cholesterol regulates vegfr-1 (flt-1) expression and...
TRANSCRIPT

1 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Cholesterol Regulates VEGFR-1 (FLT-1) Expression and Signaling in Acute
Leukemia Cells.
Casalou C. 1,2,3, Costa, A.1,2,3 , Carvalho, T.1,2,3, Gomes, A.L.1,2,3, Zhu, Z.4, Wu, Y.4
and Dias, S.1,2,3£
1 Angiogenesis Group, Instituto Português de Oncologia Franscisco Gentil de
Lisboa, EPE (CIPM/IPOLFG)
2 Neoangiogenesis Group, Instituto Gulbenkian de Ciência, Oeiras
3 CEDOC, Faculdade de Ciências Médicas, Universidade Nova de Lisboa,
Portugal.
4 Imclone Systems, New York, USA.
£ Corresponding author
Running title: FLT-1 signaling is modulated by cellular cholesterol
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

2 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Abstract
Vascular endothelial growth factor receptors -1 (FLT-1) and -2 (KDR) are
expressed on subsets of acute myeloid (AML) and lymphoid (ALL) leukemia
cells, in which they induce cell survival, proliferation and migration. However, little
is known regarding possible co-factors that regulate VEGF receptors expression
and activation on leukemia cells. Here we show that cholesterol accumulates in
leukemia-rich sites within bone marrow of xenotransplanted SCID mice.
Therefore, we hypothesized that cholesterol-rich domains might regulate FLT-1
signaling and chemotaxis of acute leukemias. We then showed that (i) FLT-1
accumulates in discrete cholesterol-rich membrane domains where it associates
with caveolin-1; and that (ii) PlGF/VEGF stimulation promoted FLT-1 localization
in such cholesterol-rich domains. Accordingly, FLT-1 localization and its
phosphorylation were abrogated by -methyl-β-cyclodextrin (MβCD) which
removes cellular cholesterol and by Nystatin, an inhibitor of lipid raft endocytosis.
Mechanistically, cholesterol increased FLT-1 expression and promoted
PlGF/VEGF-induced leukemia cells viability and also induced VEGF production
by the leukemia cells in vitro. Taken together, we conclude that cholesterol
regulates VEGF:VEGFR-1 signaling on subsets of acute leukemias, modulating
cell migration and viability, which may be crucial for disease progression. Finally,
we provide evidence obtained from Human AML samples that primary leukemia
cells accumulate significantly more cholesterol than normal cells, and that
cholesterol accumulation correlates with disease aggressiveness.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

3 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Introduction
Acute leukemia cells have been previously shown to express VEGF
receptors, which can be stimulated in a paracrine or autocrine manner, resulting
in increased cell survival, proliferation and migration (1). VEGF signaling through
VEGFR-1 (FLT-1) on acute leukemias was shown to involve p38 and Erk1/2
activation, resulting in caveolae formation (2). Others have shown that VEGF
stimulation of subsets of leukemias results in the activation of a downstream
signaling pathway which mainly involves the activation of the Pi3 kinase pathway
(3).
Concerning the function of the different VEGF receptors on leukemia cells,
we have recently shown that FLT-1 mediates leukemia migration within the bone
marrow (BM) microenvironment, promoting leukemia expansion and ultimate exit,
to colonize extramedullary sites (4). These findings led us to hypothesize that
other signals within the BM microenvironment might cooperate or promote VEGF
signaling on leukemia cells, which in turn would contribute towards favoring
leukemia migration and invasion, worsening disease outcome.
Plasma membrane lipid raft domains, which contain high concentrations of
cholesterol and sphingolipids, are known to function as centers of signaling
complexes. The ability of lipid rafts to enhance receptor signaling, has led to the
concept of a signalosome, a region where proteins are localized together to
facilitate receptor signaling. A vast body of literature is available concerning the
localization of EGF receptors in lipid rafts, and the subsequent regulatory
pathway involving the intracellular transport of EGF receptor (5). Much less is
known about the involvement of membrane rich lipid domains and VEGF
signaling (6). Our recent data showed that inhibitors of lipid raft assembly,
including nystatin, blocked VEGF-induced leukemia migration (4), which strongly
suggested that cholesterol rich domains might in fact regulate VEGF signaling on
malignant cells such as acute leukemias.
In the present report we exploited the biochemical pathways involved in
VEGF signaling in AML cells, and demonstrate that FLT-1 is modulated by
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

4 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
cellular cholesterol. We show that FLT-1 co-localizes with membrane rich
cholesterol domains, whose assembly is essential for FLT-1 expression and
activation on leukemia cells. Moreover, cholesterol content of acute leukemia
patient samples correlates with disease aggressiveness. As such, our data reveal
novel possibilities of therapeutic intervention on subsets of acute leukemias.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

