chitosan-based zaleplon nasal microparticles … · chitosan-based zaleplon nasal microparticles...
TRANSCRIPT

Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 279
Chitosan-Based Zaleplon Nasal Microparticles
Sfurti S. Sakhare, A. V. Yadav, S. S. BadadareDepartment of Pharmaceutics, Gourishankar Institute of Pharmaceutical Education and Research, Limb, Satara, Maharashtra, India
Abstract
Objectives: The purpose of present research work was to develop mucoadhesive microparticles of Zaleplon for nasal delivery with the aim to avoid hepatic first-pass metabolism, improve therapeutic efficacy and enhance residence time in the treatment of insomnia. Materials and Methods: Chitosan-based microparticles were prepared by the coacervation method by varying the drug:polymer ratio. The microparticles were evaluated for particle size and shape and surface morphology by scanning electron microscopy, drug content, drug entrapment efficiency, swelling study, in vitro mucoadhesion, differential scanning calorimetric (DSC), X-ray diffraction (XRD), Fourier transform infrared (FTIR), in vitro drug release study, and ex vivo drug permeation. Results: Zaleplon microparticles showed irregular-shaped particles and particle size was found to be in the range of 4.97–10.6 μm, which is favorable for intranasal absorption. The prepared microparticles exhibited a good swelling index. The percentage drug content and entrapment efficiency was found to be in the range between 36.36% - 80% and 37.65% - 52.88%, respectively. In vitro mucoadhesion was performed by adhesion number using goat nasal mucosa and was observed in a range from 43.33 ± 4.409 to 75 ± 2.886%. It was observed that polymer concentration enhancement led to increased mucoadhesive strength. The results of DSC and XRD studies revealed the molecular amorphous dispersion of Zaleplon into the chitosan microparticles. IR spectra of Zaleplon along with excipient showed no interaction between Zaleplon and excipients. In vitro drug release from all the formulations was biphasic, being characterized by a slight “burst” followed by slow release. At the end of 12 h, F6 (1:6) showed drug release of 81.66 ± 1.545%, indicating sustained release. The permeation data of formulation from goat nasal mucosa was found near to that obtained with dialysis membrane in vitro. Conclusion: According to the obtained results, Zaleplon loaded chitosan microparticles prepared by coacervation method proved to be capable of sustained release and could be used through nasal route as an alternative to oral administration.
Key words: Acetic acid, chitosan, nasal microparticles, sodium tripolyphosphates, Zaleplon
Address for correspondence: Sfurti S. Sakhare, Department of Pharmaceutics, Gourishankar Institute of Pharmaceutical Education and Research, Limb, Satara, affiliated to Shivaji University, Kolhapur, Maharashtra, India. Mobile: +91-9657065059. E-mail:[email protected]
Received: 19-06-2017 Revised: 11-08-2017 Accepted: 21-10-2017
INTRODUCTION
The most desirable and convenient method of drug administration are the oral route because of their ease of
administration. However, in many instances, oral administration is not desirable when the drug undergoes significant degradation through first pass effect in liver. Hence, lack of systemic absorption through the gastrointestinal tract led to research on alternate routes of drug delivery such as parenteral, intramuscular, subcutaneous, intranasal (IN), and transdermal.[1,2] IN administration is a needle-free and hence an ideal alternative to the parenteral route for systemic drug delivery. Nasal mucosa consists of a rich vasculature and a highly permeable structure for systemic absorption. Drug administration through the nasal cavity is easy and convenient. Avoidance of first-pass
metabolism is the main advantage of the nasal route of drug delivery.[1,3]
Nasal drug delivery has generated interest as an alternative route for administration of drugs and biomolecules that are susceptible to enzymatic or acidic degradation and first-pass metabolism. Possible pathways for a drug to permeate across the nasal mucosa are passive transportation carriers
RE
SEA
RC
H A
RT
ICL
E

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 280
mediated, transcytosis, and transport through tight junctions. Nasal application of drugs is suggested to be the most viable alternative to the parenteral administration.[4]
Nowadays, an increasing number of drugs are characterized by being poorly water soluble and highly lipophilic, resulting in a low and highly variable oral bioavailability. Due to this fact, many drug candidates fail to reach the market, although they exhibit potential pharmacodynamic activity.[5] On the other hand, to achieve the desired plasma level, marketed poorly water-soluble drugs are administered in higher doses than actually needed, leading to the rise of toxicity problems.[6] Therefore, suitable formulation approaches need to be developed to improve solubility and bioavailability of poorly soluble drugs.
Designing mucoadhesive drug delivery system is a novel approach in nasal drug delivery, which enhances the nasal residential time of the drug molecule and hence enhances the absorption and bioavailability of nasally administered drug products. Bioadhesion is the ability of a natural material to adhere to a biological tissue or membrane for a prolonged period of time.[7] Mucoadhesive system is the ideal choice of drug delivery system for systemic, nasal drug delivery because it improves the nasal residential time. Intimate contact of drug delivery system to the nasal mucosa not only prolongs the duration of action but also increases the extent of absorption.
Microparticles are a type of drug delivery systems where the particle size ranges from 1 μ (typically 1–1000 μm) to few mm. This microencapsulation technology allows protection of drug from the environment, stabilization of sensitive drug substances, elimination of incompatibilities, or masking of unpleasant taste. Hence, they play an important role as drug delivery systems aiming at improved bioavailability of conventional drugs and minimizing side effects.
In particulate drug delivery carriers are used for the encapsulation of drug which prevent exposure of a drug to nasal environment and improve the retention capacity in nasal cavity.[8] Microparticles may also protect the drug from enzymatic metabolism and sustain drug release, prolonging its effect.[9] Nasal drug delivery has generated interest as an alternative route for administration of drugs and biomolecules that are susceptible to enzymatic or acidic degradation and first-pass metabolism.[10,11] The formulation of poorly soluble drug for nasal delivery will be one of the biggest challenges. Among the available approaches, the mucoadhesive particulate drug delivery systems have often proven to be the most commonly used methods in improving bioavailability of the drugs[12] and mucoadhesive particulate drug delivery systems have simple method of preparation and evaluation.
