completed genomes: viruses and bacteria monday, october 20, 2003 introduction to bioinformatics...
Post on 21-Dec-2015
217 views
TRANSCRIPT

Completed Genomes:Viruses and Bacteria
Monday, October 20, 2003
Introduction to BioinformaticsME:440.714J. Pevsner

Many of the images in this powerpoint presentationare from Bioinformatics and Functional Genomicsby J Pevsner (ISBN 0-471-21004-8). Copyright © 2003 by Wiley.
These images and materials may not be usedwithout permission from the publisher.
Visit http://www.bioinfbook.org
Copyright notice

We are now beginning the last third of the course:
Today: completed genomes (Chapters 12-14)Wednesday: Fungi. Exam #2 is due at the start of class.
Next Monday: Functional genomics (Jef Boeke)Next Wednesday: Pathways (Joel Bader)
Monday Nov. 3: Eukaryotic genomesWednesday Nov. 5: Human genome
Monday Nov. 10: Human diseaseWednesday Nov. 12: Final exam (in class)
Announcements

Genome projects (Chapter 12)chronological overviewmajor issues and themes
Introduction to viruses (Chapter 13)classificationbioinformatics challenges and resources
Introduction to bacteria and archaea (Chapter 14)classificationbioinformatics challenges and resources
Outline of today’s lecture

A genome is the collection of DNA that comprisesan organism. Today we have assembled the sequenceof hundreds of genomes. We will begin by introducingthe “tree of life” in an effort to make a comprehensivesurvey of life forms.
Introduction to genomes
Page 397

Ernst Haeckel (1834-1919), a supporter of Darwin,published a tree of life (1879) including Moner(formless clumps, later named bacteria).
Chatton (1937) distinguished prokaryotes (bacteriathat lack nuclei) from eukaryotes (having nuclei).
Whittaker and others described the five-kingdomsystem: animals, plants, protists, fungi, and monera.
In the 1970s and 1980s, Carl Woese and colleaguesdescribed the archaea, thus forming a tree of lifewith three main branches.
Introduction: Systematics
Page 399

plants
animals
monera
fungi
protistsprotozoa
invertebrates
vertebrates
mammalsFive kingdom
system(Haeckel, 1879)
Page 396
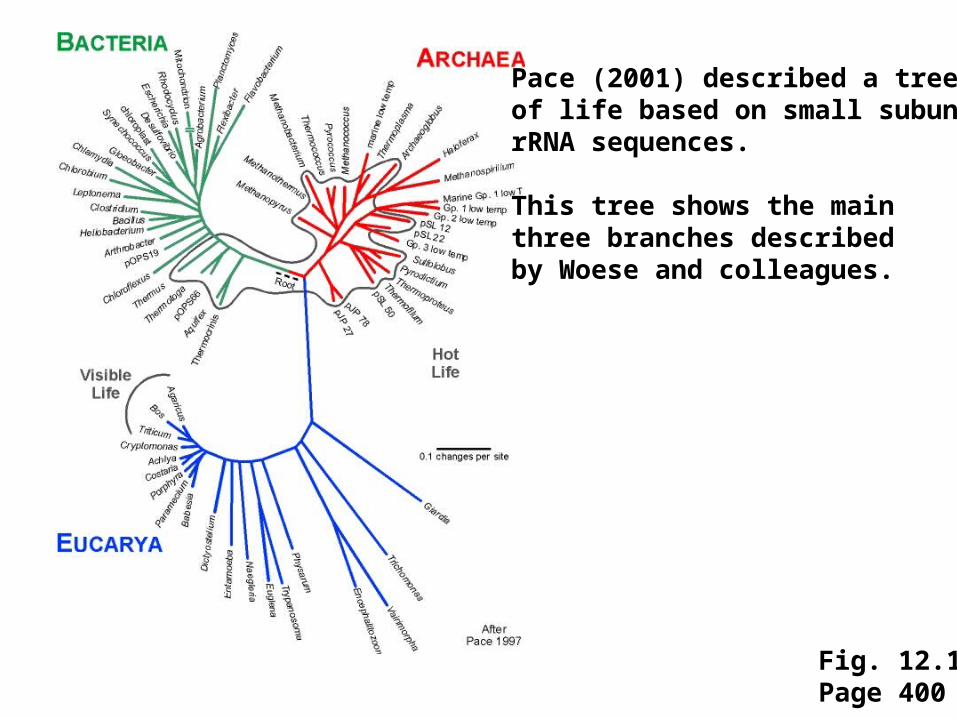
Fig. 12.1Page 400
Pace (2001) described a tree of life based on small subunit rRNA sequences.
This tree shows the mainthree branches describedby Woese and colleagues.

