (ZPE) ΔE ΔE (ZPE)
b A -16.64 -14.41 -15.25 -16.03 -13.97 -16.76 -14.53 B -12.96 -11.18 -11.87 -12.2 -10.46 -13.21 -11.43 C -9.95 -8.38 -8.92 -8.97 -7.53 -10.00 -8.42 D -8.71 -7.11 -7.86 -7.77 -6.25 -9.06 -7.45 E -7.32 -5.63 -6.37 -6.22 -4.52 -8.06 -6.37 F -6.05 -4.83 -5.26 -4.88 -3.78 -6.24 -5.02 G -5.2 -4.12 -4.58 -4.63 -3.59 -5.40 -4.32 H -5.17 -4.23 -4.60 -4.37 -3.44 -5.60 -4.66 I -4.29 -3.47 -3.73 -3.37 -2.60 -4.54 -3.71
aBSSE corrected binding energies for the cc-pVDZ, aug-cc-pVDZ and aug-cc-pVTZ
basis sets at the MP2 level of theory. bZPE correction from the MP2/aug-cc-pVDZ
calculations.

63
Figure 3.1. The calculated structures with hydrogen bond lengths (Å) and angles
(degree) of the FA– FMA dimers A - I at the MP2/ aug-cc-pVTZ level of theory.

64
The most stable FA – FMA dimer calculated is complex A with a binding energy of
-14.41 kcal/mol (MP2/aug-cc-pVTZ + ZPE), the ZPE correction is taken from the
MP2/aug-cc-pVDZ calculations. The energies of the other dimers B – I are also discussed
at this level of theory (Table 3.1).
The dimer A is stabilized by interactions (1) and (2) involving both carbonyl groups
and the N-H and O-H hydrogen atoms of the formamide and formic acid molecules. The
binding distances are 1.859 and 1.637 Å, respectively. In dimer B (-11.18 kcal/mol) the
amide hydrogen atoms of the FMA are not involved in the stabilization of the complex.
Instead, the aldehyde hydrogen atom of the FMA interacts with the carbonyl oxygen
atom of the FA at 2.304 Å (interaction 3). Cyclic dimer B is also stabilized by interaction
(2) with a binding distance of 1.663 Å (around 0.025Å longer than interaction (2) in
complex A).
The difference between the binding energies of complexes A and B is more than 3
kcal/mol. This is explained by the different hydrogen bond capabilities of the N-H vs. C-
H hydrogen atoms of FMA. Consequently, the difference between the hydrogen bond
distances of interaction (1) in A and interaction (3) in B is more than 0.4 Å.
Compared to dimer A, the FA – FMA complexes C and D are between 6 - 7 kcal/mol
less stable. The binding energy of complex C is calculated to -8.38 kcal/mol. In dimer C,
as in A and B, the carbonyl oxygen atom of FMA is interacting with the hydroxyl
hydrogen atom of FA (interaction (2)). However, with 1.735Å the hydrogen bond
distance in C is considerably larger than in A and B (Figure 3.1). In dimer C the carbonyl
oxygen atom of FA is not involved in the stabilization of the complex. Instead, one of the
amide hydrogen atoms of FMA interacts with the hydroxyl oxygen atom of FA at a
distance of 2.213 Å (interaction (4)).
Dimer D has a binding energy of -7.11 kcal/mol and it is energetically very close
(about 1 kcal/mol) to dimer C. In dimer D, again both carbonyl groups of the formamide
and formic acid molecules are involved in the stabilization of the complex via interaction
(1) (1.968Å hydrogen bond distance) and interaction (5) between the carbonyl oxygen
atom of FMA and the aldehyde hydrogen atom of FA (hydrogen bond distance 2.258 Å).
In this case, the O-H group of the FA is not involved in the stabilization of the dimer.

65
The cyclic-non planar structure of dimer E is an interesting case. The amide group is
pyramidalized, and thus makes possible interaction (6) between the nitrogen atom of
FMA and the O-H hydrogen atom of FA (2.003 Å). Complex E is also stabilized by
interaction (1) with a 2.133 Å distance. The calculated binding energy of dimer E is -5.63
kcal/mol.
The dimers F – I are weakly bound and energetically very close to each other. The
binding energies vary between -4.83 and -3.47 kcal/mol. With the exception of the non
planar dimer G, stabilized only by interaction (1), all dimers are cyclic. Dimer F is
stabilized by interactions (4) and (5) and dimer H by interactions (3) and (5). In dimer I,
interaction (5) appears together with the weakest interaction (7) between the carbonyl
hydrogen atom of FMA and the hydroxyl oxygen atom of FA resulting in a large distance
of 2.561 Å. In none of the F – I dimers the hydroxyl hydrogen atom of the formic acid
molecule interacts with other groups.
All the complexes discussed here were produced from randomly generated geometries
and not via chemical intuition. It is thus interesting to note that:
(a) The calculated geometries of the FA – FMA dimers A and B are in complete
agreement with the calculated structures of the FA – FMA complexes proposed by
Neuhauser[140] and Galvez[115] in their ab initio and DFT studies. These complexes show
also interesting analogies with the FMA – water and FMA – methanol dimers, which
have been extensively studied. These comparisons are discussed in more detail later.
(b) The most stable dimers A and B are those where both carbonyl groups of FMA and
FA are involved in the stabilization of the complex, together with the hydroxyl hydrogen
atom of FA that interacts with the carbonyl oxygen atom of FMA (interaction (2)).
(c) In the less stable complexes F – I the hydroxyl hydrogen atoms of FA are not
involved in hydrogen bonds.

66
According to the calculated geometries and binding energies of all the FA – FMA
dimers, it is possible to make some preliminary qualitative conclusions about the strength
of the different interactions:
(a) C=OFA ····H-NFMA (interaction 1) › C=OFA ···H-CFMA (interaction 3)
(b) C=OFMA ····H-OFA (interaction 2) › C=OFMA ···H-CFA (interaction 5)
(c) NHFMA…O=CFA(interaction 1) › NHFMA… (H)OCFA (interaction 4)
(d) OHFA…O=CFMA(interaction 2) › OHFA… NHFMA(interaction 6)
(e) CHFMA…O=CFA(interaction 3) › CHFMA… (H)OCFA(interaction 7)
(f) The CH group in the formic acid molecule only interacts with the O=C group of the
formamide molecule (interaction 5)
This allows for the qualitative comparison of the hydrogen-bond acceptors and donors
in the FA – FMA dimers:
(a) Donors: OH › NH › CH FMA› CHFA
(b) Acceptors: C=OFMA› C=OFA
The order of the proton donor ability of the hydrogen atoms linked to C, N and O
heteroatoms corresponds to the increase of the electronegativity from carbon to oxygen.
To compare the hydrogen bond acceptor capability of the carbonyl group of FMA with
that of the carbonyl group of FA is more complicated. In this case, the order is based on
the relative binding energies and distances in the complexes.
It is interesting to mention that for the FMA – water and FMA – methanol dimers the
binding energy to the carbonyl group of FMA is slightly more favorable than to the
amide group.[136, 137, 143] In the most stable FA – FMA dimer A both interactions are
present, and the distance between the carbonyl oxygen atom of FMA and the hydroxyl
hydrogen atom of FA is nearly 0.2 Å shorter than the hydrogen bond distance between
the amid hydrogen atom of FMA and the carbonyl oxygen atom of FA (Figure 3.1).

67
In agreement with Neuheuser’s observations[140], the weakest C-H…O interaction still
contributes significantly to the interaction energy in the FA – FMA system, for example
in dimer B.
Comparison with other dimers
The presence of carbonyl groups in both FMA and FA results in additional
stabilizations which do not exist in the water – formamide (W – FMA) and the methanol
– formamide (M – FMA) complexes. Nevertheless, there are very interesting analogies
among all the FMA complexes with water, methanol and formic acid.[136, 137, 143] (Figure
3.2).
Figure 3.2. B3LYP structures of FMA-Water, FMA-Methanol and selected FMA-
FA dimers.

68
Three stable W – FMA structures were described by Fu et al.[136] using DFT and MP2
methods with large basis sets. In all cases the main interaction is OHw…O=CFMA. FW I
and FW II are the two more stable W – FMA calculated complexes. They are cyclic
dimers with additional NHFMA…O-HW and CHFMA…O-HW interactions, respectively
(Figure 3.2). Their geometries have some similarities with the structures of some of the
FA – FMA dimers.
In the FA – FMA dimer A the carbonyl group of the formamide molecule interacts with
the hydroxyl hydrogen atom of the formic acid resembling the interaction between the
carbonyl group of the formamide and the hydroxyl hydrogen atom of water in the FW I
complex. The amide hydrogen atom of FMA interacts with the carbonyl oxygen atom of
FA in a similar way as the NHFMA…O-HW interaction in the FW I formamide – water
complex. The water – formamide dimer FW II shows the C=OFMA…HOw interaction
(similar to the C=OFMA…HOFA in complexes A and B) and the CHFMA…O-HW (similar to
the CHFMA …O=CFA interaction in complex B).
Four stable formamide – methanol (M – FMA) dimers have been studied by Fu et al.
using DFT and ab initio methods with various basis sets.[137] The two most stable M –
FMA complexes have similar geometries compared to the FA – FMA and W – FMA
dimers.
MF I is a cyclic dimer with OHM…O=CFMA and NHFMA…O-HM interactions. The MF
II dimer shows the OHM…O=CFMA and CHFMA…O-HM interactions. Both structures are
similar to the A and B FA – FMA dimers. MF IV compares very well with the FA –
FMA dimer F. They are both cyclic dimers stabilized by the NHFMA…(H)OCFA(MET)
interaction between the amide hydrogen atom of FMA and the hydroxyl oxygen atom of
the FA or methanol molecules. The second interaction is the C=OFMA…HCFA in the case
of complex F. The MF IV dimer shows the C=OFMA…HCM interaction between the
carbonyl oxygen atom of FMA and one hydrogen atom from the methyl group of the
methanol molecule.
FA – FMA dimers have been studied before using ab initio and DFT methods.
Neuheuser calculated five non cyclic and ring H-bonded FA – FMA structures as a model
for the interactions in supramolecular complexes of dicarboxylic acids and

69
dimethylformamide.[140] Pacios performed ab initio calculations of the most stable FMA
– FA dimer A.[144] Galvez and coworkers studied the variation of electron density
properties with the intermolecular distance for various cyclic dimers, including the most
stable FA – FMA complex.[115] Neuheuser, Pacios and Galvez studies corroborate our FA
– FMA dimers A and B as the most stable calculated geometries in the formic acid –
formamide system. This agreement confirms the reliability of the MMH procedure for
localizing the minima in non covalent complexes.
Methods and basis set influence on the calculated geometries and binding energies
of the FMA – FA dimers
Table 3.2 lists some selected intra- and intermolecular distances and hydrogen bond
angles at various levels of theory for selected FA – FMA dimers. Complex A is discussed
because it is the most stable calculated FA – FMA dimer. The weaker complex D has
been selected due to its very weak C=OFMA···H-CFA interaction. In addition,
intermolecular distances for complex B are presented.
The O-HFA bond lengths in dimer A are not sensitive to the method or the basis set used
for the calculations (Table 3.2). However, the N-HFMA intramolecular distances of the
interacting amide hydrogen atoms vary in both complexes A and D substantially with the
basis set. At the MP2 level of theory with the cc-pVDZ basis set, the calculated N-HFMA
bond lengths are 0.007 Å larger than with the cc-pVTZ basis set, whereas inclusion of
diffuse functions has only a minor influence. The B3LYP/cc-pVTZ calculated bond
lengths are only 0.002-0.003 Å larger than the MP2/cc-pVTZ values.
The C-HFA bond lengths in complex D behave in a similar way. In this case the
difference between the MP2 double and triple zeta basis set is even more pronounced
(0.0103-0.009 Å). The MP2/cc-pVTZ and aug-cc-pVTZ C-HFA distances are basically
the same and very similar to the B3LYP/cc-pVTZ values. The MP2/cc-pVDZ C-HFA
bond length is 0.003 Å larger than the MP2/aug-cc-pVDZ calculated value. The C=O
carbonyl bond lengths of the FMA and FA molecules in dimers A and D show a little
more dependence on the basis sets. At the MP2 level of theory the C=O distances
increase with the addition of diffuse functions (aug) in the double and triple zeta basis

70
sets. This variation is less pronounced with the triple zeta basis set. The B3LYP/cc-pVTZ
carbonyl bond lengths are 0.006- 0.003 Å shorter than the MP2/cc-pVTZ values.
TABLE 3.2: Comparison of selected intramolecular and intermolecular parameters
in the FA – FMA dimers A, B and D at the different levels of theory. Distances are
in Å and angles in degrees
B3LYP MP2 cc-pVTZ cc-pVDZ aug-cc-pVDZ cc-pVTZ aug-cc-pVTZ Monomer O-HFA 0.970 0.975 0.975 0.970 0.971 C-HFA 1.097 1.108 1.103 1.092 1.092 N-HFMA
a 1.006 1.014 1.012 1.004 1.006 C=OFA 1.197 1.209 1.215 1.203 1.205 C=OFMA 1.209 1.220 1.228 1.215 1.218 Dimer A O-HFA 1.006 1.005 1.006 1.004 1.005 N-HFMA
a 1.022 1.027 1.027 1.019 1.020 C=OFA 1.214 1.225 1.231 1.219 1.221 C=OFMA 1.229 1.237 1.245 1.232 1.235
NHFMA…O=CFA 1.879 1.872 1.871 1.853 1.859 C=OFMA…HOFA 1.643 1.661 1.657 1.634 1.637
< NHFMA…OFA 164.77 164.65 165.35 165.48 164.90 < OHFA…OFMA 175.70 173.95 173.98 173.82 174.51 Dimer D C-HFA 1.095 1.104 1.101 1.091 1.092 N-HFMA
a 1.016 1.022 1.021 1.014 1.015 C=OFA 1.207 1.219 1.225 1.213 1.214 C=OFMA 1.218 1.227 1.236 1.222 1.225 NHFMA…O=CFA 2.006 1.992 1.981 1.968 1.968
C=OFMA…HC(O)FA 2.295 2.258 2.267 2.251 2.257 < NHFMA…OFA 163.31 164.40 164.52 164.64 163.99 < CHFA…OFMA 138.65 140.70 139.51 140.57 138.88 Dimer B O-HFA 1.001 1.001 1.002 0.998 1.000 C=OFA 1.209 1.220 1.226 1.214 1.216

71
C=OFMA 1.225 1.235 1.242 1.229 1.231 C=OFMA…HOFA 1.676 1.683 1.682 1.663 1.663
(O)CHFMA ...O=CFA 2.350 2.308 2.317 2.303 2.304 aNH hydrogen atom in cis position relative to the carbonyl oxygen atom of the
formamide.
The NHFMA…O=CFA distances in both A and D dimers are 0.026 and 0.038 Å,
respectively, larger at the B3LYP level of theory compared to the MP2 calculations with
the same basis sets. In the MP2 calculations the NHFMA…O=CFA binding distances
decrease from the double to the triple zeta basis sets. The behavior of the
C=OFMA…HOFA (dimer A) and the C=OFMA ···H-CFA (dimer D) distances is very similar,
in general. The calculated hydrogen bond angles are comparable in all cases.
Intermolecular distances in complex B are basically not dependent on the augmentation
of the basis sets.
The B3LYP calculations show a tendency to give a little larger values for the
intermolecular binding distances, compared to the MP2 values with the same basis set.
However, there is no considerable difference between the B3LYP and the MP2 calculated
geometries for the FA – FMA dimers. Reliable geometries for the weak interacting FA –
FMA dimers are also calculated using the B3LYP density functional. At the MP2 level of
theory, there is basically no change of the geometries when the basis set is augmented by
adding diffuse functions.
In the FA – FMA dimers, the MP2/cc-pVDZ calculations have a tendency to
overestimate the binding energies (Table 3.1). The double zeta energies compare better to
the CCSD(T)/cc-pVTZ calculations when the augmented functions are added. At the
MP2 level, using triple zeta basis sets augmented and non-augmented, the results are very
similar to those of the CCSD(T)/cc-pVTZ single point calculations. The B3LYP/cc-
pVTZ binding energies are smaller than the MP2 and CCSD(T) energies.
The MP2 level of theory with the cc-pVTZ basis set provides a very adequate
description of the FA – FMA system. It is in general not necessary to use the expensive
aug-cc-pVTZ basis set, in agreement with the results of others studies of weakly
interacting complexes.[116, 145, 146]

72
Effect of the BSSE on the calculated geometries and binding energies
BSSE corrections have been calculated for all FA – FMA dimers at the MP2 level of
theory with the cc-pVDZ, aug-cc-pVDZ and aug-cc-pVTZ basis sets. As expected, the
BSSE decreases with increasing size of the basis sets. That can be noticed by comparing
the ΔE (binding energies without corrections) with ΔE (BSSE) (BSSE corrected binding
energies) in Table 3.1. For example, in complex A with MP2/cc-pVDZ the BSSE
correction is 6.77 kcal/mol, compared to only 2.32 and 1.39 kcal/mol at the MP2/ aug-cc-
pVDZ and MP2/ aug-cc-pVTZ levels of theory, respectively.
In addition, FA – FMA dimers A and B were optimized at the MP2/6-31G(d,p) level of
theory using the counterpoise (CP) scheme to evaluate the influence of BSSE on the
calculated energies and geometries.
The intramolecular bond distances in the FA and FMA molecules are almost not
effected by the inclusion of BSSE corrections during the optimization processes (Figure
3.3). In all cases the difference between the bond distances was in the order of 10-3 Å or
less. Only the intermolecular distances C=OFA ····H-NFMA (interaction 1), C=OFMA ····H-OFA
(interaction 2), and C=OFA ···H-CFMA (interaction 3) are significantly influenced by BSSE.
Especially for the weaker interaction 3, the BSSE-optimized distance C=OFA ···H-CFMA is
almost 0.12 Å larger than the non-BSSE optimized distance. Hydrogen bond angles are
less sensitive to BSSE corrections (Figure 3.3). Thus, the geometrical changes introduced
by BSSE corrections are very limited, and the basic geometries and interactions in FA –
FMA complexes do not depend on the inclusion of BSSE during the optimization
process, in accordance with other observations.[146]

73
Figure 3.3. MP2/6-31G(d,p) geometries with inter and intramolecular lengths(Å)
and hydrogen bond angles(degree) of dimers A and B. 1) Optimized without BSSE
corrections. 2) Optimized with BSSE corrections.
It is therefore not surprising that the binding energies of dimers A and B are almost
independent of BSSE corrections during geometry optimization. For complexes A and B
the calculated BSSE corrections are 4 – 5 kcal/mol, and the differences in binding
energies between the BSSE-optimized and the non-BSSE-optimized geometries are in the
range of 0.19 - 0.57 kcal/mol only (Table 3.3).
TABLE 3.3: Comparison of the binding energies of the calculated FA–FMA dimers
A and B at the MP2/6-31G(d,p) level of theory including BSSE corrections in the
optimization processes
MP2/6-31G(d,p) Optimization with BSSE Optimization without BSSE ΔE ΔE (BSSE) ΔE ΔE (BSSE) Dimer A -18.03 -13.39 -18.22 -13.19 Dimer B -14.88 -10.23 -14.31 -10.08
Intramolecular distances and vibrational frequencies. Calculated spectra
The vibrational frequencies of all the FA – FMA dimers have been calculated at the
B3LYP/cc-pVTZ, MP2/cc-pVDZ and MP2/aug-cc-pVDZ levels of theory. The

74
B3LYP/cc-pVTZ vibrational frequencies and selected intermolecular distances for
complexes A and B are discussed here. Based on experimental and B3LYP/cc-pVTZ
calculated vibrational frequencies of the monomers, the frequency shifts and correction
factors for some molecular vibrations of complexes A and B are estimated (Table 3.4) to
accurately match calculated with experimental frequencies (Table 3.5).
TABLE 3.4: Comparison between the experimental (Ar matrix, 10K) and the
calculated B3LYP vibrational frequencies (in cm–1) of formic acid and formamide
monomers, shift and factor of correction
Experimental Computed frequencies B3LYP/ cc-pVTZ
Shifta and factor of correction (exp/B3LYP freq)
3549.9 3722.1 172.2 (0.954) νOHb
3066.0 3043.8 -22.2 (1.007) νCHb
1766.9 1826.2 59.3 (0.967) νC=Ob
1103.5 1125.0 21.5 (0.981) νCOb
1739.1 1803.8 64.7 (0.964) νC=Oc
2882.9 2931.0 48.9 (0.984) νCHc
3547.4 3718.2 170.8 (0.954) νasNH2c
3426.6 3579.9 153.3 (0.957) νsNH2c
aShifts are calculated as the difference between the computed and the experimental
frequencies. bFormic acid fundamental modes. cFormamide fundamental modes.
Compared to the monomers, intramolecular distances and the corresponding vibrational
frequencies in the complexes are perturbed as a consequence of the intermolecular
interactions (Tables 3.8 and 3.11). In complexes A and Β the O-H stretching vibrations of
the FA molecule show the largest red shifts with -677 and -585 cm-1, respectively (Tables
3.8 and 3.11). This demonstrates the strong interaction between the OH hydrogen atom of
FA and the carbonyl oxygen atom of FMA (Figure 3.1) resulting in an elongation of the
OH bonds of 0.036 and 0.031 Å, respectively, for complexes A and B (B3LYP/cc-pVTZ,
Table 3.2).

75
TABLE 3.5: Calculated B3LYP/cc-pVTZ vibrational frequencies (in cm–1) of
dimers A and B and frequency shift in the complex, from the isolated monomer (in
parentheses). Predicted frequencies after scaling.
Monomer Dimer A Dimer B B3LYP/cc-pVTZ Calculated Predicteda Calculated Predicteda
3722.1
2983.7 3012.4 3044.8 (-677.3)
2904.7
3136.7 (-585.4)
2992.4
νOHb, c
1826.2 1779.8 (-46.4) 1721.1 1788.5 (-37.7) 1729.5 νC=Ob
1125.0 1256.5 (+131.5) 1232.6 1230.6 (+105.6) 1207.2 νCOb
1803.8 1729.9 (-73.9) 1667.6 1732.3 (-71.5) 1669.9 νC=Od
3718.2 3675.5 (-42.7) 3506.4 3715.8 (-2.4) 3544.9 νasNH2d
3579.9 3348.5 (-231.4) 3204.5 3579.4 (-0.5) 3425.5 νsNH2d
aPredicted frequencies after scaling the individual frequencies with a scaling factor
obtained by comparing calculated vs experimental frequencies of the corresponding
monomer bands (Table 3.4). bformic acid. cIn the case of complex A, there is a very
strong coupling between the νOH and the νCH vibrations of formic acid and formamide.
The same happen for the νC=O vibrations of formic acid and formamide. dFormamide.
The carbonyl stretching frequencies of the FA molecules in dimers A and B are
calculated to be shifted by -46 and -38 cm-1. The C=OFA bond lengths in A and B
increase by 0.017 and 0.012 Å compared to the monomers. The red shift for dimer B is 8
cm-1 less than for dimer A. This difference is caused by the stronger C=OFA ····H-NFMA
interaction (interaction 1) in dimer A compared to the weaker C=OFA ···H-CFMA interaction
(interaction 3) in dimer B.
The carbonyl stretching frequency shifts of the FMA molecules are -74 and -71 cm-1.
The C=OFMA bond lengths in the dimers A and B increases by 0.020 y 0.016 Å,
respectively. According to the structure of the complex, only in dimer A a significant
shift (-231 and -43 cm-1) respectively, for the symmetrical and anti-symmetrical
vibrations of the N-H group, is predicted. The intramolecular N-H bond distance of the
interacting NH group of FMA in complex A is consequently 0.016 Å larger than in the
FMA monomer.

76
Larger systems
The intermolecular interactions between FMA and FA create a very flat intermolecular
energy surface. That makes the analysis of systems larger than dimers even more
complicated. It would take huge computational efforts to get a complete description of
the possible geometries for trimers and larger aggregates. However, there are very
interesting correlations between the geometries of the FA – FMA dimers and a selection
of structures of larger FA – FMA systems preliminarily calculated at the DFT level of
theory.
1:2 Formic acid – formamide complexes
Figure 3.4 shows a selection of the most stable calculated 1:2 FA – FMA complexes T-
A to T-G and their B3LYP/cc-pVTZ binding energies with and without ZPE corrections.
It is important to remark, once again, that all the trimer structures are found starting from
a large amount of randomly generated geometries calculated with semiempirical
Hamiltonians and later refined at the B3LYP/cc-pVTZ level of theory.
T-A is the most stable calculated trimer with a binding energy of -22.22 kcal/mol. T-B
is energetically very close to T-A with -21.94 kcal/mol (Figure 3.4). It is interesting to
compare the T-A and T-B geometries with the structure of the dimers. The part of trimer
T-A where the FA and FMA molecules interact to each other, is similar to the FA – FMA
dimer B (Figure 3.1). But due to the presence of a second interacting formamide
molecule, the intermolecular C=OFA···H-CFMA and C=OFMA····H-OFA distances are 0.031
and 0.008 Å larger, respectively, compared to dimer B at the same level of theory (Table
3.2, Figure 3.4). The FMA – FMA interactions in trimer T-A reproduce the structure of
the most stable formamide homodimer.
In trimer T-B, the FA interactions with FMA disturb the structure of the FA – FMA
dimer A. The carbonyl oxygen atom of FA shows an additional interaction with one
amide hydrogen atom of the second FMA molecule. This causes an elongation of 0.102 Å
of the C=OFA ····H-NFMA distance compared to dimer A. The C=OFMA ····H-OFA distance in
trimer T-B is also 0.069 Å larger in comparison to dimer A (Table 3.2, Figure 3.4).

77
Figure 3.4. The calculated structures with hydrogen bond lengths (Å) of the 1:2
FA– FMA complexes T-A to T-G at the B3LYP/ cc-pVTZ level of theory. a
B3LYP/cc-pVTZ binding energies. b B3LYP/cc-pVTZ binding energies, ZPE
corrected.
The trimers T-C and T-D are very close energetically to each other with binding
energies of -21.35 and -21.02 kcal/mol, respectively. Again, the main interactions
between FA and FMA in T-C resemble the FA – FMA dimer B, but the intermolecular

78
distances are shorter compared to the dimer (Figure 3.4, Table 3.2). In this case, one
amide hydrogen atom of the second FMA molecule shows an additional interaction with
the carbonyl oxygen atom of the FA molecule.
T-E, T-F and T-G have calculated binding energies of -18.96, -18.59 and -18.25
kcal/mol, respectively. But in the case of complex T-G there is one imaginary out of
plane vibration at -15cm-1 that is related with the repulsive interaction at 2.306 Å between
the aldehyde hydrogen atoms of the two FMA (Figure 3.4). The geometry of the T-G
complex at the B3LYP/6-31++G (d,p) level of theory is very similar, but there is no
imaginary vibration and the distance between the aldehyde hydrogen atoms of the two
FMA molecules is 2.314 Å.
It is interesting to notice that in the trimers T-A, T-C, T-E, and T-G the interactions
between the FA and one FMA molecule resemble the geometry of the FA – FMA dimer
B. In the same way, interactions in trimers T-B, T-D and T-F resemble the structure of
dimer A. Complex T-F is the only with no direct interactions between the two FMA
molecules. Instead, the interactions between FA and the second molecule of FMA
resemble the structure of the FA – FMA dimer F (Figure 3.1). In the T-A, T-D and T-E
trimers the FA molecule interacts with only one molecule of FMA and the system is
additionally stabilized by the FMA-FMA attractions.
Compared to T-A, the trimers T-H and T-I are much less stable with binding energies
of -14.78 and -16.26 kcal/mol, respectively (Figures 3.4 and 3.5). However, they are
considered here in order to compare the geometry of T-H with the crystal structure and,
in both cases, to analyze the pairs of intermolecular interactions in the trimers.
The T-H complex is less stable compared to T-A, since the carbonyl group and the
hydroxyl hydrogen atom of the FA molecule are not directly interacting with the FMA
molecules. The stabilizing FA – FMA interactions in T-H are the same than in the FA –
FMA dimer F, the C=OFMA ···H-CFA (interaction 5) and the NHFMA…(H)OCFA (interaction
4), however in this case the FA interacts with two molecules of FMA, forming the
structure of the most stable cyclic FMA homodimer.