Zaleplon is a water-insoluble drug with a log P of 1.23. Zaleplon is a nonbenzodiazepine hypnotic from the
pyrazolopyrimidine class and is indicated for the short-term treatment of insomnia. Zaleplon is having poor water solubility (BCS class II). The oral bioavailability is approximately 30% because it undergoes significant presystemic metabolism.[13]
Zaleplon is rapidly absorbed after oral administration, its poor aqueous solubility (practically insoluble) can make its absorption dissolution rate limited and thus delay onset of action. The dissolution of drugs is a prime determinant in the absorption of poorly water-soluble drugs and also serves as a rate-limiting step.[13]
The major disadvantage of Zaleplon is high dose and also causes for occurrence of (typically short-lived) hallucinations. It is now withdrawn from the market due to the high dose and inefficient therapeutic activity. Hence, various approaches have been adopted to enhance the bioavailability of Zaleplon by improving its solubility which in turn improves the dissolution rate. These techniques in addition also aided to avoid first-pass metabolism. Many review deals with the various approaches mainly solid dispersions, complexation with cyclodextrins; lipid-based delivery systems such as proliposomes, nanoemulsifying powders, semisolid dispersions with lipid surfactants, and solid lipid nanoparticles to improve the oral bioavailability evaporation method.[14]
The present investigation relates to formulating mucoadhesive microparticles to improve in vitro release and permeation of Zaleplon using mucoadhesive drug delivery system which might increase the nasal residence time and allow more of the drug to penetrate through the nasal mucus layer.
MATERIALS AND METHODS
Materials
Zaleplon was provided ex gratis by Precise Chemipharma Pvt. Ltd. Chitosan (85% deacetylation) (Pallav Chemicals and Solvents Pvt. Ltd.), sodium tripolyphosphates (Loba Chem. Pvt. Ltd. Mumbai), and sodium hydroxide (S. D. Lab Chem. Mumbai). All other chemicals employed were of analytical grade.
Methods
Formulation of mucoadhesive microparticles
Zaleplon microparticles were prepared by coacervation method [Table 1]. Chitosan was dissolved with mild agitation in an aqueous solution of 1% v/v Glacial acetic acid. The drug was dissolved in methanol and mixed well in the polymer solution at a different concentration to yield different drug/polymer ratio on a magnetic stirrer at 500 rpm for 2 h. A solution of NaOH 2N (0.3%W/V in 100 ml) was added dropwise (1 ml/min) in above solution by micropipette

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 281
during stirring on magnetic stirrer for 1 h at 400 rpm which resulted in solidification of polymer. Sodium tripolyphosphate solution was added in above solution dropwise using a syringe no 20 gauge. Formulations were left for 20–30 min for crosslinking and hardening of microparticles. Then, microparticles were purified by centrifugation for 15 min at 1000 rpm and the obtained supernatant was separated and the supernatant was subjected to freeze-drying. Freeze dried powder was used for further studies.[15]
Characterization of microparticles
Drug content estimation
About 10 mg of the prepared microparticles were dissolved in 10 ml of methanol:phosphate buffered saline (PBS) pH 6.4 (1:9) and sonicated for 3 min. The amount of drug was estimated spectrophotometrically based on absorbance at 232 nm.[16,17]
Drug entrapment efficiency
About 10 mg microparticles were crushed in a glass mortar and pestle and the powdered microparticles were suspended in 10 ml of methanol:PBS pH 6.4 (1:9 ratio). After 24 h, the filtrate was diluted with phosphate buffer pH 6.4 and final solution was assayed spectrophotometrically at 232 nm for drug content.[16,17,18]
Estimated drug content% Entrapment efficiency = ×100Theoretical drug content
(1)
Particle size analysis
The microparticles were evaluated for the particle size. An optical microscope (Motic) was used for this purpose. The average particle size of the microparticles was expressed as the volume surface diameter; dvs (mm).[17,19,20,21]
Swelling study
The swelling ability of microparticles in physiological media was determined by swelling them to their equilibrium. Accurately weighed amount of microparticles was dispersed
in 10 ml phosphate buffer pH 6.4. Microparticles were allowed to swell for 12 h. The excess surface adhered liquid drops were removed by blotting with soft tissue papers and the swollen microparticles were weighed on weighing balance.[17,22,23]
Weight of swollen microparticles -Weight of dry microparticles% swelling study = ×100
Weight of dry microparticles (2)
Mucoadhesion study
The mucoadhesion studies described in literature were used with slight modifications. Goat nasal mucosa was obtained from a local slaughterhouse, cleaned with distilled water and then cut into 2 × 2 cm pieces for the study. The drug-loaded microparticles were immersed in 50 ml beaker at 37°C ± 0.5°C containing phosphate buffer pH 6.4 for 5 min in such a way that the solution just covered the microparticles. After the microparticles were wetted, the fresh goat nasal mucosa was placed on the microparticles surface for 5 min so as to cover all the microparticles. The nasal mucosa with attached microparticles was removed and the remaining microparticles on the glass beaker were dried at 60°C. The percentage of adhered microparticles was computed as in following Eq. 3.[24,25]
Adhered microparticles (%)= {(Wo−Wr)/Wo}×100 (3)
Where,Wo = Initial weight of the microparticlesWr = Unattached microparticles weight.