5 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Results
Acute leukemia cells localize in cholesterol-rich niches in the BM
microenvironment in vivo
We have previously observed that acute leukemia cells migrate in an FLT-1
dependent manner within the bone marrow (BM) microenvironment towards the
epiphysis, en route to colonize other organs (4). In the present study we detected
specific cholesterol accumulations in leukemia-rich BM sites in vivo. For this, we
stained smears of BM collected on days 10-12 after mice inoculation with
leukemia cells, with Nile red, to detect intracellular lipids (7, 8). We observed
major accumulations of lipids (neutral lipids, i.e. cholesterol esters and polar
lipids) around BM sinusoids, where leukemia cells also tend to accumulate (figure
1). These data suggests that leukemia cell migration within the BM
microenvironment results in their accumulation in cholesterol-rich areas.
Avidity for cholesterol of leukemia cells
We compared cholesterol content of mononucleated (MNC) cells isolated from
healthy donors with MNC obtained from AML patients (2A). Patient samples were
grouped into pediatric samples, adult samples I (AML patients with ages between
30-35 years old) and adult samples II (AML patients with ages between 64-79
years old). In peripheral blood AML patient samples (AML8 and 15) the levels of
intracellular cholesterol were increased by 2,4-3,4 fold in relation to healthy
donor’s samples. AML patients display increased intracellular cholesterol, in
particular older patients with 4 to 6 fold increase (Adult samples II). After clinical
bone marrow remission, intracellular cholesterol levels decreased to levels
compared to that obtained from healthy donors (AML3, 4 and 16-BM-R). Also,
MDS sample (patient with myelodysplasic syndrome) has lower levels of
cholesterol. Furthermore, pediatric AML samples present 3 to 4 fold increased
levels of cellular cholesterol. In figure 2B, AML cells (HEL and HL60 cell lines)
showed 2 to 4 fold significant increase of cellular cholesterol (control-1 and -2,
respectively), when compared with cells isolated from healthy donors. Next, we
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

6 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
disturbed cholesterol homeostasis of leukemia cells by exposure for 4 hours to
10μM methyl-β-cyclodextrin (MβCD), which depletes cholesterol from cellular
membranes, or by increasing its cellular cholesterol levels with the use of
cholesterol+MβCD complexes (0,4μM). Cholesterol+MβCD complex treatment
increased by 1,5-2,3 fold cellular cholesterol of leukemia cells when compared to
untreated cells. This increase was abolished by MβCD and by nystatin treatment,
which inhibits lipid raft endocytosis. Additionally, PlGF treatment also increased
intracellular cholesterol of AML cells, an effect that was reverted to control levels
(control-2; figure 2B) by the use of a neutralizing antibody against FLT-1 (6.12
Ab; p<0,02; Imclone, N.Y.). Acute leukaemia cells possess more intracellular
cholesterol when compared to normal cells. Furthermore, FLT-1 activation by
PlGF further potentiates this effect. Additionally, cellular cholesterol is highly
increased in AML patients, an effect that was reverted after clinical bone marrow
remission.
PlGF induces FLT-1 accumulation and co-localization with caveolin-1 in lipid rafts
We have previously reported that FLT-1 associates in vitro with caveolin-1, the
main component of a sub-type of lipid-rafts (caveolae), in AML cells (2). This
suggested that FLT-1 mediated signaling in acute leukemia cells might depend
on cholesterol-raft membrane domains. Therefore, in the present study we
determined a more precise membrane location of FLT-1 on leukemia cells and
asked whether it was affected by PlGF-stimulation and/or cholesterol
disturbance. We isolated lipid-rafts using sucrose-density gradients and this
analysis revealed that FLT-1 and caveolin-1 co-localize in 2 distinct regions of the
sucrose gradients; a caveolin-enriched membrane region (fractions 4 and 5; 6%-
30% sucrose) and a cytosolic region (fractions 10-11; 35% sucrose), as
assessed by caveolin-1 (a component of lipid-rafts) co-sedimentation (figure 3A).
Upon PlGF/VEGF stimulation, FLT-1 was localized preferentially into lipid-raft
fractions (fractions 4 and 5), an effect that was reverted in the presence of the
FLT-1 inhibitor IR1 (figure 3A). Also, inhibition of FLT-1 by IR1 treatment,
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

