carbonic anhydrase immobilized on encapsulated … fe 3 o 4 /sio 2 nanoparticles by using a...
TRANSCRIPT

DOI: 10.1002/chem.201201112
Carbonic Anhydrase Immobilized on Encapsulated Magnetic Nanoparticlesfor CO2 Sequestration
Mari Vinoba,[a] Margandan Bhagiyalakshmi,[b] Soon Kwan Jeong,*[a]
Sung Chan Nam,[a] and Yeoil Yoon[a]
Introduction
Anthropogenic carbon dioxide (CO2) is a major greenhousegas that acts as a blanket to absorb thermal radiation emit-ted by the earth�s surface, thereby resulting in global warm-ing and climate change. The atmospheric CO2 concentrationis predicted to rise by more than 450 ppm coupled with aglobal temperature increase of more than 2 8C in 2100 dueto CO2 emissions from the ongoing industrial revolution.[1]
Many studies have introduced new technologies for CO2
capture; however, the quest for feasible capture technolo-gies continues.[2] To this end, biology-inspired methods pres-ent innovative model systems for trapping CO2 from emis-sion sources. Recently, carbonic anhydrase (CA) was usedto convert CO2 to bicarbonate, which was then converted toCaCO3 in the presence of Ca2+ ions.[3] CA is the fastestenzyme to catalyze the conversion of one million CO2 mole-
cules to bicarbonate per second.[4] The use of free enzymesas biocatalysts for large-scale industrial processes poses sig-nificant drawbacks because the free enzymes are solubleand cannot be easily recovered and reused. The catalyticproperties of the enzymes are greatly improved by immobi-lization on nanomaterials.[5] Several techniques have beenproposed to improve the enzymatic activity of CA upon im-mobilization.[6] Surface-modified magnetic nanoparticlesprovide excellent templates for enzyme immobilization,thereby enabling their easy recovery from a reactionmedium upon application of a static magnetic field.[7] Cur-rent reports have demonstrated that SiO2 and Fe2O3/SiO2
nanoparticles are biocompatible, nontoxic, and chemicallystable toward enzyme immobilization.[8] CA immobilizationon SiO2 was achieved by amine functionalization.[9] Thepresent study describes the immobilization of bovine car-bonic anhydrase (BCA) onto amine-grafted Fe3O4/SiO2.Scattered amine groups provide a good distribution of im-mobilized enzymes over the nanomaterial surface. Accord-ingly, octa(aminophenyl)silsesquioxane (OAPS), an organic–inorganic nanostructured hybrid derivative of polyhedral oli-gomeric silsesquioxanes, was shown to be a good spacer forenzyme immobilization because it possess eight aminegroups on each edge of a cubic silicon cage structure.[9]
This study demonstrates that the OAPS-functionalizedFe3O4/SiO2 was synthesized here by using Fe2+ and Fe3+
salts in combination with tetraethyl orthosilicate (TEOS),followed by grafting with 3-chloropropyltrimethoxysilane(CPTMS). BCA was immobilized through covalent bondsbetween the amine groups of the enzyme and the OAPS-
Abstract: Bovine carbonic anhydrase(BCA) was covalently immobilizedonto OAPS (octa(aminophenyl)silses-quioxane)-functionalized Fe3O4/SiO2
nanoparticles by using glutaraldehydeas a spacer. The Fe3O4 nanoparticleswere coated with SiO2, onto which wasgrafted OAPS, and the product wascharacterized using SEM, TEM, XRD,IR, X-ray photoelectron spectroscopy(XPS), and magnetometer analysis.The enzymatic activities of the free andFe3O4/SiO2/OAPS-conjugated BCA(Fe�CA) were investigated by hydro-
lyzing p-nitrophenylacetate (p-NPA),and hydration and sequestration ofCO2 to CaCO3. The CO2 conversion ef-ficiency and reusability of the Fe�CAwere studied before and after washingthe recovered Fe�CA by applying amagnetic field and quantifying the un-reacted Ca2+ ions by using ion chroma-tography. After 30 cycles, the Fe�CA
displayed strong activity, and the CO2
capture efficiency was 26-fold higherthan that of the free enzyme. Storagestability studies suggested that Fe�CAretained nearly 82 % of its activityafter 30 days. Nucleation of the precipi-tated CaCO3 was monitored by usingpolarized light microscopy, which re-vealed the formation of two phases,calcite and valerite, at pH 10 upon ad-dition of serine. The magnetic nanobio-catalyst was shown to be an excellentreusable catalyst for the sequestrationof CO2.
Keywords: carbon dioxide · greenchemistry · magnetic properties ·nanoparticles · sequestration
[a] Dr. M. Vinoba, Dr. S. K. Jeong, Dr. S. C. Nam, Dr. Y. YoonClimate Change Technology Research DivisionKorea Institute of Energy Research102 Gajeong-ro, Yuseong-guDaejeon 305-343 (Korea)Fax: (+82) 42-860-3134E-mail : [email protected]
[b] Dr. M. BhagiyalakshmiDepartment of ChemistrySchool of Basic and Applied SciencesCentral University of Tamil NaduThiruvarur 610-004 (India)
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.201201112.
� 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2012, 18, 12028 – 1203412028

functionalized Fe3O4/SiO2 nanoparticles by using a cross-linking agent such as glutaraldehyde (GA). The activity ofthe free or immobilized BCA toward hydrolysis of p-nitro-phenylacetate (p-NPA), hydration and sequestration of CO2,the material reusability, the CO2 conversion efficiency, andthe quantity of CaCO3 generated were evaluated by usingion chromatography (IC) techniques. The formation of dif-ferent CaCO3 phases depending on the addition of aminoacid was monitored by polarized light microscopy at a con-stant pH.
