catalytically active imine-based covalent organic
TRANSCRIPT

materials
Article
Catalytically Active Imine-based Covalent OrganicFrameworks for Detoxification of Nerve AgentSimulants in Aqueous Media
Sergio Royuela 1,2 , Rodrigo Gil-San Millán 3 , María J. Mancheño 1 , M. Mar Ramos 2,José L. Segura 1,* , Jorge A. R. Navarro 3,* and Félix Zamora 4,5,6,7,*
1 Departamento de Química Orgánica I, Facultad de CC. Químicas, Universidad Complutense de Madrid,28040 Madrid, Spain; [email protected] (S.R.); [email protected] (M.J.M.)
2 Departamento de Tecnología Química y Ambiental, Universidad Rey Juan Carlos, 28933 Madrid, Spain;[email protected]
3 Departamento de Química Inorgánica, Universidad de Granada, 18071 Granada, Spain;[email protected]
4 Departamento de Inorgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain5 Institute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid,
28049 Madrid, Spain6 Condensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain7 Instituto Madrileño de Estudios Avanzados en Nanociencia (IMDEA-Nanociencia), Cantoblanco,
28049 Madrid, Spain* Correspondence: [email protected] (J.L.S.); [email protected] (J.A.R.N.); [email protected] (F.Z.)
Received: 24 May 2019; Accepted: 17 June 2019; Published: 19 June 2019�����������������
Abstract: A series of imine-based covalent organic frameworks decorated in their cavities withdifferent alkynyl, pyrrolidine, and N-methylpyrrolidine functional groups have been synthetized.These materials exhibit catalytic activity in aqueous media for the hydrolytic detoxification of nerveagents, as exemplified with nerve gas simulant diisopropylfluorophosphate (DIFP). These preliminaryresults suggest imine-based covalent organic frameworks (COFs) as promising materials fordetoxification of highly toxic molecules.
Keywords: covalent organic frameworks; catalysis; chemical warfare agents; detoxification
1. Introduction
Nerve agents are amongst the most toxic chemical compounds known to mankind as a consequenceof their easy penetration through human mucosa and ulterior damage of the central nervous system bythe inhibition of acetylcholinesterase (AChE) [1]. Some examples of nerve agents include sarin (GB) andVX (Figure 1a). Although declared illegal by international agreements [2], recent attacks with chemicalweapons against civil and military populations have been reported [3,4]. Consequently, protectionand decontamination of these toxic chemicals is a very important societal challenge. Their hydrolyticdegradation under environmental conditions (room temperature and ambient moisture) is one of themost convenient detoxification pathways, however, it will only take place in the presence of a suitablecatalyst. Ideally, such catalyst should be a porous solid combining adsorptive and catalytic properties.The high toxicity of nerve agents prevents their use in standard research laboratories, with less toxicsimulants (i.e., diisopropylfluorophosphate (DIFP), dimethylmethylphosphonate (DMMP)) (Figure 1b)being used in order to prove the suitability of a given material for decontamination purposes of thereal nerve agents.
In recent years, covalent organic frameworks (COFs) have emerged as a new family of polymericporous and crystalline materials based on the assembly of organic synthons by means of dynamic
Materials 2019, 12, 1974; doi:10.3390/ma12121974 www.mdpi.com/journal/materials

Materials 2019, 12, 1974 2 of 12
covalent bonds [5–11]. The modular nature of the COF architectures allows the fine tailoring of theirstructure and properties based on the selection of the molecular precursors [12]. Their intrinsic porosityis suited for applications related with gas adsorption and/or storage [5,7,9,10,13–17]. However,the incorporation of functional groups into their structures has envisioned different potentialapplications such as advanced materials for catalysis, solar energy collectors, optoelectronic devices,and clean energy applications [6,8,16,18–21]. Despite the fact that COFs show cavities that canbe designed for several catalytic processes, still the number of reported examples is limited [22]and restricted mainly to classical carbon–carbon coupling reactions in organic solvents, such asKnoevenagel condensation [23], Michael addition [24,25], or Diels–Alder reaction [26]. In this context,the use of “click chemistry” is a highly efficient tool to functionalize COFs and decorate their cavitieswith specific molecules [27]. Noteworthy, this strategy allows control over the composition and densityof functional groups being suitable for the incorporation of active catalytic sites [12].
Materials 2019, 12, x FOR PEER REVIEW 2 of 13
In recent years, covalent organic frameworks (COFs) have emerged as a new family of polymeric porous and crystalline materials based on the assembly of organic synthons by means of dynamic covalent bonds [5–11]. The modular nature of the COF architectures allows the fine tailoring of their structure and properties based on the selection of the molecular precursors [12]. Their intrinsic porosity is suited for applications related with gas adsorption and/or storage [5,7,9,10,13–17]. However, the incorporation of functional groups into their structures has envisioned different potential applications such as advanced materials for catalysis, solar energy collectors, optoelectronic devices, and clean energy applications [6,8,16,18–21]. Despite the fact that COFs show cavities that can be designed for several catalytic processes, still the number of reported examples is limited [22] and restricted mainly to classical carbon–carbon coupling reactions in organic solvents, such as Knoevenagel condensation [23], Michael addition [24,25], or Diels–Alder reaction [26]. In this context, the use of “click chemistry” is a highly efficient tool to functionalize COFs and decorate their cavities with specific molecules [27]. Noteworthy, this strategy allows control over the composition and density of functional groups being suitable for the incorporation of active catalytic sites [12].
Figure 1. Examples of (a) highly toxic organophosphonate nerve agents and (b) nerve agent models of lower toxicity, used in conventional laboratories. DIFP: diisopropylfluorophosphate. DMMP: dimethylmethylphosphonate. GB: Sarin.
By contrast, the field of catalysis in metal organic frameworks (MOFs) is currently much more developed as a consequence of the possibility of incorporating catalytic active sites both at the metal fragment and the organic linker [28–30]. In this regard, it has been demonstrated that the incorporation of nucleophilic sites in the structure of non-active MOFs gives rise to the hydrolytic degradation of ester bonds of DIFP nerve agent model in a 2-[tris(hydroxymethyl)-methylamino]-ethanesulfonic acid (TES)-buffered media [31]. Moreover, the incorporation of the naphthofluorescein pH sensor on a catalytically active MOF leads to the visual sensing of the easily hydrolysable diethylchlorophosphate [32].
In this work, we have considered that the porous framework structure and precise location of catalytically active sites in the porous structure of a COF can be useful for the introduction of nerve agent hydrolytic detoxification functions. Indeed, we demonstrate that an imine-based COF (Scheme 1) can be used for the hydrolytic detoxification of model nerve agent DIFP (Figure 1) in unbuffered aqueous media. Despite the relatively low hydrophilic behavior of COFs [33–35], DIFP hydrolysis takes place on the COF pore surface, representing one of the few cases of a catalytic reaction carried out in a COF cavity in water [25,36]. Moreover, we also report the functionalization of the pores with highly nucleophilic pyrrolidine residues, leading to a significant enhancement of the hydrolytic activity for the degradation of DIFP. We also show that the methylpyrrolidine moieties can be incorporated into the COF framework following two different postpolymerization functionalization strategies, leading to slightly different behavior (Scheme 1). In addition, we synthesized an
OP
O
O
F
DIFP
SP
Me
O
ON
VXGBSarin
PO
O
F
OP
O
O
DMMP
(a)
(b)
Figure 1. Examples of (a) highly toxic organophosphonate nerve agents and (b) nerve agent modelsof lower toxicity, used in conventional laboratories. DIFP: diisopropylfluorophosphate. DMMP:dimethylmethylphosphonate. GB: Sarin.
