chapter 2 syntbesis add biological activity ofpbospbate mimics boc...
TRANSCRIPT

CHAPTER 2
Syntbesis aDd Biological activity ofPbospbate mimics of Boc-Pbe-Tyr-a~mides

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
2.1 Introduction
Diabetes mellitus is a major and growing, public health problem throughout the
world. About 90-95% cases of diabetes are related to type 2 diabetes. The Type 2 diabetes is
generally associated with low secretion of insulin. In some cases the problem is associated
with inactivation of insulin receptors. The later form of the disease is referred to as insulin
resistance, where tissues develop resistance to the actions of insulin, even though in most
instances the insulin receptors in these tissues are structurally normal and are in normal
abundance. One strategy to combat this insulin resistance therapeutically may be to maintain
insulin receptors (IRs) in the active phosphorylated form by inhibiting enzymes that catalyze
IR dephosphorylation. It has been demonstrated that protein tyrosine phosphatase IB
(PTPIB) attenuates insulin signaling by catalyzing dephosphorylation of IRs. l Recently, it
was shown that PTPIB knock-out mice become more insulin sensitive and fail to gain
weight despite being fed with a fat-rich diet.2-3 Therefore, PTPIB is an attractive target for
the development of potential new drugs for treating insulin resistance.
In the past decade, rigorous efforts were made to develop agents that can selectively
inhibit PTPIB enzyme. Towards this direction, a major breakthrough was published
describing agonist and antagonist activity of CCK-S analogs with modification at phenolic
hydroxy of tyrosine for the development of selective inhibitors of PTP 1 B. Several analogues
of cholecystokinin (CCK-St-7 were found to be surprisingly potent inhibitors ofPTPlB, and
a common N-terminal tripeptide, N-acetyl-Asp-Tyr(S03H)-Nle, was shown to be necessary
for inhibition. Even simple sulfotyrosyl-containing tripeptide N-acetyl-Asp-Tyr (S03H)-Nle
NH2 (I) (Figure ·1), retains essentially all of the activity (Kj =5 IlM) and is even superior to
related octapeptide H-Asp-Tyr(S03H)-Met-Gly-Trp-Met-Asp-Phe-NH2 (CCK-S) (Kj=34
IlM).8
H + ~~eOOH _~N~eOOH ~ II
N-acetyl- Asp-Tyr(S03H)-Nle-NH2 -~ I"\A 0 - o~ oANi-? H03SO // 0 NH ~ \
~\ Hooe + eOOH
I <Ki=5 ~M) II (Kj=30 ~M) III (K j=12 ~M)
Figure 1. Structure ofO-Malonyl derivatives obtained from CCK-S. (-+) showing point of modification.
39

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
This led to the investigation of peptidomimetic derived molecules wherein the
peptidic characteristics of tripeptide were decreased and its chemical stability was increased
by replacement of the labile sulfate with a suitable bioisostere as shown in figure 1.
In this direction, the first step was the removal of the terminal amides, resulting in
simple bis-amide tyrosine sulfate II that retained significant, although reduced activity (Ki
=30 IlM). Subsequent replacement of the tyrosine sulfate moiety of II with an O-malonate
resulting in III was based on the work of Sikorski9 who had first demonstrated that 0-
malonate could serve as a mimic for phosphate, and that of Burke,IO-12 who had
demonstrated that O-Malonyltyrosine and fluoro-O-Malonyltyrosine could be successfully
incorporated as tyrosine phosphate mimic into hexapeptidic inhibitors of PTPIB. In fact,
malonate III proved to be even more effective inhibitor than II and continued to display
competitive inhibition kinetics with a Ki of 11 IlM against PTPlB and was approximately
lO-fold more selective against LAR (Leucocyte common antigen related protein), a
membrane spanning PTP. 13
It was interpreted through SAR and was confirmed with ester and amide analogues
that the carboxyl group of the N -terminus of III would be required, since it corresponded to
the aspartic acid present in the majority of the peptidic PTPIB inhibitors. However the N
terminus was amenable to further manipulation as indicated from several x-ray co-crystal
structures.8, 13 .
O-(COOH
S~ COOH
~
H 0 H HOOC~Ny)lN N~
o l~~ 0
IV (K;=O.7IJM) ...-~ 0
~ !.
Figure 2. Structures ofO-Malonyl analogues.
These modifications resulted in analogue IV which for the first time resulted in
submicromolar activity against PTPlB (Ki = 0.7 1lM) (Figure 2). Subsequent SAR quickly
determined that simple phenylalanine analogue V (Ki = 9 IlM) was more potent than III and
had the advantage of being less charged and having lower molecular weight as compared to
IV (Figure 3). 13
40

Chapter 2 Synthesis & Biological acth:ity of Phosphate mimics of Boc-Phe-Tyr-amides
H HOOC~N
o
IV (K;=O.7 ~M)
O--(COOH
1/ \ COOH
O--(COOH
S"'-\ COOH
o H N N~ H 0 .
o H 0 '/ ~ -- v (K,=9.1-IM)
y ~
VI (K;=3.4 ~M)
Figure 3. Structure of analogues of IV.
When analogue V was subjected to modification at amide bond, l.ike incorporation of
urea in place of amide, it resulted in more potent analogue VI, having Ki of 3.4 )lM. The
binding studies of the derivative VI were carried out by Larsen and co-workers.8
The proposed
model of binding sites etc. with PTPIB is shown in figure 4.
Figure 4. Schematic representation of the hydrogen-bonding interactions between VI (red) and PTPIB (black). Bound water molecules are labeled as W. All the distances are in angstroms.
41

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
2.2 Synthesis and Biological activity of Boc-Phe-Tyr(O-Malolllyl)-amides
It is evident from the above discussion that the dipeptide analog V is a highly potent
and selective inhibitor of PTPIB enzyme. This compound has also been studied for binding
at the PTPIB domain and hence can be classified as a rationally designed PTPIB inhibitor.
In spite of the promising biological data obtained from compound V, this peptide has a
severe limitation towards the possibility of clinical development, as this peptide was found
to be poorly active in cell based assay system. The compound per se, shows promising
inhibitory activity when tested in the isolated enzyme model, while it is not showing
promising activity in cell based assay system, clearly indicating that this compound has
restricted cellular uptake. To solve this problem it was decided to design new analogs of
compound V with altered lipophilicity and conformational rigidity, with the objective to
develop a compound that has better cellular permeablity as well as PTPIB inhibitory
activity. Towards this objective, three sets of compounds are designed and synthesized. The
details are presented in the following text of this chapter.
2.2.1 Basis of Work
The lipophilicity of molecule plays an important role for the expression of biological
activity as it is very crucial for pharmacokinetic, cellular transport and binding to the target
site. In this section, derivatives are synthesized where the pentyl chain in compound V has
been replaced with other short or long alkyl or heteroalkyl chains to obtain compounds with
altered lipophilicity. Development of phosphotyrosyl (PTyr) mimetics as P'fPIB inhibitors,
which are stable, yet can retain biological potency when incorporated into peptides and
peptidomimetics is an active area of drug research. It has been already discussed that the
malonate derivatives of Boc-Phe-Tyr-amide V exhibited very interesting binding properties.
Due to the relatively low molecular weight of these inhibitors, the peptidomimetic approach
for the design of new inhibitors has been quite successful. The high order of inhibitory
activity in these small molecular weight peptidomimetics may result in achieving activity in
cell based model, which is the ultimate goal of the present study. Although a number of
derivatives designed by peptidomimetic approach or otherwise are reported in literature but
compound V is of particular interest as detailed in the following text.
42

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
o
v
O--(GOOH
'I GOOH
H N~
The compound V has significantly low Ki value (9 11M) and has >90% inhibition of
PTPIB at 100 11M concentration. Beside this, the molecule has been found to exhibit
significant cellular activity, usually lacking in this series of derivatives. The compound was
able to enhance insulin stimulated uptake of 2-deoxyglucose by L6 myocytes.
Simultaneously other investigators have reported better cellular activity in compounds,
where malonyl group at tyrosine moiety was replaced with other groups (as shown in VII
and VIII) (Figure 5).
o ~~ PhoDlr~~O ~ 0 ~~ I ~
....-:
VII (~=2.0 11M) VIn (~=0.67 11M)
Figure 5. Structure of the tetrazole bearing dipeptides.
It is important to note that these modifications are generally aimed to mimic a
carboxylic group in the form of tetrazole moiety. The tetrazole moiety is reported to be the
43

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
bioisostere for the carboxylic group as carboxylic acid. Further, the tetrazole group is also
relatively more hydrophobic as compared to a carboxylic or phosphate group. These
derivatives are reported to have better cellular activity. The enhanced cellular activity may
be attributed to the enhanced lipophilicty of these derivatives.
In view of the above discussion, it can be inferred that lipophilicity plays a key role
for the PTPIB activity and their cellular activity, in particular. Therefore, we have decided
to synthesize a series of analogs of compound V, which we have selected as our lead
compound. We have introduced several alkyl chains compounds Sa-g in place of a pentyl
chain to see the effect of lipophilicity of these lipophilic derivatives on PTPIB inhibition
potential. The chemical structures of the proposed derivatives are shown in figure 6.
O--(COOH
~ \ COOH
o H 0 I ~
5a-g
Sa R=C3H7 (i) 5b R=C4H9
5c R=C6H13 5d R=C12H25 1\ 5e R= N N-
'---/
1\-0 5f R= N ,---/N- ~ /;
5g R=CsH11
Figure 6. Alkyl chain modifications at C-tenninal ofV.
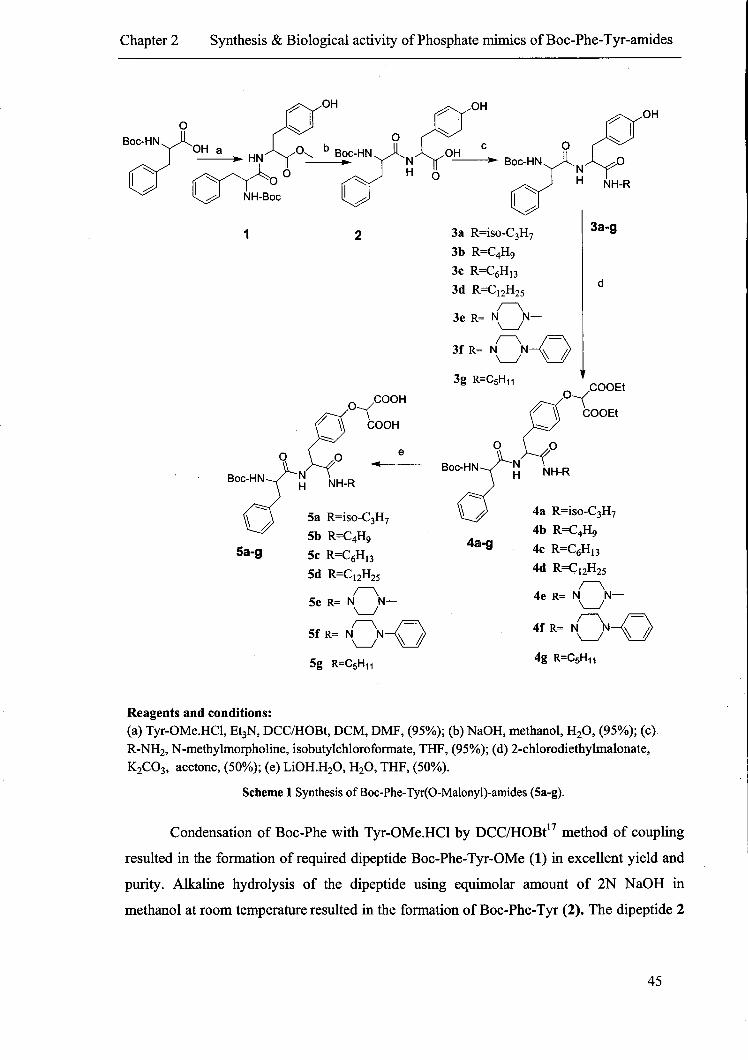
2.2.2 Chemistry
The peptidomimetic derivatives 5a-g were synthesized using solution phase method
of peptide synthesis. The key intermediate dipeptide, Boc-Phe-Tyr was synthesized starting
from Boc-Phe which was prepared by the reaction of Ditertiarybutylpyrocarbonate and
Phenylalanine according to the method reported in literature. IS Smilarly Tyr-OMe.HCI was
obtained from tyrosine by the acid catalysed esterification using thionyl chloride. 16
44
Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
:r OH :r OH
o ~ 1 ~ 1 )~OH
.. BCfC_HN 0 N~~O Boc-HN 0
"': 0 "': I "': QYCo HO
~ 1 ~ NH-Boc 1 ~
1 2
O--{COOH S COOH
Boc-HN-t~ N:.R · • a Sa R=iso-C3H7 Sb R=C4H9
5a-g Sc R=C6H\3
Reagents and conditions:
Sd R=C12H25
r\ Se R= N N-
'-.J
Sf R= N~~:}-o Sg R=CsH11
H NH-R I"': ~
3a R=iso-C3H7
3b R=C4H9 3c R=C6H13
3d R=C12H25
r\
d
3e R= N N-'-.J
r\-o 3f R= N'-.JN ~ /;
3g R=CsH11 O--{COOEt o COOEt
o ~O B""'H:-t~ NIl-R a 4a R=iso-C3H7
4b R=C4H9 4a-g 4c R=C6H13
4d R=C12H25
r\ 4e R= N N-
'-.J
4fR= N~)J-o 4g R=CsH11
(a) Tyr-OMe.HCl, Et3N, DCCIHOBt, DCM, DMF, (95%); (b) NaOH, methanol, H20, (~5%); (c)
R-NH2' N-methylmorpholine, isobutylchloroformate, THF, (95%); (d) 2-chlorodiethylmalonate,
K2C03, acetone, (50%); (e) LiOH.H20, H20, THF, (50%).
Scheme 1 Synthesis of Boc-Phe-Tyr(O-Malonyl)-amides (Sa-g).
Condensation of Boc-Phe with Tyr-OMe.HCI by DCCIHOBt17 method of coupling
resulted in the formation of required dipeptide Boc-Phe-Tyr-OMe (1) in excellent yield and
purity. Alkaline hydrolysis of the dipeptide using equimolar amount of 2N NaOH in
methanol at room temperature resulted in the formation of Boc-Phe-Tyr (2). The dipeptide 2
45

