complement protease masp-1 activates human endothelial cells
TRANSCRIPT

of April 5, 2018.This information is current as
Functiona Link between Complement and Endothelial
IsHuman Endothelial Cells: PAR4 Activation Complement Protease MASP-1 Activates
Závodszky and Péter GálDoleschall, Zoltán Prohászka, László Cervenak, Péter Márton Megyeri, Veronika Makó, László Beinrohr, Zoltán
http://www.jimmunol.org/content/183/5/3409doi: 10.4049/jimmunol.0900879August 2009;
2009; 183:3409-3416; Prepublished online 10J Immunol
Referenceshttp://www.jimmunol.org/content/183/5/3409.full#ref-list-1
, 17 of which you can access for free at: cites 48 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2009 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Complement Protease MASP-1 Activates Human EndothelialCells: PAR4 Activation Is a Link between Complement andEndothelial Function1
Marton Megyeri,2* Veronika Mako,2† Laszlo Beinrohr,* Zoltan Doleschall,‡ Zoltan Prohaszka,†
Laszlo Cervenak,§ Peter Zavodszky,* and Peter Gal3*
Activation of the complement system can induce and enhance inflammatory reaction. Mannose-binding lectin-associated serineprotease-1 (MASP-1) is an abundant protease of the complement lectin pathway; however, its physiological function is unclear.In this study, we demonstrate for the first time that MASP-1 is able to activate Ca2� signaling, NF-�B, and p38 MAPK pathwaysin cultured HUVECs. Activation was initiated by MASP-1 only; the related protease, MASP-2, had no such effect. The phenom-enon was dependent on the proteolytic activity of MASP-1, suggesting modulation of endothelial cell function through a protease-activated receptor (PAR). Using synthetic peptide substrates representing the protease-sensitive regions of PARs, we were able todemonstrate that PAR4 is a target of MASP-1. The presence of functionally active PAR4 in HUVECs was demonstrated usingPAR4 agonist peptide and mRNA quantification. Finally, we showed that the amount of membrane-bound intact PAR4 decreasesafter MASP-1 treatment. All of these results provide a novel link between the regulation of endothelial cell function and com-plement system activation, and they suggest that MASP-1-induced PAR4 activation could contribute to the development of theinflammatory reaction. The Journal of Immunology, 2009, 183: 3409–3416.
T he complement system is a part of the innate immunesystem, since it can recognize, label, and eliminate invad-ing pathogens as well as altered host cells, but it also
bridges innate and adaptive immunity (1). The central componentsof the complement system are serine protease enzymes, which ac-tivate each other in a cascade-like manner (2). The complementsystem can be activated through three different routes: the classi-cal, lectin, and alternative pathways. In the case of the classical andlectin pathways, pattern-recognition molecules bind to the activa-tor structures and this is followed by the activation of associatedserine proteases. The pattern-recognition molecules of the lectinpathway are mannose-binding lectin (MBL)4 and ficolins (H-, L-,and M-ficolin), which form supramolecular complexes with theMBL-associated serine proteases (MASPs). Three MASPs havebeen identified: MASP-1, MASP-2, and MASP-3. MASP-2 has an
unambiguous physiological role: it can autoactivate and cleave C4and C2 subcomponents, which form the C3 convertase complex(C4b2a) (3). An MBL molecule, associated with a MASP-2 dimer,is the minimal complement fixation unit of the lectin pathway (4).The substrate specificity and physiological role of MASP-1 andMASP-3 have been controversial since their discovery. There is noevidence that MASP-3 actively participates in complement acti-vation, and its role may be only regulatory. MASP-1 is the mostabundant protease of the lectin pathway. It can autoactivate andcleave synthetic substrates efficiently (5). Several protein sub-strates have been reported for MASP-1. It has been proposed thatMASP-1 can directly activate C3; however, the efficiency of thisreaction is very low (6, 7). It is also possible that MASP-1 is ableto facilitate the C3 convertase generating activity of MASP-2, ei-ther through cleavage of C2 (8) or by activating zymogen MASP-2(9). Several lines of evidence indicate that MASP-1 is a thrombin-like protease, since it cleaves fibrinogen and factor XIII (transglu-taminase) and it can be efficiently inhibited by antithrombin (5,10–12).
Endothelial cells influence leukocyte function directly by ex-pressing and producing several regulatory factors, such as cyto-kines, chemokines, lipid mediators, NO, and adhesion molecules.Control of the inflammatory reaction also requires changes in vas-cular permeability, perfusion, and coagulation. At sites of vasculardamage, endothelial cells can be activated by thrombin (13) via theprotease-activated receptors (PARs). Thrombin cleaves the N-ter-minal region of these G protein-coupled receptors, thereby un-masking a new N-terminal domain, which acts as a tethered self-activating ligand. Cleavage of endothelial PAR1 and PAR4 (14)results in morphological changes of endothelial cells and the re-lease of vasoactive substances as well as cytokines. Additionally,activation of PAR1 and PAR4 by thrombin is responsible for plate-let activation in humans (13). The cleavage of PARs in generalleads to the activation of a multitude of signaling pathways (15).Among these, intracellular calcium mobilization, NF-�B nuclear
*Institute of Enzymology, Biological Research Center, Hungarian Academy ofSciences, Budapest, Hungary; †3rd Department of Medicine, Semmelweis Uni-versity, Budapest, Hungary; ‡Department of Pathogenetics, National Institute ofOncology, Budapest, Hungary; and §Research Group of Inflammation Biology andImmunogenomics, Hungarian Academy of Sciences-Semmelweis University,Budapest, Hungary
Received for publication March 19, 2009. Accepted for publication July 2, 2009.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Anyos Jedlik grant NKFP_07_1-MASPOK07(NKTH, Hungarian National Office for Research and Technology) and by GrantsNI61915, F67937, NK77978 (to P.Z.), and NF72689 (to Z.P.) (OTKA, HungarianScientific Research Fund).2 M.M. and V.M. contributed equally to this work.3 Address correspondence and reprint requests to Dr. Peter Gal, Institute of Enzy-mology, Biological Research Center, Hungarian Academy of Sciences, H-1113Budapest, Karolina ut 29, Hungary. E-mail address: [email protected] Abbreviations used in this paper: MBL, mannose-binding lectin; C1-inh, C1 inhib-itor; MASP, mannose-binding lectin-associated serine protease; PAR, protease-acti-vated receptor.
Copyright © 2009 by The American Association of Immunologists, Inc. 0022-1767/09/$2.00
The Journal of Immunology
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0900879
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

translocation, and MAPK activation have a pivotal role in the reg-ulation of endothelial cell function. PAR-induced signaling even-tually alters the phenotype of endothelial cells through calcium/calmodulin-regulated conformational changes induced in a widerange of proteins and regulation of ion channels (15, 16), includingthe activation of endothelium-dependent NO production and va-sorelaxation (17), as well as of leukocyte rolling and adhesion tothe vascular endothelium (18). Taken together, activation of en-dothelial cells through PARs may contribute to the regulation ofvascular tone, thrombotic events, inflammation, and vascularpermeability.
Since MASP-1 has thrombin-like activity and the PARs arethe primary cellular receptors of thrombin, we hypothesizedthat MASP-1 is an activator of PARs and may have thrombin-like effects on endothelial cells. Therefore, we treated HUVECswith MASP-1 and measured the activation of several intracel-lular signaling pathways. As shown in this report, MASP-1cleaves PAR4 on the surface of HUVECs, resulting in a typicalCa2� response, as well as in the activation of NF-�B and p38MAPK pathways.
