binding of the sphingolipid s1p to htert stabilizes ... · sk1-generated intra- cellular s1p binds...
TRANSCRIPT

R E S E A R C H A R T I C L E
C A N C E R
Binding of the sphingolipid S1P to hTERTstabilizes telomerase at the nuclear periphery byallosterically mimicking protein phosphorylationShanmugam Panneer Selvam,1,2 Ryan M. De Palma,1,2 Joshua J. Oaks,1,2 Natalia Oleinik,1,2
Yuri K. Peterson,2,3 Robert V. Stahelin,4,5 Emmanuel Skordalakes,6 Suriyan Ponnusamy,1,2
Elizabeth Garrett-Mayer,2 Charles D. Smith,2,3 Besim Ogretmen1,2*
http://stkD
ownloaded from
During DNA replication, the enzyme telomerase maintains the ends of chromosomes, called telomeres.Shortened telomeres trigger cell senescence, and cancer cells often have increased telomerase activityto promote their ability to proliferate indefinitely. The catalytic subunit, human telomerase reverse transcrip-tase (hTERT), is stabilized by phosphorylation. We found that the lysophospholipid sphingosine 1-phosphate(S1P), generated by sphingosine kinase 2 (SK2), bound hTERT at the nuclear periphery in human and mousefibroblasts. Docking predictions and mutational analyses revealed that binding occurred between a hydroxylgroup (C′3-OH) in S1P and Asp684 in hTERT. Inhibiting or depleting SK2 or mutating the S1P binding site de-creased the stability of hTERT in cultured cells and promoted senescence and loss of telomere integrity. S1Pbinding inhibited the interactionof hTERTwithmakorin ring finger protein 1 (MKRN1), anE3ubiquitin ligase thattags hTERT for degradation. Murine Lewis lung carcinoma (LLC) cells formed smaller tumors in mice lackingSK2 than inwild-typemice, andknockingdownSK2 inLLCcells before implantation intomicesuppressed theirgrowth.Pharmacologically inhibitingSK2decreased thegrowthof subcutaneousA549 lungcancercell–derivedxenografts inmice, and expression of wild-type hTERT, but not an S1P-bindingmutant, restored tumor growth.Thus, our data suggest that S1P binding to hTERT allosterically mimicks phosphorylation, promoting telo-merase stability and hence telomere maintenance, cell proliferation, and tumor growth.
e.sc
on January 2, 2020iencemag.org/
INTRODUCTION
Human telomerase is an RNA-dependent DNA polymerase that contains acatalytic component, human telomerase reverse transcriptase (hTERT), andan internal RNA template, telomerase RNA (TR) (1, 2). Telomerase extendsthe ends of chromosomes and protects telomeres from replication-dependent attrition, enabling cancer cells to proliferate indefinitely by over-coming the end replication problem (3–5). Telomerase is overexpressed in>80% of all cancer types (6, 7). Inhibition of telomerase leads to telomeredamage, subsequent senescence, and tumor suppression (8–11). Lamins arekey structural components of the nuclear lamina, an intermediate filamentmeshwork that lies beneath the inner nuclear membrane, attaching chroma-tin domains to the nuclear periphery and localizing some nuclear envelopeproteins. Fibroblasts obtained from lamin B1 mutant mouse embryosdisplayed premature senescence (12). In budding yeast, telomeres are re-versibly bound to the nuclear envelope, and small ubiquitin-like modifierprotein (SUMO)–dependent association with the nuclear periphery wasproposed to restrain bound telomerase (13). Phosphorylation of hTERTincreases its stability, and protein phosphatase 2 (PP2A)–dependent de-phosphorylation of hTERT inhibits telomerase function (14).
1Department of Biochemistry and Molecular Biology, Medical University of SouthCarolina, 86 Jonathan Lucas Street, Charleston, SC 29425, USA. 2HollingsCancer Center, Medical University of South Carolina, Charleston, SC 29425,USA. 3Department of Pharmaceutical Sciences, Medical University of SouthCarolina, Charleston, SC 29425, USA. 4Department of Biochemistry and Mo-lecular Biology, Indiana University School of Medicine-South Bend, SouthBend, IN 46617, USA. 5Department of Chemistry and Biochemistry and theMike and Josie Harper Cancer Research Institute, University of Notre Dame,South Bend, IN 46556, USA. 6Gene Expression and Regulation Program, TheWistar Institute, Philadelphia, PA 19104, USA.*Corresponding author. E-mail: [email protected]
w
The bioactive sphingolipids ceramide and sphingosine 1-phosphate(S1P) exert opposing functions: ceramide is emerging as a tumor suppres-sor molecule, whereas S1P promotes tumor growth (15–19). Ceramideinhibits hTERT expression by inducing histone deacetylase 1 (HDAC1)–dependent deacetylation of Sp3 (an Sp1 family transcription factor), whichrepresses hTERT promoter function (20). S1P is generated by cytoplasmicsphingosine kinase 1 (SK1) or nuclear SK2 (21, 22). S1P generated bySK1 promotes tumor growth and metastasis (23–25). SK1-generated intra-cellular S1P binds and promotes tumor necrosis factor receptor–associatedfactor 2 (TRAF2)–dependent nuclear factor kB (NFkB) signaling (21).SK2-generated nuclear S1P directly binds and inhibits HDAC1 and HDAC2(22). SK2-generated S1P binding also induces prohibitin 2 activity, leading tocytochrome c oxidase assembly and mitochondrial respiration (26). Consid-ering S1P in the context of telomerase, we investigated how the binding ofSK2-generated S1P alters hTERTabundance and the function of telomerase.
RESULTS
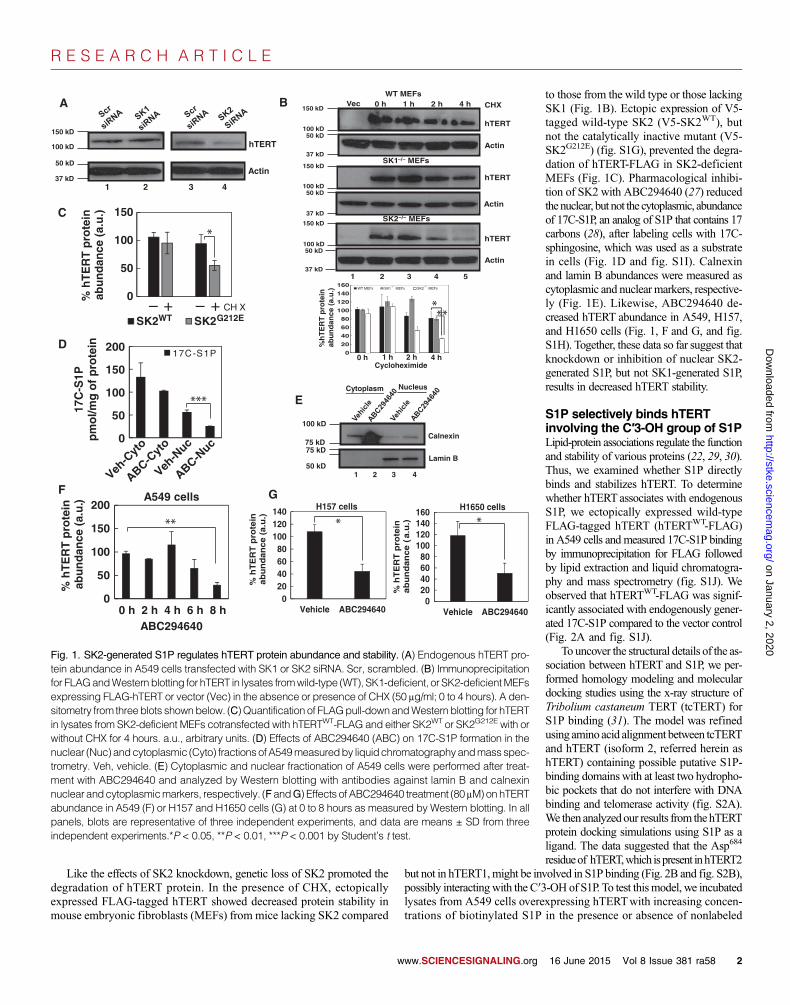
SK2-generated S1P promotes hTERT stabilityTo examine the possible roles of S1P in the regulation of hTERT, wedetermined whether down-regulation of SK1 or SK2 affected hTERTabun-dance or stability in human lung cancer cells. Small interfering RNA(siRNA)–mediated knockdown of SK2, but not SK1, decreased hTERTprotein abundancewithout affecting that of its mRNA in various human lungcancer cell lines (Fig. 1A and fig. S1, A and B). Compared with controls,stable knockdown of SK2 using one of two short hairpin RNAs (shRNAs)targeting distinct sequences decreased the abundance of hTERT in H1299andH1650 cells (fig. S1, C andD) and hTERT stability in A549 cells treatedwith cycloheximide (CHX) (fig. S1, E and F). These data suggested that SK2promotes hTERT abundance and protein stability.
ww.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 1
R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
Like the effects of SK2 knockdown, genetic loss of SK2 promoted thedegradation of hTERT protein. In the presence of CHX, ectopicallyexpressed FLAG-tagged hTERT showed decreased protein stability inmouse embryonic fibroblasts (MEFs) from mice lacking SK2 compared
www.SCIENCESIGNALING.org
to those from the wild type or those lackingSK1 (Fig. 1B). Ectopic expression of V5-tagged wild-type SK2 (V5-SK2WT), butnot the catalytically inactive mutant (V5-SK2G212E) (fig. S1G), prevented the degra-dation of hTERT-FLAG in SK2-deficientMEFs (Fig. 1C). Pharmacological inhibi-tion of SK2 with ABC294640 (27) reducedthenuclear, but not the cytoplasmic, abundanceof 17C-S1P, an analog of S1P that contains 17carbons (28), after labeling cells with 17C-sphingosine, which was used as a substratein cells (Fig. 1D and fig. S1I). Calnexinand lamin B abundances were measured ascytoplasmic and nuclear markers, respective-ly (Fig. 1E). Likewise, ABC294640 de-creased hTERT abundance in A549, H157,and H1650 cells (Fig. 1, F and G, and fig.S1H). Together, these data so far suggest thatknockdown or inhibition of nuclear SK2-generated S1P, but not SK1-generated S1P,results in decreased hTERT stability.
S1P selectively binds hTERTinvolving the C′3-OH group of S1PLipid-protein associations regulate the functionand stability of various proteins (22, 29, 30).Thus, we examined whether S1P directlybinds and stabilizes hTERT. To determinewhether hTERTassociates with endogenousS1P, we ectopically expressed wild-typeFLAG-tagged hTERT (hTERTWT-FLAG)in A549 cells and measured 17C-S1P bindingby immunoprecipitation for FLAG followedby lipid extraction and liquid chromatogra-phy and mass spectrometry (fig. S1J). Weobserved that hTERTWT-FLAG was signif-icantly associated with endogenously gener-ated 17C-S1P compared to the vector control(Fig. 2A and fig. S1J).