In vitro drug release study
The drug release was studied using modified USP type II apparatus at 37°C ± 0.5°C and at 50 rpm using 200 ml of phosphate buffer pH 6.4 as a dissolution medium. 1 ml of the sample solution was withdrawn at predetermined time intervals, filtered, diluted suitably, and analyzed spectrophotometrically at 232 nm. Equal amount of the fresh dissolution medium was replaced immediately after withdrawal of the test sample. Percentage drug dissolved at different time intervals was calculated using the Lambert-Beer’s equation. The result was obtained in triplicate and the average value reported.[20,26]
Table 1: Composition of nasal microparticlesBatch Drug (mg) Chitosan (mg) TPP (mg) NaOH 2N (ml)F1 20 20 300 50
F2 20 40 300 50
F3 20 60 300 50
F4 20 80 300 50
F5 20 100 300 50
F6 20 120 300 50
F7 20 140 300 50
F8 20 160 300 50

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 282
Analysis of in vitro drug release kinetics and mechanism
To investigate the mechanism of drug release, the data were fitted to various drug release kinetic model equations such as zero-order (cumulative % release vs. time), first-order (log of cumulative % drug remaining vs. time), Higuchi’s square root of time model (cumulative % release vs. square root of time), Hixson–Crowell cube root plot (cube root of % drug remaining vs. time), and Korsmeyer-Peppas kinetic plot (fraction release of drug vs. time). The zero-order rate Eq. (1) describes the system where the drug release rate is independent of its concentration. The first-order Eq. (2) describes the release from system where release rate is concentration dependent. Higuchi described the release of drugs from insoluble matrix as a square root of time-dependent process based on Fickian diffusion Eq. (3). The Hixson–Crowell cube root law Eq. (4) describes the release from systems where there is a change in surface area and diameter of particles. The Korsmeyer-Peppas exponential model Eq. (5) describes the drug transport mechanism.
C = K0t (1)
Where K0 is zero-order rate constant expressed in units of concentration/time and t is the time.
Log C = Log C0−Kt/2.303 (2)
Where C0 is the initial concentrwation of drug and K is the first-order rate constant.
Q = Kt1/2 (3)
Where Q is the amount of drug released at time t,K is the diffusion rate constant.
Q0 1/3 − Qt
1/3 = KHCt (4)
Where Qt is the amount of drug released in time t,Q0 is the initial amount of the drug in the microparticles,and KHC is the rate constant for Hixson–Crowell rate equation.
Mt/M8 = Ktn (5)
Where Mt/M8 is the fractional release of the drug,t is the release time,
K is a constant incorporating structural and geometric characteristic of the release device and the diffusional exponent “n” is dependent on the geometry of the device as well as the physical mechanism for release. In this context, n = 0.43 indicates Fickian (case I) release and n ≤ 0.85 indicates a purely relaxation controlled delivery which is referred to as Case II transport. Intermediate values 0.43 < n < 0.85 indicate an anomalous behavior (non-Fickian kinetics corresponding to coupled diffusion/polymer relaxation). Occasionally,
values of n > 1 have been observed, which are regarded as Super Case II kinetics.[27,28]
Ex vivo permeation study
Fresh nasal tissues were carefully removed from the nasal cavity of goat obtained from the local slaughterhouse. Goat nasal mucosa was fixed between donor and receptor compartment of Franz diffusion cell to support the microparticles. Specified quantity of microparticles was placed on the membrane in the donor compartment, the receptor compartment containing 20 ml phosphate buffer solution pH 6.4 (within the pH range in nasal cavity) maintained at 37°C ± 0.5°C was stirred constantly with the help of magnetic stirrer at about 50 rpm. At predetermined time interval of 0, 1, 2, 3, 4, 5, 6, 7, and 8 h, 1ml of sample was withdrawn, diluted, filtered, and analyzed by UV Visible spectrophotometer at 232 nm.[11,24,26,29,30]
Fourier transforms infrared spectroscopy
Fourier transform infrared (FTIR) spectra were obtained using Shimadzu FTIR-8400S spectrometer, Japan. Samples of Zaleplon, physical mixtures and optimized formulation of microparticles were taken for the study. The scanning range was 400-4000/cm and the resolution was 4/cm.[28] FTIR spectroscopy was carried out to check the compatibility between drug and polymer. The FTIR spectra of drug with polymers were compared with the standard FTIR spectrum of the pure drug.
Differential scanning calorimetric (DSC)
DSC analysis of the samples was performed on a Perkin-Elmer DSC7, USA. Samples (3.3350 mg) were heated under nitrogen atmosphere on an aluminum pan at a heating rate of 10°C/min over the temperature range of 30°C-300°C. DSC analysis was performed under nitrogen gas flow of 20 ml/min. this study is conducted for detection of melting point and heat of fusion to evaluate the crystalline nature of drugs.[22,23,27]
X-ray diffraction (XRD) studies
The crystallinities of Zaleplon and Zaleplon-loaded microparticles were determined using an X-ray diffractometer. These studies are useful to investigate crystallinity of drug in the microparticles. The samples were mounted on a sample holder and XRD patterns were recorded in the range of 5-50° at the speed of 5°/min.[21,22]
Scanning electron microscopy (SEM)
The surface morphology of the formulation was determined using a SEM. Samples were mounted on aluminum mount using double-sided adhesive tape, sputtered with gold under vacuum and scanned at an accelerating voltage of 20 KV before observation.[3-31]

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 283
RESULTS AND DISCUSSION
Characterization of microparticles
Drug content
The coacervation method is a convenient method for the preparation of mucoadhesive microparticles with good drug content. In these method drug is uniformly distributed in the polymer solution so drug can be loaded easily in the polymer. Drug content of all eight batches based on chitosan concentration was found to be in the range of 36.36 ± 1.29%–80 ± 1.823% [Table 2]. It was observed that with the increase in polymer concentration drug content also increased. This may be due to the reasons that increase in the molecular weight of the mucoadhesive polymer forms a more intact matrix network of polymer which increases drug content [Figure 1].[32]
Drug entrapment efficiency
The encapsulation efficiency determines the percentage of encapsulated drug with respect to the total drug introduced into polymer solution. Effect of polymer content on encapsulation efficiency was studied. The encapsulation efficiency in phosphate buffer pH 6.4 was found to be ranging from 37.65% to 52.88%, respectively [Table 3], as the concentration of polymer was increased, encapsulation efficiency was also found to be increased. This may be due to increase in viscosity of the polymer dispersion, which would decrease the probability of drug diffusion into the external phase during preparation, resulting in higher drug entrapment efficiency.[33] In present study as the concentration of polymer increased in a fixed volume of organic solvent the encapsulation efficiency of microparticles was also increased. This may be due to high concentration of polymers, which make a complex network of polymers and prevent the migration of drug into surrounding media.[19]
Particle size analysis
The particle size distribution of microparticles applied for nasal route is important with regard to deposition in respiratory system, as the particles size larger than 10 µm deposit at the anterior parts of nose and thus avoids ciliated absorption areas.[19] Whereas the particles size below 5 µm should be avoided because it may get inhaled in lung and those <0.5 µm are exhaled. In general, particles in the 5–10 µm range are deposited in the nostrils.[34] The particle size of each Zaleplon microparticles formulation is reported in Figure 2. The average particle size of Zaleplon microparticles ranged from 4.97 to 10.6 μm, the size distribution of the microparticles was narrow, and such particles are considered to be suitable for nasal administration. It was also noted that
increasing drug-to-polymer ratio slightly increased the size of microparticles.