7 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
removes caveolin-1 from the lipid raft fractions. Using confocal microscopy we
observed that FLT-1 (in red) and caveolin-1 (in green) co-localized in lipid
raft/caveolin-1 rich structures (figure 3B). (FLT-1 antibody specificity is shown in
additional file 1). To determine whether FLT-1 distribution in lipid-rafts was
affected by cholesterol disturbance, sucrose gradient-generated lipid-raft
fractions 4 and 5 and cytosolic fractions 10 and 11 were concentrated and
analyzed by Western-blot as before. As shown in figure 3C, cholesterol
enrichment accumulated FLT-1 in lipid-raft fractions, while MβCD extracted FLT-
1 from lipid-raft fractions (see quantification in figure 3D). β-actin was used as a
loading control (figure 3C). As determined by confocal microscopy, after
cholesterol enrichment FLT-1 co-localized with caveolin-1, whereas cholesterol
extraction with MβCD reduced this co-localization (figure. 3E). These results
indicating the preferential localization of FLT-1 in lipid-rafts after intracellular
cholesterol enrichment, suggest that FLT-1-raft interactions may regulate
PlGF/VEGF mediated signaling.
Cholesterol enrichment of acute leukemia cells induces FLT-1 activation in lipid
rafts
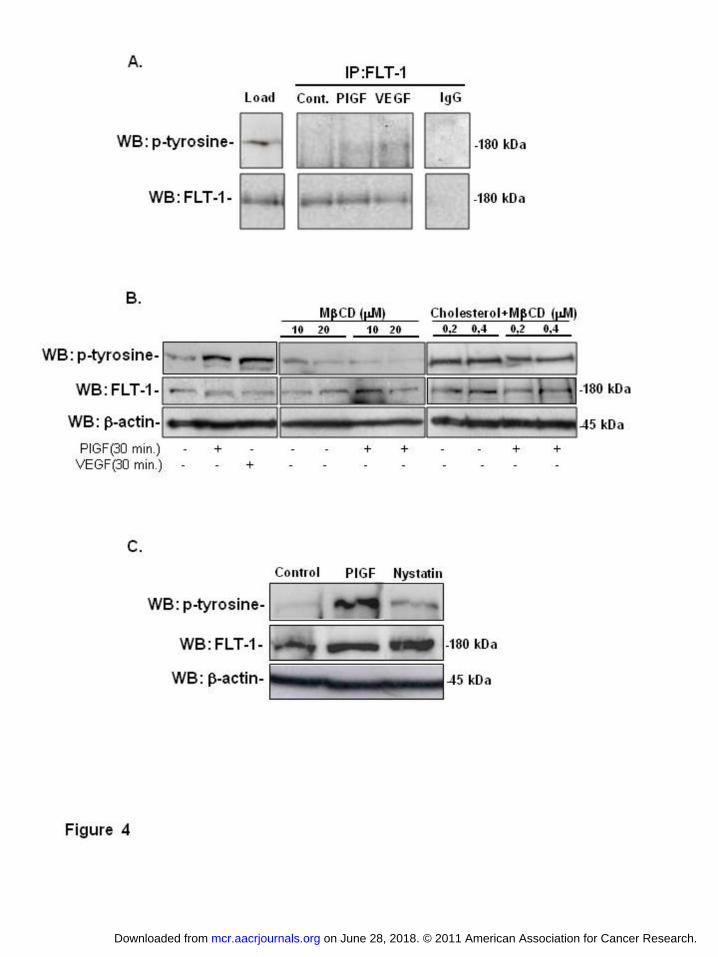
In AML cells, FLT-1 is phosphorylated by PlGF/VEGF treatment for 30 minutes
(figure 4A), as assessed by co-immunoprecipitation of FLT-1 with pan phosphor-
tyrosine antibody. We determined the consequences of disturbing cholesterol
homeostasis for FLT-1 activation on AML cells. We observed that cholesterol
extraction abolishes FLT-1 phosphorylation induced by PlGF, in a dose
dependent manner. In contrast, cholesterol enrichment increased FLT-1
phosphorylation (activation) in the absence or presence of PlGF (figure 4B).
Inhibition of FLT-1 activation was achieved by the use of nystatin, an inhibitor of
lipid-raft formation and endocytosis, which impeded PlGF-induced FLT-1
phosphorylation (figure 4C). β-actin was always used as loading control. Taken
together, these data suggests that FLT-1 activation (phosphorylation) is affected
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

8 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
by cellular cholesterol levels, and in particular by agents that perturb the
formation of lipid rafts/cholesterol rich membrane domains.
Cholesterol enrichment modulates PlGF-induced AML cells properties
To evaluate the role of cholesterol in acute leukemia cells properties, we
assessed cell viability, migration and VEGF production on cholesterol enriched or
depleted cells, alone or in the presence of PlGF. Viability was assessed after 24-
48 hours by counting cell viability with trypan exclusion dye. As previously
reported, PlGF induced a modest increase in AML cells viability. However,
leukemia cells cholesterol enrichment increased cell viability after 24hours;
interestingly, co-treatment of cholesterol enrichment with PlGF, induced a further
increase in leukemia cell viability (figure 5A). In contrast, PlGF-induced cell
viability was significantly reduced by MβCD treatment. Cholesterol enrichment of
AML cells for 4-16 hours increased their chemotactic (migratory) response
towards PlGF (figure 5B), an effect that was reverted in the presence of FLT-1
neutralizing agents. In addition, cholesterol enrichment also increased VEGF
production by AML cells in vitro (see additional file 2). Taken together, these data
suggest that cholesterol cellular levels affect FLT-1 mediated increase in cell
viability, chemotaxis and VEGF production.
FLT-1 expression on AML cells increases upon cholesterol exposure
Besides its effects in the regulation of FLT-1 activation and sub-cellular
localization, next we asked whether cholesterol levels also affected FLT-1
expression. In fact, cholesterol enrichment up-regulates FLT-1 mRNA expression
in leukemia cells (figure 6A). PlGF further increased FLT-1 expression in
cholesterol enriched cells, an effect that was reverted by the FLT-1 inhibitors.
This effect on FLT-1 mRNA expression is characteristic of leukemia cells since
FLT-1 mRNA expression remains unaltered by cholesterol disturbances in Huvec
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

9 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
cells (figure 6B). It was previously reported that CREB/ATF element regulates the
basal transcription of FLT-1 expression (18). Moreover, we observed that
increasing intracellular cholesterol on AML cells further potentiates the binding of
CREB complexes to the FLT-1 promoter region, thereby regulating FLT-1
expression (see additional figure 3). In contrast, KDR mRNA expression was not
altered by increasing cellular cholesterol in leukemia cells (figure 6C).
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