Historically, trees were generated primarily usingcharacters provided by morphological data. Molecularsequence data are now commonly used, includingsequences (such as small-subunit RNAs) that arehighly conserved.
Visit the European Small Subunit Ribosomal RNAdatabase for 20,000 SSU rRNA sequences.
Molecular sequences as basis of trees
Page 401

Genomes that span the tree of life are being sequenced at a rapid rate. There are several web-basedresources that document the progress, including:
GNN Genome News Networkhttp://www.genomenewsnetwork.org/main.shtml
GOLD Genomes Online Databasehttp://wit.integratedgenomics.com/GOLD/
PEDANT Protein Extraction, Description & Analysis Toolhttp://pedant.gsf.de/
Genome sequencing projects
Page 405

There are three main resources for genomes:
EBI European Bioinformatics Institutehttp://www.ebi.ac.uk/genomes/
NCBI National Center for Biotechnology Informationhttp://www.ncbi.nlm.nih.gov
TIGR The Institute for Genomic Researchhttp://www.tigr.org
Genome sequencing projects
Page 405

archaea
bacteria
eukaryotahttp://www.ncbi.nlm.nih.gov/Entrez/

Overview of viral complete genomes

Overview of archaea complete genomes

Overview of eukaryota genomes in NCBI’s Entez division

Overview of eukaryota genomes in NCBI’s Entrez division

We will next summarize the major achievements ingenome sequencing projects from a chronologicalperspective.
Chronology of genome sequencing projects
Page 404

1977: first viral genomeSanger et al. sequence bacteriophage X174.This virus is 5386 base pairs (encoding 11 genes).See accession J02482.
1981Human mitochondrial genome16,500 base pairs (encodes 13 proteins, 2 rRNA, 22 tRNA)Today, over 400 mitochondrial genomes sequenced
1986Chloroplast genome 156,000 base pairs (most are 120 kb to 200 kb)
Chronology of genome sequencing projects
Page 406

Fig. 12.6Page 407
Entrez nucleotide record for bacteriophage X174 (graphics display)

mitochondrion
chloroplast
Lackmitochondria (?)

1995: first genome of a free-living organism, the bacterium Haemophilus influenzae
Chronology of genome sequencing projects
Page 409

1995: genome of the bacterium Haemophilus influenzae is sequenced
Fig. 12.9Page 411