Materials and MethodsReagents
Recombinant human MASP-1 and MASP-2 catalytic fragments (CCP1-CCP2-SP) were expressed in Escherichia coli and prepared according toDobo et al. (19) and Ambrus et al. (11), respectively. We used lyophilizedplasma C1 inhibitor (C1-inh, (Berinert P; ZLB Behring), which was furtherpurified by cation-exchange chromatography (Source 15S column; GEHealthcare) and gel filtration (Sephadex 75 HiLoad 16/60; GE Healthcare).The PAR1 antagonist (SCH 79797 dihydrochloride) was purchased fromTocris Bioscience. The PAR2 agonist (SLIGKV-NH2), the PAR2 antago-nist (FSLLRY-NH2), the PAR4 agonist (AYPGKF-NH2), and the PAR4antagonist (trans-cinnamoyl-YPGKF-NH2) were purchased from PeptidesInternational. All of the other reagents were purchased from Sigma-Aldrich, unless otherwise stated.
Preparation and culture of endothelial cells (HUVECs)
Cells were harvested from fresh umbilical cords of normally delivered,healthy children, by collagenase digestion (1 mg/ml; Invitrogen), asdescribed by Oroszlan et al. (20). HUVECs were kept in gelatin-pre-coated flasks in AIM-V medium (Invitrogen) completed with 1% FCS,2 ng/ml human recombinant epidermal growth factor (R&D Systems),250 pg/ml human recombinant �-endothelial cell growth factor (Bio-Source/Invitrogen), and 7.5 U/ml heparin, hereinafter referred to asComp-AIM-V. Endotoxin contamination of the Comp-AIM-V mediumwas monitored with the Limulus amebocyte lysate assay and was alwaysbelow the 0.06 U/ml detection limit. Cells were split into a 1:3 ratiowhen they reached confluence. HUVECs were routinely checked ac-cording to morphological criteria (“cobble stone” morphology) and vonWillebrand factor positivity. To avoid genetic bias, each experimentwas performed on at least three independent primary HUVEC culturesfrom different individuals.
Quanitative real-time PCR and mRNA purification
HUVECs were lysed and stored in TRI reagent. Total RNA was purifiedfrom cells with the NucleoSpin RNA II extraction kit (Macherey-Nagel).The RNA-cDNA transcription was done with Moloney murine leukemiavirus reverse transcriptase (Promega). The LightCycler FastStart DNAMaster SYBR Green I kit (Roche) was used for quantification of cDNA ona LightCycler (Roche). The PAR4 primers were designed from database(http://medgen.ugent.be) and produced by Invitrogen. GAPDH and �-actingene-specific primers were synthesized according to the published cDNAsequences (5�-GGCATCCTCACCCTGAAGTA-3� and 5�-GGGGTGTTGAAGGTCTCAAA-3� for �-actin, 5�-AGTCCTTCCACGATACCAAA-3� and 5�-TGAACCATGAGAAGTATGACAACA-3� for GAPDH). Thesequences of the PAR4 primers were as follows: 5�-ACCATGCTGCTGATGAACCT-3� and 5�-AGCACTGAGCCATACATGTGAC-3�. Quanti-fication was performed using the second derivative maximum crossingpoint (CPSDM) by LightCycler software 3.3 (Roche). Relative values werecalculated as follows: 2(CP.actin�CP.PAR4) and 2(CP.GAPDH�CP.PAR4) for actinand GAPDH, respectively.
Fluorometric substrate assay
The peptides (PAR1 P5-P1) TLDPR-AMC, (PAR2 P5-P1) SSKGR-AMC,and (PAR4 P5-P1) LPAPR-AMC were synthesized at �90% purity by BioBasic. Assays of peptide substrates cleavage were conducted in HBSSbuffer at 25°C using final substrate concentrations within the range of 500-3000 nM. The increase of fluorescent signal was measured on a Wallac1420 (PerkinElmer) microplate reader at an excitation wavelength of 355nm and emission of 460 nm. The concentrations of thrombin and MASP-1were 2.9 and 3.8 nM, respectively. The initial reaction rates were deter-mined at early time points where the progress curves were linear. The datawere fit to the Michaelis-Menten equation and the individual kcat and KM
values were determined using GraFit (Erithacus Software). We calculatedthe kcat/KM values from these individual kcat and KM values.
In other experiments, where [S] �� KM, the kcat/KM values were directlycalculated with Origin 5.0 software (Microcal), after curve fitting by thefollowing equation: [P] � [P]� � ([P]0 � [P]�) � exp((kcatE0/KM)t). Inthese experiments the concentration of the thrombin was 2.9 nM, whereasMASP-1 was measured at both 138 nM and 69 nM.
Western blot analysis
After washing with ice-cold PBS, cells were lysed in buffer containinginhibitor cocktail (30 mM HEPES (pH 7.4), 100 mM NaCl, 1 mM EDTA,20 mM NaF, 1% Triton X-100, 1 mM PMSF, 1 mM Na3VO4, and 2%premixed protease inhibitor cocktail from BD Biosciences). Samples wereseparated with 12% SDS-PAGE and then transferred to polyvinylidenedifluoride membranes. After blocking for 2 h in TBS containing 3% BSA,membranes were washed and probed overnight at 4°C with Abs againstPAR4 (Acris Abs; 1/1000), p38 MAPK (Cell Signaling Technology,1/2000), or phospho-p38 MAPK (Cell Signaling Technology; 1/2000). Im-munoblots were washed and incubated for 2 h at room temperature withalkaline phosphatase-conjugated goat anti-rabbit or goat anti-mouse sec-ondary Abs (SouthernBiotech; 1/2,000 or 1/60,000, respectively) and thenvisualized with 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazo-lium solution. Densitometric analysis was performed using Syngene Gene-Tools software (Synoptics).
Measurement of intracellular Ca2� signaling by fluorescencemicroscopy
HUVECs were seeded onto 96-well plates at 5000 cells/well (semiconflu-ent) concentration in 100 �l of Comp-AIM-V medium for 1 day. Then, 2�M Fluo-4-AM (Molecular Probes) was added for 20 min, at 37°C, fol-lowed by a 20-min incubation in HBSS to equilibrate Fluo-4 dye concen-tration in the cytosol. Fluorescence microscopy was performed using anOlympus IX-81 inverted fluorescence microscope. Sequential images wereobtained every 10 s. At least 20 cells per image were analyzed to calculatechanges in fluorescence intensity. In experiments where antagonists wereapplied, cells were preincubated with PAR1 (10 �M), PAR2 (200 �M), orPAR4 (2 nM) antagonists for 10 min.
NF-�B nuclear translocation assay
HUVECs were seeded onto 96-well plates at 5000 cells/well (semiconflu-ent) concentration in 100 �l of Comp-AIM-V medium for 1 day. Then,cells were treated with different doses of LPS (E. coli 026:B6 serotype;Sigma-Aldrich), IL-1� (BioSource), or MASP-1 in 100 �l of Comp-AIM-V. Polymyxin had been preincubated with LPS or MASP-1 for 30min before the mixture was applied to the cells. At the end of treatment,cells were fixed with ice-cold methanol/acetone (1/1) for 10 min and thenrehydrated and stained with rabbit-anti-human NF-�B p65 (Santa CruzBiotechnology) Ab followed by Alexa 568-conjugated goat-anti-rabbit IgG(Molecular Probes/Invitrogen) and Hoechst 33342 (Molecular Probes).Samples were observed and images were recorded using an OlympusIX-81 inverted fluorescence microscope mounted with an Olympus DP70digital camera (Olympus Optical). Images were analyzed using the Analy-SIS 3.2 software (Soft Imaging System). The ratio of nuclear and perinu-clear mean red fluorescence values was calculated based on the data fromthree parallel images of at least 30 cells per image, as described earlier byDoleschall et al. (21).
Statistical analysis
Data were compared using one-way ANOVA with Tukey’s posttest andStudent’s t test (GraphPad Prism 4.02 software; GraphPad Software). A pvalue of �0.05 was considered significant. Data are presented asmeans SEM.