10 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Discussion
In this report we reveal novel molecular evidence by which cellular
cholesterol regulates the function of a receptor tyrosine kinase on malignant
leukemia cells. In detail, we show that cholesterol affects FLT-1 expression,
localization and activation, thereby modulating cellular phenotypes including
viability, chemotaxis/migration and VEGF production. The mechanisms by which
cholesterol interferes with FLT-1 signaling and cellular localization are still not
completely understood, but may involve the intracellular transport machinery and
the regulation/assembly of specific membrane domains, including vesicles and
lipid rafts. Other receptor tyrosine kinases, most notoriously EGF receptor, have
been shown to be transported intracellularly in vesicles, in and out of the cell onto
the cell nucleus where EGF receptor was shown to activate transcription (5).
Whether FLT-1 recycling involves a similar mechanism of intracellular transport is
still undisclosed. Nevertheless, the present report reveals that FLT-1 localizes
preferentially in specific lipid-enriched membrane domains. Detailed biochemical
analysis of cellular extracts, together with the use of biochemical inhibitors,
suggests these membrane domains may be considered lipid rafts. As such, our
data strongly suggests lipid rafts are essential for VEGF receptor signaling on
malignant leukemia cells.
We have previously shown that subsets of acute leukemias respond to
VEGF/PlGF gradients within the BM microenvironment, migrating towards the
epiphysis of long bones, from where the leukemia cells leave the BM onto the
peripheral blood, en route to other target organs (4). From these findings it
became clear that leukemia cells migrate towards specific “niches” within the BM
microenvironment. Recent studies have suggested leukemia cells “create”
specific niches within the BM microenvironment, where the malignant cells will
thrive and proliferate, eventually replacing the normal hematopoietic elements
(9). The signals/molecular cues involved in this “invasion” of the BM by malignant
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

11 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
cells are still largely undisclosed, but are believed to involve SDF-1:CXCR4 and
possibly integrin-mediated signaling (10-12).
Our present report suggests the BM cholesterol levels and the cholesterol
distribution throughout the BM may play an important part during leukemia
engraftment, expansion and perhaps also during leukemia spread. In detail, we
provide evidence that PlGF/VEGF signaling via FLT-1 is affected by cellular
cholesterol levels, and that specific cholesterol accumulations are seen in
leukemia-rich sites in the BM, in vivo. Interestingly, we have recently discovered
that systemic cholesterol levels also affect the levels of SDF-1 in the BM
microenvironment (13). This strongly suggests that cholesterol may affect
leukemia engraftment and expansion, by interfering with SDF-1:CXCR4 signaling
and VEGF:FLT-1 signaling respectively.
A link between cholesterol avidity and acute leukemias has been previously
suggested. In our present study AML patient samples show increased levels of
intracellular cholesterol compared to healthy donor’s samples. Moreover, the BM
cholesterol content correlates with disease aggressiveness (and stage); leukemia
patients undergoing clinical remission show a corresponding decrease in
intracellular cholesterol levels. Other studies have reported that cholesterol
uptake by leukemia cells promotes their survival and resistance to chemotherapy
(14,15). These studies led to the use of statins (cholesterol lowering agents) as
therapeutic targets for subsets of acute leukemias (16-17), with reported clinical
activity and efficacy. Nevertheless, to our knowledge, there have been no reports
explaining the importance of cholesterol in VEGF-mediated signaling on acute
leukemia cells. All together, the findings reported in the present manuscript have
implications for the understanding of the regulation of the BM microenvironment,
during the onset/engraftment and expansion/progression of acute leukemias. The
biochemical mechanisms described here, showing that lipid-enriched membrane
domains regulate VEGF receptor signaling, may be relevant also in the context of
other receptor tyrosine kinases and other tumor types.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

12 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Materials and Methods
Cell culture
HEL, HL60 and 697 cells were obtained from American Type Culture Collection.
Cells were maintained in RPMI 1640 medium supplemented with 10% fetal
bovine serum, L-glutamine 100mM and 1% penicillin-streptomycin (Invitrogen
Life Tecnologies). Huvec cells were maintained in endothelial cell growth medium
containing 5% FBS (EBM-2; Cambrex, Inc.) at 37ºC with humidified 95% air/5%
CO2.
Human samples
Sixteen patient samples (bone marrow-BM or peripheral blood-PB) were used
and grouped in 4 pediatric AML samples, 2 of them in clinical bone marrow
remission; 4 adult samples I, with ages between 30-35 years old; 7 adult samples
II, with ages between 74-79 years old, one in clinical bone marrow remission.
Also, one MDS sample was used (myelodysplasic syndrome) in the adult sample
II group. Mononuclear cells were collected from BM/PB and isolated using
Ficoll/Histopaque gradient. Dry pellets were processed for cholesterol
quantification according to Amplex red kit.
RNA extraction and RQ-PCR
Leukemia cells were analyzed for VEGF-1(FLT-1) by RQ-PCR (ABI PRISM 7700
Sequence Detection System and SYBR Green Master Mix Kit; Applied
Biosystems). Total cellular RNA was extracted using trizol protocol (Sigma-
Aldrich) and cDNA was synthesized following conventional protocols. The 18S
gene was used as a standard reference. The relative expression of FLT-1 and
KDR was obtained using comparative threshold cycle (CT) method. Primer
sequences: FLT-1 sense; 5’-CCTCGCCGGAAGTTGTAT-3’; FLT-1 anti-sense;
5’-GTCAAATAGCGAGCAGATTTCTCA-3’; KDR sense; 5’-
ATTCCTCCCCCGCATCA-3’; KDR anti-sense; 5’-GCTCGTTGGCGACTCTT-3’.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