Overview of bacterialcomplete genomes
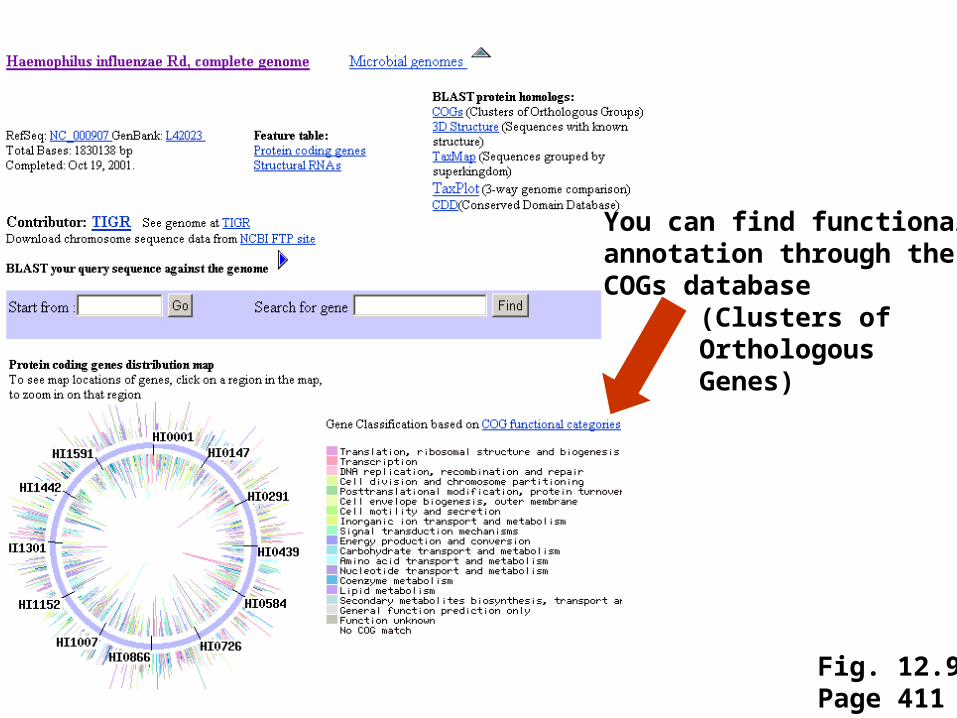
Fig. 12.9Page 411
You can find functionalannotation through theCOGs database
(Clusters ofOrthologousGenes)

Fig. 12.9Page 411
Click the circle to access the genomesequence

Fig. 12.10Page 412
Click the circle to access the genomesequence
Genes are color-codedaccording to theCOGs scheme

1996: first eukaryotic genome
The complete genome sequence of the budding yeastSaccharomyces cerevisiae was reported. We willdescribe this genome on Wednesday.
Also in 1996, TIGR reported the sequence of the firstarchaeal genome, Methanococcus jannaschii.
Chronology of genome sequencing projects
Page 413

1996: a yeast genome is sequenced

To place the sequencingof the yeast genomein context, these are theeukaryotes…

Eukaryotes(Baldauf et al. 2000)
Fungi

1997:More bacteria and archaeaEscherichia coli4.6 megabases, 4200 proteins (38% of unknown function)
1998: first multicellular organismNematode Caenorhabditis elegans 97 Mb; 19,000 genes.
1999: first human chromosomeChromosome 22 (49 Mb, 673 genes)
Chronology of genome sequencing projects
Page 413
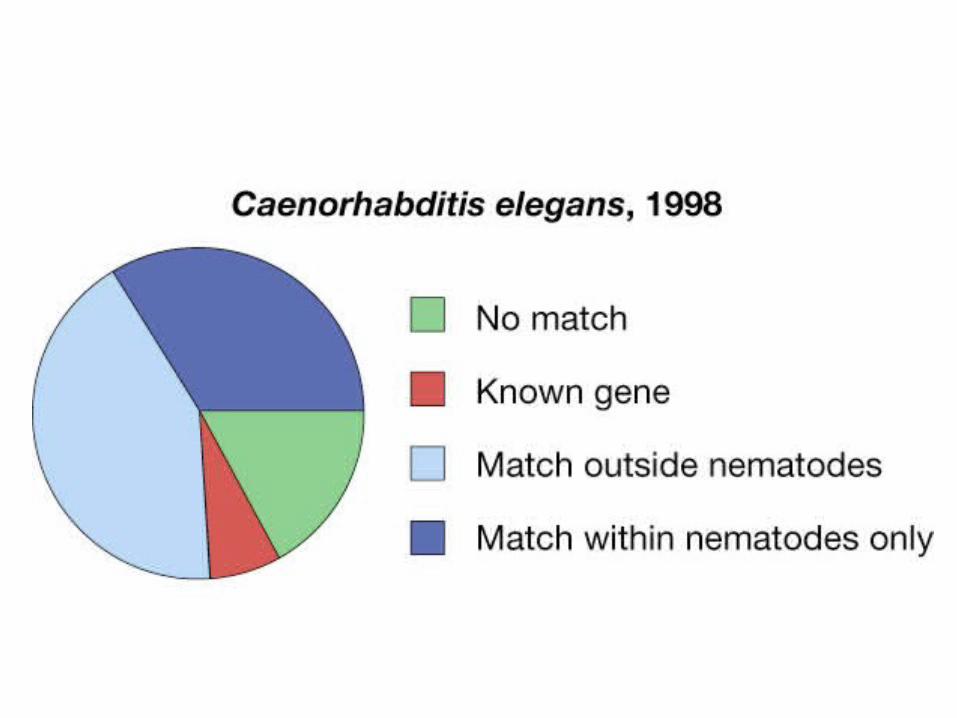