In present study as the chitosan concentration was increased in a fixed volume of aqueous phase it resulted in larger particle diameter, which may be due to the fact that higher the polymer concentration, the higher crosslinking degree. It could be postulated that higher concentration of polymer in the sample led to an enhanced frequency of collisions, resulted in fusion of semi-formed particles, and finally increased the size of the microparticles.[35] The most probable reason for increase in particle size with increase in polymer concentration may be due to increase in viscosity and hence difficulty in dispersion and subdivision of droplets.[29]
Degree of swelling
The swelling study was an important attribute of studying clearance of drug from nasal cavity. It is suggested that administration of microparticles lowers clearance of the microparticles systems which may be due to the fact that the microparticles undergo a gelation process of taking up water and swelling, which results in polymer/mucus mixture leading to reduced mucociliary clearance. It is also closely related to the nasal absorption enhancement because swelling is due to uptake of nasal fluids and responsible for tight junction cell opening.[19]
Table 2: Drug content of Zaleplon microparticlesFormulation Drug content (%)F1 36.36±1.29
F2 39.28±0.815
F3 44.51±1.17
F4 60±1.160
F5 62.52±1.068
F6 65±1.12
F7 71±1.377
F8 80±1.823Mean±SD, n=3. SD: Standard deviation
Table 3: Drug entrapment efficiency of Zaleplon microparticles
Formulation Entrapment efficiency (%)F1 56.3±1.178
F2 63.85±1.172
F3 66.5±0.851
F4 70±0.934
F5 74.5±1.178
F6 78±0.881
F7 80±0.83
F8 82.6±1.225Mean±SD, n=3. SD: Standard deviation

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 284
The degree of swelling of all the formulations is shown in Table 4. As the amount of chitosan was increased, swelling index was also found to be increased. From this, it may be concluded that when the microparticles are in contact with mucus layer, they swell rapidly and take up liquid from the mucus layer. Hence, the epithelial cells lose water and shrink
which opens the epithelial tight junctions allowing drug to be absorbed.[3,33]
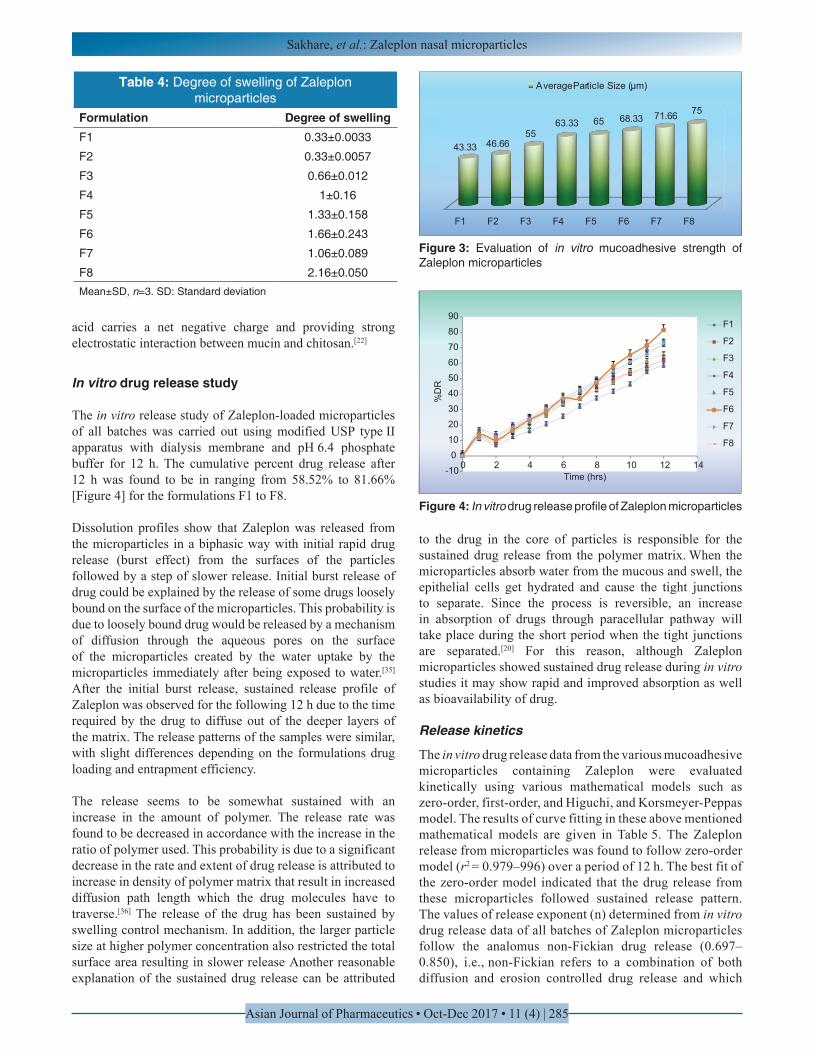
Mucoadhesion study
The results of in vitro mucoadhesion [Figure 3] showed that all the batches of microparticles had satisfactory mucoadhesive properties ranging from 43.33 ± 4.409 to 75 ± 2.886%. Percent mucoadhesion increased with the increase in concentration of mucoadhesive polymer. Excellent mucoadhesion of chitosan microparticles was from the electrostatic attraction between chitosan and mucin. Moreover, the linear molecule of chitosan expressed sufficient chain flexibility for interpenetration and entanglements.[19,34] Increase in the concentration of chitosan in the formulation increase mucoadhesion. This may be due to formation of secondary chemical bonds such as hydrogen bond or ionic bond or ionic interactions between the positively charged amino group of chitosan and the negatively charged sialic acid residue of mucus glycoproteins or mucin. Sialic
