castleman disease presenting with ophthalmic signs and symptoms
TRANSCRIPT

MELAS SYNDROME (MITOCHONDRIAL ENCEPHALOMY-
opathy, lactic acidosis, and stroke-like episodes) is amaternally inherited disorder associated with abnormalmitochondria, manifesting in tissues highly dependent onoxidative metabolism. Ocular changes in MELAS syn-drome have included reversible scotomata, ophthalmople-gia, pigmentary retinopathy, marked attenuation of thescotopic electroretinogram, myopia, and nuclear cata-ract.1,2
We highlight some of these ocular manifestations in afamily with MELAS syndrome and describe a distinctmaculopathy. A 48-year-old woman was referred for eval-uation of an asymptomatic maculopathy. Her medicalhistory included mild sensorineural hearing loss; her familyhistory included a mother with congestive heart failure,diabetes mellitus, ophthalmoplegia, and parafoveal atro-phy as well as a deceased brother who had an unknownocular disease, deafness, diabetes, and heart failure. Ocularexamination disclosed best-corrected visual acuities of RE:20/25 and LE: 20/30, normal results from Ishihara colorplate testing, significant limitation of gaze in all fields (asmuch as 70% of expected), and several patches of macularretinal pigment epithelial atrophy bilaterally, which werenoted to have become slightly larger compared withfundus photographs taken 8 years earlier. These regionsof atrophy correlated with scotomata on Amsler gridand visual field testing (Figures 1 and 2). Her electro-retinogram disclosed reduction of photopic and scotopicb-wave amplitudes with normal implicit times. Screen-ing for mitochondrial disorders was suggested but re-fused by the patient.
Several months later, the patient’s sister was diagnosedwith MELAS syndrome after presenting with stroke; shewas found to have bilateral neurosensory deafness, bicuspidaortic valve, and diabetes mellitus, and a muscle biopsy waspositive for ragged red muscle fibers. She was noted tohave ophthalmoplegia but did not have ophthalmiccomplaints. No family members had nyctalopia, waxypallor of the optic nerve, or attenuation of the retinalvessels. The four tested members of the family, includingthe sister with MELAS syndrome as well as the originalpatient and her daughter and son, were found to havethe A to G 3243 mt DNA mutation heteroplasmic withwild-type mt DNA.
This report amplifies and corroborates some of the oph-thalmic features of MELAS syndrome by describing geo-graphic atrophy of the macula in families with the syndrome.The symptoms associated with MELAS syndrome typicallyappear in childhood and often cause death by the fourthdecade.3 Two mt DNA mutations have been associated withthis syndrome and are both located in the tRNAleu (UUR)gene. In 80% of cases, the mutation is an A to G transitionat mt-3243, and in 10% of cases, the mutation is an A to Gtransition at mt-3271.4 Many maternal relatives of patientswith MELAS syndrome exhibit a scarcity of symptomssecondary to the presence of “heteroplasmy,” a mixture of
mutant and wild-type intracellular mitochondria. The mu-tant phenotype is expressed only when an intracellularthreshold of mt DNA has been reached. Mitochondrialmutation is uncommon in the general population; however,patients with the mutation were recently shown to have awider range of phenotypic diversity than previously reported.5Nutritional supplementation should be considered, althoughno large, well-designed trial has studied its efficacy.1 Atrophicmacular pigment epithelial atrophy associated with motilitydisorders and deafness should precipitate an evaluation formitochondrial disorders.
REFERENCES
1. Kuchle M, Breener PM, Englehardt A, Naumann GO. Ocularchanges in MELAS syndrome. Klin Monatsbl Augenheilkd1990;197:258–264.
2. Rummelt V, Folberg R, Ionasescu V, Yi H, Moore KC. Ocularpathology of MELAS syndrome with mitochondrial DNApoint mutation. Ophthalmology 1993;100:1756–1766.
3. Beal MF, Howell N, Bodis Wollner I. Mitochondria and freeradicals in neurodegenerative diseases. New York: Wiley-Liss,Inc, 1997.
4. Goto YI, Nonak I, Haroi S. A new mt DNA mutationassociated with mitochondrial myopathy, encephalomyopa-thy, lactic acidosis and stroke-like episodes. Biochim BiophysActa 1991;1097:238–240.