By contrast, the field of catalysis in metal organic frameworks (MOFs) is currently much moredeveloped as a consequence of the possibility of incorporating catalytic active sites both at the metalfragment and the organic linker [28–30]. In this regard, it has been demonstrated that the incorporationof nucleophilic sites in the structure of non-active MOFs gives rise to the hydrolytic degradation ofester bonds of DIFP nerve agent model in a 2-[tris(hydroxymethyl)-methylamino]-ethanesulfonicacid (TES)-buffered media [31]. Moreover, the incorporation of the naphthofluorescein pHsensor on a catalytically active MOF leads to the visual sensing of the easily hydrolysablediethylchlorophosphate [32].
In this work, we have considered that the porous framework structure and precise location ofcatalytically active sites in the porous structure of a COF can be useful for the introduction of nerveagent hydrolytic detoxification functions. Indeed, we demonstrate that an imine-based COF (Scheme 1)can be used for the hydrolytic detoxification of model nerve agent DIFP (Figure 1) in unbufferedaqueous media. Despite the relatively low hydrophilic behavior of COFs [33–35], DIFP hydrolysistakes place on the COF pore surface, representing one of the few cases of a catalytic reaction carriedout in a COF cavity in water [25,36]. Moreover, we also report the functionalization of the pores withhighly nucleophilic pyrrolidine residues, leading to a significant enhancement of the hydrolytic activityfor the degradation of DIFP. We also show that the methylpyrrolidine moieties can be incorporated intothe COF framework following two different postpolymerization functionalization strategies, leadingto slightly different behavior (Scheme 1). In addition, we synthesized an analogous polymeric materialwith similar functionalities as in the COF, but with an amorphous nature. The comparison between the

Materials 2019, 12, 1974 3 of 12
catalytic activity of the amorphous polymer and the covalent organic frameworks gives us a hint of theimportance of pore accessibility and appropriate confinement effect of the substrate in the COF cavities.Materials 2019, 12, x FOR PEER REVIEW 5 of 13
Scheme 1. Synthesis of [(S)-Py]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-COF via one-step or two-step postsynthetic modification.
Characterization of the COFs was carried out by Fourier transform infrared (FTIR) (Figures S1–S5), 13C cross-polarization magic-angle spinning NMR (13C CP/MAS NMR) spectroscopies (Figures S6–S9), and powder X-ray diffraction (PXRD) (Figure S10, Table S1). FTIR and 13C CP/MAS NMR of [HC≡C]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-COF show a similar profile (Figure 2a), which confirms that the structure of the COF skeleton was stable under the postsynthetic reaction conditions. By comparison of the aliphatic region, it was distinguished the incorporation of the methyl group by the deshielding of the signals corresponding to the -CH- and -CH2- next to the pyrrolidine nitrogen, which were shifted by 15 ppm, and the appearance of a new signal at 42 ppm corresponding to the N-CH3. PXRD of all COFs exhibited a similar diffraction pattern, corresponding to the AA stacking mode of a space group P6, corroborating their basic structural framework (Figure 2b, Table S1). PXRD of [(S)-PyMe]0.5-TPB-DMTP-COF also presented peaks related to those of the precursor COFs, indicating that the same lattice was maintained after functionalization.
The effect of the functional pendant groups on the material porosity was evaluated by N2 adsorption at 77 K (Figures 2c and S11). The Brunauer–Emmett–Teller (BET) surface area, pore volume, and pore size distribution for [HC≡C]0.5-TPB-DMTP-COF and [(S)-Py]0.5-TPB-DMTP-COF were in good agreement with the previously reported data (Figure S12, Table S2). In the case of [(S)-PyMe]0.5-TPB-DMTP-COF, the nitrogen sorption data were indicative that methylation is responsible for a significant decrease of pore accessibility to the N2 probe molecule (BET 95 m2 g−1 and pore volume of 0.108 cm3 g−1), although some degree of mesoporosity (NLDFT pore size of 2.9 nm) was maintained (Figure S13). In order to address this decrease of pore accessibility in the COF after the methylation reaction, we have carried out an alternative synthesis of [(S)-PyMe]0.5-TPB-DMTP-COF by using a one-step room-temperature post-synthetic functionalization of [HC≡C]0.5-TPB-DMTP-COF. With this aim, we have synthesized (S)-2-(azidomethyl)-1-methylpyrrolidine (2, Scheme 1) [39], in which the pyrrolidine moiety was already methylated. Subsequent copper-catalyzed Huisgen’s 1,3-dipolar cycloaddition reaction of 2
Scheme 1. Synthesis of [(S)-Py]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-COF via one-stepor two-step postsynthetic modification.
2. Materials and Methods
2.1. Characterization
All reactions with air sensitive materials were carried out under Ar using standardSchlenk techniques. Thin layer chromatography (TLC) was performed using pre-coated silicagel 60 F254 and developed in the indicated solvent system. Compounds were visualizedunder UV light (λ = 254 nm). Merck 60 (230–400 Mesh) silica gel was used for columnchromatography. The following reagents were commercially available and were used as received:2,5-dimethoxyterephthaldehyde (DMTA), CuI, o-DCB, n-butanol, NaN3 and N,N-diisopropylethylamine(DIPEA). 2,5-dihydroxyterephthaldehyde (DHTA) [25], 2,5-bis(prop-2-in-1-yloxy)terephthaldehyde(BPTA) [25], 1,3,5-tris-(4-aminophenyl)benzene (TAPB) [37], [HC≡C]0.5-TPB-DMTP-COF (TPB,triphenylbenzene; DMTP, dimethoxyterephthaldehyde) [25], [(S)-Py]0.5-TPB-DMTP-COF [25],(S)-2-(azidomethyl)pyrrolidine (1) [38], and (S)-2-(azidomethyl)-1-methylpyrrolidine (2) [39] wereprepared according to reported procedures.
1H NMR and 13C NMR spectra were recorded on a Bruker AVIII-300 MHz (Billerica, MA, USA)spectrometer. Chemical shifts were reported in ppm and referenced to the residual non-deuteratedsolvent frequencies (CDCl3: δ 7.26 ppm for 1H, 77.0 ppm for 13C). Mass spectra were recorded bymeans of matrix-assisted laser desorption/ionization with a time-of-flight (MALDI-TOF) or fast atombombardement (FAB) ionization techniques. Solids were analyzed by Fourier transform infrared(FTIR) spectroscopy on a Bruker TENSOR 27 (Billerica, MA, USA) on a diamond plate (attenuatedtotal reflectance, ATR) or as films on sodium chloride. Powder X-ray diffraction (PXRD) measurementswere carried out with X’PERT MPD (Malvern, UK), with conventional Bragg-Brentano geometry using

Materials 2019, 12, 1974 4 of 12
Cu Kα (λ = 1.5406 Å) for values of 2θ from 2◦ to 40◦. 13C CP/MAS NMR spectra were recorded ona Bruker AV-400 MHz Wide Bore (Billerica, MA, USA) spectrometer (probe: 4 mm MAS WB DVT).The sample rotation frequency was 12 kHz, and a 2.5 mm ZrO2 rotor was used. The N2 (77 K) andCO2 (273 K) adsorption isotherms were carried out on a Micromeritics Triflex equipment (Triflex,Micromeritics, Norcross, GA, USA). Prior to measurement, the materials were thermally activated at393 K under dynamic vacuum (10−5 Pa). Thermogravimetric analysis was performed on a TGA-Q50(TA Instruments, New Castle, DE, USA) instrument on a platinum plate, heating the samples undernitrogen atmosphere at a heating rate of 10 ◦C/min. Scanning electron microscopy (SEM) was carriedout using a JOEL JSM 6335F (Tokyo, Japan) scanning electron microscope. The sample was dispersedover a slice of conductive adhesive (graphite) adhered to a flat copper platform sample holder andthen coated with gold using a sputter coater before being submitted to SEM characterization.
