biodiversity, community structural shifts, and biogeography of prokaryotes … · biodiversity,...
TRANSCRIPT

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, May 2003, p. 2463–2483 Vol. 69, No. 50099-2240/03/$08.00�0 DOI: 10.1128/AEM.69.5.2463–2483.2003Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Biodiversity, Community Structural Shifts, and Biogeography ofProkaryotes within Antarctic Continental Shelf Sediment
John P. Bowman* and Robert D. McCuaig†School of Agricultural Science, University of Tasmania, Hobart, Tasmania 7001, Australia
Received 26 August 2002/Accepted 29 January 2003
16S ribosomal DNA (rDNA) clone library analysis was conducted to assess prokaryotic diversity andcommunity structural changes within a surficial sediment core obtained from an Antarctic continental shelfarea (depth, 761 m) within the Mertz Glacier Polynya (MGP) region. Libraries were created from threeseparate horizons of the core (0- to 0.4-cm, 1.5- to 2.5-cm, and 20- to 21-cm depth positions). The resultsindicated that at the oxic sediment surface (depth, 0 to 0.4 cm) the microbial community appeared to bedominated by a small subset of potentially r-strategist (fast-growing, opportunistic) species, resulting in alower-than-expected species richness of 442 operational taxonomic units (OTUs). At a depth of 1.5 to 2.5 cm,the species richness (1,128 OTUs) was much higher, with the community dominated by numerous gamma anddelta proteobacterial phylotypes. At a depth of 20 to 21 cm, a clear decline in species richness (541 OTUs)occurred, accompanied by a larger number of more phylogenetically divergent phylotypes and a decline in thepredominance of Proteobacteria. Based on rRNA and clonal abundance as well as sequence comparisons,syntrophic cycling of oxidized and reduced sulfur compounds appeared to be the dominant process in surficialMGP sediment, as phylotype groups putatively linked to these processes made up a large proportion of clonesthroughout the core. Between 18 and 65% of 16S rDNA phylotypes detected in a wide range of coastal and openocean sediments possessed high levels of sequence similarity (>95%) with the MGP sediment phylotypes,indicating that many sediment prokaryote phylotype groups defined in this study are ubiquitous in marinesediment.
Many aerobic and facultatively anaerobic isolates from coldmarine sediment have been shown to be psychrophilic (41, 60),and molecular analysis of coastal polar sediments also indi-cates the presence of a rich uncultivated prokaryotic diversityat continually low temperatures (43) and in sediment in gen-eral (34). The bacterial community of Svalbard fjord sedimentwas dominated by delta and gamma proteobacteria andsmaller numbers of many other bacterial groups (38, 39).Ravenschlag et al. (38) also used fluorescent in situ hybridiza-tion and rRNA hybridization to determine the phylogeneticcomposition of prokaryotes in the top 5 cm of Svalbard fjordsediment. A bacterium-specific fluorescent in situ hybridiza-tion probe hybridized to 65% of detectable cells on average,while fewer than 5% of cells belonged to the Archaea. Overall,about 58% of microscopically detectable cells (24% of the totaldirect count) and 45% of bacterial rRNA could be assigned toknown taxonomic groups including the Proteobacteria and Fla-vobacteria, results which compared well with clone library data(39). Results also support the contention that most benthicbacteria are autochthonous, not merely accumulating from thepelagic zone. Even in the light of these data, we still knowrelatively little about prokaryotic diversity, distribution, andfunction within oceanic sediments. By comparison, extensiveanalysis of pelagic prokaryotic communities indicates the pres-
ence of several cultured and uncultured groups, which areclearly ubiquitous (15, 18, 35). For example, marine group I ofthe Crenarchaeota, which appears to dominate bacterioplank-ton in the mesopelagic zone (200- to 1,000-m depth) of theocean (25) also may be a common community member insurficial sediment (57). Theoretically, benthic communitiesshould also contain many other ubiquitous, broadly distributedprokaryotic groups, since environmental conditions (tempera-ture, nutrient availability and supply, and pressure) and pro-cesses (sulfate, iron and nitrate reduction, and carbon miner-alization, etc.) can be considered to be generally similar overwide tracts of the oceanic seabed.
Microbial community structure analysis can be extended togive us an understanding of functional and biogeographicalrelationships (49), and such data are vital for an improvedunderstanding of benthic ecosystem processes and the role thatthe benthos plays in overall oceanic processes. The analysis ofclone libraries created from 16S ribosomal DNA (rDNA) am-plified from environmental DNA can be used to assess com-munity structure and diversity at the highest possible resolu-tion, with 16S rDNA gene data sets providing a qualitativeguide to the microbial composition of a given sample. Severalbacterial 16S rDNA clone libraries have been constructed frommarine and marine-derived sediments (6, 17, 30, 31, 34, 39, 54,56); however, many of these are quite small and provide onlya cursory examination of the microbial community present.More-detailed 16S rDNA data sets better allow the construc-tion of specific probes, as a greater part of the inherent diver-sity of subgroups has been sampled. Specific probes then couldbe used in developing techniques such as 5�-nuclease PCRassays (52) and DNA microarrays (45) for rapid detection and
* Corresponding author. Mailing address: School of AgriculturalScience, University of Tasmania, GPO Box 252-54, Hobart, Tasmania7001, Australia. Phone: 61 03 62262776. Fax: 61 03 62262642. E-mail:[email protected].
† Present address: Immunology and Cell Biology Division, JohnCurtin School of Medical Research, Australian National University,Canberra, ACT 0200, Australia.
2463
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

quantification. In addition, more-detailed information wouldalso indicate which prokaryote groups are important in surfi-cial oceanic sediment and how the abundance of these groupsvaries under different environmental conditions.
In research on Antarctic continental shelf surficial sediment(5), we demonstrated that the benthic prokaryotic communitypresent in Antarctic continental shelf sediments located belowthe Mertz Glacier Polynya (MGP) was metabolically active,contained a predominantly psychrophilic microbial commu-nity, and had well-defined and distinct structural characteris-tics. rRNA hybridization data indicated that Proteobacteria,Archaea, Flavobacteria, and Planctomycetales were abundant inMGP sediment cores and exhibited similar levels of abundanceand zonation across the MGP shelf sediment area sampled. Asubstantial proportion of the benthic microbial community wasnot covered by the RNA probes applied, suggesting that manyother prokaryote groups were also present. One hypothesis wewished to test was that community structural changes revealedby denaturing gradient gel electrophoresis (DGGE) analysiscan also be revealed by 16S rDNA clone library analysis, thusproviding a more detailed impression of community structurein MGP sediment samples. With these data available, biogeo-graphical comparisons of microbial communities of differentmarine sediment previously analyzed by clone library anal-ysis of 16S rDNA sequences can also be compared and thusused to test the hypothesis that, like the pelagic environ-ment, marine sediment contains many ubiquitous prokary-otic groups.
MATERIALS AND METHODS
Sampling. A 21- by 1-cm (diameter) core was aseptically extracted from thecentral unperturbed section of a larger sediment core (5 m in length), the geologyand paleochronology of which had been previously examined (core 10GC01[18]). The large core was obtained from the MGP (66°31.86�S, 143°38.30�E) at adepth of 761 m during the Italian-Australian geoscience research cruise AGSCOsurvey 217 on the RV Tangaroa in February 2000 by using a gravity corer with a1-metric-ton head unit. The sample was stored frozen at �20°C until processed.
DNA extraction. The frozen core was sectioned at 1-cm intervals below 3 cm.The top of the 3 cm of core was sectioned in finer intervals of about 0.4 to 0.5 cm.DNA was extracted from each slice by the method of Rochelle et al. (40). TheDNA samples were then further purified with Chroma Spin�TE-1000 columns(Clontech) and stored at �20°C before use. DNA yields were generally 50 to70% higher (with greater purity) than what was found if a bead beating protocol(6) was used.
Clone library construction and sequencing. The universal 16S rDNA primers519f (5�-CAGCMGCCGCGGTAATAC-3�) and 1492r (5�-TACGGYTACCTTGTTACGAC-3�) were used to amplify 16S rDNAs from prokaryotes from thesediment DNA. PCR and clone library construction were carried out by proce-dures described previously (6), except sequencing was performed with the Beck-man CEQ2000XL automated capillary sequencing system.
Phylogenetic analysis. The 16S rDNA sequences generated in this study werecompared to those in the National Center for Biotechnology Information nucle-otide database by using BLAST searching. Sequences closely similar to those ofclones as well as selected reference sequences were downloaded and manuallyaligned and analyzed by using BioEdit (version 5.0.9) (19). Phylogenetic treeswere created by calculation of maximum-likelihood distances and by using theneighbor-joining algorithm through the BioEdit program. The statistical signif-icance of phylotype groups within the various trees was tested by using bootstrapanalysis with the Phylip programs SEQBOOT and CONSENSE (J. Felsenstein,University of Washington, Seattle). Trees were created from the NEIGHBORoutput by using the program TREEVIEW. 16S rDNA sequences from Thermo-toga maritimum and Coprothermobacter platensis were used as outgroup refer-ences on all trees.
Clone library analysis. The depths of the subsamples taken from the core were0 to 0.4 cm, 1.5 to 2.5 cm, and 20 to 21 cm; clone data sets arising from thesamples were subsequently referred to as the 0-cm (surface), 2-cm, and 21-cm
MGP sediment libraries. No prior dereplication of clones from the libraries byrestriction enzyme-based analysis was performed before sequencing. The highdiversity within the samples and the blunt-end cloning strategy made dereplica-tion unnecessary. Chimeric sequences that were detected within the data setwere discarded unless a confirmed contiguous sequence stretched for greaterthan 50% of the 1-kb 16S rDNA region investigated. After ignoring 18S rDNA(deriving almost entirely from species of the diatom genus Chaeotoceros) andchloroplast-derived 16S rDNA sequences, 338 to 369 clones from each MGPsediment sample were analyzed. Sequences which were 98% or greater in sim-ilarity where considered the same and grouped as a phylotype. This takes intoaccount microvariations which may be induced by PCR and cloning (48) but alsovariations between 16S rDNA gene copies, which usually differ by 1% or less(29). The 16S rDNA phylotypes can in most cases be assumed to be an approx-imation of a prokaryote species as suggested by comparisons of 16S rDNAsequence divergence and genomic DNA hybridization levels (27). On the phy-logenetic trees in Fig. 1 to 12, sequences are indicated by their species or clonedesignation, followed by the GenBank accession number and, for the MGPsediments, the number of clones detected in each clone library if it exceeds one.The BioEdit alignment files created in this study are available by e-mail fromJ. P. Bowman.
Species richness analysis. Species richness was calculated by using the Rare-faction calculator, which is a web-based program written by C. J. Krebs and J.Brzustowski (University of Alberta, Edmonton, Canada) and based at internetsite http://www.biology.ualberta.ca/jbrzusto/rarefact.php. Species richness (S*1)was estimated by using the nonparametric model of Chao (10), i.e., S*1 � Sobs
� (a2/2b), in which Sobs is the number of operational taxonomic units (OTUs)(16S rDNA clones) observed, a is the number of OTUs observed just once, andb is the number of OTUs observed only twice. The variance in the Chao calcu-lations was computed by using the formula �2 � (b[{a/4b}4 � {a/b}3 � {a/2b}2])2. Ninety-five percent confidence intervals (95% CIs) were calculated bylog transforming the data in order to obtain a normal distribution of means (9).Similarity between the clone libraries was calculated by using the LIBSHUFF.PLprogram as described by Singleton et al. (44), with ActivePerl version 5.08 on aIBM PC computer as the operating environment.
Nucleotide sequence accession numbers. The 16S rDNA sequences generatedin this study are available under GenBank accession numbers AF424054 toAF424538 and AF425747 to AF425762.
RESULTS
The sediment core was sampled from the MGP, which hashigh surface primary productivity and is a major source ofAntarctic bottom water (4). Antarctic bottom water is quitenutrient rich (46) and not only plays a major role in the globalocean circulation but also contributes to enhanced biologicalactivity in Antarctic coastal regions (3, 13, 51). Analysis ofsediment cores from this area (depth range, 709 to 943 m)revealed an active, mostly psychrophilic microbial benthiccommunity, which has well-defined community structural char-acteristics. Geological, paleochrononological, and geochemicallipid marker analyses otherwise indicated that the MGP sedi-ment community had characteristics typical of modern openocean sediment (5, 20).
Phylogenetic groups. From the total of 1,046 clones ana-lyzed, 496 phylotypes were defined, with 138, 255, and 194phylotypes detected in the 0-, 2-, and 21-cm MGP sediments,respectively. A total of 76 phylotypes were found in two or allthree libraries. From phylogenetic analysis, 56 significant pro-karyotic 16S rDNA clusters (containing at least four clones[distance, �0.15; bootstrap value, �80%]) were found (Table1). Since many of the clusters do not contain cultivated pro-karyotes, they were designated arbitrarily with the names ofcorresponding 16S rDNA clones derived from earlier studies.
Gamma proteobacteria. Gamma proteobacteria were themost commonly sampled group present within the Mertz core,representing 43, 42, and 23% of clones within the 0-, 2-, and
2464 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