3410 MASP-1 ACTIVATES ENDOTHELIAL PAR4
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
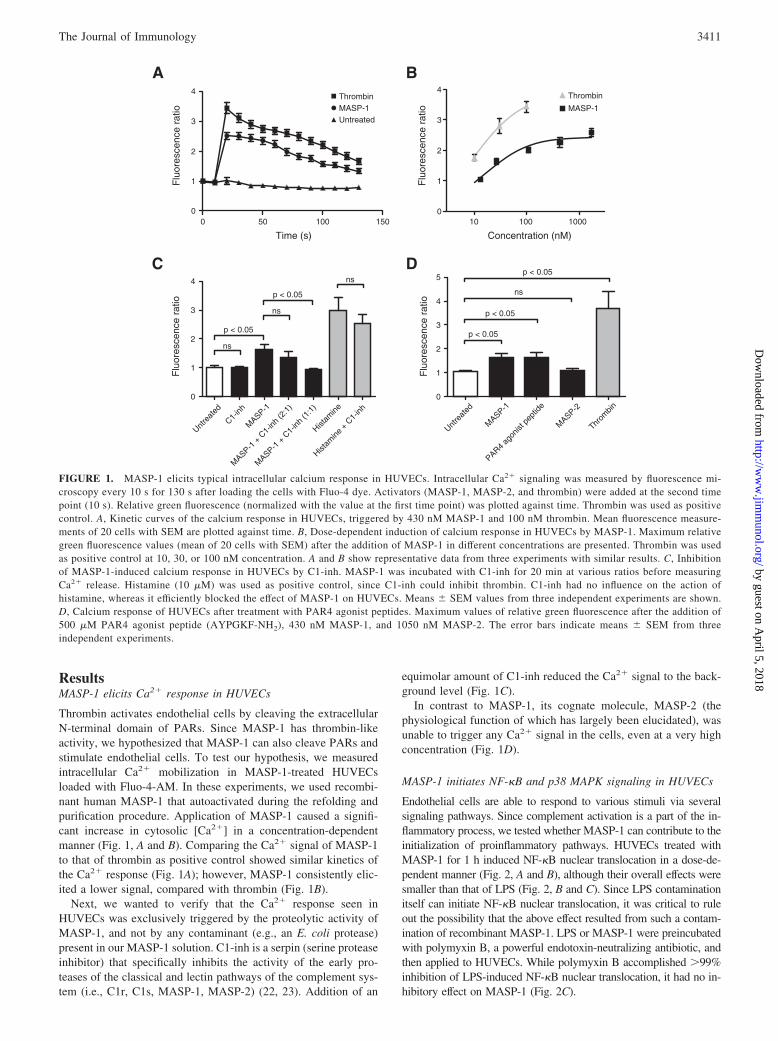
ResultsMASP-1 elicits Ca2� response in HUVECs
Thrombin activates endothelial cells by cleaving the extracellularN-terminal domain of PARs. Since MASP-1 has thrombin-likeactivity, we hypothesized that MASP-1 can also cleave PARs andstimulate endothelial cells. To test our hypothesis, we measuredintracellular Ca2� mobilization in MASP-1-treated HUVECsloaded with Fluo-4-AM. In these experiments, we used recombi-nant human MASP-1 that autoactivated during the refolding andpurification procedure. Application of MASP-1 caused a signifi-cant increase in cytosolic [Ca2�] in a concentration-dependentmanner (Fig. 1, A and B). Comparing the Ca2� signal of MASP-1to that of thrombin as positive control showed similar kinetics ofthe Ca2� response (Fig. 1A); however, MASP-1 consistently elic-ited a lower signal, compared with thrombin (Fig. 1B).
Next, we wanted to verify that the Ca2� response seen inHUVECs was exclusively triggered by the proteolytic activity ofMASP-1, and not by any contaminant (e.g., an E. coli protease)present in our MASP-1 solution. C1-inh is a serpin (serine proteaseinhibitor) that specifically inhibits the activity of the early pro-teases of the classical and lectin pathways of the complement sys-tem (i.e., C1r, C1s, MASP-1, MASP-2) (22, 23). Addition of an
equimolar amount of C1-inh reduced the Ca2� signal to the back-ground level (Fig. 1C).
In contrast to MASP-1, its cognate molecule, MASP-2 (thephysiological function of which has largely been elucidated), wasunable to trigger any Ca2� signal in the cells, even at a very highconcentration (Fig. 1D).
MASP-1 initiates NF-�B and p38 MAPK signaling in HUVECs
Endothelial cells are able to respond to various stimuli via severalsignaling pathways. Since complement activation is a part of the in-flammatory process, we tested whether MASP-1 can contribute to theinitialization of proinflammatory pathways. HUVECs treated withMASP-1 for 1 h induced NF-�B nuclear translocation in a dose-de-pendent manner (Fig. 2, A and B), although their overall effects weresmaller than that of LPS (Fig. 2, B and C). Since LPS contaminationitself can initiate NF-�B nuclear translocation, it was critical to ruleout the possibility that the above effect resulted from such a contam-ination of recombinant MASP-1. LPS or MASP-1 were preincubatedwith polymyxin B, a powerful endotoxin-neutralizing antibiotic, andthen applied to HUVECs. While polymyxin B accomplished �99%inhibition of LPS-induced NF-�B nuclear translocation, it had no in-hibitory effect on MASP-1 (Fig. 2C).
FIGURE 1. MASP-1 elicits typical intracellular calcium response in HUVECs. Intracellular Ca2� signaling was measured by fluorescence mi-croscopy every 10 s for 130 s after loading the cells with Fluo-4 dye. Activators (MASP-1, MASP-2, and thrombin) were added at the second timepoint (10 s). Relative green fluorescence (normalized with the value at the first time point) was plotted against time. Thrombin was used as positivecontrol. A, Kinetic curves of the calcium response in HUVECs, triggered by 430 nM MASP-1 and 100 nM thrombin. Mean fluorescence measure-ments of 20 cells with SEM are plotted against time. B, Dose-dependent induction of calcium response in HUVECs by MASP-1. Maximum relativegreen fluorescence values (mean of 20 cells with SEM) after the addition of MASP-1 in different concentrations are presented. Thrombin was usedas positive control at 10, 30, or 100 nM concentration. A and B show representative data from three experiments with similar results. C, Inhibitionof MASP-1-induced calcium response in HUVECs by C1-inh. MASP-1 was incubated with C1-inh for 20 min at various ratios before measuringCa2� release. Histamine (10 �M) was used as positive control, since C1-inh could inhibit thrombin. C1-inh had no influence on the action ofhistamine, whereas it efficiently blocked the effect of MASP-1 on HUVECs. Means SEM values from three independent experiments are shown.D, Calcium response of HUVECs after treatment with PAR4 agonist peptides. Maximum values of relative green fluorescence after the addition of500 �M PAR4 agonist peptide (AYPGKF-NH2), 430 nM MASP-1, and 1050 nM MASP-2. The error bars indicate means SEM from threeindependent experiments.
3411The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

p38 MAPK, another important proinflammatory signaling path-way in endothelial cells, interacts with the NF-�B pathway at dif-ferent levels. HUVECs treated with MASP-1 for 30 min inducedstrong, dose-dependent phosphorylation of p38 MAPK (Fig. 3).The effectiveness of MASP-1 was comparable with that of throm-bin used as positive control.