79
Figure 3.5. The calculated structures with hydrogen bond lengths (Å) of the 1:2 FA–
FMA complexes T-H and T-I at the B3LYP/ cc-pVTZ level of theory. aB3LYP/cc-
pVTZ binding energies. bB3LYP/cc-pVTZ binding energies, ZPE corrected.
The trimer T-I stabilizes by the NHFMA…O=CFA (interaction 1) and C=OFMA…HOFA
(interaction 2) interactions; but unlike other complexes, both interactions take place with
different molecules of FMA. This fact makes this system a very interesting case to
analyze the intermolecular interactions in the trimers. In addition, Trimer T-I is stabilized
by FMA-FMA attractions that resemble the geometry of a FMA homodimer
Analysis of the intermolecular interactions in the trimers
To quantify the contributions of intermolecular interactions in the trimers T-A, T-D, T-
F, T-H and T-I; in each of the trimers one of the three monomers (formic acid or one of
the two FMA molecules) is removed subsequently. The energies of the remaining partial
structures (remaining dimers) were calculated (B3LYP/cc-pVTZ) in the geometries of the
parent trimers (Figure 3.6). These partial structures are then compared with the optimized
dimers to analyze the influence of the third molecule in the trimer on the dimer structures.
From that, a detailed picture of the interactions in an aggregate consisting of three
components is achieved.

80
Figure 3.6. Dimers of complexes T-H and T-I. Partial structure (i): Formic acid and
FMA 1. Partial structure (ii): Formic acid and FMA 2. Partial structure (iii): FMA 1
and FMA 2.
Partial structure (i) is formed by the FA and the FMA 1 molecules; partial structure (ii)
by FA and FMA 2, and partial structure (iii) by FMA 1 and FMA 2 (Figures 3.4 and 3.6).
From trimer T-A the first partial structure is formed by removing FMA 2, and thus this
partial structure consists of FA and the remaining FMA 1 (partial structure (i)) stabilizing
via interactions (2) and (3)). Analogously, partial structure (ii) is formed by removing
FMA 1 and consists of the non-interacting FA and FMA 2. Finally, partial structure (iii)
results from removing the FA molecule and represents the most stable FMA – FMA
homodimer A stabilized via two C=OFMA…H-NFMA interactions. The partial structures (i)
– (iii) in the trimers T-D, T-F, T-H and T-I are formed analogously by subsequently
removing FMA 2, FMA 1, and the formic acid molecule (Figure 3.6).

81
In order to compare with the partial structures (iii) from the trimers, the structures of
the FMA – FMA dimers A and B were also optimized at the B3LYP/cc-pVTZ level of
theory (Figure 3.7, Table 3.6). Opposed to FMA – FMA dimer A, in the partial structures
(iii) of trimers T-A and T-H, the N-HFMA…O=CFA hydrogen bonds are not equivalent,
while one N-HFMA…O=CFA hydrogen bond is elongated; the other is shorter compared to
that in FMA – FMA dimer A. That is clearly due to effect of the interactions with the FA
molecule. Therefore, in partial structure (iii) of trimer T-A, the N-HFMA…O=CFA distance
which is more affected (0.069 Å larger) compared to FMA – FMA dimer A, is the one
corresponding to the C=OFMA group which interact in addition with the FA molecule
(Figures 3.4 and 3.7).
TABLE 3.6: Calculated binding energies (in kcal/mol) of some FMA - FMA
dimers and selected partial structures (iii) of the trimers
B3LYP/cc-pVTZ FMA – FMA dimer A -14.25 FMA – FMA partial structure (iii), Trimer T-A -13.73 FMA – FMA partial structure (iii), Trimer T-H -14.19 FMA – FMA dimer B -9.40 FMA – FMA partial structure (iii), Trimer T-D -8.64 FMA – FMA partial structure (iii), Trimer T-I -8.67
In the partial structure (iii) of trimer T-H, one N-HFMA…O=CFMA hydrogen bond
distance is larger than in the FMA – FMA dimer A. This is the one corresponding to the
side of the FMA – FMA complex that interacts with the FA molecule in the trimer. This
elongation of the hydrogen bond can be attributed not only to the attractive interaction
between one NH hydrogen atom of FMA and the OH oxygen atom of FA at 2.669 Å, but
also to the repulsion at 2.763 Å between the other amidic hydrogen atom of FMA and the
aldehyde-type hydrogen atom of FA (Figures 3.5 and 3.7).

82
Figure 3.7. Comparison between FMA – FMA dimers A and B and the partial
structures (iii) of the 1:2 FA – FMA trimers T-A, T-D, T-H, and T-I.
The partial structures (iii) of trimer T-D and T-I are similar in geometry and close in
energy to the FMA – FMA dimer B (Figure 3.7, Table 3.6). The NHFMA2…O=CFMA1
distances in partial structures (iii) are, compared to the FMA – FMA dimer B, more
affected than the C=OFMA2…H-CFMA1 distances. That is expected, since according to the
geometries of trimers T-D and T-I the carbonyl oxygen atom of FMA 1 interacts also
with the FA molecule, unlike the carbonyl group of FMA 2 (Figures 3.4, 3.5 and 3.7). In
addition, compared to FMA – FMA dimer B, the NHFMA2…O=CFMA1 distance is larger in
trimer T-I than in trimer T-D, which is explained by the interaction of one NH hydrogen
atom of FMA 2 with the carbonyl oxygen atom of FA in trimer T-I (Figure 3.5).
The partial structures (i) in trimers T-D and T-F resemble the FA – FMA dimer A with
very similar hydrogen bond distances and angles, as well as binding energies (Figure 3.8
and Table 3.7). In the same way, the partial structure (i) of trimer T-A is similar to the FA
– FMA dimer B and partial structure (ii) of trimer T-F corresponds to the FA – FMA
dimer F.

83
TABLE 3.7: Calculated binding energies (in kcal/mol) of some FMA - FA dimers
and selected partial structures (i) and (ii) of the trimers
B3LYP/cc-pVTZ FA – FMA dimer A -16.03 FA – FMA partial structure (i), Trimer T-D -15.79 FA – FMA partial structure (i), Trimer T-F -15.80 FA – FMA dimer B -12.32 FA – FMA partial structure (i), Trimer T-A -11.68 FA – FMA dimer F -4.88 FA – FMA partial structure (ii), Trimer T-F -2.88
Figure 3.8. Comparison between FA – FMA dimers A, B and F and selected partial
structures (i) y (ii) of the 1:2 FA – FMA trimers T-A, T-D, and T-F.

84
Several non-additive contributions are considered, including basis set superposition
errors and other non-conventional interactions that may contribute to the stabilization of
the trimers (Tables 3.8 and 3.9). These non-additive contributions are obtained by
subtracting the binding energies of all contributions from the trimer binding energy. As it
can be seen from the Tables 3.8 and 3.9 the non-additive contributions represent between
9.6 – 12.6 % of the total binding energy.
TABLE 3.8: B3LYP/cc-pVTZ energies of the trimers T-A, T-D, T-F, and their
partial structures (i), (ii), and (iii) (in kcal/mol). The percents of the energies of each
partial structure, compared to the total energy of the trimers, are in parenthesis.
B3LYP/cc-pVTZ
T-A T-D T-F Trimer, E(t) -25.92 -24.59 -21.75 Partial structure (i), E(i) -11.68 (45.1%) -15.79 (64.2%) -15.80 (72.6%) Partial structure (ii), E(ii) +2.69 (10.4%) +2.93 (11.9%) -2.88 (13.2%) Partial structure (iii), E(iii) -13.73 (53.0%) -8.64 (35.1%) -0.52 (2.4%) E(t) - (E(i)+ E(ii)+ E(iii)) -3.20 (12.3%) -3.09 (12.6%) -2.55 (11.7%)
TABLE 3.9: B3LYP/cc-pVTZ energies of the trimers T-H, T-I and their partial
structures (i), (ii), and (iii) (in kcal/mol)
B3LYP/cc-pVTZ
T-H T-I Trimer, E(t) -17.72 -19.22 Partial structure (i), E(i) +0.15 (0.8%) -6.67 (34.7%) Partial structure (ii), E(ii) -1.97 (11.1%) -1.59 (8.3%) Partial structure (iii), E(iii) -14.19 (80.1%) -8.76 (45.6%) E(t) - (E(i)+ E(ii)+ E(iii)) -1.71 (9.6%) -2.20 (11.4%)
In trimers T-D and T-F, partial structure (i) with 64.2 and 72.6 %, respectively,
contributes most to the trimer energies. The C=OFA…HNFMA and O-HFA…O=CFMA
interactions between the FA and FMA 1 dominate the interaction in the trimer, especially
in T-F, where the two FMA molecules basically do no interact to each other and the FA –

85
FMA 2 interactions are weaker, since neither the carbonyl oxygen atom nor the OH
hydrogen atom of FA are involved. In trimer T-D the FA – FMA 2 interactions are
repulsive, but the FMA 1 – FMA 2 interactions contribute to the stabilization of the
complex by 35.1 % (Table 3.8).
In trimer T-A, partial structure (iii) contributes 53 % to the total binding energy,
whereas the binding energy of partial structure (i) is 45.1 % and the FA – FMA 2
interactions are repulsive. This is expected, in accordance with the interactions involved
in the stabilization of partial structures (i) and (iii).
Trimers T-H and T-I are interesting complexes, since, unlike T-A, T-D and T-F, in both
cases the FA molecule interacts directly with the FMA 1 and FMA 2 molecules; and, in
each case the two FMA molecules provide an additional stabilization to the trimer by
interactions that resemble the structures of very stable FMA homodimers. Subsequently,
the energies of partial structures (iii) represent the largest contribution to the total binding
energies with 80.1 % for trimer T-H and 45.6 % in T-I. Partial structure (i) in T-I
contributes 34.7 % to the total binding energy (Table 3.9).
In trimers T-I and T-D the FMA – FMA stabilization is the same for both cases,
resembling the FMA – FMA dimer B. In addition, the interactions between the FA and
FMA molecules are of the same type ( (1) and (2) ) but, while in T-D they are both
present in the interactions of FA with FMA 1, in trimer T-I, interactions (1) and (2) occur
between the FA with the FMA 1 and FMA 2 molecules (Figures 3.4, 3.5 and 3.7).
Therefore it is interesting to point out the difference of about 20 % between the
contributions to the total energies in both cases. In T-D partial structure (i), which
contains both interactions (1) and (2), represent 64.2 % of the total binding energy; while
in T-I the sum of partial structure (i) (interaction (2)) and partial structure (ii) (interaction
(1)) represent 43 % of the total binding energy. Therefore, partial structure (iii) is the
dominating contribution to the stabilization of the complex T-I, unlike in T-D, where
partial structure (i) clearly dominates.
In T-H, the FMA – FMA interactions, which resemble the structure of the FMA
homodimer A, with 80.1 %, are contributing most to the stabilization of the complex. The
energy of the partial structure (ii) represents the 11.1 % of the total energy, whereas the

86
partial structure (i) is slightly repulsive, probably due to the repulsion at 2.763 Å between
one NH hydrogen atom of FMA 1 and the aldehyde hydrogen atom of the FA molecule
(Table 3.9, Figures 3.5 and 3.7).
1:4 Formic acid – formamide complexes
1:4 FA – FMA complexes have been calculated starting from 198 arbitrary geometries
that were optimized at the semiempirical level. A selection of complexes was refined at
the B3LYP/cc-pVTZ level of theory (Figure 3.9).
P-A is the most stable of the calculated pentamers with a binding energy of -50.54
kcal/mol. Complexes P-B and P-C are energetically close with binding energies of -46.42
and -43.95 kcal/mol, respectively. The binding energies of P-D and P-E are very similar,
with -38.64 and -38.47 kcal/mol, respectively.
The structure of the P-A complex is very interesting. Two pairs of FMA molecules are
forming two FMA cyclic homodimers which are then interacting to each other. The FA
molecule stabilizes the complex with the same type of interaction than in the FA – FMA
dimer A (Figure 3.1). The difference is that in P-A the FA molecule interacts with the
two closest FMA molecules, and the FA carbonyl oxygen atom cooperates with two NH
hydrogen atoms. In all the other complexes (P-B to P-E) the FA molecule interacts with
one FMA molecule (FMA-a) with the C=OFMA ····H-OFA and C=OFA ···H-CFMA interactions,
forming the FA – FMA dimer B (Figure 3.1).
By comparison with the 1:2 FA – FMA complexes it is easy to identify the structure of
the T-C trimer as part of the P-C, P-D and P-E complexes. The P-C pentamer is even
more interesting, since it combines the geometries of both, the T-A and the T-C trimers
(Figure 3.9). Considering the FA molecule, the FMA-a and the FMA molecule at the
right side of FMA-a, we get the geometry of trimer T-A. On a similar way, looking at the
interactions between FA, FMA-a and the FMA molecule at the left side of FMA-a, the
geometry of this subunit is very similar to trimer T-C.

87
Figure 3.9. The calculated structures with hydrogen bond lengths (Å) of the 1:4 FA–
FMA complexes P-A to P-E at the B3LYP/ cc-pVTZ level of theory. aB3LYP/cc-
pVTZ binding energies (kcal/mol) .

88
Comparison of FMA – FA complexes with the crystal structure
The complexity of the FA – FMA crystal structure can not be completely described by
a small set of FA – FMA complexes. However, interesting structural similarities are
noticed. Three sections of the FA – FMA crystal structure are presented in Figure 3.10,
whereas Figure 3.11 shows a large fragment of the FA – FMA crystal structure.[139]
Figure 3.10. Selected sections of the FMA-FA crystal structure.[139]

89
Figure 3.11. FMA-FA crystal structure. Ref [139]
The same type of interactions (1 – 4) that have been discussed above for the FA – FMA
dimers are present in the crystal structure. The geometry of dimer B is clearly reproduced
in the FA – FMA crystal interactions of fragments FA and FMA2 (Figure 3.1, Figure
3.10). In both cases the carbonyl group of the FA interacts preferentially out of plane
with another molecule.
The geometry of the trimer T-A is also very similar to the marked selection in the
FMA2 section (Figure 3.4). Trimer T-H describes the geometry of the interactions
between the FA molecule and the two FMA molecules forming the FMA cyclic
homodimer in section FA (Figure 3.10).

90
Table 3.10 lists some selected intermolecular distances in FA – FMA complexes and two
motifs of the crystal structure. For the dimers, the intermolecular distances shown were
calculated at the MP2/aug-cc-pVTZ level of theory and for trimers and pentamers at the
B3LYP/cc-pVTZ level. The C=OFMA…HOFA calculated distances of dimer B and trimer
T-A agree especially well with distances in the motif 1 of the crystal structure (CI), while
the calculated CHFMA…O=CFA distance for the pentamer P-B is closer to the one of CI
compared to the dimer and trimer. The value of the intermolecular distance for the
NHFMA…O=CFA interaction in the motif 2 of the crystal structure (CII) is very close to
the calculated one in trimers T-B and T-C, whereas the calculated NHFMA…(H)OCFA
distance in trimer T-H is similar to the value of this interaction in CII.
TABLE 3.10: Selected intermolecular distances in FMA-FA complexes and two
motifs of the crystal structure (Å)
C=OFMA…HOFA CHFMA…O=CFA NHFMA…O=CFA NHFMA…(H)OCFA
Dimers 1.663 (B) 2.304 (B) 1.859 (A) 2.213 (C)
Trimers 1.668 (T-A) 2.381 (T-A) 2.008 (T-B) 1.992 (T-C)
2.086 (T-F) 2.669 (T-H)
Pentamers 1.684 (P-A) 2.504 P-B
2.679 (P-A)a 1.987(P-D,P-E)a 2.234 (P-C) 2.228 (P-E)
Crystal structure CI[139]. 1.670 2.670 2.112 2.560 Crystal structure CII[139]. 1.760 3.084 2.057 2.643
aIn the crystal structure the carbonyl group of the FA interacts preferentially out of
plane with another molecule of FMA like in the pentamers.

91
3.4. Formic acid – dimethyl ether dimers. Results and discussion
Geometries and binding energies
Six FA – DME complexes A - F were found at the MP2 level of theory with the 6-
311++G(d,p) and cc-pVTZ basis sets (Figure 3.12). For all complexes the geometries are
almost independent of the basis sets used, therefore only hydrogen bond distances and
angles calculated at the MP2/cc-pVTZ level of theory are discussed. The calculated
binding energies (Table 3.11) predict complexes A and B as the lowest minima, both
being very close in energy. During the optimization process the second enantiomer of
complex B was also found which shows the reliability of the MMH procedure. According
to the calculated binding energies complexes C - F are much less stable.
Four basic types of interactions (1) – (4) can be differentiated in the FA-DME
complexes:
(1) HC(=O)OH…O(CH3)2 interaction between the hydroxyl hydrogen atom of FA
and the ether oxygen atom of DME.
(2) HOC(H)=O…HCH2OCH3 interaction between the carbonyl oxygen atom of FA
and the hydrogen atoms of DME.
(3) HOC(=O)H…O(CH3)2 interaction between the aldehyde H hydrogen atom of FA
and the ether oxygen atom of DME.
(4) O=C(H)O(H)…HCH2OCH3 interaction between the hydroxyl oxygen atom of FA
and the hydrogen atoms of DME.

92
Figure 3.12. The calculated structures with hydrogen bond lengths (Å) and some
bond angles (degree) of the FA – DME complexes A - F at the MP2/cc-pVTZ level of
theory.

93
TABLE 3.11: Calculated binding energies including ZPE and BSSE corrections (in
kcal/mol) of the FA – DME complexes A – F
MP2/6-311G++(d,p) ΔE ZPE BSSE ΔE (ZPE) ΔE (ZPE+BSSE) A -11.41 1.63 3.38 -9.78 -6.40 B -10.87 1.43 2.75 -9.44 -6.69 C -4.19 0.64 1.13 -3.55 -2.42 D -3.90 0.53 1.08 -3.37 -2.29 E -4.30 0.77 1.62 -3.53 -1.91 F -4.35 0.93 2.26 -3.42 -1.16 MP2/cc-pVTZ ΔE ZPE BSSE ΔE (ZPE) ΔE (ZPE+BSSE) A -12.23 1.50 2.76 -10.73 -7.97 B -11.31 1.28 2.22 -10.03 -7.81 C -5.03 0.73 1.43 -4.30 -2.87 D -4.47 0.59 1.22 -3.38 -2.66 E -5.26 0.82 1.78 -4.44 -2.66 F -5.01 0.91 1.84 -4.10 -2.26
Complexes A and B show the same type of interactions (1) and (2). The hydrogen atom
of the OH group of the FA interacts with the ether oxygen atom of DME at hydrogen
bond distances of 1.672 Å and 1.691 Å for the dimers A and B, respectively. The
difference between these complexes is caused by the interaction (2). In dimer A the C=O
group of FA interacts simultaneously with two hydrogen atoms of DME at distances of
2.663 Å, while in dimer B the C=O group of FA is approaching only one hydrogen atom
of the DME with a distance of 2.508 Å.
With the 6-311++G(d,p) and cc-pVTZ basis sets (without including ZPE and BSSE
corrections) the differences between the binding energies of complexes A and B (ΔEB –
ΔEA, ΔΔEBA) are 0.54 kcal/mol and 0.92 kcal/mol, respectively. After including BSSE
and ZPE corrections these differences are reduced to -0.29 kcal/mol (6-311++G(d,p)) and
0.16 kcal/mol(cc-pVTZ) (Table 3.11). With the 6-311++G(d,p) basis set including all
corrections the binding energies for dimers A and B are -6.40 and -6.69 kcal/mol,

94
respectively. This is due to the large BSSE error for complex A with the 6-311++G(d,p)
basis set which considerably lowers the binding energy of A compared to B.
Comparing the structures of the FA – DME complexes A and B with the DME –
methanol complex reported in literature[132, 133] reveals large similarities. Han and
Kim[133] calculated at the MP2/6-31+G** level of theory the hydrogen bond distance
CH3OH…O(CH3)2 to 1.855 Å and the OH...O hydrogen bond angle to 177.2°, in good
agreement with our OH…O hydrogen bond distances of 1.672 Å and 1.691 Å,
respectively, and OH...O bond angles of 177.3° and 176.8°, respectively, for complexes
A and B (MP2/cc-pVTZ).
A cyclic dimer of FA – water has been described experimentally by Astrand[113] and
Priem.[147] It is also interesting to compare the FA – DME complexes with the geometries
of the recently calculated FA – water complexes by Zhou.[148] The most stable FA – water
complex found by this author is a cyclic complex with both FA and water acting as
hydrogen donor and acceptor. The HC(O)OH…OH2 and HOC(H)=O…HOH distances
were calculated to 1.792 Å and 2.144 Å, respectively, at the MP2/6-311++G(d,p) level of
theory. It is remarkable how this cyclic FA – water complex shows the same type (i) and
(ii) interactions as the DME – FA complexes A and B.
Zhou reported two additional more weakly bound FA – water dimers. One of those is a
HOC(H)=O…HOH complex with a calculated C=O...H bond angle of 99.8° and an
O…H distance of 2.053 Å. Its geometry is very similar to the FA – DME dimer C, where
the calculated C=O…H distance is 2.666 Å and the COH hydrogen bond angle is 95.5°
(for comparison with Ref. [148] calculated at the MP2/6-311++G(d,p) level of theory). The
FA – DME complex C is stabilized by both interactions (2) and (3) and the C-H…O
distance is calculated to 2.257 Å.
The other FA – water complex found by Zhou is the OC(H)O(H)…HOH complex,
where the O…Hwater distance is 2.204 Å and the (H)O...Hwater angle is 147.6°. FA – DME
dimer D exhibits a similar geometry, the calculated O=C(H)O(H)…HCH2OCH3 distance
is 2.812 Å and the (H)O...H hydrogen bond angle is 154.5° (MP2/6-311++G(d,p)).
Complex D shows interactions (3) and (4), the O=C(OH)H…O(CH3)2 distance is 2.230
Å. Comparing the MP2/cc-pVTZ geometries of complexes C and D (Figure 3.12) with

95
their MP2/6-311++G(d,p) geometries (as discussed above) reveals that the structures of
these complexes are almost independent of the basis set used in the calculations.
Dimer E is stabilized by interactions (2) and (3) and dimer F by interactions (2), (3)
and (4). The calculated MP2/cc-pVTZ + ZPE + BSSE binding energies of complexes C –
F are very similar and around 5 kcal/mol smaller than the binding energies of complexes
A and B. This can be rationalized by the lack of the strongest interaction (1) in complexes
C – F.
Geometry optimization including BSSE
To investigate the influence of the basis set superposition errors (BSSE) on the
geometries of the complexes, the geometries of complexes A – E were optimized at the
MP2/6-31G(d,p) level of theory using the counterpoise (CP) scheme of Boys and
Bernardi during the optimization process. The small 6-31G(d,p) basis set was selected
since the BSSE is here more pronounced compared to larger basis sets and in addition the
calculations are less demanding. Complexes A – D are discussed here to illustrate
different types of dimers including strong and weak interactions and similar and different
binding energies (Tables 3.12 and 3.13).
The C=O and O-H bond distances in the FA part of the complexes and the C-O
distances in the DME part are almost not effected by the inclusion of BSSE during the
optimization. In all cases the difference in these bond distances was in the order of 10-3 Å
(Table 3.12). The OH…O, C=O…H, CH…O and C- O…H intermolecular distances
(interactions (1), (2), (3) and (4), respectively) are more influenced by BSSE. The weak
interactions (2) and (4), where DME hydrogen atoms are involved, exhibit the largest
changes. Thus, the C=O…H and C-O…H distances increase by about 0.2 – 0.3 Å when
BSSE is considered during the optimization. Despite these variations, the basic
geometries and interactions in the FA – DME complexes do not change.

96
Table 3.12. Comparison of selected intramolecular and intermolecular distances in
the FA – DME complexes A – D at the MP2/6-31G(d,p) level of theory. The results
from geometry optimizations without and including BSSE corrections are
compared.
MP2/6-31G (d,p) optimization with BSSE optimization without BSSE A B C D A B C D Intramolecular distances
r(C=O)FA 1.219 1.218 1.216 1.215 1.222 1.220 1.218 1.214 r(O─H)FA 0.989 0.986 0.972 0.972 0.995 0.991 0.972 0.972 r(C─O)DME 1.425 1.423 1.419 1.417 1.429 1.427 1.421 1.419
r(C─O)e 1.419 1.415 1.416 1.421 1.416 1.416 Intermolecular distances OH…Oa 1.801 1.804 _ _ 1.706 1.725 _ _ C=O…Hb 2.922 2.589 2.751 _ 2.596 2.457 2.526 _ CH…Oc _ _ 2.330 2.315 _ _ 2.260 2.252 C-O…Hd _ _ _ 2.880 _ _ _ 2.638
aInteraction (1). bInteraction (2). cInteraction (3). dInteraction (4) between FA and DME. eC-O bond distance between the DME oxygen atom and the non interacting methyl
group.
The calculated binding energies of A – D (Table 3.13) are also almost independent of
using BSSE corrections during geometry optimization, which clearly demonstrates that
consideration of BSSE during geometry optimization is not mandatory in this case (Table
3.13). The large BSSE in complex A lowers its binding energies below that of complex
B. However, both complexes are still very close in energy and more stabilized than the
other dimers. These results are similar to that obtained with the MP2/6-311++G(d,p)
level of theory described above.

97
Table 3.13. Calculated binding energies including BSSE corrections (in kcal/mol)
of the FA – DME complexes A – D at the MP2/6-31G(d,p) level of theory. The
results from geometry optimizations without and including BSSE corrections are
compared.
MP2/6-31G(d,p) optimization with BSSE optimization without BSSE ΔE BSSE ΔE + BSSE ΔE BSSE ΔE +BSSE
A -12.23 4.05 -8.18 -12.82 5.10 -7.72 B -11.60 3.26 -8.34 -11.92 3.86 -8.06 C -4.81 2.07 -2.74 -4.98 2.39 -2.59 D -4.21 1.72 -2.49 -4.35 1.99 -2.36
Intramolecular distances and vibrational frequencies
Compared to the monomers, intramolecular distances and vibrational frequencies in the
complexes are distorted as a consequence of the intermolecular interactions. For the
monomers the available experimental geometrical data are well reproduced at the
MP2/cc-pVTZ and MP2/6-311++G(d,p) levels of theory (Table 3.14).
The most perturbed vibrational modes in complexes A and Β are the O-H stretching
vibrations of the FA molecule (Tables 3.15 and 3.16). At the MP2/cc-pVTZ level of
theory the frequency shifts in the complexes A and B are -538 and -449 cm-1,
respectively. This reflects the strong interactions between the OH hydrogen atom and the
ether oxygen atom in complexes A and B (Figure 3.12) which results in an elongation of
the OH bonds of approximately 0.025 Å (MP2/cc-pVTZ, Table 3.14).