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
was converted to various amides by the reaction of isobutylchloroformate and aminolysis of
the resulting mixed anhydride18 with various amines separately to yield required amide
derivatives 3a-g in excellent yields. Diethylmalonyl derivatives 4a-g of the dipeptide 3a-g,
were obtained by nucleophilic substitution reaction of Boc-Phe-Tyr-NHR 3a-g with 2-
chlorodiethylmalonate in the presence of K2C03.13 We consistently observed the generation
of small amounts of halogenated byproducts in these alkylations that could not be easily
removed chromatographically. The products were subjected to catalytic hydrogenation to
yield O-diethymalonyl derivatives Boc-Phe-Tyr (O-diethyl malonyl)-NH-R (4a-g) in good
yields and in pure form. In the final step, hydrolysis of the 4a-g were performed with
LiOH.H20 in THF to get desired 5a-g dipeptide alkylamides with free malonate at tyrosine
residue. 13 All the steps involved in the synthesis and reagents used are presented in Scheme
1. Compound 5g is in fact a reported compound13and is sythesised to test as a positive
control.
2.2.3 Results and Discussion
The inhibitory potential of the compounds 5a-g is presented in table 1. The activity
was expressed as percentage inhibition of the test compounds at 1 00 ~. It is evident from
the data presented in table 2 that most of the newly synthesized compounds exhibited
significant PTPIB inhibition potential. Beside, these compounds also expressed high order
of selectivity. Among seven compounds, two compounds 5c and 5d, showed better
inhibition against PTPIB with reference to the reported compound V which was used as
positive control at 100 /-lM.
Table 1 Inhibition ofPTPlB and LAR (at 100 /-lM) by compounds 5a-f, V.
S.No. Compound % inhibition PTPIB % inhibition LAR
1 5a 66.60 37.87 2 5b 80.00 57.48 3 5c 93.00 68.44 4 5d 99.70 39.53 5 5e 33.00 17.27 6 5f 52.00 48.64 7 V 83.00 57.81
PTPlB enzyme is human, recombinant (residues 1-322; MW=37.4 kDa). LAR enzyme is LAR-Dl Fragment. Substrate for PTPlB is the p-nitro-phenyl phosphate (p-NPP). V was taken as test control.
46

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
PTPIB inhibition data clearly suggested that malonates with lipophilic aliphatic chain 5a,
5b, 5c, 5d and 5g were more active as compared to nitrogen bearing heterocyclic moieties
5eand 5f. Percentage inhibitions at 1 00 ~ increased with the increase in number of
aliphatic carbons at the C-terrninal. Furthermore, the data also suggested that 5d was 2.5
times more selective toward PTPIB as compared to LAR.
Compounds 5c, 5d, V were evaluated for ICso and Ki determination. Concentrations of the
compounds which inhibited enzyme activity by 50% (ICso), were obtained using four
different concentrations of the inhibitors (Table 2). Inhibition constants Ki (dissociation
constant of the [EI] complex) were measured at 37°C, at pH 7.2 (Table 2).
Table 2 PTPIB inhibitory activity of compounds 5c, 5d, V.
S.No Compound ICso (flM) Kt (flM)
1 5c 43.2 15.97
2 5d 81.2 8.06
3 V 12.9 22.38 (9)
PTPIB enzyme is human, recombinant (residues 1-322; MW=37.4 kDa). Substrate for PTPlB is the p-nitro-phenyl phosphate (P-NPP). Four independent measurements were performed for ICso and Ki determinations. V was taken as test control.
2.2.4 Experimental
Optically active amino acids L-Phe and L-Tyr were used for the study. The coupling
reactions were carried out in anhydrous solvents using pure and dry reactants. Completion of
the reactions and purity of the products were established by TLC on readymade silica gel
plates (Merck, UV active) using following solvent systems.
A
B
CHCh-MeOH-AcOH (1 drop)
CHCh-MeOH
(8:2)
(9.5:0.5)
The plates were developed either under iodine vapours or seen directly under UV-
light (254 nm). In addition to that the purity of the compounds was also detected by spraying
the TLC plates with 2N HBr/AcOH followed by 0.2 % Ninhydrin solution in acetone and
heating the plates at approx. 80-90 °C in hot air oven for 15 minutes. Amino acid with free
47

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
amino group was detected by spraying the plates with Ninhydrin solution straight away and
heating. Column chromatography was performed over silica gel (230-400 mesh).
Melting points (mp) were taken in open capillaries on Complab melting point
apparatus and are uncorrected. Characterization of all the derivatives as well as final
compounds were done with the help IR, NMR and Mass spectroscopy. The I H spectra were
obtained with Broker DPX-200 or DRX-300 MHz FT-NMR spectrometers. The chemical
shifts are reported as parts per million (0 ppm) taking tetramethylsilane (TMS) as an internal
standard. The 13C-NMR spectra were recorded on Broker DRX-300 FT-NMR spectrometer
(at 75 MHz). Infrared (IR) spectra were recorded on an FT-IR Perkin-Elmer spectrometer
and reported in wave number (cm· I). Mass spectra were obtained on JEOL-SX-102
instrument using fast atom bombardment (F AB positive) and on Micromass Quattro II,
Spectrometer using Electron spray ionization mass spectroscopy (ESI MS positive).
Boc-Phe-Tyr-OMe (1)
Boc-Phe2o (2.65 g, 10 mmole) was dissolved in dichloromethane (DCM), followed
by the addition of hydroxybenzotriazole (HOBt) (1.35 g, 10 mmole) solution in
dimethylformamide (DMF), the reaction mixture was stirred for 2 minutes at 0 °C. The
hydrochloride salt of L-tyrosine methyl ester (2.31g, 10 mmole) dissolved in DMF and
neutralized by triethylamine (2 ml, 15 mmoles) was added to above reaction mixture at O°C.
To the mixture was added dicyc1ohexylcarbodimide (DCC) (2.2 g, 11 mmole) dissolved in
DCM at 0 °C. The reaction mixture was allowed to reach at room temperature and was
stirred for 4 hrs. The dicyc10hexylurea (DCU) was filtered off and the filtrate was evaporated
under reduced pressure. The oily residue was dissolved in EtOAc, washed with 5% aqueous
sodium bicarbonate, brine, 5% citric acid solution and finally with brine. The organic layer
was dried over anhydrous Na2S04 and evaporated under reduced pressure. The crude
product was purified over silica gel column to get the pure compound 1. This compound was
obtained as a white solid. (Yield = 4.06 g, 92 %), mp 141-145 °C (144-145 0c) 19; Rf 0.5
(A); IR (KBr) 3309 cm- I , 1712 cm-I, 1666 cm- I ; IH NMR; 200 MHz, CDCh (8 ppm): 1.39
(s, 9H, (CH3)3C), 2.98-3.02 (m, 4H, Phe & Tyr ~CH2)' 3.67 (s, 3H, OCH3), 4.40 (m, IH,
Phe CuH), 4.80 (m, IH, Tyr CuH), 5.14 (br s, IH, NH), 6.48 (br s, IH, NH), 6,.65-7.23 (m,
9H, ArCH); FAB-MS m/z 443 [M+Wl
48

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
Boc-Phe-Tyr (2)
To a solution of Boc-Phe-Tyr-OMe (4.42 g, 10 mmole) in MeOH (75 ml), was added
a solution of 2N NaOH (12.5 mI, 25 mmoles). The mixture was stirred at room temperature
for 2 hrs. Solvent was evaporated and the mixture was neutralized with citric acid and
extracted with EtOAc. The organic phase was washed with brine and dried over anhydrous
Na2S04. The solvent was removed in vacuo to give 2. This compound was obtained as a
white solid (Yield = 4.19 g, 95 %), mp 227-230 DC; Rf 0.1 (B); IR (KBr) 3302 cm- I, 1722
cm-t, 1598 cm-I; IH NMR; 200 MHz, CDCh + DMSO-~ (8 ppm): 1.38 (s, 9H, (CH3)3C),
2.98 - 3.07 (m, 4H, Phe & Tyr ~CH2), 4.40 (m, 1H, Phe CuH), 4.80 (m, 1H, Tyr CuH), 5.14
(br s, 1H, NH), 6.48 (br s, 1H, NH), 6.66-7.25 (m, 9H, ArCH); FAB-MS mJz 429 [M+W].
General procedure for Boc-Phe-Tyr-amides (3a-g)
Boc-Phe-Tyr (4.28 g, 10 mmole) was dissolved in anhydrous THF and treated
successively with NMM (1.2 ml, 10 mmole) and isobutylchloroformate (1.35 ml, 10 mmole)
at -15°C under vigorous stirring. The temperature was maintained at -15 °C for 10 min.
Subsequently a solution of HOBt (1.53 g, 10 mmole) in THF was added followed by the
addition of desired amine, 20 mmole each [1.7 ml (isopropylamine), 1.99 ml (butylamine),
2.10 m1 (pentylamine), 2.32 ml (hexylamine), 4.60 ml (laurylamine), 1.280 ml (N
methylpiprazine), 2.02 ml (N-phenylpiprazine)] to the reaction mixture. The reaction was
stirred at -15°C for 15 min and then allowed to stir at room temperature for additional 2 hrs.
Solvent was then removed in vacuo, and the residue was extracted with EtOAc. The EtOAc
extract was washed successively with 5 % citric acid (except in case Of 3e where N
Methylpiprazine was used), 5% sodium bicarbonate and finally with brine. After drying over
anhydrous Na2S04, the organic layer was concentrated under reduced pressure; the crude
was purified over silica gel column to get the pure compound and the purified products
triturated with hexane to get white solids 3a-g.
It is generally believed that the activation of the dipeptide carboxylic acids generally
results in partial or complete racemisation of product. The racemisation can be suppressed or
eliminated by addition of HOBt. During this reaction an equimolar amount of HOBt was
added to the mixed anhydride solution, prior to addition of appropriate amines. This resulted
49