To uncover the structural details of the as-sociation between hTERT and S1P, we per-formed homology modeling and moleculardocking studies using the x-ray structure ofTribolium castaneum TERT (tcTERT) forS1P binding (31). The model was refinedusing aminoacid alignment between tcTERTand hTERT (isoform 2, referred herein ashTERT) containing possible putative S1P-binding domainswith at least two hydropho-bic pockets that do not interfere with DNAbinding and telomerase activity (fig. S2A).We thenanalyzedour results from thehTERTprotein docking simulations using S1P as aligand. The data suggested that the Asp684
residueof hTERT,which ispresent inhTERT2
but not in hTERT1,might be involved in S1P binding (Fig. 2B and fig. S2B),possibly interactingwith the C′3-OH of S1P. To test thismodel, we incubatedlysates from A549 cells overexpressing hTERTwith increasing concen-trations of biotinylated S1P in the presence or absence of nonlabeledA
hTERT
Actin
1 2 3 4
150 kD
100 kD
50 kD
37 kD
150 kD
50 kD
37 kD
50 kD
37 kD
50 kD
37 kD1 2 3 4 5
SK1−/−
SK2−/− MEFs
WT MEFs
hTERT
Actin
hTERT
hTERT
Actin
Actin
Vec 0 h 1 h 2 h 4 h CHX
150 kD
150 kD
100 kD
100 kD
100 kD
B
0
50
100
150
% h
TE
RT
pro
tein
ab
un
dan
ce (
a.u
.)
– + – + CH X
SK2WT SK2G212E
C
D
E
F
*
0
Veh-C
yto
Veh-N
uc
ABC-Nuc
ABC-Cyt
o
50
100
150
200
17C
-S1P
pm
ol/m
g o
f p
rote
in
17C-S1P
***
0
50
100
150
200
0 h 2 h 4 h 6 h 8 h
% h
TE
RT
pro
tein
ab
un
dan
ce (
a.u
.)
ABC294640
A549 cells
**
020406080
100120140
Vehicle ABC294640
% h
TE
RT
pro
tein
ab
un
dan
ce (
a.u
.)
H157 cells
*
020406080
100120140160
Vehicle ABC294640
% h
TE
RT
pro
tein
a
bu
nd
an
ce
(a
.u.)
H1650 cells
*
100 kD
75 kD75 kD
50 kD
Calnexin
Lamin B
Cytoplasm Nucleus
1 2 3 4
G
0
20
40
60
80
100
120
140
160WT MEFs SK1–/– MEFs SK2–/– MEFs
***
%h
TE
RT
pro
tein
abu
nd
ance
(a.
u.)
Cycloheximide0 h 1 h 2 h 4 h
MEFs
Fig. 1. SK2-generated S1P regulates hTERT protein abundance and stability. (A) Endogenous hTERT pro-tein abundance in A549 cells transfected with SK1 or SK2 siRNA. Scr, scrambled. (B) Immunoprecipitationfor FLAGandWestern blotting for hTERT in lysates fromwild-type (WT), SK1-deficient, or SK2-deficientMEFsexpressing FLAG-hTERT or vector (Vec) in the absence or presence of CHX (50 mg/ml; 0 to 4 hours). A den-sitometry from threeblots shownbelow. (C)Quantification of FLAGpull-downandWestern blotting for hTERTin lysates from SK2-deficient MEFs cotransfected with hTERTWT-FLAG and either SK2WT or SK2G212E with orwithout CHX for 4 hours. a.u., arbitrary units. (D) Effects of ABC294640 (ABC) on 17C-S1P formation in thenuclear (Nuc) andcytoplasmic (Cyto) fractionsofA549measuredby liquidchromatographyandmassspec-trometry. Veh, vehicle. (E) Cytoplasmic and nuclear fractionation of A549 cells were performed after treat-ment with ABC294640 and analyzed by Western blotting with antibodies against lamin B and calnexinnuclear andcytoplasmicmarkers, respectively. (FandG) Effects of ABC294640 treatment (80 mM)onhTERTabundance in A549 (F) or H157 and H1650 cells (G) at 0 to 8 hours as measured by Western blotting. In allpanels, blots are representative of three independent experiments, and data are means ± SD from threeindependent experiments.*P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test.
16 June 2015 Vol 8 Issue 381 ra58 2

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
stereoisomers of S1P (D-erythro-S1P, D-erythro-3-O-CH3-S1P, and L-erythro-S1P) (fig. S2C). Biotinylated S1P–bound proteins were capturedby avidin-conjugated agarose beads and examined by SDS–polyacrylamide
www.SCIENCESIGNALING.org
gel electrophoresis (SDS-PAGE) and West-ern blottingwith an antibody against hTERT.Biotinylated S1P bound hTERT effectivelycompared to biotin control, and the pres-ence of nonlabeled D-erythro-S1P, but notL-erythro-S1P or D-erythro-3-O-CH3-S1P,prevented the interaction between hTERTand biotinylated S1P (fig. S2C). Incubationof A549 cell extracts with biotinylated lyso-phosphatidic acid, which is structurally sim-ilar to S1P but lacks the C′3-OH (fig. S2D),did not result in any detectable hTERTbind-ing compared to biotinylated S1P (fig. S2D).BindingbetweenbiotinylatedS1PandhTERTwas also detected in H157 cells (fig. S2E).Thus, these data suggest that hTERTassoci-ates with S1P in vitro and in cells with highselectivity for the C′3-OH of S1P.
A hydrophobic region of hTERTthat includes Asp684 is key forS1P bindingTo examine whether the Asp684 residue isinvolved in S1P binding, we used a mutanthTERTwith D684A conversion (hTERTD684A-FLAG), which retains telomerase activity (32)comparable to hTERTWT-FLAG as measuredby the polymerase chain reaction (PCR)–based telomeric repeat amplification proto-col (TRAP) assay (fig. S2F). Then, weexamined the binding of biotinylated S1P tohTERTWT-FLAG or hTERTD684A-FLAG ex-pressed in A549 cells. The D684Amutationprevented the binding of hTERT to biotinyl-ated S1P compared to either hTERTWT-FLAGor another mutant of hTERT (hTERTR669A-FLAG) in which the mutation is in closeproximity to Asp684 but was not predictedto interfere with S1P binding (Fig. 2C). Tocontrol for endogenous hTERT, we ex-pressed hTERTWT-FLAG or hTERTD684A-FLAG in GM00847 fibroblasts that lackendogenous hTERT. hTERTWT-FLAG, butnot hTERTD684A-FLAG, associated withbiotinylated-S1P (Fig. 2D). Overall, thesedata suggest—as predicted by our molecu-lar modeling—that a hydrophobic pocket,localized between the thumb and finger do-mains and involving the Asp684 residue ofhTERT, plays a key role in S1P binding tothe C′3-OH of S1P.
S1PdirectlybindstcTERTandhTERTin vitro and in transfected cellsTo quantify the interaction between S1P andTERT, we performed surface plasmon res-onance (SPR) using S1P- or lysophosphatidic
acid (LPA)–containing lipid vesicles and purified recombinant tcTERTprotein.Although tcTERTdoes not contain the Asp684 residue, our modeling studiessuggested that both tcTERTand hTERT have similar hydrophobic pockets
**
***
A B
C D
E F
G H
–– – –
– –
Rel
ativ
e re
spo
nse
(R
U)
No
rmal
ized
sat
ura
tio
n r
esp
on
se
Time (s) Protein concentration (
S1P-biotin (nM)Bound S1P-biotin (nM)
S1P
-bio
tin
sp
ecif
ic b
ind
ing
,p
mo
l/mg
of
hT
ER
T-F
LA
G
M)
Fig. 2. SK2-generatedS1P interactswith hTERTby lipid-protein binding. (A) Ectopic expression of hTERTWT-FLAG inA549 cells (right) treatedwith 17C-sphingosinewasmeasuredby immunoprecipitation for the FLAGtag and assessed for bound 17C-S1P (data in fig. S1J). (B) Overlay of the hTERT model with the crystalstructure of tcTERT. Distinct domains of TERT are shown in color, and the S1P interacting residue Asp684
located at the interface of the palmand thumbdomains is in red stick. TRBD,TERTRBD. (C andD) Binding ofbiotinylated S1P (B-S1P) to FLAG-tagged WT or mutant hTERT in A549 cells (C) or GM00847 cells (D) as-sessed by avidin bead pull-down andWestern blotting for hTERT. (E) In vitro binding of tcTERT to either S1P[POPC/POPE/S1P (70:20:10)] or LPA [POPC/POPE/LPA (70:20:10)] vesicles relative to the control [POPC/POPE (80:20)]. (F) Kd analysis of tcTERT interaction with S1P vesicles. (G and H) In vitro binding of biotin-tagged S1P to FLAG-taggedWT or D684Amutant hTERT (0.4mg/ml) partially purified from A549 cells. In allpanels, data are means ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 byStudent’s t test.
16 June 2015 Vol 8 Issue 381 ra58 3

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
that might be involved in S1P binding (Fig. 2B). Purified tcTERT wasinjected at increasing concentrations to detect binding to S1P or LPAvesi-cles [containing the membrane phospholipids PC (phosphatidylcholine)and PE (phosphatidylethanolamine) and either S1P or LPA at a ratio of70:20:10] or control vesicles [containing POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) at a ratio of 80:20], as previously described (29, 33).Purified recombinant tcTERT bound the S1P vesicles in a concentration-dependent manner, whereas tcTERT did not bind the LPA vesicles (Fig.2E). The dissociation constant (Kd) for tcTERTwas ~4.8 mM for the S1Pvesicles (Fig. 2F). Thus, these data show the direct binding between S1PandtcTERT in vitro.
To quantify the interaction between S1P and hTERT (which, unliketcTERT, contains the integral Asp684 residue), we expressed hTERTWT-FLAGand hTERTD684A-FLAG in A549 cells and partially purified hTERT usingagarose beads recognizing FLAG. Partially purified hTERT was incubatedwith biotinylated S1P with or without increasing concentrations of un-labeled S1P. Bound biotinylated S1P was measured using a biotin-sensitiveenzyme-linked immunosorbent assay (ELISA). The Kd for hTERT
WT-FLAG was ~430 nM, whereas binding between biotinylated S1P and thehTERTD684A-FLAG was undetectable in the presence of 0.1 to 10 mM S1P(Fig. 2,G andH). The stoichiometrywas calculated as 1:1 for S1P and hTERT.Collectively, these data suggest that S1P-TERT binding involves the hydro-phobic domain between the thumb and finger domains of tcTERT andhTERTand that theAsp684 residueof hTERTis essential forS1Pbinding.UsingSPR, we also predicted that S1P may bind tcTERT in vitro at the conservedhydrophobic domain. However, this binding required much higher concentra-tions of purified tcTERTbecause theAsp684 residue is not conserved in tcTERT.