13 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Whole-cell lysate preparation and Western blotting
Acute leukemia cells were stimulated for 30 minutes with recombinant VEGF165
(50ng/mL), PlGF (100ng/mL) and/or treated with for 2-4hours with MβCD (10mM
or 20mM), Cholesterol+MβCD (0,2mM or 0,4mM), Nystatin (50μg/mL). After
stimulation/treatment, total protein extracts were obtained by suspending cell
pellets in cold buffer A (50 mM Tris-HCl, pH 7,4; 1% (v/v) Triton X-100; 150mM
NaCl; 1mM EDTA; 0,1% (v/v) SDS), in the presence of protease and
phosphatase inhibitors, for 30 minutes on ice followed by centrifugation at 12 000
g, for 15 minutes at 4 ºC. Protein concentrations were determined using the Bio-
Rad Laboratories DC protein assay kit and equal amounts were separated by
SDS-PAGE gels, transferred onto nitrocellulose membranes and processed for
Western blotting. Primary antibodies: anti-FLT-1 (0,5μg/mL; Santa Cruz
Biotechnology, Inc.), anti-caveolin-1 (1:100; BD-Biosicences), anti-phospho-
tyrosine (1:50, 0,5μg/mL; Santa Cruz Biotechnology, Inc), anti-β-actin (1:2500,
Sigma-Aldrich). Secondary antibodies HRP (Horseradish peroxidase)-conjugated
were used at 1:2500 and the enhanced chemiluminescence (ECL) detection
system and Kodak films (Amersham Pharmacia Biotech, Piscataway, NJ) were
used to visualize the presence of proteins on the nitrocellulose blots. Bands were
quantified using Image J software (rsb.info.nih.gov/ij/).
Immunoprecipitation assay
HEL lysates (900μg) were pre-cleared (1hour) with Protein G-Sepharose beads
(Sigma Aldrich) and then incubated overnight at 4ºC with anti-FLT-1 (1μg/mL;
Santa Cruz) or with rabbit IgG as a negative control of the co-
immunoprecipitation procedure. Protein G-Sepharose beads were then added
and mixed for 2 hours at 4ºC. Beads were recovered by centrifugation, washed
with cold buffer A or with buffer A supplemented with high salt concentration
(500mM Nacl), and resuspended in (20μl) Laemmli buffer. After boiling at 95ºC
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

14 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
for 5 minutes, the immunoprecipitates were analyzed by 8% SDS-Page gels
followed by western-blotting with anti-phospho tyrosine and anti-FLT-1 antibodies
(1μg/mL; Santa Cruz).
Sucrose Density Centrifugation and Isolation of Lipid Raft Fractions
Acute leukemia cells (HEL; 1x108) stimulated for 30 minutes with PlGF
(100ng/mL) or treated 1 hour with IR1 inhibitor (2μM; Calbiochem.), MβCD
(10mM) and MβCD+cholesterol (0,2mM) were washed with PBS and cell pellets
were suspended in 1.0ml of 1% (v/v) Triton X-100 in TNEV buffer (100mM Tris-
HCL (pH 7.5), 150mM NaCl, 5mM EDTA, 1mM Na2VO3, 1mM PMSF, 1x
protease inhibitors (Roche)) on ice for 60 minutes. Cells were homogenized with
10 passages through a 22-gauge needle and nuclei were removed by
centrifugation at 800 g for 8 minutes at 4ºC. The supernatants were mixed 1:1 in
85% sucrose (v/v)/TNEV buffer. The mixture (2ml) was transferred to the bottom
of the ultra-centrifuge tube and 2 solutions with different sucrose concentrations
in TNEV buffer were added sequentially (6ml of 35% (v/v) sucrose and 3,5ml of
5% (v/v) sucrose). The discontinuous gradients were separated by centrifugation
in a swing-out rotor (SW41TI) at 38,000 g during 18 hours at 4ºC in a Beckman
XL-80 Ultracentrifuge. One-milliliter fractions were collected sequentially from the
top to the bottom of the tube and Western blot analysis were done with
antibodies against FLT-1, caveolin-1. After identification by Western-blot
fractions from sucrose gradients (lipid/raft: 4 and 5; cytosolic fractions: 10 and
11) were concentrated by centrifugation (4000 g for 20 minutes at 4ºC) using
Amicon Ultra-4 devices with 10kDa cut-off membranes (millipore).
Cell viability and migration assays
Cells (1x105/mL) were cultured in serum-free RPMI medium for 48 hours in the
presence of PlGF (100ng/mL), methyl-β-cyclodextrin (MβCD; 10mM) and
cholesterol+MβCD complexes (0,4mM). When applicable, cells where previously
treated for 2 hours with 2μM IR1 (tyrosine kinase inhibitor of FLT-1;
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

15 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Calbiochem.), or with neutralizing antibodies directed to FLT-1 (1μg/mL 6.12
antibody, Imclone). Cell viability was determined at 24 hours and 48 hours by
trypan blue exclusion and cell counts (with the aid of a hemocytometer). Each
experiment was done in triplicate. Cell migration was assayed using a modified
version of a transwell migration technique described previously (Hamada et al,
1998). Serum starved cells (1x106 cell/mL) were treated for 4 hours or 18 hours
with cholesterol+MβCD. Cell aliquots (100μL) were added to 8μm-pore transwell
inserts of migration system (6.5μm in diameter, Costar) and let migrate for 4-6
hours towards PlGF in the absence/presence of neutralizing antibodies directed
to FLT-1. Cell counts were done in 7 distinct power-fields (20x magnification) with
Olympus CK2 microscope. Experiments were done in triplicate, and results are
shown as the number of migrating cells/mL.
Human VEGF Elisa and cholesterol measurement
VEGF production by leukemia cell lines used was determined by enzyme-linked
immunosorbent assay (ELISA). VEGF in serum free medium was quantified
using the human VEGF ELISA kit (Calbiochem). Cellular cholesterol was
detected using the Amplex Red Cholesterol Assay Kit according to
manufacturer´s instructions (Molecular Probes).
Immunofluorescence
Leukemia cells (5x106) were serum starved for 16 hours and further attached to
poly-L-lysine coated coverslips for 10 minutes at 37ºC. After a brief wash in PBS
cells were stimulated with PlGF (100ng/mL) for 30 minutes and/or treated with
MβCD (10mM) or MβCD+Cholesterol complexes (0,2mM cholesterol) for 2 hour-
4 hours. The cells were fixed in 2% (v/v) paraformaldehyde for 10 minutes at
room temperature, washed in PBS twice and permeabilized in 0,1% (v/v) triton
X-100 for 30 seconds. After blocking in PBS (Invitrogen Life technologies)
supplemented with 0,1% (w/v) BSA, 5%(v/v) complete goat serum, rabbit anti-
human FLT-1 (1,5 μg/mL; Santa Cruz Biotechnology, Inc), mouse anti-caveolin-1
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