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
in the formation of product without noticeable racemisation, which was confirmed by
measuring the optical rotation of the product obtained by alternate coupling reagent. .
Boc-Phe-Tyr-NH-C3H7(1) (3a)
This compound was obtained as a white solid. (Yield = 4.22 g, 90 %) , mp 180-182 °C; Rc 0.30 (B); IR (KBr) 3309 cm- I
, 1647 cm- I; IH NMR; 200 MHz, CDCh (8 ppm): 1.01-1.04
(d, J=6 Hz, 6H, isopropyl CH3), 1.32 (s, 9H, (CH3)3C), 2.80 ( m, lH, isopropyl CH), 3.03-
3.06 (m, 4H, Phe & Tyr ~CH2), 4.50 (m, IH, Phe CaH), 4.80 (m, IH, Tyr CaH), 5.14 (br s,
IH, NH), 6.28 (br s, IH, NH), 6.50 (br s, IH, NH), 6.70 -7.33 (m, 9H, Ar CH); FAB-MS
mlz 470 [M+W].
Boc-Phe-Tyr-NH-C4H9 (3b)
This compound was obtained as a white solid. (Yield = 4.35 g, 90 %), mp 174-176 °C; Rc 0.35 (B); IR (KBr) 3349 cm- I , 1649 cm-\ IH NMR; 200 MHz, CDCh (8 ppm): 0.83 - 0.91
(m, 3H, butyl CH3), 1.33 (s, 9H, (CH3)3C), 1.21-1.41 (m, 4H, butyl CH2), 2.80 (m, 2H, butyl
N-CH2), 3.02 -3.14 (m, 4H, Phe & Tyr C~H), 4.40 (m, IH, Tyr CaH), 4.80 (m, IH, Phe
CaH), 5.14 (br s, IH, NH), 6.28 (br s , IH, NH), 6.50 (br s, IH, NH) ,6.70-7.32 (m, 9H, Ar
CH); FAB-MS mlz 484 [M+W].
Boc-Phe-Tyr-NH-C6H13 (3c)
This compound was obtained as a white solid. (Yield = 4.34 g, 85%), mp 159-160 °C; Rc 0.35 (B); IR (KBr) 3340 cm- I
, 1631 cm- I ; IH NMR; 200 MHz, CDCh (8 ppm): 0.84 -0.90
(m, 3H, hexyl CH3), 1.35 (s, 9H, (CH3)3C), 1.24-1.39 (m, 8H, hexyl CH2), 2.78-2.82 (m,
2H, hexyl NCH2). 3.02 -3.13 (m, 4H, Phe & Tyr ~CH2), 4.28 (m, lH, Phe Cu.H), 4.50 (m,
IH, Tyr CaH), 4.94 (br s, lH, NH), 6.20 (br s, IH, NH), 6.4 (br s, 1H, NH),6.70 -7.32 (m,
9H, Ar CH); FAB-MS mlz 513 [M+Hl.
Boc-Phe-Tyr-NH-C12H25 (3d)
This compound was obtained as a white solid. (Yield = 5.36 g, 90 %), mp 165-167 °C; Rc 0.5
(B); IR (KBr) 3338 cm-I, 1648 cm- I; IH NMR; 200 MHz, CDCh (8 ppm): 0.83-0.90 (m,
3H, lauryl CH3), 1.24 (s, 20H, lauryl CH2), 1.32 (s, 9H, (CH3)3C), 2.80 (m, 2H, lauryl
NCH2), 3.01- 3.10 (m, 4H, Phe & Tyr ~CH2), 4.50 (m, IH, Phe CaH), 4.80 (m, IH, Tyr
50

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
CaH), 6.0 (br s, IH, NH), 6.1 (br s, IH, NH), 6.30 (br s, IH, NH), 6.68-7.32 (m, 9H, Ar
CH); FAB-MS m1z 596 [M+H+].
Boc-Phe-Tyr-Methylpiprazinamide (3 e)
This compound was obtained as a white solid. (Yield = 4.85 g, 95%), mp 108-110 °C; Rc
0.35 (A); IR (KBr) 3293 cm- I , 1630 cm- I ; IH NMR; 200 MHz, CDCh (8 ppm): 1.36 (s, 9H,
(CH3)3C), 1.9 (m, 2H, piprazine CH2), 2.19 (s, 3H, NCH3), 2.3 (m, 2H, piprazine CH2),
2.85-3.04 (m, 4H, Phe & Tyr ~CH2)' 3.4 (m, 2H, piprazine CH2), 3.6 (m, 2H, Piprazine
CH2), 4.40 (m, 1H, Phe CaH), 4.40 (m, 1H, Tyr CaH), 5.1 (br s, IH, NH), 5.2 (br s, 1H, NH),
6.64 -7.23 (m, 9H, Ar CH); FAB-MS m1z 511 [M+H+].
Boc-Phe-Tyr-Phenylpiprazinamide (3t)
This compound was obtained as a white solid. (Yield = 5.72 g, 87 %), mp 161-165 °C; Rf
0.40 (B); IR (KBr) 3291 cm- I, 1629 cm- I
; IH NMR; 200 MHz, CDCh (8 ppm): 1.37 (s, 9H,
(CH3)3C), 2.86-2.99 (m, 8H, piprazine CH2), 3.3-3.8 (m, 4H, Phe & Tyr ~CH2)' 4.40 (m,
IH, Phe CaH), 4.40 (m, IH, Tyr CaH), 6.72 -7.27 (m, 14H, Ar CH); FAB-MS m1z 573
[M+H+].
Boc-Phe-Tyr-NH-CsHll (3g)
This compound was obtained as a white solid (Yield = 4.47 g, 90 %), mp 160-162 °C; Rc
0.35 (B); IR (KBr) 3343 cm-I, 1595 cm- I ; IH NMR; 200 MHz, CDCh (8 ppm): 0.85-0.92
(m, 3H, pentyl CH3) 1.35 (s, 9H, (CH3)3C), 1.16-1.44 (m, 6H, pentyl CH2), 2.78-2.82 (m,
2H, pentyl N-CH2), 3.04-3.19 (m, 4H, Phe & Tyr ~CH2), 4.28 (m,IH, lPhe CaH), 4.57 (m,
IH, Tyr CaH), 4.94 (br s, IH, NH), 6.20 (br s,IH, NH), 6.42 (br s,IH, NH), 6.71-7.34 (m,
9H, Ar CH); FAB-MS m1z 498 [M+H+].
General procedure for the synthesis of Boc-Phe-Tyr (O-Malonyl diethyl ester)-amides
(4a-g)
To the stirring solution of dipeptide amides, 1 mmole each [3a, 469 mg; 3b, 483 mg;
3c, 511 mg; 3d, 595 mg; 3e, 511 mg; 3f, 572 mg; 3g, 497 mg] in acetone, was added K 2C03
(276 mg, 2 mmole) at room temperature. To the resulting heterogeneous mixture was added
2-chlorodiethylmalonate (300 ~l, 2 mmole), and the mixture was stirred at ambient
51

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
temperature for 36 hrs. After the completion of reaction as monitored by TLC, acetone was
evaporated and the crude was extracted with EtOAc. The organic layer was washed with
brine. After drying over anhydrous Na2S04, the organic layer was concentrated under
reduced pressure; the crudes were purified over silica gel column to get the pure compounds
4a-g.
Boc-Phe-Tyr (O-Malonyl diethyl ester)-NH-C3H, (I) (4a)
This compound was obtained as a white solid. (Yield = 301 mg, 48 %); Rf 0.45 (B); IR
(KBr) 3412 cm- I , 1664 cm- I ; IH NMR; 200 MHz, CDCb (8 ppm): 1.04-0.99 (m, 6H,
isopropyl CH3), 1.27-1.35 (m, 4H, mal.CH2), 1.33 (s, 9H, (CH3)3C), 2.78 (m, 1H, isopropyl
CH), 3.02-3.11 (m, 4H, Phe & Tyr PCH2), 4.28-4.35 (m, 6H, mal.CH3), 4.50 (m, 1H, Phe
CaH), 4.86 (m, 1H, Tyr CaH), 5.15 (s, IH, mal.CH), 5.3 (br s, 1H, NH), 6.44 (br s, 1H, NH),
6.84 -7.31 (m, 9H, Ar. CH); ESI-MS m1z 628 [M+Rl.
Boc-Phe-Tyr (O-Malonyl diethyl ester)-NH-C4H9(4b)
This compound was obtained as a white solid. (Yield = 321 mg, 50 %); Rf 0.45 (B); IR
(KBr) 3329 cm- I , 1639 cm- I ; IH NMR; 200 MHz, CDCh (8 ppm): 0,83-0.91 (m, 3H, butyl
CH3), 1.3 (s, 9H, (CH3)3C), 1.16 -1.42 (m, 10H, 6H-mal.CH3 & 4H-butyl CH2), 2.7;'2.9 (m,
2H, butyl N-CH2), 3.0-3.13 (m, 4H, Phe & Tyr PCH2), 4.24--4.35 (m, 4H, mal.CH2), 4.40
(m, 1H, Phe CaR), 4.60 (m, 1H, Tyr CaH), 5.10 (br s, 1H, NH), 5.12 (s, 1H, mal.CH), 6.28
(br s, 1H, NH), 6.60 (br s, 1H, NH), 6.70 -7.30 (m, 9H, ArCH); FAB-MS m1z ,642 [M+H+].
Boc-Phe-Tyr (O-Malonyl diethyl ester)-NH-C6H13 (4c)
This compound was obtained as a white solid. (Yield = 401 mg, 60%); Rf 0.6 (B); IR (KBr)
3425 cm- I, 1649 cm- I
; IH NMR; 200 MHz, CDCh (8 ppm): 0.84-0.87 (m, 3H, hexyl CH3),
1.26- 1.30 (m, 14H; 6H mal.CH3 & 8H hexyl CH2 ), 1.34 (s, 9H, (CH3)3C), 2.9 (m, 2H,
hexyl N-CH2), 3.03-3.09 (m, 4H, Phe & Tyr PCH2), 4.25 -4.36 (m, 5H, 1H-· Phe CaH, 4H
mal. CH2), 4.5 (m, lH, Tyr CaH), 4.8 (br s, lH, NH), 5.13 (s, lH, mal.CH), 6.0 (br s,lH,
NR), 6.2 (br s,l H, NH), 6.86 -7.29 (m, 9H, Ar CH); FAB-MS m1z 670 [M+Hl.
52

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
Boc-Phe-Tyr (O-Malonyl diethyl ester)-NH-C12H2S (4d)
This compound was obtained as a white solid. (Yield = 301 mg, 40%); Rf 0.8 (A); mp 165-
167°C; IR (KBr) 3923 em-I, 1642 em-I; IH NMR; 200 MHz, CDCh (8 ppm): 0.83-0.90 (m,
3H, lauryl CH3), 1.24-1.29 (m, 24H, 20H-lauryl CH2 & 4H-mal.CH2), 1.35 (s, 9H, (CH3)3C),
3.01-3.12 (m, 6H; 4H-CH2 Phe & Tyr ~CH2 and 2H-lauryl NCH2lauryl), 4.26-4.35 (m,4H,
mal. CH2), 4.60 (m, IH, Phe C~H), 4.90 (m, IH, Tyr CaH), 5.01(s, 1H, mal. CH), 6.1 (br s,
1H, NH), 6.50 (br s, 1H, NH), 6.80 -7.31 (m, 9H, Ar CH); FAB-MS rn/z 754 [M+H+].
Boc-Phe-Tyr (O-Malonyl diethyl ester)-Methylpiprazinamide (4e)
This compound was obtained as a white solid. (Yield = 334 mg, 50%); Rf 0.35 (A); IR
(KBr) 3296 em-I, 1638 em-I; IH NMR; 200 MHz, CDCh (8 ppm): 1.28-1.35 (m, 6H, mal.
CH3), 1.39 (s, 9H, (CH3)3C), 2.42 (s, 3H, NCH3), 2.89 -3.03 (m, 8H; 4H- Phe & Tyr ~CH2
& 4H-piprazine CH2), 3.85-3.49 (m, 4H, piprazine CH2), 3.49-3.85 (m, 4H, piprazine CH2),
4.36-4.26 (m, 5H; 1H- Phe CaH , 4H- mal.CH2), 4.98 (m, IH, Tyr CaH), 5.17 (s, IH,
mal.CH), 6.83 -7.29 (m, 9H, Ar CH); FAB-MS rn/z 669 [M+H+].
Boc-Phe-Tyr (O-Malonyl diethyl ester)-Phenylpiprazinamide (41)
This compound was obtained as a white solid. (Yield = 197 mg, 27 %); Rc 0.45 (B); IR
(KBr) 3306 em-I, 1636 em-I; IH NMR; 200 MHz, CDCh (8 ppm): 0.87 -1.35 (m, 6H, mal.
CH3), 1.42 (s, 9H, (CH3)3C), 2.63-3.05 (m, 8H, piprazine CH2), 4.24 -4.39 (m, 4H, Phe &
Tyr ~CH2),4.97 (m, IH, Phe CaR), 5.06 -5.09 (m, IH, Tyr CaH), 5.12 (m, 1H, mal.CH),
6.85-7.28 (m, 14H, Ar CH); FAB-MS rn/z 731 [M+WJ.
Boc-Phe-Tyr (O-Malonyl diethyl ester)-NH-CsHll (4g)
This compound was obtained as a white solid. (Yield = 328 mg, 50%); Rc 0.55 (B); IR (KBr)
3295 em-I, 1645 em-I; IH NMR; 200 MHz, CDCh (8 ppm): 0.86--0.91 (m, 3H, pentyl CH3),
1.3 (s, 9H, (CH3)3C), 1.16 -1.42 (m, 12H, 6H-pentyl CH2 & 6H-mal. CH2), 2.87-2.8 (m, 2H,
pentyl NCH2), 3.0-3.15 (m, 4H, Phe & Tyr ~CH2), 4.22 - 4.37 (5H, m, IH- Phe CaH & 4H
mal. CH2), 4.58-4.54 (m, 1H, Tyr CaH), 4.84 (br s, 1H, NH), 5.14 (m, 1H, mal.. CH), 6.00 (br
s, IH, NH), 6.36 (br s, IH, NH), 6.82-7.28 (m, 9H, Ar CH); FAB -MS rn/z 656 [M+W].
53