5. Morgan-Hughes JA, Sweeney MG, Cooper JM, et al. Mito-chondrial DNA (mtDNA) diseases: correlation of genotype tophenotype. Biochim Biophys Acta 1995;1271:135–140.
Castleman Disease Presenting WithOphthalmic Signs and SymptomsToru Kurokawa, MD,Sadahiro Suzuki, MD, PhD,Kenji Kawaguchi, MD,Noboru Fujisawa, MD, PhD, andNagahisa Yoshimura, MD, PhD
PURPOSE: To describe a patient with multicentric Castle-man disease who was initially examined with ophthalmicsigns and symptoms.METHODS: Case report. A 71-year-old man was initiallyexamined with swelling of both upper eyelids and diplopiaof 2 months’ duration.RESULTS: Medical evaluation and right axillary lymph nodebiopsy disclosed Castleman disease. Systemic corticosteroidtreatment temporarily resolved signs and symptoms, but the
Accepted for publication Dec 29, 1998.From the Department of Ophthalmology, Shinshu University School
of Medicine, Matsumoto, Japan (T.K., N.Y.), and the Departments ofInternal Medicine (S.S.), Pathology (K.K.), and Ophthalmology (N.F.),Shinonoi General Hospital, Nagano, Japan.
Inquiries to Nagahisa Yoshimura, MD, PhD, Department of Ophthal-mology, Shinshu University School of Medicine, Matsumoto 390-8621,Japan; fax: 81-263-32-9448; e-mail: [email protected]
AMERICAN JOURNAL OF OPHTHALMOLOGY114 JULY 1999

patient died of recurrence with cytomegalovirus and As-pergillus infection 10 months after initial examination.CONCLUSIONS: Multicentric Castleman disease is a rarebut distinct disorder that may present initially withocular signs and symptoms. This disease must be in-cluded in the differential diagnosis of orbital pseudotu-mor and lymphoma. (Am J Ophthalmol 1999;128:114–116. © 1999 by Elsevier Science Inc. All rightsreserved.)
CASTLEMAN DISEASE, AN ATYPICAL LYMPHOPROLIFERA-
tive disorder, was first reported as “localized mediasti-nal lymph node hyperplasia resembling thymoma” in19561 and is currently classified pathologically into twoforms: plasma cell type and hyaline-vascular type.2 Castle-man disease with generalized or extensive lymph nodeinvolvement is called multicentric Castleman disease.3Most patients are middle-aged or older, but the origin ofthis disease remains unknown.4 The development oflymphadenopathy or constitutional symptoms, such asfever, night sweats, weight loss, and weakness, are the mostcommon reasons that patients seek medical attention.4The prognosis of this condition is not good: 26% ofpatients die within the first year after diagnosis, andmedian survival is only 29 months.4 Although some casesof visual loss caused by cranial involvement have beenreported,4 we found only one report in the literature thatdescribed actual ocular involvement.5 We report a patientwith multicentric Castleman disease who was initiallyexamined with ophthalmic signs and symptoms.
A 71-year-old man had bilateral swelling of the uppereyelids and diplopia of 2 months’ duration. His best-corrected visual acuity was RE: 20/20 and LE: 60/200. Arelative afferent pupillary defect was noted in the left eye.
The left bulbar conjunctiva was hyperemic and showedchemosis. Left eye movement was limited in all directions.Optic disk, macula, and retina were normal bilaterally.Orbital computed tomography and magnetic resonanceimaging demonstrated bilateral enlargement of the lacri-mal glands and swelling of the tissues surrounding the leftoptic nerve (Figure 1). Right axillary lymph nodes wereswollen, and chest x-ray showed bilateral hilar lymphade-nopathy. Serum immunoglobulin G level was increased at4,260 mg per dl, and roureax formation was found. Eryth-rocyte and leukocyte counts were normal.