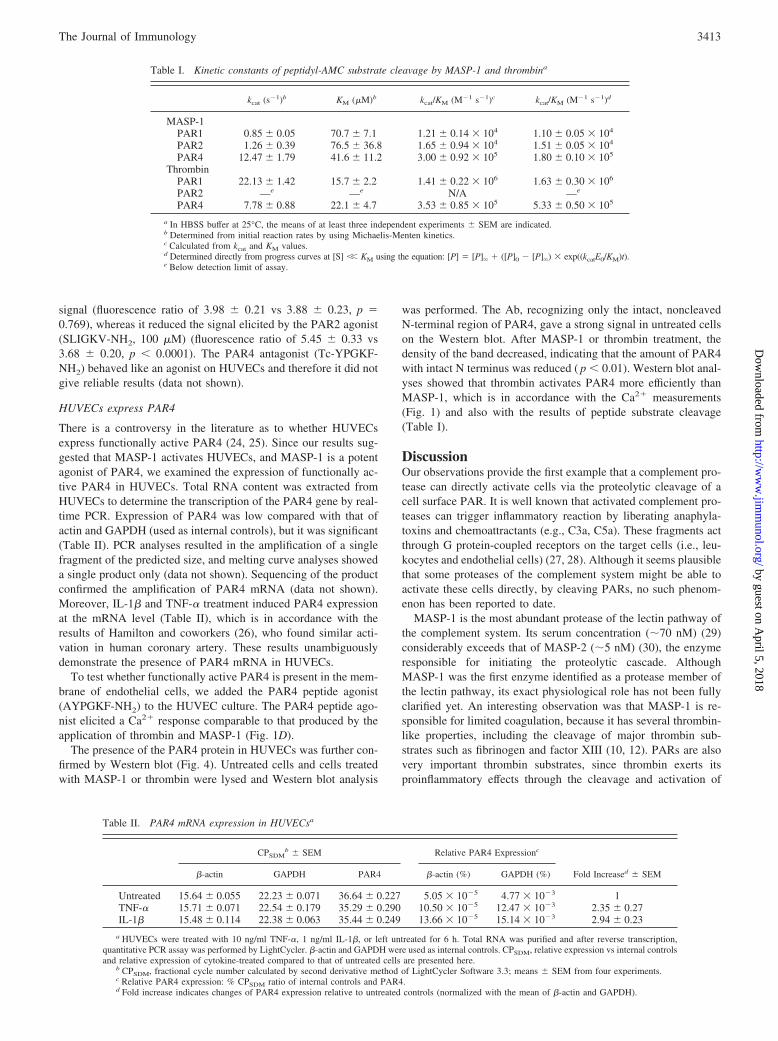
MASP-1 specifically cleaves the N-terminal sequence of PAR4
Since the Ca2� signal was triggered by the proteolytic activity ofMASP-1, it seemed plausible that the target of MASP-1 on the
endothelial cell surface might be a PAR. Because endothelial cellscan express PAR1, PAR2, and PAR4 (14), we tested the ability ofMASP-1 to cleave the N-terminal region of these receptors, usingsynthetic substrates. Fluorescent-labeled oligopeptides represent-ing the special cleavage sites of these PARs were used to test thesubstrate specificity of MASP-1, MASP-2, and thrombin. We de-termined the kcat/KM values directly and the individual kcat and KM
values as well (Table I). MASP-1 cleaved the PAR4 substrate withsubstantial efficiency (kcat/KM � 1.8 0.1 � 105 M�1 s�1),whereas it cleaved PAR1 and PAR2 peptides with low efficiencyonly (kcat/KM � 1.1 0.05 � 104 M�1 s�1 and kcat/KM � 1.5 0.05 � 104 M�1 s�1, respectively).
As expected, thrombin cleaved the PAR1 substrate very effi-ciently (kcat/KM � 1.6 0.3 � 106 M�1 s�1) and it also cleavedthe PAR4 substrate, albeit with less efficiency (kcat/KM � 5.3 0.5 � 105 M�1 s�1). This is in accordance with data in the liter-ature claiming that even a low thrombin concentration is sufficientto activate PAR1, whereas the activation of PAR4 requires ahigher concentration of thrombin.
MASP-2 did not generate any detectable signal with either ofthe PAR substrates, confirming that MASP-2 is not an agonist ofthese receptors and that the phenomenon is specific to MASP-1among the tested complement proteases.
To compare the contributions of the different receptors to thegeneration of the Ca2� signal we used PAR1, PAR2, and PAR4antagonists in our experimental system. The PAR1 antagonist(SCH 79797, 10 �M) strongly reduced the Ca2� signal elicited by100 nM thrombin (fluorescence ratio of 4.45 0.39 vs 2.14 0.28, p � 0.0001), while it had no effect on the 860 nM MASP-1-generated signal (fluorescence ratio of 4.02 0.69 vs 3.66 0.29, p � 0.664). The PAR2 antagonist (FSLLRY-NH2, 200 �M)did not significantly changed the level of the MASP-1-induced
FIGURE 2. MASP-1 induces dose-dependent NF-�B nuclear transloca-tion in HUVECs. Cells were treated with different concentrations ofMASP-1 or LPS for 1 h. Cells were fixed and incubated with anti-NF-�B(p65) followed by Alexa 568 (red)-labeled goat-anti-rabbit Ab and Hoechst33342 (blue). A, Visualization of NF-�B translocation (red, NF-�B; blue,nuclei). B, Mean red fluorescence of nuclei and perinuclear cytoplasmicregions was calculated. Effect of MASP-1 was analyzed with one-wayANOVA. Representative data out of three independent experiments areshown. C, Polymyxin B treatment of MASP-1 does not inhibit the NF-�Bnuclear translocation. Gray and black bars indicate preincubation with orwithout 10 �g/ml polymyxin B, respectively. Polymyxin B treatment wastested with Student’s t test. Representative data out of three independentexperiments are shown.
FIGURE 3. MASP-1 initiates p38 MAPK signaling. Cells were treatedwith MASP-1 (95, 268, or 860 nM) or thrombin (30, 100, or 300 nM) for30 min. Cell lysates were loaded to SDS-PAGE, transferred to nitrocellu-lose membranes, and then proteins were probed with Abs against phos-phorylated p38 or total cellular p38. A, A representative Western blot anal-ysis of phosphorylated and total p38. B, Densitometric analysis of Westernblots. Columns represent the fold increase of phosphorylation of p38,standardized by total p38 content, compared with untreated controls.Analysis with one-way ANOVA was done on the unnormalized values,but graphs represent the normalized means SEM from three inde-pendent experiments.
3412 MASP-1 ACTIVATES ENDOTHELIAL PAR4
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
signal (fluorescence ratio of 3.98 0.21 vs 3.88 0.23, p �0.769), whereas it reduced the signal elicited by the PAR2 agonist(SLIGKV-NH2, 100 �M) (fluorescence ratio of 5.45 0.33 vs3.68 0.20, p � 0.0001). The PAR4 antagonist (Tc-YPGKF-NH2) behaved like an agonist on HUVECs and therefore it did notgive reliable results (data not shown).
HUVECs express PAR4
There is a controversy in the literature as to whether HUVECsexpress functionally active PAR4 (24, 25). Since our results sug-gested that MASP-1 activates HUVECs, and MASP-1 is a potentagonist of PAR4, we examined the expression of functionally ac-tive PAR4 in HUVECs. Total RNA content was extracted fromHUVECs to determine the transcription of the PAR4 gene by real-time PCR. Expression of PAR4 was low compared with that ofactin and GAPDH (used as internal controls), but it was significant(Table II). PCR analyses resulted in the amplification of a singlefragment of the predicted size, and melting curve analyses showeda single product only (data not shown). Sequencing of the productconfirmed the amplification of PAR4 mRNA (data not shown).Moreover, IL-1� and TNF-� treatment induced PAR4 expressionat the mRNA level (Table II), which is in accordance with theresults of Hamilton and coworkers (26), who found similar acti-vation in human coronary artery. These results unambiguouslydemonstrate the presence of PAR4 mRNA in HUVECs.
To test whether functionally active PAR4 is present in the mem-brane of endothelial cells, we added the PAR4 peptide agonist(AYPGKF-NH2) to the HUVEC culture. The PAR4 peptide ago-nist elicited a Ca2� response comparable to that produced by theapplication of thrombin and MASP-1 (Fig. 1D).