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
General synthetic procedure for Boc-Phe-Tyr (O-Malonyl)-amides (Sa-g)
To the solutions of malonyl diethyl esters 4a-g, 0.4 mmole each [4a, 251 mg; 4b, 256 mg;
4c, 268 mg; 4d, 301 mg; 4e, 267 mg; 4f, 292 mg; 4g, 262 mg] in THF (10 ml), were added
solution of LiOH.H20 (200 mg, 4.8 mmole) in water (6ml). The mixtures were stirred at
room temperature for 2.5 hrs. The solvent was evaporated and the crude mixtures were
neutralized with citric acid and extracted with EtOAc. The organic phases were washed with
brine and dried over anhydrous Na2S04. The solvent was removed in vacuo to give Sa-g.
Boc-Phe-Tyr (O-Malonyl)-NH-C3H7 (I) (Sa)
This compound was obtained as a white solid. (Yield = 155 mg, 68 %), mp 121-124 °C; Rf
0.1 (A); IR (KBr) 3302 cm- I , 1654 cm- I ; IH NMR; 200 MHz, CDCh (5 ppm): 0.83-0.89 (m,
3H, isopropyl CH3), 1.33 (s, 9H, (CH3)3C), 1.42 -1.17 (m, 3H, isopropyl CH3), 2.80 (m, IH,
isopropyl CH), 3.01- 3.14 (m, 4H, Phe & Tyr ~CH2), 4.30 (m, IH, Phe CaR), 4.60 (m, IH,
Tyr CaH), 4.9 (s, IH, mal. CH), 6.28 (br s, IH, NH), 6.50 (br s, IH, NH), 6.69-7.32 (m, 9H,
Ar CH). 13C NMR; 75 MHz, DMSO-~ (5 ppm): 27.31 (2C), 33.24 (3C), 42.00, 48.33 (2C),
56.50,61.17,77.41,83.63, 119.20, 119.73, 120.13, 131.43, 133.24 (2C), 133.68, 134.31,
134.80, l35.27, l35.52, 143.09, 160.43, 173.89, 174.76, 176.65, 180.42; FAB-MS mlz 572
[M+Hl.
Boc-Phe- Tyr (O-Malonyl)-NH-C4H9 (Sb)
This compound was obtained as a white solid. (Yield = 117 mg, 50 %), mp 141-145 °C; Rf
O.I(A); IR (KBr) 3429 cm- I, 1649 cm-\ IH NMR; 300 MHz, DMSO-~ (5 ppm): 0.86 -0.90
(m, 3H, butyl CH3), 1.3 (s, 9H, (CH3)3C), 1.10 -1.42 (m, 4H, butyl CH2), 2.54-2.98 (m, 2H,
butyl NCH2), 3.0 -3.5 (m, 4H, Phe & Tyr ~CH2), 4.20 (m, IH, Phe CaH), 4.40 (IH, Tyr
CaH), 5.21 (s, IH, mal. CR), 6.74-7.2 (m, 9H, Ar CH); 13C NMR; 75 MHz, CDCh + DMSO-~ (5 ppm): 13.46, 19.78,28.00 (3C), 30.98 (2C), 38.00,39.10,54.50,56.50, 70.00,
81.5, 115.27 (2C), 126.87, 128.48 (2C), 129.16 (4C), 130.36, 136.00, 155.78 (2C), 175.40
(2C), 176.00 (2C); ESI-MS mJz 585 [M-H]-.
Boc-Phe- Tyr (O-Malonyl)-NH-C6H13 (Sc)
This compound was obtained as a white solid. (Yield = 120 mg, 50 %), mp 159-160 °C; Rf
0.1 (A); IR (KBr) 3333 cm-I, 1647 cm-I
; IH NMR; 300 MHz, DMSO-d6 (5 ppm): 0.87 (m,
3H, hexyl CH3), 1.3 (s, 9H, (CH3)3C), 1.18-1.37 (m, 8H, hexyl CH2), 2.6 -3.12 (m, 6H ; 4H-
54

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
Phe & Tyr PCH2; 2H-hexyl NCH2), 4.20 (m, IH, Phe CuH), 4.5 (m, IH, Tyr CuH), 4.9 (br s,
IH, NH), 5.13 (s, IH, mal.CH) , 6.01 (br s, IH, NH), 6.46 (br s, IH, NH), 6.84 -7.27 (m,
9H, Ar CH); BC NMR; 75 MHz, CDCh + CD30D (8 ppm): 17.79, 19.00,26.36,30.32,
31.95 (3C), 32.89 (2C), 35.27 (2C), 54.50, 56.50, 73.00, 81.50, 115.27 (2C), 130.00, 132.48
(2C), 133.12 (4C), 134.32, 140.00, 150.00 (2C), 175.40 (2C), 176.00 (2C) ; FAB-MS mlz
614 [M+H+].
Boc-Phe- Tyr (O-Malonyl)-NH-C12H23 (5d)
This compound was obtained as a white solid. (Yield = 139 mg, 50 %), mp 120-122 °C; Rf
0.1 (A); IR (KBr) 3433 em-I, 1637 em-I; IH NMR; 300 MHz, CDCh (8 ppm): 0.81-0.89 (m,
3H, lauryl CH3) 1.25 (m, 20H, lauryl CH2), 1.33 (s, 9H, (CH3)3C), 2.81-3.06 (m, 6H, 4H
CH2 Phe & Tyr PCH2 & 2H-Iauryl NCH2), 4.30 (m, IH, Phe CuH), 4.61 (m, JH, Tyr CHu),
5.24(s, IH, mal. CH), 6.84-7.62 (m, 9H, Ar CH); BC NMR; 75 MHz, CDCh (8 ppm):
12.81,21.38,25.58,26.85 (3C), 27.77, 27.98, 28.06, 28.36 (6C), 30.62 (2C), 54.50, 56.50,
73.00, 81.50, 114.27 (2C), 125.70, 127.70 (2C), 128.00 (4C), 129.50, 140.00, 150.00 (2C),
175.40 (2C), 176.00 (2C); ESI-MS mlz 698 [M+Hl.
Boc-Phe- Tyr (O-Malonyl)-Methylpiprazinamide (5e)
This compound was obtained as a a brown gummy matter. (Yield = 135 mg, 55%); IR
(KBr) 3419 em-I, 1645 em-I; IH NMR; 300 MHz, CDCh + DMSO-~(8 ppm): 1.37 (s, 9H,
(CH3)3C), 2.52 (s, 3H, NCH3), 2.51- 3.15 (m, 8H; 4H- Phe & Tyr PCR2 & 4H-piprazine
CH2), 3.5 -3.7 (m, 2H, piprazine CH2), 3.94 (m, 2H, piprazine CH2), 6.9 -7.25 (9H, m, CH
Ar); ESI-MS mlz 612 [M+W].
Boc-Phe-Tyr (O-Malonyl)-Phenylpiprazinamide (Sf)
This compound was obtained as a white solid. (Yield = 202 mg, 75 %), mp 121-124 °C; Rf
0.1 (A); IR (KBr) 3291 em-I, 1629 em-I; IH NMR; 300 MHz, CDCh + DMSO-~ (8 ppm):
1.29 (s, 9H, (CH3)3C), 2.51-3.15 (m, 8H, CH2 piprazine), 4.24-4.39 (m, 4H, Phe & Tyr
PCH2), 4.30 (m, IH, Phe CuH), 4.95 (m, IH, Tyr CuH), 4.98 (m, IH, malCH), 5.18 (m, IH,
NH), 6.76 -7.58 (m, 14H, Ar CH); BC NMR; 75 MHz, CDCh + CD30D 8 (ppm): 32.03
(3C), 33.00, 33.50, 42.30 (2C), 53.00 (2C), 59.00, 62.00, 70.60, 84.50, 119.35 (2C), 120.99
(2C), 130.69, 132.34 (4C), 133.16 (5C), 134.53 (2C), 140.00, 155.78 (2C), 175.4 (2C),
176.00 (2C); FAB-MS mlz 675 [M+H+].
55

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
Boc-Phe- Tyr (O-Malonyl)-NH-CsHll (5g)
This compound was obtained as a white solid. (Yield = 130 mg, 53 %), mp 126-128 °C; Rf
0.1 (A); IR (KBr) 3433 cm-I, 1638 cm-1
; IH NMR; 300 MHz, DMSO-~ (& ppm): 0.82·-0.87
(m, 3H, pentyl CH3), 1.3 (s, 9H, (CH3)3C), 1.18-1.37 (m, 6H, pentyl NCH2), 2.72 -3.17 (m,
6H; 4H- Phe & Tyr ~CH2, 2H-pentyl NCH2 ), 4.20 (m, IH, Phe CuH), 4.3-4.5 (m, 2H; IH
NH, IH- Tyr CuH), 5.0 (s, IH, mal. CH), 6.63-7.24 (m, 9H, Ar CH); 13C NMR; 75 MHz,
CDCh + CD30D (& ppm): 17.74, 19.00,26.08, 32.01 (3C), 32.78 (2C), 34.72 (2C), 54.50,
56.50,73.00,81.50, 115.27 (2C), 130.00, 132.28 (2C), 133.16 (4C), 134.27, 140.00, 150.00,
159.66,175.40 (2C), 176.00 (2C); FAB-MS m/z 600 [M+H+].13
Biological activity
PTPIB Drug Discovery Kit based assay is a colorimetric, non-radioactive assay
designed to measure the activity of the enzyme. This kit includes human, recombinant
PTPIB (residues 1-322; MW=37.4 kDa), expressed in E.Coli. 21 , 22 PTPIB activity is
measured as the rate of hydrolysis ofp-nitrophenyl phosphate (pNPP) in a 96-well microtiter
plate format.
All the assays were conducted at room temperature in a total volume of 0.1 mL that
contained assay buffer [Hepes buffer (50 mM, pH 7.2), EDTA (1 mM), DTT (1 mM), 0.01%
NP-40, bovine serum albumin (BSA) (0.1 mg/ml)], PTPIB (2.5 ng/mL) , pNPP (2.5 mM)
and test compounds or inhibitors (not added in control) in different concentrations as
mentioned below.
Inhibitors were dissolved in DMSO to get 10 mM stock solution and the final inhibitor
concentrations were achieved through the serial dilutions with the above buffer system. For
enzyme dilution, 0.2 mg/ml BSA was used. Appropriate volumes of assay buffer were added
to each well (Table 3). In case of control, 5111 ofPTPIB enzyme was added to each well and
for inhibitor assay, 10 ilL of test samples/inhibitors were added to appropriate wells followed
by the addition of 5 III of PTPIB enzyme to each well and in case of blank, neither enzyme
nor inhibitor was added (Table 3). The plate was warmed for 5 minutes. 50 ilL of warm
pNPP substrate was added and the air bubbles were removed. OD's of sample wells were
measured kinetically at 1 minute interval for a total of 30 minutes. The amount of product p-
nitrophenol was determined from the absorbance at 405 nm. Percentage inhibitions were
calculated as follows.
56

