chapter 1 introduction to the chemistry...
TRANSCRIPT

14
CHAPTER 1
INTRODUCTION TO THE CHEMISTRY OF N-tert-BUTANESULFINYL
IMINES, CHIRAL AMINE DRUGS AND 3-SUBSTITUTED-1,2-
BENZISOXAZOLES
1.1 General:
The discovery that chiral amines as powerful pharmacophores
[1] in new pharmaceutical drugs and the great demand for new
molecules of biological and medicinal importance has led to an
extreme significance of the development of research activities in the
area of syntheses on chiral amine drugs and chiral amine auxiliaries
like tert-butanesulfinamide [2,3].
As the chiral nitrogen-containing compounds are widely
distributed in nature and many biologically important molecules, the
asymmetric synthesis of chiral amines has long been a popular and
challenging research area [1]. The most promising and convenient
route for the synthesis of chiral nitrogen-containing carbon frame
works is through imines, as electrophiles [1].
The definition of a “chiral amine” which in particular is a “α-
chiral amine” is a nitrogen atom with an adjacent, or α-stereogenic
carbon [1]. The two generic structural features of chiral amines are
useful to point out i.e. first, the nitrogen can be primary (I), secondary
(II), tertiary (III) or even quaternary (ammonium salt) and second, the

15
α-stereogenic carbon, by necessity, can only be secondary or tertiary
[3].
C
NH2
R1
R2 R3
C
NH2
H
R1 R2
1°-aminetert-a-carbon
1°-aminesec-a-carbon
C
NHR
H
R1 R2
2°-amine
C
NR2
H
R1 R2
3°-amine
(I) (II) (III)
Of these, chiral primary amine building blocks are often sought
after because of the flexibility they lend to the synthetic design of
pharmaceutical drugs and alkaloid natural products [4].
1.2. IMPORTANCE OF CHIRALITY AND CHIRAL AMINE
FUNCTIONALITY
Chirality is a phenomenon which plays a key role in both the life
of plants and animals and also in pharmaceutical, agricultural and
other chemical industries [5]. For example, all the vital elements
necessary for the life are nucleosides, proteins, enzymes, amino acids,
carbohydrates and a number of alkaloids, hormones and almost all
natural products are chiral compounds [5].
A large number of chiral amines and its derivatives find use as
pharmaceuticals like antidepressant [6], antidiabetic [7], antimalarial
[8], antituberculosis agents [9], antihypertensive [10], anticholinergic
agents [11], calcimimetic compounds [12], antiviral derivatives [13]