1999: Human chromosome 22 sequenced

1999: Human chromosome 22 sequenced
49 MB673 genes

2000:Fruitfly Drosophila melanogaster (13,000 genes)
Plant Arabidopsis thaliana
Human chromosome 21
2001: draft sequence of the human genome(public consortium and Celera Genomics)
Chronology of genome sequencing projects
Page 415



2000

Completed genome projects (current)
Eukaryotes: 10 In progress (partial):Anopheles gambiae Danio rerio (zebrafish) Arabidopsis thaliana Glycine max (soybean) Caenorhabditis elegans Hordeum vulgare (barley) Drosophila melanogaster Leishmania major Encephalitozoon cuniculi Rattus norvegicus Guillardia theta nucleomorphMus musculusPlasmodium falciparum Saccharomyces cerevisiae (yeast)Schizosaccharomyces pombe
Viruses: 1419 Bacteria: 139Archaea: 36
Page 417

eukaryotes

[1] Selection of genomes for sequencing
[2] Sequence one individual genome, or several?
[3] How big are genomes?
[4] Genome sequencing centers
[5] Sequencing genomes: strategies
[6] When has a genome been fully sequenced?
[7] Repository for genome sequence data
[8] Genome annotation
Overview of genome analysis
Page 418
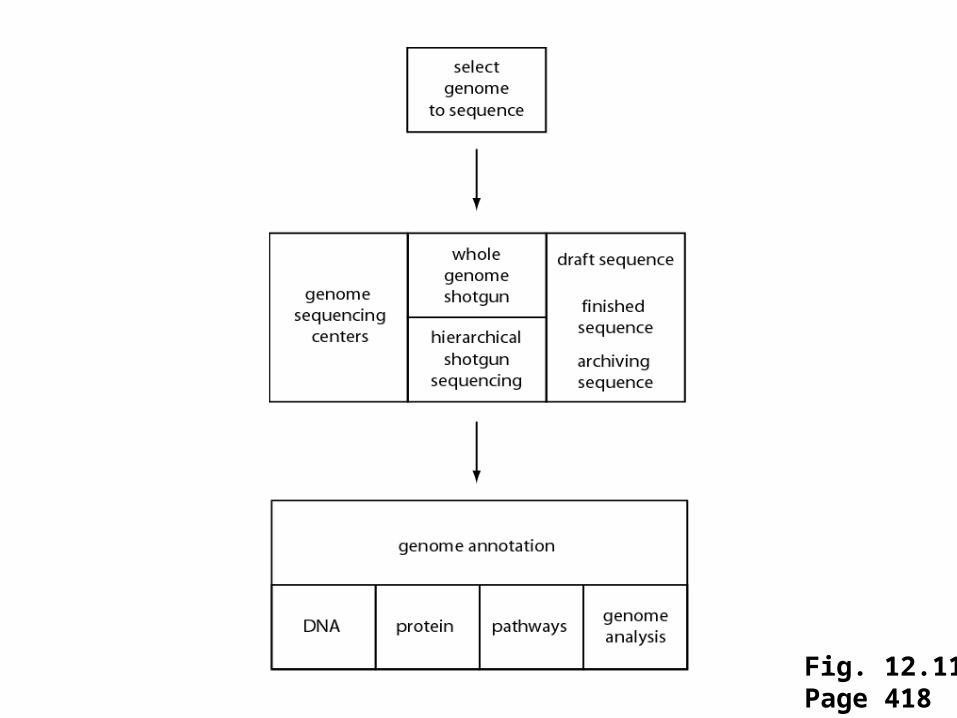
Fig. 12.11Page 418

