building phylogenies parsimony 2. methods distance-based parsimony maximum likelihood
TRANSCRIPT

Building Phylogenies
Parsimony 2

Methods
• Distance-based• Parsimony• Maximum likelihood

Searching for an MP tree
• Exhaustive search (exact)• Branch-and-bound search (exact)• Heuristic search methods
– Stepwise addition– Branch swapping– Star decomposition

Exhaustive Enumeration
• Order the taxa: s1, s2, . . . , sn
• Build (unique) unrooted tree for s1, s2, s3
• Try all possible places to add s4, and score each tree
• Try all places to add s5 to previous trees and score again . . .
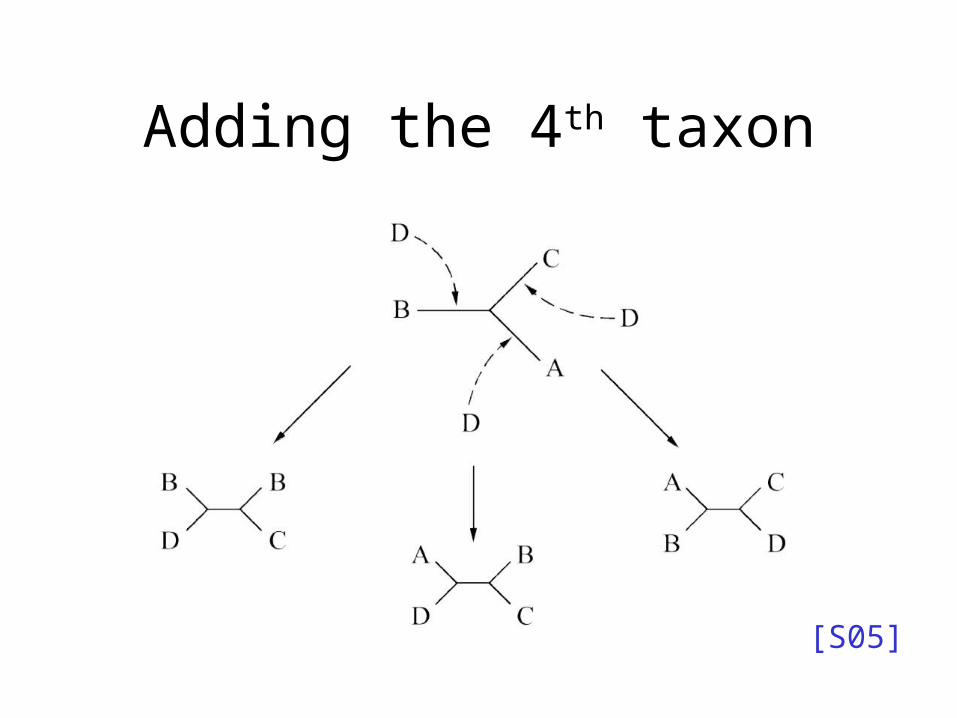
Adding the 4th taxon
[S05]
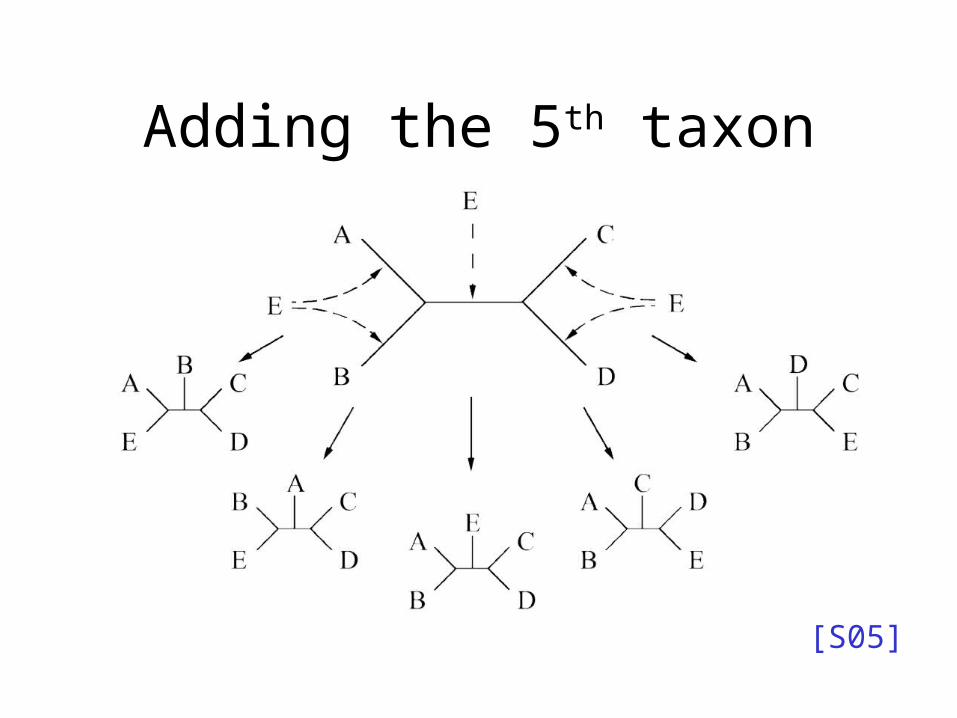
Adding the 5th taxon
[S05]
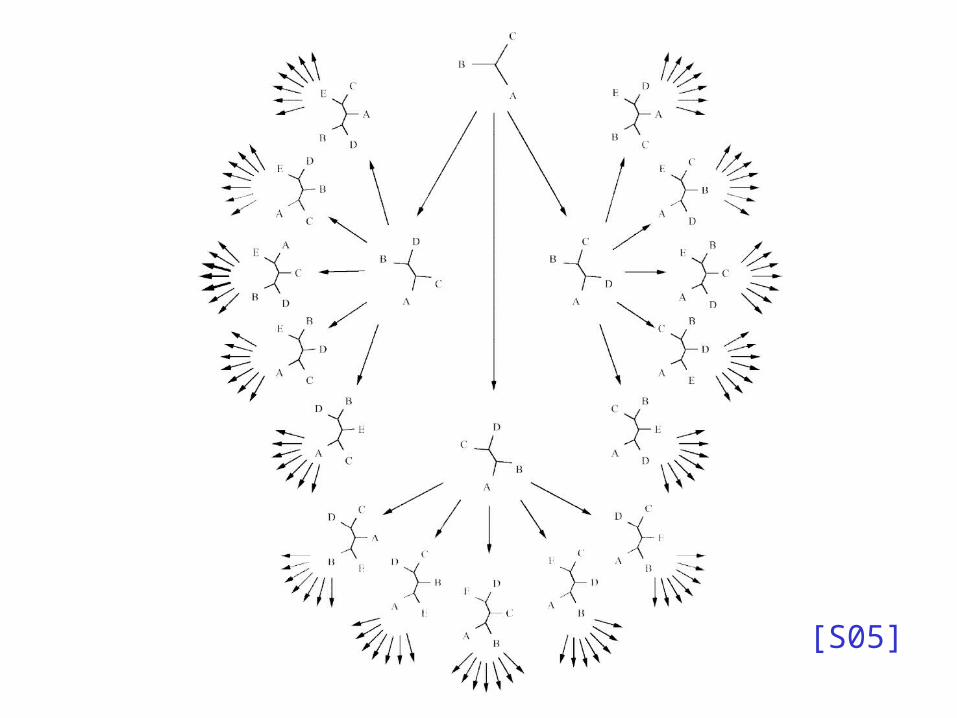
[S05]