16
and analgesics [14]. Some of the important chiral drug derivatives,
which are of commercial importance, are shown in Table-1.1
Table 1.1: Some important chiral drug derivatives:
S.No. Structure & Name
Importance
1.
NHCH3 HClCl
Cl Zoloft / Sertraline
Antidepressant agent
2.
N
NH
O
OEt
COOH
Repaglinide
Antidiabetic agent
3. N
HO
F3C
CF3
HN
H
Lariam
Antimalarial agent
4.
NH
HN
HO
OH Ethambutol
Antituberculosis agent
5. H3CO
H2NO2S NH
CH3
O
O Flomax / Tamsulosin
Antihypertensive Agent
6.
ON
NH3C
CH3 CH3
H
CH3
CH3O
Rivastigmine
Alzheimer’s [Anti cholinergic agent]
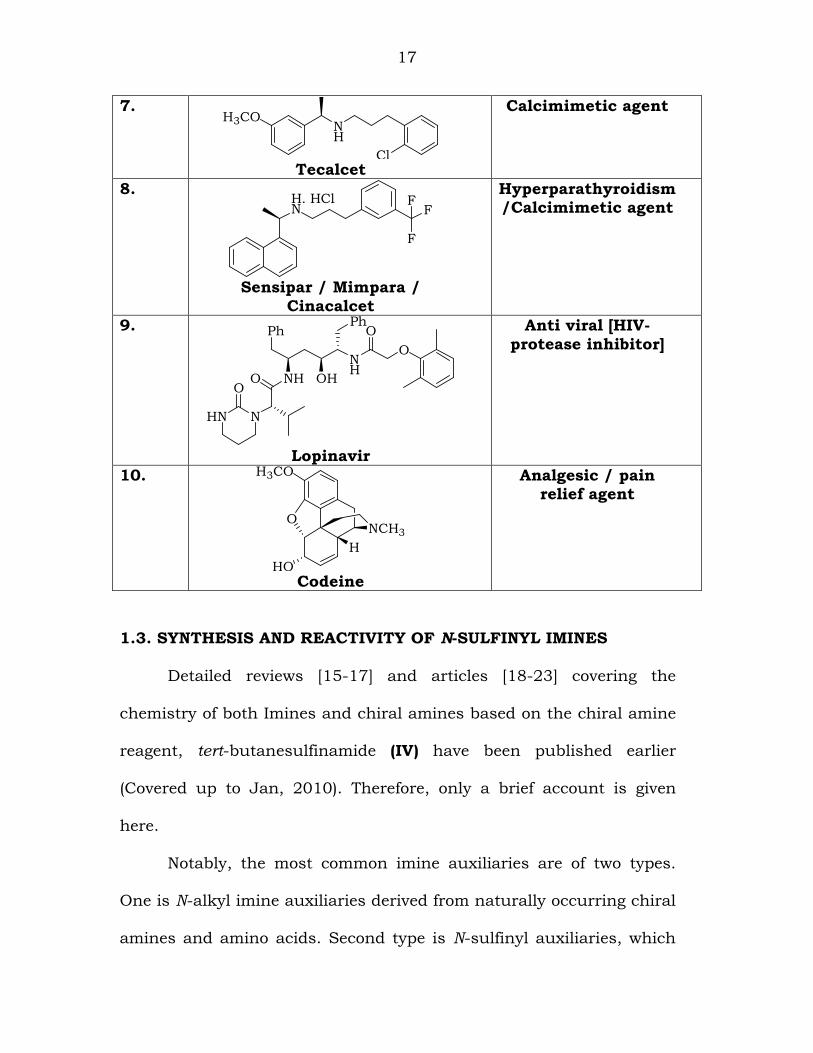
17
7. H3CO
NH
Cl Tecalcet
Calcimimetic agent
8. H. HClN F
F
F
Sensipar / Mimpara /
Cinacalcet
Hyperparathyroidism /Calcimimetic agent
9.
HN N
ONHO OH
NH
O
O
PhPh
Lopinavir
Anti viral [HIV-protease inhibitor]
10.
O
H3CO
HO
H
NCH3
Codeine
Analgesic / pain relief agent
1.3. SYNTHESIS AND REACTIVITY OF N-SULFINYL IMINES
Detailed reviews [15-17] and articles [18-23] covering the
chemistry of both Imines and chiral amines based on the chiral amine
reagent, tert-butanesulfinamide (IV) have been published earlier
(Covered up to Jan, 2010). Therefore, only a brief account is given
here.
Notably, the most common imine auxiliaries are of two types.
One is N-alkyl imine auxiliaries derived from naturally occurring chiral
amines and amino acids. Second type is N-sulfinyl auxiliaries, which

18
were developed and pioneered by Davis [16] and Ellman groups [17]
separately. N-sulfinyl imines were mainly of two types, N-tert-
butanesulfinyl aldimines (V) and N-tert-butanesulfinyl ketimines (VI),
are a special class of imines that display unique reactivity and
stereoselectivity due to the presence of a chiral and electron
withdrawing sulfinyl group (VII). They had been introduced three
decades ago, since then; they have emerged as versatile intermediates
in asymmetric synthesis of a variety of structurally diverse nitrogen
containing compounds.
S
O
NH2S
O
NH2S
O
NH2
tert-butanesulfinamide(IV)
(R)-tert-butanesulfinamide(IVa)
(S)-tert-butanesulfinamide
(IVb)
S
O
N R S
O
N R
R1
N-tert-butanesulfinyl aldimine(V)
N-tert-butanesulfinyl ketimine(VI)
S OR
Sulfinyl group(VII)
Due to its weak electron withdrawing nature, compared to
carbonyl group, the N-sulfinyl auxiliary activates the imino carbon for
nucleophilic addition, allowing reactions to proceed at lower
temperature. Thus, the sulfinyl group shows a powerful stereodirecting

19
effect, which results in the addition of organometallic reagents and
enolates to both steric and enolizable sulfinimines with high and
predictable asymmetric induction. Moreover, unlike other imine N-
auxiliaries, the sulfinyl group is easily removed under relatively mild
conditions.
N-tert-butanesulfinimines may be synthesized from:
1. condensation reaction with aldehydes
2. condensation reaction with ketones
3. condensation reaction with orthoesters
4. Oxidation of N-sulfenyl imines and
5. Miscellaneous reactions.
1.3.1. Condensation reaction with aldehydes:
The reaction between an aldehyde and tert-butanesulfinamide
was first investigated by Ellman and co-workers in 1997 [24] who,
prepared tert-butanesulfinyl aldimine (V) by condensing 2-3 equimolar
amounts of aldehyde (VIII) and tert-butanesulfinamide (IV) in the
presence of acidic catalyst pyridinium p-toluenesulfonate (PPTS) and
excess amount of MgSO4 in methylene dichloride at RT for. (Scheme -
1.1).