Results and Discussion
Synthesis and morphological characterization of the nano-materials : The magnetic nanoparticles were synthesized byco-precipitation, followed by silica coating using a modifiedStçber method. Silica-coating Fe3O4 nanoparticles was chal-lenging because of the low chemical affinity of silica towardFe3O4, which was overcome by using a modified Stçbermethod. Isopropanol has a low dielectric constant (19.9),and its use as a solvent resulted in the ready formation oflarger silica particles, in contrast to the results obtainedwhen using a high dielectric constant (24.3) solvent, etha-nol.[10] In this work, the silica coating obtained using isopro-panol as a solvent was thicker than that obtained using etha-nol. The chloropropyl moieties were grafted onto the surfa-ces of the Fe3O4/SiO2 nanoparticles by treating the Si�OHsilica groups with CPTMS. OAPS was then treated with thechloropropyl moieties of the Fe3O4/SiO2/CPTMS to yieldFe3O4/SiO2/OAPS (Scheme 1).
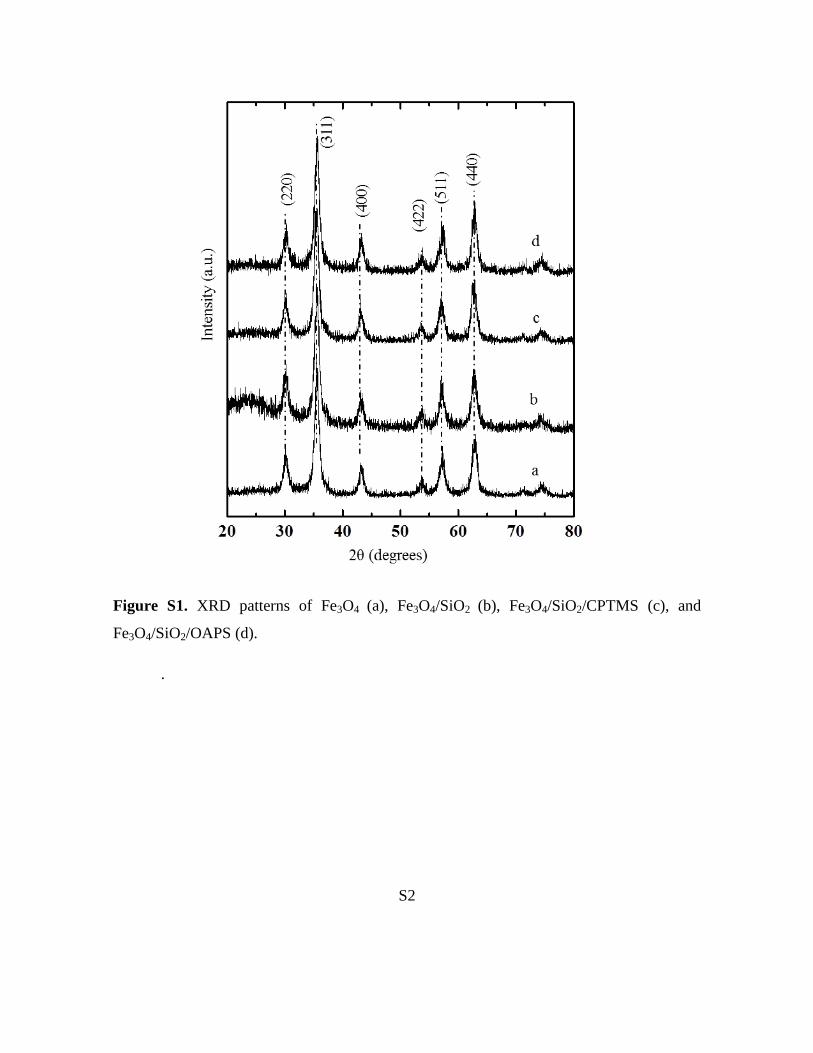
Figure S1 in the Supporting Information shows the XRDpatterns of pure, silica-coated, and functionalized magneticnanoparticles. The characteristic peaks of the pure Fe3O4 at2q= 30.1, 35.5, 43.1, 53.4, 56.97, and 62.618 were observed,marked in Figure S1a of the Supporting Information bytheir indices (220), (311), (400), (422), (511), and (440), re-spectively. These peaks coincided with those in the JCPDSdatabase for magnetic materials (82-1533), thus indicating a
crystalline cubic spinel structure for the Fe3O4 nanoparti-cles.[11] Fe3O4/SiO2 displayed a broad peak at 2q= 238, whichindicated the presence of an amorphous silica structure. Theremaining XRD peaks corresponded to structural featuresof the Fe3O4 particles (Figure S1b in the Supporting Infor-mation). Even upon functionalization with CPTMS, fol-lowed by OAPS, the phases of the Fe3O4 nanoparticles re-mained unchanged (Figure S1c,d in the Supporting Informa-tion). The average Fe3O4 nanoparticle size was measuredusing the Debye–Scherrer equation and was found to be10 nm. The XRD results agreed well with the TEM meas-urements. TEM images of Fe3O4 nanoparticles were quasi-spherical in shape with an average diameter of 10–12 nm(Figure 1). The Fe3O4/SiO2 nanoparticles were monodispersewith well-defined core–shell structures, even though thesilica shells trapped multiple Fe3O4 particles. The Fe3O4/SiO2
nanoparticles used in this case for the production of func-tionalized Fe3O4/SiO2/OAPS particles had an average diam-eter of (115�10) nm (Figure 1b).