98
Table 3.14. Comparison of the selected intramolecular distances of FA, DME, and
the FA – DME complexes A and B.
aRef. [149] bRef [131] cC-O bond between the DME oxygen atom and the non interacting
methyl group.
Table 3.15. The experimental (Ar matrix at 35 K – 45 K) and the calculated MP2/cc-
pVTZ unscaled vibrational frequencies (in cm–1) of the FA – DME complex A, along
with the frequency shift in the complex, ∆ν, from the monomer (in parentheses).
Experimental MP2/cc-pVTZ Monomer Complex A Monomer Complex A
Formic Acid 3550.5 3000.3 (-550.2) 3763.4 3225.1 (-538.3) ν (O-H) 1767.3 1735.0 (-32.3) 1818.1 1786.7 (-31.4) ν (C=O) 1103.7 1181.9 (78.2) 1136.7 1225.6 (88.9) ν (C-O) 635.4 961.6 (326.2) 686.2 1029.7 (343.5) γ (O-H) o.o.p. 629.2 679.8 (50.6) 629.2 688.4 (59.2) δ (O-C=O)
Dimethyl ether 1098.3 1077.3 (-21.0) 1138.0 1124.4 (-13.6) νas (O-C-O) 925.9 899.9 (-26.0) 970.6 943.4 (-27.2) νs (O-C-O)
Experiment MP2/cc-pVTZ MP2/6-311++G(d,p) monomer monomer A B monomer A B Formic acid
r(C=O) 1.202(10)a 1.203 1.212 1.210 1.205 1.214 1.212 r(O─H) 0.972 (5)a 0.969 0.996 0.992 0.969 0.993 0.989 r(C─O) 1.343 (10)a 1.346 1.329 1.329 1.348 1.332 1.333
Dimethyl ether r(C─O) 1.410b 1.408 1.423 1.416[c]
1.421 1.411 1.424 1.419[c]
1.424 r(C─H) in plane
1.091b 1.086 1.086 1.084 1.090 1.090 1.089
r(C─H) 1.100b 1.095 1.091 1.092 1.099 1.096 1.096

99
The carbonyl stretching frequencies of the FA molecules in A and B are predicted to be
red-shifted by 31 and 24 cm-1, respectively, again reflecting the strong CO…H interaction
(contribution (2)). The C=O bond lengths in dimers A and B compared to the monomer
increase by nearly 0.010 Å. In complex A, where the carbonyl oxygen atom of FA
interacts with two H atoms of DME, the predicted red-shift of the carbonyl stretching
frequency is 7 cm-1 larger than dimer B with only one contact.
The C-OH stretching modes of the FA molecules in A and B are blue-shifted by
89 cm-1 and 91 cm-1, respectively, and consequently the C-OH bond lengths are 0.017 Å
shorter than in the monomer. The symmetrical and antisymmetrical O-C-O and CH3
stretching modes of the DME moieties are also perturbed in A and B as listed in Tables
3.15 and 3.16
Table 3.16. The experimental (Ar matrix at 25 K – 35 K) and the calculated MP2/cc-
pVTZ unscaled vibrational frequencies (in cm–1) of the FA – DME complex B, along
with the frequency shift in the complex, ∆ν, from the monomer (in parentheses).
Experimental MP2/cc-pVTZ Monomer Complex B Monomer Complex B
Formic Acid 3550.5 3077.8 (-472.7) 3763.4 3314.6 (-448.8) ν (O-H) 1767.3 1741.2 (-26.1) 1818.1 1793.7 (-24.4) ν (C=O) 1103.7 1179.4 (75.7) 1136.7 1228.1 (91.4) ν (C-O) 635.4 852.8 (217.4) 686.2 933.6 (247.4) γ (O-H) o.o.p. 629.2 681.5 (52.3) 629.2 693.1 (63.9) δ (O-C=O) Dimethyl ether 1098.3 1092.0 (-6.3) 1138 1133.1 (-4.9) νas (O-C-O) 925.9 915.2 (-10.7) 970.6 953.2 (-17.4) ν s (O-C-O)
Comparison with matrix isolation spectroscopy results
The IR spectra of complexes A and B were assigned by comparison of the calculations
at the MP2/cc-pVTZ level of theory (Tables 3.15 and 3.16) with matrix isolation
experiments. Characteristic for the formation of complexes of FA is the red shift of the

100
C=O stretching vibration found at 1767.3 cm-1. In the symmetrical doubly bridged dimer
of FA the C=O stretching vibration is shifted by -38.6 cm-1 and in the complex with water
by -30.4 cm-1. In the complexes A and B these shifts are -32.3 and -26.1 cm-1,
respectively, and thus in the expected range. The experimental red-shifts are in excellent
agreement with the MP2 calculations which predict shifts of -31.4 and -24.4 cm-1,
respectively, for the two complexes. The red-shifts in the C=O stretching vibration reflect
the elongation of the C=O bond due to the formation of complexes. Obviously, the
interaction of the carbonyl oxygen atom in A with two DME hydrogen atoms is more
efficient than the interaction with only one hydrogen atom in complex B.
Due to a large number of absorptions in the region of the OH/CH stretching vibrations,
the assignment of bands to complexes A and B is very difficult and only tentative. For
complex A the experimental O-H stretching frequency shift of -550 cm−1 is in excellent
agreement with the calculated shift (-538 cm−1) using MP2/cc-pVTZ (Table 3.15). For
dimer B the calculated frequency shift (-449 cm−1) also agrees very well with the
experimental value (-473 cm−1) (Table 3.16). The large shifts of the O-H stretching
vibrations allows to discard the FA – DME dimers C – F where the O-H…O interaction
is lacking and thus the O-H stretching vibration of the FA molecule is much less
perturbed (Figure 3.12).
The formation of hydrogen-bonded complexes of FA results in a contraction of the C-O
bond and a blue shift of the C-O stretching vibration found at 1104 cm-1 in the
unperturbed molecule. For complex A a blue shift of 89 cm-1 is predicted at the MP2/cc-
pVTZ level of theory which matches the experimental value of 78 cm-1. For complex B a
shift of 91 cm-1 is predicted and 76 cm-1 is observed.
The DME molecule shows less pronounced shifts of IR absorptions due to the complex
formation. The strong absorptions of monomeric DME and other constituents in the
matrix do not allow to identify C-H stretching vibrations of the DME molecule in the
complexes. The asymmetric and symmetric C-O-C stretching vibration of both
complexes A and B could be identified. In complex A the symmetrical C-O-C stretching
vibration exhibits a red-shift of 25 cm-1 while the asymmetrical vibration is shifted by 15
cm-1. This might be rationalized by the smaller distortion of the weak

101
HOC(H)=O…HCH2OCH3 hydrogen bonds during the asymmetrical vibration as
compared to the symmetrical vibration. In complex B the corresponding shifts are with
15 and 6 cm-1 smaller. The experimental red-shifts are in good agreement with the MP2
calculated values (Tables 3.15 and 3.16).
3.5. Conclusion
The geometries of the FA – DME and FA – FMA complexes are calculated starting
from randomly generated molecular arrangements using the MMH procedure. Nine FA –
FMA dimers with binding energies between -2.91 and -13.02 kcal/mol (MP2/aug-cc-
pVTZ + ZPE + BSSE) are identified and seven competitive individual molecular
interactions are discussed based on the calculated geometries and binding energies of the
FA – FMA complexes. The pair contributions of each dimer to the stabilization of the 1:2
FA – FMA trimers are also analyzed.
Six different FA – DME bimolecular 1 : 1 complexes with binding energies between -
2.26 and -7.97 kcal/mol (MP2/cc-pVTZ + ZPE + BSSE) could be identified. They are
classified in two groups. One group consists of the complexes A and B, where the OH
hydrogen atom of FA forms a strong hydrogen bond with the ether oxygen atom of DME.
These two complexes are predicted to be the most stable ones and with -7.81 to -7.97
kcal/mol binding to be almost isoenergetic. Although the OH…O interaction is
dominating in complexes A and B, the secondary interaction between a methyl group
hydrogen atom of DME and the carbonyl oxygen atom of FA leads to an additional
significant stabilization of these complexes.
The second group of FA – DME complexes C – F is defined by the absence of the
strong OH…O hydrogen bond. With -2.3 to -2.9 kcal/mol the binding energy is
considerably smaller and consequently these complexes could not be identified
experimentally. The dominant interaction in these complexes is the interaction between
the aldehyde hydrogen atom of FA and the DME oxygen atom. Again, interactions
between the methyl groups of DME and oxygen atoms of FA form secondary, weak
interactions which, however, determine the geometry of the complexes.

102
The structures of the FA – FMA dimers A and B are in excellent agreement with the
geometries of the FA – FMA dimers reported in the literature. They show also interesting
analogies with the FMA – water and FMA – methanol dimers. Comparing the structures
of the FA – DME complexes A and B with the DME – methanol complex reported in
literature reveals large similarities. There are also interesting analogies between the FA –
DME and the FA – water reported dimers.
The B3LYP density functional with the cc-pVTZ basis set provides reliable geometries
for the FA – FMA complexes. At the MP2 level of theory, basically no change of the
geometries is found when the basis set is augmented by adding diffuse functions. At the
MP2 level cc-pVDZ calculations show a tendency to overestimate the binding energies,
however, triple zeta basis sets either augmented or non-augmented result in binding
energies very similar to those from CCSD(T)/cc-pVTZ single point calculations.
The geometries and energies of the FA – FMA dimers A and B do not change
considerably with the inclusion of BSSE corrections during the optimization process. For
the FA – DME dimers A – D, the intermolecular distances corresponding to weaker
interactions, where the DME hydrogen atoms are involved, are more influenced, showing
an increasing of about 0.2-0.3 Å when BSSE is considered during the geometry
optimization. Despite these variations the basic geometries and interactions in the FA –
DME complexes do not change.
The calculated geometries and binding energies of 1:2 and 1:4 FA – FMA complexes
show very interesting similarities with the FA – FMA dimers and with the FA – FMA
crystal structure. Of special interest are structural motives found in the crystal structure
that are already present in complexes of very few molecules. This could lead to in-depths
knowledge of the complex processes of molecular nucleation and crystal growth.
The distortion of the intramolecular distances and vibrational frequencies in the FA –
FMA dimers A and B compared to the monomers are discussed, and reliable vibrational
frequencies are predicted. The empirically corrected frequencies should allow for the
experimental detection of these complexes in matrix isolation or gas phase studies. For
the FA – DME dimers A and B, comparison of the experimental data from matrix

103
isolation experiments with calculated spectra indicated the formation of these two
complexes.
The identification of the FA – DME and FA – FMA complexes, as well their
comparison with matrix spectroscopy results and crystal structure data, confirms the
quality of the MMH procedure as a very useful tool for reliably localizing minima in
hydrogen bonded complexes without recurring to previous knowledge of the structure of
supramolecular complexes.

104
4. Formic Acid Complexes with π Systems
4.1. Introduction
During the past several years, weak hydrogen bonds involving a hydrogen atom bound
to a carbon atom as hydrogen donor has attracted attention from the scientific
community. It was found that these weak interactions play important roles in many
chemical and biochemical processes.[1-3] In contrast to the conventional strong hydrogen
bonds, which were extensively discussed in literature, the nature and characteristics of
weak interactions do not comprise a well-known field.[3] Thus, experimental and
theoretical studies of CH…O, and C-H…π as well as O-H…π interactions are of key
interest for the understanding of biological and chemical systems.
As already mentioned, the formic acid molecule and its interactions with other
molecules provide a good model for the study of many association processes. From early
spectroscopic studies, acetylene was recognized as a weak hydrogen bond donor and
weak acceptor through the triple bond.[1] Furan is an aromatic heterocyclic compound
with many applications in different fields of chemistry from natural product synthesis to
material science.[150]
A number of studies have been published on the weak interactions in complexes or
dimers of hydrocarbons with π systems like acetylene, benzene and ethylene.[151-157] The
dimer of acetylene was investigated both experimentally and theoretically,[158-165] and a
π–type hydrogen-bonded C2v minimum was found together with several first and second
order saddle points.[166-170] Studies of the 1:1 FA – acetylene complexes were carried out
using matrix isolation and theoretical methods.[116]
Furan, its aggregates, and complexes with small molecules have been subject to many
experimental and theoretical studies.[171-187] The complexes of furan with hydrogen
halides and alkynes were investigated by Ault using matrix isolation spectroscopy.[185, 187]
The equilibrium structures of the furan dimers and the nature of their intermolecular
interactions were studied by Pei using density functional theory and the natural bond
orbital analysis.[171] The π…π interactions in the parallel, sandwich-shaped furan dimer
were analyzed using high level ab initio theory.[188] The excited states of furan and the

105
rotational spectra of some of its complexes with halides were also investigated.[176, 177, 179]
Chan, Del Bene et al. studied the reactions of various acids with furan as part of their
research about the preferred sites of protonation and hydrogen bonding for a set of basic
substrates.[114]
Many studies of the interactions of furan with other molecules were focused on the
furan – hydrogen halide complexes.[174, 177, 185] Legon and Millen[189, 190] proposed some
general rules to predict the geometry of B…HX complexes (X: F, Cl, Br, I, CN, CCH). If
the Lewis base possesses both n pairs and π electrons, the angular geometry is determined
by the n pair rather than by the π electrons, and the Lewis acid lies along the axis of the n
pair. Since the furan molecule contains both an n pair and π electrons, several studies of
furan - HX complexes have been carried out to determine whether or not this rule is
obeyed.[174, 177, 179] Cole, Legon and Ottaviani[177] studied the rotational spectrum of furan
– HCl and HBr and showed that, while the HCl complex obeys Legon’s rule, the HBr
complex behaves differently and interacts with the π system of furan. In the same line,
the furan complexes with hydrogen halides HX were studied by Huang and Wang[174]
using ab initio calculations.
The structures of a variety of FA – furan complexes are identified here and their
binding energies and the influence of the BSSE and basis sets on their geometries are
discussed. The calculated vibrational spectra of the FA – furan dimers are compared to
experimental matrix isolation spectra. The FA – furan system exhibits both n and π
hydrogen bond interactions. Therefore, the study of the FA – furan complexes leads to a
detailed knowledge of the interactions in this type of systems.
The trimers formed by the interaction of one molecule of formic acid with two
molecules of acetylene (1:2 FA – acetylene complexes) are investigated. The
characterization of trimer complexes is a challenge both for theory and experiment. In
addition, these complexes consisting of three molecules allow to analyze in detail the
influence of a third molecule on the dimer properties, e. g. an additional acetylene
molecule on the FA – acetylene dimer or a formic acid molecule on the acetylene dimer.
An interesting feature of this system is the competition between the strongly acidic
carboxyl group, the acetylene group, and the formyl group as hydrogen bridge donors and

106
the carbonyl group, the hydroxyl group, and the acetylene π-system as hydrogen bridge
acceptors. This leads to several complexes with strong OH...O and weak CH...O or
CH...π hydrogen bridges. Due to the amount of complexes with similar binding energies
a careful search for minima on the potential energy surface is mandatory. The calculated
vibrational frequencies of the 1:2 FA – acetylene complexes are compared to data from
matrix isolation spectroscopy.
4.2. Computational methods
The Multiple Minima Hypersurface (MMH) approach was used for searching
configurational minima in the FA – furan and 1:2 FA – acetylene systems. One thousand
randomly arranged FA – furan and1:2 FA – acetylene clusters were generated as starting
point, and the resulting geometries were optimized and analyzed using PM3 and AM1
semiempirical quantum mechanical Hamiltonians. These semiempirical results provided
a preliminary overview of the FA – furan and 1:2 FA – acetylene interactions, and the
relevant configurations were further refined using ab initio methods at various levels of
theory.
After geometrical analysis both PM3 and AM1 Hamiltonians lead to nearly the same
set of minima. Compared to the PM3 results, the AM1 calculation allows for the
identification of another FA – furan minimum at all levels of theory. Another two
additional minima from AM1 starting geometries were found only when non-augmented
double zeta basis sets were used for the geometry optimizations.
The ab initio computations were performed using the Gaussian 98 and Gaussian 03
programs. The equilibrium geometries and vibrational frequencies were calculated at the
SCF level including second order Møller−Plesset perturbation theory, MP2. The tight
convergence criteria was used for the geometries optimizations and the force constants
where calculated when necessary.
The 1:2 FA – acetylene complexes were calculated in addition at the DFT level with
the B3LYP hybrid functional for initial geometry optimizations. For the 1:2 FA –
acetylene complexes A, B and C single point calculations are performed with coupled
clusters[141] of single and double substitutions (with non iterative triples) CCSD(T)/cc-

107
pVTZ, using the MOLPRO program. The DFT results for the 1:2 FA – acetylene
complexes are presented to compare with the MP2 and CCSD(T) data.
Pople’s 6-31G(d,p) basis set, the extended valence triple ζ basis set augmented with
diffuse and polarization functions 6-311++G(d,p) and the cc-pVTZ Dunning’s correlation
consistent triple ζ basis set were used. For the FA – furan dimers, in addition the
augmented and non augmented Dunning’s correlation consistent double ζ basis sets cc-
pVDZ and aug-cc-pVDZ were used. To evaluate the effect of the size of the basis set in
1:2 FA – acetylene complexes, the strongly polarized basis set 6-311++G(3df,3pd) (381
basis functions) is used and the results are compared to MP2 calculations with the cc-
pVTZ (294 basis functions) and 6-311++G(d,p) (196 basis functions) basis sets.
The stabilization energies were calculated by subtracting the energies of the monomers
from those of the complexes and including ZPE corrections. Most of the FA – furan
dimer energies were also corrected for the basis set superposition errors (BSSE) using the
counterpoise (CP) scheme of Boys and Bernardi. In the case of the 1:2 FA – acetylene
complexes, the binding energies for complexes A, B, and C were CP-BSSE corrected at
the MP2/cc-pVTZ level of theory.
To investigate the influence of the basis set superposition errors (BSSE) on the
geometries of the complexes, all FA – furan dimers were optimized at the MP2/6-
31G(d,p) level of theory using the CP corrections during the optimization process.

108
4.3. Formic acid – furan dimers. Results and discussion
Geometries and binding energies
After refining the MMH semiempirical results, nine FA – furan dimers A – I were
localized at the MP2/6-311++G(d,p) level of theory. The FA molecule exhibits two
protons that in principle can act as hydrogen bridge donor: the more acidic OH proton
and the less acidic aldehyde type CH proton. Consequently, the FA – furan dimers are
classified into two types:
• Type (i), where the acidic OH hydrogen atom of FA acts as hydrogen bond
donor (Dimers A, B, C and D1) (Figure 4.1).
• Type (ii), where the less acidic CH hydrogen atom of FA acts as hydrogen bond
donor (Dimers E, F1, G, H and I) (Figure 4.2).
The interactions between the FA and furan molecules in the complexes can be broken
down into the six basic two center interactions (1) – (6):
(1) OHFA…OF interaction between the hydroxyl hydrogen atom of FA and the
oxygen atom of furan.
(2) C=OFA…HF interaction between the carbonyl oxygen atom of FA and the
hydrogen atom of furan.
(3) O-HFA…π interaction between the hydroxyl hydrogen atom of FA and the π
system of furan.
(4) CHFA…OF interaction between the aldehyde hydrogen atom of FA and the
oxygen atom of furan.
(5) H-OFA…HF interaction between the hydroxyl oxygen atom of FA and the
hydrogen atom of furan.
(6) C-HFA…π interaction between the aldehyde hydrogen atom of FA and the π
system of furan.

109
Figure 4.1. The calculated structures with hydrogen bond lengths (Å) of the type (i)
FA – furan complexes A, B, C, and D1 at the MP2/6-311++G(d,p) level of theory.

110
Figure 4.2. The calculated structures with hydrogen bond lengths (Å) of the type
(ii) FA – furan complexes E, F1, G, H, and I at the MP2/6-311++G(d,p) level of
theory.
The complexes A – I were found being minima using double as well as triple zeta basis
sets. In addition, the five complexes D, D2, F, K, and J were localized at the double zeta
without augmentation level of theory only. These complexes do not represent minima at
higher levels of theory using larger basis sets. (Figure 4.3)

111
Figure 4.3. The calculated structures with hydrogen bond lengths (Å) of FA –
furan dimers D, D2, F, J, and K at the MP2/6-31G(d,p) level of theory. 1) Geometry
optimized without BSSE corrections. 2) Geometry optimized with BSSE corrections.
Thus, dimers showing the basic interactions (1) or (3) are classified as type (i)
complexes, while those showing interactions (4) or (6) are classified as type (ii)
complexes. In general, more than one of the basic interactions (1) – (6) contributes to the
stabilization of the complex.
Type (i) complexes
The most stable FA – furan dimer is complex A with a binding energy of -3.91
kcal/mol at the MP2/6-311++G(d,p) + BSSE + ZPE level of theory (Table 4.1). The
binding energies of all dimers are discussed at this level of theory including BSSE and
ZPE corrections unless specified differently. The influence of the basis sets on the
complexes is discussed later.

112
TABLE 4.1: Calculated binding energies of furan-Formic Acid dimers A-I at MP2
level of theory with the 6-31g(d,p) and 6-311++G(d,p) Pople’s basis set including
ZPE and BSSE corrections (in kcal/mol).
MP2 6-31g(d,p) opt with BSSE 6-31g(d,p) ΔE BSSE ZPE ΔE BSSE+ZPE ΔE BSSE ZPE ΔE BSSE+ZPE A -8.37 2.83 1.01 -4.53 -8.54 3.15 1.11 -4.27 B -5.16 1.65 0.61 -2.90 -6.05 3.11 0.88 -2.06 C -5.02 1.80 0.55 -2.67 -5.67 3.00 0.65 -2.02 D -5.11 1.61 0.48 -3.02 -5.76 2.80 0.64 -2.32 D1 D2a Da D2 -5.16 1.66 0.54 -2.96 Ba E -3.30 1.44 0.55 -1.31 -3.73 2.29 0.64 -0.8 F -2.89 1.41 0.40 -1.08 -3.17 1.91 0.65 -0.61 F1 -2.89 1.27 0.48 -1.14 -3.21 1.95 0.58 -0.68 G -2.88 1.27 0.46 -1.15 -3.21 1.96 0.58 -0.67 H -4.26 1.96 0.56 -1.74 -4.38 2.19 0.67 -1.52 I -3.40 1.56 0.47 -1.37 -3.50 1.75 0.51 -1.24 J Ha -3.37 1.99 0.64 -0.74 K —b -2.59 1.59 0.43 -0.57 6-311++G(d,p) ΔE BSSE ZPE ΔE BSSE+ZPE A -7.12 2.03 1.18 -3.91 B -5.51 2.24 1.03 -2.24 C -5.56 2.42 1.02 -2.12 D D1a D1 -5.44 2.12 0.95 -2.37 D2 D1a E -4.19 2.21 1.02 -0.96 F F1a F1 -3.76 2.01 0.93 -0.82 G -3.73 1.99 0.88 -0.86 H -3.55 1.17 0.44 -1.94 I -3.05 1.18 0.52 -1.35

113
J Ea K —b
a Indicates the dimer that it is found after geometry optimization. b Leads to a second
order saddle point.
For comparison, selected intermolecular parameters of the dimers A – D2 are shown in
Table 4.2. The dimer A is stabilized by interaction (1) that involves the OH of the FA
molecule and the oxygen atom of furan, and interaction (2) between the carbonyl oxygen
atom of FA and a α-hydrogen atom of furan. The binding distances and hydrogen bond
angles (in parentheses) for interactions (1) and (2) are 1.877 Å (177.87º) and 2.540 Å
(124.56º), respectively (Table 4.2). If dimer A is forced to Cs symmetry it shows one
imaginary out of plane vibration that after free optimization leads to a slightly distorted
equilibrium geometry with C1 symmetry (Figure 4.1). The intramolecular bond lengths
are not sensitive to this decrease of symmetry in dimer A, and the variation of the
intermolecular hydrogen bond distances and angles are less than 0.008 Å and 1º,
respectively (Table 4.2). As expected, the binding energy is also hardly affected. Since
with other basis sets the Cs symmetrical dimer is a true minimum, the distortion is
probably an artifact of the 6-311++G(d,p) basis set.
Dimers B, C, and D1 are O-H…π complexes (interaction (3)) with very similar binding
energies. Dimer D1 has binding energy -2.37 kcal/mol and the OH group of FA interacts
with the C2-C3 region of furan. The distances of the OH hydrogen atom to C2 and C3 are
2.321 Å and 2.508 Å, respectively (Figure 4.1). In the complexes B (-2.24 kcal/mol) and
C (-2.12 kcal/mol) the OH hydrogen atom of FA interacts with the C1-C2 bond of furan.
In dimer B, the O-H…C2 and O-H…C1 distances are 2.369 Å and 2.427 Å, respectively.
In dimer C the O-H…C2 and O-H…C1 distances are 2.300 Å and 2.551 Å, respectively.
The main difference between dimers B and C is the rotation of the FA molecule around
its O-H axis (Figure 4.1). Compared to dimer B the OH hydrogen atom in dimer C is
closer to atom C2.

114
TABLE 4.2: Comparison of selected intermolecular parameters in the FA – furan dimers A-D2 at various levels of theory.
Distances are in Å and angles in degrees
MP2 6-31G(d,p)BSSE 6-31G(d,p) 6-311G++(d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ Dimer A OHFA…OF 1.973 1.889 1.877 (1.885)a 1.888 1.859 1.866
C=OFA…H(C1)F 2.551 2.416 2.540 (2.536)a 2.400 2.433 2.447 < OHFA…OF 177.93 176.58 177.87(177.53)a 177.43 177.56 178.04 < C=OFA…H(C1)F 124.66 124.65 124.56(125.26)a 125.25 124.67 124.22 Dimer B OHFA…OF 3.074 3.289 2.756 3.225 2.656 2.698 OHFA…C1F 2.566 2.426 2.427 2.409 2.342 2.350 OHFA…C2F 2.432 2.354 2.369 2.328 2.294 2.305 OHFA…C3F 2.907 3.242 2.684 3.167 2.592 2.640 OHFA…C4F 3.234 3.672 2.881 3.579 2.774 2.835 <OHFA…OF 139.14 155.35 124.33 153.96 125.38 127.47 <OHFA…C2F 168.89 159.38 170.44 159.92 171.13 169.91 Dimer C OHFA…OF 3.103 2.871 2.961 2.895 2.857 2.871 OHFA…C1F 2.679 2.450 2.551 2.495 2.429 2.426 OHFA…C2F 2.441 2.303 2.300 2.297 2.236 2.253

115
MP2 6-31G(d,p)BSSE 6-31G(d,p) 6-311G++(d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ OHFA…C3F 2.772 2.672 2.610 2.617 2.596 2.638 OHFA…C4F 3.131 2.965 2.970 2.933 2.921 2.957 C=OFA…H(C4)F 3.788 3.219 3.365 3.209 3.197 3.313 <OHFA…OF 154.04 144.60 146.22 144.77 145.56 146.72 <C=OFA…H(C4)F 125.75 133.29 128.06 131.09 131.47 130.26 <OHFA…C2F 158.89 165.43 165.18 165.32 163.45 163.20 Dimer D C=OFA…H(C2)F 3.586 2.929 2.900 3.129 OHFA…OF 3.234 3.359 3.368 3.098 OHFA…C1F 2.967 3.007 3.013 2.816 OHFA…C2F 2.510 2.366 2.359 2.323 OHFA…C3F 2.510 2.366 2.359 2.323 OHFA…C4F 2.967 3.007 3.013 2.816 <C=OFA…H(C2)F 102.05 105.46 105.89 104.83 <OHFA…OF 139.35 156.72 156.57 146.91 <OHFA…C2F 163.45 156.84
D1b 156.76
D1b 159.51
Dimer D1 C=OFA…H(C3)F 3.225 3.122 3.111 OHFA…OF 2.911 2.845 3.014 OHFA…C1F 2.595 2.487 2.637 OHFA…C2F
D2b 2.321
2.230 2.244

116
OHFA…C3F 2.508 2.476 2.440 OHFA…C4F 2.846 2.818 2.890 <C=OFA…H(C3)F 111.40 110.95 108.37 <OHFA…OF 129.68 129.38 141.66 <OHFA…C2F
176.60
178.54 170.14 Dimer D2 C=OFA…H(C2)F 3.255 OHFA…OF 3.206 OHFA…C1F 2.769 OHFA…C2F 2.416 OHFA…C3F 2.704 OHFA…C4F 3.150 <C=OFA…H(C2)F 108.22 <OHFA…OF 138.07 <OHFA…C2F 176.29
Bb
D1b
Db
D1b
—c
a Geometrical parameters in the Cs symmetry dimer A b Indicates the dimer that it is found after geometry optimization. b Leads to a
saddle point structure with two imaginary frequencies. c The geometry was not optimized at this level of theory

117
Huang et al. calculated the electrostatic potential map of furan at the MP2/6-
311++G(d,p) level of theory.[174] Their results indicate that the region with the strongest
negative potential is in the vicinity of the O atom. Along the C2-C3 bond the negative
potential is also noticeable. This analysis based on electrostatic potentials agrees well
with our results of the type (i) FA – furan dimers. For the most stable dimer A the OH
hydrogen atom of FA interacts with the furan oxygen atom at the site of the strongest
negative potential. Dimer A is 1.54 kcal/mol more stable that dimer D1 where the OH
hydrogen atom of FA interacts with the C2-C3 bond of furan, which indicates a stronger
interaction of the acidic hydrogen atom with the furan oxygen atom than with the π
system.
Type (ii) complexes
The FA – furan dimers E – I are very weakly bound complexes (Table 4.1, Figure 4.2).
Dimers H (-1.94 kcal/mol) and I (-1.35kcal/mol) are both C-H…O complexes where the
CH hydrogen atom of FA interacts with the lone pairs of the furan oxygen atom
(interaction (4)) at 2.502 and 2.488 Å distances, respectively. Dimer H in addition is
stabilized by interaction (2) between the carbonyl oxygen atom of FA and the α-hydrogen
atom of furan (2.508 Å). Complex I is stabilized additionally by interaction (5) with an
intermolecular distance of 2.569 Å between the OH oxygen atom of FA and a ring
hydrogen atom of furan.
Dimers E, F1and G are energetically very close with binding energies of -0.96, -0.82
and -0.86 kcal/mol, respectively. All of them show a combination of the C-HFA…π
interaction (6) and an atypical C-H…O interaction in the plane of the π orbitals. The C-
H…O distances are 2.780 Å, 2.694 Å, and 2.682 Å for the E, F1, and G dimers,
respectively. The C-H…C1 distances take values of 2.814 Å, 2.673 Å, and 2.705 Å, in
that order. Similarly to dimers B, C, and D1, the dimers E, F1, and G show the formic
acid molecule “traveling” around the furan molecule by rotating around its CH axis. The
geometries of the type (ii) complexes suggest that not only electrostatic interactions, but
probably also orbital interactions govern the structures of the complexes.

