capsids, matrices and vesicles : structural insights into
TRANSCRIPT

CAPSIDS, MATRICES AND VESICLES – STRUCTURAL INSIGHTS INTO THE
ASSEMBLY OF PARAMYXOVIRUSES
Lassi J. P. Liljeroos
Institute of Biotechnology and Department of Biosciences, Division of Genetics Faculty of Biological and Environmental Sciences
University of Helsinki
and
Viikki Doctoral Programme in Molecular Biosciences University of Helsinki
ACADEMIC DISSERTATION
To be presented for public examination with the permission of the Faculty of Biological and Environmental Sciences of the University of Helsinki, in the lecture hall 2402 of
Biocenter 3, Viikinkaari 1, on October 11th 2013 at 12 o’clock noon.
HELSINKI 2013

Supervisor Research Director, Docent Sarah J. Butcher Institute of Biotechnology University of Helsinki Thesis committee Professor Adrian Goldman Department of Biosciences Faculty of Biological and Environmental Sciences University of Helsinki
Professor Markku Kulomaa Institute of Biomedical Technology University of Tampere
Reviewers Docent Tero Ahola Department of Food and Environmental Sciences Faculty of Agriculture and Forestry University of Helsinki
Dr. Peter B. Rosenthal Division of Physical Biochemistry MRC National Institute for Medical Research London, UK
Opponent Professor Kay Grünewald Oxford Particle Imaging Centre Division of Structural Biology Wellcome Trust Centre for Human Genetics University of Oxford, UK
Custos Professor Liisa Holm Institute of Biotechnology and Department of Biosciences Faculty of Biological and Environmental Sciences University of Helsinki
© Lassi Liljeroos 2013 Cover image: Human respiratory syncytial viruses budding from a cell. Image by Pasi Laurinmäki and Lassi Liljeroos. Micrograph courtesy of Roberta Mancini. ISBN 978-952-10-9272-5 (paperback) ISBN 978-952-10-9273-2 (PDF; http://ethesis.helsinki.fi) ISSN 1799-7372
Unigrafia Helsinki 2013

Teresalle ja Islalle
Don’t you understand that we need to be childish in order to understand? Only a child sees things with perfect clarity, because it hasn’t developed all those filters which prevent us from seeing things that we don’t expect to see.
-Douglas Adams, Dirk Gently’s Holistic Detective Agency

TABLE OF CONTENTS
Original publications ...................................................................................... i
Abbreviations ................................................................................................ ii
Summary ...................................................................................................... iii
A. INTRODUCTION ...................................................................................... 1
1. Virus structure ........................................................................................ 2
1.2. Structures of negative-sense RNA viruses ....................................... 3
1.2.1. Structures of paramyxoviruses .................................................. 8
2. Assembly of negative-sense RNA viruses ............................................ 10
2.1. Role of matrix protein in assembly ................................................ 11
2.2. Assembly of measles virus ............................................................. 13
2.3. Assembly of human respiratory syncytial virus ............................ 17
3. Entry of negative-sense RNA viruses ................................................... 19
4. Electron cryotomography as a tool in studying virus structure .......... 21
4.1. Sample preparation for electron cryotomography ........................ 23
4.2. Image formation in the electron microscope ................................24
4.3. Electron cryotomography ..............................................................26
4.4 Subvolume averaging ..................................................................... 28
B. AIMS OF THE PRESENT STUDY ........................................................... 31
C. MATERIALS AND METHODS ............................................................... 32
D. RESULTS AND DISCUSSION ................................................................ 33
1. The N0-P complex ................................................................................. 33
2. Budding of MV and HRSV and structures of the released virions ...... 35
E. CONCLUSIONS AND PERSPECTIVES ..................................................42
F. ACKNOWLEDGEMENTS........................................................................43
G. REFERENCES ......................................................................................... 45

i
ORIGINAL PUBLICATIONS
This thesis is based on the following articles, which are referred to in the text by their Roman numerals.
I Liljeroos, L., Huiskonen, J. T., Ora, A., Susi, P. and Butcher, S.J.
(2011) Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc Natl Acad Sci U S A. 108(44):18085-90
II Liljeroos, L., Krzyzaniak, M. A., Helenius, A. and Butcher, S.J. (2013)
Architecture of respiratory syncytial virus revealed by electron cryo-tomography. Proc Natl Acad Sci USA. 110(27):11133-8
III Liljeroos, L., Butcher, S.J. Expression, purification and characteriza-
tion of measles virus N0-P complex. Manuscript.
Additional unpublished data will be presented.

ii
ABBREVIATIONS
3D-cryoEM three-dimensional electron cryomicroscopy ARE apical recycling endosome ART algebraic reconstruction technique CCD charge-coupled device cryo-EM electron cryomicroscopy cryo-ET electron cryotomography DQE detective quantum efficiency EBOV Ebola virus ER endoplasmic reticulum ESCRT endosomal sorting complex required for transport F fusion protein FEG field emission gun G G glycoprotein H hemagglutinin HA influenza virus hemagglutinin HIV human immunodeficiency virus HN hemagglutinin-neuraminidase HRSV human respiratory syncytial virus HSV herpes simplex virus L-domain late domain MARV Marburg virus MCNC matrix-covered nucleocapsid MTF modulation transfer function MuV mumps virus MV measles virus MVB multivesicular body MVE multivesicular endosome N nucleoprotein NA neuraminidase NDV Newcastle disease virus NMR nuclear magnetic resonance PFU plaque-forming unit PIV parainfluenza virus PNT phosphoprotein N-terminal end RABV rabies virus RNP ribonucleoprotein SeV Sendai virus SH small hydrophobic protein SIRT simultaneous iterative reconstruction technique SNR signal-to-noise ratio -ssRNA negative single strand RNA TEM transmission electron microscope VLP virus-like particle VSV vesicular stomatitis virus WBP weighted back-projection WT wild-type

iii
SUMMARY
Paramyxoviridae constitute a family of pleomorphic, enveloped viruses includ-ing several human pathogens. Understanding of the structure and assembly of paramyxoviruses has been hindered by the lack of whole-virion three-dimensional structures. In this work, measles and human respiratory syncytial viruses were studied with three-dimensional electron microscopy and biochemi-cal analysis of recombinant proteins. The analysis revealed significant differ-ences in the structure and assembly of the two viruses. The differences were most notable in the way the matrix protein, the main factor driving budding from host cell, was organized inside the virions. In measles virions, the matrix was found to cover the genome-containing ribonucleocapsid, whereas in human respiratory syncytial virus the matrix was lining the inner surface of the mem-brane vesicle. These differences have implications on models of how each ribo-nucleoprotein complex assembles and how the viruses bud from the host cell. The early control of measles ribonucleoprotein assembly was subsequently in-vestigated to further reveal the details of the precise manner in which the intri-cate molecular ballet of viral assembly is orchestrated inside the host cell. The results presented in this thesis expand the understanding of enveloped virus structure and assembly, which is important in rational approaches to fight the pathogenic members in the group.


1
A. INTRODUCTION
Viruses are obligatory parasites that rely on the host cell for replication. They are like letters of genetic infor-mation that, in order to be duplicat-ed, require a copy machine. The copy machine is a bacterial, archaeal or eukaryotic cell. Most viruses carry components of this machine inside the viral particle in the form of struc-tural proteins encoded by genes in the viral genome that can be made of either RNA or DNA. Relying on the host cell for existence, viruses have evolved to thrive in as myriad condi-tions as living organisms. They can be found in places intuitively adverse to life, such as hot, acidic springs (Bize et al., 2008). It has been esti-mated that there are over 1030 viruses in the biosphere (Hendrix 2002) and in most environments they outnum-ber the host cells by an order of magnitude (Wommack et al., 2000). The vast majority of these viruses are bacteriophages, viruses of the bacte-ria. Eukaryotic viruses, although more modest in abundance, also come in a great variety. There are approximately 200 different viruses known to cause disease in humans of which more than one in four is con-sidered an emerging or re-emerging pathogen (Woolhouse et al., 2005). Of all the known human pathogens, viruses constitute approximately 15 %, the rest being mainly bacteria, fungi, helminths and protozoans in decreasing order of prevalence. Vi-ruses are overrepresented in the emerging and re-emerging patho-gens, RNA viruses comprising 37 % of the total 177 pathogens considered to belong to this group (Woolhouse et al., 2005).
Viral diseases cause great socio-economic burden throughout the world. Virus related death and mor-
bidity are greatest in the developing countries, but also significant in the industrialized countries. Economi-cally, prevention and preparation for possible viral threats, like pandemic influenza, causes considerable costs. In the 2009 H1N1 influenza pandem-ic, the costs were mainly incurred by vaccination programs, where many countries decided to purchase the vaccine for the whole population. In the absence of a general viral antibi-otic, vaccines are the most efficient arms to fight pathogenic viruses. Successful vaccination, however, re-quires anticipation as normally it is too late to vaccinate when disease symptoms arise. With the difficulty in predicting the vastness and pace of epidemics, preventive measures can easily be either under- or overesti-mated. Also, having to develop a new vaccine for every season for highly variant viruses, such as influenza, results in high costs for society. De-spite the efficacy of vaccines against many viruses, some have dodged all attempts to develop one. Examples include the human immunodeficien-cy (HIV) and human respiratory syn-cytial (HRSV) viruses.
The difficulty of developing a general anti-viral drug arises mainly from the parasitic and highly variable nature of viruses. They rely on the host cell for many of their functions, like replication, transport, exit and entry, and therefore blocking these functions is difficult without affect-ing uninfected cells and causing con-sequent toxicity to the whole organ-ism. Because viruses do not have similar unifying features as bacteria do (e.g. a peptidoglycan cell wall), it is difficult, if not impossible to devel-op an inhibitory molecule that would have as broad a spectrum as some of

2
the bacterial antibiotics do. One group of antivirals, effective for sev-eral different viruses, is the nucleo-side analogues that mimic the natu-ral nucleosides used in DNA synthe-sis. Acycloguanosine, for example, is effective against several members of the Herpesviridae family and is the main treatment for herpes simplex virus (HSV) infections. The mecha-nism of action of such compounds relies on the mimicry of cellular nu-cleosides, and they are preferentially incorporated into the viral DNA re-sulting in termination of DNA syn-thesis.
In the fight against viral diseas-es, it is instrumental to investigate the molecular details of the viruses and the interplay between the virus and the cell. Zooming in to the smallest atomic-level detail is re-quired in order to be able to rational-ly engineer molecules that can be used to prevent or treat infection. This level of detail for a virus in the cellular context can only be achieved with a combination of imaging tech-niques, such as x-ray crystallography, nuclear magnetic resonance (NMR),
electron cryomicroscopy (cryo-EM) and light microscopy. Enveloped pleomorphic (from the Greek: many-formed) viruses, like paramyxovirus-es, are especially amenable to a spe-cial application of cryo-EM called electron cryotomography (cryo-ET) for three dimensional studies of the whole virion.
Paramyxoviruses are envel-oped, nonsegmented, negative-sense RNA viruses that include a number of human pathogens, like HRSV, measles (MV), mumps (MuV) and parainfluenza (PIV) viruses. For MV and MuV an effective vaccine has been available for several decades and they have been largely eradicat-ed from countries with well-coordinated vaccination programs, but remain a major problem in many developing countries. Due to increas-ing reluctance to be vaccinated and the consequent decrease in herd im-munity, there have been outbreaks of these viruses recently in countries like U.S.A., Germany, France, U.K. and Finland (Kay et al., 2011, Muscat 2011). Human vaccines are yet to be developed for PIV and HRSV.
1. VIRUS STRUCTURE
Viruses are generally in the range of 20 - 1000 nm in diameter. Most known viruses have an icosahedral capsid. Icosahedral viruses include a vast number of different virus fami-lies infecting all domains of life. The genomes of icosahedral viruses are varied, being of DNA or RNA, single- or double-stranded, linear or circu-lar. Helical viruses are also common, especially within viruses infecting plants, the archetype being tobacco mosaic virus (Stubbs et al., 2012). Due to the highly ordered, symmet-rical shape of icosahedral and helical
viruses, three-dimensional electron cryomicroscopy (3D-cryoEM) cou-pled with image averaging, as well as x-ray crystallography have been the methods of choice for virus studies. Consequently, especially icosahedral viruses are overrepresented in the pool of three-dimensionally charac-terized virus structures (> 300 icosa-hedral virus structures and < 50 ple-omorphic virus structures are availa-ble in the electron microscopy data bank, accession date 09.07.2013).
An icosahedron is composed of 20 facets, thus having six five-fold,

3
10 three-fold and 15 two-fold sym-metry axes. Viral icosahedral capsids are 60-fold symmetric and the sim-plest viruses with the so-called T = 1 symmetry have 60 copies of the cap-sid protein in the shell. One triangu-lar facet of the icosahedron is there-fore composed of three copies of the capsid protein in an identical envi-ronment. Most viruses have more than one copy of the protein(s) per asymmetric unit, which increases the size of the capsid, thus allowing more space for larger genomes. In these capsids the proteins are not in exact-ly equivalent relationship to the neighboring ones and their positions in the capsid are thus referred to as quasi-equivalent (Caspar et al., 1962). Although the majority of the icosahedral virus structures solved to atomic resolution have been solved by X-ray crystallography, some have been solved by 3D-cryoEM, some by both such as that of human adenovi-rus (Liu et al., 2010, Reddy et al., 2010, Zhang et al., 2013a, Zhang et al., 2010).
Helical viruses can be found within plant viruses and bacterio-phages. Most of them have a single capsid protein that forms a helix to-gether with nucleic acid, in most cas-es ssRNA [for a review, see (Stubbs et al., 2012)]. The helix essentially comprises the whole virus. Helical nucleocapsids can also be found in many other viruses, like filoviruses, rhabdoviruses and paramyxoviruses, but they are different in that the heli-cal nucleocapsid comprises only part of the whole virion. The symmetrical, often highly ordered structure of hel-
ical viruses can be utilized in struc-ture determination and consequently helical virus structures have been solved to atomic resolution by x-ray fiber diffraction (Namba et al., 1989) and to near-atomic resolution by 3D-cryoEM (Clare et al., 2010, Sachse et al., 2007).
Outside the majority of icosa-hedral and helical viruses, most vi-ruses are enveloped and pleomorphic although some like the Alphaviridae and Flaviviridae are enveloped and icosahedral. The Herpesviridae, some Togaviridae and Cystoviridae are exceptional in that they are en-veloped and pleomorphic, but con-tain an icosahedral capsid inside the envelope. Some members of the Bunyaviridae family produce both icosahedral and pleomorphic parti-cles (Överby et al., 2008). Enveloped viruses have a host-derived lipid membrane that protects the genome and associated proteins and also provides means for cell entry via membrane fusion. The vast majority of known enveloped pleomorphic viruses are eukaryotic, although some have also been found in bacte-riophages and archaeal viruses (Ogawa et al., 1985, Pietilä et al., 2012). For pleomorphic viruses, av-eraging methods like x-ray crystal-lography, NMR or single particle 3D-cryoEM are not suitable for whole-virus structure determination. Thus, only after the relatively recent wide-scale application of cryo-ET, 3D structures of these viruses have also begun to be revealed (Subramaniam et al., 2007).
1.2. STRUCTURES OF NEGATIVE-SENSE RNA VIRUSES
Negative strand RNA (-ssRNA) vi-ruses comprise a large group of vi-ruses, many of which are significant
human pathogens. The group com-prises eight virus families: Bor-naviridae, Filoviridae, Paramyxo-

4
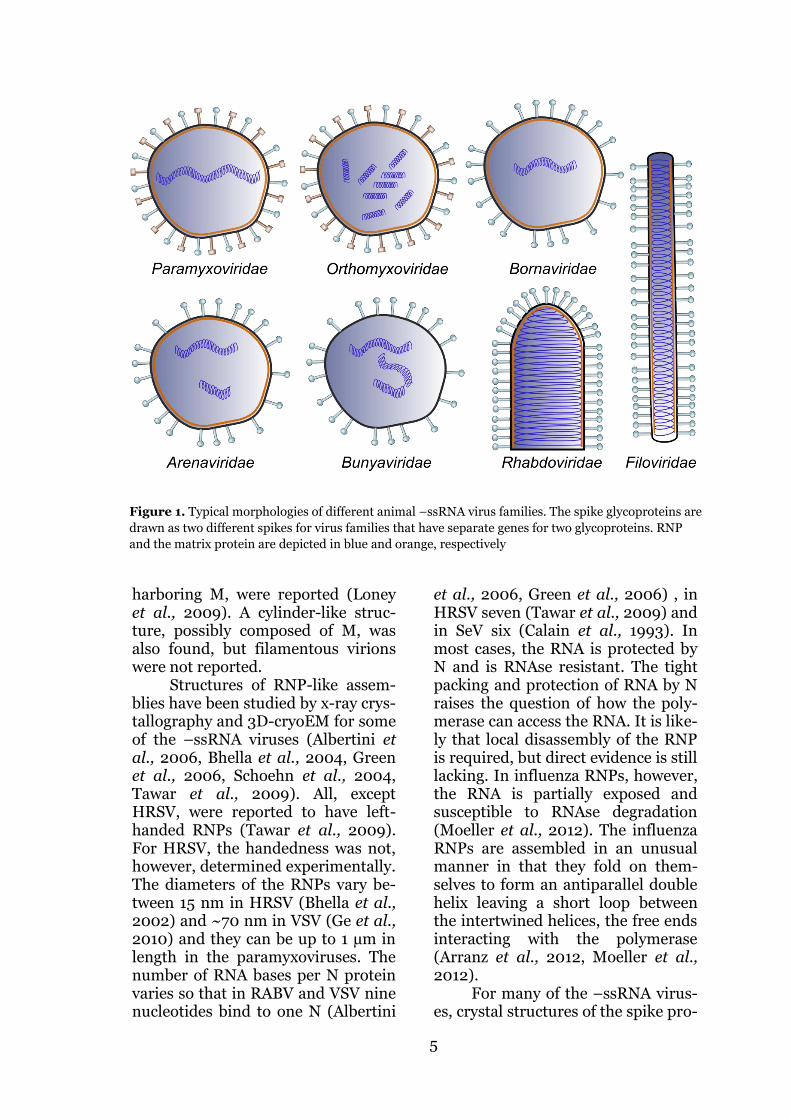
viridae and Rhabdoviridae that have a nonsegmented genome; and Are-naviridae, Bunyaviridae, Ophioviri-dae and Orthomyxoviridae that have a segmented genome. Pathogens like influenza (family Orthomyxoviri-dae), rabies (RABV, Rhabdoviridae), Ebola (EBOV, Filoviridae) viruses, MV and HRSV are members of the group. All animal –ssRNA viruses are enveloped and they are pleo-morphic, except some of the viruses in the Rhabdo- and Bunyavirus fam-ilies (Figure 1). Rhabdoviruses are bullet-shaped (Ge et al., 2010, Guichard et al., 2011) and some of the bunyaviruses have icosahedral symmetry (Freiberg et al., 2008, Överby et al., 2008).
The majority of –ssRNA viruses have a helically-arranged ribonucle-ocapsid (RNP) that contains the ge-nome wrapped either inside (Green et al., 2006) or outside (Tawar et al., 2009) a nucleoprotein (N) helix. In addition to the RNP, the host-derived lipid membrane encloses the RNA-dependent RNA polymerase(s) and the matrix proteins. In all mem-bers of the group there are one or more viral membrane-spanning gly-coproteins that are required for host cell attachment, entry and in the case of influenza and some paramyxovi-ruses, also detachment. In some vi-ruses, like influenza, filo- and rhabdoviruses, the attachment and fusion functionalities reside in the same glycoprotein (hemagglutinin, HA for influenza, G for rhabdo- and filoviruses), whereas in others these roles have been divided into two sep-arate proteins (e.g. hemagglutinin, H and fusion, F in MV). Influenza also has two glycoproteins, but the sec-ond, the neuraminidase (NA) helps in virus escape from the cell surface by cleaving off sialidase to which the HA attaches. Influenza has, in addi-tion, a proton channel forming pro-tein M2 the main role of which is to
allow acidification of the virion inte-rior during the endocytotic entry. M2 has also been shown to have a role in the final pinching off from the host cell (Rossman et al., 2010). Some of the paramyxoviruses have, in addi-tion to the attachment and fusion proteins, a third small hydrophobic (SH) membrane protein. The role of this protein is unclear, but functions in host cell TNF-α signaling inhibi-tion and as a cation selective, oligo-meric ion channel, have been sug-gested (Carter et al., 2010, Fuentes et al., 2007, Gan et al., 2008, He et al., 2001, Li et al., 2011, Wilson et al., 2006).
3D structures of several –ssRNA viruses using 3D-cryoEM have been described (Bharat et al., 2012, Ge et al., 2010, Harris et al., 2006, Loney et al., 2009, Överby et al., 2008). Influenza virion structure has now been described for both a mainly spherical X31 strain (Harris et al., 2006, Wasilewski et al., 2012) and a mainly filamentous A/Udorn/72 strain (Calder et al., 2010, Vijayakrishnan et al., 2013). In the Udorn strain the M1 matrix pro-tein was assembled in a regular, fil-amentous array directly under the membrane bilayer and could be dis-rupted with low pH treatment (Calder et al., 2010). In the X31 strain, M1 lined the inner side of the membrane but was not tubularly or-ganized (Harris et al., 2006). In Marburg virus (MARV), a member of the Filoviridae family, similar regu-larity to influenza Udorn, was detect-ed in the VP40 matrix layer (Bharat et al., 2011). In vesicular stomatitis virus (VSV), the M matrix protein formed a left-handed helical struc-ture in register with the RNP (Ge et al., 2010) and was in close proximity to both the membrane and the RNP. For Sendai virus (SeV), a member of the Paramyxoviridae, patches with thickened membrane possibly
5
harboring M, were reported (Loney et al., 2009). A cylinder-like struc-ture, possibly composed of M, was also found, but filamentous virions were not reported.
Structures of RNP-like assem-blies have been studied by x-ray crys-tallography and 3D-cryoEM for some of the –ssRNA viruses (Albertini et al., 2006, Bhella et al., 2004, Green et al., 2006, Schoehn et al., 2004, Tawar et al., 2009). All, except HRSV, were reported to have left-handed RNPs (Tawar et al., 2009). For HRSV, the handedness was not, however, determined experimentally. The diameters of the RNPs vary be-tween 15 nm in HRSV (Bhella et al., 2002) and ~70 nm in VSV (Ge et al., 2010) and they can be up to 1 µm in length in the paramyxoviruses. The number of RNA bases per N protein varies so that in RABV and VSV nine nucleotides bind to one N (Albertini
et al., 2006, Green et al., 2006) , in HRSV seven (Tawar et al., 2009) and in SeV six (Calain et al., 1993). In most cases, the RNA is protected by N and is RNAse resistant. The tight packing and protection of RNA by N raises the question of how the poly-merase can access the RNA. It is like-ly that local disassembly of the RNP is required, but direct evidence is still lacking. In influenza RNPs, however, the RNA is partially exposed and susceptible to RNAse degradation (Moeller et al., 2012). The influenza RNPs are assembled in an unusual manner in that they fold on them-selves to form an antiparallel double helix leaving a short loop between the intertwined helices, the free ends interacting with the polymerase (Arranz et al., 2012, Moeller et al., 2012).
For many of the –ssRNA virus-es, crystal structures of the spike pro-
Figure 1. Typical morphologies of different animal –ssRNA virus families. The spike glycoproteins are
drawn as two different spikes for virus families that have separate genes for two glycoproteins. RNP
and the matrix protein are depicted in blue and orange, respectively