20
R H
NS
O
R H
O+ S
O
H2N
R H
O+ S
O
H2N
R H
O+ S
O
H2N
R H
O+ S
O
H2N
R H
O+ S
O
H2N
R H
O+ S
O
H2N
R H
O+ S
O
H2N
R H
O+ S
O
H2N
CuSO4 (2 eq)
MDC, rt
MgSO4 (5 eq)
PPTS (0.5 eq)
MDC, rt
TH
F,
r t
Ti(O
Et)
4 (2 e
q)
THF, rtBnBr (
2 eq)
Zn (3 eq
)
Cs
2 CO
3 (1 eq)
MDC, 40 - 45°C
KH
SO
4 (2 e
q)
Tolu
en
e, 45° C
NaO
H (1 eq)
MeO
H, rt Yb(O
Tf) 3 (0
.1 eq)
THF, rt
(V)
(VIII) (IV)
(VIII) (IV)
(VIII) (IV)
(VIII) (IV)
(VIII) (IV)
(VIII) (IV)
(VIII) (IV)
(VIII) (IV)
. . . . . Scheme 1.1
Further studies revealed that the more Lewis acidic CuSO4 was
a more effective agent for this condensation reaction at reduced
equivalents (1.1) of aldehydes [25]. But, some substrates were found to
be unreactive with this reagent, in these cases, Ti(OEt)4 was found to
be effective both as Lewis acid catalyst and water scavenger.
All these aldimines were found to be reasonably hydrolytically
stable and were isolated by silica gel column chromatography. The
aldimines can be handled at room temperature in air, although minor

21
decomposition was observed over a period of weeks or months for
prolonged storage at room temperature in air and with exposure to
light. Thus the aldimines could be stored in closed containers at -5°C
to prevent the decomposition [25].
Since then many groups synthesized V from VIII and IV [26-29].
Nakata and co-workers, in 2004 reported a procedure using equimolar
amounts of aldehydes, sulfinamide and base. This procedure was on
the use of Cs2CO3 as an activating and dehydrating agent with gentle
heating. Another protocol was developed by Qin and co-workers, which
was mediated by KHSO4 in toluene at 45°C. These conditions were
found to be effective for a broad range of substrates, including electron
rich and electron poor aromatic aldehydes, α, β-unsaturated
aldehydes, and both sterically hindered and unhindered aliphatic
aldehydes. Thus various Lewis acids like Yb(OTf)3, Sc(OTf)3 etc.. were
used in the condensation reaction.
Another method for the synthesis of sulfinyl aldimines utilizes
NaOH or tBuOK as a base. But these conditions did not show
beneficial characteristics for the synthesis of imines derived from
enolizable aliphatic aldehydes, high yields were observed in case of α,
β-unsaturated aldehydes [30].
Recently, Fan et al [31] prepared aldimines under Barbier-type
conditions using benzyl bromide and zinc dust, which were reacted to
form BnZnBr in situ and then the aldehydes and sulfinamide are

