chapter i introduction - information and library...
TRANSCRIPT

1
CHAPTER I
Introduction
1.1. Corrosion
Corrosion is simply defined as the destruction of materials under chemical or
electrochemical action by the surrounding environment. Corrosion entails the
conversion of a metal from the atomic to ionic state, with the loss of one or more
electrons. Rusting of iron with the formation of iron oxide is a typical example of
corrosion. Corrosion of metal and alloys lead to enormous losses to the country. The
growing need of metal in every walk of life and loss of it by corrosion has put a big
question mark before the mankind regarding how to prevent it from being corroded so
that it can be used to its maximum extent. Scientists have come out with the use of
many preventive methods. The corrosion can be prevented to a large extent if not
stopped completely. A survey carried out by US Minerals Management Service [1]
which indicates that majority of the problems are due to internal corrosion, which are
more frequent than those due to external corrosion.
1.1.1 Economical loss due to corrosion
In a study of corrosion cost conducted jointly by C.C. Technologies Inc., USA
[2], Federal Highway Agencies (FHWA), USA [3] and National Association of
Corrosion Engineers [4], the direct corrosion cost was estimated to be around 276
billion US dollars, approximately 3.1% of the national gross domestic product (GDP).
Based on an extensive survey conducted by Battelle Columbus laboratories,
Columbus, Ohio, USA and National Institute of Standards and Technology (NIST), in
1975, the cost was estimated to be 82 billion US dollars, which would have exceeded
350 billion US dollars in view of price inflation over the last twenty-five years.

2
Because of the long time involved in conducting cost structure, it is not possible to
update the information every year. However, both studies show that corrosion costs
are staggering and a figure of about 350 billions US dollars appears to be a reasonable
estimate for another two to three years. At least 35% of the above amount could have
been saved by taking appropriate corrosion control measures. In UK, the corrosion
cost was estimated to be 4-5% of GDP [5]. In Japan, the cost of corrosion was
estimated about 5258 trillion Yen per year. For most industrialized nations, the
average corrosion cost is 3.5 – 4.5% of the GDP. Below are some starting figures of
corrosion losses:
The corrosion cost of gas and liquid transmission pipelines in USA exceeds
seven billion US dollars. The figure for the major oil producing countries in
the Gulf region are not known, however the cost expected to be very high
because of highly corrosive environment in the region [4].
The corrosion-free life of automobiles in the coastal regions of Arabian Gulf
is about six months only [6]
Nearly 95% of concrete damage in the Arabian Gulf coastal region is caused
by reinforcement corrosion and consequent spalling of concrete [7].
It is estimated that 10% of all aircraft maintenance in USA is spent on
corrosion remediation [8].
Major annual corrosion losses to the tune of Euro 350 million in transport,
Euro 280 million in marine, Euro 250 million in buildings and construction
and Euro 180 million in oil and chemical industries, have been reported in
UK [9]. These are uncorrected 1971 figures.
About $ 120 billion is spent on maintenance of aging and deteriorating
infrastructures in USA.

3
Automotive corrosion costs 23.4 billion US dollars annually in USA [4].
Every newborn baby in the world now has an annual corrosion debt of $ 40.
National Association of Corrosion Engineers - International India Section
(NACE) states that the loss of materials due to corrosion was about Rs.35, 000 crores
each year, nearly 2.5 - 4 % of India's GDP [10]. It was estimated that the annual cost
of all forms of corrosion to the oil and gas industries was $13.4 billion in USA for the
year 2001, of which microbially influenced corrosion accounted for about $2 billion
[11]. The importance of internal corrosion as a cause for leakage of steel tanks was
documented by various sources. A comprehensive US Environmental Protection
Agency (EPA) report documents 6-10 % of tank failures were caused by internal
corrosion. In France, it was estimated by a major company that 10 percent of
underground storage tanks leaked due to internal corrosion while other data from
France indicated 8.5% of the leakages were caused by internal corrosion. Switzerland
has reported that internal corrosion was the cause for 5% of its tank leakages. The
percentage of internal leaks was lower than that of external leakages and Sweden had
reported that half of its leaking tanks were only due to internal corrosion.
1.1.2. Theories of corrosion
Corrosion theories may be classified into 1. Homogeneous theory and
2. Heterogeneous theory
1. Homogeneous theory
Wagner and Traud [12] emphasized that the necessary condition for corrosion
is the dissolution of the metal and electronation reaction that takes place at the metal-
environment interface. Metals become unstable due to the charge transfer reaction-
taking place at the interface. Therefore it is necessary that the potential difference
across the interface be more negative than the equilibrium potential for the metal
dissolution (anodic) reaction or more positive than the equilibrium potential for the

4
electronation (cathodic) reaction. This theory is helpful in explaining the corrosion of
pure metals and uniform corrosion, as the cathodic and anodic sites interchange their
characters quite frequently.
2.Heterogeneous theory
According to this theory, the presence of impurities on the metal surface is
necessary and thus a local cell is setup between cathodic and anodic areas. Due to the
electrochemical reactions taking place at the interface between the metal and ionically
conducting films or actual electrolyte, the metal becomes unstable and hence
corrodes.
Thus a corroding metal consists of
(a) An electron sinks area where de-electronation reaction occurs.
(b) An electron source area where electronation reaction occurs.
(c) An ionic conductor to keep the ion current flowing.
This model is the basis for the local cell theory of corrosion or heterogeneous
corrosion.
1.1.3. Principles of corrosion
Principles of corrosion depends upon the following such as
1. Thermodynamic principles
2. Electrochemical principles
1. Thermodynamic principles
Thermodynamic [13] and electrochemical principles play a major role in
determining the corrosion behaviour of materials. Thermodynamics indicate the
spontaneous direction of a chemical reaction. It is used to determine whether the
corrosion is theoretically possible or not.

5
2. Electrochemical principles
Electrochemical principles are extensively used to determine the corrosion
behaviour of materials. Here the corrosion reaction can be represented by partial
reactions such as metal oxidation and reduction of some reducible species of the
environment both occurring simultaneously at equal rates at the mixed potential of the
reaction. Corrosion reaction mainly occurs at the metal-environment interface.
The electrochemical nature of corrosion can be illustrated by the attack of
iron in hydrochloric acid. When iron is dipped in acid a vigorous action occurs as a
result hydrogen gas is evolved and iron gets dissolved.
Hence the reaction is
Fe + 2H+ → Fe2+ + H2 (1.1)
The above reaction can be divided into two partial reactions
Fe → Fe2+ + 2e- oxidation (anodic reaction) (1.2)
2H+ + 2e → H2 reduction (cathodic reaction) (1.3)
Corrosion is a surface electrochemical phenomenon common to all base
metals in aqueous, or at least humid, environments. Metal dissolution takes place at
anodic sites
M M2++ 2e- (1.4)
with the electrons being accepted in a cathodic reaction at separate sites. Most
commonly the reaction at the cathodic involves oxygen and occurs at neutral or
alkaline pH values,
½ O2 +H2O + 2e- 2OH- (1.5)

6
Under acid conditions however, protons may be the cathodic reactant,
2H+ + 2e- 2H H2 (1.6)
giving rise first to atomic then to molecular hydrogen.
There are a number of ways in which such an electrolytic cell can become
established: for example, with two dissimilar metals in electrical contact, or within a
single metallic structure where different areas of the surface have developed different
electrochemical potentials. This can arise from inclusions in the metal, from surface
imperfections, or from the presence of differential concentration cells. Most
importantly these last often involve oxygen, where the differential concentrations
arise from surface deposits or growths that limit access of oxygen to the underlying
metallic surface. Areas of low oxygen, or other nutrient concentration beneath such
deposits, are anodic sites of metal dissolution. Because the corrosion resistance of,
for example, aluminium and stainless steel is due to the formation of an oxide
passivation film, oxygen depletion cells can have a further serious corrosive effect
with these metals.
Greene and Fontana [14] stated that the term ‘Corrosion’ includes the reaction
of metals, glasses, ionic solids, polymeric solids and composites with environments
that include liquids, gases and non-aqueous electrolytes. Corrosion is regarded as the
cancer of metals that arise from the thermodynamic instability [15]. It is necessary to
devote more attention to metallic corrosion now than earlier, due to the increased use
of metals in all fields of technology. The use of rare and expensive metals for special
applications such as atomic energy requires special precautions for preservation. A
more corrosive environment is due to the increasing pollution of air and water. Mild
steel is widely used in most of the chemical industries due to its low cost and easy

