basic & clinical pharmacology - psau.edu.sa...basic & clinical pharmacology edited by...
TRANSCRIPT

Chapter VIII
Chemotherapeutic Drugs
From
Basic & Clinical
Pharmacology Book


Basic & ClinicalPharmacology
Edited by
Bertram G. Katzung, MD, PhDProfessor EmeritusDepartment of Cellular & Molecular PharmacologyUniversity of California, San Francisco
Associate Editors
Susan B. Masters, PhDProfessor of Pharmacology & Academy Chair of Pharmacology EducationDepartment of Cellular & Molecular PharmacologyUniversity of California, San Francisco
Anthony J. Trevor, PhDProfessor EmeritusDepartment of Cellular & Molecular PharmacologyUniversity of California, San Francisco
Twelfth Edition
New York Chicago San Francisco Lisbon London Madrid Mexico CityMilan New Delhi San Juan Seoul Singapore Sydney Toronto
New York Chicago San Francisco Lisbon London Madrid Mexico CityMiMillann NNew DDelhlhii SSann JJu nan S Seo lul S Siningapporre S S dydnney TTorrontnto
a LANGE medical book
Katzung_FM.indd i 9/24/11 11:46:42 AM

Copyright © 2012 by The McGraw-Hill Companies, Inc. All rights reserved. Except as permitted under the United States Copyright Act of 1976, no part of this publication may be reproduced or distributed in any form or by any means, or stored in a database or retrieval system, without the prior written permission of the publisher.
ISBN: 978-0-07-176402-5
MHID: 0-07-176402-X
The material in this eBook also appears in the print version of this title: ISBN: 978-0-07-176401-8, MHID: 0-07-176401-1.
All trademarks are trademarks of their respective owners. Rather than put a trademark symbol after every occurrence of a trademarked name, we use names in an editorial fashion only, and to the benefi t of the trademark owner, with no intention of infringement of the trademark. Where such designations appear in this book, they have been printed with initial caps.
McGraw-Hill eBooks are available at special quantity discounts to use as premiums and sales promotions, or for use in corporate training programs. To contact a representative please e-mail us at [email protected].
Previous editions copyright © 2010, 2009, 2007, 2004, 2001 by McGraw-Hill Companies, Inc.; copyright © 1998, 1995, 1992, 1989, 1987 by Appleton & Lange; copyright © 1984, 1982 by Lange Medical Publications
Notice
Medicine is an ever-changing science. As new research and clinical experience broaden our knowledge, changes in treatment and drug therapy are required. The authors and the publisher of this work have checked with sources believed to be reliable in their efforts to provide information that is complete and generally in accord with the standards accepted at the time of publication. However, in view of the possibility of human error or changes in medical sciences, neither the authors nor the publisher nor any other party who has been involved in the preparation or publication of this work warrants that the information contained herein is in every respect accurate or complete, and they disclaim all responsibility for any errors or omissions or for the results obtained from use of the information contained in this work. Readers are encouraged to confi rm the information contained herein with other sources. For example and in particular, readers are advised to check the product information sheet included in the package of each drug they plan to administer to be certain that the information contained in this work is accurate and that changes have not been made in the recommended dose or in the contraindications for administration. This recommendationis of particular importance in connection with new or infrequently used drugs.
TERMS OF USE
This is a copyrighted work and The McGraw-Hill Companies, Inc. (“McGrawHill”) and its licensors reserve all rights in and to the work. Use of this work is subject to these terms. Except as permitted under the Copyright Act of 1976 and the right to store and retrieve one copy of the work, you may not decompile, disassemble, reverse engineer, reproduce, modify, create derivative works based upon, transmit, distribute, disseminate, sell, publish or sublicense the work or any part of it without McGraw-Hill’s prior consent. You may use the work for your own noncommercial and personal use; any other use of the work is strictly prohibited. Your right to use the work may be terminated if you fail to comply with these terms.
THE WORK IS PROVIDED “AS IS.” McGRAW-HILL AND ITS LICENSORS MAKE NO GUARANTEES OR WARRANTIES AS TO THE ACCURACY, ADEQUACY OR COMPLETENESS OF OR RESULTS TO BE OBTAINED FROM USING THE WORK, INCLUDING ANY INFORMATION THAT CAN BE ACCESSED THROUGH THE WORK VIA HYPERLINK OR OTHERWISE, AND EXPRESSLY DISCLAIM ANY WARRANTY, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO IMPLIED WARRANTIES OF MERCHANTABILITY OR FITNESS FOR A PARTICULAR PURPOSE. McGraw-Hill and its licensors do not warrant or guarantee that the functions contained in the work will meet your requirements or that its operation will be uninterrupted or error free. Neither McGraw-Hill nor its licensors shall be liable to you or anyone else for any inaccuracy, error or omission, regardless of cause, in the work or for any damages resulting therefrom. McGraw-Hill has no responsibility for the content of any information accessed through the work. Under no circumstances shall McGraw-Hill and/or its licensors be liable for any indirect, incidental, special, punitive, consequential or similar damages that result from the use of or inability to use the work, even if any of them has been advised of the possibility of such damages. This limitation of liability shall apply to any claim or cause whatsoever whether such claim or cause arises in contract, tort or otherwise.

S E C T I O N IBASIC PRINCIPLES 1
1. IntroductionBertram G. Katzung, MD, PhD 1
2. Drug Receptors & PharmacodynamicsMark von Zastrow, MD, PhD 15
3. Pharmacokinetics & Pharmacodynamics: Rational Dosing & the Time Course of Drug ActionNicholas H. G. Holford, MB, ChB, FRACP 37
4. Drug BiotransformationMaria Almira Correia, PhD 53
5. Development & Regulation of DrugsBertram G. Katzung, MD, PhD 69
S E C T I O N IIAUTONOMIC DRUGS 79
6. Introduction to Autonomic PharmacologyBertram G. Katzung, MD, PhD 79
7. Cholinoceptor-Activating & Cholinesterase-Inhibiting DrugsAchilles J. Pappano, PhD 97
8. Cholinoceptor-Blocking DrugsAchilles J. Pappano, PhD 115
9. Adrenoceptor Agonists & Sympathomimetic DrugsItalo Biaggioni, MD, & David Robertson, MD 129
10. Adrenoceptor Antagonist DrugsDavid Robertson, MD, & Italo Biaggioni, MD 151
S E C T I O N IIICARDIOVASCULAR-RENAL DRUGS 169
11. Antihypertensive AgentsNeal L. Benowitz, MD 169
12. Vasodilators & the Treatment of Angina PectorisBertram G. Katzung, MD, PhD 193
13. Drugs Used in Heart FailureBertram G. Katzung, MD, PhD 211
14. Agents Used in Cardiac ArrhythmiasJoseph R. Hume, PhD, & Augustus O. Grant, MD, PhD 227
15. Diuretic Agents Harlan E. Ives, MD, PhD 251
S E C T I O N IVDRUGS WITH IMPORTANT ACTIONS ON SMOOTH MUSCLE 273
16. Histamine, Serotonin, & the Ergot AlkaloidsBertram G. Katzung, MD, PhD 273
17. Vasoactive PeptidesIan A. Reid, PhD 295
18. The Eicosanoids: Prostaglandins, Thromboxanes, Leukotrienes, & Related CompoundsEmer M. Smyth, PhD, & Garret A. FitzGerald, MD 313
19. Nitric OxideSamie R. Jaffrey, MD, PhD 331
ContentsSchedule of Controlled Drugs Inside Front CoverPreface viiAuthors ixKey Features xii
iii
Katzung_FM.indd iii 9/24/11 11:46:43 AM

iv CONTENTS
20. Drugs Used in AsthmaHomer A. Boushey, MD 339
S E C T I O N VDRUGS THAT ACT IN THECENTRAL NERVOUS SYSTEM 359
21. Introduction to the Pharmacology of CNS DrugsRoger A. Nicoll, MD 359
22. Sedative-Hypnotic DrugsAnthony J. Trevor, PhD, & Walter L. Way, MD 373
23. The AlcoholsSusan B. Masters, PhD 389
24. Antiseizure DrugsRoger J. Porter, MD, &Brian S. Meldrum, MB, PhD 403
25. General AnestheticsHelge Eilers, MD, & Spencer Yost, MD 429
26. Local AnestheticsKenneth Drasner, MD 449
27. Skeletal Muscle RelaxantsMarieke Kruidering-Hall, PhD, & Lundy Campbell, MD 465
28. Pharmacologic Management of Parkinsonism & Other Movement DisordersMichael J. Aminoff, MD, DSc, FRCP 483
29. Antipsychotic Agents & LithiumHerbert Meltzer, MD, PhD 501
30. Antidepressant AgentsCharles DeBattista, MD 521
31. Opioid Analgesics & AntagonistsMark A. Schumacher, PhD, MD, Allan I. Basbaum, PhD, & Walter L. Way, MD 543
32. Drugs of AbuseChristian Lüscher, MD 565
iv CONTENTS
S E C T I O N VIDRUGS USED TO TREAT DISEASES OF THE BLOOD, INFLAMMATION, & GOUT 581
33. Agents Used in Anemias; Hematopoietic Growth FactorsSusan B. Masters, PhD 581
34. Drugs Used in Disorders of CoagulationJames L. Zehnder, MD 601
35. Agents Used in DyslipidemiaMary J. Malloy, MD, & John P. Kane, MD, PhD 619
36. Nonsteroidal Anti-Inflammatory Drugs, Disease-Modifying Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in GoutDaniel E. Furst, MD, Robert W. Ulrich, PharmD, & Shraddha Prakash, MD 635
S E C T I O N VIIENDOCRINE DRUGS 659
37. Hypothalamic & Pituitary HormonesSusan B. Masters, PhD, & Stephen M. Rosenthal, MD 659
38. Thyroid & Antithyroid DrugsBetty J. Dong, PharmD, FASHP, FCCP, & Francis S. Greenspan, MD, FACP 681
39. Adrenocorticosteroids & Adrenocortical AntagonistsGeorge P. Chrousos, MD 697
40. The Gonadal Hormones & InhibitorsGeorge P. Chrousos, MD 715
41. Pancreatic Hormones & Antidiabetic DrugsMartha S. Nolte Kennedy, MD 743
42. Agents That Affect Bone Mineral HomeostasisDaniel D. Bikle, MD, PhD 769
Katzung_FM.indd iv 9/24/11 11:46:43 AM

CONTENTS v
S E C T I O N VIIICHEMOTHERAPEUTIC DRUGS 789
43. Beta-Lactam & Other Cell Wall- & Membrane-Active AntibioticsDaniel H. Deck, PharmD, & Lisa G. Winston, MD 790
44. Tetracyclines, Macrolides, Clindamycin, Chloramphenicol, Streptogramins, & OxazolidinonesDaniel H. Deck, PharmD, & Lisa G. Winston, MD 809
45. Aminoglycosides & SpectinomycinDaniel H. Deck, PharmD, &Lisa G. Winston, MD 821
46. Sulfonamides, Trimethoprim, & QuinolonesDaniel H. Deck, PharmD, & Lisa G. Winston, MD 831
47. Antimycobacterial DrugsDaniel H. Deck, PharmD, & Lisa G. Winston, MD 839
48. Antifungal AgentsDon Sheppard, MD, & Harry W. Lampiris, MD 849
49. Antiviral AgentsSharon Safrin, MD 861
50. Miscellaneous Antimicrobial Agents; Disinfectants, Antiseptics, & SterilantsDaniel H. Deck, PharmD, & Lisa G. Winston, MD 891
51. Clinical Use of Antimicrobial AgentsHarry W. Lampiris, MD, & Daniel S. Maddix, PharmD 901
52. Antiprotozoal DrugsPhilip J. Rosenthal, MD 915
53. Clinical Pharmacology of the Antihelminthic DrugsPhilip J. Rosenthal, MD 937
54. Cancer ChemotherapyEdward Chu, MD, & Alan C. Sartorelli, PhD 949
55. ImmunopharmacologyDouglas F. Lake, PhD, Adrienne D. Briggs, MD, & Emmanuel T. Akporiaye, PhD 977
S E C T I O N IXTOXICOLOGY 1001
56. Introduction to Toxicology: Occupational & EnvironmentalDaniel T. Teitelbaum, MD 1001
57. Heavy Metal Intoxication & ChelatorsMichael J. Kosnett, MD, MPH 1013
58. Management of the Poisoned PatientKent R. Olson, MD 1027
S E C T I O N XSPECIAL TOPICS 1039
59. Special Aspects of Perinatal & Pediatric PharmacologyGideon Koren, MD 1039
60. Special Aspects of Geriatric PharmacologyBertram G. Katzung, MD, PhD 1051
61. Dermatologic PharmacologyDirk B. Robertson, MD, & Howard I. Maibach, MD 1061
62. Drugs Used in the Treatment of Gastrointestinal DiseasesKenneth R. McQuaid, MD 1081
63. Therapeutic & Toxic Potential of Over-the-Counter AgentsRobin L. Corelli, PharmD 1115
64. Dietary Supplements & Herbal MedicationsCathi E. Dennehy, PharmD, & Candy Tsourounis, PharmD 1125
CONTENTS v
Katzung_FM.indd v 9/24/11 11:46:43 AM

vi CONTENTS
65. Rational Prescribing & Prescription WritingPaul W. Lofholm, PharmD, &Bertram G. Katzung, MD, PhD 1139
66. Important Drug Interactions & Their MechanismsJohn R. Horn, PharmD, FCCP 1149
Appendix: Vaccines, Immune Globulins, & Other Complex Biologic ProductsHarry W. Lampiris, MD, & Daniel S. Maddix, PharmD 1163
Index 1171
Katzung_FM.indd vi 9/24/11 11:46:43 AM

The twelfth edition of Basic & Clinical Pharmacology continues the important changes inaugurated in the eleventh edition, with extensive use of full-color illustrations and expanded coverage of transporters, pharmacogenomics, and new drugs. Case studies have been added to several chapters and answers to questions posed in the case studies now appear at the end of each chapter. As in prior editions, the book is designed to provide a compre-hensive, authoritative, and readable pharmacology textbook for students in the health sciences. Frequent revision is necessary to keep pace with the rapid changes in pharmacology and therapeu-tics; the 2–3 year revision cycle of the printed text is among the shortest in the field and the availability of an online version pro-vides even greater currency. In addition to the full-color illustra-tions, other new features have been introduced. The Case Study Answer section at the end of chapters will make the learning pro-cess even more interesting and efficient. The book also offers special features that make it a useful reference for house officers and practicing clinicians.
Information is organized according to the sequence used in many pharmacology courses and in integrated curricula: basic principles; autonomic drugs; cardiovascular-renal drugs; drugs with important actions on smooth muscle; central nervous system drugs; drugs used to treat inflammation, gout, and diseases of the blood; endocrine drugs; chemotherapeutic drugs; toxicology; and special topics. This sequence builds new information on a founda-tion of information already assimilated. For example, early presen-tation of autonomic nervous system pharmacology allows students to integrate the physiology and neuroscience they have learned elsewhere with the pharmacology they are learning and prepares them to understand the autonomic effects of other drugs. This is especially important for the cardiovascular and central nervous system drug groups. However, chapters can be used equally well in courses and curricula that present these topics in a different sequence.
Within each chapter, emphasis is placed on discussion of drug groups and prototypes rather than offering repetitive detail about individual drugs. Selection of the subject matter and the order of its presentation are based on the accumulated experience of teach-ing this material to thousands of medical, pharmacy, dental, podiatry, nursing, and other health science students.
Major features that make this book particularly useful in inte-grated curricula include sections that specifically address the clini-cal choice and use of drugs in patients and the monitoring of their effects—in other words, clinical pharmacology is an integral part of this text. Lists of the commercial preparations available, including
trade and generic names and dosage formulations, are provided at the end of each chapter for easy reference by the house officer or practitioner writing a chart order or prescription.
Significant revisions in this edition include:• In addition to the Case Studies used to open many chapters,
Case Study Answers at the end of these chapters provide an introduction to the clinical applications of the drugs discussed.
• A Drug Summary Table is placed at the conclusion of most chapters; these provide a concise recapitulation of the most important drugs.
• Many new illustrations in full color provide significantly more information about drug mechanisms and effects and help to clarify important concepts.
• Major revisions of the chapters on sympathomimetic, sym-pathoplegic, antipsychotic, antidepressant, antidiabetic, anti-inflammatory, and antiviral drugs, prostaglandins, nitric oxide, hypothalamic and pituitary hormones, and immuno-pharmacology.
• Continued expansion of the coverage of general concepts relat-ing to newly discovered receptors, receptor mechanisms, and drug transporters.
• Descriptions of important new drugs released through August 2011.
An important related educational resource is Katzung & Trevor’s Pharmacology: Examination & Board Review, ninth edition (Trevor AJ, Katzung BG, & Masters SB: McGraw-Hill, 2010). This book provides a succinct review of pharmacology with over one thousand sample examination questions and answers. It is especially helpful to students preparing for board-type examina-tions. A more highly condensed source of information suitable for review purposes is USMLE Road Map: Pharmacology, second edition (Katzung BG, Trevor AJ: McGraw-Hill, 2006).
This edition marks the 30th year of publication of Basic & Clinical Pharmacology. The widespread adoption of the first eleven editions indicates that this book fills an important need. We believe that the twelfth edition will satisfy this need even more successfully. Spanish, Portuguese, Italian, French, Indonesian, Japanese, Korean, and Turkish translations are available. Translations into other languages are under way; the publisher may be contacted for further information.
I wish to acknowledge the prior and continuing efforts of my contributing authors and the major contributions of the staff at Lange Medical Publications, Appleton & Lange, and McGraw-Hill,
Preface
vii
Katzung_FM.indd vii 9/24/11 11:46:43 AM

viii CONTENTSviii PREFACE
and of our editors for this edition, Donna Frassetto and Rachel D’Annucci Henriquez. I also wish to thank my wife, Alice Camp, for her expert proofreading contributions since the first edition.
This edition is dedicated to the memory of James Ransom, PhD, the long-time Senior Editor at Lange Medical Publications, who provided major inspiration and invaluable guidance through the first eight editions of the book. Without him, this book would not exist.
Suggestions and comments about Basic & Clinical Pharmacology are always welcome. They may be sent to me in care of thepublisher.
Bertram G. Katzung, MD, PhDSan Francisco
December, 2011
Katzung_FM.indd viii 9/24/11 11:46:43 AM

Authors
Emmanuel T. Akporiaye, PhDAdjunct Professor, Oregon Health Sciences University, Laboratory Chief, Earle A. Chiles Research Institute, Providence Cancer Center, Portland
Michael J. Aminoff, MD, DSc, FRCPProfessor, Department of Neurology, University of California, San Francisco
Allan I. Basbaum, PhDProfessor and Chair, Department of Anatomy and W.M. Keck Foundation Center for Integrative Neuroscience, University of California, San Francisco
Neal L. Benowitz, MDProfessor of Medicine and Bioengineering & Therapeutic Science, University of California, San Francisco, San Francisco
Italo Biaggioni, MDProfessor of Pharmacology, Vanderbilt University School of Medicine, Nashville
Daniel D. Bikle, MD, PhDProfessor of Medicine, Department of Medicine, and Co-Director, Special Diagnostic and Treatment Unit, University of California, San Francisco, and Veterans Affairs Medical Center, San Francisco
Homer A. Boushey, MDChief, Asthma Clinical Research Center and Division of Allergy & Immunology; Professor of Medicine, Department of Medicine, University of California, San Francisco
Adrienne D. Briggs, MDClinical Director, Bone Marrow Transplant Program, Banner Good Samaritan Hospital, Phoenix
Lundy Campbell, MDProfessor, Department of Anesthesiology and Perioperative Medicine, University of California San Francisco, School of Medicine, San Francisco
George P. Chrousos, MDProfessor & Chair, First Department of Pediatrics, Athens University Medical School, Athens
Edward Chu, MDProfessor of Medicine and Pharmacology & Chemical Biology; Chief, Division of Hematology-Oncology, Deputy Director, University of Pittsburgh Cancer Institute, University of Pittsburgh School of Medicine, Pittsburgh
Robin L. Corelli, PharmDClinical Professor, Department of Clinical Pharmacy, School of Pharmacy, University of California, San Francisco
Maria Almira Correia, PhDProfessor of Pharmacology, Pharmaceutical Chemistry and Biopharmaceutical Sciences, Department of Cellular & Molecular Pharmacology, University of California, San Francisco
Charles DeBattista, MDProfessor of Psychiatry and Behavioral Sciences, Stanford University School of Medicine, Stanford
Daniel H. Deck, PharmDAssistant Clinical Professor, School of Pharmacy, University of California, San Francisco; Infectious Diseases Clinical Pharmacist, San Francisco General Hospital
Cathi E. Dennehy, PharmDProfessor, Department of Clinical Pharmacy, University of California, San Francisco School of Pharmacy
Betty J. Dong, PharmD, FASHP, FCCPProfessor of Clinical Pharmacy and Clinical Professor of Family and Community Medicine, Department of Clinical Pharmacy and Department of Family and Community Medicine, Schools of Pharmacy and Medicine, University of California, San Francisco
Kenneth Drasner, MDProfesor of Anesthesia and Perioperative Care, University of California, San Francisco
Helge Eilers, MDProfessor of Anesthesia and Perioperative Care, University of California, San Francisco
ix
Katzung_FM.indd ix 9/24/11 11:46:43 AM

x AUTHORS
Garret A. FitzGerald, MDChair, Department of Pharmacology; Director, Institute for Translational Medicine and Therapeutics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia
Daniel E. Furst, MDCarl M. Pearson Professor of Rheumatology, Director, Rheumatology Clinical Research Center, Department of Rheumatology, University of California, Los Angeles
Augustus O. Grant, MD, PhDProfessor of Medicine, Cardiovascular Division, Duke University Medical Center, Durham
Francis S. Greenspan, MD, FACPClinical Professor of Medicine and Radiology and Chief, Thyroid Clinic, Division of Endocrinology, Department of Medicine, University of California, San Francisco
Nicholas H. G. Holford, MB, ChB, FRACPProfessor, Department of Pharmacology and Clinical Pharmacology, University of Auckland Medical School, Auckland
John R. Horn, PharmD, FCCPProfessor of Pharmacy, School of Pharmacy, University of Washington; Associate Director of Pharmacy Services, Department of Medicine, University of Washington Medicine, Seattle
Joseph R. Hume, PhDProfessor and Chairman, Department of Pharmacology; Adjunct Professor, Department of Physiology and Cell Biology, University of Nevada School of Medicine, Reno
Harlan E. Ives, MD, PhDProfessor of Medicine, Department of Medicine, University of California, San Francisco
Samie R. Jaffrey, MD, PhDAssociate Professor of Pharmacology, Department of Pharmacology, Cornell University Weill Medical College, New York City
John P. Kane, MD, PhDProfessor of Medicine, Department of Medicine; Professor of Biochemistry and Biophysics; Associate Director, Cardiovascular Research Institute, University of California, San Francisco
Bertram G. Katzung, MD, PhDProfessor Emeritus, Department of Cellular & Molecular Pharmacology, University of California, San Francisco
Gideon Koren, MDProfessor of Pediatrics, Pharmacology, Pharmacy, Medicine and Medical Genetics; Director, Motherisk Program, University of Toronto
Michael J. Kosnett, MD, MPHAssociate Clinical Professor of Medicine, Division of Clinical Pharmacology and Toxicology, University of Colorado Health Sciences Center, Denver
Marieke Kruidering-Hall, PhDAssociate Professor, Department of Cellular and Molecular Pharmacology, University of California, San Francisco
Douglas F. Lake, PhDAssociate Professor, The Biodesign Institute, Arizona State University, Tempe
Harry W. Lampiris, MDAssociate Professor of Medicine, University of California, San Francisco
Paul W. Lofholm, PharmDClinical Professor of Pharmacy, School of Pharmacy, University of California, San Francisco
Christian Lüscher, MDDepartments of Basic and Clincial Neurosciences, Medical Faculty, University Hospital of Geneva, Geneva, Switzerland
Daniel S. Maddix, PharmDAssociate Clinical Professor of Pharmacy, University of California, San Francisco
Howard I. Maibach, MDProfessor of Dermatology, Department of Dermatology, University of California, San Francisco
Mary J. Malloy, MDClinical Professor of Pediatrics and Medicine, Departments of Pediatrics and Medicine, Cardiovascular Research Institute, University of California, San Francisco
Susan B. Masters, PhDProfessor of Pharmacology & Academy Chair of Pharmacology Education, Department of Cellular & Molecular Pharmacology, University of California, San Francisco
Kenneth R. McQuaid, MDProfessor of Clinical Medicine, University of California, San Francisco; Chief of Gastroenterology, San Francisco Veterans Affairs Medical Center
Brian S. Meldrum, MB, PhDProfessor Emeritus, GKT School of Medicine, Guy’s Campus, London
Herbert Meltzer, MD, PhDProfessor of Psychiatry and Pharmacology, Vanderbilt University, Nashville
Roger A. Nicoll, MDProfessor of Pharmacology and Physiology, Departments of Cellular & Molecular Pharmacology and Physiology, University of California, San Francisco
Katzung_FM.indd x 9/24/11 11:46:43 AM

AUTHORS xi
Martha S. Nolte Kennedy, MDClinical Professor, Department of Medicine, University of California, San Francisco
Kent R. Olson, MDClinical Professor, Departments of Medicine and Pharmacy, University of California, San Francisco; Medical Director, San Francisco Division, California Poison Control System
Achilles J. Pappano, PhDProfessor Emeritus, Department of Cell Biology and Calhoun Cardiology Center, University of Connecticut Health Center, Farmington
Roger J. Porter, MDAdjunct Professor of Neurology, University of Pennsylvania, Philadelphia; Adjunct Professor of Pharmacology, Uniformed Services University of the Health Sciences, Bethesda
Shraddha Prakash, MD Senior Fellow in Rheumatology, David Geffen School of Medicine, University of California, Los Angeles
Ian A. Reid, PhDProfessor Emeritus, Department of Physiology, University of California, San Francisco
David Robertson, MDElton Yates Professor of Medicine, Pharmacology and Neurology, Vanderbilt University; Director, Clinical & Translational Research Center, Vanderbilt Institute for Clinical and Translational Research, Nashville
Dirk B. Robertson, MDProfessor of Clinical Dermatology, Department of Dermatology, Emory University School of Medicine, Atlanta
Philip J. Rosenthal, MDProfessor of Medicine, University of California, San Francisco, San Francisco General Hospital
Stephen M. Rosenthal, MD Professor of Pediatrics, Associate Program Director, Pediatric Endocrinology; Director, Pediatric Endocrine Outpatient Services, University of California, San Francisco
Sharon Safrin, MDAssociate Clinical Professor, Department of Medicine, University of California, San Francisco; President, Safrin Clinical Research
Alan C. Sartorelli, PhDAlfred Gilman Professor of Pharmacology, Department of Pharmacology, Yale University School of Medicine, New Haven
Mark A. Schumacher, PhD, MDAssociate Professor, Department of Anesthesia and Perioperative Care, University of California, San Francisco
Don Sheppard, MDAssociate Professor, Departments of Microbiology and Immunology and Medicine, McGill University; Program Director, McGill Royal College Training Program in Medical Microbiology and Infectious Diseases, Montreal
Emer M. Smyth, PhDAssistant Professor, Department of Pharmacology, University of Pennsylvania School of Medicine, Philadelphia
Daniel T. Teitelbaum, MDProfessor, University of Colorado School of Medicine, Aurora, and Colorado School of Mines, Golden
Anthony J. Trevor, PhDProfessor Emeritus, Department of Cellular & Molecular Pharmacology, University of California, San Francisco
Candy Tsourounis, PharmDProfessor of Clinical Pharmacy, Medication Outcomes Center, University of California, San Francisco School of Pharmacy
Robert W. Ulrich, PharmDSenior Clinical Science Manager, Abbott Laboratories Inc., Covina, California
Mark von Zastrow, MD, PhDProfessor, Departments of Psychiatry and Cellular & Molecular Pharmacology, University of California, San Francisco
Walter L. Way, MD*Professor Emeritus, Departments of Anesthesia and Cellular & Molecular Pharmacology, University of California, San Francisco
Lisa G. Winston, MDAssociate Professor, Department of Medicine, Division of Infectious Diseases, University of California, San Francisco; Hospital Epidemiologist, San Francisco General Hospital
Spencer Yost, MDProfessor, Department of Anesthesia and Perioperative Care, University of California, San Francisco; Medical Director, UCSF-Mt. Zion ICU, Chief of Anesthesia, UCSF-Mt. Zion Hospital
James L. Zehnder, MDProfessor of Pathology and Medicine, Pathology Department, Stanford University School of Medicine, Stanford
∗Deceased
Katzung_FM.indd xi 9/24/11 11:46:44 AM

xii
Katzung_FM.indd xii 9/30/11 12:19:08 PM

KEY FEATURES xiii
Katzung_FM.indd xiii 9/30/11 12:19:17 PM

This page intentionally left blank

789
INTRODUCTION TO ANTIMICROBIAL AGENTS Antimicrobial agents provide some of the most dramatic examples of the advances of modern medicine. Many infectious diseases once considered incurable and lethal are now amenable to treatment with a few pills. The remarkably powerful and specific activity of antimicrobial drugs is due to their selectivity for targets that are either unique to prokaryote and fungal microorganisms or much more important in these organisms than in humans. Among these targets are bacterial and fungal cell wall-synthesizing enzymes ( Chapters 43 and 48 ), the bacterial ribosome ( Chapters 44 and 45 ), the enzymes required for nucleotide synthesis and DNA rep-lication ( Chapter 46 ), and the machinery of viral replication ( Chapter 49 ). The special group of drugs used in mycobacterial infections is discussed in Chapter 47 . The much older and less selective cytotoxic antiseptics and disinfectants are discussed in Chapter 50 . The clinical uses of all these agents are summarized in Chapter 51 .
The major problem threatening the continued success of anti-microbial drugs is the development of resistant organisms. Microorganisms can adapt to environmental pressures in a variety of effective ways, and their response to antibiotic pressure is no exception. An inevitable consequence of antimicrobial usage is the
selection of resistant microorganisms, perhaps the most obvious example of evolution in action. Overuse and inappropriate use of antibiotics in patients has fueled a major increase in prevalence of multidrug-resistant pathogens. Antibacterial antibiotics are misused by providers in a variety of ways, including use in patients who are unlikely to have bacterial infections, use over unnecessar-ily prolonged periods, and use of multiple agents or broad- spectrum agents when not needed.
As a result of antibiotic pressure, highly resistant gram-negative organisms with novel mechanisms of resistance are increasingly reported. Some of these strains have spread over vast geographic areas as a result of patients seeking medical care in different coun-tries. Much larger quantities of antibiotics have been used in agriculture to stimulate growth and prevent infection in farm animals and this has added to the selection pressure that results in resistant organisms.
Unfortunately, as the need has grown in recent years, develop-ment of novel drugs has slowed. The most vulnerable molecular targets of antimicrobial drugs have been identified and, in many cases, crystallized and characterized. Pending the identification of new targets and compounds, it seems likely that over the next decade we will have to rely on currently available families of drugs. In the face of continuing development of resistance, considerable effort will be required to maintain the effectiveness of these drug groups.
SECTION VIII CHEMOTHERAPEUTIC DRUGS
043-Katzung_Ch043_p789-808.indd 789 9/23/11 5:51:29 PM

■ BETALACTAM COMPOUNDS
PENICILLINS
The penicillins share features of chemistry, mechanism of action, pharmacology, and immunologic characteristics with cephalosporins, monobactams, carbapenems, and β-lactamase inhibitors. All are β-lactam compounds, so named because of their four-membered lactam ring.
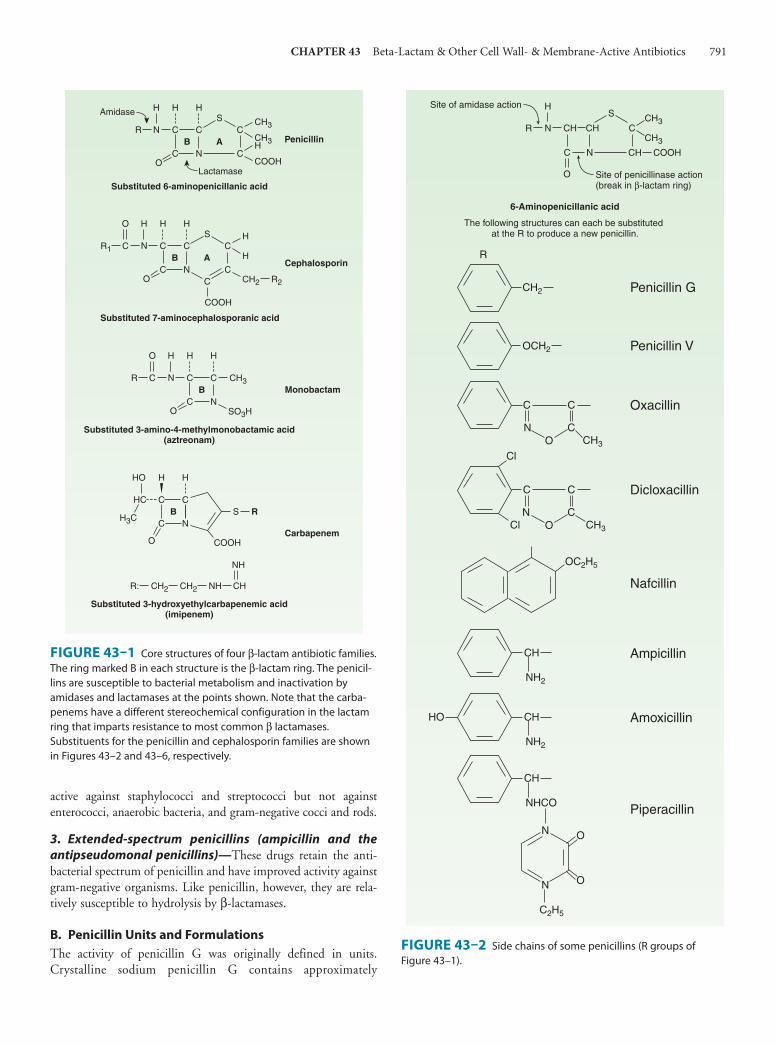
Chemistry All penicillins have the basic structure shown in Figure 43–1 . A thiazolidine ring (A) is attached to a β-lactam ring (B) that carries a secondary amino group (RNH–). Substituents (R; examples shown in Figure 43–2 ) can be attached to the amino group. Structural integrity of the 6-aminopenicillanic acid nucleus (rings A plus B) is essential for the biologic activity of these compounds.
Hydrolysis of the β-lactam ring by bacterial β-lactamases yields penicilloic acid, which lacks antibacterial activity.
A. Classification Substituents of the 6-aminopenicillanic acid moiety determine the essential pharmacologic and antibacterial properties of the result-ing molecules. Penicillins can be assigned to one of three groups (below). Within each of these groups are compounds that are rela-tively stable to gastric acid and suitable for oral administration, eg, penicillin V, dicloxacillin, and amoxicillin. The side chains of some representatives of each group are shown in Figure 43–2 , with a few distinguishing characteristics.
1. Penicillins (eg, penicillin G)— These have greatest activity against gram-positive organisms, gram-negative cocci, and non-β-lactamase producing anaerobes. However, they have little activ-ity against gram-negative rods, and they are susceptible to hydrolysis by β-lactamases.
2. Antistaphylococcal penicillins (eg, nafcillin)— These penicillins are resistant to staphylococcal β-lactamases. They are
emergency department, the man is febrile (38.7°C [101.7°F]), hypotensive (90/54 mm Hg), tachypneic (36/min), and tachycardic (110/min). He has no signs of meningismus but is oriented only to person. A stat chest x-ray shows a left lower lung consolidation consistent with pneumonia. The plan is to start empiric antibiotics and perform a lumbar puncture to rule out bacterial meningitis. What antibiotic regimen should be started to treat both pneumonia and meningitis? Does the history of amoxicillin rash affect the antibiotic choice? Why or why not?
C A S E S T U D Y
A 55-year-old man is brought to the local hospital emer-gency department by ambulance. His wife reports that he had been in his normal state of health until 3 days ago when he developed a fever and a productive cough. During the last 24 hours he has complained of a headache and is increasingly confused. His wife reports that his medical his-tory is significant only for hypertension, for which he takes hydrochlorothiazide and lisinopril, and that he is allergic to amoxicillin. She says that he developed a rash many years ago when prescribed amoxicillin for bronchitis. In the
43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics
C H A P T E R
Daniel H. Deck, PharmD & Lisa G. Winston, MD∗
∗ The authors thank Dr. Henry F. Chambers for his contributions to this chapter in previous editions.
790
043-Katzung_Ch043_p789-808.indd 790 9/23/11 5:51:29 PM
CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 791
active against staphylococci and streptococci but not against enterococci, anaerobic bacteria, and gram-negative cocci and rods.
3. Extended-spectrum penicillins (ampicillin and the
antipseudomonal penicillins)— These drugs retain the anti-bacterial spectrum of penicillin and have improved activity against gram-negative organisms. Like penicillin, however, they are rela-tively susceptible to hydrolysis by β-lactamases.
B. Penicillin Units and Formulations The activity of penicillin G was originally defined in units. Crystalline sodium penicillin G contains approximately
CS
N
CO
CH3
CH3H
COOH
C
C
C
NB
R
H H HAmidase
Lactamase
Penicillin
Substituted 6-aminopenicillanic acid
CS
N
CO
H
H
CH2
C
C
C
NB
H H H
Cephalosporin
Substituted 7-aminocephalosporanic acid
C
COOH
C
O
R1
R2
N
O SO3H
C
C
C
NB
H H H
Monobactam
Substituted 3-amino-4-methylmonobactamic acid(aztreonam)
C
O
R CH3
COOH
HC
O
C
C
C
NB
HO H H
H3CS R
CH2R:
NH
CH2 NH CH
Substituted 3-hydroxyethylcarbapenemic acid(imipenem)
Carbapenem
A
A
FIGURE 43–1 Core structures of four β-lactam antibiotic families. The ring marked B in each structure is the β-lactam ring. The penicil-lins are susceptible to bacterial metabolism and inactivation by amidases and lactamases at the points shown. Note that the carba-penems have a different stereochemical configuration in the lactam ring that imparts resistance to most common β lactamases. Substituents for the penicillin and cephalosporin families are shown in Figures 43–2 and 43–6, respectively.
S
CHCH
C N
N CRCH3
CH3
COOH
H
CH
Penicillin GCH2
CH3
R
Penicillin VOCH2
Oxacillin C C
OCN
CH3
NH2
Cl
Dicloxacillin C
CH
Cl
OC2H5
C2H5
C
OCN
Nafcillin
Ampicillin
NH2
CHHO Amoxicillin
NHCO
CH
Piperacillin
N
N
O
O
O
Site of amidase action
Site of penicillinase action(break in β-lactam ring)
6-Aminopenicillanic acid
The following structures can each be substituted at the R to produce a new penicillin.
FIGURE 43–2 Side chains of some penicillins (R groups of Figure 43–1 ).
043-Katzung_Ch043_p789-808.indd 791 9/23/11 5:51:29 PM
792 SECTION VIII Chemotherapeutic Drugs
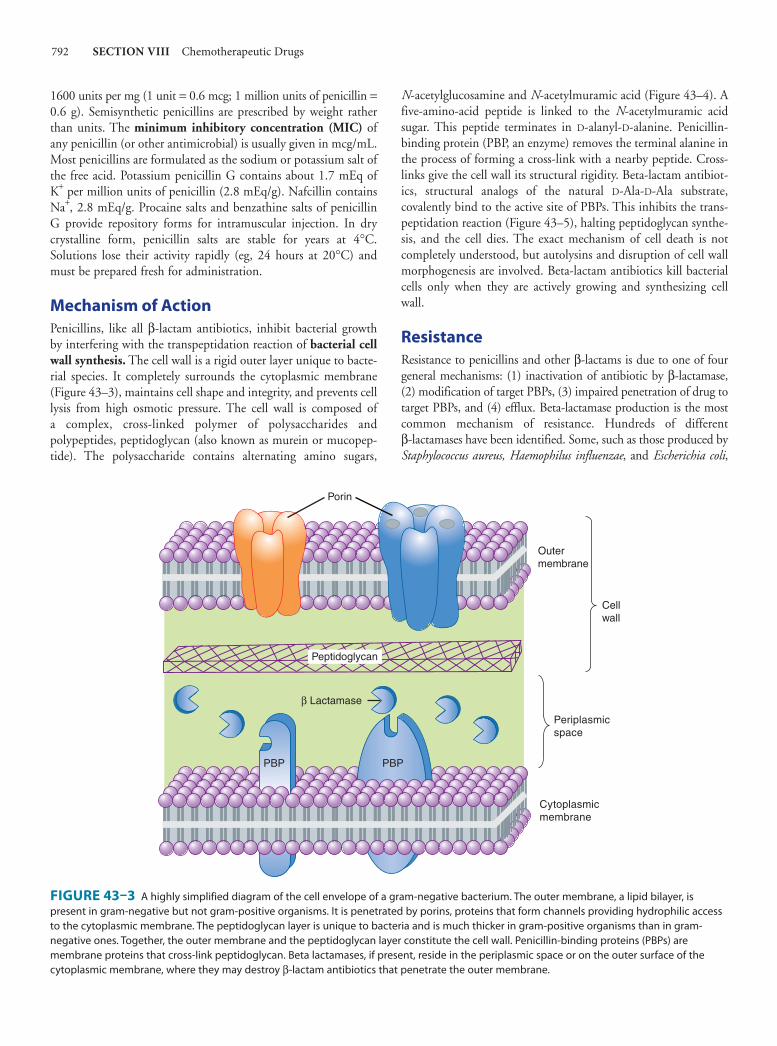
1600 units per mg (1 unit = 0.6 mcg; 1 million units of penicillin = 0.6 g). Semisynthetic penicillins are prescribed by weight rather than units. The minimum inhibitory concentration (MIC) of any penicillin (or other antimicrobial) is usually given in mcg/mL. Most penicillins are formulated as the sodium or potassium salt of the free acid. Potassium penicillin G contains about 1.7 mEq of K + per million units of penicillin (2.8 mEq/g). Nafcillin contains Na + , 2.8 mEq/g. Procaine salts and benzathine salts of penicillin G provide repository forms for intramuscular injection. In dry crystalline form, penicillin salts are stable for years at 4°C. Solutions lose their activity rapidly (eg, 24 hours at 20°C) and must be prepared fresh for administration.
Mechanism of Action Penicillins, like all β-lactam antibiotics, inhibit bacterial growth by interfering with the transpeptidation reaction of bacterial cell wall synthesis . The cell wall is a rigid outer layer unique to bacte-rial species. It completely surrounds the cytoplasmic membrane ( Figure 43–3 ), maintains cell shape and integrity, and prevents cell lysis from high osmotic pressure. The cell wall is composed of a complex, cross-linked polymer of polysaccharides and polypeptides, peptidoglycan (also known as murein or mucopep-tide). The polysaccharide contains alternating amino sugars,
N -acetylglucosamine and N -acetylmuramic acid ( Figure 43–4 ). A five-amino-acid peptide is linked to the N -acetylmuramic acid sugar. This peptide terminates in D-alanyl-D-alanine. Penicillin-binding protein (PBP, an enzyme) removes the terminal alanine in the process of forming a cross-link with a nearby peptide. Cross-links give the cell wall its structural rigidity. Beta-lactam antibiot-ics, structural analogs of the natural D-Ala-D-Ala substrate, covalently bind to the active site of PBPs. This inhibits the trans-peptidation reaction ( Figure 43–5 ), halting peptidoglycan synthe-sis, and the cell dies. The exact mechanism of cell death is not completely understood, but autolysins and disruption of cell wall morphogenesis are involved. Beta-lactam antibiotics kill bacterial cells only when they are actively growing and synthesizing cell wall.
Resistance Resistance to penicillins and other β-lactams is due to one of four general mechanisms: (1) inactivation of antibiotic by β-lactamase, (2) modification of target PBPs, (3) impaired penetration of drug to target PBPs, and (4) efflux. Beta-lactamase production is the most common mechanism of resistance. Hundreds of different β-lactamases have been identified. Some, such as those produced by Staphylococcus aureus, Haemophilus influenzae , and Escherichia coli ,
β Lactamase
PBP
Cytoplasmicmembrane
Periplasmicspace
Cellwall
Peptidoglycan
Outermembrane
PBP
Porin
FIGURE 43–3 A highly simplified diagram of the cell envelope of a gram-negative bacterium. The outer membrane, a lipid bilayer, is present in gram-negative but not gram-positive organisms. It is penetrated by porins, proteins that form channels providing hydrophilic access to the cytoplasmic membrane. The peptidoglycan layer is unique to bacteria and is much thicker in gram-positive organisms than in gram- negative ones. Together, the outer membrane and the peptidoglycan layer constitute the cell wall. Penicillin-binding proteins (PBPs) are membrane proteins that cross-link peptidoglycan. Beta lactamases, if present, reside in the periplasmic space or on the outer surface of the cytoplasmic membrane, where they may destroy β-lactam antibiotics that penetrate the outer membrane.
043-Katzung_Ch043_p789-808.indd 792 9/23/11 5:51:29 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 793
are relatively narrow in substrate specificity, preferring penicillins to cephalosporins. Other β-lactamases, eg, AmpC β-lactamase pro-duced by Pseudomonas aeruginosa and Enterobacter sp, and extended- spectrum β-lactamases (ESBLs), hydrolyze both cephalosporins and penicillins. Carbapenems are highly resistant to hydrolysis by peni-cillinases and cephalosporinases, but they are hydrolyzed by metallo-β lactamase and carbapenemases.
Altered target PBPs are the basis of methicillin resistance in staphylococci and of penicillin resistance in pneumococci and enterococci. These resistant organisms produce PBPs that have low affinity for binding β-lactam antibiotics, and consequently, they are not inhibited except at relatively high, often clinically unachievable, drug concentrations.
Resistance due to impaired penetration of antibiotic to target PBPs occurs only in gram-negative species because of their imper-meable outer cell wall membrane, which is absent in gram-positive bacteria. Beta-lactam antibiotics cross the outer membrane and enter gram-negative organisms via outer membrane protein channels called porins. Absence of the proper channel or down-regulation of its production can greatly impair drug entry into the cell. Poor penetration alone is usually not sufficient to confer resistance because enough antibiotic eventually enters the cell to inhibit growth. However, this barrier can become important in the presence of a β-lactamase, even a relatively inactive one, as long as it can hydrolyze drug faster than it enters the cell. Gram-negative organisms also may produce an efflux pump, which consists of cytoplasmic and periplasmic protein components that efficiently transport some β-lactam antibiotics from the periplasm back across the outer membrane.
Pharmacokinetics Absorption of orally administered drug differs greatly for different penicillins, depending in part on their acid stability and protein binding. Gastrointestinal absorption of nafcillin is erratic, so it is not suitable for oral administration. Dicloxacillin, ampicillin, and amoxicillin are acid-stable and relatively well absorbed, producing serum concentrations in the range of 4–8 mcg/mL after a 500-mg oral dose. Absorption of most oral penicillins (amoxicillin being an exception) is impaired by food, and the drugs should be admin-istered at least 1–2 hours before or after a meal.
Intravenous administration of penicillin G is preferred to the intramuscular route because of irritation and local pain from intra-muscular injection of large doses. Serum concentrations 30 minutes after an intravenous injection of 1 g of a penicillin (equivalent to approximately 1.6 million units of penicillin G) are 20–50 mcg/mL. Only a small amount of the total drug in serum is present as free drug, the concentration of which is determined by protein binding. Highly protein-bound penicillins (eg, nafcillin) generally achieve lower free-drug concentrations in serum than less protein-bound penicillins (eg, penicillin G or ampicillin). Protein binding becomes clinically relevant when the protein-bound percentage is approximately 95% or more. Penicillins are widely distributed in body fluids and tissues with a few exceptions. They are polar mol-ecules, so intracellular concentrations are well below those found in extracellular fluids.
L-Ala
M
G
M
G
M
G
M
G
R
L-Ala
D-Glu
L-Lys
D-Ala
D-Ala
L-Ala
D-Glu
M
G
M
G
M
G
M
G
R
+
Transpeptidase
L-Ala
R
L-Ala
D-Glu
L-Lys
D-Ala
D-Ala
L-Ala
D-Glu
L-Lys
D-Ala*
D-Ala
[Gly]5*
[Gly]5
L-Lys [Gly]5[Gly]5 D-Ala
+ D-Ala
L-Ala
R
L-Ala
R
FIGURE 43–4 The transpeptidation reaction in Staphylococcus aureus that is inhibited by β-lactam antibiotics. The cell wall of gram-positive bacteria is made up of long peptidoglycan polymer chains consisting of the alternating aminohexoses N -acetylglucosamine (G) and N -acetylmuramic acid (M) with pentapeptide side chains linked (in S aureus ) by pentaglycine bridges. The exact composition of the side chains varies among species. The diagram illustrates small segments of two such polymer chains and their amino acid side chains. These linear polymers must be cross-linked by transpeptida-tion of the side chains at the points indicated by the asterisk to achieve the strength necessary for cell viability.
043-Katzung_Ch043_p789-808.indd 793 9/23/11 5:51:29 PM

794 SECTION VIII Chemotherapeutic Drugs
Benzathine and procaine penicillins are formulated to delay absorption, resulting in prolonged blood and tissue concentra-tions. A single intramuscular injection of 1.2 million units of benzathine penicillin maintains serum levels above 0.02 mcg/mL for 10 days, sufficient to treat β-hemolytic streptococcal infection. After 3 weeks, levels still exceed 0.003 mcg/mL, which is enough to prevent β-hemolytic streptococcal infection. A 600,000 unit dose of procaine penicillin yields peak concentrations of 1–2 mcg/mL and clinically useful concentrations for 12–24 hours after a single intramuscular injection.
Penicillin concentrations in most tissues are equal to those in serum. Penicillin is also excreted into sputum and milk to levels 3–15% of those in the serum. Penetration into the eye, the prostate, and the central nervous system is poor. However, with active inflammation of the meninges, as in bacterial meningitis, penicillin concentrations of 1–5 mcg/mL can be achieved with a daily par-enteral dose of 18–24 million units. These concentrations
are sufficient to kill susceptible strains of pneumococci and meningococci.
Penicillin is rapidly excreted by the kidneys; small amounts are excreted by other routes. About 10% of renal excretion is by glomerular filtration and 90% by tubular secretion. The normal half-life of penicillin G is approximately 30 minutes; in renal failure, it may be as long as 10 hours. Ampicillin and the extended- spectrum penicillins are secreted more slowly than penicillin G and have half-lives of 1 hour. For penicillins that are cleared by the kidney, the dose must be adjusted according to renal function, with approximately one fourth to one third the normal dose being administered if creatinine clearance is 10 mL/min or less ( Table 43–1 ).
Nafcillin is primarily cleared by biliary excretion. Oxacillin, dicloxacillin, and cloxacillin are eliminated by both the kidney and biliary excretion; no dosage adjustment is required for these drugs in renal failure. Because clearance of penicillins is less efficient in
BP
BP
BP BP
BPBPBPBP
BP
PP MG PP MG MG MG PP MG MG MG MG
MGMG
MG
MGMG
MG
4
3
2
2
5
Cytoplasmicmembrane
Periplasmicspace
CytoplasmUDP
UDP G UDP M
L-Ala D-Glu L-Lys D-Ala D-Ala
D-Ala
L-Ala
P PPPcUMP
PPi
UDP
UMP
UTP
NAcGlc-1-P Glc-6-P
+PP
M PP
L-Ala
D-Glu
L-Lys
D-Ala
D-Ala
UDP M
L-Ala
D-Glu
L-Lys
D-Ala
D-AlaUDP M
L-Ala
D-Glu
L-Lys
=
1
GM PP
L-Ala
D-Glu L-Lys
D-Ala
D-Ala
GM PP
L-Ala
D-Glu
L-Lys
D-Ala
D-Ala
5-Gly tRNA
[Gly]5
1 Fosfomycin2 Cycloserine3 Bacitracin4 Vancomycin5 �-Lactams
FIGURE 43–5 The biosynthesis of cell wall peptidoglycan, showing the sites of action of five antibiotics (shaded bars; 1 = fosfomycin, 2 = cycloserine, 3 = bacitracin, 4 = vancomycin, 5 = β-lactam antibiotics). Bactoprenol (BP) is the lipid membrane carrier that transports building blocks across the cytoplasmic membrane; M, N -acetylmuramic acid; Glc, glucose; NAcGlc or G, N -acetylglucosamine.
043-Katzung_Ch043_p789-808.indd 794 9/23/11 5:51:29 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 795
the newborn, doses adjusted for weight alone result in higher sys-temic concentrations for longer periods than in the adult.
Clinical Uses Except for oral amoxicillin, penicillins should be given 1–2 hours before or after a meal; they should not be given with food to minimize binding to food proteins and acid inactivation. Blood levels of all penicillins can be raised by simultaneous administra-tion of probenecid, 0.5 g (10 mg/kg in children) every 6 hours orally, which impairs renal tubular secretion of weak acids such as β-lactam compounds. Penicillins should never be used for viral infections and should be prescribed only when there is reasonable suspicion of, or documented infection with, susceptible organisms .
A. Penicillin Penicillin G is a drug of choice for infections caused by streptococci, meningococci, some enterococci, penicillin-susceptible pneumo-cocci, non-β-lactamase-producing staphylococci, Treponema pallidum and certain other spirochetes, Clostridium species,
Actinomyces and certain other gram-positive rods, and non-β-lactamase-producing gram-negative anaerobic organisms. Depend-ing on the organism, the site, and the severity of infection, effective doses range between 4 and 24 million units per day administered intravenously in four to six divided doses. High-dose penicillin G can also be given as a continuous intravenous infusion.
Penicillin V, the oral form of penicillin, is indicated only in minor infections because of its relatively poor bioavailability, the need for dosing four times a day, and its narrow antibacterial spectrum. Amoxicillin (see below) is often used instead.
Benzathine penicillin and procaine penicillin G for intramus-cular injection yield low but prolonged drug levels. A single intra-muscular injection of benzathine penicillin, 1.2 million units, is effective treatment for β-hemolytic streptococcal pharyngitis; given intramuscularly once every 3–4 weeks, it prevents reinfec-tion. Benzathine penicillin G, 2.4 million units intramuscularly once a week for 1–3 weeks, is effective in the treatment of syphilis. Procaine penicillin G, formerly a work horse for treating uncom-plicated pneumococcal pneumonia or gonorrhea, is rarely used now because many strains are penicillin-resistant.
TABLE 43–1 Guidelines for dosing of some commonly used penicillins.
Adjusted Dose as a Percentage of Normal Dose for Renal
Failure Based on Creatinine Clearance (Clcr)
Antibiotic (Route of Administration) Adult Dose Pediatric Dose1 Neonatal Dose2
Clcr Approx
50 mL/min
Clcr Approx
10 mL/min
Penicillins
Penicillin G (IV) 1–4 × 106 units q4–6h
25,000–400,000 units/kg/d in 4–6 doses
75,000–150,000 units/kg/d in 2 or 3 doses
50–75% 25%
Penicillin V (PO) 0.25–0.5 g qid 25–50 mg/kg/d in 4 doses
None None
Antistaphylococcal penicillins
Cloxacillin, dicloxacillin (PO)
0.25–0.5 g qid 25–50 mg/kg/d in 4 doses
100% 100%
Nafcillin (IV) 1–2 g q4–6h 50–100 mg/kg/d in 4–6 doses
50–75 mg/kg/d in 2 or 3 doses
100% 100%
Oxacillin (IV) 1–2 g q4–6h 50–100 mg/kg/d in 4–6 doses
50–75 mg/kg/d in 2 or 3 doses
100% 100%
Extended-spectrum penicillins
Amoxicillin (PO) 0.25–0.5 g tid 20–40 mg/kg/d in 3 doses
66% 33%
Amoxicillin/potassium clavulanate (PO)
500/125 tid–875/125 mg bid
20–40 mg/kg/d in 3 doses
66% 33%
Piperacillin (IV) 3–4 g q4–6h 300 mg/kg/d in 4–6 doses
150 mg/kg/d in 2 doses
50–75% 25–33%
Ticarcillin (IV) 3 g q4–6h 200–300 mg/kg/d in 4–6 doses
150–200 mg/kg/d in 2 or 3 doses
50–75% 25–33%
1The total dose should not exceed the adult dose.2The dose shown is during the first week of life. The daily dose should be increased by approximately 33–50% after the first week of life. The lower dosage range should be used for neonates weighing less than 2 kg. After the first month of life, pediatric doses may be used.
043-Katzung_Ch043_p789-808.indd 795 9/23/11 5:51:29 PM

796 SECTION VIII Chemotherapeutic Drugs
B. Penicillins Resistant to Staphylococcal Beta Lactamase (Methicillin, Nafcillin, and Isoxazolyl Penicillins) These semisynthetic penicillins are indicated for infection by β-lactamase-producing staphylococci, although penicillin- susceptible strains of streptococci and pneumococci are also suscep-tible to these agents. Listeria monocytogenes , enterococci, and methicillin-resistant strains of staphylococci are resistant. In recent years the empirical use of these drugs has decreased substantially because of increasing rates of methicillin-resistance in staphylo-cocci. However, for infections caused by methicillin-susceptible and penicillin-resistant strains of staphylococci, these are consid-ered the drugs of choice.
An isoxazolyl penicillin such as oxacillin, cloxacillin, or dicloxacil-lin, 0.25–0.5 g orally every 4–6 hours (15–25 mg/kg/d for children), is suitable for treatment of mild to moderate localized staphylococcal infections. All are relatively acid-stable and have reasonable bioavail-ability. However, food interferes with absorption, and the drugs should be administered 1 hour before or after meals.
For serious systemic staphylococcal infections, oxacillin or nafcillin, 8–12 g/d, is given by intermittent intravenous infusion of 1–2 g every 4–6 hours (50–100 mg/kg/d for children).
C. Extended-Spectrum Penicillins (Aminopenicillins, Carboxypenicillins, and Ureidopenicillins) These drugs have greater activity than penicillin against gram-negative bacteria because of their enhanced ability to penetrate the gram-negative outer membrane. Like penicillin G, they are inacti-vated by many β lactamases.
The aminopenicillins, ampicillin and amoxicillin, have nearly identical spectrums of activity, but amoxicillin is better absorbed orally. Amoxicillin, 250–500 mg three times daily, is equivalent to the same amount of ampicillin given four times daily. Amoxacillin is given orally to treat urinary tract infections, sinusitis, otitis, and lower respiratory tract infections. Ampicillin and amoxicillin are the most active of the oral β-lactam antibiotics against pneumo-cocci with elevated MICs to penicillin and are the preferred β-lactam antibiotics for treating infections suspected to be caused by these strains. Ampicillin (but not amoxicillin) is effective for shigellosis. Its use to treat uncomplicated salmonella gastroenteritis is controversial because it may prolong the carrier state.
Ampicillin, at dosages of 4–12 g/d intravenously, is useful for treating serious infections caused by susceptible organisms, includ-ing anaerobes, enterococci, L monocytogenes , and β-lactamase-negative strains of gram-negative cocci and bacilli such as E coli , and Salmonella sp. Non-β-lactamase producing strains of H influenzae are generally susceptible, but strains that are resistant because of altered PBPs are emerging. Many gram-negative species produce β lactamases and are resistant, precluding use of ampicillin for empir-ical therapy of urinary tract infections, meningitis, and typhoid fever. Ampicillin is not active against Klebsiella sp, Enterobacter sp, P aeruginosa , Citrobacter sp, Serratia marcescens , indole-posi-tive proteus species, and other gram-negative aerobes that are commonly encountered in hospital-acquired infections. These organisms produce β lactamase that inactivates ampicillin.
Carbenicillin, the first antipseudomonal carboxypenicillin, is no longer used in the USA, as there are more active, better toler-ated alternatives. A carboxypenicillin with activity similar to that of carbenicillin is ticarcillin. It is less active than ampicillin against enterococci. The ureidopenicillins, piperacillin, mezlocillin, and azlocillin, are also active against selected gram-negative bacilli, such as Klebsiella pneumoniae. Although supportive clinical data are lacking for superiority of combination therapy over single-drug therapy, because of the propensity of P aeruginosa to develop resistance during treatment, an antipseudomonal penicillin is fre-quently used in combination with an aminoglycoside or fluoro-quinolone for pseudomonal infections outside the urinary tract.
Ampicillin, amoxicillin, ticarcillin, and piperacillin are also available in combination with one of several β-lactamase inhib-itors: clavulanic acid, sulbactam, or tazobactam. The addition of a β-lactamase inhibitor extends the activity of these penicil-lins to include β-lactamase-producing strains of S aureus as well as some β-lactamase-producing gram-negative bacteria (see Beta-Lactamase Inhibitors).
Adverse Reactions The penicillins are generally well tolerated, and unfortunately, this encourages their misuse and inappropriate use. Most of the serious adverse effects are due to hypersensitivity. All penicillins are cross-sensitizing and cross-reacting. The antigenic determinants are degradation products of penicillins, particularly penicilloic acid and products of alkaline hydrolysis bound to host protein. A his-tory of a penicillin reaction is not reliable; about 5–8% of people claim such a history, but only a small number of these will have an allergic reaction when given penicillin. Less than 1% of persons who previously received penicillin without incident will have an allergic reaction when given penicillin. Because of the potential for anaphylaxis, however, penicillin should be administered with cau-tion or a substitute drug given if the person has a history of serious penicillin allergy. The incidence of allergic reactions in young children is negligible.
Allergic reactions include anaphylactic shock (very rare—0.05% of recipients); serum sickness-type reactions (now rare—urticaria, fever, joint swelling, angioneurotic edema, intense pruritus, and respiratory compromise occurring 7–12 days after exposure); and a variety of skin rashes. Oral lesions, fever, interstitial nephritis (an autoimmune reaction to a penicillin-protein complex), eosino-philia, hemolytic anemia and other hematologic disturbances, and vasculitis may also occur. Most patients allergic to penicillins can be treated with alternative drugs. However, if necessary (eg, treat-ment of enterococcal endocarditis or neurosyphilis in a patient with serious penicillin allergy), desensitization can be accomplished with gradually increasing doses of penicillin.
In patients with renal failure, penicillin in high doses can cause seizures. Nafcillin is associated with neutropenia; oxacillin can cause hepatitis; and methicillin causes interstitial nephritis (and is no longer used for this reason). Large doses of penicillins given orally may lead to gastrointestinal upset, particularly nausea, vomiting, and diarrhea. Ampicillin has been associated with pseudomembranous colitis. Secondary infections such as vaginal
043-Katzung_Ch043_p789-808.indd 796 9/23/11 5:51:29 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 797
candidiasis may occur. Ampicillin and amoxicillin can cause skin rashes that are not allergic in nature. These rashes frequently occur when aminopenicillins are inappropriately prescribed for a viral illness.
■ CEPHALOSPORINS & CEPHAMYCINS
Cephalosporins are similar to penicillins, but more stable to many bacterial β lactamases and therefore have a broader spectrum of activity. However, strains of E coli and Klebsiella sp expressing extended-spectrum β lactamases that can hydrolyze most cepha-losporins are a growing clinical concern. Cephalosporins are not active against enterococci and L monocytogenes .
Chemistry The nucleus of the cephalosporins, 7-aminocephalosporanic acid ( Figure 43–6 ), bears a close resemblance to 6-aminopenicillanic acid ( Figure 43–1 ). The intrinsic antimicrobial activity of natural cephalosporins is low, but the attachment of various R 1 and R 2 groups has yielded hundreds of potent compounds of low toxicity. Cephalosporins can be classified into four major groups or gen-erations, depending mainly on the spectrum of antimicrobial activity.
FIRST-GENERATION CEPHALOSPORINS
First-generation cephalosporins include cefazolin, cefadroxil, cephalexin, cephalothin, cephapirin, and cephradine. These drugs are very active against gram-positive cocci, such as pneumo-cocci, streptococci, and staphylococci. Traditional cephalosporins are not active against methicillin-resistant strains of staphylococci; how-ever, new compounds have been developed that have activity against methicillin-resistant strains (see below). E coli, K pneumoniae , and Proteus mirabilis are often sensitive, but activity against P aeruginosa , indole-positive proteus species, Enterobacter sp, S marcescens , Citrobacter sp, and Acinetobacter sp is poor. Anaerobic cocci (eg, peptococci, peptostreptococci) are usually sensitive, but Bacteroides fragilis is not.
Pharmacokinetics & Dosage A. Oral Cephalexin, cephradine, and cefadroxil are absorbed from the gut to a variable extent. After oral doses of 500 mg, serum levels are 15–20 mcg/mL. Urine concentration is usually very high, but in most tis-sues levels are variable and generally lower than in serum. Cephalexin and cephradine are given orally in dosages of 0.25–0.5 g four times daily (15–30 mg/kg/d) and cefadroxil in dosages of 0.5–1 g twice daily. Excretion is mainly by glomerular filtration and tubular secre-tion into the urine. Drugs that block tubular secretion, eg, probenecid, may increase serum levels substantially. In patients with impaired renal function, dosage must be reduced ( Table 43–2 ).
CH3
CH3
Cephalexin
Cefadroxil
Ceftazidime
Cefepime
Ceftaroline
Cefazolin
NS
CN
H2NC
H2N
HOOC
S
N
NN NN
N
CH2
ON
SCR1 NH
O
R2
AB
R1 R2
O
O OH
OH
S
S
CH
NH2
COO–
Cefoxitin
Cefaclor
Cefprozil
Cefuroxime
Cefotetan1
Cefotaxime
Cefpodoxime1
Ceftibuten
Cefdinir
Ceftizoxime
Ceftriaxone
CH
NH2
HO
S
SSCH2
CH2
CH
HONH2
NH2O C
C C C
NOCH3
OCH3
N
N
O
S
S
S
C
C
O
O
O
O
C COOH
N
N
H2N
H2N OCH3
NS
CN
H2N OCH3
NS
CN
H2N
NS
CN
H2N OCH3
CH3
CH3
NS
S
C
CC
N
N
H2N
H2N
S
SN
N+
CH2 N+
CH2
CH2
H
H
CH
CH2 CH3O
CH3
O
NH2
CH2
OCH2
CH3
CH3
CH3
CH3
CHCH
Cl
C
O
NH2O C
CH3
O C
H3C
N
N
N
N N
NN
N
H
S
N
N
NH2N
OCH3
CH2
CH2
CH2
FIGURE 43–6 Structures of some cephalosporins. R 1 and R 2 structures are substituents on the 7-aminocephalosporanic acid nucleus pictured at the top. Other structures (cefoxitin and below) are complete in themselves. 1Additional substituents not shown.
043-Katzung_Ch043_p789-808.indd 797 9/23/11 5:51:30 PM

798 SECTION VIII Chemotherapeutic Drugs
TABLE 43–2 Guidelines for dosing of some commonly used cephalosporins and other cell-wall inhibitor antibiotics.
Adjusted Dose as a Percentage of Normal Dose for Renal Failure Based on Creatinine Clearance (Clcr)
Antibiotic (Route of Administration) Adult Dose Pediatric Dose1 Neonatal Dose2
Clcr Approx
50 mL/min
Clcr Approx
10 mL/min
First-generation cephalosporins
Cefadroxil (PO) 0.5–1 g qd–bid 30 mg/kg/d in 2 doses 50% 25%
Cephalexin, cephradine (PO)
0.25–0.5 g qid 25–50 mg/kg/d in 4 doses 50% 25%
Cefazolin (IV) 0.5–2 g q8h 25–100 mg/kg/d in 3 or 4 doses
50% 25%
Second-generation cephalosporins
Cefoxitin (IV) 1–2 g q6–8h 75–150 mg/kg/d in 3 or 4 doses
50–75% 25%
Cefotetan (IV) 1–2 g q12h 50% 25%
Cefuroxime (IV) 0.75–1.5 g q8h 50–100 mg/kg/d in 3 or 4 doses
66% 25–33%
Third- and fourth-generation cephalosporins including ceftaroline fosamil
Cefotaxime (IV) 1–2 g q6–12h 50–200 mg/kg/d in 4–6 doses 100 mg/kg/d in 2 doses
50% 25%
Ceftazidime (IV) 1–2 g q8–12h 75–150 mg/kg/d in 3 doses
100–150 mg/kg/d in 2 or 3 doses
50% 25%
Ceftriaxone (IV) 1–4 g q24h 50–100 mg/kg/d in 1 or 2 doses
50 mg/kg/d qd None None
Cefepime (IV) 0.5–2 g q12h 75–120 mg/kg/d in 2 or 3 divided doses
50% 25%
Ceftaroline fosamil (IV) 600 mg q12h 50–66% 33%
Carbapenems
Ertapenem (IM or IV) 1 g q24h 100%3 50%
Doripenem 500 mg q8h 50% 33%
Imipenem (IV) 0.25–0.5 g q6–8h 75% 50%
Meropenem (IV) 1 g q8h (2 g q8h for meningitis)
60–120 mg/kg/d in 3 doses (maximum of 2 g q8h)
66% 50%
Glycopeptides
Vancomycin (IV) 30–60 mg/kg/d in 2–3 doses
40 mg/kg/d in 3 or 4 doses 15 mg/kg load, then 20 mg/kg/d in 2 doses
40% 10%
Lipopeptides (IV)
Daptomycin 4-6 mg/kg IV daily None 50%
Telavancin 10 mg/kg IV daily 75% 50%
1The total dose should not exceed the adult dose.2The dose shown is during the first week of life. The daily dose should be increased by approximately 33–50% after the first week of life. The lower dosage range should be used for neonates weighing less than 2 kg. After the first month of life, pediatric doses may be used.350% of dose for Clcr< 30 mL/min.
043-Katzung_Ch043_p789-808.indd 798 9/23/11 5:51:30 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 799
B. Parenteral Cefazolin is the only first-generation parenteral cephalosporin still in general use. After an intravenous infusion of 1 g, the peak level of cefazolin is 90–120 mcg/mL. The usual intravenous dosage of cefazolin for adults is 0.5–2 g intravenously every 8 hours. Cefazolin can also be administered intramuscularly. Excretion is via the kidney, and dose adjustments must be made for impaired renal function.
Clinical Uses Oral drugs may be used for the treatment of urinary tract infec-tions and staphylococcal or streptococcal infections, including cellulitis or soft tissue abscess. However, oral cephalosporins should not be relied on in serious systemic infections.
Cefazolin penetrates well into most tissues. It is a drug of choice for surgical prophylaxis. Cefazolin may also be a choice in infections for which it is the least toxic drug (eg, penicillinase-producing E coli or K pneumoniae ) and in individuals with staphylococcal or streptococcal infections who have a history of penicillin allergy other than immediate hypersensitivity. Cefazolin does not penetrate the central nervous system and cannot be used to treat meningitis. Cefazolin is an alternative to an antistaphylo-coccal penicillin for patients who are allergic to penicillin.
SECOND-GENERATION CEPHALOSPORINS
Members of the second-generation cephalosporins include cefaclor, cefamandole, cefonicid, cefuroxime, cefprozil, loracarbef, and ceforanide; and the structurally related cephamycins cefoxitin, cefmetazole , and cefotetan , which have activity against anaer-obes. This is a heterogeneous group with marked individual differ-ences in activity, pharmacokinetics, and toxicity. In general, they are active against organisms inhibited by first-generation drugs, but in addition they have extended gram-negative coverage. Klebsiella sp (including those resistant to cephalothin) are usually sensitive. Cefamandole, cefuroxime, cefonicid, ceforanide, and cefaclor are active against H influenzae but not against serratia or B fragilis . In contrast, cefoxitin, cefmetazole, and cefotetan are active against B fragilis and some serratia strains but are less active against H influenzae . As with first-generation agents, none is active against enterococci or P aeruginosa . Second-generation cepha-losporins may exhibit in vitro activity against Enterobacter sp., but resistant mutants that constitutively express a chromosomal β lactamase that hydrolyzes these compounds (and third-generation cephalosporins) are readily selected, and they should not be used to treat enterobacter infections.
Pharmacokinetics & Dosage A. Oral Cefaclor, cefuroxime axetil, cefprozil, and loracarbef can be given orally. The usual dosage for adults is 10–15 mg/kg/d in two to four divided doses; children should be given 20–40 mg/kg/d up to
a maximum of 1 g/d. Except for cefuroxime axetil, these drugs are not predictably active against penicillin-non-susceptible pneumo-cocci and should be used cautiously, if at all, to treat suspected or proved pneumococcal infections. Cefaclor is more susceptible to β-lactamase hydrolysis compared with the other agents, and its usefulness is correspondingly diminished.
B. Parenteral After a 1-g intravenous infusion, serum levels are 75–125 mcg/mL for most second-generation cephalosporins. Intramuscular admin-istration is painful and should be avoided. Doses and dosing intervals vary depending on the specific agent ( Table 43–2 ). There are marked differences in half-life, protein binding, and interval between doses. All are renally cleared and require dosage adjust-ment in renal failure.
Clinical Uses The oral second-generation cephalosporins are active against β-lactamase-producing H influenzae or Moraxella catarrhalis and have been primarily used to treat sinusitis, otitis, and lower respiratory tract infections, in which these organisms have an important role. Because of their activity against anaerobes (including many B fragilis strains ), cefoxitin, cefotetan, or cefmetazole can be used to treat mixed anaero-bic infections such as peritonitis, diverticulitis, and pelvic inflamma-tory disease. Cefuroxime is used to treat community-acquired pneumonia because it is active against β-lactamase-producing H influenzae or K pneumoniae and some penicillin-non-susceptible pneumococci. Although cefuroxime crosses the blood-brain barrier, it is less effective in treatment of meningitis than ceftriaxone or cefo-taxime and should not be used.
THIRD-GENERATION CEPHALOSPORINS
Third-generation agents include cefoperazone, cefotaxime, cef-tazidime, ceftizoxime, ceftriaxone, cefixime, cefpodoxime prox-etil, cefdinir, cefditoren pivoxil, ceftibuten, and moxalactam .
Antimicrobial Activity Compared with second-generation agents, these drugs have expanded gram-negative coverage, and some are able to cross the blood-brain barrier. Third-generation drugs are active against Citrobacter , S marcescens , and Providencia (although resistance can emerge during treatment of infections caused by these species due to selection of mutants that constitutively produce cephalosporinase).They are also effective against β-lactamase-producing strains of haemophilus and neisseria. Ceftazidime and cefoperazone are the only two drugs with useful activity against P aeruginosa . Like the second-generation drugs, third-generation cephalosporins are hydrolyzed by constitutively produced AmpC β lactamase, and they are not reliably active against Enterobacter species. Serratia , Providencia , and Citrobacter also produce a chromosomally encoded cephalosporinase that, when constitutively expressed, can confer resistance to third-generation cephalosporins. Ceftizoxime and moxalactam are active against B fragilis . Cefixime, cefdinir,
043-Katzung_Ch043_p789-808.indd 799 9/23/11 5:51:30 PM

800 SECTION VIII Chemotherapeutic Drugs
ceftibuten, and cefpodoxime proxetil are oral agents possessing similar activity except that cefixime and ceftibuten are much less active against pneumococci and have poor activity against S aureus .
Pharmacokinetics & Dosage Intravenous infusion of 1 g of a parenteral cephalosporin produces serum levels of 60–140 mcg/mL. Third-generation cephalosporins penetrate body fluids and tissues well and, with the exception of cefoperazone and all oral cephalosporins, achieve levels in the cerebrospinal fluid sufficient to inhibit most susceptible pathogens.
The half-lives of these drugs and the necessary dosing intervals vary greatly: Ceftriaxone (half-life 7–8 hours) can be injected once every 24 hours at a dosage of 15–50 mg/kg/d. A single daily 1-g dose is sufficient for most serious infections, with 2 g every 12 hours recommended for treatment of meningitis. Cefoperazone (half-life 2 hours) can be infused every 8–12 hours in a dosage of 25–100 mg/kg/d. The remaining drugs in the group (half-life 1–1.7 hours) can be infused every 6–8 hours in dosages between 2 and 12 g/d, depending on the severity of infection. Cefixime can be given orally (200 mg twice daily or 400 mg once daily) for urinary tract infections and as a single 400 mg dose for uncompli-cated gonococcal urethritis and cervicitis. The adult dose for cefpodoxime proxetil or cefditoren pivoxil is 200–400 mg twice daily; for ceftibuten, 400 mg once daily; and for cefdinir, 300 mg/12 h. The excretion of cefoperazone and ceftriaxone is mainly through the biliary tract, and no dosage adjustment is required in renal insufficiency. The others are excreted by the kidney and therefore require dosage adjustment in renal insufficiency.
Clinical Uses Third-generation cephalosporins are used to treat a wide variety of serious infections caused by organisms that are resistant to most other drugs. Strains expressing extended-spectrum β lactamases, however, are not susceptible. Third-generation cephalosporins should be avoided in treatment of enterobacter infections—even if the clinical isolate appears susceptible in vitro—because of emergence of resistance. Ceftriaxone and cefotaxime are approved for treatment of meningitis, including meningitis caused by pneu-mococci, meningococci, H influenzae , and susceptible enteric gram-negative rods, but not by L monocytogenes . Ceftriaxone and cefotaxime are the most active cephalosporins against penicillin-non-susceptible strains of pneumococci and are recommended for empirical therapy of serious infections that may be caused by these strains. Meningitis caused by strains of pneumococci with penicil-lin MICs > 1 mcg/mL may not respond even to these agents, and addition of vancomycin is recommended. Other potential indica-tions include empirical therapy of sepsis of unknown cause in both the immunocompetent and the immunocompromised patient and treatment of infections for which a cephalosporin is the least toxic drug available. In neutropenic, febrile immunocom-promised patients, ceftazidime is often used in combination with other antibiotics.
FOURTH-GENERATION CEPHALOSPORINS
Cefepime is an example of a so-called fourth-generation cephalosporin. It is more resistant to hydrolysis by chromosomal β lactamases (eg, those produced by Enterobacter ). However, like the third-generation compounds, it is hydrolyzed by extended-spectrum β lactamases. Cefepime has good activity against P aeruginosa, Enterobacteriaceae , S aureus , and S pneumoniae . It is highly active against Haemophilus and Neisseria sp. It penetrates well into cerebrospinal fluid. It is cleared by the kidneys and has a half-life of 2 hours, and its pharmacokinetic properties are very similar to those of ceftazidime. Unlike ceftazidime, however, cefepime has good activity against most penicillin-non-susceptible strains of streptococci, and it is useful in treatment of entero-bacter infections.
Cephalosporins Active against Methicillin-Resistant Staphylococci Beta-lactam antibiotics with activity against methicillin-resistant staphylococci are currently under development. Ceftaroline fosamil, the prodrug of the active metabolite ceftaroline, is the first such drug to be approved for clinical use in the USA. Ceftaroline has increased binding to penicillin-binding protein 2a, which mediates methicillin resistance in staphylococci, resulting in bactericidal activity against these strains. It has some activity against enterococci and a broad gram-negative spectrum, although it is not active against extended-spectrum β-lactamase-producing strains. Since clinical experience with this and similar investiga-tional drugs is limited, their role in therapy is not yet defined.
ADVERSE EFFECTS OF CEPHALOSPORINS
A. Allergy Cephalosporins are sensitizing and may elicit a variety of hyper-sensitivity reactions that are identical to those of penicillins, including anaphylaxis, fever, skin rashes, nephritis, granulocy-topenia, and hemolytic anemia. However, the chemical nucleus of cephalosporins is sufficiently different from that of penicillins so that some individuals with a history of penicillin allergy may tolerate cephalosporins. The frequency of cross-allergenicity between the two groups of drugs is uncertain but is probably around 5–10%. Cross-allergenicity appears to be more common with penicillins and early generation cephalosporins compared with later generation cephalosporins. However, patients with a history of anaphylaxis to penicillins should not receive cephalosporins.
B. Toxicity Local irritation can produce pain after intramuscular injection and thrombophlebitis after intravenous injection. Renal toxicity, including interstitial nephritis and tubular necrosis, has been dem-onstrated with several cephalosporins and caused the withdrawal of cephaloridine from clinical use.
043-Katzung_Ch043_p789-808.indd 800 9/23/11 5:51:30 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 801
Cephalosporins that contain a methylthiotetrazole group (cefa-mandole, cefmetazole, cefotetan, and cefoperazone) may cause hypoprothrombinemia and bleeding disorders. Oral administra-tion of vitamin K 1 , 10 mg twice weekly, can prevent this. Drugs with the methylthiotetrazole ring can also cause severe disulfiram-like reactions; consequently, alcohol and alcohol-containing medications must be avoided.
■ OTHER BETALACTAM DRUGS
MONOBACTAMS
Monobactams are drugs with a monocyclic β-lactam ring ( Figure 43–1 ). Their spectrum of activity is limited to aerobic gram-negative rods (including P aeruginosa ). Unlike other β-lactam antibiotics, they have no activity against gram-positive bacteria or anaerobes. Aztreonam is the only monobactam available in the USA. It has structural similarities to ceftazidime; hence, its gram-negative spectrum is similar to that of the third-generation cepha-losporins. It is stable to many β lactamases with the notable exceptions being AmpC β lactamases and extended-spectrum β lactamases. It penetrates well into the cerebrospinal fluid. Aztreonam is given intravenously every 8 hours in a dose of 1–2 g, providing peak serum levels of 100 mcg/mL. The half-life is 1–2 hours and is greatly prolonged in renal failure.
Penicillin-allergic patients tolerate aztreonam without reaction. Occasional skin rashes and elevations of serum aminotransferases occur during administration of aztreonam, but major toxicity is uncommon. In patients with a history of penicillin anaphylaxis, aztre-onam may be used to treat serious infections such as pneumonia, meningitis, and sepsis caused by susceptible gram-negative pathogens.
BETA-LACTAMASE INHIBITORS (CLAVULANIC ACID, SULBACTAM, & TAZOBACTAM)
These substances resemble β-lactam molecules ( Figure 43–7 ), but they have very weak antibacterial action. They are potent inhibitors of many but not all bacterial β lactamases and can protect hydrolyz-able penicillins from inactivation by these enzymes. Beta-lactamase
inhibitors are most active against Ambler class A β lactamases (plasmid-encoded transposable element [TEM] β lactamases in particular), such as those produced by staphylococci, H influenzae, N gonorrhoeae, salmonella, shigella, E coli , and K pneumoniae . They are not good inhibitors of class C β lactamases, which typically are chromosomally encoded and inducible, produced by Enterobacter sp, Citrobacter sp, S marcescens , and P aeruginosa , but they do inhibit chromosomal β lactamases of B fragilis and M catarrhalis .
The three inhibitors differ slightly with respect to pharmacol-ogy, stability, potency, and activity, but these differences usually are of little therapeutic significance. Beta-lactamase inhibitors are available only in fixed combinations with specific penicillins. The antibacterial spectrum of the combination is determined by the companion penicillin, not the β-lactamase inhibitor. (The fixed combinations available in the USA are listed in Preparations Available.) An inhibitor extends the spectrum of a penicillin pro-vided that the inactivity of the penicillin is due to destruction by β lactamase and that the inhibitor is active against the β lactamase that is produced. Thus, ampicillin-sulbactam is active against β-lactamase-producing S aureus and H influenzae but not against serratia, which produces a β lactamase that is not inhibited by sulbactam. Similarly, if a strain of P aeruginosa is resistant to piperacillin, it is also resistant to piperacillin-tazobactam because tazobactam does not inhibit the chromosomal β lactamase pro-duced by P aeruginosa .
The indications for penicillin-β-lactamase inhibitor combina-tions are empirical therapy for infections caused by a wide range of potential pathogens in both immunocompromised and immu-nocompetent patients and treatment of mixed aerobic and anaero-bic infections, such as intra-abdominal infections. Doses are the same as those used for the single agents except that the recom-mended dosage of piperacillin in the piperacillin-tazobactam combination is 3–4 g every 6 hours. Adjustments for renal insuf-ficiency are made based on the penicillin component.
CARBAPENEMS
The carbapenems are structurally related to β-lactam antibiotics ( Figure 43–1 ). Doripenem, ertapenem, imipenem , and meropenem are licensed for use in the USA. Imipenem, the first drug of this class, has a wide spectrum with good activity against many gram-negative rods, including P aeruginosa , gram-positive
CO CH
C N
OCH
CH2OH
COOH
H2C
Clavulanic acid
OC N
CHH2C
Sulbactam
SO O–
COOH
CH3
CH2 R
R = H
Tazobactam
R = N N
N
FIGURE 43–7 Beta-lactamase inhibitors.
043-Katzung_Ch043_p789-808.indd 801 9/23/11 5:51:30 PM

802 SECTION VIII Chemotherapeutic Drugs
organisms, and anaerobes. It is resistant to most β lactamases but not carbapenemases or metallo-β lactamases. Enterococcus faecium , methicillin-resistant strains of staphylococci, Clostridium difficile , Burkholderia cepacia , and Stenotrophomonas maltophilia are resis-tant. Imipenem is inactivated by dehydropeptidases in renal tubules, resulting in low urinary concentrations. Consequently, it is administered together with an inhibitor of renal dehydropepti-dase, cilastatin , for clinical use. Doripenem and meropenem are similar to imipenem but have slightly greater activity against gram-negative aerobes and slightly less activity against gram- positives. They are not significantly degraded by renal dehydro-peptidase and do not require an inhibitor. Ertapenem is less active than the other carbapenems against P aeruginosa and Acinetobacter species. It is not degraded by renal dehydropeptidase.
Carbapenems penetrate body tissues and fluids well, including the cerebrospinal fluid. All are cleared renally, and the dose must be reduced in patients with renal insufficiency. The usual dosage of imipenem is 0.25–0.5 g given intravenously every 6–8 hours (half-life 1 hour). The usual adult dosage of meropenem is 0.5–1 g intravenously every 8 hours. The usual adult dosage of doripenem is 0.5 g administered as a 1- or 4-hour infusion every 8 hours. Ertapenem has the longest half-life (4 hours) and is administered as a once-daily dose of 1 g intravenously or intramuscularly. Intramuscular ertapenem is irritating, and for that reason the drug is formulated with 1% lidocaine for administration by this route.
A carbapenem is indicated for infections caused by susceptible organisms that are resistant to other available drugs, eg, P aeruginosa , and for treatment of mixed aerobic and anaerobic infections. Carbapenems are active against many penicillin-non-susceptible strains of pneumococci. Carbapenems are highly active in the treatment of enterobacter infections because they are resistant to destruction by the β lactamase produced by these organisms. Clinical experience suggests that carbapenems are also the treat-ment of choice for infections caused by extended-spectrum β-lactamase-producing gram-negative bacteria. Ertapenem is insufficiently active against P aeruginosa and should not be used to treat infections caused by that organism. Imipenem, meropenem, or doripenem, with or without an aminoglycoside, may be effec-tive treatment for febrile neutropenic patients.
The most common adverse effects of carbapenems—which tend to be more common with imipenem—are nausea, vomiting, diarrhea, skin rashes, and reactions at the infusion sites. Excessive levels of imipenem in patients with renal failure may lead to seizures. Meropenem, doripenem, and ertapenem are much less likely to cause seizures than imipenem. Patients allergic to penicil-lins may be allergic to carbapenems as well.
■ GLYCOPEPTIDE ANTIBIOTICS
VANCOMYCIN
Vancomycin is an antibiotic produced by Streptococcus orientalis and Amycolatopsis orientalis. With the exception of Flavobacterium , it is active only against gram-positive bacteria. Vancomycin is a
glycopeptide of molecular weight 1500. It is water soluble and quite stable.
Mechanisms of Action & Basis of Resistance Vancomycin inhibits cell wall synthesis by binding firmly to the D-Ala-D-Ala terminus of nascent peptidoglycan pentapeptide ( Figure 43–5 ). This inhibits the transglycosylase, preventing fur-ther elongation of peptidoglycan and cross-linking. The peptido-glycan is thus weakened, and the cell becomes susceptible to lysis. The cell membrane is also damaged, which contributes to the antibacterial effect.
Resistance to vancomycin in enterococci is due to modification of the D-Ala-D-Ala binding site of the peptidoglycan building block in which the terminal D-Ala is replaced by D-lactate. This results in the loss of a critical hydrogen bond that facilitates high-affinity binding of vancomycin to its target and loss of activity. This mechanism is also present in vancomycin-resistant S aureus strains (MIC ≥ 16 mcg/mL), which have acquired the enterococ-cal resistance determinants. The underlying mechanism for reduced vancomycin susceptibility in vancomycin-intermediate strains (MICs = 4–8 mcg/mL) of S aureus is not fully known. However these strains have altered cell wall metabolism that results in a thickened cell wall with increased numbers of D-Ala-D-Ala residues, which serve as dead-end binding sites for vanco-mycin. Vancomycin is sequestered within the cell wall by these false targets and may be unable to reach its site of action.
Antibacterial Activity Vancomycin is bactericidal for gram-positive bacteria in concentra-tions of 0.5–10 mcg/mL. Most pathogenic staphylococci, includ-ing those producing β lactamase and those resistant to nafcillin and methicillin, are killed by 2 mcg/mL or less. Vancomycin kills staphylococci relatively slowly and only if cells are actively dividing; the rate is less than that of the penicillins both in vitro and in vivo. Vancomycin is synergistic in vitro with gentamicin and streptomy-cin against Enterococcus faecium and Enterococcus faecalis strains that do not exhibit high levels of aminoglycoside resistance.
Pharmacokinetics Vancomycin is poorly absorbed from the intestinal tract and is administered orally only for the treatment of antibiotic-associated colitis caused by C difficile . Parenteral doses must be administered intravenously. A 1-hour intravenous infusion of 1 g produces blood levels of 15–30 mcg/mL for 1–2 hours. The drug is widely distributed in the body. Cerebrospinal fluid levels 7–30% of simultaneous serum concentrations are achieved if there is menin-geal inflammation. Ninety percent of the drug is excreted by glomerular filtration. In the presence of renal insufficiency, strik-ing accumulation may occur ( Table 43–2 ). In functionally anephric patients, the half-life of vancomycin is 6–10 days. A significant amount (roughly 50%) of vancomycin is removed during a standard hemodialysis run when a modern, high-flux membrane is used.
043-Katzung_Ch043_p789-808.indd 802 9/23/11 5:51:30 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 803
Clinical Uses Important indications for parenteral vancomycin are bloodstream infections and endocarditis caused by methicillin-resistant staphy-lococci. However, vancomycin is not as effective as an antistaphy-lococcal penicillin for treatment of serious infections such as endocarditis caused by methicillin-susceptible strains. Vancomycin in combination with gentamicin is an alternative regimen for treatment of enterococcal endocarditis in a patient with serious penicillin allergy. Vancomycin (in combination with cefotaxime, ceftriaxone, or rifampin) is also recommended for treatment of meningitis suspected or known to be caused by a penicillin-resis-tant strain of pneumococcus (ie, penicillin MIC > 1 mcg/mL). The recommended dosage in a patient with normal renal function is 30–60 mg/kg/d in two or three divided doses. The traditional dosing regimen in adults with normal renal function is 1 g every 12 hours (∼ 30 mg/kg/d); however, this dose will not typically achieve the trough concentrations (15–20 mcg/mL) recom-mended for serious infections. For serious infections (see below), a starting dose of 45–60 mg/kg/d should be given with titration of the dose to achieve trough levels of 15–20 mcg/mL. The dosage in children is 40 mg/kg/d in three or four divided doses. Clearance of vancomycin is directly proportional to creatinine clearance, and the dosage is reduced accordingly in patients with renal insuffi-ciency. For functionally anephric adult patients, a 1-g dose admin-istered once a week may be sufficient. For patients receiving hemodialysis, a common dosing regimen is a 1-g loading dose fol-lowed by 500 mg after each dialysis session. Patients receiving a prolonged course of therapy should have serum concentrations checked. Recommended trough concentrations are 10–15 mcg/mL for mild to moderate infections such as cellulitis and 15–20 mcg/mL for more serious infections such as endocarditis, meningitis, and necrotizing pneumonia.
Oral vancomycin, 0.125–0.25 g every 6 hours, is used to treat antibiotic-associated colitis caused by C difficile . Because of the emergence of vancomycin-resistant enterococci and the potential selective pressure of oral vancomycin for these resistant organisms, metronidazole had been preferred as initial therapy over the last two decades. However, receipt of oral vancomycin does not appear to be a significant risk factor for acquisition of vancomycin- resistant enterococci. Additionally, recent clinical data suggest that vancomycin is associated with a better clinical response than metronidazole for more severe cases of C difficile colitis. Therefore, oral vancomycin may be used as a first line treatment for severe cases or for cases that fail to respond to metronidazole.
Adverse Reactions Adverse reactions are encountered in about 10% of cases. Most reactions are minor. Vancomycin is irritating to tissue, resulting in phlebitis at the site of injection. Chills and fever may occur. Ototoxicity is rare and nephrotoxicity uncommon with current preparations. However, administration with another ototoxic or nephrotoxic drug, such as an aminoglycoside, increases the risk of these toxicities. Ototoxicity can be minimized by maintaining peak serum concentrations below 60 mcg/mL. Among the more
common reactions is the so-called “red man” or “red neck” syndrome. This infusion-related flushing is caused by release of histamine. It can be largely prevented by prolonging the infusion period to 1–2 hours or pretreatment with an antihistamine such as diphenhydramine.
TEICOPLANIN
Teicoplanin is a glycopeptide antibiotic that is very similar to vancomycin in mechanism of action and antibacterial spectrum. Unlike vancomycin, it can be given intramuscularly as well as intravenously. Teicoplanin has a long half-life (45–70 hours), per-mitting once-daily dosing. This drug is available in Europe but has not been approved for use in the United States.
TELAVANCIN
Telavancin is a semisynthetic lipoglycopeptide derived from vancomycin. Telavancin is active versus gram-positive bacteria, including strains with reduced susceptibility to vancomycin. Telavancin has two mechanisms of action. Like vancomycin, tela-vancin inhibits cell wall synthesis by binding to the D-Ala-D-Ala terminus of peptidoglycan in the growing cell wall. In addition, it disrupts the bacterial cell membrane potential and increases mem-brane permeability. The half-life of telavancin is approximately 8 hours, which supports once-daily intravenous dosing. Telavancin is approved for treatment of complicated skin and soft tissue infec-tions at a dose of 10 mg/kg IV daily. Unlike vancomycin therapy, monitoring of serum telavancin levels is not required. Telavancin is potentially teratogenic, so administration to pregnant women must be avoided.
DALBAVANCIN
Dalbavancin is a semisynthetic lipoglycopeptide derived from teicoplanin. Dalbavancin shares the same mechanism of action as vancomycin and teicoplanin but has improved activity against many gram-positive bacteria including methicillin-resistant and vancomycin-intermediate S aureus . It is not active against most strains of vancomycin-resistant enterococci. Dalbavancin has an extremely long half-life of 6–11 days, which allows for once-weekly intravenous administration. Development of dalbavancin has been put on hold pending additional clinical trials.
■ OTHER CELL WALL OR MEMBRANEACTIVE AGENTS
DAPTOMYCIN
Daptomycin is a novel cyclic lipopeptide fermentation product of Streptomyces roseosporus ( Figure 43–8 ). It was discovered decades ago but has only recently been developed as the need for drugs
043-Katzung_Ch043_p789-808.indd 803 9/23/11 5:51:30 PM

804 SECTION VIII Chemotherapeutic Drugs
active against resistant organisms has become more acute. Its spec-trum of activity is similar to that of vancomycin except that it is more rapidly bactericidal in vitro and may be active against vancomycin-resistant strains of enterococci and S aureus . The precise mechanism of action is not fully understood, but it is known to bind to the cell membrane via calcium-dependent inser-tion of its lipid tail. This results in depolarization of the cell mem-brane with potassium efflux and rapid cell death ( Figure 43–9 ). Daptomycin is cleared renally. The approved doses are 4 mg/kg/dose for treatment of skin and soft tissue infections and 6 mg/kg/dose for treatment of bacteremia and endocarditis once daily in patients with normal renal function and every other day in patients with creatinine clearance of less than 30 mL/min. For
serious infections, many experts recommend using doses of dapto-mycin higher than 6 mg/kg/dose. In clinical trials powered for noninferiority, daptomycin was equivalent in efficacy to vancomy-cin. It can cause myopathy, and creatine phosphokinase levels should be monitored weekly. Pulmonary surfactant antagonizes daptomycin, and it should not be used to treat pneumonia. Daptomycin can also cause an allergic pneumonitis in patients receiving prolonged therapy (> 2 weeks). Treatment failures have been reported in association with an increase in daptomycin MIC for clinical isolates obtained during therapy, but the relation between the increase in MIC and treatment failure is unclear at this point. Daptomycin is an effective alternative to vancomycin, and its ultimate role continues to unfold.
= O
=
L-Asp
L-Thr
L-Asp L-Asn L-Trp NH
Decanoic acid
GlyL-Orn
L-Asp D-Ala Gly D-Ser 3-MeGlu (L-theo)
L-KynO C
O
FIGURE 43–8 Structure of daptomycin. (Kyn, deaminated tryptophan.)
FIGURE 43–9 Proposed mechanism of action of daptomycin. Daptomycin first binds to the cytoplasmic membrane (step 1) and then forms complexes in a calcium-dependent manner (steps 2 and 3). Complex formation causes a rapid loss of cellular potassium, possibly by pore formation, and membrane depolarization. This is followed by arrest of DNA, RNA, and protein synthesis resulting in cell death. Cell lysis does not occur.
K+
Ca2+
Ca2+
Ca2+
Step 1
Step 2 Step 3
Daptomycin
043-Katzung_Ch043_p789-808.indd 804 9/23/11 5:51:30 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 805
FOSFOMYCIN
Fosfomycin trometamol, a stable salt of fosfomycin (phosphono-mycin), inhibits a very early stage of bacterial cell wall synthesis ( Figure 43–5 ). An analog of phosphoenolpyruvate, it is structurally unrelated to any other antimicrobial agent. It inhibits the cytoplas-mic enzyme enolpyruvate transferase by covalently binding to the cysteine residue of the active site and blocking the addition of phosphoenolpyruvate to UDP-N-acetylglucosamine. This reaction is the first step in the formation of UDP- N- acetylmuramic acid, the precursor of N -acetylmuramic acid, which is found only in bacterial cell walls. The drug is transported into the bacterial cell by glycerophosphate or glucose 6-phosphate transport systems. Resistance is due to inadequate transport of drug into the cell.
Fosfomycin is active against both gram-positive and gram-negative organisms at concentrations ≥ 125 mcg/mL. Susceptibility tests should be performed in growth medium supplemented with glucose 6-phosphate to minimize false-positive indications of resistance. In vitro synergism occurs when fosfomycin is combined with β-lactam antibiotics, aminoglycosides, or fluoroquinolones.
Fosfomycin trometamol is available in both oral and parenteral formulations, although only the oral preparation is approved for use in the USA. Oral bioavailability is approximately 40%. Peak serum concentrations are 10 mcg/mL and 30 mcg/mL following a 2-g or 4-g oral dose, respectively. The half-life is approximately 4 hours. The active drug is excreted by the kidney, with urinary concentrations exceeding MICs for most urinary tract pathogens.
Fosfomycin is approved for use as a single 3-g dose for treat-ment of uncomplicated lower urinary tract infections in women. The drug appears to be safe for use in pregnancy.
BACITRACIN
Bacitracin is a cyclic peptide mixture first obtained from the Tracy strain of Bacillus subtilis in 1943. It is active against gram-positive
microorganisms. Bacitracin inhibits cell wall formation by inter-fering with dephosphorylation in cycling of the lipid carrier that transfers peptidoglycan subunits to the growing cell wall ( Figure 43–5 ). There is no cross-resistance between bacitracin and other antimicrobial drugs.
Bacitracin is highly nephrotoxic when administered systemi-cally and is only used topically ( Chapter 61 ). Bacitracin is poorly absorbed. Topical application results in local antibacterial activity without systemic toxicity. Bacitracin, 500 units/g in an ointment base (often combined with polymyxin or neomycin), is indicated for the suppression of mixed bacterial flora in surface lesions of the skin, in wounds, or on mucous membranes. Solutions of bacitracin containing 100–200 units/mL in saline can be used for irrigation of joints, wounds, or the pleural cavity.
CYCLOSERINE
Cycloserine is an antibiotic produced by Streptomyces orchidaceous . It is water soluble and very unstable at acid pH. Cycloserine inhib-its many gram-positive and gram-negative organisms, but it is used almost exclusively to treat tuberculosis caused by strains of Mycobacterium tuberculosis resistant to first-line agents. Cycloserine is a structural analog of D-alanine and inhibits the incorporation of D-alanine into peptidoglycan pentapeptide by inhibiting alanine race-mase, which converts L-alanine to D-alanine, and D-alanyl- D-alanine ligase ( Figure 43–5 ). After ingestion of 0.25 g of cycloserine blood levels reach 20–30 mcg/mL—sufficient to inhibit many strains of mycobacteria and gram-negative bacteria. The drug is widely distributed in tissues. Most of the drug is excreted in active form into the urine. The dosage for treating tuberculosis is 0.5 to 1 g/d in two or three divided doses.
Cycloserine causes serious dose-related central nervous system toxicity with headaches, tremors, acute psychosis, and convul-sions. If oral dosages are maintained below 0.75 g/d, such effects can usually be avoided.
SUMMARY Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics
Subclass Mechanism of Action Effects Clinical ApplicationsPharmacokinetics, Toxicities, Interactions
PENICILLINS
• Penicillin G Prevents bacterial cell wall synthesis by binding to and inhibiting cell wall transpeptidases
Rapid bactericidal activity against susceptible bacteria
Streptococcal infections, meningococcal infections, neurosyphilis
IV administration • rapid renal clearance (half-life 30 min, so requires frequent dosing (every 4 h) • Toxicity: Immediate hypersensi-tivity, rash, seizures
• Penicillin V: Oral, low systemic levels limit widespread use • Benzathine penicillin, procaine penicillin: Intramuscular, long-acting formulations
• Nafcillin, oxacillin: Intravenous, added stability to staphylococcal β lactamase, biliary clearance
• Ampicillin, amoxicillin, ticarcillin, piperacillin: Greater activity versus gram-negative bacteria; addition of β-lactamase inhibitor restores activity against many β-lactamase-producing bacteria
(continued )
043-Katzung_Ch043_p789-808.indd 805 9/23/11 5:51:30 PM