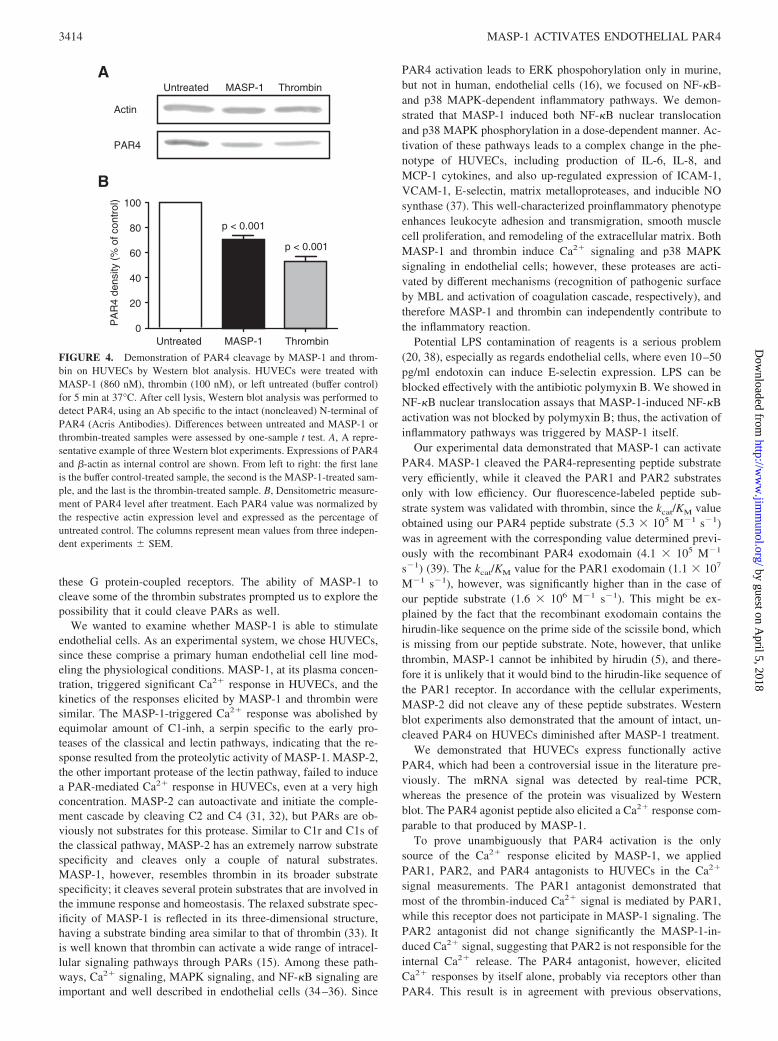
The presence of the PAR4 protein in HUVECs was further con-firmed by Western blot (Fig. 4). Untreated cells and cells treatedwith MASP-1 or thrombin were lysed and Western blot analysis
was performed. The Ab, recognizing only the intact, noncleavedN-terminal region of PAR4, gave a strong signal in untreated cellson the Western blot. After MASP-1 or thrombin treatment, thedensity of the band decreased, indicating that the amount of PAR4with intact N terminus was reduced ( p � 0.01). Western blot anal-yses showed that thrombin activates PAR4 more efficiently thanMASP-1, which is in accordance with the Ca2� measurements(Fig. 1) and also with the results of peptide substrate cleavage(Table I).
DiscussionOur observations provide the first example that a complement pro-tease can directly activate cells via the proteolytic cleavage of acell surface PAR. It is well known that activated complement pro-teases can trigger inflammatory reaction by liberating anaphyla-toxins and chemoattractants (e.g., C3a, C5a). These fragments actthrough G protein-coupled receptors on the target cells (i.e., leu-kocytes and endothelial cells) (27, 28). Although it seems plausiblethat some proteases of the complement system might be able toactivate these cells directly, by cleaving PARs, no such phenom-enon has been reported to date.
MASP-1 is the most abundant protease of the lectin pathway ofthe complement system. Its serum concentration (70 nM) (29)considerably exceeds that of MASP-2 (5 nM) (30), the enzymeresponsible for initiating the proteolytic cascade. AlthoughMASP-1 was the first enzyme identified as a protease member ofthe lectin pathway, its exact physiological role has not been fullyclarified yet. An interesting observation was that MASP-1 is re-sponsible for limited coagulation, because it has several thrombin-like properties, including the cleavage of major thrombin sub-strates such as fibrinogen and factor XIII (10, 12). PARs are alsovery important thrombin substrates, since thrombin exerts itsproinflammatory effects through the cleavage and activation of
Table I. Kinetic constants of peptidyl-AMC substrate cleavage by MASP-1 and thrombina
kcat (s�1)b KM (�M)b kcat/KM (M�1 s�1)c kcat/KM (M�1 s�1)d
MASP-1PAR1 0.85 0.05 70.7 7.1 1.21 0.14 � 104 1.10 0.05 � 104
PAR2 1.26 0.39 76.5 36.8 1.65 0.94 � 104 1.51 0.05 � 104
PAR4 12.47 1.79 41.6 11.2 3.00 0.92 � 105 1.80 0.10 � 105
ThrombinPAR1 22.13 1.42 15.7 2.2 1.41 0.22 � 106 1.63 0.30 � 106
PAR2 —e —e N/A —e
PAR4 7.78 0.88 22.1 4.7 3.53 0.85 � 105 5.33 0.50 � 105
a In HBSS buffer at 25°C, the means of at least three independent experiments SEM are indicated.b Determined from initial reaction rates by using Michaelis-Menten kinetics.c Calculated from kcat and KM values.d Determined directly from progress curves at [S] �� KM using the equation: [P] � [P]� � ([P]0 � [P]�) � exp((kcatE0/KM)t).e Below detection limit of assay.
Table II. PAR4 mRNA expression in HUVECsa
CPSDMb SEM Relative PAR4 Expressionc
Fold Increased SEM�-actin GAPDH PAR4 �-actin (%) GAPDH (%)
Untreated 15.64 0.055 22.23 0.071 36.64 0.227 5.05 � 10�5 4.77 � 10�3 1TNF-� 15.71 0.071 22.54 0.179 35.29 0.290 10.50 � 10�5 12.47 � 10�3 2.35 0.27IL-1� 15.48 0.114 22.38 0.063 35.44 0.249 13.66 � 10�5 15.14 � 10�3 2.94 0.23
a HUVECs were treated with 10 ng/ml TNF-�, 1 ng/ml IL-1�, or left untreated for 6 h. Total RNA was purified and after reverse transcription,quantitative PCR assay was performed by LightCycler. �-actin and GAPDH were used as internal controls. CPSDM, relative expression vs internal controlsand relative expression of cytokine-treated compared to that of untreated cells are presented here.
b CPSDM, fractional cycle number calculated by second derivative method of LightCycler Software 3.3; means SEM from four experiments.c Relative PAR4 expression: % CPSDM ratio of internal controls and PAR4.d Fold increase indicates changes of PAR4 expression relative to untreated controls (normalized with the mean of �-actin and GAPDH).
3413The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
these G protein-coupled receptors. The ability of MASP-1 tocleave some of the thrombin substrates prompted us to explore thepossibility that it could cleave PARs as well.