7
availability for fabrication of various vessels, tanks, pipes etc. Since mild steel suffers
from severe corrosion in aggressive environments, it needs to be protected by
appropriate methodology.
1.2. Forms of Corrosion
Fontana [16] categorized eight major forms of corrosion mainly based on
appearance. Dillon [17] considered Fontana’s basic forms of corrosion and divided
them into three groups, based on their ease of identification. The three categories used
were:
Readily identifiable by ordinary visual examination:
Uniform corrosion
Pitting corrosion
Crevice corrosion
Galvanic corrosion
May require supplementary means of examination
Erosion corrosion
Cavitation corrosion
Fretting corrosion
Intergranular corrosion.
Verification is usually required by microscopy and other chemical and
electrochemical methods:
Exfoliation
Dealloying (selective leaching)
Stress corrosion cracking
Corrosion fatigue

8
Microbial corrosion.
However, the corrosion of metals and alloys almost never follows a single law
nor is governed by a single mode. Even the simplest situation can lead to the
corrosion of a material by a combination of modes driven by intertwined mechanisms.
1.2.1 Uniform corrosion
Uniform corrosion is characterized by corrosive attack proceeding evenly over
the entire surface area, or a large fraction of the total area. General thinning takes
place until failure. On the basis of tonnage wasted, this is the most important form of
corrosion. However, uniform corrosion is relatively easily measured and predicted,
making disastrous failures relatively rare. In many cases, it is objectionable only from
an appearance standpoint. As corrosion occurs uniformly over the entire surface of
the metallic component, it can be practically controlled by cathodic protection, use of
coatings or paints, or simply by specifying a corrosion allowance.
1.2.2 Pitting corrosion
Pitting corrosion is a localized form of corrosion by which cavities or ‘holes’
are produced in the material. Pitting is considered to be more dangerous than uniform
corrosion damage, because it is more difficult to detect, predict and design against
corrosion as products often cover the pits. A small, narrow pit with minimal overall
metal loss can lead to the failure of an entire engineering system. Pitting corrosion,
which for example is almost common denominator of all types of localized corrosion
attack, may assume different shapes. Pitting is initiated by localized chemical or
mechanical damage to the protective oxide film. Water chemistry factors, which can
cause breakdown of a passive film, are acidity, low dissolved oxygen concentration
and high concentration of chloride, and localized damage or poor application of a
protective coating which also cause pitting corrosion. Besides, non-metallic inclusions

9
also cause pitting. Pitting corrosion can produce pit with their mouth open
(uncovered) or covered with a semi permeable membrane of corrosion products. Pits
can be either hemispherical or cup shaped.
1.2.3. Crevice corrosion
Crevice corrosion is a localized form of corrosion usually associated with a
stagnant solution on the micro-environmental level. Such stagnant microenvironments
tend to occur in crevices (shielded areas) such as those formed under gaskets,
washers, insulation material, fastener heads, surface deposits, disbonded coatings,
threads, cap joints and clamps. Crevice corrosion is initiated by changes in local
chemistry within the crevice such as, depletion of inhibitor in the crevice, depletion of
oxygen in the crevice, a shift to acidic conditions in the crevice; build up of
aggressive ionic species like chloride in the crevice etc.
1.2.4. Galvanic corrosion
Galvanic corrosion refers to corrosion damage induced when two dissimilar
materials are coupled in a corrosive electrolyte. It occurs when two or more
dissimilar metals are brought into electrical contact under aqueous medium such as
water. When a galvanic couple forms, one of the metals in the couple becomes the
anode and corrodes faster than it would all by itself. While the other becomes the
cathode, corrodes slower than it would alone. In a bimetallic couple, the loss of noble
material will become the anode of the corrosion cell and tend to corrode at an
accelerated rate, compared to an uncoupled condition. The more noble material will
act as a cathode in the corrosion cell. Galvanic corrosion can be one of the most
common forms of corrosion and one of the most destructive.
1.2.5. Erosion corrosion

10
Corrosion of metal or alloy can be accelerated when there is an abrasive
removal of the protective oxide layer. It is the increase in the rate of attack of a metal
because of relative movement between a corrosive medium and the metal surface.
Generally, this is removed from the surface either in the form of dissolved ions or in
the form of solid corrosion products, which are mechanically swept from the metal
surface.
1.2.6. Cavitation corrosion
Cavitation occurs when a fluid’s operational pressure drops below its vapour
pressure causing gas pockets and bubbles to form and collapse. This can occur in
what can be a rather explosive and dramatic fashion. In fact, this can actually produce
steam at the suction of a pump in a matter of minute. The most locations are likely to
occur such as the suction of a pump, at the discharge of a valve or regulator, at the
geometry-affected flow areas such as pipe elbows and expansions.
1.2.7. Fretting corrosion
Fretting corrosion refers to corrosion damage at the asperities of contact
surfaces. This damage is induced under load and in the presence of repeated relative
surface motion, as induced for e.g. by vibration. Pits or grooves and oxide debris
characteristics are the typical example of fretting damage, typically found in
machinery, bolted assemblies and ball or roller bearings.
1.2.8. Intergranular corrosion
The microstructure of metals and alloys is made up of grains, separated by
grain boundaries, intergranular corrosion is localized attack along the grain
boundaries, or immediately adjacent to grain boundaries, while the bulk of the grains
remain largely unaffected. This form of corrosion is usually associated with chemical
segregation effects or specific phases precipitated on the grain boundaries. Such

11
precipitation can produce zones of reduced corrosion resistance in the immediate
vicinity. The attack is usually related to the segregation of specific elements or the
formation of a compound in the boundary. Corrosion then occurs by preferential
attack on the grain boundary phase, or in a zone adjacent to it that has lost an element
necessary for adequate corrosion resistance thus making the grain boundary zone
anodic relative to the remainder of the surface. The attack usually progresses along a
narrow path along the grain boundary. In any case the mechanical properties of the
structure will be seriously affected.
1.2.9. Exfoliation corrosion
Exfoliation corrosion is a particular form of intergranular corrosion associated
with high strength aluminium alloys. Alloys that have been extruded or otherwise
worked heavily, with a microstructure of elongated, flattened grains are particularly
prone to this damage.
1.2.10. Dealloying
Dealloying or selective leaching refers to the selective removal of one element
from an alloy by corrosion processes. A common example is the dezincification of
unstabilized brass, whereby a weakened, porous copper structure is produced. The
selective removal of zinc can proceed in a uniform manner or on a localized scale. It
is difficult to rationalize dezincification in terms of preferential zinc dissolution out of
the brass lattice structure; rather, it is believed that brass dissolves with zinc
remaining in solution and copper replating out of the solution.
1.2.11. Stress corrosion cracking
Stress corrosion cracking (SCC) is the cracking induced from the combined
influence of tensile stress and a corrosive medium. The impact of SCC on a material
usually falls between dry cracking and the fatigue threshold of that material. The

12
required tensile stresses may be in the form of directly applied stresses or in the form
of residual stresses. One of the most important forms of stress corrosion that concerns
the nuclear industry is chloride stress corrosion.
1.2.12. Corrosion fatigue
Corrosion fatigue is the cracking as a result of the combined action of an
alternating stress and corrosive environment. The fatigue process is thought to cause
receptive or the protective passive film, upon which corrosion is accelerated. The
introduction of a corrosive environment often eliminates the normal ‘fatigue’ limit of
a ferrous alloy thereby creating a finite life regardless of stress level.
1.2.13. Microbiologically influenced corrosion (MIC)
Microorganisms, inhabitants of all natural environments, influence corrosion
phenomenon either directly by accelerating the electrochemical reactions or indirectly
by virtue of their metabolic products. However, microbiologically induced corrosion
(MIC) is not a new form of corrosion, but provides a new path way to an already
existing process [18].
Microbes can induce corrosion by creating differential aeration/concentration
cells [19], changing pH [20], depleting oxygen [21], utilizing cathodic hydrogen or by
producing hydrogen [22], removing atoms from metals [23], oxidizing or reducing
corrosion inhibitors [24], setting up of galvanic cells [25], disturbing or feeding upon
protective organic coatings and by their metabolic products [26].
1.3. Biofilms and MIC
MIC is linked to cellular adhesion and extracellular polymeric substances
(EPS) produced during biofilm formation. These adhesion processes and the
subsequent EPS production lead to an important modification of the metal solution