806 SECTION VIII Chemotherapeutic Drugs
Subclass Mechanism of Action Effects Clinical ApplicationsPharmacokinetics, Toxicities, Interactions
CEPHALOSPORINS
• Cefazolin Prevents bacterial cell wall syn-thesis by binding to and inhibit-ing cell wall transpeptidases
Rapid bactericidal activity against susceptible bacteria
Skin and soft tissue infections, urinary tract infections, surgical prophylaxis
IV administration • renal clearance (half-life 1.5 h) • dosed every 8 h • poor penetration into the central nervous system (CNS) • Toxicity: Rash, drug fever
• Cephalexin: Oral, first-generation drug, used for treating skin and soft tissue infections and urinary tract infections
• Cefuroxime: Oral and intravenous, second-generation drug, improved activity versus pneumococcus and Haemophilus influenzae
• Cefotetan, cefoxitin: Intravenous, second-generation drugs, activity versus Bacteroides fragilis allows for use in abdominal/pelvic infections
• Ceftriaxone: Intravenous, third-generation drug, mixed clearance with long half-life (6 hours), good CNS penetration, many uses including pneumonia, meningitis, pyelonephritis, and gonorrhea
• Cefotaxime: Intravenous, third-generation, similar to ceftriaxone; however, clearance is renal and half-life is 1 hour
• Ceftazidime: Intravenous, third-generation drug, poor gram-positive activity, good activity versus Pseudomonas
• Cefepime: Intravenous, fourth-generation drug, broad activity with improved stability to chromosomal β lactamase
• Ceftaroline: Intravenous, active against methicillin-resistant staphylococci, broad gram-negative activity
CARBAPENEMS • Imipenem-cilastatin Prevents bacterial cell wall
synthesis by binding to and inhibiting cell wall transpeptidases
Rapid bactericidal activity against sus-ceptible bacteria
Serious infections such as pneumonia and sepsis
IV administration • renal clearance (half-life 1 h), dosed every 6–8 h, cilastatin added to prevent hydrolysis by renal dehydropepti-dase • Toxicity: Seizures especially in renal failure or with high doses (> 2 g/d)
• Meropenem, doripenem: Intravenous, similar activity to imipenem; stable to renal dehydropeptidase, lower incidence of seizures
• Ertapenem: Intravenous, longer half-life allows for once-daily dosing, lacks activity versus Pseudomonas and Acinetobacter
MONOBACTAMS • Aztreonam Prevents bacterial cell wall
synthesis by binding to and inhibiting cell wall transpeptidases
Rapid bactericidal activity against susceptible bacteria
Infections caused by aerobic, gram-negative bacteria in patients with immediate hypersensitivity to penicillins
IV administration • renal clearance half-life 1.5 h • dosed every 8 h • Toxicity: No cross-allergenicity with penicillins
GLYCOPEPTIDE • Vancomycin Inhibits cell wall synthesis by
binding to the D-Ala-D-Ala termi-nus of nascent peptidoglycan
Bactericidal activity against susceptible bacteria, slower kill than β-lactam antibiotics
Infections caused by gram-positive bacteria including sepsis, endocarditis, and meningitis • C difficile colitis (oral formulation)
Oral, IV administration • renal clearance (half-life 6 h) • starting dose of 30 mg/kg/d in two or three divided doses in patients with normal renal function • trough concentra-tions of 10–15 mcg/mL sufficient for most infections • Toxicity: “Red man” syndrome • nephrotoxicity uncommon
• Teicoplanin: Intravenous, similar to vancomycin except that long half-life (45–75 h) permits once-daily dosing
• Dalbavancin: Intravenous, very long half-life (6–11 days) permits once-weekly dosing, more active than vancomycin
• Telavancin: Intravenous, dual mechanism of action results in improved activity against bacteria with reduced susceptibility to vancomycin, once-daily dosing
LIPOPEPTIDE • Daptomycin Binds to cell membrane, causing
depolarization and rapid cell death
Bactericidal activ-ity against suscep-tible bacteria • more rapidly bactericidal than vancomycin
Infections caused by gram-positive bacteria including sepsis and endocarditis
IV administration • renal clearance (half-life 8 h) • dosed once daily • inactivated by pul-monary surfactant so cannot be used to treat pneumonia • Toxicity: Myopathy • monitoring of weekly creatine phosphokinase levels recommended
043-Katzung_Ch043_p789-808.indd 806 9/23/11 5:51:30 PM

CHAPTER 43 Beta-Lactam & Other Cell Wall- & Membrane-Active Antibiotics 807
PENICILLINSAmoxicillin (generic, Amoxil, others)
Oral: 125, 200, 250, 400 mg chewable tablets; 500, 875 mg tablets; 250, 500 mg capsules; powder to reconstitute for 50, 125, 200, 250, 400 mg/mL solution
Amoxicillin/potassium clavulanate (generic, Augmentin)1
Oral: 250, 500, 875 mg tablets; 125, 200, 250, 400 mg chewable tablets; 1000 mg extended-release tablet; powder to reconstitute for 125, 200, 250 mg/5 mL suspension
Ampicillin (generic)Oral: 250, 500 mg capsules; powder to reconstitute for 125, 250 mg
suspensionsParenteral: powder to reconstitute for injection (125, 250, 500 mg,
1, 2 g per vial)Ampicillin/sulbactam sodium (generic, Unasyn)2
Parenteral: 1, 2 g ampicillin powder to reconstitute for IV or IM injection
Carbenicillin (Geocillin)Oral: 382 mg tablets
Dicloxacillin (generic)Oral: 250, 500 mg capsules
Nafcillin (generic)Parenteral: 1, 2 g per IV piggyback units
Oxacillin (generic)Parenteral: powder to reconstitute for injection (0.5, 1, 2, 10 g
per vial)Penicillin G (generic, Pentids, Pfizerpen)
Parenteral: powder to reconstitute for injection (1, 2, 3, 5, 10, 20 million units)
Penicillin G benzathine (Permapen, Bicillin)Parenteral: 0.6, 1.2, 2.4 million units per dose
Penicillin G procaine (generic)Parenteral: 0.6, 1.2 million units/mL for IM injection only
Penicillin V (generic, V-Cillin, Pen-Vee K, others)Oral: 250, 500 mg tablets; powder to reconstitute for 125, 250 mg/5 mL
solutionPiperacillin (Pipracil)
Parenteral: powder to reconstitute for injection (2, 3, 4 g per vial)Piperacillin and tazobactam sodium (Zosyn)3
Parenteral: 2, 3, 4 g powder to reconstitute for IV injectionTicarcillin (Ticar)
Parenteral: powder to reconstitute for injection (1, 3, 6 g per vial)Ticarcillin/clavulanate potassium (Timentin)4
Parenteral: 3 g powder to reconstitute for injection
CEPHALOSPORINS & OTHER BETA-LACTAM DRUGSNarrow-Spectrum (First-Generation) CephalosporinsCefadroxil (generic, Duricef)
Oral: 500 mg capsules; 1 g tablets; 125, 250, 500 mg/5 mL suspensionCefazolin (generic, Ancef, Kefzol)
Parenteral: powder to reconstitute for injection (0.25, 0.5, 1 g per vial or IV piggyback unit)
Cephalexin (generic, Keflex, others)Oral: 250, 500 mg capsules and tablets; 1 g tablets; 125, 250 mg/5 mL
suspension
Intermediate-Spectrum (Second-Generation) CephalosporinsCefaclor (generic, Ceclor)
Oral: 250, 500 mg capsules; 375, 500 mg extended-release tablets; powder to reconstitute for 125, 187, 250, 375 mg/5 mL suspension
Cefmetazole (Zefazone)Parenteral: 1, 2 g powder for IV injection
Cefotetan (Cefotan)Parenteral: powder to reconstitute for injection (1, 2, 10 g per vial)
Cefoxitin (Mefoxin)Parenteral: powder to reconstitute for injection (1, 2, 10 g per vial)
Cefprozil (Cefzil)Oral: 250, 500 mg tablets; powder to reconstitute 125, 250 mg/5 mL
suspensionCefuroxime (generic, Ceftin, Kefurox, Zinacef)
Oral: 125, 250, 500 mg tablets; 125, 250 mg/5 mL suspensionParenteral: powder to reconstitute for injection (0.75, 1.5, 7.5 g per
vial or infusion pack)Loracarbef (Lorabid)
Oral: 200, 400 mg capsules; powder for 100, 200 mg/5 mL suspensionBroad-Spectrum (Third- & Fourth-Generation) CephalosporinsCefdinir (Omnicef)
Oral: 300 mg capsules; 125 mg/5 mL suspensionCefditoren (Spectracef)
Oral: 200 mg tabletsCefepime (Maxipime)
Parenteral: powder for injection 0.5, 1, 2 gCefixime (Suprax)
Oral: 200, 400 mg tablets; powder for oral suspension, 100 mg/5 mL
Cefotaxime (Claforan)Parenteral: powder to reconstitute for injection (0.5, 1, 2 g per vial)
Cefpodoxime proxetil (Vantin)Oral: 100, 200 mg tablets; 50, 100 mg granules for suspension in 5 mL
Ceftaroline fosamil (Teflaro)Parenteral: powder to reconstitute for injection (0.6 g per vial)
Ceftazidime (generic, Fortaz, Tazidime)Parenteral: powder to reconstitute for injection (0.5, 1, 2 g per vial)
Ceftibuten (Cedax)Oral: 400 mg capsules; 90, 180 mg/5 mL powder for oral
suspensionCeftizoxime (Cefizox)
Parenteral: powder to reconstitute for injection and solution for injection (0.5, 1, 2 g per vial)
Ceftriaxone (Rocephin)Parenteral: powder to reconstitute for injection (0.25, 0.5, 1, 2, 10 g
per vial)Carbapenems & MonobactamAztreonam (Azactam)
Parenteral: powder to reconstitute for injection (0.5, 1, 2 g)Doripenem (Doribax)
Parenteral: powder to reconstitute for injection (500 mg per vial)Ertapenem (Invanz)
Parenteral: 1 g powder to reconstitute for IV (0.9% diluent) or IM (1% lidocaine diluent) injection
Imipenem/cilastatin (Primaxin)Parenteral: powder to reconstitute for injection (250, 500, 750 mg
imipenem per vial)
P R E P A R A T I O N S A V A I L A B L E
043-Katzung_Ch043_p789-808.indd 807 9/23/11 5:51:30 PM

808 SECTION VIII Chemotherapeutic Drugs
REFERENCES Biek D et al: Ceftaroline fosamil: A novel broad-spectrum cephalosporin with expanded
Gram-positive activity. J Antimicrob Chemother 2010;65(Suppl 4):iv9. Billeter M et al: Dalbavancin: A novel once-weekly lipoglycopeptide antibiotic.
Clin Infect Dis 2008;46:577. Bush K et al: Anti-MRSA beta-lactams in development, with a focus on ceftobiprole:
The first anti-MRSA beta-lactam to demonstrate clinical efficacy. Expert Opin Investig Drugs 2007;16:419.
Carpenter CF, Chambers HF: Daptomycin: Another novel agent for treating infec-tions due to drug-resistant gram-positive pathogens. Clin Infect Dis 2004;38:994.
Centers for Disease Control and Prevention: Vancomycin-resistant Staphylococcus aureus —Pennsylvania, 2002. JAMA 2002;288:2116.
Chow JW et al: Enterobacter bacteremia: Clinical features and emergence of anti-biotic resistance during therapy. Ann Intern Med 1991;115:585.
Fowler VG et al: Daptomycin versus standard therapy for bacteremia and endo-carditis caused by Staphylococcus aureus . N Engl J Med 2006;355:653.
Hiramatsu K et al: Methicillin resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 1997;40:135.
Hiramatsu K: Vancomycin-resistant Staphylococcus aureus : A new model of antibi-otic resistance. Lancet Infect Dis 2001;1:147.
Jacoby GA, Munoz-Price LS: The new beta-lactamases. N Engl J Med 2005;352:380.
Meropenem (Merrem IV)Parenteral: powder for injection (0.5, 1 g per vial)
OTHER DRUGS DISCUSSED IN THIS CHAPTERCycloserine (Seromycin Pulvules)
Oral: 250 mg capsulesDaptomycin (Cubicin)
Parenteral: 0.25 or 0.5 g lyophilized powder to reconstitute for IV injection
Fosfomycin (Monurol)Oral: 3 g packet
Telavancin (Vibativ)Parenteral: 0.25 and 0.75 g powder to reconstitute for IV injection
Vancomycin (generic, Vancocin, Vancoled)Oral: 125, 250 mg pulvules; powder to reconstitute for 250 mg/5 mL,
500 mg/6 mL solutionParenteral: 0.5, 1, 5, 10 g powder to reconstitute for IV injection
1Clavulanate content varies with the formulation; see package insert.2Sulbactam content is half the ampicillin content.3Tazobactam content is 12.5% of the piperacillin content.4Clavulanate content 0.1 g.
Meropenem (Merrem IV)Parenteral: powder for injection (0.5, 1 g per vial)
OTHER DRUGS DISCUSSED IN THISCHAPTERCycloserine (Seromycin Pulvules)
Oral: 250 mg capsulesDaptomycin (Cubicin)
Parenteral: 0.25 or 0.5 g lyophilized powder to reconstitute forIV injection
Fosfomycin (Monurol)Oral: 3 g packet
Telavancin (Vibativ)Parenteral: 0.25 and 0.75 g powder to reconstitute for IV injection
Vancomycin (generic, Vancocin, Vancoled)Oral: 125, 250 mg pulvules; powder to reconstitute for 250 mg/5 mL,
500 mg/6 mL solutionParenteral: 0.5, 1, 5, 10 g powder to reconstitute for IV injection
Keating GM, Perry CM: Ertapenem: A review of its use in the treatment of bacterial infections. Drugs 2005;65:2151.
Leonard SN, Rybak MJ: Telavancin: An antimicrobial with a multifunctional mechanism of action for the treatment of serious gram-positive infections. Pharmacotherapy 2008;28:458.
Mandell L: Doripenem: A new carbapenem in the treatment of nosocomial infec-tions. Clin Infect Dis 2009;49(Suppl 1):S1.
Noskin GA et al: National trends in Staphylococcus aureus infection rates: Impact on economic burden and mortality over a 6-year period. Clin Infect Dis 2007;45:1132.
Park MA, Li JT: Diagnosis and management of penicillin allergy. Mayo Clin Proc 2005;80:405.
Rybak M et al: Therapeutic monitoring of vancomycin in adults patients: A con-sensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm 2009;66:82.
Sanders EW Jr, Tenny JH, Kessler RE: Efficacy of cefepime in the treatment of infections due to multiply resistant Enterobacter species. Clin Infect Dis 1996;23:454.
Wexler HM: In vitro activity of ertapenem: Review of recent studies. J Antimicrob Chemother 2004;53(Suppl 2):ii11.
Zar FA et al: A comparison of vancomycin and metronidazole for the treat-ment of Clostridium difficile -associated diarrhea. Clin Infect Dis 2007;45:302.
C A S E S T U D Y A N S W E R
An intravenous third-generation cephalosporin (ceftriaxone or cefotaxime) with adequate penetration into inflamed meninges that is active against the common bacteria that cause community-acquired pneumonia and meningitis (pneumococcus, meningococcus, Haemophilus ) should be ordered. Vancomycin should also be administered until cul-ture and sensitivity results are available in case the patient is
infected with a resistant pneumococcus. Although the patient has a history of rash to amoxicillin, the presentation is not consistent with an anaphylactic reaction. The aminopenicillins are frequently associated with rashes that are not allergic in nature. In this instance, cross-reactivity with a cephalosporin is unlikely.
043-Katzung_Ch043_p789-808.indd 808 9/23/11 5:51:31 PM

809
Tetracyclines, Macrolides, Clindamycin, Chloramphenicol, Streptogramins, & Oxazolidinones Daniel H. Deck, PharmD & Lisa G. Winston, MD
endocervical canal. No cervical motion tenderness is present. A first-catch urine specimen is obtained to be tested for chlamydia and gonococcus. A pregnancy test is also ordered as the patient reports she “missed her last period.” Pending these results, the decision is made to treat her empirically for gonococcal and chlamydial cervicitis. What are two potential treatment options for her possible chlamydial infection? How does her potential pregnancy affect the treatment decision?
C A S E S T U D Y
A 19-year-old woman with no significant past medical history presents to her college medical clinic complaining of a 2-week history of foul-smelling vaginal discharge. She denies any fever or abdominal pain but does report vaginal bleeding after sexual intercourse. When questioned about her sexual activity, she reports having vaginal intercourse, at times unprotected, with two men in the last 6 months. A pelvic examination is per-formed and is positive for mucopurulent discharge from the
C H A P T E R
The drugs described in this chapter inhibit bacterial protein syn-thesis by binding to and interfering with ribosomes. Most are bacteriostatic, but a few are bactericidal against certain organisms.
44
Because of overuse, tetracycline and macrolide resistance is common. Except for tigecycline and the streptogramins, these antibiotics are usually administered orally.
044-Katzung_Ch044_p809-820.indd 809 9/23/11 4:21:10 PM

810 SECTION VIII Chemotherapeutic Drugs
■ TETRACYCLINES
All of the tetracyclines have the basic structure shown below:
OH
R7HOH
OH
C
OHNH2
OHO O
O
HR6 N(CH3)2R5
109 2
34
1
56
11
78
12
CH3 HCI 35
RenalClearance(mL/min)R5R6R7
ChlortetracyclineCH3 OHH 90OxytetracyclineCH3 HH 65TetracyclineH HCI 35DemeclocyclineCH2* OHH 31MethacyclineCH3* OHH 16DoxycyclineH HN(CH3)2
OH at position 6 on methacycline and doxycycline.10Minocycline
*There is no
Free tetracyclines are crystalline amphoteric substances of low solubility. They are available as hydrochlorides, which are more soluble. Such solutions are acid and, with the exception of chlortetracycline, fairly stable. Tetracyclines chelate divalent metal ions, which can interfere with their absorption and activity. A newly approved tetracycline analog, tigecycline, is a glycylcycline and a semisynthetic derivative of minocycline.
N(CH3)2 N(CH3)2
H3C
H3CCH3
NH2
OH
OOOHO
Tigecycline
O
OHHOH
NN
H
Mechanism of Action & Antimicrobial Activity Tetracyclines are broad-spectrum bacteriostatic antibiotics that inhibit protein synthesis. Tetracyclines enter microorganisms in part by passive diffusion and in part by an energy-dependent process of active transport. Susceptible organisms concentrate the drug intracellularly. Once inside the cell, tetracyclines bind revers-ibly to the 30S subunit of the bacterial ribosome, blocking the binding of aminoacyl-tRNA to the acceptor site on the mRNA-ribosome complex ( Figure 44–1 ). This prevents addition of amino acids to the growing peptide.
Tetracyclines are active against many gram-positive and gram-negative bacteria, including certain anaerobes, rickettsiae, chla-mydiae, and mycoplasmas. The antibacterial activities of most tetracyclines are similar except that tetracycline-resistant strains may be susceptible to doxycycline, minocycline, and tigecycline, all of which are poor substrates for the efflux pump that mediates resistance. Differences in clinical efficacy for susceptible organisms are minor and attributable largely to features of absorption, distri-bution, and excretion of individual drugs.
Resistance Three mechanisms of resistance to tetracycline analogs have been described: (1) impaired influx or increased efflux by an active trans-port protein pump; (2) ribosome protection due to production of proteins that interfere with tetracycline binding to the ribosome; and (3) enzymatic inactivation. The most important of these are produc-tion of an efflux pump and ribosomal protection. Tet(AE) efflux pump-expressing gram-negative species are resistant to the older tetracyclines, doxycycline, and minocycline. They are susceptible, however, to tigecycline, which is not a substrate of these pumps. Similarly, the Tet(K) efflux pump of staphylococci confers resistance to tetracycline, but not to doxycycline, minocycline, or tigecycline, none of which are pump substrates. The Tet(M) ribosomal protection protein expressed by gram-positives produces resistance to tetracycline, doxycycline, and minocycline, but not to tigecycline, which because of its bulky t -butylglycylamido substituent, has a steric hindrance effect on Tet(M) binding to the ribosome. Tigecycline is a substrate of the chromosomally encoded multidrug efflux pumps of Proteus sp and Pseudomonas aeruginosa , accounting for their intrinsic resistance to all tetracyclines including tigecycline.
Pharmacokinetics Tetracyclines differ in their absorption after oral administration and in their elimination. Absorption after oral administration is approximately 30% for chlortetracycline; 60–70% for tetracycline, oxytetracycline, demeclocycline, and methacycline; and 95–100% for doxycycline and minocycline. Tigecycline is poorly absorbed orally and must be administered intravenously. A portion of an orally administered dose of tetracycline remains in the gut lumen, alters intestinal flora, and is excreted in the feces. Absorption occurs mainly in the upper small intestine and is impaired by food (except doxycycline and minocycline); by divalent cations (Ca 2+ , Mg 2+ , Fe 2+ ) or Al 3+ ; by dairy products and antacids, which contain mul-tivalent cations; and by alkaline pH. Specially buffered tetracycline solutions are formulated for intravenous administration.
Tetracyclines are 40–80% bound by serum proteins. Oral dosages of 500 mg every 6 hours of tetracycline hydrochloride or oxytetracycline produce peak blood levels of 4–6 mcg/mL. Intravenously injected tetracyclines give somewhat higher levels, but only temporarily. Peak levels of 2–4 mcg/mL are achieved with a 200-mg dose of doxycycline or minocycline. Steady-state peak serum concentrations of tigecycline are 0.6 mcg/mL at the standard dosage. Tetracyclines are distributed widely to tissues and body fluids except for cerebrospinal fluid, where concentrations are 10–25% of those in serum. Minocycline reaches very high concen-trations in tears and saliva, which makes it useful for eradication of the meningococcal carrier state. Tetracyclines cross the placenta to reach the fetus and are also excreted in milk. As a result of chelation with calcium, tetracyclines are bound to—and damage—growing bones and teeth. Carbamazepine, phenytoin, barbiturates, and chronic alcohol ingestion may shorten the half-life of doxycycline 50% by induction of hepatic enzymes that metabolize the drug.
Tetracyclines are excreted mainly in bile and urine. Concentrations in bile exceed those in serum tenfold. Some of the drug excreted in
044-Katzung_Ch044_p809-820.indd 810 9/23/11 4:21:11 PM

CHAPTER 44 Tetracyclines, Macrolides, and Others 811
bile is reabsorbed from the intestine (enterohepatic circulation) and may contribute to maintenance of serum levels. Ten to fifty percent of various tetracyclines is excreted into the urine, mainly by glom-erular filtration. Ten to forty percent of the drug is excreted in feces. Doxycycline and tigecycline, in contrast to other tetracyclines, are eliminated by nonrenal mechanisms, do not accumulate signifi-cantly, and require no dosage adjustment in renal failure.
Tetracyclines are classified as short-acting (chlortetracycline, tet-racycline, oxytetracycline), intermediate-acting (demeclocycline and methacycline), or long-acting (doxycycline and minocycline) based on serum half-lives of 6–8 hours, 12 hours, and 16–18 hours, respectively. Tigecycline has a half-life of 36 hours. The almost com-plete absorption and slow excretion of doxycycline and minocycline allow for once-daily dosing for certain indications, but by conven-tion these two drugs are usually dosed twice daily.
Clinical Uses A tetracycline is the drug of choice in the treatment of infections caused by rickettsiae. Tetracyclines are also excellent drugs for the treatment of Mycoplasma pneumonia , chlamydiae, and some
spirochetes. They are used in combination regimens to treat gastric and duodenal ulcer disease caused by Helicobacter pylori . They may be used in various gram-positive and gram-negative bacterial infec-tions, including vibrio infections, provided the organism is not resistant. In cholera, tetracyclines rapidly stop the shedding of vibrios, but tetracycline resistance has appeared during epidemics. Tetracyclines remain effective in most chlamydial infections, including sexually transmitted infections. Tetracyclines are no lon-ger recommended for treatment of gonococcal disease because of resistance. A tetracycline—in combination with other antibiot-ics—is indicated for plague, tularemia, and brucellosis. Tetracyclines are sometimes used in the treatment or prophylaxis of protozoal infections, eg, those due to Plasmodium falciparum (see Chapter 52 ). Other uses include treatment of acne, exacerbations of bronchitis, community-acquired pneumonia, Lyme disease, relapsing fever, leptospirosis, and some nontuberculous mycobacte-rial infections (eg, Mycobacterium marinum ). Tetracyclines formerly were used for a variety of common infections, including bacterial gastroenteritis and urinary tract infections. However, many strains of bacteria causing these infections are now resistant, and other agents have largely supplanted tetracyclines.
T
C
M
12
34
1
2
3
4
5 6
6
50Sribosome
30S
Amino acid
ChargedtRNA
mRNA
t5
t5
t6
t6
Uncharged tRNA
FIGURE 44–1 Steps in bacterial protein synthesis and targets of several antibiotics. Amino acids are shown as numbered circles. The 70S ribosomal mRNA complex is shown with its 50S and 30S subunits. In step 1, the charged tRNA unit carrying amino acid 6 binds to the acceptor site on the 70S ribosome. The peptidyl tRNA at the donor site, with amino acids 1 through 5, then binds the growing amino acid chain to amino acid 6 (peptide bond formation, step 2). The uncharged tRNA left at the donor site is released (step 3), and the new 6-amino acid chain with its tRNA shifts to the peptidyl site (translocation, step 4). The antibiotic binding sites are shown schematically as triangles. Chloramphenicol (C) and macrolides (M) bind to the 50S subunit and block peptide bond formation (step 2). The tetracyclines (T) bind to the 30S subunit and prevent binding of the incoming charged tRNA unit (step 1).
044-Katzung_Ch044_p809-820.indd 811 9/23/11 4:21:12 PM

812 SECTION VIII Chemotherapeutic Drugs
Minocycline, 200 mg orally daily for 5 days, can eradicate the meningococcal carrier state, but because of side effects and resis-tance of many meningococcal strains, rifampin is preferred. Demeclocycline inhibits the action of antidiuretic hormone in the renal tubule and has been used in the treatment of inappropri-ate secretion of antidiuretic hormone or similar peptides by cer-tain tumors (see Chapter 15 ).
Tigecycline , the first glycylcycline to reach clinical practice, has several unique features that warrant its consideration apart from the older tetracyclines. Many tetracycline-resistant strains are susceptible to tigecycline because the common resistance determi-nants have no activity against it. Its spectrum is very broad. Coagulase-negative staphylococci and Staphylococcus aureus , including methicillin-resistant, vancomycin-intermediate, and vancomycin-resistant strains; streptococci, penicillin-susceptible and resistant; enterococci, including vancomycin-resistant strains; gram-positive rods; Enterobacteriaceae; multidrug-resistant strains of Acinetobacter sp; anaerobes, both gram-positive and gram- negative; rickettsiae, Chlamydia sp, and Legionella pneumophila; and rapidly growing mycobacteria all are susceptible. Proteus sp and P aeruginosa , however, are intrinsically resistant.
Tigecycline, formulated for intravenous administration only, is given as a 100-mg loading dose, then 50 mg every 12 hours. As with all tetracyclines, tissue and intracellular penetration is excellent; consequently, the volume of distribution is quite large and peak serum concentrations are low. Elimination is primarily biliary, and no dosage adjustment is needed for patients with renal insufficiency. In addition to the tetracycline class effects, the chief adverse effect of tigecycline is nausea, which occurs in up to one third of patients, and occasionally vomiting. Neither nausea nor vomiting usually requires discontinuation of the drug.
Tigecycline is Food and Drug Administration (FDA)-approved for treatment of skin and skin-structure infection, intra-abdominal infections, and community-acquired pneumonia. Because active drug concentrations in the urine are relatively low, tigecycline may not be effective for urinary tract infections and has no indica-tion for this use. Because it is active against a wide variety of multidrug-resistant nosocomial pathogens (eg, methicillin- resistant S aureus , extended- spectrum β-lactamase-producing gram-negatives, and Acinetobacter sp, tigecycline is a welcome addition to the antimicrobial drug group. However, its clinical efficacy in infections with multidrug-resistant organisms, com-pared with other agents, is largely unknown.
A. Oral Dosage The oral dosage for rapidly excreted tetracyclines, equivalent to tetracycline hydrochloride, is 0.25–0.5 g four times daily for adults and 20–40 mg/kg/d for children (8 years of age and older). For severe systemic infections, the higher dosage is indicated, at least for the first few days. The daily dose is 600 mg for demeclo-cycline or methacycline, 100 mg once or twice daily for doxycy-cline, and 100 mg twice daily for minocycline. Doxycycline is the oral tetracycline of choice because it can be given twice daily, and its absorption is not significantly affected by food. All tetracyclines chelate with metals, and none should be orally administered with
milk, antacids, or ferrous sulfate. To avoid deposition in growing bones or teeth, tetracyclines should be avoided in pregnant women and children younger than 8 years.
B. Parenteral Dosage Several tetracyclines are available for intravenous injection in doses of 0.1–0.5 g every 6–12 hours (similar to oral doses) but doxycy-cline is the usual preferred agent, at a dosage of 100 mg every 12–24 hours. Intramuscular injection is not recommended because of pain and inflammation at the injection site.
Adverse Reactions Hypersensitivity reactions (drug fever, skin rashes) to tetracyclines are uncommon. Most adverse effects are due to direct toxicity of the drug or to alteration of microbial flora.
A. Gastrointestinal Adverse Effects Nausea, vomiting, and diarrhea are the most common reasons for discontinuing tetracycline medication. These effects are attribut-able to direct local irritation of the intestinal tract. Nausea, anorexia, and diarrhea can usually be controlled by administering the drug with food or carboxymethylcellulose, reducing drug dos-age, or discontinuing the drug.
Tetracyclines alter the normal gastrointestinal flora, with sup-pression of susceptible coliform organisms and overgrowth of pseudomonas, proteus, staphylococci, resistant coliforms, clostridia, and candida. This can result in intestinal functional disturbances, anal pruritus, vaginal or oral candidiasis, or Clostridium difficile- associated colitis.
B. Bony Structures and Teeth Tetracyclines are readily bound to calcium deposited in newly formed bone or teeth in young children. When a tetracycline is given during pregnancy, it can be deposited in the fetal teeth, leading to fluorescence, discoloration, and enamel dysplasia; it can also be deposited in bone, where it may cause deformity or growth inhibition. Because of these effects, tetracyclines are generally avoided in pregnancy. If the drug is given for long periods to children younger than 8 years, similar changes can result.
C. Other Toxicities Tetracyclines can impair hepatic function, especially during pregnancy, in patients with preexisting hepatic insufficiency and when high doses are given intravenously. Hepatic necrosis has been reported with daily doses of 4 g or more intravenously.
Renal tubular acidosis and other renal injury resulting in nitrogen retention have been attributed to the administration of outdated tetracycline preparations. Tetracyclines given along with diuretics may produce nitrogen retention. Tetracyclines other than doxycycline may accumulate to toxic levels in patients with impaired kidney function.
Intravenous injection can lead to venous thrombosis. Intramuscular injection produces painful local irritation and should be avoided.
044-Katzung_Ch044_p809-820.indd 812 9/23/11 4:21:12 PM

CHAPTER 44 Tetracyclines, Macrolides, and Others 813
Systemically administered tetracycline, especially demeclocy-cline, can induce sensitivity to sunlight or ultraviolet light, par-ticularly in fair-skinned persons.
Dizziness, vertigo, nausea, and vomiting have been noted par-ticularly with doxycycline at doses above 100 mg. With dosages of 200–400 mg/d of minocycline, 35–70% of patients will have these reactions.
■ MACROLIDES
The macrolides are a group of closely related compounds charac-terized by a macrocyclic lactone ring (usually containing 14 or 16 atoms) to which deoxy sugars are attached. The prototype drug, erythromycin, which consists of two sugar moieties attached to a 14-atom lactone ring, was obtained in 1952 from Streptomyces erythreus . Clarithromycin and azithromycin are semisynthetic derivatives of erythromycin.
N(R1)2
Erythromycin (R1 = CH3, R2 = H)
Clarithromycin (R1, R2 = CH3)
Desosamine
Cladinose
Macrolidering
R1
R1
OH
R1
OH
R1
O
O
O
OO C2H5
HO
OH
O
O
O
OR1
R1
R1
R1R1
R26
R1
ERYTHROMYCIN
Chemistry The general structure of erythromycin is shown with the mac-rolide ring and the sugars desosamine and cladinose. It is poorly soluble in water (0.1%) but dissolves readily in organic solvents. Solutions are fairly stable at 4°C but lose activity rapidly at 20°C and at acid pH. Erythromycins are usually dispensed as various esters and salts.
Mechanism of Action & Antimicrobial Activity The antibacterial action of erythromycin and other macrolides may be inhibitory or bactericidal, particularly at higher concentrations, for susceptible organisms. Activity is enhanced at alkaline pH. Inhibition of protein synthesis occurs via binding to the 50S ribosomal RNA. The binding site is near the peptidyltransferase center, and peptide chain elongation (ie, transpeptidation) is prevented by blocking of the
polypeptide exit tunnel. As a result, peptidyl-tRNA is dissociated from the ribosome. Erythromycin also inhibits the formation of the 50S ribosomal subunit ( Figure 44–1 ).
Erythromycin is active against susceptible strains of gram-positive organisms, especially pneumococci, streptococci, staphylococci, and corynebacteria. Mycoplasma pneumoniae , L pneumophila , Chlamydia trachomatis , Chlamydia psittaci, Chlamydia pneumoniae , H pylori , Listeria monocytogenes, and certain mycobacteria (Mycobacterium kansasii, Mycobacterium scrofulaceum) are also susceptible. Gram-negative organisms such as Neisseria sp, Bordetella pertussis, Bartonella henselae, and Bartonella quintana as well as some Rickettsia species, Treponema pallidum , and Campylobacter species are susceptible. Haemophilus influenzae is somewhat less susceptible.
Resistance to erythromycin is usually plasmid-encoded. Three mechanisms have been identified: (1) reduced permeability of the cell membrane or active efflux; (2) production (by Enterobacteriaceae) of esterases that hydrolyze macrolides; and (3) modification of the ribosomal binding site (so-called ribosomal protection) by chromo-somal mutation or by a macrolide-inducible or constitutive methylase. Efflux and methylase production are the most important resistance mechanisms in gram-positive organisms. Cross-resistance is complete between erythromycin and the other macrolides. Constitutive methylase production also confers resistance to struc-turally unrelated but mechanistically similar compounds such as clindamycin and streptogramin B (so-called macrolide-lincosamide-streptogramin, or MLS-type B, resistance), which share the same ribosomal binding site. Because nonmacrolides are poor inducers of the methylase, strains expressing an inducible methylase will appear susceptible in vitro. However, constitutive mutants that are resistant can be selected out and emerge during therapy with clindamycin.
Pharmacokinetics Erythromycin base is destroyed by stomach acid and must be administered with enteric coating. Food interferes with absorption. Stearates and esters are fairly acid-resistant and somewhat better absorbed. The lauryl salt of the propionyl ester of erythromycin (erythromycin estolate) is the best-absorbed oral preparation. Oral dosage of 2 g/d results in serum erythromycin base and ester con-centrations of approximately 2 mcg/mL. However, only the base is microbiologically active, and its concentration tends to be similar regardless of the formulation. A 500-mg intravenous dose of eryth-romycin lactobionate produces serum concentrations of 10 mcg/mL 1 hour after dosing. The serum half-life is approximately 1.5 hours normally and 5 hours in patients with anuria. Adjustment for renal failure is not necessary. Erythromycin is not removed by dialy-sis. Large amounts of an administered dose are excreted in the bile and lost in feces, and only 5% is excreted in the urine. Absorbed drug is distributed widely except to the brain and cerebrospinal fluid. Erythromycin is taken up by polymorphonuclear leukocytes and macrophages. It traverses the placenta and reaches the fetus.
Clinical Uses Erythromycin is a drug of choice in corynebacterial infections (diphtheria, corynebacterial sepsis, erythrasma); in respiratory,
044-Katzung_Ch044_p809-820.indd 813 9/23/11 4:21:12 PM

814 SECTION VIII Chemotherapeutic Drugs
neonatal, ocular, or genital chlamydial infections; and in treatment of community-acquired pneumonia because its spectrum of activ-ity includes pneumococcus, M pneumoniae , and L pneumophila. Erythromycin is also useful as a penicillin substitute in penicillin-allergic individuals with infections caused by staphylococci (assuming that the isolate is susceptible), streptococci, or pneumo-cocci. Emergence of erythromycin resistance in strains of group A streptococci and pneumococci (penicillin-non-susceptible pneu-mococci in particular) has made macrolides less attractive as first-line agents for treatment of pharyngitis, skin and soft tissue infections, and pneumonia. Erythromycin has been recommended as prophylaxis against endocarditis during dental procedures in individuals with valvular heart disease, although clindamycin, which is better tolerated, has largely replaced it. Although eryth-romycin estolate is the best-absorbed salt, it imposes the greatest risk of adverse reactions. Therefore, the stearate or succinate salt may be preferred.
The oral dosage of erythromycin base, stearate, or estolate is 0.25–0.5 g every 6 hours (for children, 40 mg/kg/d). The dosage of erythromycin ethylsuccinate is 0.4–0.6 g every 6 hours. Oral erythromycin base (1 g) is sometimes combined with oral neomycin or kanamycin for preoperative preparation of the colon. The intravenous dosage of erythromycin gluceptate or lactobion-ate is 0.5–1.0 g every 6 hours for adults and 20–40 mg/kg/d for children. The higher dosage is recommended when treating pneu-monia caused by L pneumophila .
Adverse Reactions Anorexia, nausea, vomiting, and diarrhea are common. Gastrointestinal intolerance, which is due to a direct stimulation of gut motility, is the most common reason for discontinuing erythromycin and substitut-ing another antibiotic.
Erythromycins, particularly the estolate, can produce acute cholestatic hepatitis (fever, jaundice, impaired liver function), probably as a hypersensitivity reaction. Most patients recover from this, but hepatitis recurs if the drug is readministered. Other aller-gic reactions include fever, eosinophilia, and rashes.
Erythromycin metabolites inhibit cytochrome P450 enzymes and, thus, increase the serum concentrations of numerous drugs, including theophylline, warfarin, cyclosporine, and methylpredni-solone. Erythromycin increases serum concentrations of oral digoxin by increasing its bioavailability.
CLARITHROMYCIN
Clarithromycin is derived from erythromycin by addition of a methyl group and has improved acid stability and oral absorption compared with erythromycin. Its mechanism of action is the same as that of erythromycin. Clarithromycin and erythromycin are similar with respect to antibacterial activity except that clarithro-mycin is more active against Mycobacterium avium complex (see Chapter 47 ). Clarithromycin also has activity against Mycobacterium leprae , Toxoplasma gondii, and H influenzae . Erythromycin-resistant streptococci and staphylococci are also resistant to clarithromycin.
A 500-mg dose of clarithromycin produces serum concentra-tions of 2–3 mcg/mL. The longer half-life of clarithromycin (6 hours) compared with erythromycin permits twice-daily dosing. The recommended dosage is 250–500 mg twice daily or 1000 mg of the extended-release formulation once daily. Clarithromycin penetrates most tissues well, with concentrations equal to or exceeding serum concentrations.
Clarithromycin is metabolized in the liver. The major metabolite is 14-hydroxyclarithromycin, which also has antibacterial activity. Portions of active drug and this major metabolite are eliminated in the urine, and dosage reduction (eg, a 500-mg loading dose, then 250 mg once or twice daily) is recommended for patients with creatinine clearances less than 30 mL/min. Clarithromycin has drug interactions similar to those described for erythromycin.
The advantages of clarithromycin compared with erythromycin are lower incidence of gastrointestinal intolerance and less frequent dosing.
AZITHROMYCIN
Azithromycin, a 15-atom lactone macrolide ring compound, is derived from erythromycin by addition of a methylated nitrogen into the lactone ring. Its spectrum of activity, mechanism of action, and clinical uses are similar to those of clarithromycin. Azithromycin is active against M avium complex and T gondii . Azithromycin is slightly less active than erythromycin and clarithromycin against staphylococci and streptococci and slightly more active against H influenzae . Azithromycin is highly active against Chlamydia sp.
Azithromycin differs from erythromycin and clarithromycin mainly in pharmacokinetic properties. A 500-mg dose of azithromy-cin produces relatively low serum concentrations of approximately 0.4 mcg/mL. However, azithromycin penetrates into most tissues (except cerebrospinal fluid) and phagocytic cells extremely well, with tissue concentrations exceeding serum concentrations by 10- to 100-fold. The drug is slowly released from tissues (tissue half-life of 2–4 days) to produce an elimination half-life approaching 3 days. These unique properties permit once-daily dosing and shortening of the duration of treatment in many cases. For example, a single 1-g dose of azithromycin is as effective as a 7-day course of doxycycline for chlamydial cervicitis and urethritis. Community-acquired pneu-monia can be treated with azithromycin given as a 500-mg loading dose, followed by a 250-mg single daily dose for the next 4 days.
Azithromycin is rapidly absorbed and well tolerated orally. It should be administered 1 hour before or 2 hours after meals. Aluminum and magnesium antacids do not alter bioavailability but delay absorption and reduce peak serum concentrations. Because it has a 15-member (not 14-member) lactone ring, azithromycin does not inactivate cytochrome P450 enzymes and, therefore, is free of the drug interactions that occur with erythromycin and clarithromycin.
KETOLIDES
Ketolides are semisynthetic 14-membered-ring macrolides, differ-ing from erythromycin by substitution of a 3-keto group for the neutral sugar l-cladinose. Telithromycin is approved for limited
044-Katzung_Ch044_p809-820.indd 814 9/23/11 4:21:12 PM

CHAPTER 44 Tetracyclines, Macrolides, and Others 815
clinical use. It is active in vitro against Streptococcus pyogenes, S pneumoniae, S aureus, H influenzae, Moraxella catarrhalis , Mycoplasma sp, L pneumophila, Chlamydia sp, H pylori, Neisseria gonorrhoeae, B fragilis, T gondii, and certain nontuberculosis mycobacteria. Many macrolide-resistant strains are susceptible to ketolides because the structural modification of these compounds renders them poor substrates for efflux pump-mediated resistance, and they bind to ribosomes of some bacterial species with higher affinity than macrolides.
Oral bioavailability of telithromycin is 57%, and tissue and intracellular penetration is generally good. Telithromycin is metabolized in the liver and eliminated by a combination of biliary and urinary routes of excretion. It is administered as a once-daily dose of 800 mg, which results in peak serum concen-trations of approximately 2 mcg/mL. It is a reversible inhibitor of the CYP3A4 enzyme system and may slightly prolong the QT c interval. In the USA, telithromycin is now indicated only for treatment of community-acquired bacterial pneumonia. Other respiratory tract infections were removed as indications when it was recognized that use of telithromycin can result in hepatitis and liver failure.
■ CLINDAMYCIN
Clindamycin is a chlorine-substituted derivative of lincomycin , an antibiotic that is elaborated by Streptomyces lincolnensis .
NH
CI
CH
CH3
OH
OH
HO
Clindamycin
N
CC3H7
OO
S
CH3
CH
CH3
Mechanism of Action & Antibacterial Activity Clindamycin, like erythromycin, inhibits protein synthesis by inter-fering with the formation of initiation complexes and with amino-acyl translocation reactions. The binding site for clindamycin on the 50S subunit of the bacterial ribosome is identical with that for erythromycin. Streptococci, staphylococci, and pneumococci are inhibited by clindamycin, 0.5–5 mcg/mL. Enterococci and gram-negative aerobic organisms are resistant. Bacteroides sp and other anaerobes, both gram-positive and gram-negative, are usually sus-ceptible. Resistance to clindamycin, which generally confers cross-resistance to macrolides, is due to (1) mutation of the ribosomal receptor site; (2) modification of the receptor by a constitutively expressed methylase (see section on erythromycin resistance, above); and (3) enzymatic inactivation of clindamycin. Gram-negative aero-bic species are intrinsically resistant because of poor permeability of the outer membrane.
Pharmacokinetics Oral dosages of clindamycin, 0.15–0.3 g every 8 hours (10–20 mg/kg/d for children), yield serum levels of 2–3 mcg/mL. When administered intravenously, 600 mg of clindamycin every 8 hours gives levels of 5–15 mcg/mL. The drug is about 90% protein-bound. Clindamycin penetrates well into most tissues, with brain and cerebrospinal fluid being important exceptions. It penetrates well into abscesses and is actively taken up and concentrated by phagocytic cells. Clindamycin is metabolized by the liver, and both active drug and active metabolites are excreted in bile and urine. The half-life is about 2.5 hours in normal individuals, increasing to 6 hours in patients with anuria. No dosage adjustment is required for renal failure.
Clinical Uses Clindamycin is indicated for the treatment of skin and soft-tissue infections caused by streptococci and staphylococci. It is often active against community-acquired strains of methicillin-resistant S aureus , an increasingly common cause of skin and soft tissue infections. Clindamycin is also indicated for treatment of anaerobicinfections caused by Bacteroides sp and other anaerobes that often participate in mixed infections. Clindamycin, sometimes in com-bination with an aminoglycoside or cephalosporin, is used to treat penetrating wounds of the abdomen and the gut; infections origi-nating in the female genital tract, eg, septic abortion, pelvic abscesses, or pelvic inflammatory disease; and lung abscesses. Clindamycin is now recommended rather than erythromycin for prophylaxis of endocarditis in patients with valvular heart disease who are undergoing certain dental procedures and have significant penicillin allergies. Clindamycin plus primaquine is an effective alternative to trimethoprim-sulfamethoxazole for moderate to moderately severe Pneumocystis jiroveci pneumonia in AIDS patients. It is also used in combination with pyrimethamine for AIDS-related toxoplasmosis of the brain.
Adverse Effects Common adverse effects are diarrhea, nausea, and skin rashes. Impaired liver function (with or without jaundice) and neutrope-nia sometimes occur. Administration of clindamycin is a risk fac-tor for diarrhea and colitis due to C difficile .
■ STREPTOGRAMINS
Mechanism of Action & Antibacterial Activity Quinupristin-dalfopristin is a combination of two streptogramins—quinupristin, a streptogramin B, and dalfopristin, a streptogramin A—in a 30:70 ratio. The streptogramins share the same ribosomal binding site as the macrolides and clindamycin and thus inhibit protein synthesis in an identical manner. It is rapidly bactericidal for most susceptible organisms except Enterococcus faecium , which is killed slowly. Quinupristin-dalfopristin is active against
044-Katzung_Ch044_p809-820.indd 815 9/23/11 4:21:12 PM

816 SECTION VIII Chemotherapeutic Drugs
gram-positive cocci, including multidrug-resistant strains of strep-tococci, penicillin-resistant strains of S pneumoniae , methicillin-susceptible and -resistant strains of staphylococci, and E faecium (but not Enterococcus faecalis ). Resistance is due to modification of the quinupristin binding site (MLS-B type resistance), enzymatic inactivation of dalfopristin, or efflux.
Pharmacokinetics Quinupristin-dalfopristin is administered intravenously at a dosage of 7.5 mg/kg every 8–12 hours. Peak serum concentrations following an infusion of 7.5 mg/kg over 60 minutes are 3 mcg/mL for quinupristin and 7 mcg/mL for dalfopristin. Quinupristin and dalfopristin are rapidly metabolized, with half-lives of 0.85 and 0.7 hours, respectively. Elimination is principally by the fecal route. Dose adjustment is not necessary for renal failure, perito-neal dialysis, or hemodialysis. Patients with hepatic insufficiency may not tolerate the drug at usual doses, however, because of increased area under the concentration curve of both parent drugs and metabolites. This may necessitate a dose reduction to 7.5 mg/kg every 12 hours or 5 mg/kg every 8 hours. Quinupristin and dalfopristin significantly inhibit CYP3A4, which metabolizes warfarin, diazepam, astemizole, terfenadine, cisapride, nonnucleo-side reverse transcriptase inhibitors, and cyclosporine, among others. Dosage reduction of cyclosporine may be necessary.
Clinical Uses & Adverse Effects Quinupristin-dalfopristin is approved for treatment of infections caused by staphylococci or by vancomycin-resistant strains of E faecium , but not E faecalis , which is intrinsically resistant, prob-ably because of an efflux-type resistance mechanism. The principal toxicities are infusion-related events, such as pain at the infusion site, and an arthralgia-myalgia syndrome.
■ CHLORAMPHENICOL
Crystalline chloramphenicol is a neutral, stable compound with the following structure:
CH2OH
CHCI2
OH
C C
Chloramphenicol
N
HHH
NO2
O
C
It is soluble in alcohol but poorly soluble in water. Chlorampheni-col succinate, which is used for parenteral administration, is highly water-soluble. It is hydrolyzed in vivo with liberation of free chloramphenicol.
Mechanism of Action & Antimicrobial Activity Chloramphenicol is a potent inhibitor of microbial protein synthesis. It binds reversibly to the 50S subunit of the bacterial ribosome
( Figure 44–1 ) and inhibits peptide bond formation (step 2). Chloramphenicol is a bacteriostatic broad-spectrum antibiotic that is active against both aerobic and anaerobic gram-positive and gram-negative organisms. It is active also against Rickettsiae but not Chlamydiae . Most gram-positive bacteria are inhibited at concentra-tions of 1–10 mcg/mL, and many gram-negative bacteria are inhib-ited by concentrations of 0.2–5 mcg/mL. H influenzae, Neisseria meningitidis , and some strains of bacteroides are highly susceptible, and for these organisms, chloramphenicol may be bactericidal.
Low-level resistance to chloramphenicol may emerge from large populations of chloramphenicol-susceptible cells by selec-tion of mutants that are less permeable to the drug. Clinically significant resistance is due to production of chloramphenicol acetyltransferase, a plasmid-encoded enzyme that inactivates the drug.
Pharmacokinetics The usual dosage of chloramphenicol is 50–100 mg/kg/d. After oral administration, crystalline chloramphenicol is rapidly and completely absorbed. A 1-g oral dose produces blood levels between 10 and 15 mcg/mL. Chloramphenicol palmitate is a pro-drug that is hydrolyzed in the intestine to yield free chlorampheni-col. The parenteral formulation is a prodrug, chloramphenicol succinate, which hydrolyzes to yield free chloramphenicol, giving blood levels somewhat lower than those achieved with orally administered drug. Chloramphenicol is widely distributed to virtu-ally all tissues and body fluids, including the central nervous system and cerebrospinal fluid, such that the concentration of chloram-phenicol in brain tissue may be equal to that in serum. The drug penetrates cell membranes readily.
Most of the drug is inactivated either by conjugation with glucuronic acid (principally in the liver) or by reduction to inac-tive aryl amines. Active chloramphenicol (about 10% of the total dose administered) and its inactive degradation products (about 90% of the total) are eliminated in the urine. A small amount of active drug is excreted into bile and feces. The systemic dosage of chloramphenicol need not be altered in renal insufficiency, but it must be reduced markedly in hepatic failure. Newborns less than a week old and premature infants also clear chloramphenicol less well, and the dosage should be reduced to 25 mg/kg/d.
Clinical Uses Because of potential toxicity, bacterial resistance, and the avail-ability of many other effective alternatives, chloramphenicol is rarely used in the United States. It may be considered for treat-ment of serious rickettsial infections such as typhus and Rocky Mountain spotted fever. It is an alternative to a β-lactam antibiotic for treatment of bacterial meningitis occurring in patients who have major hypersensitivity reactions to penicillin. The dosage is 50–100 mg/kg/d in four divided doses.
Chloramphenicol is used topically in the treatment of eye infections because of its broad spectrum and its penetration of ocular tissues and the aqueous humor. It is ineffective for chla-mydial infections.
044-Katzung_Ch044_p809-820.indd 816 9/23/11 4:21:12 PM

CHAPTER 44 Tetracyclines, Macrolides, and Others 817
Adverse Reactions Adults occasionally develop gastrointestinal disturbances, includ-ing nausea, vomiting, and diarrhea. This is rare in children. Oral or vaginal candidiasis may occur as a result of alteration of normal microbial flora.
Chloramphenicol commonly causes a dose-related reversible suppression of red cell production at dosages exceeding 50 mg/kg/d after 1–2 weeks. Aplastic anemia, a rare consequence (1 in 24,000 to 40,000 courses of therapy) of chloramphenicol admin-istration by any route, is an idiosyncratic reaction unrelated to dose, although it occurs more frequently with prolonged use. It tends to be irreversible and can be fatal.
Newborn infants lack an effective glucuronic acid conjugation mechanism for the degradation and detoxification of chloram-phenicol. Consequently, when infants are given dosages above 50 mg/kg/d, the drug may accumulate, resulting in the gray baby syndrome , with vomiting, flaccidity, hypothermia, gray color, shock, and vascular collapse. To avoid this toxic effect, chloramphenicol should be used with caution in infants and the dosage limited to 50 mg/kg/d (or less during the first week of life) in full-term infants more than 1 week old and 25 mg/kg/d in premature infants.
Chloramphenicol inhibits hepatic microsomal enzymes that metabolize several drugs. Half-lives of these drugs are prolonged, and the serum concentrations of phenytoin, tolbutamide, chlorpropa-mide, and warfarin are increased. Like other bacteriostatic inhibitors of microbial protein synthesis, chloramphenicol can antagonize bac-tericidal drugs such as penicillins or aminoglycosides.
■ OXAZOLIDINONES
Mechanism of Action & Antimicrobial Activity Linezolid is a member of the oxazolidinones, a new class of synthetic antimicrobials. It is active against gram-positive organisms including staphylococci, streptococci, enterococci, gram-positive anaerobic cocci, and gram-positive rods such as corynebacteria, Nocardia sp, and L monocytogenes . It is primarily a bacteriostatic agent but is bactericidal against streptococci. It is also active against Mycobacterium tuberculosis .
Linezolid inhibits protein synthesis by preventing formation of the ribosome complex that initiates protein synthesis. Its unique binding site, located on 23S ribosomal RNA of the 50S subunit, results in no cross-resistance with other drug classes. Resistance is caused by mutation of the linezolid binding site on 23S ribosomal RNA.
Pharmacokinetics Linezolid is 100% bioavailable after oral administration and has a half-life of 4–6 hours. It is metabolized by oxidative metabolism, yielding two inactive metabolites. It is neither an inducer nor an inhibitor of cytochrome P450 enzymes. Peak serum concentra-tions average 18 mcg/mL following a 600-mg oral dose. The rec-ommended dosage for most indications is 600 mg twice daily, either orally or intravenously.
Clinical Uses Linezolid is approved for vancomycin-resistant E faecium infec-tions; nosocomial pneumonia; community-acquired pneumonia; and both complicated and uncomplicated skin and soft tissue infections caused by susceptible gram-positive bacteria. Off-label uses of linezolid include treatment of multidrug-resistant tubercu-losis and Nocardia infections.
Adverse Effects The principal toxicity of linezolid is hematologic; the effects are reversible and generally mild. Thrombocytopenia is the most common manifestation (seen in approximately 3% of treatment courses), particularly when the drug is administered for longer than 2 weeks. Anemia and neutropenia may also occur, most commonly in patients with a predisposition to or underlying bone marrow suppression. Cases of optic and peripheral neuropathy and lactic acidosis have been reported with prolonged courses of linezolid. These side effects are thought to be related to linezolid-induced inhibition of mitochondrial protein synthesis. There are case reports of serotonin syndrome occurring when linezolid is co-administered with serotonergic drugs, most frequently selective serotonin reuptake inhibitor antidepressants. The FDA issued a warning regarding the use of the drug with serotonergic agents in 2011.
044-Katzung_Ch044_p809-820.indd 817 9/23/11 4:21:12 PM

818 SECTION VIII Chemotherapeutic Drugs
SUMMARY Tetracyclines, Macrolides, Clindamycin, Chloramphenicol, Streptogramins, & Oxazolidinones
Subclass Mechanism of Action EffectsClinical Applications
Pharmacokinetics, Toxicities, Interactions
TETRACYCLINES
• Tetracycline Prevents bacterial protein synthesis by binding to the 30S ribosomal subunit
Bacteriostatic activity against susceptible bacteria
Infections caused by mycoplasma, chlamydiae, rickettsiae, some spirochetes • malaria • H pylori • acne
Oral • mixed clearance (half-life 8 h) • dosed every 6 h • divalent cations impair oral absorption • Toxicity: Gastrointestinal upset, hepatotoxicity, photosensitivity, deposition in bone and teeth
• Doxycycline: Oral and IV; longer half-life (18 h) so dosed twice daily; nonrenal elimination; absorption is minimally affected by divalent cations; used to treat community-acquired pneumonia and exacerbations of bronchitis • Minocycline: Oral; longer half-life (16 h) so dosed twice daily; frequently causes reversible vestibular toxicity • Tigecycline: IV; unaffected by common tetracycline resistance mechanisms; very broad spectrum of activity against gram-positive, gram-negative, and anaerobic bacteria; nausea and vomiting are the primary toxicities
MACROLIDES
• Erythromycin Prevents bacterial protein synthesis by binding to the 50S ribosomal subunit
Bacteriostatic activity against susceptible bacteria
Community-acquired pneumonia • pertussis • corynebacterial and chlamydial infections
Oral, IV • hepatic clearance (half-life 1.5 h) • dosed every 6 h • cytochrome P450 inhibitor • Toxicity: Gastrointestinal upset, hepatotoxicity, QTc prolongation
• Clarithromycin: Oral; longer half-life (4 h) so dosed twice daily; added activity versus M avium complex, toxoplasma, and M leprae • Azithromycin: Oral, IV; very long half-life (68 h) allows for once-daily dosing and 5-day course of therapy of community-acquired pneumonia; does not inhibit cytochrome P450 enzymes • Telithromycin: Oral; unaffected by efflux-mediated resistance so is active versus many erythromycin-resistant strains of pneumococci; rare cases of fulminant hepatic failure
LINCOSAMIDE
• Clindamycin Prevents bacterial protein synthesis by binding to the 50S ribosomal subunit
Bacteriostatic activity against susceptible bacteria
Skin and soft tissue infections • anaerobic infections
Oral, IV • hepatic clearance (half-life 2.5 h) • dosed every 6–8 hours • Toxicity: Gastrointestinal upset, C difficile colitis
STREPTOGRAMINS
• Quinupristin-dalfopristin Prevents bacterial protein synthesis by binding to the 50S ribosomal subunit
Rapid bactericidal activity against most susceptible bacteria
Infections caused by staphylococci or vancomycin-resistant strains of E faecium
IV • hepatic clearance • dosed every 8–12 h • cytochrome P450 inhibitor • Toxicity: Severe infusion-related myalgias and arthralgias
CHLORAMPHENICOL Prevents bacterial protein synthesis by binding to the 50S ribosomal subunit
Bacteriostatic activity against susceptible bacteria
Use is rare in the devel-oped world because of serious toxicities
Oral, IV • hepatic clearance (half-life 2.5 h) • dosage is 50–100 mg/kg/d in four divided doses • Toxicity: Dose-related anemia, idiosyncratic aplastic anemia, gray baby syndrome
OXAZOLIDINONES
• Linezolid Prevents bacterial protein synthesis by binding to the 23S ribosomal RNA of 50S subunit
Bacteriostatic activity against susceptible bacteria
Infections caused by methicillin-resistant staphylococci and vancomycin-resistant enterococci
Oral, IV • hepatic clearance (half-life 6 h) • dosed twice-daily • Toxicity: Duration-dependent bone marrow suppression, neuropathy, and optic neuritis • serotonin syndrome may occur when coadministered with other serotonergic drugs (eg, selective serotonin reuptake inhibitors)
044-Katzung_Ch044_p809-820.indd 818 9/23/11 4:21:13 PM

CHAPTER 44 Tetracyclines, Macrolides, and Others 819
CHLORAMPHENICOLChloramphenicol (generic, Chloromycetin)
Parenteral: 100 mg powder to reconstitute for injection
TETRACYCLINESDemeclocycline (Declomycin)
Oral: 150, 300 mg tabletsDoxycycline (generic, Vibramycin, others)
Oral: 20, 50, 75, 100 mg tablets and capsules; powder to reconstitute for 25 mg/5 mL suspension; 50 mg/5 mL syrup
Parenteral: 100, 200 mg powder to reconstitute for injectionMinocycline (generic, Minocin, various)
Oral: 20, 50, 75, 100 mg tablets and capsules; 50 mg/5 mL suspension
Tetracycline (generic, others)Oral: 250, 500 mg capsules; 125 mg/5 mL suspension
Tigecycline (Tygacil)Parenteral: 50 mg powder to reconstitute for IV administration
MACROLIDESAzithromycin (Zithromax)
Oral: 250, 500, 600 mg capsules; powder for 100, 200 mg/5 mL oral suspension
Parenteral: 500 mg powder for injectionClarithromycin (generic, Biaxin)
Oral: 250, 500 mg tablets, 500, 1000 mg extended-release tablets; granules for 125, 250 mg/5 mL oral suspension
Erythromycin (generic, others)Oral (base): 250, 333, 500 mg enteric-coated tabletsOral (base) delayed-release: 250 mg capsules, 500 mg tabletsOral (estolate): 125, 250 mg/5 mL suspensionOral (ethylsuccinate): 400 mg tablets; 200, 400 mg/5 mL suspensionOral (stearate): 250, 500 mg film-coated tabletsParenteral: lactobionate, 0.5, 1 g powder to reconstitute for
IV injection
KETOLIDESTelithromycin (Ketek)
Oral: 300, 400 mg tablets
LINCOMYCINClindamycin (generic, Cleocin)
Oral: 75, 150, 300 mg capsules; 75 mg/5 mL granules to reconstitute for solution
Parenteral: 150 mg/mL in 2, 4, 6, 60 mL vials for injection
STREPTOGRAMINSQuinupristin and dalfopristin (Synercid)
Parenteral: 30:70 formulation in 500 mg vial for reconstitution for IV injection
OXAZOLIDINONELinezolid (Zyvox)
Oral: 600 mg tablets; 100 mg powder for 5 mL suspensionParenteral: 2 mg/mL for IV infusion
P R E P A R A T I O N S A V A I L A B L E
CHLORAMPHENICOLChloramphenicol (generic, Chloromycetin)
Parenteral: 100 mg powder to reconstitute for injection
TETRACYCLINESDemeclocycline (Declomycin)
Oral: 150, 300 mg tabletsDoxycycline (generic, Vibramycin, others)
Oral: 20, 50, 75, 100 mg tablets and capsules; powder to reconstitute for 25 mg/5 mL suspension; 50 mg/5 mL syrup
Parenteral: 100, 200 mg powder to reconstitute for injectionMinocycline (generic, Minocin, various)
Oral: 20, 50, 75, 100 mg tablets and capsules; 50 mg/5 mLsuspension
Tetracycline (generic, others)Oral: 250, 500 mg capsules; 125 mg/5 mL suspension
Tigecycline (Tygacil)Parenteral: 50 mg powder to reconstitute for IV administration
MACROLIDESAzithromycin (Zithromax)
Oral: 250, 500, 600 mg capsules; powder for 100, 200 mg/5 mL oral suspension
Parenteral: 500 mg powder for injectionClarithromycin (generic, Biaxin)
Oral: 250, 500 mg tablets, 500, 1000 mg extended-release tablets;granules for 125, 250 mg/5 mL oral suspension
Erythromycin (generic, others)Oral (base): 250, 333, 500 mg enteric-coated tabletsOral (base) delayed-release: 250 mg capsules, 500 mg tabletsOral (estolate): 125, 250 mg/5 mL suspensionOral (ethylsuccinate): 400 mg tablets; 200, 400 mg/5 mL suspensionOral (stearate): 250, 500 mg film-coated tabletsParenteral: lactobionate, 0.5, 1 g powder to reconstitute for
IV injection
KETOLIDESTelithromycin (Ketek)
Oral: 300, 400 mg tablets
LINCOMYCINClindamycin (generic, Cleocin)
Oral: 75, 150, 300 mg capsules; 75 mg/5 mL granules to reconstitutefor solution
Parenteral: 150 mg/mL in 2, 4, 6, 60 mL vials for injection
STREPTOGRAMINSQuinupristin and dalfopristin (Synercid)
Parenteral: 30:70 formulation in 500 mg vial for reconstitution forIV injection
OXAZOLIDINONELinezolid (Zyvox)
Oral: 600 mg tablets; 100 mg powder for 5 mL suspensionParenteral: 2 mg/mL for IV infusion
REFERENCES De Vriese AS et al: Linezolid-induced inhibition of mitochondrial protein
synthesis. Clin Infect Dis 2006;42:1111. Fortun J et al: Linezolid for the treatment of multidrug-resistant tuberculosis.
J Antimicrob Chemother 2005;56:180. Gee T et al: Pharmacokinetics and tissue penetration of linezolid following mul-
tiple oral doses. Antimicrob Agents Chemother 2001;45:1843. Hancock RE: Mechanisms of action of newer antibiotics for gram-positive patho-
gens. Lancet Infect Dis 2005;5:209. Lawrence KR et al: Serotonin toxicity associated with the use of linezolid: A review
of postmarketing data. Clin Infect Dis 2006;42:1578. Livermore DM. Tigecycline: What is it, and where should it be used? J Antimicrob
Chemother 2005;56:611.
Moran GJ et al: Methicillin-resistant S aureus infections among patients in the emergency department. N Engl J Med 2006;355:666.
Noskin GA: Tigecycline: A new glycylcycline for treatment of serious infections. Clin Infect Dis 2005;41(Suppl 5):S303.
Schlossberg D: Azithromycin and clarithromycin. Med Clin North Am 1995;79:803.
Speer BS, Shoemaker MB, Salyers AA: Bacterial resistance to tetracycline: Mechanism, transfer, and clinical significance. Clin Microbiol Rev 1992;5:387.
Zhanel GG et al: The ketolides: A critical review. Drugs 2002;62:1771. Zuckerman JM: Macrolides and ketolides: Azithromycin, clarithromycin, telithro-
mycin. Infect Dis Clin North Am 2004;18:621.
C A S E S T U D Y A N S W E R
A tetracycline or a macrolide is effective in the treatment of chlamydial cervicitis. Doxycycline at a dose of 100 mg PO bid for 7 days is the preferred tetracycline, while azithromycin as a
single 1 g dose is the preferred macrolide. If the patient is pregnant, then tetracyclines would be contraindicated and she should receive azithromycin, which is safe in pregnancy.
044-Katzung_Ch044_p809-820.indd 819 9/23/11 4:21:13 PM

This page intentionally left blank

821
C H A P T E R
The drugs described in this chapter are bactericidal inhibitors of protein synthesis that interfere with ribosomal function. These agents are useful mainly against aerobic gram-negative microorganisms.
■ AMINOGLYCOSIDES
The aminoglycosides include streptomycin, neomycin, kanamy-cin, amikacin, gentamicin, tobramycin, sisomicin, netilmicin, and others. They are used most widely in combination with a β-lactam antibiotic in serious infections with gram-negative bac-teria, in combination with vancomycin or a β-lactam antibiotic for gram-positive endocarditis, and for treatment of tuberculosis.
General Properties of Aminoglycosides A. Physical and Chemical Properties Aminoglycosides have a hexose ring, either streptidine (in streptomy-cin) or 2-deoxystreptamine (in other aminoglycosides), to which
various amino sugars are attached by glycosidic linkages ( Figures 45–1 and 45–2 ). They are water-soluble, stable in solu-tion, and more active at alkaline than at acid pH.
B. Mechanism of Action The mode of action of streptomycin has been studied far more closely than that of other aminoglycosides, but they probably all act similarly. Aminoglycosides are irreversible inhibitors of protein synthesis, but the precise mechanism for bactericidal activity is not known. The initial event is passive diffusion via porin channels across the outer membrane (see Figure 43–3 ). Drug is then actively transported across the cell membrane into the cytoplasm by an oxygen-dependent process. The transmembrane electro-chemical gradient supplies the energy for this process, and trans-port is coupled to a proton pump. Low extracellular pH and anaerobic conditions inhibit transport by reducing the gradient. Transport may be enhanced by cell wall-active drugs such as peni-cillin or vancomycin; this enhancement may be the basis of the synergism of these antibiotics with aminoglycosides.
Inside the cell, aminoglycosides bind to specific 30S-subunit ribosomal proteins (S12 in the case of streptomycin). Protein synthesis is inhibited by aminoglycosides in at least three ways ( Figure 45–3 ): (1) interference with the initiation complex of peptide
45Aminoglycosides & Spectinomycin Daniel H. Deck, PharmD & Lisa G. Winston, MD∗
C A S E S T U D Y
∗ The authors thank Dr. Henry F. Chambers for his contributions to previous editions.
A 45-year-old man with no medical history was admitted to the intensive care unit (ICU) 10 days ago after suffering third-degree burns over 40% of his body. He had been rela-tively stable until the last 24 hours. Now he is febrile (39.5°C [103.1°F]), and his white blood cell count has risen from 8,500 to 20,000/mm 3 . He has also had an episode of hypoten-sion (86/50 mm Hg) that responded to a fluid bolus. Blood cultures were obtained at the time of his fever and results are
pending. The ICU attending physician is concerned about sepsis and decides to treat with empiric combination therapy directed against Pseudomonas . The combination therapy includes tobramycin. The patient weighs 70 kg (154 lb) and has an estimated creatinine clearance of 90 mL/min. How should tobramycin be dosed using once-daily and conven-tional dosing strategies? How should each regimen be moni-tored for efficacy and toxicity?
045-Katzung_Ch045_p821-830.indd 821 9/23/11 4:22:18 PM

822 SECTION VIII Chemotherapeutic Drugs
formation; (2) misreading of mRNA, which causes incorporation of incorrect amino acids into the peptide and results in a nonfunctional or toxic protein; and (3) breakup of polysomes into nonfunctional monosomes. These activities occur more or less simultaneously, and the overall effect is irreversible and lethal for the cell.
C. Mechanisms of Resistance Three principal mechanisms have been established: (1) production of a transferase enzyme or enzymes inactivates the aminoglycoside by adenylylation, acetylation, or phosphorylation. This is the prin-cipal type of resistance encountered clinically. (Specific transferase enzymes are discussed below.) (2) There is impaired entry of aminoglycoside into the cell. This may be genotypic, resulting from mutation or deletion of a porin protein or proteins involved in transport and maintenance of the electrochemical gradient; or phenotypic, eg, resulting from growth conditions under which the oxygen-dependent transport process described above is not func-tional. (3) The receptor protein on the 30S ribosomal subunit may be deleted or altered as a result of a mutation.
D. Pharmacokinetics and Once-Daily Dosing Aminoglycosides are absorbed very poorly from the intact gastro-intestinal tract, and almost the entire oral dose is excreted in feces after oral administration. However, the drugs may be absorbed if ulcerations are present. After intramuscular injection, aminoglyco-sides are well absorbed, giving peak concentrations in blood within 30–90 minutes. Aminoglycosides are usually administered intravenously as a 30- to 60-minute infusion; after a brief distribu-tion phase, this results in serum concentrations that are identical with those following intramuscular injection. The normal half-life of aminoglycosides in serum is 2–3 hours, increasing to 24–48 hours in patients with significant impairment of renal function. Aminoglycosides are only partially and irregularly removed by hemodialysis—eg, 40–60% for gentamicin—and even less effec-tively by peritoneal dialysis. Aminoglycosides are highly polar compounds that do not enter cells readily. They are largely excluded from the central nervous system and the eye. In the
CH3
CHO
H2N
CH3
OHO
HO HO
C
C
O O
ONH
NH
NH
NH
HO
HO
CH2OH
NH
NH2
OH
Streptidine Streptose
Streptobiosamine
N-methyl-L-glucosamine
FIGURE 45–1 Structure of streptomycin.
H2C NH2O
HO O14
5
HO OH
R
23
I
NH2
NH14
3
HO OO
2
65
II
4 3
1 2
CH2 OH
OH41
HO NH2
5
32
III
5
H2C NH2O
HO OI
NH2
NH2
HO OO
IICH2 OH
OH
HO NH2
III
HC NHO
O4
5
NH2
NH2
R3
R2
R1
I
NH2
NH3
HO OO
II
OH
HO
Gentamicin, netilmicin
Tobramycin
NH
CH32
III
Kanamycin R =H
Amikacin R =
O
C CH CH2
OH
CH2 NH2
CH3
Ring I
R1 R3R2
C4–C5bond
Ring II
Gentamicin C1Gentamicin C2Gentamicin C1aNetilmicin
CH3CH3HH
HHHC2H5
CH3HHH
SingleSingleSingleDouble
FIGURE 45–2 Structures of several important aminoglycoside antibiotics. Ring II is 2-deoxystreptamine. The resemblance between kanamycin and amikacin and between gentamicin, netilmicin, and tobramycin can be seen. The circled numerals on the kanamycin mol-ecule indicate points of attack of plasmid-mediated bacterial trans-ferase enzymes that can inactivate this drug. 1 , 2 , and 3 , acetyltransferase; 4 , phosphotransferase; 5 , adenylyltransferase. Amikacin is resistant to modification at 2 , 3 , 4 , and 5 .
045-Katzung_Ch045_p821-830.indd 822 9/23/11 4:22:20 PM

CHAPTER 45 Aminoglycosides & Spectinomycin 823
presence of active inflammation, however, cerebrospinal fluid levels reach 20% of plasma levels, and in neonatal meningitis, the levels may be higher. Intrathecal or intraventricular injection is required for high levels in cerebrospinal fluid. Even after parenteral admin-istration, concentrations of aminoglycosides are not high in most tissues except the renal cortex. Concentration in most secretions is also modest; in the bile, it may reach 30% of the blood level. With prolonged therapy, diffusion into pleural or synovial fluid may result in concentrations 50–90% of that of plasma.
Traditionally, aminoglycosides have been administered in two or three equally divided doses per day in patients with normal renal function. However, administration of the entire daily dose in a single injection may be preferred in many clinical situations. Aminoglycosides have concentration-dependent killing ; that is, increasing concentrations kill an increasing proportion of bacteria and at a more rapid rate. They also have a significant postantibiotic effect, such that the antibacterial activity persists beyond the time during which measurable drug is present. The postantibiotic effect of aminoglycosides can last several hours. Because of these proper-ties, a given total amount of aminoglycoside may have better efficacy when administered as a single large dose than when administered as multiple smaller doses. When administered with a cell wall-active antibiotic (a β lactam or vancomycin), aminogly-cosides exhibit synergistic killing against certain bacteria. The effect of the drugs in combination is greater than the anticipated effect of each individual drug, ie, the killing effect of the combina-tion is more than additive.
Adverse effects from aminoglycosides are both time- and concentration-dependent. Toxicity is unlikely to occur until a certain threshold concentration is reached, but once that concen-tration is achieved, the time beyond this threshold becomes criti-cal. This threshold is not precisely defined, but a trough concentration above 2 mcg/mL is predictive of toxicity. At clini-cally relevant doses, the total time above this threshold is greater with multiple smaller doses of drug than with a single large dose.
Numerous clinical studies demonstrate that a single daily dose of aminoglycoside is just as effective—and probably less toxic—than multiple smaller doses. Therefore, many authorities now recommend that aminoglycosides be administered as a single daily dose in many clinical situations. However, the efficacy of once-daily aminoglycoside dosing in combination therapy of enterococ-cal and staphylococcal endocarditis remains to be defined, and the standard low-dose, thrice-daily administration is still recom-mended. In contrast, there are limited data supporting once-daily dosing in streptococcal endocarditis. The role of once-daily dosing in pregnancy and in neonates also is not well defined.
Once-daily dosing has potential practical advantages. For example, repeated determinations of serum concentrations are probably unnecessary unless aminoglycoside is given for more than 3 days. A drug administered once a day rather than three times a day is less labor intensive. And once-a-day dosing is more feasible for outpatient therapy.
Aminoglycosides are cleared by the kidney, and excretion is directly proportional to creatinine clearance. To avoid accumulation
mRNA30S subunit
5´
5´
3´
3´
Nascent peptide chain50S subunitInitiationcodon
Normal bacterial cell
Aminoglycoside-treated bacterial cell
Drug (block of initiation complex)
Drug (miscoded peptide chain)
Drug (block of translocation)
30S subunitmRNA
–
FIGURE 45–3 Putative mechanisms of action of the aminoglycosides in bacteria. Normal protein synthesis is shown in the top panel. At least three aminoglycoside effects have been described, as shown in the bottom panel: block of formation of the initiation complex; miscod-ing of amino acids in the emerging peptide chain due to misreading of the mRNA; and block of translocation on mRNA. Block of movement of the ribosome may occur after the formation of a single initiation complex, resulting in an mRNA chain with only a single ribosome on it, a so-called monosome. (Reproduced, with permission, from Trevor AT, Katzung BG, Masters SB: Pharmacology: Examination & Board Review, 6th ed. McGraw-Hill, 2002.)
045-Katzung_Ch045_p821-830.indd 823 9/23/11 4:22:20 PM

824 SECTION VIII Chemotherapeutic Drugs
and toxic levels, once-daily dosing of aminoglycosides is generally avoided if renal function is impaired. Rapidly changing renal function, which may occur with acute kidney injury, must also be monitored to avoid overdosing or underdosing. Provided these pitfalls are avoided, once-daily aminoglycoside dosing is safe and effective. If the creatinine clearance is > 60 mL/min, then a single daily dose of 5–7 mg/kg of gentamicin or tobramycin is recom-mended (15 mg/kg for amikacin). For patients with creatinine clearance < 60 mL/min, traditional dosing as described below is recommended. With once-daily dosing, serum concentrations need not be routinely checked until the second or third day of therapy, depending on the stability of renal function and the anticipated duration of therapy. It is unnecessary to check peak concentrations because they will be high. The goal is to administer drug so that concentrations of less than 1 mcg/mL are present between 18 and 24 hours after dosing. This provides a sufficient period of time for washout of drug to occur before the next dose is given. Appropriate trough levels can be accurately determined by measuring serum concentrations in samples obtained 2 hours and 12 hours after dosing and then adjusting the dose based on the actual clearance of drug or by measuring the concentration in a sample obtained 8 hours after a dose. If the 8-hour concentra-tion is between 1.5 mcg/mL and 6 mcg/mL, the target trough can be achieved at 18 hours.
With traditional dosing, adjustments must be made to prevent accumulation of drug and toxicity in patients with renal insuffi-ciency. Either the dose of drug is kept constant and the interval between doses is increased, or the interval is kept constant and the dose is reduced. Nomograms and formulas have been constructed relating serum creatinine levels to adjustments in treatment regi-mens. Because aminoglycoside clearance is directly proportional to the creatinine clearance, a method for determining the aminogly-coside dose is to estimate creatinine clearance using the Cockcroft-Gault formula described in Chapter 60 . For a traditional twice- or thrice-daily dosing regimen, peak serum concentrations should be determined from a blood sample obtained 30–60 minutes after a dose, and trough concentrations from a sample obtained just before the next dose. Doses of gentamicin and tobramycin should be adjusted to maintain peak levels between 5 and 10 mcg/mL and trough levels < 2 mcg/mL (< 1 mcg/mL is optimal).
E. Adverse Effects All aminoglycosides are ototoxic and nephrotoxic. Ototoxicity and nephrotoxicity are more likely to be encountered when therapy is continued for more than 5 days, at higher doses, in the elderly, and in the setting of renal insufficiency. Concurrent use with loop diuretics (eg, furosemide, ethacrynic acid) or other nephrotoxic antimicrobial agents (eg, vancomycin or amphotericin) can poten-tiate nephrotoxicity and should be avoided if possible. Ototoxicity can manifest either as auditory damage, resulting in tinnitus and high-frequency hearing loss initially, or as vestibular damage, evi-dent by vertigo, ataxia, and loss of balance. Nephrotoxicity results in rising serum creatinine levels or reduced creatinine clearance, although the earliest indication often is an increase in trough serum aminoglycoside concentrations. Neomycin, kanamycin, and amikacin
are the most ototoxic agents. Streptomycin and gentamicin are the most vestibulotoxic. Neomycin, tobramycin, and gentamicin are the most nephrotoxic.
In very high doses, aminoglycosides can produce a curare-like effect with neuromuscular blockade that results in respira-tory paralysis. This paralysis is usually reversible by calcium gluconate (given promptly) or neostigmine. Hypersensitivity occurs infrequently.
F. Clinical Uses Aminoglycosides are mostly used against gram-negative enteric bacteria, especially when the isolate may be drug-resistant and when there is suspicion of sepsis. They are almost always used in combination with a β-lactam antibiotic to extend coverage to include potential gram-positive pathogens and to take advantage of the synergism between these two classes of drugs. Penicillin-aminoglycoside combinations also are used to achieve bactericidal activity in treatment of enterococcal endocarditis and to shorten duration of therapy for viridans streptococcal and some patients with staphylococcal endocarditis. Which aminoglycoside and what dose should be used depend on the infection being treated and the susceptibility of the isolate.
STREPTOMYCIN
Streptomycin ( Figure 45–1 ) was isolated from a strain of Streptomyces griseus. The antimicrobial activity of streptomycin is typical of that of other aminoglycosides, as are the mechanisms of resistance. Resistance has emerged in most species, severely limit-ing the current usefulness of streptomycin, with the exceptions listed below. Ribosomal resistance to streptomycin develops read-ily, limiting its role as a single agent.
Clinical Uses A. Mycobacterial Infections Streptomycin is mainly used as a second-line agent for treatment of tuberculosis. The dosage is 0.5–1 g/d (7.5–15 mg/kg/d for children), which is given intramuscularly or intravenously. It should be used only in combination with other agents to prevent emergence of resistance. See Chapter 47 for additional information regarding the use of streptomycin in mycobacterial infections.
B. Nontuberculous Infections In plague, tularemia, and sometimes brucellosis, streptomycin, 1 g/d (15 mg/kg/d for children), is given intramuscularly in combina-tion with an oral tetracycline.
Penicillin plus streptomycin is effective for enterococcal endo-carditis and 2-week therapy of viridans streptococcal endocarditis. Gentamicin has largely replaced streptomycin for these indications. Streptomycin remains a useful agent for treating enterococcal infec-tions, however, because approximately 15% of enterococcal iso-lates that are resistant to gentamicin (and therefore resistant to netilmicin, tobramycin, and amikacin) will be susceptible to streptomycin.
045-Katzung_Ch045_p821-830.indd 824 9/23/11 4:22:20 PM

CHAPTER 45 Aminoglycosides & Spectinomycin 825
Adverse Reactions Fever, skin rashes, and other allergic manifestations may result from hypersensitivity to streptomycin. This occurs most fre-quently with prolonged contact with the drug either in patients who receive a prolonged course of treatment (eg, for tuberculosis) or in medical personnel who handle the drug. Desensitization is occasionally successful.
Pain at the injection site is common but usually not severe. The most serious toxic effect with streptomycin is disturbance of ves-tibular function—vertigo and loss of balance. The frequency and severity of this disturbance are in proportion to the age of the patient, the blood levels of the drug, and the duration of admin-istration. Vestibular dysfunction may follow a few weeks of unusu-ally high blood levels (eg, in individuals with impaired renal function) or months of relatively low blood levels. Vestibular tox-icity tends to be irreversible. Streptomycin given during pregnancy can cause deafness in the newborn and, therefore, is relatively contraindicated.
GENTAMICIN
Gentamicin is an aminoglycoside ( Figure 45–2 ) isolated from Micromonospora purpurea . It is effective against both gram-positive and gram-negative organisms, and many of its properties resemble those of other aminoglycosides. Sisomicin is very similar to the C1a component of gentamicin.
Antimicrobial Activity Gentamicin sulfate, 2–10 mcg/mL, inhibits in vitro many strains of staphylococci and coliforms and other gram-negative bacteria. It is active alone, but also as a synergistic companion with β-lactam antibiotics, against Escherichia coli , Proteus , Klebsiella pneumoniae, Enterobacter, Serratia , Stenotrophomonas , and other gram-negative rods that may be resistant to multiple other antibiotics. Like all aminoglycosides, it has no activity against anaerobes.
Resistance Streptococci and enterococci are relatively resistant to gentamicin owing to failure of the drug to penetrate into the cell. However, gentamicin in combination with vancomycin or a penicillin pro-duces a potent bactericidal effect, which in part is due to enhanced uptake of drug that occurs with inhibition of cell wall synthesis. Resistance to gentamicin rapidly emerges in staphylococci during monotherapy owing to selection of permeability mutants. Ribosomal resistance is rare. Among gram-negative bacteria, resistance is most commonly due to plasmid-encoded aminoglycoside-modifying enzymes. Gram-negative bacteria that are gentamicin-resistant usu-ally are susceptible to amikacin, which is much more resistant to modifying enzyme activity. The enterococcal enzyme that modifies gentamicin is a bifunctional enzyme that also inactivates amikacin, netilmicin, and tobramycin, but not streptomycin; the latter is modified by a different enzyme. This is why some gentamicin-resistant enterococci are susceptible to streptomycin.
Clinical Uses A. Intramuscular or Intravenous Administration Gentamicin is used mainly in severe infections (eg, sepsis and pneumonia) caused by gram-negative bacteria that are likely to be resistant to other drugs, especially P aeruginosa , Enterobacter sp, Serratia marcescens , Proteus sp, Acinetobacter sp, and Klebsiella sp. It usually is used in combination with a second agent because an aminoglycoside alone may not be effective for infections outside the urinary tract. For example, gentamicin should not be used as a single agent to treat staphylococcal infections because resistance develops rapidly. Aminoglycosides also should not be used for single-agent therapy of pneumonia because penetration of infected lung tissue is poor and local conditions of low pH and low oxygen tension contribute to poor activity. Gentamicin 5–6 mg/kg/d traditionally is given intravenously in three equal doses, but once-daily administration is just as effective for some organisms and less toxic (see above).
Gentamicin, in combination with a cell wall-active antibiotic, is also indicated in the treatment of endocarditis caused by gram-positive bacteria (streptococci, staphylococci, and enterococci). The synergistic killing achieved by combination therapy may achieve bactericidal activity necessary for cure or allow for the shortening of the duration of therapy. The doses of gentamicin used for synergy against gram-positive bacteria are lower than tra-ditional doses. Typically the drug is administered at a dose of 3 mg/kg/day in three divided doses. Peak levels should be approximately 3 mcg/mL, while trough levels should be < 1 mcg/mL. There are limited data to support administering the 3-mg/kg dose as a single daily injection in the treatment of streptococcal endocarditis.
B. Topical and Ocular Administration Creams, ointments, and solutions containing 0.1–0.3% gentami-cin sulfate have been used for the treatment of infected burns, wounds, or skin lesions and in attempts to prevent intravenous catheter infections. The effectiveness of topical preparations for these indications is unclear. Topical gentamicin is partly inacti-vated by purulent exudates. Ten mg can be injected subconjuncti-vally for treatment of ocular infections.
C. Intrathecal Administration Meningitis caused by gram-negative bacteria has been treated by the intrathecal injection of gentamicin sulfate, 1–10 mg/d. However, neither intrathecal nor intraventricular gentamicin was beneficial in neonates with meningitis, and intraventricular gen-tamicin was toxic, raising questions about the usefulness of this form of therapy. Moreover, the availability of third-generation cephalosporins for gram-negative meningitis has rendered this therapy obsolete in most cases.
Adverse Reactions Nephrotoxicity is usually reversible and mild. It occurs in 5–25% of patients receiving gentamicin for longer than 3–5 days. Such toxicity requires, at the very least, adjustment of the dosing regi-men and should prompt reconsideration of the need for the drug,
045-Katzung_Ch045_p821-830.indd 825 9/23/11 4:22:20 PM

826 SECTION VIII Chemotherapeutic Drugs
particularly if there is a less toxic alternative agent. Measurement of gentamicin serum levels is essential. Ototoxicity, which tends to be irreversible, manifests itself mainly as vestibular dysfunction. Loss of hearing can also occur. The incidence of ototoxicity is in part genetically determined, having been linked to point muta-tions in mitochondrial DNA, and occurs in 1–5% for patients receiving gentamicin for more than 5 days. Hypersensitivity reac-tions to gentamicin are uncommon.
TOBRAMYCIN
This aminoglycoside ( Figure 45–2 ) has an antibacterial spec-trum similar to that of gentamicin. Although there is some cross-resistance between gentamicin and tobramycin, it is unpredictable in individual strains. Separate laboratory susceptibility tests are therefore necessary.
The pharmacokinetic properties of tobramycin are virtually identical with those of gentamicin. The daily dose of tobramycin is 5–6 mg/kg intramuscularly or intravenously, traditionally divided into three equal amounts and given every 8 hours. Monitoring blood levels in renal insufficiency is an essential guide to proper dosing.
Tobramycin has almost the same antibacterial spectrum as gentamicin with a few exceptions. Gentamicin is slightly more active against S marcescens , whereas tobramycin is slightly more active against P aeruginosa ; Enterococcus faecalis is susceptible to both gentamicin and tobramycin, but E faecium is resistant to tobramycin. Gentamicin and tobramycin are otherwise inter-changeable clinically.
Like other aminoglycosides, tobramycin is ototoxic and neph-rotoxic. Nephrotoxicity of tobramycin may be slightly less than that of gentamicin, but the difference is clinically inconsequential.
Tobramycin is also formulated in solution (300 mg in 5 mL) for inhalation for treatment of P aeruginosa lower respiratory tract infections complicating cystic fibrosis. The drug is recommended as a 300-mg dose regardless of the patient’s age or weight for administration twice daily in repeated cycles of 28 days on ther-apy, followed by 28 days off therapy. Serum concentrations 1 hour after inhalation average 1 mcg/mL; consequently, nephrotoxicity and ototoxicity rarely occur. Caution should be used when admin-istering tobramycin to patients with preexisting renal, vestibular, or hearing disorders.
AMIKACIN
Amikacin is a semisynthetic derivative of kanamycin; it is less toxic than the parent molecule ( Figure 45–2 ). It is resistant to many enzymes that inactivate gentamicin and tobramycin, and it there-fore can be used against some microorganisms resistant to the latter drugs. Many gram-negative bacteria, including many strains of Proteus , Pseudomonas , Enterobacter , and Serratia , are inhibited by 1–20 mcg/mL amikacin in vitro. After injection of 500 mg of amikacin every 12 hours (15 mg/kg/d) intramuscularly, peak levels in serum are 10–30 mcg/mL.
Strains of multidrug-resistant Mycobacterium tuberculosis , including streptomycin-resistant strains, are usually susceptible to amikacin. Kanamycin-resistant strains may be cross-resistant to amikacin. The dosage of amikacin for tuberculosis is 7.5–15 mg/kg/d as a once-daily or two to three times weekly injection and always in combination with other drugs to which the isolate is susceptible.
Like all aminoglycosides, amikacin is nephrotoxic and ototoxic (particularly for the auditory portion of the eighth nerve). Serum concentrations should be monitored. Target peak serum concen-trations for an every-12-hours dosing regimen are 20–40 mcg/mL, and troughs should be maintained between 4 and 8 mcg/mL.
NETILMICIN
Netilmicin shares many characteristics with gentamicin and tobramycin. However, the addition of an ethyl group to the 1-amino position of the 2-deoxystreptamine ring (ring II, Figure 45–2 ) sterically protects the netilmicin molecule from enzymatic degradation at the 3-amino (ring II) and 2-hydroxyl (ring III) positions. Consequently, netilmicin may be active against some gentamicin-resistant and tobramycin-resistant bacteria.
The dosage (5–7 mg/kg/d) and the routes of administration are the same as for gentamicin. Netilmicin is largely interchangeable with gentamicin or tobramycin but is no longer available in the United States.
NEOMYCIN & KANAMYCIN
Neomycin and kanamycin are closely related. Paromomycin is also a member of this group. All have similar properties.
Antimicrobial Activity & Resistance Drugs of the neomycin group are active against gram-positive and gram-negative bacteria and some mycobacteria. P aeruginosa and streptococci are generally resistant. Mechanisms of antibacterial action and resistance are the same as with other aminoglycosides. The widespread use of these drugs in bowel preparation for elec-tive surgery has resulted in the selection of resistant organisms and some outbreaks of enterocolitis in hospitals. Cross-resistance between kanamycin and neomycin is complete.
Pharmacokinetics Drugs of the neomycin group are poorly absorbed from the gas-trointestinal tract. After oral administration, the intestinal flora is suppressed or modified, and the drug is excreted in the feces. Excretion of any absorbed drug is mainly through glomerular filtration into the urine.
Clinical Uses Neomycin and kanamycin are now limited to topical and oral use. Neomycin is too toxic for parenteral use. With the advent of more
045-Katzung_Ch045_p821-830.indd 826 9/23/11 4:22:20 PM

CHAPTER 45 Aminoglycosides & Spectinomycin 827
potent and less toxic aminoglycosides, parenteral administration of kanamycin has also been largely abandoned. Paromomycin has recently been shown to be effective against visceral leishmaniasis when given parenterally (see Chapter 52 ), and this serious infec-tion may represent an important new use for this drug.
A. Topical Administration Solutions containing 1–5 mg/mL are used on infected surfaces or injected into joints, the pleural cavity, tissue spaces, or abscess cavities where infection is present. The total amount of drug given in this fashion must be limited to 15 mg/kg/d because at higher doses enough drug may be absorbed to produce systemic toxicity. Whether topical application for active infection adds anything to appropriate systemic therapy is questionable. Ointments, often formulated as a neomycin-polymyxin-bacitracin combination, can be applied to infected skin lesions or in the nares for suppression of staphylococci but they are largely ineffective.
B. Oral Administration In preparation for elective bowel surgery, 1 g of neomycin is given orally every 6–8 hours for 1–2 days, often combined with 1 g of erythromycin base. This reduces the aerobic bowel flora with little effect on anaerobes. In hepatic encephalopathy, coliform flora can be suppressed by giving 1 g every 6–8 hours together with reduced protein intake, thus reducing ammonia production. Use of neomycin for hepatic encephalopathy has been largely supplanted by lactulose and other medications that are less toxic. Use of paromomycin in the treatment of protozoal infections is discussed in Chapter 52 .
Adverse Reactions All members of the neomycin group have significant nephrotoxic-ity and ototoxicity. Auditory function is affected more than ves-tibular function. Deafness has occurred, especially in adults with impaired renal function and prolonged elevation of drug levels.
The sudden absorption of postoperatively instilled kanamycin from the peritoneal cavity (3–5 g) has resulted in curare-like neu-romuscular blockade and respiratory arrest. Calcium gluconate and neostigmine can act as antidotes.
Although hypersensitivity is not common, prolonged applica-tion of neomycin-containing ointments to skin and eyes has resulted in severe allergic reactions.
■ SPECTINOMYCIN
Spectinomycin is an aminocyclitol antibiotic that is structurally related to aminoglycosides. It lacks amino sugars and glycosidic bonds.
NH
HN
CH3
O
O
CH3
Spectinomycin
O CH3
HO
O
OH
OH
Spectinomycin is active in vitro against many gram-positive and gram-negative organisms, but it is used almost solely as an alternative treatment for drug-resistant gonorrhea or gonorrhea in penicillin-allergic patients. The majority of gonococcal isolates are inhibited by 6 mcg/mL of spectinomycin. Strains of gonococci may be resistant to spectinomycin, but there is no cross-resistance with other drugs used in gonorrhea. Spectinomycin is rapidly absorbed after intramuscular injection. A single dose of 40 mg/kg up to a maximum of 2 g is given. There is pain at the injection site and, occasionally, fever and nausea. Nephrotoxicity and anemia have been observed rarely. Spectinomycin is no longer available for use in the United States but may be available elsewhere.
045-Katzung_Ch045_p821-830.indd 827 9/23/11 4:22:20 PM