TABLE 1. 16S rDNA phylotype distribution through the MGP sediment core, with comparisons with other sedimentsfor which extensive 16S rDNA phylogenetic data are available
Taxonomic (phylogenetic)group
Phylotypic or taxonomicgroupa
No. of phylotypes (no. of clones)in the following layer:
Phylotype group presence inthe following sediment:
0–0.4 cm 1.5–2.5 cm 20–21 cm Deep seab Arcticc Antarctic in-shored
BacteriaGamma proteobacteria Photobacterium 1 (4) 1 (3) � � �
Moritella 1 (4) 1 (3) 1 (15) � � �Shewanella 2 (27) 1 (1) � � �Psychromonas 1 (1) 1 (7) 1 (17) � � �Pseudoalteromonas 1 (26) 1 (2) 1 (1) � � �Colwellia 6 (10) 3 (4) 1 (1) � � �Oceanospirillum cluster 2 (2) 3 (3) � � �Coxiella cluster 1 (1) 1 (1) 3 (3) � � �B2M60 cluster 3 (8) 7 (32) 2 (3) � � �BPC036 cluster 3 (4) 1 (1) � � �AWS98-7e cluster 3 (4) 6 (12) � � �BD3-6/JTB255 cluster 20 (42) 22 (40) 4 (12) � � �BD7-8 cluster 3 (7) 5 (11) 2 (3) � � �JTB23 cluster 1 (1) 2 (4) � � �sva0091 cluster 2 (4) 9 (10) 3 (5) � � �Ectothiorhodospiraceae 1 (1) 3 (5) 1 (1) � � �
Beta Proteobacteria Nitrosospira briensis 1 (3) 1 (2) � � �Delta proteobacteria Desulfosarcina group 4 (4) 17 (27) 15 (31) � � �
Desulfuromonas anilini group 1 (2) 3 (5) � � �Desulfobulbaceae 1 (1) 8 (9) 3 (3) � � �Desulfuromonas group 2 (2) 2 (6) 1 (1) � � �Myxobacteria 2 (2) 4 (5) 2 (3) � � �JTB38/Clear-9 group 3 (3) 4 (4) � � �Eel-TE1A4 group 1 (2) 2 (2) 5 (6) � � �
Epsilon proteobacteria Arcobacter group 2 (13) � � �Thiomicrospira group 2 (7) 1 (3) � � �
Alpha proteobacteria Olavius losiae symbiont group 1 (2) 3 (5) � � �Amaricoccus/JTB359 group 1 (1) 2 (6) 1 (4) � � �Ruegeria group 2 (7) 1 (1) 1 (2) � � �Mertz cluster A 2 (2) 1 (1) 1 (1) � � �JTB131 group 1 (1) 3 (5) � � �
Acidobacteria NKB17/sva0450 group 5 (8) 3 (5) � � �Unafilliated SAR406 group 4 (5) 3 (5) � � �OP8 Group NKB18 group 4 (5) 3 (5) � � �Flavobacteria NB1-m group 2 (2) 1 (2) 2 (2) � � �
C. fermentans group 1 (1) 5 (10) 7 (22) � � �Zobellia 2 (10) 1 (1) � � �Tenacibaculum-Polaribacter group 5 (17) 4 (6) 1 (1) � � �Cellulophaga-Psychroserpens group 8 (18) 2 (2) � � �
Spirochaetales Spirochaeta 2 (2) 3 (6) � � �Green sulfur bacteria 2 (2) 3 (5) � � �Planctomycetales BD2-16 group 2 (2) 13 (13) 13 (13) � � �
Pirellula group 2 (2) 1 (1) 10 (14) � � �ANAMMOX group 1 (1) 2 (2) 1 (1) � � �
Verrucomicrobia BD2-18 group 3 (4) 1 (1) � � �Verrucomicrobium group 3 (6) 2 (2) � � �
Actinobacteria JTB31/sva0389 group 2 (11) 3 (5) 6 (17) � � �Gram positive PAUC43f/BD2-11 group 2 (2) 4 (5) � � �
ACE-43 group 2 (4) 1 (2) � � �BURTON-30 group 1 (1) 1 (4) � � �
Green nonsulfur “Dehalococcoides” group 4 (4) 1 (1) � � �BD3-16/PENDANT-37 group 2 (2) 8 (8) 10 (16) � � �
OP11 groupe 7 (7) 13 (14) � � �
ArchaeaEuryarchaeota pBRKC84 group 1 (1) 1 (4) � �
ACE-6 group 1 (1) 1 (2) 1 (1) � �PENDANT-33 group 1 (1) 3 (4) � �
Crenarchaeota APA3-11 cm group 2 (4) 2 (4) � �Marine group I 5 (67) 2 (15) 2 (5) � �
a Phylotype groups correspond to clusters of similar 16S rDNAs shown in the phylogenetic trees in Fig. 1 to 12.b Data are from references 30 and 31. Data are combined from several small clone libraries.b Data are from reference 39. No data for archaea are available because bacterium-specific primers were used.d Data are from Powell et al. (unpublished). Data are combined from two clone libraries derived from pristine and polluted sediments.e The OP11 group is not a single-phylotype group, it was included to show clonal abundance differences with sediment depth.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2465
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