13
interface. Its partial or total coverage by strongly adherent biofilms produces a barrier
to the exchange of elements between the metal surface and the aqueous environment.
In addition, reactions between metabolites produced by bacteria and the metal take
place within the biofilm.
It is widely acknowledged in the literature that biofouling starts immediately
after metal immersion in an aqueous medium. A thin film (approximately 20-80 nm
thick), due to the depositon of inorganic ions and high relative molecular mass
organic compounds, is formed in the first stage. This initial film is able to modify the
electrostatic charge and wettability of the metal surface [27], facilitating its further
colonization by microorganisms. Hitherto, no visible biofilm will have settled at the
metal results in the development of a biofilm consisting of bacterial cells, their EPS
and, occasionally, some entrapped particulate material. Thus, a biofilm is the result of
a surface accumulation that is not necessarily uniform in time or space [28].
A dynamic system is formed at the biofouled interface, and different transport
processes will take place through the biofilm [29]. This is a consequence of the
biofilm structure, characterized by a high degree of hydration, with water comprising
nearly 90% [30].
In this way, microbial colonization of metals drastically modifies the classic
concept of the electrical interface commonly used in electrochemical studies.
Important changes in the type and concentration of ions, pH and redox condition are
induced by the biofilm, altering the passive behavior of the metal substratum and its
corrosion products, as well as the electrochemical parameters used to assess corrosion
rates [31].

14
In MIC research, the organisms mainly implicated and primarily studied are
the sulphate reducing bacteria (SRB), which are distinguished by their sulphate
reduction capacity [32]. The other important groups involved are sulphate-oxidizing
bacteria which belong to genus Thiobacillus sp. [33] that are concerned with the
oxidation of elemental sulphur and other sulphur compounds as well as the oxidation
of iron. In spite of the initial unawareness that existed about MIC, seriousness of this
menace is slowly being recognized throughout the world in the past few decades,
which is well evinced by the ever increasing case histories [34]. MIC is accused for
failures in cooling water [35], industrial process plants [36], underground sub-tanks
[37], oil storages [38], alcohol industries [39], nuclear power stations [40], ocean
going tankers, off-shore concrete storage cells [41], heat exchangers [42] etc.
1.3.1. Economic losses due to MIC
It may seem surprising to look at the percentage of contribution of MIC
towards total corrosion failures. The pipeline corrosion was estimated in 1996 to cost
the gas industry about $840 million/year [43], and in 2001 it was estimated that the
annual cost of all forms of corrosion to the oil and gas industries was $13.4 billion, of
which MIC accounted for about $2 billion [44]. It has been estimated that 40% of all
internal pipeline corrosion in the industry can be attributed to microbial corrosion
[45].
1.3.2. Microorganisms associated with MIC
Microorganisms are thought to be primarily responsible for many metallic
corrosion failures and are classified by metabolic groups as follows: Those microbes
using inorganic material as a carbon source is called autotroph and those using
organic material as a carbon source called heterotroph. Microorganisms, which use
chemical as an energy source, are called as chemotroph and those using light as an

15
energy source are called phototroph. Microbes those using inorganic compounds as
electron donor are called as lithotroph and organic compounds as an electron donor
are called as organotroph. Most of the microorganisms associated with MIC come
under the group chemotrophs referring to those which get their energy from a
chemical source rather than a light source. They are as follows:
Sulphate reducing bacteria (SRB)
Sulphur oxidizing bacteria (SOB)
Iron oxidizing/depositing bacteria (IOB)
Manganese oxidizing / depositing bacteria (MOB)
1.3.2.1 Sulphate reducing bacteria
Sulphate reducing bacteria (SRB) require strictly anaerobic conditions for their
growth. Dissimilatory sulphate reduction is always associated with a respiratory
chemiosmotic type of energy conservation. Several species of sulphate reducing
bacteria obligatorily depend on sulphate or another sulphur compound as electron
acceptor. They are generally curved rods (ranging from Vibrio, spirilloid, semi lunar
straight or coccoid) but their morphology is influenced by age and environment.
The production of hydrogen sulphide in aquatic systems was recognized as a
biologically mediated reduction of sulphate. Beijerinck [46] demonstrated microbial
sulphide production which resulted in the first isolation of a sulphate reducing
bacterium, a strict anaerobe that can be called Spirillum desulphuricans. Van Delden
[47] was the first to grow sulphate-reducing bacteria on lactate.
1.3.2.2. Sulphur bacteria
Sulphur bacteria are found in soils rich in sulphur, sulphur springs, acid mine
water and marine resources. They are small rod shaped cells (0.5 x 1.0 – 4.0 µm) with

16
some species mobile by means of polar flagella. They are gram negative and obligate
aerobes. Energy is derived from the oxidation of reduced sulphur compounds
including sulphides, elemental sulphur, thiosulphates, polythionates and sulphites and
the final oxidation product is sulphate generally. Optimum temperature ranges from
28 to 30°C and pH between 2.0 to 3.5. Upper limit of growth is near pH.6.0 and
lower limit usually near 0.5.
1.3.2.3. Iron Bacteria
The iron bacteria are a diverse group of bacteria that are able to oxidize and /
or deposit iron oxides extracellularly or, sometimes, intracellularly. Iron bacteria are
chemolithotrophs deriving energy from the oxidation of ferrous iron Fe (II) with the
formation of ferric hydroxide Fe (III). Iron reducing bacteria can modify the
protective oxide film that forms over a mild steel surface causing the surface to
depolarize [48].
1.3.2.4. Reduction
The redox cycling of iron plays a major role in the biogeochemical cycling of
many elements in natural systems [49]. Within the reductive side of the iron redox
cycle, dissimilatory microbial systematic reduction of Fe (III) oxides has an extremely
broad range of influences on the aqueous solid phase geochemistry and behaviour of
natural and contaminant compounds in non sulfidogenic subsurface sedimentary
environments. The microbiology and environmental significance of bacterial Fe (II)
oxide reduction have been extensively reviewed over the last ten years [50-58].
Facultative anaerobes of Pseudomonas sp. capable of using ferric ions and sulphide as
terminal electron acceptors for anaerobic respiration use low molecular weight
compounds such as lactate as carbon source. Some of these bacteria attached to mild

17
steel coupons and removed a passive γ - Fe2O3 film and replaced it with a biofilm
under which pits developed.
1.3.2.5. Oxidation
Iron oxidizing bacteria which oxidize ferrous ion to ferric ion, sometimes
promote the corrosion of iron and stainless steel pipes in aerobic environments.
Corrosion products composed of ferric hydroxides and other metal salts form
tubercles which accumulate on the inner surface of the pipes. The area beneath the
deposit becomes anaerobic due to the oxygen diffusion barrier created by the
precipitate and the respiratory activities of the bacteria. The oxidized iron will either
oxidize sulphur compounds to form H2SO4 or induce anodic reaction directly.
13.2.6. Manganese Oxidizing Bacteria
Manganese oxidizing bacteria has the ability to catalyze the oxidation of
divalent, soluble Mn (II) to insoluble manganese oxides of the general formula MnOx.
It resulted in the accumulation of conspicuous and easily detectable extracellular
deposits of insoluble brown-black manganese oxides. Many different organisms have
the ability to catalyse manganese oxidation, including a diverse array of bacteria,
fungi, algae and even eukaryotes [59, 60]. Among the Prokaryotes, the ability to
oxidize manganese is also quite widespread [61,62] included are members of many
physicochemical and physiological groups of Cyanobateria, a diverse heterotrophic
rods, cocci, sheathed bacteria (Leptothrix), budding bacteria (Hypomicrobium) and
Pseudomonas sp and still the controversial Metallogenium sp.
Manganese distribution and chemical specification is kinetically controlled,
thus allowing for the inter convention of microbes and microbial products into system
are shown in Fig. 1.1. The possible mechanism of Mn (II) oxidation by bacteria is