16 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
(1:100; BD biosciences) were used overnight at 4ºC. The cells were washed and
incubated with Alexa fluor 594 or 488 secondary antibodies at 1:500 (Molecular
probes) for 90 minutes and washed with PBS. Samples were mounted in
Vectashield and analyzed by Confocal microscopy in a True Confocal Scanner
Leica TCS SP2; Leica Microsystems; objectives HCX PL APOCS 63 x 1.4 oil.
Sets of optical sections with 0.5-μm intervals along the z-axis were obtained from
the top to the bottom of cells. Z-projections of the acquired images were
obtained using ImageJ software (rsb.info.nih.gov/ij/).
Bone marrow smears and Nile red staining
Ten to twelve days after 697 and HL60 cells Xenotransplantation, mice were
sacrificed and bone marrow (BalbSCID mice) was removed in toto by flushing
one femoral cavity. Bone marrow smears were prepared by streaking the
exposed bone marrow onto a glass slide. Pressure while executing the smears
was adjusted to disperse cells in a monolayer without disrupting cells and
vascular structures integrity. Bone marrow smears were air dried, fixed in cold
acetone for 10 minutes and stained with Nile Red for 15 minutes. Images were
acquired on a Zeiss Axioplan microscope with a Zeiss Axioxcam MRm
(amplification: x200, x630). Nile Red solution (1:6 diluted in 75% (v/v) glycerol)
was prepared from a stock Nile Red solution (100μg/mL in ethanol; sigma-
N3013). Nile red stained with excitation wavelength of 450–500 nm neutral lipids
(yellow‐gold emission) and 515–560 nm polar lipids (red emission) (8).
Eletrophoretic mobility shift assay (EMSA)
Nuclear extraction and electrophoretic gel mobility shift assays were performed
following standard methodology as described elsewhere (19). Briefly,
oligonucleotide probe (sequence: ACCCCTTGAGTCACCAGAAGG) was labeled
with [γ32-P] ATP using T4 polynucleotide kinase (Promega) and purified in Micro-
spin G-50 columns (Bio-Rad). For the EMSA analysis, 10 μg of nuclear proteins
were pre-incubated with EMSA binding buffer (Promega) as well as 15 ng/μl
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

17 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
poly(dI)-poly(dC) at room temperature 10 minutes before the addition of the
radiolabeled oligonucleotide for an additional 25 minutes. For Supershift studies,
before addition of the radiolabeled probe, samples were incubated for 30 minutes
with 4 μg of CREB-1 antibody (H-74; Santa Cruz).
Statistical analysis
Results are expressed as mean plus or minus SD. Data were analyzed using the
unpaired 2-tailed Student t test. P values of less than 0.05 were considered
significant.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

18 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
List of abbreviations used BM- bone marrow; PB- peripheral blood ALL- acute leukemia cells; AML- acute
myeloid leukemia; MβCD- methyl-β-cyclodextrin; HPF- high power fields
Acknowledgments This study was supported by GlaxoSmithkline. Cristina Casalou, Ana Costa, Ana
L. Gomes and Tânia Carvalho are recipients of FCT (Portuguese Government)
Fellowships. The authors would like to thank other members of the Angiogenesis
Lab. for useful discussions and for critically reading this manuscript.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

19 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
References
1. Hicklin, DJ and Ellis, LM. Role of Vascular Endothelial Growth Factor Pathway
in Tumor Growth and Angiogenesis. Journal of Clinical Oncology 2005;23:1011-
1027.
2. Casalou C, Fragoso R, Nunes, JFM, Dias, S. VEGF/PLGF induces leukemia
cell migration via P38/ERK1/2 kinase pathway, resulting in Rho GTPases
activation and caveolae formation. Leukemia 2007;21:1590-94.
3. Fragoso R, Elias AP, Dias S. Autocrine VEGF loops, signaling pathways, and
acute leukemia regulation. Leukemia & Lymphoma 2007;48:481-8.
4. Fragoso R, Pereira T, Wu Y, Zhu Z, Cabeçadas J, Dias S. VEGFR-1 (FLT-1)
activation modulates acute lymphoblastic leukemia localization and survival
within the bone marrow, determining the onset of extramedullary disease. Blood
2006;107:1608-16.
5. Patra, SK. Dissecting lipid raft facilitated cell signaling pathways in cancer.
Biochem Biophys Acta 2008;1785 (2):182-206.
6. Labrecque, L, Royal, I, Surprenant, DS, Patterson, C, Gingras, D,Béliveau, R.
Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1
and plasma membrane cholesterol. Molecular Biology of the cell2003;14:334-
347.
7. Diaz, G, Melis, M., Batetta, B., Angius, F., Falchi, A.M. Hydrophobic
characterization of intracellular lipids in situ by Nile Red/Yellow emission ratio.
Micron 2008;39:819-824.
8. Greenspan P, Fowler, SD. Spectrofluorometric studies of the lipid probe, nile
red. Journal of Lipid Research 1985;26:781-89.
9. Colmone, A, Amorim, M, Pontier, AL, Wang, S, Jacblonski, E, Sipkins, DA.
Leukemic Cells Create Bone Marrow Niches That Disrupt the Behavior of Normal
Hematopoietic Progenitor Cells. Science 2008;322:1861-1865.
10. Peled, A, Kollet, O, Ponomaryov, T, et al. The chemokine SDF-1 activates
the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