6
teins have been solved (Hashiguchi et al., 2007, Lee et al., 2008, McLellan et al., 2011, Roche et al., 2007, Varghese et al., 1983, Wilson et al., 1981, Yin et al., 2005). All, ex-cept bunya- and rhabdoviruses, have class I fusion proteins of which SeV F was the first described (Homma et al., 1973). Bunyaviruses have class II fusion proteins and spike assemblies on virions that are quite different from those of other –ssRNA viruses (Bowden et al., 2013, Huiskonen et al., 2010, Huiskonen et al., 2009). Rhabdoviruses have class III fusion proteins that combine features from both class I and class II fusion pro-teins (Roche et al., 2006, Roche et al., 2007). Class I fusion proteins are expressed in the host cell in a pretriggered, metastable, spring-loaded conformation and they un-dergo dramatic refolding upon trig-gering by either low pH or a more specific signal from binding to the host cell surface (Harrison 2008). Class I and II fusion protein undergo irreversible conformational changes upon triggering, whereas for class III fusion proteins the pH-related changes are reversible. Other than low pH, the triggering signals are not known, but they are thought to be relayed by the attachment protein after binding to the receptor (Chang et al., 2012, Navaratnarajah et al., 2011). For HRSV, a receptor for the fusion protein has also been identi-fied (Tayyari et al., 2011). Thus, it is possible that in the case of HRSV the attachment protein only indirectly enhances the perhaps lower affinity interaction between the fusion pro-tein and its receptor nucleolin. It has also been noted that HRSV lacking the attachment glycoprotein G is in-fectious, albeit at a lower level than wild-type (WT) (Techaarpornkul et al., 2001). All of the class I fusion proteins are trimeric type II trans-membrane proteins and require a
proteolytic cleavage by cellular pro-teases in order to be active. Unlike other –ssRNA viruses, HRSV re-quires two cleavages in order to be active for fusion (Gonzalez-Reyes et al., 2001).
Conversely to most of the –ssRNA virus fusion proteins that share the same general features, the attachment functionalities are highly variable. This is mainly due to the very different receptors of the virus-es. The majority, including influenza and most of the paramyxoviruses, bind to sugar moieties on the cell surface, yet some (e.g. MV, Hendra and Nipah viruses) bind to specific protein receptors. Although crystal structures of isolated ectodomains of many –ssRNA spike proteins have been solved, the in situ structures on virions have only been reported for influenza (Harris et al., 2013), Tula, Rift valley fever and Bunyamwera viruses (Bowden et al., 2013, Huiskonen et al., 2010, Huiskonen et al., 2009) from the Bunyaviridae and PIV3 (Ludwig et al., 2008) from the Paramyxoviridae. In PIV3, F, where discernible, was reported to be in the postfusion conformation (Ludwig et al., 2008). In the absence of other F in situ structures, it is not known if that is a common feature in the family. For MV and HRSV the two glycoproteins have also been suggested to form a complex on the cell surface, with the association oc-curring already in the endoplasmic reticulum (ER) for MV. It has yet to be determined if they exist as such on the virion (Low et al., 2008, Plemper et al., 2001).
Matrix proteins are the main vi-ral factors responsible for budding (Liljeroos et al., 2013). They general-ly reside in close contact with the inner side of the viral membrane. Although all –ssRNA matrix proteins share many functions, like bringing the RNP and glycoproteins together

7
at the budding sites, their structures are highly varied. Paramyxoviral, filoviral and bornaviral matrix pro-tein structures share similar mainly β-sheet folds (Figure 2) (Dessen et al., 2000, Money et al., 2009, Neumann et al., 2009), although bornaviral M is composed of only one of the two domains found in paramyxoviral and filoviral matrix proteins. Rhabdoviral M has a simi-lar secondary structure composition but is unrelated in topology (Gaudier et al., 2002, Graham et al., 2008).
Influenza M1 is completely different in structure to the others, being completely α-helical (Sha et al., 1997). Arena and bunyaviruses do not have a matrix protein per se, but arenaviruses have a small protein Z that has similar functions to the ma-trix proteins (Neuman et al., 2005, Strecker et al., 2003). Many of the matrix proteins have a propensity to self-oligomerize into sheets or heli-ces, which is important for their budding functionality.
Figure 2. Comparison of matrix protein structures of –ssRNA viruses. Ribbon diagram of the atom-
ic models from (A) HRSV M (PDB ID 2VQP) (Money et al., 2009), (B) EBOV VP40 (PDB ID 1ES6)
(Dessen et al., 2000), (C) Borna disease virus M (PDB ID 3F1J) (Neumann et al., 2009), (D) VSV M
(PDB ID 2W2R) (Gaudier et al., 2002), (E) influenza A M1 (PDB ID 1EA3) (Arzt et al., 2001) and (F)
Lassa virus Z (PDB ID 2KO5) (Volpon et al., 2010). For Lassa virus Z the structured core domain
between amino acids 26-79, is shown. The full length protein is 99 amino acids long and the termini
are unstructured. Reprinted from (Liljeroos et al., 2013) with permission from the publisher.

8
1.2.1. Structures of paramyxoviruses
The family Paramyxoviridae con-tains two subfamilies: Paramyxovir-inae and Pneumovirinae. The Para-myxovirinae subfamily has seven genera: Aquaparamyxovirus, Avula-virus, Ferlavirus, Henipavirus, Morbillivirus, Respirovirus and Rubulavirus. The Pneumovirinae subfamily is smaller having only two genera: Metapneumovirus and Pneumovirus. Of the human patho-gens, HRSV and human metapneu-movirus belong to the Pneumoviri-nae, all others to the Paramyxoviri-nae. All paramyxoviruses are envel-oped pleomorphic viruses that are highly variable in size and shape. Although the vast majority of these viruses are thought to be spherical or close to spherical in shape, filamen-tous forms have also been reported (Bächi et al., 1973, Compans et al., 1966, Yao et al., 2000). Although the family includes several human path-ogens, there was only one whole viri-on 3D paramyxovirus structure re-ported on SeV when the current study was initiated (Loney et al., 2009). SeV was reported to vary be-tween 110 and 540 nm in diameter, was densely covered by the surface glycoproteins and contained multiple copies of the helical RNP. The RNP appeared similar to what was earlier reported for purified RNPs (Bhella et al., 2002). Indications of the pres-ence of M under the membrane were derived from the thickness meas-urements of the membrane, where some regions had a thickness up to 12 nm. In the absence of other stud-ies, two of the open questions in the field were firstly, how widely these observations apply to other para-myxoviruses, and secondly, what causes some of the paramyxoviruses to adopt a filamentous shape?
Paramyxoviruses expose the ec-todomains of two membrane-spanning glycoproteins, F and the attachment protein on the mem-brane surface. The HN attachment protein of respiro-, rubula- and avulaviruses binds to sialic acids and contains hemagglutination and neu-raminidase activities. Morbillivirus-es, like MV, have protein receptors and have lost the neuraminidase ac-tivity of their attachment protein H (Muhlebach et al., 2011, Noyce et al., 2011, Tatsuo et al., 2000). Pneumo- and henipaviruses have an attach-ment protein G that lacks both he-magglutination and neuraminidase activites. The crystal structures of some paramyxoviral attachment pro-teins have been solved alone and with their receptors (Hashiguchi et al., 2011, Santiago et al., 2010, Xu et al., 2008, Zhang et al., 2013b). They all share a similar β-propeller fold in their head domain despite the vari-ous binding partners they have evolved to bind. The oligomerization state on the virions in most cases is tetrameric (Brindley et al., 2010, Yuan et al., 2008).
Crystal structures of the F pro-tein ectodomain in a prefusion state have been solved for PIV5 and HRSV and in the postfusion state for PIV3, NDV, and HRSV (McLellan et al., 2013, McLellan et al., 2011, Swanson et al., 2010, Welch et al., 2012, Yin et al., 2005, Yin et al., 2006). The pre-fusion structures resembled each other in the overall shape, but were different in certain antigenic sites and location of the fusion peptide that inserts into the target cell mem-brane (McLellan et al., 2013). The postfusion conformation was found to be highly similar in all the struc-tures solved, having the canonical six-helix bundle in the stalk and an

9
overall golf-tee-like morphology. Surprisingly, in the only analysis so far of the glycoproteins on virions, F was found in the postfusion confor-mation in PIV3 (Ludwig et al., 2008). Whether this was due to sample preparation, the fact that on-ly regions on virions with distinct spikes, were used for analysis, or that F naturally mainly is in the postfu-sion conformation on virions, has yet to be confirmed. Since all of the paramyxoviruses undergo virus-cell fusion at neutral pH, the fusion trig-gering mechanism is more complex than that for pH-dependent fusion proteins. For MV it has been shown that the fusion triggering signal is initiated by H binding to the main receptor CD150 and that a conforma-tional shift in the central stalk region of the H tetramer is required (Brindley et al., 2012).
Paramyxoviruses have a non-segmented genome typically approx-imately 15 kb in length. Consequent-ly, being approximately 15 – 20 nm in diameter, the RNPs can reach lengths of up to 1 µm. RNPs from different viruses have varying helical parameters and slightly different ap-pearance, but all have a general her-ringbone-like structure (Bhella et al., 2002). Recombinant RNPs from MV and HRSV have been reconstructed from electron micrographs and flexi-bility was detected even within a sin-gle nucleocapsid (Bhella et al., 2004, Schoehn et al., 2004, Tawar et al., 2009). A crystal structure of an RNP-like decameric ring of HRSV has been solved and in this structure the RNA was wrapped around the ring on the outside in a groove on N (Tawar et al., 2009). This structure was then modeled into a 3D electron microscopy helical reconstruction of the RNP from electron microscopy as
a left-handed helix. In general, crys-tallizing the nucleoprotein of the paramyxoviruses is exceedingly chal-lenging due to the fact that regard-less of the expression system used, they bind to cellular RNA and oli-gomerize into a heterogenous mix-ture of RNP-like helical structures. These helices cannot easily be disas-sembled as the protein-RNA binding is tight and the RNA is ribonuclease protected. Hence the early stages of assembly of the RNP have been tricky to access structurally.
The M protein of paramyxovi-ruses is generally thought to line the internal side of the lipid bilayer. It has a tendency to oligomerize as hel-ices (McPhee et al., 2011) and has been suggested to form a tubular lay-er also on the RNP in disrupted vi-rus-infected cells (Almeida et al., 1963, Brown et al., 1987). A crystal structure of HRSV M has been pub-lished and it consists of two predom-inantly β-sheet containing domains that are connected by an unstruc-tured linking region (Money et al., 2009). One side of the protein has an extensive positively-charged surface that extends to both domains. This surface was suggested to interact with the negatively-charged phos-pholipids in the virion thus placing this side towards the membrane. In the absence of the whole virus 3D structure, it is currently not known how the M is organized in HRSV vi-rions although indications of helical assemblies in the filamentous virions have been reported (Bächi et al., 1973). Additionally, it is unclear whether the M-covered RNPs found in preparations with disrupted MV (Almeida et al., 1963) are an authen-tic feature of the virion, or merely an experimental artifact.

10
2. ASSEMBLY OF NEGATIVE-SENSE RNA VIRUSES
Assembly and exit of enveloped vi-ruses is a complicated process, where all essential viral components have to be at the correct time at the place of assembly in suitable numbers. They rely on the host cell as a supply of lipids for encapsidation of their ge-netic material during the journey from one host cell to the next. They exit the host cell via an event gener-ally called “budding” that initiates by gathering the viral components to the cell membranes. Next, they begin to extrude from the cell enclosing the viral genome and viral structural proteins inside the nascent extru-sion. After a final pinching off from the cell, the viral components have become enclosed into a lipid bilayer hijacked from the host cell. During the process, the glycoproteins are incorporated into the virion mem-brane via protein-lipid and protein-protein interactions with the mem-brane and the internal viral proteins. The main role of the glycoproteins is to act as docking and entry apparat-uses to the next target cell, but they are also important in inducing mem-brane curvature, interacting with the internal viral components and in di-recting the budding to correct sites in the membrane.
Many of the –ssRNA viruses bud from specific microdomains on the plasma membrane, called lipid rafts (Manie et al., 2000, Panchal et al., 2003, Scheiffele et al., 1999). These regions are enriched in sphin-golipids, cholesterol and proteins (Lingwood et al., 2010). The budding preference from the lipid rafts has been established mainly by detecting colocalization of viral proteins and lipid raft markers in cells and the lipid composition of viruses has been found to resemble that of lipid rafts,
enriched in detergent insoluble cho-lesterol (Scheiffele et al., 1999). Some viral proteins, like MV F, when expressed alone, have a tendency to localize into lipid rafts (Pohl et al., 2007). It is enough for one of the viral proteins to have this tendency if the viral protein interaction network covers all the other components that are consequently also pulled to lipid rafts for budding. Many –ssRNA vi-ruses, like MARV, MV and HRSV also show preference for budding from one side of polarized epithelial cells (Blau et al., 1995, Roberts et al., 1995, Sänger et al., 2001). Interest-ingly, the MARV glycoprotein GP, when expressed alone, is transported to the apical side whereas viral bud-ding happens exclusively from the basolateral side (Sänger et al., 2001). Bunyaviruses, conversely to other –ssRNA viruses, bud from the Golgi apparatus (Kuismanen et al., 1982, Novoa et al., 2005).
Replication of most animal –ssRNA viruses takes place entirely in the cytoplasm, influenza and Bor-na disease viruses being the only ex-ceptions replicating in the nucleus. Most of the enzymatic activities re-quired for virus multiplication are included in the viral proteins. Yet, the viruses often rely on the host cell’s machinery for many aspects, especially for transport due to the poor diffusion of the viral compo-nents in the crowded cytoplasm. The requirement for transport includes not only the glycoproteins that go through the normal ER – Golgi ex-port route of transmembrane plasma membrane proteins, but also transport of the RNP and matrix pro-teins to the assembly sites. Addition-ally, host components have roles in regulating viral proteins by post-

11
translational modifications and in the final pinching off of the virus from the host cell. The host compo-nents include the endosomal com-plexes required for transport (ESCRT)(Bieniasz 2006), actin (Bohn et al., 1986, Carlson et al., 2010, Cudmore et al., 1995), micro-tubules (Penfold et al., 1994), and COP-coated vesicles (Yamayoshi et al., 2008).
Of the viral components, the matrix proteins are vital in assembly and budding. They act in a glue-like manner between the viral compo-nents and the membrane, bringing all the components together at the budding sites. The role of matrix pro-teins and their crucial interactions with other viral and cellular proteins are discussed next.
2.1. ROLE OF MATRIX PROTEIN IN ASSEMBLY
The important role of matrix pro-teins in virus budding was first shown with temperature sensitive mutants of SeV and VSV (Knipe et al., 1977, Yoshida et al., 1979). Bud-ding of both viruses was inefficient at elevated temperatures and altered the morphology of VSV (Schnitzer et al., 1979). Complementation with separately expressed M restored the budding ability (Lyles et al., 1996). Also, deletion of the whole M gene from RABV resulted in 5 x 105 fold reduced release from cells (Mebatsion et al., 1999). Mutations in the M gene have also been linked to altered pathogenesis. In the sub-acute sclerosing panechephalitis (SSPE) strains of MV, the M gene has undergone hypermutation of U to C residues, which results in impaired budding and consequently more cell-associated virus (Patterson et al., 2001). SSPE is a lethal neurogenera-tive disorder with an incidence of approximately 4-11 cases per 100 000 measles infections (WHO 2006).
The role of different viral pro-teins in budding has widely been as-sessed by expressing individual pro-teins in mammalian cells and analyz-ing whether vesicles with those par-ticular proteins are released as virus-like particles (VLP). With many vi-
ruses, expression of the matrix pro-tein alone is enough to cause release of VLPs (Jasenosky et al., 2001, Justice et al., 1995, Pohl et al., 2007). For some viruses, however, coex-pression of N with one of the surface glycoproteins is required or enhances VLP release (Li et al., 2009, Licata et al., 2004, Noda et al., 2002, Schmitt et al., 2002). The paramyxoviral Newcastle disease virus (NDV) ma-trix has even been shown to be able to deform and bud into phospholipid bilayer vesicles upon in vitro incuba-tion of purified components (Shnyrova et al., 2007). For influen-za virus, the role of M1 in budding is not as clear as it is for other –ssRNA viruses as conflicting reports on M1’s ability to drive VLP budding have been published (Chen et al., 2007a, Gomez-Puertas et al., 2000). It is likely, however, that M1 defines the virion shape as in the filamentous particles it is assembled as a helix and in the spherical particles as a sphere (Calder et al., 2010). Yet, in the absence of demonstrated mem-brane and glycoprotein free M1 helix, it is uncertain whether the shape de-termining factor is M1 or perhaps a helical array of surface glycoproteins.
In addition to the role in viral particle release, the matrix proteins interact with multiple other viral

12
components to be included in the budding virion. Interactions so far described include the surface glyco-protein tails (Ali et al., 2000a, Ali et al., 2000b, Ghildyal et al., 2005b) and the RNP (Ge et al., 2010, Iwasaki et al., 2009, Noton et al., 2007). For MV the interacting region on N was mapped to two C-terminal leucine residues, mutations in which result-ed in impaired virus growth (Iwasaki et al., 2009). In most cases, the in-teraction of M and N is thought to be direct, but for HRSV a mediator role for another viral protein, M2-1, has been described (Li et al., 2008).
Many viruses make use of the cellular vacuolar protein sorting (VPS) pathway in budding. Viral ma-trix proteins contain late domains (L-domains) that interact with compo-nents of this pathway. L-domains, so termed because mutations in them cause defects at a late stage in bud-ding, were first found in retroviral gag matrix protein (Göttlinger et al., 1991, Parent et al., 1995, Wills et al., 1994), but have since been found in many other viruses including filo- rhabdo- paramyxo- and arenaviruses (Harty et al., 1999, Li et al., 2009, Licata et al., 2003, Urata et al., 2006). Influenza, instead, is not de-pendent on VPS for budding and M1 does not contain L-domains (Bruce et al., 2009, Watanabe et al., 2010). The dependency on VPS for budding is generally judged based on whether dominant negative expression of VPS4, a protein functioning in the pinching-off stage of VPS, impairs budding. ESCRT complexes 0, I, II and III are components of VPS and are normally involved in sorting pro-teins to late endosomes [also termed multivesicular bodies (MVB) or mul-
tivesicular endosomes (MVE)] and in the final membrane scission step at the end of cytokinesis. Topologically, these processes are similar to virus budding from the plasma membrane. Thus, it is not surprising that viruses have evolved to hijack components of VPS to their advantage and redirect them to the plasma membrane (Fig-ure 3). Interestingly, for EBOV a role for COPI and COPII vesicles in VP40 transport and budding has been demonstrated (Yamayoshi et al., 2010, Yamayoshi et al., 2008). COPI and COPII are normally in-volved in ER-Golgi transport and are not present in the proximity of the plasma membrane. EBOV thus hi-jacks the machinery and redirects it for transport to the plasma mem-brane.
Despite the well-established role of matrix proteins in –ssRNA virus assembly and budding, it is not clear what provides the driving force for deforming the cell membrane. Evidence from the study of isolated NDV M budding into phospholipid vesicles suggested that multimeriza-tion of M provides the energy, but cellular proteins could also have a role in this. For MARV it has been shown that actin is incorporated into virions and disrupting F-actin fila-ments by cytochalasin D treatment significantly reduced VP40 VLP budding (Kolesnikova et al., 2007). In a proteomic analysis of HRSV, actin was also detected in purified virus preparations (Radhakrishnan et al., 2010), although it was not studied whether it was in the globu-lar or filamentous form. In MV, fila-mentous actin has been detected pro-truding into the viral buds with the barbed ends (Bohn et al., 1986).