broad
radica
radica
Fig. 1corro
Mn
Mn
3 M
consu
bindi
oxidi
for m
1.4. E
M
solid
micro
dly divided i
als or oxid
al.
1.1. Manganosion process
n2+ + ½ O2 +
n2+ + ¼ O2 +
Mn2+ + ½ O2
Modificat
umption and
ng compon
sing enzyme
manganese re
Ennoblemen
Microorganis
surfaces w
obes [64, 65
into two, i) i
dants produc
nese cycling s (Ghirose, 1
+ H2O
+ 3/2 H2O
2 +3H2O
tion of red
d manganese
nents like p
es favours th
eduction is 3
nt
sms slime la
when the la
5]. These bi
indirect and
ction of hyd
at steel surfa1988)
MnO2
MnOO
Mn3O
dox environ
e oxides ha
proteins, gly
he Mn (II) o
0-35 °C and
ayers known
atter are exp
iofilms are l
18
ii) direct. I
drogen pero
ace providin
(S) + 2H+
OH (S) + 2H
O4 (S) + 6H+
nments by
ad been stud
ycocalyces,
oxidation pr
d the optimum
n as biofilms
posed to aq
living entitie
In indirect m
oxide, super
ng the cathod
H+
oxygen pr
died. In dire
cell wall c
rocess. The
m pH is betw
form over a
queous envi
es that throu
methods, ther
r oxide and
dic reaction i
roduction,
ect methods,
components
optimum tem
ween 6-7 [50
a period of d
ironments c
ugh their m
re are free
d hydroxy
in
(1.7)
(1.8)
(1.9)
pH, acid
, the Mn-
and Mn-
mperature
0, 63].
days on all
containing
metabolism

19
produce major changes in the solution chemistry at an immersed stainless steel
surface in freshwater [66, 67]. Biofilms are also capable of significantly displacing the
corrosion potential of stainless steels in the noble direction. This phenomenon known
as “ennoblement” has been observed in seawater environments by several researchers.
Considering the environmental relevance of these experiments, direct oxidation of
Mn(II) to Mn(IV) by microbes [68-70] is most likely a common process in natural
environments. The biofilms formed in seawater known as natural population biofilms
because they comprise a variety of bacteria and algae, have been shown to raise the
corrosion potential up to or above the pitting potential [71-73] for types 304 and 316L
stainless steel. The ennoblement of the corrosion potential is thought to be caused by
the development of a low oxygen condition together with the formation of hydrogen
peroxide [65, 74] or by the microbial deposition of MnO2 [69,75].
Manganese dioxide deposited on stainless steel coupons, was reduced
electrochemically to divalent manganese, Mn2+, obtaining two electrons from the
metal substratum. Manganese oxyhydroxide (MnOOH) was determined as an
intermediate product in this reaction. The presence of biomineralized manganese
dioxide as a cathodic reactant may increase the corrosion rate and/or the probability of
active corrosion. The manganese oxidizing bacteria, active in the process of
manganese dioxide biomineralization, may hypothetically use the product of MnO2
reduction, Mn2+, making the cycle perpetual. Recently, the electrochemical behaviour
of manganese depositors was explained by various researchers [76-80]. Cathodic and
anodic polarization curves were explained along with manganese oxidizers. During
cathodic polarization, the reduction peaks for iron and MnO2 was routinely observed
for the n-type stainless alloy, while they were consistently absent for the p -type.
1.4.1. Theories proposed on ennoblement

20
1.4.1.1. Manganese oxidizing bacteria (MOB)
Dickinson et al [65]; Olesen et al [71] and Linhardt [81] have observed that the
first to identify the deposition of MnO2 as an agent responsible for ennoblement.
Passive film chemistry on stainless steel and conducting carbon surface is ennobled
by biomineralized manganese [75, 82]. The consequence of ennoblement on
chemistry of passive films on stainless steel was quantified using surface receptive
analytical techniques. Under well-defined laboratory conditions Stainless Steels were
ennobled to +450 mVSCE by biofilms of manganese oxidizing bacterium Leprothrix
discophra SP-6[69, 83]. Because the ennobled coupons, which may hypothetically,
contribute to their susceptibility to localized corrosion.
Many natural waters can support growth of MOB, when manganese-oxidizing
biofilms accumulate on surfaces of passive metals, there was a potential for
manganese redox cycling on the metal surface. This process was initiated by
depositing manganese oxidizing bacteria (MOB) and then reduced by the electrons
derived from anodic dissolution of the metal, which was corroding, and the
manganese oxides are reduced to divalent manganese ions [83]. However, since the
manganese, ions were immediately reoxidized, and the cycles continue.
The ennoblement of stainless steel by the manganese-depositing bacterium
Leptothrix discophora was studied to establish a chemical mechanism for
ennoblement in which manganese dioxide acts as a galvanic cathode. The relationship
among surface colonization, manganese deposition and open circuit potential for
stainless steel coupons exposed to batch cultures of L.discophora in nature. It can
cause several industrial problems, surface MnOx deposits shift Ecorr to the reduction
potential for the MnOx phase and depositions induces this process and confirm that

21
manganese biomineralization by L.discophora results were ennoblement process [83,
84].
Proposed Manganese mechanism:
The proposed half reaction that fixes Ecorr is
MnO2 +H2O +e- → MnOOH + OH-
Soluble Mn(II) microorganism⎯⎯⎯⎯⎯→ Insoluble manganese oxides(MnOx); X = 1,2.
(Insoluble brown or black manganese oxides)
Mn2+ + 1/2O2 + H2O microorganism⎯⎯⎯⎯⎯→ MnO2 + 2H+
The oxidation of Mn(II) to Mn(IV) is thermodynamically favored under aerobic conditions.
Mn2+(l) → Mn3+ (intermediate) →Mn4+
(S) (or)
Mn2+(l) → MnOOH (intermediate) →MnO2(S)(Mn4+ is adhered in metal surface)
Finally, Mn2+(l) → MnO2(S) (hydrothermal vent plume)
Reduction of deposited MnO2 takes place by electrochemical steps as given in reaction
Step1
MnO2 + H+ + e- → MnOOH (1.10)
Step 2
MnOOH + 3H+ + e- → Mn2+ + 2H2O (1.11) ----------------------------------------------------------------------------------
MnO2 + 4H+ + 2e- ⎯⎯→←⎯⎯ Mn2+ + 2H2O (Overall electrochemical reaction) (1.12)
-------------------------------------------------------
It has been reported that the manganese oxidation state for MnOx formed by L.
discophora increases as the oxide ages, from a value of +300 mV after 24 hrs to +500
mVSCE after 42 days [85, 86]. Such a change would increase the oxidizing power of
the oxide and thus shift the reduction potential to more positive values. Partitioning of
MnOx between suspended cells and sessile cells was a key issue in determining the
effectiveness of natural populations of manganese oxidizing microorganisms in

22
promoting ennoblement. However, the tendency for biofilms to form under these
conditions is undisputed, and it has been reported elsewhere that sessile growth
greatly enhances the manganese oxidation rate [87]. Manganese oxidizing bacteria
were widely distributed in nature and a variety of industrial problems ranging from
dirty drinking water to fouled heat exchangers. Numerous reports during the past 2
decades have also associated these bacteria with stainless steel corrosion, but the
biological role in the corrosion process has never been established [88]. Over
generally the same period, the ennoblement phenomenon and its corrosive
consequences have been recognized [89]. They take apart observations to a common
cause, biominerlization of MnOx and support the biological mechanism of
ennoblement. Based on these findings, ennoblement and the associated risk of
corrosion must be added to an already extensive list of industrial problems caused by
manganese oxidizing bacteria [71, 73].
1.4.1.2. Oxygen reduction theory
The oxygen plays a vital role in ennobled process on stainless steel surface. The
effect of the biofilm on a more resistant stainless steel will be to shift the corrosion
potential towards the noble direction. If the critical pitting potential for the alloy was
more active than reversible oxygen potential under exposure conditions, the corrosion
potential may shift nobly enough to initiate pitting corrosion [90]. The explanation for
the increase in cathodic activity depicted as being an increase in the exchange current
density for the oxygen reaction. An increase in current density for the oxygen reaction
was possible, considering that the extracellular slimes produced on stainless steel
surface by the bacteria known to bind heavy metals, and that some of these organo-
metallic complexes of Mn, Fe, Co, and Ni can serve as oxygen reduction catalysts
[91]. The mechanism of the noble shift in potential in the presence of bacterial film