21-cm libraries, respectively, and comprising 16 major phylo-type groups (Table 1). Several of these phylotype groups cor-responded to chemoheterotrophic species and genera inhabit-ing cold marine ecosystems (sea ice, seawater, or sediment),including members of Photobacterium, Moritella, Psychromo-nas, Shewanella, Pseudoalteromonas, Colwellia, Pseudomonas,Halomonas, and Oceanospirillum (Fig. 1). A large propor-tion of gamma proteobacterial phylotypes detected, how-ever, grouped into six clusters distinct from cultured species(Fig. 1 and 2). These groups included only clones detectedpreviously in marine sediment samples, from both coastaland deep-sea sites. The largest group (BD3-6/JTB255 group)branched deeply within the gamma subclass and included thesingle highest concentration of phylotypes and clones foundin the core samples (9% of total clones, making up 38 phylo-types). Two groups (BD7-8 and JTB148/Sva0091 groups)formed distinct lineages among predominantly phototrophic(families Chromatiaceae and Ectothiorhodospiraceae) and free-living and endosymbiotic sulfur oxidizers (Fig. 2). Two groups(B2M60 and AWS98-7e) include bacteria detected in sedimentcolonized by seagrass (12) and in seawater (15). A single phy-lotype group grouped in the beta proteobacteria and wasclosely related to the ammonia-oxidizing species Nitrosospirabriensis (Fig. 2).
Delta and epsilon proteobacteria. Levels of clone abundanceindicated that delta and epsilon proteobacteria exhibited dis-tinct zonation within MGP sediment, which concurred withRNA hybridization data (5). At the 0-cm horizon, 4% of clonesgrouped within the delta proteobacteria, but the abundanceincreased to 16 to 21% in the deeper samples. Most of thedelta proteobacterial clones fell into seven phylotype groups(Table 1), the most significant of which was allied with thegenus Desulfosarcina and relatives (6% of total clones) (Fig. 3).This extensive cluster included several clones previously de-tected in Antarctic lake sediment, Arctic fjord sediment, andmethane seeps. Several significant phylotype groups clusteredwith various sulfate-reducing bacterial species or groups, in-cluding Desulfobacterium anilini and the family Desulfobul-baceae (Fig. 4). Some clones grouped among sulfur- and iron-reducing members of the Geobacteraceae, including the generaPelobacter and Desulfuromusa. Several clones also groupedamong the Myxobacteria group, which from the studies of San-ford et al. (43) includes strains capable of anaerobic growth(Fig. 4). Three distinct clusters (the Eel-TE1A4, BD1-2/Sva0103, and JTB38/Clear-9 groups) made up only of cloneswere also found. These groups contained clones previouslyfound in a variety of other marine and marine-derived sedi-ments (Fig. 4).
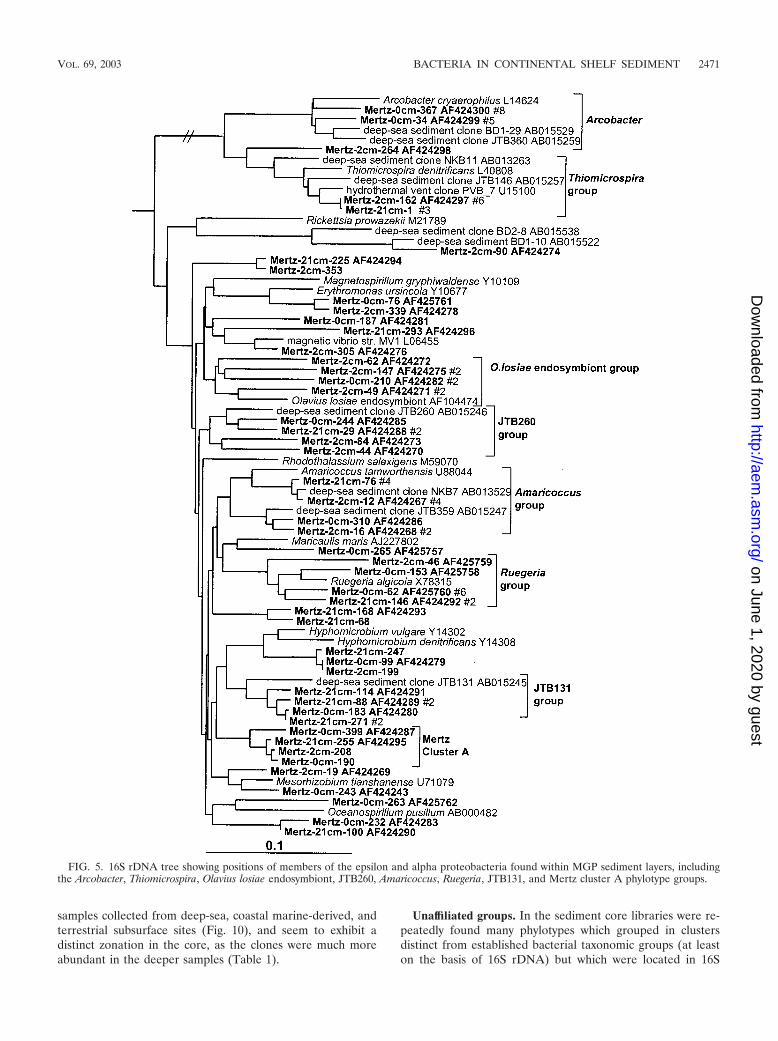
Clear zonation of epsilon proteobacteria was also evident inthe MGP sediment core (Table 1; Fig. 5), with the surface 0-cmlibrary containing several clones affiliated with the genusArcobacter. The Arcobacter group was absent in the deeperlibraries, which instead contained a group closely allied to thesulfur-reducing species Thiomicrospira denitrificans (Fig. 2).Both groups also contained a number of clones derived from avariety of deep-sea and hydrothermal sediments.
Alpha proteobacteria. The clone abundance of alpha pro-teobacteria was evenly distributed throughout the core librar-ies (6% of total clones). The biodiversity was considerable, andonly small phylotype groups were evident (Fig. 5). Three phy-
lotype groups were made up only of clones, two of whichincluded deep-sea clones (JTB131 and JTB260 groups) andanother group which was so far unique to this study (Mertzcluster A) (Fig. 5). Some phylotypes grouped near nonmarinegenera (e.g., Mesorhizobium, Amaricoccus, and Hyphomicro-bium), although the phylogenetic distances (0.05 to 0.10) sug-gest that the phylotypes represent different genera.
Acidobacteria and relatives. In this study three distinct clus-ters of Acidobacteria (Fig. 6) were detected in the anoxic por-tions of the core; none were detected in the surface 0-cmlibrary (Table 1). The most significant group (NKB17/Sva0450group) was tightly associated with other clones found in thedeep sea, a coastal Arctic fjord sample, and anoxic sedimentof a brackish Antarctic lake (Fig. 6). Mertz clones also formedtwo other smaller groups, one of which included the Arcticfjord clone sva0515. Several clones were also associated withthe Nitrospira group (Fig. 6). Within this group one clonebranched with deep-sea clone BD2-6 (30) and coastal sedimentclone KSA81 (referred to as candidate division KS-B [34]).Several phylotypes detected below the surface layer belongedto the SAR406 group (Fig. 6), which has been previously de-tected in a wide range of seawater samples (15), including theSouthern Ocean (33).
Flavobacteria, green sulfur bacteria, and spirochetes. Fla-vobacteria displayed distinct zonation throughout the sedimentcore as indicated by the distribution of phylotypes in the dif-ferent libraries as shown in Table 1. Within the 0-cm library, 45out of 48 Flavobacteria clones grouped within the family Fla-vobacteriaceae concentrated into three phylotype groups rep-resented by various marine aerobic and heterotrophic genera(Fig. 7). The incidence of Flavobacteriaceae declined with thedepth of the sample, and they were practically absent in the21-cm library. In the deeper samples several phylotype groupsassociated with predominantly anaerobic and facultativelyaerobic heterotrophic taxa of the Cytophaga fermentans group(family “Rikellanaceae”) were evident (Fig. 7), which includedseveral clones detected previously in deep-sea sediments (30).Another, more deeply branching lineage associated with deep-sea clones (JTB248/NB1-m group) included clones detectedthroughout the sediment core (Fig. 7). Two clone groups werefound to branch within the green sulfur bacteria (Fig. 7), bothof which are far removed (distance, 0.16 to 0.19) from thegenus Chlorobium and its phototrophic relatives and thuscould represent nonphototrophic members of this group. Sev-eral clones associated with the chemoheterotrophic and anaer-obic genus Spirochaeta were found in the two deeper clonelibraries (Fig. 7).
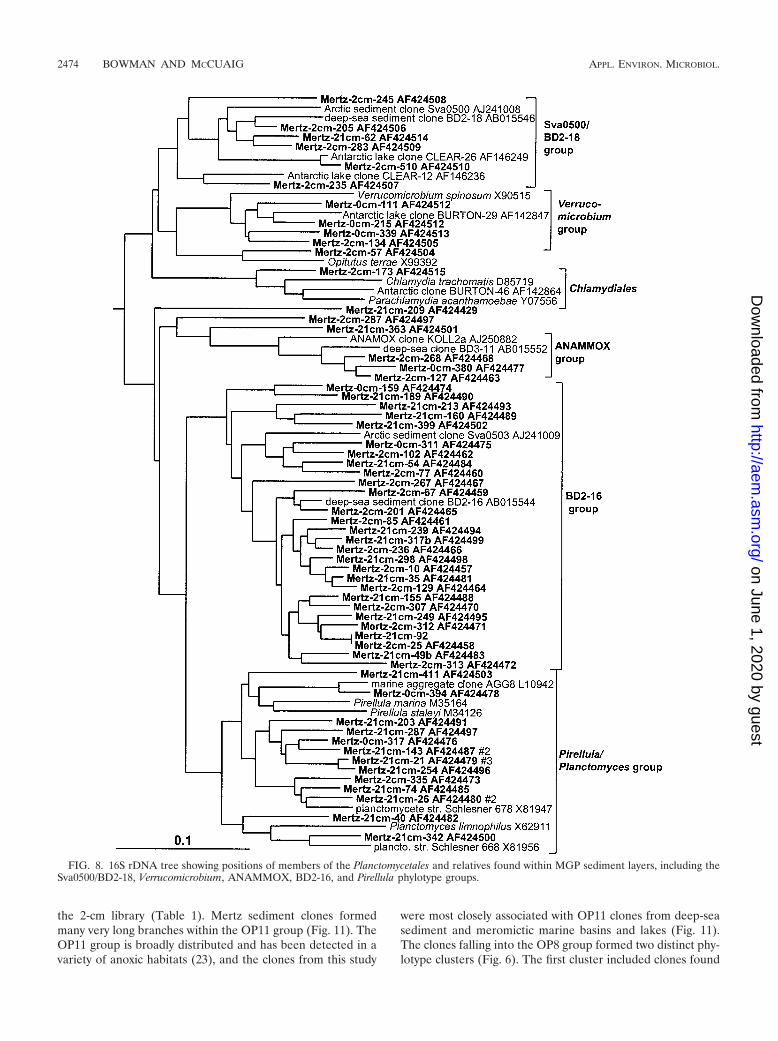
Planctomycetales and relatives. The Planctomycetales weresurprisingly diverse in the sediment core, with most of thephylotypes represented by only a single clone and formingthree broad groups (Fig. 8). The first large group consistedonly of clones, represented by deep-sea clone BD2-16 and theArctic fjord clone sva0503 (Fig. 8). The BD2-16 group domi-nated Planctomycetales in the deeper clone libraries and waspractically absent in the surface 0-cm library (Table 1). Asecond group, associated with the marine heterotrophic genusPirellula and the species Planctomyces limnophilus, was alsocommon in the 21-cm library but was overall more evenlydistributed (Table 1; Fig. 8). The third group was most closelyassociated with the anaerobic ammonia-oxidizing cluster
2466 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG. 1. 16S rDNA tree showing positions of mostly heterotrophic members of the gamma proteobacteria found within MGP sediment layers,including the AWS98-7e, B2M60, Oceanospirillum, Coxiella, Alteromonadaceae, and BPC036 phylotype groups.
2467
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG. 2. 16S rDNA tree showing positions of putatively sulfur-oxidizing and other members of the gamma proteobacteria and beta proteobac-teria found within MGP sediment layers, including the BD3-6/JTB255, Ectothiorhodospiraceae, BD7-8, and JTB148/sva0091 phylotype groups.
2468
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