22
added. The proposed mechanism for this reaction is that the BnZnBr
acts as a base to deprotonate the sulfinamide, and then the
coordinated zinc acts as a Lewis acid to activate the carbonyl to attack
via a 4-membered transition state.
1.3.2. Condensation reaction with ketones:
The condensation reaction of ketones (IX) with IV is more
challenging than that of aldimines as in most cases, the conditions
reported for the preparation of aldimines were failed to provide the
desired products with ketones as the reactant [17]. The preparation of
N-tert-butanesulfinyl ketimines was also first reported by Ellman
group in the same year [32]. After several attempts, they arrived to use
1.1 equiv of ketone, 1.0 equiv of sulfinamide and 2 equiv of Ti(OEt)4 in
THF at higher temperatures to get higher yields of the desired imine
VI. (Scheme - 1.2).
S
O
NH2O
R1
R+
(VI)(IX) (IV)
S
O
N R
R1Ti(OEt)4
THF,
. . . . . Scheme 1.2
The condensation of several aliphatic and aromatic ketones with
IV were depicted in Table-1.2.
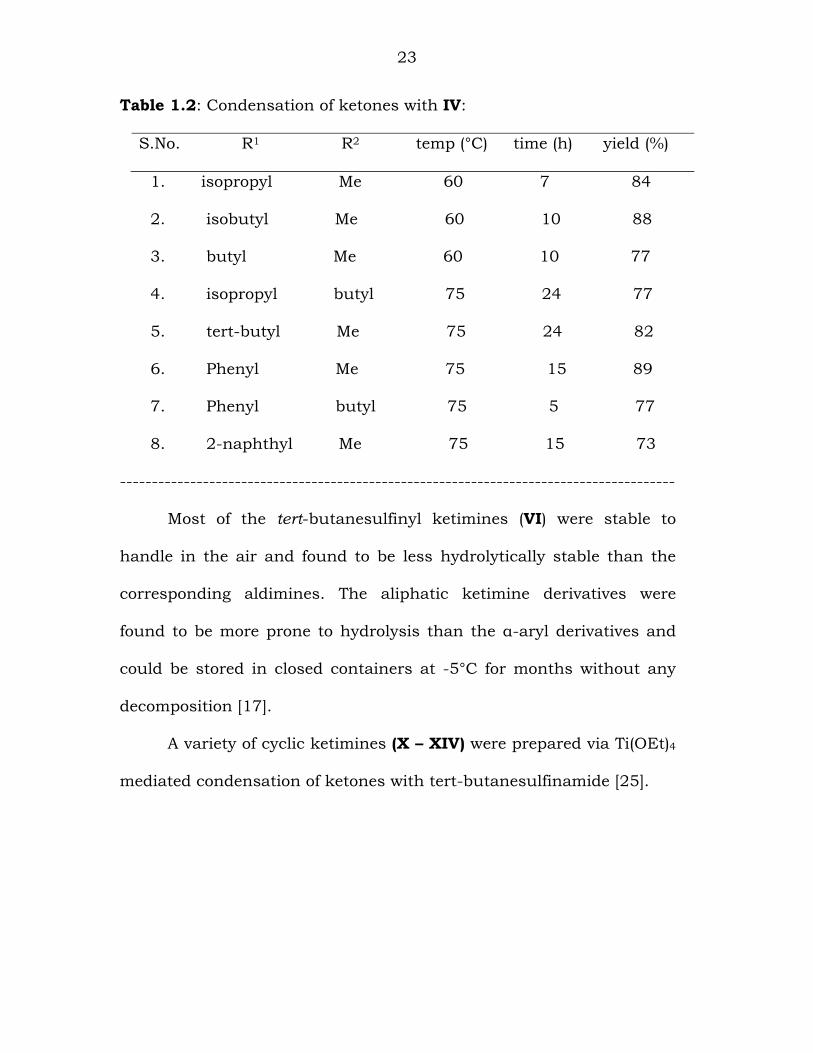
23
Table 1.2: Condensation of ketones with IV:
S.No. R1 R2 temp (°C) time (h) yield (%)
1. isopropyl Me 60 7 84
2. isobutyl Me 60 10 88
3. butyl Me 60 10 77
4. isopropyl butyl 75 24 77
5. tert-butyl Me 75 24 82
6. Phenyl Me 75 15 89
7. Phenyl butyl 75 5 77
8. 2-naphthyl Me 75 15 73
---------------------------------------------------------------------------------------
Most of the tert-butanesulfinyl ketimines (VI) were stable to
handle in the air and found to be less hydrolytically stable than the
corresponding aldimines. The aliphatic ketimine derivatives were
found to be more prone to hydrolysis than the α-aryl derivatives and
could be stored in closed containers at -5°C for months without any
decomposition [17].
A variety of cyclic ketimines (X – XIV) were prepared via Ti(OEt)4
mediated condensation of ketones with tert-butanesulfinamide [25].

24
NS
O
NO
N SO
PMB
N SO
NS
O
Y
NS
O
(X) (XI) (XII) (XIII) (XIV)
Y = NBocY = OY = S
1.3.3. Condensation reaction with orthoesters:
The condensation reaction of orthoesters (XV) with IV was
described to proceed in 83-92% yields of the N-tert-butanesulfinyl
imidate esters (XVI), under neat and mild acidic conditions [33] or in
the presence of triphenylphosphine [34]. (Scheme - 1.3).
S
O
NH2
R1
+
(XVI)(XV) (IV)
S
O
N O
p-TsOH
THF,O OO
R R
R R1
R
. . . . . Scheme 1.3
1.3.4. Oxidation of N-sulfenyl imines:
Aldehydes or ketones when reacted with S-
methylthiohydroxylamine (XVII) yields the N-sulfenimine (XVIII) which
on oxidation with a chiral oxidant, such as (+)- or (-)-N-
(phenylsulfonyl)-3,3-dichlorocamphoryloxaziridine (XIX) and achiral
oxidant, like m-CPBA yielded the desired enantiopure N-sulfinimines
V or VI in 80-95% yield [35, 36]. (Scheme - 1.4).