23
probably involves a change in the kinetics of the oxygen reaction. Assuming that a
new oxide with rate enhancing properties towards oxygen reduction developed, at
noble potentials, oxygen reduction occurred on a modified oxide surface an enhanced
rate [65]. It was speculate about the ennoblement mechanism involving catalytic
enhancement of the oxygen reduction reaction by organo-metallic complexes [90].
1.4.1.3. Peroxide theory
Another proposed theory of ennoblement process influenced by hydrogen
peroxide [66]. One of the hypothesis is based on the presence of hydrogen peroxide
(H2O2), generated by microorganisms in the native biofilm. The concentration of
H2O2 in native biofilms formed on the coupons was detected after the exposures, and
OCP for the specimens were determined. During the summer season, the
concentration of H2O2 was higher than 10 ppm and OCP were nobler than +600
mVSHE. Other hand, during the winter season, the H2O2 concentration was lower than
2 ppm and OCP were less than +300 mVSHE [74]. The ennoblement of OCP was
counteracting by the decomposing H2O2 .The value of OCP for type 316L with H2O2
free biofilms was as less noble as that with no biofilm growth. A H2O2 free state was
reached by the addition of H2O2 decomposition enzyme (catalase or peroxidase) to
water and control of the water temperature [74]. The OCP shift is due to presence and
absence of H2O2. Its hypothesis also expressed ennoblement process on stainless steel
due to not only H2O2 concentration and also biomineralization of MnO2 [86].
1.4.1.4. Enzyme theory
The process of ennoblement has been correlated with enzyme theory such as extra
cellular polysaccharides (EPS) and biogenic manganese dioxide deposition. Enzyme
theory proposed to find out the role of cations and polymers on ennoblement process.
Ethylene diamine tetra acetic acid (EDTA) has been used to eliminate all cations in

24
biofilm. The ennoblement could not be observed in EDTA treated biofilm. It reveals
that without the presence of cations in the biofilm ennoblement does not take place on
passive materials [92]. The process of ennoblement was connected to organometallic
catalysis of the oxygen reduction reaction. The ennoblement of stainless steel in
seawater was due to the role of enzyme theory, produced by the biofilm causing
microbes [93]. The biofilm matrix has both anodic inhibitor and cation like
manganese which accelerates the corrosion of metals by ennoblement. Hence, it
concludes that without biogenic cations, there is no ennoblement, which contradicts
enzyme theory proposed who pointed out that enzymes trapped in EPS of biofilm
only contribute to ennoblement. Finally, it was proposed that a contributing factor to
ennoblement comes from relations between biogenic MnO2 and bacterial polymers in
the biofilm.
1.4.1.5. n / p - type semiconducting theory
Based on photo electrochemical studies, the oxide films on the conducting
materials were classified as n-type or p-type semiconductors based on the direction in
which their OCP shifts [94]. A negative shift in OCP is characteristics of an n-type
passive film. While a positive shift indicates a p-type film. The nature of n-type films
has excess cations with an abundance of free electrons, while p-type films have excess
anions with electron holes. The nature of cathodic curves was various n/p type semi
conducting oxide films in presence of biofilm. They used variety of metal in
constraint time of exposure take place only few metals are n or p- type semiconductor.
Most iron containing alloys tend to be n-type and their surface are dominated by ferric
oxides. It is also well known that biogenic MnO2 is an n-type semiconductor. The role
of biologically deposited MnO2 is on stainless steel surface with n-type semi
conducting film [76]. And then the bacteria for maintenance of a physiological carbon

25
: phosphorus ratio(40:1) during the growth of the cell, brings out the too little carbon
and too much phosphorus from the cell in the form of glucose -6-phosphate anion and
some amount of individual sugars in microfouling material. The proposed mechanism
is that the anion of organic phosphate combines with excess cations of n-type oxide
film leading to both strengthening of oxide film and a positive shift in the corrosion
potential [94].
1.5. Corrosion control and Prevention
1.5.1. Materials selection
Stainless steel is the almost an exclusive choice of pipelines designers. This is
for pipeline systems that are used to cooling water system; is also true for those
pipelines that are used to process industries. It is also the case for piping systems that
are used to distribute natural gas, water, water-refined liquids, and so on, to the end
user [95]. Metallic corrosion can be prevented by either changing the metal (or)
altering the environment or by separating the metal from the environment (or) by
changing the electrode potential of the metal. A large number of corrosion failures are
due to improper design of equipment and the corrosion control can be therefore
warranted at the design stage itself. Corrosion can be prevented by changing the
electrode potential by taking the metal to the immune region (or) passive region.
According to Pourbaix diagram this can be accomplished by making the potential of
the cathode equal to the open circuit potential of anode.
1.5.2. Corrosion inhibitors
1.5.2.1. Introduction
An inhibitor is a chemical substance which, when added to an environment in
small concentration, effectively checks, decreases or prevents the reaction of the
metal with the environment [96]. Corrosion inhibitors are added to many systems
including, cleaning pads, cooling systems, various refinery units, chemical operations,

26
steam generators, ballast tanks, oil and gas production units. Corrosion inhibitors are
used to protect metals from corrosion, including temporary protection during storage
or transport as well as localized protection required for example to prevent corrosion
that may result from accumulation of small amounts of an aggressive phase. One
example is brine, in a nonaggressive phase, such as oil. An efficient inhibitor is
compatible with the environment, is economical for application and produces the
desired effect when present in small concentrations.
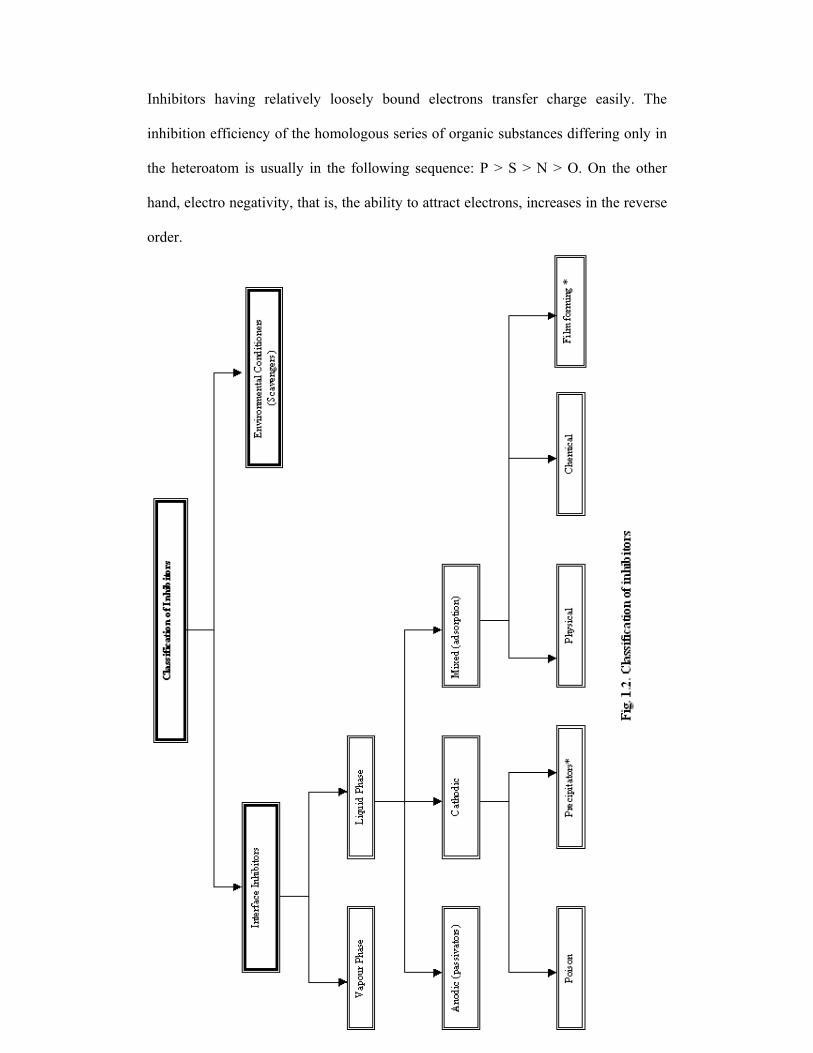
1.5.2.2. Classification of inhibitors
Inhibitor selection is based on the metal and the environment. A qualitative
classification of inhibitors is presented in Fig. 1.2. Inhibitors can be classified into
environmental conditioners and interface inhibitors.
1.5.2.3. Environmental conditioners (scavengers)
Corrosion can be controlled by removing the corrosive species from the medium.
Inhibitors that decrease corrosivity of the medium by scavenging the aggressive
substances are called environmental conditioners or scavengers. In near-neutral and
alkaline solutions, oxygen reduction is a common cathodic reaction. In such
situations, corrosion can be controlled by decreasing the oxygen content using
scavengers [97].
1.5.3. Interface inhibitors
Interface inhibitors control corrosion by forming a film at the metal /
environment interface. Interface inhibitors can be classified into liquid and vapour
phase inhibitors.
1.5.3.1. Liquid phase inhibitors