FIG. 3. 16S rDNA tree showing positions of delta proteobacteria found within MGP sediment layers, including the JTB38/Clear-9, Myxobac-teria, Desulfobacterium anilini, Desulfuromonas, Eel-TE1A4, BD1-2/sva0103, and Desulfobulbaceae phylotype groups.
2469
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

(ANAMMOX group) previously identified in wastewater sys-tems and other sites (50). Mertz sediment clones falling intothe Verrucomicrobia formed two main clusters. The first groupcontained clones mostly from the surface layer and includedthe aerobic oligotrophic and chemoheterotrophic genera Ver-rucomicrobium and Prosthecobacter. The second group con-tained only clones from anoxic deeper horizons of the core.This second group included clones from the deep sea as well ascoastal sediments.
Gram-positive bacteria. The Mertz sediment core containedan important phylotype group (3.8% of total clones) whichformed a deep, distinct branch within the Actinobacteria (Fig.9). This particular group also contained many sediment clonespreviously detected in deep-sea and coastal samples. Membersof the low-G�C gram-positive bacteria (Bacillus-Clostridiumgroup), by comparison, were much less abundant (Fig. 9).Three phylotype groups, represented by clones ACE-43, BUR-
TON-30 and BD2-10, were also found, which form distinctrRNA branches (Fig. 9) and which according to bootstrap datado not appear to be definitively associated with any of themajor gram-positive clades. The BURTON-30 group was pre-viously found to make up about one-third of clones detected inAntarctic meromictic lake and anoxic marine basin sediments(6). The lineage represented by deep-sea clone BD2-10 in-cluded several 2- and 21-cm library clones. This group wasreferred to as candidate division KS-A in a study of coastalsediment off French Guiana (25).
Green nonsulfur bacteria. Clones grouped into two distinctclusters among the green nonsulfur bacteria (Fig. 10). Thesmaller group was associated with the anaerobe “Dehalococ-coides ethenogenes” along with several clones found in a varietyof marine, freshwater, and subsurface sediments (Fig. 10). Thesecond larger group (BD3-16/PENDANT-37 group) includedonly cloned sequences, mostly deriving from anoxic sediment
FIG. 4. 16S rDNA tree showing positions of members of the Desulfosarcina group and of the genus Desulfobacula found within MGP sedimentlayers.
2470 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
samples collected from deep-sea, coastal marine-derived, andterrestrial subsurface sites (Fig. 10), and seem to exhibit adistinct zonation in the core, as the clones were much moreabundant in the deeper samples (Table 1).
Unaffiliated groups. In the sediment core libraries were re-peatedly found many phylotypes which grouped in clustersdistinct from established bacterial taxonomic groups (at leaston the basis of 16S rDNA) but which were located in 16S
FIG. 5. 16S rDNA tree showing positions of members of the epsilon and alpha proteobacteria found within MGP sediment layers, includingthe Arcobacter, Thiomicrospira, Olavius losiae endosymbiont, JTB260, Amaricoccus, Ruegeria, JTB131, and Mertz cluster A phylotype groups.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2471
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

rDNA clone groups established in other studies in which theywere designated candidate divisions; these are referred to inthis study simply as groups, as important aspects of their tax-onomy have yet to be established (8). In this study severalrepresentatives of the OP3 (Fig. 6), OP8 (Fig. 6), and OP11
(Fig. 11) groups were found. The OP series designation is fromOpal Pool, a hot spring in Yellowstone National Park, in whichthey were first discovered (23). These groups were stronglyzonated in the sediment core, absent from the surface 0-cmlibrary, and more common in the 21-cm library compared to
FIG. 6. 16S rDNA tree showing positions of members of Acidobacteria and related lineages found within MGP sediment layers, including theNKB18 (OP8 group), SAR406, and NKB17/Sva0450 phylotype groups.
2472 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
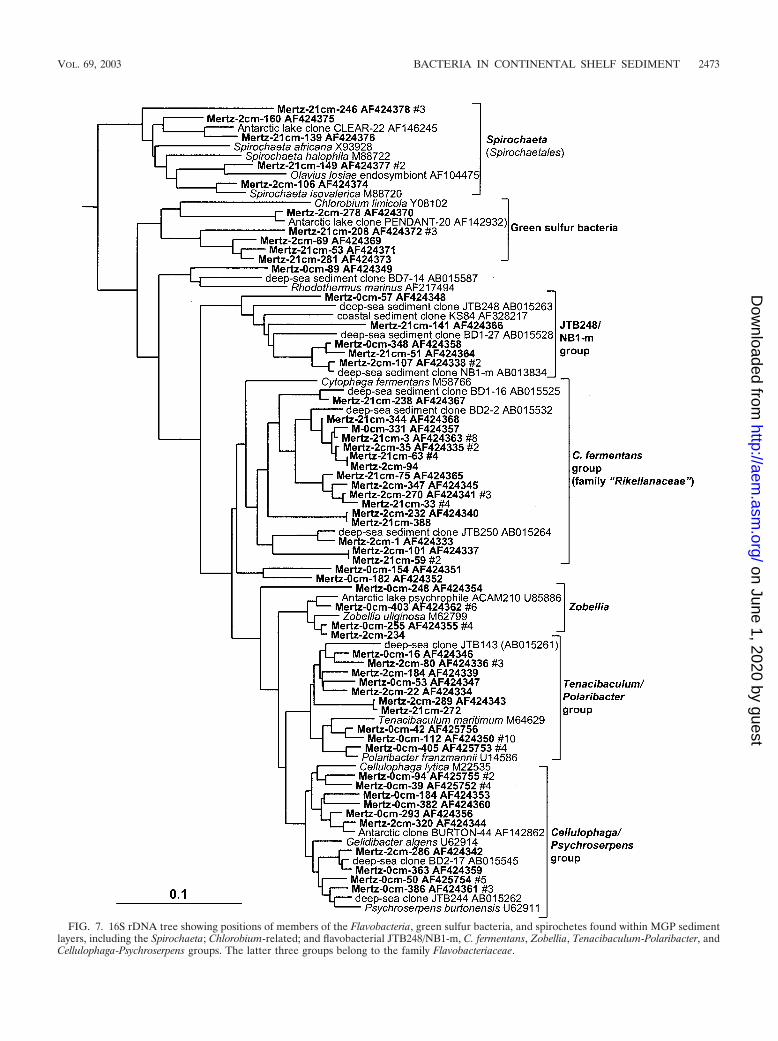
FIG. 7. 16S rDNA tree showing positions of members of the Flavobacteria, green sulfur bacteria, and spirochetes found within MGP sedimentlayers, including the Spirochaeta; Chlorobium-related; and flavobacterial JTB248/NB1-m, C. fermentans, Zobellia, Tenacibaculum-Polaribacter, andCellulophaga-Psychroserpens groups. The latter three groups belong to the family Flavobacteriaceae.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2473
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
the 2-cm library (Table 1). Mertz sediment clones formedmany very long branches within the OP11 group (Fig. 11). TheOP11 group is broadly distributed and has been detected in avariety of anoxic habitats (23), and the clones from this study
were most closely associated with OP11 clones from deep-seasediment and meromictic marine basins and lakes (Fig. 11).The clones falling into the OP8 group formed two distinct phy-lotype clusters (Fig. 6). The first cluster included clones found
FIG. 8. 16S rDNA tree showing positions of members of the Planctomycetales and relatives found within MGP sediment layers, including theSva0500/BD2-18, Verrucomicrobium, ANAMMOX, BD2-16, and Pirellula phylotype groups.
2474 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

in deep-sea environments (represented by clone NKB18), whilethe second, smaller cluster contained a clone previouslydetected in an iron-rich terrestrial subsurface sediment (Fig.6).
Archaea. Marine group I (Crenarchaeota) predominated inthe sediment core libraries, particularly in the surface 0-cmlibrary (Table 1; Fig. 12), where it contributed about 18% ofclones. A single abundant marine group I phylotype (compris-ing 79% of group I clones) was practically identical (0 to 3nucleotide differences) to hydrothermal vent clone pMC1A11(53) and was detected in all libraries. In the deeper samplesarchaeal diversity was considerably higher, with five euryar-chaeotal and four other crenarchaeotal lineages found (Table1). The CRA8-27cm/BBA6 phylotype cluster was the next mostimportant crenarchaeotal group and includes clones previouslydetected in deep-sea and shelf sediments (57, 58) (Fig. 12).Euryarchaeotal phylotypes likewise grouped with clones previ-
ously found in different sediments. Clones mostly grouped intothree long-branched lineages remote from cultivated taxa.These included an unusually deep clone group represented bybrackish sediment clone pBRKC84 (S. C. Dawson and N. R.Pace, unpublished data) and two saline Antarctic lake clonegroups (6) represented by ACE-6 and PENDANT-33 (Fig. 12).The latter groups have also been detected in a variety ofanaerobic environments, including freshwater lake sediment,subsurface sediment and waters, rice paddy soil, and hydro-thermal vents (data not shown). Only one clone each wasfound to group with marine groups II and III (Fig. 12), with themarine group III clone grouping closely with ocean sedimentclones (e.g., CRA12-27cm [57]).
MGP sediment core clone library comparisons. As the clonelibraries were created simultaneously under nearly identicalconditions, there was the opportunity for good comparativebiodiversity analysis of MGP sediment layers by using statisti-
FIG. 9. 16S rDNA tree showing positions of members of the gram-positive bacteria found within MGP sediment layers, including theJTB31/sva0389, PAUC43f/BD2-11, ACE-43, and BURTON-30 phylotype groups.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2475
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