Figure 1: Optical microscopic images of F1 to F8 formulations
F1 F2 F3 F4 F5 F6 F7 F8
4.97
6 6.717.46 7.53
8.38.8
10.6
Average Particle Size (µm)
Figure 2: Particle size analysis of Zaleplon microparticles
Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 285
acid carries a net negative charge and providing strong electrostatic interaction between mucin and chitosan.[22]
In vitro drug release study
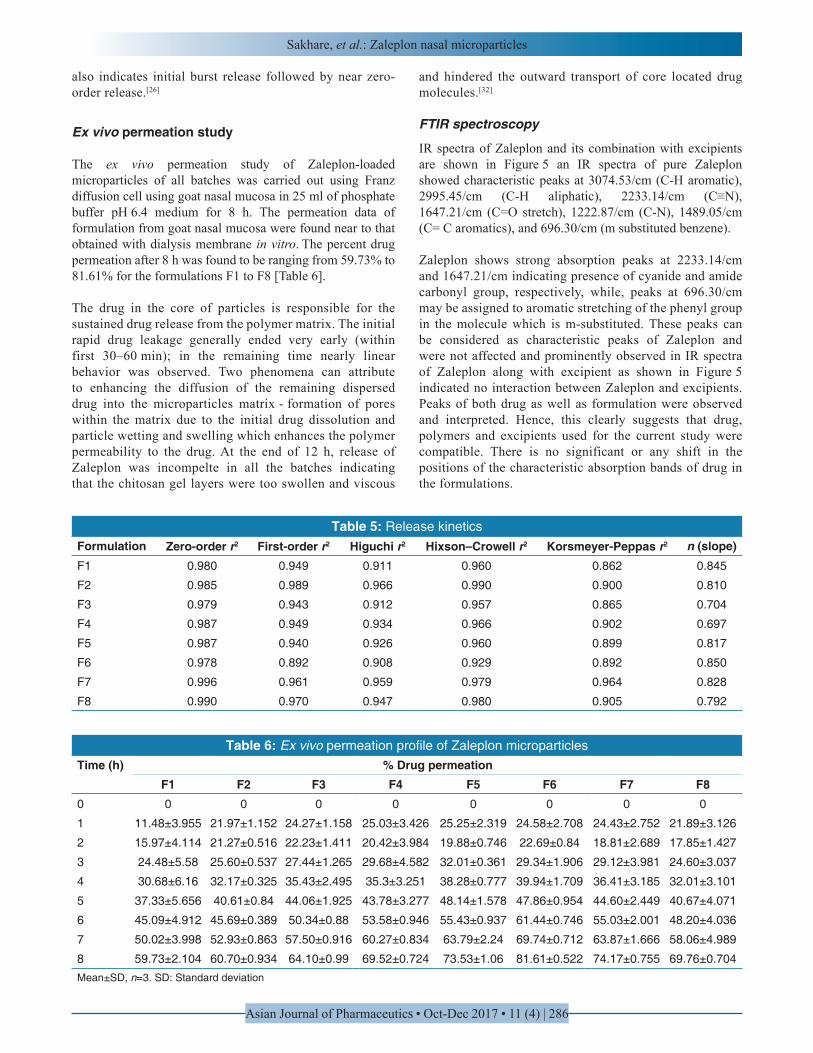
The in vitro release study of Zaleplon-loaded microparticles of all batches was carried out using modified USP type II apparatus with dialysis membrane and pH 6.4 phosphate buffer for 12 h. The cumulative percent drug release after 12 h was found to be in ranging from 58.52% to 81.66% [Figure 4] for the formulations F1 to F8.
Dissolution profiles show that Zaleplon was released from the microparticles in a biphasic way with initial rapid drug release (burst effect) from the surfaces of the particles followed by a step of slower release. Initial burst release of drug could be explained by the release of some drugs loosely bound on the surface of the microparticles. This probability is due to loosely bound drug would be released by a mechanism of diffusion through the aqueous pores on the surface of the microparticles created by the water uptake by the microparticles immediately after being exposed to water.[35] After the initial burst release, sustained release profile of Zaleplon was observed for the following 12 h due to the time required by the drug to diffuse out of the deeper layers of the matrix. The release patterns of the samples were similar, with slight differences depending on the formulations drug loading and entrapment efficiency.
The release seems to be somewhat sustained with an increase in the amount of polymer. The release rate was found to be decreased in accordance with the increase in the ratio of polymer used. This probability is due to a significant decrease in the rate and extent of drug release is attributed to increase in density of polymer matrix that result in increased diffusion path length which the drug molecules have to traverse.[36] The release of the drug has been sustained by swelling control mechanism. In addition, the larger particle size at higher polymer concentration also restricted the total surface area resulting in slower release. Another reasonable explanation of the sustained drug release can be attributed
to the drug in the core of particles is responsible for the sustained drug release from the polymer matrix. When the microparticles absorb water from the mucous and swell, the epithelial cells get hydrated and cause the tight junctions to separate. Since the process is reversible, an increase in absorption of drugs through paracellular pathway will take place during the short period when the tight junctions are separated.[20] For this reason, although Zaleplon microparticles showed sustained drug release during in vitro studies it may show rapid and improved absorption as well as bioavailability of drug.