The surface morphologies of the Fe3O4 and Fe3O4/SiO2/OAPS particles were deduced on the basis of the SEMimages. Fe3O4 nanoparticles were spherical with smooth sur-faces, as shown in Figure 2a. The SEM images of Fe3O4/SiO2/OAPS revealed well-defined spherical nanoparticles(Figure 2b). The energy-dispersive X-ray diffraction spectro-scopy (EDS) data of the Fe3O4 and Fe3O4/SiO2/OAPS nano-particles confirmed the presence of metal ions (Figure S2 inthe Supporting Information). The presence of elementalcarbon, nitrogen, oxygen, silicon, and iron in the Fe3O4/SiO2/OAPS materials indicated the successful functionaliza-tion of OAPS over Fe3O4/SiO2, and their respective elemen-tal contents were 14.96, 6.15, 34.28, 25.57, and 19.04 %. EDSelemental mapping of the Fe3O4/SiO2/OAPS revealed thatC, Si, O, and N were homogeneously distributed over thesurface of each particle (Figure S3 in the Supporting Infor-mation), thus illustrating that each particle was surface-func-tionalized by the nitrogen-containing OAPS groups.
Figure S4 in the Supporting Information shows the FTIRspectra of pristine and functionalized Fe3O4 particles. The
Scheme 1. Schematic illustrations of functionalization and carbonic anhydrase immobilization on encapsulated magnetic nanoparticles.
Chem. Eur. J. 2012, 18, 12028 – 12034 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 12029
FULL PAPER

FTIR spectrum of pure Fe3O4 displayed a characteristicpeak at 580 cm�1 due to Fe�O stretching.[12] The peaks at1423 and 3405 cm�1 were attributed to C�O, O�H bond vi-brations, which may have been on account of contact withair and water (Figure S4a in the Supporting Information).The silica coating was confirmed by the observation of anew peak at 956 cm�1, which was assigned to the Si�OHbond, and the vibrational bands at 1086 and 806 cm�1, whichcorresponded to the stretching and bending of the Si-O-Sibonds, respectively (Figure S4b in the Supporting Informa-tion). The disappearance of the peak at 956 cm�1 and theemergence of new peaks at 1403 cm�1 (dC,H) and nas =
2925 cm�1 (�CH2), as shown in Figure S4c in the SupportingInformation, indicated CPTMS grafting onto the Fe3O4/SiO2
nanoparticles. OAPS grafting onto Fe3O4/SiO2/CPTMS wasverified by the presence of the characteristic peaks at 1124,1598, 3049, and 3384 cm�1, on account of Si-O-Si, phenylC=C, phenyl C�H, and NH2, respectively (Figure S4d in theSupporting Information).[13]
Figure 3 shows the X-ray photoelectron spectroscopy(XPS) patterns of Fe3O4 and Fe3O4/SiO2/OAPS. The charac-teristic iron oxide peaks in the Fe3O4 nanoparticles, includ-
ing O 1s, Fe 2p3/2, and Fe 2p1/2, appeared at 530, 712, and725 eV, respectively, were observed in both the pristine andfunctionalized Fe3O4.
[14] The XP spectrum of Fe3O4/SiO2/OAPS revealed two new peaks at 156 and 105 eV, whichcorresponded to photoelectrons that originated from the Si2s and Si 2p energy levels, respectively. The intensity ofcharacteristic Fe 2p3/2 and Fe 2p1/2 peaks in Fe3O4/SiO2/OAPS decreases, thus indicating that all Fe3O4 nanoparticlesin the composite were confined within a SiO2 shell. Thegrafted OAPS introduced N 1s binding energies at 402 eV.The XPS and IR data reveal the confinement of Fe3O4
nanoparticles by the silica layer and the presence of aminegroups on the Fe3O4/SiO2/OAPS surfaces, respectively.
Magnetic characterization of the nanomaterials was ob-served by a vibrating sample magnetometer. Figure 4 showsthat the saturation magnetization values of Fe3O4, Fe3O4/SiO2, Fe3O4/SiO2/CPTMS, Fe3O4/SiO2/OAPS, and Fe�CAwere 77.7, 56.2, 42.3, 29.5, and 24.6 emu g�1, respectively.This descending order corresponds to silica-layer coatingfollowed by functionalization and BCA immobilization onFe3O4 nanoparticles. The magnetization values suggestedthat Fe�CA can be easily separated from reaction media inthe presence of an external magnetic field, which is demon-strated in Scheme 1, and redispersion of the catalysts occur-red rapidly with a slight shaking as the magnetic field wasremoved. These results show the efficacy of magnetic sepa-ration.
Figure 1. HRTEM images of a) Fe3O4 and b) Fe3O4/SiO2/OAPS.
Figure 2. FESEM of a) Fe3O4 and b) Fe3O4/SiO2/OAPS.
Figure 3. XPS spectrum of the Fe3O4 and Fe3O4/SiO2/OAPS particles.Inset: XPS pattern of the Fe 2p, Si 2p, Si 2s, and N 1s binding energies.
www.chemeurj.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2012, 18, 12028 – 1203412030