SK2-generated S1P binds hTERT in cellsTo determine whether the association between hTERT and S1P is physio-logically relevant, wemeasured their interaction by proximity ligation assay(PLA) using antibodies that recognize S1P (34) and hTERT, respectively.DAPI (4′,6-diamidino-2-phenylindole) staining was used to visualizenuclei. The association between S1P and hTERTwas detected in the nucleiof A549 cells transfected with scrambled control siRNA but not in thosetransfected with siRNA against SK2 (Fig. 3A). Inhibition of SK2 withABC294640 attenuated the S1P-hTERT association in A549 cells(Fig. 3A). Ectopic expression of hTERTWT-FLAG, but not hTERTD684A-FLAG, resulted in a 4.5-fold increase in S1P binding compared to vector-transfected GM00847 cells (Fig. 3B). Interactions between S1P and othernuclear proteins, such as HDAC3 or SET [Su(var), Enhancer-of-zeste,Trithorax domain–containing oncoprotein], used as negative controls, wereundetectable in A549 cells by PLA (Fig. 3C).
w
*
*
*** **
*** ***
A
B
C
D
E
Fig. 3. Interaction of hTERT with SK2-generated S1P colocalizes with laminB at the nuclear periphery. (A) PLA detection of the subcellular localization ofthe S1P-hTERT interaction in A549 cells transfected with either control (Scr)or SK2 siRNA (upper) or treated with vehicle or ABC294640 (lower). Nucleiwere counterstained with DAPI. Scale bars, 20 mm. DMSO, dimethyl sulf-oxide. (B) PLA detection of S1P-hTERT binding in GM00847 cells trans-fected with vector, hTERTWT-FLAG, or hTERTD684A-FLAG. (C) PLA for S1Pbinding to hTERT compared to HDAC3 or SET (nuclear proteins) in A549cells. (D and E) Colocalization of S1P (red) and lamin B (green) in the nu-cleus as assessed by immunofluorescence confocal microscopy (IF-CM)in WT or SK2-deficient MEFs (D) or A549 cells transfected with control(Scr) or SK2 shRNA (E). Scale bars, 100 mm. In all panels, images are rep-resentative of three independent experiments, and data are means ± SDfrom three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001by Student’s t test.
ww.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 4

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
S1P or hTERT colocalizes with a nuclear peripheryprotein, lamin BTo demonstrate the nuclear selectivity of the hTERT-S1P interaction,we examined the colocalization of S1P with the nuclear membranemarker lamin B in wild-type or SK2-deficient MEFs or A549 cellsusing immunofluorescence microscopy. S1P was colocalized with laminB in wild-type or control-transfected cells, respectively, but S1P was toodiminished in SK2-deficient cells to detect abundance at the nuclear pe-riphery (Fig. 3, D and E). Likewise, in transfected A549 cells, hTERTWT-FLAG, but not hTERTD684A-FLAG, colocalized with lamin B in thenucleus (fig. S3). Thus, these data suggest that in the presence of SK2and its product S1P, wild-type, but not mutant, hTERT localizes to thenuclear periphery.
S1P binding protects hTERT from ubiquitination andproteasomal degradationTo define whether binding by SK2-generated S1P affects hTERT stabilityby inhibiting ubiquitin-mediated proteasomal degradation, we treatedA549cells with the SK2 inhibitor ABC294640 alone or combined with the pro-teasome inhibitor lactacystin. Indeed, lactacystin prevented ABC294640-induced suppression of hTERTabundance (Fig. 4A). Likewise, exogenousS1P also prevented the ABC294640-induced loss of hTERT (Fig. 4A).Moreover, in A549 cells, ABC294640 induced the ubiquitination of en-dogenous hTERT (Fig. 4B and fig. S4A) and increased the amount ofhemagglutinin (HA)–tagged ubiquitin that had conjugated to exogenoushTERTWT-FLAG regardless of lactacystin treatment (Fig. 4C and fig. S4B).Likewise, in wild-type MEFs, the stability of ectopically expressed mutanthTERT (hTERTD684A-FLAG) was less than that of the wild-type constructin the presence of CHX, an inhibitor of de novo translation (Fig. 4D). How-ever, inhibition of proteasome activity by MG132 (N-carbobenzyloxy-L-leucyl-L-leucyl-L-leucinal) stabilized the abundance of hTERTD684A-FLAG(Fig. 4D). Accordingly, although hTERTWT-FLAG was stable in wild-typeMEFs (Fig. 4E), its abundancewas decreased in SK2-deficientMEFswhentreated with CHX, unless simultaneously treated with MG132 (Fig. 4F).Equal transfection efficiency for ectopic expression ofwild-type andmutanthTERTwithout CHX andMG132 exposure inwild-type and SK2-deficientMEFswasmeasured byWestern blotting (fig. S4, C andD). Together, thesedata suggest that hTERT is degraded through the proteasomal degradationpathway, particularly in the absence of SK2.
S1P-hTERTbindingpreventsMKRN1-mediateddegradationof hTERTThe E3 ubiquitin ligase makorin ring finger protein 1 (MKRN1) interactswith the C terminus (residues 946 to 1132) of hTERT (35), a region thatalso contains Gly932, Cys931, Phe986, His983, and Ser984 residues and is lo-cated within the putative S1P-binding domain of hTERT (Fig. 2B and fig.S2B), suggesting that S1P-hTERT bindingmay disruptMKRN1 binding tohTERT. As observed above, knocking down SK2 or inhibiting it withABC294640 decreased hTERT stability in A549 cells, but simultaneouslyknocking downMKRN1 (Fig. 5A) almost completely prevented the degra-dation of hTERT in the presence of CHX (Fig. 5, B and C). Expressing awild-type (MKRN1WT) but not a catalytically inactive RING domainmutant construct of MKRN1 (MKRN1H307E) increased the ubiquitinationof hTERT in the presence of MG132 (Fig. 5D), suggesting that MKRN1ubiquitinates hTERT. In wild-type MEFs, FLAG-tagged hTERT coimmu-noprecipitated with V5-tagged MKRN1 but was decreased in the presenceof exogenous S1P (Fig. 5E and fig. S4E). In contrast, the addition of C3-O-CH3-S1P (D-erythro-3-O-methyl S1P, an analog of S1P which lacks C′3-OH), which does not bind hTERT, did not inhibit the interaction betweenhTERT and MKRN1 (Fig. 5F and fig. S4E). The association between
w
MG132+ +
WT MEFs
0
20
40
60
80
100
120
140
% h
TE
RT
pro
tein
SK2−/− MEFs
– – + + MG132FLAG-hTERT+ + + +
CHX– – MG132
FLAG-hTERT+ +
CHX0
20
40
60
80
100
120
0 h 2 h 0 h 2 h
% h
TE
RT
stab
ility
WT MEFs
0
20
40
60
80
100
120
0 h 2 h 0 h 2 h
% h
TE
RT
sta
bili
ty
+ + + +
C D
E F
IP: hTERTIB: Ub
1 2
Ub
0
25
50
75
100
125
150
175
%h
TE
RT
pro
tein
abu
nd
ance
(a.
u.)
A B
***
* ***
* *
* **
+ + + + CHX– –
IP: FLAGIB: HA
FLAG-hTERT
HA-Ub
+ + + +
+ + + +
– – + + Lactacystin
– + – + ABC294640
(Ub)n
1 2 3 4
Fig. 4. S1P-hTERT binding prevents the ubiquitination and proteasomaldegradation of hTERT. (A)Western blotting for hTERT toassessprotein sta-bility in A549 cells pretreated with S1P, lactacystin, or vehicle followed bythe SK2 inhibitor ABC294640. (B and C) Pull-down for hTERT andWesternblotting for ubiquitin (Ub) showing the effects of ABC294640 on the ubiqui-tination of endogenous hTERT in A549 cells (B) or of FLAG-hTERT in thepresence of the protease inhibitor lactacystin in A549 cells expressingHA-Ub (C). (D) Pull-down for FLAGandWestern blotting for hTERTshowingthe stability of FLAG-taggedWT or mutant (D684A) hTERT in the presenceor absence of CHX or MG132 in WT MEFs. (E and F) Pull-down for FLAGand Western blotting for hTERT showing the stability of FLAG-hTERTWT
expressed in WT (E) or SK2-deficient MEFs (F) in the presence of CHXalone or with MG132. In all panels, blots are representative of three in-dependent experiments, and data are means ± SD from three independentexperiments. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test.
ww.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 5

R E S E A R C H A R T I C L E
www.SCIENC
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
MKRN1 and the S1P-bindingmutant hTERTD684A-FLAGwasalso detectable, and as expected, neither the presence of S1Pnorof C3-O-CH3-S1P had any effect (Fig. 5F and fig. S4E). Fur-thermore, siRNA-mediated reduction of MKRN1 abundanceblunted the suppression of A549 cell growth in soft agar thatwas induced by SK2 knockdown (Fig. 5G). Together, thesedata indicate that SK2-generated S1P promotes hTERT stabil-ity and subsequent cell proliferation by inhibiting the interac-tion of hTERTwith and ubiquitination byMKRN1 and, hence,preventing its subsequent degradation.
S1P-hTERT binding regulates endogenousMKRN1-hTERT interactionWe then determined the effects of S1P produced by SK2 on theendogenous hTERT-MKRN1 association. We expressed wild-type or a catalytically inactive mutant of SK2 (SK2G212E) inA549 and H1650 cells and assessed the interaction betweenhTERT and MKRN1 by immunoprecipitation in the presenceor absence of geldanamycin (GA), which induces MKRN1-dependent hTERT degradation (35). Ectopic expression ofwild-type, but not mutant, SK2 prevented the GA-induced in-teraction between endogenous hTERT and MKRN1 (Fig. 6Aand fig. S5, A and B). In primary normal human lung fibroblasts(NHLFs), we stably expressed vector or SK2WT and measuredhTERT-MKRN1 association using PLA. In vector-transfectedcontrols, there was a high degree of hTERT-MKRN1 associa-tion, which was primarily detected in the cytoplasm, and over-expression of SK2WT (Fig. 5, C and D) almost completelyabrogated this interaction (fig. S5C). In addition, expressionof wild-type, but not mutant, SK2 prevented the GA-inducedinhibition of the growth of A549 and H1650 cells on soft agar(Fig. 6B and fig. S5E). Thus, these data demonstrate that SK2-generated S1P inhibits the association between endogenoushTERT and MKRN1.