We wanted to examine whether MASP-1 is able to stimulateendothelial cells. As an experimental system, we chose HUVECs,since these comprise a primary human endothelial cell line mod-eling the physiological conditions. MASP-1, at its plasma concen-tration, triggered significant Ca2� response in HUVECs, and thekinetics of the responses elicited by MASP-1 and thrombin weresimilar. The MASP-1-triggered Ca2� response was abolished byequimolar amount of C1-inh, a serpin specific to the early pro-teases of the classical and lectin pathways, indicating that the re-sponse resulted from the proteolytic activity of MASP-1. MASP-2,the other important protease of the lectin pathway, failed to inducea PAR-mediated Ca2� response in HUVECs, even at a very highconcentration. MASP-2 can autoactivate and initiate the comple-ment cascade by cleaving C2 and C4 (31, 32), but PARs are ob-viously not substrates for this protease. Similar to C1r and C1s ofthe classical pathway, MASP-2 has an extremely narrow substratespecificity and cleaves only a couple of natural substrates.MASP-1, however, resembles thrombin in its broader substratespecificity; it cleaves several protein substrates that are involved inthe immune response and homeostasis. The relaxed substrate spec-ificity of MASP-1 is reflected in its three-dimensional structure,having a substrate binding area similar to that of thrombin (33). Itis well known that thrombin can activate a wide range of intracel-lular signaling pathways through PARs (15). Among these path-ways, Ca2� signaling, MAPK signaling, and NF-�B signaling areimportant and well described in endothelial cells (34–36). Since
PAR4 activation leads to ERK phospohorylation only in murine,but not in human, endothelial cells (16), we focused on NF-�B-and p38 MAPK-dependent inflammatory pathways. We demon-strated that MASP-1 induced both NF-�B nuclear translocationand p38 MAPK phosphorylation in a dose-dependent manner. Ac-tivation of these pathways leads to a complex change in the phe-notype of HUVECs, including production of IL-6, IL-8, andMCP-1 cytokines, and also up-regulated expression of ICAM-1,VCAM-1, E-selectin, matrix metalloproteases, and inducible NOsynthase (37). This well-characterized proinflammatory phenotypeenhances leukocyte adhesion and transmigration, smooth musclecell proliferation, and remodeling of the extracellular matrix. BothMASP-1 and thrombin induce Ca2� signaling and p38 MAPKsignaling in endothelial cells; however, these proteases are acti-vated by different mechanisms (recognition of pathogenic surfaceby MBL and activation of coagulation cascade, respectively), andtherefore MASP-1 and thrombin can independently contribute tothe inflammatory reaction.
Potential LPS contamination of reagents is a serious problem(20, 38), especially as regards endothelial cells, where even 10–50pg/ml endotoxin can induce E-selectin expression. LPS can beblocked effectively with the antibiotic polymyxin B. We showed inNF-�B nuclear translocation assays that MASP-1-induced NF-�Bactivation was not blocked by polymyxin B; thus, the activation ofinflammatory pathways was triggered by MASP-1 itself.
Our experimental data demonstrated that MASP-1 can activatePAR4. MASP-1 cleaved the PAR4-representing peptide substratevery efficiently, while it cleaved the PAR1 and PAR2 substratesonly with low efficiency. Our fluorescence-labeled peptide sub-strate system was validated with thrombin, since the kcat/KM valueobtained using our PAR4 peptide substrate (5.3 � 105 M�1 s�1)was in agreement with the corresponding value determined previ-ously with the recombinant PAR4 exodomain (4.1 � 105 M�1
s�1) (39). The kcat/KM value for the PAR1 exodomain (1.1 � 107
M�1 s�1), however, was significantly higher than in the case ofour peptide substrate (1.6 � 106 M�1 s�1). This might be ex-plained by the fact that the recombinant exodomain contains thehirudin-like sequence on the prime side of the scissile bond, whichis missing from our peptide substrate. Note, however, that unlikethrombin, MASP-1 cannot be inhibited by hirudin (5), and there-fore it is unlikely that it would bind to the hirudin-like sequence ofthe PAR1 receptor. In accordance with the cellular experiments,MASP-2 did not cleave any of these peptide substrates. Westernblot experiments also demonstrated that the amount of intact, un-cleaved PAR4 on HUVECs diminished after MASP-1 treatment.
We demonstrated that HUVECs express functionally activePAR4, which had been a controversial issue in the literature pre-viously. The mRNA signal was detected by real-time PCR,whereas the presence of the protein was visualized by Westernblot. The PAR4 agonist peptide also elicited a Ca2� response com-parable to that produced by MASP-1.
To prove unambiguously that PAR4 activation is the onlysource of the Ca2� response elicited by MASP-1, we appliedPAR1, PAR2, and PAR4 antagonists to HUVECs in the Ca2�
signal measurements. The PAR1 antagonist demonstrated thatmost of the thrombin-induced Ca2� signal is mediated by PAR1,while this receptor does not participate in MASP-1 signaling. ThePAR2 antagonist did not change significantly the MASP-1-in-duced Ca2� signal, suggesting that PAR2 is not responsible for theinternal Ca2� release. The PAR4 antagonist, however, elicitedCa2� responses by itself alone, probably via receptors other thanPAR4. This result is in agreement with previous observations,
FIGURE 4. Demonstration of PAR4 cleavage by MASP-1 and throm-bin on HUVECs by Western blot analysis. HUVECs were treated withMASP-1 (860 nM), thrombin (100 nM), or left untreated (buffer control)for 5 min at 37°C. After cell lysis, Western blot analysis was performed todetect PAR4, using an Ab specific to the intact (noncleaved) N-terminal ofPAR4 (Acris Antibodies). Differences between untreated and MASP-1 orthrombin-treated samples were assessed by one-sample t test. A, A repre-sentative example of three Western blot experiments. Expressions of PAR4and �-actin as internal control are shown. From left to right: the first laneis the buffer control-treated sample, the second is the MASP-1-treated sam-ple, and the last is the thrombin-treated sample. B, Densitometric measure-ment of PAR4 level after treatment. Each PAR4 value was normalized bythe respective actin expression level and expressed as the percentage ofuntreated control. The columns represent mean values from three indepen-dent experiments SEM.
3414 MASP-1 ACTIVATES ENDOTHELIAL PAR4
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

demonstrating that this PAR4 antagonist (trans-cinnamoyl-YPGKF-NH2) acts as an agonist in endothelial cells (40). Unfortu-nately, there is no selective PAR4 antagonist available at present thatcould be used in our experimental system. Taken together, theseresults suggest that neither PAR1 nor PAR2 is involved in theCa2� signal triggered by MASP-1, and PAR4 is the only protease-activated receptor that can mediate MASP-1 signaling.
We found that thrombin and MASP-1 activate HUVECs simi-larly but with slight differences. Thrombin generates a more in-tensive Ca2� signal, very probably because it cleaves both PAR1and PAR4, while MASP-1 cleaves only PAR4. Interestingly, in thecase of p38 phosphorylation, MASP-1 and thrombin elicit similareffects. Besides the differential utilization of PAR1 and PAR4 byMASP-1 and thrombin, we cannot completely exclude the involve-ment of another (non-PAR) receptor or cofactor in MASP-1signaling.
The lectin pathway of the complement system can be activatedat the site of vascular injury, where pathogenic bacteria enter thebloodstream. The proteases of the lectin pathway (MASP-1 andMASP-2) are associated with pattern-recognition molecules (i.e.,MBL and ficolins) that can recognize and bind via their C-terminallectin domains the foreign structures (e.g., carbohydrate arrays)present on the surface of pathogens. The binding of recognitionmolecules results in the activation of the associated serine pro-teases; thus, active MASP-1 is generated at the sites where bacteriainvade the body. Endothelial cells (41) and other cells involved inthe MBL-mediated clearance of self and non-self target structures(e.g., macrophages, dendritic cells) (42, 43) can bind MBL via itsN-terminal collagenous tail. A target structure, coated with multi-ple MBL-MASP complexes, affords multivalent interaction medi-ated by the collagenous domains of MBL molecules and the MBLreceptors on the host cells. This can significantly increase the localconcentration of active MASP-1 in the environment of leukocytesand endothelial cells. Active MASP-1 can then cleave PAR4 inthese cells, triggering proinflammatory reactions. Note, however,that in our substrate cleavage assay system we used short syntheticpeptides representing the P5-P1 side of the scissile bond of thetethered ligands of the PARs. Although PAR4 treated withMASP-1 or thrombin resulted in a single band with very similarmolecular mass in Western-blot (Fig. 4), we cannot exclude thatMASP-1 might cleave the PAR exodomains at other sites. In ourexperiments we used the recombinant catalytic fragment (CCP1-CCP2-SP) of MASP-1. Although this fragment is proteolyticallyequivalent to the full-length molecule, native MASP-1 is a dimerwith bigger size and it may act on the cell surface differently (e.g.,due to restricted accessibility to the cleavage site). It was shownearlier that PAR4 is a potential, active mediator in the proinflam-matory process, since it can contribute to leukocyte rolling, adher-ence, and recruitment (18). Inflammation is characterized by pro-thrombotic changes of the endothelial surface, as well as bymodified vascular permeability, vascular tone, and perfusion in theinflamed area (44). Higher permeability and perfusion jointly en-hance the extravasation of immunocompetent cells, while micro-thrombus formation can localize the inflammation and thereby pre-vent systemic infections. The Ca2� response of endothelial cellsleads to enhanced permeability and activation of the endothelialNO synthase enzyme, which results in lowered smooth muscletone and increased perfusion (45). It also leads to the degranulationof Weibel-Palade bodies, which increases thrombus formation aswell as the adhesion of leukocytes and platelets to endothelial cellsthrough von Willebrand factor and P-selectin (46). Taking ournovel observations into account, the direct endothelial cell-activat-ing property of MASP-1 may fundamentally contribute to the in-herent vascular changes that occur during the inflammatory reac-
tion, in an attempt to control and limit the damage to host tissues.Thus, endothelial PAR4 is a novel receptor linking the activationof complement system to the inflammatory reaction.