S. K. Jeong et al.
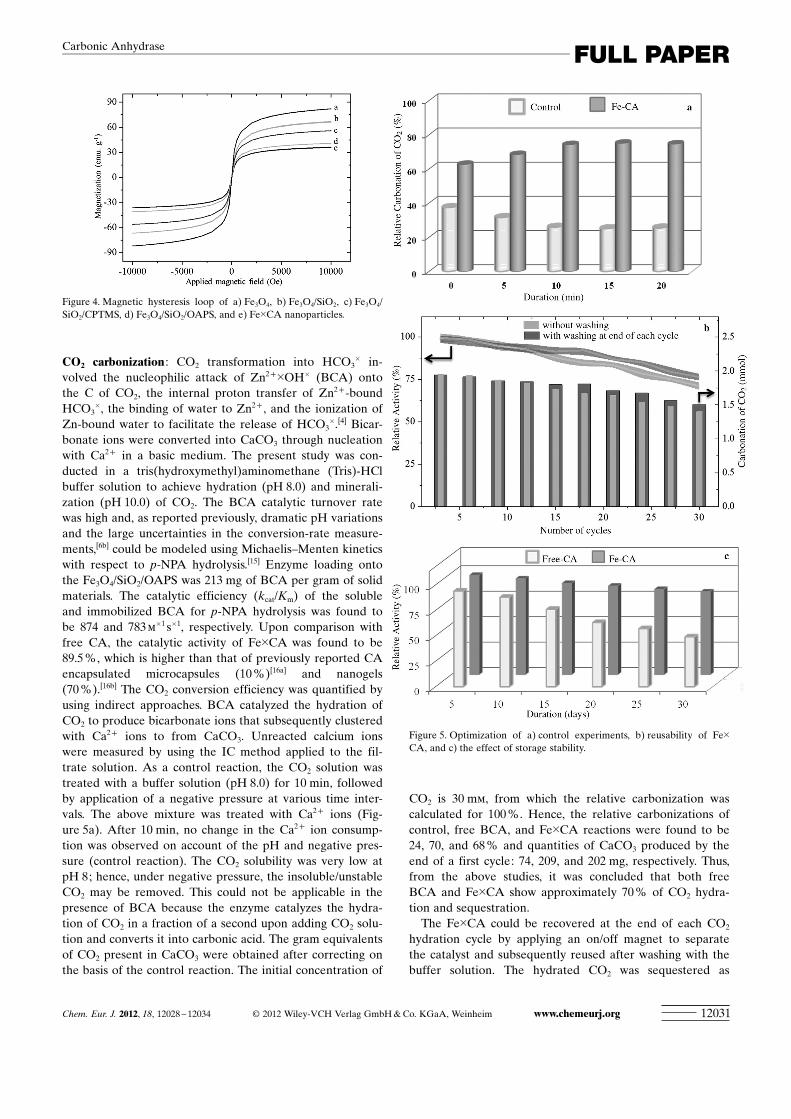
CO2 carbonization : CO2 transformation into HCO3� in-
volved the nucleophilic attack of Zn2+�OH� (BCA) ontothe C of CO2, the internal proton transfer of Zn2+-boundHCO3
�, the binding of water to Zn2+ , and the ionization ofZn-bound water to facilitate the release of HCO3
�.[4] Bicar-bonate ions were converted into CaCO3 through nucleationwith Ca2+ in a basic medium. The present study was con-ducted in a tris(hydroxymethyl)aminomethane (Tris)-HClbuffer solution to achieve hydration (pH 8.0) and minerali-zation (pH 10.0) of CO2. The BCA catalytic turnover ratewas high and, as reported previously, dramatic pH variationsand the large uncertainties in the conversion-rate measure-ments,[6b] could be modeled using Michaelis–Menten kineticswith respect to p-NPA hydrolysis.[15] Enzyme loading ontothe Fe3O4/SiO2/OAPS was 213 mg of BCA per gram of solidmaterials. The catalytic efficiency (kcat/Km) of the solubleand immobilized BCA for p-NPA hydrolysis was found tobe 874 and 783 m
�1 s�1, respectively. Upon comparison withfree CA, the catalytic activity of Fe�CA was found to be89.5 %, which is higher than that of previously reported CAencapsulated microcapsules (10 %)[16a] and nanogels(70 %).[16b] The CO2 conversion efficiency was quantified byusing indirect approaches. BCA catalyzed the hydration ofCO2 to produce bicarbonate ions that subsequently clusteredwith Ca2+ ions to from CaCO3. Unreacted calcium ionswere measured by using the IC method applied to the fil-trate solution. As a control reaction, the CO2 solution wastreated with a buffer solution (pH 8.0) for 10 min, followedby application of a negative pressure at various time inter-vals. The above mixture was treated with Ca2+ ions (Fig-ure 5a). After 10 min, no change in the Ca2+ ion consump-tion was observed on account of the pH and negative pres-sure (control reaction). The CO2 solubility was very low atpH 8; hence, under negative pressure, the insoluble/unstableCO2 may be removed. This could not be applicable in thepresence of BCA because the enzyme catalyzes the hydra-tion of CO2 in a fraction of a second upon adding CO2 solu-tion and converts it into carbonic acid. The gram equivalentsof CO2 present in CaCO3 were obtained after correcting onthe basis of the control reaction. The initial concentration of
CO2 is 30 mm, from which the relative carbonization wascalculated for 100 %. Hence, the relative carbonizations ofcontrol, free BCA, and Fe�CA reactions were found to be24, 70, and 68 % and quantities of CaCO3 produced by theend of a first cycle: 74, 209, and 202 mg, respectively. Thus,from the above studies, it was concluded that both freeBCA and Fe�CA show approximately 70 % of CO2 hydra-tion and sequestration.
The Fe�CA could be recovered at the end of each CO2
hydration cycle by applying an on/off magnet to separatethe catalyst and subsequently reused after washing with thebuffer solution. The hydrated CO2 was sequestered as
Figure 4. Magnetic hysteresis loop of a) Fe3O4, b) Fe3O4/SiO2, c) Fe3O4/SiO2/CPTMS, d) Fe3O4/SiO2/OAPS, and e) Fe�CA nanoparticles.
Figure 5. Optimization of a) control experiments, b) reusability of Fe�CA, and c) the effect of storage stability.