S1P binding at Asp684 mimics phosphorylationof hTERT at Ser921
To determine how S1P binding protects hTERT from MKRN1-mediated proteasomal degradation, we compared the stabilizingeffect of S1P binding to that of the known phosphorylationmodification (14). Molecular modeling and simulation datasuggested that binding of S1P to hTERT at the Asp684 residuemight mimic the phosphorylation of hTERTat Ser921 (fig. S6,A and B). This was tested in experiments in which S921A (anonphosphorylatable mutation) or S921D (a phosphomimeticmutation) conversions were introduced into the D684A mu-tant of hTERT to generate hTERTD684A-S921A-FLAG andhTERTD684A-S921D-FLAG double mutants. The colocalizationof the double mutants with lamin B at the nuclear periphery,their association with MKRN1, and their stability were assessed incomparison to that of the wild-type construct (hTERTWT-FLAG)in GM00847 cells. hTERTWT-FLAG, but not hTERTD684A-FLAG, was localized to the nuclear periphery, as visualizedby confocal microscopy and immunofluorescence. AlthoughhTERTD684A-S921A-FLAGwas mainly cytoplasmic, localizationof hTERT in the lamin B–containing nuclear periphery wasrestored in cells expressing the phosphomimetic and S1Pbinding–defective hTERTD684A-S921D-FLAG (Fig. 6C). More-over, hTERTS921A-FLAG, which contains intact S1P binding atAsp684, was also localized mainly in the nucleus (Fig. 6C). The
0
0.2
0.4
0.6
0.8
1
1.2
1.4
Rel
ativ
e M
KR
N1
mR
NA
lev
els
A
0255075
100125150
% h
TE
RT
stab
ility
0 h 4 h 0 h 4 h 0 h 4 h 0 h 4 h
shScr shSK2
CHX
B
siScr siMKRN1
0 5 10 20 40 µM ABC150 kD
50 kD
37 kD1 2 3 4 5 6 10
0 5 10 20 40
100 kD
7 8 9
D
WT MEFs
– – – – + + – + + – – –
– + – + – +
– – + + + +
MG132
FLAG-hTERTMKRN1WT
IP: FLAGIB: Ubiquitin
(Ub)n
+ + – – – – Vector
1 2 3 4 5 6
IB: V5-MKRN1
MKRN1H307E
IP: FLAGIB: hTERT
C
E F
G
0
50
100
150
200
250
300
No
. of
colo
nie
s
0
0.5
1
1.5
No
lip
idS
1PC
H3-
S1P
No
lip
idS
1PC
H3-
S1P
No
lip
idS
1PC
H3-
S1P
Fo
ld c
han
ge
inM
KR
N1W
T b
ind
ing
to
hT
ER
T
hTERTWT
hTERTWT
– – – – – –
– – – – – – hTERTD684A
– – –
S1PLPA
– + –– – +
0
50
100
150
% h
TE
RT
-MK
RN
1 as
soci
atio
n
+ + + + ++ + +
+ + +
+
******* *
*
* **
*** **
1IP: FLAG
2 3Vector
Fig. 5. Binding of SK2-generated S1P protects hTERT from MKRN1-mediated degrada-tion. (A) MKRN1 knockdown in A549 cells stably transfected with control (Scr) or SK2shRNA. (B and C) Western blotting for endogenous hTERT stability after MKRN1knockdown in stable shScr and shSK2 A549 cells treated with CHX (B) or ABC294640(C) for 4 hours. In (C), bottom blot is actin loading control. (D) Pull-down for FLAG andWestern blotting for ubiquitin in WT MEFs coexpressing FLAG-hTERT and either WT orRING mutant (H307E) MKRN1 and pretreated with MG132 for 2 hours. (E) Coimmuno-precipitation of FLAG-hTERTwith V5-MKRN1 in A549 cell extracts in the presence of S1Por LPA (5 mM). Samples shown are from the same representative blot but not in contigu-ous lanes. (F) Coimmunoprecipitation of FLAG-taggedWTormutant (D684A) hTERTwithMKRN1 inA549cell extracts in thepresenceof S1PorC3-O-CH3-S1P. (G) Effect of siRNA-mediatedMKRN1 knockdown on the anchorage-independent growth on soft agar ex-hibited by A549 cells stably transfected with SK2 or control (Scr) shRNA. In all panels,blots are representative of three independent experiments, and data are means ± SDfrom three independent experiments. *P< 0.05, **P< 0.01, ***P< 0.001 by Student’st test.
ESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 6

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
MKRN1-hTERT association (measured by PLA) was enhanced in cellsexpressing hTERTD684A-FLAG but was prevented by expression ofhTERTD684A-S921D-FLAG (Fig. 6D). These data are consistent with the restorationof hTERT stability that we observed after the expression of the phosphomimeticmutant, hTERTS921D-FLAG, compared to either hTERTD684A-S921A-FLAGor hTERTD684A-FLAG (Fig. 6E). Collectively, these data suggest thatSer921 phosphorylation mimetic restores hTERT stability in the absenceof S1P binding in cells expressing hTERTD684A-FLAG. These studies alsosuggest that lipid binding of hTERT by S1P allosterically mimics hTERTphosphorylation at its C terminus including Ser921, preventing hTERT-MKRN1 association and stabilizing hTERT at the nuclear periphery.
www.SCIENC
Nuclear S1P-hTERT binding prevents telomeredamage and delays senescenceWe then determined whether S1P-hTERT binding has anyimpact on telomere damage, dysfunction, or senescence—key biological processes counteracted by telomerase. Con-trol NHLFs became positive for b-galactosidase (b-gal), a cellsenescence marker, after ~10 passages in culture. ExpressingSK2WT delayed the emergence of senescence up to passage14, but concomitantly knocking down hTERT restored senes-cence on the basis of b-gal positivity at passage 10 (Fig. 7, AandB, and fig. S7, A toC).We then assessed telomere integrityin these cells by detecting the complex between phosphorylatedhistone H2AX (g-H2AX) and telomeric repeat–binding fac-tor 2 (TRF2), which is recruited to damaged or dysfunctionaltelomeres (36, 37), using a telomere dysfunction–inducedfoci (TIF) assay. Ectopic SK2 expression prevented telomeredysfunction compared to vector-transfected controls, and TERTknockdown reestablished telomere damage (Fig. 7B andfig. S7D).
Accordingly, primary SK2-deficient MEFs became senes-cent at passage 5, whereas senescence in their age-matchedwild-type or SK1-deficient MEFs was not observed untilpassage 7 (Fig. 7C). Reconstitution of hTERTWT, but nothTERTD684A, prevented telomere damage (Fig. 7D) and de-layed senescence (Fig. 7E and fig. S8A) in SK2-deficientMEFs. Telomere restriction fragment (TRF) lengthmeasure-ment by Southern blotting showed no detectable differencesin telomere lengths in wild-type or SK2-deficient MEFs atpassages 3 and 6 (fig. S8B). As controls, we used genomicDNAisolated from subcutaneous A549 xenografts expressing eithervector or hTERTWT isolated from immunocompromised miceafter 28 days of growth. Ectopic expression of hTERT length-ened telomeres in the isolated tumor cells (fig. S8C). Collective-ly, these data suggest that SK2 delays senescence and promotestelomere maintenance through its activity on hTERT.
SK2-generated S1P promotes hTERT stabilityand tumor growthBecause cellular senescence plays key roles in tumor suppres-sion (38–40), we determined the effects of systemic versuscellular S1P produced by SK2 on the regulation of tumorgrowth. We measured the enlargement of Lewis lung carcino-ma (LLC) allograft–derived tumors in wild-type and SK2-deficient mice. Knocking down SK2 in LLC cells beforeimplantation almost completely inhibited tumor growth com-pared to controls (Fig. 7F). Systemic loss of SK2 throughwhole-body deletion decreased the growth of control LLC al-lografts by ~50% compared to wild-type mice (Fig. 7F), with-out affectingmurine TERTabundance. These data indicate that
molecular targeting of tumors, rather than systemic SK2, inhibits TERTex-pression and suppresses tumor growth in vivo, but that systemic SK2 inhi-bition seems to play a role in the regulation of tumor growth by anindependent, unknown mechanism.
Finally, we measured the effects of stable expression of hTERTWT orhTERTD684A on ABC294640-mediated inhibition of A549 cell–derivedxenograft tumor growth in immunocompromised mice. hTERTWT
conferred resistance to ABC294640-mediated tumor suppression whencompared to vector-transfected controls (Fig. 7G). However, expression ofhTERTD684A had no effect on ABC294640-mediated tumor suppres-sion compared to controls (Fig. 7G). Overall, these results suggest that
hT
ER
TM
erg
eL
amin
B
Vector hTERTWT hTERTD684A hTERTS921A hTERTS921A+D684A
hTERTS921D+D684A
0
50
100
150
200
250
300
350
400
% h
TE
RT
-MK
RN
1 as
soci
atio
n
0
50
100
150
200
250
No
. of
colo
nie
s
A B
C
010203040506070
MK
RN
1-h
TE
RT
P
LA
sig
nal
/cel
l
– + – + – + – + – + – + CHX
0
50
100
150
200
% h
TE
RT
p
rote
in le
vels
D E
*
** * *
*****
*
Fig. 6. SK2-generated S1P prevents hTERT-MKRN1 interaction by mimicking hTERTphosphorylation at Ser921. (A and B) Effects of WT or catalytically inactive (G212E) SK2on endogenous hTERT-MKRN1 interaction (A) and proliferation on soft agar (B) in A549cells, eitheruntreatedor treatedwithGA. (C) Colocalization of FLAG-taggedWTormutanthTERT (red)with laminB (green) detectedby IF-CM inGM00847cells. Scale bars, 20 mm.(D) PLA detection of the interaction between MKRN1 and FLAG-tagged WT or mutanthTERT in GM00847 cells. (E) Western blotting to assess the stability of WT or mutanthTERT in the presence or absence of CHX. In all panels, images are representative ofthree independent experiments, and data are means ± SD from three independentexperiments. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test.
ESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 7

R E S E A R C H A R T I C L E
www.SCIENCE
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
pharmacologically targeting SK2 in the tumor or inhibitingS1P-hTERTbinding in cancer cellsmay suppress tumor growth.
DISCUSSION
Here, our data revealed that SK2-generated nuclear S1P direct-ly binds hTERTand regulates hTERT stability in the lamin B–positive nuclear periphery by inhibiting MKRN1-dependenthTERT ubiquitination and degradation. Binding of hTERTby S1P appears to function as an allosteric phosphomimeticof hTERTS921 at its C terminus including Ser921, whichprevents MKRN1-hTERT association, stabilizing hTERT atthe nuclear periphery. Nuclear S1P-hTERT binding plays im-portant biological functions regulated by telomerase, such asprotection of cells from telomere damage, delaying senescenceand preventing tumor suppression. Accordingly, SK2 inhibi-tion or prevention of S1P-hTERT binding accelerated senes-cence and telomere damage and suppressed tumor growth andproliferation (Fig. 7H).