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
[
Change in OD of inhibitor] % inhibition = lOO - x 100
Change in OD of control
where, Change in OD of sample = Final OD of sample - Initial OD of sample
The 50% inhibitory concentration (ICso) values were obtained from the sigmoidal dose -
response curves using non-linear regression curve fitting analysis with Graph Pad Software
Prism v. 5.0.23 Each ICso value is the result of experiments performed in duplicate. Inhibition
constants Ki (dissociation constant of the [Ell complex) were measured at 37 DC at pH 7.2.
At same substrate concentration, four inhibitor concentrations were used for initial velocity
measurements. Plots of lIVversus liS were generated and Ki values were determined. From
the same kit, we have performed the LAR assay, the only difference was the enzyme was
LAR and buffer was at pH 6.8.
Table 3 Enzyme inhibition assay.
S.No Assay Buffer Test compound in assay buffer
PTPIB pNPP (5 mM)
Background 50 III 0 o 50111 Control 47.5 III 0 2.5 III 50 III
RK-682 37.5 III 10 III 2.5 III 50 III
Test 37.5 III 10 III 2.5 III 50 III
57

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
2.3 Synthesis and Biological activity of Phe-Tyr-(O-Malonyl)-amides
and cyclic diketopiprazine derivatives
In the previous section, various derivatives of the dipeptides with malonyl tyrosine
moiety have been synthesised and evaluated for PTPIB inhibitory activity. As mentioned
earlier, the objective of the present study comprises designing of compounds with improved
cellular activity, accordingly we have attempted to evaluate the inhibition potential of free
dipeptide mimetics as well as the cyclic diketopiprazine derivatives as conformationally
constrained analogs. The details of the rational behind the design of new compounds and
synthetic details are described in the following section.
It has been described in the earlier section that the compounds 5c, 5d and 5g have
shown potent PTPIB inhibitory activities. In order to study the effect of free amino
components of these derivatives, the Boc group of the compounds 5c, 5d and 5g were
removed to generate corresponding free amino derivatives. The free amino of the compound
V was also synthesised and evaluated for the activity and was taken as the positive control.
Besides, conformationally constrained derivatives of these compounds were also
investigated. Since the malonyl group is considered to be an important surrogate phosphate
group, it was kept intact and the dipeptide was cyclised to 2, 5-diketopiprazine as
conformationally constrained prototype. The details are given below.
2.3.1 Basis of work
In the previous section, the compounds studied are bearing a protecting group at the
amino group of phenylalanine. It is quite interesting to study the effect of the compound
bearing a free amino group against PTPIB activity. It is noteworthy that dipeptide and
tripeptide mimic reported till date are bearing a Boc or acetyl group at the amino terminus.
The PTPIB inhibitory activity of the corresponding compounds with free amino group is not
available in the literature. Since peptides are generally tested for their pharmacological
activity in the free form after the removal of the protecting groups and the free peptides are
known to exhibit better interaction with the target site, this interaction is often facilitated by
the ionic interactions and hydrogen bond forming abilities of the free peptides. In view of
this, it was considered to test the selected compounds described in the previous section in
58

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
their free amino forms. The corresponding N-protected compounds have shown potent
inhibitory activity as described in the previous section. The proposed compounds are shown
in figure 7.
O--{COOH
~ ~ COOH
o H 0 I ~
--==
5c R=C6H13
5d R=C12H25
5g R=C5Hll
Figure 7. Structure of malonate analogues 6a-c with free amine derived from parent malonate analogues 5c, 5d,5g.
In addition to studies of free compounds, it was also considered important to study
the conformationally constrained peptidomimetics as potential PTPIB inhibitors. There have
been several approaches employed for the incorporation of conformational constraints in the
peptide sequence.24-30 Since this is a very small peptide (dipeptide), there are limited options
for the incorporation of conformational constraints. One of the most appropriate strategy to
make peptide highly constrained so that it is stable towards metabolic degradation seems to
be the N-C cyc1isation to yield diketopiprazine derivatives. 31-36
For the present study it was proposed to make diketopiprazines of the two dipeptides
with H2N-Phe-Tyr-OMe and H2N-Tyr-Tyr-OMe and subsequent derivatisation of phenolic
hydroxyl group to the malonyl moiety to generate novel molecules as shown in figure 8.
H HOOCyCOOH
~NW':?" 0
R~ "A ~ I o N H
R=H R = OCH(COOH)2
Figure 8. Structure of malonyl diketopiprazines.
2.3.2 Chemistry
Synthesis of Phe-Tyr-(O-Malonyl)-amides
Starting from the compounds Sa, Sb and Sg, the desired compounds 6a-c were
obtained by the acidolytic cleavage of the Boc group. The Boc group was removed by the
59

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
treatment of protected compounds 5a, 5b and 5g with 15 % HClIDioxane under anhydrous
conditions for a period of 1.5 to 2 hours at room temperature. After complete reaction as
monitored by TLC the compounds were precipitated using anhydrous ether in excess. All the
compounds were obtained as hygroscopic solid in excellent yields and purity (Scheme 2).
7 0yCOOH
o :::,..... I COOH
7 0yCOOH
o :::,..... I COOH
a H 0
H 0 5c R=CsH13 ~ --:: 5d R=C12H25
,ij 5g R=C5H11
-..;:
~ ,ij 6a R=CsH13 6b R=C12H25
6a-c 6c R=C5H11
5c,5d,5g
Reagent and Condition: (a) 15% HCVDioxane (80 %)
Scheme 2. Synthesis ofH2N-Phe-Tyr-(O-Malonyl)-amides (6a-c).
Synthesis of Malonate analogs of diketopiprazines
Thediketopiprazines were synthesized by cyc1isation of H2N-Phe-Tyr-OMe and
H2N-Tyr-Tyr-OMe employing the procedure already reported in the literature37 to obtain the
diketopiprazines Sa and Sb. Diketopiprazines were subjected to derivatisation to Phenolic
hydroxyl group by the reaction of 2-chlorodiethylmalonate under various conditions,
however the desired product could not be obtained due to severe solubility problem of the
diketopiprazine derivatives. To overcome the solubility problem, it was decided to methylate
it to get the corresponding N, N-Dimethyl diketopiprazines 12a and 12b (Scheme 3) .38 For
the synthesis of these diketopiprazines, the phenolic group of tyrosine was protected by
conversion into benzyl ether, prior to cyc1isation. Therefore cyc1isation was carried out with
H2N-Phe-Tyr(OBzl)-OMe and H2N-Tyr(OBzl)-Tyr(OBzl)-OMe. Dimethylated
diketopiprazines were successfully obtained in good yields and found to be reasonably
soluble in the solvent usually employed for derivatisation of phenolic group of tyrosine of
corresponding malonates. For the preparation of the debenzylated derivatives, the
diketopiprazines lla and llb were subjected to catalytic hydrogenation using Palladium
black as catalyst to yield diketopiprazines with free phenolic group 12a and 12b which were
60

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr··amides
then used for the next reaction with 2-chlordiethylmalonate using the similar experimental
conditions as described earlier to get the diethylmalonate derivatives of diketopiprazines 13a
and 13b.
HO
Q:ro NH
B~~ b
6 V itH
9 R 8a R=H
0X\\OEt OEt
o
~o
I~
--*- HN 0
~NH
-?' 0
~I
R a j I :=H 8a-b 8b R=OH q 7R~OH
Me{ ~o
NH
9a R=H 9b R=OBzI
B~~~ nI" Q eOOH
! 9a-b R eOOEt A b ~ EtOoC~ Hoae 6 ~o~: : "v HN~ , · 'N4· .'N~ ~NH -tr tr : I ~I 14a-b
y I 10. R~H 9 11• R~H ~ 128 R~H R 13. R~H ~ lOb R = OBzI R llb R = OBzI 12b R = OH 13b R = OCH(COOEth
IOa-b lla-b 12a-b 13a-b
Reagents and conditions:
(a) Benzyl bromide, K2C03, Acetone, (95 %); (b) 15 % HClIDioxne, TEA, n-Butanol, Toluene,
reflux (50 %); (c) NaH, CH3I, DMF (98%); (d) Pd-C,Methanol, 40 psi; (e) Diethylchloromalonate,
K2C03, Acetone, (70%).
Scheme 3 Synthesis of malonyl diketopiprazines (14a-b).
61

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
The ethyl esters were tested as such because the hydrolysis of ethyl ester to the
corresponding free malonyl group resulted in either decomposition or some other
unpredictable reaction. We could not get the free acid in spite of our efforts. However to
make these compounds, alternative synthetic methods are being explored, but not being
incorporated in thesis.
2.3.3 Results and Discussions
The compounds 6a-c and 13a-b were evaluated for their PTPIB inhibitory activity in
vitro. The assay was performed using a kit supplied by BIOMOL, USA. The details of the
experimental procedure and processing the raw data is similar to that described in the
previous section, the results of these are very interesting and summarized in table 4.
Compounds 6a-c have shown weak inhibitory activity as compared to their corresponding
Boc-protected congeners. However, it is important to note that these derivatives have shown
increase in the inhibitory activity with increase in the lipophilicity. Compound 6c was most
active with 20 % inhibition at 100 J.1M concentration. Another interesting observation is that,
the compounds are also very selective as none of the compounds has shown activity against
LAR. Similarly the diketopiprazines 13a-b are moderately active and highly selective even
at 100 J.1M concentration. Compound 13b was found to be the best compound among all with
47 % inhibition at 100 )lM concentrations. However these compounds are far less active as
compared to V but more selective towards PTPIB (Table 4) ..
Table 4 Inhibition ofPTPlB and LAR (at 100 )lM) by compounds 6a-c, 13a and 13b.
S.No. Compound % inhibition % inhibition
PTPIB LAR 6a 09.00 Inactive
2 6b 10.00 Inactive
3 6c 20.00 Inactive
4 13a 47.00 Inactive
5 13b 14.00 Inactive
6 V 83.00 57.81
PTPlB enzyme is human, recombinant (residues 1-322; MW=37.4 kDa). LAR enzyme is LAR-Dl Fragment. Substrate for PTPlB is the p-nitro-phenyl phosphate (p-NPP). V was taken as test control.
62

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
2.3.4 Experimental
Optically active amino acids L-Phe and L-Tyr were used for the study. The coupling
reactions were carried out in anhydrous solvents using pure and dry reactants. Completion of
reactions and purity of the products were established by TLC on readymade silica gel plates
(Merck, UV active) using following solvent systems.
A CHCh-MeOH-AcOH(1 drop) (8:2)
B
C
CHCh-MeOH
CHCh-MeOH
(9.5:0.5)
(9.7:0.3)
The plates were developed either under iodine vapours or seen directly under UV
light (254 nm). In addition to that the purity of the compounds was also detected by spraying
the TLC plates with 2N HBrl AcOH followed by 0.2% Ninhydrin solution in acetone and
heating the plates at approx. 80-90 °C in hot air oven for 15 minutes. Amino acid with free
amino group was detected by spraying the plates with Ninhydrin solution straight away and
heating. Column chromatography was performed over silica gel (230-400 mesh).
Melting points (mp) were taken in open capillaries on Complab melting point apparatus and
are uncorrected. Characterization of all the derivatives as well as final compounds were done
with the help IR, NMR, and Mass spectroscopy. The IH spectra were obtained with Bruker
DPX-200 or DRX-300 MHz FT-NMR spectrometers. The chemical shifts were reported as
parts per million (0 ppm) taking tetramethylsilane (TMS) as an internal standard. Infrared
(IR) spectra were recorded on an FT-IR Perkin-Elmer spectrometer and reported in wave
number (cm-I).Mass spectra were obtained on JEOL-SX-102 instrument using fast atom
bombardment (F AB positive) and on LCQ Advantage, on Micromass Quattro II,
Spectrometer using Electron spray ionization mass spectroscopy (ESI MS positive).
Synthetic procedure for H-Phe-Tyr (O-Malonyl)-amides.HCl (6a-c)
The Malonate Derivatives 0.1 mmole each [5c, 61 mg; 5d, 70 mg; 5g, 60 mg] were treated
with 15% HClIDioxane for 2 hours at room temperature. The solvent was evaporated under
reduced pressure and the residual oil precipitated from ether, filtered and dried.
Phe-Tyr(O-Malonyl)-NH-C6H13.HCl (6a)
This compound was obtained as a white solid; (Yield = 52.3 mg, 95 %), mp 182-185 °C; IR
(KBr) 1665cm-1,1595 cm-I; IH NMR; 300 MHz, CD30D (8 ppm): 0.87 (m, 3H, hexyl CH3),
63