Chem. Eur. J. 2012, 18, 12028 – 12034 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 12031
FULL PAPERCarbonic Anhydrase

CaCO3 by Ca2+ ions. The quantity of unreacted Ca2+ ionswas quantified by IC, and the CO2 conversion efficiency rel-ative to carbonization could be calculated. The CO2 conver-sion efficiency remained constant over 12 cycles both withand without washing of the catalyst. After 12 cycles, theextent of CO2 carbonization and its conversion efficiencydecreased gradually in the presence of unwashed Fe�CA.After 30 cycles without washing, the recovered Fe�CA lostits activity by around 24 % of the initial activity, and the cor-responding CO2 carbonization was 0.46 mmol. Deactivationof the enzyme on account of lack of washing led to a de-crease in Fe�CA activity; the HCO3
� species produced actsas an inhibitor by binding with the active site of enzyme(Zn2+�OH) by means of a Brønsted acid interaction,[17]
which could be solved by washing the Fe�CA catalyst withbuffer at the end of each cycle. Partial loss of the immobi-lized enzyme might have occurred during the washing stepsas well, although this loss was relatively minor. The Fe�CAwas mostly captured by using the magnetic on/off system,which prevented extensive loss of Fe�CA during washing.The retained activity of the recovered and reused Fe�CAafter washing with buffer at the end of the 30th cycle was90 %, and 1.84 mmol CO2 was found to be carbonized. ThusFe�CA exhibits excellent reusability and was 26-fold higherthan free BCA (Figure 5b). CA immobilized on chitosanbeads, on the other hand, loses its CO2 sequestration activityby about 70 % after the seventh cycle.[18] In addition to theenzyme reusability, the activities of the stored free or immo-bilized enzymes were monitored for 30 days at 5-day inter-vals by measuring the CO2 hydration and sequestration. Theresidual activities of either the free BCA or Fe�CA were 49or 82 %, respectively, after 30 days (Figure 5c). Fe�CA ex-hibit a greater stability toward CO2 conversion than CA im-mobilized on iron mesh[19a] and alginate bead[19b] materials,which retained about 70 % of its activity after 30 days.
Preparation of different CaCO3 phases : Several reportshave focused on the crystallographic structure of CaCO3(s)
obtained during precipitation from a hydrated CO2 solution.We attempted to induce formation of different CaCO3
phases, calcite or vaterite, by adding a serine at pH 10. Themechanism reported for the nucleation and growth of eachCaCO3 phase was observed by using polarized-light miACHTUNGTRENNUNGcros-ACHTUNGTRENNUNGcopy, and images are presented in Figure 6. Here, twoCaCO3 phases were obtained by the addition of serine,which interacted with the mineral-forming ions in solutionto promote nucleation or to modify the crystal growth andalter the growth rates at crystal faces at a given pH. Nuclea-tion was favored only at a critical cluster size, which was di-rectly proportional to the ratio of the energy required togrow clusters and the energy released during formation ofbonds in the aggregates. Ca2+ and CO3
2� ions are denselypacked. The most energetically stable atomic planes in therectangular rhombohedral calcite lattice result in metastablesolutions that do not undergo phase transformations overlong periods of time. Nucleation of CaCO3 involves an ini-tial burst nucleation of particles followed by reprecipitation
to form a new more stable crystallographic mononuclearphase (solid-line circle in Figure 6). Two or more particlesthen combine to form a single polynuclear crystal (dotted-line circle in Figure 6). A stable pure calcite phase formed,and the crystallographic morphology of the particles did notsubsequently change.[20] The surface morphologies of the cal-cite crystals exhibited well-defined faceted rhombohedralshapes, as confirmed by polarized-light microscopy andfield-emission scanning electron microscopy (FESEM; Fig-ure 7a, b). The organic additives were involved in nucleation
sites and induced the precipitation of stable porous sphericalvaterite particles.[21] The addition of serine influenced thegeometric shape of the CaCO3 and resulted in the formationof both vaterite and calcite phases in porous sphericalCaCO3 particles. Ca2 + ions bound to the anionic COO� moi-eties and the cationic NH3
+ groups of the zwitterionic serineto produce partially nucleated vaterite CaCO3 crystal struc-tures. Calcite and vaterite coexisted in the spherical porousCaCO3 particles, as shown in Figure 7c,d. Both CaCO3 mor-phologies were hexagonal in structure, but the vaterite wasmore complex with a packing structure that led to thegrowth of aggregates. The vaterite particles were porous,
Figure 6. Polarized-light microscopy images of CaCO3 nucleation.
Figure 7. Polarized-light microscopy and SEM images of CaCO3 precipi-tate: a, b) calcite and c,d) coexistence of calcite and vaterite.
www.chemeurj.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2012, 18, 12028 – 1203412032
S. K. Jeong et al.

whereas the thin planar calcite crystals were not porous andgrew on the external surfaces of the vaterite particles.
The XRD patterns of the CaCO3 precipitates varied for agiven pH 10.0, depending on the preparation conditions. Apure calcite phase (JCPDS 05-0586) was observed in the ab-sence of additives. The addition of a serine altered the mor-phologies of the CaCO3. Vaterite (JCPDS 33-0268) and cal-cite were found to coexist, as shown in Figure 8. Diffraction
peaks were observed at 2q= 24.8, 27.02, 32.74, 43.8, and49.988, which correspond to the vaterite crystal faces (100),(101), (102), (110), and (104), respectively. After 60 min, apure calcite phase was observed on account of the phase re-configuration of the vaterite. Diffraction peaks were ob-served at 2q= 29.5, 36.1, 39.5, and 43.38, which correspondto the calcite crystal faces (104), (110), (113), and (202), re-spectively.[22] The results described above indicated thatFe�CA satisfied important criteria for green materials: reus-ability and stability during storage. These materials arepromising candidate catalysts for the hydration of CO2 andits sequestration as CaCO3.