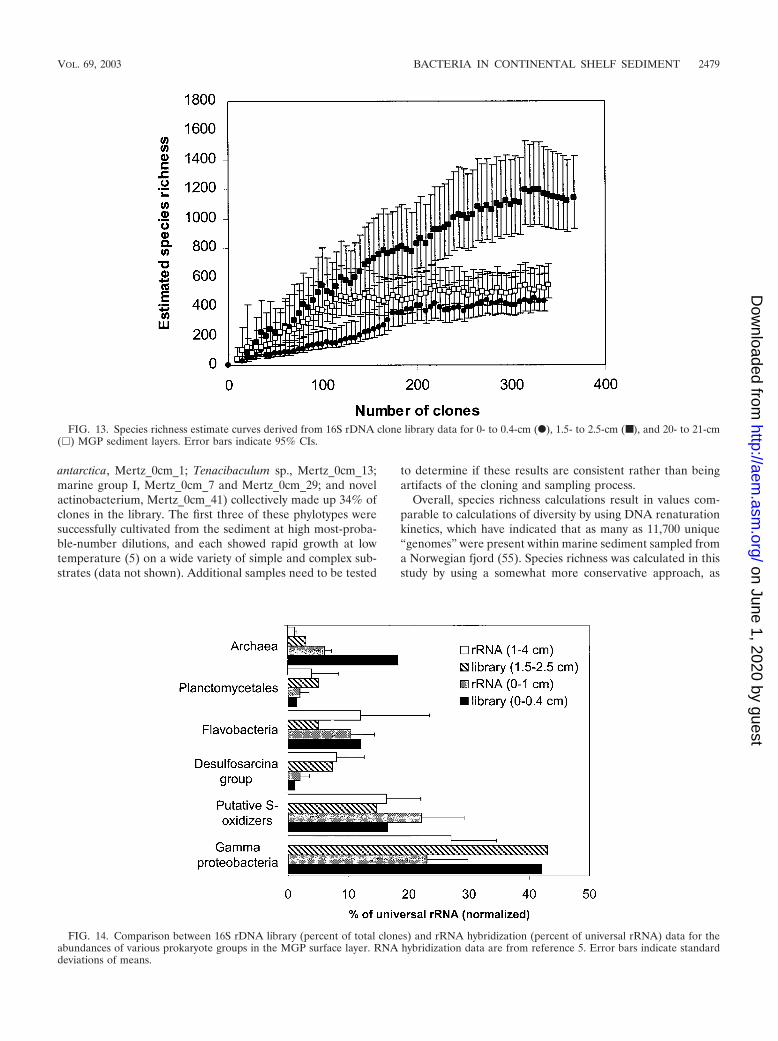
cal analyses to extrapolate species richness (24) and to quantifysimilarities or differences between the clone libraries (44). Therelatively large size of the clone libraries makes species rich-ness valid, as the species richness curves derived for eachlibrary (Fig. 13) approach an asymptote, which indicates that afinal tally of the community members can be reliably estimated.Although they are useful in this study for intersample compar-ison, such comparisons cannot be made with other 16S rDNAclone library data sets owing to the different methodologicalstrategies employed in creating them. In any case, most pub-lished data sets are too small to provide accurate values; i.e.,most obtain only a nonasymptotic species richness curve, whichmay result in serious underestimates of species diversity. Theestimated number of species was progressively compared toclone numbers in the order in which they were sampled in thelibrary. The highest estimated number of species was clearlypresent in the 2-cm-deep sediment sample (Fig. 13), whichcontained 1,128 OTUs (95% CIs, 929 and 1,426). As this spe-cies richness curve does not clearly complete an asymptote, itis possible that the species richness has been underestimated.By comparison, the 0-cm (442 OTUs [95% CIs, 370 and 622])and the 21-cm (541 OTUs [95% CIs, 455 and 691]) samplespossessed similar levels of diversity, with the respective curvesclosely approaching if not fully completing an asymptotic curve(Fig. 13).
Comparisons of homologous and heterologous coveragecurves (see reference 44 for details) between the 2-cm libraryand the 0- and 21-cm libraries resulted in high P values (P �0.69 and 0.81 [P values of �0.05 indicate that clone libraries
are significantly different]), suggesting that the 2-cm librarycontained many phylotypes in common with both the 0- and21-cm libraries. However, the reverse heterologous compari-sons produced only low P values (P � 0.08 and 0.12), whichsuggest that the 2-cm library has many sequences not presentin either of the other libraries. This result supports the speciesrichness estimations and clearly indicates that at the 2-cmdepth both surface and deeper core community members mix,resulting in a community with high taxonomic diversity andpresumably high functional diversity. The 0- and 21-cm librar-ies, when compared, were not observed as statistically different(P � 0.15 and 0.22), which may be a reflection of severalcommon phylotypes that occur throughout the core.
DISCUSSION
Methodological comparisons. MGP sediment cores librariescan be validated to some extent with rRNA hybridization data(5), in which the relative abundances of gamma proteobacteria,putative sulfide-oxidizing bacteria (phylotype clusters BD7-8,JTB255/BD3-6, and sva0091 [Fig. 2]), the Desulfosarcina group,Flavobacteria, Archaea, gram-positive bacteria, and Planctomy-cetales were estimated. The variation that was found betweenclone library and RNA hybridization data perhaps indicatesthe extent of PCR bias distortion inherent in the clone librar-ies. This is most obvious for the archaeal abundance in thesurface layer sample, which appeared to be overestimatedthreefold in the clone library analysis (Fig. 14). A positivecorrelation (�0.76) was found between the rRNA hybridiza-
FIG. 10. 16S rDNA tree showing positions of members of the green non-sulfur bacteria found within MGP sediment layers, including the“Dehalococcoides” and BD3-16/PENDANT-37 phylotype groups.
2476 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

tion values and clone library abundances when the data werepooled, although the calculated regression was not robust (r2 �0.57). Our data support the initial finding that sulfur- or sul-fate-respiring bacteria dominate the MGP sediments. rRNAprobe hybridization abundance data indicated that 16 to 27%and 2 to 9% of the community RNA (5) bound to a sulfur-oxidizer specific probe (GAM660 probe [38]) and to a probespecific to the Desulfosarcina group, respectively. Clones thathave sequences complementary to the probes were also sam-pled frequently (14 to 15% and 4 to 7%, respectively) in sam-ples of the same depth. Although the GAM660 probe suggeststhat the gamma proteobacterial phylotype groups could beinvolved in S oxidation, further cultivation analysis is required,as functionality is not always conserved between phylogeneticrelatives. The information, however, provides a hypothesis inwhich cultivation approaches could be initially tested to obtainpure cultures of what appear to be abundant uncultivatedmembers of the gamma proteobacteria.
Diversity and phylogenetic comparisons. Three areas of thecore were analyzed in an attempt to understand communitystructures within the benthos. The 0-cm library, for example,
was created to assess populations within the oxic to hypoxicsurface zone of the core. Previous PCR-DGGE analysis ofseveral MGP sediment cores indicated that the bacterial com-munity appeared to be relatively homogenous below the oxiclayer down to a depth of about 4 cm. Within this depth rangethe 2-cm level was selected, as it possessed a high level ofmetabolic activity (5). The 21-cm core section was analyzedbecause it exhibited significantly reduced metabolic activityand biomass and had distinctly different DGGE patterns com-pared to the surface layers (5). Since all three libraries werecreated at the same time, the differences in abundance foundbetween different 16S rDNAs (with PCR and cloning artifacts[48, 59] being similar) could be considered qualitatively reli-able when making comparisons between the MGP samples.The diversity within the Mertz cores was estimated to be high-est within the 2-cm layer, approximately two- to threefoldhigher than that in the other layers analyzed (Fig. 13). Thishigher level of diversity may arise from active anaerobic de-composition and transformation of deposited organic matterwhich is encouraged by faunal bioturbation and current mixingof sediment, replenishing nutrients and energy sources (47).
FIG. 11. 16S rDNA tree showing positions of members of the OP11 group found within MGP sediment layers, including known referenceclones from terrestrial and marine sediments.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2477
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

