cardiovascular autonomic regulation in systemic...
TRANSCRIPT

OULU 1999
CARDIOVASCULAR AUTONOMIC REGULATION IN SYSTEMIC HYPERTENSION
ANTTIYLITALO
Department of Internal Medicine

OULUN YLIOP ISTO, OULU 1999
CARDIOVASCULAR AUTONOMIC REGULATION IN SYSTEMIC HYPERTENSION
ANTTI YLITALO
Academic Dissertation to be presented with the assent of the Faculty of Medicine, University of Oulu, for public discussion in Auditorium 10 of the University Hospital of Oulu, on May 8th, 1999, at 2 p.m.

Copyright © 1999Oulu University Library, 1999
OULU UNIVERSITY LIBRARYOULU 1999
ALSO AVAILABLE IN PRINTED FORMAT
Manuscript received 8.4.1999Accepted 12.4.1999
Communicated by Docent Ilkka KantolaDocent Kari Niemelä
ISBN 951-42-5212-8
ISBN 951-42-5211-XISSN 0355-3221 (URL: http://herkules.oulu.fi/issn03553221/)

AbstractNeurogenic factors are known to be important in the development of hypertension. Our currentknowledge of the role of autonomic nervous system in chronic hypertension is, however, limited. Thepurpose of the present study was to evaluate the possible abnormalities in heart rate variability (HRV)and baroreflex sensitivity (BRS) in patients with long standing systemic hypertension compared tosubjects without evidence of cardiovascular disease. A particular aim was also to examine whethergenetic variation in the renin-angiotensin-aldosterone system (RAS) genes have an influence oncardiovascular autonomic regulation.
Case-control studies were carried out on a total of 280 normotensive and 214 hypertensive subjectsdrawn from a random middle-aged population originally recruited for an epidemiologic study ofcardiovascular risk factors. The possible association of BRS with the genetic polymorphisms ofrenin-angiotensin-aldosterone system genes was studied in a cross-sectional study of 315 healthycontrols. Genetic associations were also tested in a younger, independent population sample of 66subjects. The effects of intensified antihypertensive treatment on autonomic cardiovascular controlwere evaluated in 33 hypertensive patients with poor blood pressure control.
Wide interindividual variation in both HRV and BRS was observed in normotensive as well ashypertensive subjects. Overall HRV and autonomic responses to a change in body posture wereblunted in long-standing hypertension. Decreased HRV was mainly related to elevated blood pressureand obesity.
For the first time in a population-based study, it was confirmed that BRS is impaired in patients withlong-standing hypertension despite adequate antihypertensive treatment. In contrast to HRV, BRSwas reduced in hypertensive subjects also after adjustment for blood pressure and obesity. BRS alsovaried widely both between healthy and hypertensive individuals. The wide interindividual variationin the markers of autonomic cardiovascular regulation was not, however, completely explained bydemographic variables, cardiovascular risk factors or lifestyle, suggesting a genetic componentcontributing to HRV and BRS.
The polymorphism in the aldosterone synthase (CYP11B2) gene was found to strongly associate withBRS in two independent random populations of apparently healthy subjects. The association waseven stronger in the younger population. On the basis of the observations made in the olderpopulation, it seems possible that women are protected against the effect of age and blood pressureon BRS and tend to maintain the genomic influence longer.
Intensified antihypertensive combination therapy improved blood pressure control and causedregression of left ventricular hypertrophy, and resulted in significant improvements of HRV and BRS.
The present study shows that HRV and BRS are altered in long-standing systemic hypertension.Together with age, blood pressure and obesity, genetic factors seem to be important determinants ofBRS. However, abnormal autonomic cardiovascular regulation does not seem to be an irreversiblephenomenon, but can be partly restored by modern combination antihypertensive therapy.
Keywords:baroreflex sensitivity, gene polymorphism, heart variability, systemic hyper-tension.



Acknowledgements
The present research was carried out at the Department of Internal Medicine, Universityof Oulu, during 1994-1999. I wish to express my gratitude to Professor AnteroKesäniemi, M.D., head of the Department, for placing excelent facilities of the institutefor clinical and scientific work at my disposal.
I thank the official referees, Kari Niemelä, M.D. and Ilkka Kantola M.D. for theirvaluable criticism during the preparation of the final manuscript.
I wish to express my sincere thanks to professor Heikki Huikuri, M.D., for hissupervision and encouragement throughout the work. He introduced me to field of heartrate variability and taught me a lot about secrets of arrhytmias and electrofysiology.
My warmest thanks go to my second supervisor docent Juhani Airaksinen. Hiscontribution has been essential in preparation of all the papers of this manuscript.Continuous struggle for better results in research, petanque, golf etc. and supportingattitude has teached me a lot of lif e in general.
I am grateful to Docent Markku Ikäheimo for his significant contribution to the studyby examining all the echocardiographs. I am also grateful for his experienced guidance ininvasive cardiology.
Sincere gratitude is also due to Docent Markku Kupari, and to Docent AarnoHautanen, for allowing data to the paper III . I also appreciate their valuable contributionin preparation of the manuscript.
I wish to express my thanks to Perrin White and Lawrence Sellin for their prompt andexcellent work in preparation of the corresponding papers II I and IV.
I owe my special thanks to Heikki Kauma, M.D. for his advise in genetics andstatistics, and Markku Linnaluoto, M.Sc., for his uncomplaining attitude to broadspectrum of problems arised in clinical and scientific works during the years.
My warm thanks go to Sirkku Pikkujämsä, M.D., who shared all the enthusiasm andunfortunate drawbacks in planning and preparation of this work.
I wish to thank also all the other co-authors: Kari Tahvanainen, M.Sc.; Tom Kuusela,PhD.; Marion Carson, M.D.; Juha Virolainen, MD.; Markku Savolainen, M.D.; AskoRantala, M.D. and Mauno Lilja , M.D.
Thanks are due to Pirkko Huikuri, R.N., Päivi Karjalainen, R.N., Anne Lehtinen,Helena Kalliokoski, R.N., and Riitta Vanhanen, R.N. for their important contribution tothe study.

I wiss to thank Sirkka-Liisa Leinonen, Phil.Lic., for revising the English language ofthe summary and some of the papers.
I wish to thank my colleagues professor Keijo Peuhkurinen, M.D. and Matti Niemelä,M.D. for keeping me busy in many scientific project and also in other activities outsidethe work; two weeks traveling round the world was an unforgettable experience.
I wish to express my gratitude to the colleagues and the staff of the CardiovascularLaboratory, for their important support and positive attitude.
I also warmly thank my sister Sirkka Posti for her help in handling the ambulatoryblood pressure data and specially for her endless support during the years.
Finally, I wish to thank my wife Kirsi and my children Anna and Arttu for theirpatience, understanding and great support. Assi is as old as this work and Arno was bornjust before I completed this work.
This work was supported by grants from the Finnish Foundation for CardiovascularReseach, Helsinki, Finland, Medical Council of the Academy of Finland, HelsinkiFinland, the research funds of Helsinki University Central Hospital, Helsinki, Finland,and the U.S. National Institutes of Health (DK37867 and DK42169), and Astra-Finland,Masala, Finland.

Abbreviations
ACE angiotensin-converting enzymeAGT angiotensinogenANOVA analysis of varianceBMI body mass indexBRS baroreflex sensitivityBP blood pressureCYP11B1 11β-hydroxylaseCYP11B2 aldosterone synthaseECG electrocardiographyHF high frequencyHR heart rateHRV heart rate variabilityLF low frequencyLVH left ventricular hypertrophyLVM left ventricular massLVMI left ventricular mass indexPCR polymerase chain reactionRAS renin-angiotensin-aldosterone systemSD standard deviationSDANN standard deviation of RR intervals of measured segmentsSDNN standard deviation of RR intervals of 24-hour recordingVLF very low frequencyULF ultra low frequency


List of original articles
This thesis is based on the following publications, which are referred to in the text bytheir Roman numerals:
I Huikuri HV, Ylitalo A, Pikkujämsä SM, Ikäheimo MJ, Airaksinen KEJ, Rantala AO& Lilj a M, Kesäniemi YA (1996) Heart rate variability in systemic hypertension. AmJ Cardiol 77: 1073-1077.
II Ylitalo A, Airaksinen KEJ, Tahvanainen KUO, Kuusela TA, Ikäheimo MJ, RantalaA, Lilj a M & Huikuri HV (1997) Baroreflex sensitivity in drug-treated systemichypertension. Am J Cardiol 80: 1369-1372.
II I Ylitalo A, Airaksinen KEJ, Hautanen A, Kupari M, Carson M, Virolainen J,Savolainen M, Kauma H, Kesäniemi YA, White PC, Huikuri HV (1999) Baroreflexsensitivity and variants of the renin angiotensin system genes. Submitted.
IV Ylitalo A, Airaksinen KEJ, Sellin L, Huikuri HV (1999) Effects of combinationantihypertensive therapy on baroreflex sensitivity and heart rate variability in long-standing hypertension. Am J Cardiol 83: 885-889.


Contents
AbstractAcknowledgementsAbbreviationsList of original articles1. Introduction 152. Review of the literature 16
2.1. Assessment of cardiovascular autonomic function 162.1.1. Heart rate variability 16
2.1.1.1. History and general aspects 162.1.1.2. Time domain analysis of heart rate variability 162.1.1.3. Frequency domain analysis of heart rate variability 172.1.1.4. Other methods of analyzing heart rate variability 182.1.1.4. Physiological correlates of heart rate variability 18
2.1.2. Baroreflex sensitivity 202.1.2.1. General aspects 202.1.2.2. Phenylephrine test 212.1.2.3. Valsalva maneuver 212.1.2.4. Cross-spectral analysis of spontaneous baroreceptor
responsiveness 222.1.2.5. Other techniques for measuring baroreflex sensitivity 24
2.1.3. Other techniques to evaluate cardiac autonomic control 242.1.4. Reproducibility and comparability of the methods 25
2.2. Cardiovascular autonomic regulation in systemic hypertension 262.2.1. The role of autonomic function in the pathogenesis of systemic
hypertension 262.2.2. Heart rate variability and baroreflex sensitivity at the later stages
of established hypertension 272.2.3. Influence of antihypertensive therapy on heart rate variability
and baroreflex sensitivity 282.2.4. Other determinants of heart rate variability and baroreflex
sensitivity in systemic hypertension 29

2.2.5. Prognostic significance of heart rate variability and baroreflexsensitivity 30
2.3. Renin-angiotensin-aldosterone system and autonomic nervous system 312.4. Genetics of the renin-angiotensin-aldosterone system 33
2.4.1. Polymorphisms of the angiotensinogen gene 332.4.3. Polymorphisms of the angiotensin-converting enzyme gene 342.4.4. Polymorphisms of the aldosterone synthase gene 34
3. Aims of the study 364. Subjects and methods 37
4.1. Populations 374.1.1. Population 1 374.1.2. Population 2 38
4.2. Study designs 384.3. Blood pressure recordings 394.4. Echocardiographic methods 404.5. Laboratory methods 40
4.5.1. Biochemical analyses 404.5.2. DNA analyses and genotyping subjects 41
4.6. Analysis of heart rate variability 414.7. Analysis of baroreflex sensitivity 42
4.7.1. Phenylephrine test (IV) 424.7.2. Valsalva maneuver (II , III) 434.7.3. Spontaneous baroreceptor responsiveness (II) 43
4.8. Statistical analyses 445. Results 45
5.1. Heart rate variability in systemic hypertension 455.2. Baroreflex sensitivity in systemic hypertension 475.3. Determinants of autonomic cardiovascular regulation 48
5.3.1. Determinants of heart rate variability (I) 485.3.2. Determinants of baroreflex sensitivity (II) 485.3.3. Genetic influences on baroreflex sensitivity (III) 49
5.3.3.1. Population 1 495.3.3.2. Population 2 50
5.3.4. Influences of antihypertensive treatment on autonomiccardiovascular regulation (IV) 50
6. Discussion 536.1. Reduced heart rate variability in patients with systemic hypertension 536.2. Impaired baroreflex sensitivity in patients with systemic hypertension 546.3. Determinants of autonomic cardiovascular regulation in patients with
systemic hypertension 556.4. Genetic variation in renin-angiotensin-aldosterone system genes
and baroreflex sensitivity 566.5. Possible limitations of the study 57
7. Conclusions 598. References 60

1. Int roduction
Systemic hypertension is a common problem in adults, contributing in a major way to themost common causes of morbidity and mortality in developed societies (1). Whereasaging, obesity, and environmental factors contribute to the onset of hypertension,neurogenic factors are known to be important in the regulation of blood pressure (BP)and in the development of hypertension (2). Abnormal cardiovascular regulation mayalso be a potential trigger of adverse events in hypertensive patients. It is therefore easyto understand the growing enthusiasm about heart rate variability (HRV) and baroreflexsensitivity (BRS), which both indirectly reflect the autonomic input to the heart and havebeen associated with an increased cardiovascular mortality in various populations (3-7).
Untreated hypertension is known to be associated with decreased HRV (8-10) andBRS (11-13). Our current knowledge of these indices in patients with medicatedhypertension is limited, however, and the factors affecting cardiovascular autonomicregulation are largely unknown (14-18). Nor is it known whether abnormalities in HRVor BRS can be influenced or reversed by modern antihypertensive treatment.
The wide scatter in HRV and BRS observed in previous studies (17,18) suggests thatgenetic components may contribute to autonomic cardiovascular regulation (19,20). Theautonomic nervous system has multiple interactions with the renin-angiotensin-aldosterone system (RAS), which regulates BP as well as the fluid and electrolytebalance (2,21,22). Therefore, genes encoding for components of this system are attractivecandidates for the investigation of the genetic basis underlying the impairment ofautonomic cardiovascular regulation in systemic hypertension.
The present work was set out to study the possible abnormalities in HRV and BRS inpatients with medicated systemic hypertension, to evaluate the determinants of theseindices and to evaluate whether intensive antihypertensive therapy has an effect onautonomic cardiovascular regulation. A particular aim was to examine the potential riskloci in the genes encoding components of RAS.

2. Review of the literatu re
2.1. Assessment of cardiovascular function
2.1.1. Heart rate variability
2.1.1.1. History and general aspects
Heart rate (HR), BP and many other cardiovascular variables fluctuate from beat to beat.HR is principally influenced by both the intrinsic firing rate of the sinus node and theeffects of the autonomic nervous system, but various other physiological perturbationsalso alter HR. Cyclic changes in HR and BP synchronous with respiration have beennoted since ancient times, but were first systematically studied by S. Hales in 1733 (23).In 1876, S. Mayer described waves in BP which were lower than respiration. HRV refersto the amount of continuous HR fluctuation around the mean HR and reflectscomplicated interactions between the sympathetic and parasympathetic nervous systemstogether with other physiological regulatory systems modulating sinus node pacemakeractivity. HRV reflects the dynamic processes involved in the maintenance of homeostasis(24). The clinical importance of HRV was first appreciated in relation to fetal monitoring(25). Thereafter, the easy derivation of the measurement of HRV by computerizedanalysis, the availability of better algorithms to assess the oscillatory components of HRbehavior (26,27) and the prognostic significance of HRV (3), has popularized its use invarious cardiac and noncardiac pathological conditions. The variation in HR may beevaluated by a number of methods (28).
2.1.1.2. Time domain analysis of heart rate variability
In time domain methods, a simple statistical analysis of a sequence of successive RRintervals or RR interval differences is used. All measurements require accurate timing ofR waves and, for statistical time domain methods, it is essential to carefully eliminate

17
artefacts and ectopic beats from the data.The most common and simplest index is the standard deviation of normal-to-normal
RR intervals (SDNN) over a 24-hour period (estimated overall HRV). Mathematically,SDNN is the square root of variance, which equals to the total power of spectral analysis.In other words, SDNN reflects all the components responsible for variability in therecording period. However, the value of SDNN depends on the length of the recordingperiod, which has been recommended to be nominal 24 hours (28). The standarddeviation of average normal-to-normal RR intervals (SDANN, estimated long-termHRV) is calculated over short periods, usually 5 minutes. The most commonly usedmeasure derived from interval differences is the square root of the mean squareddifferences of successive normal-to-normal RR intervals (RMSSD, estimated short-termHRV). RMSSD is an estimate of HF variations in HR. The HRV triangular index (HRV-index) is a geometric measure of HRV, usually derived from a 24-hour ECG recording.In this method, the lengths of the RR intervals are plotted against the number of each RRinterval length. The HRV index represents the total number of RR intervals divided bythe largest number of the equally long RR intervals. This type of analysis is relativelyinsensitive to artefacts and ectopic beats (29).
2.1.1.3. Frequency domain analysis of heart rate variability
Spectral analysis (26) quantifies the oscillatory frequency of HR in addition to theamount of variability and thereby quantifies the different frequency domain componentsin terms of their relative intensity (power)(27). For optimal reliability of the analysis, it iseven more stringently necessary than in time domain analysis that ectopic beats andartefacts can be reliably detected and eliminated. It is essential to preview and editmanually the RR interval data. In addition, the signal should be stationary (e.g. lineartrends should be removed by detrending and filtering the data) and sufficiently long (> 5minutes).
Both a Fast Fourier transform algorithm (non-parametric) and an autoregressive model(parametric) have been used to transform RR interval signals into frequency domainmeasures (30). Autoregressive analysis has better spectral resolution and it enablesspectral estimation from shorter time periods than the Fast Fourier method (26). In mostinstances, both methods provide comparable results. A typical power spectrum in ahealthy subject is displayed in Fig. 1.
Spectral components (area under the frequency domain bands = power) are usuallyreported in absolute units (milliseconds squared) (31,32). The recommended boundariesfor different frequency bands are: total power <0.4 Hz, ultra-low frequency <0.003(ULF), very low frequency 0.003-0.04 Hz (VLF), low frequency 0.04-0.15 Hz (LF), andhigh frequency 0.15-0.4 Hz (HF). The ULF and VLF components account for 95% of thetotal power of long-term recordings (usually 24-hour), but the problem of stationarityshould be taken into account when analyzing these components from long-termrecordings (28). Temporal reciprocal changes in the different limbs of the autonomicnervous system are often analyzed from short-term recordings using normalized units(33-35). Normalized units represent the value of each power component divided by theVLF component. The value of this parameter together with the ratio of absolute LF to HF

18
power as indexes of sympathovagal balance has, however, been questioned recently (36).
Frequenzy (cycles / second)
Pow
er(m
s2 )
0.1 0.2 0.3 0.4 0.5
Fig. 1. A power spectrum of a 45-minute segment of a RR interval tachogram.
2.1.1.4. Other methods of analyzing heart rate variability
The Poincaré plot (a geometric measure) is a diagram in which each RR interval isplotted against the previous RR interval. This plot provides a visual and quantitativeanalysis of RR intervals (29,37). The Poincaré plot contains both linear and non-linearphenomena of HRV (38).
Because the cardiovascular system is not a stationary system, several new dynamicmethods have been recently developed to measure the non-linear properties of HRV.These methods provide new insight into the analysis of HRV in various cardiovasculardisorders. These analyses include 1/f scaling of Fourier spectra (39), detrendedfluctuation analysis (22), approximate entropy (40) and power-law relationship (6).
2.1.1.5. Physiological correlates of heart rate variability
Despite the vast body of literature and the extensive clinical use of HRV, thepathophysiological neural and humoral mechanisms responsible for changes in varioustime and frequency domain measures of HRV are poorly understood. It is also necessaryto bear in mind that HRV represents the net effects of several inhibitory (mostly vagal)and excitatory (sympathetic and humoral) influences, and that power spectralcomponents, are not selective for specific autonomic components. The cardiac sinusrhythm varies as a result of physical and autonomic nervous system activity, mentalstress, respiration, BP regulation, thermoregulation, RAS activity and possibly other

19
unknown factors.Time-domain measures of HRV are known to correlate with vagal activity (41,42).
The power of HF oscillation (periodicity of 2.5-7 seconds), which is know to be relatedto respiration (respiratory sinus arrhythmia), is considered a marker of vagal efferentactivity on the heart (27,30,35). The modulation of HR associated with respiration isinfluenced by BP changes secondary to respiratory movements mediated via arterialbaroreceptors or atrial stretch receptors and the reflex response to lung inflation mediatedby thoracic stretch receptors, but, most importantly, respiratory sinus arrhythmia isinfluenced by central impulses from the respiratory center (43). A faster breathing ratedecreases the HF power, but an increase in the tidal volume of respiration (34,42)increases the magnitude of HF oscillation. On the other hand, atropine and vagotomyabolish the HF component (27,33). Also, the HF component of HRV is diminished byphysical or mental stress accompanied by increased sympathetic activity (33). However,a constant and saturating influence of high vagal tone on sinus node, which might be thecase when BP is increased abruptly by phenylephrine (44), may lead, counterintuitively,to a diminished HF component of HRV (45). Taken together, the HF component can beused as an estimate of vagal activity under standard physiological conditions (45).
The opinions on the genesis of LF oscillation (periodicity of 7-25 seconds) have beencontroversial. LF power has been suggested to reflect sympathetic activity with vagalmodulation, but the results depend upon the study conditions (e.g. posture, length ofrecordings, clinical situation) (27,33,34,42). In physiological conditions, includingmoderate mental or physical stress, the LF component of HRV may reflect thesympathetic modulation of HR, and the magnitude of LF power is accordingly increased(33,46). In some clinical conditions, such as heart failure (47) or maximal exercise (48),however, LF power fails to show increased sympathetic activity and the values of LFpower are decreased. At least two potential explanations are available for theseapparently discrepant findings. One possibility is, that despite the high sympathetic tone,HR is not modulated because of a saturated influence on the sinus node (45). The otherreason for a low LF component of HRV in cases of high sympathetic tone could be thelow BRS, which is often related to these conditions (49). This theory is based on De Boermodel of the circulation. According to this model, LF power is produced by the variationof sympathetic discharge due to the resonant interaction between fast vagal and slowsympathetic responses to baroreceptor stimulation (49). This dependency of LFoscillation in the RR interval on Mayer wave BP oscillations (0.1 Hz) has beenconfirmed by some (50), but not all authors (51).
The origin of the VLF component is not clear, although contributions by RAS and thetermoregulatory system may play a role (27,52,53). VLF power is influenced bybaroreflexes, and parasympathetic activity actually seems to be the most importantmodulator of VLF oscillations (27,42,52). Vagal efferent activity is also a significantmodulating factor of the ULF component (27), but the precise origin of this component isunknown.

20
2.1.2. Baroreflex sensitivity
2.1.2.1. General aspects
Arterial baroreceptors are sensory nerve endings located primarily in the blood vesselwalls of the carotid sinuses and the aortic arch (Fig. 2) (54,55). Increased BP causesvascular stretch and activates baroreceptors, which triggers adjustments in autonomicoutflow, HR, and vascular resistance to buffer excessive fluctuations of BP (56).Conversely, decreases in BP reduce baroreceptor discharge and trigger adjustments thatcounteract the hypotension. Afferent (sensory) neurons transmit the baroreceptor activityto the central nervous system (the nucleus tractus solitarius in the medulla), where theimpulses interact with various nuclei of central neurons and the peripheral effects aremediated by the group of parasympathetic and sympathetic efferent neurons. At thecentral nervous system level, complex interactions between afferent impulses from thearterial baroreceptors and impulses from other reflex systems, e.g. cardiac baroreceptors(the Bezold-Jarisch reflex (57)), take place, which it makes it difficult to study singlespecific reflexes in humans. When interpretating studies of baroreflex responsiveness,one must be aware of the following concepts: BRS = the slope of the linear relationshipbetween the systolic BP and RR intervals; BRS equals gain = the effect of BP changesaccording to the level and situation in which they occur; set point = the minimum BPlevel at which baroreflex activation varies. Several methods are available to study theresponses to various interventions, which usually activate several reflexes simultaneously(56,58-60).
CarotidSinuses
Aortic Arch
Adrenal Medulla
Kidney Renin
Angiotensinogen
Angiotensin I
Angiotensin II
SympatheticEfferents
ParasympatheticEfferents
EpinephrineNorepinepinephrine
Heart Rate
Baroreceptor Afferents
ACEBradykinin
InactiveProducts
Fig. 2. Arterial baroreceptors, their location, the baroreflex arch and the interactionsbetween the baroreceptor-mediated regulations of BP and RAS.

21
2.1.2.2. Phenylephrine test
The phenylephrine technique has been the ”gold standard” in the quantification of arterialBRS (11,60). The advantage of this method is that all arterial baroreceptor regions areexited simultaneously and secondary buffering of one reflex to another is not a problem.The principle of this method is that an intravenous bolus of phenylephrine causes a rise inarterial BP via the vasoconstrictor effect ofα-stimulation and results in a reflex slowingdown of HR. A typical computer output of a phenylephrine test is displayed in Fig. 3.
There are some drawbacks in this method, however. The invasive technique(intravenous and occasionally intra-arterial cannulation) restricts it use in both clinicalpractice and large-scale population surveys. Also, the invasive technique may influencethe reflex responses (61). Furthermore, cardiopulmonary afferent stimuli may modulatethe baroreflex responses after the phenylephrine injection. This is, however, unlikely innormal physiological conditions in humans (62). Phenylephrine has also beenadministered with a graded infusion technique to measure BRS, but the slopes obtainedby the infusion method yield smaller BRS values and do not correlate with the slopesobtained by the bolus method (63).
2.1.2.3. Valsalva maneuver
Valsalva maneuver, i.e. forced expiration against a closed glottis, has been widely used toassess autonomic reflex mechanisms (64,65). During the procedure, there is an increasein thoracic and abdominal pressures, resulting in an accumulation of blood outside thoseareas and a consequent decrease in cardiac filling. Four typical phases of the Valsalvamaneuver have been observed (Fig. 3): Phase 1, elevation of arterial BP at the onset ofstraining for a few seconds, mainly due to mechanical propulsion of blood to the arteries;Phase 2, a fall and subsequent partial recovery of pressure above the control levels asstraining continues. HR and vascular resistance increase to compensate for the lowercardiac output (impaired venous return); Phase 3, a sudden brief further reduction of BPand elevation of HR immediately following the release of straining; and Phase 4(overshoot phase), a terminal elevation of BP above the control levels accompanied by aslowing down of HR. Sympathetic activity is minimal at phase 1, prominent during thePhases 2 and 3, and absent during and after phase 4 (65). The BP elevation during theovershoot phase is proportional to the increase of sympathetic activity during phase 2,which probably causes the increased peripheral resistance and consequent BP rise duringPhase 4 (65). Because the changes of the autonomic sensory input and neural outputtriggered by Valsalva maneuver are highly complex, the intervention must be done in astandard way (expiratory pressure, duration of strain, period of poststrain controlledbreathing, time window of the slope calculation) to obtain reproducible results.