Conclusion
BCA was immobilized onto SiO2-coated Fe3O4 magneticnanoparticles through OAPS grafting. XPS and EDS ele-mental mapping of the Fe3O4/SiO2/OAPS indicated the suc-cessful grafting of the nitrogen-containing OAPS groups.The biocatalytic activities of free BCA and Fe�CA towardthe hydrolysis of p-NPA were found to be 874 and783 m
�1 s�1, respectively. The control study showed that theCa2 + ion consumption did not vary with pH or the applica-tion of a negative pressure, which indicated that relative car-bonization of 68 % is on account of CO2 hydrated byFe�CA. Equal quantities of free or Fe�CA enzymes pro-duced equal quantities of CaCO3 by the end of the first
cycle: 209 or 202 mg, respectively. The reusability studies ofFe�CA indicated that the activity of the materials was 26-fold higher than the activity of the free BCA. The washingprotocol during recovery and reuse of Fe�CA suggestedthat the decrease in activity was on account of the lack of awashing step. Interestingly, different phases of CaCO3, in-cluding calcite and vaterite, formed upon addition of serineat pH 10, as shown by polarized-light microscopy. Fe�CAwas shown to be a reusable and stable nanobiocatalysttoward CO2 hydration and sequestration.
Experimental Section
Synthesis of OAPS magnetic nanoparticles : Magnetic nanoparticles weresynthesized by means of co-precipitation, as described in the literature,[23]
with a slight modification. Typically, the iron salt mixture in a molar ratioof 1:2 (FeCl2/FeCl3) was stirred by using a Teflon rod for 30 min at 80 8C.After addition of ammonia, the reaction mixture was stirred for 4 h. N2
was bubbled throughout the reaction. The Fe3O4 nanoparticles were fil-tered, washed, and dried at 80 8C. Thereafter, silica-coated Fe3O4 particleswere prepared by using a modified Stçber method through hydrolysisand condensation of TEOS in isopropanol with ammonium hydroxide asthe base catalyst.[24] Typically, Fe3O4 (100 mg) was dispersed in a mixtureof water and isopropanol (1:5), after which TEOS (1 mL) and ammonia(2 mL) were added to the reaction solution with constant mechanical stir-ring for 24 h at 25 8C. The obtained spherical Fe3O4/SiO2 particles werewashed with water and dried. CPTMS (50 mmol) was grafted onto Fe3O4/SiO2 particles (200 mg) dispersed in dry toluene under reflux for 24 h.The final product was filtered, washed with toluene then alcohol, anddried under vacuum at 70 8C for 8 h. The product is denoted Fe3O4/SiO2/CPTMS. Finally, OAPS was grafted onto Fe3O4/SiO2/CPTMS as per theprocedure reported previously.[9] The product was denoted Fe3O4/SiO2/OAPS.
Immobilization of BCA on Fe3O4/SiO2/OAPS and enzymatic activityassays : BCA was immobilized onto Fe3O4/SiO2/OAPS particles, as de-ACHTUNGTRENNUNGscribed previously, with slight modification.[9] Briefly, Fe3O4/SiO2/OAPSparticles (10 mg) were dispersed in a 0.1 m sodium phosphate buffer(pH 8.0) and stirred with a GA solution for 1 h. The GA-activated parti-cles were separated by magnetic decantation and subsequently washedwith water. GA acted as a spacer to produce reactive aldehyde groups onthe surface of the Fe3O4/SiO2/OAPS, which then reacted with the aminogroups of BCA to form covalent bonds. The recovered product was treat-ed with a BCA solution (2 mL) at an enzyme concentration of 3 mg mL�1
and incubated with shaking for 1 h at 25 8C. Unbound BCA was removedby applying a magnetic field and washing the BCA-immobilized particlesuntil no BCA was detected in the supernatant Tris-HCl buffer. The prod-uct was denoted Fe�CA. The quantities of immobilized and unboundBCA were calculated by using the Bradford method, and the biocatalyticactivity of the free and immobilized BCA were estimated by usingp-NPA as described.[15]
Biomimetic sequestration of CO2 : The CO2 hydration was carried out asper our recent report with modification.[9] Typically, a 30 mm CO2 solu-tion was added to the free BCA- or Fe�CA-dispersed Tris-HCl buffer(30 mL, 1.0m, pH 8). The reaction was monitored by recording the pHchange, and completion of the CO2 hydration was determined by the ab-sence of pH changes.[6b] These studies were conducted in a temperature-controlled mechanically stirred batch reactor. Upon completion, the Fe�CA nanoparticles were collected by using an on/off magnet (Kanetec,MB-BV) placed close to the wall of the reactor. The hydrated CO2 solu-tion was collected by decanting, and the recovered Fe�CA was used insubsequent reaction cycles. The hydrated CO2 solution was treated undervacuum (B�CHI R-215, V-700) for 10 min to remove any unstable or un-dissolved CO2. The solution was then treated with a 100 mm calcium-ion-containing buffer solution (100 mL, CaCl2·6H2O), with or without serine(10 mm) at pH 10.0 to enable CaCO3 precipitation. The formation of
Figure 8. XRD patterns of pure calcite, and coexistence of calcite and va-terite.
Chem. Eur. J. 2012, 18, 12028 – 12034 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 12033
FULL PAPERCarbonic Anhydrase

CaCO3 crystals was observed by polarized-light microscopy and quanti-fied by ion chromatography coupled with a conductivity detector.[9] As acontrol, the above experiment was also carried out in the absence ofenzyme. The percent efficiency of calcium-ion precipitation was obtainedafter correcting the experimental results according to the control experi-ment. The weight of calcium carbonate and gram equivalents of CO2
present in CaCO3 were also determined. Precipitated CaCO3 was driedand characterized.