27
Liquid phase inhibitors are classified as anodic, cathodic or mixed inhibitors
depending on whether they inhibit the anodic, cathodic, or both electrochemical
reactions.
1.5.3.2. Anodic inhibitors
Anodic inhibitors are usually used in near-neutral solutions where sparingly
soluble corrosion products, such as oxides, hydroxides, or salts, are formed. They
form, or facilitate the formation of passivating films that inhibit the anodic metal
dissolution reaction. Anodic inhibitors are often called passivating inhibitors.
1.5.3.3. Cathodic inhibitors
Cathodic inhibitors control corrosion by either decreasing the reduction
process (cathodic poison) or by precipitating selectively on the cathodic areas
(cathodic precipitators). Cathodic poisons, such as sulfides and selenides, are
adsorbed on the metal surface; whereas in near neutral and alkaline solutions,
inorganic anions, such as phosphates, silicates, and borates, form protective films that
decrease the cathodic reaction rate by limiting and diffusion of oxygen to the metal
surface. Cathodic precipitators increase the alkalinity at cathodic sites and precipitate
insoluble compounds on the metal surface. The most widely used cathodic
precipitators are the carbonates of calcium and magnesium.
1.5.3.4. Mixed inhibitors
About 80% of inhibitors are organic compounds that cannot be designated
specifically as anodic or cathodic and are known as mixed inhibitors. The
effectiveness of organic inhibitors is related to the extent to which they adsorb and
cover the metal surface. Adsorption depends on the structure of the inhibitor, surface
charge of the metal, and on the type of electrolyte [98]. Mixed inhibitors protect the
metal in three possible ways: physical adsorption, chemisorption and film formation.

28
Physical (or electrostatic) adsorption is a result of electrostatic attraction between the
inhibitor and metal surface. When the metal surface is positively charged (anionic)
inhibitors is facilitated (Fig. 1.3). Positively charged molecules acting in combination
with a negatively charged intermediate can inhibit a positively charged metal. Anions
such halide ions, in solution adsorb on the positively charged metal surface, and
organic cations subsequently adsorb on the dipole (Fig. 1.4a -1.4b).
Physically adsorbed inhibitors interact rapidly, but they are also easily
removed from the surface. Increase in temperature generally facilitates desorption of
physically adsorbed inhibitor molecules. The most effective inhibitors are those that
chemically adsorb (chemisorb), a process that involves charge sharing or charge
transfer between the inhibitor molecules and the metal surface. Chemisorption takes
place more slowly than physical adsorption. As temperature increases adsorption and
inhibition also increases. Chemisorption is specific and is not completely reversible
[99].
1.5.3.5. Vapour phase inhibitors
Temporary protection against atmospheric corrosion, particularly in close
environments can be achieved using vapour phase inhibitors (VPI). Substances having
low but significant pressure of vapour with inhibitive properties are effective [100].
1.5.4. Mechanistic aspects of corrosion inhibition
1.5.4.1. Environmental conditioners (scavengers)
In near-neutral solution, the common cathodic reaction is oxygen reduction:
O2 + 2H2O + 4e- ↔ 4OH- (1.13)
Scavengers deplete the oxygen by chemical reaction: for example, hydrazine removes
oxygen by the following reaction:

29
5O2 + 2(NH2-NH2) ↔ 2H2O + 4H+ + 4NO2- (1.14)
1.5.4.2 Anodic inhibitors (Passivators)
The mechanism of anodic inhibition can be explained using the polarization
diagram of an active-passive metal [101]. In the absence of inhibitors, the metal
corrodes in the active state at a rate corresponding to point A in Fig.1.4. As the
concentration of inhibitor is increased, the corrosion rate also increases until a critical
concentration and a critical corrosion rate are reached. At the critical concentration,
there is a rapid transition of the metal to the passive state, and the corrosion rate is
decreased.
1.5.4.3. Cathodic inhibitors
In acid solution, the cathodic reaction is typically the reduction of hydrogen ions
to hydrogen, which combine forming hydrogen molecules:
H+ + e- ↔ H (1.15)
2H ↔ H2 (1.16)
In alkaline solution, the cathodic reaction is typically oxygen reduction [eq.(1.13)].
Cathodic inhibitors impede reduction reactions. Substance with high over potential for
hydrogen and those that form precipitates at the cathode are effective in acid and
alkaline solution, respectively. In this case, the slope of the anodic polarization curve
is unaffected, but the slope of the cathodic polarization curve is changed [102].
1.5.4.4. Mixed inhibitors (Adsorption)
Adsorption occurs as a result of electrostatic forces between the electric charge
on the metal and the ionic charges or dipoles on the inhibitor molecules. The potential
at which there is no charge on the metal is known as the zero-charge potential (ZCP).
The charge on a metal surface in a given medium can be determined from the

30
corrosion potential (Ecorr) and zero-charge potential. When the difference, Ecorr - ZCP,
is negative, the metal is negatively charged and adsorption of cations is favored, when
Ecorr - ZCP is positive, the metal is positively charged and adsorption of anions is
favored.
The charge on inhibitors depends on the presence of loosely bound electrons,
lone pair of electrons, π-electrons cloud and functional groups containing elements of
group V and VI of the periodic table. Most organic inhibitor possesses atleast one
functional group, regarded as the reaction center or anchoring group. The strength of
adsorption depends on the charge on this anchoring group [rather on the hetero atom
(i.e., atoms other than carbon including nitrogen and sulphur) present in the anchoring
group]. The structure of the rest of the molecule influences the charge density on the
anchoring group [99]. Water molecules adsorb on the metal surface immersed in an
aqueous phase. Organic molecules adsorb by replacing the water molecules:
[Inhibitor]soln + [nH2O]adsorbed ↔ [Inhibitor] adsorbed + [nH2O]soln (1.17)
where n is the number of water molecules displaced by one inhibitor molecule.
The ability of the inhibitor to replace water molecules depends on the
electrostatic interaction between the metal and the inhibitor. On the other hand, the
number of water molecules displaced depends on the size and orientation of the
inhibitor molecule. Thus, the first interaction between inhibitor and metal surface is
nonspecific and involves low activation energy. This process, called “Physical
adsorption”, is rapid and in many cases, reversible [103].
The adsorbed molecules involved in chemical reaction (chemisorption) from
bands with the metal surface. Chemisorption is specific and is not reversible. The
bonding occurs with electron transfer or sharing between metal and inhibitor. Electron
transfer is typical for transition metals having vacant, low energy electron orbitals.
31
Inhibitors having relatively loosely bound electrons transfer charge easily. The
inhibition efficiency of the homologous series of organic substances differing only in
the heteroatom is usually in the following sequence: P > S > N > O. On the other
hand, electro negativity, that is, the ability to attract electrons, increases in the reverse
order.

32
Fig. 1.3. Adsorption of negatively charged inhibitor on a positively charged metal surface.
Fig.1.4(a). Positively charged inhibitor molecule does not interact with
positively charged metal surface.
Fig. 1.4(b). Synergistic adsorption of positively charged inhibitor molecule
and anion on a positively charged metal surface.