22
Phenylephrine test Valsalva testRRI versus time
SAP/MAP/DAP versus time
ms
mmHg
RRI(i+1) versus SAP (i)
RRI = 7 ms/mmHg x SAP + -480 msRRI = 53ms/kPa x SAP + -480 ms
r = 0.936
757799
842
884
926968
101010531095
ms
RRI versus time
SAP/MAP/DAP versus time
595693791889987
ms
mmHg
7490
107123139155174188204
732762793823854884914945975
msRRI(i+1) versus SAP (i)
220
! 2 3 4
RRI = 7 ms/mmHg x SAP + -587 msRRI = 54ms/kPa x SAP + -587 ms
r = 0.970
Fig. 3. Computer output of phenylephrine and Valsalva tests in a healthy subject. Upperpanels: beat-to-beat RR intervals and BR values. Phases (1-4) of the Valsalva maneuver areindicated by vertical lines. Lower panels: RR intervals plotted as a function of the precedingsystolic BP values. The time window used for the BRS slope calculation is indicated bybroken lines in phenylephrine test, and phase 4 is used for BRS calculation in the Valsalvatest. BRS is 7 ms/mmHg in both tests.
Although BRS calculated from the overshoot phase of a Valsalva maneuver has beencomparable with BRS calculated from a phenylephrine test (66-69), many factors operatein the early part of Phase 4. The effects of the BP rise on the arterial baroreceptor may bemodified by a sudden change in intrathoracic pressure, whereby other reflex systems,particularly those mediated by cardiopulmonary baroreceptors, are activated. In addition,stroke volume, cardiac output and left ventricular contractility are changing at this time(67). When the method has been used in a standardized way, the Valsalva-basedassessment of BRS has been moderately well reproducible (69,70). However, one mustbe cautious when interpreting the results in patients with cardiac disease, especially heartfailure (71). A failure to obtain the baroreflex slope is most commonly due to aninadequate rise in BP during the Valsalva strain (69).
2.1.2.4. Cross-spectral analysis of spontaneous baroreceptorresponsiveness
Recent studies have suggested that the spontaneous fluctuations of BP and RR intervalsoffer a useful opportunity to assess baroreflexes in natural circumstances (72-75). Thequantification of BRS by cross-spectral analysis assumes that arterial BP variationsprovoke RR interval variations. In other words, spontaneous fluctuations of BP triggerchanges of arterial baroreceptor firing, which, in turn, trigger changes of vagalmotoneuron firing (after a 2-second phase lag). The baroreflex gain is calculated as thesquare root of the ratio between the RR interval and systolic pressure variability at the

23
frequencies of physiological interest. The measuring period should be stationary withoutectopic beats and sufficiently long (about 5 minutes), to quarantee reliable results.Spectral analysis of variability in RR interval and systolic BP can be performed using afast Fourier or autoregressive method (73,76).
The baroreflex calculation is not reliable if the coherence of the transfer functionestimate between the RR interval and systolic BP is low (<0.5) (Fig. 4). Goodcorrelations between BRS calculated with cross-spectral analysis and a phenylephrinetest have been observed in healthy subjects and patients with coronary artery disease(72,73). The slopes obtained from the mid-frequency range (0.07-0.15 Hz) correlatemore closely with the slopes obtained from a phenylephrine test than the slopes obtainedfrom the respiratory frequency range (0.15-0.40 Hz). It is probable that the mid-frequency cross-correlation slopes are an estimate of simple baroreflex physiology, e.g.that pressure changes trigger RR interval changes, and could be recommended as ameasure of BRS when using this method (72). A typical computer output of cross-spectral analysis is displayed in Fig. 4.
Region 1 Region 2
Time [s]0 200 400 600 800
Region 1 Region 2
SA
P/M
AP
/DA
P[m
mH
g]
RR
I[s]
50
100
150
200
0.60
0.80
1.00
1.20
0.0 0.1 0.2 0.3 0.4 0.0 0.1 0.2 0.3 0.4-180-90
0
18090
0.0
0.5
1.0
SA
PP
SP[
mm
Hg2 /
Hz]
RR
IPS
P[m
s2 /H
z]
RR
I/SA
P[d
eg
r]R
RI/
SA
Pco
he
ren
ce
Frequenzy [Hz] Frequenzy [Hz]
Fig. 4. The computer output of cross-spectral analysis performed on single stationary periodsof around 5 minutes. Upper panels: RR interval and BP variabilities as a function of time.Lower left panel: the corresponding power spectral densities of the RR intervals and systolicBP as a function of frequency. Lower right panel: cross-spectral analysis of the couplingbetween systolic BP and RR interval, including the calculation of the coherence function(reliability of the transfer function) and the phase angle (relative timing) of the changes as afunction of frequency. The BRS is 3.3 ms/mmHg in Region 1 and 2.8 ms/mmHg in Region 2.MAP = mean arterial pressure; DAP = diastolic arterial pressure.

24
2.1.2.5. Other techniques for measuring baroreflex sensitivity
Vasoactive agents other than phenylephrine have also been used to assess BRS.Angiotensin II (77-79) as a vasoconstrictor and nitroglycerin or nitroprusside (60) asvasodilators have been used most commonly. Although these agents do not have directeffect on HR, they may have central effects on baroreflex function and effects on venousreturn and venous compliance, which may contribute to the results.
A negative pressure applied to the lower half of the body to reduce central bloodvolume (lower body suction) has been used to investigate the reflex control of humancirculation. This technique is suitable for studying reflexes of cardiopulmonary origin,but it is notably less selective with regard to arterial baroreflexes (59).
The carotid baroreceptors can be selectively stimulated by the neck chambertechnique (58,59,80), where a tightly fitting rigid collar, within which pneumatic pressurecan be reduced in a graded fashion below atmospheric pressure, is applied to the subjectsneck. A negative pressure increases carotid transmural pressure and thereby increases theactivation of the carotid baroreceptor. Deactivation of these receptors is possible bypositive pressure. The immediate response of RR interval to changes in neck chamberpressure occurring within one or two cardiac cycles is likely to be due to carotidbaroreceptor stimulation. However, this response diminishes rapidly, presumably due toadaptation of the carotid baroreceptors, but also to buffering by the resulting responseinfluencing other baroreceptors.
Apart from cross-spectral analysis, the spontaneous sequence method provides anotherpossibility to estimate spontaneous baroreceptor responsiveness (81,82). In this method,sequences of three or more beats, within which BP spontaneously increases or decreasesfollowed by changes in RR interval, are identified and baroreflex slopes are determined.Although it is not clear whether there is a causal baroreflex mediated relationshipbetween BP and HR in all measured sequences (83), a reasonable correlation between thesequence and other methods has been reported (84).
2.1.3. Other techniques to evaluate cardiac autonomic control
Plasma levels of norepinephrine have been used as a measure of the overall level ofsympathetic activity (85). They represent the net result of release, re-uptake andcatabolism of norepinephrine in many regions of the body. Cardiac norepinephrinespillover (overflow to plasma) using isotope dilution is a measure of the globalsympathetic firing rate in the heart (86). This method is invasive and requires cannulationof the coronary sinus, being therefore unsuitable for widespread use. The regionalsympathetic integrity of the heart could be assessed by single-photon emission computedtomography after administration of radiolabelled I-123 metaiodobenzylguanidine(MIBG), which acts as an analogue of norepinephrine and has a distribution reflectingthat of sympathetic nerve endings with a preserved uptake process (87).
Neural sympathetic activity can be most directly examined by peripheral nerverecordings using the microneurographic technique developed by Hagbarth and Vallbo(88). The complex effects of arterial and cardiopulmonary baroreceptors on peripheral(skin and muscle) neural sympathetic activity (65,86) could be assessed with this

25
technique (86,89). The potential limitations on the universal use of microneurography arethe time-consuming technique, the difficulties to find adequate recording sites fordetecting sympathetic impulses and the marked interindividual differences in backgroundactivity compared to the responses to different maneuvers. In most instances, it is also notpossible to perform single-fiber recordings.
Similarly to HR, short and long term BP variability, obtained from continuous BPrecordings, has been quantified by power spectrum analysis. HF oscillations of BP havebeen associated with respiration. LF oscillations are know as Mayer waves (≈ 10 soscillations), which are believed to be caused by fluctuations in vascular tone andperipheral resistance (90). High BP variability has been suggested to associate withincreased incidence of end-organ damage in hypertensive patients (90). More studies areneeded, however, to confirm the role of BP variability in evaluation of cardiac autonomiccontrol, and to establish its prognostic significance (91).
2.1.4. Reproducibility and comparability of the methods
Although reproducibility may not be so important from the prognostic point of view (92),the modification of HRV or BRS in intervention studies can be considered useful only ifthe measurements are repeatable. The reproducibility and comparability of thesemeasures depend on the length of the recordings, the measurement technique, the timebetween the measurements and the study population.
The reproducibility of 24-hour HRV measurements has turned out to be good enoughto allow interpretation of the results obtained from a single recording. Reproducibilityhas been reasonable (intraclass correlation = reproducibility within a group >0.7,coefficient of variation = within-subject SD divided by sample mean <10%) in healthysubjects (16,93,94) and even better in cardiac patients, whose HRV is usually lower andday-to-day variation smaller (95). Kleigeret al. have shown that, in addition to goodreproducibility, individual measurements from 24-hour recordings are stable in a range of3 to 65 days and lack placebo effect in healthy subjects (93). The reproducibility ofshort-term HRV measures from subsequent segments of 2-5 minutes during one sessionhas been good for time domain measures (intraclass correlation > 0.9, coefficient ofvariation <10%) and reasonable for frequency domain measures (intraclass correlation0.5-0.9) (96,97). The reproducibility of short-term HRV determined on separate days hasbeen questioned (16) and shown to be only moderate (intraclass correlation 0.6-0.8,coefficient of variation 16-64%)(98,99). To eliminate large physiological variation overtime, controlled conditions (e.g. controlled respiration) are especially important in short-term recordings (especially for the determination of the HF component of HRV) (98).Manual editing of ECG data may involve individual differences in the ability to detectectopic beats, but the interobserver reproducibility of indices of HRV has been high(100).
The comparability of indices of HRV derived from short-term and long termrecordings has been good enough (correlations ranging from 0.6 to 0.9)(92,99).Furthermore, many of the time and frequency domain measures strongly correlate witheach other, suggesting a physiological relationship (28,93,101). The correlation betweenBRS and different measures of HRV has been moderate, but not perfect (correlation

26
coefficient 0.4 - 0.6), suggesting some influence of a common physiological background(101).
The Valsalva (69,70,102), phenylephrine (69,72), cross-spectral (72) and neck suction(56,59) techniques for BRS assessment have been highly reproducible over short periods.The values obtained from the neck chamber technique have been shown to bereproducible for up to 10 weeks in healthy subjects (56). Also, no significant differenceswere found between values obtained from two phenylephrine tests up to 15 months apartin hypertensive subjects (11). The intercorrelations between the values for BRS obtainedwith different techniques vary from 0.02 to 0.9 (60,69), suggesting that these techniquesmeasure different aspects of baroreceptor function.
To summarize, the available data suggest stability and persistence of BRS and HRVmeasures derived from 24-hour recordings in healthy subjects and patients with cardiacdiseases. 24-hour indices of HRV appear to be ideal variables to assess interventiontherapies. For large-scale population studies, short-term recordings (5-15 minutes) giveimportant prognostic information and have reasonable reproducibility (28,103).
2.2. Cardiovascular autonomic regulation in systemic hypertension
2.2.1. The role of autonomic function in the pathogenesis of systemichypertension
The observations on hyperkinetic hemodynamics, including increased venous tone,increased vascular resistance, high stroke volume, high cardiac output and fast HR in theearly phases of systemic hypertension, have stimulated research on the role of theautonomic nervous system in the development of hypertension (104). An increasedsympathetic tone in borderline hypertension has been documented using severalexperimental methods: plasma norepinephrine levels have been elevated (105,106),direct microneurographic recordings from fibers of the peroneal nerve have showedincreased sympathetic activity (107), norepinephrine spillover rates have increased (108),and spectral analysis of HRV has suggested sympathetic predominance (9,109). Theearly findings of Juliuset al. (110) and Korneret al. (111) and more recent observationusing spectral analysis of HRV (112,113) suggest that increased sympathetic tone iscoupled with reduced parasympathetic activity in borderline hypertension. Since bothcomponents of neural cardiovascular autonomic control are abnormal, and the change inthe tone of these two components is usually reciprocal, it has been suggested that theprimary abnormality is of central origin and psychosomatic factors may thus play a role(22). A decreased HF component of HRV has been reported in numerous studies ofhypertensive patients (9,112,114). Guzzettiet al. reported an increased LF component(expressed as normalized units), suggesting sympathetic predominance in patients withearly stages of human hypertension (9), a finding supported by an earlier experimentalstudy (34).
There is substantial evidence to support the assumption that there could be a causalrelationship between baroreflex dysfunction and hypertension (115). Animal studies inthe 1930s and 1940s showed that the combined section of the carotid sinus and aorticnerve followed by persistent elevation in BP (116,117). Also, destruction of baroreceptor

27
inputs by a lesion in thenucleus tractus solitariiresulted in sustained hypertension inexperimental animals (118). Gordonet al. showed that decreased BRS precedes the onsetof hypertension in the rat model (119). In humans, the pioneering studies by Bristowetal. (77), Takeshitaet al. (12), Korneret al. (111), Eckberg (19) and Manciaet al. (120)documented subnormal baroreflex responses in patients with mild BP elevation,suggesting the possibility that baroreflex dysfunction may cause human arterialhypertension. In a recent study, Grassiet al. showed that diminished BRS in untreatedhypertensives is associated with a significant increase in muscle sympathetic activity(microneurography) paralleling the progressive increase in BP (121). However, inpatients with secondary hypertension (renovascular or pheochromocytoma), musclesympathetic activity was not increased despite the diminished BRS.
The aforementioned role of autonomic cardiovascular regulation in the pathogenesisof hypertension is also supported by studies of normotensive offspring of hypertensivepatients. Noll et al. first demonstrated that sympathetic nervous system activity, asassessed by plasma norepinephrine and muscle microneurography recordings, wasincreased in the offspring of hypertensive patients (122). Furthermore, Parmeret al.showed that normotensive subjects with a family history of hypertension have depressedBRS compared with subjects without a family history of hypertension (123). Furtherevidence of an autonomic influence in the pathogenesis of arterial hypertension comesfrom the prospective part of the ARIC Study, which detected an inverse relationshipbetween measures of HRV and the cumulative incidence of hypertension suggestingdiminished HRV as a potential risk factor of hypertension (124). Similar results wereobtained in the study on the Framinghampopulation: after a 4-year follow-up, 244subjects out of the originally normotensive population of 1434 subjects developedhypertension. In this study, the LF component of HRV was independently associated witha greater risk for developing hypertension in men but not in women (125).
2.2.2. Heart rate variability and baroreflex sensitivity at the later stagesof established hypertension
It has been estimated that only 30% of the patients with borderline hypertension progressto permanent established hypertension (22). At the first glance, studies on the role of theautonomic nervous system in chronic hypertension yield somewhat controversial results.Plasma norepinephrine levels, norepinephrine spillover rates, cardiac output and HRhave not been elevated in long-standing hypertension (22). However, it seems that thereis a hemodynamic state development from a hyperkinetic state to a state of increasedvascular resistance (126). Neurogenic factors could be important in this transition,although the absolute sympathetic tone is not increased and may even be decreased inlong-standing hypertension (22,85). At least three potential mechanisms by which theautonomic nervous system may contribute to the long-term regulation of arterial BP andhypertension have been suggested (127): 1) the renal sympathetic nerves promoteantinatriuresis and the release of renin to plasma, in addition to increasing renal vascularresistance; 2) the sympathetic nervous system can influence the vascular membraneproperties, resulting in augmented vasoconstrictor responsiveness to norepinephrine; and3) the sympathetic nervous system, similarily to the RAS, promotes the growth of

28
vascular and cardiac muscle, which, in turn, could maintain high BP.There has been only limited information available on the presence of abnormalities in
HRV and BRS in patients with long-standing hypertension. Furthermore, many previousstudies have not adjusted their results for the possible differences in baseline BP, age orBMI (8-10,114,120,128,129), which all have effects on autonomic cardiovascularregulation. In the cross-sectional part of the ARIC study, patients with hypertension haddecreased HRV measured as the SDNN, total power or the HF component of powerspectra. The LF component of HRV was not different compared to healthy controls, butHRV was measured from 2 minutes recording, which may partly contribute to thefindings (124). Also, Chakkoet al. (128) showed that all measures of HRV are reducedin medicated hypertensive patients with left ventricular hypertrophy (LVH). A recentstudy on the Framinham population showed in a cross-sectional analysis, that all the timeand frequency domain measures of HRV are lower in hypertensive compared tonormotensive subjects (125).
Experimental studies have documented that, in chronic hypertension, baroreflex isreset towards a higher BP range and operates with a reduced sensitivity (130). Theadvantage of resetting is that the baroreceptor pressure-activity curve shifts in thedirection of high pressure to function at a new set point (130,131). This allows thebaroreceptors to buffer fluctuations of BP at a higher level effectively because the newpressure set point remains on the steep portion of the reset pressure-activity curve.However, resetting provides positive feedback in the control of BP and thus helps tomaintain an elevated BP level and results in a vicious circle (130). At the same time thesensitivity of the baroreflex (the slope of the pressure-sensitivity curve) decreases and themagnitude of this impairment is related to the patients BP level (115).
There are several potential mechanisms to explain the decreased BRS in systemichypertension (132): 1) decreased vascular distensibility and endothelial damageassociated with atherosclerosis and aging (133-135), 2) transcellular ionic exchangesaffecting the vascular wall sodium content (136,137), 3) alterations in the release ofvarious factors from endothelium (132), 4) increase of circulating hormones, e.g.norepinephrine (106,134), angiotensin II (138,139) or aldosterone (140,141), 5)enchanced central RAS and central action of circulating angiotensin II (142) andaldosterone (143). Multiple mechanisms are most likely involved, and abnormalities inthese systems could provide a basis for the genetic component of BRS impairment inchronic hypertension.
2.2.3. Influence of antihypertensive therapy on heart rate variability andbaroreflex sensitivity
Antihypertensive therapy may augment cardiovascular autonomic regulation in multipleways. There is evidence that beta-blockers could directly improve cardiovascularautonomic regulation in hypertensive and normotensive patients (144-150). Also, thereduction of the circulating levels of angiotensin II and aldosterone by angiotensin-converting enzyme inhibitors could augment BRS and increase HRV, which has beenobserved in most (151-155), but not all studies (150). In addition to having direct effects,antihypertensive therapy could reverse the structural changes in the large arteries and

29
reverse LVH, which are known to be associated with hypertension and attenuate BRS(128,133,156,157). High BP in itself deteriorates cardiovascular autonomic control, andBP reduction could improve BRS and HRV (19,77,156,158).
Apart from beta-blockers and angiotensin-converting enzyme inhibitors, there is onlylimited information on the effects of other antihypertensive drugs on cardiac autonomicfunction. Calcium blockers may reset the baroreflex toward lower BP values (159), buttheir effects on BRS have been neutral (160) or they may augment BRS (161). Even lessis known about the effects of diuretics on HRV or BRS. However, diuretics might beexpected to attenuate baroreflex function, because they induce salt depletion and couldthereby activate RAS, at least when used as monotherapy in higher doses (162,163).
Oral pirenzepine, intravenous atropine and transdermal scopolamine have paradoxicvagomimetic (increased parasympathetic) effects in low doses and have been shown toincrease BRS (164,165). Vesalainenet al. showed that low-dose scopolamine not onlyimproves BRS and augments the HF component of HRV, but also decreases BP inpatients with mild hypertension (166). It has been shown in animal experiments,however, that despite the beneficial effect of scopolamine on HRV, this effect cannot beattributed to a decreased risk for ventricular tachycardia due to acute myocardialinfarction in association with high sympathetic activity (167). This suggests that themodification of HRV by drug interventions may not translate directly into cardiacprotection.
2.2.4. Other determinants of heart rate variability and baroreflexsensitivity in systemic hypertension
Previous studies have clearly shown that both HRV and BRS are related to severalcardiovascular risk factors (14,15,17): 1) There are significant sex-related differences inHRV and BRS (168-173), which must be taken into consideration when interpreting theresults of different studies. Although the mechanisms are unknown, the lower BRS inwomen may contribute to the observed poorer short-term prognosis of women who haveexperienced an acute myocardial infarction (174,175), 2) Age is another importantdeterminant of HRV and BRS, and both of these indices decrease with increasing age(11,176-178). Reduced arterial compliance, progressive loss of complexity in thedynamics of all physiological systems and changes in autonomic innervation andresponsiveness may contribute to age-related changes in autonomic regulation (133,179).3) Obesity and physical activity influence HRV (14,38,180-182) and BRS (183,184). Ithas been suggested that enhanced sympathetic activity may play a role in the maintenanceof elevated BP in obese subjects (185). 4) Smoking has been found to impair autonomicfunction (14,15,186). 5) Finally, other disease states often related to systemichypertension may modify cardiovascular autonomic regulation. HRV is known to bereduced in diabetic subjects (187,188), and it seems that the serum insulin levelcontribute to HRV (17). Coronary artery disease with and without myocardial infarction(78,189-191) and its most common risk factor, high cholesterol (14,15,17), may impairHRV and BRS. Patients with heart failure have shown prominent sympathoexcitation(192,193) and parasympathetic withdrawal (78). These alterations are related toabnormal HRV (188,194) and BRS (195), both of which may also have prognostic value,

30
at least in patients with mild to moderate heart failure (194,196,197).
2.2.5. Prognostic significance of heart rate variability and baroreflexsensitivity
The first observation suggesting that a lack of respiratory sinus arrhythmia predicts in-hospital cardiovascular mortality after acute myocardial infarction was published in 1978(198). Since then, these findings have been confirmed by several investigators (7).Kleiger et al. (3) showed in a prospective multicenter study that HRV measured from 24-hour Holter recordings two weeks after acute myocardial infarction is a powerfulindependent risk predictor for the first four years of follow-up. Impaired BRS measuredsoon after myocardial infarction is also a marker of high mortality (7,18,199) andvulnerability to ventricular fibrillation (200), and in combination with HRV, it could beuseful in risk stratification after myocardial infarction. A prospective multicenter study of1284 patients with a recent (<28 days) myocardial infarction (ATRAMI) showed thatboth SDNN <70 ms and BRS <3 ms/mmHg carried a significant multivariate risk ofcardiac mortality (3.2 and 2.8 risk ratio, respectively) (18). Numerous small studies havesuggested that HRV and BRS may also have prognostic value in patients with earlystages of heart failure (194,195).
The association between decreased HRV and an increased risk of mortality has alsobeen observed in various other populations. An epidemiological survey of patientsreferred for Holter monitoring for various reasons suggested that low HRV is associatedwith an increased risk of sudden death (5). A study of the Framingham cohort showedthat measures of HRV are able to predict all-cause mortality in elderly subjects (4).Dekker et al. confirm that decreased HRV predicts all-cause mortality in middle-agedpopulation (103). Another previous prospective study of elderly subjects suggested thatespecially long-term measures of HRV are not only related to cardiac death but alsoinvolve an increased risk for other, e.g. cerebrovascular, events leading to death (6). Theprognostic role of HRV or BRS has not been studied in hypertensive populations, buthypertensive subjects with depressed BRS are more likely to develop hypertensivecardiovascular disease than subjects with preserved BRS (19).
Despite the known clinical importance of depressed HRV or BRS, thepathophysiological link between impaired autonomic cardiovascular control and cardiacmortality is unknown. Animal models have suggested that decreased vagal and increasedsympathetic activity with experimental infarction predispose to ventricular fibrillation(201). Changes in HRV have also been found to precede episodes of ventriculartachycardia in humans (202,203), and some authors have suggested that especially theVLF and LF components of HRV increase the risk of spontaneous arrhythmic events inpatients with coronary artery disease (32,204-206). Also, it has been shown that bothBRS and HRV measured after myocardial infarction predict the occurrence ofpostinfarction ischemia-induced ventricular fibrillation (207), but at variance with HRV,BRS also predicts postinfarction ischemia-induced ventricular fibrillation when measuredbefore myocardial infarction in experimental models (207,208). This finding suggeststhat measures of vagal reflexes are more important than measures of vagal tone inidentifying animals at high risk in the absence of acute ischemia or recent myocardial

31
infarction (209). This concept is supported by the observation that parasympatheticactivity is an important modulator of low-frequency oscillations of HRV (52).
2.3. Renin-angiotensin-aldosterone system and autonomic nervoussystem
The RAS plays an important role in the physiological regulation of cardiovascularfunctions. It regulates sodium balance and intravascular volume and contributes to thedevelopment and maintenance of various forms of hypertension (210,211). In addition tocirculating RAS, there is evidence of the existence of local RAS in a wide variety oftissues including heart, brain, vascular smooth muscle and several endocrine glands. Atleast in certain situations, such as severe heart failure, angiotensin II may be generated bylocal tissue enzymes, such as chymases (212). The extensive interactions between RASand the autonomic nervous system, are summarized in Fig. 2 (2,21,22).
The renal enzyme renin is produced and stored in the juxtaglomerular cells located inthe wall of the afferent renal arteriole and released by the juxtaglomerular apparatus intothe circulation, where it cleaves angiotensin I from angiotensinogen (AGT). AGT issynthesized in the liver under control of estrogen, glucocorticoids, thyroid hormones andangiotensin II. Angiotensin I is rapidly cleaved by the angiotensin-converting enzyme(ACE) to form angiotensin II, which is the end product and the major effector moleculein RAS. Angiotensin I is not known to have physiological actions.
Renin release is mainly stimulated by three factors: 1) Factors that reduce volume orlower the plasma sodium or chloride levels stimulate renin release. Changes in sodiumchloride levels are detected by themacula densa, which consists of tubular epithelialcells at the distal end of Henle’s loop (213). 2) Decreases in renal perfusion pressure, aswith dehydration or hypotension, stimulate the renal baroreceptors, which, in turn,stimulate renin release. Cardiopulmonary mechanoreceptors have also been shown tohave an important role in the reflex regulation of renin in humans (22). 3) Renalsympathetic nerves secrete norepinephrine, which stimulates renin release viaβ-receptors. Propranolol blocks this mechanism and decreases renin release (214). On thecontrary, salt loading, angiotensin II, antidiuretic hormone and increased BP inhibit reninsecretion. High plasma renin activity has been observed in some hypertensive subjects(105). The high-renin state in these patients seems to reflect the generalized increase ofsympathetic drive, which seems to be the primary event and responsible for excessivevasocontriction via stimulation ofα-receptors (22). The rapid response of renin releaseinto circulation in experimental models suggests that the RAS may play an important rolein the minute-to-minute regulation of BP (215) and may also influence the VLFoscillation of HR (27,216).
In addition to renin, ACE is another key enzyme in the RAS (217). ACE is localizedin endothelial cells distributed throughout thebody, particularly in the lungs and kidneys.It catalyzes the conversion of angiotensin I to angiotensin II, but apart from angiotensin I,ACE also cleaves other peptides, most importantly bradykinin, which is a natriuretic andvasodilating peptide. The effects of bradykinin are potentiated by ACE inhibitors, but theclinical significance of this effect in the treatment of hypertensive patients is stillunknown (218). The reduction of circulating levels of angiotensin II and aldosterone by

32
ACE inhibitors has been shown to augment BRS and to increase HRV (151-153).Angiotensin II is a potent vasopressor peptide with numerous actions (219): 1) it is a
potent coronary and systemic vasoconstrictor with direct effects on the smooth muscle ofarterioles; 2) it inhibits renin release; 3) it is a potent stimulator of aldosteronebiosynthesis and release; 3) it has trophic influences on smooth muscle, cardiac myocytesand other cells of the body; 4) it augments the release of antidiuretic hormone andstimulates thirst through its effects on the central nervous system; 5) it interacts with thesympathetic nervous system and the baroreceptor reflexes to regulate BP (21).
As discussed above, the sympathetic nervous system controls renin release and is thusone of the factors that ultimately determine the circulating levels of angiotensin II. Thereare angiotensin II receptors located in the central structures, where angiotensin II acts toenhance sympathetic outflow. Angiotensin II also has an ability to stimulate the release ofcathecolamines from adrenal medulla. Furthermore, it has stimulatory effects on thesympathetic ganglia, and it could increase sympathetic activity by facilitatingneurotransmission at the adrenergic nerve terminals directly and indirectly by enhancingthe synthesis, release and reuptake of norepinephrine. Angiotensin II has been shown toattenuate baroreflex function (139,163,220). Many of the central cardiovascular effectsof angiotensin II are mediated by thearea postrema, an area of the brain lacking theblood-brain barrier and particularly sensitive to influences of the circulatingconcentrations of angiotensin II (221). This organ is located in themedulla oblongataand has interconnections with the nucleus oftractus solitarius, which is important in thebaroreflex control of circulation (222-224).
Aldosterone is synthesized in thezona glomerulosaof the adrenal cortex, where thelast steps of aldosterone biosynthesis are mediated by a single enzyme, aldosteronesynthase (CYP11B2) (210,211). Aldosterone is the most important mineralocorticoidhormone, whose main function is to promote renal sodium reabsorption in exchange forpotassium. When the sodium intake is reduced or the effective plasma volume shrinks,the increased angiotensin II stimulates aldosterone secretion from the adrenal cortex,which leads to renal retention of sodium and water. An increased potassium content isanother important stimulus for aldosterone production. Apart from the renal tubulus,aldosterone is a potent stimulator of the sodium pump in many other tissues, which maypartly explain its ability to alter the baroreflex function (140). Aldosterone may actcentrally to affect the global baroreceptor function, or it could act peripherally or have adirect effect on the carotid sinus through an endothelial cell mechanism (140). Theactions of mineralocorticoids involve binding of aldosterone to specific intracellularreceptors to induce subsequent specific protein synthesis. These receptors are found inepithelial tissues, but also in most areas of brain (143). Interestingly, progesterone has anequal affinity for mineralocorticoid receptor compared to aldosterone (225), and sexhormones may thus modulate the effects of aldosterone at the receptor level (226-228). Inaddition to having effects via the mineralocorticoid receptors, aldosterone has more rapiddirect effects on the intracellular electrolyte (Ca2+) concentration in vascular smoothmuscle cells (229). This effector system is also a potential mechanism for immediateeffects of aldosterone on baroreceptor discharge (140).