Characterization of materials : The surface morphology and elementalmapping of the amine-functionalized magnetic nanoparticles were meas-ured by FESEM by using a Hitachi S-4800 and EDS, respectively. Thesize distribution of the silica-coated magnetic nanoparticles was meas-ured by high-resolution TEM using a JEOL-2000EX operated at 120 kV.The XRD were recorded using a Rigaku Miniflex diffractometer withCuKa radiation (l= 0.154 nm). The FTIR spectra of BCA, Fe3O4, and thefunctionalized nanomaterials were recorded by the KBr pellet methodusing a Nicolet 6700 spectrometer. X-ray photoelectron spectroscopy wasused to confirm the silica coating, surface functionalization, and oxida-tion state of the iron in the oxide nanoparticles. These measurementswere recorded using a Thermo Scientific Multilab 2000 spectrometerwith a MgKa source. The ester (p-NPA) hydrolysis kinetics were moni-tored using an Optizen 2120F UV/Vis spectrophotometer. The calciumions were quantified by using an ion chromatograph (IC) with a Metro-sep C2-150 cationic column coupled to a conductivity detector (Metrohm801 Compact IC pro). CaCO3 crystal nucleation was observed by polar-ized-light microscopy, and the images were captured using an OlympusBX51M coupled with a megapixel Firewire camera and bright-field ob-jective (Olympus MPlanFL N 100 � 0.90 BD).
Acknowledgements
This work was supported by the Korea CCS R&D Center (KCRC) witha grant funded by the Korean Government (Ministry of Education, Sci-ence and Technology) (No. 2012-0008933).
[1] IPCC Fourth Assessment Report: Climate Change, 2007 (AR4).[2] a) D. M. D�Alessandro, B. Smit, J. R. Long, Angew. Chem. 2010, 122,
6194 – 6219; Angew. Chem. Int. Ed. 2010, 49, 6058 –6082; b) G. A.Olah, G. K. S. Prakash, A. Geoppert, J. Am. Chem. Soc. 2011, 133,12881 – 12898; c) J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried,R. D. Srivastava, Int. J. Greenhouse Gas Control 2008, 2, 9 –20.
[3] a) H. Miyamoto, T. Miyashita, M. Okushima, S. Nakano, T. Morita,A. Matsushiro, Proc. Natl. Acad. Sci. USA 1996, 93, 9657 –9660;b) P. Mirjafari, K. Asghari, N. Mahinpey, Ind. Eng. Chem. Res. 2007,46, 921 –926.
[4] J. M. Berg, J. L. Tymoczko, L. Stryer, Biochemistry, 6th ed., W. H.Freeman and Company, New York, 2007, pp. 254 – 259.
[5] a) Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld, I. Willner, Science2003, 299, 1877 –1881; b) U. T. Bornscheuer, Angew. Chem. 2003,115, 3458 –3459; Angew. Chem. Int. Ed. 2003, 42, 3336 – 3337.
[6] a) M. Vinoba, K. S. Lim, S. K. Jeong, S. H. Lee, M. Alagar, Lang-muir 2011, 27, 6227 –6234; b) M. Vinoba, M. Bhagiyalakshmi, S. K.Jeong, Y. Yoon, S. C. Nam, Colloids Surf. B 2012, 90, 91– 96; c) S.Wanjari, C. Prabhu, R. Yadav, T. Satyanarayana, N. Labhsetwar, S.Rayalu, Process Biochem. 2011, 46, 1010 – 1018; d) A. Z. M. Badrud-doza, K. Hidajat, M. S. Uddin, J. Colloid Interface Sci. 2010, 346,
337 – 346; e) J. D. Badjic, N. M. Kostic, Chem. Mater. 1999, 11, 3671 –3679; f) E. Ozdemir, Energy Fuels 2009, 23, 5725 –5730.
[7] a) A. K. Mukherjee, T. Satish Kumar, S. K. Rai, J. K. Roy, Biotech-nol. Bioprocess Eng. 2010, 15, 984 – 992; b) J. Huang, X. Li, Y.Zheng, Y. Zhang, R. Zhao, X. Gao, H. Yan, Macromol. Biosci.2008, 8, 508 – 515; c) N. Schultz, C. Syldatk, M. Franzreb, T. J.Hobley, J. Biotechnol. 2007, 132, 202 – 208.
[8] a) A. Kros, M. Gerritsen, V. Sprakel, N. Sommerdijk, J. Jansen, R.Nolte, Sens. Actuators B 2001, 81, 68 –75; b) L. Wang, K. G. Neoh,E. T. Kang, B. Shuter, Biomaterials 2011, 32, 2166 – 2173; c) C. Yang,J. Wu, Y. Hou, Chem. Commun. 2011, 47, 5130 –5141.
[9] M. Vinoba, M. Bhagiyalakshmi, S. K. Jeong, Y. Yoon, S. C. Nam, J.Phys. Chem. C 2011, 115, 20209 –20216.
[10] a) X. D. Wang, Z. X. Shen, T. Sang, X. B. Cheng, M. F. Li, L. Y.Chen, Z. S. Wang, J. Colloid Interface Sci. 2010, 341, 23 –29; b) H. C.Wang, C. Y. Wu, C. C. Chung, M. H. Lai, T. W. Chung, Ind. Eng.Chem. Res. 2006, 45, 8043 – 8048.
[11] R. Chen, G. Song, Y. Wei, J. Phys. Chem. C 2010, 114, 13 409 –13413.
[12] W. Xie, N. Ma, Energy Fuels 2009, 23, 1347 –1353.[13] a) J. C. Huang, C. B. He, Y. Xiao, K. Y. Mya, J. Dai, Y. P. Siow, Poly-
mer 2003, 44, 4491 – 4499; b) J. Gao, X. Li, W. Wu, H. Lin, Polym.Compos. 2011, 32, 829 –836.