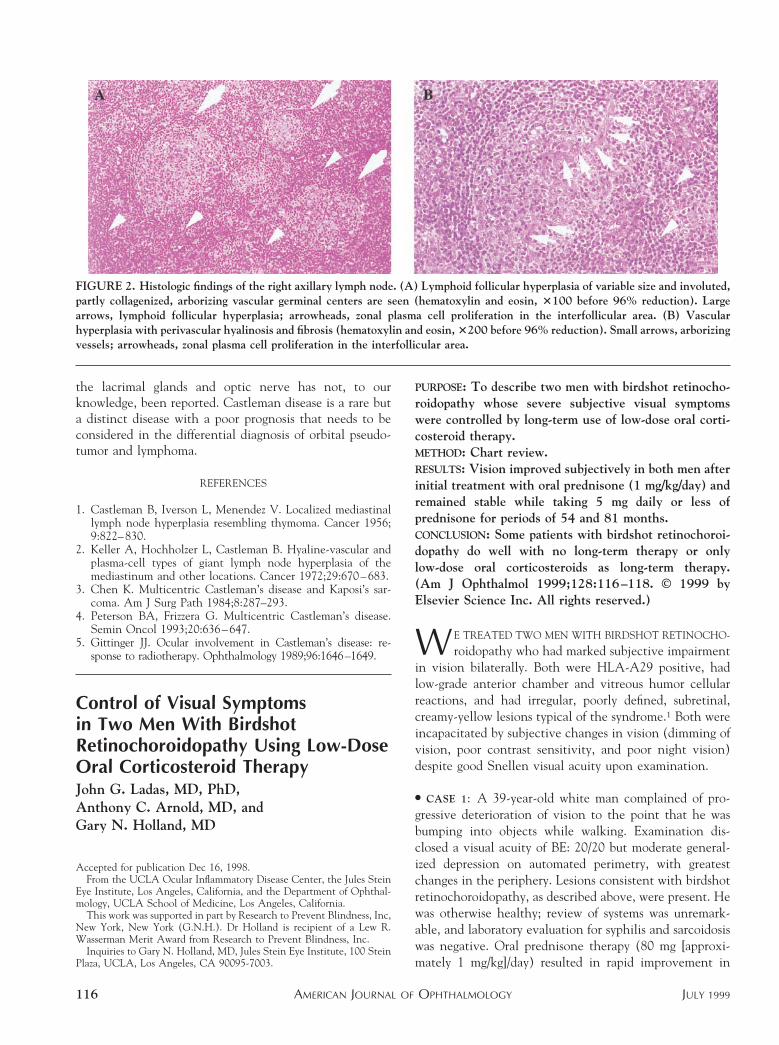
The patient refused to undergo biopsy of the lacrimalgland; however, right axillary lymph node biopsy wasperformed, and Castleman disease was diagnosed based oncharacteristic histologic findings (Figure 2). Systemic pred-nisolone acetate (60 mg/day) was given, and the patient’ssymptoms and ocular findings improved. Orbital lesionsshowed remarkable improvement on computed tomogra-phy (Figure 1) and magnetic resonance imaging. Visualacuity also improved to LE: 20/40, and systemic cortico-steroids were tapered. Ten months after initial examina-tion, the patient developed a recurrence and died becauseof systemic infection with cytomegalovirus and Aspergillus.Autopsy showed multicentric Castleman disease.
This is a rare case of Castleman disease in which ocularsigns and symptoms occurred first. This case mimics orbitalpseudotumor or lymphoma, but systemic findings, labora-tory data, and histology of the lymph nodes support thediagnosis of multicentric Castleman disease. This diseaseoccasionally involves nonlymphoid tissues,5 and the or-bital lesions seen in this patient are likely associated withCastleman disease. Although ocular complications ofCastleman disease, such as serous retinal detachment andthickening of sclera, have been reported,5 involvement of
FIGURE 1. Orbital computed tomography and magnetic resonance imaging. (A) T1-weighted image without enhancement and (B)plain computed tomography before corticosteroid treatment show enlargement of the lacrimal glands bilaterally (small arrows); theleft optic nerve, surrounding tissues, or both are swollen (large arrows). (C) After treatment, the lesions diminished.
BRIEF REPORTSVOL. 128, NO. 1 115
the lacrimal glands and optic nerve has not, to ourknowledge, been reported. Castleman disease is a rare buta distinct disease with a poor prognosis that needs to beconsidered in the differential diagnosis of orbital pseudo-tumor and lymphoma.