33
2.4. Genetics of the renin-angiotensin-aldosterone system
Approximately 30-50% of the interindividual variation in BP is attributable to geneticfactors, but the genes contributing to BP regulation are mostly unknown (230). The RASseems to be involved in the pathophysiology of hypertension by regulating BP as well asthe fluid and electrolyte balance. Consequently, each gene encoding for a component ofthe RAS (such as renin, AGT, ACE, aldosterone synthase and angiotensin II receptors)represents a potential candidate in the etiology and maintenance of hypertension (231).The genes of all these main components of RAS have been previously cloned.
The available markers of the renin gene have not been linked with cardiovasculardiseases (231). There is also limited information available about the association betweenthe angiotensin II receptor gene and cardiovascular pathologies (232).
2.4.1. Polymorphisms of the angiotensinogen gene
The AGT gene is located on chromosome 1 at q42-43 and consists of five exons and fourintrons (233). Among the 15 known molecular variants of the AGT gene, M235T hasbeen shown to associate with several cardiovascular pathologies. The mutation is locatedin exon 2. Although the mutation is not in the protein coding sequence, the plasma AGTconcentration associates with the T235 variant in such a way that subjects with the TTgenotype have the highest concentration (233). On the other hand, AGT levels appear tobe higher in young people with a genetic predisposition to high BP (234), suggesting thesignificance of AGT for the development of hypertension.
From the theoretical point of view, the M235T variant of the AGT gene (TTgenotype) leads to increased plasma or tissue AGT, which could cause a small increase inangiotensin II, accounting for a slight overreactivity of the RAS in response to sodiumand other environmental factors. This may, in turn, promote sodium retention as a resultof chronic stimulation of aldosterone secretion, vascular hypertrophy and increasedperipheral vascular resistance as a result of increased angiotensin II or abnormalstimulation of the sympathetic nervous system (233). Genetic linkage studies support therole of the AGT gene or some other gene located close to it, in linkage disequilibrium, inthe regulation of BP (231). The association between the M235T variant of the AGT geneand hypertension has been shown in some studies (233) and also in meta-analysis (235).The inconsistency between the studies may be due to differences in the geneticbackground, homogeneity and environmental circumstances of the study populations(236). Notably, no association between the M235T polymorphism of the AGT gene andBP was found in a random sample of hypertensive and control subjects matched for ageand sex (237). The regulation of AGT synthesis is strongly dependent on estrogens,which may also partly explain the controversial result obtained from studies not matchedfor sex (226,238).

34
2.4.2. Polymorphisms of the angiotensin-converting enzyme gene
The ACE gene is located on chromosome 17 at q23 and contains 26 exons (239). TheACE gene has two allelic forms, depending on the presence (insertion, I) or absence(deletion, D) of a 287 base-pair DNA sequence. The gene is highly expressed inendothelial cells and in the heart, and the level of ACE in plasma and inside cells ismodulated by the I/D polymorphism of the gene (239). Subjects with the DD genotypehave the highest plasma ACE levels (240), but the I/D polymorphism has not beenassociated with the plasma concentration of angiotensin II (231).
Two previous studies (241,242) have reported an association between the I/Dpolymorphism and hypertension. However, three random population-based studies,which avoid some of the critical points raised above concerning the former studies, havefailed to show any association between ACE polymorphism and hypertension (237),LVH (243) or ischemic heart disease (244). Two recent studies suggest that the ACE DDgenotype (245) or variation in a gene close to the ACE gene (246) may influence BP in asex-specific manner, e.g. differences were observed only in men but not in women.Differences in the populations, the sampling methods and the characterization ofphenotypes, and also the publication bias may contribute to the apparently contradictoryresults (231). Therefore, no definite conclusions concerning the role of ACEpolymorphism in cardiovascular diseases has crystallized based on these studies.
2.4.3. Polymorphisms of the aldosterone synthase gene
Aldosterone synthase (CYP11B2) is responsible for the final step of aldosteronebiosynthesis in thezona glomerulosaof the adrenal cortex (247). The gene of CYP11B2is located on chromosome 8 at q22 (Fig. 5). It is adjacent to a closely related gene thatencodes CYP11B1 (11β-hydroxylase), an enzyme required for cortisol biosynthesis(247,248). Aldosterone deficiency can be caused by mutations in the CYP11B2 gene(211,249), whereas an inherited form of hypertension, glucocorticoid suppressiblehyperaldosteronism, is caused by a genetic recombination between CYP11B1 andCYP11B2, which increases the expression of CYP11B2 and leads to inappropriatesecretion of aldosterone (250,251).
Several frequently occurring polymorphisms have recently been identified in theCYP11B2 gene (248). The relative orientations of the CYP11B2 and CYP11B1 genesand a schematic representation of two of these polymorphisms of the CYP11B2 gene areillustrated in Fig. 5. One polymorphism is located in the promoter region of CYP11B2,344 nucleotides before the start of the protein-coding sequence. This position can beeither a cytocine (-344C) or a thymidine (-344T). Persons homozygous for C,heterozygous for C and T, or homozygous for T are referred to as having the genotypes -344CC, -344CT or -344TT, respectively. The second polymorphism is in the secondintron of CYP11B2; in some individuals, the usual sequence of this intron has beenlargely replaced by the sequence typically found in the related gene, CYP11B1. Suchreplacement is termed gene conversion, and the alleles at this locus are referred to as 1(conversion) and 2 (no conversion). It has been shown that there is significant linkagedisequilibrium (e.g. nonrandom associations of alleles) between these two polymorphicloci of the CYP11B2 gene (252). Recently, Pojogaet al. reported data indicating that the
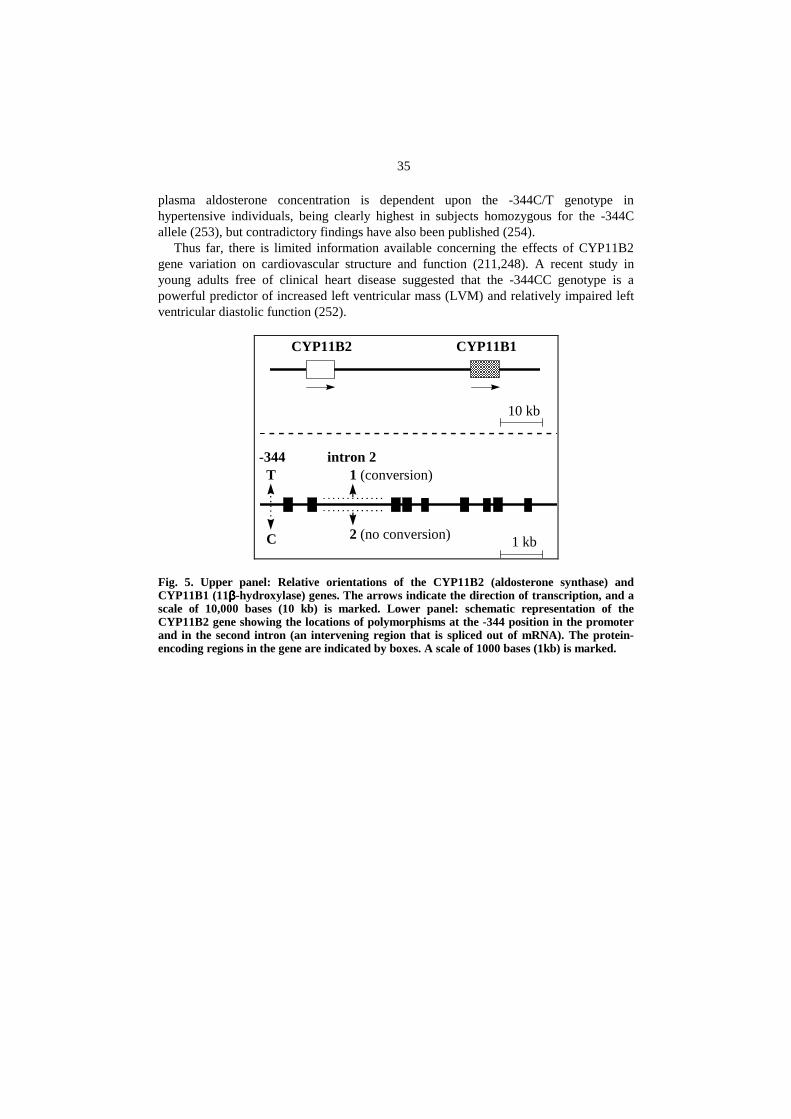
35
plasma aldosterone concentration is dependent upon the -344C/T genotype inhypertensive individuals, being clearly highest in subjects homozygous for the -344Callele (253), but contradictory findings have also been published (254).
Thus far, there is limited information available concerning the effects of CYP11B2gene variation on cardiovascular structure and function (211,248). A recent study inyoung adults free of clinical heart disease suggested that the -344CC genotype is apowerful predictor of increased left ventricular mass (LVM) and relatively impaired leftventricular diastolic function (252).
10 kb
1 kb
CYP11B2 CYP11B1
-344T
C
intron 21 (conversion)
2 (no conversion)
Fig. 5. Upper panel: Relative orientations of the CYP11B2 (aldosterone synthase) andCYP11B1 (11ββββ-hydroxylase) genes. The arrows indicate the direction of transcription, and ascale of 10,000 bases (10 kb) is marked. Lower panel: schematic representation of theCYP11B2 gene showing the locations of polymorphisms at the -344 position in the promoterand in the second intron (an intervening region that is spliced out of mRNA). The protein-encoding regions in the gene are indicated by boxes. A scale of 1000 bases (1kb) is marked.

3. Aims of the study
The purpose of the present work was to examine the potential abnormalities in autonomiccardiovascular control in patients with drug-treated hypertension compared to controlsubjects. A particular purpose was also to evaluate the determinants of HRV and BRS ina middle-aged population. The specific aims were:
1. to compare the measures of HRV in medicated hypertensive patients and controlsubjects and to assess HRV determinants in these populations (I),
2. to assess whether BRS is impaired in patients with drug-treated hypertensioncompared to control subjects, and to assess BRS determinants in these populations (II),
3. to study the possible association between BRS and candidate genes for cardiovasculardiseases encoding components of the RAS in two apparently healthy populations (III),and
4. to evaluate whether it is possible to reverse impaired HRV and BRS by intensificationof antihypertensive therapy in patients with long-standing hypertension (IV).

4. Subjects and methods
4.1. Populations
4.1.1. Population 1
The Oulu Project Elucidating the Risk of Atherosclerosis (OPERA) is a population-based, epidemiological case-control study of cardiovascular risk factors (237). Thehypertensive cohort consisted of 300 men and 300 women randomly selected from theregister for the reimbursement of antihypertensive medication maintained by the SocialInsurance Institute. At the time of randomization (September 1, 1990), the patients werefrom 40 to 59 years old and were entitled to a higher reimbursement class ofantihypertensive medication endorsed later than August 1980. The randomization wasage-stratified, i.e. for each year of birth (from 1931 to 1950), 15 hypertensive men and 15women were selected. For each hypertensive subject, an age- and sex matched controlsubject was randomly selected from the register covering the whole population of the cityof Oulu in Northern Finland (106,500 inhabitants), excluding subjects entitled to a refundfor antihypertensive medication. At the visit in the research laboratory of the Departmentof Internal Medicine, anthropometric measurements (weight, height, waist, hip) wereperformed and BP was measured. ECGs were evaluated by using the Minnesota Code(255). Past and current medical history, smoking habits, alcohol consumption andphysical activity were assessed using a standardized health questionnaire (256), andleisure-time physical activity groups (1-5) were established (257). BMI was calculated bydividing weight (kg) with the square of height (m2). The women were classified asmenopausal if at least six months had passed since their latest menstruation.Echocardiography was performed during a separate visit.
For the present study, all subjects were invited for an ambulatory ECG recording of 45minutes, Valsalva maneuver, cross-spectral analysis and 24-hour ambulatory BPmeasurement.

38
4.1.2. Population 2
The reproducibility of the genetic association study (III) was tested in anotherindependent population sample consisting of 89 subjects. Originally, the CentralPopulation Registry selected a random sample of 120 subjects from a cohort of allpersons living in Helsinki and born in 1954. The characterization of this study groupincluded an echocardiographic examination of the left ventricle, laboratory tests forblood lipids and an assessment of habitual physical activity, smoking and ethanolconsumption by 2-month daily recording. The set of tests was completed in 1990 (258).
The Valsalva tests with continuous BP recordings had been performed and stored oncomputer discs and analyzed, for the present study (III), in a blinded fashion by the Ouluinvestigators.
4.2. Study designs
The designs of the studies were approved by the Ethical Committees of the Universityof Oulu and the University of Helsinki (III), and all subjects gave their informed consent.The subjects and study designs are shown in Table 1.
Table 1. Subjects and methods in the present study
Design Methodology Setting N
I Randomized case-control HRV in hypertensive and control men 356
II Randomized case-control BRS in hypertensive and control subjects 494
III Cross-sectional BRS and variation in genes encoding RAS in healthy
subjects
315 +
66
IV Double-blind randomized parallel
group trial
Effects of combination antihypertensive therapy on
HRV and BRS in patients with poor BP control on
monotherapy
33
N=number of subjects in the final analyses of the studies
Design 1. For study I, measures of HRV were compared in normotensive andhypertensive males of the Population 1. The possible determinants (demographicvariables, cardiovascular risk factors and life style) of HRV were analyzed. Patients withsymptoms, medication, or ECG evidence of coronary artery disease, atrial fibrillation, ortechnical artefacts or frequent ectopic beats were excluded from the analyses. The finalanalyses included data from 188 normotensive and 168 hypertensive men.
Design 2.For study II, BRS was assessed in the subjects of Population 1. BRS wascompared in normotensive subjects and hypertensive patients receiving antihypertensivemedication, and the determinants of BRS were analyzed. Hypertensive subjects with nomedication and control subjects with hypertensive 24-hour BP values (>142/91) or onantihypertensive treatment were excluded. Also, subjects with diabetes mellitus, previousmyocardial infarction, atrial fibrillation, or frequent ectopic beats or inadequate Valsalva

39
maneuver were excluded. The analyses involved 214 hypertensive and 280 normotensivesubjects.
Design 3. For study III, the control subjects of Population 1 were studied for possibleassociations between BRS and polymorphisms in the genes encoding RAS. Thecandidate genes for potential risk loci for cardiovascular diseases such as angiotensin-converting enzyme (ACE), angiotensinogen (AGT) and aldosterone synthase(CYP11B2), were studied. Subjects with diabetes mellitus, coronary artery disease basedon symptoms, medication, or ECG , atrial fibrillation, frequent ectopic beats, inadequateambulatory BP monitoring or inadequate Valsalva maneuver were excluded, after whicha total of 315 individuals (161 women and 154 men, 60% of the original cohort)remained for analyses.
The study was repeated in an independent population sample (Population 2) ofyounger healthy subjects completely free of cardiac diseases. The subjects in Population2 were genotyped for ACE and CYP11B2 polymorphisms. DNA was available forgenotyping in 84 subjects, and the recordings of the Valsalva maneuver were acceptablefor BRS determination in 70 subjects. Sixty-six subjects (37 women and 29 men) hadboth types of data available for statistical analyses.
Design 4.In order to evaluate whether it is possible to affect BRS and the measures ofHRV by intensification of antihypertensive therapy, 54 patients with poor BP control onmonotherapy and 24-hour BP > 140/90 were screened for study IV (34 subjects from thehypertensive cohort of Population 1 and 20 subjects from an outpatient clinic). Patientswith, renal, hepatic, neurologic, endocrine, or cardiac diseases including atrial fibrillationor frequent ectopic beats, and patients with systolic BP >220 mmHg were excluded.Thepatients were randomized for 2 weeks of monotherapy with either 95 mg metoprolol(Group 1) as a slow-release preparation (Seloken Zoc®, Suomen Astra, Finland) or 20mg of enalapril (Renitec®, MSD)(Group 2). If, at the end of this period on randomizedmonotherapy, the mean of three sitting values of office BP measured with a mercurysphygmomanometer was > 140/90, the patient was allocated to combination treatment for10 weeks (n=33). The dose was titrated according to the study design schema to reach thetarget BP (IV, Fig. 1). The patients randomized to receive metoprolol, continued with acombination therapy of metoprolol and a the long-acting calcium antagonist felodipine(n=16) , while those who started with enalapril continued with enalapril combined withhydrochlorothiazide (n=17). All drugs were administered once daily at about 9 AM afterbreakfast. No other medications affecting BP were allowed. Standard laboratorymeasurements and a brief medical history were documented, 12-lead ECG includingcalculation of Cornell index (RaVL+ SIII ) and a physical examination was done (IV, Fig.1). All analyses were performed without knowledge of the characteristics or other data ofthe patient.
4.3. Blood pressure recordings
An automatic oscillometric recorder (Dinamap® model 18465X, Criticon Ltd., Ascot,UK) was used for the office BP measurements (I, IV). The subjects were seated for atleast 5 minutes, after which BP was measured three times at one-minute intervals. Themean of these measurements were used for the analyses. In the design III, BP was

40
measured similarly to the prosedure used in Population 2, but with a sphygmomanometer(Korotkoff phases I and IV).
Ambulatory BP (II,III,IV) was recorded using the fully automatic SpaceLabs 90207oscillometric unit (SpaceLabs Inc., Redmond, Washington, USA), which was set to takea measurement every 15 minutes from 4:00 AM till midnight and every 20 minutes frommidnight till 4:00 AM. The accuracy and reproducibility of the BP readings obtainedwith this device have been established previously (259,260). The proper positioning ofthe cuff in each case was ensured by means of the similarity (difference < 5 mmHg)between four SpaceLabs BP measurements and four auscultatory readings using a Y-connector. The subjects were asked to relax their arm during the measurement. Values ofsystolic BP <70 or > 250 mmHg, diastolic BP < 40 or > 150, and HR < 40 or >150 beatsper minute were automatically excluded from the analysis. Fewer than 3% of the BPreadings were rejected as artefacts on the basis of these criteria. The cutoff point for poorBP control was chosen to be a 24-hour BP of > 142/91 mmHg (261).
4.4. Echocardiographic methods
A Hewlett-Packard ultrasound color system Sonos 500 (Hewlett-Packard Company,Massachusetts, USA) was used for the echocardiographic examinations. Allexaminations were performed by one experienced cardiologist, who was blinded to theother data and grouping of the study subjects. M-mode measurements were obtainedunder 2-D guidance according to the recommendations of the American Society ofEchocardiography (262). A standard M-mode study was recorded and analyzed(263,264). The LVM was calculated using the formula of Troyet al. (265) (I) orDevereuxet al. (266) (II) and the left ventricular mass index (LVMI) by dividing thedifference between the LVM values by body surface area (determined by Duboisequation (267)). Fractional shortening was calculated by dividing the difference betweenthe left ventricular internal dimensions in diastole and systole by the diastolic dimensionand then multiplying it by 100. Transmitral flow velocities were measured by pulsedDoppler at the tips of the mitral leaflets from an apical transducer position, and the ratioof maximum early and late diastolic filling were calculated (IV).
4.5. Laboratory methods
4.5.1. Biochemical analyses
All the laboratory test samples were obtained after an overnight fast. The routine clinicallaboratory tests, including an oral glucose tolerance test (75g, plasma glucose determinedat 0, 60, 120 minutes) were carried out in the Central Laboratory of the Oulu UniversityHospital, and the lipid analyses were made in the research laboratory of the Departmentof Internal Medicine by using standard methods (I,II,III). For study III, laboratoryanalyses of Population 2 were carried out in the Laboratory of the Helsinki UniversityCentral Hospital.

41
4.5.2. DNA analyses and genotyping subjects (III)
DNA was extracted from whole blood. The insertion/deletion (I/D) polymorphism inintron 16 of the ACE gene (Populations 1 and 2) and the methionine/threoninepolymorphism in codon 235 of AGT (M235T) (Population 1) were detected bypolymerase chain reaction (PCR) using the conditions described previously (237). Forthe ACE polymorphism, the oligonucleotide sequences of the primers were 5’-CTGGAGACCACTCCCATCCTTTCT-3’ (sense) and 5’-GATGTGGCCATCACAT-TCGTCAGAT-3’ (antisense) (268). For the AGT polymorphism, the oligonucleotidesequences of the primers were 5’-CAGGGTGCTGTCCACACTGGACCCC-3’ (sense)and 5’-CCGTTTGTGCAGGGCCTGGCTCTCT-3’ (antisense) (269).
The subjects were also genotyped by PCR for two recently discovered polymorphismsin the aldosterone synthase (CYP11B2) gene (Population 1 and 2) (248,252). Thestructure and orientation of the CYP11B2 gene in relation to CYP11B1 and the twopolymorphisms of CYP11B2 are displayed in Fig. 5. Segments of CYP11B2 wereamplified from approximately 20 ng of each DNA sample by the PCR in 20µl reactionscontaining 0.2 U Taq DNA polymerase, 1 x concentration of the supplied buffer, 0.2 mMof each deoxynucleotide triphosphate, and 10 pmol of each primer. After initialdenaturation at 94°C for 5 min, a manual hot start at 80°C was employed, followed by 35cycles at 94°C for 1 min, annealing at 67 or 68°C for 1 min, and extension at 72°C for 2min.
The subjects were genotyped for the -344 promoter polymorphism using the primersCAGGAGGAGACCCCATGTGAC (sense) and CCTCCACCCTGTTCAGCCC(antisense). Fiveµl from each reaction (consisting mainly of a 537 bp product) wasdigested with 10 U of restriction endonuclease Hae III in the supplied buffer for 2 h at37°C. The reactions were subjected to electrophoresis in 2.5% agarose gels. The -344Tallele lacks the Hae III site (GGCC) present in the -344C allele, and the -344T alleles aretherefore detected as Hae III fragments of 273 bp and the -344C alleles as fragments of202 bp (plus smaller fragments in each case). The subjects were typed for a geneconversion polymorphism in the second intron of CYPIIB2 by allele-specific PCR. Bothreactions used the same antisense primer, AGGAACCTCTGCACGGCC. The reactionsto detect allele 1 with the gene conversion (1013 bp product) also contained the primerCAGAAAATCCCTCCCCCCTA (67°C annealing temperature), whereas the reactions todetect allele 2 lacking the gene conversion (yielding a 1017 bp product) used the senseprimer TGGAGAAAAGCCCTACCCTGT.
4.6. Analysis of heart rate variability
All subjects underwent a 45-minute recording (I) with an ambulatory ECG recorder(Dynacord Holter Recorder, Model 420, DM Scientific, Irvine, CA, USA) for 15 minutesin a supine posture, for 15 minutes in a sitting posture while breathing quietly with afrequency of 15 breaths/minute, and for 15 minutes while walking. The length of therecordings was chosen after a pilot study on 37 normotensive and 40 hypertensivesubjects by calculating the shortest period that would give a reasonable correlation(r=0.6) with the 24-hour measures of HR variability (Table 1). The ECG recordings were

42
performed between 7 AM and 3 PM, and the data were transferred from the Del MarAvionics scanner (Model 500, Del Mar Avionics, Irvine, CA, USA) to a microcomputerfor the analysis of HR variability with the use of a custom-made program (Hearts, HeartSignal Co, Kempele, Finland). A linear detrend was applied to the RR interval data insegments of 512 samples to make it more stationary. Premature beats and noise wereexcluded automatically and manually, and the gaps were refilled with an average valuecomputed in the local neighborhood. The segments with >85% qualified beats wereincluded in the analysis. An autoregressive model was used to estimate the powerdensities of four frequency bands (total power <0.4 Hz, HF power 0.15-0.4 Hz, LF power0.04 to 0.15Hz, and VLF power 0.005 to 0.04Hz) in every 15-minute period of 512beats. The SDANN (I) values from the 45-minute and SDNN from the 24-hour recordingwere used as time domain measures. The average 45-minute and 24-hour SD and thepower spectrum components of HR variability were calculated. Spectral componentswere analyzed as absolute units (ms2)(I,IV), and the low and high-frequency componentswere also analyzed as normalized units (I). Normalized units were used when the changesin HR variability in response to a change in body posture were compared.
4.7. Analysis of baroreflex sensitivity
For baroreflex sensitivity tests, noninvasive arterial BP was measured on a beat-to-beatbasis from the second finger of the left hand using the Finapress finger-cuff method, withthe monitored finger held at heart level. The reliability of the Finapress method has beenpreviously described (270). After data acquisition, the data were stored on computerdisks, and the phenylephrine test, Valsalva maneuver and spontaneous baroreceptorresponsiveness were later analyzed blinded to the other data.
4.7.1. Phenylephrine test (IV)
After a period of rest, when both the BP and HR values were stable, a bolus ofphenylephrine (80-120µg) was injected intravenously via a peripheral vein. The test wasrepeated 2-3 times at 5-minute intervals with an attempt to obtain at least a 15 mmHg risein BP. The ECG and BP data were collected and analyzed using a menu-driven softwarepackage (CAFTS, Medikro Oy, Finland). The slope of the linear relationship between thelength of the RR interval (in msec) and the preceding systolic BP value (in mmHg) fromthe analysis window (from the beat which started a sustained rise in systolic BP to thebeat following the maximal BP elevation) was calculated using linear least-mean-squaresfitting. Only the regression lines with a correlation coefficient >0.8 or significant(p<0.05) and with a BP change > 15 mmHg were accepted for analysis. The BRS wascalculated as the mean of accepted tests. A typical example of the computer output of aphenylephrine test is displayed in Fig. 3.