118
Other FA – furan geometries
Two additional type (i) and three type (ii) FA – furan dimers D, D2, F, J and K were
found with smaller basis sets than 6-311++G(d,p) (Figure 4.3). As mentioned before, at
higher levels of theory these complexes do not represent minima. Dimer D shows Cs
symmetry and at the MP2/6-31G(d,p) + ZPE + BSSE level of theory its binding energy is
-2.32 kcal/mol. The O-H…C2 distance is calculated to 2.366 Å and the O-H…C2 bond
angle to 156.84º (Tables 4.1 and 4.2, Figure 4.3). At the MP2/6-31G(d,p)BSSEopt + ZPE
+ BSSE level of theory the binding energy of dimer D is with -3.02 kcal/mol slightly
larger. The O-H…C2 distance is predicted to 2.510 Å, and the hydrogen bond angle to
163.45º.
Dimer D2 could only be localized at the MP2/6-31G(d,p) level of theory if CP-BSSE
corrections were included during the geometry optimization (Tables 4.1 and 4.2). At this
level of theory the binding energy of dimer D2, including the ZPE and BSSE corrections,
is -2.96 kcal/mol. The O-H…C2 distance is 2.416 Å and the corresponding hydrogen
bond angle is 176.29º.
By comparing the structures of dimers D, D1, and D2 (Figures 4.1 and 4.3) it is clear
that D1 and D2 are the C1 symmetrical structures that derive from the Cs symmetrical
dimer D by tilting and slightly shifting the FA molecule. Of these structures, only the D1
complex is a minimum at higher level of theory (6-311++G(d,p) and aug-cc-pVDZ basis
sets).
The type (ii) dimers F, K and J were only localized with the double zeta basis sets
without augmentation. Dimer F is a very weak interacting complex with an MP2/6-
31G(d,p)+ZPE+BSSE binding energy of -0.61 kcal/mol. At the same level of theory the
C-HFA…C2F and C=OFA…HF distances are 2.495 and 2.839 Å, respectively. Dimer F is
mainly stabilized by the weak interaction (2) between the carbonyl oxygen atom of FA
and a ring hydrogen atom of furan. Similarly, dimer J (-0.74 kcal/mol) is stabilized by
interaction (2) with an intermolecular distance of 2.477 Å and by the CH...π interaction
between the CH hydrogen atom of FA and the C1 carbon atom of furan (2.893 Å
intermolecular distance).

119
TABLE 4.3: Comparison of selected intermolecular parameters in the FA – furan dimers E-K at various levels of theory.
MP2 6-31G (d,p) BSSE 6-31G (d,p) 6- 311G ++ (d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ Dimer E CHFA…OF 2.888 2.809 2.780 2.851 2.717 2.770 CHFA…C1F 2.906 2.839 2.814 2.898 2.714 2.799 CHFA…C2F 2.967 2.934 2.911 3.022 2.751 2.868 CHFA…C3F 2.970 2.946 2.924 3.030 2.756 2.863 CHFA…C4F 2.912 2.859 2.834 2.912 2.723 2.792 <CHFA…OF 103.15 87.25 92.84 84.21 92.53 92.34 <CHFA…C1F 120.61 106.14 111.60 102.50 114.74 111.06 Dimer F
C=OFA…H(C2)F 2.815 2.495 2.430 CHFA…OF 4.227 4.103 4.349 CHFA…C1F 3.614 3.198 3.415 CHFA…C2F 2.911 2.839 2.943 CHFA…C3F 3.228 3.681 3.793 CHFA…C4F 4.010 4.322 4.516
<C=OFA…H(C2)F 94.04 95.02 96.38 <CHFA…OF 148.16 143.51 137.37 <CHFA…C2F 119.33 114.87
F1a 117.79
F1a
—b
Dimer F1 C=OFA…H(C3)F 4.299 3.886 3.850 3.880 4.099 3.742

120
MP2 6-31G (d,p) BSSE 6-31G (d,p) 6- 311G ++ (d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ CHFA…OF 2.762 2.632 2.694 2.691 2.577 2.651 CHFA…C1F 2.798 2.687 2.673 2.717 2.548 2.633 CHFA…C2F 3.043 2.957 2.857 2.971 2.764 2.844 CHFA…C3F 3.144 3.053 2.973 3.082 2.902 2.972 CHFA…C4F 2.965 2.850 2.863 2.900 2.774 2.842
<C=OFA…H(C3)F 70.61 75.61 77.88 76.83 80.96 79.47 <CHFA…OF 140.60 127.62 126.14 121.88 126.95 127.11 <CHFA…C1F 150.94 140.77 146.69 139.21 144.93 143.98 Dimer G CHFA…OF 2.716 2.637 2.682 2.710 2.561 2.643 CHFA…C1F 2.828 2.704 2.705 2.726 2.568 2.660 CHFA…C2F 3.123 2.982 2.933 2.972 2.817 2.900 CHFA…C3F 3.185 3.073 3.036 3.086 2.941 3.014 CHFA…C4F 2.934 2.859 2.876 2.916 2.775 2.847 <CHFA…OF 139.05 121.55 122.09 114.79 122.00 120.90 <CHFA…C1F 152.15 140.15 142.56 137.06 143.46 142.31 Dimer H CHFA…OF 2.558 2.440 2.502 2.467 2.424 2.462
C=OFA…H(C1)F 2.544 2.437 2.508 2.412 2.413 2.432 <CHFA…OF 131.72 129.90 127.37 129.99 134.06 131.44 <C=OFA…H(C1)F 103.50 105.56 108.39 105.07 101.56 104.16

121
Dimer I CHFA…OF 2.532 2.426 2.488 2.464 2.396 2.460 H-OFA…H(C1)F 2.646 2.533 2.569 2.504 2.505 2.543
<CHFA…OF 143.71 140.90 136.20 140.05 145.70 141.31 < H-OFA…H(C1)F 145.38 142.33 136.42 142.09 146.89 143.00
Dimer J CHFA…C1F 2.893 2.992 CHFA…C2F 3.054 3.261 CHFA…OF 3.834 3.959 C=OFA…H(C1)F 2.477 2.399 <CHFA…C1F 110.82 108.58 <CHFA…OF 119.01 114.49 <C=OFA…H(C1)F Ha 99.21
Ea 99.77
Ea
— b
Dimer K CHFA…OF 4.099 4.361
CHFA…C2F 2.902 3.021
<CHFA…C2F — c
122.16
— c
117.41
— c
— b
a Geometrical parameters in the Cs symmetry dimer A. b Indicates the dimer that it is found after geometry optimization. b The
geometry was not optimized at this level of theory. c Leads to a saddle point structure with two imaginary frequencies.

122
Dimer K (-0.57 kcal/mol) exhibits Cs symmetry and its geometry is much related to the
structure of dimer D (Figure 4.3). In dimer K the CH hydrogen atom of FA interacts with
the C2-C3 bond of furan. The C-H FA …C2 F distance is 2.902 Å. The larger A-HFA
…C2F (A = O, C) bond length of dimer K compared to dimer D and the difference
between their binding energies (dimer D is 1.75 kcal/mol more stable than dimer K) are
clearly due to the difference in the acidities of the OH hydrogen atom and the CH
hydrogen atom of the FA molecule.
It is remarkable that both structures D and K were localized after a procedure based on
a completely random exploration of the FA – furan dimers potential energy surface. The
most stable FA – furan dimer A and all the other type (i) and type (ii) dimers, as well as
their enantiomers were found using the MMH procedure.
Basis set influence on the calculated geometries of the FA – furan dimers
To analyze the influence of the basis set on the geometries of the FA – furan dimers,
the structures of the dimers were optimized using the 6-31G(d,p), 6-311++G(d,p), cc-
pVDZ, aug-cc-pVDZ and cc-pVTZ basis sets. Dimers A, B and C are minima with all
basis sets (Tables 4.1 and 4.4), but only with the 6-311++G(d,p) basis set the equilibrium
geometry of dimer A deviates from the Cs symmetry plane (Figure 4.1). Dimer D1 is
only a minimum using the triple zeta and augmented double zeta basis sets. With the 6-
31G(d,p) basis set the optimization of complex D1 leads to D, whereas with the 6-
31G(d,p) basis set including CP corrections during the geometry optimization dimer D2
is produced (Table 4.1).
The structure D was found as a minimum after geometry optimization at the MP2 level
with the 6-31G(d,p) (with and without BSSE-CP corrections) and cc-pVDZ basis sets.
However, when the basis set is augmented by diffuse functions (aug-cc-pVDZ, 6-
311++G(d,p)) it becomes a transition state that leads to the equilibrium geometry D1
(Tables 4.1 and 4.4). At the MP2/cc-pVTZ level dimer D is apparently a minimum, but
due to the huge computational requirements vibrational spectra were not calculated at this
level. Therefore, it is likely that with the cc-pVTZ basis set D would become a transition
state leading to D1, similar to the aug-cc-pVDZ and 6-311++G(d,p) basis sets.

123
TABLE 4.4: Calculated binding energies of FA – furan dimers A-I at MP2 level of
theory with the cc-pVDZ, aug-cc-pVDZ and cc-pVTZ Dunning’s basis set including
ZPE corrections (in kcal/mol).
MP2 cc-pVDZ aug-cc-pVDZ cc-pVTZ ΔE ZPE ΔE ZPE ΔE BSSE A -8.56 1.13 -7.58 1.06 -7.14 1.61 B -5.93 0.81 -6.85 0.78 -5.73 1.50 C -5.72 0.65 -6.77 0.76 -5.63 1.57 D -5.85 0.68 D1 -5.65 1.43 D1 D -6.77 0.73 -5.65 1.44 D2 D D1 —b E -3.68 0.60 -5.30 0.82 -3.75 1.15 F -3.40 0.64 F1 — b
F1 -3.14 0.57 -5.06 0.80 -3.44 1.07 G -3.17 0.57 -5.09 0.82 -3.46 1.06 H -4.67 0.68 -4.19 0.63 -3.74 1.17 I -3.73 0.52 -3.45 0.52 -2.90 0.92 J -3.61 0.64 E — b K -2.79 0.46 —a — b
a Leads to a saddle point geometry with two imaginary frequencies. b The geometry of
the complex was not optimized at this level of theory.
As already mentioned, dimer D2 could be found only at the MP2/6-31G(d,p) level of
theory when including the CP BSSE corrections during the geometry optimization. With
the augmented basis sets dimer D2 became D1 after geometry optimization. At the
MP2/6-31G(d,p) level without the CP corrections the geometry optimization of D2 lead
to structure B. With the cc-pVDZ basis set dimer D2 is converted into dimer D, which is
a minimum at this level of theory (Tables 4.1 and 4.4).
The dimers E, F1, G, H, and I are minima with all basis sets used (Tables 4.1 and 4.4).
Complex I exhibits Cs symmetry at all levels, except with the 6-311++G(d,p) basis set
where it adopts C1 symmetry (Figure 4.2). Dimer F is found only with the small double

124
zeta basis sets without augmentation. With the 6-311++G(d,p) and aug-cc-pVDZ basis
sets it transforms to dimer F1 after geometry optimization (Table 4.1).
Dimers J and K were localized only using the cc-pVDZ and 6-31G(d,p) basis sets.
When the BSSE correction was included during the geometry optimization (6-31G(d,p)
basis set), dimer J was transformed to dimer H (Table 4.1). In contrast, with the 6-
311++G(d,p) and aug-cc-pVDZ basis sets the geometry optimization of J leads to dimer
E. Geometry optimization of complex K at the 6-31G(d,p)BSSEopt, aug-cc-pVDZ and 6-
311++G(d,p) levels of theory results in a second order stationary point (Tables 4.1 and
4.4).
Tables 4.5 and 4.6 show some selected intramolecular distances at various levels of
theory for the FA – furan dimers, respectively. The C=OFA distances decrease up to 0.012
Å when increasing the size of the Pople’s basis set from the 6-31G(d,p) to 6-311++G(d,p)
whereas the O-HFA and O-CF distances decrease less, up to 0.005Å. The C-HFA and C-HF
distances increase with the size of the basis set by around 0.004 Å, and the C2-C3F
distances increase by about 0.005 Å.
With Dunning’s basis set at the double zeta level of theory the O-HFA, C2-C3F and C-
HF distances are not sensitive to the augmentation. With the augmentation the C=OFA and
O-CF bond lengths increase by only 0.006 and 0.009 Å, respectively, and the C-HFA
distances decrease by about 0.005 Å. These variations are slightly more pronounced if the
intramolecular distances calculated with Dunning’s double or triple zeta basis sets are
compared. The cc-pVTZ calculated bond lengths are in general shorter than the cc-pVDZ
values. Thus, the C2-C3F, C-HFA, and C-HF cc-pVTZ distances are 0.011, 0.015 and
0.013 Å, respectively, shorter than the cc-pVDZ values. The C=OFA, O-HFA, and O-CF
bond lengths calculated with the triple zeta basis set are also around 0.006 Å shorter than
the cc-pVDZ values.

125
TABLE 4.5: Comparison of selected intramolecular parameters in the monomer and the FA – furan dimers A - D2 at
various levels of theory. Distances are in Å and angles in degrees
MP2 Expa,b 6-31G(d,p) 6-311G++ (d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ Monomer O-HFA 0.972 0.972 0.969 0.975 0.975 0.969 C=OFA 1.202 1.213 1.205 1.209 1.215 1.203 C-OFA 1.343 1.351 1.348 1.350 1.359 1.346 C1- C2F 1.354 1.366 1.370 1.378 1.380 1.364 C2- C3F 1.440 1.427 1.432 1.436 1.438 1.425 C1-H F 1.075 1.075 1.079 1.088 1.087 1.074 O-CF 1.371 1.366 1.360 1.364 1.372 1.359 6-31G (d,p )BSSE 6-31G(d,p) 6-311G++ (d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ Dimer A O-HFA 0.979 0.980 0.977 0.983 0.984 0.978 C=OFA 1.216 1.218 1.210 1.215 1.221 1.208 C-OFA 1.342 1.339 1.338 1.339 1.347 1.335 C1- C2F 1.364 1.363 1.367 1.375 1.377 1.362 C2- C3F 1.429 1.429 1.434 1.438 1.439 1.427 C1-H F 1.075 1.075 1.079 1.088 1.087 1.075 O-C1F 1.374 1.375 1.370 1.373 1.381 1.368 O-C4F 1.371 1.372 1.367 1.370 1.377 1.364 < C1OC4 106.98 107.15 107.33 107.30 107.38 107.19

126
Dimer B O-HFA 0.977 0.978 0.974 0.981 0.982 0.977 O-C1F 1.365 1.365 1.360 1.363 1.370 1.358 O-C4F 1.366 1.366 1.360 1.364 1.372 1.359 Dimer C O-HFA 0.976 0.977 0.975 0.981 0.983 0.977 O-C4F 1.365 1.364 1.358 1.361 1.369 1.357 Dimer D O-HFA 0.976 0.977 0.980 0.976 C2-C3F 1.430 1.431
D1 1.439
D1 1.428
Dimer D1 O-HFA 0.974 0.982 0.977 C2-C3F D2
D 1.434
D 1.440 1.428
Dimer D2 O-HFA 0.976 C2-C3F 1.429
B D1 D D1 —
a FA reference[149] b Furan reference[191]

127
TABLE 4.6: Comparison of selected intramolecular parameters in the FA – furan dimers E - K at various levels of theory
MP2 6-31G (d,p) BSSE 6-31G (d,p) 6-311G++ (d,p) cc-pVDZ aug-cc-pVDZ cc-pVTZ Dimer E C-HFA 1.091 1.092 1.096 1.107 1.102 1.092 O-C1F 1.366 1.367 1.361 1.364 1.372 1.359 Dimer F C-HFA 1.092 1.092 1.107 C=OFA 1.214 1.216 1.212 C2-HF 1.077 1.077 1.090 C3-HF 1.077 1.077
F1 1.089
F1
—
Dimer F1 C-HFA 1.091 1.091 1.095 1.106 1.102 1.092 C=OFA 1.214 1.214 1.206 1.210 1.216 1.204 C2-HF 1.076 1.077 1.080 1.089 1.088 1.075 C3-HF 1.076 1.076 1.080 1.089 1.088 1.075 Dimer G O-HFA 0.972 0.972 0.969 0.976 0.975 0.970 C-HFA 1.091 1.091 1.095 1.106 1.102 1.092 O-C1F 1.367 1.367 1.362 1.364 1.373 1.360 O-C4F 1.367 1.367 1.361 1.363 1.372 1.359 Dimer H C=OFA 1.215 1.217 1.205 1.213 1.219 1.207

128
C-HFA 1.092 1.091 1.094 1.105 1.102 1.091 O-C1F 1.370 1.371 1.365 1.369 1.377 1.364 O-C4F 1.367 1.367 1.362 1.365 1.373 1.360 H-C1F 1.076 1.075 1.079 1.088 1.087 1.075
<C1OC4 106.74 106.84 107.08 107.00 107.07 106.91 Dimer I C-HFA 1.091 1.091 1.094 1.106 1.102 1.091 C-OFA 1.356 1.357 1.354 1.357 1.365 1.351 O-C1F 1.369 1.369 1.364 1.367 1.376 1.362 Dimer J C-HFA 1.092 1.107 C=OFA 1.216 1.213 C1-C2F 1.368 1.379 H
E
E
—
Dimer K C-HFA 1.091 1.107
C2-C3F —
1.428 — 1.437
—
—

129
The intermolecular bond distances and angles of dimer A are not very sensitive to the
basis set used (Table 4.2, BSSE effects on the geometries of the complexes are discussed
separately). For instance, the C=OFA…HF distance is 0.12 Å larger in the 6-311++G(d,p)
geometry than in the 6-31G(d,p) calculated structure. However, in this case it should be
taken into account that the 6-311++G(d,p) dimer A has C1 symmetry while the structures
of dimer A calculated with the other basis sets have Cs symmetry.
Most interesting is the case of dimer B where the geometry of the complex shows the
largest dependence on the basis set used (Table 4.2). While the 6-311++G(d,p), aug-cc-
pVDZ and cc-pVTZ structures are similar, the geometries calculated with the smaller 6-
31G(d,p) and cc-pVDZ basis sets show larger deviations, although not resulting in new
minima. In the structures of complex B calculated with the 6-31G(d,p) basis set
compared to that with the 6-311++G(d,p) basis set the OHFA…C2F hydrogen bond angle
is reduced by 11º and the OHFA…OF hydrogen bond angle increases by 31º. The cc-
pVTZ calculated hydrogen bond angles of dimer D1 also change considerably when
compared to the 6-311++G(d,p) and aug-cc-pVDZ calculated values (Table 4.2). Table
4.3 shows some selected intermolecular parameters for the dimers E – K at various levels
of theory.
By comparing calculated binding energies of all complexes with the different basis
sets, no general conclusion is evident (Tables 4.1 and 4.4). However, the aug-cc-pVDZ
calculated binding energies are in general higher than at the other levels of theory, except
for dimer A. The 6-311++G(d,p) and cc-pVTZ energies are in general in good agreement.
Taking into account the compromise between quality of the results and the computational
time requirements, we consider that the 6-311++G(d,p) basis set is appropriate for the
geometry optimization of these systems. In many cases the aug-cc-pVDZ basis set
provides good geometries too and compares well to the cc-pVTZ and 6-311++G(d,p)
results.
Effect of the BSSE on the calculated geometries and binding energies
BSSE corrections of the binding energies were calculated for all FA – furan dimers at
the MP2 level of theory with the cc-pVTZ, 6-311++G(d,p) and 6-31G(d,p) basis sets.
With the cc-pVTZ basis set the BSSE is smaller than with the other basis sets (Tables 4.1

130
and 4.4). With the exception of complexes F1 and G, with Pople’s basis sets the BSSE
decreases when increasing the size of the basis set from 6-31G(d,p) to 6-311++G(d,p).
However, the differences between the BSSE with the 6-311++G(d,p) and the 6-31G(d,p)
basis sets are very small (0.06 – 0.03 kcal/mol, Table 4.1).
To evaluate the influence of the BSSE on the calculated geometries the structures of all
FA – furan dimers were optimized at the MP2/6-31G(d,p) level of theory using the
counterpoise (CP) scheme during optimization (6-31G(d,p)BSSEopt). In contrast to our
previous results for other systems[146, 192] but in agreement with other studies,[90] the
BSSE effects considerably the geometry of the FA – furan complexes. As already
mentioned before, structure D2 could only be found with the 6-31G(d,p)BSSEopt
corrected geometry optimization. Furthermore, the 6-31G(d,p)BSSEopt optimizations of
dimers D1, J and K lead to entirely different structures compared to those calculated
without BSSE corrections (Tables 4.2 and 4.3). For dimer K the 6-31G(d,p)BSSEopt
optimization gave the same structure as with the larger cc-pVTZ, 6-311++G(d,p) and
aug-cc-pVDZ basis sets (Tables 4.1 and 4.4).
Here, the 6-31G(d,p)BSSE corrected geometries of the FA – furan dimers compared to
the non BSSE corrected geometries at the same level of theory are discussed. As
expected, the intramolecular parameters of FA and furan in the complexes were not
affected by the inclusion of the BSSE corrections during the geometry optimizations
(Tables 4.5 and 4.6). For the most stable dimer A, there is no significant change when
including BSSE corrections, only the OHFA…OF and C=OFA…HF hydrogen bonds are
elongated by 0.084Å and 0.135Å respectively (Table 4.2).
The geometries of other complexes, especially some angles in dimers B, C, D, F1 and
G, are more effected by the inclusion of the BSSE corrections during the geometry
optimizations (Tables 4.2 and 4.3). For complexes D and F the intermolecular distances
increase up to 0.32Å (C=OFA…HF interaction (2) of complex F) when the BSSE is
included during the geometry optimization (Figure 4.3). Our results indicate that the
BSSE corrections are more important for the weakly bound π dimers than for the relative
strongly bound dimers such as A, H, and I.

131
Comparison with other furan complexes
All geometries of the FA – furan dimers described here were localized via a random
exploration of the multiple minima hypersurface for the FA – furan system without any
previous chemical assumptions. It is therefore interesting to discuss structural similarities
between these FA – furan dimers and furan homo- and heterodimers found by other
methods described in literature.
Pei and Li[171] found four equilibrium isomers of the furan dimer at the B3LYP/6-
311G(d,p) level of theory. The most stable structure shows two equivalents CH…O
interactions at 2.547Å and resembles the geometry of the most stable FA – furan dimer
A. As expected, in dimer A the CH…O interaction is stronger than in the furan
homodimer. The dimer A is stabilized additionally by the C=OFA…HF interaction
between the α-hydrogen atom of furan and the carbonyl oxygen atom of FA.
Huang and Wang[174] found two basic geometries for the furan – hydrogen halides
dimers. One is the atom-on type where the H atom of HX interacts with the nonbonding
electron pairs of the furan oxygen atom and the HX deviates slightly from the furan ring
plane. The other geometry is the face-on type, with a hydrogen bond between the H atom
of HX and the π system of furan. For the furan – HF complexes only the atom-on
geometry is observed and for the furan –HI only the face-on type was found. Furan –HCl
and furan – HBr dimers exhibit both types of geometries.[174]
Compared to that, the FA – furan complexes A, H and I show atom-on geometries
while dimers B, C and D1 are face-on complexes. The geometries of the furan –
hydrogen halide dimers described by Huang were optimized at the same level of theory
(MP2/6-311++G(d,p)) than the FA – furan dimers. It is remarkable that while with others
basis sets the equilibrium geometries of dimers A and I are of Cs symmetry, at the
MP2/6-311++G(d,p) level of theory the geometries deviate from the Cs symmetry.
Similarly, the furan – hydrogen halide dimers deviate slightly from the C2v plane of the
furan ring.[174] With both smaller and larger basis sets a higher symmetry is found, which
indicates that the deviation from Cs symmetry might be an artifact of the 6-311++G(d,p)
basis set. At the same level of theory, the Cs geometry of the formamide molecule shows
one out of plane imaginary frequency resulting again in a slightly distorted structure.

132
Chan and Del Bene[114] carried out theoretical studies of the reactions of different acids
with furan at the MP2/aug’-cc-pVTZ//MP2/6-31+G(d,p) level of theory. They found that
hydrogen bonding is largely determined by electrostatic interactions, which generally
corresponds to hydrogen bonding to the site with the most localized negative charge.
Consequently, the furan molecule forms two hydrogen-bonded complexes with HF, one
through a lone pair at the oxygen atom, which is favored, and another dimer through the π
system at C2. They found a similar behavior for other weak acids interacting with furan.
Depending on the nature of the donor, the π or the O complex is more stable.[114]
Huang[174] obtained the atom-on geometry as the only minimum for the furan – HF dimer
and the face-on geometry exclusively for the furan – HI complex. This discrepancy
between the results of Huang and Del Bene is an evidence for the difficulties in
calculating the weak interacting complexes of furan.
Comparison with matrix isolation spectroscopy results
The experimental IR spectra of dimer A was assigned by comparison with calculations
at the MP2/aug-cc-pVDZ and MP2/6-311++G(d,p) levels of theory (Tables 4.7 and 4.8).
The red shift of the C=O stretching vibration of monomeric FA is characteristic for the
formation of complexes of FA. The experimental red-shift (-17cm-1) is in excellent
agreement with both the MP2/aug-cc-pVDZ and MP2/6-311++G (d,p) calculations for
dimer A, which predict shifts of -15.2 and -14.6 cm-1, respectively. The red-shift in the
C=O stretching vibration corresponds to an elongation of the C=O bond by 0.005 and
0.006 Å with the 6-311++G (d,p) and aug-cc-pVDZ basis sets, respectively, due to the
formation of the complex (Table 4.5). For dimer B the red shift is predicted to -12.7 cm-1,
for C to -13.2 cm-1, and for D1 to -13.5 cm-1 at the MP2/aug-cc-pVDZ level theory.