2.2. Synthesis of COFs
[HC≡C]0.5-TPB-DMTP-COF [25]. Following the procedure previously described, a Pyrex vessel(Ø = 29 mm, h = 10 cm) was charged with DMTA (215 mg, 1.10 mmol), BPTA (268 mg, 1.11 mmol),TAPB (520 mg, 1.48 mmol), and o-DCB/n-Butanol/6M acetic acid (10 mL/10 mL/2.1 mL), degassed viathree freeze-pump-thaw cycles, flame sealed, and kept at 130 ◦C for seven days. The precipitate wasfiltered, Soxhlet extracted with tetrahydrofuran (THF), and dried at 120 ◦C under vacuum, yielding866.0 mg (94%) of a yellow powder. FTIR (ATR, cm−1): 3288, 2957, 2127, 1689, 1592, 1504, 1415, 1291,1208, 1146, 1036, 878, 829, 694.
[(S)-Py]0.5-TPB-DMTP-COF [25]. To 157.1 mg of [HC≡C]0.5-TPB-DMTP-COF in 7 mL of a 3:1THF/H2O mixture were added 43.9 mg (0.230 mmol) of CuI and 138 µL of N,N-diisopropylethylamine(DIPEA). The suspension was purged with argon for 5 min, and then 64.0 mg (0.507 mmol) of(S)-2-(azidomethyl)pyrrolidine (1) was added. The mixture was stirred overnight at room temperatureunder argon and centrifuged at 6000 rpm for 5 min. After washing with acetonitrile, H2O andTHF, the solid was dried under vacuum at 120 ◦C, yielding an orange powder (215 mg, 97%).FTIR (ATR, cm−1): 2944, 1675, 1590, 1506, 1415, 1289, 1208, 1146, 1043, 830, 695.
2-step-[(S)-PyMe]0.5-TPB-DMTP-COF. 100 mg of [(S)-Py]0.5-TPB-DMTP-COF was suspended inanhydrous THF under argon together with 158 µL of DIPEA. Methyl triflate (47 µL) was subsequentlyadded, and the mixture was stirred overnight at 70 ◦C. The solid was filtered, washed with 0.5 MNaOH, H2O, and THF, and dried under vacuum at 120 ◦C, yielding 109.0 mg of an orange solid.FTIR (ATR, cm−1): 2929, 2857, 1677, 1591, 1506, 1462, 1413, 1289, 1208, 1146, 1039, 830, 696.
1-step-[(S)-Py]0.5-TPB-DMTP-COF. To 107.3 mg of [HC≡C]0.5-TPB-DMTP-COF in 4.8 mL of a 3:1THF/H2O mixture were added 30.7 mg (0.161 mmol) of CuI and 97 µL of DIPEA. The suspension waspurged with argon for 5 min, and then 50.1 mg (0.357 mmol) of (S)-2-(azidomethyl)-1-methylpyrrolidine(2) was added. The mixture was stirred overnight at room temperature under argon and centrifuged at6000 rpm for 5 min. After washing with acetonitrile, H2O and THF, the solid was dried under vacuumat 120 ◦C, yielding an orange powder (142 mg, 90%). FTIR (ATR, cm-1): 2941, 2800, 1670, 1591, 1507,1415, 1291, 1208, 1146, 1041, 830, 696.
[HC≡C]0.5-TPB-DMTP-Polym. DMTA (29.3 mg, 0.151 mmol), BPTA (36.4 mg, 0.150 mmol),and TAPB (70.1 mg, 0.200 mmol) were dissolved in 15 mL of acetone. A few minutes after the additionof 1.5 mL of 6 M acetic acid, a yellow solid appeared. The suspension was stirred for an additional2 h, filtered, and thoroughly washed with methanol and THF. The resulting solid was dried at 120 ◦Cunder vacuum to afford 86.5 mg (70%) of a yellow powder. FTIR (ATR, cm−1): 2962, 1678, 1605, 1512,1411, 1283, 1205, 1187, 1033, 879, 825, 687.
[(S)-PyMe]0.5-TPB-DMTP-Polym. To a suspension of 75 mg of [HC≡C]0.5-TPB-DMTP-Polymin 3.5 mL of a 3:1 THF/H2O mixture were added 21.3 mg (0.112 mmol) of CuI and 69 µLof DIPEA. After purging the suspension with argon for 5 min, 30.1 mg (0.215 mmol) of(S)-2-(azidomethyl)-1-methylpyrrolidine (2) was added, and the mixture was stirred overnight atroom temperature under argon. After centrifugation at 6000 rpm for 5 min, the solid was collected

Materials 2019, 12, 1974 5 of 12
by filtration, washed with acetonitrile, H2O and THF, and dried under vacuum at 120 ◦C, yieldingan orange powder (82.0 mg, 78%). FTIR (ATR, cm−1): 1679, 1609, 1512, 1412, 1286, 1207, 1186, 1036,827, 698.
2.3. DIFP Heterogeneous Catalytic Degradation Tests
The degradation of DIFP was studied by employing 0.014 mmol of the repeating block structureof each COF suspended in 0.5 mL of bi-distilled H2O (unbuffered experiments) or 0.5 mL of 0.45 MN-ethylmorpholine buffered aqueous solution. Afterwards, 2.5 µL of DMSO (used as internal reference)and 2.5 µL (0.014 mmol) of DIFP were added to the suspension. The evolution of the concentrationof DIFP was followed at room temperature by taking 0.2 µL aliquots of the supernatant solution,which were analyzed by gas chromatography (450 GC, Varian, Palo Alto, CA, USA).
3. Results and Discussion
The syntheses of the COFs endowed with pyrrolidine moieties start with the synthesis ofan alkynyl-functionalized COF ([HC≡C]0.5-TPB-DMTP-COF, Scheme 1) by following a procedure previouslydescribed [25,40]. The subsequent postsynthetic functionalization of [HC≡C]0.5-TPB-DMTP-COF wascarried out in a two-step sequence. The first step involved a copper-catalyzed Huisgen’s 1,3-dipolarcycloaddition reaction [41,42] of the alkynyl-functionalized COF with a free-pyrrolidine- functionalizedazide (1, Scheme 1) to afford a COF endowed with free pyrrolidine moieties. The second postsyntheticstep involved the treatment of the free-pyrrolidine-functionalized COF with methyl triflate at 70 ◦Cusing THF as solvent to afford the corresponding COF endowed with methylated pyrrolidine moieties([(S)-PyMe]0.5-TPB-DMTP-COF) with good yields (Scheme 1).