43
4.7.2. Valsalva maneuver (II, III)
The Valsalva maneuver was performed in a sitting position by blowing into a rubber tubeconnected to an aneroid manometer and maintaining an expiratory strain of 40 mmHg for15 seconds (69). A blow-off valve required the subjects to blow continuously to maintainthe pressure. The subjects were asked to refrain from talking or moving after strainrelease. The test was performed three times at 5 minute intervals in Population 1 and 1-2times in Population 2. The ECG and continuous BP data were stored in a digital formatand analyzed later using a menu-driven software package (CAFTS, Medikro Oy,Finland). A linear least-mean-squares fitting method was employed to calculate BRSfrom the overshoot phase after the Valsalva maneuver as the slope of the linearrelationship between the RR interval (in msec) and the preceding systolic BP value (inmmHg). The slope was determined in a time window ranging from the beat upon whichsystolic BP exceeded the systolic BP level at the end of the Valsalva strain to the beatfollowing the maximum systolic BP overshoot. Only regression lines with a correlationcoefficient >0.7 or p<0.05 and an increase in BP > 15 mmHg were accepted for analysis.BRS was calculated as a mean of the results of all accepted tests. A typical example ofthe computer output of the Valsalva test is displayed in Fig. 3.
4.7.3. Spontaneous baroreceptor responsiveness (II)
A stationary segment (without ectopic beats) of about 5 minutes was selected foranalysis. Spectral analysis of fluctuations in the RR interval and systolic BP wasperformed using a method based on fast Fourier transform (73,74). The spontaneousfluctuations in BP and the corresponding changes in RR interval were assessed by cross-spectral analysis to obtain dynamic baroreceptor responsiveness (72,146). The amount(relative power and timing) of coupling with the systolic BP as the input signal and theRR interval as the output signal was assessed over a range of frequencies of physiologicalinterest. The reliability of the transfer function estimate between the RR interval andsystolic BP signals was assessed by the coherence function, which is analogous to thecorrelation coefficient in regression analysis in the time domain during phenylephrinetesting, except that it is computed for each frequency region. The baroreflex gain(relative power) was calculated as the square root of the ratio between the RR intervaland systolic BP variabilities in the mid-frequency (0.07-0.14 Hz) and respiratoryfrequency (0.15-0.40 Hz) bands. Thus, this relative power function of frequency domainanalysis is analogous to the slope of the regression equation in the time domain analysisduring phenylephrine testing. The relative power calculation become unreliable ifcoherence is low. Hence, only the frequency regions where coherence was > 0.5 wereselected for computation. The phase angle between systolic BP and RR intervalvariabilities was calculated to evaluate the sequence and lag of the changes in the signalsat a given frequency. A negative phase angle suggests that the systolic BP signal precedesthe RR interval, e.g. -90 degrees at 0.25 Hz signifies a 1.0 s phase difference between thesignals. A typical example of the computer output of cross-spectral analysis is displayedin Fig. 4.

44
4.8. Statistical analyses
The Statistical Package for the Social Sciences (SPSS) for Windows, release 6.0, wasused for data analysis. Continuous data are given as means (SD) or medians (range). Thecorrelations between variables were evaluated by Spearman´s correlation coefficient. Achi-square test was used to establish differences in frequencies and to assess the fit of theobserved allele frequencies to the Hardy-Weinberg distribution (III). A two-tailed p value<0.05 was considered statistically significant. Unpaired t-test or Mann-Whitney U-test (incase of skewed data distribution) was used to compare the differences between groups,and paired t-test or Wilcoxon test (in case of skewed data distribution) for comparisonswithin groups. Significant differences in the baseline variables between the groups weretaken into account by means of covariance when comparing the measures of HRvariability between hypertensive and normotensive subjects (I, II).
The coefficient of gametic linkage disequilibrium was calculated by likelihoodmethods, and the coefficient (D) is reported as the ratio of the unstandardized coefficientto its maximal value (271) (III). The differences between the variables in the differentgenotype groups were tested with ANOVA (analysis of variance) or the Kruskal-Wallistest (in case of skewed data distribution). Two-way ANOVA (3x3 factorial design) withBonferroni´s corrections for multiple comparisons was used to test the potentialsynergistic effect of the ACE, AGT and CYP11B2 genes on BRS.
Stepwise linear multiple regression analysis was used to control for confoundingfactors and to assess the quantitative effects of the baseline variables (e.g. age, BP, HR,BMI, physical activity, alcohol consumption, smoking, serum cholesterol level, or the useof estrogen or progesterone therapy) on HRV or BRS. For the regression model in StudyIII, the genotype effect was assumed to be additive (or linear), dominant, or recessive.The regression analysis was made separately for men and women.

5. Results
The demographic variables, BP values, lifestyle data and metabolic measurements of thesubjects in the studies I-IV are shown in Table 2. The correlation coefficients of the 45-minute and 24-hour recordings are shown in Table 3.
Table 3. Correlation coefficients of the 45-minute and 24-hour recordings for calculating the measures of
HRV in normotensive and hypertensive subjects.
Normotensive males
(n = 37)
Hypertensive males
(n= 40)
SDANN (ms) 0.64 0.80
VLF power (ms² x 10) 0.66 0.81
LF power (ms² x 10) 0.67 0.80
HF power (ms² x 10) 0.61 0.78
P<0.001 for all .
5.1. Heart rate variabilit y in systemic hypertension
The mean values of SDANN and the frequency domain measures of HRV expressed asabsolute values are shown in I, Table III . There were significant interindividualvariations in the measures of HRV, but all the measures (except the HF component),including the ratio between the LF and HF components, were significantly lower in thehypertensive than the normotensive men.
Normotensive men showed a significant increase of the normalized LF component(from 64 (12) to 68 (10) nu; p<0.001) and a decrease of the normalized HF component(from 29 (12) to 23 (10); p<0.001) in response to an upright (sitting) posture (I, Fig. 1).In hypertensive men, the normalized LF component (from 62 (12) in the supine vs 63(12) nu in the sitting posture; NS) and the normalized HF component (29 (12) nu in thesupine and 28 (12) nu in the sitting posture; NS) did not change.

Table 2. Characteristics of study populations.
I II III IV
C
(n=188)
H
(n=168)
C
(n=280)
H
(n=214)
Pop. 1
(n=315)
Pop. 2
(n=66)
H
(n=33)
Age (yrs) 50 (6) 50 (6) 50 (6) 50 (6) 50 (6) 36-37 53 (5)
Male sex (%) 100 100 52 56 49 44 64
Body mass index (kg/m2) 26 (3) 29 (4) 26 (4) 28 (4) 26 (6) 25 (3) 28 (5)
Heart rate (bpm) 70 (10) ( 69 (11) 69 (10) 70 (8) 71 (9) 67 (10) 68 (10)
Office systolic BP (mmHg) 146 (19) 158 (18) 140 (20) 156 (19) 142 (20) 127 (14) 172 (19)
Office diastolic BP (mmHg) 89 (9) 98 (10) 84 (12) 95 (12) 85 (12) 82 (9) 100 (9)
Smokers (%) 36 26 49 50 41 41 12
Alcohol consumption (g/day) 73
(0-473)
73
(0-658)
24
(0-387)
28
(0-658)
24
(0-387)
17
(0-75)
Physical activity (†METxh/d) 3.0 (1.0)* 3.0 (0.9)* 3.1 (0.9)* 2.9 (1.0)* 3.0 (1.0)* 2.1†
(0-13.8)
Serum lipids (mmol/l)
Total cholesterol 5.8 (1.1) 5.8 (1.0) 5.6 (1.0) 5.7 (1.0) 5.6 (1.0) 5.4 (0.8) 6.0 (0.9)
HDL cholesterol 1.2 (0.3) 1.2 (0.3) 1.4 (0.4) 1.3 (0.3)‡ 1.4 (0.4) 1.8 (0.4)) 1.3 (0.3)
Triglycerides 1.6 (0.8) 1.9 (1.0)‡ 1.3 (0.6) 1.6 (0.8)‡ 1.3 (0.7) 1.1 (0.6) 2.0 (1.0)
Values are means (SD) of normally distributed variables, medians (range) of asymmetrically distributed variables
or percentage of categorical variables. C, control subjects; H, hypertensive subjects; bpm, beats per minute.
* According to Grimbyet al.(257). † METxh/d, MET indicates metabolic equivalent (energy expenditure of a person
at rest:≈ 1kcal/kg per h). ‡ p<0.05.

After adjustment for the baseline differences in systolic BP and BMI between thegroups, no significant differences persisted in the SDANN (F = 0.5, p = 0.47), absoluteunits of VLF (F = 2.3, p = 0.15), LF (F = 2.3, p = 0.13), or the changes in the normalizedunits of the LF component (F = 2.5, p = 0.11) or the HF component (F = 2.5, p = 0.11)after sitting up.
5.2. Baroreflex sensitivity in systemic hypertension
There were wide interindividual variations of BRS in both normotensive andhypertensive subjects (Fig. 6). The mean BRS assessed both by the Valsalva maneuverand by cross-spectral analysis was significantly lower in the hypertensive patients (7.0 ±4 and 6.0 ± 4 ms/mmHg, respectively) than in their normotensive counterparts (9.9 ± 5and 7.3 ± 4 ms/mmHg, p<0.001 and p<0.05, respectively). The difference in BRSremained significant after adjustment for differences in 24-hour BP, BMI, lipid valuesand echocardiographic measurements (F=17.3, p<0.0001 for Valsalva maneuver andF=5.3, p<0.05 for cross spectral analysis). Hypertensive patients had more often anabnormal BRS (<3ms/mmHg) than normotensive subjects (15% vs. 3% for Valsalva and22% vs. 9% for cross-spectral method, p<0.0001 for both, see Fig. 6). The correlationbetween the two methods used to assess baroreceptor responsiveness was moderate(r=0.38, p<0.0001).
3 5 10 15 20 25
BRS (ms/mmHg)
Prob
abil
ity
0.2
0.4
0.6hypertensives
controls
Fig. 6. BRS (assessed by Valsalva manouver) in control and hypertensive subjects. The curvesare probability density functions calculated by using the means and SDs of the BRS values.The cut-off line for very low BRS (<3 ms/mmHg) is shown.

48
5.3. Determinants of autonomic cardiovascular regulation
5.3.1. Determinants of heart rate variability (I)
The SDANN had significant inverse relations to BMI (r=-0.22, p=0.005), systolic BP(r=-0.20, p=0.008), and waist/hip ratio (r=-0.19, p=0.016) in hypertensive men, and itwas related to BMI (r=-0.20, p=0.005), systolic BP (r=-0.25, p<0.0001), age (r=-0.18,p=0.01) and fractional shortening (r=0.24, p=0.003) in normotensive subjects. Whenhypertensive men were grouped according to their use of antihypertensive medications,the absolute HF component was higher in the men receivingβ-blocking agents than inwithout β-blockers, but no other differences in HRV were seen between the groups. Thechanges in the normalized units of LF and HF components were also related to systolicBP in hypertensive men (r=-0.20, p=0.009), but not in normotensive men.
Multiple regression analysis showed the SDANN to have an independent relation onlyto systolic BP (β=-0.20, p=0.01) in hypertensive men. In normotensive men, it waspredicted by systolic BP (β=0.28, p=0.0002), fractional shortening (β=-0.23, p=0.002),and age (β=-0.21, p=0.005).
5.3.2. Determinants of baroreflex sensitivity (II)
Women with and without hypertension had significantly lower BRS assessed by theValsalva maneuver than men (5.7±3.5 and 8.7±4.6 ms/mmHg vs 8±3.9 and 11.1±4.9ms/mmHg, respectively, p<0.0001) (168), but no gender-related differences in BRSassessed by cross-spectral analysis were observed. In the univariate analysis ofhypertensive patients, age (r=-0.31, p<0.001) was the only significant determinant ofBRS assessed by Valsalva maneuver, and mean HR (r=-0.55, p<0.001) was the onlydeterminant of BRS assessed by cross-spectral analysis. Ambulatory systolic BP (r=-0.06- -0.09, NS) was not related to BRS assessed by either method. Among normotensivesubjects, BRS was inversely related to ambulatory systolic BP (r=-0.19 and -0.41,p<0.001 for both), mean HR (r=-0.30 and -0.45, p<0.001 for both) and age (r=-0.13,p<0.05 and -0.22, p<0.05 for Valsalva and cross-spectral methods, respectively). Inmultivariate regression analysis, BRS assessed by Valsalva maneuver was predicted byambulatory systolic BP (β=-0.21, p=0.0008) and HR (β=-0.16, p=0.0082), and BRSassessed by cross spectral analysis was predicted by ambulatory systolic BP (β=-0.31,p<0.0001), age (β=-0.20, p=0.0028) and HR (β=-0.16, p=0.015).
The BRS values assessed by Valsalva maneuver in quartile groups according toambulatory 24-hour systolic BP in normotensive subjects and their hypertensivecounterparts matched for ambulatory BP are displayed in II, Fig.3. BRS was significantlylower in the treated hypertensive than in the normotensive group in all except the highestBP quartile.
When the patients were grouped according to their use of beta-blockers (8.0 ± 4ms/mmHg), calcium antagonists (7.6 ± 4 mmHg), ACE inhibitors (7.7 ± 4 mmHg),diuretics (6.1 ± 4 ms/mmHg) and combination treatment (6.3 ± 4ms/mmHg), nosignificant differences in BRS (values for Valsalva maneuver) emerged between the

49
groups. Neither alcohol consumption nor smoking habits had any significant influence onBRS.
BRS tended to be lower in the subjects with no physical activity (6.9± 4 ms/mmHg)or mild activity (8.6± 5 ms/mmHg) than in those with moderate (8.8± 5 ms/mmHg) orstrenuous (9.6± 5 ms/mmHg) activity (values for Valsalva maneuver). None of theechocardiographic or metabolic measures had significant association with BRS.
5.3.3. Genetic influences on baroreflex sensitivity (III)
5.3.3.1. Population 1
The main characteristics, 24-hour BP, lifestyle data, and -344C/T genotype in men andwomen are shown in III, Table 1. There were no significant genotypic effects on anyvariables. Similarly, the intron 2 polymorphism of CYP11B2 and the ACE and AGTpolymorphisms had no influence on any of these basic data. The 24-hour systolic(129±12 vs 123±11 mmHg, p<0.001) and diastolic (81±8 vs 77±7 mmHg, p<0.001) BPswere higher in men, but there was no significant sex difference in BMI.
The genotype distributions of all polymorphisms were in agreement with the Hardy-Weinberg equilibrium both in men and in women. The data confirm the linkagedisequilibrium between the two polymorphic loci in the CYP11B2 gene (D=0.48,χ2=47.0, p<0.0001). The genotype distribution of the -344C/T polymorphism in the 72subjects with inadequate Valsalva maneuver was similar to that in the 315 subjects withadequate tests (20 (30%) in -344TT, 39 (45%) in CT and 13 (25%) in CC genotype).
BRS was significantly lower in women than in men (8.6±4.0 and 11.0±4.8 ms/mmHg,respectively, p<0.0001). In women, BRS was related to the CYP11B2 promotergenotype, averaging 10.1±4.5, 8.7±3.8 and 7.1±3.2 ms/mmHg in the -344TT, CT and CCgroups, respectively (p=0.003)(Fig. 7, panel A); a comparable association was observedbetween BRS and the intron 2 polymorphism (1/1: 11.1±4.4; 1/2 8.9±4.1; 2/2: 7.5±3.4ms/mmHg, p=0.002). In men, by contrast, BRS was unrelated to either the promotergenotype (Fig. 7, panel B) or the intron 2 genotype (data not shown) of the CYP11B2gene. In women, the association of BRS with the CYP11B2 promoter genotype wasconsistent with a gene dosage effect, such that BRS increased in a linear relationship withthe number of -344T alleles carried by each woman (F=13.1, df1, p=0.0004, R2=0.08).
There were no significant differences in BRS between the different genotypes of ACEor AGT in women or men. When ACE and AGT were added as factors in ANOVA (3x3factorial design), no significant synergistic effect of the AGT, ACE and -344C/Tgenotypes on BRS was observed.
In univariate analysis, BRS was associated with 24-hour systolic BP in both men (r=-0.22, p<0.05) and women (r=-0.30, p<0.0001). BRS was also weakly related to age inmen (r=-0.18, p<0.05), but not in women (r=-0.13, NS). The association between BRSand the different genotypes of the CYP11B2 -344C/T promoter polymorphism in the agetertiles in women is displayed in Fig. 3 (Fig. 7, panel D). Although the same trends wereseen in all age tertiles, the association was only significant among the women aged 41-46.
In multiple regression analysis of the female population, BRS was related to 24-hour

50
systolic BP (β=0.38, p=0.0067) and the -344C/T genotype of CYP11B2 (CC=1, CT=2,TT=3; β=.37, p=0.008), but was independent of age, BMI, physical activity, dailyalcohol consumption, smoking, serum cholesterol, HR, and the use of estrogen orprogesterone therapy. In men, only 24-hour systolic BP was an independent predictor(β=0.39, p=0.013), while the other variables did not enter the model. Differentassumptions about the type of the genotype effect did not alter the findings.
5.3.3.2. Population 2
There were no significant differences in the basic characteristic or BP across the -344C/T genotype groups. The -344C/T genotypes of 18 subjects with inadequateValsalva maneuver were: 3 (17%) -344TT, 8 (44%) CT and 7 (39%) CC.
In the total population of 37 women and 29 men, ANOVA revealed a significant maineffect of the -344C/T genotype on BRS (Fig. 7, panel D). The data were consistent with agene dosage effect; BRS increased in a linear relationship with the number of -344Talleles carried by each subject (F=21.1, df1, p<0.0001, R2=0.25). The association of BRSwith the -344C/T genotype was statistically significant in both men (TT: 20.2 ± 10.8; CT12.6 ± 6.5; CC: 10.0 ± 2.7 ms/mmHg, p=0.037, ANOVA) and women (TT: 18.1 ± 9.4;CT 13.0 ± 4.0; CC: 9.6 ± 5.0 ms/mmHg, p=0.017, ANOVA).
In women, BRS was also related to the intron 2 polymorphism of CYP11B2,averaging 18.9±10.5, 14.6±6.3 and 10.8±3.9 ms/mmHg in the 1/1, 1/2 and 2/2 genotypegroups, respectively (p=0.036, ANOVA). The comparable trend in men was notstatistically significant (data not shown). There were no differences in BRS between theI/D genotype groups of ACE in either women or men (data not shown). The sexdifference in BRS did not reach statistical significance (14.7±8.3 in men and 13.5±6.3ms/mmHg in women, p=0.47).
In multiple regression analysis, BRS was related to the -344C/T genotype ofCYP11B2 (CC=1, CT=2, TT=3;β=0.49, p<0.001), but was independent of sex, BMI,serum cholesterol, HR, systolic BP, LVM, average daily physical activity, smoking andaverage daily alcohol consumption.
5.3.4. Influence of antihypertensive treatment on autonomiccardiovascular regulation (IV)
The effects of intensified antihypertensive treatment on HRV and BRS were examined in33 patients. The main characteristics and casual BP values of the two study groups aresummarized in IV, Table I and the 24-hour BP values in IV, Table II. Echocardiographicdata is shown in IV, Table III. A significant reduction in LVM was observed, but the leftventricular diameter and systolic function remained unaltered by the treatment. Thereduction of LVM was also reflected as lowered ECG voltages and a reduced voltageindex of Cornell (Table 4).

51
A B
0
4
8
12
16
20
Bar
oref
lex
Sen
sitiv
ity,m
s. mm
Hg-1
-344TT -344CT -344CC
P=.003*
0
4
8
12
16
20
24
Bar
ore
flex
Se
nsiti
vity
,ms. m
mH
g-1-344TT -344CT -344CC
C D
0
10
20
30
40
50
Bar
oref
lex
Sen
sitiv
ity,m
s. mm
Hg-1
-344TT -344CT -344CC
P=.0003*
2
4
6
8
10
12
41-46 y 47-53 y 54-62 y
P=.004*
ns
ns
n=
12
n=30
n=11
n=15
n=26
n=13
n=12
n=23
n=
19
0
-344
TT
-344
TT
-344
TT
-34
4C
T
-34
4CC
-34
4C
T
-344
CC
-34
4C
T
-344
CC
Bar
ore
flex
Sen
sitiv
ity,m
s. mm
Hg-1
Fig. 7. Individual data on BRS in relation to the -344C/T polymorphism in the promoter ofCYP11B2 in women (Panel A) and in men (Panel B) in Population 1 and in Population 2(Panel C). Panel D, BRS (mean±±±±SEM) by the -344C/T polymorphism in the promoter ofCYP11B2 in age tertiles in women in Population 1. Horizontal lines indicate group meanvalues. *ANOVA.
The effects of the two combination therapies on HRV and BRS are displayed in IV,Table III. Individual BRS and mean BP values on monotherapy and after combinationtherapy in both study groups are displayed in IV, Fig. 2. The changes in BRS or themeasures of HRV were not significantly related to the initial 24-hour BP or to thereduction of 24-hour BP or regression of LVM.

52
Table 4. Electrocardiographic data of the study groups.
Group 1
(n=16)
Group2
(n=17)
Metoprolol
95 mg
Metoprolol +
felodipine
Enalapril
20mg
Enalapril +
HCTZ
Electrocardiography (mV)
RaVL 7.2 (3.5) 6.3 (3.4)* 7.8 (4.4) 6.1 (3.7)*
RV5 17.5 (6.3) 15.5 (4.5) 15.3 (5.3) 15.5 (5)
RV6 15.6 (3.8) 14.2 (3.5) 14.2 (4.8) 13.6 (4.3)
SIII 4.6 (3.8) 4.2 (3.5) 5.5 (3.6) 4.0 (3.3)*
SV1 8.8 (3.2) 7.6 (2.8)† 9.3 (2.9) 8.3 (2.9)*
SV3 6.6 (5.2) 5.3 (4.7) 7.7 (3.4) 7.9 (4.8)
Cornell index (RaVL+ SIII ) 13.8 (7.0) 11.5 (5.2)* 15.5 (4.6) 14.0 (5.6)
Values are expressed as mean (SD).
HCTZ = hydrochlorothiazide.*P<0.05,†P<0.01 between monotherapy and combination therapy.

6. Discussion
Abnormal cardiovascular neural regulation may be a potential trigger of adverse cardiacevents observed in hypertensive patients. However, there is only limited informationavailable on the presence of abnormalities in HRV or BRS in these patients. The presentstudy is the first large-scale population-based attempt to evaluate HRV and BRS inpatients on long-term antihypertensive treatment. The previous analyses of autonomiccardiovascular regulation in small numbers of nonrandomly selected hypertensivesubjects have yielded variable results (8-12,19,114,115,128,178,272). None of thesestudies adjusted their results for the differences in baseline variables, such as BP andobesity. Furthermore, the effects of age and gender have been largely ignored in thesesurveys.
The present study is the first to suggest that variation in a specific gene may influenceinterindividual variation in BRS.
This study also indicate that the abnormalities in autonomic cardiovascular regulationare not irreversible phenomenon in patients with long-standing systemic hypertension,but can be partly restored by modern antihypertensive therapy.
6.1. Reduced heart rate variabilit y in patients with systemichypertension
The present study showed that the measures of HRV are reduced in treated hypertensivesubjects relative to their age-matched normotensive counterparts. Also, hypertensivesubjects have blunted autonomic responses to changes in body posture as compared tonormotensive subjects. Because significant sex-related differences are known to occur inthe autonomic regulation of HR (168), only men were included in the study.
Concurrent with the present findings, Chakko et al. reported that HRV is reduced inhypertensive patients with evidence of LVH, and Radaelli et al. reported that theresponses of normalized LF and HF components to head-up tilting are attenuated inmedicated patients with hypertension as compared with age-matched normotensivesubjects. The reduction of the LF component observed in that study is also consistentwith the results of experimental (273) and clinical (11,12,19,115,178) studies which haveshown abnormalities in BRS in hypertensive patients. The LF component of HRV has

54
been suggested to reflect partly BRS (30,274).In contrast with the present findings, studies utilizing normalized units to determine
sympathovagal balance have shown increased normalized LF power and decreasednormalized HF power in hypertensive patients (9,109). The discrepancies between theseresult and the present findings may reflect the differences in autonomic cardiovascularregulation in recently diagnosed borderline compared to long-standing hypertension.Antihypertensive medication may also explain some of the findings. On the other hand,the value of normalized units and the ratio of LF to HF power as an index ofsympathovagal balance has recently been questioned (36).
6.2. Impaired baroreflex sensitivity in patients with systemichypertension
Previous smaller studies have suggested that BRS is lower in patients with establishedsystemic hypertension without antihypertensive medication than in normotensive subjects(11,12,106). Abnormalities in baroreflex function have also been demonstrated inborderline hypertension and it has been suggested that these abnormalities might evenprecede the onset of hypertension and contribute to its pathogenesis (19,178). Thepresent study is the first population-based study on BRS in systemic hypertension toconfirm that BRS is also impaired in the patients with long-standing hypertension despiteadequate antihypertensive treatment. The prevalence of abnormal BRS was fivefold intreated middle-aged hypertensive patients compared to their age- and sex-matchednormotensive counterparts.
There are at least two potential explanations which could contribute to the finding thatBRS is not normalized by optimal antihypertensive therapy. First, during thedevelopment and progression of hypertension, several structural alterations, such asmedial hypertrophy and endothelial damage, occur in the vessel wall (133). Thesestructural alterations may become irreversible and contribute to the permanent nature ofaltered baroreceptor sensitivity. In this respect it is noteworthy that in experimentalhypertension, an early introduction of antihypertensive therapy improves BRS more thana late initiation of treatment (275). Clinical data on the potential advantages of earlytreatment are, however, lacking. The other possibility is that defective baroreflexbuffering of BP might be a primary abnormality and one of the pathophysiologicalmechanisms leading to the development of essential hypertension (19). This concept issupported by the previous observation on the heredity of impaired BRS in hypertensivesubjects (123).
The prognostic significance of depressed BRS in patients with hypertension is notknown. It is, however, an independent marker of high mortality in patients withmyocardial infarction (7,18). Importantly, experimental data have shown that both BRSand HRV measured after myocardial infarction predict the occurrence of postinfactionischemia-induced ventricular fibrillation, but, at variance with HRV, BRS also predictspostinfarction ischemia-induced ventricular fibrillation when measured beforemyocardial infarction (207). Thus, treated hypertensive patients with abnormal BRS maybe at a higher risk for life-threatening arrhytmias and mortality during or after anischemic cardiac event.