20 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood
2000;95:3289–3296.
11. Kijowski, J, Baj-Krzyworzeka, M, Majka, M, et al. The SDF-1-CXCR4 axis
stimulates VEGF secretion and activates integrins but does not affect
proliferation and survival in lymphohematopoietic cells. Stem Cells 2001;19:453–
466.
12. Hidalgo A, Sanz-Rodriguez F, Rodriguez-Fernandez JL et al. Chemokine
stromal cell-derived factor-1alpha modulates VLA-4 integrin-dependent adhesion
to fibronectin and VCAM-1 on bone marrow hematopoietic progenitor cells.
Experimental Hematology 2001;29:345–355.
13. Gomes, A.L., Carvalho, T., Serpa, J., Torre, C., Dias, S.,
Hypercholesterolemia promotes bone marrow cell mobilization by perturbing the
SDF-1:CXCR4 axis. Blood 2010; [Epub ahead of print].
14. Li, HY, Appelbaum FR, Willman CL, Zager RA, Banker DE. Cholesterol-
modulating agents kill acute myeloid leukemia cells and sensitize them to
therapeutics by blocking adaptive cholesterol responses. Blood 2003;101: 3628-
34.
15. Banker, DE, Mayer, SJ, Li, HY, Willman, CL, Appelbaum, FR, Zager, RA.
Cholesterol synthesis and import contribute to protective cholesterol increments
in acute myeloid leukemia cells. Blood 2004;104:1816-24.
16. Sassano A, Katsoulidis E, Antico G, et al. Supressive effects of statins on
acute promyelocytic leukemia cells. Cancer Research 2007;67:4524-32.17.
17. Kornblau SM, Banker DE, Stirewalt D, et al. Blockade of adaptive defensive
changes in cholesterol uptake and synthesis in AML by the addition of
pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood
2007;109:2999-3006.
18. Morishita, K, Johnson, DE and Williams, LT. A novel promoter for Vascular
Endothelial Growth Factor Receptor (flt-1) that confers endothelial-specific gene
expression. The Journal of Biological Chemistry 1995;270:27948-53.
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

21 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
19. Santos, SC and Dias, S. Internal and external autocrine VEGF/KDR loops
regulate survival of subsets of acute leukemia through distinct signaling
pathways. Blood2004;103:3883-3889.
Figure Legends:
Figure 1: Evidence for co-localization of acute leukemia cells in cholesterol-rich
BM niches in vivo. Leukemia cells tend to gather around BM sinusoids, where
major lipid accumulation was detected (limited by the intermittent line); Nile Red
fluorescence arising from the dye–lipid interaction was selectively measured
using an excitation wavelength of 450–500 nm for neutral lipids (yellow‐gold
emission; B and F) and 515–560 nm for polar lipids (red emission; C and G).
These results were obtained from 3 independent experiments and are
representative of 4 recipients. Images were processed with Adobe Photoshop 7.0
Software.
Figure 2: Acute leukemia cells possess high avidity for cholesterol. (A) AML
primary cells display high levels of intracellular cholesterol as compared with
cholesterol content in cells obtained from healthy donors, as measured by the
Amplex Red cholesterol assay kit. Furthermore, intracellular cholesterol content
correlates with the aggressiveness of the disease. HD1/2- healthy donor sample;
AML- bone marrow acute myeloid leukemia samples; AML PB-peripheral blood
acute myeloid leukemia samples; AML BM-R- bone marrow acute myeloid
leukemia in clinical remission samples; MDS- myelodysplasic syndrome. (B)
Acute leukemia cells possess higher levels of intracellular cholesterol, an effect
potentiated by FLT-1 activation by PlGF. Leukemia cell lines (HEL and HL60)
where exposed to 0.2 μM cholesterol complex or to MβCD alone (10 μM) for 4
hours and cellular cholesterol was quantified using the Amplex red kit. Error bars
show the standard errors of two independent experiments.
Figure 3: Cholesterol enrichment of acute leukemia cells recruits FLT-1 to lipid-
raft/caveolin-1 rich domains. (A) Extracts from AML cells stimulated with PlGF,
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