828 SECTION VIII Chemotherapeutic Drugs
Amikacin (generic, Amikin)Parenteral: 50, 250 mg (in vials) for IM, IV injection
Gentamicin (generic, Garamycin)Parenteral: 10, 40 mg/mL vials for IM, IV injection
Kanamycin (Kantrex)Parenteral: 500, 1000 mg for IM, IV injection; 75 mg for pediatric
injectionNeomycin (generic, Mycifradin)
Oral: 500 mg tablets
P R E P A R A T I O N S A V A I L A B L E
Amikacin (generic, Amikin)Parenteral: 50, 250 mg (in vials) for IM, IV injection
Gentamicin (generic, Garamycin)Parenteral: 10, 40 mg/mL vials for IM, IV injection
Kanamycin (Kantrex)Parenteral: 500, 1000 mg for IM, IV injection; 75 mg for pediatric
injectionNeomycin (generic, Mycifradin)
Oral: 500 mg tablets
Paromomycin (Humatin)Oral: 250 mg capsules
Streptomycin (generic)Parenteral: 400 mg/mL for IM injection
Tobramycin (generic, Nebcin)Parenteral: 10, 40 mg/mL for IM, IV injection; powder to reconsti-
tute for injectionSolution for inhalation (TOBI): 300 mg in 5 mL sodium chloride
solution
REFERENCES Busse H-J, Wöstmann C, Bakker EP: The bactericidal action of streptomycin:
Membrane permeabilization caused by the insertion of mistranslated pro-teins into the cytoplasmic membrane of Escherichia coli and subsequent caging of the antibiotic inside the cells due to degradation of these proteins. J Gen Microbiol 1992;138:551.
Cheer SM, Waugh J, Noble S: Inhaled tobramycin (TOBI): A review of its use in the management of Pseudomonas aeruginosa infections in patients with cystic fibrosis. Drugs 2003;63:2501.
Contopoulos-Ioannidis DG et al: Extended-interval aminoglycoside administra-tion for children: A meta-analysis. Pediatrics 2004;114:111.
Kaye D: Current use for old antibacterial agents: Polymyxins, rifampin, and aminoglycosides. Infect Dis Clin North Am 2004;18:669.
Le T, Bayer AS: Combination antibiotic therapy for infective endocarditis. Clin Infect Dis 2003;36:615.
Olsen KM et al: Effect of once-daily dosing vs. multiple daily dosing of tobramycin on enzyme markers of nephrotoxicity. Crit Care Med 2004;32:1678.
Pappas G et al: Brucellosis. N Engl J Med 2005;352:2325. Paul M et al: Beta lactam monotherapy versus beta lactam-aminoglycoside combi-
nation therapy for sepsis in immunocompetent patients: Systematic review and meta-analysis of randomised trials. BMJ 2004;328:668.
Paul M, Soares-Weiser K, Leibovici L: Beta lactam monotherapy versus beta lactam-aminoglycoside combination therapy for fever with neutropenia: Systematic review and meta-analysis. BMJ 2003;326:1111.
Poole K: Aminoglycoside resistance in Pseudomonas aeruginosa . Antimicrob Agents Chemother 2005;49:479.
SUMMARY Aminoglycosides
Subclass Mechanism of Action Effects Clinical ApplicationsPharmacokinetics, Toxicities, Interactions
AMINOGLYCOSIDES & SPECTINOMYCIN • Gentamicin Prevents bacterial protein
synthesis by binding to the 30S ribosomal subunit
Bactericidal activity against susceptible bacteria • synergistic effects against gram-positive bacteria when combined with β lactams or vancomycin • concentration-dependent killing and a significant post-antibiotic effect
Sepsis caused by aerobic gram-negative bacteria • synergistic activity in endocarditis caused by streptococci, staphylococci, and enterococci
IV • renal clearance (half-life 2.5 h) • conventional dosing 1.3–1.7 mg/kg q8h with goal peak levels 5–8 mcg/mL • trough levels < 2 mcg/mL • once-daily dosing at 5–7 mg/kg as effective and may have less toxicity than conventional dosing • Toxicity: Nephrotoxicity (reversible), ototox-icity (irreversible), neuromuscular blockade
• Tobramycin: Intravenous; more active than gentamicin versus Pseudomonas; may also have less nephrotoxicity
• Amikacin: Intravenous; resistant to many enzymes that inactivate gentamicin and tobramycin; higher doses and target peaks and troughs than gentamicin and tobramycin
• Streptomycin: Intramuscular, widespread resistance limits use to specific indications such as tuberculosis and enterococcal endocarditis
• Neomycin: Oral or topical, poor bioavailability; used before bowel surgery to decrease aerobic flora; also used to treat hepatic encephalopathy
• Spectinomycin: Intramuscular; sole use is for treatment of antibiotic-resistant gonococcal infections or gonococcal infections in penicillin-allergic patients
045-Katzung_Ch045_p821-830.indd 828 9/23/11 4:22:20 PM

CHAPTER 45 Aminoglycosides & Spectinomycin 829
C A S E S T U D Y A N S W E R
The patient has normal renal function and thus qualifies for once-daily dosing. Tobramycin could be administered as a single once-daily injection at a dose of 350–490 mg (5–7 mg/kg). A serum level between 1.5 and 6 mcg/mL measured 8 hours after infusion correlates with an appropriate trough level. Alternatively, the same total daily dose could be divided
and administered every 8 hours, as a conventional dosing strategy. With conventional dosing, peak and trough con-centrations should be monitored with the target peak concentration of 5–10 mcg/mL and the target trough con-centration of < 2 mcg/mL.
045-Katzung_Ch045_p821-830.indd 829 9/23/11 4:22:21 PM

This page intentionally left blank

831
C H A P T E R
■ ANTIFOLATE DRUGS
SULFONAMIDES
Chemistry The basic formulas of the sulfonamides and their structural similar-ity to p -aminobenzoic acid (PABA) are shown in Figure 46–1 . Sulfonamides with varying physical, chemical, pharmacologic, and antibacterial properties are produced by attaching substituents to the amido group (–SO 2 –NH–R) or the amino group (–NH 2 ) of the sulfanilamide nucleus. Sulfonamides tend to be much more soluble at alkaline than at acid pH. Most can be prepared as sodium salts, which are used for intravenous administration.
Mechanism of Action & Antimicrobial Activity Sulfonamide-susceptible organisms, unlike mammals, cannot use exogenous folate but must synthesize it from PABA. This pathway ( Figure 46–2 ) is thus essential for production of purines and nucleic acid synthesis. As structural analogs of PABA, sulfon-amides inhibit dihydropteroate synthase and folate production. Sulfonamides inhibit both gram-positive and gram-negative bacteria, Nocardia sp, Chlamydia trachomatis, and some protozoa. Some enteric bacteria, such as Escherichia coli, Klebsiella pneumo-niae, Salmonella , Shigella , and Enterobacter sp are also inhibited. It is interesting that rickettsiae are not inhibited by sulfonamides but are instead stimulated in their growth. Activity is poor against anaerobes. Pseudomonas aeruginosa is intrinsically resistant to sulfonamide antibiotics.
Combination of a sulfonamide with an inhibitor of dihydro-folate reductase (trimethoprim or pyrimethamine) provides syn-ergistic activity because of sequential inhibition of folate synthesis ( Figure 46–2 ).
46Sulfonamides, Trimethoprim, & Quinolones Daniel H. Deck, PharmD, & Lisa G. Winston, MD∗
∗ The authors thank Henry F. Chambers, MD, for his contributions to previous editions.
C A S E S T U D Y
A 59-year-old woman presents to an urgent care clinic with a 4-day history of frequent and painful urination. She has had fevers, chills, and flank pain for the past 2 days. Her physician advised her to come immediately to the clinic for evaluation. In the clinic she is febrile (38.5°C [101.3°F]) but otherwise stable and states she is not experiencing any nau-sea or vomiting. Her urine dipstick test is positive for leuko-cyte esterase. Urinalysis and urine culture are ordered. Her past medical history is significant for three urinary tract
infections in the past year. Each episode was uncomplicated, treated with trimethoprim-sulfamethoxazole, and promptly resolved. She also has osteoporosis for which she takes a daily calcium supplement. The decision is made to treat her with oral antibiotics for a complicated urinary tract infection with close follow-up. Given her history, what would be a reasonable empiric antibiotic choice? Depending on the anti-biotic choice are there potential drug interactions?
046-Katzung_Ch046_p831-838.indd 831 9/23/11 4:23:48 PM

832 SECTION VIII Chemotherapeutic Drugs
Resistance Mammalian cells (and some bacteria) lack the enzymes required for folate synthesis from PABA and depend on exogenous sources of folate; therefore, they are not susceptible to sulfonamides. Sulfonamide resistance may occur as a result of mutations that (1) cause overproduction of PABA, (2) cause production of a folic acid-synthesizing enzyme that has low affinity for sulfonamides, or (3) impair permeability to the sulfonamide. Dihydropteroate synthase with low sulfonamide affinity is often encoded on a plas-mid that is transmissible and can disseminate rapidly and widely. Sulfonamide-resistant dihydropteroate synthase mutants also can emerge under selective pressure.
Pharmacokinetics Sulfonamides can be divided into three major groups: (1) oral, absorbable; (2) oral, nonabsorbable; and (3) topical. The oral, absorbable sulfonamides can be classified as short-, intermediate-, or long-acting on the basis of their half-lives ( Table 46–1 ). They are absorbed from the stomach and small intestine and distributed widely to tissues and body fluids (including the central nervous
system and cerebrospinal fluid), placenta, and fetus. Protein bind-ing varies from 20% to over 90%. Therapeutic concentrations are in the range of 40–100 mcg/mL of blood. Blood levels generally peak 2–6 hours after oral administration.
A portion of absorbed drug is acetylated or glucuronidated in the liver. Sulfonamides and inactive metabolites are then excreted into the urine, mainly by glomerular filtration. In significant renal failure, the dosage of sulfonamide must be reduced.
SO2NH2 COOH
Sulfanilamide
NH2
p-Aminobenzoic acid (PABA)
NH2
NSO2N
NO
CH3
N
CH3
Sulfadiazine
NH2
Sulfisoxazole
NH2
SO2NH
Sulfathalidine(phthalylsulfathiazole)
Sulfamethoxazole
SO2NH
NH2
SO2NH
NCOOH
S
NHCONH2
CH3ON
SO2NH
FIGURE 46–1 Structures of some sulfonamides and p- aminobenzoic acid.
−
−
Dihydropteroatesynthase
Dihydrofolatereductase
p-Aminobenzoic acid
Sulfonamides(competewith PABA)
Dihydrofolic acid
Trimethoprim
Tetrahydrofolic acid
Purines
DNA
FIGURE 46–2 Actions of sulfonamides and trimethoprim.
TABLE 46–1 Pharmacokinetic properties of some sulfonamides and pyrimidines.
Drug Half-Life Oral Absorption
Sulfonamides
Sulfacytine Short Prompt (peak levels in 1–4 hours)
Sulfisoxazole Short (6 hours) Prompt
Sulfamethizole Short (9 hours) Prompt
Sulfadiazine Intermediate (10–17 hours)
Slow (peak levels in 4–8 hours)
Sulfamethoxazole Intermediate (10–12 hours)
Slow
Sulfapyridine Intermediate (17 hours) Slow
Sulfadoxine Long (7–9 days) Intermediate
Pyrimidines
Trimethoprim Intermediate (11 hours) Prompt
Pyrimethamine Long (4–6 days) Prompt
046-Katzung_Ch046_p831-838.indd 832 9/23/11 4:23:50 PM

CHAPTER 46 Sulfonamides, Trimethoprim, & Quinolones 833
Clinical Uses Sulfonamides are infrequently used as single agents. Many strains of formerly susceptible species, including meningococci, pneu-mococci, streptococci, staphylococci, and gonococci, are now resistant. The fixed-drug combination of trimethoprim-sulfamethox- azole is the drug of choice for infections such as Pneumocystis jiroveci (formerly P carinii ) pneumonia, toxoplasmosis, nocardiosis, and occasionally other bacterial infections.
A. Oral Absorbable Agents Sulfisoxazole and sulfamethoxazole are short- to medium-acting agents used almost exclusively to treat urinary tract infections. The usual adult dosage is 1 g of sulfisoxazole four times daily or 1 g of sulfamethoxazole two or three times daily.
Sulfadiazine in combination with pyrimethamine is first-line therapy for treatment of acute toxoplasmosis. The combination of sulfadiazine with pyrimethamine, a potent inhibitor of dihydrofo-late reductase, is synergistic because these drugs block sequential steps in the folate synthetic pathway blockade ( Figure 46–2 ). The dosage of sulfadiazine is 1 g four times daily, with pyrimethamine given as a 75-mg loading dose followed by a 25-mg once-daily dose. Folinic acid, 10 mg orally each day, should also be adminis-tered to minimize bone marrow suppression.
Sulfadoxine is the only long-acting sulfonamide currently available in the USA and only as a combination formulation with pyrimethamine (Fansidar), a second-line agent in the treatment of malaria (see Chapter 52 ).
B. Oral Nonabsorbable Agents Sulfasalazine (salicylazosulfapyridine) is widely used in ulcerative colitis, enteritis, and other inflammatory bowel disease (see Chapter 62 ).
C. Topical Agents Sodium sulfacetamide ophthalmic solution or ointment is effec-tive in the treatment of bacterial conjunctivitis and as adjunctive therapy for trachoma. Another sulfonamide, mafenide acetate, is used topically but can be absorbed from burn sites. The drug and its primary metabolite inhibit carbonic anhydrase and can cause metabolic acidosis, a side effect that limits its usefulness. Silver sulfadiazine is a much less toxic topical sulfonamide and is pre-ferred to mafenide for prevention of infection of burn wounds.
Adverse Reactions All sulfonamides, including antimicrobial sulfas, diuretics, diazox-ide, and the sulfonylurea hypoglycemic agents, have been consid-ered to be partially cross-allergenic. However, evidence for this is not extensive. The most common adverse effects are fever, skin rashes, exfoliative dermatitis, photosensitivity, urticaria, nausea, vomiting, diarrhea, and difficulties referable to the urinary tract (see below). Stevens-Johnson syndrome, although relatively uncom-mon (< 1% of treatment courses), is a particularly serious and potentially fatal type of skin and mucous membrane eruption
associated with sulfonamide use. Other unwanted effects include stomatitis, conjunctivitis, arthritis, hematopoietic disturbances (see below), hepatitis, and, rarely, polyarteritis nodosa and psychosis.
A. Urinary Tract Disturbances Sulfonamides may precipitate in urine, especially at neutral or acid pH, producing crystalluria, hematuria, or even obstruction. This is rarely a problem with the more soluble sulfonamides (eg, sulfisoxazole). Sulfadiazine when given in large doses, particularly if fluid intake is poor, can cause crystalluria. Crystalluria is treated by administration of sodium bicarbonate to alkalinize the urine and fluids to increase urine flow. Sulfonamides have also been implicated in various types of nephrosis and in allergic nephritis.
B. Hematopoietic Disturbances Sulfonamides can cause hemolytic or aplastic anemia, granulocy-topenia, thrombocytopenia, or leukemoid reactions. Sulfonamides may provoke hemolytic reactions in patients with glucose-6-phosphate dehydrogenase deficiency. Sulfonamides taken near the end of pregnancy increase the risk of kernicterus in newborns.
TRIMETHOPRIM & TRIMETHOPRIM-SULFAMETHOXAZOLE MIXTURES
Mechanism of Action Trimethoprim, a trimethoxybenzylpyrimidine, selectively inhibits bacterial dihydrofolic acid reductase, which converts dihydrofolic acid to tetrahydrofolic acid, a step leading to the synthesis of purines and ultimately to DNA ( Figure 46–2 ). Trimethoprim is about 50,000 times less efficient in inhibition of mammalian dihydrofolic acid reductase. Pyrimethamine, another benzylpy-rimidine, selectively inhibits dihydrofolic acid reductase of proto-zoa compared with that of mammalian cells. As noted above, trimethoprim or pyrimethamine in combination with a sulfon-amide blocks sequential steps in folate synthesis, resulting in marked enhancement (synergism) of the activity of both drugs. The combination often is bactericidal, compared with the bacte-riostatic activity of a sulfonamide alone.
CI
C2H5
Pyrimethamine
CH2
Trimethoprim
OCH3
OCH3
OCH3
H2N
NH2
N
N
H2N
NH2
N
N
046-Katzung_Ch046_p831-838.indd 833 9/23/11 4:23:50 PM

834 SECTION VIII Chemotherapeutic Drugs
Resistance Resistance to trimethoprim can result from reduced cell permea-bility, overproduction of dihydrofolate reductase, or production of an altered reductase with reduced drug binding. Resistance can emerge by mutation, although more commonly it is due to plas-mid-encoded trimethoprim-resistant dihydrofolate reductases. These resistant enzymes may be coded within transposons on conjugative plasmids that exhibit a broad host range, accounting for rapid and widespread dissemination of trimethoprim resis-tance among numerous bacterial species.
Pharmacokinetics Trimethoprim is usually given orally, alone or in combination with sulfamethoxazole, which has a similar half-life. Trimethoprim-sulfamethoxazole can also be given intravenously. Trimethoprim is well absorbed from the gut and distributed widely in body fluids and tissues, including cerebrospinal fluid.
Because trimethoprim is more lipid-soluble than sulfamethox-azole, it has a larger volume of distribution than the latter drug. Therefore, when 1 part of trimethoprim is given with 5 parts of sulfamethoxazole (the ratio in the formulation), the peak plasma concentrations are in the ratio of 1:20, which is optimal for the combined effects of these drugs in vitro. About 30–50% of the sulfonamide and 50–60% of the trimethoprim (or their respec-tive metabolites) are excreted in the urine within 24 hours. The dose should be reduced by half for patients with creatinine clear-ances of 15–30 mL/min.
Trimethoprim (a weak base) concentrates in prostatic fluid and in vaginal fluid, which are more acidic than plasma. Therefore, it has more antibacterial activity in prostatic and vaginal fluids than many other antimicrobial drugs.
Clinical Uses A. Oral Trimethoprim Trimethoprim can be given alone (100 mg twice daily) in acute urinary tract infections. Many community-acquired organisms are susceptible to the high concentrations that are found in the urine (200–600 mcg/mL).
B. Oral Trimethoprim-Sulfamethoxazole (TMP-SMZ) A combination of trimethoprim-sulfamethoxazole is effective treatment for a wide variety of infections including P jiroveci pneumonia, shigellosis, systemic salmonella infections, urinary tract infections, prostatitis, and some nontuberculous mycobacte-rial infections. It is active against most Staphylococcus aureus strains, both methicillin-susceptible and methicillin-resistant, and against respiratory tract pathogens such as pneumococcus, Haemophilus sp, Moraxella catarrhalis, and K pneumoniae (but not Mycoplasma pneumoniae ). However, the increasing prevalence of strains of E coli (up to 30% or more) and pneumococci that are resistant to trimethoprim-sulfamethoxazole must be considered before using this combination for empiric therapy of upper uri-nary tract infections or pneumonia.
One double-strength tablet (each tablet contains trimethoprim 160 mg plus sulfamethoxazole 800 mg) given every 12 hours is effective treatment for urinary tract infections and prostatitis. One half of the regular (single-strength) tablet given three times weekly for many months may serve as prophylaxis in recurrent urinary tract infections of some women. One double-strength tablet every 12 hours is effective treatment for infections caused by susceptible strains of shigella and salmonella. The dosage for children treated for shigellosis, urinary tract infection, or otitis media is 8 mg/kg trimethoprim and 40 mg/kg sulfamethoxazole every 12 hours.
Infections with P jiroveci and some other pathogens can be treated orally with high doses of the combination (dosed on the basis of the trimethoprim component at 15–20 mg/kg/d) or can be prevented in immunosuppressed patients by one double-strength tablet daily or three times weekly.
C. Intravenous Trimethoprim-Sulfamethoxazole A solution of the mixture containing 80 mg trimethoprim plus 400 mg sulfamethoxazole per 5 mL diluted in 125 mL of 5% dextrose in water can be administered by intravenous infusion over 60–90 minutes. It is the agent of choice for moderately severe to severe pneumocystis pneumonia. It may be used for gram-negative bacterial sepsis, including that caused by some multidrug-resistant species such as enterobacter and serratia; shigellosis; typhoid fever; or urinary tract infection caused by a susceptible organism when the patient is unable to take the drug by mouth. The dosage is 10–20 mg/kg/d of the trimethoprim component.
D. Oral Pyrimethamine with Sulfonamide Pyrimethamine and sulfadiazine have been used for treatment of leishmaniasis and toxoplasmosis. In falciparum malaria, the com-bination of pyrimethamine with sulfadoxine (Fansidar) has been used (see Chapter 52 ).
Adverse Effects Trimethoprim produces the predictable adverse effects of an antifolate drug, especially megaloblastic anemia, leukopenia, and granulocy-topenia. The combination trimethoprim-sulfamethoxazole may cause all of the untoward reactions associated with sulfonamides. Nausea and vomiting, drug fever, vasculitis, renal damage, and cen-tral nervous system disturbances occasionally occur also. Patients with AIDS and pneumocystis pneumonia have a particularly high frequency of untoward reactions to trimethoprim-sulfamethoxazole, especially fever, rashes, leukopenia, diarrhea, elevations of hepatic aminotransferases, hyperkalemia, and hyponatremia.
■ DNA GYRASE INHIBITORS
FLUOROQUINOLONES
The important quinolones are synthetic fluorinated analogs of nalidixic acid ( Figure 46–3 ). They are active against a variety of gram-positive and gram-negative bacteria.
046-Katzung_Ch046_p831-838.indd 834 9/23/11 4:23:50 PM

CHAPTER 46 Sulfonamides, Trimethoprim, & Quinolones 835
Mechanism of Action Quinolones block bacterial DNA synthesis by inhibiting bacterial topoisomerase II (DNA gyrase) and topoisomerase IV. Inhibition of DNA gyrase prevents the relaxation of positively supercoiled DNA that is required for normal transcription and replication. Inhibition of topoisomerase IV interferes with separation of replicated chromo-somal DNA into the respective daughter cells during cell division.
Antibacterial Activity Earlier quinolones such as nalidixic acid did not achieve systemic antibacterial levels and were useful only in the treatment of lower urinary tract infections. Fluorinated derivatives (ciprofloxacin, levofloxacin, and others; Figure 46–3 and Table 46–2 ) have greatly improved antibacterial activity compared with nalidixic acid and achieve bactericidal levels in blood and tissues.
Fluoroquinolones were originally developed because of their excellent activity against gram-negative aerobic bacteria; they had limited activity against gram-positive organisms. Several newer agents have improved activity against gram-positive cocci. This
relative activity against gram-negative versus gram-positive species is useful for classification of these agents. Norfloxacin is the least active of the fluoroquinolones against both gram-negative and gram-positive organisms, with minimum inhibitory concentrations (MICs) fourfold to eightfold higher than those of ciprofloxacin. Ciprofloxacin, enoxacin, lomefloxacin, levofloxacin, ofloxacin, and pefloxacin comprise a second group of similar agents possessing excellent gram-negative activity and moderate to good activity against gram-positive bacteria. MICs for gram-negative cocci and bacilli, including Enterobacter sp, P aeruginosa , Neisseria menin-gitidis , Haemophilus sp, and Campylobacter jejuni , are 1–2 mcg/mL and often less. Methicillin-susceptible strains of S aureus are gener-ally susceptible to these fluoroquinolones, but methicillin-resistant strains of staphylococci are often resistant. Streptococci and entero-cocci tend to be less susceptible than staphylococci, and efficacy in infections caused by these organisms is limited. Ciprofloxacin is the most active agent of this group against gram-negative organisms, P aeruginosa in particular. Levofloxacin, the L-isomer of ofloxacin, has superior activity against gram-positive organisms, including Streptococcus pneumoniae.
Gatifloxacin, gemifloxacin, and moxifloxacin make up a third group of fluoroquinolones with improved activity against gram-positive organisms, particularly S pneumoniae and some staphylo-cocci. Gemifloxacin is active in vitro against ciprofloxacin-resistant strains of S pneumoniae, but in vivo efficacy is unproven. Although MICs of these agents for staphylococci are lower than those of ciprofloxacin (and the other compounds mentioned in the para-graph above), it is not known whether the enhanced activity is sufficient to permit use of these agents for treatment of infections caused by ciprofloxacin-resistant strains. In general, none of these agents is as active as ciprofloxacin against gram-negative organ-isms. Fluoroquinolones also are active against agents of atypical pneumonia (eg, mycoplasmas and chlamydiae) and against intra-cellular pathogens such as Legionella pneumophila and some myco-bacteria, including Mycobacterium tuberculosis and Mycobacterium avium complex. Moxifloxacin also has modest activity against anaerobic bacteria. Because of toxicity, gatifloxacin is no longer available in the United States.
Resistance During fluoroquinolone therapy, resistant organisms emerge in about one of every 10 7 –10 9 organisms, especially among staphylo-cocci, P aeruginosa , and Serratia marcescens . Resistance is due to one or more point mutations in the quinolone binding region of the target enzyme or to a change in the permeability of the organism. However, this does not account for the relative ease with which resistance develops in exquisitely susceptible bacteria. More recently two types of plasmid-mediated resistance have been described. The first type utilizes Qnr proteins, which protect DNA gyrase from the fluoroquinolones. The second is a variant of an aminoglycoside acetyltransferase capable of modifying ciprofloxacin. Both mecha-nisms confer low-level resistance that may facilitate the point muta-tions that confer high-level resistance. Resistance to one fluoroquinolone, particularly if it is of high level, generally confers cross-resistance to all other members of this class.
Gemifloxacin
NNNN
O
CO2HCH2O
H2N
FF
NNN
Levofloxacin
CH3
CH3
O
O
F COOH
NNHN
Ciprofloxacin
O
F COOH
NN
HN
Gatifloxacin
O
O
CH3CH3
F COOH
N
C2H5
HN
Norfloxacin
O
F COOH
NN N
O
Nalidixic acid
COOH
CH3
C2H5
N N
Moxifloxacin
H3C
HN
COOHFF
O
FIGURE 46–3 Structures of nalidixic acid and some fluoroquinolones.
046-Katzung_Ch046_p831-838.indd 835 9/23/11 4:23:51 PM

836 SECTION VIII Chemotherapeutic Drugs
Pharmacokinetics After oral administration, the fluoroquinolones are well absorbed (bioavailability of 80–95%) and distributed widely in body fluids and tissues ( Table 46–2 ). Serum half-lives range from 3 to 10 hours. The relatively long half-lives of levofloxacin, gemifloxacin, gati-floxacin, and moxifloxacin permit once-daily dosing. Oral absorp-tion is impaired by divalent and trivalent cations, including those in antacids. Therefore, oral fluoroquinolones should be taken 2 hours before or 4 hours after any products containing these cat-ions. Serum concentrations of intravenously administered drug are similar to those of orally administered drug. Most fluoroqui-nolones are eliminated by renal mechanisms, either tubular secre-tion or glomerular filtration ( Table 46–2 ). Dosage adjustment is required for patients with creatinine clearances less than 50 mL/min, the exact adjustment depending on the degree of renal impair-ment and the specific fluoroquinolone being used. Dosage adjust-ment for renal failure is not necessary for moxifloxacin. Nonrenally cleared fluoroquinolones are relatively contraindicated in patients with hepatic failure.
Clinical Uses Fluoroquinolones (other than moxifloxacin, which achieves rela-tively low urinary levels) are effective in urinary tract infections caused by many organisms, including P aeruginosa . These agents are also effective for bacterial diarrhea caused by Shigella , Salmonella , toxigenic E coli, and Campylobacter . Fluoroquinolones (except norfloxacin, which does not achieve adequate systemic concentra-tions) have been used in infections of soft tissues, bones, and joints and in intra-abdominal and respiratory tract infections, including those caused by multidrug-resistant organisms such as Pseudomonas and Enterobacter . Ciprofloxacin is a drug of choice for prophylaxis and treatment of anthrax, although the newer fluoroquinolones are active in vitro and very likely in vivo as well.
Ciprofloxacin and levofloxacin are no longer recommended for the treatment of gonococcal infection in the United States as resis-tance is now common. However, both drugs are effective in treat-ing chlamydial urethritis or cervicitis. Ciprofloxacin, levofloxacin, or moxifloxacin is occasionally used for treatment of tuberculosis
and atypical mycobacterial infections. These agents may be suit-able for eradication of meningococci from carriers or for prophy-laxis of infection in neutropenic cancer patients.
With their enhanced gram-positive activity and activity against atypical pneumonia agents (chlamydiae, Mycoplasma , and Legionella ), levofloxacin, gatifloxacin, gemifloxacin, and moxifloxacin—so-called respiratory fluoroquinolones—are effective and used increasingly for treatment of upper and lower respiratory tract infections.
Adverse Effects Fluoroquinolones are generally well tolerated. The most common effects are nausea, vomiting, and diarrhea. Occasionally, headache, dizziness, insomnia, skin rash, or abnormal liver function tests develop. Photosensitivity has been reported with lomefloxacin and pefloxacin. QT c prolongation may occur with gatifloxacin, levo-floxacin, gemifloxacin, and moxifloxacin, which should be avoided or used with caution in patients with known QT c interval prolon-gation or uncorrected hypokalemia; in those receiving class IA (eg, quinidine or procainamide) or class III antiarrhythmic agents (sotalol, ibutilide, amiodarone); and in patients receiving other agents known to increase the QT c interval (eg, erythromycin, tricy-clic antidepressants). Gatifloxacin has been associated with hyper-glycemia in diabetic patients and with hypoglycemia in patients also receiving oral hypoglycemic agents. Because of these serious effects (including some fatalities), gatifloxacin was withdrawn from sales in the United States in 2006; it may be available elsewhere.
Fluoroquinolones may damage growing cartilage and cause an arthropathy. Thus, these drugs are not routinely recommended for patients under 18 years of age. However, the arthropathy is revers-ible, and there is a growing consensus that fluoroquinolones may be used in children in some cases (eg, for treatment of pseudomonal infections in patients with cystic fibrosis). Tendonitis, a rare com-plication that has been reported in adults, is potentially more seri-ous because of the risk of tendon rupture. Risk factors for tendonitis include advanced age, renal insufficiency, and concur-rent steroid use. Fluoroquinolones should be avoided during preg-nancy in the absence of specific data documenting their safety.
TABLE 46–2 Pharmacokinetic properties of some fluoroquinolones.
Drug Half-Life (h)Oral Bioavailability (%)
Peak Serum Concentration (mcg/mL) Oral Dose (mg)
Primary Route of Excretion
Ciprofloxacin 3–5 70 2.4 500 Renal
Gatifloxacin 8 98 3.4 400 Renal
Gemifloxacin 8 70 1.6 320 Renal and nonrenal
Levofloxacin 5–7 95 5.7 500 Renal
Lomefloxacin 8 95 2.8 400 Renal
Moxifloxacin 9–10 > 85 3.1 400 Nonrenal
Norfloxacin 3.5–5 80 1.5 400 Renal
Ofloxacin 5–7 95 2.9 400 Renal
046-Katzung_Ch046_p831-838.indd 836 9/23/11 4:23:51 PM

CHAPTER 46 Sulfonamides, Trimethoprim, & Quinolones 837
SUMMARY Sulfonamides, Trimethoprim, and Fluoroquinolones
Subclass Mechanism of Action Effects Clinical ApplicationsPharmacokinetics, Toxicities, Interactions
FOLATE ANTAGONISTS • Trimethoprim- sulfamethoxazole
Synergistic combination of folate antagonists blocks purine production and nucleic acid synthesis
Bactericidal activity against susceptible bacteria
Urinary tract infections • P jiroveci pneumonia • toxoplasmosis • nocardiosis
Oral, IV • renal clearance (half-life 8 h) • dosed every 8–12 h • formulated in a 5:1 ratio of sulfamethoxazole to trimethoprim • Toxicity: Rash, fever, bone marrow suppression, hyperkalemia
• Sulfisoxazole: Oral; used only for lower urinary tract infections
• Sulfadiazine: Oral; first-line therapy for toxoplasmosis when combined with pyrimethamine
• Trimethoprim: Oral; used only for lower urinary tract infections; may be safely prescribed to patients with sulfonamide allergy
• Pyrimethamine: Oral; first-line therapy for toxoplasmosis when combined with sulfadiazine; coadminister with leucovorin to limit bone marrow toxicity
• Pyrimethamine-sulfadoxine: Oral; second-line malaria treatment
FLUOROQUINOLONES • Ciprofloxacin Inhibits DNA replication by
binding to DNA gyrase and topoisomerase IV
Bactericidal activity against susceptible bacteria
Urinary tract infections • gastroenteritis • osteomy-elitis • anthrax
Oral, IV • mixed clearance (half-life 4 h) • dosed every 12 h • divalent and tri-valent cations impair oral absorption • Toxicity: Gastrointestinal upset, neurotoxicity, tendonitis
• Ofloxacin: Oral; has improved pharmacokinetics and pharmacodynamics; use is limited to urinary tract infections and nongonococcal urethritis and cervicitis
• Levofloxacin: Oral, IV; L-isomer of ofloxacin; once-daily dosing; renal clearance; “respiratory” fluoroquinolone with improved activity versus pneumococcus
• Moxifloxacin: Oral, IV; “respiratory” fluoroquinolone; once-daily dosing; improved activity versus anaerobes and M tuberculosis; hepatic clearance results in lower urinary levels so use in urinary tract infections is not recommended
• Gemifloxacin: Oral; “respiratory” fluoroquinolone
GENERAL-PURPOSE SULFONAMIDESSulfadiazine (generic)
Oral: 500 mg tabletsSulfisoxazole (generic)
Oral: 500 mg tablets; 500 mg/5 mL syrup
SULFONAMIDES FOR SPECIAL APPLICATIONSMafenide (Sulfamylon)
Topical: 85 mg/g cream; 5% solutionSilver sulfadiazine (generic, Silvadene)
Topical: 10 mg/g creamSulfacetamide sodium (generic)
Ophthalmic: 1, 10, 15, 30% solutions; 10% ointment
TRIMETHOPRIMTrimethoprim (generic, Proloprim, Trimpex)
Oral: 100, 200 mg tablets
Trimethoprim-sulfamethoxazole [co-trimoxazole, TMP-SMZ] (generic, Bactrim, Septra, others)
Oral: 80 mg trimethoprim + 400 mg sulfamethoxazole per single-strength tablet; 160 mg trimethoprim + 800 mg sulfamethoxazole per double-strength tablet; 40 mg trimethoprim + 200 mg sul-famethoxazole per 5 mL suspension
Parenteral: 80 mg trimethoprim + 400 mg sulfamethoxazole per 5 mL for infusion (in 5 mL ampules and 5, 10, 20 mL vials)
PYRIMETHAMINEPyrimethamine (generic, Daraprim)
Oral: 25 mg tabletsPyrimethamine-sulfadoxine (Fansidar)
Oral: 25 mg pyrimethamine + 500 mg sulfadoxine per tablet
QUINOLONES & FLUOROQUINOLONESCiprofloxacin (generic, Cipro, Cipro I.V.)
Oral: 100, 250, 500, 750 mg tablets; 500, 1000 mg extended-release tablet; 50, 100 mg/mL suspension
Parenteral: 10 mg/mL for IV infusionOphthalmic (Ciloxan): 3 mg/mL solution; 3.3 mg/g ointment
P R E P A R A T I O N S A V A I L A B L E
GENERAL-PURPOSE SULFONAMIDESSulfadiazine (generic)
Oral: 500 mg tabletsSulfisoxazole (generic)
Oral: 500 mg tablets; 500 mg/5 mL syrup
SULFONAMIDES FOR SPECIAL APPLICATIONSMafenide (Sulfamylon)
Topical: 85 mg/g cream; 5% solutionSilver sulfadiazine (generic, Silvadene)
Topical: 10 mg/g creamSulfacetamide sodium (generic)
Ophthalmic: 1, 10, 15, 30% solutions; 10% ointment
TRIMETHOPRIMTrimethoprim (generic, Proloprim, Trimpex)
Oral: 100, 200 mg tablets
Trimethoprim-sulfamethoxazole [co-trimoxazole, TMP-SMZ] (generic, Bactrim, Septra, others)
Oral: 80 mg trimethoprim + 400 mg sulfamethoxazole per single-strength tablet; 160 mg trimethoprim + 800 mg sulfamethoxazoleper double-strength tablet; 40 mg trimethoprim + 200 mg sul-famethoxazole per 5 mL suspension
Parenteral: 80 mg trimethoprim + 400 mg sulfamethoxazole per 5mL for infusion (in 5 mL ampules and 5, 10, 20 mL vials)
PYRIMETHAMINEPyrimethamine (generic, Daraprim)
Oral: 25 mg tabletsPyrimethamine-sulfadoxine (Fansidar)
Oral: 25 mg pyrimethamine + 500 mg sulfadoxine per tablet
QUINOLONES & FLUOROQUINOLONESCiprofloxacin (generic, Cipro, Cipro I.V.)
Oral: 100, 250, 500, 750 mg tablets; 500, 1000 mg extended-release tablet; 50, 100 mg/mL suspension
Parenteral: 10 mg/mL for IV infusionOphthalmic (Ciloxan): 3 mg/mL solution; 3.3 mg/g ointment
046-Katzung_Ch046_p831-838.indd 837 9/23/11 4:23:51 PM

838 SECTION VIII Chemotherapeutic Drugs
REFERENCES Davidson R et al: Resistance to levofloxacin and failure of treatment of pneumo-
coccal pneumonia. N Engl J Med 2002;346:747. Frothingham R: Rates of torsades de pointes associated with ciprofloxacin,
ofloxacin, levofloxacin, gatifloxacin, and moxifloxacin. Pharmacotherapy 2001;21:1468.
Keating GM, Scott LJ: Moxifloxacin: A review of its use in the management of bacterial infections. Drugs 2004;64:2347.
Mandell LA et al: Update of practice guidelines for the management of community- acquired pneumonia in immunocompetent adults. Clin Infect Dis 2003;37:1405.
Mwenya DM et al: Impact of cotrimoxazole on carriage and antibiotic resistance of Streptococcus pneumoniae and Haemophilus influenzae in HIV-infected children in Zambia. Antimicrob Agents Chemother 2010;54:3756
Nouira S et al: Standard versus newer antibacterial agents in the treatment of severe acute exacerbation of chronic obstructive pulmonary disease: A randomized trial of trimethoprim-sulfamethoxazole versus ciprofloxacin. Clin Infect Dis 2010;51:143.
Robicsek A et al: The worldwide emergence of plasmid-mediated quinolone resis-tance. Lancet ID 2006;6:629.
Scheld WM: Maintaining fluoroquinolone class efficacy: Review of influencing factors. Emerg Infect Dis 2003;9:1.
Schmitz GR et al: Randomized controlled trial of trimethoprim-sulfamethoxazole for uncomplicated skin abscesses in patients at risk for community-associated methicillin-resistant Staphylococcus aureus infection. Ann Emerg Med 2010;56:283.
Strom BL et al: Absence of cross-reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics. N Engl J Med 2003;349:1628.
Talan DA et al: Prevalence of and risk factor analysis of trimethoprim-sulfamethoxazole- and fluoroquinolone-resistant E. coli infection among emergency department patients with pyelonephritis. Clin Infect Dis 2008; 47:1150.
Yoo BK et al: Gemifloxacin: A new fluoroquinolone approved for treatment of respiratory infections. Ann Pharmacother 2004;38:1226.
C A S E S T U D Y A N S W E R
A fluoroquinolone that achieves good urinary levels (ciprofloxacin or levofloxacin) would be a reasonable choice for empiric treatment of this patient’s complicated urinary tract infection. Her recent exposure to multiple courses of trimethoprim-sulfamethoxazole increases her chances of having a urinary tract infection with an isolate that is resistant
to this antibiotic, making trimethoprim-sulfamethoxazole a poor choice. The patient should be told to take the oral fluoroquinolone 2 hours before or 4 hours after her calcium supplement as divalent and trivalent cations can significantly impair the absorption of oral fluoroquinolones.
Gemifloxacin (Factive)Oral: 320 mg tablet
Levofloxacin (Levaquin)Oral: 250, 500, 750 mg tablets; 25 mg/mL solutionParenteral: 5, 25 mg/mL for IV injectionOphthalmic (Quixin): 5 mg/mL solution
Lomefloxacin (Maxaquin)Oral: 400 mg tablets
Moxifloxacin (Avelox, Avelox I.V.)Oral: 400 mg tabletsParenteral: 400 mg in IV bag
Norfloxacin (Noroxin)Oral: 400 mg tablets
Ofloxacin (Floxin)Oral: 200, 300, 400 mg tabletsOphthalmic (Ocuflox): 3 mg/mL solutionOtic (Floxin Otic): 0.3% solution
Gemifloxacin (Factive)Oral: 320 mg tablet
Levofloxacin (Levaquin)Oral: 250, 500, 750 mg tablets; 25 mg/mL solutionParenteral: 5, 25 mg/mL for IV injectionOphthalmic (Quixin): 5 mg/mL solution
Lomefloxacin (Maxaquin)Oral: 400 mg tablets
Moxifloxacin (Avelox, Avelox I.V.)Oral: 400 mg tabletsParenteral: 400 mg in IV bag
Norfloxacin (Noroxin)Oral: 400 mg tablets
Ofloxacin (Floxin)Oral: 200, 300, 400 mg tabletsOphthalmic (Ocuflox): 3 mg/mL solutionOtic (Floxin Otic): 0.3% solution
046-Katzung_Ch046_p831-838.indd 838 9/23/11 4:23:51 PM

839
C H A P T E R
Mycobacteria are intrinsically resistant to most antibiotics. Because they grow more slowly than other bacteria, antibiotics that are most active against rapidly growing cells are relatively ineffective. Mycobacterial cells can also be dormant and thus completely resis-tant to many drugs or killed only very slowly. The lipid-rich myco-bacterial cell wall is impermeable to many agents. Mycobacterial species are intracellular pathogens, and organisms residing within macrophages are inaccessible to drugs that penetrate these cells poorly. Finally, mycobacteria are notorious for their ability to develop resistance. Combinations of two or more drugs are required to overcome these obstacles and to prevent emergence of resistance during the course of therapy. The response of mycobac-terial infections to chemotherapy is slow, and treatment must be administered for months to years, depending on which drugs are used. The drugs used to treat tuberculosis, atypical mycobacterial infections, and leprosy are described in this chapter.
■ DRUGS USED IN TUBERCULOSIS
Isoniazid (INH), rifampin (or other rifamycin), pyrazinamide, ethambutol, and streptomycin are the five first-line agents for treatment of tuberculosis ( Table 47–1 ). Isoniazid and rifampin are the most active drugs. An isoniazid-rifampin combination admin-istered for 9 months will cure 95–98% of cases of tuberculosis caused by susceptible strains. The addition of pyrazinamide to an isoniazid-rifampin combination for the first 2 months allows the total duration of therapy to be reduced to 6 months without loss of efficacy ( Table 47–2 ). In practice, therapy is initiated with a four-drug regimen of isoniazid, rifampin, and pyrazinamide plus either ethambutol or streptomycin until susceptibility of the clinical isolate has been determined. Neither ethambutol nor streptomycin adds substantially to the overall activity of the regimen (ie, the duration of treatment cannot be further reduced if either drug is used), but they provide additional coverage if the isolate proves to be resistant to isoniazid, rifampin, or both. The prevalence of isoniazid resistance among clinical isolates in the United States is approximately 10%. Prevalence of resistance to both isoniazid and rifampin (ie, multidrug resistance) is about 3%.
47Antimycobacterial Drugs Daniel H. Deck, PharmD, & Lisa G. Winston, MD∗
C A S E S T U D Y
A 45-year-old homeless man presents to the emergency department complaining of a 2-month history of fatigue, weight loss (10 kg), fevers, night sweats, and a productive cough. He is currently living on the street but has spent time in homeless shelters and prison in the last several years. He reports drinking 2–3 pints of hard alcohol per day for the last 15 years, and also reports a history of intravenous drug use. In the emergency department, a chest x-ray shows a right
apical infiltrate. Given the high suspicion for pulmonary tuberculosis, the patient is placed in respiratory isolation. His first sputum smear shows many acid-fast bacilli, and a rapid HIV antibody test returns with a positive result. What drugs should be started for treatment of presumptive pulmo-nary tuberculosis? Does the patient have a heightened risk of developing medication toxicity? If so, which medication(s) would be likely to cause toxicity?
∗ The authors thank Henry F. Chambers, MD, for his contributions to previous editions.
047-Katzung_Ch047_p839-848.indd 839 9/21/11 12:09:36 PM

840 SECTION VIII Chemotherapeutic Drugs
ISONIAZID
Isoniazid is the most active drug for the treatment of tuberculosis caused by susceptible strains. It is a small molecule (MW 137) that is freely soluble in water. The structural similarity to pyridoxine is shown below.
CH3
OH
CH2OH
HOH2C
Pyridoxine
Isoniazid
CONHNH2
N
N
In vitro, isoniazid inhibits most tubercle bacilli at a concentra-tion of 0.2 mcg/mL or less and is bactericidal for actively growing tubercle bacilli. It is less effective against atypical mycobacterial species. Isoniazid penetrates into macrophages and is active against both extracellular and intracellular organisms.
Mechanism of Action & Basis of Resistance Isoniazid inhibits synthesis of mycolic acids, which are essential components of mycobacterial cell walls. Isoniazid is a prodrug that is activated by KatG, the mycobacterial catalase-peroxidase. The activated form of isoniazid forms a covalent complex with an acyl carrier protein (AcpM) and KasA, a beta-ketoacyl carrier protein synthetase, which blocks mycolic acid synthesis and kills the cell. Resistance to isoniazid is associated with mutations resulting in overexpression of inhA , which encodes an NADH-dependent acyl carrier protein reductase; mutation or deletion of the katG gene; promoter mutations resulting in overexpression of ahpC , a puta-tive virulence gene involved in protection of the cell from oxida-tive stress; and mutations in kasA . Overproducers of inhA express low-level isoniazid resistance and cross-resistance to ethionamide. KatG mutants express high-level isoniazid resistance and often are not cross-resistant to ethionamide.
Drug-resistant mutants are normally present in susceptible mycobacterial populations at about 1 bacillus in 10 6 . Since tubercu-lous lesions often contain more than 10 8 tubercle bacilli, resistant mutants are readily selected if isoniazid or any other drug is given as a single agent. The use of two independently acting drugs in com-bination is much more effective. The probability that a bacillus is initially resistant to both drugs is approximately 1 in 10 6 × 10 6 , or 1 in 10 12 , several orders of magnitude greater than the number of infecting organisms. Thus, at least two (or more in certain cases) active agents should always be used to treat active tuberculosis to prevent emergence of resistance during therapy.
Pharmacokinetics Isoniazid is readily absorbed from the gastrointestinal tract. A 300-mg oral dose (5 mg/kg in children) achieves peak plasma concentrations of 3–5 mcg/mL within 1–2 hours. Isoniazid dif-fuses readily into all body fluids and tissues. The concentration in the central nervous system and cerebrospinal fluid ranges between 20% and 100% of simultaneous serum concentrations.
Metabolism of isoniazid, especially acetylation by liver N -acetyltransferase, is genetically determined (see Chapter 4 ).
TABLE 47–1 Antimicrobials used in the treatment of tuberculosis.
Drug Typical Adult Dosage1
First-line agents (in approximate order of preference)
Isoniazid 300 mg/d
Rifampin 600 mg/d
Pyrazinamide 25 mg/kg/d
Ethambutol 15–25 mg/kg/d
Streptomycin 15 mg/kg/d
Second-line agents
Amikacin 15 mg/kg/d
Aminosalicylic acid 8–12 g/d
Capreomycin 15 mg/kg/d
Ciprofloxacin 1500 mg/d, divided
Clofazimine 200 mg/d
Cycloserine 500–1000 mg/d, divided
Ethionamide 500–750 mg/d
Levofloxacin 500 mg/d
Rifabutin 300 mg/d2
Rifapentine 600 mg once or twice weekly
1Assuming normal renal function.2150 mg/d if used concurrently with a protease inhibitor.
TABLE 47–2 Recommended duration of therapy for tuberculosis.
Regimen (in Approximate Order of Preference) Duration in Months
Isoniazid, rifampin, pyrazinamide 6
Isoniazid, rifampin 9
Rifampin, ethambutol, pyrazinamide
6
Rifampin, ethambutol 12
Isoniazid, ethambutol 18
All others ≥ 24
047-Katzung_Ch047_p839-848.indd 840 9/21/11 12:09:39 PM

CHAPTER 47 Antimycobacterial Drugs 841
the standard 300-mg adult dose. Peripheral neuropathy is more likely to occur in slow acetylators and patients with predisposing conditions such as malnutrition, alcoholism, diabetes, AIDS, and uremia. Neuropathy is due to a relative pyridoxine deficiency. Isoniazid promotes excretion of pyridoxine, and this toxicity is readily reversed by administration of pyridoxine in a dosage as low as 10 mg/d. Central nervous system toxicity, which is less com-mon, includes memory loss, psychosis, and seizures. These effects may also respond to pyridoxine.
Miscellaneous other reactions include hematologic abnormali-ties, provocation of pyridoxine deficiency anemia, tinnitus, and gastrointestinal discomfort. Isoniazid can reduce the metabolism of phenytoin, increasing its blood level and toxicity.
RIFAMPIN
Rifampin is a semisynthetic derivative of rifamycin, an antibiotic produced by Streptomyces mediterranei . It is active in vitro against gram-positive and gram-negative cocci, some enteric bacteria, mycobacteria, and chlamydiae. Susceptible organisms are inhib-ited by less than 1 mcg/mL. Resistant mutants are present in all microbial populations at approximately 1 in 10 6 organisms and are rapidly selected out if rifampin is used as a single drug, espe-cially in a patient with active infection. There is no cross-resistance to other classes of antimicrobial drugs, but there is cross-resistance to other rifamycin derivatives, eg, rifabutin and rifapentine.
Mechanism of Action, Resistance, & Pharmacokinetics Rifampin binds to the β subunit of bacterial DNA-dependent RNA polymerase and thereby inhibits RNA synthesis. Resistance results from any one of several possible point mutations in rpoB , the gene for the β subunit of RNA polymerase. These mutations result in reduced binding of rifampin to RNA polymerase. Human RNA polymerase does not bind rifampin and is not inhibited by it. Rifampin is bactericidal for mycobacteria. It readily penetrates most tissues and penetrates into phagocytic cells. It can kill organisms that are poorly accessible to many other drugs, such as intracellular organisms and those sequestered in abscesses and lung cavities.
Rifampin is well absorbed after oral administration and excreted mainly through the liver into bile. It then undergoes enterohepatic recirculation, with the bulk excreted as a deacylated metabolite in feces and a small amount excreted in the urine. Dosage adjustment for renal or hepatic insufficiency is not necessary. Usual doses result in serum levels of 5–7 mcg/mL. Rifampin is distributed widely in body fluids and tissues. The drug is relatively highly protein-bound, and adequate cerebrospinal fluid concentrations are achieved only in the presence of meningeal inflammation.
Clinical Uses A. Mycobacterial Infections Rifampin, usually 600 mg/d (10 mg/kg/d) orally, must be adminis-tered with isoniazid or other antituberculous drugs to patients with
The average plasma concentration of isoniazid in rapid acetylators is about one third to one half of that in slow acetylators, and aver-age half-lives are less than 1 hour and 3 hours, respectively. More rapid clearance of isoniazid by rapid acetylators is usually of no therapeutic consequence when appropriate doses are administered daily, but subtherapeutic concentrations may occur if drug is administered as a once-weekly dose or if there is malabsorption.
Isoniazid metabolites and a small amount of unchanged drug are excreted, mainly in the urine. The dose need not be adjusted in renal failure. Dose adjustment is not well defined in patients with severe preexisting hepatic insufficiency (isoniazid is contrain-dicated if it is the cause of the hepatitis) and should be guided by serum concentrations if a reduction in dose is contemplated.
Clinical Uses The typical dosage of isoniazid is 5 mg/kg/d; a typical adult dose is 300 mg given once daily. Up to 10 mg/kg/d may be used for serious infections or if malabsorption is a problem. A 15 mg/kg dose, or 900 mg, may be used in a twice-weekly dosing regimen in combination with a second antituberculous agent (eg, rifampin 600 mg). Pyridoxine, 25–50 mg/d, is recommended for those with conditions predisposing to neuropathy, an adverse effect of isoniazid. Isoniazid is usually given by mouth but can be given parenterally in the same dosage.
Isoniazid as a single agent is also indicated for treatment of latent tuberculosis. The dosage is 300 mg/d (5 mg/kg/d) or 900 mg twice weekly for 9 months.
Adverse Reactions The incidence and severity of untoward reactions to isoniazid are related to dosage and duration of administration.
A. Immunologic Reactions Fever and skin rashes are occasionally seen. Drug-induced sys-temic lupus erythematosus has been reported.
B. Direct Toxicity Isoniazid-induced hepatitis is the most common major toxic effect. This is distinct from the minor increases in liver aminotransferases (up to three or four times normal), which do not require cessation of the drug and which are seen in 10–20% of patients, who usu-ally are asymptomatic. Clinical hepatitis with loss of appetite, nausea, vomiting, jaundice, and right upper quadrant pain occurs in 1% of isoniazid recipients and can be fatal, particularly if the drug is not discontinued promptly. There is histologic evidence of hepatocellular damage and necrosis. The risk of hepatitis depends on age. It occurs rarely under age 20, in 0.3% of those aged 21–35, 1.2% of those aged 36–50, and 2.3% for those aged 50 and above. The risk of hepatitis is greater in individuals with alco-hol dependence and possibly during pregnancy and the postpar-tum period. Development of isoniazid hepatitis contraindicates further use of the drug.
Peripheral neuropathy is observed in 10–20% of patients given dosages greater than 5 mg/kg/d, but it is infrequently seen with
047-Katzung_Ch047_p839-848.indd 841 9/21/11 12:09:39 PM

842 SECTION VIII Chemotherapeutic Drugs
active tuberculosis to prevent emergence of drug-resistant mycobac-teria. In some short-course therapies, 600 mg of rifampin is given twice weekly. Rifampin, 600 mg daily or twice weekly for 6 months, also is effective in combination with other agents in some atypical mycobacterial infections and in leprosy. Rifampin, 600 mg daily for 4 months as a single drug, is an alternative to isoniazid for patients with latent tuberculosis who are unable to take isoniazid or who have had exposure to a case of active tuberculosis caused by an iso-niazid-resistant, rifampin-susceptible strain.
B. Other Indications Rifampin has other uses in bacterial infections. An oral dosage of 600 mg twice daily for 2 days can eliminate meningococcal car-riage. Rifampin, 20 mg/kg/d for 4 days, is used as prophylaxis in contacts of children with Haemophilus influenzae type b disease. Rifampin combined with a second agent is used to eradicate staphylococcal carriage. Rifampin combination therapy is also indicated for treatment of serious staphylococcal infections such as osteomyelitis and prosthetic valve endocarditis.
Adverse Reactions Rifampin imparts a harmless orange color to urine, sweat, and tears (soft contact lenses may be permanently stained). Occasional adverse effects include rashes, thrombocytopenia, and nephritis. Rifampin may cause cholestatic jaundice and occasionally hepatitis, and it commonly causes light-chain proteinuria. If administered less often than twice weekly, rifampin may cause a flu-like syndrome characterized by fever, chills, myalgias, anemia, and thrombocytope-nia. Its use has been associated with acute tubular necrosis. Rifampin strongly induces most cytochrome P450 isoforms (1A2, 2C9, 2C19, 2D6, and 3A4), which increases the elimination of numer-ous other drugs including methadone, anticoagulants, cyclosporine, some anticonvulsants, protease inhibitors, some nonnucleoside reverse transcriptase inhibitors, contraceptives, and a host of others (see Chapters 4 and 66 ). Co-administration of rifampin results in significantly lower serum levels of these drugs.
ETHAMBUTOL
Ethambutol is a synthetic, water-soluble, heat-stable compound, the dextro-isomer of the structure shown below, dispensed as the dihydrochloride salt.
Ethambutol
H C NH (CH2)2
CH2OH
C2H5
C2H5
C HNH
CH2OH
Mechanism of Action & Clinical Uses Susceptible strains of Mycobacterium tuberculosis and other myco-bacteria are inhibited in vitro by ethambutol, 1–5 mcg/mL. Ethambutol inhibits mycobacterial arabinosyl transferases, which
are encoded by the embCAB operon. Arabinosyl transferases are involved in the polymerization reaction of arabinoglycan, an essential component of the mycobacterial cell wall. Resistance to ethambutol is due to mutations resulting in overexpression of emb gene products or within the embB structural gene.
Ethambutol is well absorbed from the gut. After ingestion of 25 mg/kg, a blood level peak of 2–5 mcg/mL is reached in 2–4 hours. About 20% of the drug is excreted in feces and 50% in urine in unchanged form. Ethambutol accumulates in renal fail-ure, and the dose should be reduced by half if creatinine clearance is less than 10 mL/min. Ethambutol crosses the blood-brain bar-rier only when the meninges are inflamed. Concentrations in cerebrospinal fluid are highly variable, ranging from 4% to 64% of serum levels in the setting of meningeal inflammation.
As with all antituberculous drugs, resistance to ethambutol emerges rapidly when the drug is used alone. Therefore, ethambutol is always given in combination with other antituberculous drugs.
Ethambutol hydrochloride, 15–25 mg/kg, is usually given as a single daily dose in combination with isoniazid or rifampin. The higher dose is recommended for treatment of tuberculous menin-gitis. The dose of ethambutol is 50 mg/kg when a twice-weekly dosing schedule is used.
Adverse Reactions Hypersensitivity to ethambutol is rare. The most common serious adverse event is retrobulbar neuritis, resulting in loss of visual acuity and red-green color blindness. This dose-related adverse effect is more likely to occur at dosages of 25 mg/kg/d continued for several months. At 15 mg/kg/d or less, visual disturbances are very rare. Periodic visual acuity testing is desirable if the 25 mg/kg/d dosage is used. Ethambutol is relatively contraindicated in chil-dren too young to permit assessment of visual acuity and red-green color discrimination.
PYRAZINAMIDE
Pyrazinamide (PZA) is a relative of nicotinamide. It is stable and slightly soluble in water. It is inactive at neutral pH, but at pH 5.5 it inhibits tubercle bacilli at concentrations of approximately 20 mcg/mL. The drug is taken up by macrophages and exerts its activity against mycobacteria residing within the acidic environ-ment of lysosomes.
Pyrazinamide (PZA)
N
C
O
NH2N
Mechanism of Action & Clinical Uses Pyrazinamide is converted to pyrazinoic acid—the active form of the drug—by mycobacterial pyrazinamidase, which is encoded by
047-Katzung_Ch047_p839-848.indd 842 9/21/11 12:09:39 PM

CHAPTER 47 Antimycobacterial Drugs 843
pncA . The specific drug target is unknown, but pyrazinoic acid disrupts mycobacterial cell membrane metabolism and transport functions. Resistance may be due to impaired uptake of pyrazin-amide or mutations in pncA that impair conversion of pyrazin-amide to its active form.
Serum concentrations of 30–50 mcg/mL at 1–2 hours after oral administration are achieved with dosages of 25 mg/kg/d. Pyrazinamide is well absorbed from the gastrointestinal tract and widely distributed in body tissues, including inflamed meninges. The half-life is 8–11 hours. The parent compound is metabolized by the liver, but metabolites are renally cleared; therefore, pyra-zinamide should be administered at 25–35 mg/kg three times weekly (not daily) in hemodialysis patients and those in whom the creatinine clearance is less than 30 mL/min. In patients with nor-mal renal function, a dose of 40–50 mg/kg is used for thrice-weekly or twice-weekly treatment regimens.
Pyrazinamide is an important front-line drug used in conjunc-tion with isoniazid and rifampin in short-course (ie, 6-month) regimens as a “sterilizing” agent active against residual intracellular organisms that may cause relapse. Tubercle bacilli develop resis-tance to pyrazinamide fairly readily, but there is no cross-resistance with isoniazid or other antimycobacterial drugs.
Adverse Reactions Major adverse effects of pyrazinamide include hepatotoxicity (in 1–5% of patients), nausea, vomiting, drug fever, and hyperurice-mia. The latter occurs uniformly and is not a reason to halt ther-apy. Hyperuricemia may provoke acute gouty arthritis.
STREPTOMYCIN
The mechanism of action and other pharmacologic features of streptomycin are discussed in Chapter 45 . The typical adult dose is 1 g/d (15 mg/kg/d). If the creatinine clearance is less than 30 mL/min or the patient is on hemodialysis, the dose is 15 mg/kg two or three times per week. Most tubercle bacilli are inhibited by streptomycin, 1–10 mcg/mL, in vitro. Nontuberculosis species of mycobacteria other than Mycobacterium avium complex (MAC) and Mycobacterium kansasii are resistant. All large populations of tubercle bacilli contain some streptomycin-resistant mutants. On average, 1 in 10 8 tubercle bacilli can be expected to be resistant to streptomycin at levels of 10–100 mcg/mL. Resistance is due to a point mutation in either the rpsL gene encoding the S12 ribo-somal protein gene or the rrs gene encoding 16S ribosomal rRNA, which alters the ribosomal binding site.
Streptomycin penetrates into cells poorly and is active mainly against extracellular tubercle bacilli. Streptomycin crosses the blood-brain barrier and achieves therapeutic concentrations with inflamed meninges.
Clinical Use in Tuberculosis Streptomycin sulfate is used when an injectable drug is needed or desirable and in the treatment of infections resistant to other drugs. The usual dosage is 15 mg/kg/d intramuscularly or intravenously
daily for adults (20–40 mg/kg/d, not to exceed 1–1.5 g for chil-dren) for several weeks, followed by 1–1.5 g two or three times weekly for several months. Serum concentrations of approximately 40 mcg/mL are achieved 30–60 minutes after intramuscular injec-tion of a 15 mg/kg dose. Other drugs are always given in combina-tion to prevent emergence of resistance.
Adverse Reactions Streptomycin is ototoxic and nephrotoxic. Vertigo and hearing loss are the most common adverse effects and may be permanent. Toxicity is dose-related, and the risk is increased in the elderly. As with all aminoglycosides, the dose must be adjusted according to renal function (see Chapter 45 ). Toxicity can be reduced by limit-ing therapy to no more than 6 months whenever possible.
SECOND-LINE DRUGS FOR TUBERCULOSIS
The alternative drugs listed below are usually considered only (1) in case of resistance to first-line agents; (2) in case of failure of clinical response to conventional therapy; and (3) in case of serious treatment-limiting adverse drug reactions. Expert guidance to deal with the toxic effects of these second-line drugs is desirable. For many drugs listed in the following text, the dosage, emergence of resistance, and long-term toxicity have not been fully established.
Ethionamide Ethionamide is chemically related to isoniazid and similarly blocks the synthesis of mycolic acids. It is poorly water soluble and avail-able only in oral form. It is metabolized by the liver.
C
N
Ethionamide
NH2
H5C2
S
Most tubercle bacilli are inhibited in vitro by ethionamide, 2.5 mcg/mL or less. Some other species of mycobacteria also are inhibited by ethionamide, 10 mcg/mL. Serum concentrations in plasma and tissues of approximately 20 mcg/mL are achieved by a dosage of 1 g/d. Cerebrospinal fluid concentrations are equal to those in serum.
Ethionamide is administered at an initial dose of 250 mg once daily, which is increased in 250-mg increments to the recom-mended dosage of 1 g/d (or 15 mg/kg/d), if possible. The 1 g/d dosage, though theoretically desirable, is poorly tolerated because of the intense gastric irritation and neurologic symptoms that commonly occur, and one often must settle for a total daily dose of 500–750 mg. Ethionamide is also hepatotoxic. Neurologic symptoms may be alleviated by pyridoxine.
047-Katzung_Ch047_p839-848.indd 843 9/21/11 12:09:39 PM

844 SECTION VIII Chemotherapeutic Drugs
Resistance to ethionamide as a single agent develops rapidly in vitro and in vivo. There can be low-level cross-resistance between isoniazid and ethionamide.
Capreomycin Capreomycin is a peptide protein synthesis inhibitor antibiotic obtained from Streptomyces capreolus . Daily injection of 1 g intra-muscularly results in blood levels of 10 mcg/mL or more. Such concentrations in vitro are inhibitory for many mycobacteria, including multidrug-resistant strains of M tuberculosis .
Capreomycin (15 mg/kg/d) is an important injectable agent for treatment of drug-resistant tuberculosis. Strains of M tuberculosis that are resistant to streptomycin or amikacin usually are suscep-tible to capreomycin. Resistance to capreomycin, when it occurs, may be due to an rrs mutation.
Capreomycin is nephrotoxic and ototoxic. Tinnitus, deafness, and vestibular disturbances occur. The injection causes significant local pain, and sterile abscesses may occur.
Dosing of capreomycin is the same as that of streptomycin. Toxicity is reduced if 1 g is given two or three times weekly after an initial response has been achieved with a daily dosing schedule.
Cycloserine Cycloserine is an inhibitor of cell wall synthesis and is discussed in Chapter 43 . Concentrations of 15–20 mcg/mL inhibit many strains of M tuberculosis . The dosage of cycloserine in tuberculosis is 0.5–1 g/d in two divided oral doses. Cycloserine is cleared renally, and the dose should be reduced by half if creatinine clear-ance is less than 50 mL/min.
The most serious toxic effects are peripheral neuropathy and central nervous system dysfunction, including depression and psy-chotic reactions. Pyridoxine, 150 mg/d, should be given with cycloserine because this ameliorates neurologic toxicity. Adverse effects, which are most common during the first 2 weeks of therapy, occur in 25% or more of patients, especially at higher doses. Adverse effects can be minimized by monitoring peak serum concentrations. The peak concentration is reached 2–4 hours after dosing. The recommended range of peak concentrations is 20–40 mcg/mL.
Aminosalicylic Acid (PAS) Aminosalicylic acid is a folate synthesis antagonist that is active almost exclusively against M tuberculosis . It is structurally similar to p -amino-benzoic acid (PABA) and to the sulfonamides (see Chapter 46 ).
Aminosalicylic acid (PAS)
OH
NH2
COOH
Tubercle bacilli are usually inhibited in vitro by aminosalicylic acid, 1–5 mcg/mL. Aminosalicylic acid is readily absorbed from
the gastrointestinal tract. Serum levels are 50 mcg/mL or more after a 4-g oral dose. The dosage is 8–12 g/d orally for adults and 300 mg/kg/d for children. The drug is widely distributed in tissues and body fluids except the cerebrospinal fluid. Aminosalicylic acid is rapidly excreted in the urine, in part as active aminosalicylic acid and in part as the acetylated compound and other metabolic prod-ucts. Very high concentrations of aminosalicylic acid are reached in the urine, which can result in crystalluria.
Aminosalicylic acid is used infrequently because other oral drugs are better tolerated. Gastrointestinal symptoms are common and may be diminished by giving the drug with meals and with antacids. Peptic ulceration and hemorrhage may occur. Hypersensitivity reactions manifested by fever, joint pains, skin rashes, hepatosple-nomegaly, hepatitis, adenopathy, and granulocytopenia often occur after 3–8 weeks of aminosalicylic acid therapy, making it necessary to stop aminosalicylic acid administration temporarily or permanently.
Kanamycin & Amikacin The aminoglycoside antibiotics are discussed in Chapter 45 . Kanamycin has been used for treatment of tuberculosis caused by streptomycin-resistant strains, but the availability of less toxic alter-natives (eg, capreomycin and amikacin) has rendered it obsolete.
The role of amikacin in treatment of tuberculosis has increased with the increasing incidence and prevalence of multidrug-resistant tuberculosis. Prevalence of amikacin-resis-tant strains is low (< 5%), and most multidrug-resistant strains remain amikacin-susceptible. M tuberculosis is inhibited at con-centrations of 1 mcg/mL or less. Amikacin is also active against atypical mycobacteria. There is no cross-resistance between strep-tomycin and amikacin, but kanamycin resistance often indicates resistance to amikacin as well. Serum concentrations of 30–50 mcg/mL are achieved 30–60 minutes after a 15 mg/kg intravenous infusion. Amikacin is indicated for treatment of tuberculosis sus-pected or known to be caused by streptomycin-resistant or multi-drug-resistant strains. Amikacin must be used in combination with at least one and preferably two or three other drugs to which the isolate is susceptible for treatment of drug-resistant cases. The recommended dosages are the same as those for streptomycin.
Fluoroquinolones In addition to their activity against many gram-positive and gram-negative bacteria (discussed in Chapter 46 ), ciprofloxacin, levofloxa-cin, gatifloxacin, and moxifloxacin inhibit strains of M tuberculosis at concentrations less than 2 mcg/mL. They are also active against atypical mycobacteria. Moxifloxacin is the most active against M tuberculosis by weight in vitro. Levofloxacin tends to be slightly more active than ciprofloxacin against M tuberculosis , whereas ciprofloxa-cin is slightly more active against atypical mycobacteria.
Fluoroquinolones are an important addition to the drugs avail-able for tuberculosis, especially for strains that are resistant to first-line agents. Resistance, which may result from any one of several single point mutations in the gyrase A subunit, develops rapidly if a fluoroquinolone is used as a single agent; thus, the drug must be used in combination with two or more other active
047-Katzung_Ch047_p839-848.indd 844 9/21/11 12:09:39 PM

CHAPTER 47 Antimycobacterial Drugs 845
agents. The standard dosage of ciprofloxacin is 750 mg orally twice a day. The dosage of levofloxacin is 500–750 mg once a day. The dosage of moxifloxacin is 400 mg once a day.
Linezolid Linezolid (discussed in Chapter 44 ) inhibits strains of M tuberculosis in vitro at concentrations of 4–8 mcg/mL. It achieves good intra-cellular concentrations, and it is active in murine models of tuberculosis. Linezolid has been used in combination with other second- and third-line drugs to treat patients with tuberculosis caused by multidrug-resistant strains. Conversion of sputum cultures to negative was associated with linezolid use in these cases, and some may have been cured. Significant and at times treatment-limiting adverse effects, including bone marrow sup-pression and irreversible peripheral and optic neuropathy, have been reported with the prolonged courses of therapy that are necessary for treatment of tuberculosis. A 600-mg (adult) dose administered once a day (half of that used for treatment of other bacterial infections) seems to be sufficient and may limit the occurrence of these adverse effects. Although linezolid may even-tually prove to be an important new agent for treatment of tuber-culosis, at this point it should be considered a drug of last resort for infection caused by multidrug-resistant strains that also are resistant to several other first- and second-line agents.
Rifabutin Rifabutin is derived from rifamycin and is related to rifampin. It has significant activity against M tuberculosis , MAC, and Mycobacterium fortuitum (see below). Its activity is similar to that of rifampin, and cross-resistance with rifampin is virtually com-plete. Some rifampin-resistant strains may appear susceptible to rifabutin in vitro, but a clinical response is unlikely because the molecular basis of resistance, rpoB mutation, is the same. Rifabutin is both substrate and inducer of cytochrome P450 enzymes. Because it is a less potent inducer, rifabutin is indicated in place of rifampin for treatment of tuberculosis in patients with HIV infec-tion who are receiving antiretroviral therapy with a protease inhibitor or with a nonnucleoside reverse transcriptase inhibitor (eg, efavirenz), drugs that also are cytochrome P450 substrates.
The typical dosage of rifabutin is 300 mg/d unless the patient is receiving a protease inhibitor, in which case the dosage should be reduced to 150 mg/d. If efavirenz (also a cytochrome P450 inducer) is used, the recommended dosage of rifabutin is 450 mg/d.
Rifabutin is effective in prevention and treatment of dissemi-nated atypical mycobacterial infection in AIDS patients with CD4 counts below 50/μL. It is also effective for preventive therapy of tuberculosis, either alone in a 3–4 month regimen or with pyrazi-namide in a 2-month regimen.
Rifapentine Rifapentine is an analog of rifampin. It is active against both M tuberculosis and MAC. As with all rifamycins, it is a bacterial RNA polymerase inhibitor, and cross-resistance between rifampin and rifapentine is complete. Like rifampin, rifapentine is a potent inducer of cytochrome P450 enzymes, and it has the same drug
interaction profile. Toxicity is similar to that of rifampin. Rifapentine and its microbiologically active metabolite, 25-desacetylrifapentine, have an elimination half-life of 13 hours. Rifapentine, 600 mg (10 mg/kg) once weekly, is indicated for treatment of tuberculosis caused by rifampin-susceptible strains during the continuation phase only (ie, after the first 2 months of therapy and ideally after conversion of sputum cultures to negative). Rifapentine should not be used to treat patients with HIV infec-tion because of an unacceptably high relapse rate with rifampin-resistant organisms.
■ RIFAXIMIN
Rifaximin is a rifampin derivative that is not absorbed from the gastrointestinal tract. It is approved for oral administration in the prevention of hepatic encephalopathy and for the treatment of travelers’ diarrhea caused by noninvasive strains of Escherichia coli in patients 12 years of age and older. It has also been used off label for irritable bowel syndrome and for Clostridium difficile colitis not responsive to other drugs.
■ DRUGS ACTIVE AGAINST ATYPICAL MYCOBACTERIA
About 10% of mycobacterial infections seen in clinical practice in the United States are caused by nontuberculous or “atypical” mycobacteria. These organisms have distinctive laboratory charac-teristics, are present in the environment, and are generally not communicable from person to person. As a rule, these mycobacte-rial species are less susceptible than M tuberculosis to antitubercu-lous drugs. On the other hand, agents such as erythromycin, sulfonamides, or tetracycline, which are not active against M tuberculosis , may be effective for infections caused by atypical mycobacteria. Emergence of resistance during therapy is also a problem with these mycobacterial species, and active infection should be treated with combinations of drugs. M kansasii is sus-ceptible to rifampin and ethambutol, partially resistant to isoni-azid, and completely resistant to pyrazinamide. A three-drug combination of isoniazid, rifampin, and ethambutol is the con-ventional treatment for M kansasii infection. A few representative pathogens, with the clinical presentation and the drugs to which they are often susceptible, are given in Table 47–3 .
M avium complex (MAC), which includes both M avium and M intracellulare , is an important and common cause of dissemi-nated disease in late stages of AIDS (CD4 counts < 50/μL). MAC is much less susceptible than M tuberculosis to most antitubercu-lous drugs. Combinations of agents are required to suppress the infection. Azithromycin, 500 mg once daily, or clarithromycin, 500 mg twice daily, plus ethambutol, 15–25 mg/kg/d, is an effective and well-tolerated regimen for treatment of disseminated disease. Some authorities recommend use of a third agent, such as ciprofloxacin, 750 mg twice daily, or rifabutin, 300 mg once daily. Other agents that may be useful are listed in Table 47–3 . Azithromycin and clarithro-mycin are the drugs of choice for reducing the incidence of MAC
047-Katzung_Ch047_p839-848.indd 845 9/21/11 12:09:39 PM

846 SECTION VIII Chemotherapeutic Drugs
bacteremia in AIDS patients with CD4 cell counts less than 100/μL. Rifabutin in a single daily dose of 300 mg has been shown to reduce the incidence of MAC bacteremia but is less effective than macrolides.
■ DRUGS USED IN LEPROSY
Mycobacterium leprae has never been grown in vitro, but animal models, such as growth in injected mouse footpads, have permit-ted laboratory evaluation of drugs. Only those drugs with the widest clinical use are presented here. Because of increasing reports of dapsone resistance, treatment of leprosy with combinations of the drugs listed below is recommended.
DAPSONE & OTHER SULFONES
Several drugs closely related to the sulfonamides have been used effectively in the long-term treatment of leprosy. The most widely used is dapsone (diaminodiphenylsulfone). Like the sulfonamides, it
inhibits folate synthesis. Resistance can emerge in large populations of M leprae , eg, in lepromatous leprosy, if very low doses are given. Therefore, the combination of dapsone, rifampin, and clofazimine is recommended for initial therapy. Dapsone may also be used to pre-vent and treat Pneumocystis jiroveci pneumonia in AIDS patients.
O
O
S
Dapsone
NH2NH2
Sulfones are well absorbed from the gut and widely distributed throughout body fluids and tissues. Dapsone’s half-life is 1–2 days, and drug tends to be retained in skin, muscle, liver, and kidney. Skin heavily infected with M leprae may contain several times more drug than normal skin. Sulfones are excreted into bile and reab-sorbed in the intestine. Excretion into urine is variable, and most excreted drug is acetylated. In renal failure, the dose may have to be adjusted. The usual adult dosage in leprosy is 100 mg daily. For children, the dose is proportionately less, depending on weight.
Dapsone is usually well tolerated. Many patients develop some hemolysis, particularly if they have glucose-6-phosphate dehydro-genase deficiency. Methemoglobinemia is common, but usually is not a problem clinically. Gastrointestinal intolerance, fever, pruri-tus, and various rashes occur. During dapsone therapy of leproma-tous leprosy, erythema nodosum leprosum often develops. It is sometimes difficult to distinguish reactions to dapsone from manifestations of the underlying illness. Erythema nodosum lep-rosum may be suppressed by corticosteroids or by thalidomide .
RIFAMPIN
Rifampin (see earlier discussion) in a dosage of 600 mg daily is highly effective in lepromatous leprosy. Because of the probable risk of emergence of rifampin-resistant M leprae , the drug is given in com-bination with dapsone or another antileprosy drug. A single monthly dose of 600 mg may be beneficial in combination therapy.
CLOFAZIMINE
Clofazimine is a phenazine dye that can be used as an alternative to dapsone. Its mechanism of action is unknown but may involve DNA binding.
Absorption of clofazimine from the gut is variable, and a major portion of the drug is excreted in feces. Clofazimine is stored widely in reticuloendothelial tissues and skin, and its crystals can be seen inside phagocytic reticuloendothelial cells. It is slowly released from these deposits, so that the serum half-life may be 2 months.
Clofazimine is given for sulfone-resistant leprosy or when patients are intolerant to sulfones. A common dosage is 100 mg/d orally. The most prominent untoward effect is skin discoloration ranging from red-brown to nearly black. Gastrointestinal intoler-ance occurs occasionally.
TABLE 47–3 Clinical features and treatment options for infections with atypical mycobacteria.
Species Clinical Features Treatment Options
M kansasii Resembles tuberculosis
Ciprofloxacin, clarithromycin, ethambutol, isoniazid, rifampin, trimethoprim-sulfamethoxazole
M marinum Granulomatous cutaneous disease
Amikacin, clarithromycin, ethambutol, doxycycline, minocycline, rifampin, trimethoprim-sulfamethoxazole
M scrofulaceum Cervical adenitis in children
Amikacin, erythromycin (or other macrolide), rifampin, streptomycin (Surgical excision is often curative and the treatment of choice.)
M avium complex Pulmonary disease in patients with chronic lung dis-ease; disseminated infection in AIDS
Amikacin, azithromycin, clarithromycin, ciprofloxacin, ethambutol, rifabutin
M chelonae Abscess, sinus tract, ulcer; bone, joint, tendon infection
Amikacin, doxycycline, imipenem, macrolides, tobramycin
M fortuitum Abscess, sinus tract, ulcer; bone, joint, tendon infec-tion
Amikacin, cefoxitin,ciprofloxacin, doxycycline, ofloxacin, trimethoprim-sulfamethoxazole
M ulcerans Skin ulcers Isoniazid, streptomycin, rifampin, minocycline (Surgical excision may be effective.)
047-Katzung_Ch047_p839-848.indd 846 9/21/11 12:09:39 PM

CHAPTER 47 Antimycobacterial Drugs 847
SUMMARY First-Line Antimycobacterial Drugs
SubclassMechanism of Action Effects Clinical Applications
Pharmacokinetics, Toxicities, Interactions
ISONIAZID Inhibits synthesis of mycolic acids, an essen-tial component of mycobacterial cell walls
Bactericidal activity against susceptible strains ofM tuberculosis
First-line agent for tuberculosis • treatment of latent infection • less active against other mycobacteria
Oral, IV • hepatic clearance (half-life 1 h) • reduces levels of phenytoin • Toxicity: Hepatotoxic, peripheral neurop-athy (give pyridoxine to prevent)
RIFAMYCINS • Rifampin Inhibits DNA-dependent
RNA polymerase, thereby blocking production of RNA
Bactericidal activity against susceptible bacteria and mycobacteria • resistance rapidly emerges when used as a single drug in the treat-ment of active infection
First-line agent for tuberculosis • atypical mycobacterial infections • eradication of meningococcal coloni-zation, staphylococcal infections
Oral, IV • hepatic clearance (half-life 3.5 h) • potent cytochrome P450 inducer • turns body fluids orange color • Toxicity: Rash, nephritis, thrombocytopenia, cholestasis, flu-like syndrome with intermittent dosing
• Rifabutin: Oral; similar to rifampin but less cytochrome P450 induction and fewer drug interactions
• Rifapentine: Oral; long-acting analog of rifampin that may be given once weekly in the continuation phase of tuberculosis treatment
PYRAZINAMIDE Not fully understood • pyrazinamide is converted to the active pyrazinoic acid under acidic conditions in macrophage lysosomes
Bacteriostatic activity against susceptible strains of M tuberculosis • may be bactericidal against actively dividing organisms
“Sterilizing” agent used during first 2 months of therapy • allows total duration of therapy to be shortened to 6 months
Oral • hepatic clearance (half-life 9 h), but metabolites are renally cleared so use doses 3 × weekly if creatinine clearance < 30 mL/min • Toxicity: Hepatotoxic, hyperuricemia
ETHAMBUTOL Inhibits mycobacterial arabinosyl transferases, which are involved in the polymerization reac-tion of arabinoglycan, an essential component of the mycobacterial cell wall
Bacteriostatic activity against susceptible mycobacteria
Given in four-drug initial combination therapy for tuberculosis until drug sensitivities are known • also used for atypical mycobacterial infections
Oral • mixed clearance (half-life 4 h) • dose must be reduced in renal failure • Toxicity: Retrobulbar neuritis
STREPTOMYCIN Prevents bacterial protein synthesis by binding to the S12 ribosomal subunit (see also Chapter 45)
Bactericidal activity against susceptible mycobacteria
Used in tuberculosis when an injectable drug is needed or desirable and in treatment of drug-resistant strains
IM, IV • renal clearance (half-life 2.5 h) • administered daily initially, then 2 × week • Toxicity: Nephrotoxic, ototoxic
047-Katzung_Ch047_p839-848.indd 847 9/21/11 12:09:40 PM

848 SECTION VIII Chemotherapeutic Drugs
DRUGS USED IN TUBERCULOSISAminosalicylate sodium (Paser)
Oral: 4 g delayed-release granulesCapreomycin (Capastat Sulfate)
Parenteral: 1 g powder to reconstitute for injectionCycloserine (Seromycin Pulvules)
Oral: 250 mg capsulesEthambutol (Myambutol)
Oral: 100, 400 mg tabletsEthionamide (Trecator-SC)
Oral: 250 mg tabletsIsoniazid (generic)
Oral: 100, 300 mg tablets; syrup, 50 mg/5 mLParenteral: 100 mg/mL for injection
Pyrazinamide (generic)Oral: 500 mg tablets
Rifabutin (Mycobutin)Oral: 150 mg capsules
Rifampin (generic, Rifadin, Rimactane)Oral: 150, 300 mg capsulesParenteral: 600 mg powder for IV injection
Rifapentine (Priftin)Oral: 150 mg tablets
Rifaximin (Xifaxan)Oral: 200, 550 mg tablets
Streptomycin (generic)Parenteral: 1 g lyophilized for IM injection
DRUGS USED IN LEPROSYClofazimine (Lamprene)
Oral: 50 mg capsulesDapsone (generic)
Oral: 25, 100 mg tablets
P R E P A R A T I O N S A V A I L A B L E 1
DRUGS USED IN TUBERCULOSISAminosalicylate sodium (Paser)
Oral: 4 g delayed-release granulesCapreomycin (Capastat Sulfate)
Parenteral: 1 g powder to reconstitute for injectionCycloserine (Seromycin Pulvules)
Oral: 250 mg capsulesEthambutol (Myambutol)
Oral: 100, 400 mg tabletsEthionamide (Trecator-SC)
Oral: 250 mg tabletsIsoniazid (generic)
Oral: 100, 300 mg tablets; syrup, 50 mg/5 mLParenteral: 100 mg/mL for injection
Pyrazinamide (generic)Oral: 500 mg tablets
Rifabutin (Mycobutin)Oral: 150 mg capsules
Oral: 150, 300 mg capsulesParenteral: 600 mg powder for IV injection
Rifapentine (Priftin)Oral: 150 mg tablets
Rifaximin (Xifaxan)Oral: 200, 550 mg tablets
Streptomycin (generic)Parenteral: 1 g lyophilized for IM injection
DRUGS USED IN LEPROSYClofazimine (Lamprene)
Oral: 50 mg capsulesDapsone (generic)
Oral: 25, 100 mg tablets
Rifampin (generic, Rifadin, Rimactane)
REFERENCES Diagnosis and treatment of disease caused by nontuberculous mycobacteria. Am J
Respir Crit Care Med 1997;156 (2 Part 2):S1. Gillespie SH et al: Early bactericidal activity of a moxifloxacin and isoniazid com-
bination in smear-positive pulmonary tuberculosis. J Antimicrob Chemother 2005;56:1169.
Hugonnet J-E et al: Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis . Science 2009;323:1215.
Jasmer RM, Nahid P, Hopewell PC: Latent tuberculosis infection. N Engl J Med 2002;347:1860.
Kinzig-Schippers M et al: Should we use N-acetyltransferase type 2 genotyping to personalize isoniazid doses? Antimicrob Agents Chemother 2005;49:1733.
Mitnick CD et al: Comprehensive treatment of extensively drug-resistant tubercu-losis. N Engl J Med 2008;359:563.
Sulochana S, Rahman F, Paramasivan CN: In vitro activity of fluoroquinolones against Mycobacterium tuberculosis . J Chemother 2005;17:169.
Targeted tuberculin testing and treatment of latent tuberculosis infection. Am J Respir Crit Care Med 2000;161(4 Part 2):S221.
Update: Adverse event data and revised American Thoracic Society/CDC recom-mendations against the use of rifampin and pyrazinamide for treatment of latent tuberculosis infection—United States, 2003. MMWR Morb Mortal Wkly Rep 2003;52:735.
von der Lippe B, Sandven P, Brubakk O: Efficacy and safety of linezolid in multi-drug resistant tuberculosis (MDR-TB)—a report of ten cases. J Infect 2006;52:92.
Zhang Y, Yew WW: Mechanisms of drug resistance in Mycobacterium tuberculosis.Int J Tuberc Lung Dis 2009;13:1320.
C A S E S T U D Y A N S W E R
The patient should be started on four-drug therapy with rifampin, isoniazid, pyrazinamide, and ethambutol. If a protease-inhibitor-based antiretroviral regimen is used to treat his HIV, rifabutin should replace rifampin because of
the serious drug-drug interaction between rifampin and protease inhibitors. The patient is at increased risk of devel-oping hepatotoxicity from both isoniazid and pyrazinamide given his history of chronic alcohol dependence.
1Drugs used against atypical mycobacteria are listed in Chapters 43–46.
047-Katzung_Ch047_p839-848.indd 848 9/21/11 12:09:40 PM

849
C H A P T E R
Human fungal infections have increased dramatically in incidence and severity in recent years, owing mainly to advances in surgery, cancer treatment, treatment of patients with solid organ and bone marrow transplantation, the HIV epidemic, and increasing use of broad-spectrum antimicrobial therapy in critically ill patients. These changes have resulted in increased numbers of patients at risk for fungal infections.
For many years, amphotericin B was the only efficacious anti-fungal drug available for systemic use. While highly effective in many serious infections, it is also quite toxic. In the last several decades, pharmacotherapy of fungal disease has been revolution-ized by the introduction of the relatively nontoxic azole drugs (both oral and parenteral formulations) and the echinocandins (only available for parenteral administration). The new agents in these classes offer more targeted, less toxic therapy than older agents such as amphotericin B for patients with serious systemic fungal infections. Combination therapy is being reconsidered, and new formulations of old agents are becoming available. Unfortunately, the appearance of azole-resistant organisms, as well as the rise in the number of patients at risk for mycotic infections, has created new challenges.
The antifungal drugs presently available fall into the following categories: systemic drugs (oral or parenteral) for systemic infec-tions, oral systemic drugs for mucocutaneous infections, and topical drugs for mucocutaneous infections.
■ SYSTEMIC ANTIFUNGAL DRUGS FOR SYSTEMIC INFECTIONS
AMPHOTERICIN B Amphotericin A and B are antifungal antibiotics produced by Streptomyces nodosus. Amphotericin A is not in clinical use.
Chemistry & Pharmacokinetics Amphotericin B is an amphoteric polyene macrolide (polyene = containing many double bonds; macrolide = containing a large lactone ring of 12 or more atoms). It is nearly insoluble in water and is therefore prepared as a colloidal suspension of amphotericin B and sodium desoxycholate for intravenous injection. Several formulations have been developed in which amphotericin B is
48Antifungal Agents Don Sheppard, MD, & Harry W. Lampiris, MD
C A S E S T U D Y
A 55-year-old man presents to the emergency department with a 2-week history of an expanding ulcer on his left lower leg. He has a history of chronic neutropenia and transfusion-dependent anemia secondary to myelodysplastic syndrome requiring chronic therapy with deferoxamine for hepatic iron overload. He first noticed a red bump on his leg while fishing at his cabin in the woods and thought it was a bug bite. It rapidly enlarged, first as a red swollen area, and then began to ulcerate. He was given dicloxacillin orally, but with no
improvement. In the emergency department he is febrile to 39°C (102.2°F), and looks unwell. On his left leg he has a 6 by 12 cm black ulcer with surrounding swelling and erythema that is quite tender. His complete blood count demonstrates an absolute neutrophil count of 300 and a total white blood cell count of 1000. An immediate operative deb-ridement yields pathologic specimens demonstrating broad club-like nonseptate hyphae and extensive tissue necrosis. What initial medical therapy would be most appropriate?
048-Katzung_Ch048_p849-860.indd 849 9/21/11 12:09:56 PM

850 SECTION VIII Chemotherapeutic Drugs
TABLE 48–1 Properties of conventional amphotericin B and some lipid formulations.1
DrugPhysical Form
Dosing (mg/kg/d) Cmax Clearance Nephrotoxicity
Infusional Toxicity Daily Cost ($)
Conventional formulation
Fungizone Micelles 1 … … … … 24
Lipid formulations
AmBisome Spheres 3–5 ↑ ↓ ↓ ↓ 1300
Amphotec Disks 5 ↓ ↑ ↓ ↑(?) 660
Abelcet Ribbons 5 ↓ ↑ ↓ ↓(?) 570
1Changes in Cmax (peak plasma concentration), clearance, nephrotoxicity, and infusional toxicity are relative to conventional amphotericin B.
Liposomal Amphotericin B
Therapy with amphotericin B is often limited by toxicity, espe-cially drug-induced renal impairment. This has led to the devel-opment of lipid drug formulations on the assumption that lipid-packaged drug binds to the mammalian membrane less readily, permitting the use of effective doses of the drug with lower toxicity. Liposomal amphotericin preparations package the active drug in lipid delivery vehicles, in contrast to the colloidal suspensions, which were previously the only available forms. Amphotericin binds to the lipids in these vehicles with an affinity between that for fungal ergosterol and that for human choles-terol. The lipid vehicle then serves as an amphotericin reservoir, reducing nonspecific binding to human cell membranes. This preferential binding allows for a reduction of toxicity without sacrificing efficacy and permits use of larger doses. Furthermore,
some fungi contain lipases that may liberate free amphotericin B directly at the site of infection.
Three such formulations are now available and have differing pharmacologic properties as summarized in Table 48–1 . Although clinical trials have demonstrated different renal and infusion-related toxicities for these preparations compared with regular amphotericin B, there are no trials comparing the different for-mulations with each other. Limited studies have suggested at best a moderate improvement in the clinical efficacy of the lipid formulations compared with conventional amphotericin B. Because the lipid preparations are much more expensive, their use is usually restricted to patients intolerant to, or not respond-ing to, conventional amphotericin treatment.
packaged in a lipid-associated delivery system ( Table 48–1 and Box: Liposomal Amphotericin B).
Amphotericin B
NH2
OO
OO
CH3
CH3
CH3 COOH
O
CH3
OHOH
OH
OH
OH
OH
OH
OHOHHO
371
Amphotericin B is poorly absorbed from the gastrointestinal tract. Oral amphotericin B is thus effective only on fungi within the lumen of the tract and cannot be used for treatment of systemic disease. The intravenous injection of 0.6 mg/kg/d of amphotericin B results in average blood levels of 0.3–1 mcg/mL; the drug is more than 90% bound by serum proteins. Although it is mostly
metabolized, some amphotericin B is excreted slowly in the urine over a period of several days. The serum half-life is approximately 15 days. Hepatic impairment, renal impairment, and dialysis have little impact on drug concentrations, and therefore no dose adjust-ment is required. The drug is widely distributed in most tissues, but only 2–3% of the blood level is reached in cerebrospinal fluid, thus occasionally necessitating intrathecal therapy for certain types of fungal meningitis.
Mechanisms of Action & Resistance Amphotericin B is selective in its fungicidal effect because it exploits the difference in lipid composition of fungal and mammalian cell membranes. Ergosterol, a cell membrane sterol, is found in the cell membrane of fungi, whereas the predominant sterol of bacteria and human cells is cholesterol. Amphotericin B binds to ergosterol and alters the permeability of the cell by forming amphotericin B-associated pores in the cell membrane ( Figure 48–1 ). As sug-gested by its chemistry, amphotericin B combines avidly with
048-Katzung_Ch048_p849-860.indd 850 9/21/11 12:09:56 PM

CHAPTER 48 Antifungal Agents 851
lipids (ergosterol) along the double bond-rich side of its structure and associates with water molecules along the hydroxyl-rich side. This amphipathic characteristic facilitates pore formation by mul-tiple amphotericin molecules, with the lipophilic portions around the outside of the pore and the hydrophilic regions lining the inside. The pore allows the leakage of intracellular ions and mac-romolecules, eventually leading to cell death. Some binding to human membrane sterols does occur, probably accounting for the drug’s prominent toxicity.
Resistance to amphotericin B occurs if ergosterol binding is impaired, either by decreasing the membrane concentration of ergosterol or by modifying the sterol target molecule to reduce its affinity for the drug.
Antifungal Activity & Clinical Uses Amphotericin B remains the antifungal agent with the broadest spectrum of action. It has activity against the clinically significant yeasts, including Candida albicans and Cryptococcus neoformans ; the organisms causing endemic mycoses, including Histoplasma capsulatum , Blastomyces dermatitidis , and Coccidioides immitis ; and the pathogenic molds, such as Aspergillus fumigatus and the agents of mucormycosis. Some fungal organisms such as Candida lusitaniae and Pseudallescheria boydii display intrinsic amphoteri-cin B resistance.
Owing to its broad spectrum of activity and fungicidal action, amphotericin B remains a useful agent for nearly all life-threatening
mycotic infections, although newer, less toxic agents have largely replaced it for most conditions. Amphotericin B is often used as the initial induction regimen to rapidly reduce fungal burden and then replaced by one of the newer azole drugs (described below) for chronic therapy or prevention of relapse. Such induction therapy is especially important for immunosup-pressed patients and those with severe fungal pneumonia, severe cryptococcal meningitis, or disseminated infections with one of the endemic mycoses such as histoplasmosis or coccidioidomy-cosis. Once a clinical response has been elicited, these patients then often continue maintenance therapy with an azole; therapy may be lifelong in patients at high risk for disease relapse. For treatment of systemic fungal disease, amphotericin B is given by slow intravenous infusion at a dosage of 0.5–1 mg/kg/d. It is usually continued to a defined total dose (eg, 1–2 g), rather than a defined time span, as used with other antimicrobial drugs.
Intrathecal therapy for fungal meningitis is poorly tolerated and fraught with difficulties related to maintaining cerebrospinal fluid access. Thus, intrathecal therapy with amphotericin B is being increasingly supplanted by other therapies but remains an option in cases of fungal central nervous system infections that have not responded to other agents.
Local or topical administration of amphotericin B has been used with success. Mycotic corneal ulcers and keratitis can be cured with topical drops as well as by direct subconjunctival injec-tion. Fungal arthritis has been treated with adjunctive local injection
Fungal cell Fungal cell membrane and cell wall
β-glucans
Cell membranebilayer
Squalene
Squalene epoxide Lanosterol
Ergosterol
Terbinafine
Azoles
Amphotericin B,nystatin
Flucytosine
β-glucansynthase
Echinocandins
–
––
–
Proteins
Chitin
DNA, RNAsynthesis
FIGURE 48–1 Targets of antifungal drugs. Except for flucytosine (and possibly griseofulvin, not shown), all currently available antifungals target the fungal cell membrane or cell wall.
048-Katzung_Ch048_p849-860.indd 851 9/21/11 12:09:56 PM

852 SECTION VIII Chemotherapeutic Drugs
directly into the joint. Candiduria responds to bladder irrigation with amphotericin B, and this route has been shown to produce no significant systemic toxicity.
Adverse Effects The toxicity of amphotericin B can be divided into two broad categories: immediate reactions, related to the infusion of the drug, and those occurring more slowly.
A. Infusion-Related Toxicity Infusion-related reactions are nearly universal and consist of fever, chills, muscle spasms, vomiting, headache, and hypoten-sion. They can be ameliorated by slowing the infusion rate or decreasing the daily dose. Premedication with antipyretics, anti-histamines, meperidine, or corticosteroids can be helpful. When starting therapy, many clinicians administer a test dose of 1 mg intravenously to gauge the severity of the reaction. This can serve as a guide to an initial dosing regimen and premedication strategy.
B. Cumulative Toxicity Renal damage is the most significant toxic reaction. Renal impair-ment occurs in nearly all patients treated with clinically significant doses of amphotericin. The degree of azotemia is variable and often stabilizes during therapy, but it can be serious enough to necessitate dialysis. A reversible component is associated with decreased renal perfusion and represents a form of prerenal renal failure. An irreversible component results from renal tubular injury and subsequent dysfunction. The irreversible form of amphotericin nephrotoxicity usually occurs in the setting of pro-longed administration (> 4 g cumulative dose). Renal toxicity commonly manifests as renal tubular acidosis and severe potas-sium and magnesium wasting. There is some evidence that the prerenal component can be attenuated with sodium loading, and it is common practice to administer normal saline infusions with the daily doses of amphotericin B.
Abnormalities of liver function tests are occasionally seen, as is a varying degree of anemia due to reduced erythropoietin produc-tion by damaged renal tubular cells. After intrathecal therapy with amphotericin, seizures and a chemical arachnoiditis may develop, often with serious neurologic sequelae.
FLUCYTOSINE Chemistry & Pharmacokinetics Flucytosine (5-FC) was discovered in 1957 during a search for novel antineoplastic agents. Though devoid of anticancer proper-ties, it became apparent that it was a potent antifungal agent. Flucytosine is a water-soluble pyrimidine analog related to the chemotherapeutic agent 5-fluorouracil (5-FU). Its spectrum of action is much narrower than that of amphotericin B.
FN
N
H
Flucytosine
NH2
O
Flucytosine is currently available in North America only in an oral formulation. The dosage is 100–150 mg/kg/d in patients with normal renal function. It is well absorbed (> 90%), with serum concentrations peaking 1–2 hours after an oral dose. It is poorly protein-bound and penetrates well into all body fluid compart-ments, including the cerebrospinal fluid. It is eliminated by glom-erular filtration with a half-life of 3–4 hours and is removed by hemodialysis. Levels rise rapidly with renal impairment and can lead to toxicity. Toxicity is more likely to occur in AIDS patients and those with renal insufficiency. Peak serum concentrations should be measured periodically in patients with renal insuffi-ciency and maintained between 50 and 100 mcg/mL.
Mechanisms of Action & Resistance Flucytosine is taken up by fungal cells via the enzyme cytosine permease. It is converted intracellularly first to 5-FU and then to 5-fluorodeoxyuridine monophosphate (FdUMP) and fluorouridine triphosphate (FUTP), which inhibit DNA and RNA synthesis, respectively ( Figure 48–1 ). Human cells are unable to convert the parent drug to its active metabolites, resulting in selective toxicity.
Synergy with amphotericin B has been demonstrated in vitro and in vivo. It may be related to enhanced penetration of the flu-cytosine through amphotericin-damaged fungal cell membranes. In vitro synergy with azole drugs has also been seen, although the mechanism is unclear.
Resistance is thought to be mediated through altered metabo-lism of flucytosine, and, though uncommon in primary isolates, it develops rapidly in the course of flucytosine monotherapy.
Clinical Uses & Adverse Effects The spectrum of activity of flucytosine is restricted to C neofor-mans , some Candida sp, and the dematiaceous molds that cause chromoblastomycosis. Flucytosine is not used as a single agent because of its demonstrated synergy with other agents and to avoid the development of secondary resistance. Clinical use at present is confined to combination therapy, either with ampho-tericin B for cryptococcal meningitis or with itraconazole for chromoblastomycosis.
The adverse effects of flucytosine result from metabolism (possibly by intestinal flora) to the toxic antineoplastic compound fluorouracil. Bone marrow toxicity with anemia, leukopenia, and thrombocytopenia are the most common adverse effects, with derangement of liver enzymes occurring less frequently. A form of toxic enterocolitis can occur. There seems to be a narrow therapeutic window, with an increased risk of toxicity at higher drug levels and
048-Katzung_Ch048_p849-860.indd 852 9/21/11 12:09:56 PM