13
2.2. ASSEMBLY OF MEASLES VIRUS
MV is one of the most infectious hu-man pathogens known and therefore requires high coverage of vaccination in the population for herd immunity. It is uncommon in industrialized countries, but poses a significant health risk in the developing coun-tries especially to children under the age of five. In 2008, it caused the death of over 160 000 people world-wide (WHO 2009). The main reason for the mortality is the immunosup-pression that follows from the infec-tion. In countries with poor hygiene and healthcare consequent second-ary infections can lead to serious ill-ness or death. The virus replicates in the cytoplasm of lymphocytes and dendritic cells expressing the signal-ing lymphocyte activating molecule
(SLAM, CD150), which is the main receptor for MV (Tatsuo et al., 2000). Recently, nectin-4 was identi-fied as an MV receptor on epithelial cells not expressing CD150 (Muhlebach et al., 2011, Noyce et al., 2011). The laboratory-adapted vac-cine strains, but not the WT strains can also utilize the ubiquitous CD46 as their receptor. The immunosup-pression caused by the virus is mani-fold, and includes CD150-mediated lymphopenia, prolonged cytokine imbalance and silencing of peripher-al blood lymphocytes (Avota et al., 2010).
MV genome codes for eight pro-teins: N, P, C, V, M, F, H, and L in 3’ - 5’ order (Figure 4). The C and V proteins are expressed from the P
Figure 3. ESCRTs have a role in several topologically similar membrane scission events. HIV is
able to hijack the ESCRTs normally functioning in vesicle budding into late endosomes and in
cytokinesis via its gag matrix protein. Reprinted by permission from Macmillan Publishers Ltd:
Nature (Raiborg et al., 2009), © (2009).

14
gene. C is expressed from an alterna-tive initiation codon and V has the N-terminus of P, but the C-termini of the two proteins are different. This is accomplished with mRNA editing via addition of one or more untemplated G residues, which results in frame shifting (Cattaneo et al., 1989). C and
V are nonstructural proteins and their main roles have been shown to lie in inhibiting host innate immuni-ty response by interfering with inter-feron signaling (Andrejeva et al., 2004, Palosaari et al., 2003, Shaffer et al., 2003, Takeuchi et al., 2003).
MV assembly in the infected cell cytoplasm begins with the syn-thesis of a new copy of the RNA ge-nome. All viral RNA synthesis is car-ried out by the RNA-dependent RNA polymerase that is composed of the large (L) protein and its accessory phosphoprotein (P). All enzymatic activities reside on L whereas P func-tions as an adaptor between L and the template RNP. It is not known how the RNP template is read by the polymerase but a model has been suggested, where the L-P complex cartwheels around the helical RNP, with the tails of the tetrameric P con-stantly associating and disassociating from N (Longhi 2009). P is a largely intrinsically unstructured protein with a central helical tetramerization domain and a C-terminal XD domain that binds to the C-terminal part of N (N(TAIL)) in transcription and repli-cation. In addition to the mediator role between N and L, P serves a chaperone role for N until assembled onto the growing RNP (Curran et al., 1995). This is required as free N (de-noted N0 in the RNA-free form), hav-ing a high affinity for RNA, would otherwise bind unspecifically to cel-
lular RNA (Bhella et al., 2002). The N interacting regions on P have been mapped to the XD domain and to the very N-terminal end (PNT) (Harty et al., 1995). PNT has been reported to be unstructured, but partial folding could be induced with trifluoroetha-nol (Karlin et al., 2002). In the crys-tal structure of VSV N0-P the PNT folded as an α-helix, occupied a groove in a hinge region on N that was adjacent to the RNA-binding groove (Figure 5) (Green et al., 2006, Leyrat et al., 2011). N0-PNT interactions were mainly hydropho-bic, whereas N interactions with RNA were ionic. For SeV, the PNT amino acids responsible for N0 bind-ing have been mapped to residues 33-41 (Curran et al., 1995). In the absence of a paramyxoviral N0-P crystal structure, it is not known what the details of the N0-P interac-tion are. Knowing the precise region of PNT required to keep N in its RNA-free N0 form would allow for the design of peptide inhibitors as has already been shown for RABV (Castel et al., 2009).
Figure 4. Gene organization of the MV genome. Gene order is drawn in the 3’ to 5’ direction from
left to right. Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Microbiology
(Moss et al., 2006), © (2006).

15
Figure 5. Structure of VSV N in complex with PNT (A) and RNA (B). PNT prevents N from binding
to RNA by occupying a groove in a hinge region of N that is adjacent to and partly overlapping with
the RNA-binding site. Adapted from (Leyrat et al., 2011).
After replication and assembly of the RNP, the next step is to have the RNP, polymerase, M and the gly-coproteins transported to the bud-ding sites. How RNP is transported, is largely unknown, but the polymer-ase is thought to be transported as a complex with RNP. Actin and tubulin have both been reported to be im-portant in MV infection, but their precise roles are unclear. Actin has been suggested to have a role in re-lease of the virus from the cell and tubulin in replication (Bohn et al., 1986, Moyer et al., 1990, Stallcup et al., 1983). For the closely related SeV it was shown that RNPs are trans-ported along microtubules in associ-ation with intracellular vesicles (Chambers et al., 2010). Conversely to a number of other –ssRNA virus-es, MV assembly and budding is ESCRT independent (Salditt et al., 2010).
The viral M protein has been shown to have a crucial role in the transport of RNP to the budding sites on the plasma membrane, but also in transcription inhibition (Iwasaki et al., 2009, Suryanarayana et al.,
1994). In normal infection N co-localized with M to the plasma mem-brane, but formed small dots in the perinuclear area when the interac-tion was abrogated. In electron mi-croscopy of plastic-embedded sec-tions from infected cells, two mor-phologies for the RNP were detected, thinner “smooth filaments” and wid-er “granular filaments” (Dubois-Dalcq et al., 1973, Oyanagi et al., 1971). Another study reported “fuzzy nucleocapsids” in the cytoplasm of infected cells and these were shown to contain M (Brown et al., 1987). Thus, it is possible that M covers the RNP in infected cells and RNP is transported to the lipid rafts for budding as cargo, employing the same cellular machinery that transport M to the plasma mem-brane when expressed alone. If M covers the RNP in a tubular assem-bly, a transition from this assembly to the membrane would be required, as according to the present model, in the released virions M forms a layer next to the inner leaflet of the mem-brane (Figure 6) (Moss et al., 2006). Another possibility is that M

16
stays bound to the RNP inside the virion and does not form a layer di-rectly under the membrane. To shed light into this uncertainty, structural studies of the released virion are re-quired.
Figure 6. A textbook model for the MV virion.
In the model the RNP is randomly arranged
inside the virion and M is bound to the inner
leaflet of the membrane. Adapted by permis-
sion from Macmillan Publishers Ltd: Nature
Reviews Microbiology (Moss et al., 2006), ©
(2006).
M, when expressed alone, drives the release of VLPs with a sim-ilar efficiency to that of viral budding (Pohl et al., 2007). Thus, like many other matrix proteins, it has an in-herent ability to direct vesicle release from cells. In the absence of the gly-coproteins, some M is found local-
ized in membrane rafts, but raft lo-calization is enhanced in the pres-ence of F (Pohl et al., 2007). F is lo-calized in lipid rafts when expressed alone whereas H localization to lipid rafts requires the presence of F (Vincent et al., 2000). So, even though M is the main coordinator of assembly, F has an important role in taking all the components to the lipid rafts, where the budding is thought to occur (Vincent et al., 2000). The requirement of F for H translocation to lipid rafts also indicates that they have an interaction, which has also been shown by fluorescence micros-copy and pulse-chase analysis (Plemper et al., 2001). In polarized epithelial cells MV shows preference for budding from the apical side (Blau et al., 1995). The glycoproteins require expression of M in order to be transported to the apical side (Maisner et al., 1998, Naim et al., 2000). Thus, M and F have a mutual relationship in that M helps in trans-porting F to the correct side of the cell for budding and once at the membrane, F helps M to translocate into lipid rafts. The RNP and H seem to be freeloaders in the process.
What provides the final pinch-ing off functionality for the budding virion, is not known. This functional-ity could be a feature of M as purified M from the closely related NDV can bud into phospholipid vesicles (Shnyrova et al., 2007). Also, MV budding is ESCRT independent (Salditt et al., 2010) and it does not have a protein, like the M2 of influ-enza that would be capable of mem-brane scission (Rossman et al., 2010).

17
2.3. ASSEMBLY OF HUMAN RESPIRATORY SYNCYTIAL VIRUS
HRSV is a common virus that in healthy adults causes cold-like symp-toms, but can be severe in young children and the elderly (Falsey et al., 2005, Nair et al., 2010). It is re-sponsible for over 20 % of acute low-er respiratory infections world-wide and has been estimated to cause the death of 100 000 children under the age of five (Nair et al., 2010). Unlike for MV, an effective vaccine has proven difficult to develop. Hurdles for vaccine development include the early age of infection, innate immun-ity evasion by the virus, adaptive immunity incapable of preventing subsequent infections and lack of a suitable animal model (Graham 2011). Treatment is limited to passive immune-prophylaxis for neonates under high risk of severe infection (Shadman et al., 2011) and after on-set of the disease the purine analog ribavirin is sometimes used.
In addition to the proteins common for all paramyxoviruses, HRSV has genes encoding for SH, secondary matrix M2-1 and M2-2, and two nonstructural proteins, NS1 and NS2. M2-1 and M2-2 are ex-pressed from the same gene, M2-2 being expressed from an internal overlapping coding frame. M2-1 has been shown to be a transcription an-ti-terminator, enhancing transcrip-tional chain elongation and read-through at gene junctions (Collins et al., 1996, Hardy et al., 1998). It is also a structural component of the virion and has a role in transport of RNP to the budding sites (Li et al., 2008). M2-2 has been ascribed a role in the transcription-to-replication transition (Bermingham et al., 1999).
Characteristic for HRSV are the filamentous filopodium-like pro-trusions seen at the cell surface of infected cells (Mitra et al., 2012). In
purified virus preparations both spherical and filamentous particles are present (Gower et al., 2005). Currently it is not known which morphology dominates in clinical infection, but both appear to be in-fectious in vitro (Gower et al., 2005).
Similarly to MV and other paramyxoviruses, the N protein has an inherent RNA-binding activity and it readily forms RNP-like struc-tures when expressed in insect or bacterial cells (Meric et al., 1994, Murphy et al., 2003). The N-terminal part of N was shown to be responsible for the helical assembly and a deletion mutant of N that con-tained only the first 92 N-terminal amino acids of the total 391 could assemble into helices (Murphy et al., 2003). Similarly to other paramyxo-viruses, a role for P in preventing unspecific binding to cellular RNA, has been described for HRSV P (Castagne et al., 2004). The regions on P responsible for this function have not been characterized.
In polarized epithelial cells, HRSV buds from the apical side. F, when expressed alone, localizes to the apical side. F is not, however, required for sorting of the internal components to the apical side as VLPs lacking both glycoproteins still bud from the apical side (Batonick et al., 2008). RSV, similarly to MV, is thought to bud from lipid raft micro-domains in the plasma membrane (McCurdy et al., 2003). HRSV M, when expressed alone, binds to cellu-lar membranes (Henderson et al., 2002). In viral infection it is found partly in lipid rafts, but lipid raft association is dependent on F (Henderson et al., 2002). As with most of the –ssRNA viruses HRSV M is associated with the RNP during infection and also interacts with the

18
cytoplasmic tails of the glycoproteins (Ghildyal et al., 2005b, Ghildyal et al., 2002, Shaikh et al., 2012a). The M2-1 protein has also been shown to have a role in transport of the RNP from the replication sites to the plasma membrane. It was suggested to act as a mediator between the RNP and M (Li et al., 2008). The interac-tion between M and F has been shown to be crucial for the formation of viral filaments on the cell surface and this interaction was shown to be disrupted by a single phenylalanine mutation in the cytoplasmic tail of F (Oomens et al., 2006, Shaikh et al., 2012a). The M and F proteins appear to coordinate the budding so that F first initiates a bud at a lipid raft, which cannot elongate into a fila-ment before M is recruited to the site (Mitra et al., 2012). The recruitment of M occurs presumably via the in-teraction between the F cytoplasmic tail and M and further interactions with M and RNP, mediated by M2-1, guarantee the incorporation of all internal components. G and SH are dispensable for budding and quite surprisingly for the whole infection in vitro, although G does elevate in-fectivity (Techaarpornkul et al., 2001). G interacts specifically with M with its cytoplasmic tail, but has also been reported to form a complex with F on the cell surface (Ghildyal et al., 2005b, Low et al., 2008).
RSV M has been found to un-dergo nuclear-cytoplasmic shuttling. It is imported via importin β1 and exported at a late stage of infection by exportin 1 (Crm1) (Ghildyal et al., 2003, Ghildyal et al., 2009, Ghildyal et al., 2005a). Like MV M, it inhibits transcription and this function was postulated to be regulated by con-trolled export from the nuclear res-ervoir of M (Ghildyal et al., 2009). Nuclear-cytoplasmic shuttling has also been shown to occur in Nipah virus infection. In that case the ex-
port was found to be controlled by ubiquitinylation of M and depleting free cellular ubiquitin resulted in in-hibition of budding (Wang et al., 2010).
A number of cellular compo-nents have also been indicated to be important for HRSV assembly. These include actin and Rab11 family inter-acting protein 2 (FIP2), a major pro-tein associated with the apical recy-cling endosome (ARE) involved in protein trafficking in polarized cells (Burke et al., 1998). Importantly, like MV, RSV budding has been shown to be ESCRT-independent (Utley et al., 2008). Similarly to MV, it is not known how, in the absence of ESCRTs, the final pinching off from the host cell occurs. FIP2 however has been shown to control budding as a defective FIP2 mutant caused a failure in the final stage of HRSV budding (Utley et al., 2008). Fila-mentous actin has been observed at the base of viral filamentous protru-sions (Jeffree et al., 2007). Yet, it was shown that viral filaments are formed independently of host cyto-skeleton and related proteins, and actin was suggested to be important not in assembly of the filament but in anchoring the viral filaments (Shaikh et al., 2012b). HRSV activates Ras gene family member A (RhoA), a small GTPase known to regulate the actin cytoskeleton, and inhibition of RhoA compromises filament assem-bly. Inhibition resulted in blunted filaments and a shift towards spheri-cal particle production (Gower et al., 2005). Interestingly, a matrix-less HRSV mutant also showed blunted filament formation, which might in-dicate a requirement for ARE in proper M transport and assembly. How the matrix, when present at the filament assembly sites, causes elon-gation of the filaments, is not known.

19
3. ENTRY OF NEGATIVE-SENSE RNA VIRUSES
The first step in any infection process is attachment to a host cell. Viruses have evolved to use various types of molecules on the cell surface as re-ceptors for attachment. The main types of molecules used as viral re-ceptors are proteins, carbohydrate moieties on glycoproteins and lipids (Morizono et al., 2011). The type and number of receptors used by differ-ent viruses vary greatly even within viral families and receptors are the most important factors in defining tropism. Protein receptors tend to be the most specific as they can be ex-pressed in very limited cell types and thus restrict the viral infection to specific cells. Carbohydrates, like sialic acids used by many viruses, are present widely in different types of animal cells and result in more wide-spread infection of different cell types. Yet, within sialic acid binding viruses, tropism can be conferred by preferential binding to certain types of sialic acid. Human influenza, for example, binds specifically to um-brella-shaped α2-6 linked sialic acids that are present mainly in the epithe-lial cells of the respiratory tract, whereas avian influenza binds to cone-shaped α2-6 and α2-3 linked sialic acids (Chandrasekaran et al., 2008). Mutations in HA that allow binding to human-type sialic acids are thought to have a major role in influenza crossing the species barri-er.
The ligands for the receptors, the viral attachment proteins, are as varied as their receptors. In icosahe-dral viruses, the receptor binding protein is part of the capsid and in some viruses like the African horse-sickness virus it is a prominent large feature on the capsid (Manole et al., 2012). Within the –ssRNA viruses,
the receptor binding functionality is often a part of the fusion protein. Structurally the fusion protein func-tionality dominates in these proteins, as it requires much more complex mechanics than the simple binding to the receptor. Interestingly, para-myxoviruses, which have separate proteins for both functionalities, of-ten have the neuramidase activity on the attachment protein. That activity on influenza is on the separate NA protein. Thus, from an evolutionary perspective, the receptor-binding activity on influenza HA might have initially resided on the NA.
After attachment to the target cell, a virus needs to penetrate at least one lipid membrane. For envel-oped viruses, two membranes, that of the host cell and that of the virus, have to be fused. Enveloped viruses take two particular means to achieve this: endocytosis followed by fusion with the endocytotic vesicle or direct fusion with the plasma membrane. Of the –ssRNA viruses influenza, filo- rhabdo-, bunya-, arena- and some paramyxoviruses use the for-mer route, whereas most of the paramyxoviruses use the latter. The vast majority of viruses utilizing the host endocytotic machinery enters the cell via clathrin-mediated endo-cytosis and is dependent on acidifica-tion of the endosome. The lowered pH is required for triggering of the fusion protein, which results in fu-sion of the viral and endosome membranes. The degree of acid sen-sitivity of the fusion protein deter-mines the stage of endosome matu-ration, where the virus penetration occurs. VSV, for example fuses with early endosomes at around pH 6, whereas influenza and Uukuniemi virus (a member of the Bunyaviridae