Protein stability of hTERT is regulated by a direct and se-lective interaction between the C′3-OH of S1P and the Asp684
residue of hTERT, which is predicted to localize within a hy-drophobic region between the thumb and finger domains ofhTERT. The binding of S1P to purified recombinant tcTERT(31) revealed that S1P directly binds TERT in vitro. In cells,the binding of nuclear S1P to hTERT prevented MKRN1-hTERT interaction, modulating hTERTubiquitination and deg-radation. Many signaling proteins such as oncogenic RAS,hedgehog (HH), and autophagy-related Atg8 family proteinsare covalentlymodifiedwith sphingolipids (30, 41) or phospho-lipids, which is key for their correct protein localization andfunction in cells (42–44). Recently, a nuclear phospholipid,phosphatidylinositol 5-phosphate (PI5P), was shown to bindand activateUHRF1 (ubiquitin-likewith PHDandRING fingerdomains 1), a nuclear factor that maintains DNA methylationpatterns during replication (45). Our data demonstrated thatS1P binding prevents the association between MKRN1 andhTERT, thereby promoting the stability of hTERT. BecauseMKRN1 interacts with the C terminus (residues 946 to 1132)of hTERT (35), lipid binding of hTERT by S1P, mimicking thephosphorylation status at its C terminus, including Ser921,seemed to interfere physically with MKRN1-hTERT associa-tion. We believe that this is the initial discovery for a functionof protein-lipidbinding (hTERTbindingbyS1P) as an allostericphosphomimetic to regulate a protein-protein (hTERT-MKRN1)interaction and to control hTERT stability in the nuclear pe-riphery, leading to delayed senescence.
S1P binding–dependent hTERT stabilization altered se-nescence by protecting against telomere damage in primaryhuman lung fibroblasts and wild-type MEFs, whereas SK2loss in MEFs increased telomere damage and acceleratedsenescence. Furthermore, hTERTWT, and not hTERTD684A,protected SK2-deficient MEFs from telomere damage anddelayed senescence, which seemed to be independent of tel-omere length control. These data are intriguing because se-nescence inMEFsmight not be regulated solely by telomereshortening; rather, senescence in MEFs might be controlledat least in part by induction of telomere damage. However,our inability to detect any changes of telomere length in wild-type versus SK2-deficient MEFs might be due to their extra
0102030405060708090
100
P7 P8 P10 P12 P14 P16 P17
%S
A β
-gal
pos
itive
cel
ls
pCDH
pCDH-SK2WT
pCDH-SK2WT + shhTERT
A
00.20.40.60.8
11.21.4
Fo
ld c
han
ge
in t
elo
mer
e d
amag
e
0
20
40
60
80
WT SK1–/– SK2–/–
% S
A
-gal
p
osi
tive
cel
ls
00.20.40.60.8
11.2
Fo
ld c
ha
ng
e in
te
lom
ere
da
ma
ge
* *
010203040506070
SK2–/– MEFsSK2–/– MEFs
0
200
400
600
800
1000
1200
1400
1 4 7 10
Tum
or
volu
me
(mm
3 )
Days
WT shScr
WT shSK2
SK2–/– shScr
SK2–/– shSK2
B
C D
F
E
G
0
100
200
300
400
500
600
700
1 3 5 9 11 13 15 17 19 21
Tum
or v
olum
e (m
m3 )
Days
Vector
Vector + ABC
hTERTWT
hTERTWT + ABC
hTERTD684A
hTERTD684A + ABC
* ****
***
*** ** ** **
*
**
**
****
Nucleus
Sph
P
hTERT
Tumor proliferation
Telomere damageSenescence
Ubiquitination/proteasome degradationCytoplasm
Telomere damageSenescence
Tumor suppression
shRNAABC294640
hTERT
S1P
Ser921
Ser992
Lamin BS1P
hTERT
SK2
SK2-/-
E3-Ub-LMKRN1
H
β
% S
A
- gal
po
siti
ve c
ells
β
Fig. 7. SK2-generated S1P-hTERT plays key roles in the control of senescence, telomere
damage, and tumor growth. (A) b-Gal staining in primary lung fibroblasts (PLFs) [at pas-sages 7 (P7) to P17] that stably coexpress pCDH and control shRNA (shScr) or SK2 andeither shScr or shTERT. (B) Telomere damage assessed by the TIF assay (g-H2AX andTRF2 colocalization) in PLFs at P12, cotransfected as in (A). (C) b-Gal staining in SK2-deficient orWTMEFs at P5. Positive cells were counted from three to four fields. (D andE)Telomere damage as assessed by the TIF assay (D) and senescence by b-gal detection(E) in SK2-deficient MEFs transfected with WT or mutant (D684A) hTERT. (F) Volumes ofallograft tumors derived fromLLCcells stably transfectedwith shScr or shSK2 in the flanksof age-matchedWT or SK2-deficient mice. (G) Volumes of xenograft tumors derived fromA549 cells stably transfected with vector, hTERTWT, or hTERTD684A in mice treated withvehicle or ABC294640 for 21 days. In panels (A) to (E), data are means ± SD from threeindependent experiments; in (F) and (G), data are means ± SD from four mice each, witheach mouse containing two tumors. *P < 0.05, **P < 0.01, ***P < 0.001 by Student’s t test.(H) A model of our findings, revealing a nuclear lysophospholipid-mediated mechanismthrough which S1P binding to a pin-pointed region in hTERT functions as an allostericphosphomimetic and stabilizes hTERT. Various ways to reduce S1P abundance orbinding may induce the degradation of hTERT and, in turn, the acceleration of telomeredamage and senescence in tumors.SIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 8

R E S E A R C H A R T I C L E
http://stke.D
ownloaded from
long telomeres (>21 kb), which are difficult to measure accurately usingSouthern blotting. It is also possible that noncanonical roles for S1P-boundhTERT (isomer 2) might play a role in regulating telomere damage or se-nescence without affecting telomere length. This, however, is unclear andneeds further investigation. SK2-deficient mice do not exhibit any detectableobvious phenotype due to telomerase instability or accelerated senescence.This is consistent with TERT-deficient mice, which do not show any agingphenotype for the first three generations (G1 to G3) and have phenotypicchanges starting at G4 through G6 because of the particularly long telo-meres in these animals (46). It is known that even partially reduced expres-sion of telomerase has profound effects on telomere maintenance becausemice heterozygous for mTERT (mouse TERT) or mTERC (mouse telomer-ase RNA component) show haploinsufficiency in telomere maintenance(47). These data are consistent with our studies in which mTERT/hTERTinstability in response to genetic loss or molecular knockdown of SK2resulted in accelerated telomere dysfunction, senescence, and tumorsuppression.
Overall, these data suggest that lipid binding of hTERT by S1P allo-sterically mimics hTERT phosphorylation at Ser921, which then preventshTERT-MKRN1-E3 ubiquitin ligase association, enhancing hTERT sta-bility at the nuclear periphery. Increased hTERT stability at the nuclearperiphery by S1P binding has important biological implications for theregulation of telomerase-dependent control of telomere damage and cel-lular senescence, key processes involved in aging and cancer biology. Itis possible that this novel nuclear signaling mechanism mediated by in-teraction with S1P might regulate not only telomerase but also other pro-teins with diverse biological functions through protein phosphorylationmimicry.
on January 2, 2020sciencem
ag.org/
MATERIALS AND METHODS
Cell lines and culture conditionsA549, H157, and H1650 cells were cultured in Dulbecco’s modified Ea-gle’s medium (DMEM) (Cellgro) with 10% fetal bovine serum (FBS) (At-lanta Biologics) and 1% penicillin and streptomycin (Cellgro). H1341 smallcell lung cancer cells were cultured in RPMI-1640 (American Type CultureCollection) with 10% FBS and 1% penicillin and streptomycin. GM00847cellswerepurchased fromCoriellCellRepositories.Wild-type,SK1-deficient,andSK2-deficientMEFswereobtained fromK.Argraves (MedicalUniversityof South Carolina). GM00847 cells and MEFs were cultured in DMEM andincubated at 37°Cwith 5%CO2. Primary hNHLFswere purchased fromLon-za Group and cultured in fibroblast growth medium as described by themanufacturer.
PlasmidsPlasmids containing hTERTWT and hTERTD684A in pcDNA vectors wereobtained from J. Chen (Arizona State University). MKRN1WT andMKRN1H307E plasmids were obtained from M. T. Muller (University ofCentral Florida). SK2WT and SK2G212E plasmids were obtained fromS. Spiegel (Virginia Commonwealth University). pBABE-puro (plasmid#1764), pBABE-puro-hTERT (plasmid #1771), pMD2.G (plasmid#12259), and psPAX2 (plasmid #12260) were purchased fromAddgene.
Quantitative PCRTotal RNA isolation was performed using RNeasy (Qiagen), and 1 mg oftotal RNAwas used for complementary DNA (cDNA) synthesis using theiScript cDNA synthesis kit (Bio-Rad). TaqMan probes (hTERT, Hs00162669;SK1, Hs00184211; SK2, Hs00219999; and MKRN1, Hs00831972) wereused for quantitative PCR (Life Technologies).
w
AntibodiesThe antibodies used for Western blotting in this study were as follows:hTERT (1531-1, clone Y182, Epitomics), SK2 (ab37977, Abcam),MKRN1(ab72054, Abcam), rabbit V5 (ab9116, Abcam), ubiquitin (3933S, CellSignaling Technology), HA (3724, Cell Signaling Technology), calnexin(Sc6465, Santa Cruz Biotechnology), lamin B (Sc6216, Santa CruzBiotechnology), and mouse V5 (R96025, Invitrogen).
Exogenous S1P treatmentExogenous S1P (Avanti Polar Lipids) was suspended in fat-free bovineserum albumin (BSA) (4 mg/ml) in 1× phosphate-buffered saline(PBS) (pH 7.4) at 125 mM. S1P was incubated in a sonicator water bathfor 5 min followed by incubation at 37° to 55°C for 20 min. D-erythro-C3-O-CH3-S1P and L-erythro-S1P were synthesized at the LipidomicsShared Resource Facility, Medical University of South Carolina.
Stable shRNA knockdown of SK2SK2 shRNA (TRCN00000036973)– and nontargeting shRNA–containingplasmids were purchased from Open Biosystems Inc. Cells were cotrans-fected with pCMV-psPAX2 and pMD2 plasmids in 293T cells using theviral transduction protocol as described by the RNA interference (RNAi)Consortium. The viral supernatants were added to A549 cells, and selectionwas performed using puromycin (1 mg/ml) for 14 days. The followingshRNAs were purchased from Thermo Scientif ic Inc: shSK2 #1(TRC0000036973), 5′-CCGGCTACTTCTGCATCTACACCTACTC-GAGTAGGTGTAGATGCAGAAGTAGTTTTG-3′ ; shSK2 #2(TRCN0000036969), 5′-CCGGGCTTCGTGTCAGATGTGGATACTC-GAGTATCCACATCTGACACGAAGCTTTTTG-3′; and shSK2 #3(TRCN0000036970), 5′-CCGGGTTGCTCAACTGCTCACTGTTCTC-GAGAACAGTGAGCAGTTGAGCAACTTTTTG-3′.