Characterization of the COFs was carried out by Fourier transform infrared (FTIR) (Figures S1–S5),13C cross-polarization magic-angle spinning NMR (13C CP/MAS NMR) spectroscopies (Figures S6–S9),and powder X-ray diffraction (PXRD) (Figure S10 and Table S1). FTIR and 13C CP/MAS NMR of[HC≡C]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-COF show a similar profile (Figure 2a),which confirms that the structure of the COF skeleton was stable under the postsynthetic reactionconditions. By comparison of the aliphatic region, it was distinguished the incorporation of the methylgroup by the deshielding of the signals corresponding to the -CH- and -CH2- next to the pyrrolidinenitrogen, which were shifted by 15 ppm, and the appearance of a new signal at 42 ppm corresponding tothe N-CH3. PXRD of all COFs exhibited a similar diffraction pattern, corresponding to the AA stackingmode of a space group P6, corroborating their basic structural framework (Figure 2b and Table S1).PXRD of [(S)-PyMe]0.5-TPB-DMTP-COF also presented peaks related to those of the precursor COFs,indicating that the same lattice was maintained after functionalization.
The effect of the functional pendant groups on the material porosity was evaluated by N2
adsorption at 77 K (Figures 2c and S11). The Brunauer–Emmett–Teller (BET) surface area, pore volume,and pore size distribution for [HC≡C]0.5-TPB-DMTP-COF and [(S)-Py]0.5-TPB-DMTP-COF werein good agreement with the previously reported data (Figure S12 and Table S2). In the caseof [(S)-PyMe]0.5-TPB-DMTP-COF, the nitrogen sorption data were indicative that methylation isresponsible for a significant decrease of pore accessibility to the N2 probe molecule (BET 95 m2 g−1 andpore volume of 0.108 cm3 g−1), although some degree of mesoporosity (NLDFT pore size of 2.9 nm)was maintained (Figure S13). In order to address this decrease of pore accessibility in the COF after themethylation reaction, we have carried out an alternative synthesis of [(S)-PyMe]0.5-TPB-DMTP-COF byusing a one-step room-temperature post-synthetic functionalization of [HC≡C]0.5-TPB-DMTP-COF.With this aim, we have synthesized (S)-2-(azidomethyl)-1-methylpyrrolidine (2, Scheme 1) [39],in which the pyrrolidine moiety was already methylated. Subsequent copper-catalyzedHuisgen’s 1,3-dipolar cycloaddition reaction of 2 with [HC≡C]0.5-TPB-DMTP-COF afforded[(S)-PyMe]0.5-TPB-DMTP-COF. From now on, we will name this COF obtained with a one-steppostsynthetic strategy 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF, while the analogue obtained via thetwo-steps post-synthetic strategy will be named 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF.

Materials 2019, 12, 1974 6 of 12Materials 2019, 12, x FOR PEER REVIEW 7 of 13
Figure 2. Comparative of (a) 13C CP/MAS NMR spectra, (b) PXRD patterns, and (c) N2 adsorption isotherms (77 K) for [HC≡C]0.5-TPB-DMTP-COF (black line), [(S)-Py]0.5-TPB-DMTP-COF (red line), and 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue line). Open symbols correspond to the desorption branch.
Once we fully characterized the synthetized materials, we proceeded to prove the possible utility of [HC≡C]0.5-TPB-DMTP-COF, [(S)-Py]0.5-TPB-DMTP-COF, 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF, 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, and [(S)-PyMe]0.5-TPB-DMTP-Polym systems as heterogeneous catalysts of harmful molecules. With
Figure 2. Comparative of (a) 13C CP/MAS NMR spectra, (b) PXRD patterns, and (c) N2
adsorption isotherms (77 K) for [HC≡C]0.5-TPB-DMTP-COF (black line), [(S)-Py]0.5-TPB-DMTP-COF(red line), and 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue line). Open symbols correspond to thedesorption branch.

Materials 2019, 12, 1974 7 of 12
Fourier transform infrared (FTIR), 13C cross-polarization magic-angle spinning NMR(13C CP/MAS NMR) (Figure 2a) spectroscopies, and powder X-ray diffraction (PXRD)(Figure 2b) analysis showed identical patterns for 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF and2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, which indicate that the networks remained identical after thepostsynthetic steps. Nevertheless, a threefold increase in the BET surface area of 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (317 m2 g−1) was observed in comparison with that determined with the parent2-step-[(S)-PyMe]0.5-TPB-DMTP-COF (95 m2 g−1). This difference in BET surfaces might beattributed to a partial further methylation of methylpyrrolidine residues to the correspondingdimethylpyrrolidonium triflate during the two-step synthetic process, which limits pore accessibility.
In order to have a further insight about the importance of pore accessibility in thesematerials, we have also addressed the synthesis of an amorphous polymer analogue to[(S)-PyMe]0.5-TPB-DMTP-COFs. Thus, an amorphous alkynyl-functionalized polymer was obtainedby using the same monomers as those used for the synthesis of [HC≡C]0.5- TPB-DMTP-COF,but without the use of solvothermal conditions. The further reaction of this alkynyl-functionalizedamorphous polymer with azide 2 afforded an amorphous nonporous polymer endowed withmethylpyrrolidine moieties, [(S)-PyMe]0.5-TPB-DMTP-Polym. Characterization of this materialby FTIR and 13C CP/MAS NMR presented no significant differences when compared with the analogueCOFs, but it showed no diffraction peaks and its BET surface area was only 7.5 m2 g−1 (see ESI).
Finally, COFs were characterized by scanning electron microscopy (SEM), revealing that themorphology is conserved after the postsynthetic modification (Figures S14–S16). Thermal stability ofall the frameworks was determined by thermogravimetric analysis (TGA). All the COFs were stable attemperatures above 250 ◦C, with the alkynyl-functionalized material ([HC≡C]0.5-TPB-DMTP-COF)being stable up to 400 ◦C, (Figures S17–S21).
Once we fully characterized the synthetized materials, we proceeded to prove the possible utilityof [HC≡C]0.5-TPB-DMTP-COF, [(S)-Py]0.5-TPB-DMTP-COF, 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF,2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, and [(S)-PyMe]0.5-TPB-DMTP-Polym systems as heterogeneouscatalysts of harmful molecules. With this aim, we have essayed the interaction of these systems with thenerve agent simulant diisopropylfluorophosphate (DIFP). The DIFP hydrolysis tests were performedin both unbuffered aqueous media and N-ethylmorpholine-buffered media (pH = 9.2) (Figure 3 andTable S3), according to previous reports of our group [43,44].
Noteworthy, [HC≡C]0.5-TPB-DMTP-COF exhibited a rather fast initial DIFP degradation rate(t1/2 < 2 min) in unbuffered aqueous media, which was accompanied with a concomitant color changefrom light orange to dark red (Figures 3 and S22). This result can be attributed to the protonation ofthe imine residues as a consequence of the solution acidification during DIFP hydrolysis (pH changefrom initial 8.3 to 2.7). The system color change attributable to solution acidification was confirmedwith the addition of different acids (i.e., acetic acid). In order to discard the possibility of this colorchange being due to partial decomposition of the COF, we stirred a suspension of 50 mg of COFin 25 mL of a sodium acetate buffer solution (pH 4.21) at room temperature for 24 h. The materialexhibited a fast color change from yellow to red in this buffered medium. This color change was fullyreversible after simple washing with methanol. The FITR and PXRD of the material isolated exhibitedan identical profile without significant weight loss, indicating that the lattice remained unalteredafter this treatment. Similar results have been previously observed for [HC≡C]0.5-TPB-DMTP-COFunder harsher conditions (concentrated, 12 M HCl) [25]. The hydrolytic activity and color change areattributed to the Lewis basic nature of the imino residues constituting the framework. In this regard,an ethynyl synthon was observed to be nonactive in the DIFP degradation, suggesting that the catalyticactivity of the COF materials is due to the nucleophilic nature of the imine groups.