This is suggested by the fact that rates of sulfate and ironreduction, primary energy-yielding processes, appear to behighly active in marine sediment within this depth range (42).The reduced diversity observed at the 21-cm-deep layer may bedue to much lower nutrient availability and thus a greaterreliance on low-energy-yielding metabolic processes, as onlypoorer quality substrates are available (21). This occurrencecould be a good example of the energy limitation hypothesis,which posits that the amount of energy available to an ecosys-tem limits the species richness by limiting the density of itsindividual taxa (26, 37). As biomass levels at the 21-cm depthwere only 20 to 30% of those at the 2-cm depth, the hypothesishas validity in describing the reduced benthic microbial com-
munity species richness below the mixed surficial sedimentlayer. The hypothesis, however, completely fails to describe thelower diversity encountered within the oxic surface layer, whichwould be expected to have a ready supply of labile carbon andenergy sources. It is possible that the surface community struc-ture may be inhabited by various rapidly growing, nutritionallyversatile, aerobic and facultatively anaerobic species (i.e.,r strategists). This occurrence is indicated by the species rich-ness curve (Fig. 13), which increased only gradually as thepopulation was initially sampled, suggesting that a subset ofdominant species with high levels of catchability (9) was pres-ent in the sample; indeed, the six most common phylotypes(Shewanella gelidimarina, Mertz_0cm_14; Pseudoalteromonas
FIG. 12. 16S rDNA tree showing positions of members of the Archaebacteria found within MGP sediment layers, including the pBRC84,ACE-6, PENDANT-33, CRA8-27cm, and marine group I phylotype groups.
2478 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from
antarctica, Mertz_0cm_1; Tenacibaculum sp., Mertz_0cm_13;marine group I, Mertz_0cm_7 and Mertz_0cm_29; and novelactinobacterium, Mertz_0cm_41) collectively made up 34% ofclones in the library. The first three of these phylotypes weresuccessfully cultivated from the sediment at high most-proba-ble-number dilutions, and each showed rapid growth at lowtemperature (5) on a wide variety of simple and complex sub-strates (data not shown). Additional samples need to be tested
to determine if these results are consistent rather than beingartifacts of the cloning and sampling process.
Overall, species richness calculations result in values com-parable to calculations of diversity by using DNA renaturationkinetics, which have indicated that as many as 11,700 unique“genomes” were present within marine sediment sampled froma Norwegian fjord (55). Species richness was calculated in thisstudy by using a somewhat more conservative approach, as
FIG. 13. Species richness estimate curves derived from 16S rDNA clone library data for 0- to 0.4-cm (F), 1.5- to 2.5-cm (■), and 20- to 21-cm(�) MGP sediment layers. Error bars indicate 95% CIs.
FIG. 14. Comparison between 16S rDNA library (percent of total clones) and rRNA hybridization (percent of universal rRNA) data for theabundances of various prokaryote groups in the MGP surface layer. RNA hybridization data are from reference 5. Error bars indicate standarddeviations of means.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2479
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

each phylotype may actually represent a cluster of closely re-lated species that share highly similar 16S rDNA sequences.However, the combined species richness determined by pool-ing all clones for the MGP sediment core was calculated at4,350 (95% CIs, 3,715 and 5,594), which indicates that evenwith restricted sampling a potentially large proportion of thediversity present can be experimentally estimated.
Community structural shifts within the MGP core. The dif-ference in community structure within the Mertz core was moststark between the 0- and 21-cm libraries, with 27 bacterial andarchaeal groups found in either library but not in both (Table1). Only three groups were found to be present in the 0- and21-cm libraries and were not detected in the 2-cm library,suggesting that these particular groups may be spread at levelsthroughout the core at or below the resolution limits of thelibrary analysis. In the surface 0-cm library, several predomi-nant phylotype groups were found (Table 1), including thefamily Alteromonadaceae (Shewanella, Moritella, Pseudoaltero-monas, and Colwellia), the Arcobacter group, Nitrosospira, theRuegeria group, the family Flavobacteriaceae, the Verrucomi-crobium group, the actinobacterium JTB31/sva0389 group, andmarine group I Crenarchaeota. These groups are all oxygenrequiring or are closely related to aerobic taxa (as far as can beshown by phylogenetic comparisons), which have been isolatedor detected within the pelagic zone, sea ice, or surficial sedi-ment. For example, the genus Arcobacter and members ofthe Flavobacteria were found to be very abundant in Wad-den Sea surface sediment (32). The clone abundance of themajor 0-cm-depth taxa appear to decline with depth, with mostnot detected at a depth of 21 cm. The most abundant surfaceclone group, marine group I, was an exception, as it was foundthroughout the sediment core. This group has been shown torepresent a major fraction of mesopelagic bacterioplankton(25) and is also common in deep-sea sediment (57). Hetero-trophic aerobic and facultatively aerobic species found to dom-inate the surface sediment community belong to groups knownto have strong capacities for degradation of complex organicmatter (proteins and polysaccharide). For example, membersof the families Alteromonadaceae and Flavobacteriaceae, whichtogether constituted 34% of 0-cm library clones, have beenshown to be important degraders of seawater dissolved organiccarbon, have been found to be highly abundant in the SouthernOcean south of the Polar Front, and occur as the dominatebacterial populations in sea-ice algal assemblages (7, 28).
The differences between microbial communities in anoxic(2- and 21-cm libraries) and mostly oxic (0-cm library) sedi-ment layers were quite evident. For example, the clonal abun-dance of delta proteobacteria increased fivefold between the 0-and 2-cm libraries, and many clone groups (OP8 and OP11groups, SAR406 group, and ACE-43 group, etc.) (Table 1)were not detected within the 0-cm library. Previous DGGEanalysis of the sediment core suggested that distinct but grad-ual community shifts occurred below 4 cm (5). With clonelibrary analysis and its much higher level of resolution, fewdistinct gross differences between the 2- and 21-cm sampleson the phylotype group level are obvious (Table 1); however,changes in clonal abundance were obvious across many phylo-type groups. Apparent depth-related increases in clonal abun-dance occurred for the genus Psychromonas, the JTB131 group(alpha proteobacteria), the family “Rikenellaceae,” Spiro-
chaeta, Chlorobium-related groups, the JTB31/sva0389 group(Actinobacteria), the Pirellula group, the OP8 and OP11 groups,and some groups of the Euryachaeota (Table 1). The phylo-types within this list, although clustering mostly within uncul-tured lineages, appear to be associated only with anoxic con-ditions, as they were generally not detected in the 0- to 0.4-cmlayer. It is possible that the species distribution within thevarious phylotype groups changes substantially between the 2-and 21-cm depths. However, an insufficient number of cloneswere analyzed to be able discriminate differences reliably at thespecies level. At least 103 to 104 clones, as suggested by nulltheoretical models of soil bacterial diversity (14), and moresediment sampling would be required to be analyzed to ob-serve individual phylotype distributions within the MGP sedi-ments.
Biogeographical comparisons. Biogeography as applied toprokaryotic distribution in the natural environment is still anundeveloped area. Staley and Gosink (49) suggested a logicalmodel for biogeographical analysis which involves isolation ofseveral strains from different sites with confirmation of speciessimilarity by using molecular approaches such as DNA-DNAhybridization. For many environments the difficulties involvedin cultivation make it possible to make comparisons only athigher taxonomic levels for most prokaryotes.
Although the microbial community structure in marine eco-systems varies at the microscale, owing to variable nutrientdistribution (2), the same marine prokaryotic species and gen-era inhabit ecologically similar niches separated over widegeographical scales (18, 35, 62), with populations shifting inspace and time depending on environmental fluctuations. Eventhough various physiological characteristics are not necessarilyconserved within the phylogenetic groups found in this study(Table 1), the environmental conditions and energy sourcesavailable would tend not only to dictate microbial communitycomposition and species richness but also to constrain thephysiological characteristics of the community members. In thecase of the MGP sediments, most community members mustbe cold adapted and generally able to grow anaerobically andsubsist on exported primary produced carbon that is in variousstages of decomposition. As much benthic life exists underessentially the same conditions as those of the MGP sediments(hypoxia, low temperature, sulfidogenesis, and faunal mixingof sediment, etc.), it would be expected that as for seawater,dominant groups of prokaryotes are broadly represented inmarine sediment. Distributional comparisons of prokaryotes(1, 11, 22, 61) have only recently been possible as the 16SrDNA database has expanded sufficiently to cover most globalprokaryote diversity. In this study, for example, although about500 phylotypes were found, only a small minority representedtaxonomically novel groups of bacteria (similarity to GenBanksequences of �0.90), suggesting that a large proportion ofclass-and higher-level prokaryotic lineages may already havebeen discovered, at least in the marine environment.
To determine the distribution of prokaryote groups detectedin this survey, comparisons were made between the MGP li-braries and libraries derived from other sediment samples col-lected from coastal, deep-sea, and marine locations. Phylotypecomparisons (Table 2) were made at approximately the equiv-alents of intrageneric (�0.02) and intergeneric (�0.05) levels.At 16S rDNA dissimilarities of �0.02, genomic hybridization
2480 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