CHAPTER 48 Antifungal Agents 853
resistance developing rapidly at subtherapeutic concentrations. The use of drug concentration measurements may be helpful in reducing the incidence of toxic reactions, especially when flucytosine is com-bined with nephrotoxic agents such as amphotericin B.
AZOLES Chemistry & Pharmacokinetics Azoles are synthetic compounds that can be classified as either imidazoles or triazoles according to the number of nitrogen atoms in the five-membered azole ring, as indicated below. The imidazoles consist of ketoconazole, miconazole, and clotrimazole ( Figure 48–2 ). The latter two drugs are now used only in topical therapy. The triazoles include itraconazole, fluconazole, voricon-azole, and posaconazole.
R
NX X = C, imidazole
X = N, triazole
N
Azole nucleus
The pharmacology of each of the azoles is unique and accounts for some of the variations in clinical use. Table 48–2 summarizes the differences among five of the azoles.
Mechanisms of Action & Resistance The antifungal activity of azole drugs results from the reduction of ergosterol synthesis by inhibition of fungal cytochrome P450 enzymes ( Figure 48–1 ). The selective toxicity of azole drugs results
Clotrimazole
N
N
C
Cl
N
N
H2CHC O CH2 Cl
Cl
Cl
Cl
Miconazole
Itraconazole
N
N
CCl
N
CH2
Cl
O
O CH2 O
N
N N
N CH CH2 CH3
CH3O
N
N
N
H2C Cl
Cl
Ketoconazole
OO
CH2 O N N C CH3
O
F
F
N
N
N
C CH2
OH
CH2 NN
N
Fluconazole
F
F
N
N
N
CH3
NN
FHO
Voriconazole
FIGURE 48–2 Structural formulas of some antifungal azoles.
048-Katzung_Ch048_p849-860.indd 853 9/21/11 12:09:57 PM

854 SECTION VIII Chemotherapeutic Drugs
from their greater affinity for fungal than for human cytochrome P450 enzymes. Imidazoles exhibit a lesser degree of selectivity than the triazoles, accounting for their higher incidence of drug interactions and adverse effects.
Resistance to azoles occurs via multiple mechanisms. Once rare, increasing numbers of resistant strains are being reported, suggest-ing that increasing use of these agents for prophylaxis and therapy may be selecting for clinical drug resistance in certain settings.
Clinical Uses, Adverse Effects, & Drug Interactions The spectrum of action of azole medications is broad, including many species of Candida, C neoformans , the endemic mycoses (blastomycosis, coccidioidomycosis, histoplasmosis), the dermato-phytes, and, in the case of itraconazole and voriconazole, even Aspergillus infections. They are also useful in the treatment of intrinsically amphotericin-resistant organisms such as P boydii.
As a group, the azoles are relatively nontoxic. The most com-mon adverse reaction is relatively minor gastrointestinal upset. All azoles have been reported to cause abnormalities in liver enzymes and, very rarely, clinical hepatitis. Adverse effects specific to indi-vidual agents are discussed below.
All azole drugs are prone to drug interactions because they affect the mammalian cytochrome P450 system of enzymes to some extent. The most significant reactions are indicated below.
KETOCONAZOLE
Ketoconazole was the first oral azole introduced into clinical use. It is distinguished from triazoles by its greater propensity to inhibit mammalian cytochrome P450 enzymes; that is, it is less selective for fungal P450 than are the newer azoles. As a result, systemic ketoconazole has fallen out of clinical use in the USA and is not discussed in any detail here. Its dermatologic use is discussed in Chapter 61 .
ITRACONAZOLE
Itraconazole is available in oral and intravenous formulations and is used at a dosage of 100–400 mg/d. Drug absorption is increased
by food and by low gastric pH. Like other lipid-soluble azoles, it interacts with hepatic microsomal enzymes, though to a lesser degree than ketoconazole. An important drug interaction is reduced bioavailability of itraconazole when taken with rifamycins (rifampin, rifabutin, rifapentine). It does not affect mammalian steroid synthesis, and its effects on the metabolism of other hepat-ically cleared medications are much less than those of ketoconazole. While itraconazole displays potent antifungal activity, effective-ness can be limited by reduced bioavailability. Newer formula-tions, including an oral liquid and an intravenous preparation, have utilized cyclodextran as a carrier molecule to enhance solubil-ity and bioavailability. Like ketoconazole, itraconazole penetrates poorly into the cerebrospinal fluid. Itraconazole is the azole of choice for treatment of disease due to the dimorphic fungi Histoplasma, Blastomyces , and Sporothrix. Itraconazole has activity against Aspergillus sp, but it has been replaced by voriconazole as the azole of choice for aspergillosis. Itraconazole is used extensively in the treatment of dermatophytoses and onychomycosis.
FLUCONAZOLE
Fluconazole displays a high degree of water solubility and good cerebrospinal fluid penetration. Unlike ketoconazole and itracon-azole, its oral bioavailability is high. Drug interactions are also less common because fluconazole has the least effect of all the azoles on hepatic microsomal enzymes. Because of fewer hepatic enzyme interactions and better gastrointestinal tolerance, fluconazole has the widest therapeutic index of the azoles, permitting more aggres-sive dosing in a variety of fungal infections. The drug is available in oral and intravenous formulations and is used at a dosage of 100–800 mg/d.
Fluconazole is the azole of choice in the treatment and second-ary prophylaxis of cryptococcal meningitis. Intravenous flucon-azole has been shown to be equivalent to amphotericin B in treatment of candidemia in ICU patients with normal white blood cell counts. Fluconazole is the agent most commonly used for the treatment of mucocutaneous candidiasis. Activity against the dimorphic fungi is limited to coccidioidal disease, and in particu-lar for meningitis, where high doses of fluconazole often obviate the need for intrathecal amphotericin B. Fluconazole displays no activity against Aspergillus or other filamentous fungi.
TABLE 48–2 Pharmacologic properties of five systemic azole drugs.
Water Solubility AbsorptionCSF: Serum Concentration Ratio t½ (hours) Elimination Formulations
Ketoconazole Low Variable < 0.1 7–10 Hepatic Oral
Itraconazole Low Variable < 0.01 24–42 Hepatic Oral, IV
Fluconazole High High > 0.7 22–31 Renal Oral, IV
Voriconazole High High … 6 Hepatic Oral, IV
Posaconazole Low High … 25 Hepatic Oral
048-Katzung_Ch048_p849-860.indd 854 9/21/11 12:09:57 PM

CHAPTER 48 Antifungal Agents 855
Prophylactic use of fluconazole has been demonstrated to reduce fungal disease in bone marrow transplant recipients and AIDS patients, but the emergence of fluconazole-resistant fungi has raised concerns about this indication.
VORICONAZOLE
Voriconazole is available in intravenous and oral formulations. The recommended dosage is 400 mg/d. The drug is well absorbed orally, with a bioavailability exceeding 90%, and it exhibits less protein binding than itraconazole. Metabolism is predominantly hepatic. Voriconazole is a clinically relevant inhibitor of mamma-lian CYP3A4, and dose reduction of a number of medications is required when voriconazole is started. These include cyclosporine, tacrolimus, and HMG-CoA reductase inhibitors. Observed toxicities include rash and elevated hepatic enzymes. Visual dis-turbances are common, occurring in up to 30% of patients receiv-ing intravenous voriconazole, and include blurring and changes in color vision or brightness. These visual changes usually occur immediately after a dose of voriconazole and resolve within 30 minutes. Photosensitivity dermatitis is commonly observed in patients receiving chronic oral therapy.
Voriconazole is similar to itraconazole in its spectrum of action, having excellent activity against Candida sp (including fluconazole-resistant species such as Candida krusei ) and the dimorphic fungi. Voriconazole is less toxic than amphotericin B and is the treatment of choice for invasive aspergillosis.
POSACONAZOLE
Posaconazole is the newest triazole to be licensed in the USA. It is available only in a liquid oral formulation and is used at a dosage of 800 mg/d, divided into two or three doses. Absorption is improved when taken with meals high in fat. Posaconazole is rap-idly distributed to the tissues, resulting in high tissue levels but relatively low blood levels. Visual changes have not been reported, but drug interactions with increased levels of CYP3A4 substrates such as tacrolimus and cyclosporine have been documented.
Posaconazole is the broadest spectrum member of the azole family, with activity against most species of Candida and Aspergillus . It is the only azole with significant activity against the agents of mucormycosis. It is currently licensed for salvage therapy in inva-sive aspergillosis, as well as prophylaxis of fungal infections during induction chemotherapy for leukemia, and for allogeneic bone marrow transplant patients with graft-versus-host disease.
ECHINOCANDINS Chemistry & Pharmacokinetics Echinocandins are the newest class of antifungal agents to be developed. They are large cyclic peptides linked to a long-chain fatty acid. Caspofungin, micafungin, and anidulafungin are the only licensed agents in this category of antifungals, although other drugs are under active investigation. These agents are active
against Candida and Aspergillus, but not C neoformans or the agents of zygomycosis and mucormycosis.
Echinocandins are available only in intravenous formulations. Caspofungin is administered as a single loading dose of 70 mg, followed by a daily dose of 50 mg. Caspofungin is water soluble and highly protein-bound. The half-life is 9–11 hours, and the metabolites are excreted by the kidneys and gastrointestinal tract. Dosage adjustments are required only in the presence of severe hepatic insufficiency. Micafungin displays similar properties with a half-life of 11–15 hours and is used at a dose of 150 mg/d for treatment of esophageal candidiasis, 100 mg/d for treatment of candidemia, and 50 mg/d for prophylaxis of fungal infections. Anidulafungin has a half-life of 24–48 hours. For esophageal candidiasis, it is administered intravenously at 100 mg on the first day and 50 mg/d thereafter for 14 days. For candidemia, a loading dose of 200 mg is recommended with 100 mg/d thereafter for at least 14 days after the last positive blood culture.
Mechanism of Action Echinocandins act at the level of the fungal cell wall by inhibiting the synthesis of β(1–3)-glucan ( Figure 48–1 ). This results in dis-ruption of the fungal cell wall and cell death.
Clinical Uses & Adverse Effects Caspofungin is currently licensed for disseminated and mucocuta-neous candidal infections, as well as for empiric antifungal therapy during febrile neutropenia, and has largely replaced amphotericin B for the latter indication. Of note, caspofungin is licensed for use in invasive aspergillosis only as salvage therapy in patients who have failed to respond to amphotericin B, and not as primary therapy. Micafungin is licensed for mucocutaneous candidiasis, candidemia, and prophylaxis of candidal infections in bone marrow transplant patients. Anidulafungin is approved for use in esophageal candidi-asis and invasive candidiasis, including candidemia.
Echinocandin agents are extremely well tolerated, with minor gastrointestinal side effects and flushing reported infrequently. Elevated liver enzymes have been noted in several patients receiving caspofungin in combination with cyclosporine, and this combina-tion should be avoided. Micafungin has been shown to increase levels of nifedipine, cyclosporine, and sirolimus. Anidulafungin does not seem to have significant drug interactions, but histamine release may occur during intravenous infusion.
■ ORAL SYSTEMIC ANTIFUNGAL DRUGS FOR MUCOCUTANEOUS INFECTIONS
GRISEOFULVIN Griseofulvin is a very insoluble fungistatic drug derived from a species of penicillium. Its only use is in the systemic treatment of dermatophytosis (see Chapter 61 ). It is administered in a micro-crystalline form at a dosage of 1 g/d. Absorption is improved when
048-Katzung_Ch048_p849-860.indd 855 9/21/11 12:09:57 PM

856 SECTION VIII Chemotherapeutic Drugs
it is given with fatty foods. Griseofulvin’s mechanism of action at the cellular level is unclear, but it is deposited in newly forming skin where it binds to keratin, protecting the skin from new infec-tion. Because its action is to prevent infection of these new skin structures, griseofulvin must be administered for 2–6 weeks for skin and hair infections to allow the replacement of infected kera-tin by the resistant structures. Nail infections may require therapy for months to allow regrowth of the new protected nail and is often followed by relapse. Adverse effects include an allergic syn-drome much like serum sickness, hepatitis, and drug interactions with warfarin and phenobarbital. Griseofulvin has been largely replaced by newer antifungal medications such as itraconazole and terbinafine.
TERBINAFINE Terbinafine is a synthetic allylamine that is available in an oral formulation and is used at a dosage of 250 mg/d. It is used in the treatment of dermatophytoses, especially onychomycosis (see Chapter 61 ). Like griseofulvin, terbinafine is a keratophilic medi-cation, but unlike griseofulvin, it is fungicidal. Like the azole drugs, it interferes with ergosterol biosynthesis, but rather than interacting with the P450 system, terbinafine inhibits the fungal enzyme squalene epoxidase ( Figure 48–1 ). This leads to the accu-mulation of the sterol squalene, which is toxic to the organism. One tablet given daily for 12 weeks achieves a cure rate of up to 90% for onychomycosis and is more effective than griseofulvin or itraconazole. Adverse effects are rare, consisting primarily of gastrointestinal upset and headache. Terbinafine does not seem to affect the P450 system and has demonstrated no significant drug interactions to date.
■ TOPICAL ANTIFUNGAL THERAPY
NYSTATIN Nystatin is a polyene macrolide much like amphotericin B. It is too toxic for parenteral administration and is only used topically.
Nystatin is currently available in creams, ointments, suppositories, and other forms for application to skin and mucous membranes. It is not absorbed to a significant degree from skin, mucous mem-branes, or the gastrointestinal tract. As a result, nystatin has little toxicity, although oral use is often limited by the unpleasant taste.
Nystatin is active against most Candida sp and is most com-monly used for suppression of local candidal infections. Some common indications include oropharyngeal thrush, vaginal can-didiasis, and intertriginous candidal infections.
TOPICAL AZOLES The two azoles most commonly used topically are clotrimazole and miconazole; several others are available (see Preparations Available). Both are available over-the-counter and are often used for vul-vovaginal candidiasis. Oral clotrimazole troches are available for treatment of oral thrush and are a pleasant-tasting alternative to nystatin. In cream form, both agents are useful for dermatophytic infections, including tinea corporis, tinea pedis, and tinea cruris. Absorption is negligible, and adverse effects are rare.
Topical and shampoo forms of ketoconazole are also available and useful in the treatment of seborrheic dermatitis and pityriasis versicolor. Several other azoles are available for topical use (see Preparations Available).
TOPICAL ALLYLAMINES Terbinafine and naftifine are allylamines available as topical creams (see Chapter 61 ). Both are effective for treatment of tinea cruris and tinea corporis. These are prescription drugs in the USA.
048-Katzung_Ch048_p849-860.indd 856 9/21/11 12:09:57 PM

CHAPTER 48 Antifungal Agents 857
SUMMARY Antifungal Drugs
Subclass Mechanism of Action Effects Clinical ApplicationsPharmacokinetics, Toxicities, Interactions
POLYENE MACROLIDE • Amphotericin B Forms pores in fungal
membranes (which contain ergosterol) but not in mammalian (cholesterol-containing) membranes
Loss of intracellular contents through pores is fungicidal • broad spectrum of action
Localized and systemic candidemia • Cryptococcus • Histoplasma • Blastomyces • Coccidioides • Aspergillus
Oral but not absorbed • IV for systemic use • intrathecal for fungal meningitis • topical for ocular and bladder infections • duration, days • Toxicity: Infusion reactions • renal impairment • Interactions: Additive with other renal toxic drugs
• Lipid formulations: Lower toxicity, higher doses can be used
PYRIMIDINE ANALOG • Flucytosine Interferes with DNA and RNA
synthesis selectively in fungiSynergistic with amphotericin • systemic toxicity in host due to DNA and RNA effects
Cryptococcus and chromoblastomycosis infections
Oral • duration, hours • renal excretion • Toxicity: Myelosuppression
AZOLES
• Ketoconazole Blocks fungal P450 enzymes and interferes with ergosterol synthesis
Poorly selective • inter-feres with mammalian P450 function
Broad spectrum but toxic-ity restricts use to topical therapy
Oral, topical • Toxicity and interactions: Interferes with steroid hormone synthesis and phase I drug metabolism
• Itraconazole Same as for ketoconazole Much more selective than ketoconazole
Broad spectrum: Candida, Cryptococcus, blastomycosis, coccidioidomycosis, histoplasmosis
Oral and IV • duration, 1–2 d • poor entry into central nervous system (CNS) • Toxicity and interactions: Low toxicity
• Fluconazole, voriconazole, posaconazole: Fluconazole has excellent CNS penetration, used in fungal meningitis
ECHINOCANDINS • Caspofungin Blocks β-glucan synthase Prevents synthesis of
fungal cell wallFungicidal Candida sp • also used in aspergillosis
IV only • duration, 11–15 h • Toxicity: Minor gastrointestinal effects, flushing • Interactions: Increases cyclosporine levels (avoid combination)
• Micafungin, anidulafungin: Micafungin increases levels of nifedipine, cyclosporine, sirolimus; anidulafungin is relatively free of this interaction
ALLYLAMINE • Terbinafine Inhibits epoxidation of
squalene in fungi • increased levels are toxic to them
Reduces ergosterol • prevents synthesis of fungal cell membrane
Mucocutaneous fungal infections
Oral • duration, days • Toxicity: Gastrointestinal upset, headache, hepato-toxicity • Interactions: None reported
048-Katzung_Ch048_p849-860.indd 857 9/21/11 12:09:57 PM

858 SECTION VIII Chemotherapeutic Drugs
Amphotericin BParenteral: Conventional formulation (Amphotericin B, Fungizone): 50 mg
powder for injectionLipid formulations: (Abelcet): 100 mg/20 mL suspension for injection (AmBisome): 50 mg powder for injection (Amphotec): 50, 100 mg powder for injectionTopical: 3% cream, lotion, ointment
Anidulafungin (Eraxis)Parenteral: 50 mg powder for injection
Butenafine (Lotrimin Ultra, Mentax)Topical: 1% cream
Butoconazole (Gynazole-1, Mycelex-3)Topical: 2% vaginal cream
Caspofungin (Cancidas)Parenteral: 50, 70 mg powder for injection
Clotrimazole (generic, Lotrimin)Topical: 1% cream, solution, lotion; 100, 200 mg vaginal
suppositoriesEconazole (generic, Spectazole)
Topical: 1% creamFluconazole (Diflucan)
Oral: 50, 100, 150, 200 mg tablets; powder for 10, 40 mg/mL suspension
Parenteral: 2 mg/mL in 100 and 200 mL vialsFlucytosine (Ancobon)
Oral: 250, 500 mg capsulesGriseofulvin (Grifulvin, Grisactin, Fulvicin P/G)
Oral microsize: 125, 250 mg tablets; 250 mg capsule, 125 mg/5 mL suspension
Oral ultramicrosize:1 125, 165, 250, 330 mg tabletsItraconazole (Sporanox)
Oral: 100 mg capsules; 10 mg/mL solutionParenteral: 10 mg/mL for IV infusion
Ketoconazole (generic, Nizoral)Oral: 200 mg tabletsTopical: 2% cream, shampoo
Micafungin (Mycamine)Parenteral: 50 mg powder for injection
Miconazole (generic, Micatin)Topical: 2% cream, powder, spray; 100, 200 mg vaginal suppositories
Naftifine (Naftin)Topical: 1% cream, gel
Natamycin (Natacyn)Topical: 5% ophthalmic suspension
Nystatin (generic, Mycostatin)Oral: 500,000 unit tabletsTopical: 100,000 units/g cream, ointment, powder; 100,000 units
vaginal tabletsOxiconazole (Oxistat)
Topical: 1% cream, lotionPosaconazole (Noxafil)
Oral: suspension 40 mg/mLSulconazole (Exelderm)
Topical: 1% cream, solutionTerbinafine (Lamisil)
Oral: 250 mg tabletsTopical: 1% cream, gel
Terconazole (Terazol 3, Terazol 7)Topical: 0.4%, 0.8% vaginal cream; 80 mg vaginal suppositories
Tioconazole (Vagistat-1, Monistat 1)Topical: 6.5% vaginal ointment
Tolnaftate (generic, Aftate, Tinactin)Topical: 1% cream, gel, solution, aerosol powder
Voriconazole (Vfend)Oral: 50, 200 mg tablets; oral suspension 40 mg/mLParenteral: 200 mg vials, reconstituted to a 5 mg/mL solution
1Ultramicrosize formulations of griseofulvin are approximately 1.5 times more potent, milligram for milligram, than the microsize preparations.
P R E P A R A T I O N S A V A I L A B L E
Amphotericin BParenteral: Conventional formulation (Amphotericin B, Fungizone): 50 mg
powder for injectionLipid formulations: (Abelcet): 100 mg/20 mL suspension for injection (AmBisome): 50 mg powder for injection (Amphotec): 50, 100 mg powder for injectionTopical: 3% cream, lotion, ointment
Anidulafungin (Eraxis)Parenteral: 50 mg powder for injection
Butenafine (Lotrimin Ultra, Mentax)Topical: 1% cream
Butoconazole (Gynazole-1, Mycelex-3)Topical: 2% vaginal cream
Caspofungin (Cancidas)Parenteral: 50, 70 mg powder for injection
Clotrimazole (generic, Lotrimin)Topical: 1% cream, solution, lotion; 100, 200 mg vaginal
suppositoriesEconazole (generic, Spectazole)
Topical: 1% creamFluconazole (Diflucan)
Oral: 50, 100, 150, 200 mg tablets; powder for 10, 40 mg/mLsuspension
Parenteral: 2 mg/mL in 100 and 200 mL vialsFlucytosine (Ancobon)
Oral: 250, 500 mg capsulesGriseofulvin (Grifulvin, Grisactin, Fulvicin P/G)
Oral microsize: 125, 250 mg tablets; 250 mg capsule, 125 mg/5 mL suspension
Oral ultramicrosize:1 125, 165, 250, 330 mg tabletsItraconazole (Sporanox)
Oral: 100 mg capsules; 10 mg/mL solutionParenteral: 10 mg/mL for IV infusion
Ketoconazole (generic, Nizoral)Oral: 200 mg tabletsTopical: 2% cream, shampoo
Micafungin (Mycamine)Parenteral: 50 mg powder for injection
Miconazole (generic, Micatin)Topical: 2% cream, powder, spray; 100, 200 mg vaginal suppositories
Naftifine (Naftin)Topical: 1% cream, gel
Natamycin (Natacyn)Topical: 5% ophthalmic suspension
Nystatin (generic, Mycostatin)Oral: 500,000 unit tabletsTopical: 100,000 units/g cream, ointment, powder; 100,000 units
vaginal tabletsOxiconazole (Oxistat)
Topical: 1% cream, lotionPosaconazole (Noxafil)
Oral: suspension 40 mg/mLSulconazole (Exelderm)
Topical: 1% cream, solutionTerbinafine (Lamisil)
Oral: 250 mg tabletsTopical: 1% cream, gel
Terconazole (Terazol 3, Terazol 7)Topical: 0.4%, 0.8% vaginal cream; 80 mg vaginal suppositories
Tioconazole (Vagistat-1, Monistat 1)Topical: 6.5% vaginal ointment
Tolnaftate (generic, Aftate, Tinactin)Topical: 1% cream, gel, solution, aerosol powder
Voriconazole (Vfend)Oral: 50, 200 mg tablets; oral suspension 40 mg/mLParenteral: 200 mg vials, reconstituted to a 5 mg/mL solution
REFERENCES Brüggemann RJ et al: Clinical relevance of the pharmacokinetic interactions of
azole antifungal drugs with other coadministered agents. Clin Infect Dis 2009;48:1441.
Cornely OA et al: Posazonacole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med 2007;356:348.
Diekema DJ et al: Activities of caspofungin, itraconazole, posaconazole, ravucon-azole, voriconazole, and amphotericin B against 448 recent clinical isolates of filamentous fungi. J Clin Microbiol 2003;41:3623.
Groll A, Piscitelli SC, Walsh TJ: Clinical pharmacology of systemic antifungal agents: A comprehensive review of agents in clinical use, current investiga-tional compounds, and putative targets for antifungal drug development. Adv Pharmacol 1998;44:343.
Herbrecht R et al: Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med 2002;347:408.
Kim R, Khachikian D, Reboli AC: A comparative evaluation of properties and clinical efficacy of the echinocandins. Expert Opin Pharmacother 2007;8:1479.
Pasqualotto AC, Denning DW: New and emerging treatments for fungal infection. J Antimicrob Chemoth 2008;61(Suppl 1):i19.
Rogers TR: Treatment of zygomycosis: Current and new options. J Antimicrob Chemother 2008;61(Suppl 1):i35.
Wong-Beringer A, Jacobs RA, Guglielmo BJ: Lipid formulations of amphotericin B. Clinical efficacy and toxicities. Clin Infect Dis 1998;27:603.
048-Katzung_Ch048_p849-860.indd 858 9/21/11 12:09:57 PM

CHAPTER 48 Antifungal Agents 859
C A S E S T U D Y A N S W E R
The club-like nonseptate hyphae observed in cultures of intraoperative specimens from this patient are characteristic of Rhizopus , one of the agents of mucormycosis. This patient
should be treated with an initial, prolonged course of therapy with liposomal amphotericin B and caspofungin and subse-quent chronic suppressive therapy with posaconazole.
048-Katzung_Ch048_p849-860.indd 859 9/21/11 12:09:58 PM

This page intentionally left blank

861
Antiviral Agents Sharon Safrin, MD
abnormalities. White blood cell count is 5800 cells/mm 3 with a normal differential, hemoglobin is 11.8 g/dL, all liver function tests are within normal limits, CD4 cell count is 278 cells/mm 3 , and viral load (HIV RNA) is 110,000 copies/mL. What other laboratory tests should be ordered? Which antiretroviral medications would you begin?
C A S E S T U D Y
A 35-year-old white woman who recently tested seropositive for both HIV and hepatitis B virus surface antigen is referred for evaluation. She is feeling well overall but reports a 25-pack-year smoking history. She drinks 3–4 beers per week and has no known medication allergies. She has a history of heroin use and is currently receiving methadone. Physical examination reveals normal vital signs and no
C H A P T E R
Viruses are obligate intracellular parasites; their replication depends primarily on synthetic processes of the host cell. Therefore, to be effective, antiviral agents must either block viral entry into or exit from the cell or be active inside the host cell. As a corollary, non-selective inhibitors of virus replication may interfere with host cell function and result in toxicity.
Progress in antiviral chemotherapy began in the early 1950s, when the search for anticancer drugs generated several new compounds capable of inhibiting viral DNA synthesis. The two first-generation antiviral agents, 5-iododeoxyuridine and trifluorothymidine, had poor specificity (ie, they inhibited host cell DNA as well as viral DNA) that rendered them too toxic for systemic use. However, both agents are effective when used topically for the treatment of herpes keratitis.
Knowledge of the mechanisms of viral replication has provided insights into critical steps in the viral life cycle that can serve as potential targets for antiviral therapy. Recent research has focused on identifying agents with greater selectivity, higher potency, in vivo stability, and reduced toxicity. Antiviral therapy is now avail-able for herpesviruses, hepatitis C virus (HCV), hepatitis B virus (HBV), papillomavirus, influenza, and human immunodeficiency virus (HIV). Antiviral drugs share the common property of being virustatic; they are active only against replicating viruses and do not affect latent virus. Whereas some infections require monotherapy for brief periods of time (eg, acyclovir for herpes simplex virus), others require dual therapy for prolonged periods of time (inter-feron alfa/ribavirin for HCV), whereas still others require multiple drug therapy for indefinite periods (HIV). In chronic illnesses such
49
ACRONYMS & OTHER NAMES
3TC LamivudineAZT Zidovudine (previously azidothymidine)
CMV CytomegalovirusCYP Cytochrome P450d4T StavudineddC ZalcitabineddI DidanosineEBV Epstein-Barr virusFTC EmtricitabineHBeAg Hepatitis e antigenHBV Hepatitis B virusHCV Hepatitis C virusHHV-6 Human herpesvirus-6HIV Human immunodefi ciency virusHSV Herpes simplex virusNNRTI Nonnucleoside reverse transcriptase inhibitorNRTI Nucleoside/nucleotide reverse transcriptase inhibitorPI Protease inhibitorRSV Respiratory syncytial virusSVR Sustained viral responseUGT1A1 UDP-glucuronosyl transferase 1A1
VZV Varicella-zoster virus
049-Katzung_Ch049_p861-890.indd 861 9/23/11 4:25:33 PM

862 SECTION VIII Chemotherapeutic Drugs
as viral hepatitis and HIV infection, potent inhibition of viral rep-lication is crucial in limiting the extent of systemic damage.
Viral replication requires several steps ( Figure 49–1 ): (1) attach-ment of the virus to receptors on the host cell surface; (2) entry of the virus through the host cell membrane; (3) uncoating of viral nucleic acid; (4) synthesis of early regulatory proteins, eg, nucleic acid polymerases; (5) synthesis of new viral RNA or DNA; (6) syn-thesis of late, structural proteins; (7) assembly (maturation) of viral particles; and (8) release from the cell. Antiviral agents can poten-tially target any of these steps.
■ AGENTS TO TREAT HERPES SIMPLEX VIRUS HSV & VARICELLAZOSTER VIRUS VZV INFECTIONS
Three oral nucleoside analogs are licensed for the treatment of HSV and VZV infections: acyclovir, valacyclovir, and famciclovir. They have similar mechanisms of action and comparable indications for clinical use; all are well tolerated. Acyclovir has been the most exten-sively studied; it was licensed first and is the only one of the three that is available for intravenous use in the United States. Comparative trials have demonstrated similar efficacies of these three agents for the treatment of HSV but modest superiority of famciclovir and valacyclovir for the treatment of herpes zoster infections.
ACYCLOVIR
Acyclovir ( Figure 49–2 ) is an acyclic guanosine derivative with clinical activity against HSV-1, HSV-2, and VZV, but it is approx-imately 10 times more potent against HSV-1 and HSV-2 than against VZV. In vitro activity against Epstein-Barr virus (EBV), cytomegalovirus (CMV), and human herpesvirus-6 (HHV-6) is present but weaker.
Acyclovir requires three phosphorylation steps for activation. It is converted first to the monophosphate derivative by the virus-specified thymidine kinase and then to the di- and triphosphate compounds by host cell enzymes ( Figure 49–3 ). Because it requires the viral kinase for initial phosphorylation, acyclovir is selectively activated—and the active metabolite accumulates—only in infected cells. Acyclovir triphosphate inhibits viral DNA synthesis by two mechanisms: competition with deoxyGTP for the viral DNA polymerase, resulting in binding to the DNA tem-plate as an irreversible complex; and chain termination following incorporation into the viral DNA.
The bioavailability of oral acyclovir is low (15–20%) and is unaffected by food. An intravenous formulation is available. Topical formulations produce high concentrations in herpetic lesions, but systemic concentrations are undetectable by this route.
Acyclovir is cleared primarily by glomerular filtration and tubular secretion. The half-life is 2.5–3 hours in patients with normal renal function and 20 hours in patients with anuria.
Viral attachmentand entry
Viralrelease
Uncoating
Early proteinsynthesis
Nucleic acid synthesis
Late proteinsynthesis and
processingPackaging
andassembly
Mammaliancell
Blocked byamantadine,rimantadine(influenza)
Penetration
Blocked by NRTIs (HIV, HBV),
NNRTIs (HIV),acyclovir (HSV),foscarnet (CMV)
Blocked by protease inhibitors
(HIV)
Blocked byneuraminidase
inhibitors(influenza)
Blocked byenfuvirtide (HIV),maraviroc (HIV); docosanol (HSV),palivizumab (RSV)
Blocked by interferon-alfa
(HBV, HCV)
FIGURE 49–1 The major sites of antiviral drug action. Note: Interferon alfas are speculated to have multiple sites of action. (Modified and repro-
duced, with permission, from Trevor AJ, Katzung BG, Masters SB: Pharmacology: Examination & Board Review, 9th ed. McGraw-Hill, 2010.)
049-Katzung_Ch049_p861-890.indd 862 9/23/11 4:25:34 PM

CHAPTER 49 Antiviral Agents 863
Acyclovir diffuses readily into most tissues and body fluids. Cerebrospinal fluid concentrations are 20–50% of serum values.
Oral acyclovir has multiple uses. In first episodes of genital herpes, oral acyclovir shortens the duration of symptoms by approximately 2 days, the time to lesion healing by 4 days, and the
duration of viral shedding by 7 days. In recurrent genital herpes, the time course is shortened by 1–2 days. Treatment of first- episode genital herpes does not alter the frequency or severity of recurrent outbreaks. Long-term suppression with oral acyclovir in patients with frequent recurrences of genital herpes decreases the
O
NN
NNH
NH2H2N
O
HO
Acyclovir
A. Antiherpes agents
B. Anti-HIV NRTI agents
C. Anti-HBV agents
O
N
NH
NH2
O
HO
OH
Ganciclovir
OO
O
NN
NN
N
NH
NH2
O
Valacyclovir (prodrug)
O
O
N
N
NH2
Famciclovir (prodrug)
Zidovudine (AZT)
CidofovirPenciclovir
NN
NN
NH2
NH2 NH2
NH2
NH2
O
O
HO
O
O
HO NN
NNH
NH2
O
OH
O
N
N
O
NH2
N
NH
O
N3
OH
Foscarnet
O
OH
HOHO
P
O
OPHO
HO
OP
O
OH
HO
NN
NN
O
O
Stavudine (d4T)
HO N
NH
O
O
Zalcitabine (ddC)
HO N
N
O O
O
O
Didanosine (ddl)
Adefovir Entecavir
HO N
NH
N
N
OO O
OO
O
O
OP
Abacavir Tenofovir disoproxil Emtricitabine
OHN
N
NH
NN
NNH
O
N
N
N
N
O
O
N
NH2
CH2
H2N
O
O
OH
N
F
S
O
Lamivudine (3TC)
OHN
N
S
HO
H2N
O
NN
NNH
NH2
O
Valganciclovir (prodrug)
O
O
HO
HO
FIGURE 49–2 Chemical structures of some antiviral nucleoside and nucleotide analogs.
049-Katzung_Ch049_p861-890.indd 863 9/23/11 4:25:34 PM

864 SECTION VIII Chemotherapeutic Drugs
frequency of symptomatic recurrences and of asymptomatic viral shedding, thus decreasing the rate of sexual transmission. However, outbreaks may resume upon discontinuation of suppressive acyclovir. Oral acyclovir is only modestly beneficial in recurrent herpes labialis. In contrast, acyclovir therapy significantly decreases the total number of lesions, duration of symptoms, and viral shed-ding in patients with varicella (if begun within 24 hours after the onset of rash) or cutaneous zoster (if begun within 72 hours). However, because VZV is less susceptible to acyclovir than HSV, higher doses are required ( Table 49–1 ). When given prophylacti-cally to patients undergoing organ transplantation, oral or intrave-nous acyclovir prevents reactivation of HSV infection. Evidence from recent clinical trials suggests that the use of daily acyclovir (400 mg twice daily) may reduce the plasma viral load of HIV-1 and the risk of HIV-associated disease progression in individuals dually infected with HSV-2 and HIV-1.
Intravenous acyclovir is the treatment of choice for herpes simplex encephalitis, neonatal HSV infection, and serious HSV or VZV infections ( Table 49–1 ). In immunocompromised patients with VZV infection, intravenous acyclovir reduces the incidence of cutaneous and visceral dissemination.
Topical acyclovir cream is substantially less effective than oral therapy for primary HSV infection. It is of no benefit in treating recurrent genital herpes.
Resistance to acyclovir can develop in HSV or VZV through alteration in either the viral thymidine kinase or the DNA polymerase, and clinically resistant infections have been reported in immunocompromised hosts. Most clinical isolates are resistant on the basis of deficient thymidine kinase activity and thus are cross-resistant to valacyclovir, famciclovir, and ganciclovir. Agents such as
foscarnet, cidofovir, and trifluridine do not require activation by viral thymidine kinase and thus have preserved activity against the most prevalent acyclovir-resistant strains ( Figure 49–3 ).
Acyclovir is generally well tolerated. Nausea, diarrhea, and headache have occasionally been reported. Intravenous infusion may be associated with reversible renal toxicity (ie, crystalline nephropathy or interstitial nephritis) or neurologic effects (eg, tremors, delirium, seizures). However, these are uncommon with adequate hydration and avoidance of rapid infusion rates. High doses of acyclovir cause chromosomal damage and testicular atrophy in rats, but there has been no evidence of teratogenicity, reduction in sperm production, or cytogenetic alterations in peripheral blood lymphocytes in patients receiving daily suppres-sion of genital herpes for more than 10 years. A recent study found no evidence of increased birth defects in 1150 infants who were exposed to acyclovir during the first trimester.
Concurrent use of nephrotoxic agents may enhance the poten-tial for nephrotoxicity. Probenecid and cimetidine decrease acyclo-vir clearance and increase exposure. Somnolence and lethargy may occur in patients receiving concomitant zidovudine and acyclovir.
VALACYCLOVIR
Valacyclovir is the L-valyl ester of acyclovir ( Figure 49–2 ). It is rapidly converted to acyclovir after oral administration via first-pass enzymatic hydrolysis in the liver and intestine, resulting in serum levels that are three to five times greater than those achieved with oral acyclovir and approximate those achieved with intrave-nous acyclovir. Oral bioavailability is 54–70%, and cerebrospinal fluid levels are about 50% of those in serum. Elimination half-life is 2.5–3.3 hours.
Approved uses of valacyclovir include treatment of first or recurrent genital herpes, suppression of frequently recurring genital herpes, as a 1-day treatment for orolabial herpes, and as treatment for varicella and herpes zoster ( Table 49–1 ). Once-daily dosing of valacyclovir for chronic suppression in persons with recurrent genital herpes has been shown to markedly decrease the risk of sexual transmission. In comparative trials with acyclovir for the treatment of patients with zoster, rates of cutaneous healing were similar, but valacyclovir was associated with a shorter dura-tion of zoster-associated pain. Higher doses of valacyclovir (2 g four times daily) have also been shown to be effective in prevent-ing CMV disease after organ transplantation when compared with placebo.
Valacyclovir is generally well tolerated, although nausea, headache, vomiting, or rash occasionally occur. At high doses, confusion, hallucinations, and seizures have been reported. AIDS patients who received high-dosage valacyclovir chronically (ie, 8 g/d) had an increased incidence of gastrointestinal intoler-ance as well as thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome; this dose has also been associated with confusion and hallucinations in transplant patients. In a recent study, there was no evidence of increased birth defects in 181 infants who were exposed to valacyclovir during the first trimester.
Foscarnet
Acyclovirpenciclovirganciclovir
Trifluridinecidofovir
Monophosphate
Diphosphate
Competitive inhibitionof viral DNA polymerase
Triphosphate
Hostkinases
Inhibition of viral DNA synthesis
Incorporation intoviral DNA
Virus-specified
enzymes(eg, thymidinekinase, UL97)
Chaintermination
FIGURE 49–3 Mechanism of action of antiherpes agents.
049-Katzung_Ch049_p861-890.indd 864 9/23/11 4:25:34 PM

CHAPTER 49 Antiviral Agents 865
TABLE 49–1 Agents to treat or prevent herpes simplex virus (HSV) and varicella-zoster virus (VZV) infections.
Route of Administration Use Recommended Adult Dosage and Regimen
Acyclovir1 Oral First episode genital herpes treatment 400 mg tid or 200 mg 5 times daily × 7–10 days
Recurrent genital herpes treatment 400 mg tid or 200 mg 5 times daily or 800 mg bid × 3–5 days or 800 mg tid × 2 days
Genital herpes in the HIV-infected host treatment
400 mg 3–5 times daily × 5–10 days
Genital herpes suppression in the HIV-infected host
400–800 mg bid–tid
Herpes proctitis treatment 400 mg 5 times daily until healed
Orolabial herpes treatment 400 mg 5 times daily × 5 days
Varicella treatment (age ≥ 2 years) 800 mg qid × 5 days
Zoster treatment 800 mg 5 times daily × 7–10 days
Intravenous Severe HSV treatment 5 mg/kg q8h × 7–10 days
Mucocutaneous herpes in the immunocompromised host treatment
10 mg/kg q8h × 7–14 days
Herpes encephalitis treatment 10–15 mg/kg q8h × 14–21 days
Neonatal HSV infection treatment 10–20 mg/kg q8h × 14–21 days
Varicella or zoster in the immunosuppressed host treatment
10 mg/kg q8h × 7 days
Topical (5% cream) Herpes labialis treatment Thin film covering lesion 5 times daily × 4 days
Famciclovir1 Oral First episode genital herpes treatment 500 mg tid × 5–10 days
Recurrent genital herpes treatment 1000 mg bid × 1 day
Genital herpes in the HIV-infected host treatment 500 mg bid × 5–10 days
Genital herpes suppression 250 mg bid
Genital herpes suppression in the HIV-infected host
500 mg bid
Orolabial herpes treatment 1500 mg once
Orolabial or genital herpes suppression 250-500 mg bid
Zoster 500 mg tid × 7 days
Valacyclovir1 Oral First episode genital herpes treatment 1000 mg bid × 10 days
Recurrent genital herpes treatment 500 mg bid × 3 days
Genital herpes in the HIV-infected host treatment
500–1000 mg bid × 5–10 days
Genital herpes suppression 500–1000 mg once daily
Genital herpes suppression in the HIV-infected host 500 mg bid
Orolabial herpes 2000 mg bid × 1 day
Varicella (age ≥ 12 years) 20 mg/d tid × 5 days (maximum, 1 g tid)
Zoster 1 g tid × 7 days
Foscarnet1 Intravenous Acyclovir-resistant HSV and VZV infections 40 mg/kg q8h until healed
Docosanol Topical (10% cream) Recurrent herpes labialis Thin film covering lesion q2h × 4 days
Penciclovir Topical (1% cream) Herpes labialis or herpes genitalis Thin film covering lesions q2h × 4 days
Trifluridine Topical (1% solution) Acyclovir-resistant HSV infection Thin film covering lesion 5 times daily until healed
1Dosage must be reduced in patients with renal insufficiency.
HSV, herpes simplex virus; VZV, varicella-zoster virus.
049-Katzung_Ch049_p861-890.indd 865 9/23/11 4:25:34 PM

866 SECTION VIII Chemotherapeutic Drugs
FAMCICLOVIR
Famciclovir is the diacetyl ester prodrug of 6-deoxypenciclovir, an acyclic guanosine analog ( Figure 49–2 ). After oral administration, famciclovir is rapidly deacetylated and oxidized by first-pass metabo-lism to penciclovir. It is active in vitro against HSV-1, HSV-2, VZV, EBV, and HBV. As with acyclovir, activation by phosphorylation is catalyzed by the virus-specified thymidine kinase in infected cells, followed by competitive inhibition of the viral DNA polymerase to block DNA synthesis. Unlike acyclovir, however, penciclovir does not cause chain termination. Penciclovir triphosphate has lower affinity for the viral DNA polymerase than acyclovir triphosphate, but it achieves higher intracellular concentrations. The most com-monly encountered clinical mutants of HSV are thymidine kinase-deficient; these are cross-resistant to acyclovir and famciclovir.
The bioavailability of penciclovir from orally administered famciclovir is 70%. The intracellular half-life of penciclovir triphosphate is prolonged, at 7–20 hours. Penciclovir is excreted primarily in the urine.
Oral famciclovir is effective for the treatment of first and recur-rent genital herpes, for chronic daily suppression of genital herpes, for treatment of herpes labialis, and for the treatment of acute zoster ( Table 49–1 ). One-day usage of famciclovir significantly accelerates time to healing of recurrent genital herpes and of herpes labialis. Comparison of famciclovir to valacyclovir for treatment of herpes zoster in immunocompetent patients showed similar rates of cutaneous healing and pain resolution; both agents shortened the duration of zoster-associated pain compared with acyclovir.
Oral famciclovir is generally well tolerated, although headache, nausea, or diarrhea may occur. As with acyclovir, testicular toxicity has been demonstrated in animals receiving repeated doses. However, men receiving daily famciclovir (250 mg every 12 hours) for 18 weeks had no changes in sperm morphology or motility. In a recent study, there was no evidence of increased birth defects in 32 infants who were exposed to famciclovir during the first trimes-ter. The incidence of mammary adenocarcinoma was increased in female rats receiving famciclovir for 2 years.
PENCICLOVIR
The guanosine analog penciclovir, the active metabolite of famci-clovir, is available for topical use. Penciclovir cream (1%) shortens the duration of recurrent herpes labialis or genitalis ( Table 49–1 ). When applied within 1 hour of the onset of prodromal symptoms and continued every 2 hours during waking hours for 4 days, median time until healing was shortened by 17 hours compared with placebo. Adverse effects are uncommon, although applica-tion site reactions occur in about 1% of patients.
DOCOSANOL
Docosanol is a saturated 22-carbon aliphatic alcohol that inhibits fusion between the plasma membrane and the HSV envelope, thereby preventing viral entry into cells and subsequent viral
replication. Topical docosanol 10% cream is available without a prescription; application site reactions occur in approximately 2% of patients. When applied within 12 hours of the onset of prodromal symptoms, five times daily, median healing time was shortened by 18 hours compared with placebo in recurrent orolabial herpes.
TRIFLURIDINE
Trifluridine (trifluorothymidine) is a fluorinated pyrimidine nucleoside that inhibits viral DNA synthesis in HSV-1, HSV-2, CMV, vaccinia, and some adenoviruses. It is phosphorylated intra-cellularly by host cell enzymes, and then competes with thymidine triphosphate for incorporation by the viral DNA polymerase ( Figure 49–3 ). Incorporation of trifluridine triphosphate into both viral and host DNA prevents its systemic use. Application of a 1% solution is effective in treating keratoconjunctivitis and recurrent epithelial keratitis due to HSV-1 or HSV-2. Cutaneous application of trifluridine solution, alone or in combination with interferon alfa, has been used successfully in the treatment of acyclovir-resistant HSV infections.
INVESTIGATIONAL AGENTS
Valomaciclovir is an inhibitor of the viral DNA polymerase; it is currently under clinical evaluation for the treatment of patients with acute VZV infection (shingles) and acute EBV infection (infectious mononucleosis).
■ AGENTS TO TREAT CYTOMEGALOVIRUS CMV INFECTIONS
CMV infections occur primarily in the setting of advanced immu-nosuppression and are typically due to reactivation of latent infec-tion. Dissemination of infection results in end-organ disease, including retinitis, colitis, esophagitis, central nervous system disease, and pneumonitis. Although the incidence in HIV-infected patients has markedly decreased with the advent of potent anti-retroviral therapy, clinical reactivation of CMV infection after organ transplantation is still prevalent.
The availability of oral valganciclovir and the ganciclovir intraoc-ular implant has decreased the use of intravenous ganciclovir, intra-venous foscarnet, and intravenous cidofovir for the treatment of end-organ CMV disease ( Table 49–2 ). Oral valganciclovir has largely replaced oral ganciclovir because of its lower pill burden.
GANCICLOVIR
Ganciclovir is an acyclic guanosine analog ( Figure 49–2 ) that requires activation by triphosphorylation before inhibiting the viral DNA polymerase. Initial phosphorylation is catalyzed by the virus-specified protein kinase phosphotransferase UL97 in
049-Katzung_Ch049_p861-890.indd 866 9/23/11 4:25:35 PM

CHAPTER 49 Antiviral Agents 867
CMV-infected cells. The activated compound competitively inhibits viral DNA polymerase and causes termination of viral DNA elongation ( Figure 49–3 ). Ganciclovir has in vitro activity against CMV, HSV, VZV, EBV, HHV-6, and HHV-8. Its activity against CMV is up to 100 times greater than that of acyclovir.
Ganciclovir may be administered intravenously, orally, or via intraocular implant. The bioavailability of oral ganciclovir is poor. Cerebrospinal fluid concentrations are approximately 50% of serum concentrations. The elimination half-life is 4 hours, and the intracellular half-life is prolonged at 16–24 hours. Clearance of the drug is linearly related to creatinine clearance. Ganciclovir is readily cleared by hemodialysis.
Intravenous ganciclovir has been shown to delay progression of CMV retinitis in patients with AIDS. Dual therapy with foscarnet and ganciclovir is more effective in delaying progression of retinitis than either drug alone (see Foscarnet), although adverse effects are compounded. Intravenous ganciclovir is also used to treat CMV colitis, esophagitis, and pneumonitis (the latter often treated with a combination of ganciclovir and intravenous cytomegalovirus immunoglobulin) in immunocompromised patients. Intravenous ganciclovir, followed by either oral ganciclovir or high-dose oral acyclovir, reduces the risk of CMV infection in transplant recipi-ents. Oral ganciclovir is indicated for prevention of end-organ CMV disease in AIDS patients and as maintenance therapy of CMV retinitis after induction. Although less effective than intrave-nous ganciclovir, the oral form carries a diminished risk of myelo-suppression and of catheter-related complications. The risk of Kaposi’s sarcoma is reduced in AIDS patients receiving long-term ganciclovir, presumably because of activity against HHV-8.
Ganciclovir may also be administered intraocularly to treat CMV retinitis, either by direct intravitreal injection or by intraoc-ular implant. The implant has been shown to delay progression of retinitis to a greater degree than systemic ganciclovir therapy. Surgical replacement of the implant is required at intervals of
5–8 months. Concurrent therapy with a systemic anti-CMV agent is recommended to prevent other sites of end-organ CMV disease.
Resistance to ganciclovir increases with duration of usage. The more common mutation, in UL97, results in decreased levels of the triphosphorylated (ie, active) form of ganciclovir. The less common UL54 mutation in DNA polymerase results in higher levels of resistance and potential cross-resistance with cidofovir and foscarnet. Antiviral susceptibility testing is recommended in patients in whom resistance is suspected clinically, as is the substi-tution of alternative therapies and concomitant reduction in immunosuppressive therapies, if possible. The addition of CMV hyperimmune globulin may also be considered.
The most common adverse effect of systemic ganciclovir treat-ment, particularly after intravenous administration, is myelosup-pression. Myelosuppression may be additive in patients receiving concurrent zidovudine, azathioprine, or mycophenolate mofetil. Other potential adverse effects are nausea, diarrhea, fever, rash, headache, insomnia, and peripheral neuropathy. Central nervous system toxicity (confusion, seizures, psychiatric disturbance) and hepatotoxicity have been rarely reported. Ganciclovir is mutagenic in mammalian cells and carcinogenic and embryotoxic at high doses in animals and causes aspermatogenesis; the clinical signifi-cance of these preclinical data is unclear.
Levels of ganciclovir may rise in patients concurrently taking probenecid or trimethoprim. Concurrent use of ganciclovir with didanosine may result in increased levels of didanosine.
VALGANCICLOVIR
Valganciclovir is an L-valyl ester prodrug of ganciclovir that exists as a mixture of two diastereomers ( Figure 49–2 ). After oral admin-istration, both diastereomers are rapidly hydrolyzed to ganciclovir by esterases in the intestinal wall and liver.
TABLE 49–2 Agents to treat cytomegalovirus (CMV) infection.
AgentRoute of Administration Use Recommended Adult Dosage1
Valganciclovir Oral CMV retinitis treatment Induction: 900 mg bid × 21 days
Maintenance: 900 mg daily
Oral CMV prophylaxis (transplant patients) 900 mg daily
Ganciclovir Intravenous CMV retinitis treatment Induction: 5 mg/kg q12h × 14–21 days
Maintenance: 5 mg/kg/d or 6 mg/kg five times per week
Oral CMV prophylaxis 1 g tid
CMV retinitis treatment 1 g tid
Intraocular implant CMV retinitis treatment 4.5 mg every 5–8 months
Foscarnet Intravenous CMV retinitis treatment Induction: 60 mg/kg q8h or 90 mg/kg q12h × 14–21 days
Maintenance: 90–120 mg/kg/d
Cidofovir Intravenous CMV retinitis treatment Induction: 5 mg/kg/wk × 2 weeks Maintenance: 5 mg/kg every week
1Dosage must be reduced in patients with renal insufficiency.
049-Katzung_Ch049_p861-890.indd 867 9/23/11 4:25:35 PM

868 SECTION VIII Chemotherapeutic Drugs
Valganciclovir is well absorbed and rapidly metabolized in the intestinal wall and liver to ganciclovir; no other metabolites have been detected. The bioavailability of oral valganciclovir is 60%; it is recommended that the drug be taken with food. The AUC 0–24h resulting from valganciclovir (900 mg once daily) is similar to that after 5 mg/kg once daily of intravenous ganciclovir and approxi-mately 1.65 times that of oral ganciclovir. The major route of elimination is renal, through glomerular filtration and active tubular secretion. Plasma concentrations of valganciclovir are reduced approximately 50% by hemodialysis.
Valganciclovir is indicated for the treatment of CMV retinitis in patients with AIDS and for the prevention of CMV disease in high-risk kidney, heart, and kidney-pancreas transplant patients. Adverse effects, drug interactions, and resistance patterns are the same as those associated with ganciclovir.
FOSCARNET
Foscarnet (phosphonoformic acid) is an inorganic pyrophosphate analog ( Figure 49–2 ) that inhibits herpesvirus DNA polymerase, RNA polymerase, and HIV reverse transcriptase directly without requiring activation by phosphorylation. Foscarnet blocks the pyrophosphate binding site of these enzymes and inhibits cleavage of pyrophosphate from deoxynucleotide triphosphates. It has in vitro activity against HSV, VZV, CMV, EBV, HHV-6, HHV-8, HIV-1, and HIV-2.
Foscarnet is available in an intravenous formulation only; poor oral bioavailability and gastrointestinal intolerance preclude oral use. Cerebrospinal fluid concentrations are 43–67% of steady-state serum concentrations. Although the mean plasma half-life is 3–7 hours, up to 30% of foscarnet may be deposited in bone, with a half-life of several months. The clinical repercussions of this are unknown. Clearance of foscarnet is primarily renal and is directly proportional to creatinine clearance. Serum drug concentrations are reduced approximately 50% by hemodialysis.
Foscarnet is effective in the treatment of CMV retinitis, CMV colitis, CMV esophagitis, acyclovir-resistant HSV infection, and acyclovir-resistant VZV infection. The dosage of foscarnet must be titrated according to the patient’s calculated creatinine clear-ance before each infusion. Use of an infusion pump to control the rate of infusion is important to prevent toxicity, and large volumes of fluid are required because of the drug’s poor solubility. The combination of ganciclovir and foscarnet is synergistic in vitro against CMV and has been shown to be superior to either agent alone in delaying progression of retinitis; however, toxicity is also increased when these agents are administered concurrently. As with ganciclovir, a decrease in the incidence of Kaposi’s sarcoma has been observed in patients who have received long-term foscarnet.
Foscarnet has been administered intravitreally for the treat-ment of CMV retinitis in patients with AIDS, but data regarding efficacy and safety are incomplete.
Resistance to foscarnet in HSV and CMV isolates is due to point mutations in the DNA polymerase gene and is typically associated with prolonged or repeated exposure to the drug.
Mutations in the HIV-1 reverse transcriptase gene have also been described. Although foscarnet-resistant CMV isolates are typically cross-resistant to ganciclovir, foscarnet activity is usually main-tained against ganciclovir- and cidofovir-resistant isolates of CMV.
Potential adverse effects of foscarnet include renal impairment, hypo- or hypercalcemia, hypo- or hyperphosphatemia, hypokalemia, and hypomagnesemia. Saline preloading helps prevent nephrotox-icity, as does avoidance of concomitant administration of drugs with nephrotoxic potential (eg, amphotericin B, pentamidine, aminoglycosides). The risk of severe hypocalcemia, caused by chelation of divalent cations, is increased with concomitant use of pentamidine. Genital ulcerations associated with foscarnet therapy may be due to high levels of ionized drug in the urine. Nausea, vomiting, anemia, elevation of liver enzymes, and fatigue have been reported; the risk of anemia may be additive in patients receiving concurrent zidovudine. Central nervous system toxicity includes headache, hallucinations, and seizures; the risk of seizures may be increased with concurrent use of imipenem. Foscarnet caused chromosomal damage in preclinical studies.
CIDOFOVIR
Cidofovir ( Figure 49–2 ) is a cytosine nucleotide analog with in vitro activity against CMV, HSV-1, HSV-2, VZV, EBV, HHV-6, HHV-8, adenovirus, poxviruses, polyomaviruses, and human papillomavirus. In contrast to ganciclovir, phosphorylation of cidofovir to the active diphosphate is independent of viral enzymes ( Figure 49–3 ); thus activity is maintained against thymidine kinase-deficient or -altered strains of CMV or HSV. Cidofovir diphosphate acts both as a potent inhibitor of and as an alternative substrate for viral DNA polymerase, competitively inhibiting DNA synthesis and becoming incorporated into the viral DNA chain. Cidofovir-resistant isolates tend to be cross-resistant with ganciclovir but retain susceptibility to foscarnet.
Although the terminal half-life of cidofovir is approximately 2.6 hours, the active metabolite cidofovir diphosphate, has a pro-longed intracellular half-life of 17–65 hours, thus allowing infre-quent dosing. A separate metabolite, cidofovir phosphocholine, has a half-life of at least 87 hours and may serve as an intracellular reservoir of active drug. Cerebrospinal fluid penetration is poor. Elimination is by active renal tubular secretion. High-flux hemo-dialysis reduces serum levels of cidofovir by approximately 75%.
Intravenous cidofovir is effective for the treatment of CMV retinitis and is used experimentally to treat adenovirus, human papillomavirus, and poxvirus infections. Intravenous cidofovir must be administered with high-dose probenecid (2 g at 3 hours before the infusion and 1 g at 2 and 8 hours after), which blocks active tubular secretion and decreases nephrotoxicity. Before each infusion, cidofovir dosage must be adjusted for alterations in the calculated creatinine clearance or for the presence of urine protein, and aggressive adjunctive hydration is required. Initiation of cidofovir therapy is contraindicated in patients with existing renal insufficiency. Direct intravitreal administration of cidofovir is not recommended because of ocular toxicity.
049-Katzung_Ch049_p861-890.indd 868 9/23/11 4:25:35 PM

CHAPTER 49 Antiviral Agents 869
The primary adverse effect of intravenous cidofovir is a dose-dependent proximal tubular nephrotoxicity, which may be reduced with prehydration using normal saline. Proteinuria, azotemia, metabolic acidosis, and Fanconi’s syndrome may occur. Concurrent administration of other potentially nephrotoxic agents (eg, ampho-tericin B, aminoglycosides, nonsteroidal anti-inflammatory drugs, pentamidine, foscarnet) should be avoided. Prior administration of foscarnet may increase the risk of nephrotoxicity. Other poten-tial adverse effects include uveitis, ocular hypotony, and neutrope-nia (15–24%). Concurrent probenecid use may result in other toxicities or drug-drug interactions (see Chapter 36 ). Cidofovir is mutagenic, gonadotoxic, and embryotoxic, and caused mammary adenocarcinomas in rats.
■ ANTIRETROVIRAL AGENTS
Substantial advances have been made in antiretroviral therapy since the introduction of the first agent, zidovudine, in 1987 ( Table 49–3 ). Greater knowledge of viral dynamics through the use of viral load and resistance testing has made it clear that combina-tion therapy with maximally potent agents will reduce viral replica-tion to the lowest possible level and decrease the likelihood of emergence of resistance. Thus, administration of combination antiretroviral therapy, typically comprising at least three antiretro-viral agents, has become the standard of care. Viral susceptibility to specific agents varies among patients and may change with time, owing to development of resistance. Therefore, such combinations must be chosen with care and tailored to the individual, as must changes to a given regimen. In addition to potency and susceptibil-ity, important factors in the selection of agents for any given patient are tolerability, convenience, and optimization of adherence.
The retroviral genomic RNA serves as the template for synthe-sis of a double-stranded DNA copy, the provirus ( Figure 49–4 ). Synthesis of the provirus is mediated by a virus-encoded RNA-dependent DNA polymerase, or “reverse transcriptase.” The provirus is translocated to the nucleus and integrated into host DNA. Transcription of this integrated DNA is regulated primarily by cellular machinery.
Six classes of antiretroviral agents are currently available for use: nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors, CCR5 receptor antagonists, and integrase inhibitors. As new agents have become available, several older ones have had diminished usage, because of either suboptimal safety profile or inferior antiviral potency. It is important to recog-nize that the high rate of mutation of HIV-1 per replication cycle results in a great potential for genotypic variation. Genotypic resis-tance has been reported for each of the antiretroviral agents cur-rently in use. Treatment that slows or stops replication is critical in reducing the number of cumulative mutations, as is the use of combinations of agents with differing susceptibility patterns.
Discussion of antiretroviral agents in this chapter is specific to HIV-1. It should be noted that in vitro susceptibility of HIV-2 to the NRTIs is similar to that of HIV-1, albeit with a lower genetic barrier to resistance. There is innate resistance of HIV-2 to the
NNRTIs, due to a different structure of the reverse transcriptases’ NNRTI binding pockets; enfuvirtide (see below) has no activity against HIV-2. Data on the activity of PI agents and maraviroc against HIV-2 are sparse and inconclusive.
NUCLEOSIDE & NUCLEOTIDE REVERSE TRANSCRIPTASE INHIBITORS The NRTIs act by competitive inhibition of HIV-1 reverse tran-scriptase; incorporation into the growing viral DNA chain causes premature chain termination due to inhibition of binding with the incoming nucleotide ( Figure 49–4 ). Each agent requires intra-cytoplasmic activation via phosphorylation by cellular enzymes to the triphosphate form.
Typical resistance mutations include M184V, L74V, D67N, and M41L. Lamivudine or emtricitabine therapy tends to select rapidly for the M184V mutation in regimens that are not fully suppressive. While the M184V mutation confers reduced suscep-tibility to abacavir, didanosine, and zalcitabine, its presence may restore phenotypic susceptibility to zidovudine. The K65R muta-tion is associated with reduced susceptibility to tenofovir, abacavir, lamivudine, and emtricitabine.
All NRTIs may be associated with mitochondrial toxicity, prob-ably owing to inhibition of mitochondrial DNA polymerase gamma. Less commonly, lactic acidosis with hepatic steatosis may occur, which can be fatal. NRTI treatment should be suspended in the setting of rapidly rising aminotransferase levels, progressive hepatomegaly, or metabolic acidosis of unknown cause. The thymi-dine analogs zidovudine and stavudine may be particularly associ-ated with dyslipidemia and insulin resistance. Also, some evidence suggests an increased risk of myocardial infarction in patients receiving abacavir or didanosine; this bears further investigation.
ABACAVIR
Abacavir is a guanosine analog ( Figure 49–2 ) that is well absorbed following oral administration (83%) and is unaffected by food. The serum half-life is 1.5 hours. The drug undergoes hepatic glucuronidation and carboxylation. Cerebrospinal fluid levels are approximately one third those of plasma.
Abacavir is often co-administered with lamivudine, and a once-daily, fixed-dose combination formulation is available. Abacavir is also available in a fixed-dose combination with lamivudine and zidovudine.
High-level resistance to abacavir appears to require at least two or three concomitant mutations and thus tends to develop slowly.
Hypersensitivity reactions, occasionally fatal, have been reported in up to 8% of patients receiving abacavir and may be more severe in association with once-daily dosing. Symptoms, which generally occur within the first 6 weeks of therapy, include fever, fatigue, nausea, vomiting, diarrhea, and abdominal pain.
049-Katzung_Ch049_p861-890.indd 869 9/23/11 4:25:35 PM

870 SECTION VIII Chemotherapeutic Drugs
TABLE 49–3 Currently available antiretroviral agents.
AgentClass of Agent
Recommended Adult Dosage
Administration Recommendation
Characteristic Adverse Effects Comments
Abacavir NRTI1 300 mg bid or 600 mg qd
Testing to rule out the presence of the HLA-B5701 allele is recom-mended prior to the initiation of therapy
Rash, hypersensitivity reaction, nausea. Possible increase in myocardial infarction
Avoid alcohol
Atazanavir PI2 400 mg qd or 300 mg qd with ritonavir 100 mg qd. Adjust dose in hepatic insufficiency
Take with food. Separate dosing from ddI or antacids by 1 h. Separate dosing from cimetidine and other acid-reducing agents by 12 h
Nausea, vomiting, diarrhea, abdominal pain, headache, peripheral neuropathy, skin rash, indirect hyperbiliru-binemia, prolonged PR and/or QTc interval
See footnote 4 for contrain-dicated medications. Also avoid etravirine, fosampre-navir, nevirapine, and proton pump inhibitors. Avoid in severe hepatic insufficiency
Darunavir PI2 Treatment- experienced: 600 mg bid with ritona-vir 100 mg bid. Treatment-naïve: 800 mg qd with ritonavir 100 mg qd. Tablets can be dissolved in water
Take with food Diarrhea, headache, nausea, rash, hyperlipidemia, ↑ liver enzymes, ↑ serum amylase
Avoid in patients with sulfa allergy. See footnote 4 for contraindicated medications
Delavirdine NNRTI 400 mg tid Separate dosing from ddI or antacids by 1 h
Rash, ↑ liver enzymes, headache, nausea, diarrhea
See footnote 4 for contra-indicated medications. Also avoid concurrent fosampre-navir and rifabutin. Teratogenic in rats
Didanosine (ddI) NRTI1 Tablets, 400 mg qd or 200 mg bid,3 adjusted for weight. Buffered powder, 250 mg bid3
30 min before or 2 h after meals. Separate dosing from fluoroquinolones and tetracyclines by 2 h
Peripheral neuropathy, pancreatitis, diarrhea, nausea, hyperuricemia. Possible increase in myocardial infarction
Avoid concurrent neuro-pathic drugs (eg, stavudine, zalcitabine, isoniazid), ribavi-rin, and alcohol. Do not administer with tenofovir
Efavirenz NNRTI 600 mg qd Take on an empty stom-ach. Bedtime dosing recommended initially to minimize central nervous system side effects
Central nervous system effects, rash, ↑ liver enzymes, headache, nausea
See footnote 4 for contrain-dicated medications. Teratogenic in primates
Emtricitabine NRTI1 200 mg qd.3 Tablets can be dissolved in water
Oral solution should be refrigerated
Headache, diarrhea, nausea, asthenia, skin hyperpigmentation
Do not administer concur-rent lamivudine. Avoid disul-firam and metronidazole with oral solution
Enfuvirtide Fusion inhibitor
90 mg subcutaneously bid
Store at room temperature as a powder; refrigerate once reconstituted
Local injection site reactions, hypersensitivity reaction
Etravirine NNRTI 200 mg bid Take after a meal; do not take on an empty stomach
Rash, nausea, diarrhea See footnote 4 for contrain-dicated medications. Do not administer with other NNRTIs, indinavir, atazanavir-ritonavir, fosamprenavir- ritonavir, tipranavir-ritonavir, or any unboosted PI
Fosamprenavir PI2 1400 mg bid or 700 mg bid with ritonavir 100 mg bid or 1400 mg daily with ritonavir 100–200 mg qd. Adjust dose in hepatic insufficiency
Separate dosing from antacids or didanosine by ≥ 1 h. Avoid concurrent high-fat meals
Diarrhea, nausea, vomiting, hypertriglyceridemia, rash, headache, perioral paresthesias, ↑ liver enzymes
See footnote 4 for contraindi-cated medications. Do not administer with etravirine; do not administer with lopinavir/ritonavir or in severe hepatic insufficiency. Also avoid cimetidine, disulfiram, metronidazole, vitamin E, rito-navir oral solution, and alcohol when using the oral solution
(continued )
049-Katzung_Ch049_p861-890.indd 870 9/23/11 4:25:35 PM
CHAPTER 49 Antiviral Agents 871
Indinavir PI2 800 mg tid or 800 mg bid with ritonavir 100–200 mg bid. Adjust dose in hepatic insufficiency
Best on an empty stomach. Drink at least 48 oz liquid daily. Separate dosing from ddI by 1 h. Store in original container, which contains desiccant
Nephrolithiasis, nausea, indirect hyperbilirubinemia, headache, asthenia, blurred vision
See footnote 4 for contrain-dicated medications. Do not administer with etravirine
Lamivudine NRTI1 150 mg bid or 300 mg qd3
Nausea, headache, dizziness, fatigue
Do not administer with zalcitabine
Lopinavir/ritonavir PI/PI2 Treatment-experienced: 400 mg/100 mg bid. Treatment-naïve: 800 mg/200 mg qd. May need dose adjustment in hepatic insufficiency
Take with food. Separate dosing from ddI by 1 h. Store capsules and solution in refrigerator
Diarrhea, abdominal pain, nausea, hypertriglyceri-demia, headache, ↑ liver enzymes
See footnote 4 for contrain-dicated medications. Also avoid fosamprenavir. Avoid disulfiram and metronida-zole with oral solution
Maraviroc CCR5 inhibitor
300 mg bid; 150 bid with CYP3A inhibitors; 600 mg bid with CYP3A inducers3
Muscle and joint pain, diarrhea, sleep disturbance, ↑ liver enzymes
See footnote 4 for medications that must be co-administered with caution. Avoid rifampin
Nelfinavir PI2 750 mg tid or 1250 mg bid
Take with food Diarrhea, nausea, flatulence See footnote 4 for contrain-dicated medications. Do not administer with etravirine
Nevirapine NNRTI 200 mg bid. Adjust dose in hepatic insufficiency
Dose-escalate from 200 mg daily over 14 days to decrease frequency of rash
Rash, hepatitis (occasionally fulminant), nausea, headache
See footnote 4 for contrain-dicated medications. Do not administer with atazanavir
Raltegravir Integrase inhibitor
400 mg bid. Increase dose to 800 mg bid if administered with rifampin
Separate dosing from antacids by ≥ 4 h
Diarrhea, nausea, fatigue, headache, dizziness, muscle aches, ↑ creatine kinase
Avoid rifampin
Ritonavir PI2 600 mg bid Take with food. Separate dosing with ddI by 2 h. Dose-escalate from 300 mg bid over 1–2 weeks to improve tolerance. Refrigerate capsules but not oral solution
Nausea, diarrhea, paresthesias, hepatitis
See footnote 4 for contrain-dicated medications. Avoid disulfiram and metronida-zole with oral solution
Saquinavir PI2 1000 mg bid with ritonavir 100 mg bid
Take within 2 h of a full meal. Refrigeration recommended
Nausea, diarrhea, rhinitis, abdominal pain, dyspepsia, rash
See footnote 4 for contrain-dicated medications. Avoid in severe hepatic insuffi-ciency. Use sunscreen owing to an increase in photosen-sitivity. Avoid concomitant garlic capsules
Stavudine NRTI1 30–40 mg bid, depending on weight3
Peripheral neuropathy, lipodystrophy, hyperlipidemia, rapidly progressive ascending neuromuscular weakness (rare), pancreatitis
Avoid concurrent zidovu-dine and neuropathic drugs (eg, ddI, zalcitabine, isoniazid)
Tenofovir NRTI1 300 mg qd3 Take with food Nausea, diarrhea, vomiting, flatulence, headache, renal insufficiency
Avoid concurrent atazanavir, probenecid, didanosine
Tipranavir PI2 500 mg bid with ritonavir 200 mg bid. Avoid use in hepatic insufficiency
Take with food. Separate from ddI by at least 2 h. Avoid antacids. Avoid in patients with sulfa allergy. Refrigeration required
Diarrhea, nausea, vomiting, abdominal pain, rash, ↑ liver enzymes, hypercholesterolemia, hypertriglyceridemia
See footnote 4 for contrain-dicated medications. Avoid concurrent fosamprenavir, saquinavir, etravirine. Do not administer to patients at risk for bleeding
TABLE 49–3 Currently available antiretroviral agents. (Continued)
AgentClass of Agent
Recommended Adult Dosage
Administration Recommendation
Characteristic Adverse Effects Comments
(continued )
049-Katzung_Ch049_p861-890.indd 871 9/23/11 4:25:35 PM
872 SECTION VIII Chemotherapeutic Drugs
Respiratory symptoms such as dyspnea, pharyngitis, and cough may also be present, and skin rash occurs in about 50% of patients. The laboratory abnormalities of a mildly elevated serum aminotransferase or creatine kinase level may be present but are nonspecific. Although the syndrome tends to resolve quickly with discontinuation of medication, rechallenge with abacavir results in return of symptoms within hours and may be fatal. Testing for the HLA-B5701 allele before initiation of abacavir therapy is recom-mended to identify patients with an increased risk for an abacavir-associated hypersensitivity reaction. Although the positive predictive value of this test is only about 50%, it has a negative predictive value of approximately 100%.
Other potential adverse events are rash, fever, nausea, vomiting, diarrhea, headache, dyspnea, fatigue, and pancreatitis (rare). Abacavir should be used cautiously in patients with existing cardiac risk factors due to a possible increased risk of myocardial events. Since abacavir may lower methadone levels, patients receiving these two agents concurrently should be monitored for signs of opioid withdrawal and may require an increased dose of methadone.
DIDANOSINE
Didanosine (ddI) is a synthetic analog of deoxyadenosine ( Figure 49–2 ). Oral bioavailability is approximately 40%; dosing on an empty stomach is optimal, but buffered formulations are neces-sary to prevent inactivation by gastric acid ( Table 49–3 ). Cerebrospinal fluid concentrations of the drug are approximately 20% of serum concentrations. Serum half-life is 1.5 hours, but the intracellular half-life of the activated compound is as long as 20–24 hours. The drug is eliminated by both cellular metabolism and renal excretion.
The major clinical toxicity associated with didanosine therapy is dose-dependent pancreatitis. Other risk factors for pancreatitis
(eg, alcohol abuse, hypertriglyceridemia) are relative contraindications, and concurrent drugs with the potential to cause pancreatitis, includ-ing zalcitabine, stavudine, ribavirin, and hydroxyurea, should be avoided ( Table 49–3 ). The risk of peripheral distal sensory neuropa-thy, another potential toxicity, may be increased with concurrent use of stavudine, isoniazid, vincristine, or ribavirin. Other reported adverse effects include diarrhea (particularly with the buffered formu-lation), hepatitis, esophageal ulceration, cardiomyopathy, central nervous system toxicity (headache, irritability, insomnia), and hyper-triglyceridemia. Previously asymptomatic hyperuricemia may precipi-tate attacks of gout in susceptible individuals; concurrent use of allopurinol may increase levels of didanosine. Reports of retinal changes and optic neuritis in patients receiving didanosine, particularly in adults receiving high doses and in children, mandate periodic retinal examinations. Lipoatrophy appears to be more common in patients receiving didanosine or other thymidine analogs. As with abacavir, didanosine should be used cautiously in patients with cardiac risk fac-tors due to a possibly increased risk of myocardial infarction.
The buffer in didanosine tablets and powder interferes with absorption of indinavir, delavirdine, atazanavir, dapsone, itracon-azole, and fluoroquinolone agents; therefore, administration should be separated in time. Serum levels of didanosine are increased when co-administered with tenofovir or ganciclovir, and are decreased by atazanavir, delavirdine, ritonavir, tipranavir, and methadone ( Table 49–4 ).
EMTRICITABINE
Emtricitabine (FTC) is a fluorinated analog of lamivudine with a long intracellular half-life (> 24 hours), allowing for once-daily dosing ( Figure 49–2 ). Oral bioavailability of the capsules is 93% and is unaffected by food, but penetration into the cerebrospinal
Zalcitabine NRTI1 0.75 mg tid3 Administer 1 h before or 2 h after an antacid
Peripheral neuropathy; oral ulcerations, pancreatitis, headache, nausea, rash, arthralgias
Avoid concurrent cimeti-dine; avoid concurrent neuropathic drugs (eg, ddI, stavudine, isoniazid). Do not administer with lamivudine
Zidovudine NRTI1 200 mg tid or 300 mg bid3
Macrocytic anemia, neutro-penia, nausea, headache, insomnia, asthenia
Avoid concurrent stavudine and myelosuppressive drugs (eg, ganciclovir, ribavirin)
1All NRTI agents, including tenofovir, carry the risk of lactic acidosis with hepatic steatosis as a potential adverse event.2All PI agents, with the possible exception of fosamprenavir, carry the risk of hyperlipidemia, fat maldistribution, hyperglycemia, and insulin resistance as potential adverse events.3Adjust dose in renal insufficiency.4Because of altered systemic exposures, contraindicated concurrent drugs generally include antiarrhythmics (flecainide, propafenone), antihistamines (astemizole, terfenadine), sedative-hypnotics (alprazolam, diazepam, flurazepam, midazolam, triazolam, trazodone, clorazepate), neuroleptics (pimozide), ergot alkaloid derivatives, HMG-CoA reductase inhibitors (atorvastatin, simvastatin, lovastatin, rosuvastatin), anticonvulsants (phenobarbital, phenytoin), oral contraceptives (ethinyl estradiol/norethindrone acetate), cisapride, rifampin, rifapentine, and St. John’s wort. Drugs that should be used with caution owing to altered levels include amiodarone, bepridil, quinidine, lidocaine, nifedipine, nicar-dipine, felodipine, sildenafil, vardenafil, tadalafil, warfarin, levodopa, tacrolimus, cyclosporine, rapamycin, voriconazole, itraconazole, ketoconazole, carbamazepine, desipramine, bupropion, dofetilide, fluticasone, atovaquone, dapsone, dexamethasone, methadone, omeprazole, and lansoprazole. The dosages of rifabutin and clarithromycin should be decreased when administered concurrently.
NNRTI, nonnucleoside reverse transcriptase inhibitor; NRTI, nucleoside/nucleotide reverse transcriptase inhibitor; PI, protease inhibitor.
TABLE 49–3 Currently available antiretroviral agents. (Continued)
AgentClass of Agent
Recommended Adult Dosage
Administration Recommendation
Characteristic Adverse Effects Comments
049-Katzung_Ch049_p861-890.indd 872 9/23/11 4:25:35 PM

CHAPTER 49 Antiviral Agents 873
fluid is low. Elimination is by both glomerular filtration and active tubular secretion. The serum half-life is about 10 hours.
The oral solution, which contains propylene glycol, is contraindi-cated in young children, pregnant women, patients with renal or hepatic failure, and those using metronidazole or disulfiram. Also, because of its activity against HBV, patients co-infected with HIV and HBV should be closely monitored if treatment with emtricitabine is interrupted or discontinued, owing to the likelihood of hepatitis flare.
Emtricitabine is often co-administered with tenofovir, and a once-daily, fixed-dose combination formulation is available, both alone and in combination with efavirenz. In a recent placebo-controlled study, use of emtricitabine and tenofovir was effective as preexposure prophylaxis, reducing HIV acquisition in men who have sex with men.
Like lamivudine, the M184V/I mutation is most frequently associated with emtricitabine use and may emerge rapidly in
Binding
Reversetranscription
Integration
Transcription
DNA
Translation
RNA Virionassembly
Budding andmaturation
Blocked by protease and maturation inhibitors
Fusion anduncoatingBlocked by
fusion inhibitors
Blocked byNRTIs, NNRTIs
Blocked by integrase inhibitors
gp120
gp41
Chemokine receptors
Host cell membrane
Blocked by CCR5 receptor antagonists
CD4
Nucleus
RNA
FIGURE 49–4 Life cycle of HIV. Binding of viral glycoproteins to host cell CD4 and chemokine receptors leads to fusion of the viral and host cell membranes via gp41 and entry of the virion into the cell. After uncoating, reverse transcription copies the single-stranded HIV RNA genome into double-stranded DNA, which is integrated into the host cell genome. Gene transcription by host cell enzymes produces messenger RNA, which is translated into proteins that assemble into immature noninfectious virions that bud from the host cell membrane. Maturation into fully infectious virions is through proteolytic cleavage. NNRTIs, nonnucleoside reverse transcriptase inhibitors; NRTIs, nucleoside/nucleotide reverse transcriptase inhibitors.
049-Katzung_Ch049_p861-890.indd 873 9/23/11 4:25:35 PM
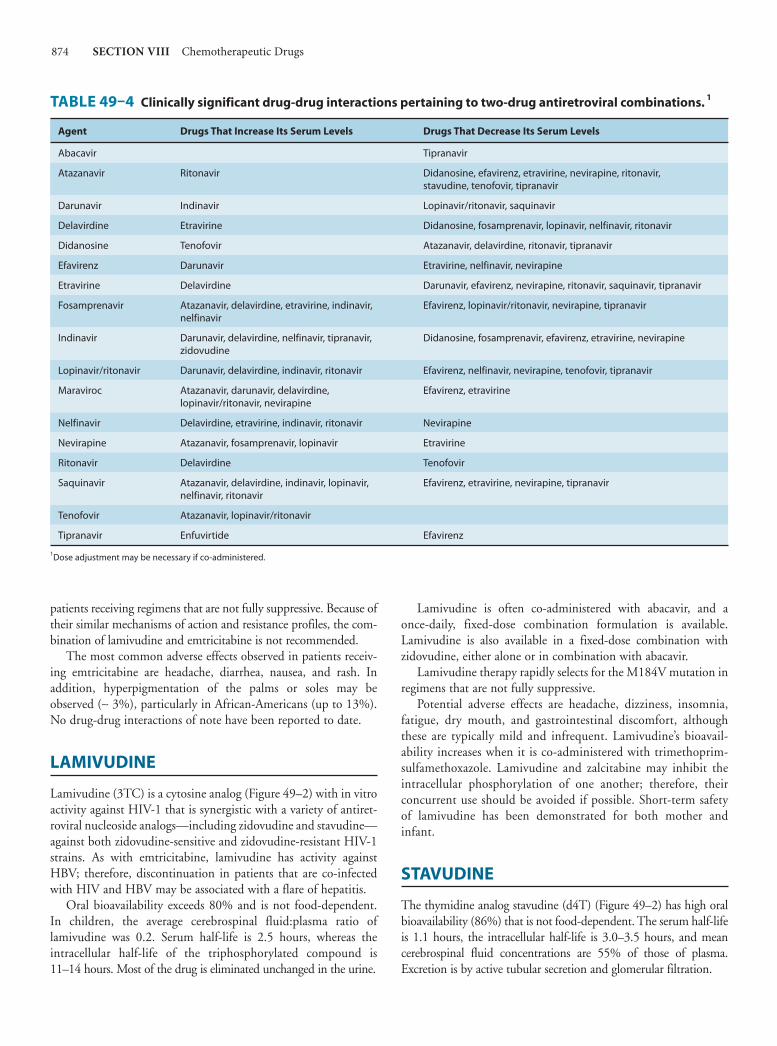
874 SECTION VIII Chemotherapeutic Drugs
patients receiving regimens that are not fully suppressive. Because of their similar mechanisms of action and resistance profiles, the com-bination of lamivudine and emtricitabine is not recommended.
The most common adverse effects observed in patients receiv-ing emtricitabine are headache, diarrhea, nausea, and rash. In addition, hyperpigmentation of the palms or soles may be observed (∼ 3%), particularly in African-Americans (up to 13%). No drug-drug interactions of note have been reported to date.
LAMIVUDINE
Lamivudine (3TC) is a cytosine analog ( Figure 49–2 ) with in vitro activity against HIV-1 that is synergistic with a variety of antiret-roviral nucleoside analogs—including zidovudine and stavudine—against both zidovudine-sensitive and zidovudine-resistant HIV-1 strains. As with emtricitabine, lamivudine has activity against HBV; therefore, discontinuation in patients that are co-infected with HIV and HBV may be associated with a flare of hepatitis.
Oral bioavailability exceeds 80% and is not food-dependent. In children, the average cerebrospinal fluid:plasma ratio of lamivudine was 0.2. Serum half-life is 2.5 hours, whereas the intracellular half-life of the triphosphorylated compound is 11–14 hours. Most of the drug is eliminated unchanged in the urine.
Lamivudine is often co-administered with abacavir, and a once-daily, fixed-dose combination formulation is available. Lamivudine is also available in a fixed-dose combination with zidovudine, either alone or in combination with abacavir.
Lamivudine therapy rapidly selects for the M184V mutation in regimens that are not fully suppressive.
Potential adverse effects are headache, dizziness, insomnia, fatigue, dry mouth, and gastrointestinal discomfort, although these are typically mild and infrequent. Lamivudine’s bioavail-ability increases when it is co-administered with trimethoprim- sulfamethoxazole. Lamivudine and zalcitabine may inhibit the intracellular phosphorylation of one another; therefore, their concurrent use should be avoided if possible. Short-term safety of lamivudine has been demonstrated for both mother and infant.
STAVUDINE
The thymidine analog stavudine (d4T) ( Figure 49–2 ) has high oral bioavailability (86%) that is not food-dependent. The serum half-life is 1.1 hours, the intracellular half-life is 3.0–3.5 hours, and mean cerebrospinal fluid concentrations are 55% of those of plasma. Excretion is by active tubular secretion and glomerular filtration.
TABLE 49–4 Clinically significant drug-drug interactions pertaining to two-drug antiretroviral combinations. 1
Agent Drugs That Increase Its Serum Levels Drugs That Decrease Its Serum Levels
Abacavir Tipranavir
Atazanavir Ritonavir Didanosine, efavirenz, etravirine, nevirapine, ritonavir, stavudine, tenofovir, tipranavir
Darunavir Indinavir Lopinavir/ritonavir, saquinavir
Delavirdine Etravirine Didanosine, fosamprenavir, lopinavir, nelfinavir, ritonavir
Didanosine Tenofovir Atazanavir, delavirdine, ritonavir, tipranavir
Efavirenz Darunavir Etravirine, nelfinavir, nevirapine
Etravirine Delavirdine Darunavir, efavirenz, nevirapine, ritonavir, saquinavir, tipranavir
Fosamprenavir Atazanavir, delavirdine, etravirine, indinavir, nelfinavir
Efavirenz, lopinavir/ritonavir, nevirapine, tipranavir
Indinavir Darunavir, delavirdine, nelfinavir, tipranavir, zidovudine
Didanosine, fosamprenavir, efavirenz, etravirine, nevirapine
Lopinavir/ritonavir Darunavir, delavirdine, indinavir, ritonavir Efavirenz, nelfinavir, nevirapine, tenofovir, tipranavir
Maraviroc Atazanavir, darunavir, delavirdine, lopinavir/ritonavir, nevirapine
Efavirenz, etravirine
Nelfinavir Delavirdine, etravirine, indinavir, ritonavir Nevirapine
Nevirapine Atazanavir, fosamprenavir, lopinavir Etravirine
Ritonavir Delavirdine Tenofovir
Saquinavir Atazanavir, delavirdine, indinavir, lopinavir, nelfinavir, ritonavir
Efavirenz, etravirine, nevirapine, tipranavir
Tenofovir Atazanavir, lopinavir/ritonavir
Tipranavir Enfuvirtide Efavirenz
1Dose adjustment may be necessary if co-administered.
049-Katzung_Ch049_p861-890.indd 874 9/23/11 4:25:35 PM

CHAPTER 49 Antiviral Agents 875
The major toxicity is a dose-related peripheral sensory neuropathy. The incidence of neuropathy may be increased when stavudine is administered with other neuropathy-inducing drugs such as didanosine, zalcitabine, vincristine, isoniazid, or ribavirin, or in patients with advanced immunosuppression. Symptoms typically resolve upon discontinuation of stavudine; in such cases, a reduced dosage may be cautiously restarted. Other potential adverse effects are pancreatitis, arthralgias, and elevation in serum aminotransferases. Lactic acidosis with hepatic steatosis, as well as lipodystrophy, appear to occur more frequently in patients receiv-ing stavudine than in those receiving other NRTI agents. Moreover, because the co-administration of stavudine and didanos-ine may increase the incidence of lactic acidosis and pancreatitis, concurrent use should be avoided. This combination has been implicated in several deaths in HIV-infected pregnant women. A rare adverse effect is a rapidly progressive ascending neuromuscu-lar weakness. Since zidovudine may reduce the phosphorylation of stavudine, these two drugs should not be used together. There is no evidence of human teratogenicity in those taking stavudine.
TENOFOVIR
Tenofovir is an acyclic nucleoside phosphonate (ie, nucleotide) analog of adenosine ( Figure 49–2 ). Like the nucleoside analogs, tenofovir competitively inhibits HIV reverse transcriptase and causes chain termination after incorporation into DNA. However, only two rather than three intracellular phosphorylations are required for active inhibition of DNA synthesis. Tenofovir is also approved for the treatment of patients with HBV infection.
Tenofovir disoproxil fumarate is a water-soluble prodrug of active tenofovir. The oral bioavailability in fasted patients is approximately 25% and increases to 39% after a high-fat meal. The prolonged serum (12–17 hours) and intracellular half-lives allow once-daily dosing. Elimination occurs by both glomerular filtration and active tubular secretion.
Tenofovir is often co-administered with emtricitabine, and a once-daily, fixed-dose combination formulation is available, either alone or in combination with efavirenz. A recent placebo-controlled study found that use of emtricitabine and tenofovir was effective as preexposure prophylaxis, reducing HIV acquisi-tion in men who have sex with men. In another placebo-con-trolled study, use of the experimental 1% tenofovir gel as a vaginal microbicide was effective in decreasing the incidence of heterosexual HIV acquisition.
The primary mutation associated with resistance to tenofovir is K65R.
Gastrointestinal complaints (eg, nausea, diarrhea, vomiting, flatulence) are the most common adverse effects but rarely require discontinuation of therapy. Since tenofovir is formulated with lactose, these may occur more frequently in patients with lactose intolerance. Other potential adverse effects include headache and asthenia. Tenofovir-associated proximal renal tubulopathy causes excessive renal phosphate and calcium losses and 1-hydroxylation defects of vitamin D, and preclinical studies in several animal spe-cies have demonstrated bone toxicity (eg, osteomalacia). Monitoring
of bone mineral density should be considered with long-term use in those with risk factors for or with known osteoporosis, as well as in children. Reduction of renal function over time, as well as cases of acute renal failure and Fanconi’s syndrome, have been reported in patients receiving tenofovir alone or in combination with emtricit-abine. For this reason, tenofovir should be used with caution in patients at risk for renal dysfunction. Tenofovir may compete with other drugs that are actively secreted by the kidneys, such as cidofovir, acyclovir, and ganciclovir. Concurrent use of atazanavir or lopinavir/ritonavir may increase serum levels of tenofovir ( Table 49–4 ).
Tenofovir is associated with decreased fetal growth and reduc-tion in fetal bone porosity in monkeys. There is significant placental passage in humans.
ZALCITABINE
Zalcitabine (ddC) is a cytosine analog with high oral bioavailabil-ity (87%) and a serum half-life of 1–2 hours ( Figure 49–2 ). An intracellular half-life of 2.6 hours necessitates thrice-daily dosing, which limits its usefulness. Plasma levels decrease by 25–39% when the drug is administered with food or antacids. The drug is excreted renally. Cerebrospinal fluid concentrations are approxi-mately 20% of those in the plasma.
Although a variety of mutations associated with in vitro resis-tance to zalcitabine have been described, phenotypic resistance appears to be rare.
Zalcitabine therapy is associated with a dose-dependent periph-eral neuropathy that can be treatment-limiting in 10–20% of patients but appears to be slowly reversible if treatment is stopped promptly. The potential for causing peripheral neuropathy consti-tutes a relative contraindication to use with other drugs that may cause neuropathy, including stavudine, didanosine, isoniazid, vincristine, and ribavirin. Decreased creatinine clearance or concur-rent use of potential nephrotoxins (eg, amphotericin B, foscarnet, and aminoglycosides) may increase the risk of zalcitabine neuropa-thy, as does more advanced immunosuppression. The other major reported toxicity is oral and esophageal ulceration. Pancreatitis occurs less frequently than with didanosine administration, but co-administration of other drugs that cause pancreatitis may increase the frequency of this adverse effect. Headache, nausea, rash, and arthralgias may occur but tend to be mild or to resolve during therapy. Zalcitabine causes thymic lymphoma in rodents, as well as hydrocephalus at high doses; clinical relevance is unclear. The AUC of zalcitabine increases when co-administered with probenecid or cimetidine, and bioavailability decreases with concurrent antacids or metoclopramide. Lamivudine inhibits the phosphorylation of zalcitabine in vitro, potentially interfering with its efficacy.
ZIDOVUDINE
Zidovudine ( azidothymidine; AZT ) is a deoxythymidine analog ( Figure 49–2 ) that is well absorbed (63%) and distributed to most body tissues and fluids, including the cerebrospinal fluid, where drug levels are 60–65% of those in serum. Although the serum
049-Katzung_Ch049_p861-890.indd 875 9/23/11 4:25:35 PM

876 SECTION VIII Chemotherapeutic Drugs
half-life averages 1 hour, the intracellular half-life of the phospho-rylated compound is 3–4 hours, allowing twice-daily dosing. Zidovudine is eliminated primarily by renal excretion following glucuronidation in the liver.
Zidovudine is available in a fixed-dose combination formulation with lamivudine, either alone or in combination with abacavir.
Zidovudine was the first antiretroviral agent to be approved and has been well studied. The drug has been shown to decrease the rate of clinical disease progression and prolong survival in HIV-infected individuals. Efficacy has also been demonstrated in the treatment of HIV-associated dementia and thrombocytope-nia. In pregnancy ( Table 49–5 ), a regimen of oral zidovudine beginning between 14 and 34 weeks of gestation, intravenous zidovudine during labor, and zidovudine syrup to the neonate from birth through 6 weeks of age has been shown to reduce the rate of vertical (mother-to-newborn) transmission of HIV by up to 23%.
High-level zidovudine resistance is generally seen in strains with three or more of the five most common mutations: M41L, D67N, K70R, T215F, and K219Q. However, the emergence of certain mutations that confer decreased susceptibility to one drug (eg, L74V for didanosine and M184V for lamivudine) may enhance zidovudine susceptibility in previously zidovudine- resistant strains. Withdrawal of zidovudine may permit the rever-sion of zidovudine-resistant HIV-1 isolates to the susceptible wild-type phenotype.
The most common adverse effect of zidovudine is myelosup-pression, resulting in macrocytic anemia (1–4%) or neutropenia (2–8%). Gastrointestinal intolerance, headaches, and insomnia may occur but tend to resolve during therapy. Lipoatrophy appears to be more common in patients receiving zidovudine or other thymidine analogs. Less common toxicities include throm-bocytopenia, hyperpigmentation of the nails, and myopathy. High doses can cause anxiety, confusion, and tremulousness. Zidovudine causes vaginal neoplasms in mice; however, no human cases of genital neoplasms have been reported to date. Short-term safety has been demonstrated for both mother and infant.
Increased serum levels of zidovudine may occur with concomi-tant administration of probenecid, phenytoin, methadone, flucon-azole, atovaquone, valproic acid, and lamivudine, either through inhibition of first-pass metabolism or through decreased clearance. Zidovudine may decrease phenytoin levels. Hematologic toxicity may be increased during co-administration of other myelosuppres-sive drugs such as ganciclovir, ribavirin, and cytotoxic agents. Combination regimens containing zidovudine and stavudine should be avoided due to in vitro antagonism.
NONNUCLEOSIDE REVERSE TRANSCRIPTASE INHIBITORS The NNRTIs bind directly to HIV-1 reverse transcriptase ( Figure 49–4 ), resulting in allosteric inhibition of RNA- and DNA-dependent DNA polymerase activity. The binding site of NNRTIs is near to but distinct from that of NRTIs. Unlike the NRTI agents, NNRTIs neither compete with nucleoside triphosphates nor require phosphorylation to be active.
Baseline genotypic testing is recommended prior to initiating NNRTI treatment because primary resistance rates range from approximately 2% to 8%. NNRTI resistance occurs rapidly with monotherapy and can result from a single mutation. The K103N and Y181C mutations confer resistance across the entire class of NNRTIs, with the exception of the newest agent, etravirine. Other mutations (eg, L100I, Y188C, G190A) may confer cross-resistance among the NNRTI class. However, there is no cross-resistance between the NNRTIs and the NRTIs; in fact, some nucleoside-resistant viruses display hypersusceptibility to NNRTIs.
As a class, NNRTI agents tend to be associated with varying levels of gastrointestinal intolerance and skin rash, the latter of which may infrequently be serious (eg, Stevens-Johnson syndrome). A further limitation to use of NNRTI agents as a component of antiretroviral therapy is their metabolism by the CYP450 system, leading to innumerable potential drug-drug interactions ( Tables 49–3 and 49–4 ). All NNRTI agents are substrates for CYP3A4 and can act as inducers (nevirapine), inhibitors (delavirdine), or mixed inducers and inhibitors (efavirenz, etravirine). Given the large number of non-HIV medications that are also metabolized by this pathway (see Chapter 4 ), drug-drug interactions must be expected and looked for; dosage adjustments are frequently required and some combinations are contraindicated.
DELAVIRDINE
Delavirdine has an oral bioavailability of about 85%, but this is reduced by antacids or H 2 -blockers. It is extensively bound (∼ 98%) to plasma proteins and has correspondingly low cerebrospi-nal fluid levels. Serum half-life is approximately 6 hours.
Skin rash occurs in up to 38% of patients receiving delavirdine; it typically occurs during the first 1–3 weeks of therapy and does not preclude rechallenge. However, severe rash such as erythema multiforme and Stevens-Johnson syndrome have rarely been reported. Other possible adverse effects are headache, fatigue, nausea, diarrhea, and increased serum aminotransferase levels.
TABLE 49–5 The use of antiretroviral agents in pregnancy. 1
Recommended Agents Alternate Agents
Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs)
Lamivudine, zidovudine Abacavir, didanosine, emtricitabine, stavudine
Nonnucleoside reverse transcriptase inhibitors (NNRTIs)
Nevirapine
Protease inhibitors (PIs)
Lopinavir/ritonavir Atazanavir/ritonavir, indinavir/ritonavir, nelfinavir, ritonavir, saquinavir
1Data are insufficient to recommend the use of entry inhibitors or integrase inhibitors in pregnancy at the present time.
049-Katzung_Ch049_p861-890.indd 876 9/23/11 4:25:35 PM