Our results also confirm the evolutionary and functional rela-tionship between the two major proteolytic cascade systems in theblood: the coagulation and complement systems. Cross-talk be-tween the two cascades can take place by several means. For ex-ample, the proteases of the lectin pathway can initiate coagulation(12, 47), whereas thrombin can serve as a C5 convertase in C3-deficient mice (48). MASP-1 seems to be a protease with proper-ties typical for both complement and coagulation proteases (2).MASP-1 is part of the complement system, since it binds to thesame recognition molecules (MBL, ficolins) as MASP-2, and itautoactivates when the recognition molecules bind to the surfaceof foreign cells. The structure, evolutionary status, and enzymaticpropeties of MASP-1, however, differ from those of typical, earlycomplement proteases (i.e., C1r, C1s, MASP-2). The fact thatMASP-1 cleaves and activates PAR4 substantiates our previousfindings about the thrombin-like properties of this protease, and itpoints to a yet undisclosed, novel connection between the com-plement and coagulation systems during the inflammatory process.
AcknowledgmentsWe thank Andras Szilagyi for critical reading of the manuscript, as well asKaroly Liliom and Attila Baksa for their advice on the experiments.
DisclosuresThe authors have no financial conflicts of interest.
References1. Walport, M. J. 2001. Complement: first of two parts. N. Engl. J. Med. 344:
1058–1066.2. Gal, P., L. Barna, A. Kocsis, and P. Zavodszky. 2007. Serine proteases of the
classical and lectin pathways: similarities and differences. Immunobiology 212:267–277.
3. Thiel, S., T. Vorup-Jensen, C. M. Stover, W. Schwaeble, S. B. Laursen,K. Poulsen, A. C. Willis, P. Eggleton, S. Hansen, U. Holmskov, et al. 1997. Asecond serine protease associated with mannan-binding lectin that activates com-plement. Nature 386: 506–510.
4. Chen, C. B., and R. Wallis. 2001. Stoichiometry of complexes between mannose-binding protein and its associated serine proteases: defining functional units forcomplement activation. J. Biol. Chem. 276: 25894–25902.
5. Presanis, J. S., K. Hajela, G. Ambrus, P. Gal, and R. B. Sim. 2004. Differentialsubstrate and inhibitor profiles for human MASP-1 and MASP-2. Mol. Immunol.40: 921–929.
6. Matsushita, M., S. Thiel, J. C. Jensenius, I. Terai, and T. Fujita. 2000. Proteolyticactivities of two types of mannose-binding lectin-associated serine protease.J. Immunol. 165: 2637–2642.
7. Rossi, V., S. Cseh, I. Bally, N. M. Thielens, J. C. Jensenius, and G. J. Arlaud.2001. Substrate specificities of recombinant mannan-binding lectin-associatedserine proteases-1 and -2. J. Biol. Chem. 276: 40880–40887.
8. Møller-Kristensen, M., S. Thiel, A. Sjöholm, M. Matsushita, and J. C. Jensenius.2007. Cooperation between MASP-1 and MASP-2 in the generation of C3 con-vertase through the MBL pathway. Int. Immunol. 19: 141–149.
9. Takahashi, M., D. Iwaki, K. Kanno, Y. Ishida, J. Xiong, M. Matsushita, Y. Endo,S. Miura, N. Ishii, K. Sugamura, and T. Fujita. 2008. Mannose-binding lectin(MBL)-associated serine protease (MASP)-1 contributes to activation of the lec-tin complement pathway. J. Immunol. 180: 6132–6138.
10. Hajela, K., M. Kojima, G. Ambrus, K. H. Wong, B. E. Moffatt, J. Ferluga,S. Hajela, P. Gal, and R. B. Sim. 2002. The biological functions of MBL-asso-ciated serine proteases (MASPs). Immunobiology 205: 467–475.
11. Ambrus, G., P. Gal, M. Kojima, K. Szilagyi, J. Balczer, J. Antal, L. Graf,A. Laich, B. E. Moffatt, W. Schwaeble, et al. 2003. Natural substrates and in-hibitors of mannan-binding lectin-associated serine protease-1 and -2: a study onrecombinant catalytic fragments. J. Immunol. 170: 1374–1382.
12. Krarup, A., K. C. Gulla, P. Gal, K. Hajela, and R. B. Sim. 2008. The action ofMBL-associated serine protease 1 (MASP1) on factor XIII and fibrinogen. Bio-chim. Biophys. Acta 1784: 1294–1300.
13. Coughlin, S. R. 2000. Thrombin signalling and protease-activated receptors. Na-ture 407: 258–264.
14. Coughlin, S. R. 2005. Protease-activated receptors in hemostasis, thrombosis andvascular biology. J. Thromb. Haemost. 3: 1800–1814.
15. Steinhoff, M., J. Buddenkotte, V. Shpacovitch, A. Rattenholl, C. Moormann,N. Vergnolle, T. A. Luger, and M. D. Hollenberg. 2005. Proteinase-activatedreceptors: transducers of proteinase-mediated signaling in inflammation and im-mune response. Endocr. Rev. 26: 1–43.
3415The Journal of Immunology
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

16. Kataoka, H., J. R. Hamilton, D. D. McKemy, E. Camerer, Y. W. Zheng,A. Cheng, C. Griffin, and S. R. Coughlin. 2003. Protease-activated receptors 1and 4 mediate thrombin signaling in endothelial cells. Blood 102: 3224–3231.
17. Hamilton, J. R., and T. M. Cocks. 2000. Heterogeneous mechanisms of endo-thelium-dependent relaxation for thrombin and peptide activators of protease-activated receptor-1 in porcine isolated coronary artery. Br. J. Pharmacol. 130:181–188.
18. Vergnolle, N., C. K. Derian, M. R. D’Andrea, M. Steinhoff, andP. Andrade-Gordon. 2002. Characterization of thrombin-induced leukocyte roll-ing and adherence: a potential proinflammatory role for proteinase-activated re-ceptor-4. J. Immunol. 169: 1467–1473.
19. Dobo, J., V. Harmat, E. Sebestyen, L. Beinrohr, P. Zavodszky, and P. Gal. 2008.Purification, crystallization and preliminary X-ray analysis of human mannose-binding lectin-associated serine protease-1 (MASP-1) catalytic region. Acta Crys-tallogr. Sect. F Struct. Biol. Cryst. Commun. 64: 781–784.
20. Oroszlan, M., E. Herczenik, S. Rugonfalvi-Kiss, A. Roos, A. J. Nauta,M. R. Daha, I. Gombos, I. Karadi, L. Romics, Z. Prohaszka, et al. 2006. Proin-flammatory changes in human umbilical cord vein endothelial cells can be in-duced neither by native nor by modified CRP. Int. Immunol. 18: 871–878.