Materials 2019, 12, 1974 8 of 12
Materials 2019, 12, x FOR PEER REVIEW 8 of 13
this aim, we have essayed the interaction of these systems with the nerve agent simulant diisopropylfluorophosphate (DIFP). The DIFP hydrolysis tests were performed in both unbuffered aqueous media and N-ethylmorpholine-buffered media (pH = 9.2) (Figure 3, Table S3), according to previous reports of our group [43,44].
Noteworthy, [HC≡C]0.5-TPB-DMTP-COF exhibited a rather fast initial DIFP degradation rate (t1/2 < 2 min) in unbuffered aqueous media, which was accompanied with a concomitant color change from light orange to dark red (Figures 3 and S22). This result can be attributed to the protonation of the imine residues as a consequence of the solution acidification during DIFP hydrolysis (pH change from initial 8.3 to 2.7). The system color change attributable to solution acidification was confirmed with the addition of different acids (i.e., acetic acid). In order to discard the possibility of this color change being due to partial decomposition of the COF, we stirred a suspension of 50 mg of COF in 25 mL of a sodium acetate buffer solution (pH 4.21) at room temperature for 24 h. The material exhibited a fast color change from yellow to red in this buffered medium. This color change was fully reversible after simple washing with methanol. The FITR and PXRD of the material isolated exhibited an identical profile without significant weight loss, indicating that the lattice remained unaltered after this treatment. Similar results have been previously observed for [HC≡C]0.5-TPB-DMTP-COF under harsher conditions (concentrated, 12 M HCl) [25]. The hydrolytic activity and color change are attributed to the Lewis basic nature of the imino residues constituting the framework. In this regard, an ethynyl synthon was observed to be nonactive in the DIFP degradation, suggesting that the catalytic activity of the COF materials is due to the nucleophilic nature of the imine groups.
Figure 3. (a) Diisopropylfluorophosphate (DIFP) hydrolytic degradation reaction; (b) Reaction profiles for [HC≡C]0.5-TPB-DMTP-COF (black), [(S)-Py]0.5-TPB-DMTP-COF (red), and 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue) in unbuffered aqueous media. The inset in b shows the change of color exhibited by [HC≡C]0.5-TPB-DMTP-COF concomitant to solution acidification after DIFP degradation; (c) Reaction profiles for 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue), 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF (purple), and amorphous [(S)-PyMe]0.5-TPB-DMTP-Polym (green) in unbuffered aqueous media.
However, despite the very fast degradation rate, the reaction was not completed because of the rapid poisoning of the catalyst, as a probable consequence of the protonation of the framework imine residues by DIFP acidic degradation products (HF, H3PO4). This limitation was circumvented with pyrrolidine and N-methylpyrrolidine functionalized COFs, namely [(S)-Py]0.5-TPB-DMTP-COF, 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF, and 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, in which pyrrolidine nitrogen atoms acted as basic groups leading to the completion of DIFP degradation. As expected, the tertiary amine-doped COFs exhibited higher reaction rates than the secondary amine-doped COF because of their increased
Figure 3. (a) Diisopropylfluorophosphate (DIFP) hydrolytic degradation reaction; (b) Reaction profilesfor [HC≡C]0.5-TPB-DMTP-COF (black), [(S)-Py]0.5-TPB-DMTP-COF (red), and 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue) in unbuffered aqueous media. The inset in b shows the changeof color exhibited by [HC≡C]0.5-TPB-DMTP-COF concomitant to solution acidification afterDIFP degradation; (c) Reaction profiles for 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue),2-step-[(S)-PyMe]0.5-TPB-DMTP-COF (purple), and amorphous [(S)-PyMe]0.5-TPB-DMTP-Polym(green) in unbuffered aqueous media.
However, despite the very fast degradation rate, the reaction was not completed because ofthe rapid poisoning of the catalyst, as a probable consequence of the protonation of the frameworkimine residues by DIFP acidic degradation products (HF, H3PO4). This limitation was circumventedwith pyrrolidine and N-methylpyrrolidine functionalized COFs, namely [(S)-Py]0.5-TPB-DMTP-COF,1-step-[(S)-PyMe]0.5-TPB-DMTP-COF, and 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, in which pyrrolidinenitrogen atoms acted as basic groups leading to the completion of DIFP degradation. As expected,the tertiary amine-doped COFs exhibited higher reaction rates than the secondary amine-dopedCOF because of their increased basicity. Indeed, the final pH after hydrolysis was 4.8 forthe secondary amine, and above 5 for the COFs based on the tertiary amine. With regard tothe amorphous [(S)-PyMe]0.5-TPB-DMTP-Polym, the final pH after a similar treatment was 4.2.Noteworthy, the catalytic activity of the crystalline porous 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF washigher than the related 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-Polymexhibiting lower pore accessibilities. It can be observed (Figure 3c) that the reaction wasfaster and reached completion at shorter times for the most nucleophilic and accessible1-step-[(S)-PyMe]0.5-TPB-DMTP-COF material. This behavior can be explained in terms of theconfinement of the reactants in the cavity of the crystalline porous material, given that the chemicalfunctionalities are similar in both materials.
It is also worth pointing out that the catalytic process was heterogeneous since the filtration of thecatalyst led to the halting of the degradation reaction, which is indicative of the heterogeneity of theDIFP degradation process (Figure 4).
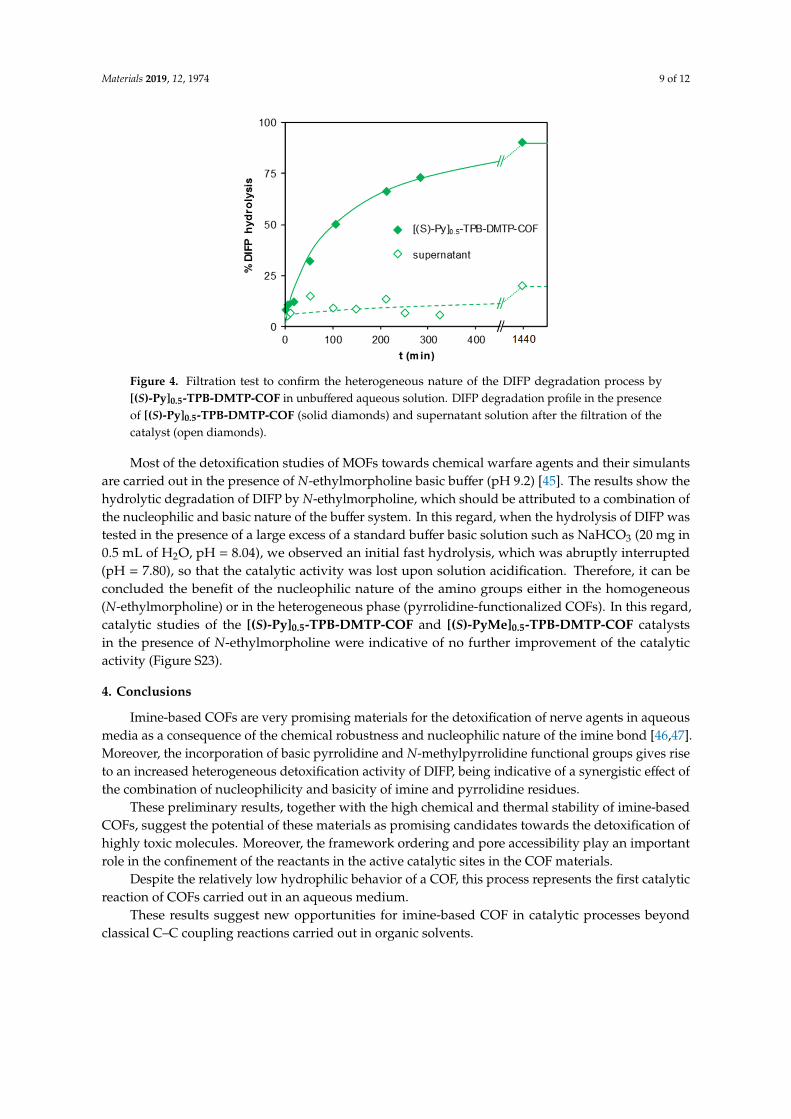
Materials 2019, 12, 1974 9 of 12
Materials 2019, 12, x FOR PEER REVIEW 9 of 13
basicity. Indeed, the final pH after hydrolysis was 4.8 for the secondary amine, and above 5 for the COFs based on the tertiary amine. With regard to the amorphous [(S)-PyMe]0.5-TPB-DMTP-Polym, the final pH after a similar treatment was 4.2. Noteworthy, the catalytic activity of the crystalline porous 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF was higher than the related 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-Polym exhibiting lower pore accessibilities. It can be observed (Figure 3c) that the reaction was faster and reached completion at shorter times for the most nucleophilic and accessible 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF material. This behavior can be explained in terms of the confinement of the reactants in the cavity of the crystalline porous material, given that the chemical functionalities are similar in both materials.