Release kinetics
The in vitro drug release data from the various mucoadhesive microparticles containing Zaleplon were evaluated kinetically using various mathematical models such as zero-order, first-order, and Higuchi, and Korsmeyer-Peppas model. The results of curve fitting in these above mentioned mathematical models are given in Table 5. The Zaleplon release from microparticles was found to follow zero-order model (r2 = 0.979–996) over a period of 12 h. The best fit of the zero-order model indicated that the drug release from these microparticles followed sustained release pattern. The values of release exponent (n) determined from in vitro drug release data of all batches of Zaleplon microparticles follow the analomus non-Fickian drug release (0.697–0.850), i.e., non-Fickian refers to a combination of both diffusion and erosion controlled drug release and which
F1 F2 F3 F4 F5 F6 F7 F8
43.33 46.6655
63.33 65 68.33 71.66 75
AverageParticle Size (µm)
Figure 3: Evaluation of in vitro mucoadhesive strength of Zaleplon microparticles
Table 4: Degree of swelling of Zaleplon microparticles
Formulation Degree of swellingF1 0.33±0.0033
F2 0.33±0.0057
F3 0.66±0.012
F4 1±0.16
F5 1.33±0.158
F6 1.66±0.243
F7 1.06±0.089
F8 2.16±0.050Mean±SD, n=3. SD: Standard deviation
-100
102030405060708090
0 2 4 6 8 10 12 14
% D
R
Time (hrs)
F1
F2
F3
F4
F5
F6
F7
F8
Figure 4: In vitro drug release profile of Zaleplon microparticles
Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 286
also indicates initial burst release followed by near zero-order release.[26]
Ex vivo permeation study
The ex vivo permeation study of Zaleplon-loaded microparticles of all batches was carried out using Franz diffusion cell using goat nasal mucosa in 25 ml of phosphate buffer pH 6.4 medium for 8 h. The permeation data of formulation from goat nasal mucosa were found near to that obtained with dialysis membrane in vitro. The percent drug permeation after 8 h was found to be ranging from 59.73% to 81.61% for the formulations F1 to F8 [Table 6].
The drug in the core of particles is responsible for the sustained drug release from the polymer matrix. The initial rapid drug leakage generally ended very early (within first 30–60 min); in the remaining time nearly linear behavior was observed. Two phenomena can attribute to enhancing the diffusion of the remaining dispersed drug into the microparticles matrix - formation of pores within the matrix due to the initial drug dissolution and particle wetting and swelling which enhances the polymer permeability to the drug. At the end of 12 h, release of Zaleplon was incompelte in all the batches indicating that the chitosan gel layers were too swollen and viscous
and hindered the outward transport of core located drug molecules.[32]
FTIR spectroscopy
IR spectra of Zaleplon and its combination with excipients are shown in Figure 5 an IR spectra of pure Zaleplon showed characteristic peaks at 3074.53/cm (C-H aromatic), 2995.45/cm (C-H aliphatic), 2233.14/cm (C≡N), 1647.21/cm (C=O stretch), 1222.87/cm (C-N), 1489.05/cm (C= C aromatics), and 696.30/cm (m substituted benzene).
Zaleplon shows strong absorption peaks at 2233.14/cm and 1647.21/cm indicating presence of cyanide and amide carbonyl group, respectively, while, peaks at 696.30/cm may be assigned to aromatic stretching of the phenyl group in the molecule which is m-substituted. These peaks can be considered as characteristic peaks of Zaleplon and were not affected and prominently observed in IR spectra of Zaleplon along with excipient as shown in Figure 5 indicated no interaction between Zaleplon and excipients. Peaks of both drug as well as formulation were observed and interpreted. Hence, this clearly suggests that drug, polymers and excipients used for the current study were compatible. There is no significant or any shift in the positions of the characteristic absorption bands of drug in the formulations.
Table 5: Release kineticsFormulation Zero-order r2 First-order r2 Higuchi r2 Hixson–Crowell r2 Korsmeyer-Peppas r2 n (slope)F1 0.980 0.949 0.911 0.960 0.862 0.845
F2 0.985 0.989 0.966 0.990 0.900 0.810
F3 0.979 0.943 0.912 0.957 0.865 0.704
F4 0.987 0.949 0.934 0.966 0.902 0.697
F5 0.987 0.940 0.926 0.960 0.899 0.817
F6 0.978 0.892 0.908 0.929 0.892 0.850
F7 0.996 0.961 0.959 0.979 0.964 0.828
F8 0.990 0.970 0.947 0.980 0.905 0.792
Table 6: Ex vivo permeation profile of Zaleplon microparticlesTime (h) % Drug permeation
F1 F2 F3 F4 F5 F6 F7 F80 0 0 0 0 0 0 0 0
1 11.48±3.955 21.97±1.152 24.27±1.158 25.03±3.426 25.25±2.319 24.58±2.708 24.43±2.752 21.89±3.126
2 15.97±4.114 21.27±0.516 22.23±1.411 20.42±3.984 19.88±0.746 22.69±0.84 18.81±2.689 17.85±1.427
3 24.48±5.58 25.60±0.537 27.44±1.265 29.68±4.582 32.01±0.361 29.34±1.906 29.12±3.981 24.60±3.037
4 30.68±6.16 32.17±0.325 35.43±2.495 35.3±3.251 38.28±0.777 39.94±1.709 36.41±3.185 32.01±3.101
5 37.33±5.656 40.61±0.84 44.06±1.925 43.78±3.277 48.14±1.578 47.86±0.954 44.60±2.449 40.67±4.071
6 45.09±4.912 45.69±0.389 50.34±0.88 53.58±0.946 55.43±0.937 61.44±0.746 55.03±2.001 48.20±4.036
7 50.02±3.998 52.93±0.863 57.50±0.916 60.27±0.834 63.79±2.24 69.74±0.712 63.87±1.666 58.06±4.989
8 59.73±2.104 60.70±0.934 64.10±0.99 69.52±0.724 73.53±1.06 81.61±0.522 74.17±0.755 69.76±0.704Mean±SD, n=3. SD: Standard deviation

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 287
DSC
The DSC thermogram of Zaleplon exhibited a single sharp endothermic peak at 185°C corresponding to its melting transition temperature.[37] Zaleplon has high melting point, which is indicative of strong crystal lattice energy. This high melting point is one of the factors responsible for lower aqueous solubility.[37] This endothermic peak was not observed in the thermogram of optimized drug-loaded microparticles (F6). The broadened and shifted peaks suggest that the drug was present as molecular dispersion in the polymer matrix.[24] Further, the decrease in sharpness of Zaleplon endothermic peak in drug-loaded microparticles may be due to decreased crystallinity of Zaleplon [Figure 6].[31]