21. Doleschall, M., B. Mayer, J. Cervenak, L. Cervenak, and I. Kacskovics. 2007.Cloning, expression and characterization of the bovine p65 subunit of NF�B.Dev. Comp. Immunol. 31: 945–961.
22. Davis, A. E., 3rd, P. Mejia, and F. Lu. 2008. Biological activities of C1 inhibitor.Mol. Immunol. 45: 4057–4063.
23. Beinrohr, L., J. Dobo, P. Zavodszky, and P. Gal. 2008. C1, MBL-MASPs andC1-inhibitor: novel approaches for targeting complement-mediated inflammation.Trends Mol. Med. 14: 511–521.
24. O’Brien, P. J., N. Prevost, M. Molino, M. K. Hollinger, M. J. Woolkalis,D. S. Woulfe, and L. F. Brass. 2000. Thrombin responses in human endothelialcells: contributions from receptors other than PAR1 include the transactivation ofPAR2 by thrombin-cleaved PAR1. J. Biol. Chem. 275: 13502–13509.
25. Fujiwara, M., E. Jin, M. Ghazizadeh, and O. Kawanami. 2004. Differential ex-pression of protease-activated receptors 1, 2, and 4 on human endothelial cellsfrom different vascular sites. Pathobiology 71: 52–58.
26. Hamilton, J. R., A. G. Frauman, and T. M. Cocks. 2001. Increased expression ofprotease-activated receptor-2 (PAR2) and PAR4 in human coronary artery byinflammatory stimuli unveils endothelium-dependent relaxations to PAR2 andPAR4 agonists. Circ. Res. 89: 92–98.
27. Albrecht, E. A., A. M. Chinnaiyan, S. Varambally, C. Kumar-Sinha,T. R. Barrette, J. V. Sarma, and P. A. Ward. 2004. C5a-induced gene expressionin human umbilical vein endothelial cells. Am. J. Pathol. 164: 849–859.
28. Ward, P. A. 2009. Functions of C5a receptors. J. Mol. Med. 87: 375–378.29. Terai, I., K. Kobayashi, M. Matsushita, and T. Fujita. 1997. Human serum man-
nose-binding lectin (MBL)-associated serine protease-1 (MASP-1): determina-tion of levels in body fluids and identification of two forms in serum. Clin. Exp.Immunol. 110: 317–323.
30. Møller-Kristensen, M., J. C. Jensenius, L. Jensen, N. Thielens, V. Rossi,G. Arlaud, and S. Thiel. 2003. Levels of mannan-binding lectin-associated serineprotease-2 in healthy individuals. J. Immunol. Methods 282: 159–167.
31. Harmat, V., P. Gal, J. Kardos, K. Szilagyi, G. Ambrus, B. Vegh, G. Naray-Szabo,and P. Zavodszky. 2004. The structure of MBL-associated serine protease-2 re-veals that identical substrate specificities of C1s and MASP-2 are realizedthrough different sets of enzyme-substrate interactions. J. Mol. Biol. 342:1533–1546.
32. Gal, P., V. Harmat, A. Kocsis, T. Bian, L. Barna, G. Ambrus, B. Vegh, J. Balczer,R. B. Sim, G. Naray-Szabo, and P. Zavodszky. 2005. A true autoactivating en-zyme: structural insight into mannose-binding lectin-associated serine protease-2activations. J. Biol. Chem. 280: 33435–33444.
33. Dobo, J., V. Harmat, L. Beinrohr, E. Sebestyen, P. Zavodszky, and Peter Gal.2009. MASP-1, a promiscuous complement protease: structure of its catalyticregion reveals the basis of its broad specificity. J. Immunol. 183: 1207–1214.
34. Bair, A. M., P. B. Thippegowda, M. Freichel, N. Cheng, R. D. Ye, S. M. Vogel,Y. Yu, V. Flockerzi, A. B. Malik, and C. Tiruppathi. 2009. Ca2� entry via TRPCchannels is necessary for thrombin-induced NF-�B activation in endothelial cellsthrough AMP-activated protein kinase and protein kinase C�. J. Biol. Chem. 284:563–574.
35. Dauphinee, S. M., and A. Karsan. 2006. Lipopolysaccharide signaling in endo-thelial cells. Lab. Invest. 86: 9–22.
36. Madge, L. A., and J. S. Pober. 2001. TNF signaling in vascular endothelial cells.Exp. Mol. Pathol. 70: 317–325.
37. Viemann, D., M. Goebeler, S. Schmid, K. Klimmek, C. Sorg, S. Ludwig, andJ. Roth. 2004. Transcriptional profiling of IKK2/NF-�B- and p38 MAP kinase-dependent gene expression in TNF-�-stimulated primary human endothelialcells. Blood 103: 3365–3373.
38. Taylor, K. E., J. C. Giddings, and C. W. van den Berg. 2005. C-reactive protein-induced in vitro endothelial cell activation is an artefact caused by azide andlipopolysaccharide. Arterioscler. Thromb. Vasc. Biol. 25: 1225–1230.
39. Nieman, M. T., and A. H. Schmaier. 2007. Interaction of thrombin with PAR1and PAR4 at the thrombin cleavage site. Biochemistry 46: 8603–8610.
40. Hollenberg, M. D., M. Saifeddine, S. Sandhu, S. Houle, and N. Vergnolle. 2004.Proteinase-activated receptor-4: evaluation of tethered ligand-derived peptides asprobes for receptor function and as inflammatory agonists in vivo.Br. J. Pharmacol. 143: 443–454.
41. Oroszlan, M., M. R. Daha, L. Cervenak, Z. Prohaszka, G. Fust, and A. Roos.2007. MBL and C1q compete for interaction with human endothelial cells. Mol.Immunol. 44: 1150–1158.
42. Ogden, C. A., A. deCathelineau, P. R. Hoffmann, D. Bratton, B. Ghebrehiwet,V. A. Fadok, and P. M. Henson. 2001. C1q and mannose binding lectin engage-ment of cell surface calreticulin and CD91 initiates macropinocytosis and uptakeof apoptotic cells. J. Exp. Med. 194: 781–795.
43. Nauta, A. J., G. Castellano, W. Xu, A. M. Woltman, M. C. Borrias, M. R. Daha,C. van Kooten, and A. Roos. 2004. Opsonization with C1q and mannose-bindinglectin targets apoptotic cells to dendritic cells. J. Immunol. 173: 3044–3050.
44. Martorell, L., J. Martinez-Gonzalez, C. Rodriguez, M. Gentile, O. Calvayrac, andL. Badimon. 2008. Thrombin and protease-activated receptors (PARs) in athero-thrombosis. Thromb. Haemost. 99: 305–315.
45. Busse, R., and A. Mulsch. 1990. Calcium-dependent nitric oxide synthesis inendothelial cytosol is mediated by calmodulin. FEBS Lett. 265: 133–136.
46. Cleator, J. H., W. Q. Zhu, D. E. Vaughan, and H. E. Hamm. 2006. Differentialregulation of endothelial exocytosis of P-selectin and von Willebrand factor byprotease-activated receptors and cAMP. Blood 107: 2736–2744.
47. Krarup, A., R. Wallis, J. S. Presanis, P. Gal, and R. B. Sim. 2007. Simultaneousactivation of complement and coagulation by MBL-associated serine protease 2.PLoS ONE 2: e623.
48. Huber-Lang, M., J. V. Sarma, F. S. Zetoune, D. Rittirsch, T. A. Neff,S. R. McGuire, J. D. Lambris, R. L. Warner, M. A. Flierl, L. M. Hoesel, et al.2006. Generation of C5a in the absence of C3: a new complement activationpathway. Nat. Med. 12: 682–687.
3416 MASP-1 ACTIVATES ENDOTHELIAL PAR4
by guest on April 5, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from