[14] a) A. L. Morel, S. I. Nikitenko, K. Gionnet, A. Wattiaux, J. Lai-Kee-Him, C. Labrugere, B. Chevalier, G. Deleris, C. Petibois, A. Brisson,M. Simonoff, ACS Nano 2008, 2, 847 –856; b) Y. Qin, H. Ren, F.Zhu, L. Zhang, C. Shang, Z. Wei, M. Luo, Eur. Polym. J. 2011, 47,853 – 860.
[15] M. Vinoba, D. H. Kim, K. S. Lim, S. K. Jeong, S. W. Lee, M. Alagar,Energy Fuels 2011, 25, 438 – 445.
[16] a) R. C. Boguslaski, A. M. Janik, Biochim. Biophys. Acta Enzymol.1971, 250, 266 –269; b) M. Yan, Z. Liu, D. Lu, Z. Liu, Biomacromo-lecules 2007, 8, 560 –565.
[17] P. Pocker, T. L. Deits, J. Am. Chem. Soc. 1983, 105, 980 – 986.[18] A. Sharma, A. Bhattacharya, A. Shrivastava, Enzyme Microb. Tech-
nol. 2011, 48, 416 – 426.[19] a) S. Bhattacharya, M. Schiavone, S. Chakrabarti, S. K. Bhattachar-
ya, Biotechnol. Appl. Biochem. 2003, 38, 111 – 117; b) R. R. Yadava,S. N. Mudliarb, A. Y. Shekha, A. B. Fulkea, S. S. Devia, K. Krishna-murthia, A. Juwarkarc, T. Chakrabartia, Process Biochem. 2012, 47,585 – 590.
[20] a) L. A. Estroff, I. Cohen, Nat. Mater. 2011, 10, 810 –811; b) D. Ge-bauer, A. Volkel, H. Colfen, Science 2008, 322, 1819 –1822; c) J.Xiao, Z. Wang, Y. Tang, S. Yang, Langmuir 2010, 26, 4977 – 4983.
[21] a) S. Mann, Nature 1988, 332, 119 – 124; b) S. Mann, R. Heywood, S.Rajam, J. D. Bichall, Nature 1988, 334, 692 –695; c) H. Wei, Q. Shen,Y. Zhao, D.-J. Wang, D.-F. Xu, J. Cryst. Growth 2003, 250, 516 – 524;d) A. Ahmad, D. Rautaray, M. Sastry, Adv. Funct. Mater. 2004, 14,1075 – 1080; e) Q. Shen, Y. Chen, H. Wei, Y. Zhao, D. Wang, D. Xu,Cryst. Growth Des. 2005, 5, 1387 –1391; f) X. An, C. Cho, J. Phys.Chem. C 2008, 112, 6526 –6530; g) S. Lee, S. G. Lee, D. Kwak, J. H.Park, K. Cho, J. Phys. Chem. C 2011, 115, 2026 –2029.
[22] Q. Shen, L. Wang, Y. Huang, J. Sun, H. Wang, Y. Zhou, D. Wang, J.Phys. Chem. B 2006, 110, 23148 –23153.
[23] X. Wang, P. Dou, P. Zhao, C. Zhao, Y. Ding, P. Xu, ChemSusChem2009, 2, 947 –950.
[24] a) W. Stçber, A. Fink, J. Colloid Interface Sci. 1968, 26, 62– 69; b) T.Ung, L. M. Liz-Marz�n, P. Mulvaney, J. Phys. Chem. B 2001, 105,3441 – 3452.
Received: April 2, 2012Published online: August 9, 2012
www.chemeurj.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2012, 18, 12028 – 1203412034
S. K. Jeong et al.

Supporting Information� Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2012
Carbonic Anhydrase Immobilized on Encapsulated Magnetic Nanoparticlesfor CO2 Sequestration
Mari Vinoba,[a] Margandan Bhagiyalakshmi,[b] Soon Kwan Jeong,*[a]
Sung Chan Nam,[a] and Yeoil Yoon[a]
chem_201201112_sm_miscellaneous_information.pdf

S1
Materials. Bovine carbonic anhydrase (BCA), tetraethylorthosilicate (TEOS), 3-
chloropropyltrimethoxysilane (CPTMS), FeCl3.6H2O, FeCl2.4H2O, tetrahydrofuran (THF),
isopropanol, aqueous ammonia, glutaraldehyde (GA), 2-amino 2-(hydroxymethyl)-1,3-
propanediol (Tris base), p-nitrophenol (p-NP), p-nitrophenyl acetate (p-NPA), acetonitrile,
CaCl2·6H2O, and protein assay reagents for the Bradford assay were purchased from
Aldrich. OAPS was obtained from Hybrid Polymer (AM0280) and was used without
further purification. All solutions were prepared using ultrapure water obtained from a
Milli-Q water purification system.
S2
Figure S1. XRD patterns of Fe3O4 (a), Fe3O4/SiO2 (b), Fe3O4/SiO2/CPTMS (c), and
Fe3O4/SiO2/OAPS (d).
.

S3
Figure S2. EDS spectrum of Fe3O4 (a) and Fe3O4/SiO2/OAPS (b).

S4
Figure S3. Mapping of Fe3O4/SiO2/OAPS

S5
4000 3500 3000 2500 2000 1500 1000 500
Wave numbers (cm-1)
% T
ran
smit
tance
a
b
c
d
e
58
0
16
24
34
02
34
40
10
86
95
6 80
6
58
6
34
23 32
22 29
25
14
03
59
1
34
40
33
84
32
31 30
49
29
53
15
98
14
86
14
39
12
74
11
24
99
77
88
70
0
33
90 3
22
8
29
34
10
97
16
07
30
51
59
7
Figure S4. FT-IR spectrum of Fe3O4 (a), Fe3O4/SiO2 (b), Fe3O4/SiO2/CPTMS (c),
Fe3O4/SiO2/OAPS (d), and OAPS (e).