25
But this methodology was found to have limited synthetic value
due to the complex nature of both the chiral oxidants and N-
sulfenimines.
RS RL
NS
R
ClCl
O N O2SPh
RS RL
NS
O
RCl
Cl
O N O2SPh
RS RL
NS
O
R
(XVIII)
RS RL
O+ R
SH2N
(VIII) or (IX) (XVII)
m-CPBAR=OR1
S
RS RL
NS
O
R
RS RL
NS
O
R
+
83-99%72:28-99:1 dr
(RS)-(V) or (VI) (SS)-(V) or (VI)
80-95%>97ee
59-80%>97ee
(XIX) (XIX)
. . . . . Scheme 1.4
1.3.5. Miscellaneous reactions:
In addition to N-tert-butanesulfinyl aldimines and ketimines,
several aliphatic and aromatic imines bearing α-heteroatom
functionality, including α-alkoxy aldimines (XX) [37] and ketimines
(XXI) [38], α-halo aldimines (XXII) [39] and ketimines (XXIII) [40] and
α-amino aldimines (XXIV) [41] and ketimines (XXV) [42] have been
synthesized.

26
S
O
N
H
OR S
O
N
R
OR1 S
O
N
H
X
(XX) (XXI) (XXII)
S
O
N
H
R
S
O
NNHBoc
S
O
N
R1
X
NBn2
F
R R
(XXIV) (XXV)(XXIII)
Ellman et al reported the first preparation of N-tert-
butanesulfinyl imino esters (XXVII) by condensing methyl glyoxalate
(XXVI) with IV in the presence of CuSO4 at RT, in 65% yield [25].
(Scheme - 1.5).
S
O
NH2
O
+
(XXVII)(XXVI) (IV)
S
O
N
HCuSO4
MDC, rtOH CH3
OCH3
O
O
. . . . . Scheme 1.5
Similarly, tert-butyl ester (XXVIII) [43]and benzyl ester (XXIX)
[44] have also been synthesized under these conditions, which are
more stable enough to be purified by column chromatography and can
be stored in closed containers for long period when compared to N-
sulfonyl and N-Boc imino esters.

27
(XXVIII)
S
O
N
H
O
O
CCH3
CH3
CH3
(XXIX)
S
O
N
H
O
O
1.3.6. Reactivity of N-tert-butanesulfinyl imines:
Due to its weak electron withdrawing nature, N-tert-
butanesulfinyl imines favor the nucleophilic addition reaction at very
low temperatures. In particular, N-tert-butanesulfinyl aldimines are
prone to conjugate and 1,2-additions and N-tert-butanesulfinyl
ketimines favors reduction of imine bond with borohydride reagents
and transition metal catalyzed reductions.
a) 1,2-Additions to N-tert-butanesulfinyl aldimines :
Conjugate addition of Copper reagents to α,β-unsaturated N-tert-
butanesulfinyl imines (XXX) was first developed by Ellman and
McMahon [45]. The highest yields (68-76%) with diastereoselectivities
(≥ 92:8) were obtained by the addition of a butyl cuprate with n-BuLi,
CuCN and BF3.OEt2 in THF. The facial selectivity of conjugate addition
of copper reagent was rationalized by the transition state XXXI, where
coordination of the cuprate to the sulfinyl oxygen and delivery of the
nucleophile occurs on the face opposite to the tert-butyl group.
(Scheme - 1.6).

28
S
O
N
R
R1
R2[Cu]
n-BuLi, CuCN
BF3.OEt2, THF
-78°C
R
NS
O
R1
Li
CuLmXn
R2 O
R
N
R1
R2
(XXX) (XXXI) (XXXII)
S
. . . . . Scheme 1.6
1,2-Addition of organometallic reagents, like Grignard reagents
and organolithium reagents to N-tert-butanesulfinyl aldimines V in the
presence of a Lewis acid (e.g. AlMe3 or Ti(OET)4) was first developed in
1997 by Ellman and co-workers [24]. The addition afforded chiral
sulfinamides XXXV and XXXVI, via a six-membered ring transition
states XXXIII and XXXIV respectively, with the metal cation
coordinated to the oxygen atom of the sulfinyl group and the Lewis
acid coordinated to the nitrogen lone pair, in high yields with excellent
stereoselectivity. (Scheme - 1.7).

29
S
O
N R
H
(V)
(XXXIII) (XXXV)R1 MgBr
AlMe 3
or Ti(O
ET) 4
R 2Li
S
OMg
R1
N R S
O
NH
R1
H
R
NO
R H R2Li
(XXXVI)
S
O
NH
R
H
R2
(XXXIV)
. . . . . Scheme 1.7
It is interesting to know that Grignard reagents have shown
better selectivity when compared to organolithium reagents.
1,2-Addition of cyanide nucleophile across N-sulfinyl imines is
an important method for the synthesis of Strecker α-amino nitriles
(XXXVIII), and the corresponding α-amino acids (XXXIX) [46]. Cordi
and Mabic [47] presented the first asymmetric Strecker reaction of N-
tert-butanesulfinyl aldimine (XXXVII) with ethylaluminium
cyanoisopropoxide [EtAl(OiPr)CN] or TMSCN in the presence of a Lewis
acid and CH2Cl2. The highest diastereoselectivity (98:2) with higher
yield (90%) was obtained with Y(OTf)3. Hydrolysis of the α-amino nitrile
intermediate with acids such as HCl underwent deprotection of the N-
sulfinyl group and hydrolysis of the nitrile, giving rise to XXXIX in
moderate to good yields. (Scheme - 1.8).