CHAPTER 49 Antiviral Agents 877
Delavirdine has been shown to be teratogenic in rats, causing ventricular septal defects and other malformations at dosages not unlike those achieved in humans. Thus, pregnancy should be avoided when taking delavirdine.
Delavirdine is extensively metabolized by the CYP3A and CYP2D6 enzymes and also inhibits CYP3A4 and 2C9. Therefore, there are numerous potential drug-drug interactions to consider ( Tables 49–3 and 49–4 ). The concurrent use of delavirdine with fosamprenavir and rifabutin is not recom-mended because of decreased delavirdine levels. Other medica-tions likely to alter delavirdine levels include didanosine, lopinavir, nelfinavir, and ritonavir. Co-administration of dela-virdine with indinavir or saquinavir prolongs the elimination half-life of these protease inhibitors, thus allowing them to be dosed twice rather than thrice daily.
EFAVIRENZ
Efavirenz can be given once daily because of its long half-life (40–55 hours). It is moderately well absorbed following oral administration (45%). Since toxicity may increase owing to increased bioavailability after a high-fat meal, efavirenz should be taken on an empty stomach. Efavirenz is principally metabolized by CYP3A4 and CYP2B6 to inactive hydroxylated metabolites; the remainder is eliminated in the feces as unchanged drug. It is highly bound to albumin (∼ 99%), and cerebrospinal fluid levels range from 0.3% to 1.2% of plasma levels.
The principal adverse effects of efavirenz involve the central nervous system. Dizziness, drowsiness, insomnia, nightmares, and headache tend to diminish with continued therapy; dosing at bedtime may also be helpful. Psychiatric symptoms such as depres-sion, mania, and psychosis have been observed and may necessi-tate discontinuation. Skin rash has also been reported early in therapy in up to 28% of patients; the rash is usually mild to moderate in severity and typically resolves despite continuation. Rarely, rash has been severe or life-threatening. Other potential adverse reactions are nausea, vomiting, diarrhea, crystalluria, ele-vated liver enzymes, and an increase in total serum cholesterol by 10–20%. High rates of fetal abnormalities occurred in pregnant monkeys exposed to efavirenz in doses roughly equivalent to the human dosage; several cases of congenital anomalies have been reported in humans. Therefore, efavirenz should be avoided in pregnant women, particularly in the first trimester.
As both an inducer and an inhibitor of CYP3A4, efavirenz induces its own metabolism and interacts with the metabolism of many other drugs ( Tables 49–3 and 49–4 ). Since efavirenz may lower methadone levels, patients receiving these two agents con-currently should be monitored for signs of opioid withdrawal and may require an increased dose of methadone.
ETRAVIRINE
In 2008, etravirine was approved in the United States for use in treatment-experienced patients with HIV infection. Etravirine
may be effective against strains of HIV that have developed resis-tance to first-generation NNRTIs, depending on the number of mutations present. Although etravirine has a higher genetic barrier to resistance than the other NNRTIs, mutations selected by etravirine usually are associated with resistance to efavirenz, nevirapine, and delavirdine.
The most common adverse effects of etravirine are rash, nausea, and diarrhea. The rash is typically mild and usually resolves after 1–2 weeks without discontinuation of therapy. Rarely, rash has been severe or life-threatening. Laboratory abnormalities include elevations in serum cholesterol, triglyceride, glucose, and hepatic transaminase levels. Transaminase elevations are more common in patients with HBV or HCV co-infection.
Etravirine is a substrate as well as an inducer of CYP3A4 and an inhibitor of CYP2C9 and CYP2C19; it has many therapeuti-cally significant drug-drug interactions ( Tables 49–3 and 49–4 ). Some of the interactions are difficult to predict. For example, etravirine may decrease itraconazole and ketoconazole concentra-tions but increase voriconazole concentrations.
NEVIRAPINE
The oral bioavailability of nevirapine is excellent (> 90%) and is not food-dependent. The drug is highly lipophilic and achieves cerebrospinal fluid levels that are 45% of those in plasma. Serum half-life is 25–30 hours. It is extensively metabolized by the CYP3A isoform to hydroxylated metabolites and then excreted, primarily in the urine.
A single dose of nevirapine (200 mg) is effective in the preven-tion of transmission of HIV from mother to newborn when administered to women at the onset of labor and followed by a 2 mg/kg oral dose to the neonate within 3 days after delivery. There is no evidence of human teratogenicity. However, resistance has been documented after this single dose.
Rash, usually a maculopapular eruption that spares the palms and soles, occurs in up to 20% of patients, usually in the first 4–6 weeks of therapy. Although typically mild and self-limited, rash is dose-limiting in about 7% of patients. Women appear to have an increased incidence of rash. When initiating therapy, gradual dose escalation over 14 days is recommended to decrease the incidence of rash. Severe and life-threatening skin rashes have been rarely reported, including Stevens-Johnson syndrome and toxic epidermal necrolysis. Nevirapine therapy should be immedi-ately discontinued in patients with severe rash and in those with accompanying constitutional symptoms; since rash may accom-pany hepatotoxicity, liver function tests should be assessed. Symptomatic liver toxicity may occur in up to 4% of patients and is more frequent in those with higher pretherapy CD4 cell counts (ie, > 250 cells/mm 3 in women and > 400 cells/mm 3 in men), in women, and in those with HBV or HCV co-infection. Fulminant, life-threatening hepatitis has been reported, typically within the first 18 weeks of therapy. Other adverse effects include fever, nau-sea, headache, and somnolence.
Nevirapine is a moderate inducer of CYP3A metabolism, resulting in decreased levels of amprenavir, indinavir, lopinavir,
049-Katzung_Ch049_p861-890.indd 877 9/23/11 4:25:36 PM

878 SECTION VIII Chemotherapeutic Drugs
saquinavir, efavirenz, and methadone. Drugs that induce the CYP3A system, such as rifampin, rifabutin, and St. John’s wort, can decrease levels of nevirapine, whereas those that inhibit CYP3A activity, such as fluconazole, ketoconazole, and clarithro-mycin, can increase nevirapine levels. Since nevirapine may lower methadone levels, patients receiving these two agents concurrently should be monitored for signs of opioid withdrawal and may require an increased dose of methadone.
PROTEASE INHIBITORS During the later stages of the HIV growth cycle, the gag and gag - pol gene products are translated into polyproteins, and these become immature budding particles. The HIV protease is respon-sible for cleaving these precursor molecules to produce the final structural proteins of the mature virion core. By preventing post-translational cleavage of the Gag-Pol polyprotein, protease inhibi-tors (PIs) prevent the processing of viral proteins into functional conformations, resulting in the production of immature, nonin-fectious viral particles ( Figure 49–4 ). Unlike the NRTIs, PIs do not need intracellular activation.
Specific genotypic alterations that confer phenotypic resistance are fairly common with these agents, thus contraindicating mono-therapy. Some of the most common mutations conferring broad resistance to PIs are substitutions at the 10, 46, 54, 82, 84, and 90 codons; the number of mutations may predict the level of phe-notypic resistance. The I50L substitution emerging during atazana-vir therapy has been associated with increased susceptibility to other PIs. Darunavir and tipranavir appear to have improved virologic activity in patients harboring HIV-1 resistant to other PIs.
A syndrome of redistribution and accumulation of body fat that results in central obesity, dorsocervical fat enlargement (buffalo hump), peripheral and facial wasting, breast enlargement, and a cushingoid appearance has been observed in patients receiv-ing antiretroviral therapy. These abnormalities may be particularly associated with the use of PIs, although the recently licensed atazanavir appears to be an exception (see below). Concurrent increases in triglyceride and low-density lipoprotein levels, along with hyperglycemia and insulin resistance, have also been noted. The cause is not yet known.
Whether PI agents are associated with bone loss and osteoporosis after long-term use is controversial and under investigation. PIs have been associated with increased spontaneous bleeding in patients with hemophilia A or B; an increased risk of intracranial hemorrhage has been reported in patients receiving tipranavir with ritonavir.
The concurrent use of saquinavir and ritonavir has recently been found to be associated with QT and PR interval prolonga-tion, and is contraindicated. QT prolongation may result in life-threatening torsades de pointes arrhythmia.
All of the antiretroviral PIs are extensively metabolized by CYP3A4, with ritonavir having the most pronounced inhibitory effect and saquinavir the least. Some PI agents, such as amprenavir and ritonavir, are also inducers of specific CYP isoforms. As a result, there is enormous potential for drug-drug interactions with other antiretroviral agents and other commonly used medications
( Tables 49–3 and 49–4 ). Expert resources about drug-drug inter-actions should be consulted, as dosage adjustments are frequently required and some combinations are contraindicated. It is note-worthy that the potent CYP3A4 inhibitory properties of ritonavir are used to clinical advantage by having it “boost” the levels of other PI agents when given in combination, thus acting as a phar-macokinetic enhancer rather than an antiretroviral agent. Ritonavir boosting increases drug exposure, thereby prolonging the drug’s half-life and allowing reduction in frequency; in addition, the genetic barrier to resistance is raised.
ATAZANAVIR
Atazanavir is an azapeptide PI with a pharmacokinetic profile that allows once-daily dosing. It should be taken with a light meal to enhance bioavailability. Atazanavir requires an acidic medium for absorption and exhibits pH-dependent aqueous solubility; there-fore, separation of ingestion from acid-reducing agents by at least 12 hours is recommended and concurrent proton pump inhibitors are contraindicated. Atazanavir is able to penetrate both the cerebro-spinal and seminal fluids. The plasma half-life is 6–7 hours, which increases to approximately 11 hours when co-administered with ritonavir. The primary route of elimination is biliary; atazanavir should not be given to patients with severe hepatic insufficiency.
Resistance to atazanavir has been associated with various known PI mutations as well as with the novel I50L substitution. Whereas some atazanavir resistance mutations have been associated in vitro with decreased susceptibility to other PIs, the I50L mutation has been associated with increased susceptibility to other PIs.
The most common adverse effects in patients receiving ataza-navir are diarrhea and nausea; vomiting, abdominal pain, head-ache, peripheral neuropathy, and skin rash may also occur. As with indinavir, indirect hyperbilirubinemia with overt jaundice may occur in approximately 10% of patients, owing to inhibition of the UGT1A1 glucuronidation enzyme. Elevation of hepatic enzymes has also been observed, usually in patients with underly-ing HBV or HCV co-infection. Nephrolithiasis has recently been described in association with atazanavir use. In contrast to the other PIs, atazanavir does not appear to be associated with dys-lipidemia, fat redistribution, or the metabolic syndrome. Atazanavir may be associated with prolongation of the electrocardiographic PR interval, which is usually inconsequential but may be exacer-bated by other causative agents such as calcium channel blockers and may result in AV block QT prolongation is another potential electrocardiographic effect of atazanavir but is rarely clinically significant.
As an inhibitor of CYP3A4 and CYP2C9, the potential for drug-drug interactions with atazanavir is great ( Tables 49–3 and 49–4 ). Atazanavir AUC is reduced by up to 76% when combined with a proton pump inhibitor; thus, these combinations are to be avoided. In addition, co-administration of atazanavir with other drugs that inhibit UGT1A1, such as irinotecan, may increase its levels. Tenofovir and efavirenz should not be co-administered with atazanavir unless ritonavir is added to boost levels.
049-Katzung_Ch049_p861-890.indd 878 9/23/11 4:25:36 PM

CHAPTER 49 Antiviral Agents 879
DARUNAVIR
Darunavir is licensed as a PI that must be co-administered with ritonavir. It was initially licensed for use in treatment-experienced patients only; thus there is less clinical experience with its use in treatment-naïve patients. Darunavir may be administered once daily in treatment-naïve patients.
Symptomatic adverse effects of darunavir include diarrhea, nausea, headache, and rash. Laboratory abnormalities include dyslipidemia (though possibly less frequent than with other boosted PI regimens) and increases in amylase and hepatic transaminase levels. Liver toxicity, including severe hepatitis, has been reported in some patients taking darunavir; the risk of hepatotoxicity may be higher for persons with HBV, HCV, or other chronic liver disease.
Darunavir contains a sulfonamide moiety and should be used cautiously in patients with sulfonamide allergy.
Darunavir both inhibits and is metabolized by the CYP3A enzyme system, conferring many possible drug-drug interactions ( Tables 49–3 and 49–4 ). In addition, the co-administered ritona-vir is a potent inhibitor of CYP3A and CYP2D6, and an inducer of other hepatic enzyme systems.
FOSAMPRENAVIR
Fosamprenavir is a prodrug of amprenavir that is rapidly hydro-lyzed by enzymes in the intestinal epithelium. Because of its sig-nificantly lower daily pill burden, fosamprenavir tablets have replaced amprenavir capsules for adults. Fosamprenavir is most often administered in combination with low-dose ritonavir.
Amprenavir is rapidly absorbed from the gastrointestinal tract, and its prodrug can be taken with or without food. However, high-fat meals decrease absorption and thus should be avoided. The plasma half-life is relatively long (7–11 hours). Amprenavir is metabolized in the liver by CYP3A4 and should be used with caution in the setting of hepatic insufficiency.
The most common adverse effects of fosamprenavir are headache, nausea, diarrhea, perioral paresthesias, depression, and rash. Up to 3% of patients may experience rashes (including Stevens-Johnson syndrome) severe enough to warrant drug discontinuation.
Amprenavir is both an inducer and an inhibitor of CYP3A4 and is contraindicated with numerous drugs ( Tables 49–3 and 49–4 ). The oral solution, which contains propylene glycol, is con-traindicated in young children, pregnant women, patients with renal or hepatic failure, and those using metronidazole or disul-firam. Also, the oral solutions of amprenavir and ritonavir should not be co-administered because the propylene glycol in one and the ethanol in the other may compete for the same metabolic pathway, leading to accumulation of either. Because the oral solu-tion also contains vitamin E at several times the recommended daily dosage, supplemental vitamin E should be avoided. Amprenavir, a sulfonamide, is contraindicated in patients with a history of sulfa allergy. Lopinavir/ritonavir should not be co- administered with amprenavir owing to decreased amprenavir and
altered lopinavir exposures. An increased dosage of amprenavir is recommended when co-administered with efavirenz (with or without the addition of ritonavir to boost levels).
INDINAVIR
Indinavir requires an acidic environment for optimum solubility and therefore must be consumed on an empty stomach or with a small, low-fat, low-protein meal for maximal absorption (60–65%). The serum half-life is 1.5–2 hours, protein binding is approxi-mately 60%, and the drug has a high level of cerebrospinal fluid penetration (up to 76% of serum levels). Excretion is primarily fecal. An increase in AUC by 60% and in half-life to 2.8 hours in the setting of hepatic insufficiency necessitates dose reduction.
The most common adverse effects of indinavir are indirect hyperbilirubinemia and nephrolithiasis due to urinary crystalliza-tion of the drug. Nephrolithiasis can occur within days after initiat-ing therapy, with an estimated incidence of approximately 10%. Consumption of at least 48 ounces of water daily is important to maintain adequate hydration. Thrombocytopenia, elevations of serum aminotransferase levels, nausea, diarrhea, insomnia, dry throat, dry skin, and indirect hyperbilirubinemia have also been reported. Insulin resistance may be more common with indinavir than with the other PIs, occurring in 3–5% of patients. There have also been rare cases of acute hemolytic anemia. In rats, high doses of indinavir are associated with development of thyroid adenomas.
Since indinavir is an inhibitor of CYP3A4, numerous and complex drug interactions can occur ( Tables 49–3 and 49–4 ). Combination with ritonavir (boosting) allows for twice-daily rather than thrice-daily dosing and eliminates the food restriction associated with use of indinavir. However, there is potential for an increase in nephrolithiasis with this combination compared with indinavir alone; thus, a high fluid intake (1.5–2 L/d) is advised.
LOPINAVIR
Lopinavir is currently formulated only in combination with ritonavir, which inhibits the CYP3A-mediated metabolism of lopinavir, thereby resulting in increased exposure to lopinavir. In addition to improved patient compliance due to reduced pill burden, lopinavir/ritonavir is generally well tolerated.
Lopinavir should be taken with food to enhance bioavailability. The drug is highly protein bound (98–99%), and its half-life is 5–6 hours. Lopinavir is extensively metabolized by CYP3A, which is inhibited by ritonavir. Serum levels of lopinavir may be increased in patients with hepatic impairment.
The most common adverse effects of lopinavir are diarrhea, abdominal pain, nausea, vomiting, and asthenia. Elevations in serum cholesterol and triglycerides are common. Potential drug-drug inter-actions are extensive ( Tables 49–3 and 49–4 ). Increased dosage of lopinavir/ritonavir is recommended when co-administered with efavirenz or nevirapine, which induce lopinavir metabolism. Concurrent use of fosamprenavir should be avoided owing to altered
049-Katzung_Ch049_p861-890.indd 879 9/23/11 4:25:36 PM

880 SECTION VIII Chemotherapeutic Drugs
exposure to lopinavir with decreased levels of amprenavir. Also, con-comitant use of lopinavir/ritonavir and rifampin is contraindicated due to an increased risk for hepatotoxicity. Since the oral solution of lopinavir/ritonavir contains alcohol, concurrent disulfiram and metronidazole are contraindicated. There is no evidence of human teratogenicity of lopinavir/ritonavir; short-term safety in pregnant women has been demonstrated for mother and infant.
NELFINAVIR
Nelfinavir has high absorption in the fed state (70–80%), under-goes metabolism by CYP3A, and is excreted primarily in the feces. The plasma half-life in humans is 3.5–5 hours, and the drug is more than 98% protein-bound.
The most common adverse effects associated with nelfinavir are diarrhea and flatulence. Diarrhea often responds to anti-diarrheal medications but can be dose-limiting. Nelfinavir is an inhibitor of the CYP3A system, and multiple drug interactions may occur ( Tables 49–3 and 49–4 ). An increased dosage of nelfi-navir is recommended when co-administered with rifabutin (with a decreased dose of rifabutin), whereas a decrease in saquinavir dose is suggested with concurrent nelfinavir. Co-administration with efavirenz should be avoided due to decreased nelfinavir levels. Nelfinavir has a favorable safety and pharmacokinetic profile for pregnant women compared with that of other PIs ( Table 49–5 ); there is no evidence of human teratogenicity.
RITONAVIR
Ritonavir has a high bioavailability (about 75%) that increases with food. It is 98% protein-bound and has a serum half-life of 3–5 hours. Metabolism to an active metabolite occurs via the CYP3A and CYP2D6 isoforms; excretion is primarily in the feces. Caution is advised when administering the drug to persons with impaired hepatic function.
Potential adverse effects of ritonavir, particularly when adminis-tered at full dosage, are gastrointestinal disturbances, paresthesias (circumoral or peripheral), elevated serum aminotransferase levels, altered taste, headache, and elevations in serum creatine kinase. Nausea, vomiting, diarrhea, or abdominal pain typically occur during the first few weeks of therapy but may diminish over time or if the drug is taken with meals. Dose escalation over 1–2 weeks is recommended to decrease the dose-limiting side effects. Liver adenomas and carcinomas have been induced in male mice receiv-ing ritonavir; no similar effects have been observed to date in humans.
Ritonavir is a potent inhibitor of CYP3A4, resulting in many potential drug interactions ( Tables 49–3 and 49–4 ). However, this characteristic has been used to great advantage when ritonavir is administered in low doses (100–200 mg twice daily) in combina-tion with any of the other PI agents, in that increased blood levels of the latter agents permit lower or less frequent dosing (or both) with greater tolerability as well as the potential for greater efficacy against resistant virus. Therapeutic levels of digoxin and theophyl-line should be monitored when co-administered with ritonavir
owing to a likely increase in their concentrations. The concurrent use of saquinavir and ritonavir is contraindicated due to an increased risk of QT prolongation (with torsades de pointes arrhythmia) and PR interval prolongation. There is limited experience with full-dose ritonavir during pregnancy to date; however, low-dose ritonavir as a “booster” has appeared to be well tolerated in mother and infant.
SAQUINAVIR
In its original formulation as a hard gel capsule (saquinavir-H; Invirase), oral saquinavir was poorly bioavailable (only about 4% after food). However, reformulation of saquinavir-H for once-daily dosing in combination with low-dose ritonavir has both improved antiviral efficacy and decreased gastrointestinal adverse effects.
Saquinavir should be taken within 2 hours after a fatty meal for enhanced absorption. Saquinavir is 97% protein-bound, and serum half-life is approximately 2 hours. Saquinavir has a large volume of distribution, but penetration into the cerebrospinal fluid is negligible. Excretion is primarily in the feces. Reported adverse effects include gastrointestinal discomfort (nausea, diarrhea, abdominal discomfort, dyspepsia) and rhinitis. When administered in combination with low-dose ritonavir, there appears to be less dyslipidemia or gastrointestinal toxicity than with some of the other boosted PI regimens. However, the concur-rent use of saquinavir and ritonavir is newly recognized to confer an increased risk of QT prolongation (with torsades de pointes arrhythmia) and PR interval prolongation.
Saquinavir is subject to extensive first-pass metabolism by CYP3A4 and functions as a CYP3A4 inhibitor as well as a sub-strate; thus, there are many potential drug-drug interactions ( Tables 49–3 and 49–4 ). A decreased dose of saquinavir is recom-mended when co-administered with nelfinavir. Increased saquina-vir levels when co-administered with omeprazole necessitate close monitoring for toxicities. Digoxin levels may increase if co- administered with saquinavir and should therefore be monitored. Liver function tests should be monitored if saquinavir is co- administered with delavirdine or rifampin. There is no evidence of human teratogenicity from saquinavir; there is short-term safety data for both mother and infant.
TIPRANAVIR
Tipranavir is a newer PI indicated for use in treatment-experienced HIV-1-infected patients who harbor strains resistant to other PI agents. It is used in combination with ritonavir to achieve effective serum levels and is not approved for treatment-naïve patients.
Bioavailability is poor but is increased when taken with a high-fat meal. The drug is metabolized by the liver microsomal system and is contraindicated in patients with hepatic insufficiency. Tipranavir contains a sulfonamide moiety and should not be administered to patients with known sulfa allergy.
The most common adverse effects from tipranavir are diarrhea, nausea, vomiting, and abdominal pain. An urticarial or maculo-papular rash is more common in women and may be accompanied
049-Katzung_Ch049_p861-890.indd 880 9/23/11 4:25:36 PM

CHAPTER 49 Antiviral Agents 881
by systemic symptoms or desquamation. Liver toxicity, including life-threatening hepatic decompensation, has been observed and is more common in patients with chronic HBV or HCV infection. Tipranavir should be discontinued in patients who have increased serum transaminase levels that are more than 10 times the upper limit of normal or more than 5 times normal in combination with increased serum bilirubin. Because of an increased risk for intra-cranial hemorrhage in patients receiving tipranavir/ritonavir, the drug should be avoided in patients with head trauma or bleeding diathesis. Other potential adverse effects include depression, eleva-tion in amylase, and decreased white blood cell count.
Tipranavir both inhibits and induces the CYP3A4 system. When used in combination with ritonavir, its net effect is inhibi-tion. Tipranavir also induces P-glycoprotein transporter and thus may alter the disposition of many other drugs ( Table 49–4 ). Concurrent administration of tipranavir with fosamprenavir or saquinavir should be avoided owing to decreased blood levels of the latter drugs. Tipranavir/ritonavir may also decrease serum levels of valproic acid and omeprazole. Levels of lovastatin, simva-statin, atorvastatin, and rosuvastatin may be increased, increasing the risk for rhabdomyolysis and myopathy. Tipranavir contains a sulfonamide moiety and should be used cautiously in patients with sulfonamide allergy.
ENTRY INHIBITORS The process of HIV-1 entry into host cells is complex; each step presents a potential target for inhibition. Viral attachment to the host cell entails binding of the viral envelope glycoprotein com-plex gp160 (consisting of gp120 and gp41) to its cellular receptor CD4. This binding induces conformational changes in gp120 that enable access to the chemokine receptors CCR5 or CXCR4. Chemokine receptor binding induces further conformational changes in gp120, allowing exposure to gp41 and leading to fusion of the viral envelope with the host cell membrane and sub-sequent entry of the viral core into the cellular cytoplasm.
ENFUVIRTIDE
Enfuvirtide is a synthetic 36-amino-acid peptide fusion inhibitor that blocks HIV entry into the cell ( Figure 49–4 ). Enfuvirtide binds to the gp41 subunit of the viral envelope glycoprotein, pre-venting the conformational changes required for the fusion of the viral and cellular membranes. Enfuvirtide, which must be admin-istered by subcutaneous injection, is the only parenterally adminis-tered antiretroviral agent. Metabolism appears to be by proteolytic hydrolysis without involvement of the CYP450 system. Elimination half-life is 3.8 hours.
Resistance to enfuvirtide can result from mutations in gp41; the frequency and significance of this phenomenon are being investigated. However, enfuvirtide lacks cross-resistance with the other currently approved antiretroviral drug classes.
The most common adverse effects associated with enfuvirtide therapy are local injection site reactions, consisting of painful erythematous nodules. Although frequent, these are typically mild
to moderate and rarely lead to discontinuation. Other symptom-atic side effects may include insomnia, headache, dizziness, and nausea. Hypersensitivity reactions may rarely occur, are of varying severity, and may recur on rechallenge. Eosinophilia is the primary laboratory abnormality seen with enfuvirtide administration. In prospective clinical trials, an increased rate of bacterial pneumonia was noted in patients receiving enfuvirtide. No drug-drug interac-tions have been identified that would require the alteration of the dosage of concomitant antiretroviral or other drugs.
MARAVIROC
Maraviroc binds specifically and selectively to the host protein CCR5, one of two chemokine receptors necessary for entrance of HIV into CD4+ cells. Maraviroc is approved for adults with CCR5-tropic (also known as R5) HIV-1 infection who are expe-riencing virologic failure due to resistance to other antiretroviral agents. Studies have shown that 52–60% of patients in whom at least two antiviral regimens had failed were infected with R5 HIV. Since maraviroc is active against HIV that uses the CCR5 co-receptor exclusively, and not against HIV strains with CXCR4, dual, or mixed tropism, tropism testing should be per-formed before initiating treatment with maraviroc. Clinical experience with the use of maraviroc in treatment-naïve patients is limited.
The absorption of maraviroc is rapid but variable, with the time to maximum absorption generally being 1–4 hours after ingestion of the drug. Most of the drug (≥ 75%) is excreted in the feces, whereas approximately 20% is excreted in urine. The recom-mended dose of maraviroc varies according to renal function and the concomitant use of CYP3A inducers or inhibitors. Maraviroc is contraindicated in patients with severe or end-stage renal impairment who are taking concurrent CYP3A inhibitors or inducers, and caution is advised when used in patients with preex-isting hepatic impairment and in those co-infected with HBV or HCV. Maraviroc has been shown to have excellent penetration into the cervicovaginal fluid, with levels almost four times higher than the corresponding concentrations in blood plasma.
Resistance to maraviroc is associated with one or more mutations in the V3 loop of gp120. There appears to be no cross-resistance with drugs from any other class, including the fusion inhibitor enfu-virtide. However, virologic failure of regimens containing maraviroc may potentially be caused by emergence of non–CCR5-tropic virus (eg, CXCR4-tropic virus) or by changes in viral tropism, owing to the development of multiple mutations throughout gp160.
Maraviroc is a substrate for CYP3A4 and therefore requires adjustment in the presence of drugs that interact with these enzymes ( Tables 49–3 and 49–4 ). It is also a substrate for P-glycoprotein, which limits intracellular concentrations of the drug. The dosage of maraviroc must be decreased if it is co- administered with strong CYP3A inhibitors (eg, delavirdine, ketoconazole, itraconazole, clarithromycin, or any protease inhibi-tor other than tipranavir) and must be increased if co-administered with CYP3A inducers (eg, efavirenz, etravirine, rifampin, car-bamazepine, phenytoin, or St. John’s wort).
049-Katzung_Ch049_p861-890.indd 881 9/23/11 4:25:36 PM

882 SECTION VIII Chemotherapeutic Drugs
Potential adverse effects include cough, upper respiratory tract infections, postural hypotension (particularly in the setting of renal insufficiency), muscle and joint pain, abdominal pain, diarrhea, and sleep disturbance. Due to reports of hepatotoxicity, which may be preceded by evidence of a systemic allergic reaction (ie, pruritic rash, eosinophilia, or elevated IgE), discontinuation of maraviroc should be considered promptly if this constellation of signs occurs. Myocardial ischemia and infarction have been observed in patients receiving maraviroc; therefore caution is advised in patients at increased cardiovascular risk.
There has been some concern that blockade of the chemokine CCR5 receptor—a human protein—may result in decreased immune surveillance, with a subsequent increased risk of malig-nancy (eg, lymphoma) or infection. To date, however, there has been no evidence of an increased risk of either malignancy or infection in patients receiving maraviroc.
INTEGRASE STRAND TRANSFER INHIBITORS RALTEGRAVIR
Raltegravir is a pyrimidinone analog that binds integrase, a viral enzyme essential to the replication of both HIV-1 and HIV-2. By doing so, it inhibits strand transfer, the third and final step of provirus integration, thus interfering with the integration of reverse-transcribed HIV DNA into the chromosomes of host cells ( Figure 49–4 ). It was initially licensed for use in treatment-experi-enced adult patients infected with strains of HIV-1 resistant to multiple other agents, but more recently has received approval for use in initial therapy as well. However, clinical experience is lim-ited in treatment-naïve patients.
Absolute bioavailability of raltegravir has not been established but does not appear to be food-dependent. The drug is metabo-lized by glucuronidation and does not interact with the cyto-chrome P450 system; therefore, it is expected to have fewer drug-drug interactions than many of the other antiretroviral agents. However, in combination with rifampin, a strong inducer of UDP-glucuronosyl transferase 1A1 (UGT1A1), the dose of raltegravir should be increased from 400 mg twice daily to 800 mg twice daily. Since polyvalent cations (eg, magnesium, calcium, and iron) may bind integrase inhibitors and interfere with their activ-ity, antacids should be used cautiously and taken separately from raltegravir.
Although virologic failure has been uncommon in clinical trials of raltegravir to date, in vitro resistance requires only a single point mutation (eg, at codons 148 or 155). The low genetic barrier to resistance emphasizes the importance of combination therapies and of adherence. Integrase mutations are not expected to affect sensitivity to other classes of antiretroviral agents.
Potential adverse effects of raltegravir include insomnia, headache, diarrhea, nausea, dizziness, and fatigue. Increases in cre-atine kinase may occur, with potential myopathy or rhabdomyolysis; there is minimal effect on serum lipids.
NEW & INVESTIGATIONAL ANTIRETROVIRAL AGENTS New therapies are continually being sought that exploit other HIV targets, have activity against resistant viral strains, have a lower incidence of adverse effects, and offer convenient dosing. Newly approved agents and those currently in advanced stages of clinical development include the NRTI agents elvucitabine , racivir , and apricitabine; the NNRTI agent rilpivirine ; entry inhibitors such as the CCR5 receptor antagonists vicriviroc and PRO 140, the fusion inhibitor TNX-355 ( ibalizumab ); and integrase inhibitors such as elvitegravir . In addition, new drug classes such as maturation inhibitors ( bevirimat ) are under investigation.
■ ANTIHEPATITIS AGENTS
Several agents effective against HBV and HCV are now available ( Table 49–6 ). Current treatment is suppressive rather than cura-tive and the high prevalence of these infections worldwide, with their concomitant morbidity and mortality, reflect a critical need for improved therapeutics.
INTERFERON ALFA
Interferons are host cytokines that exert complex antiviral, immuno-modulatory, and antiproliferative actions (see Chapter 55 ). Interferon alfa appears to function by induction of intracellular signals following binding to specific cell membrane receptors, resulting in inhibition of viral penetration, translation, transcrip-tion, protein processing, maturation, and release, as well as increased host expression of major histocompatibility complex antigens, enhanced phagocytic activity of macrophages, and aug-mentation of the proliferation and survival of cytotoxic T cells.
Injectable preparations of interferon alfa are available for treat-ment of both HBV and HCV infections ( Table 49–6 ). Interferon alfa-2a and interferon alfa-2b may be administered either subcu-taneously or intramuscularly; interferon alfacon-1 is administered subcutaneously. Elimination half-life is 2–5 hours for interferon alfa-2a and -2b, depending on the route of administration. The half-life of interferon alfacon-1 in patients with chronic HCV ranges from 6 to 10 hours. Alfa interferons are filtered at the glomerulus and undergo rapid proteolytic degradation during tubular reabsorption, such that detection in the systemic circula-tion is negligible. Liver metabolism and subsequent biliary excre-tion are considered minor pathways.
A meta-analysis of clinical trials in patients with chronic HBV infection showed that treatment with interferon alfa is associated with a higher incidence of hepatitis e antigen (HBeAg) serocon-version and undetectable HBV DNA levels compared with placebo. The addition of the pegylated moiety results in further increases in the proportion of patients with HBeAg seroconver-sion (∼ 30%) and a decline by approximately 4 log copies/mL (a 99.99% reduction) in HBV DNA after 1 year.
049-Katzung_Ch049_p861-890.indd 882 9/23/11 4:25:36 PM

CHAPTER 49 Antiviral Agents 883
TABLE 49–6 Drugs used to treat viral hepatitis.
Agent Indication Recommended Adult Dosage Route of Administration
Hepatitis B
Lamivudine1 Chronic hepatitis B 100 mg once daily (150 mg once daily if co-infection with HIV is present)
Oral
Adefovir1 Chronic hepatitis B 10 mg once daily Oral
Entecavir1 Chronic hepatitis B 0.5–1 mg once daily Oral
Tenofovir1 Chronic hepatitis B 300 mg once daily Oral
Telbivudine1 Chronic hepatitis B 600 mg once daily Oral
Interferon alfa-2b Chronic hepatitis B 5 million units once daily or 10 million units three times weekly
Subcutaneous or intramuscular
Pegylated interferon alfa-2a1 Chronic hepatitis B 180 mcg once weekly Subcutaneous
Hepatitis C
Pegylated interferon alfa-2a1 Chronic hepatitis C 180 mcg once weekly plus ribavirin (800–1200 mg/d)
Subcutaneous
Pegylated interferon alfa-2b1 Chronic hepatitis C 1.5 mcg/kg once weekly with ribavirin (800–1200 mg/d)
Subcutaneous
Interferon alfa-2b1 Acute hepatitis C 5 million units once daily for 3–4 weeks, then 5 million units three times weekly
Subcutaneous or intramuscular
1Dose must be reduced in patients with renal insufficiency.
The use of pegylated (polyethylene glycol-complexed) interferon alfa-2a and pegylated interferon alfa-2b results in slower clearance and longer terminal half-lives and steadier concentrations, which allows for less frequent dosing in patients with chronic HCV infec-tion. Renal elimination accounts for about 30% of clearance, and clearance is approximately halved in subjects with impaired renal function; dosage must therefore be adjusted.
The adverse effects of interferon alfa include a flu-like syndrome (ie, headache, fevers, chills, myalgias, and malaise) that typically occurs within 6 hours after dosing; this syndrome occurs in more than 30% of patients during the first week of therapy and tends to resolve upon continued administration. Transient hepatic enzyme elevations may occur in the first 8–12 weeks of therapy and appear to be more common in responders. Potential adverse effects during chronic therapy include neuro-toxicities (mood disorders, depression, somnolence, confusion, seizures), myelosuppression, profound fatigue, weight loss, rash, cough, myalgia, alopecia, tinnitus, reversible hearing loss, retin-opathy, pneumonitis, and possibly cardiotoxicity. Induction of autoantibodies may occur, causing exacerbation or unmasking of autoimmune disease (particularly thyroiditis). The polyethylene glycol molecule is a nontoxic polymer that is readily excreted in the urine.
Contraindications to interferon alfa therapy include hepatic de-compensation, autoimmune disease, and history of cardiac arrhyth-mia. Caution is advised in the setting of psychiatric disease, epilepsy, thyroid disease, ischemic cardiac disease, severe renal insufficiency, and cytopenia. Alfa interferons are abortifacient in primates and should not be administered in pregnancy. Potential drug-drug interac-tions include increased theophylline and methadone levels. Co-administration with didanosine is not recommended because of a
risk of hepatic failure, and co-administration with zidovudine may exacerbate cytopenias.
TREATMENT OF HEPATITIS B VIRUS INFECTION The goals of chronic HBV therapy are to sustain suppression of HBV replication, resulting in slowing of progression of hepatic disease (with retardation of hepatic fibrosis and even reversal of cirrhosis), prevention of complications (ie, cirrhosis, hepatic failure, and hepa-tocellular carcinoma), and reduction of the need for liver transplanta-tion. This translates into suppression of HBV DNA to undetectable levels, seroconversion of HBeAg (or more rarely, HBsAg) from posi-tive to negative, and reduction in elevated hepatic transaminase lev-els. These end points are correlated with improvement in necroinflammatory disease, a decreased risk of hepatocellular carci-noma and cirrhosis, and a decreased need for liver transplantation. All the currently licensed therapies achieve these goals. However, because current therapies suppress HBV replication without eradi-cating the virus, initial responses may not be durable. The covalently closed circular (ccc) viral DNA exists in stable form indefinitely within the cell, serving as a reservoir for HBV throughout the life of the cell and resulting in the capacity to reactivate. Relapse is more common in patients co-infected with HBV and hepatitis D virus.
As of 2010 seven drugs were approved for treatment of chronic HBV infection in the United States: five oral nucleoside/nucleotide analogs (lamivudine, adefovir dipivoxil, tenofovir, entecavir, telbivudine) and two injectable interferon drugs (interferon alfa-2b, pegylated interferon alfa-2a) ( Table 49–6 ). The use of interferon has been supplanted by long-acting pegylated interferon, allowing
049-Katzung_Ch049_p861-890.indd 883 9/23/11 4:25:36 PM

884 SECTION VIII Chemotherapeutic Drugs
once-weekly rather than daily or thrice-weekly dosing. In general, nucleoside/nucleotide analog therapies have better tolerability and ultimately produce a higher response rate than the interferons (though less rapid); however, response may be less sustained after discontinuation of the nucleoside/nucleotide therapies, and emer-gence of resistance may be problematic. The nucleotides are effec-tive in nucleoside resistance and vice versa. In addition, oral agents may be used in patients with decompensated liver disease, and the therapy is chronic rather than finite as with interferon therapy.
Several anti-HBV agents have anti-HIV activity as well, includ-ing lamivudine, adefovir dipivoxil, and tenofovir. Emtricitabine, an antiretroviral NRTI, is under clinical evaluation for HBV treat-ment in combination with tenofovir, which may be particularly suited to individuals co-infected with HIV and HBV. Because NRTI agents may be used in patients co-infected with HBV and HIV, it is important to note that acute exacerbation of hepatitis may occur upon discontinuation or interruption of these agents.
ADEFOVIR DIPIVOXIL
Although initially and abortively developed for treatment of HIV infection, adefovir dipivoxil gained approval, at lower and less toxic doses, for treatment of HBV infection. Adefovir dipivoxil is the diester prodrug of adefovir, an acyclic phosphonated adenine nucleotide analog ( Figure 49–2 ). It is phosphorylated by cellular kinases to the active diphosphate metabolite and then competi-tively inhibits HBV DNA polymerase and causes chain termina-tion after incorporation into viral DNA. Adefovir is active in vitro against a wide range of DNA and RNA viruses, including HBV, HIV, and herpesviruses.
Oral bioavailability of adefovir dipivoxil is about 59% and is unaffected by meals; it is rapidly and completely hydrolyzed to the parent compound by intestinal and blood esterases. Protein bind-ing is low (< 5%). The intracellular half-life of the diphosphate is prolonged, ranging from 5 to 18 hours in various cells; this makes once-daily dosing feasible. Adefovir is excreted by a combination of glomerular filtration and active tubular secretion and requires dose adjustment for renal dysfunction; however, it may be admin-istered to patients with decompensated liver disease.
Of the oral agents, adefovir may be slower to suppress HBV DNA levels and the least likely to induce HBeAg seroconversion. Although emergence of resistance is rare during the first year of therapy, it approaches 30% at the end of 4 years. Naturally occur-ring (ie, primary) adefovir-resistant rt233 HBV mutants have recently been described. There is no cross-resistance between adefovir and lamivudine.
Adefovir dipivoxil is well tolerated. A dose-dependent nephro-toxicity has been observed in clinical trials, manifested by increased serum creatinine with decreased serum phosphorous and more common in patients with baseline renal insufficiency and those receiving high doses (60 mg/d). Other potential adverse effects are headache, diarrhea, asthenia, and abdominal pain. As with other NRTI agents, lactic acidosis and hepatic steatosis are considered a risk owing to mitochondrial dysfunction. No clini-cally important drug-drug interactions have been recognized to
date; however, co-administration with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of adefovir or the co-administered drug. Pivalic acid, a by-product of adefovir dipivoxil metabolism, can esterify free carnitine and result in decreased carnitine levels. However, it is not felt necessary to administer carnitine supple-mentation with the low doses used to treat patients with HBV (10 mg/d). Adefovir is embryotoxic in rats at high doses and is genotoxic in preclinical studies.
ENTECAVIR
Entecavir is an orally administered guanosine nucleoside analog ( Figure 49–2 ) that competitively inhibits all three functions of HBV DNA polymerase, including base priming, reverse transcrip-tion of the negative strand, and synthesis of the positive strand of HBV DNA. Oral bioavailability approaches 100% but is decreased by food; therefore, entecavir should be taken on an empty stomach. The intracellular half-life of the active phosphorylated compound is 15 hours. It is excreted by the kidney, undergoing both glomerular filtration and net tubular secretion.
Comparison with lamivudine in patients with chronic HBV infection demonstrated similar rates of HBeAg seroconversion but significantly higher rates of HBV DNA viral suppression with entecavir, normalization of serum alanine aminotransferase levels, and histologic improvement in the liver. Entecavir appears to have a higher barrier to the emergence of resistance than lamivudine. Although selection of resistant isolates with the S202G mutation has been documented during therapy, clinical resistance is rare (< 1% at 4 years). Also, decreased susceptibility to entecavir may occur in association with lamivudine resistance.
Entecavir is well tolerated. The most frequent adverse events are headache, fatigue, dizziness, and nausea. Lung adenomas and carci-nomas in mice, hepatic adenomas and carcinomas in rats and mice, vascular tumors in mice, and brain gliomas and skin fibromas in rats have been observed at varying exposures. Co-administration of entecavir with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of either entecavir or the co-administered drug.
LAMIVUDINE
The pharmacokinetics of lamivudine are described earlier in this chapter (see section, Nucleoside and Nucleotide Reverse Transcriptase Inhibitors). The more prolonged intracellular half-life in HBV cell lines (17–19 hours) than in HIV-infected cell lines (10.5–15.5 hours) allows for lower doses and less frequent administration. Lamivudine can be safely administered to patients with decompensated liver disease.
Lamivudine inhibits HBV DNA polymerase and HIV reverse transcriptase by competing with deoxycytidine triphosphate for incorporation into the viral DNA, resulting in chain termination. Lamivudine achieves 3–4 log decreases in viral replication in most patients and suppression of HBV DNA to undetectable levels in about 44% of patients. Seroconversion of HBeAg from positive to
049-Katzung_Ch049_p861-890.indd 884 9/23/11 4:25:36 PM

CHAPTER 49 Antiviral Agents 885
negative occurs in about 17% of patients and is durable at 3 years in about 70% of responders. Continuation of treatment for 4–8 months after seroconversion may improve the durability of response. Response in HBeAg-negative patients is initially high but less durable.
Although lamivudine initially results in rapid and potent virus suppression, chronic therapy may ultimately be limited by the emer-gence of lamivudine-resistant HBV isolates (eg, L180M or M204I/V), estimated to occur in 15–30% of patients at 1 year and 70% at 5 years of therapy. Resistance has been associated with flares of hepati-tis and progressive liver disease. Cross-resistance between lamivudine and emtricitabine or entecavir may occur; however, adefovir main-tains activity against lamivudine-resistant strains of HBV.
In the doses used for HBV infection, lamivudine has an excel-lent safety profile. Headache, nausea, and dizziness are rare. Co-infection with HIV may increase the risk of pancreatitis. No evidence of mitochondrial toxicity has been reported.
TELBIVUDINE
Telbivudine is a thymidine nucleoside analog with activity against HBV DNA polymerase. It is phosphorylated by cellular kinases to the active triphosphate form, which has an intracellular half-life of 14 hours. The phosphorylated compound competitively inhibits HBV DNA polymerase, resulting in incorporation into viral DNA and chain termination. It is not active in vitro against HIV-1.
Oral bioavailability is unaffected by food. Plasma protein-binding is low (3%) and distribution wide. The serum half-life is approximately 15 hours and excretion is renal. There are no known metabolites and no known interactions with the CYP450 system or other drugs.
In a comparative trial against lamivudine in patients with chronic HBV infection, significantly more patients receiving telbivudine achieved the combined end point of suppression of HBV DNA to less than 5 log copies/mL plus loss of serum HBeAg. The mean reduction in HBV DNA from baseline, the proportion with ALT normalization, and HBeAg seroconversion all were greater in those receiving telbivudine. Liver biopsies per-formed 1 year later showed less scarring. However, emergence of resistance, typically due to the M204I mutation, may occur in up to 22% of patients with durations of therapy exceeding 1 year, and may result in virologic rebound.
Adverse effects are mild; they include fatigue, headache, abdominal pain, upper respiratory infection, increased creatine kinase levels, and nausea and vomiting. Both uncomplicated myalgia and myopathy have been reported, as has peripheral neuropathy. As with other nucleoside analogs, lactic acidosis and severe hepatomegaly with steatosis may occur during therapy as well as flares of hepatitis after discontinuation.
TENOFOVIR
Tenofovir, a nucleotide analog of adenosine in use as an antiretro-viral agent, has recently gained licensure for the treatment of
patients with chronic HBV infection. The characteristics of teno-fovir are described earlier in this chapter. Tenofovir maintains activity against lamivudine- and entecavir-resistant hepatitis virus isolates but has reduced activity against adefovir-resistant strains. Although similar in structure to adefovir dipivoxil, comparative trials showed a significantly higher rate of complete response, defined as serum HBV DNA levels less than 400 copies/mL, as well as of histologic improvement, in patients with chronic HBV infection receiving tenofovir than in those receiving adefovir dipivoxil. The emergence of resistance appears to be substantially less frequent during therapy with tenofovir than with adefovir.
INVESTIGATIONAL AGENTS
Compounds in clinical development for the treatment of patients with HBV infection include the nucleoside analogs emtricitabine and clevudine , as well as the immunologic modulator thymosin alpha-1, agents that facilitate uptake by the liver using conjuga-tion to ligands, and RNA interference compounds.
TREATMENT OF HEPATITIS C INFECTION In contrast to the treatment of patients with chronic HBV infection, the primary goal of treatment in patients with HCV infection is viral eradication. In clinical trials, the primary efficacy end point is typi-cally achievement of sustained viral response (SVR), defined as the absence of detectable viremia for 6 months after completion of therapy. SVR is associated with improvement in liver histology, reduction in risk of hepatocellular carcinoma, and, occasionally, with regression of cirrhosis as well. Late relapse occurs in less than 5% of patients who achieve SVR.
In acute hepatitis C, the rate of clearance of the virus without therapy is estimated at 15–30%. In one (uncontrolled) study, treatment of acute infection with interferon alfa-2b, in doses higher than those used for chronic hepatitis C, resulted in a sus-tained rate of clearance of 95% at 6 months. Therefore, if HCV RNA testing documents persistent viremia 12 weeks after initial seroconversion, antiviral therapy is recommended.
Treatment of patients with chronic HCV infection is recom-mended for those with an increased risk for progression to cirrhosis. The parameters for selection are complex. In those who are to be treated, the current standard of treatment is once-weekly pegylated interferon alfa in combination with daily oral ribavirin. Pegylated interferon alfa-2a and -2b have replaced their unmodified inter-feron alfa counterparts because of superior efficacy in combination with ribavirin, regardless of genotype. It is also clear that combina-tion therapy with oral ribavirin is more effective than monother-apy with either interferon or ribavirin alone. Therefore, monotherapy with pegylated interferon alfa is recommended only in patients who cannot tolerate ribavirin. Factors associated with a favorable therapeutic response include HCV genotype 2 or 3, absence of cirrhosis on liver biopsy, and low pretreatment HCV RNA levels.
049-Katzung_Ch049_p861-890.indd 885 9/23/11 4:25:36 PM

886 SECTION VIII Chemotherapeutic Drugs
RIBAVIRIN
Ribavirin is a guanosine analog that is phosphorylated intracellu-larly by host cell enzymes. Although its mechanism of action has not been fully elucidated, it appears to interfere with the synthesis of guanosine triphosphate, to inhibit capping of viral messenger RNA, and to inhibit the viral RNA-dependent polymerase of certain viruses. Ribavirin triphosphate inhibits the replication of a wide range of DNA and RNA viruses, including influenza A and B, parainfluenza, respiratory syncytial virus, paramyxoviruses, HCV, and HIV-1.
The absolute oral bioavailability of ribavirin is 45–64%, increases with high-fat meals, and decreases with co-administration of antacids. Plasma protein binding is negligible, volume of distri-bution is large, and cerebrospinal fluid levels are about 70% of those in plasma. Ribavirin elimination is primarily through the urine; therefore, clearance is decreased in patients with creatinine clearances less than 30 mL/min.
Higher doses of ribavirin (ie, 1000–1200 mg/d, according to weight, rather than 800 mg/d) or a longer duration of therapy or both may be more efficacious in those with a lower likelihood of response to therapy (eg, those with genotype 1 or 4) or in those who have relapsed. This must be balanced with an increased likeli-hood of toxicity. A dose-dependent hemolytic anemia occurs in 10–20% of patients. Other potential adverse effects are depres-sion, fatigue, irritability, rash, cough, insomnia, nausea, and pruritus. Contraindications to ribavirin therapy include anemia, end-stage renal failure, ischemic vascular disease, and pregnancy. Ribavirin is teratogenic and embryotoxic in animals as well as mutagenic in mammalian cells. Patients exposed to the drug should not conceive children for at least 6 months thereafter.
NEW & INVESTIGATIONAL AGENTS
Among the agents for the treatment of HCV infection, those holding the greatest promise currently are the HCV NS3 protease inhibitors telaprevir and boceprevir . These highly potent agents are likely to decrease the overall duration of therapy, with possibly greater tolerability than current regimens. Another class of prom-ising agents is the HCV NS5B polymerase inhibitors, including both nucleoside and nonnucleoside analogs.
■ ANTIINFLUENZA AGENTS
Influenza virus strains are classified by their core proteins (ie, A, B, or C), species of origin (eg, avian, swine), and geographic site of isolation. Influenza A, the only strain that causes pandemics, is classified into 16 H (hemagglutinin) and 9 N (neuraminidase) known subtypes based on surface proteins. Although influenza B viruses usually infect only people, influenza A viruses can infect a variety of animal hosts. Current influenza A subtypes that are circulating among worldwide populations include H1N1, H1N2, and H3N2. Fifteen subtypes are known to infect birds, providing an extensive reservoir. Although avian influenza subtypes are
typically highly species-specific, they have on rare occasions crossed the species barrier to infect humans and cats. Viruses of the H5 and H7 subtypes (eg, H5N1, H7N7, and H7N3) may rapidly mutate within poultry flocks from a low to high patho-genic form and have recently expanded their host range to cause both avian and human disease. Of particular concern is the avian H5N1 virus, which first caused human infection (including severe disease and death) in 1997 and has become endemic in Southeast Asian poultry since 2003. To date, the spread of H5N1 virus from person to person has been rare, limited, and unsustained. However, the emergence of the 2009 H1N1 influenza virus (pre-viously called “swine flu”) in 2009–2010 caused the first influenza pandemic (ie, global outbreak of disease caused by a new flu virus) in more than 40 years.
Although antiviral drugs available for influenza have activity against influenza A, many or most of the circulating strains of avian H5N1, as well as H1 and H3 strains causing seasonal influenza in the United States, are resistant to amantadine and rimantadine. Resistance to oseltamivir has also increased dramatically.
OSELTAMIVIR & ZANAMIVIR
The neuraminidase inhibitors oseltamivir and zanamivir, analogs of sialic acid, interfere with release of progeny influenza virus from infected host cells, thus halting the spread of infection within the respiratory tract. These agents competitively and reversibly interact with the active enzyme site to inhibit viral neuraminidase activity at low nanomolar concentrations. Inhibition of viral neuraminidase results in clumping of newly released influenza virions to each other and to the membrane of the infected cell. Unlike amantadine and rimantadine, oseltami-vir and zanamivir have activity against both influenza A and influenza B viruses. Early administration is crucial because repli-cation of influenza virus peaks at 24–72 hours after the onset of illness. When a 5-day course of therapy is initiated within 36–48 hours after the onset of symptoms, the duration of illness is decreased by 1–2 days compared with those on placebo, severity is diminished, and the incidence of secondary complications in children and adults decreases. Once-daily prophylaxis is 70–90% effective in preventing disease after exposure. Oseltamivir is approved by the Food and Drug Administration (FDA) for patients 1 year and older, whereas zanamivir is approved in patients 7 years or older.
Oseltamivir is an orally administered prodrug that is activated by hepatic esterases and widely distributed throughout the body. The dosage is 75 mg twice daily for 5 days for treatment and 75 mg once daily for prevention; dosage must be modified in patients with renal insufficiency. Oral bioavailability is approximately 80%, plasma protein binding is low, and concentrations in the middle ear and sinus fluid are similar to those in plasma. The half-life of oseltamivir is 6–10 hours, and excretion is by glomerular filtration and tubular secretion in the urine. Probenecid reduces renal clearance of oseltamivir by 50%. Serum concentrations of oseltamivir carboxylate, the active metabolite of oseltamivir, increase with declining renal function; therefore, dosage should be
049-Katzung_Ch049_p861-890.indd 886 9/23/11 4:25:36 PM

CHAPTER 49 Antiviral Agents 887
adjusted in such patients. Potential adverse effects include nausea, vomiting, and abdominal pain, which occur in 5–10% of patients early in therapy but tend to resolve spontaneously. Taking oselta-mivir with food does not interfere with absorption and may decrease nausea and vomiting. Headache, fatigue, and diarrhea have also been reported and appear to be more common with prophylactic use. Rash is rare. Transient neuropsychiatric events (self-injury or delirium) have been reported, particularly in adoles-cents and adults living in Japan.
Zanamivir is delivered directly to the respiratory tract via inhalation. Ten to twenty percent of the active compound reaches the lungs, and the remainder is deposited in the oropharynx. The concentration of the drug in the respiratory tract is estimated to be more than 1000 times the 50% inhibitory concentration for neuraminidase, and the pulmonary half-life is 2.8 hours. Five to fifteen percent of the total dose (10 mg twice daily for 5 days for treatment and 10 mg once daily for prevention) is absorbed and excreted in the urine with minimal metabolism. Potential adverse effects include cough, bronchospasm (occasionally severe), revers-ible decrease in pulmonary function, and transient nasal and throat discomfort. Zanamivir administration is not recommended for patients with underlying airway disease.
Resistance to oseltamivir may be associated with point muta-tions in the viral hemagglutinin or neuraminidase (eg, the H275Y mutation) genes. Rates of resistance to oseltamivir among seasonal H1N1 viruses have risen abruptly and dramatically worldwide, reaching 97.4% in tested strains in the United States from 2008 to 2009. Resistance to oseltamivir in pandemic H1N1 viruses and resistance to zanamivir in seasonal and pandemic H1N1 viruses are rare. All influenza A (H3N2) and influenza B viruses were susceptible to both oseltamivir and zanamivir. Swine-origin influenza A (H1N1) viruses are nearly always susceptible to both oseltamivir and zanamivir.
AMANTADINE & RIMANTADINE
Amantadine (1-aminoadamantane hydrochloride) and its α-methyl derivative, rimantadine, are tricyclic amines of the adamantane family that block the M2 proton ion channel of the virus particle and inhibit uncoating of the viral RNA within infected host cells, thus preventing its replication. They are active against influenza A only. Rimantadine is four to ten times more active than amanta-dine in vitro. Amantadine is well absorbed and 67% protein-bound. Its plasma half-life is 12–18 hours and varies by creatinine clearance. Rimantadine is about 40% protein-bound and has a half-life of 24–36 hours. Nasal secretion and salivary levels approximate those in the serum, and cerebrospinal fluid levels are 52–96% of those in the serum; nasal mucus concentrations of rimantadine average 50% higher than those in plasma. Amantadine is excreted unchanged in the urine, whereas rimantadine under-goes extensive metabolism by hydroxylation, conjugation, and glucuronidation before urinary excretion. Dose reductions are required for both agents in the elderly and in patients with renal insufficiency, and for rimantadine in patients with marked hepatic insufficiency.
In the absence of resistance, both amantadine and rimantadine, at 100 mg twice daily or 200 mg once daily, are 70–90% protective in the prevention of clinical illness when initiated before exposure. When begun within 1–2 days after the onset of illness, the duration of fever and systemic symptoms is reduced by 1–2 days.
The primary target for both agents is the M2 protein within the viral membrane, incurring both influenza A specificity and a mutation-prone site that results in the rapid development of resis-tance in up to 50% of treated individuals. Resistant isolates with single-point mutations are genetically stable, retain pathogenicity, can be transmitted to close contacts, and may be shed chronically by immunocompromised patients. The marked increase in the prevalence of resistance to both agents in clinical isolates over the last decade, in influenza A H1N1 as well as H3N2, has limited the usefulness of these agents for either the treatment or the pre-vention of influenza. Cross-resistance to zanamivir and oseltamivir does not occur.
The most common adverse effects are gastrointestinal (nausea, anorexia) and central nervous system (nervousness, difficulty in concentrating, insomnia, light-headedness); side effects are dose-related and may diminish or disappear after the first week of treatment despite continued drug ingestion. More serious side effects (eg, marked behavioral changes, delirium, hallucinations, agitation, and seizures) may be due to alteration of dopamine neurotransmission (see Chapter 28 ); are less frequent with riman-tadine than with amantadine; are associated with high plasma concentrations; may occur more frequently in patients with renal insufficiency, seizure disorders, or advanced age; and may increase with concomitant antihistamines, anticholinergic drugs, hydro-chlorothiazide, and trimethoprim-sulfamethoxazole. Clinical manifestations of anticholinergic activity tend to be present in acute amantadine overdose. Both agents are teratogenic and embryotoxic in rodents, and birth defects have been reported after exposure during pregnancy.
INVESTIGATIONAL AGENTS
The neuraminidase inhibitor peramivir , a cyclopentane analog, has activity against both influenza A and B viruses. Peramivir received temporary emergency use authorization by FDA for intravenous administration in November 2009 due to the H1N1 pandemic. The drug is marketed in South Korea but has not yet been licensed in the United States. Clinical data on cross-resistance to oseltamivir and zanamivir are not yet available. Reported side effects include diarrhea, nausea, vomiting, and neutropenia.
■ OTHER ANTIVIRAL AGENTS
INTERFERONS
Interferons have been studied for numerous clinical indications. In addition to HBV and HCV infections (see Antihepatitis Agents), intralesional injection of interferon alfa-2b or alfa-n3 may be used for treatment of condylomata acuminata (see Chapter 61 ).
049-Katzung_Ch049_p861-890.indd 887 9/23/11 4:25:36 PM

888 SECTION VIII Chemotherapeutic Drugs
RIBAVIRIN
In addition to oral administration for HCV infection in combina-tion with interferon alfa (see Antihepatitis Agents), aerosolized ribavirin is administered by nebulizer (20 mg/mL for 12–18 hours per day) to children and infants with severe respiratory syncytial virus (RSV) bronchiolitis or pneumonia to reduce the severity and dura-tion of illness. Aerosolized ribavirin has also been used to treat influ-enza A and B infections but has not gained widespread use. Systemic absorption is low (< 1%). Aerosolized ribavirin is generally well toler-ated but may cause conjunctival or bronchial irritation. Health care workers should be protected against extended inhalation exposure. The aerosolized drug may precipitate on contact lenses.
Intravenous ribavirin decreases mortality in patients with Lassa fever and other viral hemorrhagic fevers if started early. High con-centrations inhibit West Nile virus in vitro, but clinical data are lacking. Clinical benefit has been reported in cases of severe measles pneumonitis and certain encephalitides, and continuous infusion of ribavirin has decreased virus shedding in several patients with severe lower respiratory tract influenza or parainfluenza infections. At steady state, cerebrospinal fluid levels are about 70% of those in plasma.
PALIVIZUMAB
Palivizumab is a humanized monoclonal antibody directed against an epitope in the A antigen site on the F surface protein of RSV. It is licensed for the prevention of RSV infection in high-risk infants and children, such as premature infants and those with bronchopul-monary dysplasia or congenital heart disease. A placebo-controlled
trial using once-monthly intramuscular injections (15 mg/kg) for 5 months beginning at the start of the RSV season demonstrated a 55% reduction in the risk of hospitalization for RSV in treated patients, as well as decreases in the need for supplemental oxygen, the illness severity score, and the need for intensive care. Although resistant strains have been isolated in the laboratory, no resistant clinical isolates have yet been identified. Potential adverse effects include upper respiratory tract infection, fever, rhinitis, rash, diarrhea, vomiting, cough, otitis media, and elevation in serum aminotransferase levels.
A number of other agents are under investigation for the treat-ment or prophylaxis of patients with RSV infection, including the RNA interference (RNAi) therapeutic ALN-RSV01, the human-ized monoclonal antibody motavizumab, and the benzodiazepine RSV604.
IMIQUIMOD
Imiquimod is an immune response modifier shown to be effective in the topical treatment of external genital and perianal warts (ie, condyloma acuminatum; see Chapter 61 ). The 5% cream is applied three times weekly and washed off 6–10 hours after each application. Recurrences appear to be less common than after ablative therapies. Imiquimod may also be effective against mol-luscum contagiosum but is not licensed in the United States for this indication. Local skin reactions are the most common adverse effect; these tend to resolve within weeks after therapy. However, pigmentary skin changes may persist. Systemic adverse effects such as fatigue and influenza-like syndrome have occasionally been reported.
AbacavirOral (Ziagen): 300 mg tablets; 20 mg/mL solutionOral (Epzicom): 600 mg plus 300 mg lamivudineOral (Trizivir): 300 mg tablets in combination with 150 mg
lamivudine and 300 mg zidovudineAcyclovir (generic, Zovirax)
Oral: 200 mg capsules; 400, 800 mg tablets; 200 mg/5 mL suspensionParenteral: 50 mg/mL; powder to reconstitute for injection (500,
1000 mg/vial)Topical: 5% ointment
Adefovir (Hepsera)Oral: 10 mg tablets
Amantadine (generic, Symmetrel)Oral: 100 mg capsules, tablets; 50 mg/5 mL syrup
Atazanavir (Reyataz)Oral: 100, 150, 200 mg capsules
Boceprivir (Victrelis)Oral: 375 mg tablets
Cidofovir (Vistide)Parenteral: 375 mg/vial (75 mg/mL) for IV injection
Darunavir (Prezista)Oral: 300 mg tablets (must be taken with ritonavir)
Delavirdine (Rescriptor)Oral: 100, 200 mg tablets
Didanosine (dideoxyinosine, ddI)Oral (Videx): 25, 50, 100, 150, 200 mg tablets; 100, 167, 250 mg
powder for oral solution; 2, 4 g powder for pediatric solutionOral (Videx-EC): 125, 200, 250, 400 mg delayed-release capsules
Docosanol (Abreva) (over-the-counter)Topical: 10% cream
Efavirenz (Sustiva)Oral: 50, 100, 200 mg capsules; 600 mg tablets
EmtricitabineOral (Emtriva): 200 mg tabletsOral (Truvada): 200 mg plus 300 mg tenofovir tabletsOral (Atripla): 200 mg plus 300 mg tenofovir plus 600 mg efavirenz
Enfuvirtide (Fuzeon)Parenteral: 90 mg/mL for injection
Entecavir (Baraclude)Oral: 0.5, 1 mg tablets; 0.05 mg/mL oral solution
P R E P A R A T I O N S A V A I L A B L E
AbacavirOral (Ziagen): 300 mg tablets; 20 mg/mL solutionOral (Epzicom): 600 mg plus 300 mg lamivudineOral (Trizivir): 300 mg tablets in combination with 150 mg
lamivudine and 300 mg zidovudineAcyclovir (generic, Zovirax)
Oral: 200 mg capsules; 400, 800 mg tablets; 200 mg/5 mL suspensionParenteral: 50 mg/mL; powder to reconstitute for injection (500,
1000 mg/vial)Topical: 5% ointment
Adefovir (Hepsera)Oral: 10 mg tablets
Amantadine (generic, Symmetrel)Oral: 100 mg capsules, tablets; 50 mg/5 mL syrup
Atazanavir (Reyataz)Oral: 100, 150, 200 mg capsules
Boceprivir (Victrelis)Oral: 375 mg tablets
Cidofovir (Vistide)Parenteral: 375 mg/vial (75 mg/mL) for IV injection
Darunavir (Prezista)Oral: 300 mg tablets (must be taken with ritonavir)
Delavirdine (Rescriptor)Oral: 100, 200 mg tablets
Didanosine (dideoxyinosine, ddI)Oral (Videx): 25, 50, 100, 150, 200 mg tablets; 100, 167, 250 mg
powder for oral solution; 2, 4 g powder for pediatric solutionOral (Videx-EC): 125, 200, 250, 400 mg delayed-release capsules
Docosanol (Abreva) (over-the-counter)Topical: 10% cream
Efavirenz (Sustiva)Oral: 50, 100, 200 mg capsules; 600 mg tablets
EmtricitabineOral (Emtriva): 200 mg tabletsOral (Truvada): 200 mg plus 300 mg tenofovir tabletsOral (Atripla): 200 mg plus 300 mg tenofovir plus 600 mg efavirenz
Enfuvirtide (Fuzeon)Parenteral: 90 mg/mL for injection
Entecavir (Baraclude)Oral: 0.5, 1 mg tablets; 0.05 mg/mL oral solution
049-Katzung_Ch049_p861-890.indd 888 9/23/11 4:25:36 PM

CHAPTER 49 Antiviral Agents 889
Etravirine (Intelence)Oral: 100 mg tablets
Famciclovir (Famvir)Oral: 125, 250, 500 mg tablets
Fosamprenavir (Lexiva)Oral: 700 mg tablets
Foscarnet (Foscavir)Parenteral: 24 mg/mL for IV injection
Ganciclovir (Cytovene)Oral: 250, 500 mg capsulesParenteral: 500 mg/vial for IV injectionIntraocular implant (Vitrasert): 4.5 mg ganciclovir/implant
Imiquimod (Aldara)Topical: 5% cream
Indinavir (Crixivan)Oral: 100, 200, 333, 400 mg capsules
Interferon alfa-2a (Roferon-A)Parenteral: 3, 6, 9, 36 million IU vials
Interferon alfa-2b (Intron A)Parenteral: 3, 5, 10, 18, 25, and 50 million IU vials
Interferon alfa-2b (Rebetron)Parenteral: 3 million IU vials (supplied with oral ribavirin, 200 mg
capsules)Interferon alfa-n3 (Alferon N)
Parenteral: 5 million IU/vialInterferon alfacon-1 (Infergen)
Parenteral: 9 and 15 mcg vialsLamivudine
Oral (Epivir): 150, 300 mg tablets; 10 mg/mL oral solutionOral (Epivir-HBV): 100 mg tablets; 5 mg/mL solutionOral (Combivir): 150 mg tablets in combination with 300 mg
zidovudineOral (Trizivir): 150 mg tablets in combination with 300 mg abacavir
and 300 mg zidovudineLopinavir/ritonavir (Kaletra)
Oral: 133.3 mg/33.3 mg capsules; 80 mg/20 mg per mL solutionMaraviroc (Selzentry)
Oral: 150, 300 mg tabletsNelfinavir (Viracept)
Oral: 250, 625 mg tablets; 50 mg/g powderNevirapine (Viramune)
Oral: 200 mg tablets; 50 mg/5 mL suspensionOseltamivir (Tamiflu)
Oral: 75 mg capsules; powder to reconstitute as suspension (12 mg/mL)
Palivizumab (Synagis)Parenteral: 50, 100 mg/vial
Peginterferon alfa-2a (pegylated interferon alfa-2a, Pegasys)Parenteral: 180 mcg/mL
Peginterferon alfa-2b (pegylated interferon alfa-2b, PEG-Intron)Parenteral: powder to reconstitute as 100, 160, 240, 300 mcg/mL
injection
Penciclovir (Denavir)Topical: 1% cream
Raltegravir (Isentress)Oral: 400 mg tablets
RibavirinAerosol (Virazole): powder to reconstitute for aerosol; 6 g/100 mL vialOral (Rebetol, generic): 200 mg capsules, tablets; 40 mg/mL oral
solutionOral (Rebetron): 200 mg in combination with 3 million units
interferon alfa-2b (Intron-A)Rilpivirine
Oral (Edurant): 25 mg tabletsOral (Complera): 200 mg emtricitabine, 25 mg rilpivirine, 300 mg
tenofovir) tabletsRimantadine (Flumadine)
Oral: 100 mg tablets; 50 mg/5 mL syrupRitonavir (Norvir)
Oral: 100 mg capsules; 80 mg/mL oral solutionSaquinavir
Oral (Invirase): 200 mg hard gel capsules, 500 mg tabletsStavudine
Oral (Zerit): 15, 20, 30, 40 mg capsules; powder for 1 mg/mL oral solution
Oral extended-release (Zerit XR): 37.5, 50, 75, 100 mg capsulesTelaprivir (Incivik)
Oral: 200 mg capsulesTelbivudine (Tyzeka)
Oral: 600 mg tabletsTenofovir (Viread)
Oral: 300 mg tabletsTipranavir (Aptivus)
Oral: 250 mg capsulesTrifluridine (Viroptic)
Topical: 1% ophthalmic solutionValacyclovir (Valtrex)
Oral: 500, 1000 mg tabletsValganciclovir (Valcyte)
Oral: 450 mg capsulesZalcitabine (dideoxycytidine, ddC) (Hivid)
Oral: 0.375, 0.75 mg tabletsZanamivir (Relenza)
Inhalational: 5 mg/blisterZidovudine (azidothymidine, AZT) (Retrovir)
Oral: 100 mg capsules, 300 mg tablets, 50 mg/5 mL syrupOral (Combivir): 300 mg tablets in combination with 150 mg
lamivudineOral (Trizivir): 300 mg tablets in combination with 150 mg
lamivudine and 300 mg zidovudineParenteral: 10 mg/mL
Etravirine (Intelence)Oral: 100 mg tablets
Famciclovir (Famvir)Oral: 125, 250, 500 mg tablets
Fosamprenavir (Lexiva)Oral: 700 mg tablets
Foscarnet (Foscavir)Parenteral: 24 mg/mL for IV injection
Ganciclovir (Cytovene)Oral: 250, 500 mg capsulesParenteral: 500 mg/vial for IV injectionIntraocular implant (Vitrasert): 4.5 mg ganciclovir/implant
Imiquimod (Aldara)Topical: 5% cream
Indinavir (Crixivan)Oral: 100, 200, 333, 400 mg capsules
Interferon alfa-2a (Roferon-A)Parenteral: 3, 6, 9, 36 million IU vials
Interferon alfa-2b (Intron A)Parenteral: 3, 5, 10, 18, 25, and 50 million IU vials
Interferon alfa-2b (Rebetron)Parenteral: 3 million IU vials (supplied with oral ribavirin, 200 mg
capsules)Interferon alfa-n3 (Alferon N)
Parenteral: 5 million IU/vialInterferon alfacon-1 (Infergen)
Parenteral: 9 and 15 mcg vialsLamivudine
Oral (Epivir): 150, 300 mg tablets; 10 mg/mL oral solutionOral (Epivir-HBV): 100 mg tablets; 5 mg/mL solutionOral (Combivir): 150 mg tablets in combination with 300 mg
zidovudineOral (Trizivir): 150 mg tablets in combination with 300 mg abacavir
and 300 mg zidovudineLopinavir/ritonavir (Kaletra)
Oral: 133.3 mg/33.3 mg capsules; 80 mg/20 mg per mL solutionMaraviroc (Selzentry)
Oral: 150, 300 mg tabletsNelfinavir (Viracept)
Oral: 250, 625 mg tablets; 50 mg/g powderNevirapine (Viramune)
Oral: 200 mg tablets; 50 mg/5 mL suspensionOseltamivir (Tamiflu)
Oral: 75 mg capsules; powder to reconstitute as suspension (12 mg/mL)
Palivizumab (Synagis)Parenteral: 50, 100 mg/vial
Peginterferon alfa-2a (pegylated interferon alfa-2a, Pegasys)Parenteral: 180 mcg/mL
Peginterferon alfa-2b (pegylated interferon alfa-2b, PEG-Intron)Parenteral: powder to reconstitute as 100, 160, 240, 300 mcg/mL
injection
Penciclovir (Denavir)Topical: 1% cream
Raltegravir (Isentress)Oral: 400 mg tablets
RibavirinAerosol (Virazole): powder to reconstitute for aerosol; 6 g/100 mL vialOral (Rebetol, generic): 200 mg capsules, tablets; 40 mg/mL oral
solutionOral (Rebetron): 200 mg in combination with 3 million units
interferon alfa-2b (Intron-A)Rilpivirine
Oral (Edurant): 25 mg tabletsOral (Complera): 200 mg emtricitabine, 25 mg rilpivirine, 300 mg
tenofovir) tabletsRimantadine (Flumadine)
Oral: 100 mg tablets; 50 mg/5 mL syrupRitonavir (Norvir)
Oral: 100 mg capsules; 80 mg/mL oral solutionSaquinavir
Oral (Invirase): 200 mg hard gel capsules, 500 mg tabletsStavudine
Oral (Zerit): 15, 20, 30, 40 mg capsules; powder for 1 mg/mL oralsolution
Oral extended-release (Zerit XR): 37.5, 50, 75, 100 mg capsulesTelaprivir (Incivik)
Oral: 200 mg capsulesTelbivudine (Tyzeka)
Oral: 600 mg tabletsTenofovir (Viread)
Oral: 300 mg tabletsTipranavir (Aptivus)
Oral: 250 mg capsulesTrifluridine (Viroptic)
Topical: 1% ophthalmic solutionValacyclovir (Valtrex)
Oral: 500, 1000 mg tabletsValganciclovir (Valcyte)
Oral: 450 mg capsulesZalcitabine (dideoxycytidine, ddC) (Hivid)
Oral: 0.375, 0.75 mg tabletsZanamivir (Relenza)
Inhalational: 5 mg/blisterZidovudine (azidothymidine, AZT) (Retrovir)
Oral: 100 mg capsules, 300 mg tablets, 50 mg/5 mL syrupOral (Combivir): 300 mg tablets in combination with 150 mg
lamivudineOral (Trizivir): 300 mg tablets in combination with 150 mg
lamivudine and 300 mg zidovudineParenteral: 10 mg/mL
049-Katzung_Ch049_p861-890.indd 889 9/23/11 4:25:37 PM

890 SECTION VIII Chemotherapeutic Drugs
C A S E S T U D Y A N S W E R
Combination antiviral therapy against both HIV and hepatitis B virus (HBV) is indicated in this patient, given the high viral load and low CD4 cell count. However, the use of methadone and possibly excessive alcohol consumption necessitate cau-tion. Tenofovir and emtricitabine (two nucleoside/nucleotide reverse transcriptase inhibitors) would be a potentially excel-lent choice as components of an initial regimen, since both are active against HIV-1 and HBV, do not interact with methadone, and are available in a once-daily, fixed-dose
combination. Efavirenz, a nonnucleoside reverse tran-scriptase inhibitor, could be added and still maintain a once-daily regimen. Prior to initiation of this regimen, renal function should be checked, and a bone mineral density test should be considered. Pregnancy should be ruled out, and the patient should be counseled that efavirenz should not be taken during pregnancy. The potential for lowered metha-done levels with efavirenz necessitates close monitoring and possibly an increased dose of methadone.
REFERENCES Centers for Disease Control and Prevention: Sexually transmitted diseases treat-
ment guidelines, 2006. MMWR 2006;55(RR-11):16. Drugs for non-HIV viral infections. Med Lett Drugs Ther 2010;98:71. Ghany MG et al: Diagnosis, management, and treatment of hepatitis C: An
update. Hepatology 2009;49:1335. Hirsch MS: Antiviral drug resistance testing in adult HIV-1 infection: 2008 rec-
ommendations of an international AIDS society-USA panel. Clin Infect Dis 2008;47:266.
Lok ASF, McMahon BJ: Chronic hepatitis B: Update 2009. Hepatology 2009;50:1.
Moss RB et al: Targeting pandemic influenza: A primer on influenza antivirals and drug resistance. J Anitimicrob Chemother 2010;5:1086.
Panel on Treatment of HIV-Infected Pregnant Women and Prevention of Perinatal Transmission: Recommendations for Use of Antiretroviral Drugs in Pregnant
HIV-1-Infected Women for Maternal Health and Interventions to Reduce Perinatal HIV Transmission in the United States. May 24, 2010;1. Available at http://aidsinfo.nih.gov/ContentFiles/PerinatalGL.pdf .
Thompson MA et al: Antiretroviral treatment of adult HIV infection: 2010 recom-mendations of the International AIDS Society USA panel. JAMA 2010;304:321.
RELEVANT WEB SITES http://www.aidsinfo.nih.gov http://www.hiv-druginteractions.org http://www.hivinsite.com http://www.iasusa.org
049-Katzung_Ch049_p861-890.indd 890 9/23/11 4:25:37 PM

891
C H A P T E R
■ METRONIDAZOLE, MUPIROCIN, POLYMYXINS, & URINARY ANTISEPTICS
METRONIDAZOLE
Metronidazole is a nitroimidazole antiprotozoal drug (see Chapter 52 ) that also has potent antibacterial activity against anaerobes, including Bacteroides and Clostridium species. Metronidazole is selectively absorbed by anaerobic bacteria and sensitive protozoa. Once taken up by anaerobes, it is nonenzymatically reduced by reacting with reduced ferredoxin. This reduction results in prod-ucts that are toxic to anaerobic cells, and allows for their selective accumulation in anaerobes. The metabolites of metronidazole are taken up into bacterial DNA, forming unstable molecules. This
action only occurs when metronidazole is partially reduced, and because this reduction usually happens only in anaerobic cells, it has relatively little effect on human cells or aerobic bacteria.
Metronidazole is well absorbed after oral administration, is widely distributed in tissues, and reaches serum levels of 4–6 mcg/mL after a 250-mg oral dose. It can also be given intravenously or by rectal suppository. The drug penetrates well into the cerebrospinal fluid and brain, reaching levels similar to those in serum. Metronidazole is metabolized in the liver and may accumulate in hepatic insuf-ficiency.
Metronidazole is indicated for treatment of anaerobic or mixed intra-abdominal infections (in combination with other agents with activity against aerobic organisms), vaginitis (trichomonas infection, bacterial vaginosis), Clostridium difficile colitis, and brain abscess. The typical dosage is 500 mg three times daily orally or intravenously (30 mg/kg/d). Vaginitis may respond to a single 2 g dose. A vaginal gel is available for topical use.
Adverse effects include nausea, diarrhea, stomatitis, and peripheral neuropathy with prolonged use. Metronidazole has a disulfiram-like effect, and patients should be instructed to avoid
50Miscellaneous Antimicrobial Agents; Disinfectants, Antiseptics, & Sterilants Daniel H. Deck, PharmD, & Lisa G. Winston, MD∗
C A S E S T U D Y
A 66-year-old man is admitted to the intensive care unit of a hospital for treatment of community-acquired pneumonia. He receives ceftriaxone and azithromycin upon admission, rapidly improves, and is transferred to a semiprivate ward room. On day 7 of his hospitalization, he develops copious diarrhea with eight bowel movements that day but is otherwise clinically
stable. Clostridium difficile- associated colitis is suspected and a toxin assay is sent to confirm this diagnosis. What is an accept-able treatment for the patient’s diarrhea? The patient is transferred to a single-bed room the following day. The house-keeping staff asks if the old room should be cleaned with alcohol or bleach. Which product should be chosen? Why?
∗ The authors thank Henry F. Chambers, MD, the author of this chapter in previous editions, for his contributions.
050-Katzung_Ch050_p891-900.indd 891 9/21/11 12:10:32 PM

892 SECTION VIII Chemotherapeutic Drugs
alcohol. Although teratogenic in some animals, metronidazole has not been associated with this effect in humans. Other properties of metronidazole are discussed in Chapter 52 .
A structurally similar agent, tinidazole , is a once-daily drug approved for treatment of trichomonas infection, giardiasis, and amebiasis. It also is active against anaerobic bacteria, but is not approved by the Food and Drug Administration (FDA) for treat-ment of anaerobic infections.
MUPIROCIN
Mupirocin (pseudomonic acid) is a natural substance produced by Pseudomonas fluorescens. It is rapidly inactivated after absorption, and systemic levels are undetectable. It is available as an ointment for topical application.
Mupirocin is active against gram-positive cocci, including methicillin-susceptible and methicillin-resistant strains of Staphylococcus aureus . Mupirocin inhibits staphylococcal isoleucyl tRNA synthetase. Low-level resistance, defined as a minimum inhibitory concentration (MIC) of up to 100 mcg/mL, is due to point mutation in the gene of the target enzyme. Low-level resis-tance has been observed after prolonged use. However, local con-centrations achieved with topical application are well above this MIC, and this level of resistance does not lead to clinical failure. High-level resistance, with MICs exceeding 1000 mcg/mL, is due to the presence of a second isoleucyl tRNA synthetase gene, which is plasmid-encoded. High-level resistance results in complete loss of activity. Strains with high-level resistance have caused hospital-associated (nosocomial) outbreaks of staphylococcal infection and colonization. Although higher rates of resistance are encountered with intensive use of mupirocin, more than 95% of staphylococcal isolates are still susceptible.
Mupirocin is indicated for topical treatment of minor skin infections, such as impetigo (see Chapter 61 ). Topical application over large infected areas, such as decubitus ulcers or open surgical wounds, is an important factor leading to emergence of mupiro-cin-resistant strains and is not recommended. Mupirocin effec-tively eliminates S aureus nasal carriage by patients or health care workers, but results are mixed with respect to its ability to prevent subsequent staphylococcal infection.
POLYMYXINS
The polymyxins are a group of basic peptides active against gram-negative bacteria and include polymyxin B and polymyxin E (colistin) . Polymyxins act as cationic detergents. They attach to and disrupt bacterial cell membranes. They also bind and inacti-vate endotoxin. Gram-positive organisms, Proteus sp, and Neisseria sp are resistant.
Owing to their significant toxicity with systemic administra-tion, polymyxins are largely restricted to topical use. Ointments containing polymyxin B, 0.5 mg/g, in mixtures with bacitracin or neomycin (or both) are commonly applied to infected superficial
skin lesions. Emergence of strains of Acinetobacter baumannii , Pseudomonas aeruginosa, and Enterobacteriaceae that are resistant to all other agents has renewed interest in polymyxins as a paren-teral agents for salvage therapy of infections caused by these organisms.
FIDAXOMICIN
Fidaxomicin is a narrow-spectrum, macrocyclic antibiotic that is active against gram-positive aerobes and anaerobes but lacks activity against gram-negative bacteria. Fidaxomicin inhibits bac-terial protein synthesis by binding to the sigma subunit of RNA polymerase. When administered orally, systemic absorption is negligible but fecal concentrations are high. Fidaxomicin has been approved by the FDA for the treatment for C difficile colitis in adults. Preliminary data have demonstrated it is as effective as oral vancomycin and may be associated with lower rates of relapsing disease. The FDA has granted orphan drug designation to all for-mulations of fidaxomicin for the treatment of C difficile infection in pediatric patients aged 16 years and younger. Fidaxomicin is administered as 200 mg orally twice daily.
URINARY ANTISEPTICS
Urinary antiseptics are oral agents that exert antibacterial activity in the urine but have little or no systemic antibacterial effect. Their usefulness is limited to lower urinary tract infections. Prolonged suppression of bacteriuria with urinary antiseptics may be desirable in chronic or recurrent urinary tract infections in which eradication of infection by short-term systemic therapy has not been possible.
Nitrofurantoin At therapeutic doses, nitrofurantoin is bactericidal for many gram-positive and gram-negative bacteria; however, P aeruginosa and many strains of Proteus are inherently resistant. Nitrofurantoin has a complex mechanism of action that is not fully understood. Antibacterial activity appears to correlate with rapid intracellular conversion of nitrofurantoin to highly reactive intermediates by bacterial reductases. These intermediates react nonspecifically with many ribosomal proteins and disrupt the synthesis of pro-teins, RNA, DNA, and metabolic processes. It is not known which of the multiple actions of nitrofurantoin is primarily responsible for its bactericidal activity.
There is no cross-resistance between nitrofurantoin and other antimicrobial agents, and resistance emerges slowly. As resistance to trimethoprim-sulfamethoxazole and fluoroquinolones has become more common in Escherichia coli , nitrofurantoin has become an important alternative oral agent for treatment of uncomplicated urinary tract infection.
Nitrofurantoin is well absorbed after ingestion. It is metabo-lized and excreted so rapidly that no systemic antibacterial action is achieved. The drug is excreted into the urine by both glomerular
050-Katzung_Ch050_p891-900.indd 892 9/21/11 12:10:33 PM

CHAPTER 50 Miscellaneous Antimicrobial Agents; Disinfectants, Antiseptics, & Sterilants 893
filtration and tubular secretion. With average daily doses, concen-trations of 200 mcg/mL are reached in urine. In renal failure, urine levels are insufficient for antibacterial action, but high blood levels may cause toxicity. Nitrofurantoin is contraindicated in patients with significant renal insufficiency (creatinine clearance < 60 mL/min).
The dosage for urinary tract infection in adults is 100 mg orally taken four times daily. The drug should not be used to treat upper urinary tract infection. Oral nitrofurantoin can be given for months for the suppression of chronic urinary tract infection. It is desirable to keep urinary pH below 5.5, which greatly enhances drug activity. A single daily dose of nitrofurantoin, 100 mg, can prevent recurrent urinary tract infections in some women.
Anorexia, nausea, and vomiting are the principal side effects of nitrofurantoin. Neuropathies and hemolytic anemia occur in patients with glucose-6-phosphate dehydrogenase deficiency. Nitrofurantoin antagonizes the action of nalidixic acid. Rashes, pulmonary infiltration and fibrosis, and other hypersensitivity reactions have been reported.
Methenamine Mandelate & Methenamine Hippurate Methenamine mandelate is the salt of mandelic acid and methe-namine and possesses properties of both of these urinary antisep-tics. Methenamine hippurate is the salt of hippuric acid and methenamine. Below pH 5.5, methenamine releases formalde-hyde, which is antibacterial (see Aldehydes, below). Mandelic acid or hippuric acid taken orally is excreted unchanged in the urine, in which these drugs are bactericidal for some gram-negative bacteria when pH is less than 5.5.
Methenamine mandelate, 1 g four times daily, or methenamine hippurate, 1 g twice daily by mouth (children, 50 mg/kg/d or 30 mg/kg/d, respectively), is used only as a urinary antiseptic to sup-press, not treat, urinary tract infection. Acidifying agents (eg, ascorbic acid, 4–12 g/d) may be given to lower urinary pH below 5.5. Sulfonamides should not be given at the same time because they may form an insoluble compound with the formaldehyde released by methenamine. Persons taking methenamine mandelate may exhibit falsely elevated tests for catecholamine metabolites.
■ DISINFECTANTS, ANTISEPTICS, & STERILANTS
Disinfectants are strong chemical agents that inhibit or kill micro-organisms ( Table 50–1 ). Antiseptics are disinfecting agents with sufficiently low toxicity for host cells that they can be used directly on skin, mucous membranes, or wounds. Sterilants kill both veg-etative cells and spores when applied to materials for appropriate times and temperatures. Some of the terms used in this context are defined in Table 50–2 .
Disinfection prevents infection by reducing the number of potentially infective organisms by killing, removing, or diluting them. Disinfection can be accomplished by application of chemi-cal agents or use of physical agents such as ionizing radiation, dry or moist heat, or superheated steam (autoclave, 120°C) to kill microorganisms. Often a combination of agents is used, eg, water and moderate heat over time (pasteurization); ethylene oxide and moist heat (a sterilant); or addition of disinfectant to a detergent. Prevention of infection also can be achieved by washing, which dilutes the potentially infectious organism, or by establishing a
TABLE 50–1 Activities of disinfectants.
Bacteria Viruses Other
Gram-
Positive
Gram-
Negative
Acid-
Fast Spores Lipophilic Hydrophilic Fungi
Amebic
Cysts Prions
Alcohols (isopropa-nol, ethanol)
HS HS S R S V … … R
Aldehydes (glutaral-dehyde, formalde-hyde)
HS HS MS S (slow) S MS S … R
Chlorhexidine gluconate
HS MS R R V R … … R
Sodium hypochlo-rite, chlorine dioxide
HS HS MS S (pH 7.6) S S (at high conc)
MS S MS (at high conc)
Hexachlorophene S (slow) R R R R R R R R
Povidone, iodine HS HS S S (at high conc)
S R S S R
Phenols, quaternary ammonium compounds
HS HS MS R S R S … R
HS, highly susceptible; S, susceptible; MS, moderately susceptible; R, resistant; V, variable; …, no data.
050-Katzung_Ch050_p891-900.indd 893 9/21/11 12:10:33 PM

894 SECTION VIII Chemotherapeutic Drugs
barrier, eg, gloves, condom, or respirator, which prevents the pathogen from gaining entry to the host.
Hand hygiene is the most important means of preventing transmission of infectious agents from person to person or from regions of high microbial load, eg, mouth, nose, or gut, to poten-tial sites of infection. Soap and warm water efficiently and effec-tively remove bacteria. Skin disinfectants along with detergent and water are usually used preoperatively as a surgical scrub for sur-geons’ hands and the patient’s surgical incision.
Evaluation of effectiveness of antiseptics, disinfectants, and sterilants, although seemingly simple in principle, is very complex. Factors in any evaluation include the intrinsic resistance of the microorganism, the number of microorganisms present, mixed populations of organisms, amount of organic material present (eg, blood, feces, tissue), concentration and stability of disinfectant or sterilant, time and temperature of exposure, pH, and hydration and binding of the agent to surfaces. Specific, standardized assays of activity are defined for each use. Toxicity for humans also must be evaluated. In the United States, the Environmental Protection Agency (EPA) regulates disinfectants and sterilants and the FDA regulates antiseptics.
Users of antiseptics, disinfectants, and sterilants need to con-sider their short-term and long-term toxicity because they may have general biocidal activity and may accumulate in the environ-ment or in the body of the patient or caregiver using the agent. Disinfectants and antiseptics may also become contaminated by resistant microorganisms—eg, spores, P aeruginosa , or Serratia marcescens —and actually transmit infection. Most topical antisep-tics interfere with wound healing to some degree. Simple cleansing of wounds with soap and water is less damaging than the applica-tion of antiseptics. Topical antibiotics with a narrow spectrum of action and low toxicity (eg, bacitracin and mupirocin) can be used for temporary control of bacterial growth and are generally pre-ferred to antiseptics. Methenamine-containing agents release
formaldehyde in a low antibacterial concentration at acid pH and can be an effective urinary antiseptic for long-term control of urinary tract infections.
Some of the chemical classes of antiseptics, disinfectants, and sterilants are described briefly in the text that follows. The reader is referred to the general references for descriptions of physical disinfection and sterilization methods.
ALCOHOLS
The two alcohols most frequently used for antisepsis and disinfec-tion are ethanol and isopropyl alcohol ( isopropanol ). They are rapidly active, killing vegetative bacteria, Mycobacterium tuberculosis , and many fungi, and inactivating lipophilic viruses. The optimum bactericidal concentration is 60–90% by volume in water. They probably act by denaturation of proteins. They are not used as sterilants because they are not sporicidal, do not penetrate protein-containing organic material, may not be active against hydrophilic viruses, and lack residual action because they evaporate com-pletely. The alcohols are useful in situations in which sinks with running water are not available for washing with soap and water. Their skin-drying effect can be partially alleviated by addition of emollients to the formulation. Use of alcohol-based hand rubs has been shown to reduce transmission of health care-associated bacte-rial pathogens and is recommended by the Centers for Disease Control and Prevention (CDC) as the preferred method of hand decontamination. Alcohol-based hand rubs are ineffective against spores of C difficile, and assiduous handwashing with soap and water is still required for decontamination after caring for a patient with infection from this organism.
Alcohols are flammable and must be stored in cool, well-ventilated areas. They must be allowed to evaporate before cau-tery, electrosurgery, or laser surgery. Alcohols may be damaging if applied directly to corneal tissue. Therefore, instruments such as tonometers that have been disinfected in alcohol should be rinsed with sterile water, or the alcohol should be allowed to evaporate before they are used.
CHLORHEXIDINE
Chlorhexidine is a cationic biguanide with very low water solubil-ity. Water-soluble chlorhexidine digluconate is used in water-based formulations as an antiseptic. It is active against vegetative bacteria and mycobacteria and has moderate activity against fungi and viruses. It strongly adsorbs to bacterial membranes, causing leakage of small molecules and precipitation of cytoplasmic proteins. It is active at pH 5.5–7.0. Chlorhexidine gluconate is slower in its action than alcohols, but, because of its persistence, it has residual activity when used repeatedly, producing bactericidal action equivalent to alcohols. It is most effective against gram-positive cocci and less active against gram-positive and gram-negative rods. Spore germination is inhibited by chlorhexidine. Chlorhexidine digluconate is resistant to inhibition by blood and organic materials. However, anionic and nonionic agents in
TABLE 50–2 Commonly used terms related to chemical and physical killing of microorganisms.
Antisepsis Application of an agent to living tissue for the purpose of preventing infection
Decontamination Destruction or marked reduction in number or activity of microorganisms
Disinfection Chemical or physical treatment that destroys most vegetative microbes or viruses, but not spores, in or on inanimate surfaces
Sanitization Reduction of microbial load on an inanimate surface to a level considered acceptable for public health purposes
Sterilization A process intended to kill or remove all types of microorganisms, including spores, and usually including viruses, with an acceptably low probability of survival
Pasteurization A process that kills nonsporulating micro-organisms by hot water or steam at 65–100°C
050-Katzung_Ch050_p891-900.indd 894 9/21/11 12:10:33 PM

CHAPTER 50 Miscellaneous Antimicrobial Agents; Disinfectants, Antiseptics, & Sterilants 895
moisturizers, neutral soaps, and surfactants may neutralize its action. Chlorhexidine digluconate formulations of 4% concentra-tion have slightly greater antibacterial activity than newer 2% formulations. The combination of chlorhexidine gluconate in 70% alcohol, available in some countries including the United States, is the preferred agent for skin antisepsis in many surgical and percutaneous procedures. The advantage of this combination over povidone-iodine may derive from its more rapid action after application, its retained activity after exposure to body fluids, and its persistent activity on the skin. Chlorhexidine has a very low skin-sensitizing or irritating capacity. Oral toxicity is low because it is poorly absorbed from the alimentary tract. Chlorhexidine must not be used during surgery on the middle ear because it causes sensorineural deafness. Similar neural toxicity may be encountered during neurosurgery.
HALOGENS
Iodine Iodine in a 1:20,000 solution is bactericidal in 1 minute and kills spores in 15 minutes. Tincture of iodine USP contains 2% iodine and 2.4% sodium iodide in alcohol. It is the most active antiseptic for intact skin. It is not commonly used because of serious hyper-sensitivity reactions that may occur and because of its staining of clothing and dressings.
Iodophors Iodophors are complexes of iodine with a surface-active agent such as polyvinyl pyrrolidone (PVP; povidone-iodine) . Iodophors retain the activity of iodine. They kill vegetative bacteria, myco-bacteria, fungi, and lipid-containing viruses. They may be spori-cidal upon prolonged exposure. Iodophors can be used as antiseptics or disinfectants, the latter containing more iodine. The amount of free iodine is low, but it is released as the solution is diluted. An iodophor solution must be diluted according to the manufacturer’s directions to obtain full activity.
Iodophors are less irritating and less likely to produce skin hypersensitivity than tincture of iodine. They require drying time on skin before becoming active, which can be a disadvantage. Although iodophors have a somewhat broader spectrum of activ-ity than chlorhexidine, including sporicidal action, they lack its persistent activity on skin.
Chlorine Chlorine is a strong oxidizing agent and universal disinfectant that is most commonly provided as a 5.25% sodium hypochlorite solution, a typical formulation for household bleach . Because formulations may vary, the exact concentration should be verified on the label. A 1:10 dilution of household bleach provides 5000 ppm of available chlorine. The CDC recommends this concentration for disinfection of blood spills. Less than 5 ppm kills vegetative bac-teria, whereas up to 5000 ppm is necessary to kill spores. A con-centration of 1000–10,000 ppm is tuberculocidal. One hundred
ppm kills vegetative fungal cells in 1 hour, but fungal spores require 500 ppm. Viruses are inactivated by 200–500 ppm. Dilutions of 5.25% sodium hypochlorite made up in pH 7.5–8.0 tap water retain their activity for months when kept in tightly closed, opaque containers. Frequent opening and closing of the container reduces the activity markedly.
Because chlorine is inactivated by blood, serum, feces, and protein-containing materials, surfaces should be cleaned before chlorine disinfectant is applied. Undissociated hypochlorous acid (HOCl) is the active biocidal agent. When pH is increased, the less active hypochlorite ion, OCl – , is formed. When hypochlorite solutions contact formaldehyde, the carcinogen bischloromethyl is formed. Rapid evolution of irritating chlorine gas occurs when hypochlorite solutions are mixed with acid and urine. Solutions are corrosive to aluminum, silver, and stainless steel.
Alternative chlorine-releasing compounds include chlorine dioxide and chloramine T . These agents retain chlorine longer and have a prolonged bactericidal action.
PHENOLICS
Phenol itself (perhaps the oldest of the surgical antiseptics) is no longer used even as a disinfectant because of its corrosive effect on tissues, its toxicity when absorbed, and its carcinogenic effect. These adverse actions are diminished by forming derivatives in which a functional group replaces a hydrogen atom in the aromatic ring. The phenolic agents most commonly used are o -phenylphenol, o -benzyl- p -chlorophenol , and p -tertiary amylphenol . Mixtures of phenolic derivatives are often used. Some of these are derived from coal tar distillates, eg, cresols and xylenols. Skin absorption and skin irritation still occur with these derivatives, and appropri-ate care is necessary in their use. Detergents are often added to formulations to clean and remove organic material that may decrease the activity of a phenolic compound.
Phenolic compounds disrupt cell walls and membranes, pre-cipitate proteins, and inactivate enzymes. They are bactericidal (including mycobacteria) and fungicidal and are capable of inacti-vating lipophilic viruses. They are not sporicidal. Dilution and time of exposure recommendations of the manufacturer must be followed.
Phenolic disinfectants are used for hard surface decontamina-tion in hospitals and laboratories, eg, floors, beds, and counter or bench tops. They are not recommended for use in nurseries and especially in bassinets, where their use has been associated with hyperbilirubinemia. Use of hexachlorophene as a skin disinfec-tant has caused cerebral edema and convulsions in premature infants and, occasionally, in adults.
QUATERNARY AMMONIUM COMPOUNDS
The quaternary ammonium compounds (“quats”) are cationic surface-active detergents. The active cation has at least one long water-repellent hydrocarbon chain, which causes the molecules to
050-Katzung_Ch050_p891-900.indd 895 9/21/11 12:10:33 PM