20
family) fuse with late endosomes at a pH closer to 5 (Lozach et al., 2010, White et al., 1981).
Some of the –ssRNA viruses en-ter via macropinocytosis, a ligand-induced clathrin-independent pro-cess normally used for uptake of fluid and solutes (Mercer et al., 2012). Most of the viruses dependent on macropinocytotis for entry are also dependent on acidification of the macropinosome. However, HRSV and possibly others rely on proteolyt-ic activation by cellular proteases in the macropinosome and do not re-quire acidic conditions (Krzyzaniak et al., 2013). The filoviruses are ex-ceptional in that they require both low pH and proteolytic cleavage by host pH-dependent proteases for fusion (Chandran et al., 2005).
Viruses that are independent of endocytosis and acidic pH for entry fuse directly with the cell membrane and have evolved alternative ways for ensuring suitable time and place for fusion triggering. These include the requirement for binding to a co-receptor in addition to the primary receptor for HIV and relay of recep-tor binding signal from the attach-ment protein to the fusion protein by altered lateral contacts for paramyx-oviruses. For most paramyxoviruses the signaling is thought to occur by either dissociation or formation of a complex between the attachment and fusion protein (Chang et al., 2012). In most cases the interaction is virus specific as substituting the attach-ment protein to that of another virus from the family does not produce infectious virions (Das et al., 2000, Hu et al., 1992). MV H and F form a complex already in ER of the host cell before budding and are likely to remain as a complex until F trigger-ing for fusion (Plemper et al., 2001). With most paramyxoviruses (ones with HN as the attachment protein), however, the attachment and fusion
proteins do not form a complex in-side the cell and are thought to inter-act only after attachment protein binding to the target cell (Paterson et al., 1997).
It is well established that the stalk region of the attachment pro-tein plays a key role in triggering F for fusion (Ader et al., 2012, Apte-Sengupta et al., 2013, Bose et al., 2011, Brindley et al., 2013, Maar et al., 2012, Melanson et al., 2004). Based on two different crystal forms observed for MV H in complex with the receptor CD150 , it was suggested that a sliding movement between the two dimers of the tetramer results in a conformational change of the stalk that then causes triggering of F (Hashiguchi et al., 2011, Nakashima et al., 2013). The two states of the H tetramer have also been detected in native polyacrylamide gel electro-phoresis of free and receptor-bound H (Brindley et al., 2012). For para-myxoviruses with HN as the attach-ment protein an additional mecha-nism for control has been observed. In crystal structures of HN the large head domains were found in two dis-tinct orientations; bent down to block any possible interaction be-tween F and the HN stalk, or bent up exposing the trunk (Welch et al., 2013, Yuan et al., 2011). It was sug-gested that HN binding to the recep-tor would shift the heads up and al-low F to interact with the trunk, a prerequisite for F triggering (Welch et al., 2013). Similar large conforma-tional shifts have not been observed for paramyxoviruses with H or G as the attachment protein. As in most of these viruses F is thought to form a complex with the attachment protein already before target cell attachment, they are unlikely to employ such a mechanism.
The actual mechanism of fusion is incompletely understood although multiple structures of the fusion pro-

21
tein ectodomains in both pre- and postfusion conformations have been solved (Bullough et al., 1994, McLellan et al., 2011, Roche et al., 2006, Roche et al., 2007, Swanson et al., 2010, Welch et al., 2012, Wilson et al., 1981, Yin et al., 2005, Yin et al., 2006). In the current model for Class 1 fusion protein mediated fu-sion (Harrison 2008, Plemper 2011), triggering of F first results in expo-sure of the hydrophobic fusion pep-tides that get casted out in a spring-like manner. The fusion peptides then insert into the target membrane connecting the two membranes. Next, F refolds back into a compact structure bringing the two mem-branes in close apposition. The re-folding is driven by the formation of a highly stable six-helix bundle by the so-called heptad repeats present in both ends of each F monomer of the trimer. Finally, the two mem-branes fuse opening a fusion pore, probably via a hemifusion interme-diate, and the virion cargo gets deliv-ered to the cytoplasm. Fusion of the two membranes is an energetically
unfavorable process to which the re-folding of F is thought to provide the energy. Despite the wealth of infor-mation already obtained, the lack of intermediate state structures leaves great gaps to the model. The struc-ture of the fusion pore, for example, has not been described and it is not known how many copies of the F trimer are required to construct a fusion pore.
After fusion of the viral and host membranes, uncoating of the RNP from the matrix protein is re-quired before another round of infec-tion can begin. How and where this occurs, is poorly understood for most –ssRNA viruses. For influenza it is known that a drop in pH results in disassembly of the M1 from the viral membrane possibly preceded by a conformational change and thinning of the M1 layer (Calder et al., 2010, Fontana et al., 2012). M1 then disso-ciates from the RNP prior to RNP transport to the nucleus where repli-cation takes place (Martin et al., 1991).
4. ELECTRON CRYOTOMOGRAPHY AS A TOOL IN
STUDYING VIRUS STRUCTURE
Eukaryotic cells are typically over 10 µm in size, smallest bacteria approx-imately 1 µm and viruses in the order of tens to hundreds of nanometers in diameter. Eukaryotic and to some extent also bacterial cells can be studied using visible light microsco-py. Resolution in any imaging tech-nique is limited by the wavelength of the radiation used to inspect the specimen, which for visible light ranges between 400 and 700 nm. Therefore, in order to be able to vis-ualize details within viruses, visible light microscopy cannot be used. In-
stead, electrons at voltages of 200 – 300 kV used in modern electron mi-croscopes have a wavelength of ~2 pm which is more than adequate for virus studies even at atomic resolu-tion. X-ray crystallography can sometimes be used, but by definition, only for samples that can be crystal-lized and crystallization of even large regular viruses is often difficult. Thus, electron microscopy is usually the method of choice.
When the electrons hit the sample inside the vacuum of a transmission electron microscope

22
(TEM), the vast majority of the elec-trons pass through the sample unaf-fected. Those that interact with the sample scatter either elastically or inelastically. In elastic scattering, the energy of the incident electrons does not change, only the direction. This change in electron direction corre-sponds to a change in the phase of the diffracted electron wave and can be used in phase contrast imaging. In inelastic scattering, some of the en-ergy is transferred to the specimen, which results not only in damage to the sample by ionization, chemical bond rearrangements or free radical formation, but also results in noise in the image (Orlova et al., 2011). For heavy metal stained specimens the changes are tolerable and such sam-ples can be imaged with a bright electron beam resulting in high con-trast. Also the high scattering cross sections of the heavy metal atoms provide higher contrast than the light atoms in biological samples. In order to preserve the sample in its un-stained native state, cryo-fixation is used. In the absence of heavy-metal stain the radiation damage becomes a significant hurdle. It is indeed the most significant factor preventing straightforward imaging of biological samples in atomic detail. In order to avoid excessive damage to the sam-ple, dim electron beam and short exposure of the sample have to be used. Due to the low number of elec-trons passing through the sample and contributing to the collected im-age, the images are noisy and poor in contrast. To overcome this issue, av-eraging of multiple low-contrast im-ages of an object present in multiple copies can be used to enhance the contrast. Verifying that one object
indeed is a copy of the other is often unreliable or impossible by eye and therefore computational methods are normally used.
For 3D studies of biological ob-jects in transmission electron mi-croscopy, multiple images of the same object in different orientations have to be obtained. This is done by one of two means: by collecting mul-tiple images of an object present in multiple copies in different orienta-tions in the sample (e.g. a suspension of an icosahedral virus) or by tilting the sample inside the microscope and collecting images of the same object from multiple orientations. The former is called single particle reconstruction and the latter tomog-raphy. Of the two, tomography is more versatile as it can be used to image unique objects unlike single particle reconstruction. Tomography is therefore ideal for 3D imaging of pleomorphic viruses and is currently the method of choice not only for pleomorphic virus studies but also for structural studies of the cell (Gan et al., 2012, Guerrero-Ferreira et al., 2013, Subramaniam et al., 2007). Using single particle methods com-bined with the powerful utilization of icosahedral symmetry of some virus-es, atomic resolution reconstructions have been obtained (Liu et al., 2010, Zhang et al., 2010). Resolutions from electron cryotomographic studies are currently more modest (typically 3 - 4 nm), but no fundamental re-strictions exist for obtaining atomic resolution from tomography when combined with averaging of repeat-ing structures within the tomograms. Challenges in averaging of tomo-graphic data are discussed in section 4.4.

23
4.1. SAMPLE PREPARATION FOR ELECTRON CRYOTOMOGRAPHY
All biological reactions take place in aqueous solutions. Thus, in order to view biological molecules in native or close-to-native state they should be imaged in such conditions. Electrons have a rest mass which results in poor penetration in air and therefore a high vacuum must be maintained in the electron microscope column. To prevent dissipation of the water from the sample, the sample can be fixed by cooling it to low enough temperatures for the water to solidi-fy. Ice at atmospheric temperatures is crystalline and composed of a hex-agonal lattice of water molecules. Formation of water crystals destroys the intricate biological structures and thus fixation by ordinary freezing is not useful for electron cryomicrosco-py. However, water, when cooled rapidly to temperatures below -140 °C, forms a metastable, amorphous state called vitrified water (Figure 7). Fast enough cooling can be achieved with rapid immersion into liquid ethane cooled by liquid nitro-gen (Adrian et al., 1984, Dubochet et al., 1988) normally using a gravity-driven guillotine. Warming up the sample causes an irreversible phase transformation to cubically or hex-agonally crystallized water, which destroys the biological sample (Fig-ure 7). Thus, the sample has to be maintained at all times under -150 °C normally with the help of liquid ni-trogen, the boiling point of which is -196 °C at atmospheric pressure.
The cooling rate close to the surface of the specimen is fast enough for vitrification, but quickly slows down towards the interior. Therefore, only thin samples of up to maximally a few micrometers in thickness can be vitrified using plunge-freezing. For thicker samples,
Figure 7. Three forms of solid water viewed with an electron microscope. Corresponding diffractograms are shown in the middle. In A) hexagonal ice, in B) cubic ice, and in C) vitre-ous water are shown. In C) the absence of in-tensity peaks in the diffractogram indicates that the water molecules are not ordered, whereas in A) and B) the intensity peaks or rings indicate an ordered state. Reprinted from (Dubochet et al., 1988) with permission from the publisher.
such as mammalian cells, high pres-sure can be used to prevent crystalli-zation of water. In this high-pressure freezing the sample is frozen at pres-sures up to 200 MPa, and this way samples as thick as 200 µm can be vitrified (Vanhecke et al., 2008). The method is based on freezing point depression, reduction in the rate of

24
ice crystal formation and reduction of ice crystal growth at high pressure. However, due to the poor penetra-tion of the electron beam through the sample, the optimal sample thickness for TEM is less than 200 nm. Thus, high-pressure frozen samples have to be processed into sections, preferably under 200 nm thick that can then be viewed with the microscope. Cryo-electron microscopy of vitrified sec-tions (CEMOVIS) is a rising tech-nique that currently requires skilled hands to be successful (Al-Amoudi et al., 2004).
For viruses in suspension, plunge-freezing is normally used. In practice, ~3 µl of sample is pipetted onto a holey carbon film supported by a copper mesh grid. The carbon layer is normally first ionized with a glow-discharger or a plasma cleaner to make the carbon layer charged and thus hydrophilic for even distri-
bution of the sample on the grid. Next, the excess sample is removed from the grid by blotting with a piece of filter paper to leave only a thin layer of sample on the grid. Immedi-ately after blotting, the grid is dropped in the tip of tweezers to liq-uid ethane using a guillotine. The vitrified samples can then be stored in liquid nitrogen for years until use. To preserve the vitrified state, the sample has to be cooled also during transfer to the microscope and dur-ing imaging. For this, a special liquid nitrogen cooled sample holder is used. For samples to be used for elec-tron tomography, colloidal gold beads are normally added to the sample before plunge-freezing. Those provide high contrast features in the images that are useful in alignment of the tomographic tilt series.
4.2. IMAGE FORMATION IN THE ELECTRON MICROSCOPE
Similarly to a conventional light mi-croscope, a TEM is composed of a source of electrons, a lens system to condense, focus and project the beam, and a viewing screen, film or a camera to view and record the formed image. As electrons are nega-tively charged particles, electromag-netic lenses instead of refractive glass lenses are used to focus the beam. The electron source has typi-cally been a thermionic heated tung-sten filament or a lanthanium hexa-boride (LaB6) crystal. Of these two, LaB6 as an electron source provides a more coherent, brighter beam. Yet, the most coherent and brightest beam is provided by field emission gun (FEG) electron sources. The FEG electrons also are also closer to mon-
ochromatic than the electrons from thermionic guns and are used in all modern TEMs for high resolution analysis.
In the FEG the electrons are ex-tracted from the emitter surface of a Zr02 coated sharp tungsten crystal tip by a voltage gradient and subse-quently accelerated with a potential of typically 200 – 300 kV (Orlova et al., 2011). Throughout the TEM col-umn the beam is guided by electro-magnetic beam deflectors, parallel-ized or focused by electromagnetic lenses and restricted by metallic ap-ertures with small holes in them. First the divergent electron beam is parallelized and focused by a con-denser lens system with apertures. Next, the beam passes through the

25
sample, mostly unaffected, but some of the electrons undergo elastic or inelastic scattering events. The image and initial magnification is then formed by the objective lens which is the most critical part for high resolu-tion imaging and where most image aberrations originate in. After the objective lens, the beam passes through an aperture, which is used to prevent inelastically scattered elec-trons with high deflection angles from contributing noise to the image. The formed image is further magni-fied by a magnifying lens system which is adjusted to obtain different final magnifications. Finally, the formed image is viewed from a phos-phorus screen or recorded on film or a camera. The camera has commonly been a charge-coupled device (CCD) connected to an electron scintillator, but direct electron detectors (DED) without a scintillator, are becoming more popular due to their high detec-tive quantum efficiencies (DQE) and the resulting low level of noise (Bammes et al., 2012). To reduce the noise in the formed image, it is also possible to filter the electrons after they have passed through the sample
based on their energy (Angert et al., 2000, Schroder 1992). This energy-filtering can remove inelastically scattered electrons deflected with low angles that have passed through the objective aperture. Energy filters are routinely used in modern cryo-TEMs to enhance contrast.
Contrast in TEM is formed by two different principles. Amplitude contrast results from absorption of electrons to the sample via inelastic interactions and in essence means that some parts of the sample do not let electrons pass through as well as other parts and therefore show as darker areas in the image. Amplitude contrast for unstained biological specimens with mainly light ele-ments, such as hydrogen, carbon, nitrogen and oxygen, only contrib-utes approximately 7 % of the total contrast (Toyoshima et al., 1988). The main fraction of the contrast comes from the interference between the unaffected electron wave gone through the sample and the diffract-ed wave with elastically scattered electrons. Transfer of contrast in TEM is described by the contrast transfer function (Baker et al., 1999):
( ) ( )
( ( )) ( ( )) ( )
where ( ) ( ) , v is the spatial fre-quency per Å, Famp is the fraction of amplitude contrast, λ is the electron wavelength in Ångströms, Δf is the underfocus in micrometers and Cs is the spherical aberration of the mi-croscope objective lens in millime-ters. CTF is a periodic function that is modulated by components of both amplitude and phase contrast and is attenuated at high spatial frequen-cies by the envelope component
( ) (where δ describes beam co-herence) of the CTF originating from the instabilities and incoherence of
the electron source. Thus an FEG provides much more subtle attenua-tion of high frequencies than thermi-onic guns. The phase contrast com-ponent of the CTF is a periodic sine function that is affected by defocus and the spherical aberration of the objective lens. Thus, the inherent spherical aberration has a phase shifting effect on the function and the effect varies with the defocus of the lens. Different defoci result in high signal at different spatial fre-quencies depending on the positions of the minima and maxima of the CTF. Focusing slightly under the ex-

26
act focus enhances low frequency components and is normally used for imaging. Overfocusing would in principle do the same, but in overfo-cus the amplitude and phase contrast components of CTF have opposite signs at low frequencies, which re-sults in destructive interference and consequently low signal.
As the phase contrast compo-nent of CTF is a sine function, its values vary between 1 and -1. In or-der to have human interpretable da-ta, the negative regions of the func-tion need to be phase flipped. The positions of CTF zeros can be calcu-lated from the function if the spheri-cal aberration coefficient and defocus are known. The defocus of an image
can be accurately estimated from the positions of the so-called Thon rings visible in the power spectrum of the image that correspond to CTF zeros. Since there are zeros in the CTF, in order to cover the full spatial fre-quency range, multiple images at different defoci are required. Cor-recting for the effects of CTF are al-ways used in high-resolution cryo-EM, but are beginning to be used also in cryo-ET. Previously the data were often lowpass filtered to the first CTF zero to avoid artefacts from the negative CTF values. This howev-er often limits the obtainable resolu-tion to ~3 – 4 nm and is to be avoid-ed if high resolutions are strived for.
4.3. ELECTRON CRYOTOMOGRAPHY
Cryo-ET has been widely used for ultrastructural studies of pleo-morphic viruses (Briggs et al., 2006, Cyrklaff et al., 2005, Grünewald et al., 2006, Harris et al., 2006, Pietilä et al., 2012), cellular structures (Al-Amoudi et al., 2007, Brandt et al., 2010, Maimon et al., 2012, Psencik et al., 2009) and virus-cell interactions (Carlson et al., 2010, Ibiricu et al., 2011). Samples for cryo-ET are pre-pared either by plunge vitrification or cryo-sectioning. Typically, colloi-dal gold of 5 - 10 nm in diameter is included in the sample suspension before vitrification. For cryo-sections of cells, this cannot however be done, but quantum dots in organic solvents can be added directly on the vitrified sections and used in a similar man-ner for alignment of the tilt series (Masich et al., 2006).
After having successfully vitri-fied the sample and inserted it into a TEM, regions of interest and of thin enough ice are searched with low
magnification. Low magnification is not only useful for easy location of the positions to be imaged, but it also minimizes the electron dose on the sample before the actual data collec-tion. Once the microscope is aligned, the region of interest in the sample is positioned to the center of the field, the beam brightness is set, and a tilt series is collected. Typically the tilt series is collected from -60° to +60° and an image recorded every two degrees. Thus in total approximately 60 images of the same object are rec-orded. For optimally thin regions (~100 nm in thickness) with an ener-gy-filter equipped TEM, tilting the sample up to ±70 ° can yield useful data. The reason for not tilting to higher tilts is that because the sam-ple has slab geometry, the more it is tilted the longer distance the elec-trons have to travel in the ice layer. The thicker the ice is the higher is the probability that the electrons under-go inelastic scattering or scatter

27
more than once which leads to noise in the image. The mean free path describing the mean distance of two consecutive scattering events for 200 kV accelerated electrons is approxi-mately 270 nm in vitreous water (Diebolder et al., 2012).
The thickness of the sample varies with 1 / cos α, where α is the tilt angle. What follows is that at 60° the relative sample thickness is twice and at 70° almost three times that at 0°. To obtain approximately even contrast in the images at all tilts, the beam intensity is often varied ac-cordingly throughout the tilt series. Like in all cryo-EM, the electron dose that the sample can tolerate without destruction of fine details is a major limiting factor. For reasons poorly understood, it appears that when the electron dose is divided to multiple separate exposures, higher total dose can be used. In single particle cryo-EM the dose per image is normally limited to ~10-20 e / Å2 depending on the coherence and the resolution aimed for, whereas in cryo-ET total doses of ~100 e / Å2 are commonly used. In order to limit the total dose in cryo-ET, focusing and tracking of the position are done in a region that is a few micrometers off the actual tilt series collection site.
Once the tilt series is collected, a 3D tomogram can be calculated. Multiple program suites are available for this, but they generally rely on the same principles (Heymann et al., 2008, Korinek et al., 2011, Kremer et al., 1996). First, due to the inaccura-cies in the microscope stage and go-niometer, the precise tilt angle and position of the individual images are determined in a process called alignment. Here, the gold beads can be used as fiducial markers. As they have high density and are therefore
clearly visible even in the high tilt images, their trajectories through the tilt series can be used in alignment. Alternatively, a cross correlation based fiducialless alignment can be performed, but it tends to be less ro-bust and accurate than that using fiducial markers. Once the positional and angular relationship of each im-age to the next in the tilt series is de-termined, a 3D reconstruction can be calculated. This is most commonly done using weighted back-projection (WBP), but also alternative methods like simultaneous iterative recon-struction technique (SIRT) or alge-braic reconstruction technique (ART) are sometimes used. The principle of electron cryotomography is summa-rized in Figure 8.
According to the central section theorem, for weak phase objects such as thin biological samples, the 2D Fourier transform of a projection of a 3D object corresponds to a central section in the 3D Fourier transform of the object (Bracewell 1956, Crowther et al., 1970, De Rosier et al., 1968). The thickness of the sec-tion is inversely proportional to the thickness of the sample. According to Crowther et al., for an ideal object the maximal resolution of the recon-struction is determined by the num-ber of projections and the diameter of the object by the following formula (Crowther et al., 1970):
where dx is the resolution, D is the object diameter and N is the number of projections. In order to reproduce a completely faithful reconstruction of the 3D object, the entire Fourier space should be evenly sampled. The limited tilting range in electron

28
Figure 8. Principle of electron cryotomography. (A) Vitrified specimen is tilted inside the micro-
scope to collect 2D projection images of the object of interest in different orientations. The tilting
range typically varies from ±60° to ±70° with increments of 1.5° to 3°. The images are recorded with
a CCD or a DED. As a result, a series of projections of the specimen is obtained (B). After alignment
of the projection images, a 3D model of the original specimen can be reconstructed with WBP (C),
SIRT or ART. Reprinted from Biophysical Chemistry, 100/1-3, Grünewald et al., Prospects of electron
cryotomography to visualize macromolecular complexes inside cellular compartments: implications
of crowding, Pages 577-591, © (2003), with permission from Elsevier.
tomography results in a missing wedge-shaped region of information in the Fourier space, commonly re-ferred to in the literature as the “missing wedge”. The missing wedge causes anisotropic resolution in the reconstruction, which is poorest along the beam direction and best in the directions perpendicular to the beam. Consequently, the tomograms are poorly interpretable in the direc-
tion along the beam. The missing region of information can be reduced from a wedge to a cone by collecting two orthogonal tilt series of the same region of interest. This is however rarely done in cryo-ET due to the limited acceptable electron dose.
4.4 SUBVOLUME AVERAGING
To enhance contrast and alleviate missing wedge artifacts, multiple copies of a particular object in differ-ent orientations in the tomograms can be averaged together. Averaging
results in enhanced contrast because the random noise in the reconstruc-tions in proportion to the nonran-dom signal from biological features decreases with an increasing number