The presence of detectable peaks of Zaleplon in physical mixture is indication of uniform mixing of excipients. The additional peak was due to loss of moisture (hydrate form) and the other degradation peak. The absence of detectable crystalline domains of Zaleplon in drug-loaded microparticles clearly indicates that Zaleplon encapsulated in microparticles is in the amorphous-crystalline phase or in the solid state solubilized form in the polymer matrix.[29]
XRD studies
XRD study was performed to determine the physical state of the Zaleplon. The corresponding patterns are displayed in Figure 7. The Zaleplon has crystalline characteristics which are represented by peaks in X-ray diffractograms, and the most evident and intense peaks appear at 2θ = 10.4, 14.06, 16.7, 17.42, 19.52, 25.37, 26.6. Zaleplon has high melting point, which is indicative of strong crystal lattice energy.[37] These characteristic peaks of the drug still existed in physical mixture and optimized batch F6 systems but with lower intensity. Whereas, the intensity of characteristic peaks of the drug was greatly minimized and broadening of peaks appear in the case of optimized batch F6, which could be attributed to the destruction of the drug crystal lattice because of progressive amorphization. It is obvious that poorly water-soluble pharmaceuticals with lower crystallinity usually have higher dissolution rate and bioavailability.[37] Accordingly, the decrease in crystallinity of prepared Zaleplon microparticles is expected to improve its dissolution rate and bioavailability. This indicates that drug particles are dispersed at molecular level in the polymer matrices since no indication about the crystalline nature of the drugs was observed in the drug-loaded microparticles.
SEM
Studies using SEM provided a better understanding of the morphological characteristics of the microparticles. Zaleplon microparticles showed irregular-shaped particles with a wide particle size distribution. The pores at the surface of Zaleplon microparticles may be due to the rapid evaporation of solvent. During the drying process, crust that is first formed on the surface of the droplets prevents the evaporation of the solvent
causing the building up of the vapor pressure as a result small eruptions, opening is formed. Surface indentations could be attributed to the subsequent shrinking of the microparticles after solid crust is formed.[38]
The SEM microphotographs [Figure 8] of the prepared microparticles showed the particles are irregular-shaped and rough texture with shrinkage which is due to removal of water from microparticles during drying. Thus, the rate of removal of water from microparticles exerts an influence on the morphology of final product.[39]
Figure 5: Fourier transforms infrared spectra of Zaleplon, chitosan, physical mixture, optimized formulation (batch-F6)
Figure 6: Differential scanning calorimetric thermograms of Zaleplon, chitosan, physical mixture, optimized formulation (batch-F6)
Figure 7: X-ray diffraction spectra’s of Zaleplon, Chitosan, physical mixture, optimized formulation (batch-F6)

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 288
CONCLUSION
From the study, we successfully developed microparticulate drug delivery system of Zaleplon using mucoadhesive polymer chitosan. It can be prepared using the coacervation method. It can be concluded that chitosan is better mucoadhesive polymer for the formulation of mucoadhesive microparticles of Zaleplon for IN administration. Thus, the formulated microparticles seem to be a potential candidate as IN sustained drug delivery system for symptomatic therapy of insomnia. Further, in vivo investigation is required to establish efficiency and IVIVC of this formulation.
ACKNOWLEDGMENT
The generosity of Precise Chemipharma Pvt. Ltd is gratefully acknowledged for providing gift sample of Zaleplon. The authors are also thankful to the Management and Principal of Gourishankar Institute of Pharmaceutical Education and Research, Limb, Satara, for providing all the necessary laboratory facilities.
REFERENCES
1. Panchal DR, Patel UL, Bhimani BV, Daslaniya DJ, Patel GV. Nasal in situ gel: A novel drug delivery system. Int J Pharm Res Sch 2012;1:457-73.
2. More BA, Mene HR, Pawar RK. A review on in situ nasal gel drug delivery system. Int J Pharm Sci Rev Res 2015;33:199-207.
3. Rao V, Gangadi JR, Kumar S, Venu K, Jayaveera KN. Formulation and evaluation of mucoadhesive microspheres for intra-nasal delivery. Int J Pharm Technol 2010;2:1158-98.
4. Tyagi S, Sharma N, Sharma PK. A review on application of natural bioadhesive polysaccharides for intranasal drug delivery. Int J.A.PS.BMS.2012;1:80-94.
5. Mehta K, Borade G, Rasve G, Bendre A. Self-emulsifying
drug delivery system: Formulation and evaluation. Int J Pharm Biol Sci 2011;2:P-398.
6. Abdalla A, Ma¨der K. Preparation and characterization of a self-emulsifying pellet formulation. Eur J Pharm Biopharm 2007;66:220-6.
7. Bhise SB, Yadav AV, Avachat AM. Bioavailability of intranasal drug delivery system. Asian J Pharm 2008;2:201-15.
8. Pagar S, Shinkar D. A review on intranasal drug delivery system. J Adv Pharm Edu Res 2013;3:333-46.
9. Kashyap N, Mishra A, Pathak A. preparation and evaluation of mucoadhesive microspheres of proprqanolol HCL for nala delivery. Int J Adv Pharm 2015;4:49-54.
10. Nandgude T, Thube R, Jaiswal N, Deshmukh P. Formulation and evaluation of pH induced in situ nasal gel of salbutamo sulphate. Int J Pharm Sci Nanotechnol 2008;1:177-83.