896 SECTION VIII Chemotherapeutic Drugs
concentrate as an oriented layer on the surface of solutions and colloidal or suspended particles. The charged nitrogen portion of the cation has high affinity for water and prevents separation out of solution. The bactericidal action of quaternary compounds has been attributed to inactivation of energy-producing enzymes, denaturation of proteins, and disruption of the cell membrane. These agents are fungistatic and sporistatic and also inhibit algae. They are bactericidal for gram-positive bacteria and moderately active against gram-negative bacteria. Lipophilic viruses are inacti-vated. They are not tuberculocidal or sporicidal, and they do not inactivate hydrophilic viruses. Quaternary ammonium com-pounds bind to the surface of colloidal protein in blood, serum, and milk and to the fibers in cotton, mops, cloths, and paper towels used to apply them, which can cause inactivation of the agent by removing it from solution. They are inactivated by anionic detergents (soaps), by many nonionic detergents, and by calcium, magnesium, ferric, and aluminum ions.
Quaternary compounds are used for sanitation of noncritical surfaces (floors, bench tops, etc). Their low toxicity has led to their use as sanitizers in food production facilities. The CDC recom-mends that quaternary ammonium compounds such as benzalko-nium chloride not be used as antiseptics because several outbreaks of infections have occurred that were due to growth of Pseudomonas and other gram-negative bacteria in quaternary ammonium anti-septic solutions.
ALDEHYDES
Formaldehyde and glutaraldehyde are used for disinfection or sterilization of instruments such as fiberoptic endoscopes, respira-tory therapy equipment, hemodialyzers, and dental handpieces that cannot withstand exposure to the high temperatures of steam sterilization. They are not corrosive for metal, plastic, or rubber. These agents have a broad spectrum of activity against microor-ganisms and viruses. They act by alkylation of chemical groups in proteins and nucleic acids. Failures of disinfection or sterilization can occur as a result of dilution below the known effective concen-tration, the presence of organic material, and the failure of liquid to penetrate into small channels in the instruments. Automatic circulating baths are available that increase penetration of alde-hyde solution into the instrument while decreasing exposure of the operator to irritating fumes.
Formaldehyde is available as a 40% weight per volume solution in water (100% formalin) . An 8% formaldehyde solution in water has a broad spectrum of activity against bacteria, fungi, and viruses. Sporicidal activity may take as long as 18 hours. Its rapid-ity of action is increased by solution in 70% isopropanol. Formaldehyde solutions are used for high-level disinfection of hemodialyzers, preparation of vaccines, and preservation and embalming of tissues. The 4% formaldehyde (10% formalin) solutions used for fixation of tissues and embalming may not be mycobactericidal.
Glutaraldehyde is a dialdehyde (1,5-pentanedial). Solutions of 2% weight per volume glutaraldehyde are most commonly used. The solution must be alkalinized to pH 7.4–8.5 for activation.
Activated solutions are bactericidal, sporicidal, fungicidal, and virucidal for both lipophilic and hydrophilic viruses. Glutaraldehyde has greater sporicidal activity than formaldehyde, but its tubercu-locidal activity may be less. Lethal action against mycobacteria and spores may require prolonged exposure. Once activated, solutions have a shelf life of 14 days, after which polymerization reduces activity. Other means of activation and stabilization can increase the shelf life. Because glutaraldehyde solutions are frequently reused, the most common reason for loss of activity is dilution and exposure to organic material. Test strips to measure residual activ-ity are recommended.
Formaldehyde has a characteristic pungent odor and is highly irritating to respiratory mucous membranes and eyes at concentra-tions of 2–5 ppm. The United States Occupational Safety and Health Administration (OSHA) has declared that formaldehyde is a potential carcinogen and has established an employee exposure standard that limits the 8-hour time-weighted average (TWA) exposure to 0.75 ppm. Protection of health care workers from exposure to glutaraldehyde concentrations greater than 0.2 ppm is advisable. Increased air exchange, enclosure in hoods with exhausts, tight-fitting lids on exposure devices, and use of protec-tive personal equipment such as goggles, respirators, and gloves may be necessary to achieve these exposure limits.
Ortho-phthalaldehyde (OPA) is a phenolic dialdehyde chem-ical sterilant with a spectrum of activity comparable to glutaralde-hyde, although it is several times more rapidly bactericidal. OPA solution typically contains 0.55% OPA. Its label claim is that high-level disinfection can be achieved in 12 minutes at room temperature compared with 45 minutes for 2.4% glutaraldehyde. Unlike glutaraldehyde, OPA requires no activation, is less irritat-ing to mucous membranes, and does not require exposure moni-toring. It has good materials compatibility and an acceptable environmental safety profile. OPA is useful for disinfection or sterilization of endoscopes, surgical instruments, and other medical devices.
SUPEROXIDIZED WATER
Electrolysis of saline yields a mixture of oxidants, primarily hypochlorous acid and chlorine, with potent disinfectant and ste-rilant properties. The solution generated by the process, which has been commercialized and marketed as Sterilox for disinfection of endoscopes and dental materials, is rapidly bactericidal, fungicidal, tuberculocidal, and sporicidal. High-level disinfection is achieved with a contact time of 10 minutes. The solution is nontoxic and nonirritating and requires no special disposal precautions.
PEROXYGEN COMPOUNDS
The peroxygen compounds, hydrogen peroxide and peracetic acid , have high killing activity and a broad spectrum against bac-teria, spores, viruses, and fungi when used in appropriate concen-tration. They have the advantage that their decomposition products are not toxic and do not injure the environment. They
050-Katzung_Ch050_p891-900.indd 896 9/21/11 12:10:33 PM

CHAPTER 50 Miscellaneous Antimicrobial Agents; Disinfectants, Antiseptics, & Sterilants 897
are powerful oxidizers that are used primarily as disinfectants and sterilants.
Hydrogen peroxide is a very effective disinfectant when used for inanimate objects or materials with low organic content such as water. Organisms with the enzymes catalase and peroxidase rapidly degrade hydrogen peroxide. The innocuous degradation products are oxygen and water. Concentrated solutions containing 90% weight per volume H 2 O 2 are prepared electrochemically. When diluted in high-quality deionized water to 6% and 3% and put into clean containers, they remain stable. Hydrogen peroxide has been proposed for disinfection of respirators, acrylic resin implants, plastic eating utensils, soft contact lenses, and cartons intended to contain milk or juice products. Concentrations of 10–25% hydrogen peroxide are sporicidal. Vapor phase hydrogen peroxide (VPHP) is a cold gaseous sterilant that has the potential to replace the toxic or carcinogenic gases ethylene oxide and formaldehyde. VPHP does not require a pressurized chamber and is active at temperatures as low as 4°C and concentrations as low as 4 mg/L. It is incompatible with liquids and cellulose products. It penetrates the surface of some plastics. Automated equipment using vaporized hydrogen peroxide (eg, Sterrad) or hydrogen peroxide mixed with formic acid (Endoclens) is available for sterilizing endoscopes.
Peracetic acid (CH 3 COOOH) is prepared commercially from 90% hydrogen peroxide, acetic acid, and sulfuric acid as a catalyst. It is explosive in the pure form. It is usually used in dilute solution and transported in containers with vented caps to prevent increased pressure as oxygen is released. Peracetic acid is more active than hydrogen peroxide as a bactericidal and sporicidal agent. Concentrations of 250–500 ppm are effective against a broad range of bacteria in 5 minutes at pH 7.0 at 20°C. Bacterial spores are inactivated by 500–30,000 ppm peracetic acid. Only slightly increased concentrations are necessary in the presence of organic matter. Viruses require variable exposures. Enteroviruses require 2000 ppm for 15–30 minutes for inactivation.
An automated machine (Steris) that uses buffered peracetic acid liquid of 0.1–0.5% concentration has been developed for steriliza-tion of medical, surgical, and dental instruments. Peracetic acid sterilization systems have also been adopted for hemodialyzers. The food processing and beverage industries use peracetic acid exten-sively because the breakdown products in high dilution do not produce objectionable odor, taste, or toxicity. Because rinsing is not necessary in this use, time and money are saved.
Peracetic acid is a potent tumor promoter but a weak carcino-gen. It is not mutagenic in the Ames test.
HEAVY METALS
Heavy metals, principally mercury and silver, are now rarely used as disinfectants. Mercury is an environmental hazard, and some pathogenic bacteria have developed plasmid-mediated resistance to mercurials. Hypersensitivity to thimerosal is common, possibly
in up to 40% of the population. These compounds are absorbed from solution by rubber and plastic closures. Nevertheless, thimerosal 0.001–0.004% is still used safely as a preservative of vaccines, antitoxins, and immune sera. Although a causative link to autism has never been established, thimerosal-free vaccines are available for use in children and pregnant woman.
Inorganic silver salts are strongly bactericidal. Silver nitrate , 1:1000, has been most commonly used, particularly as a preven-tive for gonococcal ophthalmitis in newborns. Antibiotic oint-ments have replaced silver nitrate for this indication. Silver sulfadiazine slowly releases silver and is used to suppress bacterial growth in burn wounds (see Chapter 46 ).
STERILANTS
For many years, pressurized steam (autoclaving) at 120°C for 30 minutes has been the basic method for sterilizing instruments and other heat-resistant materials. When autoclaving is not possible, as with lensed instruments and materials containing plastic and rub-ber, ethylene oxide —diluted with either fluorocarbon or carbon dioxide to diminish explosive hazard—is used at 440–1200 mg/L at 45–60°C with 30–60% relative humidity. The higher concentra-tions have been used to increase penetration.
Ethylene oxide is classified as a mutagen and carcinogen. The OSHA permissible exposure limit (PEL) for ethylene oxide is 1 ppm calculated as a time-weighted average. Alternative sterilants now being used increasingly include vapor phase hydrogen peroxide, peracetic acid, ozone, gas plasma, chlorine dioxide, formaldehyde, and propylene oxide. Each of these sterilants has potential advan-tages and problems. Automated peracetic acid systems are being used increasingly for high-level decontamination and sterilization of endoscopes and hemodialyzers because of their effectiveness, automated features, and the low toxicity of the residual products of sterilization.
PRESERVATIVES
Disinfectants are used as preservatives to prevent the overgrowth of bacteria and fungi in pharmaceutical products, laboratory sera and reagents, cosmetic products, and contact lenses. Multi-use vials of medication that may be reentered through a rubber dia-phragm, and eye and nose drops, require preservatives. Preservatives should not be irritating or toxic to tissues to which they will be applied, they must be effective in preventing growth of microor-ganisms likely to contaminate them, and they must have sufficient solubility and stability to remain active.
Commonly used preservative agents include organic acids such as benzoic acid and salts, the parabens , (alkyl esters of p -hydroxybenzoic acid), sorbic acid and salts, phenolic compounds, quaternary ammonium compounds, alcohols, and mercurials such as thimerosal in 0.001–0.004% concentration.
050-Katzung_Ch050_p891-900.indd 897 9/21/11 12:10:33 PM

898 SECTION VIII Chemotherapeutic Drugs
SUMMARY Miscellaneous Antimicrobials
SubclassMechanism of Action Effects
Clinical Applications
Pharmacokinetics, Toxicities, Interactions
NITROIMIDAZOLE • Metronidazole Disruption of electron
transport chainBactericidal activity against susceptible anaerobic bacteria and protozoa
Anaerobic infec-tions • vaginitis • C difficile colitis
Oral or IV • hepatic clearance (half-life = 8 h) • disulfiram-like reaction when given with alcohol • Toxicity: Gastrointestinal upset • metallic taste • neuropathy • seizures
• Tinidazole: Oral; similar to metronidazole but dosed once daily; approved for trichomonas, giardiasis, and amebiasis
URINARY ANTISEPTICS • Nitrofurantoin Not fully understood
• disrupts protein synthesis and inhibits multiple bacterial enzyme systems
Bacteriostatic or bacteri-cidal activity against susceptible bacteria
Uncomplicated uri-nary tract infec-tions • long-term prophylaxis
Oral • rapid renal clearance (half-life = 0.5 h) • blood levels are negligible • contraindicated in renal failure • Toxicity: Gastrointestinal upset • neuropathies • hypersensitivity in patients with pulmonary fibrosis
• Methenamine hippurate and methenamine mandelate: Oral; release formaldehyde at acidic pH in the urine; used only for suppression, not treatment, of urinary tract infections
MACROLIDE • Fidaxomicin Inhibits bacterial RNA
polymeraseBactericidal in gram posi-tive bacteria
C difficile colitis Oral • blood levels negligible
MISCELLANEOUS ANTIMICROBIAL DRUGSColistimethate sodium (Coly-Mycin M)
Parenteral: 150 mg for injectionFidaxomicin (Dificid)
Oral: 200 mg tabletsMethenamine hippurate (Hiprex, Urex)
Oral: 1.0 g tabletsMethenamine mandelate (generic)
Oral: 0.5, 1 g tablets; 0.5 g/5 mL suspensionMetronidazole (generic, Flagyl)
Oral: 250, 500 mg tablets; 375 mg capsules; 750 mg extended-release tablets
Topical: 0.75% gelParenteral: 5 mg/mL; 500 mg for injection
Mupirocin (Bactroban)Topical: 2% ointment, cream
Nitrofurantoin (generic, Macrodantin)Oral: 25, 50, 100 mg capsules, 25 mg/5 mL suspension
Polymyxin B (Polymyxin B Sulfate)Parenteral: 500,000 units per vial for injectionOphthalmic: 500,000 units per vial
DISINFECTANTS, ANTISEPTICS, & STERILANTSBenzalkonium (generic, Zephiran)
Topical: 17% concentrate; 50% solution; 1:750 solution
Benzoyl peroxide (generic)Topical: 2.5%, 5%, 10% liquid; 5%, 5.5%, 10% lotion; 5%, 10% cream;
2.5%, 4%, 5%, 6%, 10%, 20% gelChlorhexidine gluconate (Hibiclens, Hibistat, others)
Topical: 2%, 4% cleanser, sponge; 0.5% rinse in 70% alcoholOral rinse (Peridex, Periogard): 0.12%
Glutaraldehyde (Cidex)Instruments: 2%, 3.2% solution
Hexachlorophene (pHisoHex)Topical: 3% liquid; 0.23% foam
Iodine aqueous (generic, Lugol’s Solution)Topical: 2–5% in water with 2.4% sodium iodide or 10% potassium
iodideIodine tincture (generic)
Topical: 2% iodine or 2.4% sodium iodide in 47% alcohol, in 15, 30, 120 mL and in larger quantities
Nitrofurazone (generic, Furacin)Topical: 0.2% solution, ointment, and cream
Ortho-phthalaldehyde (Cidex OPA)Instruments: 0.55% solution
Povidone-iodine (generic, Betadine)Topical: available in many forms, including aerosol, ointment,
antiseptic gauze pads, skin cleanser (liquid or foam), solution, and swabsticks
Silver nitrate (generic)Topical: 10, 25, 50% solution
Thimerosal (generic, Mersol)Topical: 1:1000 tincture and solution
P R E P A R A T I O N S A V A I L A B L E
MISCELLANEOUS ANTIMICROBIAL DRUGSColistimethate sodium (Coly-Mycin M)
Parenteral: 150 mg for injectionFidaxomicin (Dificid)
Oral: 200 mg tabletsMethenamine hippurate (Hiprex, Urex)
Oral: 1.0 g tabletsMethenamine mandelate (generic)
Oral: 0.5, 1 g tablets; 0.5 g/5 mL suspensionMetronidazole (generic, Flagyl)
Oral: 250, 500 mg tablets; 375 mg capsules; 750 mg extended-release tablets
Topical: 0.75% gelParenteral: 5 mg/mL; 500 mg for injection
Mupirocin (Bactroban)Topical: 2% ointment, cream
Nitrofurantoin (generic, Macrodantin)Oral: 25, 50, 100 mg capsules, 25 mg/5 mL suspension
Polymyxin B (Polymyxin B Sulfate)Parenteral: 500,000 units per vial for injectionOphthalmic: 500,000 units per vial
DISINFECTANTS, ANTISEPTICS, & STERILANTSBenzalkonium (generic, Zephiran)
Topical: 17% concentrate; 50% solution; 1:750 solution
Benzoyl peroxide (generic)Topical: 2.5%, 5%, 10% liquid; 5%, 5.5%, 10% lotion; 5%, 10% cream;
2.5%, 4%, 5%, 6%, 10%, 20% gelChlorhexidine gluconate (Hibiclens, Hibistat, others)
Topical: 2%, 4% cleanser, sponge; 0.5% rinse in 70% alcoholOral rinse (Peridex, Periogard): 0.12%
Glutaraldehyde (Cidex)Instruments: 2%, 3.2% solution
Hexachlorophene (pHisoHex)Topical: 3% liquid; 0.23% foam
Iodine aqueous (generic, Lugol’s Solution)Topical: 2–5% in water with 2.4% sodium iodide or 10% potassium
iodideIodine tincture (generic)
Topical: 2% iodine or 2.4% sodium iodide in 47% alcohol, in 15, 30,120 mL and in larger quantities
Nitrofurazone (generic, Furacin)Topical: 0.2% solution, ointment, and cream
Ortho-phthalaldehyde (Cidex OPA)Instruments: 0.55% solution
Povidone-iodine (generic, Betadine)Topical: available in many forms, including aerosol, ointment,
antiseptic gauze pads, skin cleanser (liquid or foam), solution, andswabsticks
Silver nitrate (generic)Topical: 10, 25, 50% solution
Thimerosal (generic, Mersol)Topical: 1:1000 tincture and solution
050-Katzung_Ch050_p891-900.indd 898 9/21/11 12:10:33 PM

CHAPTER 50 Miscellaneous Antimicrobial Agents; Disinfectants, Antiseptics, & Sterilants 899
REFERENCES Bischoff WE et al: Handwashing compliance by health care workers: The impact
of introducing an accessible, alcohol-based hand antiseptic. Arch Intern Med 2000;160:1017.
Chambers HF, Winston LG: Mupirocin prophylaxis misses by a nose. Ann Intern Med 2004;140:484.
Humphreys PN: Testing standards for sporicides. J Hosp Infect 2011;77:193. Louie TJ et al. Fidaxomicin versus vancomycin for Clostridium difficile infection.
N Engl J Med 2011;364:422. Gordin FM et al: Reduction in nosocomial transmission of drug-resistant bacteria
after introduction of an alcohol-based hand-rub. Infect Control Hosp Epidemiol 2005;26:650.
Noorani A et al: Systematic review and meta-analysis of preoperative antisepsis with chlorhexidine versus povidone-iodine in clean-contaminated surgery. Br J Surg 2010;97:1614.
Rutala WA, Weber DJ: New disinfection and sterilization methods. Emerg Infect Dis 2001;7:348.
Tinidazole. Med Lett Drugs Ther 2004;46:70. Widmer AF, Frei R: Decontamination, disinfection, and sterilization. In: Murray
PR et al (editors). Manual of Clinical Microbiology , 7th ed. American Society for Microbiology, 1999.
C A S E S T U D Y A N S W E R
The patient should be treated with oral metronidazole, which is considered the preferred drug for mild to moderate cases of C difficile -associated colitis. The room should not be
cleaned with alcohol since it does not kill C difficile spores. A concentrated bleach solution (5000 ppm) would be preferred because it is sporicidal.
050-Katzung_Ch050_p891-900.indd 899 9/21/11 12:10:33 PM

This page intentionally left blank

901
C H A P T E R
The development of antimicrobial drugs represents one of the most important advances in therapeutics, both in the control or cure of serious infections and in the prevention and treatment of infectious complications of other therapeutic modalities such as cancer chemotherapy, immunosuppression, and surgery. However, evidence is overwhelming that antimicrobial agents are vastly overprescribed in outpatient settings in the United States, and the availability of antimicrobial agents without prescription in many developing countries has—by facilitating the development of resistance—already severely limited therapeutic options in the treatment of life-threatening infections. Therefore, the clinician should first determine whether antimicrobial therapy is warranted for a given patient. The specific questions one should ask include the following:
1. Is an antimicrobial agent indicated on the basis of clinical find-ings? Or is it prudent to wait until such clinical findings become apparent?
2. Have appropriate clinical specimens been obtained to establish a microbiologic diagnosis?
3. What are the likely etiologic agents for the patient’s illness? 4. What measures should be taken to protect individuals exposed
to the index case to prevent secondary cases, and what mea-sures should be implemented to prevent further exposure?
5. Is there clinical evidence (eg, from well-executed clinical trials) that antimicrobial therapy will confer clinical benefit for the patient?
Once a specific cause is identified based on specific microbio-logic tests, the following further questions should be considered:
1. If a specific microbial pathogen is identified, can a narrower-spectrum agent be substituted for the initial empiric drug?
2. Is one agent or a combination of agents necessary? 3. What are the optimal dose, route of administration, and dura-
tion of therapy? 4. What specific tests (eg, susceptibility testing) should be under-
taken to identify patients who will not respond to treatment? 5. What adjunctive measures can be undertaken to eradicate the
infection? For example, is surgery feasible for removal of devi-talized tissue or foreign bodies—or drainage of an abscess—into which antimicrobial agents may be unable to penetrate?
51Clinical Use of Antimicrobial Agents Harry W. Lampiris, MD, & Daniel S. Maddix, PharmD
C A S E S T U D Y
A 51-year-old alcoholic patient presents to the emergency department with fever, headache, neck stiffness, and altered mental status for 12 hours. Vital signs are blood pressure 90/55 mm Hg, pulse 120/min, respirations 30/min, tempera-ture 40°C [104°F] rectal. The patient is minimally responsive to voice and does not follow commands. Examination is sig-nificant for a right third cranial nerve palsy and nuchal rigid-ity. Laboratory results show a white blood cell count of 24,000/mm 3 with left shift, but other hematologic and chem-istry values are within normal limits. An emergency CT scan
of the head is normal. Blood cultures are obtained, and a lumbar puncture reveals the following cerebrospinal fluid (CSF) values: white blood cells 5000/mm 3 , red blood cells 10/mm 3 , protein 200 mg/dL, glucose 15 mg/dL (serum glu-cose 96 taken at same time). CSF Gram stain reveals gram-positive cocci in pairs. What is the most likely diagnosis in this patient? What organisms should be treated empirically? Are there other pharmacologic interventions to consider before initiating antimicrobial therapy?
051-Katzung_Ch051_p901-914.indd 901 9/21/11 12:10:50 PM

902 SECTION VIII Chemotherapeutic Drugs
Is it possible to decrease the dosage of immunosuppressive therapy in patients who have undergone organ transplanta-tion? Is it possible to reduce morbidity or mortality due to the infection by reducing host immunologic response to the infec-tion (eg, by the use of corticosteroids for the treatment of se-vere Pneumocystis jiroveci pneumonia or meningitis due to Streptococcus pneumoniae )?
■ EMPIRIC ANTIMICROBIAL THERAPY
Antimicrobial agents are frequently used before the pathogen responsible for a particular illness or the susceptibility to a par-ticular antimicrobial agent is known. This use of antimicrobial agents is called empiric (or presumptive) therapy and is based on experience with a particular clinical entity. The usual justification for empiric therapy is the hope that early intervention will improve the outcome; in the best cases, this has been established by placebo-controlled, double-blind prospective clinical trials. For example, treatment of febrile episodes in neutropenic cancer patients with empiric antimicrobial therapy has been demon-strated to have impressive morbidity and mortality benefits even though the specific bacterial agent responsible for fever is deter-mined for only a minority of such episodes.
Finally, there are many clinical entities, such as certain epi-sodes of community-acquired pneumonia, in which it is diffi-cult to identify a specific pathogen. In such cases, a clinical response to empiric therapy may be an important clue to the likely pathogen.
Frequently, the signs and symptoms of infection diminish as a result of empiric therapy, and microbiologic test results become available that establish a specific microbiologic diagnosis. At the time that the pathogenic organism responsible for the illness is identified, empiric therapy is optimally modified to definitive therapy, which is typically narrower in coverage and is given for an appropriate duration based on the results of clinical trials or experience when clinical trial data are not available.
Approach to Empiric Therapy Initiation of empiric therapy should follow a specific and system-atic approach.
A. Formulate a Clinical Diagnosis of Microbial Infection Using all available data, the clinician should determine that there is anatomic evidence of infection (eg, pneumonia, cellulitis, sinusitis).
B. Obtain Specimens for Laboratory Examination Examination of stained specimens by microscopy or simple examination of an uncentrifuged sample of urine for white blood cells and bacteria may provide important etiologic clues in a very short time. Cultures of selected anatomic sites (blood, sputum, urine, cerebrospinal fluid, and stool) and nonculture methods (antigen testing, polymerase chain reaction, and serology) may also confirm specific etiologic agents.
C. Formulate a Microbiologic Diagnosis The history, physical examination, and immediately available laboratory results (eg, Gram stain of urine or sputum) may pro-vide highly specific information. For example, in a young man with urethritis and a Gram-stained smear from the urethral meatus demonstrating intracellular gram-negative diplococci, the most likely pathogen is Neisseria gonorrhoeae . In the latter instance, however, the clinician should be aware that a significant number of patients with gonococcal urethritis have uninformative Gram stains for the organism and that a significant number of patients with gonococcal urethritis harbor concurrent chlamydial infection that is not demonstrated on the Gram-stained smear.
D. Determine the Necessity for Empiric Therapy Whether or not to initiate empiric therapy is an important clinical decision based partly on experience and partly on data from clini-cal trials. Empiric therapy is indicated when there is a significant risk of serious morbidity if therapy is withheld until a specific pathogen is detected by the clinical laboratory.
In other settings, empiric therapy may be indicated for public health reasons rather than for demonstrated superior outcome of therapy in a specific patient. For example, urethritis in a young sexually active man usually requires treatment for N gonorrhoeae and Chlamydia trachomatis despite the absence of microbiologic confirmation at the time of diagnosis. Because the risk of noncom-pliance with follow-up visits in this patient population may lead to further transmission of these sexually transmitted pathogens, empiric therapy is warranted.
E. Institute Treatment Selection of empiric therapy may be based on the microbiologic diagnosis or a clinical diagnosis without available microbiologic clues. If no microbiologic information is available, the antimicro-bial spectrum of the agent or agents chosen must necessarily be broader, taking into account the most likely pathogens responsible for the patient’s illness.
Choice of Antimicrobial Agent Selection from among several drugs depends on host factors that include the following: (1) concomitant disease states (eg, AIDS, neutropenia due to the use of cytotoxic chemotherapy, organ transplantation, severe chronic liver or kidney disease) or the use of immunosuppressive medications; (2) prior adverse drug effects; (3) impaired elimination or detoxification of the drug (may be genetically predetermined but more frequently is associated with impaired renal or hepatic function due to underlying disease); (4) age of the patient; (5) pregnancy status; and (6) epidemiologic exposure (eg, exposure to a sick family member or pet, recent hospitalization, recent travel, occupational exposure, or new sexual partner).
Pharmacologic factors include (1) the kinetics of absorption, distribution, and elimination; (2) the ability of the drug to be delivered to the site of infection; (3) the potential toxicity of an agent; and (4) pharmacokinetic or pharmacodynamic interactions with other drugs.
051-Katzung_Ch051_p901-914.indd 902 9/21/11 12:10:51 PM

CHAPTER 51 Clinical Use of Antimicrobial Agents 903
Knowledge of the susceptibility of an organism to a specific agent in a hospital or community setting is important in the selec-tion of empiric therapy. Pharmacokinetic differences among agents with similar antimicrobial spectrums may be exploited to reduce the frequency of dosing (eg, ceftriaxone may be conveniently given once every 24 hours). Finally, increasing consideration is being given to the cost of antimicrobial therapy, especially when multiple agents with comparable efficacy and toxicity are available for a specific infection. Changing from intravenous to oral antibiotics for prolonged administration can be particularly cost-effective.
Brief guides to empiric therapy based on presumptive microbial diagnosis and site of infection are given in Tables 51–1 and 51–2 .
■ ANTIMICROBIAL THERAPY OF INFECTIONS WITH KNOWN ETIOLOGY
INTERPRETATION OF CULTURE RESULTS
Properly obtained and processed specimens for culture frequently yield reliable information about the cause of infection. The lack of a confirmatory microbiologic diagnosis may be due to the following:
1. Sample error, eg, obtaining cultures after antimicrobial agents have been administered, or contamination of specimens sent for culture
2. Noncultivable or slow-growing organisms ( Histoplasma capsula-tum, Bartonella or Brucella species ), in which cultures are often discarded before sufficient growth has occurred for detection
3. Requesting bacterial cultures when infection is due to other organisms
4. Not recognizing the need for special media or isolation tech-niques (eg, charcoal yeast extract agar for isolation of legion-ella species, shell-vial tissue culture system for rapid isolation of cytomegalovirus)
Even in the setting of a classic infectious disease for which isolation techniques have been established for decades (eg, pneu-mococcal pneumonia, pulmonary tuberculosis, streptococcal pharyngitis), the sensitivity of the culture technique may be inad-equate to identify all cases of the disease.
GUIDING ANTIMICROBIAL THERAPY OF ESTABLISHED INFECTIONS
Susceptibility Testing Testing bacterial pathogens in vitro for their susceptibility to anti-microbial agents is extremely valuable in confirming susceptibility, ideally to a narrow-spectrum nontoxic antimicrobial drug. Tests measure the concentration of drug required to inhibit growth of the organism ( minimal inhibitory concentration [MIC] ) or to kill the organism ( minimal bactericidal concentration [MBC] ). The results of these tests can then be correlated with known drug concentrations in various body compartments. Only MICs are
routinely measured in most infections, whereas in infections in which bactericidal therapy is required for eradication of infection (eg, meningitis, endocarditis, sepsis in the granulocytopenic host), MBC measurements occasionally may be useful.
Specialized Assay Methods A. Beta-Lactamase Assay For some bacteria (eg, Haemophilus species), the susceptibility pat-terns of strains are similar except for the production of β lacta-mase. In these cases, extensive susceptibility testing may not be required, and a direct test for β lactamase using a chromogenic β-lactam substrate (nitrocephin disk) may be substituted.
B. Synergy Studies Synergy studies are in vitro tests that attempt to measure synergis-tic, additive, indifferent, or antagonistic drug interactions. In general, these tests have not been standardized and have not cor-related well with clinical outcome. (See section on Antimicrobial Drug Combinations for details.)
MONITORING THERAPEUTIC RESPONSE: DURATION OF THERAPY
The therapeutic response may be monitored microbiologically or clinically. Cultures of specimens taken from infected sites should eventually become sterile or demonstrate eradication of the patho-gen and are useful for documenting recurrence or relapse. Follow-up cultures may also be useful for detecting superinfec-tions or the development of resistance. Clinically, the patient’s systemic manifestations of infection (malaise, fever, leukocytosis) should abate, and the clinical findings should improve (eg, as shown by clearing of radiographic infiltrates or lessening hypox-emia in pneumonia).
The duration of definitive therapy required for cure depends on the pathogen, the site of infection, and host factors (immunocom-promised patients generally require longer courses of treatment). Precise data on duration of therapy exist for some infections (eg, streptococcal pharyngitis, syphilis, gonorrhea, tuberculosis, and cryptococcal meningitis). In many other situations, duration of therapy is determined empirically. For recurrent infections (eg, sinus-itis, urinary tract infections), longer courses of antimicrobial therapy or surgical intervention are frequently necessary for eradication.
Clinical Failure of Antimicrobial Therapy When the patient has an inadequate clinical or microbiologic response to antimicrobial therapy selected by in vitro susceptibility testing, systematic investigation should be undertaken to deter-mine the cause of failure. Errors in susceptibility testing are rare, but the original results should be confirmed by repeat testing. Drug dosing and absorption should be scrutinized and tested directly using serum measurements, pill counting, or directly observed therapy.
The clinical data should be reviewed to determine whether the patient’s immune function is adequate and, if not, what can be
051-Katzung_Ch051_p901-914.indd 903 9/21/11 12:10:51 PM

904 SECTION VIII Chemotherapeutic Drugs
TABLE 51–1 Empiric antimicrobial therapy based on microbiologic etiology.
Suspected or Proven Disease or Pathogen Drugs of First Choice Alternative Drugs
Gram-negative cocci (aerobic)
Moraxella (Branhamella) catarrhalis TMP-SMZ,1 cephalosporin (second- or third-generation)2
Quinolone,3 macrolide4
Neisseria gonorrhoeae Ceftriaxone, cefixime Spectinomycin, azithromycin
Neisseria meningitides Penicillin G Chloramphenicol, ceftriaxone, cefotaxime
Gram-negative rods (aerobic)
E coli, Klebsiella, Proteus Cephalosporin (first- or second-generation),2 TMP-SMZ1
Quinolone,3 aminoglycoside5
Enterobacter, Citrobacter, Serratia TMP-SMZ,1 quinolone,3 carbapenem6
Antipseudomonal penicillin,7 aminoglycoside,5 cefepime
Shigella Quinolone3 TMP-SMZ,1 ampicillin, azithromycin, ceftriaxone
Salmonella Quinolone,3 ceftriaxone Chloramphenicol, ampicillin, TMP-SMZ1
Campylobacter jejuni Erythromycin or azithromycin Tetracycline, quinolone3
Brucella species Doxycycline + rifampin or aminoglycoside5
Chloramphenicol + aminoglycoside5 or TMP-SMZ1
Helicobacter pylori Proton pump inhibitor + amoxi-cillin + clarithromycin
Bismuth + metronidazole + tetracycline + proton pump inhibitor
Vibrio species Tetracycline Quinolone,3 TMP-SMZ1
Pseudomonas aeruginosa Antipseudomonal penicillin ± aminoglycoside5
Antipseudomonal penicillin ± quinolone,3 cefepime, ceftazidime, antipseudomonal carbapenem,6 or aztreonam ± aminoglycoside5
Burkholderia cepacia (formerly Pseudomonas cepacia)
TMP-SMZ1 Ceftazidime, chloramphenicol
Stenotrophomonas maltophilia (formerly Xanthomonas maltophilia)
TMP-SMZ1 Minocycline, ticarcillin-clavulanate, tigecycline, ceftazidime, quinolone3
Legionella species Azithromycin or quinolone3 Clarithromycin, erythromycin
Gram-positive cocci (aerobic)
Streptococcus pneumoniae Penicillin8 Doxycycline, ceftriaxone, antipneumococcal quinolone,3
macrolide,4 linezolid
Streptococcus pyogenes (group A) Penicillin, clindamycin Erythromycin, cephalosporin (first-generation)2
Streptococcus agalactiae (group B) Penicillin (± aminoglycoside5) Vancomycin
Viridans streptococci Penicillin Cephalosporin (first- or third-generation),2 vancomycin
Staphylococcus aureus
β-Lactamase negative Penicillin Cephalosporin (first-generation),2 vancomycin
β-Lactamase positive Penicillinase-resistant penicillin9 As above
Methicillin-resistant Vancomycin TMP-SMZ,1 minocycline, linezolid, daptomycin, tigecycline
Enterococcus species10 Penicillin ± aminoglycoside5 Vancomycin ± aminoglycoside5
Gram-positive rods (aerobic)
Bacillus species (non-anthracis) Vancomycin Imipenem, quinolone,3 clindamycin
Listeria species Ampicillin (± aminoglycoside5) TMP-SMZ1
Nocardia species Sulfadiazine, TMP-SMZ1 Minocycline, imipenem, amikacin, linezolid
Anaerobic bacteria
Gram-positive (clostridia, Peptococcus, Actinomyces, Peptostreptococcus)
Penicillin, clindamycin Vancomycin, carbapenem,6 chloramphenicol
Clostridium difficile Metronidazole Vancomycin, bacitracin
Bacteroides fragilis Metronidazole Chloramphenicol, carbapenem,6 β-lactam–β-lactamase-inhibitor combinations, clindamycin
(continued )
051-Katzung_Ch051_p901-914.indd 904 9/21/11 12:10:51 PM

CHAPTER 51 Clinical Use of Antimicrobial Agents 905
TABLE 51–1 Empiric antimicrobial therapy based on microbiologic etiology.
Suspected or Proven Disease or Pathogen Drugs of First Choice Alternative Drugs
Fusobacterium, Prevotella, Porphyromonas Metronidazole, clindamycin, penicillin
As for B fragilis
Mycobacteria
Mycobacterium tuberculosis Isoniazid + rifampin + ethambutol + pyrazinamide
Streptomycin, moxifloxacin, amikacin, ethionamide, cycloserine, PAS, linezolid
Mycobacterium leprae
Multibacillary Dapsone + rifampin + clofazimine
Paucibacillary Dapsone + rifampin
Mycoplasma pneumoniae Tetracycline, erythromycin Azithromycin, clarithromycin, quinolone3
Chlamydia
C trachomatis Tetracycline, azithromycin Clindamycin, ofloxacin
C pneumoniae Tetracycline, erythromycin Clarithromycin, azithromycin
C psittaci Tetracycline Chloramphenicol
Spirochetes
Borrelia recurrentis Doxycycline Erythromycin, chloramphenicol, penicillin
Borrelia burgdorferi
Early Doxycycline, amoxicillin Cefuroxime axetil, penicillin
Late Ceftriaxone
Leptospira species Penicillin Tetracycline
Treponema species Penicillin Tetracycline, azithromycin, ceftriaxone
Fungi
Aspergillus species Voriconazole Amphotericin B, itraconazole, caspofungin
Blastomyces species Amphotericin B Itraconazole, fluconazole
Candida species Amphotericin B, echinocandin11 Fluconazole, itraconazole, voriconazole
Cryptococcus Amphotericin B ± flucytosine (5-FC)
Fluconazole, voriconazole
Coccidioides immitis Amphotericin B Fluconazole, itraconazole, voriconazole, posaconazole
Histoplasma capsulatum Amphotericin B Itraconazole
Mucoraceae (Rhizopus, Absidia) Amphotericin B Posaconazole
Sporothrix schenckii Amphotericin B Itraconazole
1Trimethoprim-sulfamethoxazole (TMP-SMZ) is a mixture of one part trimethoprim plus five parts sulfamethoxazole.2First-generation cephalosporins: cefazolin for parenteral administration; cefadroxil or cephalexin for oral administration. Second-generation cephalosporins: cefuroxime for parenteral administration; cefaclor, cefuroxime axetil, cefprozil for oral administration. Third-generation cephalosporins: ceftazidime, cefotaxime, ceftriaxone for parenteral administration; cefixime, cefpodoxime, ceftibuten, cefdinir, cefditoren for oral administration. Fourth-generation cephalosporin: cefepime for parenteral administration. Cephamycins: cefoxitin and cefotetan for parenteral administration.3Quinolones: ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, norfloxacin, ofloxacin. Norfloxacin is not effective for the treatment of systemic infections. Gemifloxacin, levofloxacin, and moxifloxacin have excellent activity against pneumococci. Ciprofloxacin and levofloxacin have good activity against Pseudomonas aeruginosa.4Macrolides: azithromycin, clarithromycin, dirithromycin, erythromycin.5Generally, streptomycin and gentamicin are used to treat infections with gram-positive organisms, whereas gentamicin, tobramycin, and amikacin are used to treat infections with gram-negatives.6Carbapenems: doripenem, ertapenem, imipenem, meropenem. Ertapenem lacks activity against enterococci, Acinetobacter, and P aeruginosa.7Antipseudomonal penicillin: piperacillin, piperacillin/tazobactam, ticarcillin/clavulanic acid.8See footnote 3 in Table 51–2 for guidelines on the treatment of penicillin-resistant pneumococcal meningitis.9Parenteral nafcillin or oxacillin; oral dicloxacillin.10There is no regimen that is reliably bactericidal for vancomycin-resistant enterococcus for which there is extensive clinical experience; daptomycin has bactericidal activity in vitro. Regimens that have been reported to be efficacious include nitrofurantoin (for urinary tract infection); potential regimens for bacteremia include daptomycin, linezolid, and dalfopristin/quinupristin.11Echinocandins: anidulafungin, caspofungin, micafungin.
(Continued)
051-Katzung_Ch051_p901-914.indd 905 9/21/11 12:10:51 PM

906 SECTION VIII Chemotherapeutic Drugs
TABLE 51–2 Empiric antimicrobial therapy based on site of infection.
Presumed Site of Infection Common Pathogens Drugs of First Choice Alternative Drugs
Bacterial endocarditis
Acute Staphylococcus aureus Vancomycin + gentamicin Penicillinase-resistant penicillin1 + gentamicin
Subacute Viridans streptococci, enterococci Penicillin + gentamicin Vancomycin + gentamicin
Septic arthritis
Child H influenzae, S aureus, β-hemolytic streptococci
Ceftriaxone Ampicillin-sulbactam
Adult S aureus, Enterobacteriaceae Cefazolin Vancomycin, quinolone
Acute otitis media, sinusitis
H influenzae, S pneumoniae, M catarrhalis
Amoxicillin Amoxicillin-clavulanate, cefuroxime axetil, TMP-SMZ
Cellulitis S aureus, group A streptococcus Penicillinase-resistant penicillin, cephalosporin (first-generation)2
Vancomycin, clindamycin, linezolid, daptomycin
Meningitis
Neonate Group B streptococcus, E coli, Listeria Ampicillin + cephalosporin (third-generation)
Ampicillin + aminoglycoside, chloramphenicol, meropenem
Child H influenzae, pneumococcus, meningococcus
Ceftriaxone or cefotaxime ± vancomycin3
Chloramphenicol, meropenem
Adult Pneumococcus, meningococcus Ceftriaxone, cefotaxime Vancomycin + ceftriaxone or cefotaxime3
Peritonitis due to ruptured viscus
Coliforms, B fragilis Metronidazole + cephalosporin (third-generation), piperacillin/tazobactam
Carbapenem, tigecycline
Pneumonia
Neonate As in neonatal meningitis
Child Pneumococcus, S aureus, H influenzae Ceftriaxone, cefuroxime, cefotaxime Ampicillin-sulbactam
Adult (community- acquired)
Pneumococcus, Mycoplasma, Legionella, H influenzae, S aureus, C pneumonia, coliforms
Outpatient: Macrolide,4 amoxicillin, tetracycline
Outpatient: Quinolone
Inpatient: Macrolide4 + cefotaxime, ceftriaxone, ertapenem, or ampicillin
Inpatient: Doxycycline + cefotaxime, ceftriaxone, ertapenem, or ampicillin; respiratory quinolone5
Septicemia6 Any Vancomycin + cephalosporin (third-generation) or piperacillin/tazobactam or imipenem or meropenem
Septicemia with granulocytopenia
Any Antipseudomonal penicillin + aminoglycoside; ceftazidime; cefepime; imipenem or meropenem; consider addition of systemic antifungal therapy if fever persists beyond 5 days of empiric therapy
1See footnote 9, Table 51–1.2See footnote 2, Table 51–1.3When meningitis with penicillin-resistant pneumococcus is suspected, empiric therapy with this regimen is recommended.4Erythromycin, clarithromycin, or azithromycin (an azalide) may be used.5Quinolones used to treat pneumonococcal infections include levofloxacin, moxifloxacin, and gemifloxacin.6Adjunctive immunomodulatory drugs such as drotrecogin-alfa can also be considered for patients with severe sepsis.
done to maximize it. For example, are adequate numbers of granulocytes present and are HIV infection, malnutrition, or underlying malignancy present? The presence of abscesses or for-eign bodies should also be considered. Finally, culture and suscep-tibility testing should be repeated to determine whether superinfection has occurred with another organism or whether the original pathogen has developed drug resistance.
ANTIMICROBIAL PHARMACODYNAMICS
The time course of drug concentration is closely related to the anti-microbial effect at the site of infection and to any toxic effects. Pharmacodynamic factors include pathogen susceptibility testing, drug bactericidal versus bacteriostatic activity, drug synergism, antag-onism, and postantibiotic effects. Together with pharmacokinetics,
051-Katzung_Ch051_p901-914.indd 906 9/21/11 12:10:51 PM

CHAPTER 51 Clinical Use of Antimicrobial Agents 907
pharmacodynamic information permits the selection of optimal anti-microbial dosage regimens.
Bacteriostatic versus Bactericidal Activity Antibacterial agents may be classified as bacteriostatic or bacteri-cidal ( Table 51–3 ). For agents that are primarily bacteriostatic, inhibitory drug concentrations are much lower than bactericidal drug concentrations. In general, cell wall-active agents are bacteri-cidal, and drugs that inhibit protein synthesis are bacteriostatic.
The classification of antibacterial agents as bactericidal or bac-teriostatic has limitations. Some agents that are considered to be bacteriostatic may be bactericidal against selected organisms. On the other hand, enterococci are inhibited but not killed by vanco-mycin, penicillin, or ampicillin used as single agents.
Bacteriostatic and bactericidal agents are equivalent for the treatment of most infectious diseases in immunocompetent hosts. Bactericidal agents should be selected over bacteriostatic ones in circumstances in which local or systemic host defenses are impaired. Bactericidal agents are required for treatment of endo-carditis and other endovascular infections, meningitis, and infec-tions in neutropenic cancer patients.
Bactericidal agents can be divided into two groups: agents that exhibit concentration-dependent killing (eg, aminoglycosides and quinolones) and agents that exhibit time-dependent killing (eg, β lactams and vancomycin). For drugs whose killing action is concentration-dependent, the rate and extent of killing increase with increasing drug concentrations. Concentration-dependent killing is one of the pharmacodynamic factors responsible for the efficacy of once-daily dosing of aminoglycosides.
For drugs whose killing action is time-dependent, bactericidal activity continues as long as serum concentrations are greater than the MBC. Drug concentrations of time-dependent killing agents
that lack a postantibiotic effect should be maintained above the MIC for the entire interval between doses.
Postantibiotic Effect Persistent suppression of bacterial growth after limited exposure to an antimicrobial agent is known as the postantibiotic effect (PAE). The PAE can be expressed mathematically as follows:
PAE = T – C
where T is the time required for the viable count in the test (in vitro) culture to increase tenfold above the count observed imme-diately before drug removal and C is the time required for the count in an untreated culture to increase tenfold above the count observed immediately after completion of the same procedure used on the test culture. The PAE reflects the time required for bacteria to return to logarithmic growth.
Proposed mechanisms include (1) slow recovery after reversible nonlethal damage to cell structures; (2) persistence of the drug at a binding site or within the periplasmic space; and (3) the need to synthesize new enzymes before growth can resume. Most antimi-crobials possess significant in vitro PAEs (≥ 1.5 hours) against susceptible gram-positive cocci ( Table 51–4 ). Antimicrobials with significant PAEs against susceptible gram-negative bacilli are lim-ited to carbapenems and agents that inhibit protein or DNA synthesis.
In vivo PAEs are usually much longer than in vitro PAEs. This is thought to be due to postantibiotic leukocyte enhancement (PALE) and exposure of bacteria to subinhibitory antibiotic con-centrations. The efficacy of once-daily dosing regimens is in part due to the PAE. Aminoglycosides and quinolones possess concen-tration-dependent PAEs; thus, high doses of aminoglycosides given once daily result in enhanced bactericidal activity and extended PAEs. This combination of pharmacodynamic effects allows aminoglycoside serum concentrations that are below the MICs of target organisms to remain effective for extended periods of time.
PHARMACOKINETIC CONSIDERATIONS
Route of Administration Many antimicrobial agents have similar pharmacokinetic proper-ties when given orally or parenterally (ie, tetracyclines, trimetho-prim-sulfamethoxazole, quinolones, chloramphenicol, metronid-azole, clindamycin, rifampin, linezolid, and fluconazole). In most cases, oral therapy with these drugs is equally effective, is less costly, and results in fewer complications than parenteral therapy.
The intravenous route is preferred in the following situations: (1) for critically ill patients; (2) for patients with bacterial menin-gitis or endocarditis; (3) for patients with nausea, vomiting, gast-rectomy, or diseases that may impair oral absorption; and (4) when giving antimicrobials that are poorly absorbed following oral administration.
TABLE 51–3 Bactericidal and bacteriostatic antibacterial agents.
Bactericidal Agents Bacteriostatic Agents
Aminoglycosides Chloramphenicol
Bacitracin Clindamycin
β-Lactam antibiotics Ethambutol
Daptomycin Macrolides
Glycopeptide antibiotics Nitrofurantoin
Isoniazid Novobiocin
Ketolides Oxazolidinones
Metronidazole Sulfonamides
Polymyxins Tetracyclines
Pyrazinamide Tigecycline
Quinolones Trimethoprim
Rifampin
Streptogramins
051-Katzung_Ch051_p901-914.indd 907 9/21/11 12:10:51 PM

908 SECTION VIII Chemotherapeutic Drugs
Drug Concentrations in Body Fluids Most antimicrobial agents are well distributed to most body tissues and fluids. Penetration into the cerebrospinal fluid is an exception. Most do not penetrate uninflamed meninges to an appreciable extent. In the presence of meningitis, however, the cerebrospinal fluid concentrations of many antimicrobials increase ( Table 51–6 ).
Monitoring Serum Concentrations of Antimicrobial Agents For most antimicrobial agents, the relation between dose and therapeutic outcome is well established, and serum concentration monitoring is unnecessary for these drugs. To justify routine serum concentration monitoring, it should be established (1) that a direct relationship exists between drug concentrations and effi-cacy or toxicity; (2) that substantial interpatient variability exists in serum concentrations on standard doses; (3) that a small differ-ence exists between therapeutic and toxic serum concentrations; (4) that the clinical efficacy or toxicity of the drug is delayed or difficult to measure; and (5) that an accurate assay is available.
In clinical practice, serum concentration monitoring is rou-tinely performed on patients receiving aminoglycosides. Despite the lack of supporting evidence for its usefulness or need, serum vancomycin concentration monitoring is also widespread. Flucytosine serum concentration monitoring has been shown to reduce toxicity when doses are adjusted to maintain peak concen-trations below 100 mcg/mL.
■ MANAGEMENT OF ANTIMICROBIAL DRUG TOXICITY
Owing to the large number of antimicrobials available, it is usually possible to select an effective alternative in patients who develop serious drug toxicity ( Table 51–1 ). However, for some infections there are no effective alternatives to the drug of choice. For example, in patients with neurosyphilis who have a history of
TABLE 51–4 Antibacterial agents with in vitro postantibiotic effects ≥ 1.5 hours.
Against Gram-Positive Cocci Against Gram-Negative Bacilli
Aminoglycosides Aminoglycosides
Carbapenems Carbapenems
Cephalosporins Chloramphenicol
Chloramphenicol Quinolones
Clindamycin Rifampin
Daptomycin Tetracyclines
Glycopeptide antibiotics Tigecycline
Ketolides
Macrolides
Oxazolidinones
Penicillins
Quinolones
Rifampin
Streptogramins
Sulfonamides
Tetracyclines
Tigecycline
Trimethoprim
TABLE 51–5 Antimicrobial agents that require dosage adjustment or are contraindicated in patients with renal or hepatic impairment.
Dosage Adjustment Needed in Renal ImpairmentContraindicated in Renal Impairment
Dosage Adjustment Needed in Hepatic Impairment
Acyclovir, amantadine, aminoglycosides, aztreonam, carbapenems, cephalosporins,1 clarithromycin, colistin, cycloserine, daptomycin, didanosine, emtricitabine, ethambutol, ethionamide, famciclovir, fluconazole, flucytosine, foscarnet, ganciclovir, lamivudine, penicillins,3 pyrazinamide, quinolones, 4 rimantadine, stavudine, telavancin, telbivudine, telithromycin, tenofovir, terbinafine, trimethoprim-sulfamethoxazole, valacyclovir, vancomycin, zidovudine
Cidofovir, methenamine, nalidixic acid, nitrofurantoin, sulfonamides (long-acting), tetracyclines2
Amprenavir, atazanavir, chloram-phenicol, clindamycin, erythromycin, fosamprenavir, indinavir, metronida-zole, rimantadine, tigecycline
1Except ceftriaxone.2Except doxycycline and possibly minocycline.3Except antistaphylococcal penicillins (eg, nafcillin and dicloxacillin).4Except moxifloxacin.
Conditions That Alter Antimicrobial Pharmacokinetics Various diseases and physiologic states alter the pharmacokinetics of antimicrobial agents. Impairment of renal or hepatic function may result in decreased elimination. Table 51–5 lists drugs that require dosage reduction in patients with renal or hepatic insuffi-ciency. Failure to reduce antimicrobial agent dosage in such patients may cause toxic effects. Conversely, patients with burns, cystic fibrosis, or trauma may have increased dosage requirements for selected agents. The pharmacokinetics of antimicrobials is also altered in the elderly, in neonates, and in pregnancy.
051-Katzung_Ch051_p901-914.indd 908 9/21/11 12:10:51 PM

CHAPTER 51 Clinical Use of Antimicrobial Agents 909
anaphylaxis to penicillin, it is necessary to perform skin testing and desensitization to penicillin. It is important to obtain a clear history of drug allergy and other adverse drug reactions. A patient with a documented antimicrobial allergy should carry a card with the name of the drug and a description of the reaction. Cross-reactivity between penicillins and cephalosporins is less than 10%. Cephalosporins may be administered to patients with penicillin-induced maculopapular rashes but should be avoided in patients with a history of penicillin-induced immediate hypersensitivity reactions. On the other hand, aztreonam does not cross-react with penicillins and can be safely administered to patients with a his-tory of penicillin-induced anaphylaxis. For mild reactions, it may be possible to continue therapy with use of adjunctive agents or dosage reduction.
Adverse reactions to antimicrobials occur with increased frequency in several groups, including neonates, geriatric patients, renal failure patients, and AIDS patients. Dosage adjustment of the drugs listed in Table 51–5 is essential for the prevention of adverse effects in patients with renal failure. In addition, several agents are contraindicated in patients with renal impairment because of increased rates of serious toxicity ( Table 51–5 ). See the preceding chapters for discussions of specific drugs.
Polypharmacy also predisposes to drug interactions. Although the mechanism is not known, AIDS patients have an unusually high incidence of toxicity to a number of drugs, including clin-damycin, aminopenicillins, and sulfonamides. Many of these reac-tions, including rash and fever, may respond to dosage reduction or
TABLE 51–6 Cerebrospinal fluid (CSF) penetration of selected antimicrobials.
Antimicrobial Agent
CSF Concentration (Uninflamed Meninges) as % of Serum Concentration
CSF Concentration (Inflamed Meninges) as % of Serum Concentration
Ampicillin 2–3 2–100
Aztreonam 2 5
Cefepime 0–2 4–12
Cefotaxime 22.5 27–36
Ceftazidime 0.7 20–40
Ceftriaxone 0.8–1.6 16
Cefuroxime 20 17–88
Ciprofloxacin 6–27 26–37
Imipenem 3.1 11–41
Meropenem 0–7 1–52
Nafcillin 2–15 5–27
Penicillin G 1–2 8–18
Sulfamethoxazole 40 12–47
Trimethoprim < 41 12–69
Vancomycin 0 1–53
treatment with corticosteroids and antihistamines. Other examples are discussed in the preceding chapters and in Chapter 66 .
■ ANTIMICROBIAL DRUG COMBINATIONS
RATIONALE FOR COMBINATION ANTIMICROBIAL THERAPY
Most infections should be treated with a single antimicrobial agent. Although indications for combination therapy exist, anti-microbial combinations are often overused in clinical practice. The unnecessary use of antimicrobial combinations increases tox-icity and costs and may occasionally result in reduced efficacy due to antagonism of one drug by another. Antimicrobial combina-tions should be selected for one or more of the following reasons:
1. To provide broad-spectrum empiric therapy in seriously ill patients.
2. To treat polymicrobial infections (such as intra-abdominal abscesses, which typically are due to a combination of anaero-bic and aerobic gram-negative organisms, and enterococci). The antimicrobial combination chosen should cover the most common known or suspected pathogens but need not cover all possible pathogens. The availability of antimicrobials with excellent polymicrobial coverage (eg, β-lactamase inhibitor combinations or carbapenems) may reduce the need for com-bination therapy in the setting of polymicrobial infections.
3. To decrease the emergence of resistant strains. The value of combination therapy in this setting has been clearly demon-strated for tuberculosis.
4. To decrease dose-related toxicity by using reduced doses of one or more components of the drug regimen. The use of flucyto-sine in combination with amphotericin B for the treatment of cryptococcal meningitis in non–HIV-infected patients allows for a reduction in amphotericin B dosage with decreased amphotericin B-induced nephrotoxicity.
5. To obtain enhanced inhibition or killing. This use of antimicro-bial combinations is discussed in the paragraphs that follow.
SYNERGISM & ANTAGONISM
When the inhibitory or killing effects of two or more antimicrobi-als used together are significantly greater than expected from their effects when used individually, synergism is said to result. Synergism is marked by a fourfold or greater reduction in the MIC or MBC of each drug when used in combination versus when used alone. Antagonism occurs when the combined inhibitory or killing effects of two or more antimicrobial drugs are significantly less than observed when the drugs are used individually.
Mechanisms of Synergistic Action The need for synergistic combinations of antimicrobials has been clearly established for the treatment of enterococcal endocarditis. Bactericidal activity is essential for the optimal management of
051-Katzung_Ch051_p901-914.indd 909 9/21/11 12:10:51 PM

910 SECTION VIII Chemotherapeutic Drugs
bacterial endocarditis. Penicillin or ampicillin in combination with gentamicin or streptomycin is superior to monotherapy with a penicillin or vancomycin. When tested alone, penicillins and van-comycin are only bacteriostatic against susceptible enterococcal isolates. When these agents are combined with an aminoglycoside, however, bactericidal activity results. The addition of gentamicin or streptomycin to penicillin allows for a reduction in the duration of therapy for selected patients with viridans streptococcal endocardi-tis. Some evidence exists that synergistic combinations of antimi-crobials may be of benefit in the treatment of gram-negative bacillary infections in febrile neutropenic cancer patients and in systemic infections caused by Pseudomonas aeruginosa .
Other synergistic antimicrobial combinations have been shown to be more effective than monotherapy with individual components. Trimethoprim-sulfamethoxazole has been success-fully used for the treatment of bacterial infections and P jiroveci ( carinii ) pneumonia. ∗ β-Lactamase inhibitors restore the activ-ity of intrinsically active but hydrolyzable β lactams against organisms such as Staphylococcus aureus and Bacteroides fragilis . Three major mechanisms of antimicrobial synergism have been established: 1. Blockade of sequential steps in a metabolic sequence:
Trimethoprim-sulfamethoxazole is the best-known example of this mechanism of synergy (see Chapter 46 ). Blockade of the two sequential steps in the folic acid pathway by trimethoprim-sulfamethoxazole results in a much more complete inhibition of growth than achieved by either component alone.
2. Inhibition of enzymatic inactivation: Enzymatic inactivation of β-lactam antibiotics is a major mechanism of antibiotic resistance. Inhibition of β lactamase by β-lactamase inhibitor drugs (eg, sulbactam) results in synergism.
3. Enhancement of antimicrobial agent uptake: Penicillins and other cell wall-active agents can increase the uptake of amino-glycosides by a number of bacteria, including staphylococci, enterococci, streptococci, and P aeruginosa . Enterococci are thought to be intrinsically resistant to aminoglycosides because of permeability barriers. Similarly, amphotericin B is thought to enhance the uptake of flucytosine by fungi.
Mechanisms of Antagonistic Action There are few clinically relevant examples of antimicrobial antago-nism. The most striking example was reported in a study of patients with pneumococcal meningitis. Patients who were treated with the combination of penicillin and chlortetracycline had a mortality rate of 79% compared with a mortality rate of 21% in patients who received penicillin monotherapy (illustrating the first mechanism set forth below).
The use of an antagonistic antimicrobial combination does not preclude other potential beneficial interactions. For example, rifampin may antagonize the action of anti-staphylococcal penicil-lins or vancomycin against staphylococci. However, the aforemen-tioned antimicrobials may prevent the emergence of resistance to rifampin.
Two major mechanisms of antimicrobial antagonism have been established:
1. Inhibition of cidal activity by static agents: Bacteriostatic agents such as tetracyclines and chloramphenicol can antago-nize the action of bactericidal cell wall-active agents because cell wall-active agents require that the bacteria be actively grow-ing and dividing.
2. Induction of enzymatic inactivation: Some gram-negative bacilli, including enterobacter species, P aeruginosa, Serratia marcescens, and Citrobacter freundii, possess inducible β lacta-mases. β-Lactam antibiotics such as imipenem, cefoxitin, and ampicillin are potent inducers of β-lactamase production. If an inducing agent is combined with an intrinsically active but hydrolyzable β lactam such as piperacillin, antagonism may result.
■ ANTIMICROBIAL PROPHYLAXIS
Antimicrobial agents are effective in preventing infections in many settings. Antimicrobial prophylaxis should be used in circum-stances in which efficacy has been demonstrated and benefits outweigh the risks of prophylaxis. Antimicrobial prophylaxis may be divided into surgical prophylaxis and nonsurgical prophylaxis.
Surgical Prophylaxis Surgical wound infections are a major category of nosocomial infections. The estimated annual cost of surgical wound infections in the United States is $1.5 billion.
The National Research Council (NRC) Wound Classification Criteria have served as the basis for recommending antimicrobial prophylaxis. NRC criteria consist of four classes [see Box: National Research Council (NRC) Wound Classification Criteria].
The Study of the Efficacy of Nosocomial Infection Control (SENIC) identified four independent risk factors for postopera-tive wound infections: operations on the abdomen, operations lasting more than 2 hours, contaminated or dirty wound classifi-cation, and at least three medical diagnoses. Patients with at least two SENIC risk factors who undergo clean surgical procedures have an increased risk of developing surgical wound infections and should receive antimicrobial prophylaxis.
Surgical procedures that necessitate the use of antimicrobial prophylaxis include contaminated and clean-contaminated opera-tions, selected operations in which postoperative infection may be catastrophic such as open heart surgery, clean procedures that involve placement of prosthetic materials, and any procedure in an immunocompromised host. The operation should carry a signifi-cant risk of postoperative site infection or cause significant bacte-rial contamination.
General principles of antimicrobial surgical prophylaxis include the following:
1. The antibiotic should be active against common surgical wound pathogens; unnecessarily broad coverage should be avoided.
2. The antibiotic should have proved efficacy in clinical trials. * Pneumocystis jiroveci is a fungal organism found in humans ( P carinii infects animals) that responds to antiprotozoal drugs. See Chapter 52 .
051-Katzung_Ch051_p901-914.indd 910 9/21/11 12:10:52 PM

CHAPTER 51 Clinical Use of Antimicrobial Agents 911
TABLE 51–7 Recommendations for surgical antimicrobial prophylaxis.
Type of Operation Common Pathogens Drug of Choice
Cardiac (with median sternotomy) Staphylococci, enteric gram-negative rods Cefazolin
Noncardiac, thoracic Staphylococci, streptococci, enteric gram-negative rods
Cefazolin
Vascular (abdominal and lower extremity) Staphylococci, enteric gram-negative rods Cefazolin
Neurosurgical (craniotomy) Staphylococci Cefazolin
Orthopedic (with hardware insertion) Staphylococci Cefazolin
Head and neck (with entry into the oropharynx) S aureus, oral flora Cefazolin + metronidazole
Gastroduodenal (high-risk patients1) S aureus, oral flora, enteric gram-negative rods Cefazolin
Biliary tract (high-risk patients2) S aureus, enterococci, enteric gram-negative rods Cefazolin
Colorectal (elective surgery) Enteric gram-negative rods, anaerobes Oral erythromycin plus neomycin3
Colorectal (emergency surgery or obstruction) Enteric gram-negative rods, anaerobes Cefoxitin, cefotetan, or cefazolin + metronidazole
Appendectomy, nonperforated Enteric gram-negative rods, anaerobes Cefoxitin or cefazolin + metronidazole
Hysterectomy Enteric gram-negative rods, anaerobes, enterococci, group B streptococci
Cefazolin or cefoxitin
Cesarean section Enteric gram-negative rods, anaerobes, enterococci, group B streptococci
Cefazolin4
1Gastric procedures for cancer, ulcer, bleeding, or obstruction; morbid obesity; suppression of gastric acid secretion.2Age > 60, acute cholecystitis, prior biliary tract surgery, common duct stones, jaundice, or diabetes mellitus.3In conjunction with mechanical bowel preparation.4Administer immediately following cord clamping.
National Research Council (NRC) Wound Classification Criteria
Clean: Elective, primarily closed procedure; respiratory, gas-trointestinal, biliary, genitourinary, or oropharyngeal tract not entered; no acute inflammation and no break in technique; expected infection rate ≤ 2%. Clean contaminated: Urgent or emergency case that is oth-erwise clean; elective, controlled opening of respiratory, gas-trointestinal, biliary, or oropharyngeal tract; minimal spillage or minor break in technique; expected infection rate ≤ 10%. Contaminated: Acute nonpurulent inflammation; major technique break or major spill from hollow organ; penetrat-ing trauma less than 4 hours old; chronic open wounds to be grafted or covered; expected infection rate about 20%. Dirty: Purulence or abscess; preoperative perforation of respiratory, gastrointestinal, biliary, or oropharyngeal tract; penetrating trauma more than 4 hours old; expected infec-tion rate about 40%.
3. The antibiotic must achieve concentrations greater than the MIC of suspected pathogens, and these concentrations must be present at the time of incision.
4. The shortest possible course—ideally a single dose—of the most effective and least toxic antibiotic should be used.
5. The newer broad-spectrum antibiotics should be reserved for therapy of resistant infections.
6. If all other factors are equal, the least expensive agent should be used.
The proper selection and administration of antimicrobial pro-phylaxis are of utmost importance. Common indications for sur-gical prophylaxis are shown in Table 51–7 . Cefazolin is the prophylactic agent of choice for head and neck, gastroduodenal, biliary tract, gynecologic, and clean procedures. Local wound infection patterns should be considered when selecting antimicro-bial prophylaxis. The selection of vancomycin over cefazolin may be necessary in hospitals with high rates of methicillin-resistant S aureus or S epidermidis infections. The antibiotic should be present in adequate concentrations at the operative site before incision and throughout the procedure; initial dosing is dependent on the volume of distribution, peak levels, clearance, protein binding, and bioavailability. Parenteral agents should be administered dur-ing the interval beginning 60 minutes before incision; administra-tion up to the time of incision is preferred. In cesarean section, the antibiotic is administered after umbilical cord clamping. If short-acting agents such as cefoxitin are used, doses should be repeated if the procedure exceeds 3–4 hours in duration. Single-dose pro-phylaxis is effective for most procedures and results in decreased toxicity and antimicrobial resistance.
Improper administration of antimicrobial prophylaxis leads to excessive surgical wound infection rates. Common errors in anti-biotic prophylaxis include selection of the wrong antibiotic,
051-Katzung_Ch051_p901-914.indd 911 9/21/11 12:10:52 PM

912 SECTION VIII Chemotherapeutic Drugs
TABLE 51–8 Recommendations for nonsurgical antimicrobial prophylaxis.
Infection to Be Prevented Indication(s) Drug of Choice Efficacy
Anthrax Suspected exposure Ciprofloxacin or doxycycline Proposed effective
Cholera Close contacts of a case Tetracycline Proposed effective
Diphtheria Unimmunized contacts Penicillin or erythromycin Proposed effective
Endocarditis Dental, oral, or upper respiratory tract procedures1 in at-risk patients2
Amoxicillin or clindamycin Proposed effective
Genital herpes simplex Recurrent infection (≥ 4 episodes per year) Acyclovir Excellent
Perinatal herpes simplex type 2 infection
Mothers with primary HSV or frequent recurrent genital HSV Acyclovir Proposed effective
Group B streptococcal (GBS) infection
Mothers with cervical or vaginal GBS colonization and their newborns with one or more of the following: (a) onset of labor or membrane rupture before 37 weeks’ gestation, (b) prolonged rupture of membranes (> 12 hours), (c) mater-nal intrapartum fever, (d) history of GBS bacteriuria during pregnancy, (e) mothers who have given birth to infants who had early GBS disease or with a history of streptococcal bac-teriuria during pregnancy
Ampicillin or penicillin Excellent
Haemophilus influenzae type B infection
Close contacts of a case in incompletely immunized children (> 48 months old)
Rifampin Excellent
HIV infection Health care workers exposed to blood after needle-stick injury
Tenofovir/emtricitabine ± lopinavir/ritonavir
Good
Pregnant HIV-infected women who are at ≥ 14 weeks of ges-tation; newborns of HIV-infected women for the first 6 weeks of life, beginning 8–12 hours after birth
HAART3 Excellent
Influenza A and B Unvaccinated geriatric patients, immunocompromised hosts, and health care workers during outbreaks
Oseltamivir Good
Malaria Travelers to areas endemic for chloroquine-susceptible disease Chloroquine Excellent
Travelers to areas endemic for chloroquine-resistant disease Mefloquine, doxycycline, or atovaquone/proguanil
Excellent
Meningococcal infection Close contacts of a case Rifampin, ciprofloxacin, or ceftriaxone
Excellent
Mycobacterium avium complex HIV-infected patients with CD4 count < 75/μL Azithromycin, clarithromy-cin, or rifabutin
Excellent
Otitis media Recurrent infection Amoxicillin Good
Pertussis Close contacts of a case Azithromycin Excellent
Plague Close contacts of a case Tetracycline Proposed effective
Pneumococcemia Children with sickle cell disease or asplenia Penicillin Excellent
Pneumocystis jiroveci pneumo-nia (PCP)
High-risk patients (eg, AIDS, leukemia, transplant) Trimethoprim-sulfamethoxazole, dapsone, or atovaquone
Excellent
Rheumatic fever History of rheumatic fever or known rheumatic heart disease Benzathine penicillin Excellent
Toxoplasmosis HIV-infected patients with IgG antibody to Toxoplasma and CD4 count < 100/μL
Trimethoprim-sulfamethoxazole
Good
Tuberculosis Persons with positive tuberculin skin tests and one or more of the following: (a) HIV infection, (b) close contacts with newly diagnosed disease, (c) recent skin test conversion, (d) medical conditions that increase the risk of developing tuberculosis, (e) age < 35
Isoniazid, rifampin, or pyrazinamide
Excellent
Urinary tract infections (UTI) Recurrent infection Trimethoprim-sulfamethoxazole
Excellent
1Prophylaxis is recommended for the following: dental procedures that involve manipulation of gingival tissue or the periapical region of teeth or perforation of the oral mucosa, and invasive procedure of the respiratory tract that involves incision or biopsy of the respiratory mucosa, such as tonsillectomy and adenoidectomy.2Prophylaxis should be targeted to those with the following risk factors: prosthetic heart valves, previous bacterial endocarditis, congenital cardiac malformations, cardiac trans-plantation patients who develop cardiac valvulopathy.3Highly active antiretroviral therapy. See http://www.hivatis.org/ for updated guidelines.
051-Katzung_Ch051_p901-914.indd 912 9/21/11 12:10:52 PM

CHAPTER 51 Clinical Use of Antimicrobial Agents 913
administering the first dose too early or too late, failure to repeat doses during prolonged procedures, excessive duration of prophy-laxis, and inappropriate use of broad-spectrum antibiotics.
Nonsurgical Prophylaxis Nonsurgical prophylaxis includes the administration of antimicro-bials to prevent colonization or asymptomatic infection as well as the administration of drugs following colonization by or inocula-tion of pathogens but before the development of disease. Nonsurgical prophylaxis is indicated in individuals who are at high risk for temporary exposure to selected virulent pathogens and in patients who are at increased risk for developing infection because of under-lying disease (eg, immunocompromised hosts). Prophylaxis is most effective when directed against organisms that are predictably sus-ceptible to antimicrobial agents. Common indications and drugs for nonsurgical prophylaxis are listed in Table 51–8 .
REFERENCES American Thoracic Society: Guidelines for the management of adults with hospital-
acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005;171:388.
Antimicrobial prophylaxis for surgery. Treat Guidel Med Lett 2009;7:47. Baddour LM et al: Infective endocarditis: Diagnosis, antimicrobial therapy, and
management of complications. Circulation 2005;111:3167. Blumberg HM et al: American Thoracic Society/Centers for Disease Control and
Prevention/Infectious Diseases Society of America: Treatment of tuberculo-sis. Am J Respir Crit Care Med 2003;167:603.
Bochner BS et al: Anaphylaxis. N Engl J Med 1991;324:1785. Bratzler DW et al: Antimicrobial prophylaxis for surgery: An advisory statement
from the National Surgical Infection Prevention Project. Clin Infect Dis 2004;38:1706.
Gonzales R et al: Principles of appropriate antibiotic use for treatment of acute respiratory tract infections in adults: Background, specific aims, and meth-ods. Ann Intern Med 2001;134:479.
Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents. Recommendations of the National Institutes of Health (NIH), the Centers for Disease Control and Prevention (CDC), and the HIV Medicine Association of the Infectious Diseases Society of America (HIVMA/IDSA). AIDSinfo April 10, 2009. http://AIDSinfo.nih.gov
Jones RN, Pfaller MA: Bacterial resistance: A worldwide problem. Diagn Microbiol Infect Dis 1998;31:379.
Kaye KS, Kaye D: Antibacterial therapy and newer agents. Infect Dis Clin North Am 2009;23:757.
Mandell LA et al: Infectious Diseases Society of America/American Thoracic Society Consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007;44:S27.
Mazuski JE: Surgical infections. Surg Clin North Am 2009;89:295. National Nosocomial Infections Surveillance (NNIS) System Report, Data
Summary from January 1992–June 2004, issued October 2004. Am J Infect Control 2004;32:470.
Sexually transmitted diseases treatment guidelines 2006. Centers for Disease Control and Prevention. MMWR Morb Mortal Wkly Rep 2006;54(RR-11):1.
Tunkel AR et al: Practice guidelines for the management of bacterial meningitis. Clin Infect Dis 2004;39:1267.
Wilson W et al: Prevention of infective endocarditis: Guidelines from the American Heart Association. Circulation 2007;116:1736.
C A S E S T U D Y A N S W E R
The most likely diagnosis for this patient is Streptococcus pneumoniae meningitis, the most common bacterial cause of meningitis in adults. Other possible microbiologic etiologies include Neisseria meningitidis, Listeria monocytogenes, and enteric gram-negative bacilli. Intravenous antimicrobials to
which local strains of these organisms are sensitive should be started while awaiting culture and sensitivity results. In addi-tion, the use of dexamethasone has also been demonstrated to reduce mortality in adults with pneumococcal meningitis in conjunction with appropriate antimicrobial therapy.
051-Katzung_Ch051_p901-914.indd 913 9/21/11 12:10:52 PM

This page intentionally left blank

915
C H A P T E R
■ MALARIA
Malaria is the most important parasitic disease of humans and causes hundreds of millions of illnesses per year. Four species of plasmo-dium typically cause human malaria: Plasmodium falciparum, P vivax, P malariae, and P ovale. A fifth species, P knowlesi, is pri-marily a pathogen of monkeys, but has recently been recognized to cause illness, including severe disease, in humans in Asia. Although all of the latter species may cause significant illness, P falciparum is responsible for the majority of serious complications and deaths. Drug resistance is an important therapeutic problem, most notably with P falciparum.
PARASITE LIFE CYCLE
An anopheline mosquito inoculates plasmodium sporozoites to initiate human infection ( Figure 52–1 ). Circulating sporozoites rapidly invade liver cells, and exoerythrocytic stage tissue schizonts mature in the liver. Merozoites are subsequently released from the liver and invade erythrocytes. Only erythrocytic parasites cause clinical illness. Repeated cycles of infection can lead to the infec-tion of many erythrocytes and serious disease. Sexual stage game-tocytes also develop in erythrocytes before being taken up by mosquitoes, where they develop into infective sporozoites.
In P falciparum and P malariae infection, only one cycle of liver cell invasion and multiplication occurs, and liver infection ceases spontaneously in less than 4 weeks. Thus, treatment that eliminates erythrocytic parasites will cure these infections. In P vivax and P ovale infections, a dormant hepatic stage, the hyp-nozoite, is not eradicated by most drugs, and subsequent relapses can therefore occur after therapy directed against erythrocytic parasites. Eradication of both erythrocytic and hepatic parasites is required to cure these infections and usually requires two or more drugs.
DRUG CLASSIFICATION
Several classes of antimalarial drugs are available ( Table 52–1 and Figure 52–2 ). Drugs that eliminate developing or dormant liver forms are called tissue schizonticides; those that act on erythro-cytic parasites are blood schizonticides; and those that kill sexual stages and prevent transmission to mosquitoes are gametocides. No single available agent can reliably effect a radical cure, ie, eliminate both hepatic and erythrocytic stages. Few available agents are causal prophylactic drugs, ie, capable of preventing erythro-cytic infection. However, all effective chemoprophylactic agents kill erythrocytic parasites before they increase sufficiently in num-ber to cause clinical disease.
52Antiprotozoal Drugs Philip J. Rosenthal, MD
C A S E S T U D Y
A 5-year-old American girl presents with a 1-week history of intermittent chills, fever, and sweats. She had returned home 2 weeks earlier after leaving the United States for the first time to spend 3 weeks with her grandparents in Nigeria. She received all standard childhood immunizations, but no addi-tional treatment before travel, since her parents have returned to their native Nigeria frequently without medical conse-quences. Three days ago, the child was seen in an outpatient
clinic and diagnosed with a viral syndrome. Examination reveals a lethargic child, with a temperature of 39.8°C (103.6°F) and splenomegaly. She has no skin rash or lymphadenopathy. Initial laboratory studies are remarkable for hematocrit 29.8%, platelets 45,000/mm 3 , creatinine 2.5 mg/dL (220 μmol/L), and mildly elevated bilirubin and transaminases. A blood smear shows ring forms of Plasmodium falciparum at 1.5% para-sitemia. What treatment should be started?
052-Katzung_Ch052_p915-936.indd 915 9/23/11 4:27:47 PM

916 SECTION VIII Chemotherapeutic Drugs
which are widely available internationally (and, in the case of Coartem ∗∗ , in the USA); Malarone; mefloquine; and quinine. Most authorities do not recommend routine terminal chemopro-phylaxis with primaquine to eradicate dormant hepatic stages of P vivax and P ovale after travel, but this may be appropriate in some circumstances, especially for travelers with major exposure to these parasites.
Multiple drugs are available for the treatment of malaria that presents in the USA ( Table 52–3 ). Most nonfalciparum infections and falciparum malaria from areas without known resistance should be treated with chloroquine. For vivax malaria from areas with suspected chloroquine resistance, including Indonesia and Papua New Guinea, other therapies effective against falciparum malaria may be used. Vivax and ovale malaria should subsequently be treated with primaquine to eradicate liver forms. Uncomplicated falciparum malaria from most areas is typically treated with Malarone or oral quinine, but new artemisinin-based combina-tions are increasingly the international standard of care, and one combination, Coartem, is now available in the USA. Other agents that are generally effective against resistant falciparum malaria include mefloquine and halofantrine, both of which have toxicity concerns at treatment dosages. Severe falciparum malaria is treated with intravenous artesunate, quinidine, or quinine (intravenous quinine is not available in the USA).
CHLOROQUINE
Chloroquine has been the drug of choice for both treatment and chemoprophylaxis of malaria since the 1940s, but its usefulness against P falciparum has been seriously compromised by drug resistance. It remains the drug of choice in the treatment of sensi-tive P falciparum and other species of human malaria parasites.
Chemistry & Pharmacokinetics Chloroquine is a synthetic 4-aminoquinoline ( Figure 52–2 ) for-mulated as the phosphate salt for oral use. It is rapidly and almost completely absorbed from the gastrointestinal tract, reaches maxi-mum plasma concentrations in about 3 hours, and is rapidly dis-tributed to the tissues. It has a very large apparent volume of distribution of 100–1000 L/kg and is slowly released from tissues and metabolized. Chloroquine is principally excreted in the urine with an initial half-life of 3–5 days but a much longer terminal elimination half-life of 1–2 months.
Antimalarial Action & Resistance When not limited by resistance, chloroquine is a highly effective blood schizonticide. It is also moderately effective against gameto-cytes of P vivax, P ovale, and P malariae but not against those of P falciparum. Chloroquine is not active against liver stage para-sites. Chloroquine probably acts by concentrating in parasite food
∗ Malarone is a proprietary formulation of atovaquone plus proguanil. ∗∗ Coartem is a proprietary formulation of artemether and lumefantrine.
Bloodschizonticide
Tissueschizonticide
Blood
Gametocide
Liver
Schizonts
Hypnozoites
FIGURE 52–1 Life cycle of malaria parasites. Only the asexual erythrocytic stage of infection causes clinical malaria. All effective antimalarial treatments are blood schizonticides that kill this stage. (Reproduced, with permission, from Baird JK: Effectiveness of antimalarial drugs.
N Engl J M 2005;352:1565.)
CHEMOPROPHYLAXIS & TREATMENT
When patients are counseled on the prevention of malaria, it is imperative to emphasize measures to prevent mosquito bites (eg, with insect repellents, insecticides, and bed nets), because parasites are increasingly resistant to multiple drugs and no chemoprophy-lactic regimen is fully protective. Current recommendations from the Centers for Disease Control and Prevention (CDC) include the use of chloroquine for chemoprophylaxis in the few areas infested by only chloroquine-sensitive malaria parasites (princi-pally the Caribbean and Central America west of the Panama Canal), mefloquine or Malarone ∗ for most other malarious areas, and doxycycline for areas with a very high prevalence of multidrug-resistant falciparum malaria (principally border areas of Thailand) ( Table 52–2 ). CDC recommendations should be checked regu-larly (Phone: 770-488-7788; Internet: http://www.cdc.gov/malaria ), because these may change in response to changing resis-tance patterns and increasing experience with new drugs. In some circumstances, it may be appropriate for travelers to carry supplies of drugs with them in case they develop a febrile illness when medical attention is unavailable. Regimens for self-treatment include new artemisinin-based combination therapies (see below),
052-Katzung_Ch052_p915-936.indd 916 9/23/11 4:27:48 PM

CHAPTER 52 Antiprotozoal Drugs 917
vacuoles, preventing the biocrystallization of the hemoglobin breakdown product, heme, into hemozoin, and thus eliciting parasite toxicity due to the buildup of free heme.
Resistance to chloroquine is now very common among strains of P falciparum and uncommon but increasing for P vivax. In P falciparum, mutations in a putative transporter, PfCRT, have been correlated with resistance. Chloroquine resistance can be reversed by certain agents, including verapamil, desipramine, and chlor-pheniramine, but the clinical value of resistance-reversing drugs is not established.
Clinical Uses A. Treatment Chloroquine is the drug of choice in the treatment of nonfalci-parum and sensitive falciparum malaria. It rapidly terminates fever (in 24–48 hours) and clears parasitemia (in 48–72 hours) caused by sensitive parasites. It is still used to treat falciparum malaria in some areas with widespread resistance, in particular much of Africa, owing to its safety, low cost, antipyretic proper-ties, and partial activity, but continued use of chloroquine for this purpose is discouraged, especially in nonimmune individuals. Chloroquine has been replaced by other drugs, principally artemisinin-based combination therapies, as the standard therapy to treat falciparum malaria in most endemic countries.
Chloroquine does not eliminate dormant liver forms of P vivax and P ovale, and for that reason primaquine must be added for the radical cure of these species.
B. Chemoprophylaxis Chloroquine is the preferred chemoprophylactic agent in malari-ous regions without resistant falciparum malaria. Eradication of P vivax and P ovale requires a course of primaquine to clear hepatic stages.
C. Amebic Liver Abscess Chloroquine reaches high liver concentrations and may be used for amebic abscesses that fail initial therapy with metronidazole (see below).
Adverse Effects Chloroquine is usually very well tolerated, even with prolonged use. Pruritus is common, primarily in Africans. Nausea, vomit-ing, abdominal pain, headache, anorexia, malaise, blurring of vision, and urticaria are uncommon. Dosing after meals may reduce some adverse effects. Rare reactions include hemolysis in glucose-6-phosphate dehydrogenase (G6PD)-deficient persons, impaired hearing, confusion, psychosis, seizures, agranulocytosis, exfoliative dermatitis, alopecia, bleaching of hair, hypotension,
TABLE 52–1 Major antimalarial drugs.
Drug Class Use
Chloroquine 4-Aminoquinoline Treatment and chemoprophylaxis of infection with sensitive parasites
Amodiaquine1 4-Aminoquinoline Treatment of infection with some chloroquine-resistant P falciparum strains and in fixed combination with artesunate
Piperaquine1 Bisquinoline Treatment of P falciparum infection in fixed combination with dihydroartemisinin
Quinine Quinoline methanol Oral and intravenous1 treatment of P falciparum infections
Quinidine Quinoline methanol Intravenous therapy of severe infections with P falciparum
Mefloquine Quinoline methanol Chemoprophylaxis and treatment of infections with P falciparum
Primaquine 8-Aminoquinoline Radical cure and terminal prophylaxis of infections with P vivax and P ovale; alternative chemoprophylaxis for all species
Sulfadoxine-pyrimethamine (Fansidar)
Folate antagonist combination Treatment of infections with some chloroquine-resistant P falciparum, including combination with artesunate; intermittent preventive therapy in endemic areas
Atovaquone-proguanil (Malarone)
Quinone-folate antagonist combination Treatment and chemoprophylaxis of P falciparum infection
Doxycycline Tetracycline Treatment (with quinine) of infections with P falciparum; chemoprophylaxis
Halofantrine1 Phenanthrene methanol Treatment of P falciparum infections
Lumefantrine2 Amyl alcohol Treatment of P falciparum malaria in fixed combination with artemether (Coartem)
Artemisinins (artesunate, artemether,2 dihydro-artemisinin1)
Sesquiterpene lactone endoperoxides Treatment of P falciparum infections; oral combination therapies for uncomplicated disease; intravenous artesunate for severe disease
1Not available in the USA.2Available in the USA only as the fixed combination Coartem.
052-Katzung_Ch052_p915-936.indd 917 9/23/11 4:27:49 PM

918 SECTION VIII Chemotherapeutic Drugs
CH3O
CH
N
HO N
CH2
CH2CH CH2
Quinine
NCl
NH CH CH2 CH2 CH2 NC2H5
C2H5
CH3
Chloroquine
N
N
N
N
N
N
NN
NH
CH2 NC2H5
C2H5
Amodiaquine
Piperaquine
OH
Cl
Cl
Cl
N
NH CH CH2 CH2 CH2 NH2
CH3
Primaquine
CH3O
CF3
HC
N
HO
NH
Mefloquine
CF3
Cl
C2H5
NH2
H2N
Pyrimethamine
N
N
Cl NH
HC N
N
H2N
NH2
H3C CH3
Proguanil
Quinoline methanols4-Aminoquinolines
8-Aminoquinoline
Folate antagonists
CH2CH2CH2CH3
CH2CH2CH2CH3
CH3
CH3
HOCHCH2CH2N
Cl
Cl
ClCl
HO
ClF3C Halofantrine
Lumefantrine
Quinone
Phenanthrene methanol
H2N
NH
N N
OCH3
OCH3
SulfadoxineO
O
OH
Cl
Atovaquone
Endoperoxides
O
O
O
O
R2 R1
Artemisinins(multiple structures)
CH3
H3C
CH3
S
OO
FIGURE 52–2 Structural formulas of antimalarial drugs.
052-Katzung_Ch052_p915-936.indd 918 9/23/11 4:27:49 PM

CHAPTER 52 Antiprotozoal Drugs 919
TABLE 52–2 Drugs for the prevention of malaria in travelers. 1
Drug Use2 Adult Dosage3
Chloroquine Areas without resistant P falciparum 500 mg weekly
Malarone Areas with chloroquine-resistant P falciparum 1 tablet (250 mg atovaquone/100 mg proguanil) daily
Mefloquine Areas with chloroquine-resistant P falciparum 250 mg weekly
Doxycycline Areas with multidrug-resistant P falciparum 100 mg daily
Primaquine4 Terminal prophylaxis of P vivax and P ovale infections; alternative for primary prevention
52.6 mg (30 mg base) daily for 14 days after travel; for primary pre-vention 52.6 mg (30 mg base) daily
1Recommendations may change, as resistance to all available drugs is increasing. See text for additional information on toxicities and cautions. For additional details and pedi-atric dosing, see CDC guidelines (phone: 877-FYI-TRIP; http://www.cdc.gov). Travelers to remote areas should consider carrying effective therapy (see text) for use if they develop a febrile illness and cannot reach medical attention quickly.2Areas without known chloroquine-resistant P falciparum are Central America west of the Panama Canal, Haiti, Dominican Republic, Egypt, and most malarious countries of the Middle East. Malarone or mefloquine are currently recommended for other malarious areas except for border areas of Thailand, where doxycycline is recommended.3For drugs other than primaquine, begin 1–2 weeks before departure (except 2 days before for doxycycline and Malarone) and continue for 4 weeks after leaving the endemic area (except 1 week for Malarone). All dosages refer to salts.4Screen for glucose-6-phosphate dehydrogenase (G6PD) deficiency before using primaquine.
TABLE 52–3 Treatment of malaria.
Clinical Setting Drug Therapy1 Alternative Drugs
Chloroquine-sensitive P falciparum and P malariae infections
Chloroquine phosphate, 1 g, followed by 500 mg at 6, 24, and 48 hours
orChloroquine phosphate, 1 g at 0 and 24 hours, then 0.5 g at 48 hours
P vivax and P ovale infections Chloroquine (as above), then (if G6PD normal) primaquine, 52.6 (30 mg base) for 14 days
For infections from Indonesia, Papua New Guinea, and other areas with suspected resistance: therapies listed for uncomplicated chloroquine-resistant P falciparum plus primaquine
Uncomplicated infections with chloroquine-resistant P falciparum
Coartem (artemether, 20 mg, plus lumefantrine, 120 mg), four tablets twice daily for 3 days
Malarone, four tablets (total of 1 g atovaquone, 400 mg proguanil) daily for 3 days
orMefloquine, 15 mg/kg once or 750 mg, then 500 mg in 6–8 hours
orQuinine sulfate, 650 mg 3 times daily for 3 days, plus doxycycline, 100 mg twice daily for 7 days, or clindamycin, 600 mg twice daily for 7 daysorOther artemisinin-based combination regimens (see Table 52–4)
Severe or complicated infections with P falciparum
Artesunate,2 2.4 mg/kg IV, every 12 hours for 1 day, then daily for 2 additional days; follow with 7-day oral course of doxycycline or clindamycin or full treatment course of Coartem, Malarone, or mefloquine
Artemether,3 3.2 mg/kg IM, then 1.6 mg/kg/d IM; follow with oral therapy as for artesunate
or orQuinidine gluconate,4,5 10 mg/kg IV over 1–2 hours, then 0.02 mg/kg IV/min
Quinine dihydrochloride,3–5 20 mg/kg IV, then 10 mg/kg every 8 hours
orQuinidine gluconate4,5 15 mg/kg IV over 4 hours, then 7.5 mg/kg IV over 4 hours every 8 hours
1All dosages are oral and refer to salts unless otherwise indicated. See text for additional information on all agents, including toxicities and cautions. See CDC guidelines (phone: 770-488-7788; http://www.cdc.gov) for additional information and pediatric dosing.2Available in the United States only on an investigational basis through the CDC (phone: 770-488-7788).3Not available in the USA.4Cardiac monitoring should be in place during intravenous administration of intravenous quinidine or quinine. Change to an oral regimen as soon as the patient can tolerate it.5Avoid loading doses in persons who have received quinine, quinidine, or mefloquine in the prior 24 hours.
G6PD, glucose-6-phosphate dehydrogenase.
052-Katzung_Ch052_p915-936.indd 919 9/23/11 4:27:49 PM

920 SECTION VIII Chemotherapeutic Drugs
and electrocardiographic changes (QRS widening, T-wave abnor-malities). The long-term administration of high doses of chloro-quine for rheumatologic diseases (see Chapter 36 ) can result in irreversible ototoxicity, retinopathy, myopathy, and peripheral neuropathy. These abnormalities are rarely if ever seen with stan-dard-dose weekly chemoprophylaxis, even when given for pro-longed periods. Large intramuscular injections or rapid intravenous infusions of chloroquine hydrochloride can result in severe hypotension and respiratory and cardiac arrest. Parenteral admin-istration of chloroquine is best avoided, but if other drugs are not available for parenteral use, it should be infused slowly.
Contraindications & Cautions Chloroquine is contraindicated in patients with psoriasis or por-phyria, in whom it may precipitate acute attacks of these diseases. It should generally not be used in those with retinal or visual field abnormalities or myopathy. Chloroquine should be used with cau-tion in patients with a history of liver disease or neurologic or hematologic disorders. The antidiarrheal agent kaolin and cal-cium- and magnesium-containing antacids interfere with the absorption of chloroquine and should not be co-administered with the drug. Chloroquine is considered safe in pregnancy and for young children.
OTHER QUINOLINES
Amodiaquine is closely related to chloroquine, and it probably shares mechanisms of action and resistance with that drug. Amodiaquine has been widely used to treat malaria because of its low cost, limited toxicity, and, in some areas, effectiveness against chloroquine-resistant strains of P falciparum. Reports of toxicities of amodiaquine, including agranulocytosis, aplastic anemia, and hepatotoxicity, have limited use of the drug in recent years. However, recent reevaluation has shown that serious toxicity from amodiaquine is rare, and it may be used as a replacement for chlo-roquine in areas with high rates of resistance but limited resources. The most important current use of amodiaquine is in combination therapy. The World Health Organization (WHO) lists amodiaquine plus artesunate as a recommended therapy for falciparum malaria in areas with resistance to older drugs ( Table 52–4 ). This combina-tion is now available as a single tablet (ASAQ, Arsucam, Coarsucam) and is the first-line therapy for the treatment of uncomplicated falciparum malaria in many countries in Africa. Another combina-tion, amodiaquine plus sulfadoxine-pyrimethamine, remains reasonably effective for the treatment of falciparum malaria in many areas with some resistance to the individual drugs, and it is a preferred alternative treatment if artemisinin-containing thera-pies are unavailable. Chemoprophylaxis with amodiaquine is best avoided because of its apparent increased toxicity with long-term use.
Piperaquine is a bisquinoline that was used widely to treat chloroquine-resistant falciparum malaria in China in the 1970s through the 1980s, but its use waned after resistance became widespread. Recently, piperaquine has been combined with
dihydroartemisinin in co-formulated tablets (Artekin, Duocotecxin) that have shown excellent efficacy and safety for the treatment of falciparum malaria, without apparent drug resistance. Piperaquine has a longer half-life (∼ 28 days) than amodiaquine (∼ 14 days), mefloquine (∼ 14 days), or lumefan-trine (∼ 4 days), leading to a longer period of post-treatment prophylaxis with dihydroartemisinin-piperaquine than with the other leading artemisinin-based combinations; this feature should be particularly advantageous in high transmission areas. Dihydroartemisinin-piperaquine is now the first-line therapy for the treatment of uncomplicated malaria in Vietnam.
ARTEMISININ & ITS DERIVATIVES
Artemisinin (qinghaosu) is a sesquiterpene lactone endoperoxide ( Figure 52–2 ), the active component of an herbal medicine that has been used as an antipyretic in China for over 2000 years. Artemisinin is insoluble and can only be used orally. Analogs have been synthesized to increase solubility and improve antimalarial efficacy. The most important of these analogs are artesunate (water-soluble; useful for oral, intravenous, intramuscular, and rectal administration), artemether (lipid-soluble; useful for oral, intramuscular, and rectal administration), and dihydroartemisinin (water-soluble; useful for oral administration).
Chemistry & Pharmacokinetics Artemisinin and its analogs are rapidly absorbed, with peak plasma levels occurring in 1–2 hours and half-lives of 1–3 hours after oral administration. Artemisinin, artesunate, and artemether are rapidly metabolized to the active metabolite dihydroartemisi-nin. Drug levels appear to decrease after a number of days of therapy.
TABLE 52–4 WHO recommendations for the treatment of falciparum malaria.
Regimen Notes
Artemether-lumefantrine (Coartem, Riamet)
Co-formulated; first-line therapy in many countries; approved in the USA
Artesunate-amodiaquine (ASAQ, Arsucam, Coarsucam)
Co-formulated; first-line therapy in many African countries
Artesunate-mefloquine Co-formulated; first-line therapy in parts of Southeast Asia and South America
Dihydroartemisinin-piperaquine (Artekin, Duocotecxin)
Co-formulated; first-line therapy in some countries in Southeast Asia
Artesunate-sulfadoxine-pyrimethamine
First-line therapy in some countries, but efficacy lower than other regi-mens in most areas
World Health Organization: Guidelines for the Treatment of Malaria. World Health Organization. Geneva, 2010.
052-Katzung_Ch052_p915-936.indd 920 9/23/11 4:27:49 PM

CHAPTER 52 Antiprotozoal Drugs 921
Antimalarial Action & Resistance The artemisinins are now widely available around the world. However, artemisinin monotherapy for the treatment of uncompli-cated malaria is now strongly discouraged. Rather, co-formulated artemisinin-based combination therapies are recommended to improve efficacy and prevent the selection of artemisinin-resistant parasites. The oral combination regimen Coartem (artemether-lumefantrine) was approved by the Food and Drug Administration (FDA) in 2009, and may be considered the new first-line therapy in the USA for uncomplicated falciparum malaria, although the drug may not be widely available. Intravenous artesunate was made available by the CDC in 2007; use of the drug can be initi-ated by contact with the CDC, which will release the drug for appropriate indications (falciparum malaria with signs of severe disease or inability to take oral medications) from stocks stored around the USA.
Artemisinin and its analogs are very rapidly acting blood sch-izonticides against all human malaria parasites. Artemisinins have no effect on hepatic stages. The antimalarial activity of artemisinins may result from the production of free radicals that follows the iron-catalyzed cleavage of the artemisinin endoperoxide bridge in the parasite food vacuole or from inhibition of a parasite calcium ATPase. Artemisinin resistance is not yet an important problem, but P falciparum isolates with diminished in vitro susceptibility to arte-mether have recently been described. In addition, increasing rates of treatment failure and increases in parasite clearance times after use of artesunate or artesunate-mefloquine in parts of Cambodia may be early signs of a worrisome decrease in artesunate efficacy .
Clinical Uses Artemisinin-based combination therapy is now the standard for treatment of uncomplicated falciparum malaria in nearly all areas endemic for falciparum malaria. These regimens were developed because the short plasma half-lives of the artemisinins led to unac-ceptably high recrudescence rates after short-course therapy, which were reversed by inclusion of longer-acting drugs. Combination therapy also helps to protect against the selection of artemisinin resistance. However, with completion of dosing after 3 days, the artemisinin components are rapidly eliminated, and so selection of resistance to partner drugs is of concern.
The WHO recommends five artemisinin-based combinations for the treatment of uncomplicated falciparum malaria ( Table 52–4 ). One of these, artesunate-sulfadoxine-pyrimethamine is not rec-ommended in many areas owing to unacceptable levels of resis-tance to sulfadoxine-pyrimethamine, but it is the first-line therapy in some countries in Asia, South America, and North Africa. The other four recommended regimens are now all available as combi-nation formulations, although manufacturing standards may vary. Artesunate-mefloquine is highly effective in Southeast Asia, where resistance to many antimalarials is common; it is the first-line therapy in some countries in Southeast Asia and South America. This regimen is less practical for other areas, particularly Africa, because of its relatively high cost and poor tolerability. Either artesunate-amodiaquine or artemether-lumefantrine is now the
standard treatment for uncomplicated falciparum malaria in most countries in Africa and some additional endemic countries on other continents. Dihydroartemisinin-piperaquine is a newer regi-men that has shown excellent efficacy; it is the first-line therapy for falciparum malaria in Vietnam.
The relative efficacy and safety of artemisinin-based combina-tion therapies are now under active investigation. In general, the leading regimens are highly efficacious, safe, and well tolerated, and they are the new standard of care for the treatment of uncom-plicated falciparum malaria.
Artemisinins are also proving to have outstanding efficacy in the treatment of complicated falciparum malaria. Large random-ized trials and meta-analyses have shown that intramuscular artemether has an efficacy equivalent to that of quinine and that intravenous artesunate is superior to intravenous quinine in terms of parasite clearance time and—most important—patient sur-vival. Intravenous artesunate also has a superior side-effect profile compared with that of intravenous quinine or quinidine. Thus, intravenous artesunate will likely replace quinine as the standard of care for the treatment of severe falciparum malaria, although it is not yet widely available in most areas. Artesunate and arte-mether have also been effective in the treatment of severe malaria when administered rectally, offering a valuable treatment modality when parenteral therapy is not available.
Adverse Effects & Cautions Artemisinins are generally very well tolerated. The most commonly reported adverse effects are nausea, vomiting, diarrhea, and dizzi-ness, and these may often be due to underlying malaria rather than the medications. Rare serious toxicities include neutropenia, ane-mia, hemolysis, elevated liver enzymes, and allergic reactions. Irreversible neurotoxicity has been seen in animals, but only after doses much higher than those used to treat malaria. Artemisinins have been embryotoxic in animal studies, but rates of congenital abnormalities, stillbirths, and abortions were not elevated, com-pared with those of controls, in women who received artemisinins during pregnancy. Based on this information and the significant risk of malaria during pregnancy, the WHO recommends artemisinin-based combination therapies for the treatment of uncomplicated falciparum malaria during the second and third trimesters of preg-nancy, intravenous artesunate or quinine for the treatment of severe malaria during the first trimester, and intravenous artesunate for treatment of severe malaria during the second and third trimesters.
QUININE & QUINIDINE
Quinine and quinidine remain important therapies for falciparum malaria—especially severe disease—although toxicity may compli-cate therapy. Resistance to quinine is uncommon but may be increasing.
Chemistry & Pharmacokinetics Quinine is derived from the bark of the cinchona tree, a tradi-tional remedy for intermittent fevers from South America. The
052-Katzung_Ch052_p915-936.indd 921 9/23/11 4:27:49 PM