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
1.25-1.39 (m, 8H, hexyl CH2), 2.8-3.2 (m, 6H; 4H- Phe & Tyr ~CH2; 2H-hexyl. CH2), 4.20
(m, IH, Phe CaH), 4.5 (m, IH, Tyr CaH), 5.13 (s, IH, mal.CH), 6.8-7.33 (m, 9H, Ar CH);
ESI-MS m1z 514 [M+H+].
H-Phe-Tyr(O-Malonyl)-NH-C12H2soHCI (6b)
This compound was obtained as a white solid (Yield = 61 mg, 95 %), mp 192-195 °C; IR
(KBr) 1667cm- I ,1604 cm- I ; IH NMR; 300 MHz, CD30D (0 ppm): 0.86-0.89 (m, 3H, lauryl
CH3), 1.25 (s, 20H, lauryl CH2), 2.83-3.15 (m, 6H; 4H, Phe & Tyr ~CH2; 2H-Iauryl CH2),
4.30 (m, IH, Phe CaH), 4.5 (m, IH, Tyr CaH), 5.24 (s, IH, mal. CH), 6.86-7.36 (m, 9H, Ar
CH); ESI-MS m1z 598 [M+Hl.
H-Phe-Tyr(O-Malonyl)-NH-CsHlloHCI (6c)
This compound was obtained as a white solid; (Yield = 51.0 mg, 95 %); mp 182-185 °C; IR
(KBr) 1682cm-I ,1600 cm- I ; IH NMR; 300 MHz, CD30D (0 ppm): 0.96-1.00 (m, 3H, pentyl
CH3), 1.29- 1.59 (m, 6H, pentyl CH2), 2.94-3.25 (m, 6H; 4H, Phe & Tyr ~CH2' 2H- pentyl
CH2), 4.20 (m, IH, Phe CaH), 4.3-4.5 (m, 2H; IH-NH, IH, Tyr CaH), 5.0 (s, IH, mal.CH),
6.97-7.24 (m, 9H, Ar CH); ESI-MS m1z 500 [M+H+].
Synthesis of Boc-Tyr-Tyr-OMe (7)
Boc-Tyr39 (2.71g, 10 mmole) was dissolved in DCM, followed by the addition of
HOBt (1.35 g, 10 mmole) solution in DMF, the reaction mixture was stirred for 2 minutes at
o °C. The hydrochloride salt of L-Tyrosine methyl ester (2.31g, 10 mmole) dissolved in
DMF and neutralized by triethylamine (2 ml, 15 mmoles) was added to the above reaction
mixture at O°C. To the mixture, was added DCC (2.2 g, 11 mmole) dissolved in DCM at 0
°c. The reaction mixture was allowed to reach at room temperature and was stirred for 4 hrs.
The DCU was filtered off and the filtrate was evaporated under reduced pressure. The oily
residue was dissolved in EtOAc, washed with 5% aqueous sodium bicarbonate, brine, 5%
citric acid solution and finally with brine. The organic layer was dried over anhydrous
Na2S04 and evaporated under reduced pressure. The crude product was purified over silica
gel column to get the pure compound 7. This compound was obtained as white solid. (Yield
= 4.12 g, 90 %), mp 85-88 °c (87-90 °C),40 Rc 0.5 (A); IR (KBr) 3340 cm-], 1670 cm- I ; IH
NMR; 200 MHz, CDCh (0 ppm): 1.42 (s, 9H, (CH3)3C), 2.91-2.99 (m, 4H, Tyr C~H), 3.74
64

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
(s, 3H, OCH3), 4.32 (m, IH, Tyr CaH), 4.75 (m, IH, Tyr CaH), 5.4 (m, 2H, NH), 6.65 -6.95
(m, 8H, Ar CH); ESI-MS mlz 459 [M+H+], 481 [M+Na+].
Synthesis of Diketopiprazines (8a and 8b)
The dipeptide, 10 mmole each (1, 4.42 g and 7, 4.58) were treated with 15%
HClIDioxane (20 ml) for 2 hours at room temperature. The solvent was evaporated under
reduced pressure and the residual oil precipitated from ether, filtered and dried .The
hydrochloride thus obtained was dissolved in 2-butanol (20 mL), toluene (5 mL) and Et3N (1
ml, 1 mmol) and was refluxed for 5 hrs. Most of the 2, 5-piprazinediones crystallized from
the reaction mixture and were collected by filtration and washed with MeOH. Analytical
samples were obtained by drying the crystals in vacuo over P20 S.
3-Benzyl-6-(4-hydroxy-benzyl)-piprazine-2, 5-dione (8a)
This compound was obtained as a white solid. (Yield = 1.5 g, 50 %); Rf 0.1 (B); IH NMR;
300 MHz, DMSO-<4(o ppm): 2.18 (m, 2H, Tyr ~CH2), 2.51-2.62 (m, 2H, Phe ~CH2), 3.88
(m, 2H, Phe & Tyr CaH), 6.60-6.86 (m, 4H, Ar CH), 7.03-7.31 (m, 5H, Ar CH), 7.83 (m,
2H, NH); FAB-MS mlz 311 [M+W].
3,6-Bis-( 4-hydroxy-benzyl)-piprazine-2, 5-dione (8b)
This compound was obtained as a white solid. (Yield = 1.6 g, 50 %); Rf 0.1 (B); IH NMR;
300 MHz, DMSO-<4 (0 ppm): 2.16-2.26 (m, 4H, Tyr ~CH2), 3.88 (m, 2H, Tyr CaH), 6.60-
6.86 (m, 4H, Ar CH), 7.03-7.31 (m, 4H, Ar CH), 7.83 (m, 2H, NH); F'AB-MS mlz 327
[M+Hl.
Benzylation of 8a and 8b (9a and 9b)
To the stirring solution of esters 10 mmole each [1, 4.42 g and 7, 4.58 g) in acetone,
was added K2C03 (2.76 g, 20 mmole) at room temperature. To the resulting heterogeneous
mixture, was added Benzyl bromide (3 ml, 20 mmole), and the mixture was stirred at
ambient temperature for 36 hrs. After the completion of reaction as monitored by TLC,
acetone was evaporated and the crude was extracted with EtOAc. The organic layer was
washed with brine. After drying over anhydrous Na2S04, the organic layer was concentrated
65

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
under reduced pressure; the crudes were purified over silica gel column to get the pure
compounds 9a and 9b.
Boc-Phe-Tyr(OBzI)-OMe (9a)
This compound was obtained as a white solid. (Yield = 5.06 g, 95 %); Rr 0.5 (B); IR (KBr)
3426 cm- I, 1678 cm- I
; IH NMR; 200 MHz, CDCb (8 ppm): 1.39 (s, 9H, (CH3)3C) 2.97-3.03
(m, 4H, Phe & Tyr ~CH2), 3.65 (s, 3H, OCH3), 4.40 (m, IH, Phe CaH), 4.80 (m, IH, Tyr
CaH), 4.99 (s, 2H, CH20BzI), 6.48 (br s, IH, NH), 6.80 -7.40 (m, 14H, Ar CH); ESI-MS
m1z 533 [M+H+], 555 [M+Na+].
Boc- Tyr(OBzI)-Tyr(OBzI)-OMe (9b)
This compound was obtained as a white solid. (Yield = 6.1 g, 95 %); RrO.55 (B); IR (KBr)
3350 cm- I, 1665 cm- I
; IH NMR; 200 MHz, CDCb (8 ppm): 1.44 (s, 9H, (CH3)3C), 2.96-3.1
(m, 4H, Tyr ~CH2), 3.72 (s, 3H, OCH3), 4.32 (IH, m, Tyr CaH), 4.60 (IH, m, Tyr CaH), 5.03
(s, 4H, CH2 OBzI), 6.32 (m, IH, NH), 6.86 -7.35 (m, 18H, Ar CH); ESI-MS m1z 639
[M+Hl, 661 [M+Na+].
Synthesis of diketopiprazines (lOa and lOb)
The benzylated products 10 mmole each [9a, 5.32 g and 9b, 6.38 g] were treated with
15% HClIDioxane (20 ml) for 2 hours at room temperature. The solvent was evaporated
under reduced pressure and the residual oil precipitated from ether, filtered and dried .The
hydrochloride thus obtained was dissolved in 2-butanol (20 mL), tolue;ne (5 mL) and Et3N (1
ml, 1 mmol) and was refluxed for 5 hrs. Most of 2, 5-piprazinediones were crystallized from
the reaction mixture and were collected by filtration and washed with MeOH. Analytical
samples were obtained by drying the crystals in vacuo over P205.
3-Benzyl-6-( 4' -benzyloxy-benzyl)-2, 5-piprazinedione (lOa)
This compound was obtained as a white solid. (Yield = 1.8 g, 45 %), mp-did not melt below
250 DC; Rr 0.1 (B); IR (KBr)1669 cm- I; IH NMR; 300 MHz, CDCb + DMSO-c4 (8 ppm):
2.15 -2.25 (m, 2H, Phe C~H), 2.2 - 2.5 (m, 2H, Tyr ~CH2)' 3.92 (m, IH, Phe CaH), 3.98 (m,
66

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
IH, Tyr CuH), 5.06 (s, 2H, CH20BzI), 6.90-6.93 (m, 4H, Ar CH), 7.04-7.39 (m, 10H, Ar
CH), 7.91 (m, 2H, NH); FAB-MS mJz 400 [M+H+].
3, 6-Bis(4'-benzyloxy-benzyl)-2, 5-piprazinedione (lOb)
This compound was obtained as a white solid. (Yield = 3.2 g, 62 %); Rf 0.1 (B); IR (KBr)
1672 cm,l; IH NMR; 300 MHz, DMSO-c4 (8 ppm): 1.12-1.17 (m, 2H, Tyr ~CH2)' 2.18 (m,
2H, Tyr ~CH2)' 3.90 (br s, IH, Tyr CuH), 3.92 (br s, IH, Tyr CuH), 5.05 (s, 4H, CH2 OBzl),
6.67-7.89 (m, I8H, Ar CH); FAB-MS m/z 506 [M+H+].
N-Methylation of diketopiprazines (lla and llb)
I,4-Dihydro-2,5- piprazinediones, 0.5 mmole each [lOa, 193 mg and lOb, 253 mg]
were suspended in DMF (10 mL) and cooled in an ice bath. NaH (50%, 400 mg, 8 mmole)
was added and the mixture was stirred for 0.5 hr followed by addition of Mel (0.3 ml, 3
mmole). The reaction mixture was allowed to warm up to room temperature for 3 hrs. Water
(30 ml) was added and the reaction mixture extracted with EtOAc and washed sequentially
with water and brine. The organic layer was dried over anhydrous Na2S04 and concentrated.
Chromatography afforded the desired 1, 4-Dimethyl-2, 5-piprazinediones.
1, 4-Dimethyl-3-benzyl-6-( 4' -benzyloxy-benzyl)-2, 5-piprazinedione (Ita)
This compound was obtained as a light yellow syrup. (Yield = 203 mg, 98 %); Rf 0.8 (B);
IR (Neat) 1510 cm,l; IH NMR; 300 MHz, CDCh (8 ppm): 2.18 - 2.30 (m, 2H, Phe ~CH2),
2.78-3.01 (m, 8H; 6H-2-NCH3 & 2H- Tyr ~CH2), 4.02 - 4.11 (m, 2H, Tyr & Phe CuH), 5:03
(s, 2H, CH20BzI), 6.86 -7.14 (m, 6H, Ar CH), 7.25-7.46 (m, 8H, Ar CH); ESI-MS mJz 429
[M+H+].
1,4-Dimethyl-3,6-bis( 4' -benzyloxy-benzyl)-2, 5-piprazinedione (lib)
This compound was obtained as a light yellow syrup. (Yield = 262 mg, 98 %); Rf 0.7 (B);
IR (Neat) 1500 cm,l; IH NMR; 300 MHz, CDCh (8 ppm): 2.17 (m, 2H, Tyr ~CH2), 2.50 (m,
IH, Tyr ~CH2), 2.70 (m, 6H, 2-NCH3), 3.98 (m, IH, Tyr CuH), 4.03 (m, lH, Tyr CuH), 5.03
(s, 4H, 2-CH20Bzl), 6.90 -7.42 (m, 18H, Ar CH); ESI-MS m/z 536 [M+Hl.
67