133
TABLE 4.7: The experimental (Ar matrix at 30 K – 35 K) and the calculated MP2/aug-cc-pVDZ and MP2/6-311++G(d.p)
vibrational frequencies (unscaled, in cm–1) of the FA – furan dimer A, along with the frequency shift in the complex, ∆ν, from
the monomer (in parentheses).
MP2 /aug-cc-pVDZ MP2 /6-311++G(d,p) M Experimental M Complex A M Complex A
Formic Acid
3550.5 3375.2 3365.3 3364.2
(-175.3) (-185.1) (-186.3)
3726.7 3543.9 (-182.8) 3797.7 3637.7 (-160.0) ν(O-H)
1767.3 1750.3 (-17.0) 1771.0 1755.8 (-15.2) 1807.6 1793.0 (-14.6) ν(C=O)
1103.5 1150.4 1153.0
(46.9) (49.5)
1115.7 1169.2 (53.5) 1142.7 1193.9 (51.2) ν(C-O)
Furan 1177.7 1167.7 (-10.0) 1218.9 1207.9 (-11.0) 1249.9 1236.2 (-13.7) νas(C-O-C) 1065.0 1051.6 (-13.4) 1094.0 1081.4 (-12.6) 1112.5 1095.8 (-16.7) νs(C-C)+ νs(C-O) 993.6 988.3 (-5.3) 1011.4 1006.2 (-5.3) 1025.5 1021.7 (-3.8) δ(CC-H) 869.1 874.8 (5.7) 865.6 873.3 (7.7) 879.9 885.9 (6.0) δs(C-O-C)
744.1 769.7 767.9 764.8
(25.6) (23.8) (20.7)
742.6 758.3 (15.7) 732.5 748.9 (16.4) γ(CC-H)

134
TABLE 4.8: Calculated vibrational frequencies (in cm–1) of FA – furan
complexes B - D1 by MP2/aug-cc-pVDZ level theory, along with the frequency shifts
in the complexes ∆ν, from the monomer (in parentheses).
MP2 /aug-cc-pVDZ M Complex B Complex C Complex D1
Formic Acid 3726.7 3580.8 (-145.9) 3565.7 (-161.0) 3577.4 (-149.3) ν(O-H) 1771.0 1758.2 (-12.8) 1757.8 (-13.2) 1757.4 (-13.6) ν(C=O) 1115.7 1134.8 (19.1) 1140.7 (25.0) 1141.8 (26.1) ν(C-O) Furan 1218.9 1218.4 (-0.5) 1222.8 (3.9) 1217.3 (-1.6) νas(C-O-C)
1094.0 1094.7 (0.7) 1099.2 (5.2) 1094.5 (0.5) νs(C-C)+ νs(C-O)
1011.45 1011.47 (0.02) 1013.3 (1.9) 1011.6 (0.2) δ(CC-H) 865.6 865.5 (0.1) 865.9 (0.3) 865.6 (0) δs(C-O-C) 742.6 746.9 (4.3) 757.7 (15.1) 744.5 (1.9) γ(CC-H) 770.0 27.4
For the OH stretching vibration of FA, the calculations at the MP2/aug-cc-pVDZ and
MP2/6-311++G (d,p) levels of theory for dimer A predict red shifts of -182.7 cm-1 and
-159.9 cm-1, respectively. In particular the results obtained with the aug-cc-pVDZ basis
are in excellent agreement with the experiment (-185 cm-1) (Table 4.7). The formation of
a complex results in an elongation of the O-HFA bond length in dimer A (compared to the
FA monomer) by 0.008 and 0.009 Å with the 6-311++G(d,p) and aug-cc-pVDZ basis
sets, respectively (Table 4.5). For dimer B the red-shift is calculated to -145.8 cm-1, for C
to -160.9 cm-1, and for D1 to -149.4 cm-1 (aug-cc-pVDZ basis, Table 4.8). The large
shifts of the OH stretching vibrations allow to discard the FA – furan complexes type (ii)
where the OH…O and O-H…π interactions are lacking and thus the OH stretching
vibration of the FA molecule is much less perturbed (Table 4.7).
The formation of hydrogen-bonded complexes of FA results in a contraction of the C-
OH bond compared to the monomer (0.010 and 0.012 Å with the 6-311++G(d,p) and
aug-cc-pVDZ basis sets, respectively, Table 4.5 ) and a blue shift of the corresponding C-

135
OH stretching vibration (observed at 1103.5 cm-1 in the unperturbed FA). For complex A
the predicted blue shift of 53.4 cm-1 (aug-cc-pVDZ) nicely matches the experimental
value of 46.9 cm-1. For complex B the shift is calculated to 19.1cm-1, for complex C to
24.9 cm-1, and for complex D1 to 26.1 cm-1 (Table 4.8), in much less agreement with the
experimental value.
The IR spectrum of the furan molecule in dimer A is less affected by the formation of
intermolecular complexes. The symmetrical and asymmetrical C-O-C stretching
vibrations of the furan ring are red-shifted by -13.4 cm-1 and by -10 cm-1, respectively.
These experimental shifts are in excellent agreement with the aug-cc-pVDZ calculated
values (Table 4.7). The CCH deformation mode δ(CCH) of monomeric furan at 993.6
cm-1 is shifted to 988.5 cm-1 in dimer A, which again is in good agreement with the
calculation (Table 4.7). The δ(COC) and γ(CCH) vibration modes are blue-shifted by 5.7
and 23.8 cm-1, respectively. The aug-cc-pVDZ calculations of complex A predict shifts of
7.7 and 15.7, respectively for these vibrations.

136
4.4. 1:2 Formic acid – acetylene complexes. Results and
discussion
Geometries and binding energies
Six complexes (A – G) corresponding to local minima between one molecule of formic
acid and two molecules of acetylene were found at the MP2/cc-pVTZ level of theory.
Four additional structures B1, E1, G1 and H1 were located using DFT theory (B3LYP/6-
311++G(d,p)) or MP2 with a smaller basis set (MP2/6-311++G(d,p)), but are not minima
at higher levels of theory (Figures 4.4 and 4.5). At all levels of theory complex A is
predicted to be the global minimum, followed by complexes B and C (Tables 4.9 – 4.11).
At the CCSD(T)/cc-pVTZ//MP2/cc-pVTZ level of theory after ZPE corrections the
calculated binding energy for complex A is -7.44 kcal/mol and the binding energies for
complexes B and C are -6.85 and -6.47 kcal/mol, respectively. Thus, three stable
complexes with binding energies within 1 kcal/mol are predicted.
The binding in complexes between formic acid and acetylene shows contributions from
the following five basic binding motives:
(1) OH…π interaction between the acidic H atom of formic acid and the π system of
acetylene;
(2) CH… π interaction between the formyl H atom of formic acid and the π system of
acetylene;
(3i) CH… π interaction between one acetylene H atom and the π system of the second
acetylene molecule;
(4) CH…O interaction between one acetylene H atom and the carbonyl oxygen atom
of formic acid;
(5) CH…O interaction between one acetylene H atom and the hydroxyl oxygen atom
of formic acid.
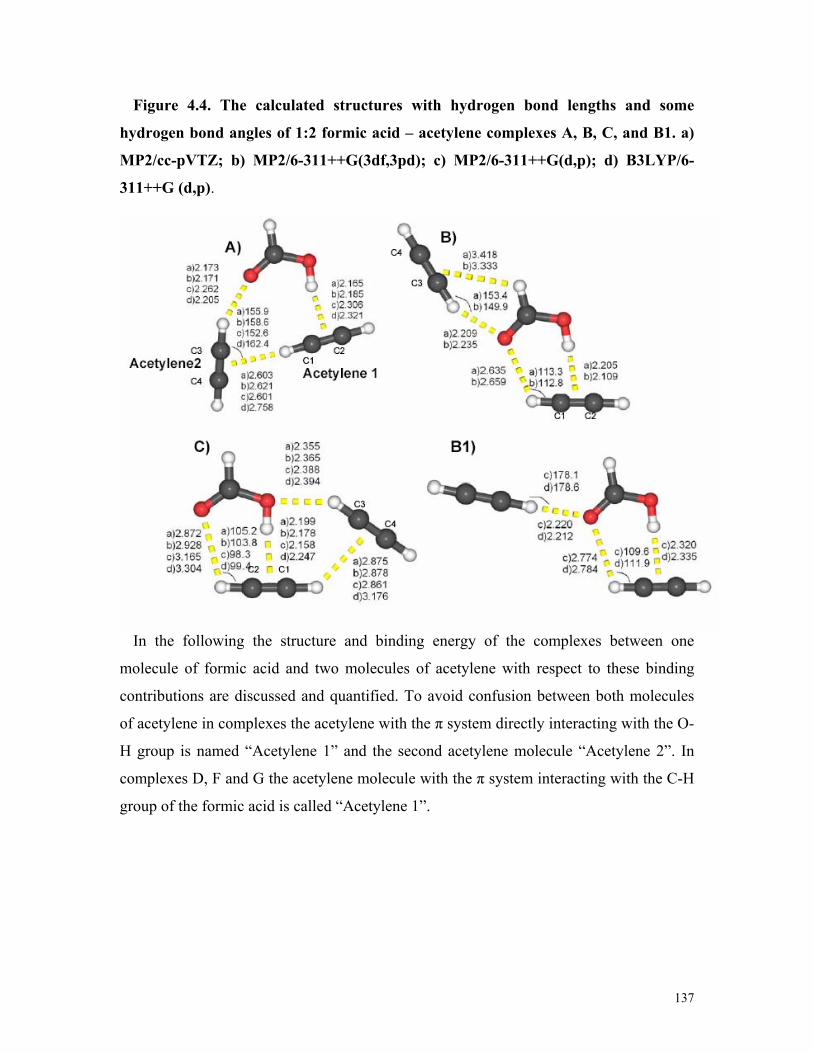
137
Figure 4.4. The calculated structures with hydrogen bond lengths and some
hydrogen bond angles of 1:2 formic acid – acetylene complexes A, B, C, and B1. a)
MP2/cc-pVTZ; b) MP2/6-311++G(3df,3pd); c) MP2/6-311++G(d,p); d) B3LYP/6-
311++G (d,p).
In the following the structure and binding energy of the complexes between one
molecule of formic acid and two molecules of acetylene with respect to these binding
contributions are discussed and quantified. To avoid confusion between both molecules
of acetylene in complexes the acetylene with the π system directly interacting with the O-
H group is named “Acetylene 1” and the second acetylene molecule “Acetylene 2”. In
complexes D, F and G the acetylene molecule with the π system interacting with the C-H
group of the formic acid is called “Acetylene 1”.

138
Figure 4.5. The calculated structures with hydrogen bond lengths and some
hydrogen bond angles of 1:2 formic acid – acetylene complexes: D, F, G, E1, G1 and
H1. a) MP2/cc-pVTZ; c) MP2/6-311++G(d,p); d) B3LYP/6-311++G(d,p).

139
At the MP2 level of theory the calculated binding energies and geometries are not very
dependent of the size of the basis set. For complex A, using the 6-311G++(d,p), cc-
pVTZ, and 6-311G++(3df,3pd) basis sets, the binding energies after ZPE corrections are
-7.64, -7.98, and -8.29 kcal/mol, respectively. B3LYP computations predict larger
binding energies compared to MP2. At all levels of theory and in accordance with
qualitative expectations, complexes D, F and G are less stable than the complexes A, B
and C (Table 4.9 and Figures 4.4 and 4.5). The MP2 geometries and binding energies
calculated with the cc-pVTZ and the 6-311++G(3df,3pd) basis sets are very similar.
However, the geometries obtained with the smaller 6-311++G(d,p) basis set differs
significantly from the cc-pVTZ results. The MP2/cc-pVTZ level of theory has been
established as very adequate for calculating weakly interacting systems as has been
confirmed by calculations of molecular systems with similar types interactions, like the
1:1 formic acid – acetylene complexes[116] and the acetylene dimers studies by
Karpfen.[166] The good performance of the cc-pVTZ basis set in these systems also
suggests to use this more economical basis set instead of the 6-311++G(3df,3pd) basis
set. For complexes A, B, and C the BSSE corrected binding energies at the MP2/cc-
pVTZ level of theory are also listed (Table 4.9).
TABLE 4.9: Calculated binding energies and ZPE corrected values of the 1:2
formic acid–acetylene complexes A – D, F, and G (in kcal/mol)
B3LYP/6-311++G(d,p) MP2/6-311G++(d,p) MP2/cc-pVTZ
ΔE ΔE (ZPE) ΔE ΔE (ZPE) ΔE ΔE (ZPE) ΔE (BSSE) A -6.58 -4.95 -9.23 -7.64 -9.52 -7.98 -7.87 B - -8.77 -7.18 -6.84 C -5.12 -3.80 -8.03 -6.63 -8.42 -7.05 -6.73 D -4.43 -2.98 -7.00 -5.27 -6.79 -5.47 - F -3.06 -1.85 -6.02 -4.33 -5.37 -4.32 - G - - -5.01 -3.65 -5.06 -3.93 -

140
TABLE 4.10: MP2 and CCSD(T) calculated binding energies for the complexes A,
B and Ca
MP2/6-311G++(3df,3pd) CCSD(T)/cc-pVTZ // MP2/cc-pVTZ
ΔE ΔE (ZPE) ΔE ΔE (ZPE)
A -9.83 -8.29 -8.98 -7.44 B -8.62 -7.03 -8.44 -6.85 C -8.53 -7.16 -7.84 -6.47
a ZPE corrections are from the MP2/cc-pVTZ calculations.
TABLE 4.11: Calculated binding energies and ZPE corrected values of 1:2 formic
acid–acetylene complexes B1, E1, G1, and H1 (in kcal/mol)
B3LYP/6-311++G(d,p) MP2/6-311G++(d,p) MP2/cc-pVTZ Complex
ΔE ΔE (ZPE) ΔE ΔE (ZPE) ΔE ΔE (ZPE)
B1 -6.08 -4.55 -7.91 -6.06 B E1 -4.15 -2.92 -6.63 -4.94 C G1 -3.12 -1.98 G H1 -4.48 -3.24 -5.03 -3.00 D
In complex A the acidic hydrogen atom of the formic acid molecule interacts with the π
system of one molecule of acetylene (contribution (1)). The MP2 calculated distance of
the C1 carbon atom of acetylene to the acidic hydrogen atom of formic acid (OH…C1
distance) is 2.293 Å (2.301 Å), and that of C2 (OH…C2 distance) is 2.297 Å (2.307 Å)
using a cc-pVTZ (6-311++G(3df,3pd)) basis set. The carbonyl oxygen atom of formic
acid interacts with one hydrogen atom of the second acetylene molecule (contribution
(4)) with a CH…O distance of 2.173 Å (cc-pVTZ, 2.171 Å with the 6-311++G(3df,3pd)
basis). Additional stabilization of the complex is due to the C-H…π interaction between
the two acetylene molecules (contribution (3), which corresponds to the “T” shape of the
acetylene dimer.[166] In this case, the T is distorted by interactions of both acetylene
molecules with the formic acid molecule. The acetylene – acetylene interaction is
characterized by a CH…C3 distance of 2,505 Å (2.496 Å) and a CH…C4 distances of
2,836 Å (2.815 Å) at the MP2/cc-pVTZ (MP2/6-311++G(3df,3pd)) level of theory. These

141
distances are in agreement with the expectations for weak hydrogen bonds[1] and compare
well with that of the undisturbed “T” shape acetylene dimer.[166, 167]
Complexes B and C provide further evidence for O-H…π, C-H…π and CH…O
interactions (Figure 4.4). In complex B the two molecules of acetylene do not interact
directly (thus, contribution (3) is absent), but both interact with the formic acid molecule
via contribution (4) and contribution (1). A type (4) interaction is found between the C1
H atom and the carbonyl oxygen atom. However, the long C1H…O distance of 2.635 Å
indicates that this interaction is rather weak. Even weaker is, with a distance of 3.418 Å,
the CH… π interaction of type (2) between the CH hydrogen atom of formic acid and the
acetylene π system.
Complex C shows an additional weak CH…O interaction between the hydroxyl oxygen
atom of the formic acid molecule and the hydrogen atom of one of the acetylene
molecules (contribution (5)). The other complexes - D, F and G - show C-H…π and
CH…O interactions; however, here the acidic hydroxyl hydrogen atom of the formic acid
molecule is not involved, resulting in weak overall binding energies.
The structures B1, E1, G1 and H1 are artifacts of the inability of the B3LYP functional
to deal with weak C-H…π dispersive interactions. Geometry optimization at the MP2/cc-
pVTZ level of theory results in the transformation of B1 to B, E1 to C, G1 to G and H1 to
D (Table 4.11, Figure 4.5). Thus, by comparison of complex B with structure B1 it is
evident that weak C-H…π interactions between the CH hydrogen atom of formic acid
and the π system of acetylene are not reproduced by B3LYP. This has been referred in
the study of the 1:1 formic acid- acetylene complexes.[116]
Intramolecular distances and vibrational frequencies
The intermolecular interactions in the complexes result in a distortion of the monomer
intramolecular distances and vibrational frequencies. Table 4.12 lists some selected bond
distances of the formic acid and acetylene monomer and the A, B, and C 1:2 formic acid–
acetylene complexes. For the monomers the experimental values [149, 193] are well
reproduced by calculations at the MP2/cc-pVTZ and MP2/6-311++G(3df,3pd) levels of
theory. The vibrational frequencies of the A, B and C complexes calculated at various

142
levels of theory are listed in Tables 4.13 – 4.15. The Dunning basis sets are expected to
be most reliable, since for the 1:1 complexes at this level an excellent agreement between
theory and experiment was found. [116]
TABLE 4.12: Comparison of selected intramolecular distances of the formic acid
and acetylene monomers (M), and 1:2 formic acid–acetylene complexes (A, B and
C)a
Exp MP2/cc-pVTZ MP2/6-311++G(3df,3pd) M M A B C M A B C
HCOOH r(C=O) 1.202(10)b 1.203 1.209 1.211 1.205 1.202 1.207 1.209 1.204 r(O─H) 0.972 (5)b 0.969 0.978 0.977 0.979 0.968 0.976 0.975 0.977 r(C─O) 1.343(10)b 1.346 1.333 1.332 1.342 1.343 1.331 1.331 1.339
C2H2 (acetylene1) r(C1≡C2) 1.203c 1.211 1.214 1.213 1.214 1.211 1.213 1.213 1.213 r(C1─H) 1.062c 1.061 1.067 1.064 1.064 1.062 1.067 1.064 1.064 r(C2─H) 1.063 1.063 1.064 1.063 1.063 1.064
C2H2 (acetylene2) r (C3≡C4) 1.213 1.213 1.213 1.213 1.212 1.212
r(C3─H) 1.066 1.067 1.064 1.067 1.067 1.065 r(C4─H) 1.062 1.062 1.062 1.062 1.062 1.062
a The distances are given in Å. b Reference[149] c Reference [193]

TABLE 4.13: Calculated vibrational frequencies (in cm–1) of the complex A and
frequency shifts relative to the isolated monomer M (in parentheses)
B3LYP/6-311++G(d,p) MP2/6-311++G(d,p) MP2/cc-pVTZ
M A M A M A 3738.0 3583.2 (-155) 3797.2 3672.7 (–125) 3763.4 3604.9 (-159) νO─Ha
1816.2 1794.0 (-22) 1807.5 1795.3 (–12) 1818.1 1800.1 (-18) νC=Oa 1125.3 1167.3 (+42) 1142.6 1184.0 (+41) 1136.7 1183.9 (+47) νC─Oa
3057.5 3054.3 (-3) 3132.3 3126.4 (-6) 3125.1 3119.2 (-6) νC─Ha 3419.8 3379.2 (-41)
3375.2 (-45) 3455.2 3420.4 (–35)
3429.8 (-25) 3446.1 3397.0 (-49)
3409.3 (-37) νC─Hb
772.7 801.3 (+29) 819.5 (+47) 832.1 (+59) 835.0 (+62)
766.3 789.7 (+23) 795.2 (+29) 809.9 (+44) 828.8 (+63)
753.0 786.9 (+34) 791.8 (+39) 802.0 (+49) 806.3 (+53)
δCCHb
a Formic acid modes in the complex. b Acetylene modes in the complex.
TABLE 4.14: Calculated vibrational frequencies (in cm–1) and frequency shifts
relative to the isolated monomer (in parentheses) of the complexes B and B1
B3LYP/6-311++G(d,p) MP2/6-311++G(d,p) MP2/cc-pVTZ
M B1 M B1 M B 3738.0 3586.4 (-152) 3797.2 3681.6 (–116) 3763.4 3618.4 (-145) νO─Ha
1816.2 1786.1 (-30) 1807.5 1788.6 (–19) 1818.1 1787.3 (-31) νC=Oa 1125.3 1172.8 (+48) 1142.6 1187.6 (+45) 1136.7 1186.7 (+50) νC─Oa
3057.5 3057.7 (+0.2) 3132.3 3128.3 (-4) 3125.1 3121.4 (-4) νC─Ha 3419.8 3370.6 (-49)
3404.1 (-16) 3455.2 3417.9 (–37)
3442.0 (-13) 3446.1 3393.7 (-52)
3429.4 (-17) νC─Hb
772.7 791.7 (+19) 795.8 (+23) 838.5 (+66) 841.0 (+68)
766.3 780.4 (+14) 782.6 (+16) 869.2 (+101) 872.6 (+106)
753.0 767.6 (+15) 785.7 (+33) 796.2 (+43) 816.9 (+64)
δCCHb
a Formic acid modes in the complex. b Acetylene modes in the complex.

144
TABLE 4.15: Calculated vibrational frequencies (in cm–1) of complex C and
frequency shifts relative to the isolated monomer (in parentheses)
B3LYP/6-311++G(d,p) MP2/6-311++G(d,p) MP2/cc-pVTZ
M C M C M C 3738.0 3562.6 (-175) 3797.2 3650.7 (–147) 3763.4 3580.4 (-183) νO─Ha
1816.2 1806.2 (-10) 1807.5 1798.8 (–9) 1818.1 1804.2 (-14) νC=Oa 1125.3 1153.0 (+28) 1142.6 1168.9 (+26) 1136.7 1171.5 (+35) νC─Oa
3057.5 3048.1 (-9) 3132.3 3121.2 (-11) 3125.1 3114.9 (-10) νC─Ha 3419.8 3396.0 (-24)
3400.0 (-20) 3455.2 3434.4 (-21)
3435.7 (–20) 3446.1 3422.6 (-24)
3424.2 (-22) νC─Hb
772.7 790.0 (+17) 797.4 (+25) 805.2 (+33) 813.4 (+41)
766.3 779.9 (+14) 788.2 (+22) 800.9 (+35) 802.2 (+36)
753.0 767.5 (+15) 771.2 (+18) 789.9 (+37) 794.3 (+41)
δCCH b
a Formic acid modes in the complex. b Acetylene modes in the complex.
The most perturbed vibrational modes in complexes A, B and C are the O-H stretching
vibrations of the formic acid molecules (Tables 4.13 – 4.15). At the MP2/cc-pVTZ level
of theory the frequency shifts in the complexes are -159, -145, and -183 cm-1, for the
trimers A, B, and C, respectively. This reflects the structures of complexes A, B and C
(Fig. 4.4) which all exhibit strong interactions between the OH hydrogen atom and the π
system of Acetylene 1. In complex C, with the largest frequency shift (-183 cm-1), an
additional interaction of the OH oxygen atom with one hydrogen atom of Acetylene 2 is
found. The formation of the complexes results in an elongation of the OH bonds of nearly
0.01 Å (MP2/cc-pVTZ, Table 4.12)
The carbonyl stretching frequencies of the formic acid molecules are predicted to be
shifted by -18, -31, and -14 cm-1 in the complexes A, B, and C, respectively. This again
reflects the CO…H interaction between the carbonyl oxygen atom and the acetylene H
atom (contribution (4)). In complex B, with a red shift (-31 cm-1) almost twice as large as
in complexes A and C, there is an additional CO…H interaction of the carbonyl group
with the H atom of Acetylene 2. This interaction results in a larger increase of the C=O
bond length in B compared to A and C.

145
The C-OH stretching modes of formic acid are blue-shifted in the complexes, and the
C-OH formic acid bond lengths are consequently shorter with respect to the monomer.
Again, trimer B shows a larger blue shift (+50 cm-1) than complexes A and C ( +47 cm-1
and +35 cm-1, respectively) which can be associated with the very weak CH…π
interaction between the CH group of the formic acid and the π system of Acetylene 2
(contribution (2)).
The C-H stretching modes of the acetylene moieties are also perturbed in the
complexes. At the MP2/cc-pVTZ level of theory these bands in trimer A are red-shifted
by -49 and -37 cm-1, in complex B by -52 and -17 cm-1, and in complex C by -24 and -22
cm-1.
Comparison with matrix isolation spectroscopy results
A mixture of aggregates was found in the matrix isolation experiments with FA –
acetylene. The main constitutes of these mixtures had been identified previously: the
acetylene dimer,[194],[195],[196] formic acid dimer[197],[7],[198] and 1:1 complex between
acetylene and formic acid.[116] However, the careful analysis of the IR absorptions
revealed the formation of several new bands which could not be assigned to any of the
previously known species (Table 4.16).
At higher acetylene concentrations two new peaks were observed in the O─H stretching
region at 3395.0 and 3384.0 cm−1. Since the intensity of these bands increase with
increasing acetylene concentration more than that of the 1:1 complexes they are
tentatively assigned to the O─H stretching vibration of formic acid in a 1:2 complex of
formic acid and acetylene. The experimental frequency shift (-155cm−1) is in excellent
agreement with the calculated frequency shifts (-159 cm−1 with MP2/cc-pVTZ and -155
with B3LYP/6-311++G(d,p)) for complex A (Table 4.16).