922 SECTION VIII Chemotherapeutic Drugs
alkaloid quinine was purified from the bark in 1820, and it has been used in the treatment and prevention of malaria since that time. Quinidine, the dextrorotatory stereoisomer of quinine, is at least as effective as parenteral quinine in the treatment of severe falciparum malaria. After oral administration, quinine is rapidly absorbed, reaches peak plasma levels in 1–3 hours, and is widely distributed in body tissues. The use of a loading dose in severe malaria allows the achievement of peak levels within a few hours. The pharmacokinetics of quinine varies among populations. Individuals with malaria develop higher plasma levels of the drug than healthy controls, but toxicity is not increased, apparently because of increased protein binding. The half-life of quinine also is longer in those with severe malaria (18 hours) than in healthy controls (11 hours). Quinidine has a shorter half-life than qui-nine, mostly as a result of decreased protein binding. Quinine is primarily metabolized in the liver and excreted in the urine.
Antimalarial Action & Resistance Quinine is a rapid-acting, highly effective blood schizonticide against the four species of human malaria parasites. The drug is gametocidal against P vivax and P ovale but not P falciparum. It is not active against liver stage parasites. The mechanism of action of quinine is unknown.
Increasing in vitro resistance of parasites from a number of areas suggests that quinine resistance will be an increasing prob-lem. Resistance to quinine is already common in some areas of Southeast Asia, especially border areas of Thailand, where the drug may fail if used alone to treat falciparum malaria. However, qui-nine still provides at least a partial therapeutic effect in most patients.
Clinical Uses A. Parenteral Treatment of Severe Falciparum Malaria For many years, quinine dihydrochloride or quinidine gluconate have been the treatments of choice for severe falciparum malaria, although intravenous artesunate now provides an alternative for this indication. Quinine can be administered slowly intravenously or, in a dilute solution, intramuscularly, but parenteral prepara-tions of this drug are not available in the USA. Quinidine has been the standard therapy in the USA for the parenteral treatment of severe falciparum malaria. The drug can be administered in divided doses or by continuous intravenous infusion; treatment should begin with a loading dose to rapidly achieve effective plasma concentrations. Because of its cardiac toxicity and the rela-tive unpredictability of its pharmacokinetics, intravenous quini-dine should be administered slowly with cardiac monitoring. Therapy should be changed to an effective oral agent as soon as the patient has improved and can tolerate oral medications.
B. Oral Treatment of Falciparum Malaria Quinine sulfate is appropriate therapy for uncomplicated falci-parum malaria except when the infection was transmitted in an area without documented chloroquine-resistant malaria. Quinine is commonly used with a second drug (most often doxycycline or,
in children, clindamycin) to shorten quinine’s duration of use (usually to 3 days) and limit toxicity. Quinine is less effective than chloroquine against other human malarias and is more toxic. Therefore, it is not used to treat infections with these parasites.
C. Malarial Chemoprophylaxis Quinine is not generally used in chemoprophylaxis owing to its toxicity, although a daily dose of 325 mg is effective.
D. Babesiosis Quinine is first-line therapy, in combination with clindamycin, in the treatment of infection with Babesia microti or other human babesial infections.
Adverse Effects Therapeutic dosages of quinine and quinidine commonly cause tinnitus, headache, nausea, dizziness, flushing, and visual distur-bances, a constellation of symptoms termed cinchonism. Mild symptoms of cinchonism do not warrant the discontinuation of therapy. More severe findings, often after prolonged therapy, include more marked visual and auditory abnormalities, vomiting, diarrhea, and abdominal pain. Hypersensitivity reactions include skin rashes, urticaria, angioedema, and bronchospasm. Hematologic abnormalities include hemolysis (especially with G6PD defi-ciency), leukopenia, agranulocytosis, and thrombocytopenia. Therapeutic doses may cause hypoglycemia through stimulation of insulin release; this is a particular problem in severe infections and in pregnant patients, who have increased sensitivity to insulin. Quinine can stimulate uterine contractions, especially in the third trimester. However, this effect is mild, and quinine and quinidine remain drugs of choice for severe falciparum malaria even during pregnancy. Intravenous infusions of the drugs may cause throm-bophlebitis.
Severe hypotension can follow too-rapid intravenous infusions of quinine or quinidine. Electrocardiographic abnormalities (QT interval prolongation) are fairly common with intravenous quini-dine, but dangerous arrhythmias are uncommon when the drug is administered appropriately in a monitored setting.
Blackwater fever is a rare severe illness that includes marked hemolysis and hemoglobinuria in the setting of quinine therapy for malaria. It appears to be due to a hypersensitivity reaction to the drug, although its pathogenesis is uncertain.
Contraindications & Cautions Quinine (or quinidine) should be discontinued if signs of severe cinchonism, hemolysis, or hypersensitivity occur. It should be avoided if possible in patients with underlying visual or auditory problems. It must be used with great caution in those with under-lying cardiac abnormalities. Quinine should not be given concur-rently with mefloquine and should be used with caution in a patient with malaria who has previously received mefloquine chemoprophylaxis. Absorption may be blocked by aluminum-containing antacids. Quinine can raise plasma levels of warfarin and digoxin. Dosage must be reduced in renal insufficiency.
052-Katzung_Ch052_p915-936.indd 922 9/23/11 4:27:49 PM

CHAPTER 52 Antiprotozoal Drugs 923
MEFLOQUINE
Mefloquine is effective therapy for many chloroquine-resistant strains of P falciparum and against other species. Although toxicity is a concern, mefloquine is one of the recommended chemopro-phylactic drugs for use in most malaria-endemic regions with chloroquine-resistant strains.
Chemistry & Pharmacokinetics Mefloquine hydrochloride is a synthetic 4-quinoline methanol that is chemically related to quinine. It can only be given orally because severe local irritation occurs with parenteral use. It is well absorbed, and peak plasma concentrations are reached in about 18 hours. Mefloquine is highly protein-bound, extensively distrib-uted in tissues, and eliminated slowly, allowing a single-dose treat-ment regimen. The terminal elimination half-life is about 20 days, allowing weekly dosing for chemoprophylaxis. With weekly dos-ing, steady-state drug levels are reached over a number of weeks; this interval can be shortened to 4 days by beginning a course with three consecutive daily doses of 250 mg, although this is not stan-dard practice. Mefloquine and acid metabolites of the drug are slowly excreted, mainly in the feces. The drug can be detected in the blood for months after the completion of therapy.
Antimalarial Action & Resistance Mefloquine has strong blood schizonticidal activity against P falciparum and P vivax, but it is not active against hepatic stages or gametocytes. The mechanism of action of mefloquine is unknown. Sporadic resistance to mefloquine has been reported from many areas. At present, resistance appears to be uncommon except in regions of Southeast Asia with high rates of multidrug resistance (especially border areas of Thailand). Mefloquine resis-tance appears to be associated with resistance to quinine and halofantrine but not with resistance to chloroquine.
Clinical Uses A. Chemoprophylaxis Mefloquine is effective in prophylaxis against most strains of P falciparum and probably all other human malarial species. Mefloquine is therefore among the drugs recommended by the CDC for chemoprophylaxis in all malarious areas except for those with no chloroquine resistance (where chloroquine is preferred) and some rural areas of Southeast Asia with a high prevalence of mefloquine resistance. As with chloroquine, eradication of P vivax and P ovale requires a course of primaquine.
B. Treatment Mefloquine is effective in treating most falciparum malaria. The drug is not appropriate for treating individuals with severe or complicated malaria, since quinine, quinidine, and artemisinins are more rapidly active, and since drug resistance is less likely with those agents. The combination of artesunate plus mefloquine showed excellent antimalarial efficacy in regions of Southeast Asia with some resistance to mefloquine, and this regimen is now one
of the combination therapies recommended by the WHO for the treatment of uncomplicated falciparum malaria ( Table 52–4 ). Artesunate-mefloquine is the first-line therapy for uncomplicated malaria in a number of countries in Asia and South America.
Adverse Effects Weekly dosing with mefloquine for chemoprophylaxis may cause nausea, vomiting, dizziness, sleep and behavioral disturbances, epigastric pain, diarrhea, abdominal pain, headache, rash, and diz-ziness. Neuropsychiatric toxicities have received a good deal of publicity, but despite frequent anecdotal reports of seizures and psychosis, a number of controlled studies have found the fre-quency of serious adverse effects from mefloquine to be no higher than that with other common antimalarial chemoprophylactic regimens. Leukocytosis, thrombocytopenia, and aminotransferase elevations have been reported.
The latter adverse effects are more common with the higher dosages required for treatment. These effects may be lessened by administering the drug in two doses separated by 6–8 hours. The incidence of neuropsychiatric symptoms appears to be about ten times more common than with chemoprophylactic dosing, with widely varying frequencies of up to about 50% being reported. Serious neuropsychiatric toxicities (depression, confusion, acute psychosis, or seizures) have been reported in less than 1 in 1000 treatments, but some authorities believe that these toxicities are actually more common. Mefloquine can also alter cardiac conduc-tion, and arrhythmias and bradycardia have been reported.
Contraindications & Cautions Mefloquine is contraindicated in a patient with a history of epi-lepsy, psychiatric disorders, arrhythmia, cardiac conduction defects, or sensitivity to related drugs. It should not be co-administered with quinine, quinidine, or halofantrine, and caution is required if quinine or quinidine is used to treat malaria after mefloquine chemoprophylaxis. Theoretical risks of mefloquine must be bal-anced with the risk of contracting falciparum malaria. The CDC no longer advises against mefloquine use in patients receiving β-adrenoceptor antagonists. Mefloquine is also now considered safe in young children. Available data suggest that mefloquine is safe throughout pregnancy, although experience in the first trimes-ter is limited. An older recommendation to avoid mefloquine use in those requiring fine motor skills (eg, airline pilots) is controver-sial. Mefloquine chemoprophylaxis should be discontinued if sig-nificant neuropsychiatric symptoms develop.
PRIMAQUINE
Primaquine is the drug of choice for the eradication of dormant liver forms of P vivax and P ovale and can also be used for chemo-prophylaxis against all malarial species.
Chemistry & Pharmacokinetics Primaquine phosphate is a synthetic 8-aminoquinoline ( Figure 52–2 ). The drug is well absorbed orally, reaching peak plasma levels in
052-Katzung_Ch052_p915-936.indd 923 9/23/11 4:27:49 PM

924 SECTION VIII Chemotherapeutic Drugs
1–2 hours. The plasma half-life is 3–8 hours. Primaquine is widely distributed to the tissues, but only a small amount is bound there. It is rapidly metabolized and excreted in the urine. Its three major metabolites appear to have less antimalarial activity but more potential for inducing hemolysis than the parent compound.
Antimalarial Action & Resistance Primaquine is active against hepatic stages of all human malaria parasites. It is the only available agent active against the dormant hypnozoite stages of P vivax and P ovale. Primaquine is also gameto-cidal against the four human malaria species. Primaquine acts against erythrocytic stage parasites, but this activity is too weak to play an important role. The mechanism of antimalarial action is unknown.
Some strains of P vivax in New Guinea, Southeast Asia, Central and South America, and other areas are relatively resistant to primaquine. Liver forms of these strains may not be eradicated by a single standard treatment with primaquine and may require repeated therapy. Because of decreasing efficacy, the standard dos-age of primaquine for radical cure of P vivax infection was recently doubled to 30 mg base daily for 14 days.
Clinical Uses A. Therapy (Radical Cure) of Acute Vivax and Ovale Malaria Standard therapy for these infections includes chloroquine to eradi-cate erythrocytic forms and primaquine to eradicate liver hypnozo-ites and prevent a subsequent relapse. Chloroquine is given acutely, and therapy with primaquine is withheld until the G6PD status of the patient is known. If the G6PD level is normal, a 14-day course of primaquine is given. Prompt evaluation of the G6PD level is helpful, since primaquine appears to be most effective when insti-tuted before completion of dosing with chloroquine.
B. Terminal Prophylaxis of Vivax and Ovale Malaria Standard chemoprophylaxis does not prevent a relapse of vivax or ovale malaria, because the hypnozoite forms of these parasites are not eradicated by chloroquine or other available blood schizonti-cide agents. To markedly diminish the likelihood of relapse, some authorities advocate the use of primaquine after the completion of travel to an endemic area.
C. Chemoprophylaxis of Malaria Primaquine has been studied as a daily chemoprophylactic agent. Daily treatment with 30 mg (0.5 mg/kg) of base provided good levels of protection against falciparum and vivax malaria. However, potential toxicities of long-term use remain a concern, and pri-maquine is generally recommended for this purpose only when mefloquine, Malarone, and doxycycline cannot be used.
D. Gametocidal Action A single dose of primaquine (45 mg base) can be used as a control measure to render P falciparum gametocytes noninfective to mos-quitoes. This therapy is of no clinical benefit to the patient but will disrupt transmission.
E. Pneumocystis jiroveci Infection The combination of clindamycin and primaquine is an alternative regimen in the treatment of pneumocystosis, particularly mild to moderate disease. This regimen offers improved tolerance com-pared with high-dose trimethoprim-sulfamethoxazole or pentami-dine, although its efficacy against severe pneumocystis pneumonia is not well studied.
Adverse Effects Primaquine in recommended doses is generally well tolerated. It infrequently causes nausea, epigastric pain, abdominal cramps, and headache, and these symptoms are more common with higher dosages and when the drug is taken on an empty stomach. More serious but rare adverse effects are leukopenia, agranulocytosis, leukocytosis, and cardiac arrhythmias. Standard doses of pri-maquine may cause hemolysis or methemoglobinemia (manifested by cyanosis), especially in persons with G6PD deficiency or other hereditary metabolic defects.
Contraindications & Cautions Primaquine should be avoided in patients with a history of granu-locytopenia or methemoglobinemia, in those receiving potentially myelosuppressive drugs (eg, quinidine), and in those with disor-ders that commonly include myelosuppression. It is never given parenterally because it may induce marked hypotension.
Patients should be tested for G6PD deficiency before pri-maquine is prescribed. When a patient is deficient in G6PD, treat-ment strategies may consist of withholding therapy and treating subsequent relapses, if they occur, with chloroquine; treating patients with standard dosing, paying close attention to their hema-tologic status; or treating with weekly primaquine (45 mg base) for 8 weeks. G6PD-deficient individuals of Mediterranean and Asian ancestry are most likely to have severe deficiency, whereas those of African ancestry usually have a milder biochemical defect. This difference can be taken into consideration in choosing a treatment strategy. In any event, primaquine should be discontinued if there is evidence of hemolysis or anemia. Primaquine should be avoided in pregnancy because the fetus is relatively G6PD-deficient and thus at risk of hemolysis.
ATOVAQUONE
Atovaquone, a hydroxynaphthoquinone ( Figure 52–2 ), was ini-tially developed as an antimalarial agent, and as a component of Malarone is recommended for treatment and prevention of malaria. Atovaquone has also been approved by the FDA for the treatment of mild to moderate P jiroveci pneumonia.
The drug is only administered orally. Its bioavailability is low and erratic, but absorption is increased by fatty food. The drug is heavily protein-bound and has a half-life of 2–3 days. Most of the drug is eliminated unchanged in the feces. Atovaquone acts against plasmodia by disrupting mitochondrial electron transport. It is active against tissue and erythrocytic schizonts, allowing
052-Katzung_Ch052_p915-936.indd 924 9/23/11 4:27:49 PM

CHAPTER 52 Antiprotozoal Drugs 925
chemoprophylaxis to be discontinued only 1 week after the end of exposure (compared with 4 weeks for mefloquine or doxycycline, which lack activity against tissue schizonts).
Initial use of atovaquone to treat malaria led to disappointing results, with frequent failures, apparently due to the selection of resistant parasites during therapy. In contrast, Malarone, a fixed combination of atovaquone (250 mg) and proguanil (100 mg), is highly effective for both the treatment and chemoprophylaxis of falciparum malaria, and it is now approved for both indications in the USA. For chemoprophylaxis, Malarone must be taken daily ( Table 52–2 ). It has an advantage over mefloquine and doxycy-cline in requiring shorter periods of treatment before and after the period at risk for malaria transmission, but it is more expensive than the other agents. It should be taken with food.
Atovaquone is an alternative therapy for P jiroveci infection, although its efficacy is lower than that of trimethoprim-sulfamethoxazole. Standard dosing is 750 mg taken with food twice daily for 21 days. Adverse effects include fever, rash, nausea, vomiting, diarrhea, headache, and insomnia. Serious adverse effects appear to be mini-mal, although experience with the drug remains limited. Atovaquone has also been effective in small numbers of immunocompromised patients with toxoplasmosis unresponsive to other agents, although its role in this disease is not yet defined.
Malarone is generally well tolerated. Adverse effects include abdominal pain, nausea, vomiting, diarrhea, headache, and rash, and these are more common with the higher dosage required for treatment. Reversible elevations in liver enzymes have been reported. The safety of atovaquone in pregnancy is unknown. Plasma concentrations of atovaquone are decreased about 50% by co-administration of tetracycline or rifampin.
INHIBITORS OF FOLATE SYNTHESIS
Inhibitors of enzymes involved in folate metabolism are used, generally in combination regimens, in the treatment and preven-tion of malaria.
Chemistry & Pharmacokinetics Pyrimethamine is a 2,4-diaminopyrimidine related to trimetho-prim (see Chapter 46 ). Proguanil is a biguanide derivative ( Figure 52–2 ). Both drugs are slowly but adequately absorbed from the gastrointestinal tract. Pyrimethamine reaches peak plasma lev-els 2–6 hours after an oral dose, is bound to plasma proteins, and has an elimination half-life of about 3.5 days. Proguanil reaches peak plasma levels about 5 hours after an oral dose and has an elimination half-life of about 16 hours. Therefore, proguanil must be administered daily for chemoprophylaxis, whereas pyrimethamine can be given once a week. Pyrimethamine is extensively metabolized before excretion. Proguanil is a prodrug; only its triazine metabolite, cycloguanil, is active. Fansidar, a fixed combination of the sulfon-amide sulfadoxine (500 mg per tablet) and pyrimethamine (25 mg per tablet), is well absorbed. Its components display peak plasma levels within 2–8 hours and are excreted mainly by the kidneys. The average half-life of sulfadoxine is about 170 hours.
Antimalarial Action & Resistance Pyrimethamine and proguanil act slowly against erythrocytic forms of susceptible strains of all four human malaria species. Proguanil also has some activity against hepatic forms. Neither drug is adequately gametocidal or effective against the persistent liver stages of P vivax or P ovale. Sulfonamides and sulfones are weakly active against erythrocytic schizonts but not against liver stages or gametocytes. They are not used alone as antimalarials but are effective in combination with other agents.
The mechanism of action of pyrimethamine and proguanil involves selective inhibition of plasmodial dihydrofolate reductase, a key enzyme in the pathway for synthesis of folate. Sulfonamides and sulfones inhibit another enzyme in the folate pathway, dihy-dropteroate synthase. As described in Chapter 46 and shown in Figure 46–2 , combinations of inhibitors of these two enzymes provide synergistic activity.
Resistance to folate antagonists and sulfonamides is common in many areas for P falciparum and less common for P vivax. Resistance is due primarily to mutations in dihydrofolate reductase and dihydropteroate synthase, with increasing numbers of muta-tions leading to increasing levels of resistance. At present, resis-tance seriously limits the efficacy of sulfadoxine-pyrimethamine (Fansidar) for the treatment of malaria in most areas, but in Africa most parasites exhibit an intermediate level of resistance, such that antifolates may continue to offer some preventive efficacy against malaria. Because different mutations may mediate resistance to different agents, cross-resistance is not uniformly seen.
Clinical Uses A. Chemoprophylaxis Chemoprophylaxis with single folate antagonists is no longer rec-ommended because of frequent resistance, but a number of agents are used in combination regimens. The combination of chloroquine (500 mg weekly) and proguanil (200 mg daily) was previously widely used, but with increasing resistance to both agents it is no longer recommended. Fansidar and Maloprim (the latter is a com-bination of pyrimethamine and the sulfone dapsone) are both effective against sensitive parasites with weekly dosing, but they are no longer recommended because of resistance and toxicity. Considering protection of populations in endemic regions, trimethoprim-sulfamethoxazole, an antifolate combination that is more active against bacteria than malaria parasites, is increas-ingly used as a daily prophylactic therapy for HIV-infected patients in developing countries. Although it is administered primarily to prevent typical HIV opportunistic and bacterial infections, this regimen offers partial preventive efficacy against malaria in Africa.
B. Intermittent Preventive Therapy A new strategy for malaria control is intermittent preventive therapy, in which high-risk patients receive intermittent treatment for malaria, regardless of their infection status, typically with Fansidar, which benefits from simple dosing and prolonged activ-ity. Considering the two highest risk groups for severe malaria in
052-Katzung_Ch052_p915-936.indd 925 9/23/11 4:27:49 PM

926 SECTION VIII Chemotherapeutic Drugs
Africa, this strategy is best validated in pregnant women and is increasingly studied in young children. Typical schedules include single doses of Fansidar during the second and third trimesters of pregnancy and monthly doses whenever children present for scheduled immunizations or, in areas with seasonal malaria, monthly doses during the transmission season. However, optimal preventive dosing schedules have not been established.
C. Treatment of Chloroquine-Resistant Falciparum Malaria Fansidar is commonly used to treat uncomplicated falciparum malaria and until recently it was a first-line therapy for this indica-tion in some tropical countries. Advantages of Fansidar are ease of administration (a single oral dose) and low cost. However, rates of resistance are increasing, and Fansidar is no longer a recom-mended therapy. In particular, Fansidar should not be used for severe malaria, since it is slower-acting than other available agents. Fansidar is also not reliably effective in vivax malaria, and its use-fulness against P ovale and P malariae has not been adequately studied. A new antifolate-sulfone combination, chlorproguanil-dapsone (Lapdap), was until recently available in some African countries for the treatment of uncomplicated falciparum malaria, and the combination of chlorproguanil-dapsone and artesunate (Dacart) was under development. However, this project was dis-continued in 2008 as a result of concerns about hematologic tox-icity in those with G6PD deficiency, and chlorproguanil-dapsone will no longer be marketed.
D. Toxoplasmosis Pyrimethamine, in combination with sulfadiazine, is first-line therapy in the treatment of toxoplasmosis, including acute infec-tion, congenital infection, and disease in immunocompromised patients. For immunocompromised patients, high-dose therapy is required followed by chronic suppressive therapy. Folinic acid is included to limit myelosuppression. Toxicity from the combina-tion is usually due primarily to sulfadiazine. The replacement of sulfadiazine with clindamycin provides an effective alternative regimen.
E. Pneumocystosis P jiroveci is the cause of human pneumocystosis and is now recog-nized to be a fungus, but this organism is discussed in this chapter because it responds to antiprotozoal drugs, not antifungals. (The related species P carinii is now recognized to be the cause of ani-mal infections.) First-line therapy of pneumocystosis is trimetho-prim plus sulfamethoxazole (see also Chapter 46 ). Standard treatment includes high-dose intravenous or oral therapy (15 mg trimethoprim and 75 mg sulfamethoxazole per day in three or four divided doses) for 21 days. High-dose therapy entails signifi-cant toxicity, especially in patients with AIDS. Important toxici-ties include nausea, vomiting, fever, rash, leukopenia, hyponatremia, elevated hepatic enzymes, azotemia, anemia, and thrombocytope-nia. Less common effects include severe skin reactions, mental status changes, pancreatitis, and hypocalcemia. Trimethoprim-sulfamethoxazole is also the standard chemoprophylactic drug for
the prevention of P jiroveci infection in immunocompromised individuals. Dosing is one double-strength tablet daily or three times per week. The chemoprophylactic dosing schedule is much better tolerated than high-dose therapy in immunocompromised patients, but rash, fever, leukopenia, or hepatitis may necessitate changing to another drug.
Adverse Effects & Cautions Most patients tolerate pyrimethamine and proguanil well. Gastrointestinal symptoms, skin rashes, and itching are rare. Mouth ulcers and alopecia have been described with proguanil. Fansidar is no longer recommended for chemoprophylaxis because of uncommon but severe cutaneous reactions, including erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. Severe reactions appear to be much less common with single-dose or intermittent therapy, and use of the drug has been justified by the risks associated with falciparum malaria.
Rare adverse effects with a single dose of Fansidar are those associated with other sulfonamides, including hematologic, gas-trointestinal, central nervous system, dermatologic, and renal toxicity. Maloprim is no longer recommended for chemoprophy-laxis because of unacceptably high rates of agranulocytosis. Folate antagonists should be used cautiously in the presence of renal or hepatic dysfunction. Although pyrimethamine is teratogenic in animals, Fansidar has been safely used in pregnancy for therapy and as an intermittent chemoprophylactic regimen to improve pregnancy outcomes. Proguanil is considered safe in pregnancy. Folate supplements should be routinely administered during preg-nancy, but in women receiving Fansidar preventive therapy, high-dose folate supplementation (eg, 5 mg daily) should probably be avoided because it may limit preventive efficacy. The standard recommended dosage of 0.4–0.6 mg daily is less likely to affect Fansidar’s protective efficacy.
ANTIBIOTICS
A number of antibiotics in addition to the folate antagonists and sulfonamides are modestly active antimalarials. The antibiotics that are bacterial protein synthesis inhibitors appear to act against malaria parasites by inhibiting protein synthesis in a plasmodial prokaryote-like organelle, the apicoplast. None of the antibiotics should be used as single agents in the treatment of malaria because their action is much slower than that of standard antimalarials.
Tetracycline and doxycycline (see Chapter 44 ) are active against erythrocytic schizonts of all human malaria parasites. They are not active against liver stages. Doxycycline is used in the treat-ment of falciparum malaria in conjunction with quinine, allowing a shorter and better-tolerated course of that drug. Doxycycline is also used to complete treatment courses after initial treatment of severe malaria with intravenous quinine, quinidine, or artesunate. In all of these cases a 1-week treatment course of doxycycline is carried out. Doxycycline has also become a standard chemopro-phylactic drug, especially for use in areas of Southeast Asia with high rates of resistance to other antimalarials, including mefloquine.
052-Katzung_Ch052_p915-936.indd 926 9/23/11 4:27:49 PM

CHAPTER 52 Antiprotozoal Drugs 927
Doxycycline adverse effects include gastrointestinal symptoms, candidal vaginitis, and photosensitivity. Its safety in long-term chemoprophylaxis has not been extensively evaluated.
Clindamycin (see Chapter 44 ) is slowly active against erythro-cytic schizonts and can be used after treatment courses of quinine, quinidine, or artesunate in those for whom doxycycline is not recommended, such as children and pregnant women. Azithromycin (see Chapter 44 ) also has antimalarial activity and is now under study as an alternative chemoprophylactic drug. Antimalarial activity of fluoroquinolones has been demonstrated, but efficacy for the therapy or chemoprophylaxis of malaria has been suboptimal.
Antibiotics also are active against other protozoans. Tetracycline and erythromycin are alternative therapies for the treatment of intestinal amebiasis. Clindamycin, in combination with other agents, is effective therapy for toxoplasmosis, pneumocystosis, and babesiosis. Spiramycin is a macrolide antibiotic that is used to treat primary toxoplasmosis acquired during pregnancy. Treatment lowers the risk of the development of congenital toxoplasmosis.
HALOFANTRINE & LUMEFANTRINE
Halofantrine hydrochloride, a phenanthrene-methanol, is effec-tive against erythrocytic (but not other) stages of all four human malaria species. Oral absorption is variable and is enhanced with food. Because of toxicity concerns, it should not be taken with meals. Plasma levels peak 16 hours after dosing, and the half-life is about 4 days. Excretion is mainly in the feces. The mechanism of action of halofantrine is unknown. The drug is not available in the USA (although it has been approved by the FDA), but it is widely available in malaria-endemic countries.
Halofantrine (three 500-mg doses at 6-hour intervals, repeated in 1 week for nonimmune individuals) is rapidly effective against most strains of P falciparum, but its use is limited by irregular absorption and cardiac toxicity. It should not be used for chemopro-phylaxis. Halofantrine is generally well tolerated. The most com-mon adverse effects are abdominal pain, diarrhea, vomiting, cough, rash, headache, pruritus, and elevated liver enzymes. Of greater concern, the drug alters cardiac conduction, with dose-related pro-longation of QT and PR intervals. This effect is seen with standard doses and is worsened by prior mefloquine therapy. Rare instances of dangerous arrhythmias and deaths have been reported. The drug is contraindicated in patients who have cardiac conduction defects or who have recently taken mefloquine. Halofantrine is embryo-toxic in animals and therefore contraindicated in pregnancy.
Lumefantrine, an aryl alcohol related to halofantrine, is avail-able only as a fixed-dose combination with artemether (Coartem), which is now the first-line therapy for uncomplicated falciparum malaria in many countries. In addition, Coartem is approved in many nonendemic countries, including the USA. The half-life of lumefantrine, when used in combination, is approximately 4 days. Drug levels may be altered by interactions with other drugs, including those that affect CYP3A4 metabolism, but this area has not been well studied. As with halofantrine, oral absorption is highly variable and improved when the drug is taken with food.
Since lumefantrine does not engender the dangerous toxicity con-cerns of halofantrine, Coartem should be administered with fatty food to maximize antimalarial efficacy. Coartem is highly effective in the treatment of falciparum malaria when administered twice daily for 3 days. Coartem can cause minor prolongation of the QT interval, but this appears to be clinically insignificant, and the drug does not carry the risk of dangerous arrhythmias seen with halofantrine and quinidine. Indeed, Coartem is very well toler-ated. The most commonly reported adverse events in drug trials have been gastrointestinal disturbances, headache, dizziness, rash, and pruritus, and in many cases these toxicities may have been due to underlying malaria or concomitant medications rather than to Coartem.
■ AMEBIASIS
Amebiasis is infection with Entamoeba histolytica. This organism can cause asymptomatic intestinal infection, mild to moderate colitis, severe intestinal infection (dysentery), ameboma, liver abscess, and other extraintestinal infections. The choice of drugs for amebiasis depends on the clinical presentation ( Table 52–5 ).
Treatment of Specific Forms of Amebiasis A. Asymptomatic Intestinal Infection Asymptomatic carriers generally are not treated in endemic areas, but in nonendemic areas they are treated with a luminal amebi-cide. A tissue amebicidal drug is unnecessary. Standard luminal amebicides are diloxanide furoate, iodoquinol, and paromomycin. Each drug eradicates carriage in about 80–90% of patients with a single course of treatment. Therapy with a luminal amebicide is also required in the treatment of all other forms of amebiasis.
B. Amebic Colitis Metronidazole plus a luminal amebicide is the treatment of choice for amebic colitis and dysentery. Tetracyclines and erythromycin are alternative drugs for moderate colitis but are not effective against extraintestinal disease. Dehydroemetine or emetine can also be used, but are best avoided because of toxicity.
C. Extraintestinal Infections The treatment of choice for extraintestinal infections is metron-idazole plus a luminal amebicide. A 10-day course of metronida-zole cures over 95% of uncomplicated liver abscesses. For unusual cases in which initial therapy with metronidazole has failed, aspi-ration of the abscess and the addition of chloroquine to a repeat course of metronidazole should be considered. Dehydroemetine and emetine are toxic alternative drugs.
METRONIDAZOLE & TINIDAZOLE
Metronidazole, a nitroimidazole ( Figure 52–3 ), is the drug of choice in the treatment of extraluminal amebiasis. It kills tropho-zoites but not cysts of E histolytica and effectively eradicates
052-Katzung_Ch052_p915-936.indd 927 9/23/11 4:27:49 PM

928 SECTION VIII Chemotherapeutic Drugs
intestinal and extraintestinal tissue infections. Tinidazole, a related nitroimidazole available in the USA since 2004, appears to have similar activity and a better toxicity profile than metronidazole. It offers simpler dosing regimens and can be substituted for the indications listed below.
Pharmacokinetics & Mechanism of Action Oral metronidazole and tinidazole are readily absorbed and permeate all tissues by simple diffusion. Intracellular concentrations rapidly approach extracellular levels. Peak plasma concentrations are reached in 1–3 hours. Protein binding of both drugs is low (10–20%); the half-life of unchanged drug is 7.5 hours for metronidazole and 12–14 hours for tinidazole. Metronidazole and its metabolites are excreted mainly in the urine. Plasma clearance of metronidazole is decreased in patients with impaired liver function.
The nitro group of metronidazole is chemically reduced in anaerobic bacteria and sensitive protozoans. Reactive reduction products appear to be responsible for antimicrobial activity. The mechanism of tinidazole is assumed to be the same.
Clinical Uses A. Amebiasis Metronidazole or tinidazole is the drug of choice in the treatment of all tissue infections with E histolytica . Neither drug is reliably effective against luminal parasites and so must be used with a luminal amebicide to ensure eradication of the infection.
B. Giardiasis Metronidazole is the treatment of choice for giardiasis. The dosage for giardiasis is much lower—and the drug thus better tolerated—than that for amebiasis. Efficacy after a single treatment is about 90%. Tinidazole is at least equally effective.
C. Trichomoniasis Metronidazole is the treatment of choice. A single dose of 2 g is effective. Metronidazole-resistant organisms can lead to treatment failures. Tinidazole may be effective against some of these resistant organisms.
TABLE 52–5 Treatment of amebiasis. Not all preparations are available in the USA.1
Clinical Setting Drugs of Choice and Adult Dosage Alternative Drugs and Adult Dosage
Asymptomatic intesti-nal infection
Luminal agent: Diloxanide furoate,2 500 mg 3 times daily for 10 days
or
Iodoquinol, 650 mg 3 times daily for 21 days
or
Paromomycin, 10 mg/kg 3 times daily for 7 days
Mild to moderate intestinal infection
Metronidazole, 750 mg 3 times daily (or 500 mgIV every 6 hours) for 10 days
Luminal agent (see above)
or
Tinidazole, 2 g daily for 3 days
plus
Luminal agent (see above)
plus either
Tetracycline, 250 mg 3 times daily for 10 days
or
Erythromycin, 500 mg 4 times daily for 10 days
Severe intestinal infection
Metronidazole, 750 mg 3 times daily (or 500 mgIV every 6 hours) for 10 days
or
Tinidazole, 2 g daily for 3 days
plus
Luminal agent (see above)
Luminal agent (see above)
plus either
Tetracycline, 250 mg 3 times daily for 10 days
or
Dehydroemetine2 or emetine,2 1 mg/kg SC or IM for 3–5 days
Hepatic abscess, ame-boma, and other extraintestinal disease
Metronidazole, 750 mg 3 times daily (or 500 mg IV every 6 hours) for 10 days
or
Dehydroemetine2 or emetine,2 1 mg/kg SC or IM for 8–10 days, followed by (liver abscess only) chloroquine, 500 mg twice daily for 2 days, then 500 mg daily for 21 days
Tinidazole, 2 g daily for 5 days
plus plus
Luminal agent (see above) Luminal agent (see above)
1Route is oral unless otherwise indicated. See text for additional details and cautions.2Not available in the USA.
052-Katzung_Ch052_p915-936.indd 928 9/23/11 4:27:50 PM

CHAPTER 52 Antiprotozoal Drugs 929
Adverse Effects & Cautions Nausea, headache, dry mouth, or a metallic taste in the mouth occurs commonly. Infrequent adverse effects include vomiting, diarrhea, insomnia, weakness, dizziness, thrush, rash, dysuria, dark urine, vertigo, paresthesias, and neutropenia. Taking the drug with meals lessens gastrointestinal irritation. Pancreatitis and severe central nervous system toxicity (ataxia, encephalopathy, seizures) are rare. Metronidazole has a disulfiram-like effect, so that nausea and vomiting can occur if alcohol is ingested during therapy. The drug should be used with caution in patients with central nervous system disease. Intravenous infusions have rarely caused seizures or peripheral neuropathy. The dosage should be adjusted for patients with severe liver or renal disease. Tinidazole has a similar adverse-effect profile, although it appears to be some-what better tolerated than metronidazole.
Metronidazole has been reported to potentiate the anticoagu-lant effect of coumarin-type anticoagulants. Phenytoin and phe-nobarbital may accelerate elimination of the drug, whereas cimetidine may decrease plasma clearance. Lithium toxicity may occur when the drug is used with metronidazole.
Metronidazole and its metabolites are mutagenic in bacteria. Chronic administration of large doses led to tumorigenicity in mice. Data on teratogenicity are inconsistent. Metronidazole is
thus best avoided in pregnant or nursing women, although con-genital abnormalities have not clearly been associated with use in humans.
IODOQUINOL
Iodoquinol (diiodohydroxyquin) is a halogenated hydroxyquino-line. It is an effective luminal amebicide that is commonly used with metronidazole to treat amebic infections. Pharmacokinetic data are incomplete but 90% of the drug is retained in the intes-tine and excreted in the feces. The remainder enters the circula-tion, has a half-life of 11–14 hours, and is excreted in the urine as glucuronides.
The mechanism of action of iodoquinol against trophozoites is unknown. It is effective against organisms in the bowel lumen but not against trophozoites in the intestinal wall or extraintestinal tissues.
Infrequent adverse effects include diarrhea—which usually stops after several days—anorexia, nausea, vomiting, abdominal pain, headache, rash, and pruritus. The drug may increase protein-bound serum iodine, leading to a decrease in measured 131 I uptake that persists for months. Some halogenated hydroxyquinolines can produce severe neurotoxicity with prolonged use at greater than recommended doses. Iodoquinol is not known to produce these
Metronidazole
I
I
Paromomycin
Pentamidine
3Na
Sodium stibogluconate
N
NC
C
C CH3
CH2CH2OH
O2N
H
Iodoquinol
OH
CH2OH
OH
O
OHHO
NH2
O
H2N
O
NH2
O
OH OCH2OH
O
H2N
OH
OH
CH2NH2 OCOO N
CH3
COCHCl2
Diloxanide furoate
C
HN
H2NOCH2(CH2)3CH2O C
NH
NH2
CH2OH
CHOH
CHO
CHO
CHO
COO–
+Sb
OH
O Sb
O–
HOH2C
HOHC
OHC
OHC
OHC
OOC–
N
FIGURE 52–3 Structural formulas of other antiprotozoal drugs.
052-Katzung_Ch052_p915-936.indd 929 9/23/11 4:27:50 PM

930 SECTION VIII Chemotherapeutic Drugs
effects at its recommended dosage, and this dosage should never be exceeded.
Iodoquinol should be taken with meals to limit gastrointestinal toxicity. It should be used with caution in patients with optic neu-ropathy, renal or thyroid disease, or nonamebic hepatic disease. The drug should be discontinued if it produces persistent diarrhea or signs of iodine toxicity (dermatitis, urticaria, pruritus, fever). It is contraindicated in patients with intolerance to iodine.
DILOXANIDE FUROATE
Diloxanide furoate is a dichloroacetamide derivative. It is an effec-tive luminal amebicide but is not active against tissue trophozo-ites. In the gut, diloxanide furoate is split into diloxanide and furoic acid; about 90% of the diloxanide is rapidly absorbed and then conjugated to form the glucuronide, which is promptly excreted in the urine. The unabsorbed diloxanide is the active antiamebic substance. The mechanism of action of diloxanide furoate is unknown.
Diloxanide furoate is considered by many the drug of choice for asymptomatic luminal infections. It is not available commercially in the USA, but can be obtained from some compounding phar-macies. It is used with a tissue amebicide, usually metronidazole, to treat serious intestinal and extraintestinal infections. Diloxanide furoate does not produce serious adverse effects. Flatulence is com-mon, but nausea and abdominal cramps are infrequent and rashes are rare. The drug is not recommended in pregnancy.
PAROMOMYCIN SULFATE
Paromomycin sulfate is an aminoglycoside antibiotic (see also Chapter 45 ) that is not significantly absorbed from the gastroin-testinal tract. It is used only as a luminal amebicide and has no effect against extraintestinal amebic infections. The small amount absorbed is slowly excreted unchanged, mainly by glomerular fil-tration. However, the drug may accumulate with renal insuffi-ciency and contribute to renal toxicity. Paromomycin is an effective luminal amebicide that appears to have similar efficacy and probably less toxicity than other agents; in a recent study, it was superior to diloxanide furoate in clearing asymptomatic infec-tions. Adverse effects include occasional abdominal distress and diarrhea. Parenteral paromomycin is now used to treat visceral leishmaniasis and is discussed separately in the text that follows.
EMETINE & DEHYDROEMETINE
Emetine, an alkaloid derived from ipecac, and dehydroemetine, a synthetic analog, are effective against tissue trophozoites of E histolytica, but because of major toxicity concerns their use is limited to unusual circumstances in which severe amebiasis requires effective therapy and metronidazole cannot be used. Dehydroemetine is preferred because of its somewhat better toxic-ity profile. The drugs should be used for the minimum period needed to relieve severe symptoms (usually 3–5 days) and should be administered subcutaneously (preferred) or intramuscularly in a supervised setting. Emetine and dehydroemetine should not be used intravenously. Adverse effects, which are generally mild with use for 3–5 days, increase over time and include pain, tenderness, and sterile abscesses at the injection site; diarrhea, nausea, and vomiting; muscle weakness and discomfort; and minor electrocar-diographic changes. Serious toxicities include cardiac arrhythmias, heart failure, and hypotension. The drugs should not be used in patients with cardiac or renal disease, in young children, or in pregnancy unless absolutely necessary.
■ OTHER ANTIPROTOZOAL DRUGS
The primary drugs used to treat African trypanosomiasis are set forth in Table 52–6 , and those for other protozoal infections are listed in Table 52–7 . Important drugs that are not covered else-where in this or other chapters are discussed below.
PENTAMIDINE
Pentamidine has activity against trypanosomatid protozoans and against P jiroveci, but toxicity is significant.
Chemistry & Pharmacokinetics Pentamidine is an aromatic diamidine ( Figure 52–3 ) formulated as an isethionate salt. Pentamidine is only administered parenter-ally. The drug leaves the circulation rapidly, with an initial half-life of about 6 hours, but it is bound avidly by tissues. Pentamidine thus accumulates and is eliminated very slowly, with a terminal elimination half-life of about 12 days. The drug can be detected in urine 6 or more weeks after treatment. Only trace amounts of pentamidine appear in the central nervous system, so it is not
TABLE 52–6 Treatment of African trypanosomiasis.
Disease Stage First-Line Drugs Alternative Drugs
West African Early Pentamidine Suramin, eflornithine
CNS involvement Eflornithine Melarsoprol,1 eflornithine-nifurtimox1
East African Early Suramin1 Pentamidine
CNS involvement Melarsoprol1
1Available in the USA from the Drug Service, CDC, Atlanta, Georgia (phone: 404-639-3670; http://www.cdc.gov/laboratory/drugservice/index.html).
052-Katzung_Ch052_p915-936.indd 930 9/23/11 4:27:50 PM

CHAPTER 52 Antiprotozoal Drugs 931
TABLE 52–7 Treatment of other protozoal infections. Not all preparations are available in the USA.1
Organism or Clinical Setting Drugs of Choice2 Alternative Drugs
Babesia species Clindamycin, 600 mg 3 times daily for 7 days Atovaquone or azithromycin
plus
Quinine, 650 mg for 7 days
Balantidium coli Tetracycline, 500 mg 4 times daily for 10 days Metronidazole, 750 mg 3 times daily for 5 days
Cryptosporidium species Paromomycin, 500–750 mg 3 or 4 times daily for 10 days Azithromycin, 500 mg daily for 21 days
Cyclospora cayetanensis Trimethoprim-sulfamethoxazole, one double-strength tablet 4 times daily for 7–14 days
Dientamoeba fragilis Iodoquinol, 650 mg 3 times daily for 20 days Tetracycline, 500 mg 4 times daily for 10 days
or
Paromomycin, 500 mg 3 times daily for 7 days
Giardia lamblia Metronidazole, 250 mg 3 times daily for 5 days Furazolidone, 100 mg 4 times daily for 7 days
or or
Tinidazole, 2 g once Albendazole, 400 mg daily for 5 days
Isospora belli Trimethoprim-sulfamethoxazole, one double-strength tablet 4 times daily for 10 days, then twice daily for 21 days
Pyrimethamine, 75 mg daily for 14 days
plus
Folinic acid, 10 mg daily for 14 days
Microsporidia Albendazole, 400 mg twice daily for 20–30 days
Leishmaniasis
Visceral (L donovani, L chagasi, L infantum) or mucosal (L braziliensis)
Sodium stibogluconate, 20 mg/kg/d IV or IM for 28 days Meglumine antimoniate
or
Pentamidine
or
Amphotericin
or
Miltefosine
or
Paromomycin
Cutaneous (L major, L tropica, L mexicana, L braziliensis)
Sodium stibogluconate, 20 mg/kg/d IV or IM for 20 days Meglumine antimoniate
or
Amphotericin
or
Pentamidine
or
Topical or intralesional therapies
Pneumocystis jiroveci, P carinii3 Trimethoprim-sulfamethoxazole, 15–20 mg trimethoprim component/kg/d IV, or two double-strength tablets every 8 hours for 21 days
Pentamidine
or
Trimethoprim-dapsone
or
Clindamycin plus primaquine
or
Atovaquone
Toxoplasma gondii
(continued)
052-Katzung_Ch052_p915-936.indd 931 9/23/11 4:27:50 PM

932 SECTION VIII Chemotherapeutic Drugs
effective against central nervous system African trypanosomiasis. Pentamidine can also be inhaled as a nebulized powder for the prevention of pneumocystosis. Absorption into the systemic circu-lation after inhalation appears to be minimal. The mechanism of action of pentamidine is unknown.
Clinical Uses A. Pneumocystosis Pentamidine is a well-established alternative therapy for pulmo-nary and extrapulmonary disease caused by P jiroveci. The drug has somewhat lower efficacy and greater toxicity than trimetho-prim-sulfamethoxazole. The standard dosage is 3 mg/kg/d intrave-nously for 21 days. Significant adverse reactions are common, and with multiple regimens now available to treat P jiroveci infection, pentamidine is best reserved for patients with severe disease who cannot tolerate or fail other drugs.
Pentamidine is also an alternative agent for primary or second-ary prophylaxis against pneumocystosis in immunocompromised individuals, including patients with advanced AIDS. For this indi-cation, pentamidine is administered as an inhaled aerosol (300 mg inhaled monthly). The drug is well tolerated in this form. Its effi-cacy is very good but clearly less than that of daily trimethoprim-sulfamethoxazole. Because of its cost and ineffectiveness against nonpulmonary disease, it is best reserved for patients who cannot tolerate oral chemoprophylaxis with other drugs.
B. African Trypanosomiasis (Sleeping Sickness) Pentamidine has been used since 1940 and is the drug of choice to treat the early hemolymphatic stage of disease caused by Trypanosoma brucei gambiense (West African sleeping sickness). The drug is infe-rior to suramin for the treatment of early East African sleeping sick-ness. Pentamidine should not be used to treat late trypanosomiasis
with central nervous system involvement. A number of dosing regi-mens have been described, generally providing 2–4 mg/kg daily or on alternate days for a total of 10–15 doses. Pentamidine has also been used for chemoprophylaxis against African trypanosomiasis, with dosing of 4 mg/kg every 3–6 months.
C. Leishmaniasis Pentamidine is an alternative to sodium stibogluconate in the treatment of visceral leishmaniasis, with similar efficacy, although resistance has been reported. The drug has been successful in some cases that have failed therapy with antimonials. The dosage is 2–4 mg/kg intramuscularly daily or every other day for up to 15 doses, and a second course may be necessary. Pentamidine has also shown success against cutaneous leishmaniasis, but it is not rou-tinely used for this purpose.
Adverse Effects & Cautions Pentamidine is a highly toxic drug, with adverse effects noted in about 50% of patients receiving 4 mg/kg/d. Rapid intravenous administration can lead to severe hypotension, tachycardia, dizzi-ness, and dyspnea, so the drug should be administered slowly (over 2 hours), and patients should be recumbent and monitored closely during treatment. With intramuscular administration, pain at the injection site is common, and sterile abscesses may develop.
Pancreatic toxicity is common. Hypoglycemia due to inappro-priate insulin release often appears 5–7 days after onset of treat-ment, can persist for days to several weeks, and may be followed by hyperglycemia. Reversible renal insufficiency is also common. Other adverse effects include rash, metallic taste, fever, gastro-intestinal symptoms, abnormal liver function tests, acute pancrea-titis, hypocalcemia, thrombocytopenia, hallucinations, and cardiac arrhythmias. Inhaled pentamidine is generally well tolerated but may cause cough, dyspnea, and bronchospasm.
TABLE 52–7 Treatment of other protozoal infections. Not all preparations are available in the USA.1
Organism or Clinical Setting Drugs of Choice2 Alternative Drugs
Acute, congenital, immunocompro- mised
Pyrimethamine plus clindamycin plus folinic acid Pyrimethamine plus sulfadiazine plus folinic acid
Pregnancy Spiramycin, 3 g daily until delivery
Trichomonas vaginalis Metronidazole, 2 g once or 250 mg 3 times daily for 7 days
or
Tinidazole, 2 g once
Trypanosoma cruzi Nifurtimox
or
Benznidazole
1Additional information may be obtained from the Parasitic Disease Drug Service, Parasitic Diseases Branch, CDC, Atlanta, Georgia (phone: 404-639-3670; http://www.cdc.gov/laboratory/drugservice/index.html).2Established, relatively simple dosing regimens are provided. Route is oral unless otherwise indicated. See text for additional information, toxicities, cautions, and discussions of dosing for the more rarely used drugs, many of which are highly toxic.3P jiroveci (carinii in animals) has traditionally been considered a protozoan because of its morphology and drug sensitivity, but recent molecular analyses have shown it to be most closely related to fungi.
(Continued)
052-Katzung_Ch052_p915-936.indd 932 9/23/11 4:27:50 PM

CHAPTER 52 Antiprotozoal Drugs 933
SODIUM STIBOGLUCONATE
Pentavalent antimonials, including sodium stibogluconate (pen-tostam; Figure 52–3 ) and meglumine antimoniate, are generally considered first-line agents for cutaneous and visceral leishmania-sis except in parts of India, where the efficacy of these drugs has diminished greatly. The drugs are rapidly absorbed and distributed after intravenous (preferred) or intramuscular administration and eliminated in two phases, with short initial (about 2-hour) half-life and much longer terminal (> 24-hour) half-life. Treatment is given once daily at a dosage of 20 mg/kg/d intravenously or intra-muscularly for 20 days in cutaneous leishmaniasis and 28 days in visceral and mucocutaneous disease.
The mechanism of action of the antimonials is unknown. Their efficacy against different species may vary, possibly based on local drug resistance patterns. Cure rates are generally quite good, but resistance to sodium stibogluconate is increasing in some endemic areas, notably in India where other agents (eg, amphot-ericin or miltefosine) are generally recommended.
Few adverse effects occur initially, but the toxicity of stibogluconate increases over the course of therapy. Most common are gastrointestinal symptoms, fever, headache, myalgias, arthralgias, and rash. Intramuscular injections can be very painful and lead to sterile abscesses. Electrocardiographic changes may occur, most commonly T-wave changes and QT prolongation. These changes are generally reversible, but continued therapy may lead to dangerous arrhythmias. Thus, the electrocardiogram should be monitored during therapy. Hemolytic anemia and serious liver, renal, and cardiac effects are rare.
NITAZOXANIDE
Nitazoxanide is a nitrothiazolyl-salicylamide prodrug. Nitazoxanide was recently approved in the USA for use against Giardia lamblia and Cryptosporidium parvum. It is rapidly absorbed and converted to tizoxanide and tizoxanide conjugates, which are subsequently excreted in both urine and feces. The active metabolite, tizox-anide, inhibits the pyruvate-ferredoxin oxidoreductase pathway. Nitazoxanide appears to have activity against metronidazole-resistant protozoal strains and is well tolerated. Unlike metronida-zole, nitazoxanide and its metabolites appear to be free of mutagenic effects. Other organisms that may be susceptible to nitazoxanide include E histolytica, Helicobacter pylori, Ascaris lum-bricoides, several tapeworms, and Fasciola hepatica. The recom-mended adult dosage is 500 mg twice daily for 3 days.
OTHER DRUGS FOR TRYPANOSOMIASIS & LEISHMANIASIS
Available therapies for all forms of trypanosomiasis are seriously deficient in efficacy, safety, or both. Availability of these therapies is also a concern, since they are supplied mainly through donation or nonprofit production by pharmaceutical companies. For vis-ceral leishmaniasis, three new promising therapies are liposomal amphotericin, miltefosine, and paromomycin.
A. Suramin Suramin is a sulfated naphthylamine that was introduced in the 1920s. It is the first-line therapy for early hemolymphatic East African trypanosomiasis ( T brucei rhodesiense infection), but because it does not enter the central nervous system, it is not effec-tive against advanced disease. Suramin is less effective than pent-amidine for early West African trypanosomiasis. The drug’s mechanism of action is unknown. It is administered intravenously and displays complex pharmacokinetics with very tight protein binding. Suramin has a short initial half-life but a terminal elimi-nation half-life of about 50 days. The drug is slowly cleared by renal excretion.
Suramin is administered after a 200-mg intravenous test dose. Regimens that have been used include 1 g on days 1, 3, 7, 14, and 21 or 1 g each week for 5 weeks. Combination therapy with pen-tamidine may improve efficacy. Suramin can also be used for chemoprophylaxis against African trypanosomiasis. Adverse effects are common. Immediate reactions can include fatigue, nausea, vomiting, and, more rarely, seizures, shock, and death. Later reac-tions include fever, rash, headache, paresthesias, neuropathies, renal abnormalities including proteinuria, chronic diarrhea, hemo-lytic anemia, and agranulocytosis.
B. Melarsoprol Melarsoprol is a trivalent arsenical that has been available since 1949 and is first-line therapy for advanced central nervous system East African trypanosomiasis, and second-line therapy (after eflornithine) for advanced West African trypanosomiasis. After intravenous administration it is excreted rapidly, but clinically relevant concentrations accumulate in the central nervous system within 4 days. Melarsoprol is administered in propylene glycol by slow intravenous infusion at a dosage of 3.6 mg/kg/d for 3–4 days, with repeated courses at weekly intervals, if needed. A new regi-men of 2.2 mg/kg daily for 10 days had efficacy and toxicity similar to what was observed with three courses over 26 days. Melarsoprol is extremely toxic. The use of such a toxic drug is justified only by the severity of advanced trypanosomiasis and the lack of available alternatives. Immediate adverse effects include fever, vomiting, abdominal pain, and arthralgias. The most impor-tant toxicity is a reactive encephalopathy that generally appears within the first week of therapy (in 5–10% of patients) and is probably due to disruption of trypanosomes in the central nervous system. Common consequences of the encephalopathy include cerebral edema, seizures, coma, and death. Other serious toxicities include renal and cardiac disease and hypersensitivity reactions. Failure rates with melarsoprol appear to have increased recently in parts of Africa, suggesting the possibility of drug resistance.
C. Eflornithine Eflornithine (difluoromethylornithine), an inhibitor of ornithine decarboxylase, is the only new drug registered to treat African trypanosomiasis in the last half-century. It is now the first-line drug for advanced West African trypanosomiasis, but is not effec-tive for East African disease. Eflornithine is less toxic than melar-soprol but not as widely available. The drug had very limited
052-Katzung_Ch052_p915-936.indd 933 9/23/11 4:27:50 PM

934 SECTION VIII Chemotherapeutic Drugs
availability until recently, when it was developed for use as a topi-cal depilatory cream, leading to donation of the drug for the treat-ment of trypanosomiasis. Eflornithine is administered intravenously, and good central nervous system drug levels are achieved. The elimination half-life is about 3 hours. The usual regimen is 100 mg/kg intravenously every 6 hours for 7–14 days (14 days was superior for a newly diagnosed infection). Eflornithine appears to be as effective as melarsoprol against advanced T brucei gambiense infection, but its efficacy against T brucei rhodesiense is limited by drug resistance. Toxicity from eflornithine is signifi-cant, but considerably less than that from melarsoprol. Adverse effects include diarrhea, vomiting, anemia, thrombocytopenia, leukopenia, and seizures. These effects are generally reversible. Increased experience with eflornithine and increased availability of the compound in endemic areas may lead to its replacement of suramin, pentamidine, and melarsoprol in the treatment of T brucei gambiense infection.
D. Nifurtimox Nifurtimox, a nitrofuran, is the most commonly used drug for American trypanosomiasis (Chagas’ disease). Nifurtimox is also under study in the treatment of African trypanosomiasis, particu-larly in combination with eflornithine. Nifurtimox is well absorbed after oral administration and eliminated with a plasma half-life of about 3 hours. The drug is administered at a dosage of 8–10 mg/kg/d (divided into three to four doses) orally for 3–4 months in the treatment of acute Chagas’ disease. Nifurtimox decreases the sever-ity of acute disease and usually eliminates detectable parasites, but it is often ineffective in fully eradicating infection. Thus, it often fails to prevent progression to the gastrointestinal and cardiac syndromes associated with chronic infection that are the most important clini-cal consequences of Trypanosoma cruzi infection. Efficacy may vary in different parts of South America, possibly related to drug resis-tance in some areas. Nifurtimox has not been proved effective in the treatment of chronic Chagas’ disease. Toxicity related to nifurtimox is common. Adverse effects include nausea, vomiting, abdominal pain, fever, rash, restlessness, insomnia, neuropathies, and seizures. These effects are generally reversible but often lead to cessation of therapy before completion of a standard course.
E. Benznidazole Benznidazole is an orally administered nitroimidazole that appears to have efficacy similar to that of nifurtimox in the treatment of acute Chagas’ disease. Availability of the drug is currently limited. Important toxicities include peripheral neuropathy, rash, gastro-intestinal symptoms, and myelosuppression.
F. Amphotericin This important antifungal drug (see Chapter 48 ) is an alternative therapy for visceral leishmaniasis, especially in parts of India with
high-level resistance to sodium stibogluconate. Liposomal ampho-tericin has shown excellent efficacy at a dosage of 3 mg/kg/d intravenously on days 1–5, 14, and 21. Nonliposomal amphoteri-cin (1 mg/kg intravenously every other day for 30 days) is much less expensive, also efficacious, and widely used in India. Amphotericin is also used for cutaneous leishmaniasis in some areas. The use of amphotericin, and especially liposomal prepara-tions, is limited in developing countries by difficulty of adminis-tration, cost, and toxicity.
G. Miltefosine Miltefosine is an alkylphosphocholine analog that is the first effec-tive oral drug for visceral leishmaniasis. It has recently shown excellent efficacy in the treatment of visceral leishmaniasis in India, where it is administered orally (2.5 mg/kg/d with varied dosing schedules) for 28 days. It was also recently shown to be effective in regimens including a single dose of liposomal ampho-tericin followed by 7–14 days of miltefosine. A 28-day course of miltefosine (2.5 mg/kg/d) was also effective for the treatment of New World cutaneous leishmaniasis. Vomiting and diarrhea are common but generally short-lived toxicities. Transient elevations in liver enzymes and nephrotoxicity are also seen. The drug should be avoided in pregnancy (or in women who may become pregnant within 2 months of treatment) because of its teratogenic effects. Miltefosine is registered for the treatment of visceral leishmaniasis in India and some other countries, and—considering the serious limitations of other drugs, including parenteral administration, toxicity, and resistance—it may become the treatment of choice for that disease. Resistance to miltefosine develops readily in vitro. To circumvent this problem, various drug combinations, includ-ing miltefosine with antimonials, amphotericin, or paromomycin, are under study.
H. Paromomycin Paromomycin sulfate is an aminoglycoside antibiotic that until recently was used in parasitology only for oral therapy of intestinal parasitic infections (see previous text). It has recently been devel-oped for the treatment of visceral leishmaniasis. A phase 3 trial in India showed excellent efficacy for this disease, with a daily intra-muscular dosage of 11 mg/kg for 21 days yielding a 95% cure rate, and noninferiority compared with amphotericin. The drug was registered for the treatment of visceral leishmaniasis in India in 2006. However, a recent trial showed poorer efficacy in Africa, with the cure rate for paromomycin significantly inferior to that with sodium stibogluconate. In initial studies, paromomycin was well tolerated, with common mild injection pain, uncommon ototoxicity and reversible liver enzyme elevations, and no nephro-toxicity. Paromomycin is much less expensive than liposomal amphotericin or miltefosine, the other promising new therapies for visceral leishmaniasis.
052-Katzung_Ch052_p915-936.indd 934 9/23/11 4:27:50 PM

CHAPTER 52 Antiprotozoal Drugs 935
REFERENCES General
Drugs for parasitic infections. Med Lett Drugs Ther 2010;Supplement.
Malaria
Baird JK: Effectiveness of antimalarial drugs. N Engl J Med 2005;352:1565. Baird JK: Resistance to therapies for infection by Plasmodium vivax . Clin
Microbiol Rev 2009;22:508. Barnes KI et al: Efficacy of rectal artesunate compared with parenteral quinine in
initial treatment of moderately severe malaria in African children and adults: A randomised study. Lancet 2004;363:1598.
Boggild AK et al: Atovaquone-proguanil: Report from the CDC expert meeting on malaria chemoprophylaxis (II). Am J Trop Med Hyg 2007;76:208.
Dondorp AM et al: Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2009;361:455.
Dondorp A et al: Artesunate versus quinine for treatment of severe falciparum malaria: A randomised trial. Lancet 2005;366:717.
Dondorp AM et al: Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): An open-label, randomised trial. Lancet 2010;376:1647.
Dorsey G et al: Combination therapy for uncomplicated falciparum malaria in Ugandan children: A randomized trial. JAMA 2007;297:2210.
Efferth T, Kaina B: Toxicity of the antimalarial artemisinin and its derivatives. Crit Rev Toxicol 2010;40:405.
Freedman DO: Malaria prevention in short-term travelers. N Engl J Med 2008;359:603.
Greenwood BM et al: Malaria. Lancet 2005;365:1487.
Hill DR et al: Primaquine: Report from CDC expert meeting on malaria chemo-prophylaxis I. Am J Trop Med Hyg 2006;75:402.
Nosten F, White NJ: Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg 2007;77(Suppl 6):181.
Nosten F et al: Antimalarial drugs in pregnancy: A review. Curr Drug Saf 2006;1:1.
Rosenthal PJ: Artesunate for the treatment of severe falciparum malaria. N Engl J Med 2008;358:1829.
Stepniewska K, White NJ: Pharmacokinetic determinants of the window of selec-tion for antimalarial drug resistance. Antimicrob Agents Chemother 2008;52:1589.
Taylor WR, White NJ: Antimalarial drug toxicity: A review. Drug Saf 2004;27:25.
White NJ: Cardiotoxicity of antimalarial drugs. Lancet Infect Dis 2007;7:549. White NJ: Qinghaosu (artemisinin): The price of success. Science 2008;320:330. Whitty CJ, et al: Malaria: An update on treatment of adults in non-endemic coun-
tries. Br Med J 2006;333:241. World Health Organization: Guidelines for the treatment of malaria. Geneva.
2010. http://www.who.int/malaria/publications/atoz/9789241547925/en/index.html
Intestinal Protozoal Infections
Blessmann J et al: Treatment of amoebic liver abscess with metronidazole alone or in combination with ultrasound-guided needle aspiration: A comparative, prospective and randomized study. Trop Med Int Health 2003;8:1030.
Fox LM, Saravolatz LD: Nitazoxanide: A new thiazolide antiparasitic agent. Clin Infect Dis 2005;40:1173.
Haque R et al: Amebiasis. N Engl J Med 2003;348:1565.
*Available in the USA only from the Drug Service, CDC, Atlanta, Georgia (phone: 404-639-3670; http://www.cdc.gov/laboratory/drugservice/index.html).
Albendazole (Albenza)Oral: 200 mg tablets
Artemether/lumefantrine (Coartem)Oral: 20 mg artemether/120 mg lumefantrine tablets
Artesunate*Atovaquone (Mepron)
Oral: 750 mg/5 mL suspensionAtovaquone-proguanil (Malarone)
Oral: 250 mg atovaquone + 100 mg proguanil tablets; pediatric 62.5 mg atovaquone + 25 mg proguanil tablets
Chloroquine (generic, Aralen)Oral: 250, 500 mg tablets (equivalent to 150, 300 mg base,
respectively)Parenteral: 50 mg/mL (equivalent to 40 mg/mL base) for injection
Clindamycin (generic, Cleocin)Oral: 75, 150, 300 mg capsules; 75 mg/5 mL suspensionParenteral: 150 mg/mL for injection
Doxycycline (generic, Vibramycin)Oral: 20, 50, 100 mg capsules; 50, 100 mg tablets; 25 mg/5 mL
suspension; 50 mg/5 mL syrupParenteral: 100, 200 mg for injection
Eflornithine (Ornidyl)Parenteral: 200 mg/mL for injection
Iodoquinol (Yodoxin)Oral: 210, 650 mg tablets
Mefloquine (generic, Lariam)Oral: 250 mg tablets
P R E P A R A T I O N S A V A I L A B L E
Albendazole (Albenza)Oral: 200 mg tablets
Artemether/lumefantrine (Coartem)Oral: 20 mg artemether/120 mg lumefantrine tablets
Artesunate*Atovaquone (Mepron)
Oral: 750 mg/5 mL suspensionAtovaquone-proguanil (Malarone)
Oral: 250 mg atovaquone + 100 mg proguanil tablets; pediatric 62.5 mg atovaquone + 25 mg proguanil tablets
Chloroquine (generic, Aralen)Oral: 250, 500 mg tablets (equivalent to 150, 300 mg base,
respectively)Parenteral: 50 mg/mL (equivalent to 40 mg/mL base) for injection
Clindamycin (generic, Cleocin)Oral: 75, 150, 300 mg capsules; 75 mg/5 mL suspensionParenteral: 150 mg/mL for injection
Doxycycline (generic, Vibramycin)Oral: 20, 50, 100 mg capsules; 50, 100 mg tablets; 25 mg/5 mL
suspension; 50 mg/5 mL syrupParenteral: 100, 200 mg for injection
Eflornithine (Ornidyl)Parenteral: 200 mg/mL for injection
Iodoquinol (Yodoxin)Oral: 210, 650 mg tablets
Mefloquine (generic, Lariam)Oral: 250 mg tablets
Melarsoprol (Mel B)*Metronidazole (generic, Flagyl)
Oral: 250, 500 mg tablets; 375 mg capsules; extended-release 750 mg tablets
Parenteral: 5 mg/mLNifurtimox*Nitazoxanide (Alinia)
Oral: 500 mg tablets, powder for 100 mg/5 mL oral solutionParomomycin (Humatin)
Oral: 250 mg capsulesPentamidine (Pentam 300, Pentacarinat, pentamidine isethionate)
Parenteral: 300 mg powder for injectionAerosol (Nebupent): 300 mg powder
Primaquine (generic)Oral: 26.3 mg (equivalent to 15 mg base) tablet
Pyrimethamine (Daraprim)Oral: 25 mg tablets
Quinidine gluconate (generic)Parenteral: 80 mg/mL (equivalent to 50 mg/mL base) for injection
Quinine (generic)Oral: 260 mg tablets; 200, 260, 325 mg capsules
Sodium stibogluconate*Sulfadoxine and pyrimethamine (Fansidar)
Oral: 500 mg sulfadoxine plus 25 mg pyrimethamine tabletsSuramin*Tinidazole (Tindamax)
Oral: 250, 500 mg tablets
052-Katzung_Ch052_p915-936.indd 935 9/23/11 4:27:50 PM

936 SECTION VIII Chemotherapeutic Drugs
C A S E S T U D Y A N S W E R
This child has acute falciparum malaria, and her lethargy and abnormal laboratory tests are consistent with progres-sion to severe disease. She should be hospitalized and treated urgently with intravenous artesunate or, if this is unavailable, intravenous quinine or quinidine. She should be followed
closely for progression of severe malaria, in particular neuro-logic, renal, or pulmonary complications, and if treated with quinine or quinidine should have cardiac monitoring for potential toxicities.
Petri WA: Therapy of intestinal protozoa. Trends Parasitol 2003;19:523. Pierce KK et al: Update on human infections caused by intestinal protozoa. Curr
Opin Gastroenterol 2009;25:12. Pritt BS, Clark DG: Amebiasis. Mayo Clin Proc 2008;83:1154. Rossignol JF: Cryptosporidium and Giardia : Treatment options and prospects for
new drugs. Exp Parasitol 2010;124:45.
Other Protozoal Infections
Amato VS et al: Treatment of mucosal leishmaniasis in Latin America: Systematic review. Am J Trop Med Hyg 2007;77:266.
Aronson NE et al: A randomized controlled trial of local heat therapy versus intra-venous sodium stibogluconate for the treatment of cutaneous Leishmania major infection. PLoS Negl Trop Dis 2010;4:e628.
Bhattacharya SK et al: Phase 4 trial of miltefosine for the treatment of Indian vis-ceral leishmaniasis. J Infect Dis 2007;196:591.
Bisser S et al: Equivalence trial of melarsoprol and nifurtimox monotherapy and combination therapy for the treatment of second-stage Trypanosoma brucei gambiense sleeping sickness. J Infect Dis 2007;195:322.
Brun R et al: Human African trypanosomiasis. Lancet 2010;375:148. Chappuis F et al: Visceral leishmaniasis: What are the needs for diagnosis, treat-
ment and control? Nat Rev Microbiol 2007;5:873. Croft SL, Barrett MP, Urbina JA: Chemotherapy of trypanosomiases and leishma-
niasis. Trends Parasitol 2005;21:508. Hailu A et al: Geographical variation in the response of visceral leishmaniasis to
paromomycin in East Africa: A multicentre, open-label, randomized trial. PLoS Negl Trop Dis 2010;4:e709.
Jackson Y et al: Tolerance and safety of nifurtimox in patients with chronic Chagas disease. Clin Infect Dis 2010;51:e69.
Lescure FX et al: Chagas disease: Changes in knowledge and management. Lancet Infect Dis 2010;10:556.
Murray HW et al: Advances in leishmaniasis. Lancet 2005;366:1561. Priotto G et al: Nifurtimox-eflornithine combination therapy for second-stage
African Trypanosoma brucei gambiense trypanosomiasis: A multicentre, ran-domised, phase III, non-inferiority trial. Lancet 2009;374:56.
Rassi A et al: Chagas disease. Lancet. 2010;375:1388. Reithinger R et al: Cutaneous leishmaniasis. Lancet Infect Dis 2007;7:581. Schmid C et al: Effectiveness of a 10-day melarsoprol schedule for the treatment
of late-stage human African trypanosomiasis: Confirmation from a multina-tional study (IMPAMEL II). J Infect Dis 2005;191:1922.
Soto J et al: Efficacy of miltefosine for Bolivian cutaneous leishmaniasis. Am J Trop Med Hyg 2008;78:210.
Sundar S et al: Injectable paromomycin for visceral leishmaniasis in India. N Engl J Med 2007;356:2571.
Sundar S et al: New treatment approach in Indian visceral leishmaniasis: Single-dose liposomal amphotericin B followed by short-course oral miltefosine. Clin Infect Dis 2008;47:1000.
Sundar S et al: Oral miltefosine for Indian visceral leishmaniasis. N Engl J Med 2002;347:1739.
Tuon FF et al: Treatment of New World cutaneous leishmaniasis—a systematic review with a meta-analysis. Int J Dermatol 2008;47:109.
van Griensven J, et al: Combination therapy for visceral leishmaniasis. Lancet Infect Dis 2010;10:184.
Vélez I et al: Efficacy of miltefosine for the treatment of American cutaneous leishmaniasis. Am J Trop Med Hyg 2010;83:351.
Wortmann G et al: Liposomal amphotericin B for treatment of cutaneous leishma-niasis. Am J Trop Med Hyg 2010;83:1028.
052-Katzung_Ch052_p915-936.indd 936 9/23/11 4:27:51 PM

937
Clinical Pharmacology of the Antihelminthic Drugs Philip J. Rosenthal, MD
ago, the patient emigrated to the United States from a rural area of Peru where his family trades in sheepskins. His father and sister have undergone resection of abdominal masses, but details of their diagnoses are unavailable. What is your differential diagnosis? What are your diagnostic and thera-peutic plans?
C A S E S T U D Y
A 29-year-old Peruvian man presents with the incidental finding of a 10 by 8 by 8 cm liver cyst on an abdominal com-puted tomography (CT) scan. The patient had noted 2 days of abdominal pain and fever, and his clinical evaluation and CT scan were consistent with appendicitis. His clinical find-ings resolved after laparoscopic appendectomy. Ten years
C H A P T E R
■ CHEMOTHERAPY OF HELMINTHIC INFECTIONS
Helminths (worms) are multicellular organisms that infect very large numbers of humans and cause a broad range of diseases. Over 1 billion people are infected with intestinal nematodes, and many millions are infected with filarial nematodes, flukes, and tape-worms. They are an even greater problem in domestic animals. Many drugs, which are directed against a number of different tar-gets, are available to treat helminthic infections. In many cases, especially in the developing world, the goal is control of infection, with elimination of most parasites controlling disease symptoms and decreasing the transmission of infection. In other cases, com-plete elimination of parasites is the goal of therapy, although this goal can be challenging with certain helminthic infections, because of both limited efficacy of drugs and frequent reinfection after therapy in endemic areas.
Table 53–1 lists the major helminthic infections and provides a guide to the drug of choice and alternative drugs for each infec-tion. In the text that follows, these drugs are arranged alphabeti-cally. In general, parasites should be identified before treatment is started.
53
ALBENDAZOLE
Albendazole, a broad-spectrum oral antihelminthic, is the drug of choice and is approved in the USA for treatment of hydatid disease and cysticercosis. It is also used in the treatment of pinworm and hookworm infections, ascariasis, trichuriasis, and strongyloidiasis.
Basic Pharmacology Albendazole is a benzimidazole carbamate. After oral administra-tion, it is erratically absorbed (increased with a fatty meal) and then rapidly undergoes first-pass metabolism in the liver to the active metabolite albendazole sulfoxide. It reaches variable maxi-mum plasma concentrations about 3 hours after a 400-mg oral dose, and its plasma half-life is 8–12 hours. The sulfoxide is mostly protein-bound, distributes well to tissues, and enters bile, cerebrospinal fluid, and hydatid cysts. Albendazole metabolites are excreted in the urine.
Benzimidazoles are thought to act against nematodes by inhibiting microtubule synthesis. Albendazole also has larvicidal effects in hydatid disease, cysticercosis, ascariasis, and hookworm infection and ovicidal effects in ascariasis, ancylostomiasis, and trichuriasis.
053-Katzung_Ch053_p937-948.indd 937 9/21/11 12:11:27 PM

938 SECTION VIII Chemotherapeutic Drugs
Clinical Uses Albendazole is administered on an empty stomach when used against intraluminal parasites but with a fatty meal when used against tissue parasites.
A. Ascariasis, Trichuriasis, and Hookworm and Pinworm Infections
For adults and children older than 2 years of age with ascariasis and hookworm infections, the treatment is a single dose of 400 mg
TABLE 53–1 Drugs for the treatment of helminthic infections. 1
Infecting Organism Drug of Choice Alternative Drugs
Roundworms (nematodes)
Ascaris lumbricoides (roundworm) Albendazole or pyrantel pamoate or mebendazole
Ivermectin, piperazine
Trichuris trichiura (whipworm) Mebendazole or albendazole Ivermectin
Necator americanus (hookworm); Ancylostoma duodenale (hookworm)
Albendazole or mebendazole or pyrantel pamoate
Strongyloides stercoralis (threadworm) Ivermectin Albendazole or thiabendazole
Enterobius vermicularis (pinworm) Mebendazole or pyrantel pamoate Albendazole
Trichinella spiralis (trichinosis) Mebendazole or albendazole; add corticosteroids for severe infection
Trichostrongylus species Pyrantel pamoate or mebendazole Albendazole
Cutaneous larva migrans (creeping eruption) Albendazole or ivermectin Thiabendazole (topical)
Visceral larva migrans Albendazole Mebendazole
Angiostrongylus cantonensis Albendazole or mebendazole
Wuchereria bancrofti (filariasis); Brugia malayi (filariasis); tropical eosinophilia; Loa loa (loiasis)
Diethylcarbamazine Ivermectin
Onchocerca volvulus (onchocerciasis) Ivermectin
Dracunculus medinensis (guinea worm) Metronidazole Thiabendazole or mebendazole
Capillaria philippinensis (intestinal capillariasis) Albendazole Mebendazole
Flukes (trematodes)
Schistosoma haematobium (bilharziasis) Praziquantel Metrifonate
Schistosoma mansoni Praziquantel Oxamniquine
Schistosoma japonicum Praziquantel
Clonorchis sinensis (liver fluke); Opisthorchis species Praziquantel Albendazole
Paragonimus westermani (lung fluke) Praziquantel Bithionol
Fasciola hepatica (sheep liver fluke) Bithionol or triclabendazole
Fasciolopsis buski (large intestinal fluke) Praziquantel or niclosamide
Heterophyes heterophyes; Metagonimus yokogawai (small intestinal flukes)
Praziquantel or niclosamide
Tapeworms (cestodes)
Taenia saginata (beef tapeworm) Praziquantel or niclosamide Mebendazole
Diphyllobothrium latum (fish tapeworm) Praziquantel or niclosamide
Taenia solium (pork tapeworm) Praziquantel or niclosamide
Cysticercosis (pork tapeworm larval stage) Albendazole Praziquantel
Hymenolepis nana (dwarf tapeworm) Praziquantel Niclosamide, nitazoxanide
Echinococcus granulosus (hydatid disease); Echinococcus multilocularis
Albendazole
1Additional information may be obtained from the Parasitic Disease Drug Service, Parasitic Diseases Branch, Centers for Disease Control and Prevention, Atlanta, Georgia, 30333. Telephone: (404) 639-3670. Some of the drugs listed are not generally available in the USA.
053-Katzung_Ch053_p937-948.indd 938 9/21/11 12:11:27 PM

CHAPTER 53 Clinical Pharmacology of the Antihelminthic Drugs 939
orally (repeated for 2–3 days for heavy ascaris infections and in 2 weeks for pinworm infections). These treatments typically achieve good cure rates and marked reduction in egg counts in those not cured. For trichuriasis, three daily 400-mg oral doses of albendazole are now recommended. A recent meta-analysis showed albendazole to be superior to mebendazole or pyrantel pamoate for treatment of hookworm infection; other studies showed that three doses of mebendazole and albendazole increased stool clearance of eggs compared with single treatments, with albendazole superior to mebendazole. Cure rates for trichuriasis with single-dose albenda-zole or mebendazole were less than 30%, which suggests that the three-dose regimen just noted, or other drugs (eg, ivermectin), might be superior.
B. Hydatid Disease Albendazole is the treatment of choice for medical therapy and is a useful adjunct to surgical removal or aspiration of cysts. It is more active against Echinococcus granulosus than against E mul-tilocularis . Dosing is 400 mg twice daily with meals for 1 month or longer. Daily therapy for up to 6 months has been well toler-ated. One reported therapeutic strategy is to treat with albenda-zole and praziquantel, to assess response after 1 month or more, and, depending on the response, to then manage the patient with continued chemotherapy or combined surgical and drug therapy.
C. Neurocysticercosis Indications for medical therapy for neurocysticercosis are contro-versial, since antihelminthic therapy is not clearly superior to therapy with corticosteroids alone and may exacerbate neurologic disease. Therapy is probably most appropriate for symptomatic parenchymal or intraventricular cysts. Corticosteroids are usually given with the antihelminthic drug to decrease inflammation caused by dying organisms. Albendazole is now generally consid-ered the drug of choice over praziquantel because of its shorter course, lower cost, improved penetration into the subarachnoid space, and increased drug levels (as opposed to decreased levels of praziquantel) when administered with corticosteroids. Albendazole is given in a dosage of 400 mg twice daily for up to 21 days.
D. Other Infections Albendazole is the drug of choice in the treatment of cutaneous larva migrans (400 mg daily for 3 days), visceral larva migrans (400 mg twice daily for 5 days), intestinal capillariasis (400 mg daily for 10 days), microsporidial infections (400 mg twice daily for 2 weeks or longer), and gnathostomiasis (400 mg twice daily for 3 weeks). It also has activity against trichinosis (400 mg twice daily for 1–2 weeks) and clonorchiasis (400 mg twice daily for 1 week). There have been reports of some effectiveness in treat-ment of opisthorchiasis, toxocariasis, and loiasis, and conflicting reports of effectiveness in giardiasis and taeniasis. Albendazole is included in programs to control lymphatic filariasis, but it appears to be less active than diethylcarbamazine or ivermectin for this purpose. Albendazole has been recommended as empiric therapy to treat those who return from the tropics with persistent unex-plained eosinophilia.
Adverse Reactions, Contraindications, & Cautions When used for 1–3 days, albendazole is nearly free of significant adverse effects. Mild and transient epigastric distress, diarrhea, headache, nausea, dizziness, lassitude, and insomnia can occur. In long-term use for hydatid disease, albendazole is well tolerated, but it can cause abdominal distress, headaches, fever, fatigue, alo-pecia, increases in liver enzymes, and pancytopenia.
Blood counts and liver function studies should be monitored during long-term therapy. The drug should not be given to patients with known hypersensitivity to other benzimidazole drugs or to those with cirrhosis. The safety of albendazole in pregnancy and in children younger than 2 years of age has not been established.
BITHIONOL
Bithionol is an alternative to triclabendazole for the treatment of fascioliasis (sheep liver fluke). Bithionol is also an alternative drug in the treatment of pulmonary paragonimiasis.
Basic Pharmacology After ingestion, bithionol reaches peak blood levels in 4–8 hours. Excretion appears to be mainly via the kidney.
Clinical Uses For treatment of paragonimiasis and fascioliasis, the dosage of bithionol is 30–50 mg/kg in two or three divided doses, given orally after meals on alternate days for 10–15 doses. For pulmo-nary paragonimiasis, cure rates are over 90%. For cerebral parago-nimiasis, repeat courses of therapy may be necessary.
Adverse Reactions, Contraindications, & Cautions Adverse effects, which occur in up to 40% of patients, are gener-ally mild and transient, but occasionally their severity requires interruption of therapy. These problems include diarrhea, abdom-inal cramps, anorexia, nausea, vomiting, dizziness, and headache. Skin rashes may occur after a week or more of therapy, suggesting a reaction to antigens released from dying worms.
Bithionol should be used with caution in children younger than 8 years of age because there has been limited experience in this age group.
DIETHYLCARBAMAZINE CITRATE
Diethylcarbamazine is a drug of choice in the treatment of filari-asis, loiasis, and tropical eosinophilia. It has been replaced by ivermectin for the treatment of onchocerciasis.
Basic Pharmacology Diethylcarbamazine, a synthetic piperazine derivative, is marketed as a citrate salt. It is rapidly absorbed from the gastrointestinal
053-Katzung_Ch053_p937-948.indd 939 9/21/11 12:11:28 PM

940 SECTION VIII Chemotherapeutic Drugs
tract; after a 0.5 mg/kg dose, peak plasma levels are reached within 1–2 hours. The plasma half-life is 2–3 hours in the presence of acidic urine but about 10 hours if the urine is alkaline, a Henderson-Hasselbalch trapping effect (see Chapter 1 ). The drug rapidly equilibrates with all tissues except fat. It is excreted, prin-cipally in the urine, as unchanged drug and the N-oxide metabo-lite. Dosage may have to be reduced in patients with persistent urinary alkalosis or renal impairment.
Diethylcarbamazine immobilizes microfilariae and alters their surface structure, displacing them from tissues and making them more susceptible to destruction by host defense mechanisms. The mode of action against adult worms is unknown.
Clinical Uses The drug should be taken after meals.
A. Wuchereria bancrofti, Brugia malayi, Brugia timori,
and Loa loa
Diethylcarbamazine is the drug of choice for treatment of infections with these parasites because of its efficacy and lack of serious toxicity. Microfilariae of all species are rapidly killed; adult parasites are killed more slowly, often requiring several courses of treatment. The drug is highly effective against adult L loa . The extent to which W bancrofti and B malayi adults are killed is not known, but after appropriate therapy microfilariae do not reappear in the majority of patients.
These infections are treated for 2 or (for L loa ) 3 weeks, with initial low doses to reduce the incidence of allergic reactions to dying microfilariae. This regimen is 50 mg (1 mg/kg in children) on day 1, three 50 mg doses on day 2, three 100 mg doses (2 mg/kg in children) on day 3, and then 2 mg/kg three times daily to complete the 2–3 week course.
Antihistamines may be given for the first few days of therapy to limit allergic reactions, and corticosteroids should be started and doses of diethylcarbamazine lowered or interrupted if severe reactions occur. Cures may require several courses of treatment. For patients with high L loa worm burdens (more than 2500 cir-culating parasites/mL), strategies to decrease risks of severe toxicity include apheresis, if available, to remove microfilariae before treat-ment with diethylcarbamazine or therapy with albendazole, which is slower acting and better tolerated, before therapy with diethyl-carbamazine or ivermectin.
Diethylcarbamazine may also be used for chemoprophylaxis (300 mg weekly or 300 mg on 3 successive days each month for loiasis; 50 mg monthly for bancroftian and Malayan filariasis).
B. Other Uses For tropical eosinophilia, diethylcarbamazine is given orally at a dosage of 2 mg/kg three times daily for 7 days. Diethylcarbamazine is effective in Mansonella streptocerca infections, since it kills both adults and microfilariae. Limited information suggests that the drug is not effective, however, against adult M ozzardi or M per-stans and that it has limited activity against microfilariae of these parasites. An important application of diethylcarbamazine has been mass treatment to reduce the prevalence of W bancrofti infec-tion, generally in combination with ivermectin or albendazole.
This strategy has led to excellent progress in disease control in a number of countries.
Adverse Reactions, Contraindications, & Cautions Reactions to diethylcarbamazine, which are generally mild and transient, include headache, malaise, anorexia, weakness, nausea, vomiting, and dizziness. Adverse effects also occur as a result of the release of proteins from dying microfilariae or adult worms. Reactions are particularly severe with onchocerciasis, but diethyl-carbamazine is no longer commonly used for this infection, because ivermectin is equally efficacious and less toxic. Reactions to dying microfilariae are usually mild in W bancrofti , more intense in B malayi , and occasionally severe in L loa infections. Reactions include fever, malaise, papular rash, headache, gastroin-testinal symptoms, cough, chest pain, and muscle or joint pain. Leukocytosis is common. Eosinophilia may increase with treat-ment. Proteinuria may also occur. Symptoms are most likely to occur in patients with heavy loads of microfilariae. Retinal hemor-rhages and, rarely, encephalopathy have been described.
Between the third and twelfth days of treatment, local reactions may occur in the vicinity of dying adult or immature worms. These include lymphangitis with localized swellings in W bancrofti and B malayi , small wheals in the skin in L loa , and flat papules in M streptocerca infections. Patients with attacks of lymphangitis due to W bancrofti or B malayi should be treated during a quiescent period between attacks.
Caution is advised when using diethylcarbamazine in patients with hypertension or renal disease.
DOXYCYCLINE
This tetracycline antibiotic is described in more detail in Chapter 44 . Doxycycline has recently been shown to have significant macro-filaricidal activity against W bancrofti , suggesting better activity than any other available drug against adult worms. Activity is also seen against onchocerciasis. Doxycycline acts indirectly, by killing Wolbachia, an intracellular bacterial symbiont of filarial parasites. It may prove to be an important drug for filariasis, both for treat-ment of active disease and in mass chemotherapy campaigns.
IVERMECTIN
Ivermectin is the drug of choice in strongyloidiasis and onchocer-ciasis. It is also an alternative drug for a number of other helmin-thic infections.
Basic Pharmacology Ivermectin, a semisynthetic macrocyclic lactone, is a mixture of avermectin B 1a and B 1b . It is derived from the soil actinomycete Streptomyces avermitilis .
Ivermectin is used only orally in humans. The drug is rapidly absorbed, reaching maximum plasma concentrations 4 hours after a
053-Katzung_Ch053_p937-948.indd 940 9/21/11 12:11:28 PM

CHAPTER 53 Clinical Pharmacology of the Antihelminthic Drugs 941
12 mg dose. The drug has a wide tissue distribution and a volume of distribution of about 50 L. Its half-life is about 16 hours. Excretion of the drug and its metabolites is almost exclusively in the feces.
Ivermectin appears to paralyze nematodes and arthropods by intensifying γ-aminobutyric acid (GABA)–mediated transmission of signals in peripheral nerves. In onchocerciasis, ivermectin is microfilaricidal. It does not effectively kill adult worms but blocks the release of microfilariae for some months after therapy. After a single standard dose, microfilariae in the skin diminish rapidly within 2–3 days, remain low for months, and then gradually increase; microfilariae in the anterior chamber of the eye decrease slowly over months, eventually clear, and then gradually return. With repeated doses of ivermectin, the drug appears to have a low-level macrofilaricidal action and to permanently reduce microfi-larial production.
Clinical Uses A. Onchocerciasis Treatment is with a single oral dose of ivermectin, 150 mcg/kg, with water on an empty stomach. Doses are repeated; regimens vary from monthly to less frequent (every 6–12 months) dosing schedules. After acute therapy, treatment is repeated at 12-month intervals until the adult worms die, which may take 10 years or longer. With the first treatment only, patients with microfilariae in the cornea or anterior chamber may be treated with corticoster-oids to avoid inflammatory eye reactions.
Ivermectin also now plays a key role in onchocerciasis control. Annual mass treatments have led to major reductions in disease transmission. However, evidence of diminished responsiveness after mass administration of ivermectin has raised concern regard-ing selection of drug-resistant parasites.
B. Strongyloidiasis Treatment consists of two daily doses of 200 mcg/kg. In immuno-suppressed patients with disseminated infection, repeated treat-ment is often needed, and cure may not be possible. In this case, suppressive therapy—ie, once monthly—may be helpful.
C. Other Parasites Ivermectin reduces microfilariae in B malayi and M ozzardi infec-tions but not in M perstans infections. It has been used with dieth-ylcarbamazine and albendazole for the control of W bancrofti , but it does not kill adult worms. In loiasis, although the drug reduces microfilaria concentrations, it can occasionally induce severe reac-tions and appears to be more dangerous in this regard than dieth-ylcarbamazine. Ivermectin is also effective in controlling scabies, lice, and cutaneous larva migrans and in eliminating a large pro-portion of ascarid worms.
Adverse Reactions, Contraindications, & Cautions In strongyloidiasis treatment, infrequent adverse effects include fatigue, dizziness, nausea, vomiting, abdominal pain, and rashes. In onchocerciasis treatment, adverse effects are principally from
the killing of microfilariae and can include fever, headache, dizzi-ness, somnolence, weakness, rash, increased pruritus, diarrhea, joint and muscle pains, hypotension, tachycardia, lymphadenitis, lymphangitis, and peripheral edema. This reaction starts on the first day and peaks on the second day after treatment. This reac-tion occurs in 5–30% of persons and is generally mild, but it may be more frequent and more severe in individuals who are not long-term residents of onchocerciasis-endemic areas. A more intense reaction occurs in 1–3% of persons and a severe reaction in 0.1%, including high fever, hypotension, and bronchospasm. Corticosteroids are indicated in these cases, at times for several days. Toxicity diminishes with repeated dosing. Swellings and abscesses occasionally occur at 1–3 weeks, presumably at sites of adult worms.
Some patients develop corneal opacities and other eye lesions several days after treatment. These are rarely severe and generally resolve without corticosteroid treatment.
It is best to avoid concomitant use of ivermectin with other drugs that enhance GABA activity, eg, barbiturates, benzodiaz-epines, and valproic acid. Ivermectin should not be used during pregnancy. Safety in children younger than 5 years has not been established.
MEBENDAZOLE
Mebendazole is a synthetic benzimidazole that has a wide spec-trum of antihelminthic activity and a low incidence of adverse effects.
Basic Pharmacology Less than 10% of orally administered mebendazole is absorbed. The absorbed drug is protein-bound (> 90%), is rapidly converted to inactive metabolites (primarily during its first pass in the liver), and has a half-life of 2–6 hours. It is excreted mostly in the urine, principally as decarboxylated derivatives. In addition, a portion of absorbed drug and its derivatives are excreted in the bile. Absorption is increased if the drug is ingested with a fatty meal.
Mebendazole probably acts by inhibiting microtubule synthe-sis; the parent drug appears to be the active form. Efficacy of the drug varies with gastrointestinal transit time, with intensity of infection, and perhaps with the strain of parasite. The drug kills hookworm, ascaris, and trichuris eggs.
Clinical Uses Mebendazole is indicated for use in ascariasis, trichuriasis, hook-worm and pinworm infections, and certain other helminthic infections. It can be taken before or after meals; the tablets should be chewed before swallowing. For pinworm infection, the dose is 100 mg once, repeated at 2 weeks. For ascariasis, trichuriasis, hookworm, and trichostrongylus infections, a dosage of 100 mg twice daily for 3 days is used for adults and for children older than 2 years of age. Cure rates are good for pinworm infections and ascariasis, but have been disappointing in recent studies of trichu-riasis. Cure rates are also lower for hookworm infections, but a
053-Katzung_Ch053_p937-948.indd 941 9/21/11 12:11:28 PM

942 SECTION VIII Chemotherapeutic Drugs
marked reduction in the worm burden occurs in those not cured. For intestinal capillariasis, mebendazole is used at a dosage of 200 mg twice daily for 21 or more days. In trichinosis, limited reports suggest efficacy against adult worms in the intestinal tract and tis-sue larvae. Treatment is three times daily, with fatty foods, at 200–400 mg per dose for 3 days and then 400–500 mg per dose for 10 days; corticosteroids should be coadministered for severe infections.
Adverse Reactions, Contraindications, & Cautions Short-term mebendazole therapy for intestinal nematodes is nearly free of adverse effects. Mild nausea, vomiting, diarrhea, and abdom-inal pain have been reported infrequently. Rare side effects, usually with high-dose therapy, are hypersensitivity reactions (rash, urti-caria), agranulocytosis, alopecia, and elevation of liver enzymes.
Mebendazole is teratogenic in animals and therefore contrain-dicated in pregnancy. It should be used with caution in children younger than 2 years of age because of limited experience and rare reports of convulsions in this age group. Plasma levels may be decreased by concomitant use of carbamazepine or phenytoin and increased by cimetidine. Mebendazole should be used with cau-tion in patients with cirrhosis.
METRIFONATE (TRICHLORFON)
Metrifonate is a safe, low-cost alternative drug for the treatment of Schistosoma haematobium infections. It is not active against S mansoni or S japonicum . It is not available in the USA.
Basic Pharmacology Metrifonate, an organophosphate compound, is rapidly absorbed after oral administration. After the standard oral dose, peak blood levels are reached in 1–2 hours; the half-life is about 1.5 hours. Clearance appears to be through nonenzymatic transformation to dichlorvos, its active metabolite. Metrifonate and dichlorvos are well distributed to the tissues and are completely eliminated in 24–48 hours.
The mode of action is thought to be related to cholinesterase inhibition. This inhibition temporarily paralyzes the adult worms, resulting in their shift from the bladder venous plexus to small arterioles of the lungs, where they are trapped, encased by the immune system, and die. The drug is not effective against S haematobium eggs; live eggs continue to pass in the urine for several months after all adult worms have been killed.
Clinical Uses In the treatment of S haematobium , an oral dose of 7.5–10 mg/kg is given three times at 14-day intervals. Cure rates on this schedule are 44–93%, with marked reductions in egg counts in those not cured. Metrifonate was also effective as a prophylactic agent when given monthly to children in a highly endemic area, and it has been used in mass treatment programs. In mixed infections with S haematobium and S mansoni , metrifonate has been successfully combined with oxamniquine.
Adverse Reactions, Contraindications, & Cautions Some studies note mild and transient cholinergic symptoms, including nausea and vomiting, diarrhea, abdominal pain, bron-chospasm, headache, sweating, fatigue, weakness, dizziness, and vertigo. These symptoms may begin within 30 minutes and persist up to 12 hours.
Metrifonate should not be used after recent exposure to insec-ticides or drugs that might potentiate cholinesterase inhibition. Metrifonate is contraindicated in pregnancy.
NICLOSAMIDE
Niclosamide is a second-line drug for the treatment of most tape-worm infections, but it is not available in the USA.
Basic Pharmacology Niclosamide is a salicylamide derivative. It appears to be mini-mally absorbed from the gastrointestinal tract—neither the drug nor its metabolites have been recovered from the blood or urine.
Adult worms (but not ova) are rapidly killed, presumably due to inhibition of oxidative phosphorylation or stimulation of ATPase activity.
Clinical Uses The adult dose of niclosamide is 2 g once, given in the morning on an empty stomach. The tablets must be chewed thoroughly and then swallowed with water.
A. Taenia saginata (Beef Tapeworm), T solium (Pork Tapeworm), and Diphyllobothrium latum (Fish Tapeworm)
A single 2 g dose of niclosamide results in cure rates of over 85% for D latum and about 95% for T saginata . It is probably equally effective against T solium . Cysticercosis can theoretically occur after treatment of T solium infections, because viable ova are released into the gut lumen after digestion of segments, but no such cases have been reported.
B. Other Tapeworms Most patients treated with niclosamide for Hymenolepsis diminuta and Dipylidium caninum infections are cured with a 7-day course of treatment; a few require a second course. Praziquantel is supe-rior for Hymenolepis nana (dwarf tapeworm) infection. Niclosamide is not effective against cysticercosis or hydatid disease.
C. Intestinal Fluke Infections Niclosamide can be used as an alternative drug in the treatment of Fasciolopsis buski , Heterophyes heterophyes , and Metagonimus yokogawai infections. The standard dose is given every other day for three doses.
053-Katzung_Ch053_p937-948.indd 942 9/21/11 12:11:28 PM