33
1.6. Biocides
Biocides are the most underused, misunderstood, and misapplied chemical
products in the process industry for many reasons. Biocides are used to combat a
problem that is subtle and difficult to detect. In general, biocides are needed to control
the activity of the bacteria in a system. However, biocides alone cannot solve a
microbiological problem. Very few sample and reliable means of monitoring are
available to the supplier or the end user, and the benefits of biocide use take a long
time to become evident. Finally, once bacteria are well established it is nearly
impossible to control without drastic measures [104]. Hence, identification of
microbes and also the role of inhibitors on microbial growth are to be studied in
detail.
1.7. Summary
This chapter mainly deals with the definition of corrosion followed by various forms
of corrosion. Special emphasize is given to microbiologically influenced corrosion
and the economic losses caused by MIC. The effect of various corrosion causing
microorganisms such as sulphate reducing bacteria, iron oxidizing bacteria and
manganese oxidizing bacteria and their morphology, biochemistry of each
microorganisms are discussed in detail. The mechanistic aspects of corrosion of
passive metals caused by ennoblement process are also dealt with. Some recent
research in the area of microbiologically influenced corrosion and some open
problems for further research have been also discussed in this chapter. Besides,
control the corrosion, an efficient corrosion inhibitor should be able to bind
irreversibly to the metal surface, provide good surface coverage and at the same time
with very good biocidel effect. It explains the brief history of inhibitor of corrosion
and the importance of corrosion inhibitors along with their potential applications in
cooling water / transporting pipelines.

34
References
[1] R.M. Atlas, Microbial. Rev. 45 (1981) 180. [2] C.C. Technologies Laboratories, Inc. (2001). Cost of corrosion and prevention
strategies in the United States, Ohio: Dublin, USA. [3] Federal High way Administration (FHWA), office of the Infrastructure and
development (2001). Report FHWA-RD-01-156. [4] National Association of Corrosion Engineers (NACE) (2002). Material
Performance, Special Issue, Houston, Texas, USA, July.Jointly with C.C.Technologies and FHWA.
[5] H.H. Uhlig, Corrosion and Corrosion control, 3rd ed. New York: John Wiley and
sons. (1985). [6] Z. Ahmad, British Corrosion Journal, 31 (2) (1996) 191. [7] S. Rashid-uz-Zafar, G.J. Al-Sulaiman, A.S. Al-Gahtani, Symp. Corrosion and the
control, Riyadh, Saudi Arabia, May, (1992) 110. [8] M.A.A. Tullmin, P.R. Roberge, L. Grenier, M.A. Little, Canadian Aeronautics
and Space Journal, 42 (2) (1990) 272. [9] T.P. Hoar, Report of the committee on Corrosion and Protection, London: HMSO. (1971). [10] a) http://www.naceindia.org/ b) http://www.sreechem.com/corrosion%202.htm. [11] G.H. Koch, M.P.H. Brongers, N.G. Thompson, Y.P. Virmani, and J.H. Payer.
2001. Corrosion costs and preventive strategies in the United States. FHWA-RD-01–156 [Online].Federal Highway Administration, Washington, D.C. http://www.corrosioncost.com.
[12] C. Wagner, W. Traud.Zeit.Electrochem.44 (1938) 391.
[13] J.O.M. Bockris, A.K.N. Reddy, Modern Electrochemistry, Plenum Press, (1970) 1091.
[14] N.D. Greene, M.G. Fontana, A critical analysis of pitting corrosion,Corrosion 15(1959) 25. [15] K.A. Chandler, D.A. Bayless, Corrosion protection of steel structures, Elsevier Applied Science Publication Ltd.(1985). [16] M.G. Fontana, Corrosion Engineering, New York, McGraw Hill(1986). [17] C.P. Dillon, Forms of corrosion, Recognition and Prevention, NACE International, Houston, TX (1982).

35
[18] G.H. Booth (ed.), Microbiological corrosion, M and B monographs CE11, Mills and Boon, London (1971). [19] R.N. Miller, W.C. Herron, A.G. Krigens, Mat. Prot. 9 (1964) 60. [20] A.K. Tiller, Aspects of Microbial corrosion in corrosion processes (RN Purkiss, ed.), Appl.Sci.Pub.NewYork (1982)115. [21] K. Chidambaram, K. Balakrishnan, Sulphate reducing bacterial attack on corrosion preventive and corrosion resistant materials, Proc. 6th Asian pacific corrosion control conference, Singapore, 13-19 (1989). [22] I.P. Pankhania, Biofouling 1 (1988)27. [23] H.G. Hedrick, R.J. Reynolds, M.G. Crum, Electrochem. Technol. 79 (1967) 3. [24] W.P. Iverson, Biological Corrosion Advan. Corr. Sci.andTech., 2, (M.G.Fontana, R.W, St Aechle, ed) Plenum Press, London (1972). [25] R.A. King, JDA. Miller, Nature 283 (1971) 491. [26] W.P. Iverson, Mater.Perf. 23 (1984) 28. [27] SC. Dexter, Influence of substrate wettability on the formation of bacterial slime films on solid surfaces immersed in natural seawater. In proceedings of the 4thinternational congress on Marine Corrosion and Fouling, Juan les Pins, Antibes, France (1976) 137. [28] W.G. Characklis, K.C. Marshall Biofilms: a basis for an interdisciplinary approach. In Biofilms, ed. Wiley Interscience, New York (1990) 3. [29] W.G. Characklis, Biotechnology and Bioengineering, 23(1981)1923. [30] G.G. Geesey, Microbial exopolymers: ecological and economic considerations, American Society for Microbiology News 48 (1982) 9. [31] H.A. Videla, Metal dissolution/ redox in biofilms. In structure and function of Biofilm, ed, W. G. Characklis and P. A. Wilderer, Wiley Interscience, Chichester (1989a) p. 301. [32] W.P. Iverson, Advan. Appl. Microbiol. 32 (1987) 1. [33] C.O. Obuekwe, Microbial corrosion of crude oil pipelines, Ph.D. thesis, University of Alberta, Edmonton (1980). [34] A.C. Sequeira, Electrochemical techniques for studying microbial corrosion. Microbial Corrosion, 1 (C.A.C Sequeira and A.K.Tiller eds.), Elsevier Appl.Sci, England (1988) 99. [35] C.R. Schmitt, Anomalous microbiological tuberculation and AC pitting

36
corrosion case histories, Biologically induced corrosion, (S.C.Dexter ed), NACE, TX (1985)69. [36] P.F. Sanders, W.A. Hamilton (1985). Biological and corrosion activities of SRB in industrial process plant, Biologically induced corrosion, (SC Dexter, ed.) NACE, TX (1985) 47. [37] G. Kobrin, Reflections on MIC of SS, Biologically induced corrosion (SC Dexter Ed.) NACE, TX (1985) 33. [38] Anon, The role of bacteria in the corrosion of oil field equipments, Technical publication committee release No.3, NACE, TX (1976). [39] A.J.N. Silva, J.N. Tanis, J.O. Silva, Alcohol industry biofilms and their effect on corrosion of 304 SS. Biologically induced corrosion (SC Dexter, ed.) NACE, TX (1985) 76. [40] M. Bibb, Bacterial corrosion in S. African Power industry, Biologically induced corrosion (SC Dexter, ed.) NACE, TX,(1985) 96. [41] W.A. Hamilton, Annu. Rev. Microbiol. 39(1985) 195. [42] K.D. Efird, Mat. Perf. 15 (1976)16. [43] E. Buck, G.C. Maddux, R.L. Sullivan, Intern Corrosion Cost Impact Study – United States natural gas exploration and production industry. GRI-96/0056 document No 96-1466. Gas Research Institute, Des Plaines (1996). [44] G.H. Koch, M.P.H. Brongers, N.G. Thompson, Y.P. Virmani, J.H. Payer, Corrosion costs and preservative strategies in the United States FHWA-RD-01- 156 (on-line), Federal Highway Administration, Washington, D.C (2001). http://corrosioncost.com. [45] B.G. Pound, Gap analysis of the Pipeline Research Committee International (PRCI)/Gas Research Institute (GRI) research program on internal corrosion. GRI contract 6008. Topical report SF26363.000/AOTO/1198/BP02. Gas Research Institute, Des Plaines, Ill. (1998). [46] M.W. Beijerinck,Uber Spirillum desulfuricans als Ursache von Sulfat- reduktion, Centr. Bakteriol. Abt. 1 (1895)1, 49, 104. [47] A. Van Delden, BeitragzurKenntnis der Sulfatreduktiondurch Baketereien. Centralbl.Bakteriol.II.abt.11 (1903) (81-94), 113-119. [48] C.O. Obuekwe, D.W.S. Westlake, F.D. Cook, J.W. Costerton, Appl. Envir. Microbiol. 41(1981) 766. [49] WL.Stumm, SB.Sulzberger, The role of coordination at the surface of aquatic particles, In. Chemical and biological regulation of aquatic systems, Buffle J, De vitre RR (eds), Lewis Publishers(1994) p.45.