55
6.3. Determinants of autonomic cardiovascular regulation in patientswith systemic hypertension
Since reduced overall HRV and BRS are associated with an increased risk of overall andcardiac mortality in various populations (3,4,6,7), it is important to know which factorsare responsible for these alterations. After adjustment for systolic BP and BMI, nodifferences were observed in measures of HRV between the hypertensive andnormotensive subjects (I), showing that poorly controlled hypertension and obesity arethe major determinants of reduced overall HRV in the OPERA population. However, thecorrelations were relatively weak, suggesting that the wide interindividual variation in themeasures of HRV is partly also due to other factors (not measured here). Notably, HRVwas not related to aging, leisure-time physical activity, smoking or LVM, all of whichhave been previously proposed to influence the cardiovascular autonomic regulation inthe hypertensive population (14,128,176,180,186).
At variance with HRV, BRS was also reduced in hypertensive subjects afteradjustment for BP and obesity, suggesting that impaired BRS may be a primary featurerelated to systemic hypertension (II). Earlier studies in untreated hypertensive subjectshave shown that BRS decreases with increasing BP and advancing age (11,115,178). Inthe present study, a similar association was demonstrated for age in both hypertensiveand normotensive subjects, but for BP only in the normotensive group. Thus,antihypertensive therapy may fade out the direct effect of high BP on BRS. Previousstudies have also revealed weak associations between cardiovascular risk factors, such assmoking habits and lipid status, and autonomic modulation of HR (14,15,17), but wewere unable to demonstrate such correlations with BRS. Interestingly, one marker of end-organ damage, LVH, was not associated with blunting of BRS. Physical activity may alsoimprove BRS (184), but the present cross-sectional study did not demonstrate significantdifferences in BRS between the groups representing different intensities of leisure-timephysical activity.
Antihypertensive medication may contribute to the observed reductions in HRV (I)and BRS (II) in hypertensive patients. According to previous studies, however,medication should improve rather than attenuate HRV or BRS (144-147,151,153,276). Inthe present study (I), the patients receivingβ-blocking medication had a somewhat higherHF component of HRV. This may explain why the HF-component did not differ betweenhypertensive and normotensive subjects. No differences were observed in HRV or BRSbetween patients with other types of medication, although the number of patients in thetreatment groups may have been too small for statistically valid comparisons of drugeffects. Intensification of the treatment by two commonly used drug combinations (IV),however, improved significantly HRV and BRS in patients with long-standinghypertension and poor BP control on monotherapy. The observed effect tended to bemore obvious in the patients with the lowest BRS values at baseline (IV, Fig. 2). There isalso increasing evidence to suggest that impaired cardiovascular autonomic regulationmay predict mortality and adverse events in patients without documented ischemic heartdisease (4,6). The recent HOT study showed that the substantial BP reduction achievedwith the antihypertensive agents used in the present study was associated with a reducedincidence of cardiovascular events (277). It is plausible that the improved BRS and HRVachieved by combination therapy may partly contribute to the beneficial prognostic

56
effects in patients with systemic hypertension.
6.4. Genetic variation in renin-angiotensin-aldosterone system genesand baroreflex sensitivity
Thus far, there is limited information available concerning the effects of CYP11B2 genevariation on cardiovascular structure and function (211,248). A recent report of studies inPopulation 2 suggested that the -344CC genotype is a powerful predictor of increasedLVM and relatively impaired left ventricular diastolic function (252). The relationshipbetween BRS and the CYP11B2 genotypes observed in the present study was, however,independent of LVM. Thus, the -344CC genotype may associate independently with lowBRS and LVH, both of which predict increased cardiovascular mortality (7,18,278-280).Data from previous clinical studies have not provided definitive answers concerning therole of polymorphisms of the ACE and AGT genes in BP regulation or othercardiovascular disorders, such as LVH or cardiac events (239,281). In line with thesestudies, we did not find any significant association between either ACE or AGTgenotypes and BRS.
The association of BRS with CYP11B2 gene variation was initially observed inPopulation 1. Since multiple testing for genetic associations may notoriously lead tochance findings even in non-mixed populations, we repeated the study in a fullyindependent population-based sample of younger healthy subjects (Population 2). Insupport of our primary data, an even stronger association of BRS with the CYP11B2genotypes was observed, this time not only in women, but also in men. These findings arephysiologically relevant, since aging and BP have independent major effects on BRS(11,18,60,177,199), and the influence of polymorphisms on BRS may be filtered outupon aging in subjects with higher BP values (281).
The genotypic effects were analyzed separately for both sexes, because severalprevious studies have shown significant gender-related differences in autonomiccardiovascular regulation in various populations (94,168,169,171,282). In Population 1,an association between BRS and the -344C/T genotypes was observed only amongwomen and, furthermore, mainly in those younger than 46 years, who werepredominantly premenopausal. The observed sex difference in the genomic effect amongmiddle-aged subjects is physiologically plausible in the light of the prior studies andanalogous to the well-established gender difference in BRS and BP regulation (168-171,282). Sex hormones may modulate the effects of circulating angiotensin II,aldosterone and cortisol at the receptor level in the central nervous system and interactwith components of tissue RAS in the brain, heart, vascular bed and adrenals (225-228).We do not know why there was a sex difference in the association between BRS and the -344C/T genotypes in Population 1, but not in Population 2. However, the subjects inPopulation 1 were older and heterogeneous for age, and their BP and BMI were higher,which differences may have modified the genetic influences on BRS.
The molecular mechanisms underlying the association between BRS and CYP11B2polymorphism cannot be deduced from this study. Unfortunately, plasma concentrationsof RAS hormones were not available for the populations we studied. However, it isreasonable to speculate that one of the CYP11B2 polymorphisms we examined, or an

57
additional polymorphism in linkage disequilibrium with it, alters CYP11B2 geneexpression, i.e. plasma aldosterone synthesis, while aldosterone levels, in turn, affectBRS. The -344C/T polymorphism lies in an element that is essential for the expression ofthe corresponding murine and bovine genes (283-285), but it is not known whether thiselement affects the expression of CYP11B2 in humans. Recently, however, Pojogaet al.reported data indicating that the plasma aldosterone concentration is dependent upon the-344C/T genotype in hypertensive individuals, being clearly highest in subjectshomozygous for the -344C allele (253). Interestingly, intravenous infusion of aldosteroneimpairs BRS in both animal models and man (140,141).
6.5. Possible limitations of the study
Due to exclusions made in Designs 1,2 and 4, the populations in the present study maynot purely represent the large original populations of the OPERA. Also, the influence ofantihypertensive medication on the results might be unpredictable in these studies. On theother hand, one of the main purposes was to evaluate whether patients onantihypertensive medication have abnormalities in cardiovascular autonomic regulation.Furthermore, based on previous studies, the medication should improve rather thanattenuate HRV and BRS in hypertensive patients (144,145,149,151-154,286).
Design 1. A 45-minute standardized period was used to calculate the differentmeasures of HRV. Although previous prognostic studies have been performed bycalculating the 24-hour HRV, in line with previous studies (92,99), we found areasonably good correlation between the 45-minute and 24-hour measurements (Table 3).Furthermore, Dekkeret al. has showed that decreased HRV determined from short-termrecording predicts mortality in middle-aged population (103). Our method of recordingdata in different positions served as an effort to mimic the habitual activities during theday.
Design 2 and 3.Prognostic information on the role of baroreflex function is based onphenylephrine-based BRS measurements (7,18). The predictive significance of BRS dataobtained from a Valsalva maneuver is unknown, but the measurements by means of theValsalva maneuver and cross-spectral analysis are reproducible and correlate closelywith indexes derived using the invasive phenylephrine method (66-69,72,73). Indeed, thestimulus to arterial baroreceptors obtained by the relatively high expiratory pressure (40mmHg) in the Valsalva maneuver is close to the stimulus obtained by a phenylephrineinjection (68), and when the baroreflex slope is analyzed, as in the present study, fromthe overshoot phase of the Valsalva maneuver (67), it is likely that the results of thesetwo methods are comparable. It is important to point out that we only analyzed subjectswithout apparent cardiac diseases, which improves the accuracy and eliminates pitfalls ofthe Valsalva test (71). Both measurements of BRS used in the present study represent amore physiological challenge for the baroreflex than the pharmacological methods and,are, owing to their noninvasive nature, more suitable for large-scale population surveys.However, the results of the Valsalva maneuver and cross-spectral analysis may not befully interchangeable, because the intrathoracic stretch receptors and increased venousreturn in the Valsalva maneuver may modify the cardiovascular reflexes independent ofthe carotid baroreflexes (60).

58
Design 3.Theoretically, it is possible that the medication used before randomizationmay cause bias to the results by carry-over effect. However, it is likely that 2 weeksrandomized monotherapy in the parallel design is long enough to avoid this problem inthe study III (287).
Design 4.It is possible that the CYP11B2 polymorphisms we examined are merelymarkers for one or more additional nearby polymorphisms that actually mediate theobserved effects on BRS. There might be a nonrandom association between the ”BRS”allele of such a hypothetical locus and the -344C allele of CYP11B2, a condition termedgenetic linkage disequilibrum. It is, however, even theoretically possible thatpolymorphisms in CYP11B2 are responsible for the observed findings (250,288).

7. Conclusions
1. Low-frequency spectral components (LF and VLF) of HRV are reduced in treatedhypertensive patients.
2. Despite adequate antihypertensive therapy, BRS is impaired in subjects with long-standing systemic hypertension as compared with age-matched normotensive subjects.
3. Poorly controlled hypertension and obesity are the major determinants of HRV in ahypertensive population. In contrast, BRS is reduced in hypertensive subjects even afteradjustment for BP and obesity, suggesting that impaired BRS is a primary feature relatedto hypertension and probably partly determined by genetic factors.
4. The common genetic polymorphisms in the aldosterone synthase gene (CYP11B2)predict interindividual variation in BRS, explaining up to 25% of the variation of BRS. Itseems that the observed associations are stronger in younger subjects and diminish earlierin aging men.
5. In spite of genetic influences, abnormal cardiovascular autonomic regulation is not anirreversible phenomenon in patients with long-standing systemic hypertension. It wasshown that appropriate modern antihypertensive combination therapy not only improvespoor BP control and induces regression of LVH, but also improves cardiovascularautonomic regulation.

8. References
1. MacMahon S, Peto R, Cutler J, Collins R, Sorlie P, Neaton J, Abbott R, Godwin J, Dyer A &Stamler J (1990) Blood pressure, stroke, and coronary heart disease. Part 1, Prolongeddifferences in blood pressure: prospective observational studies corrected for the regressiondilution bias. Lancet 335: 765-774.
2. Julius S (1991) Autonomic nervous system dysregulation in human hypertension. Am JCardiol 67: 3B-7B.
3. Kleiger RE, Miller JP, Bigger JT, Jr., Moss AJ & The multicenter post-infarction researchgroup. (1987) Decreased heart rate variability and its association with increased mortalityafter acute myocardial infarction. Am J Cardiol 59: 256-262.
4. Tsuji H, Venditti FJ, Jr., Manders ES, Evans JC, Larson MG, Feldman CL & Levy D (1994)Reduced heart rate variability and mortality risk in an elderly cohort. The Framingham HeartStudy. Circulation 90: 878-883.
5. Algra A, Tijssen JGP, Roelandt JRTC, Pool J & Lubsen J (1993) Heart rate variability from24-hour electrocardiography and the 2-year risk for sudden death. Circulation 88: 180-185.
6. Huikuri HV, Mäkikallio TH, Airaksinen KEJ, Seppänen T, Puukka P, Räihä IJ & SouranderLB (1998) Pover-law relationship of heart rate variability as a predictor of mortality in theelderly. Circulation 97: 2031-2036.
7. La Rovere MT, Bigger JT, Jr., Marcus FI, Mortara A, Schwartz PJ & for ATRAMIInvestigators (1998) Baroreflex sensitivity and heart-rate variability in prediction of totalcardiac mortality after myocardial infarction. Lancet 351: 478-484.
8. Mancia G, Ferrari A, Gregorini L, Parati G, Pomidossi G, Bertinieri G, Grassi G, Di RienzoM, Pedotti A & Zanchetti A (1983) Blood pressure and heart rate variabilities innormotensive and hypertensive human subjects. Circ Res 53: 96-104.
9. Guzzetti S, Piccaluga E, Casati R, Cerutti S, Lombardi F, Pagani M & Malliani A (1988)Sympathetic predominance in essential hypertension: a study employing spectral analysis ofheart rate variability. J Hypertens 6: 711-717.
10. Furlan R, Guzzetti S, Crivellaro W, Dassi S, Tinelli M, Baselli G, Cerutti S, Lombardi F,Pagani M & Malliani A (1990) Continuous 24-hour assessment of the neural regulation ofsystemic arterial pressure and RR variabilities in ambulant subjects. Circulation 81: 537-547.
11. Gribbin B, Pickering TG, Sleight P & Peto R (1971) Effect of age and high blood pressureon baroreflex sensitivity in man. Circ Res 29: 424-431.
12. Takeshita A, Tanaka S, Kuroiwa A & Nakamura M (1975) Reduced baroreceptor sensitivityin borderline hypertension. Circulation 51: 738-742.
13. Eckberg DL & Orshan CR (1977) Respiratory and baroreceptor reflex interactions in man. JClin Invest 59: 780-785.
14. Molgaard H, Hermansen K & Bjerregaard P (1994) Spectral components of short-term RRinterval variability in healthy subjects and effects of risk factors. Eur Heart J 15: 1174-1183.
15. Kupari M, Virolainen J, Koskinen P & Tikkanen MJ (1993) Short-term heart rate variabilityand factors modifying the risk of coronary artery disease in a population sample. Am JCardiol 72: 897-903.

61
16. Huikuri HV, Kessler KM, Terracall E, Castellanos A, Linnaluoto MK & Myerburg RJ (1990)Reproducibility and circadian rhythm of heart rate variability in healthy subjects. Am JCardiol 65: 391-393.
17. Pikkujämsä SM, Huikuri HV, Ikäheimo MJ, Airaksinen KEJ, Rantala AO, Lilja M,Savolainen MJ, Reunanen A & Kesäniemi YA (1996) Relationship between heart ratevariability and cardiovascular risk factors in middle-aged males. A N E 1: 354-362.
18. La Rovere MT, Specchia G, Mortara A & Schwartz PJ (1988) Baroreflex sensitivity, clinicalcorrelates, and cardiovascular mortality among patients with a first myocardial infarction. Aprospective study. Circulation 78: 816-824.
19. Eckberg DL (1979) Carotid baroreflex function in young men with borderline blood pressureelevation. Circulation 59: 632-636.
20. Schwartz PJ & La Rovere MT (1998) ATRAMI: a mark in the quest for the prognostic valueof autonomic markers. Eur Heart J 19: 1593-1595.
21. Reid IA (1992) Interactions between ANG II, sympathetic nervous system, and baroreceptorreflexes in regulation of blood pressure. Am J Physiol 262: E763-E778.
22. Julius S (1988) Interaction between renin and the autonomic nervous system in hypertension.Am Heart J 116: 611-616.
23. Hales S. (1733) Haemostaticks. Statistical Essays, vol II. London, Innings & Manby &Woodward.
24. Appel ML, Berger RD, Saul JP, Smith JM & Cohen RJ (1989) Beat to beat variability incardiovascular variables: noise or music? J Am Coll Cardiol 14: 1139-1148.
25. Hon EH (1998) The electronic evaluation of fetal heart rate. Preliminary report. Am J ObstetGynecol 75: 1215-1230.
26. Kay SM & Marple SL (1981) Spectrum analysis-a modern perspective. Proc IEEE 69: 1380-1384.
27. Akselrod S, Gordon D, Ubel FA, Shannon DC, Berger AC & Cohen RJ (1981) Powerspectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascularcontrol. Science 213: 220-222.
28. Task Force of the European Society of Cardiology and the North American Society of Pacingand Electrofysiology (1996) Heart rate variability: standards of measurement, physiologicalinterpretation, and clinical use. Circulation 93: 1043-1065.
29. Malik M, Farrell T, Cripps T & Camm AJ (1989) Heart rate variability in relation toprognosis after myocardial infarction: selection of optimal processing techniques. Eur Heart J10: 1060-1074.
30. Öri Z, Monir G, Weiss J, Sayhouni X & Singer DH (1992) Heart rate variability. Frequencydomain analysis. Cardiol Clin 10: 499-537.
31. Saul JP, Berger RD, Albrecht P, Stein SP, Chen MH & Cohen RJ (1991) Transfer functionanalysis of the circulation: unique insights into cardiovascular regulation. Am J Physiol 261:H1231-H1245.
32. Bigger JT, Jr., Fleiss JL, Steinman RC, Rolnitzky LM, Kleiger RE & Rottman JN (1992)Frequency domain measures of heart period variability and mortality after myocardialinfarction. Circulation 85: 164-171.
33. Pomeranz B, Macaulay RJB, Caudill MA, Kutz I, Adam D, Gordon D, Kilborn KM, BargerAC, Shannon DC, Cohen RJ & Benson H (1985) Assessment of autonomic function inhumans by heart rate spectral analysis. Am J Physiol 248: H151-H153.
34. Pagani M, Lombardi F, Guzzetti S, Rimoldi O, Furlan R, Pizzinelli P, Sandrone G, MalfattoG, Dell'Orto S, Piccaluga E, Turiel M, Baselli G, Cerutti S & Malliani A (1986) Powerspectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagalinteraction in man and conscious dog. Circ Res 59: 178-193.
35. Malliani A, Pagani M, Lombardi F & Cerutti S (1991) Cardiovascular neural regulationexplored in the frequency domain. Circulation 84: 482-492.
36. Eckberg DL (1997) Sympathovagal balance: a critical appraisal. Circulation 96: 3224-3232.37. Huikuri HV, Seppänen T, Koistinen MJ, Airaksinen KEJ, Ikäheimo MJ, Castellanos A &
Myerburg RJ (1996) Abnormalities in beat-to-beat dynamics of heart rate before thespontaneous onset of life-threatening ventricular tachyarrhytmias in patients with priormyocardial infarction. Circulation 312: 170-177.
38. Tulppo M, Mäkikallio TH, Takala TES, Seppänen T & Huikuri HV (1996) Quantitative beat-to-beat analysis of heart rate dynamics during exercise. Am J Physiol 271: H244-H252.
39. Goldberger AL & West BJ (1987) Applications of nonlinear dynamics to clinical cardiology.Ann New York Acad Sci 504: 155-212.

62
40. Mäkikallio TH, Seppänen T, Niemelä M, Airaksinen KE, Tulppo M & Huikuri HV (1996)Abnormalities in beat to beat complexity of heart rate dynamics in patients with a previousmyocardial infarction. J Am Coll Cardiol 28: 1005-1011.
41. Eckberg DL (1983) Human sinus arrhythmia as an index of vagal cardiac outflow. J ApplPhysiol 54: 961-966.
42. Hayano J, Sakakibara Y, Yamada A, Yamada M, Mukai S, Fujinami T, Yokoyama K,Watanabe Y & Takata K (1991) Accuracy of assessment of cardiac vagal tone by heart ratevariability in normal subjects. Am J Cardiol 67: 199-204.
43. Hirsch JA & Bishop B (1981) Respiratory sinus arrhythmia in humans: how breathingpattern modulates heart rate. Am J Physiol 241: H620-H629.
44. Goldberger JJ, Ahmed MW, Parker MA & Kadish AH (1994) Dissociation of heart ratevariability from parasympathetic tone. Am J Physiol 266: H2152-H2157.
45. Malik M & Camm AJ (1993) Components of heart rate variability - what they really meanand what we really measure. Am J Cardiol 72: 821-822.
46. Pagani M, Mazzuero G, Ferrari A, Liberati D, Cerutti S, Vaitl D, Tavazzi L & Malliani A(1991) Sympathovagal interaction during mental stress. A study using spectral analysis ofheart rate variability in healthy control subjects and patients with a prior myocardialinfarction. Circulation 83: II43-II51.
47. Van de Borne P, Montano N, Pagani M, Oren R & Somers VK (1997) Absense of lowfrequency variability of sympathetic nerve activity in severe heart failure. Circulation 95:1449-1454.
48. Arai Y, Saul JP, Albrecht P, Hartley LH, Lilly LS, Cohen RJ & Colucci WS (1989)Modulation of cardiac autonomic activity during and immediately after exercise. Am JPhysiol 256: H132-H141.
49. Sleight P, La Rovere MT, Mortara A, Pinna G, Maestri R, Leuzzi S, Bianchini B, Tavazzi L& Bernardi L (1995) Physiology and pathophysiology of heart rate and blood pressurevariability in humans: is power spectral analysis largely an index of baroreflex gain? Clin Sci88: 103-109.
50. Piepoli M, Sleight P, Leuzzi S, Valle F, Spadacini G, Passino C, Johnston J & Bernardi L(1997) Origin of respiratory sinus arrhythmia in conscious humans. An important role forarterial carotid baroreceptors. Circulation 95:1813-1821.
51. Cooley RL, Montano N, Cogliati C, Van de Borne P, Richenbacher W, Oren R & SomersVK (1998) Evidence for a central origin of the low-frequency oscillation in RR-intervalvariability. Circulation 98: 556-561.
52. Taylor JA, Carr DL, Myers CW & Eckberg DL (1998) Mechanisms underlying very-low-frequency RR-interval oscillations in humans. Circulation 98: 547-555.
53. Kitney RI (1975) An analysis of the nonlinear behaviour of the human thermal vasomotorcontrol system. J Theor Biol 52: 231-248.
54. Kirchheim HR (1976) Systemic arterial baroreceptor reflexes. Physiol Rev 56:100-176.55. Brown AM (1980) Receptorsunder pressure: an update on baroreceptors. Circ Res 46: 156. Eckberg DL & Sleight P (1992) Human baroreflexes in health and disease. Oxford, England:
Clarendon Press57. Mark AL (1983) The Bezold-Jarisch reflex revisited: clinical implications of inhibitory
reflexes originating in the heart. J Am Coll Cardiol 1: 90-102.58. Eckberg DL (1977) Baroreflex inhibition of the human sinus node: importance of stimulus
intensity, duration, and rate of pressure change. J Physiol 269: 561-577.59. Mancia G, Ferrari A, Gregorini L, Parati G, Ferrari MC, Pomidossi G & Zanchetti A (1979)
Control of blood pressure by carotid sinus baroreceptors in human beings. Am J Cardiol 44:895-902.
60. Goldstein DS, Horwitz D & Keiser HR (1982) Comparison of techniques for measuringbaroreflex sensitivity in man. Circulation 66: 432-439.
61. Stevens PM (1960) Cardiovascular dunamics during orthostasis and the influence ofintravascular instrumentation. Am J Cardiol 17: 211-218.
62. Takeshita A, Mark AL, Eckberg DL & Abboud FM (1979) Effect of central venous pressureon arterial baroreflex control of heart rate. Am J Physiol 236: H42-H47.
63. Sullebarger JT, Liang CS, Woolf PD, Willick AE & Richeson JF (1990) Comparison ofphenylephrine bolus and infusion methods in baroreflex measurements. J Appl Physiol 69:962-967.
64. Sarnoff SJ, Hardenbergh E & Whittenberger JL (1948) Mechanism of the arterial pressureresponse to the Valsalva test: the basis for its use as an indicator of the intactness of thesympathetic outflow. Am J Physiol 154: 316-327.

63
65. Smith ML, Beightol LA, Fritsch-Yelle JM, Ellenbogen KA, Porter TR & Eckberg DL (1996)Valsalva's maneuver revisited: a quantitative method yielding insights into human autonomiccontrol. Am J Physiol 271: H1240-H1249.
66. Palmero HA, Caeiro TF, Iosa DJ & Bas J (1981) Baroreceptor reflex sensitivity indexderived from phase 4 of the Valsalva manoeuvre. Hypertension 3: 134-137.
67. Smith SA, Stallard TJ, Salih MM & Littler WA (1987) Can sinoaortic baroreseptor heart ratereflex sensitivity be determined from phase IV of the Valsalva manoeuvre? Cardiovasc Res21: 422-427.
68. Trimarco B, Volpe M, Ricciardelli B, Vigorito C, De Luca N, Sacca L & Condorelli M(1983) Valsalva maneuver in the assessment of baroreflex responsiveness in borderlinehypertensives. Cardiology 70: 6-14.
69. Airaksinen KE, Hartikainen JE, Niemelä MJ, Huikuri HV, Mussalo HM & Tahvanainen KU(1993) Valsalva manoeuvre in the assessment of baroreflex sensitivity in patients withcoronary artery disease. Eur Heart J 14: 1519-1523.
70. Kautzner J, Hartikainen JE, Camm AJ & Malik M (1996) Arterial baroreflex sensitivityassessed from phase IV of the Valsalva maneuver. Am J Cardiol 78: 575-579.
71. Eckberg DL (1980) Parasympathetic cardiovascular control in human disease: a criticalreview of methods and results. Am J Physiol 239: H581-H593.
72. Airaksinen KEJ, Tahvanainen KUO, Kuusela TA, Huikuri HV, Niemelä MJ, Karjalainen P &Eckberg DL (1997) Cross spectral analysis in assessment of baroreflex gain in patients withcoronary artery disease. A N E 2: 229-235.
73. Robbe HW, Mulder LJ, Ruddel H, Langewitz WA, Veldman JB & Mulder G (1987)Assessment of baroreceptor reflex sensitivity by means of spectral analysis. Hypertension 10:538-543.
74. deBoer RW, Karemaker JM & Strackee J (1987) Hemodynamic fluctuations and baroreflexsensitivity in humans: a beat-to-beat model. Am J Physiol 253: H680-H689.
75. Blaber AP, Yamamoto Y & Hughson RL (1995) Methodology of spontaneus baroreflexrelationship assessed by surrogate data analysis. Am J Physiol 268: H1682-H1687.
76. Patton DJ, Triedman JK, Perrott MH, Vidian AA & Saul JP (1996) Baroreflex gain:characterization using autoregressive moving average analysis. Am J Physiol 270: H1240-H1249.
77. Bristow JD, Honour AJ, Pickering GW, Sleight P & Smyth HS (1969) Diminished baroreflexsensitivity in high blood pressure. Circulation 39: 48-54.
78. Eckberg DL, Drabinsky M & Braunwald E (1971) Defective cardiac parasympathetic controlin patients with heart disease. N Engl J Med 285: 877-883.
79. Smyth HS, Sleight P & Pickering GW (1969) Reflex regulation of arterial pressure duringsleep in man. A quantitative method of assessing baroreflex sensitivity. Circ Res 24: 109-121.
80. Eckberg DL, Cavanaugh MS, Mark AL & Abboud FM (1975) A simplified neck suctiondevice for activation of carotid baroreceptors. J Lab Clin Med 85:167-173.
81. Steptoe A & Vögele C (1990) Cardiac baroreflex function during postural change assessedusing non-invasive spontaneous sequence analysis in young men. Cardiovasc Res 24: 627-632.
82. Parati G, Di Rienzo M, Bertinieri G, Pomidossi G, Casadei R, Groppelli A, Pedotti A,Zanchetti A & Mancia G (1988) Evaluation of the baroreceptor-heart rate reflex by 24-hourintra-arterial blood pressure monitoring in humans. Hypertension 12: 214-222.
83. Taylor JA & Eckberg DL (1996) Fundamental relations between short-term RR interval andarterial pressure oscillations in humans. Circulation 93: 1527-1532.
84. Parlow J, Viale JP, Annat G, Hughson R & Quintin L (1995) Spontaneous cardiac baroreflexin humans. Comparison with drug-induced responses. Hypertension 25: 1058-1068.
85. Chang PC, Kriek E & van Brummelen P (1994) Sympathetic activity and presynapticadrenoceptor function in patients with longstanding essential hypertension. J Hypertens 12:179-190.
86. Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL & Esler MD (1994)Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerveactivity during human sympathetic nervous activation and failure. Circulation 90: 234-240.
87. Mäntysaari M, Kuikka J, Mustonen J, Tahvanainen K, Vanninen E, Länsimies E & UusitupaM (1992) Noninvasive detection of cardiac sympathetic nervous dysfunction in diabeticpatients using [1231]metaiodobenzylguanidine. Diabetes 41: 1069-1075.
88. Hagbarth KE & Vallbo ÅB (1968) Pulse and respiratory grouping of sympathetic impulses inhuman peripheral nerves. Acta Physiol Scand 74: 96-108.