29
of averaged volumes. Before averag-ing, the subvolumes are extracted and aligned together by methods adopted from the single particle field. The main difference is that in subvolume averaging, all procedures are carried out on 3D volumes in-stead of the 2D images in single par-ticle reconstruction. Subvolume av-erages of an increasing number of cellular and viral structures have been published and include the her-pes simplex virus capsid (Grünewald
et al., 2003), glycoprotein spike and gag of HIV (Briggs et al., 2009, White et al., 2010), influenza HA (Harris et al., 2013), Chlamydomo-nas flagellum microtubule doublets (Pigino et al., 2011), nuclear pore complexes (Beck et al., 2007, Maimon et al., 2012) and COPI coat-ed vesicles (Faini et al., 2012). A basic workflow for subvolume aver-aging is illustrated in Figure 9.
Figure 9. A simplified workflow for subvolume averaging.
The most commonly used method for alignment is a rotational and translational search of a sub-volume against a template using cross correlation as a similarity measure. To avoid template bias in the final reconstruction, the initial template should be an object lacking fine features. Typically, a low-pass filtered random average of the ex-tracted subvolumes is used as an ini-tial template. To validate the final
reconstruction, multiple runs with different initial templates can be per-formed and they all should converge to the same final average. In sub-volume alignment, the missing wedge is problematic because the subvolumes have a tendency to align to their missing wedges. To avoid this, many of the programs only in-clude regions with information in Fourier space in the analysis, or re-scale the similarity measure accord-

30
ing to the fraction of overlap of the missing wedge (Bartesaghi et al., 2008, Forster et al., 2008, Heumann et al., 2011, Schmid et al., 2008). For objects with large protruding fea-tures, alignment to the missing wedge can be prevented well, but it tends to be more difficult for objects with fine features.
To obtain a faithful reconstruc-tion of the original object by simple averaging, the subvolumes would have to evenly cover the orientation space. Because a perfectly even dis-tribution of subvolumes in all orien-tations is practically impossible to obtain, the average volume is never a perfect estimate of the true object. The imbalances in the data distribu-tion can, however be compensated for by dividing the average with the average of the missing wedges of all subvolumes included in the average. Another approach used for sub-volume averaging that does not rely on cross correlation is based on max-imum-likelihood refinement and is also widely used in the field (Scheres 2012, Sorzano et al., 2004).
For high resolution subvolume averages, correction for the CTF is required. Two main factors compli-cate the correction in comparison to the CTF correction in single particle processing. First, as the sample is tilted in most of the images, a gradi-ent of defoci is present in each image. Second, the individual images have extremely low signal-to-noise ratios (SNR) as a total dose of ~100 e / Å2
has to be distributed across the whole tilt series. These are, however not fundamental unsolvable issues and methods have been developed that can relatively well correct the CTF effects in the images (Xiong et al.,
2009, Zanetti et al., 2009). Due to the low DQE of the CCD cameras used, the modulation transfer func-tion (MTF) of the camera dampens the signal at high resolutions. Thus, often careful boosting of the high frequency signal with an inverse of the MTF is required to bring out de-tail (Xiong et al., 2009). DEDs should reduce this need as their MTFs fall off at higher spatial fre-quencies than those of CCD cameras.
In any averaging method it is important to average together only homogenous particles. In cryo-tomograms SNR is higher than in 2D micrographs and the three-dimensionality helps in identifying identical particles by eye, but often a computational classification method is required for objectivity. Classifica-tion is widely used in single particle processing, but has only recently been adapted to subvolume analysis. Similarly to alignment of the sub-volumes, the missing wedge causes some difficulties in classification as well and easily leads to classification to groups differing in the orientation of the missing wedge. Ways to cir-cumvent this problem have however been described (Forster et al., 2008, Frank et al., 2012, Heumann et al., 2011, Stolken et al., 2011, Winkler et al., 2009, Xu et al., 2012, Yu et al., 2011) and these methods have been applied to sort out heterogeneity in structures like influenza spikes (Fontana et al., 2012), HIV spikes (Liu et al., 2008), and axonemes (Heuser et al., 2012).

31
B. AIMS OF THE PRESENT STUDY
At the start of this study in 2008, no 3D whole virus structures of paramyxovi-ruses had been published. We chose two members of the family for cryo-ET analysis. The viruses were selected based on their significance as human patho-gens and the potential benefit of the results for helping to fight the viruses. The specific aims were:
1) To characterize the ultrastructure of measles virus, determine how the RNP is organized and solve the controversy of how the matrix protein is assembled in released virions (Study I).
2) To characterize the ultrastructure of human respiratory syncytial virus, investigate how the matrix protein is assembled in spherical and filamen-tous viruses, and how the glycoproteins and the RNP are organized in re-leased virions. To combine the obtained structural data with previous bi-ochemical knowledge for an assembly model (Study II).
3) To express, purify and characterize a measles virus RNA-free N-P com-plex. To develop a method for producing the complex in pure, large enough quantities for crystallization (Study III).

32
C. MATERIALS AND METHODS
The main method in the studies was cryo-ET combined with subvolume averaging (Study I and II). This method was chosen as currently it is the only method suitable for studies on detailed 3D-stuctures of envel-oped, pleomorphic viruses. All imag-ing for cryo-ET was done on plunge-vitrified virus samples using an FEI F20 electron microscope. Typically, ±60° tilt series were collected with 2° steps at a nominal magnification of 39400x resulting in a sampling of 0.38 nm / pixel on a Gatan Ultrascan 4000 CCD camera. Tomograms were calculated in IMOD (Kremer et al., 1996) using WBP and typically binned to have a final sampling of 0.77 nm / pixel. From the tomo-
grams repeating features of interest were extracted, iteratively aligned and averaged to obtain the final av-erage structure. In study I Jsubtomo (Huiskonen et al., 2010) and in study II PEET (Nicastro et al., 2006) pro-gram packages were used for sub-volume processing. The rationale for using different program packages in study I and study II was that of the two packages only PEET offered a classification option required for sorting out the spike heterogeneity in study II. For classification the prin-cipal component decomposition and clustering features in PEET were used. The imaging and image pro-cessing details are described in the methods section of each study.
Table 1 Methods used in the study
Method Used in study
virus culture and purification I, II
molecular cloning I, III
recombinant protein expression I, III
electron cryotomography I, II
subvolume averaging I, II
3D classification II
electron microscopy of negatively stained samples I, III
electron microscopy of plastic embedded stained sections II
immunosorbent electron microscopy I
fluorescence microscopy II
size exclusion chromatography and multi-angle light scat-tering
III
SDS-polyacrylamide gel electrophoresis III
blue native polyacrylamide gel electrophoresis III
limited proteolysis and mass spectrometry III

33
D. RESULTS AND DISCUSSION
One of the main aims in this study was to shed light on the way para-myxoviruses assemble in the host cell and how the viral components are organized in the virion released from the cell (studies I and II). We also looked at an early stage of assembly for MV by characterizing a pre-
assembly intermediate of the RNP by recombinant protein production and in vitro biochemical analysis (study III). The results are summarized in a model for budding in Figure 10 and will be discussed in the text in the order in which the events are thought to occur during virus assembly.
1. THE N0-P COMPLEX
To address the very first step in MV budding, the encapsidation of the genomic RNA, we set out to produce an intermediate RNA-free N0-P complex in E. coli (Study III). The two proteins were co-expressed and purified as a complex using an N-terminal hexahistidine tag on P. The complex was verified to be RNA-free and did not form helical RNPs which confirmed that co-expression of the two proteins leads to formation of a soluble RNA-free N0. Due to the highly protease-sensitive nature of P, and the fact that major parts of the protein are intrinsically unstructured (Bourhis et al., 2006), we construct-ed a number of C-terminally truncat-ed versions of P that were co-expressed with N. This was done for two reasons: to map the region on P that is required to keep N in the N0
form and to obtain a stable N0-P con-struct amenable for biochemical characterization and crystallization. We expressed some of the shortest P constructs as GST fusion proteins as they could not be expressed without a bulky tag and determined that the first 49 N-terminal amino acids of P were enough to bind N and prevent N assembly on RNA. The results were consistent with an earlier report
where deletion of the first 10 N-terminal amino acids of P led to over a 90 % decrease in signal in a mam-malian two hybrid system (Harty et al., 1995). Also, for SeV and VSV the critical regions for N0-P interactions have been mapped to amino acids 33-41 and 11-30, respectively (Chen et al., 2007b, Curran et al., 1995).
The C-terminal domain of N is known to be unstructured in the ab-sence of a binding partner (Longhi et al., 2003) and thus might prevent crystallization of the protein. In search of a solid structured con-struct, guided by limited proteolysis and mass spectrometry of the cleav-age products, we cloned an N con-struct lacking the protease sensitive C-terminus. This construct was com-posed of the 408 N-terminal amino acids on N. We co-expressed this construct together with GST-hexahistidine-P1-49 and purified the complex with Ni-NTA resin followed by on-beads enterokinase digestion and size-exclusion chromatography (SEC). In SEC-multi-angle light scat-tering (SEC-MALS) analysis, the pu-rified complex was determined to be a 52±2 kDa 1:1 heterodimer, which is similar to the crystallized VSV N in complex with VSV P1-60. In

34
Figure 10. Assembly of MV and HRSV. Initially, the newly synthesized genomic RNA is encapsi-
dated with N into a helical RNP (green box). Before assembly onto the helix, N is prevented from
binding to cellular RNA by P. For MV the first 49 N-terminal amino acids of P are enough for this
chaperone function. This N0-P complex is then thought to be dissociated by the polymerase (blue) to
assemble N on the RNP. In MV the assembled RNP is then covered by M to form a matrix-covered
nucleocapsid (MCNC), which is required for transport of the RNP to the budding sites. At the bud-
ding site, the MCNC are organized as bundles that are incorporated into the nascent bud on the
membrane (blue box). After the final pinching off from the membrane, the RNP remains covered by
M and has become enveloped in membrane that also carries the viral glycoproteins on the outside.
The spikes are transported to the budding sites independently of the MCNC via the ER-Golgi route.
Unlike in MV, HRSV RNPs are not covered by M, although the transport of RNP to the budding sites
requires M. The interaction between RNP and M is mediated by M2-1. For poorly understood rea-
sons, HRSV M undergoes nuclear-cytoplasmic shuttling before being transported to the budding site
with M2-1 and RNP. At the budding site HRSV budding is initiated by F, after which M assembly into
a tubular structure elongates the bud into a filament (yellow box). The RNP is incorporated into the
virion via the interaction with M, mediated by M2-1. After pinching off from the cell, in some of the
particles F converts into the postfusion conformation and M disassembles from the membrane caus-
ing the particle to adopt a spherical form.

35
addition to SEC-MALS we studied the negatively-stained complex in electron microscopy and the sample appeared to mainly contain a mono-disperse complex clearly different from the helical assembly observed in the Ni-NTA resin flow-through. This further confirmed that we had produced an N0-P complex (Study III).
To characterize the main mode of interaction in the MV N1-408-P1-49 complex, we screened for release of N1-408 from GST-hexahistidine-P1-49 bound on Ni-NTA beads in different conditions. We found the interaction to be mainly hydrophobic as Triton-X 100 caused release of N1-408 from the complex, but neither high NaCl nor KCl concentrations (up to 2 M) resulted in N1-408 release. Thus, it is
likely that the PNT in MV binds to N0 in a similar manner to that in VSV. This could be confirmed by crystalliz-ing and solving the structure of the N1-408-P1-49 complex. Knowing the precise nature of the interaction would lead to a better understanding of how N assembly on RNA is con-trolled and how it could potentially be prevented. Also, being able to break the complex by mild detergent treatment provides a means to obtain pure N0, a form of the protein long sought for RNP assembly studies (Bourhis et al., 2006).
2. BUDDING OF MV AND HRSV AND STRUCTURES OF THE
RELEASED VIRIONS
The association of the matrix protein with the RNP in transport of the RNP to the budding sites has been well established for both MV and HRSV (Ghildyal et al., 2002, Iwasaki et al., 2009, Li et al., 2008). Yet, to what extent M is present in the transport-ed complex, had not been well de-scribed. For MV, early electron mi-croscopic studies indicated that M is present on the RNP in significant enough numbers to form a “fuzzy” layer on the RNP (Brown et al., 1987), contrary to the commonly held view reported in many review articles/text books. To confirm the presence of M on the surface of RNPs in infected cells and for a more de-tailed picture of the complex, we car-ried out immunosorbent electron microscopy on MV infected cell ly-sates (Study I). On MV anti-M grids, approximately 30 % of the RNPs ob-
served had an additional layer of density on the outside and the RNPs were found in large aggregates. On anti-N grids, only 0.8 % of RNPs had a similar covering, and the RNPs were not aggregated. Thus, the addi-tional layer observed was composed of M and it resulted in aggregation of the RNPs. The appearance of these matrix-covered nucleocapsids (MCNC) was similar to the RNPs with an additional covering detected earlier in MV-infected cells (Almeida et al., 1963).
Having verified that the tubular structure covering MV RNPs was composed of M, it was of interest to see whether this assembly was re-tained in the budded virus, or wheth-er M was relocated to the inner sur-face of the membrane, or perhaps both. Because both HRSV and SeV M had been reported to form tubular

36
structures (Heggeness et al., 1982, McPhee et al., 2011) it was essential to compare HRSV with MV and SeV (Study I and II). In a cryo-ET study of 11 released SeV virions MCNCs were not reported (Loney et al., 2009). As in MV only ~20 % of the particles contained MCNC, it is pos-sible that analysis of a larger number of SeV virions could reveal these structures in SeV too. In general in tomography, getting a comprehen-sive description of the sample is dif-ficult as in diluted samples such as many viral preparations one tilt se-ries might have only one virion in it. In the future automated data collec-tion and processing should help in overcoming this issue.
We produced MV (Helsinki strain, isolated from a patient in 1964 and the Edmonston vaccine strain) and HRSV (A2 strain) in cell culture and purified the viruses from the cell culture supernatants. Initial-ly, only supernatant viruses were used to allow analysis of the released fraction of virions. Later, for HRSV, we compared the morphology of the released fraction and a fraction re-leased from cells by freeze-thawing and found it to be essentially similar. The titers of the preparations from supernatants were on the order of 109 and 108 plaque-forming units (PFU) / ml for MV Helsinki strain (study I) and HRSV (study II), re-spectively. We considered these titers high as generally both are thought to be viruses difficult to produce in high titers. Yet, the preparations were di-lute for cryo-ET as often only one or a few virions could be included in a tilt series.
In cryo-ET of the released viri-ons, the most striking difference be-tween the MV and HRSV was that in MV virions the RNP was indeed mainly covered by M, whereas in HRSV this was not the case. MV viri-ons were spherical or close-to spheri-
cal, but HRSV also formed filamen-tous virions although 95 % were close-to-spherical. In the filamentous HRSV, two layers of density were observed under the membrane, which was not the case in MV. The one closest to the membrane was most likely formed by M as it is known to be capable of forming heli-cal structures on membranes and is required for filamentous virion elon-gation in infected cells (McPhee et al., 2011, Mitra et al., 2012). The identity of the innermost layer was less clear, but it was likely to be com-posed of the M2-1 protein as it is known to mediate the M-RNP inter-action and is a structural component of the virion (Li et al., 2008). We subjected the M and RNPs of both viruses to subvolume alignment and averaging to analyze more closely the nature of the M and RNP assemblies.
As the MCNC were readily visi-ble in the tomograms of MV, we manually defined the initial positions of the structures for subvolume anal-ysis. Next, partially overlapping seg-ments of the tubular structures were extracted and averaged and this av-erage used as a refinement template. In iterative refinement of the average structure, we noticed that the M lay-er outside the RNP did not refine to any distinct structure, but remained blurry as if the subvolumes were randomly aligned. The RNP, however rapidly started to acquire a left-handed single-start helical structure similar to that which had earlier been published for recombinant RNP-like helices (Bhella et al., 2004, Schoehn et al., 2004). Fortunately, excluding the inner helix from the refinement by masking resulted in rapid refine-ment of the M-layer to a left-handed five-start helix with a ridge-to-ridge distance of 7.2 nm. Thus, the RNP and M both form left-handed helices but have different symmetry. Form-ing such a complex requires flexibil-

37
ity in the interactions between M and N. Such flexibility could be conferred by the intrinsically unstructured C-terminus of N in which the interac-tion with M is known to occur (Iwasaki et al., 2009).
After having determined that M forms a helix around the RNP in MV we studied how the MCNC are orga-nized in the virions. This was done by template matching using the sub-volume averages of the individual helices as templates. The MCNC were found to form bundles where the M-layers of adjacent helices were in close contact. The adjacent RNPs were often anti-parallel whereas the M-helices did not appear to have di-rectionality at the resolution of the reconstruction (4.4 nm). The pres-ence of a dyad axis in the M helix was shown by the almost perfect correla-tion of the reconstruction with its 180° rotated copy (rotation around the axis perpendicular to the helical axis). As the individual helix seg-ments in a bundle were on average only approximately 100 nm in length, multiple segments would be required to accommodate a full ge-nome. Thus, even though we could not detect the connections, possibly due to thin connections or the effects
of the missing wedge in the tomo-grams, they were likely to be linked. Subsequently, in a tomogram of an aggregate of material released from infected cells, we detected a connec-tion between two MCNC tubes that based on its thickness appeared to be naked RNA (Figure 11). Therefore the linkages between the MCNC in the virions could be formed of naked RNA too thin to detect in the crowd-ed environment of the virion interior. To summarize, our results suggest that in MV the matrix coats the RNP already inside the host cell and that state is maintained in the released virion. Thus deforming the host membrane in MV budding is unlikely to be a result of a sheet-like assembly of M under the plasma membrane that would cause curvature to the membrane. However, the MCNC as-sembly does not exclude a possible role for M in shaping the membrane as the edges of the MCNC bundles were often in close proximity to the membrane. It is also possible that not all of the M is in MCNC and some of the protein could be bound to the membrane in small patches not read-ily visible in the tomograms.
Figure 11. MCNC are possibly connected by
naked RNA. In a slice from a tomogram of a
broken MV virion, thin and long linkers can
be seen. The appearance and width of the
linker suggests that it is composed of free
RNA. Alternatively it could be RNA decorated
with N that has been unwound from the typi-
cally helical RNP. In the inset a piece of the
structure where RNP has unwound from
inside of the M helix is shown.