37
[50] K.R. Nealson, C.R. Myers, Appl. Environ. Microbiol.58(1991) 439. [51] D.R. Lovely. Microbiol Rev. 55(1991) 259. [52] D.R. Lovely, Microbial oxidation of organic matter coupled to the reduction of Fe (III) and Mn (IV) oxides, In Biomineralization processes of iron and manganese, Skinner MCW, Fitzpatrick RW (eds.); Catena Verlag (1992) 101. [53] K.R. Nealson, Saffarini,Annual.Rev.Microbiol. 48 (1994) 311. [54] D.R. Lovely, Microbial reduction of iron, manganese and other metals, In Advances in Agronomy, Sparks DL (ed.), Academic Press Inc.54 (1995) 175. [55] D.R. Lovely, Fe (III) and Mn (IV) reduction, In Environmental metal-microbe interactions, Lovely DR (ed.) ASM Press (2000) 3. [56] JK. Fredrickson, Y.A. Gorby ,Curr.Opin. Biotechnol. 7 (1996) 287. [57] B.Thamdrup, Adv. Micro. Ecol. 16 (2000) 41. [58] A. Rajasekar, T. Ganesh Babu, S. KaruthaPandian, S. Maruthamuthu, N. Palaniswamy, A. Rajendran, Corros. Sci. 49 (2007) 2694. [59] W.C. Ghirose, Ann.Rev. Microbiol. 38 (1984) 515. [60] W.C. Ghirose, The biology of manganese transforming microorganisms in Soil, In: P.Graham et al., (ed.) manganese in soil and plants, Kheyer Academic Pub.Delft, Netherlands (1988) 75. [61] Marshall KC, Biogeochemistry of Manganese minerals, In: P.A. Trudinger and D.J. Swaine (ed.) Biogeochemistry of mineral forming elements. Elsevier, Amsterdam.(1979) 252. [62] K.H. Nealson, Microbial oxidation and reduction of manganese and iron; In: P.Westbrock and E.W. dejong (ed), Biomineralisation and biological metal accumulation, Reidel Pub. Co. Amsterdam (1983) 459. [63] G.I. Karavaiko, V.A. Yurchenko, V.I. Remizov, T.M. Klyushnikova, Microbiology 55 (1987)553. [64] S.Mododa, Y. Suzuki, T. Shinohara, Corros. Sci. 31 (1990) 515. [65] W.H. Dickison,Z. Lewandowski, and R.D.Geer, Corrosion 52(1996) 911. [66] V.Scotto and M.E.Lai, Corrosion science 40(1996) 1007. [67] K.Mattila, L. Carpen, L Raaska, H-L Alakomi, T. Hakkarainen, M.S Salkinoja Salonen, Journal of industrial microbiology and biotechnology 24 (2000) 410.

38
[68] Paul W. Baker, Kimioito, K. Watanabe, Environmental microbiology 5(2003) 925. [69] S. Campbell, G. Geesey, Z.Lewandowski, G. Jackson, Corrosion 60 (2004)670. [70] W.Wei, W. Jia, X. Haibo, L.Xiangbo, Material and corrosion 5(2005) 56. [71] B. H. Olesen, R.Avci, Z.Lewandowski, Corrosion science 42 (2000)211. [72] T.J. Hakkarainen, Material and corrosion 54(2003) 503. [73] P. Linhardt, Electrochemicaacta 51 (2006) 6081. [74] N.Washizu, Y.Katada, T.Kodama, Corrosion science 46 (2004) 1291. [75] K. R Braughton, R.L. Lafond, Z. Lewandowski, Biofouling 17 (2001) 241. [76] S.C. Dexter, S. Maruthamuthu, Corrosion 2001 paper No. 01256. [77] S. Palaniswamy, S. Maruthamuthu, S.T. Manickam, A. Rajendran, Curr.Sci. 82(2002) 865. [78] X. Shi, R. Avci, Z. Lewandowski, Corrosion (2002) paper 02456. [79] Z. Lewandowski, W.A. Hamilton, Corrosion (2002) Paper No. 02474. [80] B. Anandkumar, S. Maruthamuthu, Curr. Sci. 94(2008) 891. [81] P. Linhardt,Material Science forum 289(1998) 1267. [82] SC. Dexter, K Xu and G. L. Luther III, Biofouling19 (2003) 139. [83] Wayne H. Dickinson, Frank Caccavo, JR., Bo Olesen, and Z.Lewandowski, Applied and environmental microbiology (1997) 2502. [84] Jan kielmoes, Isabelle bultinck, Hedwig Storms, Nico boon, Willy Verstraete, FEMS microbiology Ecology 39 (2002) 41. [85] K.Mattila, L.Carpen, T.Hakkarainen, M.S.Salkinoja-Salonen, International Biodeterioration and biodegradation 40 (1997) 1. [86] C. Marconnet, Y. Wouters, F. Miserque, C. Dagbert, J.P. Petit, A. Galerie, D. Feron, ElectrochimicaActa 54 (2008) 123. [87] Wayne H. Dickinson , Z. Lewandowski, Biodegradation 9 (1998) 11. [88] F.Mansfeld, Materials and Corrosion 54 (2003) 489.

39
[89] IwonaB. Beech,International biodeterioration and biodegradation 53 (2004)177.
[90] F. Mansfeld, S. Lin, S.Kim, H. Shih, Mat. Sci. Forum44 (1989) 83. [91] S.C.Dexter and G.Y.Gao, Corrosion 44 (1988)717. [92] S.Maruthamuthu, S. Sathiyanaranyanan ,R.Palaniappan and N.Palaiswamy,
Corrosion (2002) Paper No. 02468. [93] V.L. Hostis, C.Dagbert, D.Feron, ElectrochemicaActa 48 (2003) 1451. [94] S.Maruthamuthu, G.Rajagapal, S.Sathiyanarayanan, S.Angappan, M.Eashwar and K. Balakrishnan, Current science 71(1996) 25. [95] L. Garverick (Ed.), Corrosion in the petrochemical industry, ASM international, Materials Park, OH 44073-0002. (1994) 13. [96] O.L. Riggs, Jr., in C.C.Nathan (Ed.), Corrosion Inhibitors, NACE, Houston, TX,
(1973) 11. [97] M.G. Noack. Mater. Perform. 21 (1982) 26. [98] A. Frignani, G. Trabanelli, F. Zucchi, M. Zucchini, Proceeding of 5th European
Symposium of corrosion inhibitors, Ferrara, Italy (1980) 1185. [99] V.S. Sastri, Corrosion Inhibitors: Principles and Applications, J.Wiley, New
York(1998) 39. [100] S.A. Levin, S.A. Gintzbergy, I.S. Dinner, and V.N. Kuchinsky, Proceedings of
Second European Symposium on Corrosion inhibitors, Ferrara, Italy(1965) 765. [101] M.G. Fontana, Modern theory- Principles in Corrosion Engineering, McGraw-
Hill, New York, (1986) 445. [102] N.Hackerman and E.S. Snavely, in L.S.V Delinder (Ed.)., Corrosion Basics,
NACE Houston, TX (1984) 127. [103] W. Lorenz, Z.Phys.Chem.219(1962) 421.and 244 (1970) 65. [104] E. Widdel, J. Appl. Bacterial.(1980) 271.