64
89. Delius W, Hagbarth KE, Hongell A & Wallin BG (1972) Manoeuvres affecting sympatheticoutflow in human muscle nerves. Acta Physiol Scand 84: 82-94.
90. Parati G, Ravogli A, Frattola A, Groppelli A, Ulian L, Santucciu C & Mancia G (1994)Blood pressure variability: clinical implications and effects of antihypertensive treatment. JHypertens 12: S35-40.
91. Floras JS, Hassan MO, Jones JV, Osikowska BA, Sever PS & Sleight P (1988) Factorsinfluencing blood pressure and heart rate variability in hypertensive humans. Hypertension11: 273-281.
92. Bigger JT, Fleiss JL, Rolnitzky LM & Steinman RC (1993) The ability of several short-termmeasures of RR variability to predict mortality after myocardial infarction. Circulation 88:927-934.
93. Kleiger RE, Bigger JT, Bosner MS, Chung MK, Cook JR, Rolnitzky LM, Steinman R &Fleiss JL (1991) Stability over time of variables measuring heart rate variability in normalsubjects. Am J Cardiol 68: 626-630.
94. Van Hoogenhuyze D, Weinstein N, Martin GJ, Weiss JS, Schaad JW, Sahyoni N, Fintel D,Remme WJ & Singer DH (1991) Reproducibility and relation to mean heart rate of heart ratevariability in normal subjects and in patients with congestive heart failure secondary tocoronary artery disease. Am J Cardiol 68: 1668-1676.
95. Bigger JT, Fleiss JL, Rolnitzky LM, Steinman RC & with data contributed by the CAPS andESVEM Investigators (1992) Stability over time of heart period variability in patients withprevious myocardial infarction and ventricular arrhytmias. Am J Cardiol 69: 718-723.
96. Freed LA, Stein KM, Gordon M, Urban M & Kligfield P (1994) Reproducibility of powerspectral measures of heart rate variability obtained from short-term sampling periods. Am JCardiol 74: 972-973.
97. Ahmed MW, Kadish AH, Parker MA & Goldberger JJ (1994) Effect of physiologic andpharmacologic adrenergic stimulation on heart rate variability. J Am Coll Cardiol 24: 1082-1090.
98. Pitzalis MV, Mastropasqua F, Massari F, Forleo C, Di Maggio M, Passantio A, Colombo R,Di Biase M & Rizzon P (1996) Short- and long-term reproducibility of time and frequencydomain heart rate variability measurements in normal subjects. Cardiovasc Res 32: 226-233.
99. Dekker JM, de Vries EL, Lengton RR, Schouten EG, Swenne CA & Maan A (1996)Reproducibility and comparability of short- and long-term heart rate variability measures inhealthy young men. A N E 1: 287-292.
100. Kroll DJ, Freed LA, Stein KM, Borer JS & Kligfield P (1996) Rhythm annotation andinterobserver reproducibility of measures of heart rate variability. Am J Cardiol 78: 1050-1057.
101. Bigger JT, La Rovere MT, Steinman RC, Fleiss JL, Rottman JN, Rolnitzky LM & SchwartzPJ (1989) Comparison of baroreflex sensitivity and heart period variability after myocardialinfarction. J Am Coll Cardiol 14: 1511-1518.
102. Levin AB (1966) A simple test of cardiac function based upon the heart rate changes inducedby the Valsalva maneuver. Am J Cardiol 18: 90-99.
103. Dekker JM, Schouten EG, Kloowijk P, Pool J, Swenne CA & Kromhout D (1997) Heart ratevariability from short electrocardiographic recordings predicts mortality from all causes inmiddle-aged and elderly men. Am J Epidemiol 145: 899-908.
104. Julius S & Conway J (1968) Hemodynamic studies in patients with borderline blood pressureelevation. Circulation 38: 282-288.
105. Esler M, Julius S, Zweifler A, Randall O, Harburg E, Gardiner H & DeQuattro V (1977)Mild high-renin essential hypertension. Neurogenic human hypertension? N Engl J Med 296:405-411.
106. Goldstein DS (1983) Arterial baroreflex sensitivity, plasma catecholamines, and pressorresponsiveness in essential hypertension. Circulation 68: 234-240.
107. Anderson EA, Sinkey CA, Lawton WJ & Mark AL (1989) Elevated sympathetic nerveactivity in borderline hypertensive humans. Evidence from direct intraneural recordings.Hypertension 14: 177-183.
108. Esler M, Jennings G, Biviano B, Lambert G & Hasking G (1986) Mechanism of elevatedplasma noradrenaline in the course of essential hypertension. J Cardiovasc Pharmacol 8: S39-S43.
109. Guzzetti S, Dassi S, Balsama M, Ponti GB, Pagani M & Malliani A (1994) Altered dynamicsof the circadian relationship between systemic arterial pressure and cardiac sympathetic driveearly on in mild hypertension. Clin Sci 86: 209-215.

65
110. Julius S, Pascoe L & London R (1971) Role of parasympathetic inhibition in the hyperkinetictype of borderline hypertension. Circulation 44: 413-418.
111. Korner PI, Shaw J, Uther JB, West MJ, McRitchie RJ & Richards JG (1972) Autonomic andnon-autonomic circulatory components in essential hypertension in man. Circulation 48:107-111.
112. Langewitz W, Ruddel H & Schachinger H (1994) Reduced parasympathetic cardiac controlin patients with hypertension at rest and under mental stress. Am Heart J 127: 122-128.
113. Pagani M, Furlan R, Pizzinelli P, Crivellaro W, Cerutti S & Malliani A (1989) Spectralanalysis of R-R and arterial pressure variabilities to assess sympatho-vagal interaction duringmental stress in humans. J Hypertens 7: S14-5.
114. Dassi S, Balsama M, Guzzetti S, Ponti GB, Magni L, Pagani M & Malliani A (1991)Twenty-four-hour power spectral analysis of heart rate variability and of arterial pressurevalues in normotensive and hypertensive subjects. J Hypertens 9: S72-S73.
115. Sleight P (1979) Reflex control of the heart. Am J Cardiol 44: 889-894.116. Nowak SJ (1940) Chronic hypertension produced by carotid sinus and aortic nerve section.
Ann Surg 111: 102-111.117. Green MF, DeGroat AF & McDonald CH (1935) Observations on denervation of the carotid
sinuses and section of the depressor nerves as a method of producing arterial hypertension.Am J Physiol 110: 513-520.
118. Carey RM, Dacey RG, Jane JA, Winn HR, Ayers CR & Tyson GW (1979) Production ofsustained hypertension by lesions in the nucleus tractus solitarii of the American foxhound.Hypertension 1: 246-254.
119. Gordon FJ, Matsuguchi H & Mark AL (1981) Abnormal baroreflex control of heart rate inprehypertensive and hypertensive Dahl genetically salt-sensitive rats. Hypertension 3: I135-41.
120. Mancia G, Ludbrook J, Ferrari A, Gregorini L & Zanchetti A (1978) Baroreceptor reflexes inhuman hypertension. Circ Res 43: 170-177.
121. Grassi G, Cattaneo BM, Seravalle G, Lanfranchi A & Mancia G (1998) Baroreflex control ofsympathetic nerve activity in essential and secondary hypertension. Hypertension 31: 68-72.
122. Noll G, Wenzel RR, Schneider M, Oesch V, Binggeli C, Shaw S, Weidmann P & Luscher TF(1996) Increased activation of sympathetic nervous system and endothelin by mental stress innormotensive offspring of hypertensive parents. Circulation 93: 866-869.
123. Parmer RJ, Cervenka JH & Stone RA (1992) Baroreflex sensitivity and heredity in essentialhypertension. Circulation 85: 497-503.
124. Liao D, Cai J, Barnes RW, Tyroler HA, Rautaharju P, Holme I & Heiss G (1996) Associationof cardiac autonomic function and the development of hypertension. The ARIC Study. Am JHypertens 9: 1147-1156.
125. Singh JP, Larson MG, Tsuji H, Evans JC, O'Donnel CJ & Levy D (1998) Reduced heart ratevariability and new-onset hypertension: insights into pathogenesis of hypertension: theFramingham Heart Study. Hypertension 32: 293-297.
126. Eich RH, Cuddy RP, Smulyan H & Lyons RH (1966) Hemodynamics in labile hypertension:a follow-up study. Circulation 34: 299-307.
127. Mark AL (1996) The sympathetic nervous system in hypertension: a potential longtermregulator of arterial pressure. J Hypertens 14: S159-S165.
128. Chakko S, Mulingtapang RF, Huikuri HV, Kessler KM, Materson BJ & Myerburg RJ (1993)Alterations in heart rate variability and its circadian rhythm in hypertensive patients with leftventricular hypertrophy free of coronary artery disease. Am Heart J 126: 1364-1372.
129. Odemuyiwa O, Farrell TG, Malik M, Bashir Y, Millane T, Cripps T, Poloniecki J, Bennett D& Camm AJ (1992) Influence of age on the relation between heart rate variability, leftventricular ejection fraction, frequency of ventricular extrasystoles, and sudden death aftermyocardial infarction. Br Heart J 67: 387-391.
130. Andresen MC & Yang M (1989) Arterial baroreceptor resetting: contributions of chronic andacute processes. Clin Exp Pharmacol Physiol Suppl 15: 19-30.
131. Andresen MC & Yang MY (1989) Rapid baroreceptor resetting is unaltered by chronichypertension in rats. Am J Physiol 256: H1228-H1235.
132. Chapleau MW, Hauduczok G & Abboud F (1988) Mechanisms of resetting of arterialbaroreceptors: an overview. Am J Med Sci295: 327-334.
133. Kingwell BA, Cameron JD, Gillies KJ, Jennings GL & Dart AM (1995) Arterial compliancemay influence baroreflex function in athletes and hypertensives. Am J Physiol 268: H411-H418.

66
134. Hartikainen J, Fyhrquist F, Tahvanainen K, Länsimies E & Pyörälä K (1995) Baroreflexsensitivity and neurohormonal activation in patients with acute myocardial infarction. BrHeart J 74: 21-26.
135. Carretta R, Bardelli M, Cominotto F, Ussi D, Fazio M, Fabris B, Fischetti F & Campanacci L(1996) Relationship between mechanical properties of the carotid artery wall and baroreflexfunction in acutely treated hypertensive patients. J Hypertens 14: 1105-1110.
136. Canessa M, Adragna N, Solomon HS, Connolly TM & Tosteson DC (1980) Increasedsodium-lithium countertransport in red cells of patients with essential hypertension. N Engl JMed 302: 772-776.
137. Herrera VL & Ruiz-Opazo N (1990) Alteration of alpha 1 Na+,K(+)-ATPase 86Rb+ influxby a single amino acid substitution. Science 249: 1023-1026.
138. Brooks VL & Reid IA (1986) Interaction between angiotensin II and the baroreceptor reflexin the control of adrenocorticotropic hormone secretion and heart rate in conscious dogs.Circ Res 58: 816-828.
139. Segar JL, Merrill DC & Robillard JE (1994) Role of endogenous ANG II on resetting arterialbaroreflex during development. Am J Physiol 266: H52-H59.
140. Wang W (1994) Chronic administration of aldosterone depresses baroreceptor reflexfunction in the dog. Hypertension 24: 571-575.
141. Yee KM& Struthers AD. Aldosterone causes baroreceptor dysfunction in man. (Abstract). JAm Coll Cardiol 1998;31:507A
142. Brooks VL, Ell KR & Wright RM (1993) Pressure-independent baroreflex resettingproduced by chronic infusion of angiotensin II in rabbits. Am J Physiol 265: H1275-H1282.
143. Funder JW (1996) Mineralocorticoid receptors in the central nervous system. J SteroidBiochem Mol Biol 56: 179-183.
144. Eckberg DL, Abboud FM & Mark AL (1976) Modulation of carotid baroreflexresponsiveness in man: effects of posture and propranolol. J Appl Physiol 41: 383-387.
145. Krediet RT & Dunning AJ (1979) Baroreflex sensitivity in hypertension during beta-adrenergic blockade. Br Heart J 41: 106-110.
146. Lucini D, Pagani M, Mela GS & Malliani A (1994) Sympathetic restraint of baroreflexcontrol of heart period in normotensive and hypertensive subjects. Clin Sci 86: 547-556.
147. Lucini D, Pagani M & Malliani A (1993) Improved baroreflex control of the heart rate withchronic beta-adrenergic blockade in mild hypertension. J Hypertens 11: S156-S157.
148. Keeley EC, Page RL, Lange RA, Willard JE, Landau C & Hillis LD (1996) Influence ofmetoprolol on heart rate variability in survivors of remote myocardial infarction. Am JCardiol 77: 557-560.
149. Parati G, Mutti E, Frattola A, Castiglioni P, Di Rienzo M & Mancia G (1994) Beta-adrenergic blocking treatment and 24-hour baroreflex sensitivity in essential hypertensivepatients. Hypertension 23: 992-996.
150. Vesalainen RK, Kantola IM, Airaksinen KE, Tahvanainen KU & Kaila TJ (1998) Vagalcardiac activity in essential hypertension: the effects of metoprolol and ramipril. Am JHypertens 11: 649-658.
151. Mancia G, Parati G, Pomidossi G, Grassi G, Bertinieri G, Buccino N, Ferrari A, Gregorini L,Rupoli L & Zanchetti A (1982) Modification of arterial baroreflexes by captopril in essentialhypertension. Am J Cardiol 49: 1415-1419.
152. Bonaduce D, Marciano F, Petretta M, Migaux ML, Morgano G, Bianchi V, Salemme L,Valva G & Condorelli M (1994) Effects of converting enzyme inhibition on heart periodvariability in patients with acute myocardial infarction. Circulation 90: 108-113.
153. Ebert TJ (1985) Captopril potentiates chronotropic baroreflex responses to carotid stimuli inhumans. Hypertension 7: 602-606.
154. Egan BM, Fleissner MJ, Stepniakowski K, Neahring JM, Sagar KB & Ebert TJ (1993)Improved baroreflex sensitivity in elderly hypertensives on lisinopril is not explained byblood pressure reduction alone. J Hypertens 11: 1113-1120.
155. Dibner-Dunlap ME, Smith ML, Kinugawa T & Thames MD (1996) Enalaprilat augmentsarterial and cardiopulmonary baroreflex control of sympathetic nerve activity in patients withheart failure. J Am Coll Cardiol 27: 358-364.
156. Chapleau MW, Cunningham JT, Sullivan MJ, Wachtel RE & Abboud FM (1995) Structuralversus functional modulation of the arterial baroreflex. Hypertension 26: 341-347.
157. Kingwell BA, Krause L & Julius S (1994) The effect of hypertensive episodes and cardiachypertrophy on the canine cardiac baroreflex. Clin Exp Pharmacol Physiol 21: 31-39.

67
158. Ichikawa M, Suzuki H, Kumagai K, Ryuzaki M, Kumagai H, Nishizawa M & Saruta T(1995) Baroreceptor function is restored by antihypertensive therapy through lowering ofblood pressure in adult SHR. Clin Exp Pharmacol Physiol 22: S67-S69.
159. Parati G & Mancia G (1993) Calcium antagonists in the treatment of arterial hypertension.Am Heart J 125: 642-648.
160. Goldsmith SR (1995) Effect of amlodipine and felodipine on sympathetic activity andbaroreflex function in normal humans. Am J Hypertens 8: 902-908.
161. Kassis E & Amtorp O (1987) Cardiovascular and neurohumoral postural responses andbaroreceptor abnormalities during a course of adjunctive vasodilator therapy with felodipinefor congestive heart failure. Circulation 75: 1204-1213.
162. Nader PC, Thompson JR & Alpern RJ (1988) Complications of diuretic use. Semin Nephrol8: 364-387.
163. Guo GB & Abboud FM (1984) Angiotensin II attenuates baroreflex control of heart rate andsympathetic activity. Am J Physiol 246: H80-H89.
164. Pedretti RFE, Colombo E, Braga SS, Ballardini L & Caru B (1995) Effects of oralpirenzepine on heart rate variability and baroreceptor reflex sensitivity after myocardialinfarction. J Am Coll Cardiol 4: 915-921.
165. Casadei R, Conway J, Forfar C & Sleight P (1996) Effect of low doses of scopolamine onRR interval variability, baroreflex sensitivity, and exercise performance in patients withchronic heart failure. Heart 75: 274-280.
166. Vesalainen RK, Kaila TJ, Kantola IM, Tahvanainen KU, Airaksinen KEJ, Kuusela TA &Eckberg DL (1998) Low-dose transdermal scopolamine decreases blood pressure in mildessential hypertension. J Hypertens 16: 321-329.
167. Hull SS, Jr., Vanoli E, Adamson PB, De Ferrari GM, Foreman RD & Schwartz PJ (1995) Doincreases in markers of vagal activity imply protection from sudden death? The case ofscopolamine. Circulation 91: 2516-2519.
168. Huikuri HV, Pikkujämsä SM, Airaksinen KE, Ikäheimo MJ, Rantala AO, Kauma H, Lilja M& Kesäniemi YA (1996) Sex-related differences in autonomic modulation of heart rate inmiddle-aged subjects. Circulation 94: 122-125.
169. Cerati D, Mortara A, La Rovere MT, Marcus FI, Malik M, Nohara R& Mazzuero G.Influence of gender on autonomic markers and risk after myocardial infarction in theATRAMI study. (Abstract). Circulation 1996;94:I-490
170. Chen YF (1996) Sexual dimorphism of hypertension. Curr Opin Nephrol Hypertens 5: 181-185.
171. Abdel-Rahman AR, Merrill RH & Wooles WR (1994) Gender-related differences in thebaroreceptor reflex control of heart rate in normotensive humans. J Appl Physiol 77:606-613.
172. Liao D, Barnes RW, Chambless LE, Simpson RJJ, Sorlie P, Heiss G & for the ARICInvestigators (1995) Age, race, and sex differences in autonomic cardiac function measuredby spectral analysis of heart rate variability: the ARIC study. Am J Cardiol 76: 906-912.
173. Laitinen T, Hartikainen J, Vanninen E, Niskanen L, Geelen G & Länsimies E (1998) Age andgender dependency of baroreflex sensitivity in healthy subjects. J Appl Physiol 84: 576-583.
174. Greenland P, Reicher-Reiss H, Goldbourt U, Behar S & and Israeli SPRINT Investigators(1991) In-hospital and 1-year mortality in 1524 women after myocardial infarction:comparison with 4315 men. Circulation 83: 484-491.
175. Marrugat J, Sala J, Masia R, Pavesi M, Sanz G, Valle V, Molina L, Seres L, Elosua R & forthe RESCATE Investigators (1998) Mortality differences between men and women followingfirst myocardial infarction. JAMA 280: 1405-1409.
176. O'Brien IAD, O'Hare P & Corall RJM (1986) Heart rate variability in healthy subjects: effectof age and the deviation of normal ranges for tests of autonomic function. Br Heart J 55: 248-354.
177. Hartikainen J, Mäntysaari M, Mussalo H, Tahvanainen K, Länsimies E & Pyörälä K (1994)Baroreflex sensitivity in men with recent myocardial infarction; impact of age. Eur Heart J15: 1512-1519.
178. Bristow JD, Gribbin B, Honour AJ, Pickering TG & Sleight P (1969) Diminished baroreflexsensitivity in high blood pressure and ageing man. J Physiol 202: P45-P46.
179. Lipsitz LA & Goldberger AL (1992) Loss of "complexity" and ageing. Potential applicationsof fractals and chaos theory to senescence. JAMA 267: 1806-1809.
180. Goldsmith RL, Bigger JT, Steinman RC & Fleiss JL (1992) Comparison of 24-hourparasympathetic activity in endurance-trained and auntrained young men. J Am Coll Cardiol20: 552-558.

68
181. Zahorska-Markiewicz B, Kuagowska E, Kucio C & Klin M (1993) Heart rate variability inobesity. Int J Obes Relat Metab Disord 17: 21-23.
182. Tulppo MP, Mäkikallio TH, Seppänen T, Laukkanen RJ & Huikuri HV (1998) Vagalmodulation of heart rate during exercise: effects of age and physical fitness. Am J Physiol274: H424-H429.
183. Barney JA, Ebert TJ, Groban L, Farrell PA, Hughes CV & Smith JJ (1988) Carotidbaroreflex responsiveness in high-fit and sedentary young men. J Appl Physiol 65: 2190-2194.
184. Somers VK, Conway J, Johnston J & Sleight P (1991) Effects of endurance training onbaroreflex sensitivity and blood pressure in borderline hypertension. Lancet 337: 1363-1368.
185. Sowers JR, Whitfield LA, Catania RA, Stern N, Tuck ML, Dornfeld L & Maxwell M (1982)Role of the sympathetic nervous system in blood pressure maintenance in obesity. ClinEndocrinol Metab 54: 1181-1186.
186. Hayano J, Yamada M, Sakakibara Y, Fujinami T, Yokoyama K, Watanabe Y & Takata K(1990) Short- and long-term effects of cigarette smoking on heart rate variability. Am JCardiol 65: 84-88.
187. Murray A, Ewing DJ, Campbell IW, Neilson JM & Clarke BF (1975) RR interval variationsin young male diabetics. Br Heart J 37: 882-885.
188. Ewing DJ, Campbell JIW & Clarke BF (1980) Assessment of cardiovascular effects indiabetic neuropathy and prognostic implications. Ann Intern Med 92: 308-311.
189. Huikuri HV (1995) Heart rate variability in coronary artery disease. J Inter Med 237: 349-357.
190. Airaksinen KE, Ikäheimo MJ, Linnaluoto MK, Niemelä MJ & Takkunen JT (1987) Impairedvagal heart rate control in coronary artery disease. Br Heart J 58: 592-597.
191. Niemelä MJ, Airaksinen KE, Ikäheimo MJ, Groundstroem K, Linnaluoto MK & TakkunenJT (1989) Impaired parasympathetic control of heart rate after myocardial infarction. Int JCardiol 24: 305-309.
192. Cohn JN, Levine TB, Olivari MT, Gartberg V, Lura D, Francis GS, Simon AB & Rector T(1984) Plasma norepinephrine as a guide to prognosis in patients with chronic congestiveheart failure. N Engl J Med 311: 819-823.
193. Dzau VJ, Colucci WS, Hollenberg NK & Williams GH (1981) Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 63:645-651.
194. Brouwer J, van Veldhuisen DJ, Man in 't Veld AJ, Haaksma J, Dijk WA, Visser KR,Boomsma F, Dunselman PHJM, Lie KI & for the Dutch ibopamine multicenter trial studygroup (1996) Prognostic value of heart rate variability during long-term follow-up in patientswith mild to moderate heart failure. J Am Coll Cardiol 28: 1183-1189.
195. Osterziel KJ, Hanlein D, Willenbrock R, Eichhorn C, Luft F & Dietz R (1995) Baroreflexsensitivity and cardiovascular mortality in patients with mild to moderate heart failure. BrHeart J 73: 517-522.
196. Lombardi F & Mortara A (1998) Heart rate variability and cardiac failure. Heart 80: 213-214.
197. Wijbenga JAM, Balk AHMM, Meij SH, Simoons ML & Malik M (1998) Heart ratevariability index in congestive heart failure: relation to clinical variables and prognosis. EurHeart J 19: 1719-1724.
198. Wolf MM, Varigos GA, Hunt D & Sloman JG (1978) Sinus arrhythmia in acute myocardialinfarction. Med J Austral 2: 52-53.
199. Farrell TG, Odemuyiwa O, Bashir Y, Cripps TR, Malik M, Ward DE & Camm AJ (1992)Prognostic value of baroreflex sensitivity testing after acute myocardial infarction. Br Heart J67: 129-137.
200. Hohnloser SH, Klingenheben T, van de Loo A, Hablawetz E, Just H & Schwartz PJ (1994)Reflex versus tonic vagal activity as a prognostic parameter in patients with sustainedventricular tachycardia or ventricular fibrillation. Circulation 89: 1068-1073.
201. Lown B & Verrier RL (1976) Neural activity and ventricular fibrillation. N Engl J Med 294:1165-1170.
202. Valkama JO, Huikuri HV, Airaksinen KE, Linnaluoto MK & Takkunen JT (1993) Changesin frequency domain measures of heart rate variability in relation to the onset of ventriculartachycardia in acute myocardial infarction. Int J Cardiol 38: 177-182.
203. Huikuri HV, Valkama JO, Airaksinen KE, Seppänen T, Kessler KM, Takkunen JT &Myerburg RJ (1993) Frequency domain measures of heart rate variability before the onset of

69
nonsustained and sustained ventricular tachycardia in patients with coronary artery disease.Circulation 87: 1220-1228.
204. Valkama JO, Huikuri HV, Koistinen MJ, Yli-Mayry S, Airaksinen KE & Myerburg RJ(1995) Relation between heart rate variability and spontaneous and induced ventriculararrhythmias in patients with coronary artery disease. J Am Coll Cardiol 25: 437-443.
205. Huikuri HV, Koistinen MJ, Yli-Mäyry S, Airaksinen KE, Seppänen T, Ikäheimo MJ &Myerburg RJ (1995) Impaired low-frequency oscillations of heart rate in patients with prioracute myocardial infarction and life-threatening arrhythmias. Am J Cardiol 76: 56-60.
206. Bigger JT, Jr., Fleiss JL, Kleiger R, Miller JP, Rolnitzky LM & The MulticenterPostinfarction Research Group. (1984) The relationships among ventricular arrhythmias, leftventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation69: 250-258.
207. Schwartz PJ, Vanoli E, Stramba-Badiale M, De Ferrari GM, Billman GE & Foreman RD(1988) Autonomic mechanisms and sudden death. New insights from analysis ofbaroreceptor reflexes in conscious dogs with and without a myocardial infarction. Circulation78: 969-979.
208. Hull SS, Jr., Evans JC, Vanoli E, Adamson PB, Stramba-Badiale M, Albert DE, Foreman RD& Schwartz PJ (1990) Heart rate variability before and after myocardial infarction inconscious dogs at high and low risk of sudden cardiac death. J Am Coll Cardiol 16: 978-985.
209. De Ferrari GM, Landolina M, Mantica M, Manfredini R, Schwartz PJ & Lotto A (1995)Baroreflex sensitivity, but not heart rate variability, is reduced in patients with life-threatening ventricular arrhythmias long after myocardial infarction. Am Heart J 130: 473-480.
210. White PC (1996) Inherited forms of mineralocorticoid hypertension. Hypertension 28: 927-936.
211. White PC (1994) Disorders of aldosterone biosynthesis and action. N Engl J Med 331: 250-258.
212. Dzau VJ (1988) Circulating versus local renin-angiotensin system in cardiovascularhomeostasis. Circulation 77: I4-I13.
213. Harris RC (1996) The macula densa: recent developments. J Hypertens 14:815-822.214. Bunag RD, Page IH & McCubbin JW (1966) Neural stimulation of release of renin. Circ Res
19: 851-858.215. Gutmann FD, Tagawa H, Haber E & Barger AC (1973) Renal arterial pressure, renin
secretion, and blood pressure control in trained dogs. Am J Physiol 224: 66-72.216. Duprez DA, De Sutter JH, De Buyzere ML, Rietzschel ER, Rimbaut S, Kaufman JM, Van
Hoecke MJ & Clement DL (1995) Renin-angiotensin-aldosterone system, RR interval, andblood pressure variability during postural changes in borderline arterial hypertension. Am JHypertens 8: 683-688.
217. Erdös EG (1990) Angiotensin I converting enzyme and the changes in our concepts throughthe years. Hypertension 16: 363-370.
218. Ferrario CM, Jaiswal N, Yamamoto K, Diz DI & Schiavone MT (1991) Hypertensivemechanisms and converting enzyme inhibitors. Clin Cardiol 14: S56-S62.
219. Fyhrquist F, Metsärinne K & Tikkanen I (1995) Role of angiotensin II in blood pressureregulation and in the pathophysiology of cardiovascular disorders. J Hum Hypertens 9: S19-S24.
220. Bishop VS, Ryuzaki M, Cai Y, Nishida Y & Cox BF (1995) Angiotensin II-dependenthypertension and the arterial baroreflex. Clin Exp Hypertens 17: 29-38.
221. Joy MD & Lowe RD (1970) Evidence that the area postrema mediates the centralcardiovascular response to angiotensin II. Nature 228: 1303-1304.
222. Matsukawa S & Reid IA (1990) Role of the area postrema in the modulation of thebaroreflex control of heart rate by angiotensin II. Circ Res 67: 1462-1473.
223. Phillips MI (1987) Functions of angiotensin in the central nervous system. Annu Rev Physiol49: 413-435.
224. Diz DI, Barnes KL & Ferrario CM (1986) Contribution of the vagus nerve to angiotensin IIbinding sites in the canine medulla. Brain Res Bull 17: 497-505.
225. Myles K & Funder JW (1996) Progesterone binding to mineralocorticoid receptors: in vitroand in vivo studies. Am J Physiol 270: E601-E607.
226. Fisher ND, Ferri C, Bellini C, Santucci A, Gleason R, Williams GH, Hollenberg NK & SeelyEW (1997) Age, gender, and non-modulation. A sexual dimorphism in essentialhypertension. Hypertension 29: 980-985.