38
We generated radial profiles by aligning and averaging subvolumes extracted from the surfaces of spher-ical MV and HRSV as well as fila-mentous HRSV particles. In the av-erages of spherical particle sub-volumes no other layers than the membrane were discernible. In the average of filamentous particles the M-layer was at 6.1 nm from the membrane and the putative M2-1 layer at 6.9 nm from the M layer. Although it was possible that M formed a helical assembly under the filamentous HRSV membrane, we could not confirm this most likely due to the fact that most of the fila-mentous virions were flattened in the thin ice required for high resolution tomograms. However, a repeating feature at approximately 8 nm fre-quency could be detected in the lay-er. Similar observations have been reported for influenza M1 and MARV VP40 (Bharat et al., 2011, Calder et al., 2010). In a number of filamen-tous virions, the M-layer continued as a curved sheet when it had de-tached from the membrane. This in-dicated that M had an inherent abil-ity to form tubular structures and that these structures were stable in the absence of a membrane. Thus, it is likely that the filamentous shape in HRSV is determined by the assembly of M into tubes. This is supported by the fact that the presence of M is re-quired for formation of long fila-ments at the cell surface (Mitra et al., 2012). The filamentous regions on virions varied in diameter between 70 nm and 190 nm, which indicated that M assembly into tubular struc-tures could accommodate multiple different symmetries. The flexibility extended to even more extreme cases as exemplified by a virion that con-tained an inverted M-tube with the membrane and spikes inside the tube. We reconstructed this tube in subvolume averaging and found it to
be composed of a seven-start helix with a ridge-to-ridge distance of 7.8 nm. The repeat distance corresponds to that observed in the Fourier trans-forms of the filament surface. It is therefore possible that a similar alt-hough inverted assembly of M was present in both.
In a study published while our work was in progress, the ultrastruc-ture of NDV by cryo-ET was de-scribed (Battisti et al., 2012). Mor-phologically, most NDV particles were spherical without an apparent matrix layer under the membrane, but also elongated particles were ob-served. In the elongated particles part of the inner side of the mem-brane had an M-covering similar in appearance to the one we detected in HRSV filaments. In the same article also a crystal structure of M was solved and it could be fitted into a subvolume average of the M-layer. M appeared to form a sheet with 7 nm repeat distance in orthogonal direc-tions, which is approximately similar to what we saw in HRSV. The ultra-structures of MV, HRSV, SeV and NDV are shown for comparison in Figure 12.
The RNPs in HRSV and regions of MV RNP without M-layer were more curved and randomly oriented than the MCNC. This was indicative of M’s role in making the MV RNPs straight and rigid. In HRSV the viri-ons were packed to a varying extent with the RNP material and in fila-mentous virions long stretches of RNP could often be seen aligned along the longitudinal axis. In NDV, the RNPs in elongated virions were aligned similarly to HRSV and were in close contact with the M-layer (Battisti et al., 2012). As HRSV viri-ons were more tightly packed with the RNPs, it was difficult to judge whether the RNPs were specifically bound to the M/M2-1-layer or whether the adjacent positioning was

39
due to the crowded interior of the virion.
We analyzed the structure of the HRSV RNP in essentially a simi-lar manner to that used for MV RNP and found the helix to be left-handed. This was in direct contradic-tion with a previously published model (Tawar et al., 2009). To verify that the tomogram calculation re-tained the correct absolute handed-ness, we collected tilt series and cal-culated tomograms of left-handed nanogold decorated DNA-origami structures (Kuzyk et al., 2012). While our article was in press, the authors of the conflicting article published a second article correcting their origi-nal mistake in the handedness (Bakker et al., 2013). Their mistake arose from not experimentally de-termining the handedness, but decid-ing on it based on modeling the RNP-like ring X-ray structure as a helix (Tawar et al., 2009). Correction of the handedness is important in this case because it affects the direc-tion of the RNA in the model. That in turn has implications on models of how the polymerase could access the RNA in the structure (Bakker et al., 2013). It has also been suggested that all the –ssRNA RNPs are left handed (Bharat et al., 2011). Yet, for NDV,
the RNP was reported to be right-handed based on experimenting with different helical averaging of the tomographic structure and estimat-ing cross-correlations with the origi-nal structure. The data were, howev-er, not shown (Battisti et al., 2012). Helical averaging per se does not give the absolute hand, but use of a control tomographic sample would have done.
The spike glycoproteins on MV and SeV virions formed a densely packed layer from which it was diffi-cult to discern individual proteins (Loney et al., 2009). In NDV, the spikes were packed to similar densi-ties as in MV and SeV, but showed preferential lateral location to the regions with M-layer under the membrane (Battisti et al., 2012). In HRSV the individual spikes, alt-hough densely packed in the mem-brane could be discerned and ana-lyzed by subvolume averaging. We attempted subvolume averaging also for MV, but were unable to classify the subvolumes for each of the two spikes. In HRSV spherical virions the spikes appeared to be of one single type with a golf-tee like shape re-sembling the HRSV F in a postfusion conformation (PDB ID 3RRT) (McLellan et al., 2011). The same
Figure 12. Tomographic slices of paramyxoviruses. In MV the RNP is covered by M, whereas in
HRSV and NDV the RNP is not covered, but M forms a layer next to the internal side of the mem-
brane. In SeV M does not cover the RNP nor form a continuous layer under the membrane. MCNC
are shown with white arrowheads and RNPs without M with black arrowheads. Star denotes an area
of the NDV membrane free of glycoprotein spikes and M. Adapted from study I; study II; Loney et
al. 2009; Battisti et al. 2012 with permission from the publishers.

40
type of spike could also be detected on the filamentous regions, but there also another smaller spike was pre-sent. Of all the virions, only those with the small spike appeared to have some local order in the spike layer. The small spikes were ar-ranged roughly in lines on the sur-face and the lines were often approx-imately perpendicular to the longitu-dinal axis of the filament.
Because HRSV has two glyco-protein spikes, we found ourselves facing a problem of sorting which type of spike was G, which F, and whether or not F was perhaps pre-sent in more than one conformation. To begin with, we cultured and puri-fied a mutant of HRSV that lacked SH and G on the surface. This rgΔGΔSH strain has been shown to be infectious and in general F on the surface is enough for a productive HRSV infection in vitro (Techaarpornkul et al., 2001). On the mutant virions, the majority of the spikes appeared to be of the golf-tee type thus identifying it as F. Initially, we inferred that the other smaller spike should then be G. To be objec-tive, we did computational classifica-tion analysis on the spike sub-volumes. The classification resulted in two well-defined classes one of which was similar to F and the other similar to the small spike. Surpris-ingly, some of the subvolumes classi-fied into the small spike class origi-nated in the rgΔGΔSH tomograms. Additionally, an average calculated from only the rgΔGΔSH subvolumes in the small spike class clearly re-sembled more the small spike than the golf-tee spike. Thus, both classes
most likely represented F. As the golf-tee shaped class was reminiscent of the HRSV postfusion F crystal structure, and the small spike re-sembled in shape the prefusion structure of PIV5, we attempted to fit the crystal structures into the aver-ages. Initially, we fitted the prefusion F from PIV5 (PDB ID 4GIP) (Welch et al., 2012) into the small spike den-sity (Study II). Subsequently, a crys-tal structure of the prefusion ecto-domain of HRSV F was published (PDB ID 4JHW) (McLellan et al., 2013). As this structure was biologi-cally more relevant to our study, we fitted that into the small spike densi-ty instead of the PIV5 F (Figure 13A). We also fitted the postfusion HRSV F into the golf-tee spike densi-ty (PDB ID 3RRT) (McLellan et al., 2011). The dimensions of the sub-volume average and the crystal struc-ture were similar and the crystal structure appeared to fit well into the subvolume average density (Figure 13B). Although the limited resolu-tion of the subvolume averages pre-vented reliable numerical assessment of goodness of the fits, it was clear that the small spike corresponded to the prefusion and the golf-tee spike to the postfusion F. From our analy-sis, the structure and presence of G was left unresolved. It is possible that G was similar enough in shape to one of the F conformations that it did not separate into its own class or it was not prominent enough or pre-sent in too low numbers to be includ-ed in the analysis. Using G-specific antibodies for labeling the virus prior to analysis could help in clearing this uncertainty.

41
Figure 13. Crystal structures of
HRSV F in prefusion (A) and postfu-
sion (B) states fitted into the sub-
volume average densities. The isosur-
faces in A and B were rendered at 2 σ
and at 5 σ from the mean, respectively.
HRSV F is exceptional in that it requires two cleavages by a furin-like protease to be fusion active (Zimmer et al., 2001). It has recently been shown that HRSV entry involves macropinocytosis and that the sec-ond cleavage occurs inside the target cell after internalization of the virion (Krzyzaniak et al., 2013). Thus, also in the virus preparation used in our study, F was uncleaved at the second site and regardless of this the majori-ty had converted into the postfusion state. Currently it is unclear why the conversion should happen before attaching to the target cell, but it is possible that the virions with the converted F are inactive as the parti-cle-to-infectivity ratio of HRSV prep-arations have been reported to be high, which was also the case in our preparations (50 ± 10:1). Additional-ly, we noticed that the fraction of fil-amentous virions in the preparation went down on freezing and incuba-tion at 37 °C (5.2 %, 1.1 % and 0.9 % for fresh, -80 °C frozen and 37 ° C incubated virus, respectively) and this correlated with a drop in infec-tivity (90 % drop in -80 °C frozen compared to fresh virus). Although not causally proven in our study, the
following scenario is likely: Initially, most of the virions were filamentous. With time, during preparation or storage, the matrix layer disassem-bled from some virions’ membrane making the particles spherical in shape. In the same process, F con-verted into the postfusion confor-mation rendering the virions inactive (Figure 10). The conversion of F and disassembly of M are correlated because prefusion F was not present in spherical particles, but appear not to be strictly temporally connected as postfusion F was also present in fil-amentous virions. Accordingly, it is rather the conversion of F than dis-assembly of M that begins the trans-formation process. Thus, it is possi-ble that during entry, triggering of F for fusion leads to disassembly of M. Disassembly of M is likely to be re-quired before the next round of repli-cation can begin.

42
E. CONCLUSIONS AND PERSPECTIVES
In this study we have conducted cryo-ET studies on two important pathogens from the Paramyxoviri-dae family. We also produced and characterized a complex of MV N and P that is important in the early stages of virus assembly. The results indi-cated clear differences in the virion structure between MV, HRSV and other paramyxoviruses. The differ-ences were most noticeable in the way the matrix protein was assem-bled and how it defined the shape of the whole virion. For MV we were able to rectify a commonly adopted model for the virus architecture by showing that M did not line the in-ternal side of the membrane but was wrapped as a helix around the RNP. In HRSV we analyzed in detail all the main components of the virion. This analysis showed that M and F were responsible for the filamentous mor-phology observed for a fraction of the virions. We also corrected a mistake in the earlier literature regarding the handedness of the RNP and showed that F is present in both pre- and postfusion conformations in the viri-ons.
Although we mainly studied vi-ruses isolated from the cell, a fair amount of information on the as-sembly of the viruses could also be derived. As structure is most useful when it can be interpreted in terms of biological functionality, we com-bined our structural models with da-ta from biochemical and cell biologi-cal literature into models for virus assembly.
As is typical for any scientific study, the studies reported here raise
at least as many new questions as they were able to answer. The most intriguing ones regarding the RNP assembly are: How does P of para-myxoviruses prevent N from binding to RNA and at least equally im-portantly, how is P then released from N to bind RNA when it is re-quired? Phosphorylation of P could possibly have a role in this. It would be of great interest to study if the RNP could be assembled in vitro and whether the sequence or structure of RNA has a role in efficient encapsi-dation. Could the N-P interaction be prevented with PNT mimicking pep-tides or drug compounds? Trials with RABV demonstrate that this might be possible (Castel et al., 2009).
The greatest gaps in knowledge on enveloped virus assembly lie in understanding the virus-host inter-actions required for assembly and release of the virion and spread to adjacent cells. What are the compo-nents required from the cell for transport of viral proteins, shaping the cellular membrane and final scis-sion of the virion off the membrane? Addressing these topics will require careful analysis of all aspects com-bining structural biology, cell biology and biochemistry of the isolated complexes. Undoubtedly, future technological development combined with the true medical need will evi-dently lead to answers to these cur-rently unresolved questions and open paths to new currently un-mapped territories.

43
F. ACKNOWLEDGEMENTS
The work for this thesis was carried out between 2008 and 2013 at the Finnish Centre of Excellence in Virus Research (2006-2011), at the Insti-tute of Biotechnology at the Universi-ty of Helsinki under the supervision of Docent Sarah Butcher. The work was funded by Finnish Academy, Viikki Doctoral Programme in Mo-lecular Biosciences (VGSB) and Eu-ropean Molecular Biology Organiza-tion (EMBO).
I want to express my warmest thanks to Sarah. You have guided me through this project. This project that sometimes led me to misty dark mires of slow progress, and even there I often managed to hit a wall. You have taught me to find light where I could barely notice a spark. You have not held my hand, but al-ways been available and offered a signpost at a crossing or a ladder over a wall when I have needed help. I especially would like to acknowledge you for seeing your stu-dents and employees not only as working force, but humans who you treat with respect.
I want to express my gratitude to our collaborators Dr Juha Huis-konen, Docent Petri Susi, Dr Magda-lena Krzyzaniak and Professor Ari Helenius. Without your input, this work would not have been possible.
Thanks to Juha for teaching me the principles and more of electron tomography. Thank you for making me feel welcome to visit both Mar-tinsried and Oxford. Also, thanks for your never-ending patience in sitting with me through several nights at the microscope during my visit to Mar-tinsried.
I would like to thank all the present members of the Butcher lab as well as you who have already
moved on. It has been a real pleasure to work in an environment with so many colorful personalities.
I want to acknowledge Ari O. for the unbelievable enthusiasm and energy that you spread to the people around you. Thanks for teaching me so many of your practical “old tricks” in the lab to help the everyday work. Thanks for the interesting discus-sions of various kinds during these years. You are a walking encyclope-dia of everything. Furthermore, I am extremely grateful for the initial virus preparations you made for the mea-sles project. Without those this pro-ject would have ended before it even really begun.
Thanks to Shabih for being such a great and friendly officemate. You have always found the time to answer my questions and help with things you master and I don’t.
Thanks to Violeta for keeping me aware of the superiority of gar-dening over work. It has been a real pleasure to get to know you. I have really enjoyed all the discussions on various topics unrelated to work dur-ing the years.
Thanks to Pasi for teaching me how to do electron microscopy and to Eevakaisa for a lot of help in data collection for my projects.
I want to thank Professors Adrian Goldman and Markku Kulo-maa for the advice you have given during the thesis committee meet-ings. Thanks also to Tero Ahola and Peter Rosenthal for reviewing this thesis. Your comments were very helpful.
I want to thank my mom and dad for the great support throughout my studies and life in general. Raija I want to thank for providing the calm

44
environment of Nokian mökki where I wrote a major part of this thesis.
My deepest gratitude goes to my beloved wife Teresa. You have been the best support imaginable. You have helped me stand up when I have felt like giving up. You have pa-tiently read through almost every text and presentation I have pro-duced during these years. You have always shown interest in what I have been doing, or at least pretended
well. You have also kept in check that my work-life balance has all the time been on a sound basis.
Finally, I want to thank our wonderful baby daughter Isla for the joy and happiness you have brought into our lives in these past few months. You have helped me com-plete this work much faster than I would have done would I not have expected such a nice change in life.
Lassi Liljeroos Helsinki, September 2013

45
G. REFERENCES
Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, and Plattet P (2012) Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J Biol Chem 287(20):16324-16334.
Adrian M, Dubochet J, Lepault J, and McDowall AW (1984) Cryo-electron microscopy of viruses. Nature 308(5954):32-36.
Al-Amoudi A, Diez DC, Betts MJ, and Frangakis AS (2007) The molecular architecture of cadherins in native epidermal desmosomes. Nature 450(7171):832-837.
Al-Amoudi A, Norlen LP, and Dubochet J (2004) Cryo-electron microscopy of vitreous sections of native biological cells and tissues. J Struct Biol 148(1):131-135.
Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, and Ruigrok RW (2006) Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313(5785):360-363.
Ali A, Avalos RT, Ponimaskin E, and Nayak DP (2000a) Influenza virus assembly: effect of influenza virus glycoproteins on the membrane association of M1 protein. J Virol 74(18):8709-8719.
Ali A and Nayak DP (2000b) Assembly of Sendai virus: M protein interacts with F and HN proteins and with the cytoplasmic tail and transmembrane domain of F protein. Virology 276(2):289-303.
Almeida JD and Howatson AF (1963) A negative staining method for cell-associated virus. J Cell Biol 16:616-620.
Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, and Randall RE (2004) The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A 101(49):17264-17269.
Angert I, Majorovits E, and Schroder RR (2000) Zero-loss image formation and modified contrast transfer theory in EFTEM. Ultramicroscopy 81(3-4):203-222.
Apte-Sengupta S, Navaratnarajah CK, and Cattaneo R (2013) Hydrophobic and charged residues in the central segment of the measles virus hemagglutinin stalk mediate transmission of the fusion-triggering signal. J Virol 87(18):10401-10404.
Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, and Martin-Benito J (2012) The structure of native influenza virion ribonucleoproteins. Science 338(6114):1634-1637.
Arzt S, Baudin F, Barge A, Timmins P, Burmeister WP, and Ruigrok RW (2001) Combined results from solution studies on intact influenza virus M1 protein and from a new crystal form of its N-terminal domain show that M1 is an elongated monomer. Virology 279(2):439-446.
Avota E, Gassert E, and Schneider-Schaulies S (2010) Measles virus-induced immunosuppression: from effectors to mechanisms. Med Microbiol Immunol 199(3):227-237.
Baker TS, Olson NH, and Fuller SD (1999) Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol Rev 63(4):862-922. Erratum in: Microbiol Mol Biol Rev 2000;64(1):237.
Bakker SE, Duquerroy S, Galloux M, Loney C, Conner E, Eleouet JF, Rey FA, and Bhella D (2013) The Respiratory Syncytial Virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J Gen Virol 94(Pt 8):1734-1738.
Bammes BE, Rochat RH, Jakana J, Chen DH, and Chiu W (2012) Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency. J Struct Biol 177(3):589-601.

46
Bartesaghi A, Sprechmann P, Liu J, Randall G, Sapiro G, and Subramaniam S (2008) Classification and 3D averaging with missing wedge correction in biological electron tomography. J Struct Biol 162(3):436-450.
Batonick M, Oomens AGP, and Wertz GW (2008) Human respiratory syncytial virus glycoproteins are not required for apical targeting and release from polarized epithelial cells. J Virol 82(17):8664-8672.
Battisti AJ, Meng G, Winkler DC, McGinnes LW, Plevka P, Steven AC, Morrison TG, and Rossmann MG (2012) Structure and assembly of a paramyxovirus matrix protein. Proc Natl Acad Sci U S A
Beck M, Lucic V, Forster F, Baumeister W, and Medalia O (2007) Snapshots of nuclear pore complexes in action captured by cryo-electron tomography. Nature 449(7162):611-615.
Bermingham A and Collins PL (1999) The M2-2 protein of human respiratory syncytial virus is a regulatory factor involved in the balance between RNA replication and transcription. Proc Natl Acad Sci U S A 96(20):11259-11264.
Bharat TA, Noda T, Riches JD, Kraehling V, Kolesnikova L, Becker S, Kawaoka Y, and Briggs JA (2012) Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc Natl Acad Sci U S A 109(11):4275-4280.
Bharat TA, Riches JD, Kolesnikova L, Welsch S, Krahling V, Davey N, Parsy ML, Becker S, and Briggs JA (2011) Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol 9(11):e1001196.
Bhella D, Ralph A, Murphy LB, and Yeo RP (2002) Significant differences in nucleocapsid morphology within the Paramyxoviridae. J Gen Virol 83(Pt 8):1831-1839.
Bhella D, Ralph A, and Yeo RP (2004) Conformational flexibility in recombinant measles virus nucleocapsids visualised by cryo-negative stain electron microscopy and real-space helical reconstruction. J Mol Biol 340(2):319-331.
Bieniasz PD (2006) Late budding domains and host proteins in enveloped virus release. Virology 344(1):55-63.
Bize A, Peng X, Prokofeva M, Maclellan K, Lucas S, Forterre P, Garrett RA, Bonch-Osmolovskaya EA, and Prangishvili D (2008) Viruses in acidic geothermal environments of the Kamchatka Peninsula. Res Microbiol 159(5):358-366.
Blau DM and Compans RW (1995) Entry and Release of Measles Virus Are Polarized in Epithelial Cells. Virology 210(1):91-99.
Bohn W, Rutter G, Hohenberg H, Mannweiler K, and Nobis P (1986) Involvement of actin filaments in budding of measles virus: studies on cytoskeletons of infected cells. Virology 149(1):91-106.
Bose S, Welch BD, Kors CA, Yuan P, Jardetzky TS, and Lamb RA (2011) Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J Virol 85(24):12855-12866.
Bourhis J-M, Canard B, and Longhi S (2006) Structural disorder within the replicative complex of measles virus: Functional implications. Virology 344(1):94-110.
Bowden TA, Bitto D, McLees A, Yeromonahos C, Elliott RM, and Huiskonen JT (2013) Orthobunyavirus ultrastructure and the curious tripodal glycoprotein spike. PLoS Pathog 9(5):e1003374.
Bracewell RN (1956) Strip integration in radio astronomy. Austr J Phys (9):198-217. Brandt F, Carlson LA, Hartl FU, Baumeister W, and Grunewald K (2010) The three-
dimensional organization of polyribosomes in intact human cells. Mol Cell 39(4):560-569.
Briggs JA, Grunewald K, Glass B, Forster F, Krausslich HG, and Fuller SD (2006) The mechanism of HIV-1 core assembly: insights from three-dimensional reconstructions of authentic virions. Structure 14(1):15-20.
Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, and Krausslich HG (2009) Structure and assembly of immature HIV. Proc Natl Acad Sci U S A 106(27):11090-11095.