11. Shah V, Sharma M, Parmar V, Upadhyay U. Formulation of sildenafil citrate loaded nasal microsphers: An in vitro, ex vivo characterization. Int J Drug Deliv 2010;2:213-20.
12. Carvalho FC, Bruschi ML, Evangelista RC. Mucoadhesive drug delivery systems. Braz J Pharm Sci 2010;46:1984-8250.
13. Waghmare A, Pore Y, Kuchekar B. Development and characterization of zaleplon solid dispersion systems: A technical note. AAPS PharmSciTech 2008;9:536-43.
14. Dudhipala N. A review of novel formulation strategy to enhance oral delivery of zaleplon. J Bioequiv Availab 2016;8:211-3.
15. Ozbas S, Akbuga J, Aral C. Controlled release of interleukin-2 from chitosan microspheres. J Pharm Sci 2002;91:1245-51.
16. Mali D, Bedse S, Dighe R, Talele S. Formulation evaluation of chitosan microspheres for intranasal delivery of zolmitriptan. Am J Pharm Health Res 2014;2:239-55.
17. Khan MS, Doharey V. A review on nasal microsphere. Int J Plast Surg 2014;4:496-506.
18. Patil S, Babbar A, Mathur R. Mucoadhesive chitosan microspheres of carvedilol for nasal administration. J Drug Target 2010;18:321-31.
19. Mahajan HS, Tatiya BV, Nerkar PP. Ondansetron loaded pectin based microspheres for nasal administration: In vitro and in vivo studies. Power Tech 2012;221:168-76.
20. Jain SA, Chauk DS, Mahajan HS, Tekade AR, Gattani SG. Formulation and evaluation of nasal mucoadhesive microspheres of sumatriptan succinate. J Microencapsul 2009;26:711-21.
21. Tatia GD, Basarkar GD. Formulation development and in vitro evaluation of mucoadhesive microspheres of simvastatin for nasal delivery. Int J Pharm Sci Rev Res 2013;21:334-41.
22. Khom TC, Yadav HK, Manne N. Development of mucoadhesive nanoparticulate system of ebastine for nasal drug delivery. Trop J Pharm Res 2014;13:1013-9.
23. Khan MS, Pandey SC, Sidiqui AR. Development and
Figure 8: Scanning electron photograph of drug loaded microparticles

Sakhare, et al.: Zaleplon nasal microparticles
Asian Journal of Pharmaceutics • Oct-Dec 2017 • 11 (4) | 289
evaluation of nasal mucoadhesive nanoparticles of an analgesic drug. Sch Res Libr 2012;4:1846-54.
24. Dandagi PM, Sharma R, Gadad AP. Thermosensitive gel containing carbamazepin microspheres for intranasal brain targeting. World J Pharm Res 2014;3:716-30.
25. Mufassir MM, Saifee M, Khan S. Effect of solvents: Prepration and characterization of sustained release intranasal microspheres of rizatriptan benzoate. J Innov Pharm Biol Sci 2014;1:68-71.
26. Patil SB, Sawant KK. Development, optimization and in vitro evaluation of alginate mucoadhesive microspheres of carvedilol for nasal delivery. J Microencapsul 2009;26:432-43.
27. Prajapati ST, Pathak SP, Thakkar JH, Patel CN. Nanoemulsion based transnasal delevery of risperidone for nose to brain targeting. Bull Pharm Res 2015;5:6-13.
28. Mahajan H, Gattani S, Surana S. Spray dried mucoadhesive microspheres of ondansetron for nasal administration. Int J Pharm Sci Nanotechnol 2008;1:267-74.
29. Patil S, Babbar A, Mathur R. Mucoadhesive chitosan microspheres of carvedilol for nasal administration. J Drug Target 2010;18:321-31.
30. Pawar D, Goyal AK, Mangal S, Mishra N, Tiwari S. Evaluation of mucoadhesive PLGA microparticles for nasal immunization. Am Assoc Pharm Sci 2010;12:130-7.
31. Mouez M, Zaki NM, Mansour S. Bioavailability enhancement of verapamil Hcl via intranasal chitosan microspheres. Eur J Pharm Sci 2014;51:59-66.
32. Pilicheva B, Zagorchev P, Uzunova Y, Kassarova M. Development and in vitro evaluation of mucoadhesive
microsphere carriers for intranasal delivery of betahistine dihydrochloride. Indian J Drug Dermatol 2013;5:389-401.
33. Dhakar RC, Maurya SD, Saluja V. From formulation variables to drug entrapment efficiency of microspheres: A technical review. J Drug Deliv Ther 2012;2:128-33.
34. Bissera P, Plamen Z, Yordanka U, Margarita K. Development and in vitro evaluation of mucoadhesive microspheres carriers for intranasal delivery of betahistidine dihydrochloride. Int J Drug Deliv 2013;5:389-401.
35. Gungor S, Okyar A, Baktir G. Ondansetron-loaded biodegradable microspheres as a nasal sustained delivery system: In vitro/in vivo studies. Pharm Dev Technol 2010;15:258-65.
36. Swamy NG, Abbas Z. Preparation and in vitro characterization of mucoadhesive hydroxypropyl guar microspheres containing amlodipine besylate for nasal administration. Indian J Pharm Sci 2011;73:608-14.
37. El-bary AA, El-gazayerly. Prepration of zaleplon microparticles using emulsion solvent diffusion technique. J Pharm Drug Deliv Res 2012;1:1-7.
38. Dandagi PM, Sharma R, Gadad AP. Thermosensitive gel containing carbamazepin microspheres for intranasal brain targeting. World J Pharm Res 2014;3:716-30.
39. Kumar V, Sekhar C, Rao S. Preparation and in vitro characterization of mucoadhesive microcapsules of the pantoprazole sodium for oral controlled delivery. International Journal of Asia Pac Stud 2016;7:976-3090.
Source of Support: Nil. Conflict of Interest: None declared.