Immunoprecipitation and Western blottingFLAG-hTERTwas expressed in cells, which were then treated with CHX(Sigma-Aldrich) (50 mg/ml) for up to 4 hours. The cells were lysed with theFLAG lysis buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA,and 1% Triton X-100] with protease inhibitor cocktail. Immunoprecipitationwas performed using beads conjugated with antibody against FLAG (anti-FLAG M2 affinity gel; A2220, Sigma). Immunoprecipitation and elution ofbound proteins with FLAG beads were carried out according to the manu-facturer’s instructions. Immunoprecipitation with hTERTantibody (4 mg/ml)was carried out using 250 mg of total protein lysate made up to 500-ml volumewith FLAG lysis buffer. Lysates were precleared with 30 ml of protein A/Gbeads and incubated in a rotary shaker for 1 hour at 4°C. Sampleswere spunat 1000 rpm for 1 min, and the supernatant was transferred to a new micro-centrifuge tube and incubatedwith an hTERTantibody (4 mg/ml) overnight,followed by the addition of protein A/G beads and incubation at 4°C for1 hour. Samples were centrifuged at 1000 rpm for 1 min and washed twicewith lysis buffer and 1× PBS, followed by the addition of gel loading dye.Samples were boiled using heating block and centrifuged to collect elutedproteins, followed by Western blotting with an antibody against hTERT. Arabbit immunoglobulinG (IgG) controlwas used alongside hTERTpull-down.
For assays inA549 cells, cells were plated at 200,000 cells per well in asix-well plate for 18 hours before treatment with either DMSO (vehiclecontrol) or ABC294640 (Apogee Inc.) at 80 mM for 8 hours. After treat-ment, cells were centrifuged at 1300 rpm for 3.5 min and washed with1× PBS (pH 7.4) (Gibco). Cells were lysed with 1× CHAPS lysis buffer[10mMtris-HCl (pH7.5), 1mMMgCl2, 1mMEDTA,0.1mMbenzamidine,5 mM b-mercaptoethanol, 0.5% CHAPS, 10% glycerol] including a prote-ase inhibitor cocktail (Sigma-Aldrich) for 20 min on ice. Cell lysates werecentrifuged at 12,000g for 15 min at 4°C, and supernatants were used for
ww.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 9

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
Western blotting. SDS-PAGE (4 to 20%) was carried out using the Bio-RadCriterion apparatus, followed by semi-dry transfer onto a polyvinylidenedifluoride membrane. The membrane was blocked with 5% milk + 0.1%Tween 20 in 1× PBS (pH 7.4). Primary antibodies were used at 1:1000 di-lution overnight at 4°C, followed by rabbit or mouse secondary antibodies(Jackson Research Laboratories) conjugated with horseradish peroxidase atroom temperature for 1 hour. The rabbit secondary antibody was used at1:2500 dilution for detecting hTERT and at 1:5000 dilution for the rest ofthe antibodies. Actin was used as an internal control for Western blotting.The membranes were washed with either 1× PBS with Tween 20 or 1× tris-buffered saline with Tween 20 and developed using ECL Plus chemi-luminescence detection kit (GE Healthcare).
17C-Sphingosine labeling and measurement of S1PA549 cells were treated with 17C-sphingosine (5 mM) for 30 min, and thecytoplasmic and nuclear fractions were analyzed for 17C-S1P formation usingliquid chromatography and mass spectrometry, as previously described (28).
Homology modeling of hTERTCurrently, there is no experimental structural information available forhTERT. Amodel of a complete hTERT-RNA-DNA complex has been pub-lished (PMID: 21606328) and was optimized for mechanistic interactionswith the RNA template and DNA. The goal of our modeling and simulationstudies was to better understand the topology and chemistry of potentialTERT lipid interaction sites, not to predict the overall structure or catalyticmechanism of the full-length enzyme complex. Basic Local AlignmentSearch Tool (BLAST) searching of hTERT (accession number O14746)amino acid sequence against the Protein Data Bank (PDB) database indi-cated a highly similar sequence from tcTERT containing the RNA-bindingdomain (RBD) and reverse transcriptase (RT) domain. The hTERT aminoacid sequence has 41% coverage and 27% identity to the tcTERT structure.To begin homology modeling, the amino acid sequence from hTERTwasaligned with the amino acid sequence of the x-ray crystal structure oftcTERT. Pairwise sequence alignment was performed with BioEdit v7.0using standard Clustal alignment parameters and the BLOSUM62 matrix.Alignment of tcTERT and hTERT indicated 127 identities and 104 simila-rities, giving an overall similarity of 31% (231 of 744). Before homologymodeling, the tcTERT structure was protonated at pH 7.5 and the structurewas energy-minimized, with heavy atoms constrained.
Alignment homology modeling was performed using the MolecularOperating Environment (MOE) HomologyModel. Tenmodels were gener-ated, and the best model was selected using fine-grain intermediates,generalized born/volume integral (GB/VI) scoring, and all atom optimizedpotential for liquid simulations (OPLS-AA) force field. OPLS was chosenbecause it tolerates small molecules like S1P better than most other energyfields [Assisted Model Building with Energy Refinement (AMBER),Chemistry at HarvardMacromolecular Mechanics (CHARMM), andMerckMolecular Force Field (MMFF)]. There is a series of short inserts in hTERTthat are not present in tcTERT. A large insertion is located right before the RTdomain. The insert is a region of low confidence in the predicted model.Using MOE, we refined this region using a loop library. Comparison ofthe homology models of hTERTwith tcTERT revealed that the overall rootmean square deviation divergence of the crystal structures from the homologymodel was 1.27 Å. As expected, the highest structural divergence wascentered on the largest insert of hTERTbetween theRBDand theRTdomain.
Molecular docking of hTERT with S1PTo further understand the potential regulatory mechanism of TERT, S1Pwas docked to hTERT. Before the simulation, S1P chirality was formalizedand the molecule was protonated at pH 7.5. Docking was set to probe the
ww
entire protein surface. Initial placement calculated 500 poses for S1P usingTriangle Matching with London dG scoring. The top 250 poses were thenrefined using force field–based refinement and alpha sphere and excludedvolume (ASE) scoring. Four of the top five poses showedAsp684within 20Åof S1P. The large number of poses and the two-stage docking were done tofully explore the entire surface and to place scoring emphasis on the shapeand hydrophobicity of the interaction. The best pose was then subjected tobimolecular energy minimization of the S1P-hTERT complex.
Detection of S1P-hTERT bindingA549 cells were lysed in 1× CHAPS lysis buffer. One milligram of totalprotein was incubated with biotinylated S1P (5 mM). For cold competitionassays, nonbiotinylated sphingolipids were preincubated with cell lysatesbefore binding reactions were carried out using biotinylated S1P. Streptavi-din beads were added to the reaction mixture, and the assay was performedas described by the manufacturer (Miltenyi Biotec) (29, 30).
A549 cells transfected with the hTERTWT-FLAG or hTERTD684A-FLAG(or empty vector) were lysed by freeze-thawing in a lysis buffer containing50 mM tris (pH 7.4), 150 mMNaCl, 1 mMEDTA, and 0.5%NP-40 with aprotease inhibitor cocktail (1:500). Cell lysates (400 mg of protein) were in-cubated with 150 ml of beads conjugated with an antibody against FLAG(Sigma-Aldrich) for 18 hours at 4°C with agitation. Then, the beads werewashed and incubated with or without unlabeled S1P in the presence ofincreasing concentrations of biotinylated S1P in 150 ml of binding buffercontaining 50 mM tris (pH 7.5), 137 mM NaCl, 1 mM MgCl2, 2.7 mMKCl, 15 mMNaF, and 0.5 mMNaV3O4 for 30 min at 30°C. BiotinylatedS1P–bound hTERT proteins were eluted with 40 ml of FLAG peptide(250 mg/ml) and quantified using biotin-ELISA (Eagle Biosciences). Theresults were analyzed using GraphPad Prism 4 software.
Binding of S1P with tcTERT using SPRSPRwas used tomeasure the kinetics of S1P-tcTERTbinding invitro. Lipidvesicles containing S1P (PC/PE/S1P, 70:20:10), LPA (PC/PE/LPA,70:20:10), or control vesicles (POPC/POPE, 80:20) were injected with pur-ified tcTERT (250 nM to 6 mM) at a flow rate of 30 ml/min in 10mMHepes(pH 7.4) containing 0.16MKCl. All SPRmeasurements were performed at25°C as previously described (29). For each experiment, POPC/POPE (80:20),used as a control surface, andS1PorLPA(10mol%S1PorLPA)vesicles, usedas active surfaces, were injected with tcTERT. Each data set was repeatedthree times to verify the binding of tcTERT to control or active surfaces.
Quantitative detection of S1P-hTERT bindingA549 cells transfected with FLAG-tagged hTERTWTand hTERTD684Avec-tors or an empty vector were lysed by freeze-thawing in buffer containing50mM tris (pH 7.4), 150mMNaCl, 1 mMEDTA, 0.5%NP-40, and 1:500protease inhibitor cocktail. Lysates (400 mg of protein) were incubated with150 ml of agarose-conjugated antibodies specific for FLAG (Sigma-Aldrich)at 4°C for 18 hours with agitation. Then, the beads were extensively washedand incubated without or with unlabeled S1P in the presence of increasingconcentrations of biotinylated S1P (starting at 250 nM) in 150 ml of bindingbuffer. Bound hTERT proteins were eluted with 40 ml of FLAG peptide(250 mg/ml). Biotinylated S1P bound to the eluted proteins was quantifiedusing biotin-ELISA (Eagle Biosciences). The results were analyzed usingGraphPad Prism 4 software. The stoichiometry was determined using thefollowing equation: n = NKd/(L + N), in which N = L/M (L,free ligandconcentration; M, protein concentration).
ImmunofluorescenceA549 cells (50,000 perwell)were plated on glass coverslips in a six-well platefor 18 hours. Cellswere fixed and permeabilized using 4%paraformaldehyde
w.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 10

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
(20 min) and 0.1% Triton X-100 in 1× PBS (pH 7.4) for 10 min. The cellswere thenblockedwith 1%BSAanddissolved in 1×PBS (pH7.4) for 1 hour.Cells were incubated for 18 hours at 4°Cwith antibodies specific for S1P(Sphingomab, LT1002; 20 mg/ml), mouse IgG, lamin B, or hTERT(1:1000) in blocking solution followed by Alexa Fluor 488–, AlexaFluor 594–, or Cy5-conjugated secondary antibodies (1:500) for 1 hour.Immunofluorescence was performed using a Leica TSC SP2 AOBS TCSconfocal microscope or an Olympus FV10i microscope with 543- and488-nm channels for visualizing red and green fluorescence. Images weretaken at ×63 magnification. At least three random fields were selectedfor images.
Visualization of S1P in nuclear membranes using PLAA549 cells or wild-type and SK2-deficient MEFs were grown inDMEM growth medium. Cells were then fixed with formalin. Fixedand permeabilized cells were incubated with an antibody specific for S1P(Sphingomab, LT1002; 20 mg/ml) and an antibody that recognizes hTERTat 4°C for 18 hours. PLA was then performed and visualized by IF-CM,using the Duolink in situ hybridization kit as described by the manufac-turer (Olink Biosciences).