47
Brindley MA and Plemper RK (2010) Blue native PAGE and biomolecular complementation reveal a tetrameric or higher-order oligomer organization of the physiological measles virus attachment protein H. J Virol 84(23):12174-12184.
Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, and Plemper RK (2013) A Stabilized Headless Measles Virus Attachment Protein Stalk Efficiently Triggers Membrane Fusion. J Virol
Brindley MA, Takeda M, Plattet P, and Plemper RK (2012) Triggering the measles virus membrane fusion machinery. Proc Natl Acad Sci U S A 109(44):E3018-3027.
Brown HR, Goller N, Thormar H, and Norrby E (1987) Fuzzy material surrounding measles virus nucleocapsids identified as matrix protein. Arch Virol 94(1):163-168.
Bruce EA, Medcalf L, Crump CM, Noton SL, Stuart AD, Wise HM, Elton D, Bowers K, and Digard P (2009) Budding of filamentous and non-filamentous influenza A virus occurs via a VPS4 and VPS28-independent pathway. Virology 390(2):268-278.
Bullough PA, Hughson FM, Skehel JJ, and Wiley DC (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371(6492):37-43.
Burke E, Dupuy L, Wall C, and Barik S (1998) Role of cellular actin in the gene expression and morphogenesis of human respiratory syncytial virus. Virology 252(1):137-148.
Bächi T and Howe C (1973) Morphogenesis and ultrastructure of respiratory syncytial virus. J Virol 12(5):1173-1180.
Calain P and Roux L (1993) The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol 67(8):4822-4830.
Calder LJ, Wasilewski S, Berriman JA, and Rosenthal PB (2010) Structural organization of a filamentous influenza A virus. Proc Natl Acad Sci U S A 107(23):10685-10690.
Carlson LA, de Marco A, Oberwinkler H, Habermann A, Briggs JA, Krausslich HG, and Grunewald K (2010) Cryo electron tomography of native HIV-1 budding sites. PLoS Pathog 6(11):e1001173.
Carter SD, Dent KC, Atkins E, Foster TL, Verow M, Gorny P, Harris M, Hiscox JA, Ranson NA, Griffin S, and Barr JN (2010) Direct visualization of the small hydrophobic protein of human respiratory syncytial virus reveals the structural basis for membrane permeability. FEBS Lett 584(13):2786-2790.
Caspar DL and Klug A (1962) Physical principles in the construction of regular viruses. Cold Spring Harb Symp Quant Biol 27:1-24.
Castagne N, Barbier A, Bernard J, Rezaei H, Huet JC, Henry C, Da Costa B, and Eleouet JF (2004) Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J Gen Virol 85(Pt 6):1643-1653.
Castel G, Chteoui M, Caignard G, Prehaud C, Mehouas S, Real E, Jallet C, Jacob Y, Ruigrok RW, and Tordo N (2009) Peptides that mimic the amino-terminal end of the rabies virus phosphoprotein have antiviral activity. J Virol 83(20):10808-10820.
Cattaneo R, Kaelin K, Baczko K, and Billeter MA (1989) Measles virus editing provides an additional cysteine-rich protein. Cell 56(5):759-764.
Chambers R and Takimoto T (2010) Trafficking of Sendai virus nucleocapsids is mediated by intracellular vesicles. PLoS One 5(6):e10994.
Chandran K, Sullivan NJ, Felbor U, Whelan SP, and Cunningham JM (2005) Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308(5728):1643-1645.
Chandrasekaran A, Srinivasan A, Raman R, Viswanathan K, Raguram S, Tumpey TM, Sasisekharan V, and Sasisekharan R (2008) Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotech 26(1):107-113.
Chang A and Dutch RE (2012) Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4(4):613-636.

48
Chen BJ, Leser GP, Morita E, and Lamb RA (2007a) Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol 81(13):7111-7123.
Chen M, Ogino T, and Banerjee AK (2007b) Interaction of vesicular stomatitis virus P and N proteins: identification of two overlapping domains at the N terminus of P that are involved in N0-P complex formation and encapsidation of viral genome RNA. J Virol 81(24):13478-13485.
Clare DK and Orlova EV (2010) 4.6A Cryo-EM reconstruction of tobacco mosaic virus from images recorded at 300 keV on a 4k x 4k CCD camera. J Struct Biol 171(3):303-308.
Collins PL, Hill MG, Cristina J, and Grosfeld H (1996) Transcription elongation factor of respiratory syncytial virus, a nonsegmented negative-strand RNA virus. Proc Natl Acad Sci U S A 93(1):81-85.
Compans RW, Holmes KV, Dales S, and Choppin PW (1966) An electron microscopic study of moderate and virulent virus-cell interactions of the parainfluenza virus SV5. Virology 30(3):411-426.
Crowther RA, DeRosier DJ, and Klug A (1970) The reconstruction of a three-dimensional structure from projections and its application to electron microscopy. Proc R Soc A 317(1530):319-340.
Cudmore S, Cossart P, Griffiths G, and Way M (1995) Actin-based motility of vaccinia virus. Nature 378(6557):636-638.
Curran J, Marq JB, and Kolakofsky D (1995) An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J Virol 69(2):849-855.
Cyrklaff M, Risco C, Fernandez JJ, Jimenez MV, Esteban M, Baumeister W, and Carrascosa JL (2005) Cryo-electron tomography of vaccinia virus. Proc Natl Acad Sci U S A 102(8):2772-2777.
Das SC, Baron MD, and Barrett T (2000) Recovery and characterization of a chimeric rinderpest virus with the glycoproteins of peste-des-petits-ruminants virus: homologous F and H proteins are required for virus viability. J Virol 74(19):9039-9047.
De Rosier DJ and Klug A (1968) Reconstruction of three dimensional structures from electron micrographs. Nature 217(5124):130-134.
Dessen A, Volchkov V, Dolnik O, Klenk HD, and Weissenhorn W (2000) Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J 19(16):4228-4236.
Diebolder CA, Koster AJ, and Koning RI (2012) Pushing the resolution limits in cryo electron tomography of biological structures. J Microsc 248(1):1-5.
Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, and Schultz P (1988) Cryo-electron microscopy of vitrified specimens. Q Rev Biophys 21(2):129-228.
Dubois-Dalcq M and Barbosa LH (1973) Immunoperoxidase stain of measles antigen in tissue culture. J Virol 12(4):909-918.
Faini M, Prinz S, Beck R, Schorb M, Riches JD, Bacia K, Brugger B, Wieland FT, and Briggs JA (2012) The structures of COPI-coated vesicles reveal alternate coatomer conformations and interactions. Science 336(6087):1451-1454.
Falsey AR, Hennessey PA, Formica MA, Cox C, and Walsh EE (2005) Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 352(17):1749-1759.
Fontana J, Cardone G, Heymann JB, Winkler DC, and Steven AC (2012) Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J Virol 86(6):2919-2929.
Forster F, Pruggnaller S, Seybert A, and Frangakis AS (2008) Classification of cryo-electron sub-tomograms using constrained correlation. J Struct Biol 161(3):276-286.
Frank GA, Bartesaghi A, Kuybeda O, Borgnia MJ, White TA, Sapiro G, and Subramaniam S (2012) Computational separation of conformational heterogeneity using cryo-electron tomography and 3D sub-volume averaging. J Struct Biol 178(2):165-176.

49
Freiberg AN, Sherman MB, Morais MC, Holbrook MR, and Watowich SJ (2008) Three-dimensional organization of Rift Valley fever virus revealed by cryoelectron tomography. J Virol 82(21):10341-10348.
Fuentes S, Tran KC, Luthra P, Teng MN, and He B (2007) Function of the respiratory syncytial virus small hydrophobic protein. J Virol 81(15):8361-8366.
Gan L and Jensen GJ (2012) Electron tomography of cells. Q Rev Biophys 45(1):27-56. Gan SW, Ng L, Lin X, Gong X, and Torres J (2008) Structure and ion channel activity of the
human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci 17(5):813-820.
Gaudier M, Gaudin Y, and Knossow M (2002) Crystal structure of vesicular stomatitis virus matrix protein. EMBO J 21(12):2886-2892.
Ge P, Tsao J, Schein S, Green TJ, Luo M, and Zhou ZH (2010) Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 327(5966):689-693.
Ghildyal R, Baulch-Brown C, Mills J, and Meanger J (2003) The matrix protein of Human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol 148(7):1419-1429.
Ghildyal R, Ho A, Dias M, Soegiyono L, Bardin PG, Tran KC, Teng MN, and Jans DA (2009) The respiratory syncytial virus matrix protein possesses a Crm1-mediated nuclear export mechanism. J Virol 83(11):5353-5362.
Ghildyal R, Ho A, Wagstaff KM, Dias MM, Barton CL, Jans P, Bardin P, and Jans DA (2005a) Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin beta1 independent of importin alpha. Biochemistry 44(38):12887-12895.
Ghildyal R, Li D, Peroulis I, Shields B, Bardin PG, Teng MN, Collins PL, Meanger J, and Mills J (2005b) Interaction between the respiratory syncytial virus G glycoprotein cytoplasmic domain and the matrix protein. J Gen Virol 86(Pt 7):1879-1884.
Ghildyal R, Mills J, Murray M, Vardaxis N, and Meanger J (2002) Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J Gen Virol 83(Pt 4):753-757.
Gomez-Puertas P, Albo C, Perez-Pastrana E, Vivo A, and Portela A (2000) Influenza virus matrix protein is the major driving force in virus budding. J Virol 74(24):11538-11547.
Gonzalez-Reyes L, Ruiz-Arguello MB, Garcia-Barreno B, Calder L, Lopez JA, Albar JP, Skehel JJ, Wiley DC, and Melero JA (2001) Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc Natl Acad Sci U S A 98(17):9859-9864.
Gower TL, Pastey MK, Peeples ME, Collins PL, McCurdy LH, Hart TK, Guth A, Johnson TR, and Graham BS (2005) RhoA signaling is required for respiratory syncytial virus-induced syncytium formation and filamentous virion morphology. J Virol 79(9):5326-5336.
Graham BS (2011) Biological challenges and technological opportunities for respiratory syncytial virus vaccine development. Immunol Rev 239(1):149-166.
Graham SC, Assenberg R, Delmas O, Verma A, Gholami A, Talbi C, Owens RJ, Stuart DI, Grimes JM, and Bourhy H (2008) Rhabdovirus matrix protein structures reveal a novel mode of self-association. PLoS Pathog 4(12):e1000251.
Green TJ, Zhang X, Wertz GW, and Luo M (2006) Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313(5785):357-360.
Grünewald K and Cyrklaff M (2006) Structure of complex viruses and virus-infected cells by electron cryo tomography. Curr Opin Microbiol 9(4):437-442.
Grünewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, and Steven AC (2003) Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302(5649):1396-1398.
Guerrero-Ferreira RC and Wright ER (2013) Cryo-electron tomography of bacterial viruses. Virology 435(1):179-186.

50
Guichard P, Krell T, Chevalier M, Vaysse C, Adam O, Ronzon F, and Marco S (2011) Three dimensional morphology of rabies virus studied by cryo-electron tomography. J Struct Biol 176(1):32-40.
Göttlinger HG, Dorfman T, Sodroski JG, and Haseltine WA (1991) Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A 88(8):3195-3199.
Hardy RW and Wertz GW (1998) The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J Virol 72(1):520-526.
Harris A, Cardone G, Winkler DC, Heymann JB, Brecher M, White JM, and Steven AC (2006) Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc Natl Acad Sci U S A 103(50):19123-19127.
Harris AK, Meyerson JR, Matsuoka Y, Kuybeda O, Moran A, Bliss D, Das SR, Yewdell JW, Sapiro G, Subbarao K, and Subramaniam S (2013) Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc Natl Acad Sci U S A 110(12):4592-4597.
Harrison SC (2008) Viral membrane fusion. Nat Struct Mol Biol 15(7):690-698. Harty RN and Palese P (1995) Measles virus phosphoprotein (P) requires the NH2- and
COOH-terminal domains for interactions with the nucleoprotein (N) but only the COOH terminus for interactions with itself. J Gen Virol 76(11):2863-2867.
Harty RN, Paragas J, Sudol M, and Palese P (1999) A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J Virol 73(4):2921-2929.
Hashiguchi T, Kajikawa M, Maita N, Takeda M, Kuroki K, Sasaki K, Kohda D, Yanagi Y, and Maenaka K (2007) Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc Natl Acad Sci U S A 104(49):19535-19540.
Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, and Yanagi Y (2011) Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18(2):135-141.
He B, Lin GY, Durbin JE, Durbin RK, and Lamb RA (2001) The SH integral membrane protein of the paramyxovirus simian virus 5 is required to block apoptosis in MDBK cells. J Virol 75(9):4068-4079.
Heggeness MH, Smith PR, and Choppin PW (1982) In vitro assembly of the nonglycosylated membrane protein (M) of Sendai virus. Proc Natl Acad Sci USA 79(20):6232-6236.
Henderson G, Murray J, and Yeo RP (2002) Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology 300(2):244-254.
Hendrix RW (2002) Bacteriophages: evolution of the majority. Theor Popul Biol 61(4):471-480.
Heumann JM, Hoenger A, and Mastronarde DN (2011) Clustering and variance maps for cryo-electron tomography using wedge-masked differences. J Struct Biol 175(3):288-299.
Heuser T, Dymek EE, Lin J, Smith EF, and Nicastro D (2012) The CSC connects three major axonemal complexes involved in dynein regulation. Mol Biol Cell 23(16):3143-3155.
Heymann JB, Cardone G, Winkler DC, and Steven AC (2008) Computational resources for cryo-electron tomography in Bsoft. J Struct Biol 161(3):232-242.
Homma M and Ouchi M (1973) Trypsin action on the growth of Sendai virus in tissue culture cells. 3. Structural difference of Sendai viruses grown in eggs and tissue culture cells. J Virol 12(6):1457-1465.
Hu XL, Ray R, and Compans RW (1992) Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J Virol 66(3):1528-1534.

51
Huiskonen JT, Hepojoki J, Laurinmaki P, Vaheri A, Lankinen H, Butcher SJ, and Grunewald K (2010) Electron cryotomography of Tula hantavirus suggests a unique assembly paradigm for enveloped viruses. J Virol 84(10):4889-4897.
Huiskonen JT, Överby AK, Weber F, and Grunewald K (2009) Electron cryo-microscopy and single-particle averaging of Rift Valley fever virus: evidence for GN-GC glycoprotein heterodimers. J Virol 83(8):3762-3769.
Ibiricu I, Huiskonen JT, Dohner K, Bradke F, Sodeik B, and Grunewald K (2011) Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog 7(12):e1002406.
Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, and Yanagi Y (2009) The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J Virol 83(20):10374-10383.
Jasenosky LD, Neumann G, Lukashevich I, and Kawaoka Y (2001) Ebola virus VP40-induced particle formation and association with the lipid bilayer. J Virol 75(11):5205-5214.
Jeffree CE, Brown G, Aitken J, Su-Yin DY, Tan BH, and Sugrue RJ (2007) Ultrastructural analysis of the interaction between F-actin and respiratory syncytial virus during virus assembly. Virology 369(2):309-323.
Justice PA, Sun W, Li Y, Ye Z, Grigera PR, and Wagner RR (1995) Membrane vesiculation function and exocytosis of wild-type and mutant matrix proteins of vesicular stomatitis virus. J Virol 69(5):3156-3160.
Karlin D, Longhi S, Receveur V, and Canard B (2002) The N-terminal domain of the phosphoprotein of morbilliviruses belongs to the natively unfolded class of proteins. Virology 296(2):251-262.
Kay D, Roche M, Atkinson J, Lamden K, and Vivancos R (2011) Mumps outbreaks in four universities in the North West of England: Prevention, detection and response. Vaccine 29(22):3883-3887.
Knipe DM, Baltimore D, and Lodish HF (1977) Maturation of viral proteins in cells infected with temperature-sensitive mutants of vesicular stomatitis virus. J Virol 21(3):1149-1158.
Kolesnikova L, Bohil AB, Cheney RE, and Becker S (2007) Budding of Marburg virus is associated with filopodia. Cell Microbiol 9(4):939-951.
Korinek A, Beck F, Baumeister W, Nickell S, and Plitzko JM (2011) Computer controlled cryo-electron microscopy--TOM(2) a software package for high-throughput applications. J Struct Biol 175(3):394-405.
Kremer JR, Mastronarde DN, and McIntosh JR (1996) Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116(1):71-76.
Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, and Helenius A (2013) Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9(4):e1003309.
Kuismanen E, Hedman K, Saraste J, and Pettersson RF (1982) Uukuniemi virus maturation: accumulation of virus particles and viral antigens in the Golgi complex. Mol Cell Biol 2(11):1444-1458.
Kuzyk A, Schreiber R, Fan Z, Pardatscher G, Roller EM, Hogele A, Simmel FC, Govorov AO, and Liedl T (2012) DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 483(7389):311-314.
Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, and Saphire EO (2008) Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454(7201):177-182.
Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA, Jr., Jensen MR, Blackledge M, Ruigrok RW, and Jamin M (2011) Structure of the vesicular stomatitis virus N(0)-P complex. PLoS Pathog 7(9):e1002248.

52
Li D, Jans DA, Bardin PG, Meanger J, Mills J, and Ghildyal R (2008) Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J Virol 82(17):8863-8870.
Li M, Schmitt PT, Li Z, McCrory TS, He B, and Schmitt AP (2009) Mumps virus matrix, fusion, and nucleocapsid proteins cooperate for efficient production of virus-like particles. J Virol 83(14):7261-7272.
Li Z, Xu J, Patel J, Fuentes S, Lin Y, Anderson D, Sakamoto K, Wang LF, and He B (2011) Function of the small hydrophobic protein of J paramyxovirus. J Virol 85(1):32-42.
Licata JM, Johnson RF, Han Z, and Harty RN (2004) Contribution of ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J Virol 78(14):7344-7351.
Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, and Harty RN (2003) Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J Virol 77(3):1812-1819.
Liljeroos L and Butcher SJ (2013) Matrix proteins as centralized organizers of negative-sense RNA virions. Front Biosci 18:696-715.
Lingwood D and Simons K (2010) Lipid rafts as a membrane-organizing principle. Science 327(5961):46-50.
Liu H, Jin L, Koh SB, Atanasov I, Schein S, Wu L, and Zhou ZH (2010) Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329(5995):1038-1043.
Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, and Subramaniam S (2008) Molecular architecture of native HIV-1 gp120 trimers. Nature 455(7209):109-113.
Loney C, Mottet-Osman G, Roux L, and Bhella D (2009) Paramyxovirus ultrastructure and genome packaging: cryo-electron tomography of Sendai virus. J Virol 83(16):8191-8197.
Longhi S (2009) Nucleocapsid structure and function. Curr Top Microbiol Immunol 329:103-128.
Longhi S, Receveur-Bréchot V, Karlin D, Johansson K, Darbon H, Bhella D, Yeo R, Finet S, and Canard B (2003) The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem 278(20):18638-18648.
Low KW, Tan T, Ng K, Tan BH, and Sugrue RJ (2008) The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem Biophys Res Commun 366(2):308-313.
Lozach PY, Mancini R, Bitto D, Meier R, Oestereich L, Overby AK, Pettersson RF, and Helenius A (2010) Entry of bunyaviruses into mammalian cells. Cell Host Microbe 7(6):488-499.
Ludwig K, Schade B, Bottcher C, Korte T, Ohlwein N, Baljinnyam B, Veit M, and Herrmann A (2008) Electron cryomicroscopy reveals different F1+F2 protein States in intact parainfluenza virions. J Virol 82(7):3775-3781.
Lyles DS, McKenzie MO, Kaptur PE, Grant KW, and Jerome WG (1996) Complementation of M gene mutants of vesicular stomatitis virus by plasmid-derived M protein converts spherical extracellular particles into native bullet shapes. Virology 217(1):76-87.
Maar D, Harmon B, Chu D, Schulz B, Aguilar HC, Lee B, and Negrete OA (2012) Cysteines in the stalk of the nipah virus G glycoprotein are located in a distinct subdomain critical for fusion activation. J Virol 86(12):6632-6642.
Maimon T, Elad N, Dahan I, and Medalia O (2012) The human nuclear pore complex as revealed by cryo-electron tomography. Structure 20(6):998-1006.
Maisner A, Klenk HD, and Herrler G (1998) Polarized budding of measles virus is not determined by viral surface glycoproteins. J Virol 72(6):5276-5278.