It is also worth pointing out that the catalytic process was heterogeneous since the filtration of the catalyst led to the halting of the degradation reaction, which is indicative of the heterogeneity of the DIFP degradation process (Figure 4).
Figure 4. Filtration test to confirm the heterogeneous nature of the DIFP degradation process by [(S)-Py]0.5-TPB-DMTP-COF in unbuffered aqueous solution. DIFP degradation profile in the presence of [(S)-Py]0.5-TPB-DMTP-COF (solid diamonds) and supernatant solution after the filtration of the catalyst (open diamonds).
Most of the detoxification studies of MOFs towards chemical warfare agents and their simulants are carried out in the presence of N-ethylmorpholine basic buffer (pH 9.2) [45]. The results show the hydrolytic degradation of DIFP by N-ethylmorpholine, which should be attributed to a combination of the nucleophilic and basic nature of the buffer system. In this regard, when the hydrolysis of DIFP was tested in the presence of a large excess of a standard buffer basic solution such as NaHCO3 (20 mg in 0.5 mL of H2O, pH= 8.04), we observed an initial fast hydrolysis, which was abruptly interrupted (pH= 7.80), so that the catalytic activity was lost upon solution acidification. Therefore, it can be concluded the benefit of the nucleophilic nature of the amino groups either in the homogeneous (N-ethylmorpholine) or in the heterogeneous phase (pyrrolidine-functionalized COFs). In this regard, catalytic studies of the [(S)-Py]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-COF catalysts in the presence of N-ethylmorpholine were indicative of no further improvement of the catalytic activity (Figure S23).
4. Conclusions
Imine-based COFs are very promising materials for the detoxification of nerve agents in aqueous media as a consequence of the chemical robustness and nucleophilic nature of the imine bond [46,47]. Moreover, the incorporation of basic pyrrolidine and N-methylpyrrolidine functional groups gives rise to an increased heterogeneous detoxification activity of DIFP, being indicative of a
Figure 4. Filtration test to confirm the heterogeneous nature of the DIFP degradation process by[(S)-Py]0.5-TPB-DMTP-COF in unbuffered aqueous solution. DIFP degradation profile in the presenceof [(S)-Py]0.5-TPB-DMTP-COF (solid diamonds) and supernatant solution after the filtration of thecatalyst (open diamonds).
Most of the detoxification studies of MOFs towards chemical warfare agents and their simulantsare carried out in the presence of N-ethylmorpholine basic buffer (pH 9.2) [45]. The results show thehydrolytic degradation of DIFP by N-ethylmorpholine, which should be attributed to a combination ofthe nucleophilic and basic nature of the buffer system. In this regard, when the hydrolysis of DIFP wastested in the presence of a large excess of a standard buffer basic solution such as NaHCO3 (20 mg in0.5 mL of H2O, pH = 8.04), we observed an initial fast hydrolysis, which was abruptly interrupted(pH = 7.80), so that the catalytic activity was lost upon solution acidification. Therefore, it can beconcluded the benefit of the nucleophilic nature of the amino groups either in the homogeneous(N-ethylmorpholine) or in the heterogeneous phase (pyrrolidine-functionalized COFs). In this regard,catalytic studies of the [(S)-Py]0.5-TPB-DMTP-COF and [(S)-PyMe]0.5-TPB-DMTP-COF catalystsin the presence of N-ethylmorpholine were indicative of no further improvement of the catalyticactivity (Figure S23).
4. Conclusions
Imine-based COFs are very promising materials for the detoxification of nerve agents in aqueousmedia as a consequence of the chemical robustness and nucleophilic nature of the imine bond [46,47].Moreover, the incorporation of basic pyrrolidine and N-methylpyrrolidine functional groups gives riseto an increased heterogeneous detoxification activity of DIFP, being indicative of a synergistic effect ofthe combination of nucleophilicity and basicity of imine and pyrrolidine residues.
These preliminary results, together with the high chemical and thermal stability of imine-basedCOFs, suggest the potential of these materials as promising candidates towards the detoxification ofhighly toxic molecules. Moreover, the framework ordering and pore accessibility play an importantrole in the confinement of the reactants in the active catalytic sites in the COF materials.
Despite the relatively low hydrophilic behavior of a COF, this process represents the first catalyticreaction of COFs carried out in an aqueous medium.
These results suggest new opportunities for imine-based COF in catalytic processes beyondclassical C–C coupling reactions carried out in organic solvents.

Materials 2019, 12, 1974 10 of 12
Supplementary Materials: The following are available online at http://www.mdpi.com/1996-1944/12/12/1974/s1, Figure S1: FTIR (ATR) spectrum of [HC≡C]0.5-TPB-DMTP-COF, Figure S2: FTIR (ATR) spectrumof [(S)-Py]0.5-TPB-DMTP-COF, Figure S3: FTIR (ATR) spectrum of 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF,Figure S4: FTIR (ATR) spectrum of 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, Figure S5: FTIR (ATR)spectrum of [(S)-PyMe]0.5-TPB-DMTP-Polym, Figure S6: 13C CP/MAS NMR of [HC≡C]0.5-TPB-DMTP-COF,Figure S7: 13C CP/MAS NMR of [(S)-Py]0.5-TPB-DMTP-COF, Figure S8: 13C CP/MAS NMR of2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, Figure S9: 13C CP/MAS NMR of 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF,Figure S10: PXRD of 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue), 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF(purple) and [(S)-PyMe]0.5-TPB-DMTP-Polym (green), Figure S11: N2 (77 K) adsorption isothermsfor 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue), 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF (purple) and[(S)-PyMe]0.5-TPB-DMTP-Polym (green), Figure S12: NLDFT pore size distribution for the essayed COF materials[HC≡C]0.5-TPB-DMTP-COF (a), [(S)-Py]0.5-TPB-DMTP-COF (b), 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF (c)and 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (d), Figure S13: CO2 (298 K) adsorption isotherms for[HC≡C]0.5-TPB-DMTP-COF (black), [(S)-Py]0.5-TPB-DMTP-COF (red) and [(S)-PyMe]0.5-TPB-DMTP-COF(blue), Figure S14: SEM image of [(S)-Py]0.5-TPB-DMTP-COF, Figure S15: SEM image of2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, Figure S16: SEM image of 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF, FigureS17: TGA profile of [HC≡C]0.5-TPB-DMTP-COF, Figure S18: TGA profile of [(S)-Py]0.5-TPB-DMTP-COF,Figure S19: TGA profile of 2-step-[(S)-PyMe]0.5-TPB-DMTP-COF, Figure S20: TGA profile of1-step-[(S)-PyMe]0.5-TPB-DMTP-COF, Figure S21: TGA profile of [(S)-PyMe]0.5-TPB-DMTP-Polym, FigureS22: Effect of DIFP of addition to the essayed COF materials suspension on the pH and color in both buffer andunbuffered conditions, Figure S23: DIFP degradation studies in basic N-ethylmorpholine buffered media (pH = 9.2).Reaction profiles for 1-step-[(S)-PyMe]0.5-TPB-DMTP-COF (blue) and [(S)-PyMe]0.5-TPB-DMTP-Polym (green),Table S1: Lattice parameters of the synthesized COFs, Table S2: Summary of textural properties of COF materialsfrom N2 adsorption measurements at 77 K, Table S3: Catalytic degradation of diisopropylfluorophosphate (DIFP)nerve agent simulant.