30
H
NS
O
TMSCN
Y(OTf)3 (20 mol%)
CH2Cl2, 10°C
CN
HNS
O
CN
HNS
O
+
98% 2%(XXXVIII)(XXXVII)
HCl, MeOH
CO2H
NH2
CO2H
NH2
+
98% 2%(XXXIX)
. . . . . Scheme 1.8
Similarly, 1,2-addition of ester enolates (XL) to N-tert-
butanesulfinyl imines has been reported by Ellman et al in 1999 [48],
for the synthesis of β-amino esters (XLII) in highest yields and
diastereoselectivities at low temperature in THF. (Scheme - 1.9).
RL RS
NS
O + R3
OR4
OClTi(OiPr)3 (2 eq)
LDA (2.1 eq)
THF, -78°C RL
NH
R3
RS O
OR4
SO
O
TiNRS
RL
R3
OR4
SBut O
(V) or (VI) (XL)
(XLI)
(XLII)
. . . . . Scheme 1.9
b) Reductions of N-tert-butanesulfinyl ketimines :
The reduction of N-tert-butanesulfinyl ketimines with metal
borohydride reagents (XLIII) was first investigated by Ellman and co-
workers [49] who, prepared α-branched amines (XLV) in the best yield

31
(83%) and diastereoselectivity (91:9 dr) by adding NaBH4 at low
temperature in THF. (Scheme - 1.10).
R R2
NS
O +Ti(OEt)4 (2 eq)
THF, -48°CR2R
NHS
O
(XLIII)
(XLIV)
(XLV)(VI)
NaBH4
O
SN
HM
RR2
. . . . . Scheme 1.10
The delivery of the hydride was occurring from the si-face of the
imine which was evident by the proposed coordination (XLIV) of the
sulfinyl oxygen to the metal atom. Conversely, an open transition state
(XLVII) was proposed for the L-Selectride (XLVI) reduction. The
addition of the hydride occurring from the least hindered face (re-face)
in THF at -78°C. (Scheme - 1.11).
R R2
NS
O +THF, -78°C
R2R
HN
(XLVI)
(XLVII)
(XLV)(VI)
L-Selectride
N
OR
R2
H-M
SO
. . . . . Scheme 1.11