146
TABLE 4.16: Experimental (Ar matrix at 30 K – 10 K) and calculated (MP2/cc-
pVTZ and B3LYP/6-311++G(d,p)) vibrational frequencies (in cm–1) of the formic
acid–acetylene 1:2 complex A and frequency shifts ∆ν in the complex relative to the
isolated monomers (in parentheses)
Calculated frequencies Experimental frequencies MP2/ cc-pVTZ B3LYP/6-
311++G(d,p) M Complex A M Complex A M Complex A
3550.4 3395.0 (-155) 3763.4 3604.9 (-159) 3738.0 3583.2 (-155) νO─Ha
1767.1 1747.1 (-20) 1818.1 1800.1 (-18) 1816.2 1794.0 (-22) νC=O a 1103.5 1145.4 (+42) 1136.7 1183.9 (+47) 1125.5 1167.3(+42) νC─Oa
3288.8 3244.5 (-44) 3236.9 (-52)
3446.1 3409.3 (-37) 3397.0 (-49)
3419.8 3379.2 (-41) 3375.2 (-45)
νC─H b
a Formic acid modes in the complex. b Acetylene modes in the complex.
Figure 4.6. Matrix isolation IR spectra in the O─H stretching region of formic
acid. a: HCOOH : Ar = 1:600. b: HCOOH : C2H2 : Ar = 1:1:600. c: HCOOH :
C2H2 : Ar = 1:2:600. [145]
In the C=O stretching region increasing acetylene concentration results in the increase
of the intensity of the band at 1747 cm−1 assigned to the less stable, unsymmetrical

147
formic acid dimer. In addition, this absorption broadens significantly. This indicates that
in addition to the formic acid dimer a new species assigned to the 1:2 complex of formic
acid and acetylene is formed under these conditions. The intensity of this band depends
on the acetylene concentration, in agreement with the assignment to the C=O stretching
vibration of the 1:2 complex A. The good agreement between the experimental frequency
shift (-20 cm−1) and the calculated values (-18 cm−1 with MP2/cc-pVTZ and -22 with
B3LYP/6-311++G(d,p)) further confirms the formation of complex A.
Figure 4.7. Matrix isolation IR spectra in the C=O stretching region of formic
acid. a: HCOOH : Ar = 1:400. b: HCOOH : C2H2 : Ar = 1:1:600. c: HCOOH :
C2H2–Ar (1/2/600). FAD: non-symmetrical (acyclic) formic acid dimer; FCD: cyclic
formic acid dimer. [145]
In the C-O stretching region a new band appears at 1145.4 cm−1 at higher acetylene
concentration. By comparing with the calculated frequencies this band is also assigned to
the 1 : 2 complex A. The experimental frequency shift (+42 cm−1) is in excellent
agreement with the B3LYP/6-311++G(d,p) value (+42) and the MP2/cc-pVTZ (+47)
value.

148
Figure 4.8. Matrix isolation IR spectra in the C─O stretching region of formic
acid. a: HCOOH : Ar = 1:600. b: HCOOH : C2H2 : Ar = 1:1:600. c: HCOOH :
C2H2 : Ar = 1:2:600.[145]
The experimental frequency shifts for the acetylene streching (-44 and -52 cm−1) are in
reasonable agreement with the calculated shifts for complex A (-37, -49 cm−1 with
MP2/cc-pVTZ and -41, -45 cm−1 with B3LYP/6-311++G(d,p), Table 4.16). In the CCH
bending region of acetylene the 1 : 2 complex absorptions are too weak to be observed.
Analysis of the intermolecular interactions in the trimers
To quantify the contributions of intermolecular interactions in the trimers A, B, and C
in each of the trimers one of the three monomers (formic acid or one of the two acetylene
molecules) is subsequently removed. The energies of the remaining partial structures
(remaining dimers) were calculated (MP2/cc-pVTZ) in the geometries of the parent
trimers (Figure 4.9). These partial structures are then compared with the optimized
dimers to analyze the influence of the third molecule in the trimer on the dimer structures.
From that, a detailed picture of the non-covalent interactions in an aggregate consisting of
three components is achieved.

149
Figure 4.9. Dimers of complex A. Partial structure (i): Formic acid and Acetylene 1.
Partial structure (ii): Formic acid and Acetylene 2. Partial structure (iii): Acetylene1
and Acetylene 2.
In trimer A the first partial structure is formed by removing Acetylene 2 and thus
consists of formic acid and the remaining Acetylene1 (partial structure (i)) interacting via
noncovalent bond contribution (1). Analogously, partial structure (ii) is formed by
removing Acetylene 1 and consists of formic acid and Acetylene 2 interacting via
contribution (4). Finally, partial structure (iii) results from removing the formic acid
molecule and represents the (more or less distorted) “T” shaped acetylene dimer
interacting via contribution (3). The partial structures (i) – (iii) in the trimers B and C are
formed analogously by subsequently removing Acetylene 2, Acetylene 1, and the formic
acid molecule.
Several non-additive contributions have to be taken in to account, including basis set
superposition errors and other non-conventional interactions that may contribute to the
stabilization of the trimers (Table 4.16). These non-additive contributions are obtained by
subtracting all dimer binding energies from the trimer binding energy. As can be seen

150
from the Table 4.16 the non-additive contributions are small and never exceed 5% of the
total binding energy.
TABLE 4.16: MP2/cc-pVTZ energies of the trimers A, B, and C and their partial
structures (i), (ii), and (iii) (in kcal/mol)
MP2/cc-pVTZ
A B C FA – acetylene 2:1 Complex E(t) -9.52 -8.77 -8.42
Partial structure (i), E(i) -4.62 (48.5%) -5.18 (59.1%) -4.97 (59.0%) Partial structure (ii), E(ii) -2.67 (28.1%) -3.33 (38.0%) -1.73 (20.5%) Partial structure (iii), E(iii) -1.39 (14.6%) +0.11 (1.2%) -1.35 (16.0%) E(t) - (E(i)+ E(ii)+ E(iii)) -0.84 (8.8%) -0.37 (4.2%) -0.37 (4.4%)
In all complexes partial structure (i) contributes most to the trimer energies. The
“strongest” O-H…π interaction between the formic acid and Acetylene1 (contribution
(1)) dominates the interaction in the trimer. In trimers B and C, where partial structure (i)
contributes 59% to the total binding energy, an additional CH…O interaction between the
carbonyl oxygen atom and one of the H atoms of Acetylene 1 can be found (contribution
(4)). This increases the binding energy of this partial structure in the trimers B and C
compared to A (48.5%).
Partial structure (ii) contributes differently to each trimer: 28.1% in complex A (only
contribution 4), 38.0% in complex B and 20.5% in complex C. In B the interaction of
type (4) is most important, but in this case it is accompanied by a very weak interaction
of type (2).
In complex B a type (3) interaction between the acetylene molecules is not possible,
only a very small repulsive interaction (1.2%) is observed. This is a consequence of
repulsions between the closest hydrogen atoms in the two acetylene molecules.
For complex A it is interesting to compare the calculated binding energies and
geometries of the acetylene dimer from partial structure (iii) with the well known C2v
acetylene dimer (Table 4.17).[166] The presence of formic acid results in a small
destabilization of the partial structure (iii) compared to the non distorted acetylene dimer

151
by around 0.25 kcal/mol. For the C2v acetylene dimer the CH…C3 (and CH…C4)
distance is 2.727Å at the MP2/cc-pVTZ level of theory, while for the partial structure (iii)
the CH…C3 distance is 2.505Å and the CH…C4 distance 2.836Å.
TABLE 4.17: Calculated binding energies (in kcal/mol) of the acetylene dimer
(partial structure (iii) from complex A) and the C2v acetylene dimer
MP2/6-311++G(3df,3pd) MP2/cc-pVTZ Acetylene dimer (complex A, partial structure (iii))
-1.61 -1.39
C2v acetylene dimera -1.86 -1.63 a Reference[166]
Figure 4.10 shows the FA – acetylene dimers (left hand side) compared to several
partial structures (i) and (ii) (right hand side). A comparison between these structures
reveals interesting similarities between the partial structures and the dimers. Thus, the O-
H…π bidentate 1:1 complex is very similar to the partial structure (i), and the structures
of the CH…O=C bidentate and monodentate 1:1 complexes agree well with the partial
structures (ii) of the B and B1 trimers, respectively.

152
Figure 4.10. Comparison of 1:1 complexes of formic acid – acetylene with the partial
structures of the 1:2 complexes. Left side: 1:1 complexes (MP2/6-311++G(d,p); right
side: Partial structures (i) and (ii) from the 1:2 complex B (MP2/cc-pVTZ) and
partial structure (ii) from the 1:2 complex B1 (MP2/6-311++G(d,p)).
4.5. Conclusion
Furan is a heterocyclic system that is able to accept hydrogen bonds both in the
molecular plane at the basic oxygen atom and perpendicular to the molecular plane at its
electron rich π-system. Formic acid, on the other hand, can act as hydrogen bridge donor
either via its acidic OH group or via the less acidic CH group. In addition, it can serve as
hydrogen bridge acceptor via its two oxygen atoms. The combination of furan and formic
acid thus results in a variety of non-covalently bound dimers which were systematically
studied.
FA – acetylene is an interesting system to study non-covalent interactions, since a
variety of “non-classical” hydrogen bonds can be formed where four acidic hydrogen
atoms (OH and CH at formic acid, two CH at acetylene) compete for the two oxygen
atoms in formic acid and the acetylene π-system as hydrogen bond acceptors.
The MMH method was used to localize and characterize the FA – furan and 1:2 FA –
acetylene structures. Six 1:2 FA – acetylene complexes with binding energies between -

153
3.93 and -7.98 kcal/mol (MP2/cc-pVTZ + ZPE) are identified. Since the introduction of a
further acetylene molecule in the trimer complexes results in a number of energetically
close lying complexes, the three most strongly bound 1:2 FA – acetylene complexes are
found within a range of 1 kcal/mol. The binding interactions in these complexes are O-
H…π, C-H…π and CH…O interactions which can be classified as weak hydrogen bonds.
Nine FA – furan complexes with binding energies between -3.91 and -0.82 kcal/mol
were identified as minima at the MP2/6-311++G(d,p) + ZPE + BSSE level of theory.
Another five weaker bound complexes are minima at lower level of theory only. The FA
– furan dimers are classified into two types: type (i) with the OH hydrogen atom of FA
interacting with the furan molecule and type (ii) where the main interactions are via the
CH hydrogen atom of formic acid. Due to the lower acidity of the CH hydrogen atom,
type (ii) complexes are less stable than type (i) complexes.
The binding energy of the most stable complex A is -3.91 kcal/mol (MP2/6-
311++G(d,p) + ZPE+ BSSE). Although the OH…O interaction (1) is dominating in
dimer A, the secondary C=OFA…HF interaction (2) between the carbonyl oxygen atom of
FA and the hydrogen atom of furan leads to an additional significant stabilization of this
complex. The matrix isolation experiments reveal that dimer A is the major – if not only
– complex formed if the two monomers are allowed to diffuse slowly in solid argon. The
additional IR absorptions that appear in the matrix spectra under these conditions nicely
match the theoretical predictions for complex A.
FA – furan complexes B, C, and D1 are π complexes defined by the absence of the
strong in-plane OH…O hydrogen bond. These dimers are stabilized by the O-HFA…π
interaction (2) between the OH hydrogen atom of FA and the π system of furan. With
-2.24, -2.12 and -2.37 kcal/mol (MP2/6-311++G(d,p) + ZPE+ BSSE, Table 1) the
binding energies are considerably smaller than that of A, and consequently these
complexes are not identified experimentally.
The second group of FA – furan dimers is mainly stabilized by the very weak CH…O
or CH…π interaction. The most stable dimers in this group are H and I with binding
energies of -1.94 and -1.35 kcal/mol, respectively at the MP2/6-311++G(d,p) + ZPE+
BSSE level of theory. The higher stability of dimer H can be attributed to an additional

154
C=OFA…HF interaction between the carbonyl oxygen atom of FA and one of the
hydrogen atoms of furan. Dimer I exhibits the less stabilizing interaction HOFA…HF
between the OH oxygen atom of FA and one of the hydrogen atoms of furan. Dimers E,
F1, and G are very weakly bound CH…π complexes with binding energies between -0.96
and -0.82 kcal/mol (MP2/6-311++G(d,p) + ZPE+ BSSE).
The MP2 level of theory with the 6-311++G(d,p) and aug-cc-pVDZ basis sets provides
reliable geometries for FA – furan complexes. With the small double zeta basis sets
without augmentation the structures of five additional very weak FA – furan complexes
could be localized. Introducing the BSSE corrections during the geometry optimization at
the MP2/6-31G(d,p) level of theory leads to large changes of the calculated geometries of
some of the very weak FA – furan complexes. Here, clearly, BSSE and variations of the
basis sets have the largest effects on the dimers.
1:2 FA – acetylene complexes were analyzed at the B3LYP and MP2 levels of theory
with large basis sets. The deficit of the B3LYP method and MP2 calculations with
smaller basis sets to account for intermediate range dispersive interactions results in
producing artificial minima B1, E1, G1, and H1 which disappear at the higher level of
theory.
In 1:2 FA – acetylene complexes the interaction between the acidic H atom of formic
acid and the acetylene π-system (partial structure (i)) provides 50 – 60% of the binding
energies of the most stable complexes A – C, and thus dominates the non-covalent
interactions in these systems. Partial structures (ii) and (iii) add between 15 and 29% to
the total binding energy. Only in complex B, where no “T-acetylene” interaction is
present, contribution (iii) is slightly repulsive. Other interactions are less important for
the stabilization of the complexes, but might influence the structure. Thus, complex B is
only slightly stabilized by an additional very weak CH...π interaction compared to B1.
This interaction results in a considerable structural change. The calculated vibrational
frequencies of the most stable 1:2 complex A are in good agreement with the
experimental values, indicating that under the conditions of matrix isolation indeed the
complex A is formed.

155
5. Acetylene Complexes with Oxygen Heterocycles. An Outlook
5.1. Introduction
The interactions of the formic acid molecule with acetylene, as well as the structure of
the FA – furan dimers, were already discussed in a previous chapter. The investigation of
the interaction between acetylene and furan is the next logical step to provide a deeper
insight into the characterization of weak interacting complexes. Therefore, the
interactions of acetylene with different types of oxygen heterocycles like furan,
tetrahydrofuran (THF), and 1,4-dioxane are investigated. The study of the acetylene –
furan complexes is a very interesting and challenging task due to the competition
between very weak CH…π and CH…O interactions. The acetylene – furan dimers are
compared to the acetylene – THF and acetylene – 1,4-dioxane complexes.
“Twist” and “envelope” conformers have been identified for the THF molecule[199-201]
(Figure 5.1). The twist conformation is described by Cremer and Pople as the most stable
form with a low barrier to pseudorotation into an envelope form[199] .
Figure 5.2. Twist and envelope conformers of THF [199]
X-ray analysis indicated that THF has a twist conformation in the crystal structure,
according to Luger and Buschmann[200] (Figure 5.2). The microwave spectrum of THF

156
has been studied by Engerholm and coworkers and their results indicated that the twisted
conformation is at lower energy than the bent conformer.[201]
Figure 5.2. Twist conformer of the THF in crystal structure. Figure taken from
“Twist conformation of tetrahydrofuran in the crystal form” by P Luger and J.
Buschmann.[200]
Cadioli et al. studied one twisted (C2), one envelope (Cs), the C2v and two asymmetric
(C1) conformations of THF at the HF and MP2 levels of theory with different basis sets.
Their most reliable computations show the twist conformation being the absolute energy
minimum, the envelope structure being a transition state, only 0.3 kcal/mol higher, and
the C2v, being an energy maximum, 4.7 kcal/mol high.[202] However, in a recent ab initio
study on the pseudorotation motion in tetrahydrofuran, Rayón and Sordo extensively
explored the conformational potential energy surface of tetrahydrofuran at the MP2 level,
using double and triple ζ Dunning´s correlation consistent basis sets. They predict that the
equilibrium conformation of tetrahydrofuran is an envelope Cs structure.[203]
Alonso, Lopez et al. studied seven isotopomers of the hydrogen bonded heterodimer
THF…HF using Fourier transform microwave spectroscopy.[204] They concluded that
there is a pseudorotation of the THF subunit of the complex. The spectroscopic
parameters are interpreted in terms of a geometry in which THF has a conformation close
to the twisted ring form, with HF lying in the plane bisected by the COC ring angle.[204]

157
Ten stationary points as energy minima or transition states for the 1,4-dioxane were
characterized by Chapman and Hester at the HF and DFT levels of theory using the 6-
31G* basis set (Figure 5.3).[205] They found that the chair conformation is the lowest in
energy, followed by the two twist-boats. A half-chair structure, in which four of the ring
atoms lie in a plane, was found to be the transition state connecting the chair and the
twist-boats.
Figure 5.3. Eight conformations of the 1,4-dioxane and their calculated energies.
Figure taken from “Ab initio conformational analysis of 1,4-dioxane” by D.M
Chapman and R.E Hester.[205]

158
5.2. Computational methods
The Multiple Minima Hypersurface (MMH) approach was used for searching
configurational minima in the acetylene – furan, acetylene – THF and acetylene – 1,4-
dioxane dimers. In each case, one thousand randomly arranged structures were generated
as starting points, and the resulting geometries were optimized and analyzed using the
PM3 semiempirical quantum mechanical Hamiltonians. For the acetylene – furan dimers,
the AM1 semiempirical geometries were also analyzed. In all cases, the relevant
configurations were further refined using ab initio methods at various levels of theory.
The ab initio computations were performed using the Gaussian 98 and Gaussian 03
programs. The equilibrium geometries and vibrational frequencies were calculated with
tight convergence criteria at the SCF level including second order Møller−Plesset
perturbation theory, MP2. The force constants where calculated when necessary. Pople’s
6-31G(d,p) and 6-311++G(d,p) basis set as well as augmented and non augmented
Dunning’s correlation consistent double and triple ζ basis sets (cc-pVDZ, aug-cc-pVDZ
and cc-pVTZ) were used. Here, only the results with the 6-31G(d,p), 6-311++G(d,p) and
cc-pVTZ basis sets are discussed. Vibrational frequencies were calculated at all levels of
theory, except at the MP2/cc-pVTZ for the acetylene – THF and acetylene – 1,4-dioxane
dimers.
The stabilization energies were calculated by subtracting the energies of the monomers
from those of the complexes and ZPE corrections are included. The energies were also
corrected for the basis set superposition errors (BSSE) using the counterpoise (CP)
scheme of Boys and Bernardi.
To investigate the influence of the basis set superposition errors (BSSE) on the
geometries of the complexes, the acetylene – furan dimers were optimized at the MP2/6-
31G(d,p) level of theory using CP corrections during the optimization process. In
addition, the geometries were optimized without BSSE at the same level of theory to
compare the influence of the BSSE on the binding energies as well as on the geometries.

159
5.3. Acetylene – furan dimers. Results and discussion
After refining the MMH results, five acetylene – furan dimers A – E were localized at
the MP2/6-311++G(d,p) level of theory with binding energies between -0.75 and -1.77
kcal/mol (MP2/6-311++G(d,p) + BSSE) (Table 5.1). Here the geometries and binding
energies of the complexes are discussed at this level of theory.
TABLE 5.1: Calculated binding energies of the acetylene – furan dimers A – E at
the MP2 level of theory with the 6-311++G(d,p) and cc-pVTZ basis sets, including
BSSE and ZPE corrections (in kcal/mol)
MP2 6-311++G(d,p cc-pVTZ ΔE ΔE BSSE ΔE BSSE+ZPE ΔE ΔE BSSE ΔE BSSE+ZPE
A -3.51 -1.77 -0.55 -3.11 -2.45 -2.00
B -2.71 -1.76 -1.31 -2.60 -1.95 -1.46 B1 -a - a C -2.29 -1.08 -0.51 C1 b C1 C b -1.92 -1.45 -1.09 D -1.98 -0.83 -0.35 -1.39 -1.09 -0.83 E -1.91 -0.75 -0.28 C1 b
a Transition state b Geometry that was found after optimization
Three basic types of interactions (1) – (3) are differentiated in the acetylene – furan
complexes: (Figure 5.4)
(1) C-Hacet…OF interaction between the hydrogen atom of acetylene and the oxygen
atom of furan.
(2) C-Hacet…π interaction between the hydrogen atom of acetylene and the π system
of furan.
(3) C-HF…π interaction between the hydrogen atom of furan and the π system of
acetylene.

160
Figure 5.4. The calculated structures with hydrogen bond lengths (Å) of the
acetylene – furan complexes A – E at the (a) MP2/6-311++G(d,p) and (b) MP2/cc-
pVTZ levels of theory
Dimer A is the most stable complex (binding energy -1.77 kcal/mol (BSSE corrected,
Table 5.1)) and the only structure that was found as a minimum at all levels of theory
(including the cc-pVDZ and aug-cc-pVDZ basis set, which are not discussed here).
Dimer A is stabilized by interaction (2) between one hydrogen atom of acetylene and the
π system of furan (Figure 5.5). This hydrogen atom shows a distance of 2.725 Å to the
oxygen atom of furan (interaction (1)) (Figure 5.4). The geometry of dimer A resembles
the benzene – acetylene dimer described by Steiner el at in their crystallographic,

161
spectroscopic and quantum mechanical studies on the C-H…π hydrogen bonding in
terminal alkynes[206] (Figure 5.5). Dimer A shows an additional stabilization, not present
in the benzene – acetylene system, due to the C-H…O interaction (1). Consequently, the
acetylene molecule is not perpendicular to the aromatic ring.
Figure 5.5. Acetylene – Benzene and acetylene – furan dimers. The acetylene –
benzene figure was taken from “An Introduction to Hydrogen Bonding” by G.A Jeffrey[1]
and the original source is ref[206]. The acetylene – furan dimer parameters are referred to
the furan plane at the MP2/6-311++G(d,p) level of theory.
The binding energy of dimer B is -1.76 kcal/mol and it is basically the same that for
dimer A (at this level of theory and BSSE corrected). Dimer B has a planar Cs geometry
and is stabilized by interaction (1) with C-H…O distance 2.387 Å and interaction (3)
between the α-hydrogen atom of furan and the π system of acetylene at C-HF…Cacet 2.959
Å distance. With the cc-pVTZ basis set, including BSSE corrections, dimer B is 0.5
kcal/mol less stable than dimer A (Table 5.1). The geometry of complex B was localized
after following the imaginary vibration of dimer B1, which is a transition state with both,
the 6-311++G(d,p) and cc-pVTZ basis sets. Compared to B, dimer B1 has C2v symmetry
and is stabilized only by interaction (1) with shorter C-H…O hydrogen bond distances
and a hydrogen bond angle of 180º (Figure 5.4). At a lower level of theory, with the 6-
31G(d,p) basis set, in both, the standard and the BSSE free PES, dimer B could not be
located as a minimum. Instead, complex B1 was identified as the most stable minimum at
this level (Table 5.2).
Dimers C, D and E are stabilized only by interaction (3) between one hydrogen atom of
furan and the π system of acetylene and thus they are less stable. Dimer C (-1.08

162
kcal/mol) was located as a minimum only with the 6-311++g(d,p) basis set. The C-
HF…Cacet distance is large, even for a weak hydrogen bond (3.054 Å) and its geometry
suggests a stabilization via π…π stacking between the furan and the acetylene π systems.
With the cc-pVTZ basis set, dimer C is a transition state which leads to the geometry of
C1, where the acetylene molecule is not in a symmetric position with respect to the C1-
C2 bond of furan (Figure 5.4). The optimization of C1 with the 6-311++G(d,p) basis set
leads to the dimer C. With the 6-31G(d,p) basis set, the optimization of both C and C1
produces the dimer A. When BSSE corrections are introduced in the geometry
optimization, dimer C is a transition state and the optimization of C1 produces dimer A
(Tables 5.1 and 5.2).
TABLE 5.2: Calculated binding energies of the acetylene – furan dimers A – E at
the MP2 level of theory with the 6-31g(d,p) basis set, including (or not) BSSE
corrections during the geometry optimizations
MP2 6-31g(d,p)opt BSSE 6-31g(d,p) ΔE ΔE BSSE ΔE ΔE BSSE
A -2.73 -1.64 -2.97 -1.39
B B1b B1 b B1 -2.99 -1.76 -3.04 -1.71 C - a A b C1 A A b D -1.52 -0.81 -1.62 -0.72 E -1.35 -0.68 -1.45 -0.58
a Transition state b Geometry that was found after optimization
Dimers D and E are very weakly interacting complexes with binding energies of -0.83
and -0.75 kcal/mol, respectively. They show very similar geometries, in dimer D the C-
HF…Cace distance is 2.796 Å and in dimer E, 2.823 Å (Figure 5.4).The main difference
between these two complexes is which hydrogen atom of furan interacts with the
acetylene; in the case of dimer D, it is the α-hydrogen atom of furan; for dimer E, the β-

163
hydrogen of furan. With the cc-pVTZ basis set, the geometry optimization of dimer E
leads to the structure of dimer C1.
The similarity between the two most stable FA – furan complexes and the A and B
acetylene – furan dimers is noticeable. The FA – furan dimer A is stabilized by the
interactions of the oxygen and the α-hydrogen atoms of furan with the FA molecule,
similar to the acetylene – furan dimer B (Figures 5.4 and 5.6). In FA – furan dimer B, the
O-H hydrogen of FA interacts with the π system of furan; in acetylene – furan dimer A it
is the hydrogen atom of acetylene which interacts with the π system of furan.
Figure 5.6. FA – furan dimers A and B.
Therefore, at the same level of theory the comparison between the binding energies and
geometries in both systems provides preliminary conclusions about the relative strengths
of the interactions involved. In the FA – furan system, the O-HFA…OF and the
C=OFA…HF interactions define the dimer A as more stable compared to dimer B,
stabilized by the O-H…π interaction, whereas in the acetylene – furan system, dimer A
(C-Hacet…πF (interaction (2) and C-Hacet…OF ( interaction (1)) is slightly favored over
dimer B, stabilized by the C-Hacet…OF and C-HF…π interactions (1) and (3).
However, when ZPE corrections are also included at the MP2/6-311++G(d,p) level of
theory, the acetylene – furan dimer B is more stable than dimer A. The very low BSSE
and ZPE corrected binding energy of dimer A at this level of theory may be due to the

164
overestimation of the ZPE corrections by the model (harmonic oscillator) used, which
provides an unexpected high value of ZPE for dimer A. At this level of theory, the ZPE
calculated correction for dimer A is 1.75 kcal/mol, which represent 50% of the
uncorrected binding energy. At the MP2/cc-pVTZ level of theory the values of the
ZPE+BSSE corrected binding energies behave “normally” and dimer A is ~0.5 kcal/mol
more stable than dimer B.
The acetylene – furan dimers were optimized at the MP2/6-31G(d,p) level of theory
using CP corrections during the optimization process to investigate the influence of the
basis set superposition errors (BSSE) on the geometries of the complexes. The results
were similar to the standard optimization at the same level of theory (Tables 5.2 and 5.3).
The BSSE corrected binding energies for the BSSE geometry-corrected dimers are 0.05 –
0.25 kcal/mol larger compared to the standard values. The BSSE-corrected hydrogen
bond distances are 0.1 – 0.3 Å larger than the standard ones (Table 5.3)
TABLE 5.3: Selected intermolecular distances and angles for the acetylene – furan
dimers A, B1, D and E at the MP2 level of theory with the 6-31G(d,p) basis set,
including BSSE-CP corrections during the geometry optimizations
MP2/6-31G (d,p) optimization with BSSE optimization without BSSE A B1 D E A B1 D E C-Hacet…OF 2.981 2.358 – – 2.688 2.261 – –
C-Hacet…C1F 2.922 – – – 2.671 – – – Cacet…H-CF – – 3.008 3.073 – – 2.810 2.855 <C-Hacet…OF 134.6 180.0 – – 135.0 180.0 – –
<C-Hacet…C1F 143.5 – – – 143.9 – – – <Cacet…H-CF – – 165.5 168.6 – – 166.2 167.7
5.4. Acetylene – THF dimers. Results and discussion
Two acetylene – THF dimers were found after refining the MMH results (Figure 5.7).
In this case (and for the acetylene – 1, 4-dioxane dimers, too) only the PM3 results were