[1] Selection of genomes for sequencing is basedon criteria such as:
• genome size (some plants are >>>human genome)• cost• relevance to human disease (or other disease)• relevance to basic biological questions• relevance to agriculture
Overview of genome analysis
Page 419

[1] Selection of genomes for sequencing is basedon criteria such as:
• genome size (some plants are >>>human genome)• cost• relevance to human disease (or other disease)• relevance to basic biological questions• relevance to agriculture
Ongoing projects:Chicken Fungi (many)Chimpanzee Honey beeCow Sea urchinDog (recent publication) Rhesus macaque
Overview of genome analysis
Page 419

[2] Sequence one individual genome, or several?
Try one…
--Each genome center may study one
chromosome from an organism
--It is necessary to measure polymorphisms
(e.g. SNPs) in large populations (November 5)
For viruses, thousands of isolates may be sequenced.
For the human genome, cost is the impediment.
Overview of genome analysis
Page 419

[3] How big are genomes?
Viral genomes: 1 kb to 350 kb (Mimivirus: 800 kb)
Bacterial genomes: 0.5 Mb to 13 Mb
Eukaryotic genomes: 8 Mb to 686 Mb(discussed further on Monday, November 3)
Overview of genome analysis
Page 420

viruses
plasmids
bacteria
fungi
plants
algae
insects
mollusks
reptiles
birds
mammals
Genome sizes in nucleotide base pairs
104 108105 106 107 10111010109
The size of the humangenome is ~ 3 X 109 bp;almost all of its complexityis in single-copy DNA.
The human genome is thoughtto contain ~30,000-40,000 genes.
bony fish
amphibians
http://www3.kumc.edu/jcalvet/PowerPoint/bioc801b.ppt
[4] 20 Genome sequencing centers contributedto the public sequencing of the human genome.
Many of these are listed at the Entrez genomes site.(See Table 17.6, page 625.)
Overview of genome analysis
Page 421

[5] There are two main stragies for sequencing genomes
Whole Genome Shotgun (from the NCBI website)
An approach used to decode an organism's genome by shredding it into smaller fragments of DNA which can be sequenced individually. The sequences of thesefragments are then ordered, based on overlaps in the genetic code, and finally reassembled into the complete sequence. The 'whole genome shotgun' (WGS) method isapplied to the entire genome all at once, while the 'hierarchical shotgun' method is applied to large, overlapping DNA fragments of known location in the genome.
Overview of genome analysis
Page 421

Hierarchical shotgun methodAssemble contigs from various chromosomes, then sequence and assemble them. A contig is a set of overlapping clones or sequences from which a sequence can be obtained. The sequence may be draft or finished.
A contig is thus a chromosome map showing the locations of those regions of a chromosome where contiguous DNA segments overlap. Contig maps are important because they provide the ability to study a complete, and often large segment of the genome by examining a series of overlapping clones which then provide an unbroken succession of information about that region.
Overview of genome analysis
Page 421