REFERENCES
1. Castleman B, Iverson L, Menendez V. Localized mediastinallymph node hyperplasia resembling thymoma. Cancer 1956;9:822–830.
2. Keller A, Hochholzer L, Castleman B. Hyaline-vascular andplasma-cell types of giant lymph node hyperplasia of themediastinum and other locations. Cancer 1972;29:670–683.
3. Chen K. Multicentric Castleman’s disease and Kaposi’s sar-coma. Am J Surg Path 1984;8:287–293.
4. Peterson BA, Frizzera G. Multicentric Castleman’s disease.Semin Oncol 1993;20:636–647.
5. Gittinger JJ. Ocular involvement in Castleman’s disease: re-sponse to radiotherapy. Ophthalmology 1989;96:1646–1649.
Control of Visual Symptomsin Two Men With BirdshotRetinochoroidopathy Using Low-DoseOral Corticosteroid TherapyJohn G. Ladas, MD, PhD,Anthony C. Arnold, MD, andGary N. Holland, MD
PURPOSE: To describe two men with birdshot retinocho-roidopathy whose severe subjective visual symptomswere controlled by long-term use of low-dose oral corti-costeroid therapy.METHOD: Chart review.RESULTS: Vision improved subjectively in both men afterinitial treatment with oral prednisone (1 mg/kg/day) andremained stable while taking 5 mg daily or less ofprednisone for periods of 54 and 81 months.CONCLUSION: Some patients with birdshot retinochoroi-dopathy do well with no long-term therapy or onlylow-dose oral corticosteroids as long-term therapy.(Am J Ophthalmol 1999;128:116–118. © 1999 byElsevier Science Inc. All rights reserved.)
WE TREATED TWO MEN WITH BIRDSHOT RETINOCHO-
roidopathy who had marked subjective impairmentin vision bilaterally. Both were HLA-A29 positive, hadlow-grade anterior chamber and vitreous humor cellularreactions, and had irregular, poorly defined, subretinal,creamy-yellow lesions typical of the syndrome.1 Both wereincapacitated by subjective changes in vision (dimming ofvision, poor contrast sensitivity, and poor night vision)despite good Snellen visual acuity upon examination.
● CASE 1: A 39-year-old white man complained of pro-gressive deterioration of vision to the point that he wasbumping into objects while walking. Examination dis-closed a visual acuity of BE: 20/20 but moderate general-ized depression on automated perimetry, with greatestchanges in the periphery. Lesions consistent with birdshotretinochoroidopathy, as described above, were present. Hewas otherwise healthy; review of systems was unremark-able, and laboratory evaluation for syphilis and sarcoidosiswas negative. Oral prednisone therapy (80 mg [approxi-mately 1 mg/kg]/day) resulted in rapid improvement in
Accepted for publication Dec 16, 1998.From the UCLA Ocular Inflammatory Disease Center, the Jules Stein
Eye Institute, Los Angeles, California, and the Department of Ophthal-mology, UCLA School of Medicine, Los Angeles, California.
This work was supported in part by Research to Prevent Blindness, Inc,New York, New York (G.N.H.). Dr Holland is recipient of a Lew R.Wasserman Merit Award from Research to Prevent Blindness, Inc.
Inquiries to Gary N. Holland, MD, Jules Stein Eye Institute, 100 SteinPlaza, UCLA, Los Angeles, CA 90095-7003.
FIGURE 2. Histologic findings of the right axillary lymph node. (A) Lymphoid follicular hyperplasia of variable size and involuted,partly collagenized, arborizing vascular germinal centers are seen (hematoxylin and eosin, 3100 before 96% reduction). Largearrows, lymphoid follicular hyperplasia; arrowheads, zonal plasma cell proliferation in the interfollicular area. (B) Vascularhyperplasia with perivascular hyalinosis and fibrosis (hematoxylin and eosin, 3200 before 96% reduction). Small arrows, arborizingvessels; arrowheads, zonal plasma cell proliferation in the interfollicular area.
AMERICAN JOURNAL OF OPHTHALMOLOGY116 JULY 1999