70
227. Hopkins PN, Lifton RP, Hollenberg NK, Jeunemaitre X, Hallouin MC, Skuppin J, WilliamsCS, Dluhy RG, Lalouel JM, Williams RR & Williams GH (1996) Blunted renal vascularresponse to angiotensin II is associated with a common variant of the angiotensinogen geneand obesity. J Hypertens 14: 199-207.
228. Funder JW (1995) Mineralocorticoid receptors and hypertension. J Steroid Biochem MolBiol 53: 53-55.
229. Wehling M, Neylon CB, Fullerton M, Bobik A & Funder JW (1995) Nongenomic effects ofaldosterone on intracellular Ca2+ in vascular smooth muscle cells. Circ Res 76: 973-979.
230. Mongeau J (1989) Heridity and blood pressure. Semin Nephrol 9: 208-216.231. Harrap SB (1996) Cardiovascular disease and genetics of the renin-angiotensin system. Heart
76: 13-17.232. Bonnardeaux A, Davies E, Jeunemaitre X, Fery I, Charru A, Clauser E, Tiret L, Cambien F,
Corvol P & Soubrier F (1994) Angiotensin II type 1 receptor gene polymorphisms in humanessential hypertension. Hypertension 24: 63-69.
233. Jeunemaitre X, Soubrier F, Kotelevtsev YV, Lifton RP, Williams CS, Charru A, Hunt SC,Hopkins PN, Williams RR, Lalouel JM & et al. (1992) Molecular basis of humanhypertension: role of angiotensinogen. Cell 71: 169-180.
234. Watt GC, Harrap SB, Foy CJ, Holton DW, Edwards HV, Davidson HR, Connor JM, LeverAF & Fraser R (1992) Abnormalities of glucocorticoid metabolism and the renin-angiotensinsystem: a four-corners approach to the identification of genetic determinants of bloodpressure. J Hypertens 10: 473-482.
235. Kunz R, Kreutz R, Beige J, Distler A & Sharma AM (1997) Association between theangiotensinogen 235T-variant and essential hypertension in whites: a systematic review andmethodological appraisal. Hypertension 30: 1331-1337.
236. Harrap SB (1994) Hypertension: genes versus environment. Lancet 344: 169-171.237. Kiema T, Kauma H, Rantala AO, Lilja M, Reunanen A, Kesäniemi YA & Savolainen MJ
(1996) Variation at the angiotensin-converting enzyme gene and angiotensinogen gene lociin relation to blood pressure. Hypertension 28: 1070-1075.
238. Schunkert H, Danser AH, Hense HW, Derkx FH, Kurzinger S & Riegger GA (1997) Effectsof estrogen replacement therapy on the renin-angiotensin system in postmenopausal women.Circulation 95: 39-45.
239. Soubrier F, Nadaud S & Williams TA (1994) Angiotensin I converting enzyme gene:regulation, polymorphism and implications in cardiovascular diseases. Eur Heart J 15: S24-S29.
240. Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P & Soubrier F (1990) Aninsertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting forhalf the variance of serum enzyme levels. J Clin Invest 86: 1343-1346.
241. Zee RYL, Lou Y, Griffiths LR & Morris BJ (1992) Association of a polymorphism of theangiotensin I-converting enzyme gene with essential hypertension. Biochem Biophys ResCommun 184: 9-15.
242. Morris BJ, Zee RYL & Schrader AP (1994) Different frequencies of angiotensin-convertingenzyme genotypes in older hypertensive individuals. J Clin Invest 94: 1085-1089.
243. Kupari M, Perola M, Koskinen P, Virolainen J & Karhunen PJ (1994) Left ventricular size,mass, and function in relation to angiotensin-converting enzyme gene polymorphism inhumans. Am J Physiol 267: H1107-H1111.
244. Agerholm-Larsen B, Nordestgaard BG, Steffensen R, Sorensen TIA, Jensen G & Tybaerg-Hansen A (1997) ACE gene polymorphism: Ischemic heart disease and longevity in 10150individuals. A case-referent and retrospective cohort study based on the Copenhagen CityHeart Study. Circulation 95: 2358-2367.
245. O'Donnel CJ, Lindpaintner K, Larson MG, Rao VS, Ordovas JM, Schaefer EJ, Myers RH &Levy D (1998) Evidence for association and genetic linkage of the angiotensin-convertingenzyme logus with hypertension and blood pressure in men but not women in theFramingham heart study. Circulation 97: 1766-1772.
246. Fornage M, Amos CI, Kardia S, Sing CF, Turner ST & Boerwinkle E (1998) Variation in theregion of the angiotensin-converting enzyme gene influences interindividual differences inblood pressure levels in young white males. Circulation 97: 1773-1779.
247. White PC, Curnow KM & Pascoe L (1994) Disorders of steroid 11 beta-hydroxylaseisozymes. Endocr Rev 15: 421-438.
248. White PC & Slutsker L (1995) Haplotype analysis of CYP11B2. Endocrine Res 21: 437-442.

71
249. Pascoe L, Curnow KM, Slutsker L, Rosler A & White PC (1992) Mutations in the humanCYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency.Proc Natl Acad Sci USA 89: 4996-5000.
250. Lifton RP, Dluhy RG, Powers M, Rich GM, Gutkin M, Fallo F, Gill JRJ, Feld L, Ganguly A& Laidlaw JC (1992) Hereditary hypertension caused by chimaeric gene duplications andectopic expression of aldosterone synthase. Nat Genet 2: 66-74.
251. Pascoe L & Curnow KM (1995) Genetic recombination as a cause of inherited disorders ofaldosterone and cortisol biosynthesis and a contributor to genetic variation in blood pressure.Steroids 60: 22-27.
252. Kupari M, Hautanen A, Lankinen L, Koskinen P, Virolainen J, Nikkilä H & White PC(1998) Associations between human aldosterone synthase (CYP11B2) gene polymorphismsand left ventricular size, mass, and function. Circulation 97: 569-575.
253. Pojoga L, Gautier S, Blanc H, Guyene TT, Poirier O, Cambien F & Benetos A (1998)Genetic determination of plasma aldosterone levels in essential hypertension. Am JHypertens 11: 856-860.
254. Hautanen A, Lankinen L, Kupari M, Jänne OA, Adlercreutz H, Nikkilä H & White PC(1998) Association between aldosterone synthase gene polymorphism and the adrenocorticalfunction in males. J Inter Med 224: 11-18.
255. Prineas R, Crown RS, Blackburn H. (1982) The Minnesota code manual ofelectrocardiographic findings: standards and prosedures for measurements and classification.Littleton, John Wright/PSG.
256. Rose G, Blackburn H, Gillum R, Prineas R. (1982) Cardiovascular survey methods. WHOmonograph, World Health Organization.
257. Grimby G. (1992) Physical activity and muscle training in the elderly. Acta Med Scand 711:233-237.
258. Kupari M, Koskinen P & Virolainen J (1994) Correlates of left ventricular mass in apopulation sample aged 36 to 37 years. Focus on lifestyle and salt intake. Circulation 89:1041-1050.
259. O'Brien E, Mee F, Atkins N, Halligan A & O'Malley K (1993) Accuracy of the SpaceLabs90207 ambulatory blood pressure measuring system in normotensive pregnant womendetermined by the British Hypertension Society protocol. J Hypertens 11: S282-3.
260. O'Brien E, Atkins N, Mee F & O'Malley K (1993) Comparativeaccuracy of six ambulatorydevices according to blood pressure levels. J Hypertens 11:673-675.
261. Staessen JA, Fagard RH, Lijnen PJ, Thijs L, Van Hoof R & Amery AK (1991) Mean andrange of the ambulatory pressure in normotensive subjects from a meta-analysis of 23studies. Am J Cardiol 67: 723-727.
262. Sahn DJ, DeMaria A, Kisslo J & Weyman A (1978) The committee on M-modestandardization of the American Society of Echocardiography: recommendations regardingquantitation in M-mode echocardiography: results of a survey of echocardiographicmeasurements. Circulation 58: 1072-1083.
263. Airaksinen KEJ, Ikäheimo MJ, Salmela PI, Kirkinen P, Linnaluoto MK & Takkunen JT(1989) Impaired cardiac adjustment to pregnancy in type I diabetes. Diabetes Care 9: 376-383.
264. Airaksinen KE, Koistinen MJ, Ikäheimo MJ, Huikuri HV, Korhonen U, Pirttiaho H,Linnaluoto MK & Takkunen JT (1989) Augmentation of atrial contribution to left ventricularfilling in IDDM subjects as assessed by Doppler echocardiography. Diabetes Care 12: 159-161.
265. Troy BL, Pombo J & Rackley CE (1972) Measurement of left ventricular wall thickness andmass by echocardiography. Circulation 45: 602-611.
266. Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Gampo E, Sachs I & Reichek N (1986)Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsyfindings. Am J Cardiol 57: 450-458.
267. Du Bois D & Du Bois EF (1916) A formula to estimate the approximate surface area ifheight and weight be known. Arch Intern Med 17: 863-871.
268. Rigat B, Hubert C, Corvol P & Soubrier F (1992) PCR detection of the insertion/deletionpolymorphism of the human angiotensin converting enzyme gene (DCP1) (dipeptidylcarboxypeptidase 1). Nucleic Acids Res 20: 1433.
269. Russ AP, Maerz W, Ruzicka V, Stein U & Gross W (1993) Rapid detection of thehypertension-associated Met235-->Thr allele of the human angiotensinogen gene. Hum MolGenet 2: 609-610.

72
270. Imholz BPM, van Montfrans GA, Settels JJ, van der Hoeven GMA, Karemaker JM &Wieling W (1988) Continuous non-invasive blood pressure monitoring: reliability ofFinapres device during the Valsalva manoeuvre. Cardiovasc Res 22: 390-397.
271. Hartl DL (1988) A primer of population genetics. Sinauer Associates, Sunderland, Mass pp40-47.
272. Radaelli A, Bernardi L, Valle F, Leuzzi S, Salvucci F, Pedrotti L, Marchesi E, Finardi G &Sleight P (1994) Cardiovascular autonomic modulation in essential hypertension. Effect oftilting. Hypertension 24: 556-563.
273. McCubbin JW, Green JH & Page IH (1956) Baroreceptor function in chronic renalhypertension. Circ Res 4: 205-211.
274. Koh J, Brown TE, Beightol LA, Ha CY & Eckberg DL (1994) Human autonomic rhythms:vagal cardiac mechanisms in tetraplegic subjects. J Physiol 474: 483-495.
275. Kumagai K, Suzuki H, Ichikawa M, Jimbo M, Nishizawa M, Ryuzaki M & Saruta T (1996)Comparison of early and late start of antihypertensive agents and baroreceptor reflexes.Hypertension 27: 209-218.
276. Bonaduce D, Petretta M, Morgano G, Attisano T, Bianchi V, Arrichiello P, Rotondi F &Condorelli M (1992) Effects of converting enzyme inhibition on baroreflex sensitivity inpatients with myocardial infarction. J Am Coll Cardiol 20: 587-593.
277. Hansson L, Zanchetti A, Carruthers SG, Dahlöf B, Elmfeldt D, Julius S, Menard J, Rahn KH,Wedel H, Westerling S & for the HOT Study Group (1998) Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of theHypertension Optimal Trial (HOT) randomized trial. Lancet 351: 1755-1762.
278. Levy D, Garrison RJ, Savage DD, Kannel WB & Castelli WP (1990) Prognostic implicationsof echocardiographically determined left ventricular mass in the Framingham heart study. NEngl J Med 322: 1561-1566.
279. Sullivan JM, Vander Zwaag RV, el-Zeky F, Ramanathan KB & Mirvis DM (1993) Leftventricular hypertrophy: effect on survival. J Am Coll Cardiol 22: 508-513.
280. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, Reboldi G &Porcellati C (1998) Prognostic significance of serial changes in left ventricular mass inessential hypertension. Circulation 97: 48-54.
281. Montgomery H (1997) Should the contribution of ACE gene polymorphism to leftventricular hypertrophy be reconsidered? Heart 77: 489-490.
282. Airaksinen KE, Ikäheimo MJ, Linnaluoto M, Tahvanainen KU & Huikuri HV (1998) Genderdifference in autonomic and hemodynamic reactions to abrupt coronary occlusion. J Am CollCardiol 31: 301-306.
283. Rice DA, Mouw AR, Bogerd AM & Parker KL (1991) A shared promoter element regulatesthe expression of three steroidogenic enzymes. Mol Endocrinol 5: 1552-1561.
284. Clyne CD, Zhang Y, Slutsker L, Mathis JM, White PC & Rainey WE (1997) Angiotensin IIand potassium regulate human CYP11B2 transcription through common cis elements. MolEndocrinol 11: 638-649.
285. Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C, Moalic JM, Swynghedauw B& Delcayre C (1998) Myocardial production of aldosterone and corticosterone in the rat.Physiological regulation. J Biol Chem 273: 4883-4891.
286. Cook JR, Bigger JT, Jr., Kleiger RE, Fleiss JL, Steinman RC & Rolnitzky LM (1991) Effectof atenolol and diltiazem on heart period variability in normal persons. J Am Coll Cardiol 17:480-484.
287. Cleopas TJ (1993) Carry-over biases in clinical pharmacology. Eur J Clin Chem ClinBiochem 31: 803-809.
288. Globerman H, Rosler A, Theodor R, New MI & White PC (1988) An inherited defect inaldosterone biosynthesis caused by a mutation in or near the gene for steroid 11-hydroxylase.N Engl J Med 319: 1193-1197.

Origina l papers

1
Baroreflex sensitivity and variants of the renin angiotensin system genes
Antti Ylitalo, MD; K.E. Juhani Airaksinen, MD; Aarno Hautanen, MD; Markku Kupari, MD; Marion Carson,MD; Juha Virolainen, MD; Markku Savolainen, MD; Heikki Kauma, MD; Y Antero Kesäniemi, MD; Perrin C.
White, MD and Heikki V. Huikuri, MD.
AbstractBackgroundBaroreflex sensitivity (BRS) varies widely between healthy individuals. This is not well explained
by demographic variables, cardiovascular risk factors or life style, suggesting a genetic componentresponsible for the interindividual variation of BRS. Because the renin-angiotensin-aldosterone system (RAS)modifies cardiovascular autonomic regulation, we studied the possible associations between BRS andpolymorphisms in the angiotensin-converting enzyme (ACE), angiotensinogen (AGT) and aldosteronesynthase (CYP11B2) genes.
Methods and ResultsBRS, as measured from the overshoot phase of the Valsalva maneuver, and geneticpolymorphisms were examined first in a random sample of 161 women and 154 men aged 41-61 years andthereafter in an independent random cohort of 29 men and 37 women aged 36-37 years. An insertion/deletion(I/D) polymorphism of ACE, M235T variants of AGT and two diallelic polymorphisms in the gene encodingCYP11B2, one in the promoter (-344C/T) and the other in the second intron (gene conversion/non-conversion, 1/2), were identified by polymerase chain reaction. In the older population, BRS (mean±SD)differed significantly across CYP11B2 genotype groups in women (10.1±4.5, 8.7±3.8 and 7.1±3.2 ms.
mmHg-1 in genotypes -344TT, CT and CC, respectively, P=.003, and 11.1±4.4, 8.9±4.1 and 7.5±3.4 ms.
mmHg-1 in intron 2 genotypes 1/1, 1/2 and 2/2, respectively, P=.002), but not in men. No comparableassociations were found for BRS with the I/D polymorphism of ACE or the M235T variant of AGT in eithersex. In the youngerpopulation, BRS was strongly related to the CYP11B2 promoter genotype, averaging19.2±9.9, 12.9±5.1 and 9.7±4.1 ms. mmHg-1 in the -344TT, CT and CC groups, respectively, (P=.0003). Theassociation was statistically significant both in men (P=.015) and in women (P=.03). BRS was unrelated tothe I/D polymorphism of ACE even in the youngerpopulation.
ConclusionsCommon genetic polymorphisms in the aldosterone synthase (CYP11B2) gene is associatedwith interindividual variation in BRS. Because low BRS is a marker of increased risk ofcardiovascular morbidity and mortality, the clinical and prognostic significance of CYP11B2polymorphism merits further investigation.
Key words: baroreflex sensitivity, renin-angiotensin system, aldosterone synthase, gene polymorphism,gender
Impaired baroreflex sensitivity (BRS) is a predictorof cardiovascular mortality,1,2 and experimental3
and clinical4 data suggest that BRS associates withthe development of high blood pressure (BP) andmay be an important hereditary component in thepathogenesis of essential hypertension.5 BRS varieswidely between healthy individuals.6,7 This is notwell explained by demographic variables,cardiovascular risk factors or life style, suggesting agenetic component contributing to BRS.7
The renin-angiotensin-aldosterone system(RAS) has an important role in cardiovascularhomeostasis. It regulates sodium balance andintravascular volume8 and, in addition, interactswith other BP control mechanisms, including thesympathetic nervous system and baroreceptorreflexes.9 Many of these actions are mediated byangiotensin II and aldosterone, which both havebeen shown to impair BRS.10-12
From division of Cardiology (A.Y., K.E.J.A., H.V.H.); Atherosclerosis Research Group (M.S., H.K., Y.A.K.),Department of Internal Medicine, and Biocenter Oulu, University of Oulu, Oulu; Finland; Division ofCardiology, Helsinki University Central Hospital (A.H., M.K., M.C., J.V.), Helsinki, Finland, and Department ofPediatrics (P.C.W.), University of Texas Southwestern Medical Center, Dallas, TX, USA
This work was supported by grants from the Finnish Foundation for Cardiovascular Research, Helsinki, Finland,the research funds of Helsinki University Central Hospital, Helsinki, Finland, and the U.S. National Institutes ofHealth (DK37867 and DK42169).
Address for correspondence: Heikki Huikuri, MD, Division of Cardiology, Department of Medicine, Universityof Oulu, Kajaanintie 50, FIN-90220 Oulu, Finland. Tel: +358 8 3153550, fax +358 8 3155599.
Genes encoding components of RAS, such asthe angiotensin-converting enzyme (ACE),
angiotensinogen (AGT) and aldosterone synthase(CYP11B2), are potential risk loci for

2
cardiovascular diseases.8,13,14We, therefore, set outto study the possible associations between BRS andpolymorphisms in these genes in two groups ofhealthy subjects randomly selected from ethnicallyhomogeneous populations.
MethodsSubjectsPopulation 1. OPERA (Oulu Project ElucidatingRisk of Atherosclerosis) is a population-based,epidemiological study of cardiovascular riskfactors. 14 Subjects with no treatment forhypertension were randomly selected by agestratification from the register of the SocialInsurance Institution covering the whole populationof the City of Oulu in Northern Finland (106 500inhabitants). Two-hundred and fifty-nine men and267 women volunteered to take part, the overallparticipation rate being 87%.
All subjects were invited to be studied byValsalva maneuver and 24-hour ambulatory BPmeasurement and 483 participated. Participantswith diabetes mellitus (n=22), coronary arterydisease based on symptoms, medication, or ECG(n=35), atrial fibrillation (n=12), frequent ectopicbeats (n=8), inadequate ambulatory BP monitoring(n=19) or inadequate Valsalva maneuver (n=72)were excluded, after which a total of 315individuals (161 women and 154 men, 60% of theoriginal cohort) remained for analyses. Of thewomen, 76 were classified as postmenopausal, sinceat least 6 months had passed after their latestmenstruation, but 28 of them were on estrogen orprogesterone replacement therapy. Nine of the 85premenopausal women were taking these hormones.Past and current medical history, smoking habits,alcohol consumption and physical activity wereassessed with a standardized health questionnaire,15
and leisure-time physical activity groups (1-5) wereformed as described.16 A clinical examination andstandard 12-lead electrocardiography wereperformed. Routine blood tests, including an oralglucose tolerance test, were performed on eachsubject.Population 2. The reproducibility of theassociations between BRS and geneticpolymorphisms of CYP11B2 and ACE was testedin another population sample consisting of 89subjects aged 36-37 years apparently free ofcardiovascular disease. The characterization of thisstudy group, including echocardiographicexamination of the left ventricle, laboratory tests forblood lipids and assessment of habitual physicalactivity, smoking and ethanol consumption by 2-month daily recording, has been detailed earlier.17,18 The subjects were also characterized for theirheart rate and BP responses to the Valsalvamaneuver. The ECG and BP recordings wereanalyzed for BRS by two of the authors (A.Y. and
K.E.J.A.) blinded to the genotypes and other data.DNA was available for genotyping in 84 subjects,and the recordings of the Valsalva maneuver wereacceptable for BRS determination in 70 subjects; 66subjects (37 women and 29 men) had both types ofdata available for statistical analyses.
The design of the tests was approved by theEthical Committees of the respective institutions,and all subjects gave their informed consent.Baroreflex SensitivityFor the assessment of BRS, the subjects performedthe Valsalva maneuver in a sitting position byblowing into a rubber tube connected to an aneroidmanometer and maintaining an expiratory strain of40 mmHg for 15 seconds. The test was performedthree times at 5 minute intervals in Population 1 and1-2 times in Population 2. Otherwise, the testmethod and the data analysis19 were identical in thetwo study groups. In short, noninvasive arterial BPwas recorded on a beat-to-beat basis, using theFinapres finger-cuff method (Ohmeda Inc.,USA)20
with the monitored finger held at heart level. TheECG and continuous BP data were stored in adigital format and analyzed later using a menu-driven software package (CAFTS, Medikro Oy,Finland). A linear least-mean-squares fitting methodwas employed to calculate BRS from the overshootphase after the Valsalva maneuver as the slope ofthe linear relationship between the RR interval (inmsec) and the preceding systolic BP value (inmmHg). 19 Only regression lines with a correlationcoefficient >.7 or P<.05 and an increase in BP > 15mmHg were accepted. BRS was calculated as amean of the results of all the accepted tests.Genotyping of SubjectsThe insertion/deletion (I/D) polymorphism in intron16 of the ACE (DCP1) gene (chromosome 17q23)21and the methionine/threonine polymorphism(M235T) in codon 235 of the angiotensinogen gene(AGT, chromosome 1q42)22 were detected bypolymerase chain reaction (PCR), using theconditions described previously.14 The subjectswere also genotyped by PCR for two recentlydiscovered polymorphisms in the aldosteronesynthase gene (CYP11B2, chromosome 8q22). Thedetails of the technique and the structure of the genehave been described elsewhere.18,23 One of thepolymorphisms is located in the 5`flanking region,or promoter, of CYP11B2, 344 nucleotides beforethe start of the protein-coding sequence. Thisposition can be either a cytosine (-344C) or athymidine (-344T). The other is in the second intronof the gene. In some individuals, the usual sequenceof this intron has been almost entirely replaced by asequence typically found in the related gene,CYP11B1. Such replacement is termed geneconversion, and the alleles at this locus will bereferred to as 1 (conversion) and 2 (no conversion).In Population 1, genotype data were missing in the

3
case of two men for CYP11B2, two women forACE, and one women for AGT polymorphism.Blood Pressure MeasurementsIn Population 1, ambulatory BP was recorded usingthe fully automatic SpaceLabs 90207 oscillometricunit (SpaceLabs Inc., Redmond, Washington,USA), which was set to take a measurement every15 minutes from 4:00 AM till midnight and every20 minutes from midnight till 4:00 AM. Theaccuracy and reproducibility of BP readingsobtained with this device have been establishedpreviously.24 The proper positioning of the cuff ineach case was ensured by means of the similarity(difference < 5 mmHg) between four SpaceLabs BPmeasurements and four auscultatory readings usinga Y-connector.
In Population 2, resting brachial artery cuff BPwas averaged over three measurements.Statistical AnalysesThe Statistical Package for the Social Sciences forWindows, release 6.0, was used for data analysis. Achi-square test was used to analyze the frequencydata and to assess the fit of the observed allelefrequencies to the Hardy-Weinberg distribution.The coefficient of gametic linkage disequilibriumwas calculated by likelihood methods, and thecoefficient (D) is reported as the ratio of theunstandardized coefficient to its maximal value.25
Continuous data are given as means (SD) accordingto the genotypes. Gender differences were assessedby unpaired t-test or Mann-Whitney test (skeweddata distribution). The differences between thevariables in the different genotype groups weretested with ANOVA or the Kruskal-Wallis test(skewed data distribution). A two-way ANOVA(3x3 factorial design) with Bonferroni´s correctionsfor multiple comparisons was used to test thepotential synergistic effect of the ACE and AGTgenes on BRS. Multiple regression analysis wasused to assess the quantitative effects of age, BP,heart rate, body mass index, physical activity (log-transformed), alcohol consumption (log-transformed), smoking, serum cholesterol level, andthe use of estrogen or progesterone therapy on BRS.For the regression model, the genotype effect wasassumed to be additive (or linear) (with scores 1, 2and 3 assigned for -344TT, -344CT and -344CC,respectively), dominant (with scores 1 for -344TTand 2 for -344CT and -344CC combined), orrecessive (with scores 1 for -344TT and -344CTcombined and 2 for -344CC). Similar models wereused for the polymorphism in the intron region ofCYP11B2, the ACE gene polymorphism and theAGT genotypes. The regression analysis was madeseparately for men and women. Predictive values of
different genotypes for BRS tertiles werecalculated. The correlations between variables wereevaluated by Spearman´s correlation coefficient. Avalue of P<.05 was considered statisticallysignificant.
ResultsPopulation 1Main characteristics, 24-hour BP, lifestyle data, and-344C/T genotype in men and women are shown inTable 1. There were no significant genotypic effectson any variables, including BP. Similarly, the ACEand AGT polymorphisms had no influence on anyof these basic data (not shown). The 24-hoursystolic (129±12 vs 123±11 mmHg, P<.001) anddiastolic (81±8 vs 77±7 mmHg, P<.001) BPs werehigher in men, but there was no significant sexdifference in body mass index. Men were moreoften smokers and their total cholesterol andtriglyceride levels were higher and HDL cholesterollower those of women.
The genotype distributions of all polymorphismswere in agreement with the Hardy-Weinbergequilibrium both in men and in women. Thegenotype prevalences and allele frequencies of thepromoter -344C/T and intron 2 conversionpolymorphisms by sex are shown in Table 2. Thedata confirm linkage disequilibrium between thetwo polymorphic loci in the CYP11B2 gene(D=0.48, χ2=47.0, P<.0001). The genotypedistribution of the -344C/T polymorphism in 72subjects with inadequate Valsalva maneuver wassimilar to that in 315 subjects with adequate tests(20 (30%) in -344TT, 39 (45%) in CT and 13(25%) in CC genotype).
BRS was significantly lower in women than inmen (8.6 ± 4.0 and 11.0 ± 4.8 ms. mmHg-1,respectively, P<.0001). In women, BRS was relatedto the CYP11B2 promoter genotype, averaging10.1±4.5, 8.7±3.8 and 7.1±3.2 ms. mmHg-1 in the -344TT, CT and CC groups, respectively(P=.003)(Figure 1); a comparable association wasobserved between BRS and the intron 2polymorphism (1/1: 11.1 ± 4.4; 1/2 8.9 ± 4.1; 2/2:7.5 ± 3.4 ms . mmHg-1, P=.002). In men, bycontrast, BRS was unrelated to either the promotergenotype (Figure 1) or the intron 2 genotype (datanot shown) of the CYP11B2 gene. In women, theassociation of BRS with the CYP11B2 promotergenotype was consistent with a gene dosage effect,such that BRS increased in a linear relationship withthe number of -344T alleles carried by each woman(F=13.1, df1, P=.0004, R2=.08).