Branch and bound
• Similar to exhaustive search, except that we maintain– Score of best tree obtained so far– A lower bound on score of best tree that can be obtained
from this point forward.
• If score of current tree exceeds the current best score, backtrack and takes the next available path.
• When a tip of the search tree is reached the tree is either optimal (and hence retained) or suboptimal (and rejected).
• When all paths leading from the initial 3-taxon tree have been explored, the algorithm terminates, and all most-parsimonious trees will have been identified.


Branch Swapping
• Local search approach:– Define a “neighborhood” for a tree– Neighbors are obtained by
rearranging branches: cut and paste– Instead of exhaustive exploration of
tree space, just try neighbors.

Branch Swapping
• Nearest-Neighbor Interchange (NNI)
• Subtree Pruning and Regrafting (SPR)
• Tree Bisection and Reconnection (TBR)

Nearest-Neighbor Interchange

All 15 5-taxon trees, connected by NNIs

Subtree Pruning and Regrafting
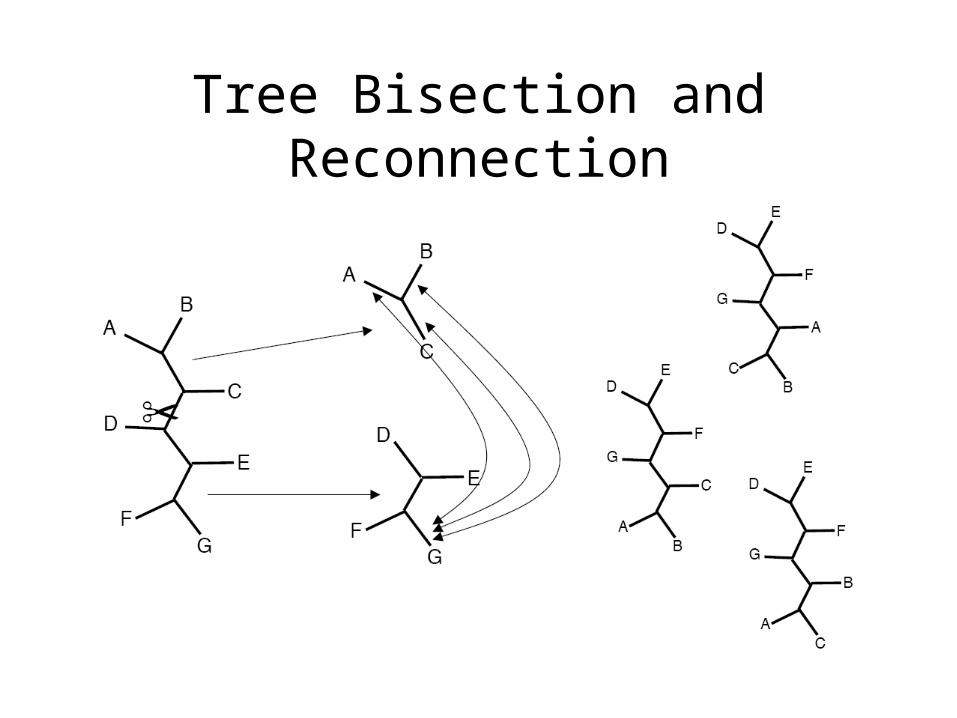
Tree Bisection and Reconnection

Stepwise Addition
• A greedy method• Start with 3-taxon tree• Add taxa one at a time.• Keep only the best tree found so far• No guarantee of optimality, but may
provide good starting point for search

A problem with parsimony: Long branch attraction
Convergent evolution along long branches can confuse parsimony
G G
A A
G
G
A
A
Incorrect!

Compatibility
a0111
ABCD
c0011
e0001
f1000
b0111
A B C D
f
a, b
c
e
A set of characters is compatible if there exixts a tree where each character state emerges exactly once.

Consistency index
• Homoplasy: Multiple emergence of the same state in a phylogeny
• Perfect fit (= compatible characters) no homoplasy
• Let mi = min #(steps possible for site i) and si = min #(steps for site i given the tree)
• The consistency index is CI = mi / si (0 CI 1)
• CI measures amount of homoplasy in tree

The bootstrap
• A bootstrap sample is obtained by sampling sites randomly with replacement– Obtain a data matrix with same number of taxa and number
of characters as original one
• Construct phylogenies for samples• For each branch in original tree, compute fraction of
bootstrap samples in which that branch appears– Assigns a bootstrap support value to each branch.
• Idea: If a grouping has a lot of support, it will be supported by at least some positions in most of the bootstrap samples
• Can be applied to other methods of phylogenetic reconstruction