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
Synthesis of 12a and 12b
Debenzylation was carried out in 10 ml MeOH at 40 psi H2 pressure for 6 hrs with
10% Pd-C as catalyst. After filtration through celite, the filtrate was concentrated under
reduced pressure and the residue was recrystallized from hexane/ether to afford the desired
debenzylated compounds.
3-Benzyl-6-( 4-hydroxy-benzyl)-1, 4-dimethyl-piprazine-2, 5-dione (12a)
This compound was obtained as a white solid. (Yield = 165 mg, 95 %); Rf 0.2 (B); IH
NMR; 300 MHz, CDCh (8 ppm): 2.18 -2.30 (m, 2H, Phe BCH2), 2.78 -3.01 (m, 8H; 2H
NCH3 & 2H- Tyr CpH), 4.02-4.11 (m, 2H, Tyr & Phe CuH), 6.68 -7.12 (m, 3H, Ar CH), 7.22
-7.34 (m, 6H, Ar CH); ESI-MS m/z 339 [M+H+].
3,6-Bis-( 4-hydroxy-benzyl)-1,4-dimethyl-piprazine-2, 5-dione (12b)
This compound was obtained as a white solid. (Yield = 157 mg, 95 %); Rf 0.2 (B); IR (KBr)
1509 cm- I; IH NMR; 300 MHz, CDCh (8 ppm): 2.18 (m, 2H, Tyr BCH2), 2.52 (m, 2H, Tyr
BCH2), 2.66-2.99 (m, 6H, 2-NCH3), 3.99 (m, IH, Tyr CuH), 4.13 (m, IH, Tyr CuH), 6.91-
7.37 (m, 8H, Ar CH); ESI-MS m/z 355 [M+H+].
Synthesis of diethylmalonyl derivatives of N-methylated diketopiprazines (13a and 13b)
To the stirring solution of diketopiprazines, 0.1 mmole each [12a, 35.4 mg and 12b,
33.8 mg] in acetone, was added K2C03 (276 mg, 2 mmole) at room temperature. To the
resulting heterogeneous mixture was added 2-chlorodiethylmalonate (300 Ill, 2 mmole), and
the mixture was stirred at ambient temperature for 36 hrs. After the completion of reaction as
monitored by TLC, acetone was evaporated and the crude was extracted with EtOAc. The
organic layer was washed with brine. After drying over anhydrous Na2S04, the organic layer
was concentrated under reduced pressure; the crudes were purified over silica gel column to
get the pure compounds 13a and 13b.
6-Benzyl-2-( 4-0-diethylmalonyl) benzyloxy-1, 4-dimethyl-piprazine-3, 6-dione (13a)
This compound was obtained as a gummy matter. (Yield = 46.9 mg, 70 %); m (KBr) 1509
cm-I; IH NMR; 300 MHz, CDCh (8 ppm): 1.32-1.6 (m, 6H, mal. CH3), 2.0 ( m, IH, Phe
68

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
~CH), 2.6 (m, 1H, Phe ~CH), 2.7 -3.01 (m, 6H, 2-NCH3), 3.98 (m, 2H, Tyr ~CH2), 4.12 (m,
2H, Tyr & Phe CaH), 4.22-4.25 (m, 2H, mal. CH2), 5.1 (s, IH, mal. CH), 6.91-7.37 (m, 9H,
Ar CH); ESI-MS m/z 498 [M+H+].
Bis[(4-0-diethylmalonyl) benzyloxy]-1, 4-dimethyl-piprazine-3, 6-dioHle (13b)
This compound was obtained as a gummy matter. (Yield = 33 mg, 70%); IR (KBr) 1510 cm-
1; IH NMR; 300 MHz, CDCh (0 ppm): 1.23-1.35 (m, 12H, mal.CH3). 2.3-2.4 (m, 2H, Tyr
C~H), 2.7 -2.9 (m, 8H; 6H, 2-NCH3 & 2H- Tyr ~CH2), 4.02 (m, IH, Tyr CaH), 4.22-4.33 (m,
8H, mal.CH2), 4.85(m, IH, Tyr CaH), 5.13 (s, 2H, mal.CH), 6.91-7.28 (m, 8H, Ar CH);
ESI.,MS m/z 671 [M+H+].
Biological activity
The ability of the compounds 6a-c, 13a and 13 b for PTP I B inhibition was studied in vitro
using a kit based assay as per the procedure described in the previous section.
69

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
2.4 Synthesis and Biological activity of Boc-Phe-Tyr(O-
Carhoxymethyl)-amides
2.4.1 Basis of work
In the previous section all the compounds synthesised are bearing a malonate group
at the phenolic hydroxy of tyrosine, which served as surrogate phosphate group to make
these compounds as phosphatase inhibitors particularly of PTPIB. The malonyl moiety
although serves as useful phosphate substitute, but is generally associated with poor cellular
transport. To address this issue, it is proposed to replace the malonyl gToup with the acetate
to reduce the ionic and acidic property of the resulting derivatives. For the present study,
compounds already showing promising PTPIB inhibition were chosen for the modification
of the malonyl group to the acetate functionality. The structure of the proposed compounds
and their similarity to the lead compound (compound V) is shown in figure 9.
O--{COOH
~ \ COOH
H 0 . H
o H 0 ~ ~
v 16a-c
Figure 9. Structure of Carboxy methyl derivatives derived from parent molecules that are malonate derivatives.
2.4.2 Chemistry
Ethylcarboxymethyl derivatives 15a-c were obtained by simple nucleophilic
substitution reaction of Boc-Phe-Tyr-amides 3a, 3d and 3g with I-Bromoethylacetate in the
presence ofK2C03.13 In the final step, hydrolysis of the diamides 15a-c were performed with
LiOH.H20 in THF to get the desired 16a-c dipeptide alkylamides with free acetate at
tyrosine residue.13 All the steps involved in the synthesis and reagents used are presented in
scheme 4.
70

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
y-' O,-/COOEt eOH
~I
Bore-HN 0 ~ NH-R d
"'" ° 1.0
~I
.. H ° -..0::
~ ~ ISa R=C6H I9
e °,-/COOH
~I
e BOd_HN ° NH-R ---l"'~ N
H ° ~ -: 16a R=CsH19
3c,3d,3g 15a-c
Reagents and conditions:
ISb R=C12H2S
ISc R=CsHII 16a-c
(d) Ethylbromoacetate, K2C03, Acetone, (90 %); (e) LiOH, H20, THF, (97 %)
16b R=C12H2S
16c R=CsH11
Scheme 4. Synthesis ofBoc-Phe-Tyr(O-Carboxymethyl)-amides (16a-c).
2.4.3 Results and discussions
The acetate derivatives synthesized were tested for their PTPIB inhibitory activity in
vitro. The assay was performed using a kit supplied by BIOMOL, USA. The inhibitory data
is presented in Table 5.
Table 5 Inhibition ofPTPIB and LAR (at 100 JlM) by compounds 16a---c
S.No. Compound % inhibition % inhibition PTP1B LAR
1 16a 13.00 Inactive
2 16b 18.00 Inactive
3 16c 86.00 Inactive
4 V 83.00 57.81
PTPIB enzyme is human, recombinant (residues 1-322; MW=37.4 kDa). LAR enzyme is LAR-Dl Fragment. Substrate for PTPIB is the p-nitrophenyl phosphate (p-NPP). V was taken as test control.
As evident from the data, all the acetate derivatives have shown very interesting
activity. We have observed similar correlation with activity and lipophilicity in this series
also, as observed with the series with free amino group. Compound 16c has shown 86 %
inhibition at 100 JlM concentration which is equal to or slightly better than compound V.
Another striking feature of this class of compounds is that all the compounds are highly
selective at 100 JlM concentration as all the three compounds did not show any inhibition of
71

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
LAR activity. Allthough the lead compound (compound V) has shown potent PTPIB
inhibitory activity, but has shown reduced inhibition towards LAR. In view of this, it can be
concluded that the lipophilic acetate derivative 16c is the best compound of this series and
also more selective compared as compared to compound V. This is an important lead for the
discovery of novel highly selective PTPIB inhibitors.
2.4.4 Experimental
Optically active amino acids L-Phe and L-Tyr were used for the study. The coupling
reactions were carried out in anhydrous solvents using pure and dry reactants. Completion of
the reactions and purity of the products were established by TLC on readymade silica gel
plates (Merck, UV active) using following solvent systems.
A. CHCh-MeOH (8:2)
B. CHCh-MeOH
C. CHCh-MeOH
(9:1)
(9.5:0.5)
The plates were developed either under iodine vapours or seen directly under UV-
light (254 nrn). In addition to that the purity of the compounds was also detected by spraying
the TLC plates with 2N HBr/AcOH followed by 0.2% Ninhydrin solution in acetone and
heating the plates at approx. 80-90 °C in hot air oven for 15 minutes. Column
chromatography was performed over silica gel (230-400 mesh).
Melting points (mp) were taken in open capillaries on Complab melting point
apparatus and are uncorrected. Characterization of all the derivatives as well as final
compounds were done with the help IR, NMR and Mass spectroscopy. The IH spectra were
obtained with Bruker DPX-200 or DRX-300 MHz FT-NMR spectrometers. The chemical
shifts were reported as parts per million (& ppm) taking tetramethylsilane (TMS) as an
internal standard. The 13C-NMR spectra were recorded on Bruker DRX-300 FT-NMR
spectrometer (at 75 MHz). Infrared (IR) spectra were recorded on an FT-IR Perkin-Elmer
spectrometer and reported in wave number (cm-I). Mass spectra were obtained on
Micromass Quattro II, Spectrometer using Electron spray ionization mass spectroscopy (ESI
MS positive).
72

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
Synthetic procedure for Boc-Phe-Tyr(O-Ethylcarboxymethyl)-amides (15a-c)
To the stirring solution of amides, 0.5 mmole each [3c, 256 mg; 3d, 298 mg; 3g, 249
mg] in acetone, was added K 2C03 (175 mg, 0.5 mmole) at room temperature. To the
resulting heterogeneous mixture was added 2-chlorodiethylmalonate (55.6 Ill, 0.5 mmole),
and the mixture was stirred at ambient temperature for 36 hrs. After the completion of
reaction as monitored by TLC, acetone was evaporated and the crude was extracted with
EtOAc. The organic layer was washed with brine. After drying over anhydrous Na2S04, the
organic layer was concentrated under reduced pressure; the crudes were purified over silica
gel column to get the pure compounds 15a-c.
Boc-Phe-Tyr(O-Ethylcarboxymethyl)-NH-C6H13 (15a)
This compound was obtained as a white solid. (Yield = 270 mg, 90 %); RfO.7 (C), IR (KBr)
3326 cm- I, 1635 cm- I
; IH NMR; 300 MHz, CDCh (8 ppm): 0.87-0.91 (m, 3H, hexyl CH3),
1.24-1.31 (m, llH; 8H-hexyl CH2, 3H- acetate CH3), 1.34 (s, 9H, (CH3)3C), 2.80-2.85 (m,
2H, hexyl CH2), 3.04-3.14 (m, 4H, Phe & Tyr PCH2), 4.24 (m, IH, Phe CaH), 4.27 (m, 2H,
OCH2), 4.58 (s, 2H, CH2CO), 4.75 (m, IH, Tyr CaH), 5.97 (br s, IH, NH), 6.31 (br s, IH,
NH), 6.79 - 7.37 (m, 9H, Ar CH); ESI-MS m1z 598 [M+Hl.
Boc-Phe-Tyr(O-Ethylcarboxymethyl)-NH-C12H2s (15b)
This compound was obtained as a white solid. (Yield = 290 mg, 85 %), mp 128-130 °C; Rf
0.9 (A); IR (KBr) 3315 cm- I, 1645 cm- I
; IH NMR; 300 MHz, CDCh (8 ppm): 0.87-0.92
(m, 3H, lauryl CH3), 1.27 (m, 23H; 20H-lauryl CH2, 3H-acetate CH3), 1.35 (s, 9H, (CH3)3C),
2.78-2.85 (m, 2H, lauryl CH2), 3.03 -3.18 (m, 4H, Phe & Tyr PCH2), 4.24 (m, 2H,
OCH2CH3), 4.59 (m, 2H, CH2COOEt), 4.71 (m, IH, Phe CaH), 4.72 (m, IH, Tyr CaH), 6.0
(br s, IH, NH), 5.96 (br s, IH, NH), 6.28 (br s, IH, NH), 6.78 -7.32 (m, 9H, Ar CH); ESI
MS m1z 654 [M+H+].
Boc-Phe-Tyr(O-Ethylcarboxymethyl)-NH-CsHll (15c)
This compound was obtained as a white solid. (Yield = 281 mg, 95 %); RfO.7 (C), IR (KBr)
3021 cm- I, 1665 cm-l; IH NMR; 300 MHz, CDCh (8 ppm): 0.85-0.91 (m, 3H, pentyl CH3),
1.20-1.27 (m, 6H, pentyl CH2), 1.28 (m, 3H, acetate CH3), 1.35 (s, 9H, (CH3)3C), 2.73-2.88
(m, 2H, pentyl CH2), 3.04-3.27 (m, 4H, Phe & Tyr PCH2), 4.24-4.29 (m, 2H, OCH2), 4.38
73