levels are often quite high, sometimes above the point whichdenotes prokaryote species boundaries (60 to 70%) and wellwithin generic boundaries; however, owing to quantum evolu-tion of 16S rDNAs (8), it is generally not possible to makeaccurate species-level comparisons by using 16S rDNA dataalone. Regardless of this limitation, comparisons of this sortcan reveal and define potentially ubiquitous bacterial groups.
Compared to the combined MPG sediment phylotype list,the highest number of similar phylotypes (similarity at �0.02,17 to 32%; similarity at �0.05, 36 to 65%) was found in sedi-ment from Antarctic in-shore (S. M. Powell et al., unpublisheddata), Arctic fjord (39), and various deep-sea (30, 31) sites.Although ranging widely in depth, these sediments shared withMertz sediment similar low temperatures (�3°C) and a dom-inance of sulfidogenic processes as suggested by the high abun-dance of gamma, delta, and epsilon proteobacteria. The simi-larity was much lower (Table 2) in environments that havecharacteristics not typical of most oceanic sediment. TheFrench Guiana coastal sediments were nonsulfidogenic andsubject to very rapid mixing (34), while the Antarctic marine-derived meromictic lake and marine basin sediments werechemically stratified stagnant water bodies, sulfide rich butsulfate depleted, separated from the ocean for 8,000 to 10,000years (6). MGP sediments were also quite different from ex-tensively characterized Guaymas Basin sediments, which arethermally active, hydrocarbon rich, and exposed to high terri-genous input (54).
Similarities between the MGP sediments and other sedi-ments were found to coincide to a large degree in certaintaxonomic groups, especially the Proteobacteria (Table 1). Forexample, a large proportion of delta proteobacteria found inmethane-oxidizing sediments of the Eel River Basin (36) werequite similar to phylotypes in the MGP sediments (Fig. 3 and4). The apparent high population of many of the gamma anddelta proteobacteria in sediment, especially those involved insulfur cycling, appears to increase the probability of them be-ing detected in wide-ranging locations. On the other hand,certain phylogenetic groups do not show much if any similaritybetween different sediment samples (e.g., most clones of thePlanctomycetales and Verrucomicrobia and many of the un-affiliated taxa [e.g., OP11 group and Euryarchaeota]), eventhough numerous phylotypes were detected in these groups.This may indicate that the distribution of certain prokaryotes
is potentially endemic, with localized populations specificallyadapted to the idiosyncratic properties of the given environ-ment. Another equally likely interpretation is that these groupshave not been sufficiently sampled. Finlay (16) suggested thatthe high abundance, short generation time, small size, and highdispersal rates of prokaryotes makes them unlikely to be en-demic, as they are able to overcome large geographical barri-ers. Thus, only with extremely large clone libraries or, betterstill, by using a more specific targeting strategy (e.g., group-specific primers) will the actual distribution of rarer phylotypesbe ascertained.
Conclusions. This study examined closely the microbial com-munity in a specific location on the ocean seafloor, with thecommunity found to be very diverse and complex. The diversitywas almost equivalent to what has been found in terrestrialsoils (14). Local environmental conditions and cultivation stud-ies (5) indicate that the community is highly cold adapted, andthus psychrophilic adaptations are present within many bac-terial classes. Community structural shifts were evident be-tween the thin oxic layer and the anoxic zone below; however,changes within the anoxic zone were more subtle, with thesuggestion of different groups gradually become more dom-inant or, alternatively, declining with depth. The changes incommunity structure in the anoxic zone are possibly driven bynutrient availability, with lower-quality sources of carbon andenergy leading to the attrition of prokaryotic groups commonin the top layers. Comparisons made between the MGP sedi-ment library and clone library data obtained from other sedi-ments suggested that many prokaryote groups, taxons equiva-lent to the species to family level, were ubiquitously present indeep-ocean and trench sediments as well as coastal regions.(Table 1 and 2). Several prokaryote phylotype groups werefound to be broadly distributed in disparate marine sediments(Table 1) found both in shallow and deep waters. The paucityof data means that many groups have been detected in only afew sediment samples so far, and thus more samples need toanalyzed in detail before we can determine the true ubiquityand distribution of specific prokaryotic groups in the benthos.Further definition of major benthic prokaryotic groups will aidin the better understanding of microbial processes and inter-actions in marine sediment. Improved and more useful target-ing and quantification of these groups will be achieved withmolecular ecological techniques only now emerging.
TABLE 2. Comparison of individual Mertz sediment 16S rDNA phylotype distributions in sediment samples from other regions
Sedimentclone librarydesignationa
Sediment characteristics Location Depth(m)
No. ofphylotypescompared
% Similarity at:
�0.02 �0.05
OB3/BB2 Pristine and polluted (hydrocarbons, heavy metals) Brown Bay, O’Briens Bay, East Antarctica 15–25 124 35 65sva Sulfidogenic Svalbard, Arctic (Hornsund fjord) 155 69 32 61JTB Calyptogena colony, cold methane seep Japan Trench, Pacific Ocean 6,437 38 32 47Eelb Cold methane seep Eel River Basin, United States 500 17 29 65BD Deep-sea trough and trench surficial sediment Pacific Ocean trenches and troughs 1,200–7,400 58 17 36Guaymas Hydrothermally active, methane seep Guaymas Basin, Mexico 2,000 73 8 19BURTON Meromictic, seasonally mixed, marine basin Vestfold Hills, Antarctica 16 56 1 23ACE SO4-depleted, saline meromictic lake Vestfold Hills, Antarctica 21 45 9 18KSA Highly mobile, Fe reducing Coastal French Guiana 1 61 3 25
a MGP clone data were compared with those for other sediment clone libraries analyzed: BD (30), JTB (31), sva (39), OB3/BB2 (Powell et al., unpublished data),Eel (36), SA (56), BURTON and ACE (6), and KSA (34). DNA was extracted from sediment grabs, except for the sva (integrated DNA samples from sediment depthsof 0 to 2, 3 to 6, and 8 to 11 cm), Eel (3-cm layers extruded from push or piston cores down to 22 cm), and KSA (10- to 30-cm section of a 1.2-m core) libraries.
b Only delta proteobacterial clones were compared from the Eel library, as methanogens were not detected in MGP surficial sediment.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2481
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