[6] When has a genome been fully sequenced?
A typical goal is to obtain five to ten-fold coverage.
Finished sequence: a clone insert is contiguouslysequenced with high quality standard of error rate0.01%. There are usually no gaps in the sequence.
Draft sequence: clone sequences may contain severalregions separated by gaps. The true order andorientation of the pieces may not be known.
Overview of genome analysis
Page 422

[7] Repository for genome sequence data
Raw data from many genome sequencing projectsare stored at the trace archive at NCBI or EBI
(main NCBI page, bottom right)
Overview of genome analysis
Page 425

Fig. 12.14Page 426

Fig. 12.14Page 426

[8] Genome annotation
Information content in genomic DNA includes:
-- repetitive DNA elements
-- nucleotide composition (GC content)
-- protein-coding genes, other genes
These topics will be discussed in detail on
November 3 (eukaryotic genomes)
Overview of genome analysis
Page 425

20 30 40 50 60 70 80
GC content (%)
Vertebrates
Invertebrates
Plants
Bacteria
3
5
10
Nu
mb
er o
f sp
ecie
sin
eac
h G
C c
lass
5
10
5
GC content varies across genomes
Fig. 12.16Page 428


Viruses are small, infectious, obligate intracellularparasites. They depend on host cells to replicate.Because they lack the resources for independentexistence, they exist on the borderline of the definitionof life.
The virion (virus particle) consists of a nucleic acidgenome surrounded by coat proteins (capsid) that maybe enveloped in a host-derived lipid bilayer.
Viral genomes consist of either RNA or DNA. They may be single-, double, or partially double stranded. The genomes may be circular, linear, or segmented.
Introduction to viruses
Page 437

Viruses have been classified by several criteria:
-- based on morphology (e.g. by electron microscopy)
-- by type of nucleic acid in the genome
-- by size (rubella is about 2 kb; HIV-1 about 9 kb; poxviruses are several hundred kb). Mimivirus (for Mimicking microbe) has a double-stranded circular genome of 800 kb.
-- based on human disease
Page 438
Introduction to viruses

Fig. 13.1Page 439

Fig. 13.2Page 440
The International Committee on Taxonomy of Viruses(ICTV) offers a website, accessible via NCBI’s Entrez site
http://www.ncbi.nlm.nih.gov/ICTVdb/

Vaccine-preventable viral diseases include:
Hepatitis A Hepatitis BInfluenzaMeaslesMumpsPoliomyelitisRubellaSmallpox
Page 441
Introduction to viruses

Some of the outstanding problems in virology include:
-- Why does a virus such as HIV-1 infect one species (human) selectively?
-- Why do some viruses change their natural host? In 1997 a chicken influenza virus killed six people.
-- Why are some viral strains particularly deadly?
-- What are the mechanisms of viral evasion of the host immune system?
-- Where did viruses originate?
Bioinformatic approaches to viruses
Page 439-441

The unique nature of viruses presents special challengesto studies of their evolution.
• viruses tend not to survive in historical samples• viral polymerases of RNA genomes typically lack proofreading activity• viruses undergo an extremely high rate of replication• many viral genomes are segmented; shuffling may occur• viruses may be subjected to intense selective pressures (host immune respones, antiviral therapy)• viruses invade diverse species• the diversity of viral genomes precludes us from making comprehensive phylogenetic trees of viruses
Diversity and evolution of viruses
Page 441

Herpesviruses are double-stranded DNA viruses thatinclude herpes simplex, cytomegalovirus, and Epstein-Barr.
Phylogenetic analysis suggests three major groups that originated about 180-220 MYA.
Bioinformatic approaches to herpesvirus
Page 442

Fig. 13.3Page 443

Consider human herpesvirus 9 (HHV-8). Its genome isabout 140,000 base pairs and encodes about 80 proteins.
We can explore this virus at the NCBI website.Try NCBI Entrez Genomes viruses dsDNA
Bioinformatic approaches to herpesvirus
Page 442