4
TABLE 1. Main characteristics of Men and Women in Different Genotype Groups by the Promoter -344 C/T Genotypes ofCYP11B2: Population 1
CYP11B2 GenotypeTT CT CC
Men n=34 n=72 n=46Age, y 50 (6) 49 (6) 50 (6)Heart rate, bpm 69 (9) 70 (9) 70 (10)Average 24-hour systolic BP, mmHg 129 (10) 128 (12) 131 (15)Average 24-hour diastolic BP, mmHg 82 (7) 81 (8) 82 (8)Body mass index, kg/m² 26 (3) 26 (3) 26 (3)Smoking, n of smokers/all 15/34 24/72 14/46Physical activity score‡ 2.9 (1.0) 3.0 (0.9) 3.2 (1.0)Alcohol consumption, g/day 13.5 (14.9) 13.8 (12.2) 10.1 (11.7)Total cholesterol, mmol/L 5.8 (1.0) 5.7 (1.1) 5.7 (0.9)HDL cholesterol, mmol/L 1.3 (0.3) 1.2 (0.3) 1.2 (0.3)Triglycerides, mmol/L 1.4 (0.7) 1.5 (0.8) 1.5 (0.6)
Women n=39 n=79 n=43Age, y 51 (6) 50 (6) 52 (6)Heart rate, bpm 73 (9) 72 (8) 73 (9)Average 24-hour systolic BP, mmHg 121 (12) 124 (11) 123 (9)†Average 24-hour diastolic BP, mmHg 76 (8) 77 (7) 76 (7)†Body mass index, kg/m² 26 (4) 26 (4) 24 (3)Smoking, n of smokers/all 9/39 25/79 12/43*Physical activity score‡ 3.2 (0.9) 3.2 (0.9) 3.1 (0.8)Alcohol consumption, g/wk 3.7 (6.9) 3.0 (3.8) 3.8 (5.7)†Total cholesterol, mmol/L 5.5 (1.1) 5.5 (1.1) 5.4 (1.0)*HDL cholesterol, mmol/L 1.6 (0.4) 1.5 (0.4) 1.5 (0.3)†Triglycerides, mmol/L 1.1 (0.4) 1.1 (0.5) 1.1 (0.4)†
BP indicates blood pressure. All values means (SD) except for smoking. *P<0.05 and †P<0.001 between men and women. ‡Score 1-5,where 5 = highly active and 1 = sedentary. No differences were found between genotypes analysed by sex.
TABLE 2. Prevalence of Promoter -344 C/T and Intron 2 Conversion Genotypes of Aldosterone Synthase in Menand Women: Population 1
Men WomenGenotypes n(%) Expected* n(%) Expected*
Promoter -344 C/T TT 34(23) 32 39(24) 38CT 72(47) 76 79(49) 80CC 46(30) 44 43(27) 42
Intron 2 † 1/1 24(16) 19 21(13) 211/2 58(38) 69 74(46) 742/2 69(46) 64 65(41) 65
*Expected number of genotypes by Hardy-Weinberg equilibrum. †Allele 1 carries a gene conversion from correspondingintron of CYP11B1; allele 2 does not carry the conversion.
TABLE 3. Prevalence of -344C/T Genotypes in Tertiles of Baroreflex Sensitivity in Women: Population 1Baroreflex Sensitivity (ms.mmHg-1)
1.4-5.9 6.0-10.0 10.1-19.6 N-344 CC 23 11 9 43-344 CT 24 27 28 79-344 TT 6 15 18 39N 53 53 55 161
χ2=14.3 (df=4), p=0.006.
TABLE 4. Prevalence of -344C/T Genotypes in Tertiles of Baroreflex Sensitivity:Population 2
Baroreflex Sensitivity (ms.mmHg-1)2.7-10.7 10.9-13.9 14.1-47.3 N
-344 CC 9 3 1 13-344 CT 11 13 10 34-344 TT 1 7 11 19N 21 23 22 66
χ2=16.7 (df=4), p=0.002.
The distribution of the female population intotertiles of BRS by C/T genotypes is shown in Table3. Calculated from the data, the predictive value ofthe -344CC genotype for low BRS (i.e. value in thelowest tertile of the study group) was 53% inwomen. By contrast, the predictive value of the -
344TT genotype for low BRS was only 15%.Instead, this genotype predicted high BRS (i.e. avalue in the highest tertile of the study group) withan accuracy of 49%.
There were no significant differences in BRSbetween the different genotypes of ACE or AGT inwomen or men. When ACE and AGT were added

5
as factors in ANOVA (3x3 factorial design), nosignificant synergistic effect of the AGT and -344C/T genotypes on BRS was observed.
In univariate analysis, BRS was associated with24-hour systolic BP in both men (r=-.22, P<.05) andwomen (r=-.30, P<.0001). BRS was also weaklyrelated to age in men (r=-.18, P<.05), but not inwomen (r=-.13, ns). The association between BRS
and the different genotypes of the CYP11B2 -344C/T promoter polymorphism in age tertiles inwomen is displayed in Figure 1. Although the sametrends were seen in all age tertiles, the associationwas only significant among the women aged 41-46.All but two women in this tertile werepremenopausal.
A B
0
4
8
12
16
20
Bar
ore
flex
Sen
sitiv
ity,m
s. mm
Hg-1
-344TT -344CT -344CC
P=.003*
0
4
8
12
16
20
24
Bar
oref
lex
Se
nsiti
vity
,ms. m
mH
g-1
-344TT -344CT -344CC
C D
0
10
20
30
40
50
Bar
ore
flex
Sen
sitiv
ity,m
s. mm
Hg-1
-344TT -344CT -344CC
P=.0003*
2
4
6
8
10
12
41-46 y 47-53 y 54-62 y
P=.004*
ns
ns
n=
12
n=30
n=11
n=15
n=26
n=13
n=12
n=23
n=
19
0
-344
TT
-344
TT
-344
TT
-34
4C
T
-34
4CC
-34
4C
T
-344
CC
-34
4C
T
-344
CC
Bar
ore
flex
Sen
sitiv
ity,m
s. mm
Hg-1
Fig. 1. Individual data on BRS in relation to the -344C/T polymorphism in the promoter of CYP11B2 in women (Panel A) andin men (Panel B) in Population 1 and in Population 2 (Panel C). Panel D, BRS (mean±±±±SEM) by the -344C/T polymorphism in thepromoter of CYP11B2 in age tertiles in women in Population 1. Horizontal lines indicate group mean values. *ANOVA.
In multiple regression analysis of the femalepopulation, BRS was related to 24-hour systolic BP(β=.38, P=.0067) and the -344C/T genotype ofCYP11B2 (CC=1, CT=2, TT=3;β=.37, P=.008),but was independent of age (β=-.10, P=.45), bodymass index (β=-.14, P=.33), physical activity (score1-5; β=.03, P=.81), daily alcohol consumption(β=.03, P=.82), smoking (nonsmoker=1,
smokers=2;β=.07, P=.63), serum cholesterol (β=-.20, P=.12), heart rate (β=-.01, P=.99), and the useof estrogen or progesterone therapy (no therapy=1,on therapy=2;β=-23, P=.07). In men, only 24-hoursystolic BP was an independent predictor (β=.39,P=.013), while the other variables did not enter themodel. Different assumptions about the type of thegenotype effect did not alter the findings.

6
Population 2
The genotype distributions and baselinecharacteristics of the study population have beendescribed in full detail previously.17,18 There wereno significant differences in sex distribution, bodymass index, heart rate, BP, physical activity,smoking, ethanol consumption or blood lipidsacross the -344C/T genotype groups.18 Nor did theACE I/D polymorphisms have any influence onthese basic data.17 The -344C/T genotypes of 18subjects with inadequate Valsalva maneuver were 3(17%) -344TT, 8 (44%) CT and 7 (39%) CC.In the total population of 37 women and 29 men,ANOVA revealed a significant main effect of the -344C/T genotype on BRS (Figure 1). The data wereconsistent with a gene dosage effect; BRS increased
in a linear relationship with the number of -344Talleles carried by each subject (F=21.1, df1,P<.0001, R2=0.25). The association of BRS withthe -344C/T genotype was statistically significant inboth men (TT: 20.2 ± 10.8; CT 12.6 ± 6.5; CC:10.0 ± 2.7 ms. mmHg-1, P=.037, ANOVA) andwomen (TT: 18.1 ± 9.4; CT 13.0 ± 4.0; CC: 9.6 ±5.0 ms. mmHg-1, P=.017, ANOVA). Thedistribution of the subjects into tertiles of BRS bythe C/T genotype is shown in Table 4. Calculatedfrom the data, the predictive value of the -344CCgenotype for low BRS (i.e. a value in the lowesttertile of the study group) was 69%. The predictivevalue of the -344TT genotype for low BRS wasonly 5%, but this genotype predicted high BRS (i.e.a value in the highest tertile of the study group) withan accuracy of 58%.
In women, BRS was also related to the intron 2polymorphism of CYP11B2, averaging 18.9 ± 10.5,14.6 ± 6.3 and 10.8 ± 3.9 ms. mmHg-1 in the 1/1,1/2 and 2/2 genotype groups, respectively (P=.036,ANOVA). The comparable trend in men was notstatistically significant (data not shown). There wereno differences in BRS between the I/D genotypegroups of ACE in either women or men (data notshown). The sex difference in BRS did not reachstatistical significance (14.7 ± 8.3 in men and 13.5± 6.3 ms. mmHg-1 in women, P=0.47).
In multiple regression analysis, BRS was relatedto the -344C/T genotype of CYP11B2 (CC=1,CT=2, TT=3;β=.49, P<.001), but was independentof sex (male=1, female=2,β=.12, P=.34), bodymass index (β=.02, P=.92), serum cholesterol(β=.11, P=.40), heart rate (β=-.15, P=.32), systolicBP (β=-,09, P=.56), left ventricular mass (β=-.02,P=.91), average daily physical activity (expenditureof metabolic equivalents;β=.11, P=.37), smoking(nonsmoker=0, 1-10 cigarettes/day=1, >10cigarettes/day=2;β=.13, P=.32) and average dailyalcohol consumption (β=.05, P=.69).
DiscussionThe present study is the first to suggest thatvariation in a specific gene may influenceinterindividual variation in BRS. The observedassociations between polymorphisms of thealdosterone synthase gene (CYP11B2) and BRSwere not explained by any potentially confoundingfactor and were particularly clear in a highlyhomogenous youngerpopulation. Our findingsagree well with the earlier experimental and clinicaldata suggesting a hereditary background for thewide interindividual variation of BRS. 3-5,26-33
Thus far, there is limited information availableconcerning the effects of CYP11B2 gene variationon cardiovascular structure and function.8,23 Arecent report of studies in Population 2 suggestedthat the -344CC genotype is a powerful predictor of
increased left ventricular mass and relativelyimpaired left ventricular diastolic function.18 Therelationship between BRS and the CYP11B2genotypes observed in the present study was,however, independent of left ventricular mass.Thus, the -344CC genotype may associateindependently with low BRS and left ventricularhypertrophy, both of which predict increasedcardiovascular mortality.1,2,34,35Data from previousclinical studies have not provided definitive answersconcerning the role of polymorphisms of the ACEand AGT genes in BP regulation or othercardiovascular disorders, such as left ventricularhypertrophy or cardiac events.13,36 In line withthese studies, we did not find any significantassociation between either ACE or AGT genotypesand BRS.
The association of BRS with CYP11B2 genevariation initially came out in a study of more than500 randomly selected middle-aged non-hypertensive subjects (Population 1). Since multipletesting for genetic associations may notoriously leadto chance findings even in non-mixed populations,we repeated the study in a fully independentpopulation-based sample of younger healthysubjects (Population 2). In support of our primarydata, an even stronger association of BRS with theCYP11B2 genotypes was observed, this time notonly in women, but also in men. Although onlysystolic BP and not age was related to BRS inmultivariate models in Population 1, aging and BPhave had independent effects on BRS in otherstudies, 1,6,37-39 suggesting that the influence ofpolymorphisms on BRS may be filtered out uponaging in subjects with higher BP values.36
The genotypic effects were analyzed separatelyfor both sexes, because several previous studieshave shown significant gender-related differences inthe baroreceptor reflex control of heart rate invarious populations.40-43 In Population 1, anassociation between BRS and the -344C/Tgenotypes was observed only among women and,

7
furthermore, mainly in those younger than 46 years,who were predominantly premenopausal. Theobserved sex difference in the genomic effectamong the middle-aged subjects is physiologicallyplausible in the light of the prior studies andanalogous to the well-established gender differencein BRS and BP regulation.40-44 Sex hormones maymodulate the effects of circulating angiotensin II,aldosterone and cortisol at the receptor level in thecentral nervous system and interact withcomponents of tissue RAS in the brain, heart,vascular bed and adrenals.45-48 We do not knowwhy there was a sex difference in the associationbetween BRS and the -344C/T genotypes inPopulation 1, but not in Population 2. However, thesubjects in Population 1 were older, and their BPand body mass index were higher, which differencesmay have modified the genetic influences on BRS.
The molecular mechanisms underlying theassociation between BRS and CYP11B2polymorphism cannot be deduced from this study.Unfortunately, plasma concentrations of RAShormones were not available for the populations westudied. However, it is reasonable to speculate thatone of the CYP11B2 polymorphisms we examined,or an additional polymorphism in linkagedisequilibrium with it, alters CYP11B2 geneexpression in the adrenal cortex and thus influencesplasma aldosterone levels which in turn affect BRS.Indeed, intravenous infusion of aldosterone impairsBRS in both animal models and man.11,12 The -344position is within an element that binds thetranscriptional regulatory factor, SF-149 and the -344C/T polymorphism influences binding of SF-1to this element. 23 However, current data onassociations of the -344C/T polymorphism withaldosterone levels is contradictory with significantgenotype effects reported in both directions indifferent populations.50-52 Alternatively, CYP11B2is known to be expressed at low levels in both theheart 53 and the vasculature,54 and autocrine orparacrine effects of locally synthesized aldosteronemay account for the observed genotype effects.
Prognostic information on the role of baroreflexfunction is based on phenylephrine-based BRSmeasurements.1,2 The predictive significance ofBRS data obtained from a Valsalva maneuver isunknown, but the measurements are reproducibleand correlate closely with indexes derived using theinvasive phenylephrine method.19,55-57 Indeed, thestimulus to arterial baroreceptors obtained by therelatively high expiratory pressure (40 mmHg) inthe Valsalva maneuver is close to the stimulusobtained by a phenylephrine injection,55 and whenthe baroreflex slope is analyzed, as in the presentstudy, from the overshoot phase of the Valsalvamaneuver,56 it is likely that the results of these twomethods are comparable. Importantly, we onlyanalyzed healthy subjects without apparent cardiac
diseases, which improves the accuracy andeliminates pitfalls of the Valsalva test.58
ConclusionsGenetic variation in the aldosterone synthase(CYP11B2) gene is associated with interindividualvariation in BRS. Because low BRS is a marker ofincreased risk of cardiovascular morbidity andmortality, the clinical and prognostic significance ofCYP11B2 polymorphism merits furtherinvestigation.
AcknowledgmentsThe skillful technical assistance of Mrs Saija Eirolais gratefully acknowledged.
References1. La Rovere MT, Specchia G, Mortara A, Schwartz PJ.Baroreflex sensitivity, clinical correlates, andcardiovascular mortality among patients with a firstmyocardial infarction. A prospective study.Circulation.1988;78:816-824.2. La Rovere MT, Bigger JT, Jr., Marcus FI, Mortara A,Schwartz PJ, for ATRAMI Investigators. Baroreflexsensitivity and heart-rate variability in prediction of totalcardiac mortality after myocardial infarction.Lancet.1998;351:478-484.3. Gordon FJ, Mark AL. Mechanism of impairedbaroreflex control in prehypertensive Dahl salt-sensitiverats.Circ Res. 1984;54:378-387.4. Hayes CG, Tyroler HA, Cassel JC. Family aggregationof blood pressure in Evans County, Georgia.Arch InternMed. 1971;128:965-975.5. Parmer RJ, Cervenka JH, Stone RA. Baroreflexsensitivity and heredity in essential hypertension.Circulation. 1992;85:497-503.6. Goldstein DS, Horwitz D, Keiser HR. Comparison oftechniques for measuring baroreflex sensitivity in man.Circulation. 1982;66:432-439.7. Ylitalo A, Airaksinen KEJ, Tahvanainen KUO,Kuusela TA, Ikäheimo MJ, Rantala A, Lilja M, HuikuriHV. Baroreflex sensitivity in drug-treated systemichypertension.Am J Cardiol. 1997;80:1369-1372.8. White PC. Disorders of aldosterone biosynthesis andaction.N Engl J Med. 1994;331:250-258.9. Reid IA. Interactions between ANG II, sympatheticnervous system, and baroreceptor reflexes in regulationof blood pressure.Am J Physiol. 1992;262:E763-E778.10. Guo GB, Abboud FM. Angiotensin II attenuatesbaroreflex control of heart rate and sympathetic activity.Am J Physiol. 1984;246:H80-H89.11. Wang W. Chronic administration of aldosteronedepresses baroreceptor reflex function in the dog.Hypertension. 1994;24:571-575.12. Yee KM, Struthers AD. Aldosterone causesbaroreceptor dysfunction in man.J Am Coll Cardiol1998;31:507A (Abstract).13. Soubrier F, Nadaud S, Williams TA. Angiotensin Iconverting enzyme gene: regulation, polymorphism andimplications in cardiovascular diseases.Eur Heart J.1994;15:S24-S29.

8
14. Kiema T, Kauma H, Rantala AO, Lilja M, ReunanenA, Kesäniemi YA, Savolainen MJ. Variation at theangiotensin-converting enzyme gene and angiotensinogengene loci in relation to blood pressure.Hypertension.1996;28:1070-1075.15. Rose G, Blackburn H, Gillum R, Prineas R.Cardiovascular survey methods.WHO monograph,World Health Organization. 1982.16. Grimby G. Physical activity and muscle training inthe elderly.Acta Med Scand. 1992;711:233-237.17. Kupari M, Perola M, Koskinen P, Virolainen J,Karhunen PJ. Left ventricular size, mass, and function inrelation to angiotensin-converting enzyme genepolymorphism in humans. Am J Physiol.1994;267:H1107-H1111.18. Kupari M, Hautanen A, Lankinen L, Koskinen P,Virolainen J, Nikkilä H, White PC. Associations betweenhuman aldosterone synthase (CYP11B2) genepolymorphisms and left ventricular size, mass, andfunction.Circulation. 1998;97:569-575.19. Airaksinen KE, Hartikainen JE, Niemelä MJ, HuikuriHV, Mussalo HM, Tahvanainen KU. Valsalvamanoeuvre in the assessment of baroreflex sensitivity inpatients with coronary artery disease.Eur Heart J.1993;14:1519-1523.20. Imholz BPM, van Montfrans GA, Settels JJ, van derHoeven GMA, Karemaker JM, Wieling W. Continuousnon-invasive blood pressure monitoring: reliability ofFinapres device during the Valsalva manoeuvre.Cardiovasc Res. 1988;22:390-397.21. Rigat B, Hubert C, Corvol P, Soubrier F. PCRdetection of the insertion/deletion polymorphism of thehuman angiotensin converting enzyme gene (DCP1)(dipeptidyl carboxypeptidase 1).Nucleic Acids Res.1992;20:1433.22. Russ AP, Maerz W, Ruzicka V, Stein U, Gross W.Rapid detection of the hypertension-associated Met235-->Thr allele of the human angiotensinogen gene.Hum MolGenet. 1993;2:609-610.23. White PC, Slutsker L. Haplotype analysis ofCYP11B2.Endocrine Res. 1995;21:437-442.24. O'Brien E, Atkins N, Mee F, O'Malley K.Comparative accuracy of six ambulatory devicesaccording to blood pressure levels.J Hypertens.1993;11:673-675.25. Hartl DL. A primer of population genetics.SinauerAssociates, Sunderland, Mass. 1988;pp 40-47.26. Carey RM, Dacey RG, Jane JA, Winn HR, Ayers CR,Tyson GW. Production of sustained hypertension bylesions in the nucleus tractus solitarii of the Americanfoxhound.Hypertension. 1979;1:246-254.27. Green MF, DeGroat AF, McDonald CH.Observations on denervation of the carotid sinuses andsection of the depressor nerves as a method of producingarterial hypertension.Am J Physiol. 1935;110:513-520.28. Schwartz PJ, Vanoli E, Stramba-Badiale M, DeFerrari GM, Billman GE, Foreman RD. Autonomicmechanisms and sudden death. New insights fromanalysis of baroreceptor reflexes in conscious dogs withand without a myocardial infarction.Circulation.1988;78:969-979.29. Eckberg DL. Carotid baroreflex function in youngmen with borderline blood pressure elevation.Circulation. 1979;59:632-636.
30. Takeshita A, Tanaka S, Kuroiwa A, Nakamura M.Reduced baroreceptor sensitivity in borderlinehypertension.Circulation. 1975;51:738-742.31. Sleight P. Reflex control of the heart.Am J Cardiol.1979;44:889-894.32. Gordon FJ, Matsuguchi H, Mark AL. Abnormalbaroreflex control of heart rate in prehypertensive andhypertensive Dahl genetically salt-sensitive rats.Hypertension. 1981;3:I135-41.33. Schwartz PJ, La Rovere MT. ATRAMI: a mark in thequest for the prognostic value of autonomic markers.EurHeart J. 1998;19:1593-1595.34. Levy D, Garrison RJ, Savage DD, Kannel WB,Castelli WP. Prognostic implications ofechocardiographically determined left ventricular mass inthe Framingham heart study.N Engl J Med.1990;322:1561-1566.35. Sullivan JM, Vander Zwaag RV, el-Zeky F,Ramanathan KB, Mirvis DM. Left ventricularhypertrophy: effect on survival.J Am Coll Cardiol.1993;22:508-513.36. Montgomery H. Should the contribution of ACE genepolymorphism to left ventricular hypertrophy bereconsidered?Heart. 1997;77:489-490.37. Hartikainen J, Mäntysaari M, Mussalo H,Tahvanainen K, Länsimies E, Pyörälä K. Baroreflexsensitivity in men with recent myocardial infarction;impact of age.Eur Heart J. 1994;15:1512-1519.38. Gribbin B, Pickering TG, Sleight P, Peto R. Effect ofage and high blood pressure on baroreflex sensitivity inman.Circ Res. 1971;29:424-431.39. Farrell TG, Odemuyiwa O, Bashir Y, Cripps TR,Malik M, Ward DE, Camm AJ. Prognostic value ofbaroreflex sensitivity testing after acute myocardialinfarction.Br Heart J. 1992;67:129-137.40. Abdel-Rahman AR, Merrill RH, Wooles WR.Gender-related differences in the baroreceptor reflexcontrol of heart rate in normotensive humans.J ApplPhysiol. 1994;77:606-613.41. Huikuri HV, Pikkujämsä SM, Airaksinen KE,Ikäheimo MJ, Rantala AO, Kauma H, Lilja M, KesäniemiYA. Sex-related differences in autonomic modulation ofheart rate in middle-aged subjects.Circulation.1996;94:122-125.42. Cerati D, Mortara A, La Rovere MT, Marcus FI,Malik M, Nohara R, Mazzuero G. Influence of gender onautonomic markers and risk after myocardial infarction inthe ATRAMI study. Circulation 1996;94:I-490(Abstract).43. Airaksinen KE, Ikäheimo MJ, Linnaluoto M,Tahvanainen KU, Huikuri HV. Gender difference inautonomic and hemodynamic reactions to abruptcoronary occlusion.J Am Coll Cardiol. 1998;31:301-306.44. Chen YF. Sexual dimorphism of hypertension.CurrOpin Nephrol Hypertens. 1996;5:181-185.45. Fisher ND, Ferri C, Bellini C, Santucci A, Gleason R,Williams GH, Hollenberg NK, Seely EW. Age, gender,and non-modulation. A sexual dimorphism in essentialhypertension.Hypertension. 1997;29:980-985.46. Hopkins PN, Lifton RP, Hollenberg NK, JeunemaitreX, Hallouin MC, Skuppin J, Williams CS, Dluhy RG,Lalouel JM, Williams RR, Williams GH. Blunted renalvascular response to angiotensin II is associated with acommon variant of the angiotensinogen gene and obesity.J Hypertens. 1996;14:199-207.

9
47. Myles K, Funder JW. Progesterone binding tomineralocorticoid receptors: in vitro and in vivo studies.Am J Physiol. 1996;270:E601-E607.48. Funder JW. Mineralocorticoid receptors andhypertension.J Steroid Biochem Mol Biol. 1995;53:53-55.49. Clyne CD, Zhang Y, Slutsker L, Mathis JM, WhitePC, Rainey WE. Angiotensin II and potassium regulatehuman CYP11B2 transcription through common ciselements.Mol Endocrinol. 1997;11:638-649.50. Pojoga L, Gautier S, Blanc H, Guyene TT, Poirier O,Cambien F, Benetos A. Genetic determination of plasmaaldosterone levels in essential hypertension.Am JHypertens. 1998;11:856-860.51. Hautanen A, Lankinen L, Kupari M, Jänne OA,Adlercreutz H, Nikkilä H, White PC. Associationbetween aldosterone synthase gene polymorphism andthe adrenocortical function in males.J Inter Med.1998;224:11-18.52. Brand E, Chatelain N, Mulatero P, Fery I, CurnowKM, Jeunemaitre X, Corvol P, Pascoe L, Soubrier F.Structural analysis and evaluation of the aldosteronesynthase gene in hypertension.Hypertension.1998;32:198-204.53. Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B,Mouas C, Moalic JM, Swynghedauw B, Delcayre C.Myocardial production of aldosterone and corticosteronein the rat. Physiological regulation.J Biol Chem.1998;273:4883-4891.54. Takeda Y, Miyamori I, Yoneda M, Hatakeyama H,Inaba S, Furukawa K, Mabuchi H, Takeda R. Regulationof aldosterone synthase in human vascular endothelialcells by angiotensin II and adrenocorticotropin.J ClinEndocrinol Metab. 1996;81:2797-2800.55. Trimarco B, Volpe M, Ricciardelli B, Vigorito C, DeLuca N, Sacca L, Condorelli M. Valsalva maneuver inthe assessment of baroreflex responsiveness in borderlinehypertensives.Cardiology. 1983;70:6-14.56. Smith SA, Stallard TJ, Salih MM, Littler WA. Cansinoaortic baroreseptor heart rate reflex sensitivity bedetermined from phase IV of the Valsalva manoeuvre?Cardiovasc Res. 1987;21:422-427.57. Palmero HA, Caeiro TF, Iosa DJ, Bas J. Baroreceptorreflex sensitivity index derived from phase 4 of theValsalva manoeuvre.Hypertension. 1981;3:134-137.58. Eckberg DL. Parasympathetic cardiovascular controlin human disease: a critical review of methods andresults.Am J Physiol. 1980;239:H581-H593.