Author Contributions: Conceptualization, J.L.S., J.A.R.N. and F.Z.; methodology, M.J.M.; investigation, S.R. andR.G.-S.M.; supervision, M.M.R.; writing—original draft preparation, S.R., J.L.S. and J.A.R.N.; writing—review andediting, S.R., J.L.S., J.A.R.N. and F.Z.
Funding: This work was financially supported by MINECO (MAT2016-77608-C3-1-P and 2-P, CTQ2017-84692-R)and EU FEDER funding.
Conflicts of Interest: The authors declare no conflict of interest.
References
1. Worek, F.; Thiermann, H.; Szinicz, L.; Eyer, P. Kinetic analysis of interactions between humanacetylcholinesterase, structurally different organophosphorus compounds and oximes. Biochem. Pharmacol.2004, 68, 2237–2248. [CrossRef] [PubMed]
2. Organisation for the Prohibition of Chemical Weapons Website. Available online: https://www.opcw.org/
(accessed on 27 June 2017).3. Okumura, T.; Takasu, N.; Ishimatsu, S.; Miyanoki, S.; Mitsuhashi, A.; Kumada, K.; Tanaka, K.; Hinohara, S.
Report on 640 Victims of the Tokyo Subway Sarin Attack. Ann. Emerg. Med. 1996, 28, 129–135. [CrossRef]4. United Nations Mission to Investigate Allegations of the Use of Chemical Weapons in the Syrian Arab Republic;
United Nations: New York, NY, USA, 2013.5. Cote, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, crystalline, covalent
organic frameworks. Science 2005, 310, 1166–1170. [CrossRef] [PubMed]6. Wan, S.; Guo, J.; Kim, J.; Ihee, H.; Jiang, D.L. A Belt-Shaped, Blue Luminescent, and Semiconducting Covalent
Organic Framework. Angew. Chem. Int. Ed. 2008, 47, 8826–8830. [CrossRef] [PubMed]7. Tilford, R.W.; Gemmill, W.R.; zur Loye, H.C.; Lavigne, J.J. Facile synthesis of a highly crystalline, covalently
linked porous boronate network. Chem. Mater. 2006, 18, 5296–5301. [CrossRef]8. Spitler, E.L.; Dichtel, W.R. Lewis acid-catalysed formation of two-dimensional phthalocyanine covalent
organic frameworks. Nat. Chem. 2010, 2, 672–677. [CrossRef]9. Tilford, R.W.; Mugavero, S.J.; Pellechia, P.J.; Lavigne, J.J. Tailoring microporosity in covalent organic
frameworks. Adv. Mater. 2008, 20, 2741–2746. [CrossRef]10. El-Kaderi, H.M.; Hunt, J.R.; Mendoza-Cortes, J.L.; Cote, A.P.; Taylor, R.E.; O’Keeffe, M.; Yaghi, O.M. Designed
synthesis of 3D covalent organic frameworks. Science 2007, 316, 268–272. [CrossRef]11. Jiang, J.X.; Cooper, A.I. Microporous Organic Polymers: Design, Synthesis, and Function. Top. Curr. Chem.
2010, 293, 1–33. [CrossRef]

Materials 2019, 12, 1974 11 of 12
12. Diercks, C.S.; Yaghi, O.M. The atom, the molecule, and the covalent organic framework. Science 2017,355, eaal1585. [CrossRef]
13. Cote, A.P.; El-Kaderi, H.M.; Furukawa, H.; Hunt, J.R.; Yaghi, O.M. Reticular synthesis of microporous andmesoporous 2D covalent organic frameworks. J. Am. Chem. Soc. 2007, 129, 12914–12915. [CrossRef]
14. Jiang, J.X.; Su, F.; Trewin, A.; Wood, C.D.; Niu, H.; Jones, J.T.A.; Khimyak, Y.Z.; Cooper, A.I. Syntheticcontrol of the pore dimension and surface area in conjugated microporous polymer and copolymer networks.J. Am. Chem. Soc. 2008, 130, 7710–7720. [CrossRef] [PubMed]
15. Uribe-Romo, F.J.; Hunt, J.R.; Furukawa, H.; Klock, C.; O’Keeffe, M.; Yaghi, O.M. A Crystalline Imine-Linked3-D Porous Covalent Organic Framework. J. Am. Chem. Soc. 2009, 131, 4570–4571. [CrossRef]
16. Furukawa, H.; Yaghi, O.M. Storage of Hydrogen, Methane, and Carbon Dioxide in Highly Porous CovalentOrganic Frameworks for Clean Energy Applications. J. Am. Chem. Soc. 2009, 131, 8875–8883. [CrossRef][PubMed]
17. Doonan, C.J.; Tranchemontagne, D.J.; Glover, T.G.; Hunt, J.R.; Yaghi, O.M. Exceptional ammonia uptake bya covalent organic framework. Nat. Chem. 2010, 2, 235–238. [CrossRef] [PubMed]
18. Kuhn, P.; Antonietti, M.; Thomas, A. Porous, covalent triazine-based frameworks prepared by ionothermalsynthesis. Angew. Chem. Int. Ed. 2008, 47, 3450–3453. [CrossRef]
19. Spitler, E.L.; Giovino, M.R.; White, S.L.; Dichtel, W.R. A mechanistic study of Lewis acid-catalyzed covalentorganic framework formation. Chem. Sci. 2011, 2, 1588–1593. [CrossRef]
20. Ding, S.Y.; Gao, J.; Wang, Q.; Zhang, Y.; Song, W.G.; Su, C.Y.; Wang, W. Construction of Covalent OrganicFramework for Catalysis: Pd/COF-LZU1 in Suzuki-Miyaura Coupling Reaction. J. Am. Chem. Soc. 2011, 133,19816–19822. [CrossRef]
21. Feng, X.A.; Chen, L.; Dong, Y.P.; Jiang, D.L. Porphyrin-based two-dimensional covalent organic frameworks:Synchronized synthetic control of macroscopic structures and pore parameters. Chem. Commun. 2011, 47,1979–1981. [CrossRef]
22. Lohse, M.S.; Bein, T. Covalent Organic Frameworks: Structures, Synthesis, and Applications. Adv. Funct.Mater. 2018, 28, 1705553. [CrossRef]
23. Fang, Q.; Gu, S.; Zheng, J.; Zhuang, Z.; Qiu, S.; Yan, Y. 3D Microporous Base-Functionalized Covalent OrganicFrameworks for Size-Selective Catalysis. Angew. Chem. Int. Ed. 2014, 53, 2878–2882. [CrossRef] [PubMed]
24. Xu, H.; Chen, X.; Gao, J.; Lin, J.; Addicoat, M.; Irle, S.; Jiang, D. Catalytic covalent organic frameworks viapore surface engineering. Chem. Commun. 2014, 50, 1292–1294. [CrossRef] [PubMed]
25. Xu, H.; Gao, J.; Jiang, D. Stable, crystalline, porous, covalent organic frameworks as a platform for chiralorganocatalysts. Nat. Chem. 2015, 7, 905. [CrossRef] [PubMed]
26. Wu, Y.; Xu, H.; Chen, X.; Gao, J.; Jiang, D. A π-electronic covalent organic framework catalyst: π-walls ascatalytic beds for Diels–Alder reactions under ambient conditions. Chem. Commun. 2015, 51, 10096–10098.[CrossRef]
27. Nagai, A.; Guo, Z.Q.; Feng, X.; Jin, S.B.; Chen, X.; Ding, X.S.; Jiang, D.L. Pore surface engineering in covalentorganic frameworks. Nat. Commun. 2011, 2, 536. [CrossRef] [PubMed]
28. Corma, A.; García, H.; Llabrés i Xamena, F.X. Engineering Metal Organic Frameworks for HeterogeneousCatalysis. Chem. Rev. 2010, 110, 4606–4655. [CrossRef] [PubMed]
29. Zhu, L.; Liu, X.-Q.; Jiang, H.-L.; Sun, L.-B. Metal–Organic Frameworks for Heterogeneous Basic Catalysis.Chem. Rev. 2017, 117, 8129–8176. [CrossRef]
30. García, H.; Navalón, S. Metal-Organic Frameworks: Applications in Separations and Catalysis; Wiley VCH:Weinheim, Germany, 2018.