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
(m, IH, Phe CuH), 4.58 (s, 2H, CH2COOH), 4.81 (m, IH, Tyr CuH), 5.96 (br s, JH, NH),
6.29 (br s, IH, NH), 6.79-7.38 (m, 9H, Ar CH); ESI-MS mlz 584 [M+W].
Synthetic procedure for Boc-Phe-Tyr(O-Carboxymethyl)-amides (16a-c)
To the solutions of solid 15a-c, 0.25 mmole each [15a, 150 mg; 15b, 171 mg; 15c,
146 mg] in THF (5 ml), added solution of LiOH.H20 (l05 mg, 2.5 mmole) (Sml). The
mixtures were stirred at room temperature for 2.5 hrs. The solvent was evaporated and the
crude mixtures were neutralized with citric acid and extracted with EtOAc. The organic
phases were washed with brine and dried over anhydrous Na2S04. The solvent was removed
in vacuo to give 16a-c.
Boc-Phe-Tyr (O-Carboxymethyl)-NH-C6Hl3 (16a)
This compound was obtained as a white solid. (Yield = 140 mg, 97 %), mp 125-128 °C; Rf
0.3 (A); IR (KBr) 3326 cm- I, 1635 cm- I
; IH NMR; 300 MHz, CDCh + DMSO-<4 (0 ppm):
0.82-0.87 (m, 3H, hexyl CH3), 1.19-1.26 (m, 8H, hexyl CH2), 1.32(s, 9H, (CH3)3C), 2.78-
2.89 (m, 2H, hexyl CH2), 3.03-3.16 (m, 4H, Phe & Tyr ~CH2), 4.25 (m, IH, Phe CuH), 4.54
(m, 2H, CH2COOH), 5.25 (m, IH, Tyr CuH), 6.54 (br s, IH, NH), 6.76 - 7.28 (m, 9H, Ar
CH); ESI-MS mlz 570 [M+H+].
Boc-Phe-Tyr (O-Carboxymethyl}-NH-C12H2S (16b)
This compound was obtained as a white solid. (Yield = 161 mg, 98 %), mp 128-·130 °C; Rf
0.2 (A); IR (KBr) 3020 cm- I, 1665 cm- I
; IH NMR; 300 MHz, CDCh + CD30D (0 ppm):
0.86-0.88 (m, 3H, lauryl CH3), 1.27 (s, 23H, lauryl CH2), 1.35 (s, 9H, (CH3)3C), 2.78-2.89
(m, 2H, lauryl CH2), 3.00-3.08 (m, 4H, Phe & Tyr ~CH2), 4.56 (m, 2H, CH2COOH) , 4.62
(m, IH, Phe CuH), 5.38 (m, IH, Tyr CuH), 6.80-7.29 (m, 9H, Ar CH); l3e NMR; 75 MHz,
CDCh + CD30D (0 ppm): 13.92,22.55,26.72,27.99 (3C), 29.22, 29.43 (7C), 29.52, 31.79
(2C), 42.71 65.00, 72.79 (2C), 81.50, 114.58 (2C), 128.49 (2C), 129.16 (4C), 130.31 (2C),
156.76 (2C), 172.32 (2C), 175.55. ESI-MS m/z 654 [M+H+].
Boc-Phe-Tyr (O-Carboxymethyl}-NH-CsHll (16c)
This compound was obtained as a white solid. (Yield = 135 mg, 97 %), mp 115-118 °C; Rf
0.3 (B); IR (KBr) 3021 cm- I, 1665 cm- I
; IH NMR; 300 MHz, CDCh + DMSO-~ (0 ppm):
0.86-0.91 (m, 3H, pentyl CH3), 1.20-1.26 (s, 6H, pentyl CH2), 1.35 (s, 9H, (CH3)3C), 2.78-
74

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
2.89 (m, 2H, pentyl CH2), 3.03-3.16 (m, 4H, Phe & Tyr ~CH2) 4.38 (m, IH, Phe CaH), 4.55
(m, 2H, CH2COOH), 5.30 (br s, IH, NH), 6.44 (m, IH, Tyr CaH), 6.81-7.38 (m, 9H, Ar
CH); 13C NMR; 75 MHz, CDCh + CD30D (8 ppm): 17.70,26.08,31.97,32.73 (3C), 34.14
(2C), 42.50, 48.00, 52.90, 53.18, 73.00, 81.50, 115.27 (2C), 131.06, 133.08 (2C), 135.10
(4C), 134.27, 145.00, 160.00 (2C), 175.40 (2C), 177.01(2C); ESI-MS m/z 556 [M+H+].
Biological activity The ability of the compounds 16a-c for PTPIB inhibition was studied in vitro using a kit
based assay as per the procedure described in the previous section.
75

Chapter 2 Synthesis & Biological activity of Phosphate mimics of Boc-Phe-Tyr-amides
References 1. Saltiel, A.R. and Kahn, C.R. Nature 2001, 414, 799.
2. Elchebly, M. Science 1999, 283, 1544.
3. Klaman, L.D. Mol. Cell. BioI. 2000,20,5479.
4. Tsai, T. D.; Shuck, M. E.; Thompson, D. P.; Bienkowski, M. J.; and Lee, K. S. Am. J.
Physiol. 1995,268, Cl173.
5. Songyang, Z.; Carraway, K. L.; Eck, M. J.; Harrison, S. C.; Feldman, R. A.;
Mohammadi, M.; Schlessinger, J.; Hubbard, S. R.; Smith, D. P.; Eng, C.; Lorenzo,
M. J.; Ponder, B. A. J.; Mayer, B. J.; Cantley, L. C. Nature 1995, 373, 536.
6. Jia, Z. c.; Barford, D.; Flint, A. J.; Tonks, N. K. Science 1995,268, 1754.
7. Otwinoski, Z. and Minor, W. Methods Enzyrnol. 1997,277,307.
8. Bleasdale, J. E.; Ogg, D.; Palazuk, B. J.; Jacob, C. S.; Swanson, M. L.; Wang, W.-Y.;
Thompson, D. P.; Conradi, R. A.; Mathews, W. R.; Laborde, A. L.; Stuchly, C. W.;
Heijbel, A.; Bergdahl, K.; Bannow, C. A.; Smith, C. W.; Liljebris, C.; Schostarez, H.
J.; May, P. D.; Stevens, F. C.; Larsen, S. D. Biochemistry 2001,40,5642.
9. Miller, M. J.; Anderson, K. S.; Braccolino, D. S.; Cleary, D. G.; Gruys, K. J.; Han, C.
y.; Lin, K.-c.; Pansegrau, P. D.; Ream, J. E.; Sammons, R. D.; Sikorski, J. A. Biorg.
Med. Chern. Lett. 1993,3, 1435.
10. Kole, H. K.; Akamatsu, M.; Ye, B.; Yan, X.; Barford, D.; Roller, P. P.; Burke, T. R.
Biochern. Biophys. Res. Cornrnun., 1995,209,817.
11. Akamatsu, M.; Roller, P. P.; Chen, L.; Zhang, Z. Y.; Ye, B.; Burke, T. R., Jr. Bioorg.
Med. Chern. 1997,5, 157.
12. Roller, P. P.; Wu, L.; Zhang, Z. Y.; Burke, T. R., Jr. Bioorg. Med. Chern. Lett. 1998,
8,2149.
13. Larsen, S. D.; Barf, T.; Liljebris, C.; May, P. D.; Ogg, D.; O'Sullivan, T. J.; Palazuk,
B. J.; Schostarez, H. J.; Stevens, F. C.; Bleasdale, J. E. J. Med. Chern, 2002, 45,598.
14. Larsen, S. D.; Stevens, F. C.; Lindberg, T. J.; Bodnar, P. M.; O'Sullivan, T. J.;
Schostarez, H. J.; Palazuka, B. J.; Bleasdale, J. E. Biorg. Med. Chern. Lett. 2003, 13,
971.
15. Tarbell, D. S.; Yamamoto, Y.; Pale, B. N. Proc. Nat!. Acad. Sci.1972, 69, 730.
16. Brenner, M. and Huber, W. Helv. Chirn. Acta. 1953,36, 1109.
17. Konig, W. and Geiger, R. Chern. Ber. 1970, 103, 788.
18. Vaughen, J. R. J. Am. Chern. Soc. 1951, 73, 3547.
76

Chapter 2 Synthesis & Biological activity of Phosphate mimics ofBoc-Phe-Tyr-amides
19. Kenji, 0.; Kochi, Y.; Kazuyoshi, F.; Shinichi, K; Kenichi, A. Chemical and
Pharmaceutical Bulletin, English 1984, 32, 430.
20. Moroder, L.; Hallett, A.; Wunsch, E.; Keller, 0.; Wersin, G. Z. Physiol. Chem. 1976,
357, 1651.
21. Puius, Y. A.; Zhao, Y.; Sullivan, M.; Lawrence, D.; Almo, S. C.; Zhang, Z. Proc.
Natl. Acad. Sci. 1997, 94, 13420.
22. Liu, F.; Hill, D. E.; Chernoff, J. J. Bioi. Chem. 1996,271,31290.
23. GraphPad Prism,version 5.0, GraphPad Software Inc.,10855 Sorrento Valley
Rd.#203, San Diego, CA 92121.
24. Schiller, P.W. The peptides: Analysis, Synthesis, Biology, 1984, 6, Acadmic press,
Orlando, FL.
25. Sawer, T. K; Hruby, V. J.; Darman, P. S.; Hadley, M. E. Proc. Natl. Acad. Sci. USA,
1982, 79, 1751.
26. Ishikawa, K; Fukami, T.; Nagase, T.; Hayama, T.; Niiiyama, T.; Yano, M. J. Med.
Chem. 1992,35,2139.
27. Marshall, G. Tetrahedron 1993,49,3547.
28. Venkatraman, 1.; Shakaramanna, S. C.; Balram, P. Chem. Rev. 2001, 101,3131.
29. Hruby, V. J.; BaIse, P. M. Curro Med. Chem. 2000, 7, 945.
30.7 Hruby, V. J. Acc. Chem. Res. 2001, 34, 389.
31. Rosenmund, P.; Kaiser, K. Angew. Chem. Int. Ed. Engl. 1970,9, 162.
32. Nitecki, D. E.; Halpern, B.; Westley, J. W. J. argo Chem. 1968,33, 864.
33. Kopple, K. D.; Ghazarian, H.G. J. argo Chem. 1968, 33, 862.
34. Veda, T.; Saito, M.; Kato, T.; Izumiya, N. Bull. Chem. Soc. Jpn 1983, 56, 568.
35. Suzuki, K; Sasaki, Y.; Endo, N.; Mihara, Y. Chem. Pharm. Bull. 1981,29,233.
36. Bergeron, R, J.; Phanstiel, 0.; Yao, G.W.; Milstein, S.; Weimar, W. R. J. Am. Chem.
Soc. 1994, 116,8479.
37. Zeng, Y.; Li, Q.; Hanzlika, R. P.; Aube, J. Biorg. Med. Chem. Lett .. 2005, 15,3034.
38. Coggins, 1. R.; Benoiton, N. L. Can. J. Chem. 1971,49, 1968.
39. Schnabel, E. Justus Liebigs Ann. Chem. 1967, 702, 188.
40. Jung, M. E.; Rohloff, 1. C. J. argo Chem. 1985, 50, 4909.
77