ACKNOWLEDGMENTS
This research was supported by Australian Research Council largegrant A09905709 and by Antarctic Science Advisory Committee grants1012 and 1065.
We thank Lisette Robertson and Peter Harris for providing themarine sediment core that was analyzed in this study.
REFERENCES
1. Andrews, J. H., and R. F. Harris. 2000. The ecology and biogeography ofmicroorganisms of plant surfaces. Annu. Rev. Phytopathol. 38:145–180.
2. Azam, F., and R. A. Long. 2001. Oceanography—sea snow microcosms.Nature 414:495–497.
3. Bates, N. R., D. A. Hansell, C. A. Carlson, and L. I. Gordon. 1998. Distri-bution of CO2 species, estimates of net community production, and air-seaCO2 exchange in the Ross Sea polynya. J. Geophys. Res. Oceans 103:2883–2896.
4. Bindoff, N. L., G. D. Williams, and I. Allison. 2002. Sea-ice growth and watermass modification in the Mertz Glacier Polynya during winter. Ann. Glaciol.33:399–406.
5. Bowman, J. P., S. A. McCammon, J. A. E. Gibson, L. Robertson, and P. D.Nichols. 2003. Prokaryotic metabolic activity and community structure inAntarctic continental shelf sediment. Appl. Environ. Microbiol. 69:2448–2462.
6. Bowman, J. P., S. M. Rea, S. A. McCammon, and T. A. McMeekin. 2000.Diversity and community structure within anoxic sediment from marinesalinity meromictic lakes and a coastal meromictic marine basin, VestfoldHills, Eastern Antarctica. Environ. Microbiol. 2:227–237.
7. Brown, M. V., and J. P. Bowman. 2001. A molecular phylogenetic survey ofsea-ice microbial communities (SIMCO). FEMS Microbiol. Ecol. 35:267–275.
8. Cavalier-Smith, T. 2002. The neomuran origin of archaebacteria, the negi-bacterial root of the universal tree and bacterial megaclassification. Int. J.Syst. Evol. Microbiol. 52:7–76.
9. Chao, A. 1987. Estimating the population size for capture-recapture datawith unequal catchability. Biometrics 43:783–791.
10. Chao, A. 1984. Non-parametric estimation of the number of classes in apopulation. Scand. J. Stat. 11:265–270.
11. Cho, J. C., and J. M. Tiedje. 2000. Biogeography and degree of endemicityof fluorescent Pseudomonas strains in soil. Appl. Environ. Microbiol. 66:5448–5456.
12. Cifuentes, A., J. Anton, S. Benlloch, A. Donnelly, R. A. Herbert, and F.Rodriguez-Valera. 2000. Prokaryotic diversity in Zostera noltii-colonized ma-rine sediments. Appl. Environ. Microbiol. 66:1715–1719.
13. DiTullio, G. R., J. M. Grebmeier, K. R. Arrigo, M. P. Lizotte, D. H. Robin-son, A. Leventer, J. B. Barry, M. L. VanWoert, and R. B. Dunbar. 2000.Rapid and early export of Phaeocystis antarctica blooms in the Ross Sea,Antarctica. Nature 404:595–598.
14. Dunbar, J., S. M. Barns, L. O. Ticknor, and C. R. Kuske. 2002. Empiricaland theoretical bacterial diversity in four Arizona soils. Appl. Environ. Mi-crobiol. 68:3035–3045.
15. Eilers, H., J. Pernthaler, J. Peplies, F. O. Glockner, G. Gerdts, and R.Amann. 2001. Isolation of novel pelagic bacteria from the German Bight andtheir seasonal contributions to surface picoplankton. Appl. Environ. Micro-biol. 67:5134–5142.
16. Finlay, B. J. 2002. Global dispersal of free-living microbial eukaryote species.Science 296:1061–1063.
17. Gray, J. P., and R. P. Herwig. 1996. Phylogenetic analysis of the bacterialcommunities in marine sediments. Appl. Environ. Microbiol. 62:4049–4059.
18. Hagstrom, A., J. Pinhassi, and U. L. Zweifel. 2000. Biogeographical diversityamong marine bacterioplankton. Aquat. Microb. Ecol. 21:231–244.
19. Hall, T. A. 1999. A user-friendly biological sequence alignment editor forWindows 95/98/NT. Nucleic Acids Symp. Ser. 41:95–98.
20. Harris, P. T., G. Brancolini, L. Armand, M. Busetti, R. J. Beaman, G.Giorgetti, M. Presti, and F. Trincardi. 2001. Continental shelf drift depositindicates non-steady state Antarctic bottom water production in the Holo-cene. Mar. Geol. 179:1–8.
21. Hoehler, T. M., M. J. Alperin, D. B. Albert, and C. S. Martens. 2001.Apparent minimum free energy requirements for methanogenic Archaeaand sulfate-reducing bacteria in an anoxic marine sediment. FEMS Micro-biol. Ecol. 38:33–41.
22. Hollibaugh, J. T., N. Bano, and H. W. Ducklow. 2002. Widespread distribu-tion in polar oceans of a 16S rRNA gene sequence with affinity to Ni-trosospira-like ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 68:1478–1484.
23. Hugenholtz, P., B. M. Goebel, and N. R. Pace. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity.J. Bacteriol. 180:4765–4774.
24. Hughes, J. B., J. J. Hellmann, T. H. Ricketts, and B. J. M. Bohannan. 2001.Counting the uncountable: statistical approaches to estimating microbialdiversity. Appl. Environ. Microbiol. 67:4399–4406.
25. Karner, M. B., E. F. DeLong, and D. M. Karl. 2001. Archaeabacterial
dominance in the mesopelagic zone of the Pacific Ocean. Nature 409:507–510.
26. Kaspari, M., S. O’Donnell, and J. Kercher. 2000. Energy, density, andconstraints to species richness: ant assemblages along a productivity gradi-ent. Am. Nat. 155:280–293.
27. Keswani, J., and W. B. Whitman. 2001. Relationship of 16S rRNA sequencesimilarity to DNA hybridization in prokaryotes. Int. J. Syst. Evol. Microbiol.51:667–678.
28. Kirchman, D. L. 2002. The ecology of Cytophaga-Flavobacteria in aquaticenvironments. FEMS Microbiol. Ecol. 137:1–10.
29. Kroes, I., P. W. Lepp, and D. A. Relman. 1999. Bacterial diversity within thehuman subgingival crevice. Proc. Natl. Acad. Sci. USA 96:14547–14552.
30. Li, L., C. Kato, and K. Horikoshi. 1999. Bacterial diversity in deep-seasediments from different depths. Biodivers. Conserv. 8:659–677.
31. Li, L., C. Kato, and K. Horikoshi. 1999. Microbial diversity in sedimentscollected from the deepest cold-seep area, the Japan Trench. Mar. Biotech-nol. 1:391–400.
32. Llobet-Brossa, E., R. Rosello-Mora, and R. Amann. 1998. Microbial com-munity composition of Wadden Sea sediments as revealed by fluorescence insitu hybridization. Appl. Environ. Microbiol. 64:2691–2696.
33. Lopez-Garcia, P., A. Lopez-Lopez, D. Moreira, and F. Rodriguez-Valera.2001. Diversity of free-living prokaryotes from a deep-sea site at the Ant-arctic Polar Front. FEMS Microbiol. Ecol. 36:193–202.
34. Madrid, V. M., J. Y. Aller, R. C. Aller, and A. Y. Chistoserdov. 2001. Highprokaryote diversity and analysis of community structure in mobile muddeposits off French Guiana: identification of two new bacterial candidatedivisions. FEMS Microbiol. Ecol. 37:197–209.
35. Mullins, T. D., T. B. Britschgi, R. I. Krest, and S. J. Giovannoni. 1995.Genetic comparisons reveal the same unknown lineages in Atlantic andPacific bacterioplankton communities. Limnol. Oceanogr. 40:148–158.
36. Orphan, V. J., K.-U. Hinrichs, W. Ussler III, C. K. Paull, L. T. Taylor, S. P.Sylva, J. M. Hayes, and E. F. Delong. 2001. Comparative analysis of meth-ane-oxidizing Archaea and sulfate-reducing bacteria in anoxic marine sedi-ments. Appl. Environ. Microbiol. 67:1922–1934.
37. Paine, R. T. 1966. Food wed complexity and species diversity. Am. Nat.100:65–75.
38. Ravenschlag, K., K. Sahm, and R. Amann. 2001. Quantitative molecularanalysis of the microbial community in marine Arctic sediments (Svalbard).Appl. Environ. Microbiol. 67:387–395.
39. Ravenschlag, K., K. Sahm, J. Pernthaler, and R. Amann. 1999. High bacte-rial diversity in permanently cold marine sediments. Appl. Environ. Micro-biol. 65:3982–3989.
40. Rochelle, P. A., B. A. Cragg, J. C. Fry, R. J. Parkes, and A. J. Weightman.1994. Effect of sample handling on estimation of bacterial diversity in marinesediments by 16S rRNA gene sequence analysis. Bact. Genet. Ecol. 15:215–226.
41. Ruger, H.-J. 1989. Benthic studies of the northwest African upwelling region:psychrophilic and psychrotrophic bacterial communities from areas withdifferent upwelling intensities. Mar. Ecol. Prog. Ser. 57:45–52.
42. Rysgaard, S., B. Thamdrup, N. Risgaard-Petersen, H. Fossing, P. Berg, P. B.Christensen, and T. Dalsgaard. 1999. Seasonal carbon and nutrient miner-alization in a high-Arctic coastal marine sediment, Young Sound, NortheastGreenland. Mar. Ecol. Prog. Ser. 175:261–276.
43. Sanford, R. A., J. R. Cole, and J. M. Tiedje. 2002. Characterization anddescription of Anaeromyxobacter dehalogenans gen. nov., sp. nov., an aryl-halorespiring facultative anaerobic myxobacterium. Appl. Environ. Micro-biol. 68:893–900.
44. Singleton, D. A., M. A. Furlong, S. L. Rathbun, and W. B. Whitman. 2001.Quantitative comparisons of 16S rDNA gene sequence libraries from envi-ronmental samples. Appl. Environ. Microbiol. 67:4374–4376.
45. Small, J., R. C. Douglas, F. J. Brockman, T. M. Straub, and D. P. Chandler.2001. Direct detection of 16S rRNA in soil extracts by using oligonucleotidemicroarrays. Appl. Environ. Microbiol. 67:4708–4716.
46. Sohrin, Y., S. Iwamoto, M. Matsui, H. Obata, E. Nakayama, K. Suzuki, N.Handa, and M. Ishii. 2000. The distribution of Fe in the Australian sector ofthe Southern Ocean. Deep-Sea Res. I 47:55–84.
47. Soltwedel, T., and K. Vopel. 2001. Bacterial abundance and biomass inresponse to organism-generated habitat heterogeneity in deep-sea sedi-ments. Mar. Ecol. Prog. Ser. 219:291–298.
48. Speksnijder, A. G. C. L., G. A. Kowalchuk, S. De Jong, E. Kline, J. R.Stephen, and H. J. Laanbroek. 2001. Microvariation artifacts introduced byPCR and cloning of closely related 16S rRNA gene sequences. Appl. Envi-ron. Microbiol. 67:469–472.
49. Staley, J. T., and J. J. Gosink. 1999. Poles apart: biodiversity and biogeog-raphy of sea ice bacteria. Annu. Rev. Microbiol. 53:189–215.
50. Strous, M., J. A. Fuerst, E. H. Kramer, S. Logemann, G. Muyzer, K. T. vande Pas-Schoonen, R. Webb, J. G. Kuenen, and M. S. Jetten. 1999. Missinglithotroph identified as new planctomycete. Nature 400:446–449.
51. Strutton, P. G., F. B. Griffiths, R. L. Waters, S. W. Wright, and N. L. Bindoff.2000. Primary productivity off the coast of East Antarctica (80–150°E): Jan-uary to March 1996. Deep-Sea Res. Part II 47:2327–2362.
52. Suzuki, M. T., L. T. Taylor, and E. F. DeLong. 2000. Quantitative analysis of
2482 BOWMAN AND MCCUAIG APPL. ENVIRON. MICROBIOL.
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from

small-subunit rRNA genes in mixed microbial populations via 5�-nucleaseassays. Appl. Environ. Microbiol. 66:4605–4614.
53. Takai, K., and K. Horikoshi. 1999. Genetic diversity of archaea in deep-seahydrothermal vent environments. Genetics 152:1285–1297.
54. Teske, A., K. U. Hinrichs, V. Edgcomb, A. de Vera Gomez, D. Kysela, S. P.Sylva, M. L. Sogin, and H. W. Jannasch. 2002. Microbial diversity of hydro-thermal sediments in the Guaymas Basin: evidence for anaerobic meth-anotrophic communities. Appl. Environ. Microbiol. 68:1994–2007.
55. Torsvik, V., R. Sorheim, and J. Goksoyr. 1996. Total bacterial diversity insoil and sediment communities—a review. J. Indust. Microbiol. 17:170–178.
56. Urakawa, H., K. Kita-Tsukamoto, and K. Ohwada. 1999. Microbial diversityin marine sediments from Sagami Bay and Tokyo Bay, Japan, as determinedby 16S rRNA gene analysis. Microbiology 145:3305–3315.
57. Vetriani, C., H. W. Jannasch, B. J. MacGregor, D. A. Stahl, and A. L.Reysenbach. 1999. Population structure and phylogenetic characterization ofmarine benthic Archaea in deep-sea sediments. Appl. Environ. Microbiol.65:4375–4384.
58. Vetriani, C., A. L. Reysenbach, and J. Dore. 1998. Recovery and phylogeneticanalysis of archaeal rRNA sequences from continental shelf sediments.FEMS Microbiol. Lett. 161:83–88.
59. von Wintzingerode, F., U. B. Gobel, and E. Stackebrandt. 1997. Determina-tion of microbial diversity in environmental samples: pitfalls of PCR-basedrRNA analysis. FEMS Microbiol. Rev. 21:213–229.
60. Xiao, C., P. Zhou, and D. Wang. 1990. Identification and growth temperaturecharacteristics of psychrophiles from Antarctic sediments. Acta Microbiol.Sin. 30:239–242.
61. Zehr, J. P., and M. Voytek. 1999. Molecular ecology of aquatic communities:reflections and future directions. Hydrobiologia 401:1–8.
62. Zwart, G., W. D. Hiorns, B. A. Methe, M. P. Van Agterveld, R. Huismans,S. C. Nold, J. P. Zehr, and H. J. Laanbroek. 1998. Nearly identical 16S rRNAsequences recovered from lakes in North America and Europe indicate theexistence of clades of globally distributed freshwater bacteria. Syst. Appl.Microbiol. 21:546–556.
VOL. 69, 2003 BACTERIA IN CONTINENTAL SHELF SEDIMENT 2483
on June 1, 2020 by guesthttp://aem
.asm.org/
Dow
nloaded from