31. Bromberg, L.; Klichko, Y.; Chang, E.P.; Speakman, S.; Straut, C.M.; Wilusz, E.; Hatton, T.A.Alkylaminopyridine-Modified Aluminum Aminoterephthalate Metal-Organic Frameworks As Componentsof Reactive Self-Detoxifying Materials. ACS Appl. Mater. Interfaces 2012, 4, 4595–4602. [CrossRef]
32. Moon, S.-Y.; Howarth, A.J.; Wang, T.; Vermeulen, N.A.; Hupp, J.T.; Farha, O.K. A visually detectable pHresponsive zirconium metal-organic framework. Chem. Commun. 2016, 52, 3438–3441. [CrossRef]
33. Mullangi, D.; Shalini, S.; Nandi, S.; Choksi, B.; Vaidhyanathan, R. Super-hydrophobic covalent organicframeworks for chemical resistant coatings and hydrophobic paper and textile composites. J. Mater. Chem. A2017, 5, 8376–8384. [CrossRef]

Materials 2019, 12, 1974 12 of 12
34. Sun, Q.; Fu, C.-W.; Aguila, B.; Perman, J.; Wang, S.; Huang, H.-Y.; Xiao, F.-S.; Ma, S. Pore Environment Controland Enhanced Performance of Enzymes Infiltrated in Covalent Organic Frameworks. J. Am. Chem. Soc. 2018,140, 984–992. [CrossRef] [PubMed]
35. Halder, A.; Karak, S.; Addicoat, M.; Bera, S.; Chakraborty, A.; Kunjattu, S.H.; Pachfule, P.; Heine, T.;Banerjee, R. Ultrastable Imine-Based Covalent Organic Frameworks for Sulfuric Acid Recovery: An Effect ofInterlayer Hydrogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 5797–5802. [CrossRef] [PubMed]
36. Johnson, E.M.; Haiges, R.; Marinescu, S.C. Covalent-Organic Frameworks Composed of Rhenium Bipyridineand Metal Porphyrins: Designing Heterobimetallic Frameworks with Two Distinct Metal Sites. ACS Appl.Mater. Interfaces 2018, 10, 37919–37927. [CrossRef] [PubMed]
37. de la Peña Ruigómez, A.; Rodríguez-San-Miguel, D.; Stylianou, K.C.; Cavallini, M.; Gentili, D.; Liscio, F.;Milita, S.; Roscioni, O.M.; Ruiz-González, M.L.; Carbonell, C.; et al. Direct On-Surface Patterning ofa Crystalline Laminar Covalent Organic Framework Synthesized at Room Temperature. Chem. Eur. J. 2015,21, 10666–10670. [CrossRef] [PubMed]
38. Luo, S.; Xu, H.; Mi, X.; Li, J.; Zheng, X.; Cheng, J.-P. Evolution of Pyrrolidine-Type Asymmetric Organocatalystsby “Click” Chemistry. J. Org. Chem. 2006, 71, 9244–9247. [CrossRef] [PubMed]
39. Jin, H.; Cho, S.M.; Lee, J.; Ryu, D.H. Role of Configuration at C6 in Catalytic Activity of l-Proline-DerivedBifunctional Organocatalysts. Org. Lett. 2017, 19, 2434–2437. [CrossRef] [PubMed]
40. Meri-Bofi, L.; Royuela, S.; Zamora, F.; Ruiz-Gonzalez, M.L.; Segura, J.L.; Munoz-Olivas, R.; Mancheno, M.J.Thiol grafted imine-based covalent organic frameworks for water remediation through selective removal ofHg(ii). J. Mater. Chem. A 2017, 5, 17973–17981. [CrossRef]
41. Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by RegiospecificCopper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67,3057–3064. [CrossRef]
42. Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process:Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002,41, 2596–2599. [CrossRef]
43. López-Maya, E.; Montoro, C.; Rodríguez-Albelo, L.M.; Aznar Cervantes, S.D.; Lozano-Pérez, A.A.; Cenís, J.L.;Barea, E.; Navarro, J.A.R. Textile/Metal–Organic-Framework Composites as Self-Detoxifying Filters forChemical-Warfare Agents. Angew. Chem. Int. Ed. 2015, 54, 6790–6794. [CrossRef]
44. Gil-San-Millan, R.; López-Maya, E.; Hall, M.; Padial, N.M.; Peterson, G.W.; DeCoste, J.B.;Rodríguez-Albelo, L.M.; Oltra, J.E.; Barea, E.; Navarro, J.A.R. Chemical Warfare Agents DetoxificationProperties of Zirconium Metal–Organic Frameworks by Synergistic Incorporation of Nucleophilic and BasicSites. ACS Appl. Mater. Interfaces 2017, 9, 23967–23973. [CrossRef] [PubMed]
45. Liu, Y.; Howarth, A.J.; Vermeulen, N.A.; Moon, S.-Y.; Hupp, J.T.; Farha, O.K. Catalytic degradation ofchemical warfare agents and their simulants by metal-organic frameworks. Coord. Chem. Rev. 2017, 346,101–111. [CrossRef]
46. Ma, T.; Kapustin, E.A.; Yin, S.X.; Liang, L.; Zhou, Z.; Niu, J.; Li, L.-H.; Wang, Y.; Su, J.; Li, J.; et al. Single-crystalx-ray diffraction structures of covalent organic frameworks. Science 2018, 361, 48–52. [CrossRef] [PubMed]
47. Navarro, J.A.R. The dynamic art of growing COF crystals. Science 2018, 361, 35. [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open accessarticle distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/).