32
1.4 Recent work towards the synthesis of N-tert-butanesulfinyl
imine derivatives:
Das et al made the concise formal syntheses of the dendrobate
alkaloid (+)-241D and its C-4 epimer by means of a highly
diastereoselective Barbier-type indium mediated allylation strategy
involving an (S)-(N-tert-butanesulfinyl) imine [50]. (Scheme – 1.12).
OH
1.PCC, CH2Cl2, r.t, 2 h, 90%
2.(S)-tert-butanesulfinamide
CuSO4 (anhyd), CH2Cl2 r.t, 24 h, 88%
H
NS
O
CH2=CHCH2Br, In
THF, 66°C, 4 h, 86% NS
O
1) 4M HCl-dioxane, MeOH, r.t, 30 min
2) CBzCl, sat. aqNaHCO3
1 h, 67%
HNCBz
NH
C9H19
OH
(XLVIII) (XLIX)
(L)
(LI) (LII)
. . . . . Scheme 1.12
Rodriguez and co-workers reported the asymmetric synthesis of
1-(9-anthracenyl)ethylamine and its trifluoromethyl analogue via
nucleophilic addition to an N-(tert-butylsulfinyl)imine [51]. (Scheme -
1.13).
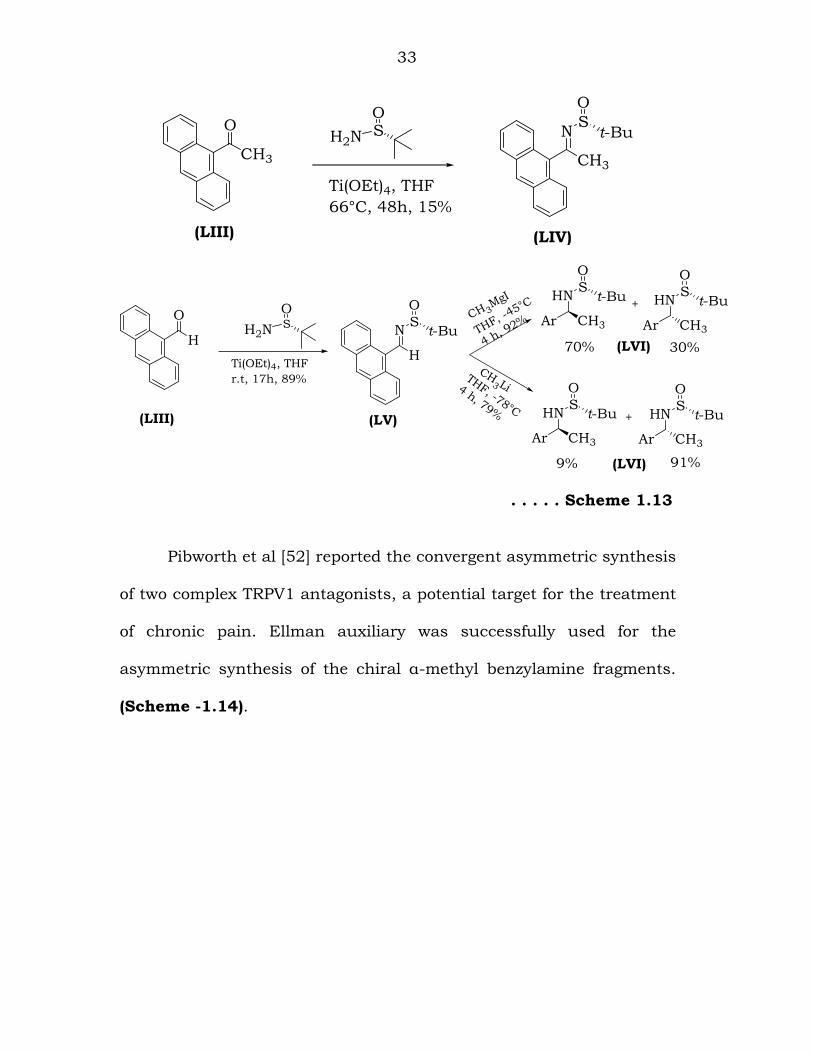
33
CH3
O SO
H2N
Ti(OEt)4, THF
66°C, 48h, 15%
CH3
NS
t-Bu
O
(LIII) (LIV)
H
OS
O
H2N
Ti(OEt)4, THF
r.t, 17h, 89%
H
NS
t-Bu
O
(LV)(LIII)
CH 3MgI
THF, -45°C
4 h, 92%
CH3 Li
THF, -78°C
4 h, 79%
(LVI)
Ar CH3
HNS
t-Bu
O
(LVI)
Ar CH3
HNS
t-Bu
O
Ar CH3
HNS
t-Bu
O
Ar CH3
HNS
t-Bu
O
+
+
70% 30%
9% 91%
. . . . . Scheme 1.13
Pibworth et al [52] reported the convergent asymmetric synthesis
of two complex TRPV1 antagonists, a potential target for the treatment
of chronic pain. Ellman auxiliary was successfully used for the
asymmetric synthesis of the chiral α-methyl benzylamine fragments.
(Scheme -1.14).

34
O
Cl
NH
SO
O SO
H2N
1) Ti(OEt)4, THF
66°C, 16h+
2) NaBH4, THF
-20°C, 12h, 42% Cl
NH
S
N
O
O
S
O
Cl
NH
S
NH
O
O
S
O
1.25 M HCl/MeOH
r.t, 86%Cl
NH
S
NH2.HCl
O
O
N CF3
O
HO
50% 1-Propylphosphonic acid cyclic anhydride in EtOAc, DIPEA, THF, 50°C, 2 h, 77%
Cl
NH
S
NH
O
O
O
NCF3
(LVII) (R)-IV(LVIII)
(LIX) (LX)
(LXI)
(LXII)
. . . . . Scheme 1.14
1.5. CHIRAL AMINE DRUGS 1.5.1. INTRODUCTION:
A large number of drugs and drug candidates have an
asymmetric carbon centre attached to amine functionality [1].
Therefore, the asymmetric synthesis of optically pure chiral amine
drugs is of great value and their synthesis remains a challenge. Hence,
there is high demand for the development and production of new drug
candidates.
(S)-Cetrizine hydrochloride (LXV), Manzacidin C (LXX), (S)-
Salsolidine & (R)-Salsolidine (LXXIII), (S)-O-Methylbharatamine