22 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
control cells or cells treated with FLT-1 tyrosine kinase inhibitor (IR1) were
separated by sucrose gradient fractionation and analyzed by Western blot for
FLT-1 and caveolin-1 distribution. FLT-1 was mainly localized in caveolin-1
enriched fractions (4 and 5) upon PlGF stimulation; an effect reverted by FLT-1
tyrosine kinase inhibition. (B) Co-localization of FLT-1 with caveolin-1 was also
observed by confocal microscopy. (C) After AML exposure to cholesterol
disturbing agents, cell extracts were fractionated by sucrose gradients and
fractions enriched for lipid raft/caveolin-1 (fractions 4 and 5) and cytosolic
(fractions 10 and 11) were concentrated using Amicon ultra devices and
analysed by SDS-Page. Results are representative of three independent
experiments. (D) The bands obtained via western-blot for FLT-1 distribution upon
cholesterol disturbance (C) were quantified with ImageJ software based analysis
(http://rsb.info.nih.gov/ij/). (E) Extraction of FLT-1 from caveolin-1-enriched rafts
was observed after MβCD treatment the reverse effect of cholesterol+MβCD cell
treatment (see confocal microscopy analysis). Scale bars= 10μm.
Figure 4: Cholesterol enrichment promotes FLT-1 signaling in lipid rafts. FLT-1
receptor activation was tested by addition of PlGF, MβCD, Cholesterol+MβCD
and Nystatin as indicated. Total cell extracts were analyzed using anti-
phosphotyrosine, anti-FLT-1 and anti-β-actin antibodies. (A) Immunoprecipitation
of FLT-1 in AML lysates showed that PlGF/VEGF treatment for 30 minutes
activates FLT-1 receptor. (B) Leukemia cell treatment with MβCD abolishes FLT-
1 phosphorylation mediated by VEGF/PlGF. In contrast, AML intracellular
cholesterol increase activates FLT-1. (C) Inhibition of lipid-raft
formation/endocytosis by nystatin impedes FLT-1 activation. The
autoradiographs are representative of similar results obtained from three
independent experiments.
Figure 5: Cholesterol enrichment promotes PLGF/VEGF-induced leukemia cell
viability and migration. Viability of AML cells was assessed for 48 hours by trypan
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

23 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
blue exclusion dye. Cholesterol+MβCD acted synergistically with PlGF inducing
significantly (P=0,002) cell viability whereas cholesterol extraction by MβCD cell
treatment had the opposite effect (P=0,01). The chemotactic capacity of leukemia
cells towards PlGF was evaluated using a transwell migration system after their
treatment with Cholesterol+MβCD complexes (4-16 hours). A significant increase
in cell migration was obtained, an effect reverted by FLT-1-signaling blockage
(6.12 neutralizing antibody-P<0,01; tyrosine kinase inhibitor IR1-P=0,08). Results
are representative of 3 independent experiments. Error bars depict the standard
error of the mean.
Figure 6: Acute leukemia cell exposure to cholesterol up-regulates FLT-1
expression. (A) As determined by real-time PCR, FLT-1 expression after AML
cell treatment with cholesterol+MβCD complexes (4 hours) in the
presence/absence of PlGF is significantly up-regulated (without PlGF: P=0,048;
with PlGF: P<0,025). Inhibition of FLT-1 signaling (6.12 neutralizing antibody; IR1
chemical inhibitor) significantly abolishes the up-regulated FLT-1 expression
obtained after cholesterol cell exposure (P<0,03). In contrast, FLT-1 mRNA
expression was not affected by cellular cholesterol disturbance in Huvec cells
(B). KDR (also expressed in these cells) mRNA expression remains unaltered by
enriched cellular cholesterol (C). Experiments were done in triplicate and
represented the mean (n=3).
Additional files
Additional file 1- FLT-1 antibody specificity was assessed by confocal
microscopy using cells that express distinct FLT-1 amounts (HEL; 697). FLT-1
immunolocalization on HEL cells (A and C) is distinct when compared with FLT-1
staining obtained with the same antibody on 697 cells (E and F). B and D are DIC
images of A and C, obtained for HEL cells. As negative control HEL cells were
incubated only with secondary antibody (G, H).
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

24 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.
Copyright © 2010 American Association for Cancer Research
Additional file 2- Acute leukemia cells produce significantly higher amounts of
VEGF after exposure to cholesterol, an effect FLT-1 dependent as observed by
FLT-1 blockage with 6.12 neutralizing antibody and with tyrosine kinase inhibitor
IR1. ELISA quantification of VEGF levels produced by acute leukemia cells after
treatment with Cholesterol+MβCD complexes for 4 hours.
Additional file 3- Cholesterol treatment (0,2μM cholesterol complex) increases
CREB complex formation at FLT-1 promoter. (A) Electrophoretic mobility shift
assay (EMSA) revealed the binding of CREB to the human FLT-1 proximal
promoter containing 2 putative FLT-1 binding sites. Nuclear extracts of untreated
(lane 2 and 3) and cholesterol+MβCD treated cells (lanes 4 and 5) were
incubated with labeled probe of wild-type FLT-1 binding site. Additionally, FLT-1
phosphorylation was induced by PlGF treatment for 30 minutes (lanes 3 and 5).
Excess of cold competitor (lane 1) inhibits CREB complex formation. In lane 6
untreated AML nuclear extracts (10μg) were incubated with 4 μg of anti-CREB-1
antibody for 1 hour at 4ºC before the addition of 32P-labeled probe. A- Band of
CREB complex; SS- Supershifted band with anti-CREB-1 antibody. (B)
Quantification of the bands obtained in A using ImageJ software based analysis
(http://rsb.info.nih.gov/ij/).
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155

Published OnlineFirst January 5, 2011.Mol Cancer Res Cristina Casalou, Ana Costa, Tania Carvalho, et al. Signaling in Acute Leukemia Cells.Cholesterol Regulates VEGFR-1 (FLT-1) Expression and
Updated version
10.1158/1541-7786.MCR-10-0155doi:
Access the most recent version of this article at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mcr.aacrjournals.org/content/early/2011/01/05/1541-7786.MCR-10-0155To request permission to re-use all or part of this article, use this link
on June 28, 2018. © 2011 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on January 5, 2011; DOI: 10.1158/1541-7786.MCR-10-0155