Stable expression of vector, hTERTWT, and hTERTD684A
in A549 cellspBabe-puro, pBabe-WT, and pBabe-hTERTD684A plasmids were obtainedfrom Addgene. Plat-A amphotropic cells were plated at 70% confluenceand transfected with the plasmids using the Effectene transfection reagentfor 48 hours. After centrifugation of viral supernatant at 1250 rpm for 5min,it was filtered through a 0.45-mm filter, and viral transduction of A549 cellswere performed using polybrene (8 mg/ml). The selection of transfectedcells was performed using puromycin (1 mg/ml) for 14 days. Westernblotting was performed to assess the expression of hTERT.
Detection of MKRN1-hTERT interactionColocalization of MKRN1 and hTERT in primary human lung fibroblastswas carried out by PLA using the Duolink in situ hybridization kit (OlinkBiosciences). Antibodies against MKRN1 (ab119096; 1:50) and hTERT(ab32020; 1:50) were used for 18 hours at 4°C, and PLA signals were de-tected using IF-CM and the Duolink ImageTool (Olink Biosciences). Asso-ciation of MKRN1 and hTERTwas also detected by immunoprecipitationand Western blotting of A549 and H1650 cells expressing vector, SK2WT,and SK2G212E in the absence or presence of 5 mM GA for 4 hours.
TIF assayA549 cells and primary human lung fibroblasts were fixed using 4% form-aldehyde for 20 min and blocked with 1% goat serum in 0.3M glycine, 1%BSA, and 0.1% Tween 20 in 1× PBS (pH 7.4) for 2 hours. The fixed cellswere then incubatedwith primary antibodies that recognize g-H2AX (5 mg/ml;ab2893, Abcam) and TRF2 (5 mg/ml; IMG-124A, Imgenex) for 18 hours.They were then incubated with secondary antibodies containing red (Alexa568) and blue (Cy5) fluorophores against g-H2AX and TRF2, respectively(colocalization resulted in purple images). Imageswere captured using IF-CM,and colocalization was quantified using ImageJ Fiji software (48, 49).
TRAP assayCellswere lysed in 1×CHAPSbuffer, and total proteinwas quantified usingBradford assay. Onemicrogram of total protein was used per assay reaction.Assay conditions were followed as per the manufacturer’s instructions(Millipore), and the reaction products were separated on a 12.5% acrylam-ide gel under nonreducing conditions. The TRAP products on gels werethen stained using SYBR Green (Invitrogen).
ww
TRF length measurementTRF length analysis using genomic DNAs was performed using the TRFassay kit (Roche) and the Telo TAGGG probe by Southern blotting as de-scribed by the manufacturer.
Detection of senescence by senescence-associatedb-gal stainingPrimary human lung fibroblasts andMEFswere used up to 17 and 6 passages,respectively, for the detection of senescence using the senescence-associatedb-gal assay kit as described by themanufacturer (Cell Signaling Technology).
Anchorage-independent growth on soft agarCells were grown in 2× DMEM and 5% agar at a ratio of 9:1. Around20,000 cells per well were grown in agar medium mix and incubated at37°C. DMEM was changed for cells every 3 days for 14 to 21 days.
Animal xenograft studiesSevere combined immunodeficient (SCID) mice were purchased fromHarlan Laboratories. Age- and sex-matched mice were used. All animalprotocols were approved by the Institutional Animal Care and Use Com-mittee at the Medical University of South Carolina. A549 cells (2 × 106)stably transfected with control shRNA or shRNA targeting SK2 were im-planted into the flanks of SCID mice (n = 4 mice, each containing twotumors on both flanks).When the tumors were palpable, the mice weretreated daily with either vehicle control or ABC294640 by oral gavageat 100 mg/kg for 21 days. Tumor volume was measured using digitalcalipers. At the end of the 21-day treatment, the mice were euthanizedand tumor tissues were collected. Similarly, LLC cells stably transfectedwith shRNA against SK2 or control (scrambled) shRNA were injectedsubcutaneously into either wild-type or SK2-deficient C57/BL6 mice (n =4 mice, each containing two tumors on both flanks). Allografted tumorvolumes were measured using digital calipers every 3 days for 14 days.
Statistical analysisAll data are presented as means ± SD, and group comparisons were per-formed with a two-tailed Student’s t test. P < 0.05 was considered statisti-cally significant.
SUPPLEMENTARY MATERIALSwww.sciencesignaling.org/cgi/content/full/8/381/ra58/DC1Fig. S1. Roles of SK1 versus SK2 on hTERT abundance and 17C-S1P binding.Fig. S2. S1P-TERT binding is mediated by the C′3-OH of S1P and the hydrophobic regionof TERT between the thumb and finger domains.Fig. S3. hTERT colocalizes with lamin B at the nuclear periphery.Fig. S4. Detection of stably expressed hTERT in MEFs.Fig. S5. Effects of S1P-hTERT binding on hTERT-MKRN1 interaction and growth inhibi-tion in response to GA treatment.Fig. S6. S1P binding might mimic protein phosphorylation of hTERT at Ser921.Fig. S7. Effects of SK2-generated S1P on hTERT-dependent senescence.Fig. S8. Regulation of senescence by S1P-hTERT binding in wild-type or SK2-deficient MEFs.
REFERENCES AND NOTES1. E. H. Blackburn, K. Collins, Telomerase: An RNP enzyme synthesizes DNA. Cold
Spring Harb. Perspect. Biol. 3, a003558 (2011).2. C. W. Greider, E. H. Blackburn, The telomere terminal transferase of Tetrahymena is
a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51, 887–898(1987).
3. C. M. Counter, W. C. Hahn, W. Wei, S. D. Caddle, R. L. Beijersbergen, P. M. Lansdorp,J. M. Sedivy, R. A. Weinberg, Dissociation among in vitro telomerase activity, telomeremaintenance, and cellular immortalization. Proc. Natl. Acad. Sci. U.S.A. 95, 14723–14728(1998).
w.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 11

R E S E A R C H A R T I C L E
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from
4. T. de Lange, Telomeres and senescence: Ending the debate. Science 279, 334–335(1998).
5. J. W. Shay, W. E. Wright, Senescence and immortalization: Role of telomeres andtelomerase. Carcinogenesis 26, 867–874 (2005).
6. S. B. Cohen, M. E. Graham, G. O. Lovrecz, N. Bache, P. J. Robinson, R. R. Reddel,Protein composition of catalytically active human telomerase from immortal cells. Science315, 1850–1853 (2007).
7. C. Günes, K. L. Rudolph, The role of telomeres in stem cells and cancer. Cell 152,390–393 (2013).
8. A. G. Bodnar, M. Ouellette, M. Frolkis, S. E. Holt, C. P. Chiu, G. B. Morin, C. B. Harley,J. W. Shay, S. Lichtsteiner, W. E. Wright, Extension of life-span by introduction oftelomerase into normal human cells. Science 279, 349–352 (1998).
9. B. Fu, J. Quintero, C. C. Baker, Keratinocyte growth conditions modulate telomeraseexpression, senescence, and immortalization by human papillomavirus type 16 E6and E7 oncogenes. Cancer Res. 63, 7815–7824 (2003).
10. W.C.Hahn,S.A.Stewart,M.W.Brooks,S.G.York,E.Eaton,A.Kurachi,R. L.Beijersbergen,J. H. Knoll, M. Meyerson, R. A. Weinberg, Inhibition of telomerase limits the growth ofhuman cancer cells. Nat. Med. 5, 1164–1170 (1999).
11. J. W. Shay, I. B. Roninson, Hallmarks of senescence in carcinogenesis and cancertherapy. Oncogene 23, 2919–2933 (2004).
12. B. Burke, C. L. Stewart, The nuclear lamins: Flexibility in function. Nat. Rev. Mol. CellBiol. 14, 13–24 (2013).
13. H. C. Ferreira, B. Luke, H. Schober, V. Kalck, J. Lingner, S. M. Gasser, The PIAShomologue Siz2 regulates perinuclear telomere position and telomerase activity inbudding yeast. Nat. Cell Biol. 13, 867–874 (2011).
14. H. Li, L. L. Zhao, J. W. Funder, J. P. Liu, Protein phosphatase 2A inhibits nucleartelomerase activity in human breast cancer cells. J. Biol. Chem. 272, 16729–16732(1997).
15. 15.Y. A. Hannun, L. M. Obeid, Principles of bioactive lipid signalling: Lessons fromsphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 (2008).
16. R. Kolesnick, Z. Fuks, Radiation and ceramide-induced apoptosis. Oncogene 22,5897–5906 (2003).
17. B. Ogretmen, Y. A. Hannun, Biologically active sphingolipids in cancer pathogenesisand treatment. Nat. Rev. Cancer 4, 604–616 (2004).
18. N. J. Pyne, S. Pyne, Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 10,489–503 (2010).
19. S. Spiegel, S. Milstien, Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat.Rev. Mol. Cell Biol. 4, 397–407 (2003).
20. L. G. Wooten-Blanks, P. Song, C. E. Senkal, B. Ogretmen, Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via de-acetylation of Sp3 by histone deacetylase 1. FASEB J. 21, 3386–3397 (2007).
21. S. E. Alvarez, K. B. Harikumar, N. C. Hait, J. Allegood, G. M. Strub, E. Y. Kim,M. Maceyka, H. Jiang, C. Luo, T. Kordula, S. Milstien, S. Spiegel, Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465,1084–1088 (2010).
22. N. C. Hait, J. Allegood, M. Maceyka, G. M. Strub, K. B. Harikumar, S. K. Singh, C. Luo,R. Marmorstein, T. Kordula, S. Milstien, S. Spiegel, Regulation of histone acetylation inthe nucleus by sphingosine-1-phosphate. Science 325, 1254–1257 (2009).
23. N. Hirata, S. Yamada, T. Shoda, M. Kurihara, Y. Sekino, Y. Kanda, Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat. Commun. 5, 4806 (2014).
24. J. Liang, M. Nagahashi, E. Y. Kim, K. B. Harikumar, A. Yamada, W. C. Huang, N. C. Hait,J. C. Allegood, M. M. Price, D. Avni, K. Takabe, T. Kordula, S. Milstien, S. Spiegel,Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflam-mation, and development of colitis-associated cancer. Cancer Cell 23, 107–120(2013).
25. S. Ponnusamy, S. P. Selvam, S. Mehrotra, T. Kawamori, A. J. Snider, L. M. Obeid,Y. Shao, R. Sabbadini, B. Ogretmen, Communication between host organism andcancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol. Med. 4, 761–775(2012).
26. G. M. Strub, M. Paillard, J. Liang, L. Gomez, J. C. Allegood, N. C. Hait, M. Maceyka,M. M. Price, Q. Chen, D. C. Simpson, T. Kordula, S. Milstien, E. J. Lesnefsky, S. Spiegel,Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interactswith prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25,600–612 (2011).