53
Manie SN, de Breyne S, Vincent S, and Gerlier D (2000) Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J Virol 74(1):305-311.
Manole V, Laurinmaki P, Van Wyngaardt W, Potgieter CA, Wright IM, Venter GJ, van Dijk AA, Sewell BT, and Butcher SJ (2012) Structural insight into African horsesickness virus infection. J Virol 86(15):7858-7866.
Martin K and Helenius A (1991) Transport of incoming influenza virus nucleocapsids into the nucleus. J Virol 65(1):232-244.
Masich S, Ostberg T, Norlen L, Shupliakov O, and Daneholt B (2006) A procedure to deposit fiducial markers on vitreous cryo-sections for cellular tomography. J Struct Biol 156(3):461-468.
McCurdy LH and Graham BS (2003) Role of plasma membrane lipid microdomains in respiratory syncytial virus filament formation. J Virol 77(3):1747-1756.
McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, and Graham BS (2013) Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340(6136):1113-1117.
McLellan JS, Yang Y, Graham BS, and Kwong PD (2011) Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol 85(15):7788-7796.
McPhee HK, Carlisle JL, Beeby A, Money VA, Watson SM, Yeo RP, and Sanderson JM (2011) Influence of lipids on the interfacial disposition of respiratory syncytical virus matrix protein. Langmuir 27(1):304-311.
Mebatsion T, Weiland F, and Conzelmann KK (1999) Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J Virol 73(1):242-250.
Melanson VR and Iorio RM (2004) Amino acid substitutions in the F-specific domain in the stalk of the newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J Virol 78(23):13053-13061.
Mercer J and Helenius A (2012) Gulping rather than sipping: macropinocytosis as a way of virus entry. Curr Opin Microbiol 15(4):490-499.
Meric C, Spehner D, and Mazarin V (1994) Respiratory syncytial virus nucleocapsid protein (N) expressed in insect cells forms nucleocapsid-like structures. Virus Res 31(2):187-201.
Mitra R, Baviskar P, Duncan-Decocq RR, Patel D, and Oomens AG (2012) The human respiratory syncytial virus matrix protein is required for maturation of viral filaments. J Virol 86(8):4432-4443.
Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, and Wilson IA (2012) Organization of the influenza virus replication machinery. Science 338(6114):1631-1634.
Money VA, McPhee HK, Mosely JA, Sanderson JM, and Yeo RP (2009) Surface features of a Mononegavirales matrix protein indicate sites of membrane interaction. Proc Natl Acad Sci U S A 106(11):4441-4446.
Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, and Chen IS (2011) The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 9(4):286-298.
Moss WJ and Griffin DE (2006) Global measles elimination. Nat Rev Microbiol 4(12):900-908.
Moyer SA, Baker SC, and Horikami SM (1990) Host cell proteins required for measles virus reproduction. J Gen Virol 71 ( Pt 4):775-783.
Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB, Jr., Cichutek K, von Messling V, Lopez M, and Cattaneo R (2011) Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480(7378):530-533.

54
Murphy LB, Loney C, Murray J, Bhella D, Ashton P, and Yeo RP (2003) Investigations into the amino-terminal domain of the respiratory syncytial virus nucleocapsid protein reveal elements important for nucleocapsid formation and interaction with the phosphoprotein. Virology 307(1):143-153.
Muscat M (2011) Who gets measles in Europe? J Infect Dis 204(suppl 1):S353-S365. Naim HY, Ehler E, and Billeter MA (2000) Measles virus matrix protein specifies apical
virus release and glycoprotein sorting in epithelial cells. EMBO J 19(14):3576-3585.
Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, and Campbell H (2010) Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375(9725):1545-1555.
Nakashima M, Shirogane Y, Hashiguchi T, and Yanagi Y (2013) Mutations in the putative dimer-dimer interfaces of the measles virus hemagglutinin head domain affect membrane fusion triggering. J Biol Chem 288(12):8085-8091.
Namba K, Pattanayek R, and Stubbs G (1989) Visualization of protein-nucleic acid interactions in a virus. Refined structure of intact tobacco mosaic virus at 2.9 A resolution by X-ray fiber diffraction. J Mol Biol 208(2):307-325.
Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VH, Braun W, and Cattaneo R (2011) The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat Struct Mol Biol 18(2):128-134.
Neuman BW, Adair BD, Burns JW, Milligan RA, Buchmeier MJ, and Yeager M (2005) Complementarity in the supramolecular design of arenaviruses and retroviruses revealed by electron cryomicroscopy and image analysis. J Virol 79(6):3822-3830.
Neumann P, Lieber D, Meyer S, Dautel P, Kerth A, Kraus I, Garten W, and Stubbs MT (2009) Crystal structure of the Borna disease virus matrix protein (BDV-M) reveals ssRNA binding properties. Proc Natl Acad Sci U S A 106(10):3710-3715.
Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, and McIntosh JR (2006) The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313(5789):944-948.
Noda T, Sagara H, Suzuki E, Takada A, Kida H, and Kawaoka Y (2002) Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J Virol 76(10):4855-4865.
Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, and Digard P (2007) Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol 88(Pt 8):2280-2290.
Novoa RR, Calderita G, Cabezas P, Elliott RM, and Risco C (2005) Key Golgi factors for structural and functional maturation of bunyamwera virus. J Virol 79(17):10852-10863.
Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, and Richardson CD (2011) Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7(8):e1002240.
Ogawa HI and Nakamura M (1985) Characterization of a mycoplasma virus (MV-O1) derived from and infecting Acholeplasma oculi. J Gen Microbiol 131(11):3117-3126.
Oomens AG, Bevis KP, and Wertz GW (2006) The cytoplasmic tail of the human respiratory syncytial virus F protein plays critical roles in cellular localization of the F protein and infectious progeny production. J Virol 80(21):10465-10477.
Orlova EV and Saibil HR (2011) Structural analysis of macromolecular assemblies by electron microscopy. Chem Rev 111(12):7710-7748.

55
Oyanagi S, ter Meulen V, Katz M, and Koprowski H (1971) Comparison of subacute sclerosing panencephalitis and measles viruses: an electron microscope study. J Virol 7(1):176-187.
Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, and Horvath CM (2003) STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol 77(13):7635-7644.
Panchal RG, Ruthel G, Kenny TA, Kallstrom GH, Lane D, Badie SS, Li L, Bavari S, and Aman MJ (2003) In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc Natl Acad Sci U S A 100(26):15936-15941.
Parent LJ, Bennett RP, Craven RC, Nelle TD, Krishna NK, Bowzard JB, Wilson CB, Puffer BA, Montelaro RC, and Wills JW (1995) Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J Virol 69(9):5455-5460.
Paterson RG, Johnson ML, and Lamb RA (1997) Paramyxovirus fusion (F) protein and hemagglutinin-neuraminidase (HN) protein interactions: intracellular retention of F and HN does not affect transport of the homotypic HN or F protein. Virology 237(1):1-9.
Patterson JB, Cornu TI, Redwine J, Dales S, Lewicki H, Holz A, Thomas D, Billeter MA, and Oldstone MB (2001) Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology 291(2):215-225.
Penfold ME, Armati P, and Cunningham AL (1994) Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc Natl Acad Sci U S A 91(14):6529-6533.
Pietilä MK, Atanasova NS, Manole V, Liljeroos L, Butcher SJ, Oksanen HM, and Bamford DH (2012) Virion architecture unifies globally distributed pleolipoviruses infecting halophilic archaea. J Virol 86(9):5067-5079.
Pigino G, Bui KH, Maheshwari A, Lupetti P, Diener D, and Ishikawa T (2011) Cryoelectron tomography of radial spokes in cilia and flagella. J Cell Biol 195(4):673-687.
Plemper RK (2011) Cell entry of enveloped viruses. Curr Opin Virol 1(2):92-100. Plemper RK, Hammond AL, and Cattaneo R (2001) Measles virus envelope glycoproteins
hetero-oligomerize in the endoplasmic reticulum. J Biol Chem 276(47):44239-44246.
Pohl C, Duprex WP, Krohne G, Rima BK, and Schneider-Schaulies S (2007) Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J Gen Virol 88(Pt 4):1243-1250.
Psencik J, Collins AM, Liljeroos L, Torkkeli M, Laurinmaki P, Ansink HM, Ikonen TP, Serimaa RE, Blankenship RE, Tuma R, and Butcher SJ (2009) Structure of chlorosomes from the green filamentous bacterium Chloroflexus aurantiacus. J Bacteriol 191(21):6701-6708.
Radhakrishnan A, Yeo D, Brown G, Myaing MZ, Iyer LR, Fleck R, Tan BH, Aitken J, Sanmun D, Tang K, Yarwood A, Brink J, and Sugrue RJ (2010) Protein analysis of purified respiratory syncytial virus particles reveals an important role for heat shock protein 90 in virus particle assembly. Mol Cell Proteomics 9(9):1829-1848.
Raiborg C and Stenmark H (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458(7237):445-452.
Reddy VS, Natchiar SK, Stewart PL, and Nemerow GR (2010) Crystal structure of human adenovirus at 3.5 A resolution. Science 329(5995):1071-1075.
Roberts SR, Compans RW, and Wertz GW (1995) Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J Virol 69(4):2667-2673.
Roche S, Bressanelli S, Rey FA, and Gaudin Y (2006) Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313(5784):187-191.
Roche S, Rey FA, Gaudin Y, and Bressanelli S (2007) Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315(5813):843-848.

56
Rossman JS, Jing X, Leser GP, and Lamb RA (2010) Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142(6):902-913.
Sachse C, Chen JZ, Coureux PD, Stroupe ME, Fandrich M, and Grigorieff N (2007) High-resolution electron microscopy of helical specimens: a fresh look at tobacco mosaic virus. J Mol Biol 371(3):812-835.
Salditt A, Koethe S, Pohl C, Harms H, Kolesnikova L, Becker S, and Schneider-Schaulies S (2010) Measles virus M protein-driven particle production does not involve the endosomal sorting complex required for transport (ESCRT) system. J Gen Virol 91(Pt 6):1464-1472.
Santiago C, Celma ML, Stehle T, and Casasnovas JM (2010) Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat Struct Mol Biol 17(1):124-129.
Scheiffele P, Rietveld A, Wilk T, and Simons K (1999) Influenza viruses select ordered lipid domains during budding from the plasma membrane. J Biol Chem 274(4):2038-2044.
Scheres SH (2012) RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180(3):519-530.
Schmid MF and Booth CR (2008) Methods for aligning and for averaging 3D volumes with missing data. J Struct Biol 161(3):243-248.
Schmitt AP, Leser GP, Waning DL, and Lamb RA (2002) Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J Virol 76(8):3952-3964.
Schnitzer TJ, Dickson C, and Weiss RA (1979) Morphological and biochemical characterization of viral particles produced by the tsO45 mutant of vesicular stomatitis virus at restrictive temperature. J Virol 29(1):185-195.
Schoehn G, Mavrakis M, Albertini A, Wade R, Hoenger A, and Ruigrok RW (2004) The 12 A structure of trypsin-treated measles virus N-RNA. J Mol Biol 339(2):301-312.
Schroder RR (1992) Zero-loss energy-filtered imaging of frozen-hydrated proteins: model calculations and implications for future developments. J Microsc 166(Pt 3):389-400.
Sha B and Luo M (1997) Structure of a bifunctional membrane-RNA binding protein, influenza virus matrix protein M1. Nat Struct Biol 4(3):239-244.
Shadman KA and Wald ER (2011) A review of palivizumab and emerging therapies for respiratory syncytial virus. Expert Opin Biol Ther 11(11):1455-1467.
Shaffer JA, Bellini WJ, and Rota PA (2003) The C protein of measles virus inhibits the type I interferon response. Virology 315(2):389-397.
Shaikh FY, Cox RG, Lifland AW, Hotard AL, Williams JV, Moore ML, Santangelo PJ, and Crowe JE, Jr. (2012a) A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. MBio 3(1)
Shaikh FY, Utley TJ, Craven RE, Rogers MC, Lapierre LA, Goldenring JR, and Crowe JE, Jr. (2012b) Respiratory syncytial virus assembles into structured filamentous virion particles independently of host cytoskeleton and related proteins. PLoS One 7(7):e40826.
Shnyrova AV, Ayllon J, Mikhalyov, II, Villar E, Zimmerberg J, and Frolov VA (2007) Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. J Cell Biol 179(4):627-633.
Sorzano CO, Marabini R, Velazquez-Muriel J, Bilbao-Castro JR, Scheres SH, Carazo JM, and Pascual-Montano A (2004) XMIPP: a new generation of an open-source image processing package for electron microscopy. J Struct Biol 148(2):194-204.
Stallcup KC, Raine CS, and Fields BN (1983) Cytochalasin B inhibits the maturation of measles virus. Virology 124(1):59-74.
Stolken M, Beck F, Haller T, Hegerl R, Gutsche I, Carazo JM, Baumeister W, Scheres SH, and Nickell S (2011) Maximum likelihood based classification of electron tomographic data. J Struct Biol 173(1):77-85.

57
Strecker T, Eichler R, Meulen J, Weissenhorn W, Dieter Klenk H, Garten W, and Lenz O (2003) Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J Virol 77(19):10700-10705.
Stubbs G and Kendall A (2012) Helical viruses. Adv Exp Med Biol 726:631-658. Subramaniam S, Bartesaghi A, Liu J, Bennett AE, and Sougrat R (2007) Electron
tomography of viruses. Curr Opin Struct Biol 17(5):596-602. Suryanarayana K, Baczko K, ter Meulen V, and Wagner RR (1994) Transcription inhibition
and other properties of matrix proteins expressed by M genes cloned from measles viruses and diseased human brain tissue. J Virol 68(3):1532-1543.
Swanson K, Wen X, Leser GP, Paterson RG, Lamb RA, and Jardetzky TS (2010) Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402(2):372-379.
Sänger C, Mühlberger E, Ryabchikova E, Kolesnikova L, Klenk HD, and Becker S (2001) Sorting of Marburg virus surface protein and virus release take place at opposite surfaces of infected polarized epithelial cells. J Virol 75(3):1274-1283.
Takeuchi K, Kadota SI, Takeda M, Miyajima N, and Nagata K (2003) Measles virus V protein blocks interferon (IFN)-alpha/beta but not IFN-gamma signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett 545(2-3):177-182.
Tatsuo H, Ono N, Tanaka K, and Yanagi Y (2000) SLAM (CDw150) is a cellular receptor for measles virus. Nature 406(6798):893-897.
Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet J, and Rey FA (2009) Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326(5957):1279-1283.
Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, and Hegele RG (2011) Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med 17(9):1132-1135.
Techaarpornkul S, Barretto N, and Peeples ME (2001) Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol 75(15):6825-6834.
Toyoshima C and Unwin N (1988) Contrast transfer for frozen-hydrated specimens: determination from pairs of defocused images. Ultramicroscopy 25(4):279-291.
Urata S, Noda T, Kawaoka Y, Yokosawa H, and Yasuda J (2006) Cellular factors required for Lassa virus budding. J Virol 80(8):4191-4195.
Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, Goldenring JR, and Crowe JE, Jr. (2008) Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc Natl Acad Sci U S A 105(29):10209-10214.
Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg AN, and Lee B (2010) Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog 6(11):e1001186.
Vanhecke D, Graber W, and Studer D (2008) Close-to-native ultrastructural preservation by high pressure freezing. Methods Cell Biol 88:151-164.
Varghese JN, Laver WG, and Colman PM (1983) Structure of the influenza virus glycoprotein antigen neuraminidase at 2.9 Å resolution. Nature 303(5912):35-40.
Wasilewski S, Calder LJ, Grant T, and Rosenthal PB (2012) Distribution of surface glycoproteins on influenza A virus determined by electron cryotomography. Vaccine 30(51):7368-7373.
Watanabe R and Lamb RA (2010) Influenza virus budding does not require a functional AAA+ ATPase, VPS4. Virus Res 153(1):58-63.
Welch BD, Liu Y, Kors CA, Leser GP, Jardetzky TS, and Lamb RA (2012) Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc Natl Acad Sci U S A 109(41):16672-16677.

58
Welch BD, Yuan P, Bose S, Kors CA, Lamb RA, and Jardetzky TS (2013) Structure of the Parainfluenza Virus 5 (PIV5) Hemagglutinin-Neuraminidase (HN) Ectodomain. PLoS Pathog 9(8):e1003534.
White J, Matlin K, and Helenius A (1981) Cell fusion by Semliki Forest, influenza, and vesicular stomatitis viruses. J Cell Biol 89(3):674-679.
White TA, Bartesaghi A, Borgnia MJ, Meyerson JR, de la Cruz MJ, Bess JW, Nandwani R, Hoxie JA, Lifson JD, Milne JL, and Subramaniam S (2010) Molecular architectures of trimeric SIV and HIV-1 envelope glycoproteins on intact viruses: strain-dependent variation in quaternary structure. PLoS Pathog 6(12):e1001249.
WHO (2006) Weekly Epidemiological Record, No 81.13-20. WHO (2009) Weekly Epidemiological Record, No 49. 84:509-516. Vijayakrishnan S, Loney C, Jackson D, Suphamungmee W, Rixon FJ, and Bhella D (2013)
Cryotomography of budding influenza A virus reveals filaments with diverse morphologies that mostly do not bear a genome at their distal end. PLoS Pathog 9(6):e1003413.
Wills JW, Cameron CE, Wilson CB, Xiang Y, Bennett RP, and Leis J (1994) An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J Virol 68(10):6605-6618.
Wilson IA, Skehel JJ, and Wiley DC (1981) Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289(5796):366-373.
Wilson RL, Fuentes SM, Wang P, Taddeo EC, Klatt A, Henderson AJ, and He B (2006) Function of small hydrophobic proteins of paramyxovirus. J Virol 80(4):1700-1709.
Vincent S, Gerlier D, and Manie SN (2000) Measles virus assembly within membrane rafts. J Virol 74(21):9911-9915.
Winkler H, Zhu P, Liu J, Ye F, Roux KH, and Taylor KA (2009) Tomographic subvolume alignment and subvolume classification applied to myosin V and SIV envelope spikes. J Struct Biol 165(2):64-77.
Volpon L, Osborne MJ, Capul AA, de la Torre JC, and Borden KL (2010) Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc Natl Acad Sci U S A 107(12):5441-5446.
Wommack KE and Colwell RR (2000) Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev 64(1):69-114.
Woolhouse ME and Gowtage-Sequeria S (2005) Host range and emerging and reemerging pathogens. Emerg Infect Dis 11(12):1842-1847.
Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, and Mastronarde DN (2009) CTF determination and correction for low dose tomographic tilt series. J Struct Biol 168(3):378-387.
Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, and Nikolov DB (2008) Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc Natl Acad Sci U S A 105(29):9953-9958.
Xu M, Beck M, and Alber F (2012) High-throughput subtomogram alignment and classification by Fourier space constrained fast volumetric matching. J Struct Biol 178(2):152-164.
Yamayoshi S, Neumann G, and Kawaoka Y (2010) Role of the GTPase Rab1b in ebolavirus particle formation. J Virol 84(9):4816-4820.
Yamayoshi S, Noda T, Ebihara H, Goto H, Morikawa Y, Lukashevich IS, Neumann G, Feldmann H, and Kawaoka Y (2008) Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe 3(3):168-177.
Yao Q and Compans RW (2000) Filamentous particle formation by human parainfluenza virus type 2. J Gen Virol 81(5):1305-1312.

59
Yin HS, Paterson RG, Wen X, Lamb RA, and Jardetzky TS (2005) Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A 102(26):9288-9293.
Yin HS, Wen X, Paterson RG, Lamb RA, and Jardetzky TS (2006) Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439(7072):38-44.
Yoshida T, Nagai Y, Maeno K, Iinuma M, Hamaguchi M, Matsumoto T, Nagayoshi S, and Hoshino M (1979) Studies on the role of M protein in virus assembly using a ts mutant of HVJ (Sendai virus). Virology 92(1):139-154.
Yu Z and Frangakis AS (2011) Classification of electron sub-tomograms with neural networks and its application to template-matching. J Struct Biol 174(3):494-504.
Yuan P, Leser GP, Demeler B, Lamb RA, and Jardetzky TS (2008) Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378(2):282-291.
Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, and Jardetzky TS (2011) Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc Natl Acad Sci U S A 108(36):14920-14925.
Zanetti G, Riches JD, Fuller SD, and Briggs JA (2009) Contrast transfer function correction applied to cryo-electron tomography and sub-tomogram averaging. J Struct Biol 168(2):305-312.
Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, and Zhou ZH (2013a) Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat Struct Mol Biol 20(1):105-110.
Zhang X, Jin L, Fang Q, Hui WH, and Zhou ZH (2010) 3.3 A cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 141(3):472-482.
Zhang X, Lu G, Qi J, Li Y, He Y, Xu X, Shi J, Zhang CW, Yan J, and Gao GF (2013b) Structure of measles virus hemagglutinin bound to its epithelial receptor nectin-4. Nat Struct Mol Biol 20(1):67-72.
Zimmer G, Budz L, and Herrler G (2001) Proteolytic activation of respiratory syncytial virus fusion protein. Cleavage at two furin consensus sequences. J Biol Chem 276(34):31642-31650.
Överby AK, Pettersson RF, Grünewald K, and Huiskonen JT (2008) Insights into bunyavirus architecture from electron cryotomography of Uukuniemi virus. Proc Natl Acad Sci U S A 105(7):2375-2379.