35
(LXXVIII), DPP-4 inhibitor for type 2 diabetes (LXXXII), Sibutramine
(LXXXIII) and Sertraline (LXXXIV) etc., are some of the most
commonly used chiral amine drugs synthesized through N-tert-
butanesulfinyl imine intermediates [17].
a) Asymmetric synthesis of (S)-Cetrizine Dihydrochloride:
Recemic Cetrizine hydrochloride, a histidine H1-receptor
antagonist used for the treatment of allergies, is marketed in the U.S.
as Zyrtec® [53]. The asymmetric synthesis of (S)-cetrizine 2HCl was
first reported by Senanayake [54], by the addition of an aryl Grignard
reagent to 2-methyl-propane-2-sulfinic acid 4-chloro-benzylideneamide
(LXIII) in toluene at -20° to 0°C. (Scheme – 1.15).
Cl
H
NS
O
MgBr
(in Et2O)
toluene-20 to0°C Cl
HNS
O
Cl
N
N
OOH
O
.2HCl
(LXIII) (LXIV) (LXV)
. . . . . Scheme 1.15
b) Asymmetric synthesis of Manzacidin C:
Lanter and co-workers introduced the 1,3-diamine framework in
their elegant asymmetric synthesis of manzacidin C [55]. Coupling of
2-methyl-propane-2-sulfinic acid (2-benzyloxy-1-methyl-
ethylidene)amide (LXVI) and 2-methyl-propane-2-sulfonic acid (3-
phenyl-allylidene)amide (LXVII) in the presence of LHMDS in THF at -

36
78°C provided 2-methylpropane-2-sulfonic acid [4-benzyloxy-3-(2-
methyl-propane-2-sulfinyl imino)-1-styryl-butyl]amide (LXVIII) in 85%
yield. Grignard reagent was added to LXVIII to get the tertiary
carbinamine (LXIX) as a single diastereomer, followed by the removal
of sulfinyl group yielded the desired natural product LXX in ten linear
steps and 28% overall yield. (Scheme – 1.16).
N
BnO
SO + H
N LHMDS(1.1 equiv)
THF, -78°C
N
BnO
SO
HN
NH
Br
O
O
HN N
CO2H
(LXVI) (LXVII) (LXVIII)
(LXX)
SO
OS
O
O
2. HCldioxane
1. MeMgBrTHF, 0°C H2N
BnO
HN
(LXIX)
SO
O
. . . . . Scheme 1.16
c) Asymmetric synthesis of (S)-Salsolidine:
Asymmetric synthesis of the isoquinoline alkaloid LXXIII, was
reported by Kasciolowicz [56] by the stereoselective addition of
MeMgBr to aldimine (LXXI) in THF to provide LXXII in 89% yield with
97:3 dr, and further crystallization increased to 99:1 dr. Subsequent

37
transformations yielded the desired product with 98% ee in 24%
overall yield. (Scheme – 1.17).
MeO
H
NS
O MeMgBr
(in Et2O)
MeO
NS
O
MeOOMe OMe
CH2Cl2, -48°C to rt
or THF, -27°C
NH
OMe
(LXXI) (LXXII) (LXXIII)
. . . . . Scheme 1.17
d) Asymmetric synthesis of (S)-(-)-O-Methylbharatamine:
LXXVIII is an isoquinoline alkaloid, synthesized by Grajewska
[57]. The addition of laterally lithiated o-toluamide (LXXIV) to N-tert-
butanesulfinyl aryl imine (LXXV) in the presence of tBuLi in THF at -
72°C yielded (LXXVI) in 93% yield. Subsequent cleavage of sulfinyl
group followed by cyclisation provided (LXXVII) with 85% ee, which on
further transformations yielded the desired product. (Scheme – 1.18).

38
O
N + H
NS
O
OCH3
OCH3
tBuLi
THF, -72°CH3CO
OCH3
HNS
O
N
O
1. HCl/MeOHbasic workup
2. BuLi (1.0 eq)THF, -78°C
HN
O
OCH3
H3CO
N
OCH3
H3CO
(LXXIV) (LXXV) (LXXVI)
(LXXVII) (LXXVIII)
. . . . . Scheme 1.18
e) Scalable asymmetric synthesis of DPP-4 Inhibitor:
Dipeptidyl peptidase 4 (DPP-4) inhibitors are used for the
treatment of type 2 diabetes. Shieh and co-workers [58] at Novartis
Pharma developed an optimized, scalable process of chiral amine
(LXXXII) a novel and potent inhibitor of DPP-4 in clinical trials for the
treatment of type 2 diabetes. The process involves the addition of a
benzylic Grignard reagent (LXXIX) to N-tert-butanesulfinyl aldimine
(LXXX) to get chiral amine intermediate (LXXXI) in methylene
dichloride and cyclopentyl methyl ether (CPME) as a stabilizer at -70°C
with diastereoselectivity of 85:15. Pure diastereomer was isolated by
recrystallization of the crude product from methanol. Further
transformations yielded the desired DPP-4 inhibitor. (Scheme – 1.19).

39
BrMg
F
F
F+ N
H
N SO
BzC CPME (1.0 eq)
CH2Cl2, -70°C
N HN SO
BzC
F
F
F
N NH2
F
F
F
O
SN
O
O
O
(LXXIX) (LXXX)(LXXXI)
(LXXXII)
. . . . . Scheme 1.19