165
analyzed. With -3.66 and -3.48 kcal/mol binding energies, respectively (MP2/6-
311++G(d,p) + BSSE), acetylene – THF dimers A and B are very close in energy (Table
5.4). The THF subunit in dimer A shows the twist conformation, while in dimer B
resembles a slightly distorted “envelope”. Both dimers A and B are stabilized by the
OTHF…C-Hacet interaction with similar hydrogen bond distances and angles (Figure 5.7).
However, despite the hydrogen atoms which are closer to the acetylene molecule show C-
HTHF…Cacet distances larger than 3 Å, the position of the acetylene subunit in the
complex suggest an additional C-HTHF…π stabilizing interaction.
At the MP2/cc-pVTZ level of theory, the geometries and binding energies of dimers A
and B are very similar to the MP2/6-311++G(d,p) results. In both cases, dimer A is
slightly more stable than dimer B, which is in agreement with previous results that state
that the “twist conformation” of THF is more stable than the “envelope”.[199, 202] By our
calculations, the “envelope” monomer of THF is a transition state at both levels of theory
and is 0.17 and 0.14 kcal/mol less stable than the “twist” form. (MP2/6-311++G(d,p) and
MP2/cc-pVTZ, respectively). Therefore, it is noticeable how the formation of the
complex leads to the stabilization of the “envelope” form in the dimer, compared to the
isolated monomer. Another point to remark is that both geometries were localized after
optimization of the semiempirical geometries, which were produced without any previous
considerations of the conformation equilibrium of the THF monomer.
TABLE 5.4: Calculated binding energies of the acetylene – THF dimers A and B at
MP2 level of theory with the 6-311++G(d,p) and cc-pVTZ basis sets, including BSSE
corrections (in kcal/mol)
MP2 6-311++G(d,p cc-pVTZ ΔE ΔE BSSE ΔE ΔE BSSE
A -5.23 -3.66 -5.23 -3.92
B -5.23 -3.48 -5.14 -3.76

166
Figure 5.7. The calculated structures with hydrogen bond lengths (Å) of the
acetylene – THF complexes A and B at the (a)MP2/6-311++G(d,p) and (b)MP2/cc-
pVTZ levels of theory
TABLE 5.5: Selected intramolecular parameters, at the MP2/cc-pVTZ level of
theory, of the THF as isolated monomer and in the acetylene – THF dimers
MP2/cc-pVTZ THF “twist” THF “envelope” M Dimer A M Dimer B
Bond lengths
O-C1 1.431 1.435 1.420 1.425 C1-C2 1.522 1.519 1.533 1.526 C2-C3 1.526 1.527 1.545 1.543 Angles
<C1OC4 109.3 109.2 104.0 104.5 <C1C2C3 101.0 100.9 103.1 102.9 <C2C3C4 101.0 101.4 103.1 103.5

167
Compared to the monomer at the same level of theory, the “twist” and “envelope”
conformers of THF in the complexes show a distortion of the C2 and Cs symmetry,
respectively (Table 5.5).
For both complexes, vibrational frequencies were calculated at the MP2/6-311++G(d,p)
level of theory. The structures of the acetylene – THF dimers A and B are minima also
with others basis sets. (cc-pVDZ and aug-cc-pVDZ)
5.5. Acetylene – 1,4-dioxane dimers. Results and discussion
Two acetylene – 1,4-dioxane dimers A and B were found at the MP2/cc-pVTZ level of
theory, with BSSE corrected binding energies -3.22 and -3.06 kcal/mol, respectively.
(Figure 5.8, Table 5.6) At the MP2/6-311++G(d,p) level of theory, only the structure of
dimer A was found, since the geometry optimization of B leads to dimer A geometry.
Both dimers stabilize by the OTHF…C-Hacet interaction between the oxygen atom of 1,4-
dioxane and one hydrogen atom of acetylene. The main difference between both
complexes is the orientation of the acetylene molecule.
Figure 5.8. The calculated structures with hydrogen bond lengths (Å) of the
acetylene – 1,4-dioxane complexes A and B at the (a) MP2/6-311++G(d,p) and (b)
MP2/cc-pVTZ levels of theory

168
TABLE 5.6: Calculated binding energies of the acetylene – 1,4-dioxane dimers A
and B at MP2 level of theory with the 6-311++G(d,p) and cc-pVTZ basis sets,
including BSSE corrections (in kcal/mol)
MP2 6-311++G(d,p cc-pVTZ ΔE ΔE BSSE ΔE ΔE BSSE
A -4.54 -2.96 -4.46 -3.22
B A -4.33 -3.06
5.6. Conclusion
The geometries of the acetylene – furan, acetylene – THF and acetylene – 1,4- dioxane
dimers are calculated starting from randomly generated molecular arrangements using the
MMH procedure. Five acetylene – furan dimers with binding energies between -0.75 and
-1.77 kcal/mol are identified (MP2/6-311++G(d,p)+ BSSE). Two dimers of acetylene –
THF were found with binding energies -3.66 and -3.48 kcal/mol, (MP2/6-311++G(d,p)+
BSSE) respectively and the THF subunit in the “twist” and “envelope” conformations. At
the same level of theory, only one acetylene – 1,4-dioxane dimer (-2.96 kcal/mol) was
localized. However with other basis sets (e.g. cc-pVTZ) a second complex with a
different orientation of the acetylene molecule but very close in energy was identified.
In the acetylene – furan dimers two types of interactions are found: the C-H…O and
the C-H…π interaction. The C-H…π interaction appears in two variations, depending on
which molecule provides the hydrogen atom and which molecule the π system. The
results indicate that the C-H…π interaction between one hydrogen atom of acetylene and
the π system of furan is the stronger interaction, also compared to the in-plane C-H…O
interaction of the second more stable acetylene – furan dimer B. Acetylene – THF and
acetylene – 1,4-dioxane dimers are stabilized by the C-H…O interaction between the
oxygen atom of the heterocycle and the hydrogen atom of acetylene, however, the
position of the acetylene subunit in the complex suggests an additional C-HTHF…π
stabilizing interaction.

169
6. General Conclusion
Molecular interactions in formic acid complexes
The complexes formed by the s-trans conformer of the formic acid molecule with
formamide, dimethyl ether, furan and acetylene were studied. The variety of functional
groups shown in these molecules allows for a more detailed analysis of the
intermolecular interactions in formic acid complexes, from the moderate-“strong”
C=OFA… H-N and O-HFA…O=C interactions in FA – FMA dimers to the weak C-
HFA…π in FA – furan and 1:2 FA – acetylene complexes (Table 6.1).
The geometries of the most stable complexes in all cases show that the FA molecule
interacts strongly via its OH hydrogen atom and the carbonyl oxygen via O-HFA…O,
C=OFA…H and the O-HFA…π interaction which gets especial relevance in the
stabilization of the FA – furan and 1:2 FA – acetylene complexes. The O-HFA…O and
C=OFA…H interactions are responsible of the geometries of the most stable FA – FMA,
FA – DME and FA – furan dimers and the different stabilities of these complexes allow
to suggest a preliminary scale of strength of the interactions:
a) O-HFA…O=CFMA > O-HFA…ODME > O-HFA…OF
b) C=OFA...H-NFMA> C=OFA...HDME > C=OFA...HF
In accordance to that, the interactions of the OH hydrogen atom of FA are stronger with
a carbonyl oxygen atom than with an open-chain ether oxygen atom, and the weakest is
the O-H…OF interaction with the oxygen atom of an aromatic heterocycle like furan.
For the C=O…H interactions the most favored is the amide hydrogen atom, followed
by the methyl hydrogen of DME and the hydrogen atom of furan. Therefore, the FA
interacts weaker with the aromatic system than with the others. This can be
corroborated by an outlook into the acetylene – furan, acetylene – THF and acetylene –
1,4-dioxane dimers, which shows that the acetylene molecule also interacts more
strongly with saturated oxygen heterocycles than with the aromatic furan.

170
TABLE 6.1: Calculated binding energies of the most stable FA dimers at the MP2
level of theory with the cc-pVTZ Dunning’s basis set including BSSE corrections (in
kcal/mol).
MP2/cc-pVTZ FA – FMA ΔE ΔE (BSSE)
A -16.90 -14.21
B -13.21 -10.93
FA – DME A -12.23 -9.47
B -11.31 -9.09
FA - Furan A -7.14 -5.53
B -5.73 -4.23
The geometries of all the FA complexes show that not only the OH hydrogen atom and
the carbonyl oxygen atom of FA play an important role in the stabilization of the FA
aggregates. The aldehyde hydrogen and the OH oxygen atoms of FA contribute also to
the geometries of the FA – FMA, FA – DME, FA – furan and 1:2 FA – acetylene

171
complexes with their H-OFA…H, C-HFA…O (clearly not found in acetylene complexes)
and C-HFA…π interactions.
MMH for localizing the minima
The MMH approach, in combination with high level ab initio theory, was used to
localize and characterize the FA – FMA, FA – DME, FA – furan, 1:2 FA – acetylene,
acetylene – furan, acetylene – THF and acetylene – 1,4-dioxane structures. The
comparison with matrix spectroscopy, crystal structure data, and the analysis of studies of
similar complexes from the literature, confirms the quality of the MMH procedure as a
very useful tool for reliably localizing minima in hydrogen bonded complexes without
recurring to previous knowledge of the structure of supramolecular complexes.
One question is the reliability of using a semiempirical Hamiltonian for the preliminary
calculations of geometries, and if starting from semiempirical minima might result in
dropping some structures which are not equilibrium geometries at the semiempirical
level. The refinement requires the calculation of a large number of starting geometries
and to carefully select and analyze the semiempirical geometries. The comparison of
semiempirical an ab initio structures shows that many ab initio minima, which are not
minima at the semiempirical level, are found. In addition, in general, the amount of
minima decreases by increasing the level of theory, since many geometries, which are a
consequence of a poor optimization, converge into a limited set of final structures.
There are interesting similarities between the semiempirical and ab initio structures
(Figure 6.1). In many cases the “global” minimum with ab initio calculations was also
found at the semiempirical level as one of the most stable geometries. In the PM3
structures the artificial stabilization of the complex due to H…H interactions at ~1.7 –
1.75 Ǻ was found.[61, 62] For the FA – FMA dimers, the amide group was also optimized
to an out of plane geometry with the PM3 method. The FA – FMA PI and PII PM3
minima differ by the hydrogen bonding angles, which demonstrates the irregularities of
PM3 in reproducing the Cs symmetry of the complex. In this case, the AM1 method
shows better results, e.g. for the FA – furan complexes AM1 is able to predict the most
stable dimer as the “global” semiempirical minimum and also predicts the geometries of
others ab initio minima. But some hydrogen bond distances are much better described by

172
the PM3 hamiltonian, like in FA – FMA dimers PI and AI. The PM3 method is also able
to find as one of the more stable local minima, the structure of FA – furan dimer D1. All
of this corroborate the fact that there is not a conclusive criterion to select the
semiempirical method, but semiempirical Hamiltonians are adequate for a previous
discrimination of geometries in the MMH approach.
Figure 6.1. Selected PM3, AM1 and ab initio structures of the FA – furan, FA– FMA
and 1:2 FA – acetylene complexes.

173
BSSE effect on the geometry of the complex. Effect of the method and basis set
selection
The geometries of FA – FMA, FA – DME, FA – furan and acetylene – furan dimers
were optimized at the MP2/6-31G(d,p) level of theory using the CP scheme during the
optimization process to investigate the influence of the basis set superposition errors
(BSSE) on the geometries of the complexes. It is found that the geometries of the
“stronger” interacting complexes like FA – FMA are not as sensitive to the BSSE as the
weakly interacting complexes like the FA – furan dimers. For the FA – furan dimers the
BSSE corrected PES was closer to the PES calculated at higher level of theory than the
non-BSSE corrected, since the inclusion of the CP scheme during the optimization
process lead in many cases to geometries different to the non-BSSE ones. However, that
is critical for the weakest interacting complexes, which are also very sensitive to the basis
set used. The most stable complexes are not so affected by the inclusion of the CP
scheme during the optimization process. In general, the BSSE-free complexes show
larger hydrogen bond distances compared to the standard geometries at the same level of
theory.
Weakly interacting complexes are also more dependent on the method and basis set
used. In general, the cc-pVTZ basis set provides very good results, but with increasing
complexity of the system it becomes computationally very demanding. In this cases, the
use and comparison of the results with the aug-cc-pVDZ and the 6-311++G(d,p) basis
sets is an alternative.

174
Summary
To find out a representative and large set of the possible molecular arrangements
(minima) of hydrogen bonded and weakly interacting complexes is quite often one of the
most complicated questions. Therefore, the structural analysis application of the Multiple
Minima Hypersurface (MMH) approach[4-6] as a tool for localizing minima is introduced
here. Randomly arranged clusters are generated as starting points and subsequently
optimized. The results are processed with programs especially written for this purpose
and the geometries are afterward re-optimized at higher level of theory.
There are several steps in the MMH procedure:
• Generation of starting geometries: Random geometries are generated with the
GRANADA program,[4] especially written for this purpose. In most cases,
around 1000 randomly arranged clusters or complexes are generated as starting
points.
• Preliminary calculation of the energy: To calculate the preliminary energies of
all the generated molecular arrangements, PM3 and AM1 semiempirical
Hamiltonians are used.
• First discrimination and similarity analysis: All optimized structures which are
degenerated are discarded. Two types of degeneracy are considered. The first
consists of clusters which are identical, which means that they have both the
same energy and molecular geometry (SD). The second consists of clusters with
different molecular geometry but the same energy (VD). A subroutine called
Tanimoto is introduced in the program to analyze the similarity among
molecular arrangements, to discard the SD and keep the VD. The Tanimoto
procedure uses the Tanimoto similarity index to calculate the similarity between
structures pair by pair.
• Refinement of the geometries: The semiempirical results provide just a
preliminary overview of the interactions in the complex. For this reason, the set
of relevant semiempirical local minima are refined using DFT and MP2

175
methods. The ab initio and DFT computations are performed using the
Gaussian 98,[141] Gaussian 03,[141] and MOLPRO[142] programs. The
equilibrium geometries and vibrational frequencies are calculated using second
order Møller−Plesset perturbation theory (MP2). Pople’s 6-31G(d,p), 6-
311++G(d,p) and 6-311++G(3df,3pd) basis set as well of augmented and non
augmented Dunning’s correlation consistent double and triple ζ basis sets (cc-
pVDZ, aug-cc-pVDZ, cc-pVTZ and aug-cc-pVTZ) are used. In some cases
single point calculations are done with coupled clusters of single and double
substitutions (with non iterative triples) CCSD(T) and Dunning’s correlation
consistent triple ζ basis sets. The B3LYP density functional and Dunning’s
correlation consistent triple ζ basis sets are used also for the DFT calculations.
Formic acid – formamide (FA – FMA)
Nine FA – FMA dimers A – I with binding energies between -2.91 and -13.02 kcal/mol
(MP2/aug-cc-pVTZ + ZPE + BSSE) are identified after MMH search and refinement
with both DFT and MP2 calculations. The B3LYP density functional with the cc-pVTZ
basis set provides reliable geometries for the FA – FMA complexes. At the MP2 level of
theory, basically no change of the geometries when the basis set is augmented by adding
diffuse functions is found. At the MP2 level cc-pVDZ calculations show a tendency to
overestimate the binding energies, however, triple zeta basis sets either augmented or
non-augmented result in binding energies very similar to those from CCSD(T)/cc-pVTZ
single point calculations. The geometries and energies of the FA – FMA dimers A and B
do not change considerably with the inclusion of BSSE corrections during the
optimization process.
Seven basic types of interactions can be differentiated in the FA – FMA complexes.
They are the NHFMA…O=CFA interaction between the amide hydrogen atom of FMA and
the carbonyl oxygen atom of FA; the C=OFMA…HOFA interaction between the carbonyl
oxygen atom of FMA and the hydroxyl hydrogen atom of FA; the (O)CHFMA…O=CFA
interaction between the aldehyde hydrogen atom of FMA and the carbonyl oxygen atom
of FA; the NHFMA…(H)OCFA interaction between the amide hydrogen atom of FMA and
the hydroxyl oxygen atom of FA; the C=OFMA…HC(O)FA interaction between the

176
carbonyl oxygen atom of FMA and the aldehyde hydrogen atom of FA; the
HN(H)FMA…HOFA interaction between the nitrogen atom of FMA and the hydroxyl
hydrogen atom of FA; and the (O)CHFMA…(H)OCFA interaction between the aldehyde
hydrogen atom of FMA and the hydroxyl oxygen atom of FA.
The most stable dimers A and B are those where both carbonyl groups of FMA and FA
are involved in the stabilization of the complex, together with the hydroxyl hydrogen
atom of FA that interacts with the carbonyl oxygen atom of FMA. In the less stable
complexes F – I the hydroxyl hydrogen atoms of FA are not involved in hydrogen bonds.
Since all the geometries of the complexes were produced from randomly generated
geometries and not via chemical intuition, it is interesting to note that the structures of the
FA – FMA dimers A and B are in excellent agreement with the geometries of the FA –
FMA dimers reported in the literature. They show also interesting analogies with the
FMA – water and FMA – methanol dimers. The calculated geometries and binding
energies of 1:2 and 1:4 FA – FMA complexes show very interesting similarities with the
FA – FMA dimers and with the FA – FMA crystal structure. Of special interest are
structural motives found in the crystal structure that are already present in complexes of
very few molecules. The pair contributions of each dimer to the stabilization of the 1:2
FA – FMA trimers are also analyzed.
Formic acid – dimethyl ether (FA – DME)
Six FA – DME complexes with binding energies between -2.26 and -7.97 kcal/mol
(MP2/cc-pVTZ + ZPE+ BSSE) are identified. The two strongest bound complexes are
within a range of 0.3 kcal/mol isoenergetic. The binding in these six dimers can be
described in terms of OH…O, C=O…H, C-O…H and CH…O interactions. In the most
stable complexes A and B, the OH hydrogen atom of FA forms a strong hydrogen bond
with the ether oxygen atom of DME. Although the OH…O interaction is dominating in
complexes A and B, the secondary interaction between a methyl group hydrogen atom of
DME and the carbonyl oxygen atom of FA leads to an additional significant stabilization
of these complexes. The difference between these complexes is that in dimer A the C=O
group of FA interacts simultaneously with two hydrogen atoms of DME, while in dimer
B the C=O group of FA is approaching only one hydrogen atom of the DME. These two

177
complexes are predicted to be the most stable ones and with -7.81 to -7.97 kcal/mol
binding to be almost isoenergetic. During the optimization process the second enantiomer
of complex B was also found which shows the reliability of the MMH procedure.
The second group of FA – DME complexes C – F is defined by the absence of the
strong hydrogen bond. With -2.3 to -2.9 kcal/mol the binding energy is considerably
smaller and consequently these complexes could not be identified experimentally. The
dominant interaction in these complexes is the interaction between the aldehyde
hydrogen atom of FA and the DME oxygen atom. Again, interactions between the methyl
groups of DME and oxygen atoms of FA form secondary, weak interactions which,
however, determine the geometry of the complexes.
For all complexes the geometries are almost independent of the basis sets used. When
BSSE corrections are included in the optimization the intermolecular distances
corresponding to weaker interactions, where the DME hydrogen atoms are involved are
more influenced, showing an increasing of about 0.2-0.3 Å. Despite these variations the
basic geometries and interactions in the FA – DME complexes do not change.
Comparing the structures of the FA – DME complexes A and B with the DME –
methanol complex reported in literature reveals large similarities. There are also
interesting analogies between the FA – DME and the FA – water reported dimers. The
calculated vibrational frequencies are compared to matrix isolation IR spectra and the two
strongest bound complexes are identified.
Formic acid – furan (FA – furan)
Nine FA – furan complexes with binding energies between -3.91 and -0.82 kcal/mol
(MP2/6-311G(d,p) + ZPE+ BSSE) are identified. Another five weaker bound complexes
are localized at lower level of theory only. The binding in the furan – FA dimers can be
described in terms of OH…O, C=O…H, HO…H, CH…O, OH…π and CH…π
interactions. Therefore, the furan – FA complexes are classified in two types: (i) the
dimers where the hydroxyl group of formic acid interacts with the furan molecule and (ii)
the dimers where the main interactions of FA with the furan molecule are via the less

178
acidic C-H group. Due to the lower acidity of the CH hydrogen atom, type (ii) complexes
are less stable than type (i) complexes.
The most stable complex A has binding energy -3.91 kcal/mol (MP2/6-311++G(d,p) +
ZPE+ BSSE). Although the OH…O interaction is dominating in dimer A, the secondary
C=OFA…HF interaction between the carbonyl oxygen atom of FA and the hydrogen atom
of furan leads to an additional significant stabilization of this complex. The experimental
matrix isolation IR vibrational frequencies agree very well with the calculated IR spectra
of dimer A.
FA – furan complexes B, C, and D1 are π complexes defined by the absence of the
strong OH…O hydrogen bond. These dimers are stabilized by the O-HFA…π interaction
between the OH hydrogen atom of FA and the π system of furan. With -2.24, -2.12 and -
2.37 kcal/mol (MP2/6-311++G(d,p) + ZPE+ BSSE, Table 1) the binding energies are
considerably smaller than that of A, and consequently these complexes are not identified
experimentally.
The second group of FA – furan dimers is mainly stabilized by the very weak CH…O
or CH…π interaction. The most stable dimers in this group are H and I with binding
energies of -1.94 and -1.35 kcal/mol, respectively at the MP2/6-311++G(d,p) + ZPE+
BSSE level of theory. The higher stability of dimer H can be attributed to an additional
C=OFA…HF interaction between the carbonyl oxygen atom of FA and one of the
hydrogen atoms of furan. Dimer I exhibits the less stabilizing interaction HOFA…HF
between the OH oxygen atom of FA and one of the hydrogen atoms of furan. Dimers E,
F1, and G are very weakly bound CH…π complexes with binding energies between -0.96
and -0.82 kcal/mol (MP2/6-311++G(d,p) + ZPE+ BSSE).
The MP2 level of theory with the 6-311++G(d,p) and aug-cc-pVDZ basis sets provides
reliable geometries for FA – furan complexes. With the small double zeta basis sets
without augmentation the structures of five additional very weak FA – furan complexes
could be localized. Introducing the BSSE corrections during the geometry optimization at
the MP2/6-31G(d,p) level of theory leads to large changes of the calculated geometries of
some of the very weak FA – furan complexes. Here, clearly, BSSE and variations of the
basis sets have the largest effects on the dimers.

179
1:2 Formic acid – Acetylene Complexes (1:2 FA – acetylene)
An interesting feature of this system is the competition between the strongly acidic
carboxyl group, the acetylene group, and the formyl group as hydrogen bridge donors and
the carbonyl group, the hydroxyl group, and the acetylene π-system as hydrogen bridge
acceptors.
Six complexes with binding energies between -3.93 and -7.98 kcal/mol (MP2/cc-pVTZ
+ ZPE) are identified. Four additional structures B1, E1, G1 and H1 were located using
DFT theory (B3LYP/6-311++G(d,p)) or MP2 with the MP2/6-311++G(d,p) basis set, but
are not minima at higher levels of theory. Three stable complexes with binding energies
within 1 kcal/mol are predicted. The binding in complexes between formic acid and
acetylene shows contributions from the CH...O, OH...π and CH...π interactions.
In 1:2 FA – acetylene complexes the interaction between the acidic H atom of formic
acid and the acetylene π-system (partial structure (i)) provides 50 – 60% of the binding
energies of the most stable complexes A – C, and thus dominates the non-covalent
interactions in these systems. Partial structures (ii) and (iii) add between 15 and 29% to
the total binding energy. Only in complex B, where no “T-acetylene” interaction is
present, contribution (iii) is slightly repulsive. Other interactions are less important for
the stabilization of the complexes, but might influence the structure. Thus, complex B is
only slightly stabilized by an additional very weak CH...π interaction compared to B1.
This interaction results in a considerable structural change. The calculated vibrational
frequencies of the most stable 1:2 complex A are in good agreement with the
experimental values, indicating that under the conditions of matrix isolation indeed the
complex A is formed.
Acetylene complexes with oxygen heterocycles
Five acetylene – furan dimers with binding energies between -0.75 and -1.77 kcal/mol
are identified (MP2/6-311++G(d,p)+ BSSE). Two dimers of acetylene – THF were found
with binding energies -3.66 and -3.48 kcal/mol, (MP2/6-311++G(d,p)+ BSSE)
respectively and the THF subunit in the “twist” and “envelope” conformations. At the
same level of theory, only one acetylene – 1,4-dioxane dimer (-2.96 kcal/mol) was

180
localized. However, with other basis sets (e.g. cc-pVTZ) a second complex with a
different orientation of the acetylene molecule but very close in energy was identified.
In the acetylene – furan dimers two types of interactions are found: the C-H…O and
the C-H…π interaction. The C-H…π interaction appears in two variations, depending on
which molecule provides the hydrogen atom and which molecule the π system. The
results indicate that the C-H…π interaction between one hydrogen atom of acetylene and
the π system of furan is the stronger interaction, also compared to the in-plane C-H…O
interaction found in the second more stable acetylene – furan dimer B. Acetylene – THF
and acetylene – 1,4-dioxane dimers are stabilized by the C-H…O interaction between the
oxygen atom of the heterocycle and the hydrogen atom of acetylene, however, the
position of the acetylene subunit in the complex suggests an additional very weak C-
HTHF(DIOXANE)…π stabilizing interaction.
For the different systems studied, the comparison with matrix spectroscopy, crystal
structure data, and the analysis of studies of similar complexes from the literature,
confirms the quality of the MMH procedure as a very useful tool for reliably localizing
minima in hydrogen bonded complexes without recurring to previous knowledge of the
structure of supramolecular complexes.

181
References
[1] G. A. Jeffrey, An Introduction to Hydrogen Bonding, New York, 1997. [2] G. A. Jeffrey, Journal of Molecular Structure 1994, 322, 21. [3] M. Nishio, M. Hirota, Y. Umezawa, CH/p Interaction: Evidence, Nature, and
Consequences, 1998. [4] L. A. Montero, GRANADA and Q programs for PC computers ed., 1996. [5] L. A. Montero, A. M. Esteva, J. Molina, A. Zapardiel, L. Hernandez, H. Marquez,
A. Acosta, Journal of the American Chemical Society 1998, 120, 12023. [6] L. A. Montero, J. Molina, J. Fabian, International Journal of Quantum Chemistry
2000, 79, 8. [7] M. Gantenberg, M. Halupka, W. Sander, Chemistry--A European Journal 2000, 6,
1865. [8] A. K. Roy, A. J. Thakkar, in Chemical Physics, Vol. 312, 2005, pp. 119. [9] W. H. Hocking, Zeitschrift fuer Naturforschung, Teil A: Astrophysik, Physik und
Physikalische Chemie 1976, 31A, 1113. [10] J. Karle, L. O. Brockway, Journal of the American Chemical Society 1944, 66,
574. [11] R. C. Millikan, K. S. Pitzer, Journal of Chemical Physics 1957, 27, 1305. [12] E. Bjarnov, W. H. Hocking, Zeitschrift fuer Naturforschung, Teil A: Astrophysik,
Physik und Physikalische Chemie 1978, 33A, 610. [13] M. Pettersson, J. Lundell, L. Khriachtchev, M. Raesaenen, J. Am. Chem. Soc.
1997, 119, 11715. [14] M. D. Taylor, J. Bruton, Journal of the American Chemical Society 1952, 74,
4151. [15] I. Nahringbauer, Acta Crystallographica, Section B: Structural Crystallography
and Crystal Chemistry 1978, B34, 315. [16] I. Bako, G. Schubert, T. Megyes, G. Palinkas, G. I. Swan, J. Dore, M.-C.
Bellisent-Funel, Chemical Physics 2004, 306, 241. [17] E. Spinner, Spectrochimica Acta, Part A: Molecular and Biomolecular
Spectroscopy 1999, 55A, 1819. [18] D. Chapman, Journal of the Chemical Society 1956, 225. [19] S. Scheiner, C. W. Kern, in Journal of the American Chemical Society, Vol. 101,
1979, pp. 4081. [20] Y. J. Zheng, K. M. Merz, Jr., Journal of Computational Chemistry 1992, 13,
1151. [21] I. Rozas, I. Alkorta, J. Elguero, Journal of Physical Chemistry B 2004, 108, 3335. [22] J. W. Keller, in Journal of Physical Chemistry A, Vol. 108, 2004, pp. 4610. [23] P. Pfeiffer, G. Birencweig, A. Hofmann, C. Windheuser, Ber. 1914, 47, 1580. [24] A. Werner, Liebigs Annalen der Chemie 1902, 261. [25] T. S. Moore, T. F. Winmill, Journal of the Chemical Society, Transactions 1912,
101, 1635. [26] W. M. Latimer, W. H. Rodebush, Journal of the American Chemical Society
1920, 42, 1419. [27] L. Pauling, The Nature of the Chemical Bond., Cornell University Press, Ithaca,
NY, 1939.