Fig. 13.4Page 444

Fig. 13.5Page 445

Fig. 13.10Page 449

Consider human herpesvirus 9 (HHV-8). Its genome isabout 140,000 base pairs and encodes about 80 proteins.
Microarrays have been used to define changes in viral geneexpression at different stages of infection (Paulose-Murphyet al., 2001). Conversely, gene expression changes havebeen measured in human cells following viral infection.
Bioinformatic approaches to herpesvirus
Page 442

Fig. 13.11Page 450
Paulose-Murphy et al. (2001)described HHV-8 viral genesthat are expressed at different times post infection

Human Immunodeficiency Virus (HIV) is the cause ofAIDS. At the end of the year 2002, 42 million people wereinfected. HIV-1 and HIV-2 are primate lentiviruses.
The HIV-1 genome is 9181 bases in length. Note that there are almost 100,000 Entrez nucleotide recordsfor this genome (but only one RefSeq entry).
Phylogenetic analyses suggest that HIV-2 appeared asa cross-species contamination from a simian virus,SIVsm (sooty mangebey). Similarly, HIV-1 appeared from simian immunodeficiency virus of the chimpanzee(SIVcpz).
Bioinformatic approaches to HIV
Page 446

Fig. 13.6Page 446

Two major resources are NCBI and the Los AlamosNational Laboratory (LANL) databases.
See http://hiv-web.lanl.gov/
LANL offers-- an HIV BLAST server-- Synonymous/non-synonymous analysis program-- a multiple alignment program-- a PCA-like tool-- a geography tool
Bioinformatic approaches to HIV
Page 453

Fig. 13.13Page 452

Fig. 13.6Page 446

Bacteria and archaea constitute two of the three mainbranches of life. Together they are the prokaryotes.
We can classify prokaryotes based on six criteria:[1] morphology[2] genome size[3] lifestyle[4] relevance to human disease[5] molecular phylogeny (rRNA)[6] molecular phylogeny (other molecules)
Bacteria and archaea: genome analysis
Page 466

Fig. 14.1Page 468

Fig. 14.2Page 470
M. genitalium hasone of the smallestbacterial genomesizes. View itsgenome atwww.tigr.org

We may distinguish six prokaryotic lifestyles:[1] Extracellular (e.g. E. coli)[2] Facultatively intracellular (Mycobacterium tuberculosis)[3] Extremophilic (e.g. M. jannaschi)[4] epicellular bacteria (e.g. Mycoplasma pneumoniae)[5] obligate intracellular and symbiotic (B. aphidicola)[6] obligate intracellular and parasitic (Rickettsia)
Bacteria and archaea: lifestyles
Page 472

Fig. 14.4Page 477

Fig. 14.5Page 478
Revisedfigure

Fig. 14.6Page 479

DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae
Nature 406, 477- 483 (2000)






Four main features of genomic DNA are useful:[1] Open reading frame length[2] Consensus for ribosome binding (Shine-Dalgarno)[3] Pattern of codon usage[4] Homology of putative gene to other genes
Bacteria and archaea: finding genes
Page 480

Fig. 14.7Page 482
GLIMMER for gene-findingin bacteria (www.tigr.org)

Fig. 14.8Page 484
Lateral gene transfer occurs in stages

COGs database: organisms and tools

COGs database: functional annotation


COGs database: distribution of COGs by number of species
COGs database: distribution of COGs by number of clades...


How can whole genomes be compared?
-- molecular phylogeny
-- You can BLAST (or PSI-BLAST) all the DNA and/or
protein in one genome against another
-- TaxPlot and COG for bacterial (and for
some eukaryotic) genomes
-- PipMaker, MUMmer and other programs align large
stretches of genomic DNA from multiple species

Fig. 14.16Page 493

Fig. 14.16Page 493

Fig. 14.17Page 494

Fig. 14.18Page 495