27. P. Gao, Y. K. Peterson, R. A. Smith, C. D. Smith, Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLOS One 7, e44543 (2012).
28. S. Spassieva, J. Bielawski, V. Anelli, L. M. Obeid, Combination of C17 sphingoid basehomologues and mass spectrometry analysis as a new approach to study sphingo-lipid metabolism. Methods Enzymol. 434, 233–241 (2007).
29. S. A. Saddoughi, S. Gencer, Y. K. Peterson, K. E. Ward, A. Mukhopadhyay, J. Oaks,J. Bielawski, Z. M. Szulc, R. J. Thomas, S. P. Selvam, C. E. Senkal, E. Garrett-Mayer,
ww
R. M. De Palma, D. Fedarovich, A. Liu, A. A. Habib, R. V. Stahelin, D. Perrotti,B. Ogretmen, Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediateslung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBOMol. Med. 5, 105–121 (2013).
30. R. D. Sentelle, C. E. Senkal, W. Jiang, S. Ponnusamy, S. Gencer, S. P. Selvam,V. K. Ramshesh, Y. K. Peterson, J. J. Lemasters, Z. M. Szulc, J. Bielawski, B. Ogretmen,Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat.Chem. Biol. 8, 831–838 (2012).
31. A. J. Gillis, A. P. Schuller, E. Skordalakes, Structure of the Tribolium castaneum telomer-ase catalytic subunit TERT. Nature 455, 633–637 (2008).
32. M. Xie, J. D. Podlevsky, X. Qi, C. J. Bley, J. J. Chen, A novel motif in telomerasereverse transcriptase regulates telomere repeat addition rate and processivity. NucleicAcids Res 38, 1982–1996 (2010).
33. K. E. Ward, N. Bhardwaj, M. Vora, C. E. Chalfant, H. Lu, R. V. Stahelin, The molecularbasis of ceramide-1-phosphate recognition by C2 domains. J. Lipid Res. 54, 636–648(2013).
34. B. Visentin, J. A. Vekich, B. J. Sibbald, A. L. Cavalli, K. M. Moreno, R. G. Matteo,W. A. Garland, Y. Lu, S. Yu, H. S. Hall, V. Kundra, G. B. Mills, R. A. Sabbadini, Validationof an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducinggrowth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 9, 225–238(2006).
35. J. H. Kim, S. M. Park, M. R. Kang, S. Y. Oh, T. H. Lee, M. T. Muller, I. K. Chung,Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis ofhTERT. Genes Dev. 19, 776–781 (2005).
36. K. Fujita, I. Horikawa, A. M. Mondal, L. M. Jenkins, E. Appella, B. Vojtesek, J. C. Bourdon,D. P. Lane, C. C. Harris, Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat. Cell Biol. 12, 1205–1212 (2010).
37. M. Fumagalli, F. Rossiello, M. Clerici, S. Barozzi, D. Cittaro, J. M. Kaplunov, G. Bucci,M. Dobreva, V. Matti, C. M. Beausejour, U. Herbig, M. P. Longhese, F. d’Adda di Fagagna,Telomeric DNA damage is irreparable and causes persistent DNA-damage-responseactivation. Nat. Cell Biol. 14, 355–365 (2012).
38. M. Braig, S. Lee, C. Loddenkemper, C. Rudolph, A. H. Peters, B. Schlegelberger,H. Stein, B. Dörken, T. Jenuwein, C. A. Schmitt, Oncogene-induced senescence as aninitial barrier in lymphoma development. Nature 436, 660–665 (2005).
39. Z. Chen, L. C. Trotman, D. Shaffer, H. K. Lin, Z. A. Dotan, M. Niki, J. A. Koutcher,H. I. Scher, T. Ludwig, W. Gerald, C. Cordon-Cardo, P. P. Pandolfi, Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature436, 725–730 (2005).
40. C. Michaloglou, L. C. Vredeveld, M. S. Soengas, C. Denoyelle, T. Kuilman, C. M. van der Horst,D. M. Majoor, J. W. Shay, W. J. Mooi, D. S. Peeper, BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 (2005).
41. C. F. Snook, J. A. Jones, Y. A. Hannun, Sphingolipid-binding proteins. Biochim. Biophys.Acta 1761, 927–946 (2006).
42. J. Briscoe, P. P. Thérond, The mechanisms of Hedgehog signalling and its roles indevelopment and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 (2013).
43. M. H. Gelb, Protein prenylation, et cetera: Signal transduction in two dimensions. Science275, 1750–1751 (1997).
44. Y. Ichimura, T. Kirisako, T. Takao, Y. Satomi, Y. Shimonishi, N. Ishihara, N. Mizushima,I. Tanida, E. Kominami, M. Ohsumi, T. Noda, Y. Ohsumi, A ubiquitin-like system med-iates protein lipidation. Nature 408, 488–492 (2000).
45. K. A. Gelato, M. Tauber, M. S. Ong, S. Winter, K. Hiragami-Hamada, J. Sindlinger,A. Lemak, Y. Bultsma, S. Houliston, D. Schwarzer, N. Divecha, C. H. Arrowsmith,W. Fischle, Accessibility of different histone H3-binding domains of UHRF1 is allosteri-cally regulated by phosphatidylinositol 5-phosphate. Mol. Cell 54, 905–919 (2014).
46. M. A. Blasco, H. W. Lee, M. P. Hande, E. Samper, P. M. Lansdorp, R. A. DePinho,C. W. Greider, Telomere shortening and tumor formation by mouse cells lacking telomer-ase RNA. Cell 91, 25–34 (1997).
47. C. W. Greider, Telomerase RNA levels limit the telomere length equilibrium. ColdSpring Harb. Symp. Quant. Biol. 71, 225–229 (2006).
48. U. Herbig, W. A. Jobling, B. P. Chen, D. J. Chen, J. M. Sedivy, Telomere shorteningtriggers senescence of human cells through a pathway involving ATM, p53, andp21CIP1, but not p16INK4a. Mol. Cell 14, 501–513 (2004).
49. H. Takai, A. Smogorzewska, T. de Lange, DNA damage foci at dysfunctional telo-meres. Curr. Biol. 13, 1549–1556 (2003).
Acknowledgments: Sphingomab was provided to us by R. Sabbadini (Lpath Inc.). Wethank J. J. Chen (Arizona State University) and S. Spiegel (Virginia Commonwealth Univer-sity) for providing us with hTERT and SK2 constructs, respectively. We also thank Z. M. Szulc(Lipidomics Shared Resource Facility, Medical University of South Carolina) for providing uswith S1P analogs. Funding: This work was supported by research funding from the NIH(CA088932, CA173687, and DE016572 to B.O.), as was the construction of the core facilitiesused in this study (C06 RR015455). Additional funding was provided by the BiostatisticsShared Resource, Hollings Cancer Center, Medical University of South Carolina (P30CA138313 to E.G.-M.). Author contributions: S.P.S. performed experiments and helped
w.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 12

R E S E A R C H A R T I C L E
with study design, data analysis, preparation of figures, and writing of the manuscript;R.M.D.P., J.J.O., N.O., and S.P. performed experiments; Y.K.P and E.S. performedmolecular modeling and docking studies; R.V.S. conducted SPR studies; E.G.-M.performed statistical analyses; and C.D.S. provided ABC294640 and contributed tothe design of the experiments. B.O. developed concepts and hypotheses, designedexperiments, analyzed data, and wrote the manuscript. Competing interests: C.D.S. isthe founder of Apogee Inc, which developed ABC294640. The other authors declare thatthey have no competing financial interests. Data and materials availability: A materialstransfer agreement is required for the Sphingomab (S1P) antibody.
ww
Submitted 15 December 2014Accepted 29 May 2015Final Publication 16 June 201510.1126/scisignal.aaa4998Citation: S. Panneer Selvam, R. M. De Palma, J. J. Oaks, N. Oleinik, Y. K. Peterson,R. V. Stahelin, E. Skordalakes, S. Ponnusamy, E. Garrett-Mayer, C. D. Smith,B. Ogretmen, Binding of the sphingolipid S1P to hTERT stabilizes telomerase at thenuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 8,ra58 (2015).
w.SCIENCESIGNALING.org 16 June 2015 Vol 8 Issue 381 ra58 13
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from

allosterically mimicking protein phosphorylationBinding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by
Emmanuel Skordalakes, Suriyan Ponnusamy, Elizabeth Garrett-Mayer, Charles D. Smith and Besim OgretmenShanmugam Panneer Selvam, Ryan M. De Palma, Joshua J. Oaks, Natalia Oleinik, Yuri K. Peterson, Robert V. Stahelin,
DOI: 10.1126/scisignal.aaa4998 (381), ra58.8Sci. Signal.
xenografts in mice.telomere maintenance and promoted senescence in cultured cells and decreased the growth of lung cancer cell the interaction between S1P and TERT by either depleting S1P or mutating the interaction site in TERT impairedbinding of the phospholipid S1P to TERT mimicked protein phosphorylation and prevented TERT degration. Disrupting
. found thatet alsubunit (called TERT) of this enzyme. In both normal fibroblasts and lung cancer cells, Panneer Selvam senescent. The enzyme telomerase maintains the integrity of telomeres, and phosphorylation stabilizes the catalyticshorter with each replication cycle. When telomeres get too short or damaged, the cell stops dividing and becomes
In normal adult cells, the structure at the ends of chromosomes, called the telomere, becomes progressivelyTelomerase stabilized by a sphingolipid
ARTICLE TOOLS http://stke.sciencemag.org/content/8/381/ra58
MATERIALSSUPPLEMENTARY http://stke.sciencemag.org/content/suppl/2015/06/12/8.381.ra58.DC1
CONTENTRELATED
http://stke.sciencemag.org/content/sigtrans/10/496/eaap9030.fullhttp://stke.sciencemag.org/content/sigtrans/9/456/ec283.abstracthttp://science.sciencemag.org/content/sci/336/6088/1549.fullhttp://science.sciencemag.org/content/sci/336/6088/1519.fullhttp://science.sciencemag.org/content/sci/350/6260/aab4070.fullhttp://stke.sciencemag.org/content/sigtrans/8/389/pc20.fullhttp://stke.sciencemag.org/content/sigtrans/8/389/ra79.fullhttp://science.sciencemag.org/content/sci/348/6238/1036.fullhttp://science.sciencemag.org/content/sci/347/6225/1006.fullhttp://stke.sciencemag.org/content/sigtrans/5/230/ec173.abstract
REFERENCES
http://stke.sciencemag.org/content/8/381/ra58#BIBLThis article cites 49 articles, 14 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.Science SignalingYork Avenue NW, Washington, DC 20005. The title (ISSN 1937-9145) is published by the American Association for the Advancement of Science, 1200 NewScience Signaling
Copyright © 2015, American Association for the Advancement of Science
on January 2, 2020http://stke.sciencem
ag.org/D
ownloaded from