182
[28] G. C. Pimentel, A. L. McClellan, The Hydrogen Bond, Freeman, San Francisco, 1960.
[29] G. R. Desiraju, T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, 1999.
[30] L. Hunter, Ann. Repts. Progress Chem. (Chem. Soc. London) 1947, 43, 141. [31] G. R. Desiraju, Accounts of Chemical Research 2002, 35, 565. [32] T. W. Panunto, Z. Urbanczyk-Lipkowska, R. Johnson, M. C. Etter, Journal of the
American Chemical Society 1987, 109, 7786. [33] W. Saenger, T. Steiner, Conference Proceedings - Italian Physical Society 1993,
43, 55. [34] W. Saenger, Nature (London, United Kingdom) 1979, 279, 343. [35] G. Gilli, F. Bellucci, V. Ferretti, V. Bertolasi, Journal of the American Chemical
Society 1989, 111, 1023. [36] J. Del Bene, J. A. Pople, Journal of Chemical Physics 1970, 52, 4858. [37] J. L. Koenig, Accounts of Chemical Research 1981, 14, 171. [38] E. Whittle, D. A. Dows, G. C. Pimentel, J. Chem. Phys. 1954, 22, 1943. [39] I. Norman, G. Porter, Nature 1954, 174, 508. [40] http://www.orch.ruhr-uni-bochum.de/sander/. [41] M. Squillacote, R. S. Sheridan, O. L. Chapman, F. A. L. Anet, J. Am. Chem. Soc.
1975, 97, 3244. [42] M. J. Almond, K. S. Wiltshire, Annual Reports on the Progress of Chemistry,
Section C: Physical Chemistry 2001, 97, 3. [43] S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University
Press, New York, 1997. [44] C. J. Cramer, Essentials of Computational Chemistry : Theories and Models, John
Wiley and Sons, Ltd, 2004. [45] J. M. Foster, S. F. Boys, Reviews of Modern Physics 1960, 32, 300. [46] J. A. Pople, M. Head-Gordon, K. Raghavachari, Journal of Chemical Physics
1987, 87, 5968. [47] R. Ahlrichs, Computer Physics Communications 1979, 17, 31. [48] P. Carsky, I. Hubac, V. Staemmler, Theoretica Chimica Acta 1982, 60, 445. [49] J. Cizek, J. Paldus, Physica Scripta 1980, 21, 251. [50] J. A. Pople, R. Krishnan, H. B. Schlegel, J. S. Binkley, International Journal of
Quantum Chemistry 1978, 14, 545. [51] G. D. Purvis, III, R. J. Bartlett, Journal of Chemical Physics 1982, 76, 1910. [52] J. S. Binkley, J. A. Pople, International Journal of Quantum Chemistry 1975, 9,
229. [53] J. A. Pople, J. S. Binkley, R. Seeger, International Journal of Quantum
Chemistry, Symposium 1976, 10, 1. [54] A. C. Whal, G. Das, in Methods of Electronic Structure Theory, Vol. 3 (Ed.: H. F.
Schaefer), Plenum, New York, 1977. [55] B. O. Roos, P. R. Taylor, E. M. Siegbahn, Chemical Physics 1980, 48, 157. [56] F. Jensen, Introduction to Computational Chemistry, John Wiley and Sons, 1999. [57] W. Thiel, in Modern Methods and Algorithms of Quantum Chemistry (Ed.: J.
Grotendorst), John von Neumann Institute for Computing, Jülich, 2000, pp. 233.

183
[58] D. Hadzi, G. Koller, in Theoretical Treatments of Hydrogen Bonding (Ed.: D. Hadzi), John Wiley and Sons, 1997.
[59] M. J. S. Dewar, E. G. Zoebisch, E. F. Healy, J. J. P. Stewart, Journal of the American Chemical Society 1985, 107, 3902.
[60] J. J. P. Stewart, Journal of Computational Chemistry 1989, 10, 221. [61] G. I. Csonka, Journal of Computational Chemistry 1993, 14, 895. [62] G. I. Csonka, J. G. Angyan, Theochem 1997, 393, 31. [63] M. J. S. Dewar, C. Jie, J. Yu, Tetrahedron 1993, 49, 5003. [64] L. Turi, J. J. Dannenberg, Journal of Physical Chemistry 1993, 97, 12197. [65] L. Turi, J. J. Dannenberg, Journal of Physical Chemistry 1993, 97, 7899. [66] P. Hohenberg, W. Kohn, Phys. Rev. 1964, 136, B864. [67] W. Kohn, L. J. Sham, Phys. Rev. 1965, 140, A1133. [68] J. B. Foresman, A. Frisch, Exploring Chemistry with Electronic Structure
Methods, Second ed., Gaussian, Inc., Pittsburgh, 1996. [69] S. J. Vosko, L. Wilk, M. Nusair, Can. J. Phys 1980, 58, 1200. [70] H. Guo, S. Sirois, E. I. Proynov, D. R. Salahub, in Theoretical Treatments of
Hydrogen Bonding (Ed.: D. Hadzi), John Wiley and Sons, 1997. [71] A. D. Becke, Phys. Rev. A: Gen. Phys. 1988, 38, 3098. [72] J. P. Perdew, Y. Wang, Phys. Rev. 1986, 8800. [73] C. Lee, W. Yang, R. G. Parr, Physical Review B: Condensed Matter and
Materials Physics 1988, 37, 785. [74] A. D. Becke, Journal of Chemical Physics 1993, 98, 5648. [75] Y. Zhao, O. Tishchenko, D. G. Truhlar, Journal of Physical Chemistry B 2005,
109, 19046. [76] Y. Zhao, D. G. Truhlar, Physical Chemistry Chemical Physics 2005, 7, 2701. [77] E. R. Davidson, D. Feller, Chemical Reviews (Washington, DC, United States)
1986, 86, 681. [78] T. H. Dunning, P. J. Hay, in Methods of Electronic Structure Theory, Vol. 3 (Ed.:
H. F. Schaeffer), Plenum, New York, 1977. [79] W. J. Hehre, R. F. Stewart, J. A. Pople, Journal of Chemical Physics 1969, 51,
2657. [80] R. Ditchfield, W. J. Hehre, J. A. Pople, Journal of Chemical Physics 1971, 54,
724. [81] R. Krishnan, J. S. Binkley, R. Seeger, J. A. Pople, in Journal of Chemical
Physics, Vol. 72, 1980, pp. 650. [82] T. H. Dunning, Jr., Journal of Chemical Physics 1989, 90, 1007. [83] S. F. Boys, F. Bernardi, Mol Phys 1970, 19, 553. [84] S. Simon, J. Bertran, M. Sodupe, Journal of Physical Chemistry A 2001, 105,
4359. [85] P. Salvador, S. Simon, M. Duran, J. J. Dannenberg, Journal of Chemical Physics
2000, 113, 5666. [86] P. Salvador, B. Paizs, M. Duran, S. Suhai, Journal of Computational Chemistry
2001, 22, 765. [87] P. Salvador, M. M. Szczesniak, Journal of Chemical Physics 2003, 118, 537. [88] M. C. Daza, J. A. Dobado, J. M. Molina, P. Salvador, M. Duran, J. L. Villaveces,
Journal of Chemical Physics 1999, 110, 11806.

184
[89] S. Simon, M. Duran, J. J. Dannenberg, Journal of Chemical Physics 1996, 105, 11024.
[90] R. Crespo-Otero, L. A. Montero, W.-D. Stohrer, J. M. Garcia de la Vega, Journal of Chemical Physics 2005, 123, 134107/1.
[91] P. Hobza, Z. Havlas, Theoretical Chemistry Accounts 1998, 99, 372. [92] I. Mayer, International Journal of Quantum Chemistry 1983, 23, 341. [93] A. Bende, A. Vibok, G. J. Halasz, S. Suhai, International Journal of Quantum
Chemistry 2001, 84, 617. [94] P. Salvador, M. Duran, X. Fradera, in Journal of Chemical Physics, Vol. 116,
2002, pp. 6443. [95] I. Mayer, A. Vibok, G. Halasz, P. Valiron, International Journal of Quantum
Chemistry 1996, 57, 1049. [96] I. Mayer, P. R. Surjan, International Journal of Quantum Chemistry 1989, 36,
225. [97] I. Mayer, A. Vibok, Chemical Physics Letters 1987, 140, 558. [98] I. Mayer, A. Vibok, Molecular Physics 1997, 92, 503. [99] A. Hamza, A. Vibok, G. J. Halasz, I. Mayer, Theochem 2000, 501-502, 427. [100] L. Turi, J. J. Dannenberg, Journal of Physical Chemistry 1993, 97, 2488. [101] P. Valiron, I. Mayer, Chemical Physics Letters 1997, 275, 46. [102] B. H. Wells, S. Wilson, Chemical Physics Letters 1983, 101, 429. [103] P. Willett, J. M. Barnard, G. M. Downs, Journal of Chemical Information and
Computer Sciences 1998, 38, 983. [104] W. Fisanick, A. H. Lipkus, A. Rusinko, III, Journal of Chemical Information and
Computer Sciences 1994, 34, 130. [105] D. Butina, Journal of Chemical Information and Computer Sciences 1999, 39,
747. [106] E. S. Garcia, L. A. Montero, J. M. Hermida, R. Cruz, G. Gonzalez, Revista
Cubana de Fisica 2000, 17, 41. [107] J. J. P. Stewart, Journal of Computational Chemistry 1989, 10, 209. [108] J. J. P. Stewart, MOPAC v 6.0 ed. [109] J. J. P. Stewart, Journal of Computer-Aided Molecular Design 1990, 4, 1. [110] J. M. Stewart, Frank J. Seiler Res. Lab.,United States Air Force Acad.,Colorado
Springs,CO,USA., 1988, p. 183 pp. [111] P. Culot, G. Dive, V. H. Nguyen, J. M. Ghuysen, Theoretica Chimica Acta 1992,
82, 189. [112] J. Baker, Journal of Computational Chemistry 1986, 7, 385. [113] P. O. Astrand, G. Karlstrom, A. Engdahl, B. Nelander, Journal of Chemical
Physics 1995, 102, 3534. [114] B. Chan, J. E. Del Bene, J. Elguero, L. Radom, Journal of Physical Chemistry A
2005, 109, 5509. [115] O. Galvez, P. C. Gomez, L. F. Pacios, Journal of Chemical Physics 2003, 118,
4878. [116] L. George, E. Sanchez-Garcia, W. Sander, Journal of Physical Chemistry A 2003,
107, 6850. [117] L. George, W. Sander, Spectrochimica Acta, Part A: Molecular and Biomolecular
Spectroscopy 2004, 60A, 3225.

185
[118] I. Agranat, N. V. Riggs, L. Radom, in Journal of the Chemical Society, Chemical Communications, 1991, pp. 80.
[119] P. R. L. Markwick, N. L. Doltsinis, D. Marx, Journal of Chemical Physics 2005, 122, 054112/1.
[120] C. S. Tautermann, M. J. Loferer, A. F. Voegele, K. R. Liedl, Journal of Chemical Physics 2004, 120, 11650.
[121] N. R. Brinkmann, G. S. Tschumper, G. Yan, H. F. Schaefer, Journal of Physical Chemistry A 2003, 107, 10208.
[122] F. Madeja, M. Havenith, Journal of Chemical Physics 2002, 117, 7162. [123] H. Ushiyama, K. Takatsuka, in Journal of Chemical Physics, Vol. 115, 2001, pp.
5903. [124] J. Kohanoff, S. Koval, D. A. Estrin, D. Laria, Y. Abashkin, in Journal of
Chemical Physics, Vol. 112, 2000, pp. 9498. [125] S. J. Grabowski, T. M. Krygowski, in Chemical Physics Letters, Vol. 305, 1999,
pp. 247. [126] T. Loerting, K. R. Liedl, Journal of the American Chemical Society 1998, 120,
12595. [127] Y. Kim, Journal of the American Chemical Society 1996, 118, 1522. [128] C. Zhu, Y. Ling, W. Y. Feng, C. Lifshitz, International Journal of Mass
Spectrometry 2000, 194, 93. [129] G. P. Ayers, A. D. E. Pullin, Spectrochimica Acta, Part A: Molecular and
Biomolecular Spectroscopy 1976, 32A, 1641. [130] M. L. H. Jeng, B. S. Ault, Journal of Molecular Structure 1991, 246, 33. [131] A. Engdahl, B. Nelander, J. Chem. Soc., Faraday Trans. 1992, 88, 177. [132] B. C. Bricknell, T. M. Letcher, T. A. Ford, South African Journal of Chemistry
1995, 48, 142. [133] S. W. Han, K. Kim, Journal of Molecular Structure 1999, 475, 43. [134] J. Goebel, B. S. Ault, J. E. D. Bene, Journal of Physical Chemistry A 2000, 104,
2033. [135] A. Loutellier, L. Schriver, A. Burneau, J. P. Perchard, Journal of Molecular
Structure 1982, 82, 165. [136] A. Fu, D. Du, Z. Zhou, Theochem 2003, 623, 315. [137] A. Fu, D. Du, Z. Zhou, International Journal of Quantum Chemistry 2004, 97,
865. [138] E. L. Coitino, K. Irving, J. Rama, A. Iglesias, M. Paulino, O. N. Ventura,
Theochem 1990, 69, 405. [139] I. Nahringbauer, G. Larsson, Arkiv foer Kemi 1968, 30, 91. [140] T. Neuheuser, B. A. Hess, C. Reutel, E. Weber, Journal of Physical Chemistry
1994, 98, 6459. [141] G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, , J. R. C. M. A. Robb, J. A.
Montgomery, Jr., T. Vreven, , J. C. B. K. N. Kudin, J. M. Millam, S. S. Iyengar, J. Tomasi, , B. M. V. Barone, M. Cossi, G. Scalmani, N. Rega, , H. N. G. A. Petersson, M. Hada, M. Ehara, K. Toyota, , J. H. R. Fukuda, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, , M. K. H. Nakai, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, , J. J. C. Adamo, R. Gomperts, R. E. Stratmann, O. Yazyev, , R. C. A. J. Austin, C. Pomelli, J. W. Ochterski, P. Y. Ayala, , G. A. V.

186
K. Morokuma, P. Salvador, J. J. Dannenberg, , S. D. V. G. Zakrzewski, A. D. Daniels, M. C. Strain, , D. K. M. O. Farkas, A. D. Rabuck, K. Raghavachari, , J. V. O. J. B. Foresman, Q. Cui, A. G. Baboul, S. Clifford, , B. B. S. J. Cioslowski, G. Liu, A. Liashenko, P. Piskorz, , R. L. M. I. Komaromi, D. J. Fox, T. Keith, M. A. Al-Laham, , A. N. C. Y. Peng, M. Challacombe, P. M. W. Gill, , W. C. B. Johnson, M. W. Wong, C. Gonzalez, J. A. Pople., Gaussian 03, Revision B.03, Pittsburgh PA, 2003.
[142] H.-J. K. Werner, P. J.; Schu¨tz, M.; Lindh, R.; Celani, P.;, T. R. Korona, G.; Manby, F. R.; Amos, R. D.; Bernhardsson, A.;, A. C. Berning, D. L.; Deegan, M. J. O.; Dobbyn, A. J.; Eckert, F.;, C. H. Hampel, G.; Lloyd, A. W.; McNicholas, S. J.; Meyer, W.; Mura,, A. P. M. E.; Nicklass, P.; Pitzer, R.; Schumann, U.; Stoll, H.; Stone,, R. T. A. J.; Tarroni, T. MOLPRO; Universita¨t Stuttgart:, G. Stuttgart.
[143] P. G. Jansien, Stevens, WalterJ, J.Chem. Phys 1986, 84, 3271. [144] L. F. Pacios, Journal of Physical Chemistry A 2004, 108, 1177. [145] E. Sanchez-Garcia, L. George, L. A. Montero, W. Sander, Journal of Physical
Chemistry A 2004, 108, 11846. [146] E. Sanchez-Garcia, M. Studentkowski, L. A. Montero, W. Sander,
ChemPhysChem 2005, 6, 618. [147] D. Priem, T.-K. Ha, A. Bauder, Journal of Chemical Physics 2000, 113, 169. [148] Z. Zhou, Y. Shi, X. Zhou, Journal of Physical Chemistry A 2004, 108, 813. [149] M. D. Harmony, V. W. Laurie, R. L. Kuczkowski, R. H. Schwendeman, D. A.
Ramsay, F. J. Lovas, W. J. Lafferty, A. G. Maki, Journal of Physical and Chemical Reference Data 1979, 8, 619.
[150] A. R. Katritzky, J. M. Lagowski, Heterocyclic Chemistry, 1960. [151] M. Hartmann, S. D. Wetmore, L. Radom, Journal of Physical Chemistry A 2001,
105, 4470. [152] P. Hobza, C. Riehn, A. Weichert, B. Brutschy, Chemical Physics 2002, 283, 331. [153] M. L. H. Jeng, B. S. Ault, Journal of Physical Chemistry 1990, 94, 4851. [154] M. L. H. Jeng, B. S. Ault, Journal of Physical Chemistry 1990, 94, 1323. [155] S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami, K. Tanabe, Journal of Physical
Chemistry A 1999, 103, 8265. [156] P. Hobza, H. L. Selzle, E. W. Schlag, Collection of Czechoslovak Chemical
Communications 1992, 57, 1186. [157] H. Petrusova, Z. Havlas, P. Hobza, R. Zahradnik, Collection of Czechoslovak
Chemical Communications 1988, 53, 2495. [158] G. Maier, C. Lautz, European Journal of Organic Chemistry 1998, 769. [159] V. Lukes, M. Breza, Petroleum and Coal 2002, 44, 51. [160] J. S. Muenter, Journal of Chemical Physics 1991, 94, 2781. [161] J. S. Craw, M. A. C. Nascimento, M. N. Ramos, Spectrochimica Acta, Part A:
Molecular and Biomolecular Spectroscopy 1991, 47A, 69. [162] S. Scheiner, S. J. Grabowski, in Journal of Molecular Structure, Vol. 615, 2002,
pp. 209. [163] S. M. Resende, W. B. De Almeida, Chemical Physics 1996, 206, 1. [164] C. E. Dykstra, Journal of the American Chemical Society 1990, 112, 7540. [165] K. Shuler, C. E. Dykstra, Journal of Physical Chemistry A 2000, 104, 11522. [166] A. Karpfen, Journal of Physical Chemistry A 1999, 103, 11431.

187
[167] D. G. Prichard, R. N. Nandi, J. S. Muenter, Journal of Chemical Physics 1988, 89, 115.
[168] J. S. Craw, W. B. De Almeida, A. Hinchliffe, Theochem 1989, 60, 69. [169] T. Aoyama, O. Matsuoka, N. Nakagawa, Chemical Physics Letters 1979, 67, 508. [170] I. L. Alberts, T. W. Rowlands, N. C. Handy, Journal of Chemical Physics 1988,
88, 3811. [171] K. Pei, H. Li, in Journal of Molecular Structure, Vol. 693, 2004, pp. 141. [172] A. C. Legon, P. Ottaviani, Physical Chemistry Chemical Physics 2004, 6, 488. [173] H. Koppel, E. V. Gromov, A. B. Trofimov, Chemical Physics 2004, 304, 35. [174] D.-M. Huang, Y.-B. Wang, L. M. Visco, F.-M. Tao, in Journal of Physical
Chemistry A, Vol. 108, 2004, pp. 11375. [175] E. V. Gromov, A. B. Trofimov, N. M. Vitkovskaya, H. Koppel, J. Schirmer, H. D.
Meyer, L. S. Cederbaum, Journal of Chemical Physics 2004, 121, 4585. [176] E. V. Gromov, A. B. Trofimov, N. M. Vitkovskaya, J. Schirmer, H. Koppel,
Journal of Chemical Physics 2003, 119, 737. [177] G. C. Cole, A. C. Legon, P. Ottaviani, Journal of Chemical Physics 2002, 117,
2790. [178] S. A. Cooke, G. K. Corlett, A. C. Legon, Chemical Physics Letters 1998, 291,
269. [179] S. A. Cooke, G. K. Corlett, J. H. Holloway, A. C. Legon, Journal of the Chemical
Society, Faraday Transactions 1998, 94, 2675. [180] L. A. Montero, R. Gonzalez-Jonte, L. A. Diaz, J. R. Alvarez-Idaboy, Journal of
Physical Chemistry 1994, 98, 5607. [181] A. C. Legon, Faraday Discussions 1994, 97, 19. [182] J. R. Alvarez-Idaboy, L. A. Montero, Theochem 1992, 85, 243. [183] J. R. Alvarez, L. Montero, R. Martinez, Makromolekulare Chemie 1990, 191,
281. [184] L. Radom, R. H. Nobes, D. J. Underwood, W. K. Li, Pure Appl. Chem. 1986, 58,
75. [185] B. S. Ault, in Journal of Molecular Structure, Vol. 127, 1985, pp. 343. [186] Z. Latajka, H. Ratajczak, W. J. Orville-Thomas, E. Ratajczak, J. Mol. Struct.
(THEOCHEM) 1981, 85, 303. [187] A. M. DeLaat, B. S. Ault, Journal of the American Chemical Society 1987, 109,
4232. [188] B. W. Hopkins, G. S. Tschumper, in Journal of Physical Chemistry A, Vol. 108,
2004, pp. 2941. [189] A. C. Legon, D. J. Millen, Faraday Discussions of the Chemical Society 1982, 73,
71. [190] A. C. Legon, D. J. Millen, Chemical Society Reviews 1987, 16, 467. [191] B. Bak, L. Hansen, J. Rastrup-Andersen, Discussions of the Faraday Society
1955, No. 19, 30. [192] E. Sanchez-Garcia, L. A. Montero, W. Sander, Journal of Physical Chemistry A,
in press. [193] A. Baldacci, S. Ghersetti, S. C. Hurlock, K. N. Rao, Journal of Molecular
Spectroscopy 1976, 59, 116.

188
[194] E. S. Kline, Z. H. Kafafi, R. H. Hauge, J. L. Margrave, Journal of the American Chemical Society 1985, 107, 7559.
[195] S. A. McDonald, G. L. Johnson, B. W. Keelan, L. Andrews, Journal of the American Chemical Society 1980, 102, 2892.
[196] E. D. Jemmis, K. T. Giju, K. Sundararajan, K. Sankaran, V. Vidya, K. S. Viswanathan, J. Leszczynski, Journal of Molecular Structure 1999, 510, 59.
[197] M. Halupka, W. Sander, in Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy, Vol. 54A, 1998, pp. 495.
[198] I. D. Reva, A. M. Plokhotnichenko, E. D. Radchenko, G. G. Sheina, Y. P. Blagoi, Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy 1994, 50A, 1107.
[199] D. Cremer, J. A. Pople, Journal of the American Chemical Society 1975, 97, 1358.
[200] P. Luger, J. Buschmann, Angewandte Chemie 1983, 95, 423. [201] G. G. Engerholm, A. C. Luntz, W. D. Gwinn, D. O. Harris, Journal of Chemical
Physics 1969, 50, 2446. [202] B. Cadioli, E. Gallinella, C. Coulombeau, H. Jobic, G. Berthier, Journal of
Physical Chemistry 1993, 97, 7844. [203] M. Rayon Victor, A. Sordo Jose, The Journal of chemical physics 2005, 122,
204303. [204] J. L. Alonso, J. C. Lopez, S. Blanco, A. Lesarri, F. J. Lorenzo, Journal of
Chemical Physics 2000, 113, 2760. [205] D. M. Chapman, R. E. Hester, Journal of Physical Chemistry A 1997, 101, 3382. [206] T. Steiner, E. B. Starikov, A. M. Amado, J. J. C. Teixeira-Dias, Journal of the
Chemical Society, Perkin Transactions 2: Physical Organic Chemistry 1995, 1321.

189
Acknowledgments
I am very much indebted to Prof Dr. Luis A. Montero for his support and guidance
over all these years and to Prof. Dr. Martina Havenith-Newen for her assistance and
encouragement.
Many thanks to Dr. Holger Bettinger for the help and advices in Computational
Chemistry and to Dr.Lisa George, Arthur Mardyukov and Marc Studentkowski, who did
the matrix isolation experiments. I also wish to thank Herr Torsten Haenschke for
keeping the computers running and his support. Thanks to Dr. Friedrich Scheidt and Frau
Ulrike Steger for their always kind attention, and to all the members of the Organic
Chemistry II research group for the nice working atmosphere and their support. Many
thanks to Dr. Thomas Koch and Frau Gundula Talbot of the Graduate School of
Chemistry and Biochemistry. I thank Prof. Dr Silvia Bravslasky and the members of the
Bravslasky group in the Max Planck Institute of Bioinorganic Chemistry.
I am very grateful to Rachel Crespo of the Computational Chemistry Laboratory in
Havana, for her friendship and invaluable unconditional support since many years. I
specially thank my friends Dr. Victor Martinez and Denise Larrieux, for their total
support. Thanks Loli and Vir for your help.

190
Curriculum Vitae
Name Elsa Sánchez García
Born 15th October 1976, Havana, Cuba
Nationality Cuban, with residence in Germany
Education Since Sept. 2002 Postgraduate studies of Chemistry, University of Bochum, Germany
1999-2002 Postgraduate studies of Chemistry at the Laboratory of Theoretical
and Computational Chemistry, Havana University
1999-2001 Study of English Language, Abraham Lincoln School, Havana
1999 Diploma for Chemistry at the University of Havana. Title: "Efectos
del ambiente molecular sobre procesos y estructuras moleculares en
solventes puros"
1994- 1999 Study of Chemistry at the University of Havana
1991-1994 Pre-University training in Havana
1988-1991 Secondary School in Havana
1981-1988 Primary School in Havana
Professional Career September 2002 - till present
Research Scientist at the Ruhr University of Bochum, Germany, with
Prof. Dr. W. Sander
April 2002 - July 2002
Research Scientist, Max Planck Institute für Strahlenchemie (now
Max Planck Institute für Bioanorganische Chemie), Mülheim an der
Ruhr, Germany, with Prof Dr. S. Bravslasky
1999-2002 Novel Lecturer, Faculty of Chemistry, Havana University

191
Awards 2002 Award of a Scholarship of the DAAD (German Academic Exchange
Service).
2002 Best Novel Lecturer (Reserva Científica), Department of General
Chemistry, Faculty of Chemistry, University of Havana
1999-2000 Award in the category of "Best Novel Lecturer (Reserva Científica)".
Faculty of Chemistry, University of Havana
1998-1999 Best graduated student of the University of Havana.
1999 Suma cum Laude Diploma.
1998-1999 Best graduated student of the Chemistry Faculty
1998 Award in the Student’s Scientific Meeting. Commission of Physical
Chemistry, Faculty of Chemistry, University of Havana
1998 Award in the Student’s Scientific Meeting. Commission of
Computational Sciences, University of Havana
1998 Outstanding student of the Havana University
1993-1994 Bronze Medal in the National Competition of Chemistry for Pre-
Universitary students, Cuba
1992-1993 Gold Medal in the National Competition of Spanish Language and
Literature for Pre-Universitary students.
1992-1993 Bronze Medal in the National Competition of Chemistry for Pre-
Universitary students, Cuba
1991-1992 Gold Medal in the National Competition of Chemistry for Pre-
Universitary students, Cuba