CHAPTER 53 Clinical Pharmacology of the Antihelminthic Drugs 943
Adverse Reactions, Contraindications, & Cautions Infrequent, mild, and transitory adverse events include nausea, vomiting, diarrhea, and abdominal discomfort. The consumption of alcohol should be avoided on the day of treatment and for 1 day afterward. The safety of the drug has not been established in preg-nancy or for children younger than 2 years of age.
OXAMNIQUINE
Oxamniquine is an alternative to praziquantel for the treatment of S mansoni infections. It has also been used extensively for mass treatment. It is not effective against S haematobium or S japoni-cum . It is not available in the USA.
Basic Pharmacology Oxamniquine, a semisynthetic tetrahydroquinoline, is readily absorbed orally; it should be taken with food. Its plasma half-life is about 2.5 hours. The drug is extensively metabolized to inactive metabolites and excreted in the urine—up to 75% in the first 24 hours. Intersubject variations in serum concentration have been noted, which may explain some treatment failures.
Oxamniquine is active against both mature and immature stages of S mansoni but does not appear to be cercaricidal. The mechanism of action is unknown. Contraction and paralysis of the worms results in detachment from terminal venules in the mesentery and transit to the liver, where many die; surviving females return to the mesenteric vessels but cease to lay eggs. Strains of S mansoni in dif-ferent parts of the world vary in susceptibility. Oxamniquine has been effective in instances of praziquantel resistance.
Clinical Uses Oxamniquine is safe and effective in all stages of S mansoni dis-ease, including advanced hepatosplenomegaly. In the acute (Katayama) syndrome, treatment results in disappearance of acute symptoms and clearance of the infection. The drug is generally less effective in children, who require higher doses than adults. It is better tolerated with food.
Optimal dosage schedules vary for different regions of the world. In the western hemisphere and western Africa, the adult oxamniquine dosage is 12–15 mg/kg given once. In northern and southern Africa, standard schedules are 15 mg/kg twice daily for 2 days. In eastern Africa and the Arabian peninsula, standard dos-age is 15–20 mg/kg twice in 1 day. Cure rates are 70–95%, with marked reduction in egg excretion in those not cured. In mixed schistosome infections, oxamniquine has been successfully used in combination with metrifonate.
Adverse Reactions, Contraindications, & Cautions Mild symptoms, starting about 3 hours after a dose and lasting for several hours, occur in more than one third of patients. Central
nervous system symptoms (dizziness, headache, drowsiness) are most common; nausea and vomiting, diarrhea, colic, pruritus, and urticaria also occur. Infrequent adverse effects are low-grade fever, an orange to red discoloration of the urine, proteinuria, micro-scopic hematuria, and a transient decrease in leukocytes. Seizures have been reported rarely.
Since the drug makes many patients dizzy or drowsy, it should be used with caution in patients whose work or activity requires mental alertness (eg, no driving for 24 hours). It should be used with caution in those with a history of epilepsy. Oxamniquine is contraindicated in pregnancy.
PIPERAZINE
Piperazine is an alternative for the treatment of ascariasis, with cure rates over 90% when taken for 2 days, but it is not recom-mended for other helminth infections. Piperazine is available as the hexahydrate and as a variety of salts. It is readily absorbed, and maximum plasma levels are reached in 2–4 hours. Most of the drug is excreted unchanged in the urine in 2–6 hours, and excre-tion is complete within 24 hours.
Piperazine causes paralysis of ascaris by blocking acetylcholine at the myoneural junction; unable to maintain their position in the host, live worms are expelled by normal peristalsis.
For ascariasis, the dosage of piperazine (as the hexahydrate) is 75 mg/kg (maximum dose, 3.5 g) orally once daily for 2 days. For heavy infections, treatment should be continued for 3–4 days or repeated after 1 week.
Occasional mild adverse effects include nausea, vomiting, diar-rhea, abdominal pain, dizziness, and headache. Neurotoxicity and allergic reactions are rare. Piperazine compounds should not be given to women during pregnancy, to patients with impaired renal or hepatic function, or to those with a history of epilepsy or chronic neurologic disease.
PRAZIQUANTEL
Praziquantel is effective in the treatment of schistosome infections of all species and most other trematode and cestode infections, including cysticercosis. The drug’s safety and effectiveness as a single oral dose have also made it useful in mass treatment of sev-eral infections.
Basic Pharmacology Praziquantel is a synthetic isoquinoline-pyrazine derivative. It is rapidly absorbed, with a bioavailability of about 80% after oral administration. Peak serum concentrations are reached 1–3 hours after a therapeutic dose. Cerebrospinal fluid concentrations of prazi-quantel reach 14–20% of the drug’s plasma concentration. About 80% of the drug is bound to plasma proteins. Most of the drug is rapidly metabolized to inactive mono- and polyhydroxylated prod-ucts after a first pass in the liver. The half-life is 0.8–1.5 hours. Excretion is mainly via the kidneys (60–80%) and bile (15–35%). Plasma concentrations of praziquantel increase when the drug is
053-Katzung_Ch053_p937-948.indd 943 9/21/11 12:11:28 PM

944 SECTION VIII Chemotherapeutic Drugs
taken with a high-carbohydrate meal or with cimetidine; bioavail-ability is markedly reduced with some antiepileptics (phenytoin, carbamazepine) or with corticosteroids.
Praziquantel appears to increase the permeability of trematode and cestode cell membranes to calcium, resulting in paralysis, dislodgement, and death. In schistosome infections of experimen-tal animals, praziquantel is effective against adult worms and immature stages, and it has a prophylactic effect against cercarial infection.
Clinical Uses Praziquantel tablets are taken with liquid after a meal; they should be swallowed without chewing because their bitter taste can induce retching and vomiting.
A. Schistosomiasis Praziquantel is the drug of choice for all forms of schistosomiasis. The dosage is 20 mg/kg per dose for two ( S mansoni and S haema-tobium ) or three ( S japonicum and S mekongi ) doses at intervals of 4–6 hours. High cure rates (75–95%) are achieved when patients are evaluated at 3–6 months; there is marked reduction in egg counts in those not cured. The drug is effective in adults and children and is generally well tolerated by patients in the hepatos-plenic stage of advanced disease. There is no standard regimen for acute schistosomiasis (Katayama syndrome), but standard doses as described above, often with corticosteroids to limit inflammation from the acute immune response and dying worms, are recom-mended. Increasing evidence indicates rare S mansoni drug resis-tance, which may be countered with extended courses of therapy (eg, 3–6 days at standard dosing) or treatment with oxamniquine. Effectiveness of praziquantel for chemoprophylaxis has not been established.
B. Clonorchiasis, Opisthorchiasis, and Paragonimiasis Standard dosing is 25 mg/kg three times daily for 2 days for each of these fluke infections.
C. Taeniasis and Diphyllobothriasis A single dose of praziquantel, 5–10 mg/kg, results in nearly 100% cure rates for T saginata , T solium , and D latum infections. Because praziquantel does not kill eggs, it is theoretically possible that larvae of T solium released from eggs in the large bowel could penetrate the intestinal wall and give rise to cysticercosis, but this hazard is probably minimal.
D. Neurocysticercosis Albendazole is now the preferred drug, but when it is not appropri-ate or available, praziquantel has similar efficacy. Indications for praziquantel are similar to those for albendazole. The praziquantel dosage is 100 mg/kg/d in three divided doses for 1 day, then 50 mg/kg/d to complete a 2- to 4-week course. Clinical responses to ther-apy vary from dramatic improvements of seizures and other neuro-logic findings to no response and even progression of the disease. Praziquantel—but not albendazole—has diminished bioavailability
when taken concurrently with a corticosteroid. Recommendations on use of both antihelminthics and corticosteroids in neurocysticer-cosis vary.
E. Hymenolepis nana Praziquantel is the drug of choice for H nana infections and the first drug to be highly effective. A single dose of 25 mg/kg is taken initially and repeated in 1 week.
F. Hydatid Disease In hydatid disease, praziquantel kills protoscoleces but does not affect the germinal membrane. Praziquantel is being evaluated as an adjunct with albendazole pre- and postsurgery. In addition to its direct action, praziquantel enhances the plasma concentration of albendazole.
G. Other Parasites Limited trials at a dosage of 25 mg/kg three times daily for 1–2 days indicate effectiveness of praziquantel against fasciolopsia-sis, metagonimiasis, and other forms of heterophyiasis. Praziquantel was not effective for fascioliasis, however, even at dosages as high as 25 mg/kg three times daily for 3–7 days.
Adverse Reactions, Contraindications, & Cautions Mild and transient adverse effects are common. They begin within several hours after ingestion of praziquantel and may persist for about 1 day. Most common are headache, dizziness, drowsiness, and lassitude; others include nausea, vomiting, abdominal pain, loose stools, pruritus, urticaria, arthralgia, myalgia, and low-grade fever. Mild and transient elevations of liver enzymes have been reported. Several days after starting praziquantel, low-grade fever, pruritus, and skin rashes (macular and urticarial), sometimes asso-ciated with worsened eosinophilia, may occur, probably due to the release of proteins from dying worms rather than direct drug tox-icity. The intensity and frequency of adverse effects increase with dosage such that they occur in up to 50% of patients who receive 25 mg/kg three times in 1 day.
In neurocysticercosis, neurologic abnormalities may be exac-erbated by inflammatory reactions around dying parasites. Common findings in patients who do not receive corticoster-oids, usually presenting during or shortly after therapy, are head-ache, meningismus, nausea, vomiting, mental changes, and seizures (often accompanied by increased cerebrospinal fluid pleocytosis). More serious findings, including arachnoiditis, hyperthermia, and intracranial hypertension, may also occur. Corticosteroids are commonly used with praziquantel in the treatment of neurocysticercosis to decrease the inflammatory reaction, but this is controversial and complicated by knowledge that corticosteroids decrease the plasma level of praziquantel up to 50%. Praziquantel is contraindicated in ocular cysticercosis, because parasite destruction in the eye may cause irreparable damage. Some workers also caution against use of the drug in spinal neurocysticercosis.
053-Katzung_Ch053_p937-948.indd 944 9/21/11 12:11:28 PM

CHAPTER 53 Clinical Pharmacology of the Antihelminthic Drugs 945
The safety of praziquantel in children younger than age 4 years is not established, but no specific problems in young children have been documented. Indeed, the drug appears to be better tolerated in children than in adults. Praziquantel increased abortion rates in rats and therefore should be avoided in pregnancy if possible. Because the drug induces dizziness and drowsiness, patients should not drive during therapy and should be warned regarding activities requiring particular physical coordination or alertness.
PYRANTEL PAMOATE
Pyrantel pamoate is a broad-spectrum antihelminthic highly effec-tive for the treatment of pinworm, ascaris, and Trichostrongylus orientalis infections. It is moderately effective against both species of hookworm. It is not effective in trichuriasis or strongyloidiasis. Oxantel pamoate, an analog of pyrantel not available in the USA, has been used successfully in the treatment of trichuriasis; the two drugs have been combined for their broad-spectrum antihelmint-hic activity.
Basic Pharmacology Pyrantel pamoate is a tetrahydropyrimidine derivative. It is poorly absorbed from the gastrointestinal tract and active mainly against luminal organisms. Peak plasma levels are reached in 1–3 hours. Over half of the administered dose is recovered unchanged in the feces.
Pyrantel is effective against mature and immature forms of susceptible helminths within the intestinal tract but not against migratory stages in the tissues or against ova. The drug is a neuro-muscular blocking agent that causes release of acetylcholine and inhibition of cholinesterase; this results in paralysis of worms, which is followed by expulsion.
Clinical Uses The standard dose is 11 mg (base)/kg (maximum, 1 g), given orally once with or without food. For pinworm, the dose is repeated in 2 weeks, and cure rates are greater than 95%. The drug is available in the USA without prescription for this indication.
For ascariasis, a single dose yields cure rates of 85–100%. Treatment should be repeated if eggs are found 2 weeks after treat-ment. For hookworm infections, a single dose is effective against light infections; but for heavy infections, especially with Necator americanus , a 3-day course is necessary to reach 90% cure rates. A course of treatment can be repeated in 2 weeks.
Adverse Reactions, Contraindications, & Cautions Adverse effects are infrequent, mild, and transient. They include nausea, vomiting, diarrhea, abdominal cramps, dizziness,
drowsiness, headache, insomnia, rash, fever, and weakness. Pyrantel should be used with caution in patients with liver dys-function, because low, transient aminotransferase elevations have been noted in a small number of patients. Experience with the drug in pregnant women and children younger than 2 years of age is limited.
THIABENDAZOLE
Thiabendazole is an alternative to ivermectin or albendazole for the treatment of strongyloidiasis and cutaneous larva migrans.
Basic Pharmacology Thiabendazole is a benzimidazole compound. Although it is a chelating agent that forms stable complexes with a number of metals, including iron, it does not bind calcium.
Thiabendazole is rapidly absorbed after ingestion. With a stan-dard dose, drug concentrations in plasma peak within 1–2 hours; the half-life is 1.2 hours. The drug is almost completely metabo-lized in the liver to the 5-hydroxy form; 90% is excreted in the urine in 48 hours, largely as the glucuronide or sulfonate conju-gate. Thiabendazole can also be absorbed from the skin.
The mechanism of action of thiabendazole is probably the same as that of other benzimidazoles (see above). The drug has ovicidal effects against some parasites.
Clinical Uses The standard dosage, 25 mg/kg (maximum 1.5 g) twice daily, should be given after meals. Tablets should be chewed. For strongyloides infection, treatment is for 2 days. Cure rates are reportedly 93%. A course can be repeated in 1 week if indicated. In patients with hyperinfection syndrome, the standard dose is continued twice daily for 5–7 days. For cutaneous larva migrans, thiabendazole cream can be applied topically, or the oral drug can be given for 2 days (although albendazole is less toxic and there-fore preferred).
Adverse Reactions, Contraindications, & Cautions Thiabendazole is much more toxic than other benzimidazoles and more toxic than ivermectin, so other agents are now preferred for most indications. Common adverse effects include dizziness, anorexia, nausea, and vomiting. Less common problems are epi-gastric pain, abdominal cramps, diarrhea, pruritus, headache, drowsiness, and neuropsychiatric symptoms. Irreversible liver failure and fatal Stevens-Johnson syndrome have been reported.
Experience with thiabendazole is limited in children weighing less than 15 kg. The drug should not be used in pregnancy or in the presence of hepatic or renal disease
053-Katzung_Ch053_p937-948.indd 945 9/21/11 12:11:28 PM

946 SECTION VIII Chemotherapeutic Drugs
REFERENCES Bagheri H et al: Adverse drug reactions to anthelmintics. Ann Pharmacother
2004;38:383. Basáñez MG et al: Effect of single-dose ivermectin on Onchocerca volvulus : A sys-
tematic review and meta-analysis. Lancet Infect Dis 2008;8:310. Bethony J et al: Soil-transmitted helminth infections: Ascariasis, trichuriasis, and
hookworm. Lancet 2006;367:1521. Bockarie MJ et al: Efficacy of single-dose diethylcarbamazine compared with
diethylcarbamazine combined with albendazole against Wuchereria bancroftiinfection in Papua New Guinea. Am J Trop Med Hyg 2007;76:62.
Bockarie MJ et al: Mass treatment to eliminate filariasis in Papua New Guinea. N Engl J Med 2002;347:1841.
Craig P, Ito A: Intestinal cestodes. Curr Opin Infect Dis 2007;20:524. Danso-Appiah A et al: Drugs for treating urinary schistosomiasis. Cochrane
Database Syst Rev 2008:CD000053 Dayan AD: Albendazole, mebendazole and praziquantel. Review of non-clinical
toxicity and pharmacokinetics. Acta Trop 2003;86:141. Drugs for parasitic infections. Med Lett Drugs Ther 2007;(Suppl 1). Flohr C et al: Low efficacy of mebendazole against hookworm in Vietnam: Two
randomized controlled trials. Am J Trop Med Hyg 2007;76:732. Fox LM: Ivermectin: Uses and impact 20 years on. Curr Opin Infect Dis
2006;19:588. Garcia HH et al: Current consensus guidelines for treatment of neurocysticercosis.
Clin Microbiol Rev 2002;15:747. Garcia HH et al: A trial of antiparasitic treatment to reduce the rate of seizures due
to cerebral cysticercosis. N Engl J Med 2004;350:249. Horton J: Albendazole: A broad spectrum anthelminthic for treatment of indi-
viduals and populations. Curr Opin Infect Dis 2002;15:599.
Keiser J, Utzinger J: Efficacy of current drugs against soil-transmitted helm-inth infections: Systematic review and meta-analysis. JAMA 2008;299:1937.
Matthaiou DK et al: Albendazole versus praziquantel in the treatment of neuro-cysticercosis: A meta-analysis of comparative trials. PLoS Negl Trop Dis 2008;2:e194.
Meltzer E et al: Eosinophilia among returning travelers: A practical approach. Am J Trop Med Hyg 2008;78:702.
Osei-Atweneboana MY: Prevalence and intensity of Onchocerca volvulus infection and efficacy of ivermectin in endemic communities in Ghana: A two-phase epidemiological study. Lancet 2007;369:2021.
Ramzy RM et al: Effect of yearly mass drug administration with diethylcarbamaz-ine and albendazole on bancroftian filariasis in Egypt: A comprehensive assessment. Lancet 2006;367:992.
Reddy M et al: Oral drug therapy for multiple neglected tropical diseases: A sys-tematic review. JAMA 2007;298:1911.
Smego RA Jr, Sebanego P: Treatment options for hepatic cystic echinococcosis. Int J Infect Dis 2005;9:69.
Supali T et al: Doxycycline treatment of Brugia malayi -infected persons reduces microfilaremia and adverse reactions after diethylcarbamazine and albenda-zole treatment. Clin Infect Dis 2008;46:1385.
Taylor MJ et al: Macrofilaricidal activity after doxycycline treatment of Wuchereria bancrofti : A double-blind, randomised placebo-controlled trial. Lancet 2005;365:2116.
Tisch DJ, Michael E, Kazura JW: Mass chemotherapy options to control lym-phatic filariasis: A systematic review. Lancet Infect Dis 2005;5:514.
Udall DN: Recent updates on onchocerciasis: Diagnosis and treatment. Clin Infect Dis 2007;44:53.
Albendazole (Albenza)Oral: 200 mg tablets; 100 mg/5 mL suspensionNote: Albendazole is approved in the USA for the treatment of
cysticercosis and hydatid disease.Bithionol (Bitin)1
Diethylcarbamazine (Hetrazan)Oral: 50 mg tablets
Ivermectin (Mectizan, Stromectol)Oral: 3, 6 mg tabletsNote: Ivermectin is approved for use in the USA for the treatment
of onchocerciasis and strongyloidiasis. See Chapter 65 for com-ment on the unlabeled use of drugs.
Mebendazole (generic, Vermox)Oral: 100 mg chewable tablets; outside the USA, 100 mg/5 mL
suspensionMetrifonate (trichlorfon, Bilarcil)1
Niclosamide (Niclocide)1
Oxamniquine (Vansil, Mansil)1
Oxantel pamoate (Quantrel); oxantel/pyrantel pamoate (Telopar)
Oral: tablets containing 100 mg (base) of each drug; suspensions containing 20 or 50 mg (base) per mL
Note: Oxantel pamoate and oxantel/pyrantel pamoate are not avail-able in the USA.
Piperazine (generic, Vermizine)1
Praziquantel (Biltricide; others outside the USA)Oral: 600 mg tablets (other strengths outside the USA)
Pyrantel pamoate (Antiminth, Combantrin, Pin-rid, Pin-X)Oral: 50 mg (base)/mL suspension; 180 mg; 62.5 mg (base) capsules
(available without prescription in the USA)Suramin (Bayer 205, others)1
Thiabendazole (Mintezol)Oral: 500 mg chewable tablets; suspension, 500 mg/mL
P R E P A R A T I O N S A V A I L A B L E 1
Albendazole (Albenza)Oral: 200 mg tablets; 100 mg/5 mL suspensionNote: Albendazole is approved in the USA for the treatment of
cysticercosis and hydatid disease.Bithionol (Bitin)1
Diethylcarbamazine (Hetrazan)Oral: 50 mg tablets
Ivermectin (Mectizan, Stromectol)Oral: 3, 6 mg tabletsNote: Ivermectin is approved for use in the USA for the treatment
of onchocerciasis and strongyloidiasis. See Chapter 65 for com-ment on the unlabeled use of drugs.
Mebendazole (generic, Vermox)Oral: 100 mg chewable tablets; outside the USA, 100 mg/5 mL
suspensionMetrifonate (trichlorfon, Bilarcil)1
Niclosamide (Niclocide)1
Oxamniquine (Vansil, Mansil)1
Oxantel pamoate (Quantrel); oxantel/pyrantel pamoate(Telopar)
Oral: tablets containing 100 mg (base) of each drug; suspensions containing 20 or 50 mg (base) per mL
Note: Oxantel pamoate and oxantel/pyrantel pamoate are not avail-able in the USA.
Piperazine (generic, Vermizine)1
Praziquantel (Biltricide; others outside the USA)Oral: 600 mg tablets (other strengths outside the USA)
Pyrantel pamoate (Antiminth, Combantrin, Pin-rid, Pin-X)Oral: 50 mg (base)/mL suspension; 180 mg; 62.5 mg (base) capsules
(available without prescription in the USA)Suramin (Bayer 205, others)1
Thiabendazole (Mintezol)Oral: 500 mg chewable tablets; suspension, 500 mg/mL
1Additional information may be obtained from the Parasitic Disease Drug Service, Parasitic Diseases Branch, Centers for Disease Control and Prevention, Atlanta, Georgia, 30333. Telephone: (404) 639-3670.
053-Katzung_Ch053_p937-948.indd 946 9/21/11 12:11:28 PM

CHAPTER 53 Clinical Pharmacology of the Antihelminthic Drugs 947
C A S E S T U D Y A N S W E R
The presentation is highly suggestive of cystic hydatid dis-ease (infection with Echinococcus granulosis ), which is trans-mitted by eggs from the feces of dogs in contact with livestock. Other causes of liver fluid collections include ame-bic and pyogenic abscesses, but these are usually not cystic in appearance. For echinococcosis, a typical cystic lesion and
positive serology support the diagnosis, and treatment gen-erally entails albendazole in conjunction with cautious sur-gery or percutaneous aspiration. One approach entails treatment with albendazole followed by aspiration to con-firm the diagnosis and, if it is confirmed, to remove most of the infecting worms.
053-Katzung_Ch053_p937-948.indd 947 9/21/11 12:11:28 PM

This page intentionally left blank

949
Cancer Chemotherapy Edward Chu, MD, & Alan C. Sartorelli, PhD
he receive adjuvant chemotherapy? The patient receives a combination of 5-fluorouracil (5-FU), leucovorin, and oxali-platin as adjuvant therapy. One week after receiving the first cycle of therapy, he experiences significant toxicity in the form of myelosuppression, diarrhea, and altered mental sta-tus. What is the most likely explanation for this increased toxicity? Is there any role for genetic testing to determine the etiology of this level of toxicity?
C A S E S T U D Y
A 55-year-old man presents with increasing fatigue, 15-pound weight loss, and a microcytic anemia. Colonoscopy identifies a mass in the ascending colon, and biopsy specimens reveal well-differentiated colorectal cancer (CRC). He undergoes surgical resection and is found to have high-risk stage III CRC with five positive lymph nodes. After surgery, he feels entirely well with no symptoms. Of note, he has no other comorbid illnesses. What is this patient’s prognosis? Should
C H A P T E R
Cancer continues to be the second leading cause of mortality from disease in the USA, accounting for nearly 500,000 deaths in 2008. Cancer is a disease characterized by a loss in the normal control mechanisms that govern cell survival, proliferation, and differen-tiation. Cells that have undergone neoplastic transformation usu-ally express cell surface antigens that may be of normal fetal type, may display other signs of apparent immaturity, and may exhibit qualitative or quantitative chromosomal abnormalities, including various translocations and the appearance of amplified gene sequences. It is now well established that a small subpopulation of cells, referred to as tumor stem cells, reside within a tumor mass. They retain the ability to undergo repeated cycles of proliferation as well as to migrate to distant sites in the body to colonize various organs in the process called metastasis. Such tumor stem cells thus can express clonogenic (colony-forming) capability, and they are characterized by chromosome abnormalities reflecting their genetic instability, which leads to progressive selection of subclones that can survive more readily in the multicellular environment of the host. This genetic instability also allows them to become resistant to chemotherapy and radiotherapy. The invasive and metastatic processes as well as a series of metabolic abnormalities associated with the cancer result in tumor-related symptoms and eventual death of the patient unless the neoplasm can be eradicated with treatment.
54
CAUSES OF CANCER
The incidence, geographic distribution, and behavior of specific types of cancer are related to multiple factors, including sex, age, race, genetic predisposition, and exposure to environmental car-cinogens. Of these factors, environmental exposure is probably most important. Exposure to ionizing radiation has been well documented as a significant risk factor for a number of cancers, including acute leukemias, thyroid cancer, breast cancer, lung cancer, soft tissue sarcoma, and basal cell and squamous cell skin cancers. Chemical carcinogens (particularly those in tobacco smoke) as well as azo dyes, aflatoxins, asbestos, benzene, and radon have all been well documented as leading to a wide range of human cancers.
Several viruses have been implicated in the etiology of various human cancers. For example, hepatitis B and hepatitis C are asso-ciated with the development of hepatocellular cancer; HIV is associated with Hodgkin’s and non-Hodgkin’s lymphomas; human papillomavirus is associated with cervical cancer and head and neck cancer; and Ebstein-Barr virus is associated with nasopharyn-geal cancer. Expression of virus-induced neoplasia may also depend on additional host and environmental factors that modu-late the transformation process. Cellular genes are known that are homologous to the transforming genes of the retroviruses, a family
054-Katzung_Ch054_p949-976.indd 949 9/21/11 12:11:46 PM

950 SECTION VIII Chemotherapeutic Drugs
of RNA viruses, and induce oncogenic transformation. These mammalian cellular genes, known as oncogenes, have been shown to code for specific growth factors and their corresponding recep-tors. These genes may be amplified (increased number of gene copies) or mutated, both of which can lead to constitutive overex-pression in malignant cells. The bcl -2 family of genes represents a series of pro-survival genes that promotes survival by directly inhibiting apoptosis, a key pathway of programmed cell death.
Another class of genes, known as tumor suppressor genes, may be deleted or mutated, which gives rise to the neoplastic phenotype. The p53 gene is the best-established tumor suppressor gene identi-fied to date, and the normal wild-type gene appears to play an important role in suppressing neoplastic transformation. Of note, p53 is mutated in up to 50% of all human solid tumors, including liver, breast, colon, lung, cervix, bladder, prostate, and skin.
CANCER TREATMENT MODALITIES
With present methods of treatment, about one third of patients are cured with local treatment strategies, such as surgery or radio-therapy, when the tumor remains localized at the time of diagno-sis. Earlier diagnosis might lead to increased cure rates with such local treatment. In the remaining cases, however, early microme-tastasis is a characteristic feature, indicating that a systemic approach with chemotherapy is required for effective cancer man-agement. In patients with locally advanced disease, chemotherapy
is often combined with radiotherapy to allow for surgical resection to take place, and such a combined modality approach has led to improved clinical outcomes. At present, about 50% of patients who are initially diagnosed with cancer can be cured. In contrast, chemotherapy alone is able to cure less than 10% of all cancer patients when the tumor is diagnosed at an advanced stage.
Chemotherapy is presently used in three main clinical settings: (1) primary induction treatment for advanced disease or for can-cers for which there are no other effective treatment approaches, (2) neoadjuvant treatment for patients who present with localized disease, for whom local forms of therapy such as surgery or radia-tion, or both, are inadequate by themselves, (3) adjuvant treat-ment to local methods of treatment, including surgery, radiation therapy, or both.
Primary induction chemotherapy refers to chemotherapy administered as the primary treatment in patients who present with advanced cancer for which no alternative treatment exists. This has been the main approach in treating patients with advanced metastatic disease, and in most cases, the goals of ther-apy are to relieve tumor-related symptoms, improve overall quality of life, and prolong time to tumor progression. Studies in a wide range of solid tumors have shown that chemotherapy in patients with advanced disease confers survival benefit when compared with supportive care, providing sound rationale for the early ini-tiation of drug treatment. However, cancer chemotherapy can be curative in only a small subset of patients who present with advanced disease. In adults, these curable cancers include Hodgkin’s and non-Hodgkin’s lymphoma, acute myelogenous leukemia, germ cell cancer, and choriocarcinoma, while the curable child-hood cancers include acute lymphoblastic leukemia, Burkitt’s lymphoma, Wilms’ tumor, and embryonal rhabdomyosarcoma.
Neoadjuvant chemotherapy refers to the use of chemotherapy in patients who present with localized cancer for which alternative local therapies, such as surgery, exist but which are less than com-pletely effective. At present, neoadjuvant therapy is most often administered in the treatment of anal cancer, bladder cancer, breast cancer, esophageal cancer, laryngeal cancer, locally advanced non-small cell lung cancer, and osteogenic sarcoma. For some of these diseases, such as anal cancer, gastroesophageal cancer, laryn-geal cancer, and non-small cell lung cancer, optimal clinical ben-efit is derived when chemotherapy is administered with radiation therapy either concurrently or sequentially.
One of the most important roles for cancer chemotherapy is as an adjuvant to local treatment modalities such as surgery or radia-tion therapy, and this has been termed adjuvant chemotherapy. The goal of chemotherapy in this setting is to reduce the incidence of both local and systemic recurrence and to improve the overall survival of patients. In general, chemotherapy regimens with clinical activity against advanced disease may have curative poten-tial following surgical resection of the primary tumor, provided the appropriate dose and schedule are administered. Adjuvant chemotherapy is effective in prolonging both disease-free survival (DFS) and overall survival (OS) in patients with breast cancer, colon cancer, gastric cancer, non-small cell lung cancer, Wilms’ tumor, anaplastic astrocytoma, and osteogenic sarcoma. Patients with primary malignant melanoma at high risk of local recurrence
ACRONYMS
ABVD Doxorubicin (Adriamycin, hydroxydaunorubicin), bleo-mycin, vinblastine, dacarbazine
CHOP Cyclophosphamide, doxorubicin (hydroxydaunorubi-cin, Adriamycin), vincristine (Oncovin), prednisone
CMF Cyclophosphamide, methotrexate, fl uorouracil
COP Cyclophosphamide, vincristine (Oncovin), prednisone
FAC 5-Fluorouracil, doxorubicin (Adriamycin, hydroxydauno-rubicin), cyclophosphamide
FEC 5-Fluorouracil, epirubicin, cyclophosphamide
5-FU 5-Fluorouracil
FOLFIRI 5-Fluorouracil, leucovorin, irinotecan
FOLFOX 5-Fluorouracil, leucovorin, oxaliplatin
MP Melphalan, prednisone
6-MP 6-Mercaptopurine
MOPP Mechlorethamine, vincristine (Oncovin), procarbazine, prednisone
MTX Methotrexate
PCV Procarbazine, lomustine, vincristine
PEB Cisplatin (platinum), etoposide, bleomycin
6-TG 6-Thioguanine
VAD Vincristine, doxorubicin (Adriamycin), dexamethasone
XELOX Capecitabine, oxaliplatin
054-Katzung_Ch054_p949-976.indd 950 9/21/11 12:11:46 PM

CHAPTER 54 Cancer Chemotherapy 951
or systemic metastases derive clinical benefit from adjuvant treat-ment with the biologic agent α-interferon, although this treat-ment must be given for 1 year’s duration for maximal clinical efficacy. Finally, the antihormonal agents tamoxifen, anastrozole, and letrozole are effective in the adjuvant therapy of postmeno-pausal women with early-stage breast cancer whose breast tumors express the estrogen receptor (see Chapter 40 for additional detail). However, because these agents are cytostatic rather than cytocidal, they must be administered on a long-term basis, with the standard recommendation being 5 years’ duration.
ROLE OF CELL CYCLE KINETICS & ANTICANCER EFFECT
The key principles of cell cycle kinetics were initially developed using the murine L1210 leukemia as the experimental model system ( Figure 54–1 ). However, drug treatment of human cancers requires a clear understanding of the differences between the char-acteristics of this rodent leukemia and of human cancers, as well as an understanding of the differences in growth rates of normal target tissues between mice and humans. For example, L1210 is a rapidly growing leukemia with a high percentage of cells synthesiz-ing DNA, as measured by the uptake of tritiated thymidine (the labeling index). Because L1210 leukemia has a growth fraction of 100% (ie, all its cells are actively progressing through the cell cycle), its life cycle is consistent and predictable. Based on the murine L1210 model, the cytotoxic effects of anticancer drugs follow log cell-kill kinetics. As such, a given agent would be predicted to kill a constant fraction of cells as opposed to a constant number.
Thus, if a particular dose of an individual drug leads to a 3 log kill of cancer cells and reduces the tumor burden from 10 10 to 10 7 cells, the same dose used at a tumor burden of 10 5 cells reduces the tumor mass to 10 2 cells. Cell kill is, therefore, proportional, regardless of tumor burden. The cardinal rule of chemotherapy—the invariable inverse relation between cell number and curability—was established with this model, and this relationship is applicable to other hematologic malignancies.
Although growth of murine leukemias simulates exponential cell kinetics, mathematical modeling data suggest that most human solid tumors do not grow in such an exponential manner. Taken together, the experimental data in human solid cancers support a Gompertzian model of tumor growth and regression. The critical distinction between Gompertzian and exponential growth is that the growth fraction of the tumor is not constant with Gompertzian kinetics but instead decreases exponentially with time (exponential growth is matched by exponential retarda-tion of growth, due to blood supply limitations and other fac-tors). The growth fraction peaks when the tumor is approximately one third its maximum size. Under the Gompertzian model, when a patient with advanced cancer is treated, the tumor mass is larger, its growth fraction is low, and the fraction of cells killed is, there-fore, small. An important feature of Gompertzian growth is that response to chemotherapy in drug-sensitive tumors depends, in large measure, on where the tumor is in its particular growth curve.
Information on cell and population kinetics of cancer cells explains, in part, the limited effectiveness of most available anti-cancer drugs. A schematic summary of cell cycle kinetics is pre-sented in Figure 54–2 . This information is relevant to the mode of action, indications, and scheduling of cell cycle-specific (CCS) and cell cycle-nonspecific (CCNS) drugs. Agents falling into these two major classes are summarized in Table 54–1 .
The Role of Drug Combinations With rare exceptions (eg, choriocarcinoma and Burkitt’s lym-phoma), single drugs at clinically tolerable doses have been unable to cure cancer. In the 1960s and early 1970s, drug combination regimens were developed based on the known biochemical actions
1012
1010
108
106
104
102
100
Num
ber
of c
ance
r ce
lls (
log
scal
e)
Time
Symptoms
Death
Diagnosis
Sub
clin
ical
Surgery
FIGURE 54–1 The log-kill hypothesis. Relationship of tumor cell number to time of diagnosis, symptoms, treatment, and survival. Three alternative approaches to drug treatment are shown for comparison with the course of tumor growth when no treatment is given (dashed line). In the protocol diagrammed at top, treatment (indicated by the arrows) is given infrequently, and the result is manifested as prolongation of survival but with recurrence of symptoms between courses of treatment and eventual death of the patient. The combination chemotherapy treatment diagrammed in the middle section is begun earlier and is more intensive. Tumor cell kill exceeds regrowth, drug resistance does not develop, and “cure” results. In this example, treatment has been continued long after all clinical evidence of cancer has disappeared (1–3 years). This approach has been established as effective in the treatment of childhood acute leukemia, testicular cancers, and Hodgkin’s lymphoma. In the treatment diagrammed near the bottom of the graph, early surgery has been employed to remove the primary tumor and intensive adjuvant chemotherapy has been administered long enough (up to 1 year) to eradicate the remaining tumor cells that comprise the occult micrometastases.
054-Katzung_Ch054_p949-976.indd 951 9/21/11 12:11:46 PM

952 SECTION VIII Chemotherapeutic Drugs
of available anticancer drugs rather than on their clinical efficacy. Such regimens were, however, largely ineffective. The era of effec-tive combination chemotherapy began when a number of active drugs from different classes became available for use in combina-tion in the treatment of the acute leukemias and lymphomas. Following this initial success with hematologic malignancies, com-bination chemotherapy was extended to the treatment of solid tumors.
The use of combination chemotherapy is important for several reasons. First, it provides maximal cell kill within the range of toxicity tolerated by the host for each drug as long as dosing is not compromised. Second, it provides a broader range of interaction between drugs and tumor cells with different genetic abnormali-ties in a heterogeneous tumor population. Finally, it may prevent or slow the subsequent development of cellular drug resistance. The same principles apply to the therapy of chronic infections, such as HIV and tuberculosis.
Certain principles have guided the selection of drugs in the most effective drug combinations, and they provide a paradigm for the development of new drug therapeutic programs.
1. Efficacy: Only drugs known to be somewhat effective when used alone against a given tumor should be selected for use in combination. If available, drugs that produce complete remis-sion in some fraction of patients are preferred to those that produce only partial responses.
2. Toxicity: When several drugs of a given class are available and are equally effective, a drug should be selected on the basis of toxicity that does not overlap with the toxicity of other drugs in the combination. Although such selection leads to a wider range of adverse effects, it minimizes the risk of a lethal effect
caused by multiple insults to the same organ system by differ-ent drugs and allows dose intensity to be maximized.
3. Optimum scheduling: Drugs should be used in their optimal dose and schedule, and drug combinations should be given at consistent intervals. Because long intervals between cycles negatively affect dose intensity, the treatment-free interval between cycles should be the shortest time necessary for recov-ery of the most sensitive normal target tissue, which is usually the bone marrow.
4. Mechanism of interaction: There should be a clear under-standing of the biochemical, molecular, and pharmacokinetic mechanisms of interaction between the individual drugs in a given combination, to allow for maximal effect. Omission of a drug from a combination may allow overgrowth by a tumor clone sensitive to that drug alone and resistant to other drugs in the combination.
DN
A synthesis
Synthesisof cellular
componentsfor mitosis
Synthesisof cellular
componentsneeded for
DNA synthesis
Replicationof DNA genome
Differentiation
G0
2%Mitosis
40%
39%
19%
G2
S
G1
M
The cellcycle
FIGURE 54–2 The cell cycle and cancer. A conceptual depiction of the cell cycle phases that all cells—normal and neoplastic—must traverse before and during cell division. The percentages given represent the approximate percentage of time spent in each phase by a typical malignant cell; the duration of G 1 , however, can vary markedly. Many of the effective anticancer drugs exert their action on cells traversing the cell cycle and are called cell cycle-specific (CCS) drugs (see Table 54–1 ). A second group of agents called cell cycle-nonspecific (CCNS) drugs can sterilize tumor cells whether they are cycling or resting in the G 0 compartment. CCNS drugs can kill both G 0 and cycling cells (although cycling cells are more sensitive).
TABLE 54–1 Cell cycle effects of major classes of anticancer drugs.
Cell Cycle-Specific (CCS) Agents
Cell Cycle-Nonspecific (CCNS) Agents
Antimetabolites (S phase) Alkylating agents
Capecitabine Altretamine
Cladribine Bendamustine
Clofarabine Busulfan
Cytarabine (ara-C) Carmustine
Fludarabine Chlorambucil
5-Fluorouracil (5-FU) Cyclophosphamide
Gemcitabine Dacarbazine
6-Mercaptopurine (6-MP) Lomustine
Methotrexate (MTX) Mechlorethamine
Nelarabine Melphalan
Pralatrexate Temozolomide
6-Thioguanine (6-TG) Thiotepa
Epipodophyllotoxin (topoisomerase II inhibitor) (G1–S phase)
Etoposide
Taxanes (M phase)
Albumin-bound paclitaxel
Cabazitaxel
Paclitaxel
Vinca alkaloids (M phase)
Vinblastine
Vincristine
Vinorelbine
Antimicrotubule inhibitor (M phase)
Ixabepilone
Antitumor antibiotics (G2–M phase)
Bleomycin
Antitumor antibiotics
Dactinomycin
Mitomycin
Camptothecins (topoi-somerase I inhibitors)
Irinotecan
Topotecan
Platinum analogs
Carboplatin
Cisplatin
Oxaliplatin
Anthracyclines
Daunorubicin
Doxorubicin
Epirubicin
Idarubicin
Mitoxantrone
054-Katzung_Ch054_p949-976.indd 952 9/21/11 12:11:46 PM

CHAPTER 54 Cancer Chemotherapy 953
5. Avoidance of arbitrary dose changes: An arbitrary reduction in the dose of an effective drug in order to add other less effec-tive drugs may reduce the dose of the most effective agent below the threshold of effectiveness and destroy the ability of the combination to cure disease in a given patient.
Dosage Factors Dose intensity is one of the main factors limiting the ability of chemotherapy or radiation therapy to achieve cure. The dose- response curve in biologic systems is usually sigmoidal in shape, with a threshold, a linear phase, and a plateau phase. For chemo-therapy, therapeutic selectivity is dependent on the difference between the dose-response curves of normal and tumor tissues. In experimental animal models, the dose-response curve is usually steep in the linear phase, and a reduction in dose when the tumor is in the linear phase of the dose-response curve almost always results in a loss in the capacity to cure the tumor effectively before a reduction in the antitumor activity is observed. Although com-plete remissions continue to be observed with dose reduction as low as 20%, residual tumor cells may not be entirely eliminated, thereby allowing for eventual relapse. Because anticancer drugs are associated with toxicity, it is often appealing for clinicians to avoid acute toxicity by simply reducing the dose or by increasing the time interval between each cycle of treatment. However, such empiric modifications in dose represent a major cause of treatment failure in patients with drug-sensitive tumors.
A positive relationship between dose intensity and clinical effi-cacy has been documented in several solid tumors, including advanced ovarian, breast, lung, and colon cancers, as well as in hematologic malignancies, such as the lymphomas. At present, there are three main approaches to dose-intense delivery of chemotherapy. The first approach, dose escalation, involves increasing the doses of the respective anticancer agents. The second strategy is administra-tion of anticancer agents in a dose-intense manner by reducing the interval between treatment cycles, while the third approach involves sequential scheduling of either single agents or of combination regimens. Each of these strategies is presently being applied to a wide range of solid cancers, including breast, colorectal, and non-small cell lung, and in general, such dose-intense regimens have significantly improved clinical outcomes.
DRUG RESISTANCE
A fundamental issue in cancer chemotherapy is the development of cellular drug resistance. Some tumor types, eg, malignant mela-noma, renal cell cancer, and brain cancer, exhibit primary resis-tance, ie, absence of response on the first exposure, to currently available agents. The presence of inherent drug resistance was first proposed by Goldie and Coleman in the early 1980s and was thought to result from the genomic instability associated with the development of most cancers. For example, mutations in the p53 tumor suppressor gene occur in up to 50% of all human tumors. Preclinical and clinical studies have shown that loss of p53 func-tion leads to resistance to radiation therapy as well as resistance to
a wide range of anticancer agents. Defects in the mismatch repair enzyme family, which are tightly linked to the development of familial and sporadic colorectal cancer, lead to resistance to several unrelated anticancer agents, including the fluoropyrimidines, the thiopurines, and cisplatin/carboplatin. In contrast to primary resistance, acquired resistance develops in response to exposure to a given anticancer agent. Experimentally, drug resistance can be highly specific to a single drug and is usually based on a specific change in the genetic machinery of a given tumor cell with ampli-fication or increased expression of one or more genes. In other instances, a multidrug-resistant phenotype occurs, associated with increased expression of the MDR1 gene, which encodes a cell surface transporter glycoprotein (P-glycoprotein, see Chapter 1 ). This form of drug resistance leads to enhanced drug efflux and reduced intracellular accumulation of a broad range of structurally unrelated anticancer agents, including the anthracyclines, vinca alkaloids, taxanes, camptothecins, epipodophyllotoxins, and even small molecule inhibitors, such as imatinib.
■ BASIC PHARMACOLOGY OF CANCER CHEMOTHERAPEUTIC DRUGS
ALKYLATING AGENTS The major clinically useful alkylating agents ( Figure 54–3 ) have a structure containing a bis(chloroethyl)amine, ethyleneimine, or nitrosourea moiety, and they are classified in several different groups. Among the bis(chloroethyl)amines, cyclophosphamide, mechlorethamine, melphalan, and chlorambucil are the most use-ful. Ifosfamide is closely related to cyclophosphamide but has a somewhat different spectrum of activity and toxicity. Thiotepa and busulfan are used to treat breast and ovarian cancer, and chronic myeloid leukemia, respectively. The major nitrosoureas are carmustine (BCNU) and lomustine (CCNU).
Mechanism of Action As a class, the alkylating agents exert their cytotoxic effects via transfer of their alkyl groups to various cellular constituents. Alkylations of DNA within the nucleus probably represent the major interactions that lead to cell death. However, these drugs react chemically with sulfhydryl, amino, hydroxyl, carboxyl, and phosphate groups of other cellular nucleophiles as well. The general mechanism of action of these drugs involves intramolecular cycliza-tion to form an ethyleneimonium ion that may directly or through formation of a carbonium ion transfer an alkyl group to a cellular constituent ( Figure 54–4 ). In addition to alkylation, a secondary mechanism that occurs with nitrosoureas involves carbamoylation of lysine residues of proteins through formation of isocyanates.
The major site of alkylation within DNA is the N7 position of guanine; however, other bases are also alkylated albeit to lesser degrees, including N1 and N3 of adenine, N3 of cytosine, and O6 of guanine, as well as phosphate atoms and proteins associated with DNA. These interactions can occur on a single strand or on
054-Katzung_Ch054_p949-976.indd 953 9/21/11 12:11:47 PM
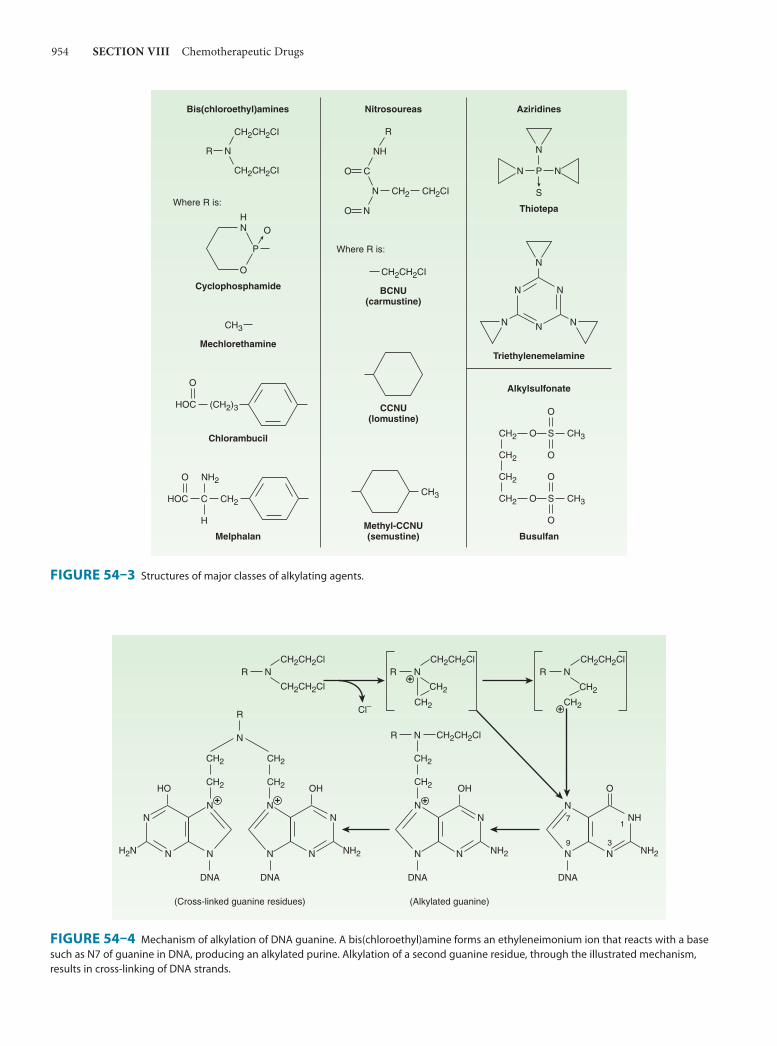
954 SECTION VIII Chemotherapeutic Drugs
R N
CH2CH2CI
CH2CH2CI
P
O
OHN
CH3
Cyclophosphamide
Mechlorethamine
(CH2)3HOC
O
Chlorambucil
CH2HOC
O
Melphalan
NH2
C
H
Where R is:
Bis(chloroethyl)amines
CH2CH2CI
BCNU(carmustine)
CCNU(lomustine)
CH3
Methyl-CCNU(semustine)
Where R is:
Nitrosoureas
O C
NH
N
O N
CH2 CH2CI
R
Thiotepa
Aziridines
Alkylsulfonate
Busulfan
SO
O
O
CH3CH2
CH2
CH2
SO
O
O
CH3CH2
N
P
S
N N
Triethylenemelamine
N
NN
N
N N
FIGURE 54–3 Structures of major classes of alkylating agents.
OH
N
R
HO
N
NH2N N N
N
N
DNA
(Cross-linked guanine residues)
DNA
OH
N N
N
DNA
+
(Alkylated guanine)
NR
CH2
NRNR NR
N N
NH
DNA
O
3
17
9
CH2CH2Cl
CH2CH2Cl CH2CH2Cl CH2CH2Cl
CH2CH2Cl
CH2 CH2
CH2
N
CH2
CH2
N
CH2
CH2
NN+
CH2
CH2
+
+
+
Cl–
NH2 NH2 NH2
FIGURE 54–4 Mechanism of alkylation of DNA guanine. A bis(chloroethyl)amine forms an ethyleneimonium ion that reacts with a base such as N7 of guanine in DNA, producing an alkylated purine. Alkylation of a second guanine residue, through the illustrated mechanism, results in cross-linking of DNA strands.
054-Katzung_Ch054_p949-976.indd 954 9/21/11 12:11:47 PM

CHAPTER 54 Cancer Chemotherapy 955
both strands of DNA through cross-linking, as most major alky-lating agents are bifunctional, with two reactive groups. Alkylation of guanine can result in miscoding through abnormal base pairing with thymine or in depurination by excision of guanine residues. The latter effect leads to DNA strand breakage through scission of the sugar-phosphate backbone of DNA. Cross-linking of DNA appears to be of major importance to the cytotoxic action of alky-lating agents, and replicating cells are most susceptible to these drugs. Thus, although alkylating agents are not cell cycle specific, cells are most susceptible to alkylation in late G 1 and S phases of the cell cycle.
Resistance The mechanism of acquired resistance to alkylating agents may involve increased capability to repair DNA lesions, decreased trans-port of the alkylating drug into the cell, and increased expression or activity of glutathione and glutathione-associated proteins, which are needed to conjugate the alkylating agent, or increased glutathione S -transferase activity, which catalyzes the conjugation.
Adverse Effects The adverse effects usually associated with alkylating agents are generally dose-related and occur primarily in rapidly growing tis-sues such as bone marrow, gastrointestinal tract, and reproductive system. Nausea and vomiting can be a serious issue with a number of these agents. In addition, they are potent vesicants and can damage tissues at the site of administration as well as produce
systemic toxicity. As a class, alkylating agents are carcinogenic in nature, and there is an increased risk of secondary malignancies, especially acute myelogenous leukemia.
Cyclophosphamide is one of the most widely used alkylating agents. One of the potential advantages of this compound relates to its high oral bioavailability. As a result, it can be administered via the oral and intravenous routes with equal clinical efficacy. It is inactive in its parent form, and must be activated to cytotoxic forms by liver microsomal enzymes ( Figure 54–5 ). The cyto-chrome P450 mixed-function oxidase system converts cyclophos-phamide to 4-hydroxycyclophosphamide, which is in equilibrium with aldophosphamide. These active metabolites are delivered to both tumor and normal tissue, where nonenzymatic cleavage of aldophosphamide to the cytotoxic forms—phosphoramide mus-tard and acrolein—occurs. The liver appears to be protected through the enzymatic formation of the inactive metabolites 4-ketocyclophosphamide and carboxyphosphamide.
The major toxicities of the individual alkylating agents are outlined in Table 54–2 and discussed below.
NITROSOUREAS
These drugs appear to be non-cross-resistant with other alkylating agents; all require biotransformation, which occurs by nonenzy-matic decomposition, to metabolites with both alkylating and carbamoylating activities. The nitrosoureas are highly lipid-soluble and are able to cross the blood-brain barrier, making them effective
NH
P
O
O
N(CH2CH2Cl)2
N(CH2CH2Cl)2
N(CH2CH2Cl)2
Cyclophosphamide
NH
P
O
O
OH
4-Hydroxycyclophosphamide(active)
NH
P
O
O
O
4-Ketocyclophosphamide(inactive)
CH
O
CH2
CH2 O
P
H2N O
Aldophosphamide(active)
Aldehyde oxidase
O
P
H2N O
Carboxyphosphamide(inactive)
2 CH2CHHOC
O
CH CHOCH2
Acrolein(cytotoxic)
+
HO
P
H2N O
Phosphoramide mustard(cytotoxic)
Nonenzymatic
Liver cytochromeP450 oxidase
N(CH2CH2Cl)2
N(CH2CH2Cl)2
N(CH2CH2Cl)2
FIGURE 54–5 Cyclophosphamide metabolism.
054-Katzung_Ch054_p949-976.indd 955 9/21/11 12:11:47 PM

956 SECTION VIII Chemotherapeutic Drugs
TABLE 54–2 Alkylating agents and platinum analogs: Clinical activity and toxicities.
Alkylating Agent Mechanism of Action Clinical Applications Acute Toxicity Delayed Toxicity
Mechlorethamine Forms DNA cross-links, resulting in inhibition of DNA synthesis and function
Hodgkin’s and non-Hodgkin’s lymphoma
Nausea and vomiting Moderate depression of peripheral blood count; excessive doses produce severe bone marrow depression with leuko-penia, thrombocytope-nia, and bleeding; alopecia and hemor-rhagic cystitis occasion-ally occur with cyclophosphamide; cys-titis can be prevented with adequate hydra-tion; busulfan is associated with skin pigmentation, pulmo-nary fibrosis, and adrenal insufficiency
Chlorambucil Same as above CLL and non-Hodgkin’s lymphoma Nausea and vomiting
Cyclophosphamide Same as above Breast cancer, ovarian cancer, non-Hodgkin’s lymphoma, CLL, soft tissue sarcoma, neuroblas-toma, Wilms’ tumor, rhabdomyo-sarcoma
Nausea and vomiting
Bendamustine Same as above CLL, non-Hodgkin’s lymphoma Nausea and vomiting
Melphalan Same as above Multiple myeloma, breast cancer, ovarian cancer
Nausea and vomiting
Thiotepa Same as above Breast cancer, ovarian cancer, superficial bladder cancer
Nausea and vomiting
Busulfan Same as above CML Nausea and vomiting
Carmustine Same as above Brain cancer, Hodgkin’s and non-Hodgkin’s lymphoma
Nausea and vomiting Myelosuppression; rarely interstitial lung disease and interstitial nephritis
Lomustine Same as above Brain cancer Nausea and vomiting
Altretamine Same as above Ovarian cancer Nausea and vomiting Myelosuppression, periph-eral neuropathy, flu-like syndrome
Temozolomide Methylates DNA and inhibits DNA synthesis and function
Brain cancer, melanoma Nausea and vomiting, headache and fatigue
Myelosuppression, mild elevation in liver function tests, photosensitivity
Procarbazine Methylates DNA and inhibits DNA synthesis and function
Hodgkin’s and non-Hodgkin’slymphoma, brain tumors
Central nervous system depression
Myelosuppression, hypersensitivity reactions
Dacarbazine Methylates DNA and inhibits DNA synthesis and function
Hodgkin’s lymphoma, melanoma, soft tissue sarcoma
Nausea and vomiting Myelosuppression, central nervous system toxicity with neuropathy, ataxia, lethargy, andconfusion
Cisplatin Forms intrastrand and inter-strand DNA cross-links; binding to nuclear and cytoplasmic proteins
Non-small cell and small cell lung cancer, breast cancer, bladder can-cer, cholangiocarcinoma, gastroe-sophageal cancer, head and neck cancer, ovarian cancer, germ cell cancer
Nausea and vomiting Nephrotoxicity, peripheral sensory neuropathy, ototoxicity, nervedysfunction
Carboplatin Same as cisplatin Non-small cell and small cell lung cancer, breast cancer, bladder cancer, head and neck cancer, ovarian cancer
Nausea and vomiting Myelosuppression; rarely peripheral neuropathy, renal toxicity, hepatic dysfunction
Oxaliplatin Same as cisplatin Colorectal cancer, gastroeso-phageal cancer, pancreatic cancer
Nausea and vomiting, laryngopharyngeal dysesthesias
Myelosuppression, periph-eral sensory neuropathy, diarrhea
CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia.
054-Katzung_Ch054_p949-976.indd 956 9/21/11 12:11:47 PM

CHAPTER 54 Cancer Chemotherapy 957
in the treatment of brain tumors. Although the majority of alkyla-tions by the nitrosoureas are on the N7 position of guanine in DNA, the critical alkylation responsible for cytotoxicity appears to be on the O6 position of guanine, which leads to G-C crosslinks in DNA. After oral administration of lomustine, peak plasma levels of metabolites appear within 1–4 hours; central nervous system concentrations reach 30–40% of the activity present in the plasma. Urinary excretion appears to be the major route of elimination from the body. One naturally occurring sugar-containing nitrosou-rea, streptozocin, is interesting because it has minimal bone mar-row toxicity. This agent has activity in the treatment of insulin-secreting islet cell carcinoma of the pancreas.
NONCLASSIC ALKYLATING AGENTS
Several other compounds have mechanisms of action that involve DNA alkylation as their cytotoxic mechanism of action. These agents include procarbazine, dacarbazine, and bendamus-tine. Their respective clinical activities and associated toxicities are listed in Table 54–2 .
Procarbazine Procarbazine is an orally active methylhydrazine derivative, and in the clinical setting, it is used in combination regimens for Hodgkin’s and non-Hodgkin’s lymphoma as well as brain tumors.
The precise mechanism of action of procarbazine is uncertain; however, it inhibits DNA, RNA, and protein biosynthesis; pro-longs interphase; and produces chromosome breaks. Oxidative metabolism of this drug by microsomal enzymes generates azopro-carbazine and H 2 O 2 , which may be responsible for DNA strand scission. A variety of other drug metabolites are formed that may be cytotoxic. One metabolite is a weak monoamine oxidase (MAO) inhibitor, and adverse events can occur when procarbazine is given with other MAO inhibitors as well as with sympathomimetic agents, tricyclic antidepressants, antihistamines, central nervous system depressants, antidiabetic agents, alcohol, and tyramine-containing foods.
There is an increased risk of secondary cancers in the form of acute leukemia, and its carcinogenic potential is thought to be higher than that of most other alkylating agents.
Dacarbazine Dacarbazine is a synthetic compound that functions as an alkylat-ing agent following metabolic activation in the liver by oxidative N -demethylation to the monomethyl derivative. This metabolite spontaneously decomposes to diazomethane, which generates a methyl carbonium ion that is believed to be the key cytotoxic spe-cies. Dacarbazine is administered parenterally and is used in the treatment of malignant melanoma, Hodgkin’s lymphoma, soft tis-sue sarcomas, and neuroblastoma. In terms of safety profile, the main dose-limiting toxicity is myelosuppression, but nausea and vomiting can be severe in some cases. This agent is a potent vesi-cant, and care must be taken to avoid extravasation during drug administration.
Bendamustine Bendamustine is a bifunctional alkylating agent consisting of a purine benzimidazole ring and a nitrogen mustard moiety. As with other alkylating agents, it forms cross-links with DNA resulting in single- and double-stranded breaks, leading to inhibition of DNA synthesis and function. This molecule also inhibits mitotic check-points and induces mitotic catastrophe, which leads to cell death. Of note, the cross-resistance between bendamustine and other alkylating agents is only partial, thereby providing a rationale for its clinical activity despite the development of resistance to other alkylating agents. This agent is approved for use in chronic lym-phocytic leukemia, with activity also observed in Hodgkin’s and non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer. The main dose-limiting toxicities include myelosuppression and mild nausea and vomiting. Hypersensitivity infusion reactions, skin rash, and other skin reactions occur rarely.
PLATINUM ANALOGS
Three platinum analogs are currently used in clinical practice: cis-platin, carboplatin, and oxaliplatin. Cisplatin (cis-diamminedichlo-roplatinum [II]) is an inorganic metal complex that was initially discovered through a serendipitous observation that neutral plati-num complexes inhibited division and filamentous growth of Escherichia coli. Several platinum analogs were subsequently synthe-sized. Although the precise mechanism of action of the platinum analogs is unclear, they are thought to exert their cytotoxic effects in the same manner as alkylating agents. As such, they kill tumor cells in all stages of the cell cycle and bind DNA through the formation of intrastrand and interstrand cross-links, thereby leading to inhibi-tion of DNA synthesis and function. The primary binding site is the N7 position of guanine, but covalent interaction with the N3 posi-tion of adenine and O6 position of cytosine can also occur. In addi-tion to targeting DNA, the platinum analogs have been shown to bind to both cytoplasmic and nuclear proteins, which may also contribute to their cytotoxic and antitumor effects. The platinum complexes appear to synergize with certain other anticancer drugs, including alkylating agents, fluoropyrimidines, and taxanes.
Cl
Pt
ClH3N
H3N
Cisplatin
Cisplatin has major antitumor activity in a broad range of solid tumors, including non-small cell and small cell lung cancer, esophageal and gastric cancer, cholangiocarcinoma, head and neck cancer, and genitourinary cancers, particularly testicular, ovarian, and bladder cancer. When used in combination regimens, cispla-tin-based therapy has led to the cure of nonseminomatous testicu-lar cancer. Cisplatin and the other platinum analogs are extensively cleared by the kidneys and excreted in the urine. As a result, dose modification is required in patients with renal dysfunction.
Carboplatin is a second-generation platinum analog whose mechanisms of cytotoxic action, mechanisms of resistance, and clinical pharmacology are identical to those described for cisplatin.
054-Katzung_Ch054_p949-976.indd 957 9/21/11 12:11:47 PM

958 SECTION VIII Chemotherapeutic Drugs
As with cisplatin, carboplatin has broad-spectrum activity against a wide range of solid tumors. However, in contrast to cisplatin, it exhibits significantly less renal toxicity and gastrointestinal toxicity. Its main dose-limiting toxicity is myelosuppression. It has therefore been widely used in transplant regimens to treat refractory hema-tologic malignancies. Moreover, since vigorous intravenous hydra-tion is not required for carboplatin therapy, carboplatin is viewed as an easier agent to administer to patients, and as such, it has replaced cisplatin in various combination chemotherapy regimens.
Oxaliplatin is a third-generation diaminocyclohexane plati-num analog. Its mechanism of action and clinical pharmacology are identical to those of cisplatin and carboplatin. However, tumors that are resistant to cisplatin or carboplatin on the basis of mismatch repair defects are not cross-resistant to oxaliplatin, and this finding may explain the activity of this platinum compound in colorectal cancer. Oxaliplatin was initially approved for use as second-line therapy in combination with the fluoropyrimidine 5-fluorouracil (5-FU) and leucovorin, termed the FOLFOX regi-men, for metastatic colorectal cancer. The FOLFOX regimen was subsequently (2005) approved for the first-line treatment of meta-static colorectal cancer. At this time, oxaliplatin-based chemo-therapy has also been approved in the adjuvant therapy of high-risk stage II and stage III colon cancer. Clinical activity has been observed in other gastrointestinal cancers, such as pancreatic, gastroesophageal, and hepatocellular cancer. In the side effect profile, neurotoxicity is the main dose-limiting toxicity, and is manifested by a peripheral sensory neuropathy. There are two forms of neurotoxicity, an acute form that is often triggered and worsened by exposure to cold, and a chronic form that is dose-dependent. Although this chronic form is dependent on the cumulative dose of drug administered, it tends to be reversible, in contrast to cisplatin-induced neurotoxicity.
The major toxicities of the individual platinum analogs are outlined in Table 54–2 .
ANTIMETABOLITES The development of drugs with actions on intermediary metabolism of proliferating cells has been important both conceptually and clini-cally. While biochemical properties unique to all cancer cells have yet to be discovered, there are a number of quantitative differences in metabolism between cancer cells and normal cells that render cancer cells more sensitive to the antimetabolites. Many of these agents have been rationally designed and synthesized based on knowledge of critical cellular processes involved in DNA biosynthesis.
The individual antimetabolites and their respective clinical spectrum and toxicities are presented in Table 54–3 and are dis-cussed below.
ANTIFOLATES
Methotrexate Methotrexate (MTX) is a folic acid analog that binds with high affin-ity to the active catalytic site of dihydrofolate reductase (DHFR). This results in inhibition of the synthesis of tetrahydrofolate (THF),
the key one-carbon carrier for enzymatic processes involved in de novo synthesis of thymidylate, purine nucleotides, and the amino acids serine and methionine. Inhibition of these various metabolic processes thereby interferes with the formation of DNA, RNA, and key cellular proteins (see Figure 33–3 ). Intracellular formation of polyglutamate metabolites, with the addition of up to 5–7 glutamate residues, is critically important for the therapeutic action of MTX, and this process is catalyzed by the enzyme folylpolyglutamate syn-thase (FPGS). MTX polyglutamates are selectively retained within cancer cells, and they display increased inhibitory effects on enzymes involved in de novo purine nucleotide and thymidylate biosynthesis, making them important determinants of MTX’s cytotoxic action.
N
H
C
NH2
NN
N N
C
O
N
H
CH
CH2
CH2
COOH
COOH
Folic acid
H2
OH
N
CH3
C
NH2
NN
N N
C
O
N
H
CH
CH2
CH2
COOH
COOH
Methotrexate
H2
NH2
Resistance to MTX has been attributed to (1) decreased drug trans-port via the reduced folate carrier or folate receptor protein, (2) decreased formation of cytotoxic MTX polyglutamates, (3) increased levels of the target enzyme DHFR through gene amplification and other genetic mechanisms, and (4) altered DHFR protein with reduced affinity for MTX. Recent studies have suggested that decreased accumulation of drug through activation of the multidrug resistance transporter P170 glycoprotein may also result in drug resistance.
MTX is administered by the intravenous, intrathecal, or oral route. However, oral bioavailability is saturable and erratic at doses greater than 25 mg/m 2 . Renal excretion is the main route of elimination and is mediated by glomerular filtration and tubular secretion. As a result, dose modification is required in the setting of renal dysfunction. Care must also be taken when MTX is used in the presence of drugs such as aspirin, nonsteroidal anti-inflam-matory agents, penicillin, and cephalosporins, as these agents inhibit the renal excretion of MTX. The biologic effects of MTX can be reversed by administration of the reduced folate leucovorin (5-formyltetrahydrofolate) or by L -leucovorin, which is the active enantiomer. Leucovorin rescue is used in conjunction with high-dose MTX therapy to rescue normal cells from undue toxicity, and it has also been used in cases of accidental drug overdose. The main adverse effects are listed in Table 54–3 .
054-Katzung_Ch054_p949-976.indd 958 9/21/11 12:11:47 PM

CHAPTER 54 Cancer Chemotherapy 959
Pemetrexed Pemetrexed is a pyrrolopyrimidine antifolate analog with activity in the S phase of the cell cycle. As in the case of MTX, it is trans-ported into the cell via the reduced folate carrier and requires activation by FPGS to yield higher polyglutamate forms. While this agent targets DHFR and enzymes involved in de novo purine nucleotide biosynthesis, its main mechanism of action is inhibition of thymidylate synthase. At present, this antifolate is approved for use in combination with cisplatin in the treatment of mesothe-lioma, as a single agent in the second-line therapy of non-small cell lung cancer, and in combination with cisplatin for the first-line treatment of non-small cell lung cancer. As with MTX, pemetrexed is mainly excreted in the urine, and dose modification is required in the setting of renal dysfunction. The main adverse effects include myelosuppression, skin rash, mucositis, diarrhea, fatigue, and hand-foot syndrome. Of note, vitamin supplementation with
folic acid and vitamin B 12 appear to reduce the toxicities associated with pemetrexed, while not interfering with clinical efficacy. The hand-foot syndrome is manifested by painful erythema and swell-ing of the hands and feet, and dexamethasone treatment has been shown to be effective in reducing the incidence and severity of this toxicity.
Pralatrexate Pralatrexate is a 10-deaza-aminopterin antifolate analog, and as in the case of MTX, it is transported into the cell via the reduced folate carrier (RFC) and requires activation by FPGS to yield higher polyglutamate forms. However, this molecule was designed to be a more potent substrate for the RFC-1 carrier protein as well as an improved substrate for FPGS. It inhibits DHFR, inhibits enzymes involved in de novo purine nucleotide biosynthesis, and also inhibits thymidylate synthase. Although pralatrexate was
TABLE 54–3 Antimetabolites: Clinical activity and toxicities.
Drug Mechanism of Action Clinical Applications Toxicity
Capecitabine Inhibits TS; incorporation of FUTP into RNA resulting in alteration in RNA processing; incorporation of FdUTP into DNA resulting in inhibition of DNA synthesis and function
Breast cancer, colorectal cancer, gas-troesophageal cancer, hepatocellular cancer, pancreatic cancer
Diarrhea, hand-foot syndrome, myelosuppression, nausea and vomiting
5-Fluorouracil Inhibits TS; incorporation of FUTP into RNA result-ing in alteration in RNA processing; incorporation of FdUTP into DNA resulting in inhibition of DNA synthesis and function
Colorectal cancer, anal cancer, breast cancer, gastroesophageal cancer, head and neck cancer, hepatocellular cancer
Nausea, mucositis, diarrhea, bone marrow depression, neurotoxicity
Methotrexate Inhibits DHFR; inhibits TS; inhibits de novo purine nucleotide synthesis
Breast cancer, head and neck cancer, osteogenic sarcoma, primary central nervous system lymphoma, non-Hodg-kin’s lymphoma, bladder cancer, chori-ocarcinoma
Mucositis, diarrhea, myelosup-pression with neutropenia and thrombocytopenia
Pemetrexed Inhibits TS, DHFR, and purine nucleotide synthesis Mesothelioma, non-small cell lung cancer
Myelosuppression, skin rash, mucositis, diarrhea, fatigue, hand-foot syndrome
Cytarabine Inhibits DNA chain elongation, DNA synthesis and repair; inhibits ribonucleotide reductase with reduced formation of dNTPs; incorporation of cytarabine triphosphate into DNA
AML, ALL, CML in blast crisis Nausea and vomiting, myelosup-pression with neutropenia and thrombocytopenia, cerebellar ataxia
Gemcitabine Inhibits DNA synthesis and repair; inhibits ribonucleotide reductase with reduced formation of dNTPs; incorporation of gemcitabine triphos-phate into DNA resulting in inhibition of DNA synthesis and function
Pancreatic cancer, bladder cancer, breast cancer, non-small cell lung can-cer, ovarian cancer, non-Hodgkin’s lym-phoma, soft tissue sarcoma
Nausea, vomiting, diarrhea, myelosuppression
Fludarabine Inhibits DNA synthesis and repair; inhibits ribonu-cleotide reductase; incorporation of fludarabine triphosphate into DNA; induction of apoptosis
Non-Hodgkin’s lymphoma, CLL Myelosuppression, immunosuppression, fever, myalgias, arthralgias
Cladribine Inhibits DNA synthesis and repair; inhibits ribonucleotide reductase; incorporation of cladrib-ine triphosphate into DNA; induction of apoptosis
Hairy cell leukemia, CLL, non-Hodgkin’s lymphoma
Myelosuppression, nausea and vomiting, and immunosuppression
6-Mercaptopu-rine (6-MP)
Inhibits de novo purine nucleotide synthesis; incorporation of triphosphate into RNA; incorporation of triphosphate into DNA
AML Myelosuppression, immunosuppression, and hepatotoxicity
6-Thioguanine Same as 6-MP ALL, AML Same as above
ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; DHFR, dihydrofolate reductase; dNTP, deoxyribonucleotide triphosphate; FdUTP, 5-fluorodeoxyuridine-5’-triphosphate; FUTP, 5-fluorouridine-5’-triphosphate; TS, thymidine synthase.
054-Katzung_Ch054_p949-976.indd 959 9/21/11 12:11:47 PM

960 SECTION VIII Chemotherapeutic Drugs
originally developed for non-small lung cancer, it is presently approved for use in the treatment of relapsed or refractory periph-eral T-cell lymphoma. As with the other antifolate analogs, prala-trexate is mainly excreted in the urine, and dose modification is required in the setting of renal dysfunction. The main adverse effects include myelosuppression, skin rash, mucositis, diarrhea, and fatigue. Vitamin supplementation with folic acid and vitamin B 12 appear to reduce the toxicities associated with pralatrexate, while not interfering with clinical efficacy.
FLUOROPYRIMIDINES
5-Fluorouracil 5-Fluorouracil (5-FU) is inactive in its parent form and requires activation via a complex series of enzymatic reactions to ribosyl and deoxyribosyl nucleotide metabolites. One of these metabo-lites, 5-fluoro-2′-deoxyuridine-5′-monophosphate (FdUMP), forms a covalently bound ternary complex with the enzyme thy-midylate synthase and the reduced folate 5,10-methylenetetrahy-drofolate, a reaction critical for the de novo synthesis of thymidylate. This results in inhibition of DNA synthesis through “thymineless death.” 5-FU is converted to 5-fluorouridine-5′-triphosphate (FUTP), which is then incorporated into RNA, where it interferes with RNA processing and mRNA translation. 5-FU is also converted to 5-fluorodeoxyuridine-5′-triphosphate (FdUTP), which can be incorporated into cellular DNA, resulting in inhibition of DNA synthesis and function. Thus, the cytotoxic-ity of 5-FU is thought to be the result of combined effects on both DNA- and RNA-mediated events.
5-FU is administered intravenously, and the clinical activity of this drug is highly schedule-dependent. Because of its extremely short half-life, on the order of 10–15 minutes, infusional schedules of administration have been generally favored over bolus schedules. Up to 80–85% of an administered dose of 5-FU is catabolized by the enzyme dihydropyrimidine dehydrogenase (DPD). Of note, a pharmacogenetic syndrome involving partial or complete defi-ciency of the DPD enzyme is seen in up to 5% of cancer patients; in this setting, severe toxicity in the form of myelosuppression, diarrhea, nausea and vomiting, and neurotoxicity is observed.
Uracil
NH
HN
O
O
H
5-FU
NH
HN
O
O
F
5-FU remains the most widely used agent in the treatment of colorectal cancer, both as adjuvant therapy and for advanced disease. It also has activity against a wide variety of solid tumors, including cancers of the breast, stomach, pancreas, esophagus, liver, head and neck, and anus. Major toxicities include myelosuppression, gastro-intestinal toxicity in the form of mucositis and diarrhea, skin toxic-ity manifested by the hand-foot syndrome, and neurotoxicity.
Capecitabine Capecitabine is a fluoropyrimidine carbamate prodrug with 70–80% oral bioavailability. It undergoes extensive metabolism in the liver by the enzyme carboxylesterase to an intermediate, 5′-deoxy-5-fluorocytidine. This metabolite is then converted to 5′-deoxy-5-fluorouridine by the enzyme cytidine deaminase. These two initial steps occur mainly in the liver. The 5′-deoxy-5-fluorouridine metabolite is finally hydrolyzed by thymidine phos-phorylase to 5-FU directly in the tumor. The expression of thymidine phosphorylase has been shown to be significantly higher in a broad range of solid tumors than in corresponding normal tissue, particularly in breast cancer and colorectal cancer.
This oral fluoropyrimidine is used in the treatment of meta-static breast cancer either as a single agent or in combination with other anticancer agents, including docetaxel, paclitaxel, lapatinib, ixabepilone, and trastuzumab. It is also approved for use in the adjuvant therapy of stage III and high-risk stage II colon cancer as well as for treatment of metastatic colorectal cancer as monother-apy. At this time, significant efforts are directed at combining this agent with other active cytotoxic agents, including irinotecan and oxaliplatin. In Europe and Asia, the capecitabine/oxaliplatin (XELOX) regimen is approved for the first-line treatment of meta-static colorectal cancer, and this regimen is now widely used in the USA. The main toxicities of capecitabine include diarrhea and the hand-foot syndrome. While myelosuppression, nausea and vomit-ing, and mucositis are also observed with this agent, their incidence is significantly less than that observed with intravenous 5-FU.
DEOXYCYTIDINE ANALOGS
Cytarabine Cytarabine (ara-C) is an S phase-specific antimetabolite that is con-verted by deoxycytidine kinase to the 5'-mononucleotide (ara-CMP). Ara-CMP is further metabolized to the diphosphate and triphos-phate metabolites, and the ara-CTP triphosphate is felt to be the main cytotoxic metabolite. Ara-CTP competitively inhibits DNA polymerase-α and DNA polymerase-β, thereby resulting in blockade of DNA synthesis and DNA repair, respectively. This metabolite is also incorporated into RNA and DNA. Incorporation into DNA leads to interference with chain elongation and defective ligation of fragments of newly synthesized DNA. The cellular retention of ara-CTP appears to correlate with its lethality to malignant cells.
Cytosinedeoxyriboside
N
N
O
NH2
O
HO
HOCH2
Cytosinearabinoside(cytarabine)
N
N
O
NH2
O
HO
HOCH2
HO
054-Katzung_Ch054_p949-976.indd 960 9/21/11 12:11:47 PM

CHAPTER 54 Cancer Chemotherapy 961
After intravenous administration, the drug is cleared rapidly, with most of an administered dose being deaminated to inactive forms. The stoichiometric balance between the level of activation and catabolism of cytarabine is important in determining its even-tual cytotoxicity.
The clinical activity of cytarabine is highly schedule-dependent and because of its rapid degradation, it is usually administered via continuous infusion over a 5–7 day period. Its activity is limited exclusively to hematologic malignancies, including acute myelog-enous leukemia and non-Hodgkin’s lymphoma. This agent has absolutely no activity in solid tumors. The main adverse effects associated with cytarabine therapy include myelosuppression, mucositis, nausea and vomiting, and neurotoxicity when high-dose therapy is administered.
Gemcitabine Gemcitabine is a fluorine-substituted deoxycytidine analog that is phosphorylated initially by the enzyme deoxycytidine kinase to the monophosphate form and then by other nucleoside kinases to the diphosphate and triphosphate nucleotide forms. The antitu-mor effect is considered to result from several mechanisms: inhibi-tion of ribonucleotide reductase by gemcitabine diphosphate, which reduces the level of deoxyribonucleoside triphosphates required for DNA synthesis; inhibition by gemcitabine triphos-phate of DNA polymerase-α and DNA polymerase-β, thereby resulting in blockade of DNA synthesis and DNA repair; and incorporation of gemcitabine triphosphate into DNA, leading to inhibition of DNA synthesis and function. Following incorpora-tion of the gemcitabine triphosphate into DNA, only one addi-tional nucleotide can be added to the growing DNA strand, resulting in chain termination.
N
N
O
NH2
OHOCH2
OH F
F
Gemcitabine
This nucleoside analog was initially approved for use in advanced pancreatic cancer but is now widely used to treat a broad range of malignancies, including non-small cell lung cancer, blad-der cancer, ovarian cancer, soft tissue sarcoma, and non-Hodgkin’s lymphoma. Myelosuppression in the form of neutropenia is the principal dose-limiting toxicity. Nausea and vomiting occur in 70% of patients and a flu-like syndrome has also been observed. In rare cases, renal microangiopathy syndromes, including hemo-lytic-uremic syndrome and thrombotic thrombocytopenic pur-pura have been reported.
PURINE ANTAGONISTS
6-Thiopurines 6-Mercaptopurine (6-MP) was the first of the thiopurine analogs found to be effective in cancer therapy. This agent is used primarily in the treatment of childhood acute leukemia, and a closely related analog, azathioprine, is used as an immunosuppressive agent (see Chapter 55 ). As with other thiopurines, 6-MP is inactive in its parent form and must be metabolized by hypoxanthine-guanine phosphoribosyl transferase (HGPRT) to form the monophosphate nucleotide 6-thioinosinic acid, which in turn inhibits several enzymes of de novo purine nucleotide synthesis ( Figure 54–6 ). The monophosphate form is eventually metabolized to the triphosphate form, which can then be incorporated into both RNA and DNA. Significant levels of thioguanylic acid and 6-methylmercaptopurine ribotide (MMPR) are also formed from 6-MP. These metabolites may contribute to its cytotoxic action.
6-Thioguanine (6-TG) also inhibits several enzymes in the de novo purine nucleotide biosynthetic pathway ( Figure 54–6 ). Various metabolic lesions result, including inhibition of purine nucleotide interconversion; decrease in intracellular levels of gua-nine nucleotides, which leads to inhibition of glycoprotein synthe-sis; interference with the formation of DNA and RNA; and incorporation of thiopurine nucleotides into both DNA and RNA. 6-TG has a synergistic action when used together with cytarabine in the treatment of adult acute leukemia.
6-MP is converted to an inactive metabolite (6-thiouric acid) by an oxidation reaction catalyzed by xanthine oxidase, whereas 6-TG undergoes deamination. This is an important issue because the purine analog allopurinol, a potent xanthine oxidase inhibitor, is frequently used as a supportive care measure in the treatment of acute leukemias to prevent the development of hyperuricemia that often occurs with tumor cell lysis. Because allopurinol inhibits xan-thine oxidase, simultaneous therapy with allopurinol and 6-MP would result in increased levels of 6-MP, thereby leading to excessive
Inosinate(IMP)
Adenylosuccinate Adenylate(AMP)
Xanthylate(XMP)
Guanylate(GMP)
DNA
MercaptopurineThioguanine
Mercaptopurine
Hydroxyurea
–
–
–
–
FIGURE 54–6 Mechanism of action of 6-mercaptopurine and 6-thioguanine.
054-Katzung_Ch054_p949-976.indd 961 9/21/11 12:11:48 PM

962 SECTION VIII Chemotherapeutic Drugs
toxicity. In this setting, the dose of mercaptopurine must be reduced by 50–75%. In contrast, such an interaction does not occur with 6-TG, which can be used in full doses with allopurinol.
Hypoxanthine
N
N
NH
N
OH
6-Mercaptopurine
N
N
NH
N
SH
Allopurinol
N
N
NH
N
OH
Guanine
N
N
NH
N
OH
H2N
6-Thioguanine
N
N
NH
N
SH
H2N
The thiopurines are also metabolized by the enzyme thiopurine methyltransferase (TPMT), in which a methyl group is attached to the thiopurine ring. Patients who have a pharmacogenetic syndrome involving partial or complete deficiency of this enzyme are at increased risk for developing severe toxicities in the form of myelosuppression and gastrointestinal toxicity with mucositis and diarrhea.
Fludarabine Fludarabine phosphate is rapidly dephosphorylated to 2-fluoro-arab-inofuranosyladenosine and then phosphorylated intracellularly by deoxycytidine kinase to the triphosphate. The triphosphate metabo-lite interferes with the processes of DNA synthesis and DNA repair through inhibition of DNA polymerase-α and DNA polymerase-β. The triphosphate form can also be directly incorporated into DNA, resulting in inhibition of DNA synthesis and function. The diphos-phate metabolite of fludarabine inhibits ribonucleotide reductase, leading to inhibition of essential deoxyribonucleotide triphosphates. Finally, fludarabine induces apoptosis in susceptible cells through as yet undetermined mechanisms. This purine nucleotide analog is used mainly in the treatment of low-grade non-Hodgkin’s lymphoma and chronic lymphocytic leukemia (CLL). It is given parenterally, and up to 25–30% of parent drug is excreted in the urine. The main dose-limiting toxicity is myelosuppression. This agent is a potent immuno-suppressant with inhibitory effects on CD4 and CD8 T cells. Patients are at increased risk for opportunistic infections, including fungi, herpes, and Pneumocystis jiroveci pneumonia (PCP). Patients should receive PCP prophylaxis with trimethoprim-sulfamethoxazole (dou-ble strength) at least three times a week, and this should continue for up to 1 year after stopping fludarabine therapy.
Cladribine Cladribine (2-chlorodeoxyadenosine) is a purine nucleoside analog with high specificity for lymphoid cells. Inactive in its parent form, it is initially phosphorylated by deoxycytidine kinase to the mono-phosphate form and eventually metabolized to the triphosphate
form, which can then be incorporated into DNA. The triphos-phate metabolite can also interfere with DNA synthesis and DNA repair by inhibiting DNA polymerase-α and DNA polymerase-β, respectively. Cladribine is indicated for the treatment of hairy cell leukemia, with activity in other low-grade lymphoid malignancies such as CLL and low-grade non-Hodgkin’s lymphoma. It is nor-mally administered as a single continuous 7-day infusion; under these conditions, it has a very manageable safety profile with the main toxicity consisting of transient myelosuppression. As with other purine nucleoside analogs, it has immunosuppressive effects, and a decrease in CD4 and CD8 T cells, lasting for over 1 year, is observed in patients.
NATURAL PRODUCT CANCER CHEMOTHERAPY DRUGS VINCA ALKALOIDS
Vinblastine Vinblastine is an alkaloid derived from the periwinkle plant Vinca rosea . Its mechanism of action involves inhibition of tubulin polym-erization, which disrupts assembly of microtubules, an important part of the cytoskeleton and the mitotic spindle. This inhibitory effect results in mitotic arrest in metaphase, bringing cell division to a halt, which then leads to cell death. Vinblastine and other vinca alkaloids are metabolized by the liver P450 system, and the majority of the drug is excreted in feces via the hepatobiliary system. As such, dose modification is required in the setting of liver dysfunction. The main adverse effects are outlined in Table 54–4 , and they include nausea and vomiting, bone marrow suppression, and alopecia. This agent is also a potent vesicant, and care must be taken in its admin-istration. It has clinical activity in the treatment of Hodgkin’s and non-Hodgkin’s lymphomas, breast cancer, and germ cell cancer.
Vincristine Vincristine is another alkaloid derivative of V rosea and is closely related in structure to vinblastine. Its mechanism of action, mechanism of resistance, and clinical pharmacology are identical to those of vinblastine. Despite these similarities to vinblastine, vincristine has a strikingly different spectrum of clinical activity and safety profile.
R: CH3
NH N
CH3O N
R
HOCOCH3H
OH
C O CH3O
CH2CH3
H
N
H
C
CH3O
OHO
CH2CH3
Vinblastine
R: O
Vincristine
C H
054-Katzung_Ch054_p949-976.indd 962 9/21/11 12:11:48 PM
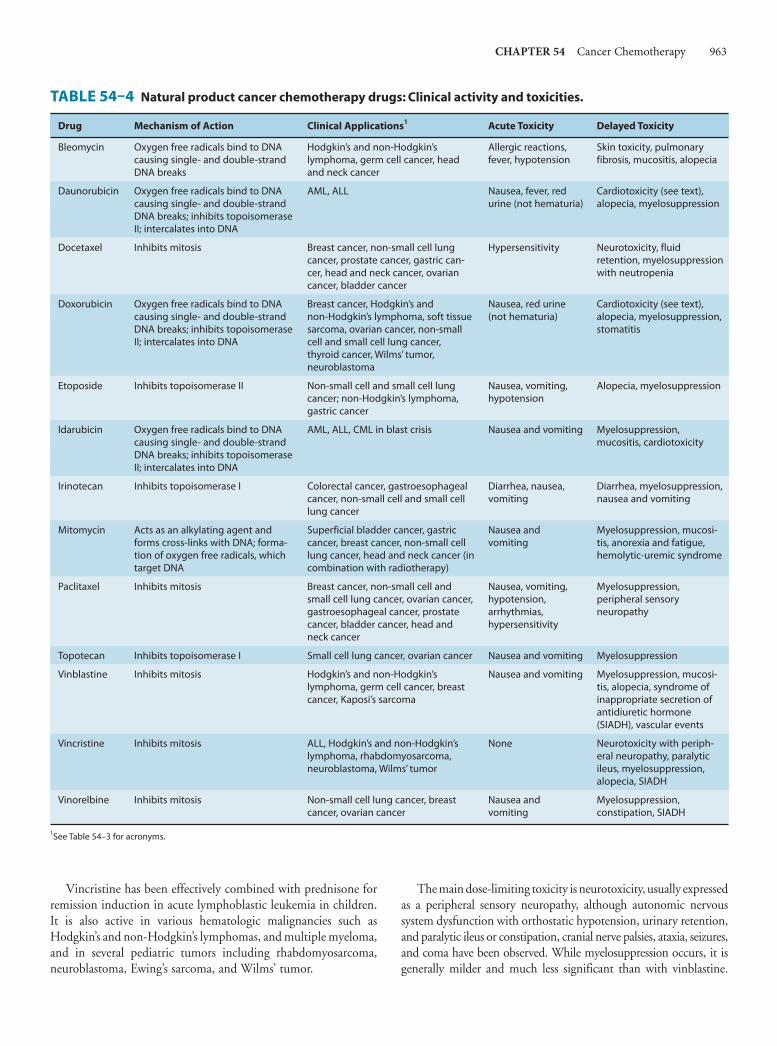
CHAPTER 54 Cancer Chemotherapy 963
Vincristine has been effectively combined with prednisone for remission induction in acute lymphoblastic leukemia in children. It is also active in various hematologic malignancies such as Hodgkin’s and non-Hodgkin’s lymphomas, and multiple myeloma, and in several pediatric tumors including rhabdomyosarcoma, neuroblastoma, Ewing’s sarcoma, and Wilms’ tumor.
The main dose-limiting toxicity is neurotoxicity, usually expressed as a peripheral sensory neuropathy, although autonomic nervous system dysfunction with orthostatic hypotension, urinary retention, and paralytic ileus or constipation, cranial nerve palsies, ataxia, seizures, and coma have been observed. While myelosuppression occurs, it is generally milder and much less significant than with vinblastine.
TABLE 54–4 Natural product cancer chemotherapy drugs: Clinical activity and toxicities.
Drug Mechanism of Action Clinical Applications1 Acute Toxicity Delayed Toxicity
Bleomycin Oxygen free radicals bind to DNA causing single- and double-strand DNA breaks
Hodgkin’s and non-Hodgkin’s lymphoma, germ cell cancer, head and neck cancer
Allergic reactions, fever, hypotension
Skin toxicity, pulmonary fibrosis, mucositis, alopecia
Daunorubicin Oxygen free radicals bind to DNA causing single- and double-strand DNA breaks; inhibits topoisomerase II; intercalates into DNA
AML, ALL Nausea, fever, red urine (not hematuria)
Cardiotoxicity (see text), alopecia, myelosuppression
Docetaxel Inhibits mitosis Breast cancer, non-small cell lung cancer, prostate cancer, gastric can-cer, head and neck cancer, ovarian cancer, bladder cancer
Hypersensitivity Neurotoxicity, fluid retention, myelosuppression with neutropenia
Doxorubicin Oxygen free radicals bind to DNA causing single- and double-strand DNA breaks; inhibits topoisomerase II; intercalates into DNA
Breast cancer, Hodgkin’s and non-Hodgkin’s lymphoma, soft tissue sarcoma, ovarian cancer, non-small cell and small cell lung cancer, thyroid cancer, Wilms’ tumor, neuroblastoma
Nausea, red urine (not hematuria)
Cardiotoxicity (see text), alopecia, myelosuppression, stomatitis
Etoposide Inhibits topoisomerase II Non-small cell and small cell lung cancer; non-Hodgkin’s lymphoma, gastric cancer
Nausea, vomiting, hypotension
Alopecia, myelosuppression
Idarubicin Oxygen free radicals bind to DNA causing single- and double-strand DNA breaks; inhibits topoisomerase II; intercalates into DNA
AML, ALL, CML in blast crisis Nausea and vomiting Myelosuppression, mucositis, cardiotoxicity
Irinotecan Inhibits topoisomerase I Colorectal cancer, gastroesophageal cancer, non-small cell and small cell lung cancer
Diarrhea, nausea, vomiting
Diarrhea, myelosuppression, nausea and vomiting
Mitomycin Acts as an alkylating agent and forms cross-links with DNA; forma-tion of oxygen free radicals, which target DNA
Superficial bladder cancer, gastric cancer, breast cancer, non-small cell lung cancer, head and neck cancer (in combination with radiotherapy)
Nausea and vomiting
Myelosuppression, mucosi-tis, anorexia and fatigue, hemolytic-uremic syndrome
Paclitaxel Inhibits mitosis Breast cancer, non-small cell and small cell lung cancer, ovarian cancer, gastroesophageal cancer, prostate cancer, bladder cancer, head and neck cancer
Nausea, vomiting, hypotension, arrhythmias, hypersensitivity
Myelosuppression, peripheral sensory neuropathy
Topotecan Inhibits topoisomerase I Small cell lung cancer, ovarian cancer Nausea and vomiting Myelosuppression
Vinblastine Inhibits mitosis Hodgkin’s and non-Hodgkin’s lymphoma, germ cell cancer, breast cancer, Kaposi’s sarcoma
Nausea and vomiting Myelosuppression, mucosi-tis, alopecia, syndrome of inappropriate secretion of antidiuretic hormone (SIADH), vascular events
Vincristine Inhibits mitosis ALL, Hodgkin’s and non-Hodgkin’s lymphoma, rhabdomyosarcoma, neuroblastoma, Wilms’ tumor
None Neurotoxicity with periph-eral neuropathy, paralytic ileus, myelosuppression, alopecia, SIADH
Vinorelbine Inhibits mitosis Non-small cell lung cancer, breast cancer, ovarian cancer
Nausea and vomiting
Myelosuppression, constipation, SIADH
1See Table 54–3 for acronyms.
054-Katzung_Ch054_p949-976.indd 963 9/21/11 12:11:48 PM

964 SECTION VIII Chemotherapeutic Drugs
The other adverse effect that may develop is the syndrome of inap-propriate secretion of antidiuretic hormone (SIADH).
Vinorelbine Vinorelbine is a semisynthetic derivative of vinblastine whose mechanism of action is identical to that of vinblastine and vincris-tine, ie, inhibition of mitosis of cells in the M phase through inhibition of tubulin polymerization. This agent has activity in non-small cell lung cancer, breast cancer, and ovarian cancer. Myelosuppression with neutropenia is the dose-limiting toxicity, but other adverse effects include nausea and vomiting, transient elevations in liver function tests, neurotoxicity, and SIADH.
TAXANES & RELATED DRUGS
Paclitaxel is an alkaloid ester derived from the Pacific yew ( Taxus brevifolia ) and the European yew ( Taxus baccata ). The drug func-tions as a mitotic spindle poison through high-affinity binding to microtubules with enhancement of tubulin polymerization. This promotion of microtubule assembly by paclitaxel occurs in the absence of microtubule-associated proteins and guanosine triphos-phate and results in inhibition of mitosis and cell division.
Paclitaxel has significant activity in a broad range of solid tumors, including ovarian, advanced breast, non-small cell and small cell lung, head and neck, esophageal, prostate, and bladder cancers and AIDS-related Kaposi’s sarcoma. It is metabolized extensively by the liver P450 system, and nearly 80% of the drug is excreted in feces via the hepatobiliary route. Dose reduction is required in patients with liver dysfunction. The primary dose-limiting toxicities are listed in Table 54–4 . Hypersensitivity reac-tions may be observed in up to 5% of patients, but the incidence is significantly reduced by premedication with dexamethasone, diphenhydramine, and an H 2 blocker.
A novel albumin-bound paclitaxel formulation (Abraxane) is approved for use in metastatic breast cancer. In contrast to paclitaxel, this formulation is not associated with hypersensitivity reactions, and premedication to prevent such reactions is not required. Moreover, this agent has significantly reduced myelosuppressive effects com-pared with paclitaxel, and the neurotoxicity that results appears to be more readily reversible than is typically observed with paclitaxel.
Docetaxel is a semisynthetic taxane derived from the European yew tree. Its mechanism of action, metabolism, and elimination are identical to those of paclitaxel. It is approved for use as second-line therapy in advanced breast cancer and non-small cell lung cancer, and it also has major activity in head and neck cancer, small cell lung cancer, gastric cancer, advanced platinum-refrac-tory ovarian cancer, and bladder cancer. Its major toxicities are listed in Table 54–4 .
Cabazitaxel is a semisynthetic taxane produced from a precursor extracted from the yew tree. Its mechanism of action, metabolism, and elimination are identical to those of the other taxanes. However, unlike other taxanes, cabazitaxel is a poor substrate for the multi-drug resistance P-glycoprotein efflux pump and may therefore be useful for treating multidrug-resistant tumors. It is approved for use
in combination with prednisone in the second-line therapy of hor-mone-refractory metastatic prostate cancer previously treated with a docetaxel-containing regimen. Its major toxicities include myelo-suppression, neurotoxicity, and allergic reactions.
Although not strictly a taxane, ixabepilone is a semisynthetic epothilone B analog that functions as a microtubule inhibitor and binds directly to β-tubulin subunits on microtubules, leading to inhibition of normal microtubule dynamics. As such, it is active in the M phase of the cell cycle. This agent is presently approved for metastatic breast cancer in combination with the oral fluoropy-rimidine capecitabine or as monotherapy. Of note, this agent continues to have activity in drug-resistant tumors that overex-press P-glycoprotein or tubulin mutations. The main adverse effects include myelosuppression, hypersensitivity reactions, and neurotoxicity in the form of peripheral sensory neuropathy.
EPIPODOPHYLLOTOXINS
Etoposide is a semisynthetic derivative of podophyllotoxin, which is extracted from the mayapple root ( Podophyllum peltatum ). Intravenous and oral formulations of etoposide are approved for clinical use in the USA. The oral bioavailability is about 50%, requiring the oral dose to be twice that of an intravenous dose. Up to 30–50% of drug is excreted in the urine, and dose reduction is required in the setting of renal dysfunction. The main site of action is inhibition of the DNA enzyme topoisomerase II. Etoposide has clinical activity in germ cell cancer, small cell and non-small cell lung cancer, Hodgkin’s and non-Hodgkin’s lym-phomas, and gastric cancer. In addition, it is effective in high-dose regimens in the transplant setting for breast cancer and lympho-mas. Major toxicities are listed in Table 54–4 .
CAMPTOTHECINS
The camptothecins are natural products derived from the Camptotheca acuminata tree originally found in China; they inhibit the activity of topoisomerase I, the key enzyme responsible for cutting and religating single DNA strands. Inhibition of this enzyme results in DNA damage. Topotecan and irinotecan are the two camptothecin analogs used in clinical practice in the USA. Although they both inhibit the same molecular target, their spec-trum of clinical activity is quite different. Topotecan is indicated in the treatment of advanced ovarian cancer as second-line therapy following initial treatment with platinum-based chemotherapy. It is also approved as second-line therapy of small cell lung cancer. The main route of elimination is renal excretion, and dosage must be adjusted in patients with renal impairment.
Irinotecan is a prodrug that is converted mainly in the liver by the carboxylesterase enzyme to the SN-38 metabolite, which is 1000-fold more potent as an inhibitor of topoisomerase I than the parent compound. In contrast to topotecan, irinotecan and SN-38 are mainly eliminated in bile and feces, and dose reduction is required in the setting of liver dysfunction. Irinotecan was originally approved as second-line monotherapy in patients with metastatic
054-Katzung_Ch054_p949-976.indd 964 9/21/11 12:11:48 PM

CHAPTER 54 Cancer Chemotherapy 965
colorectal cancer who had failed fluorouracil-based therapy. It is now approved as first-line therapy when used in combination with 5-FU and leucovorin. Myelosuppression and diarrhea are the two most common adverse events ( Table 54–4 ). There are two forms of diarrhea: an early form that occurs within 24 hours after administra-tion and is thought to be a cholinergic event effectively treated with atropine, and a late form that usually occurs 2–10 days after treat-ment. The late diarrhea can be severe, leading to significant electro-lyte imbalance and dehydration in some cases.
ANTITUMOR ANTIBIOTICS Screening of microbial products has led to the discovery of a num-ber of growth-inhibiting compounds that have proved to be clini-cally useful in cancer chemotherapy. Many of these antibiotics bind to DNA through intercalation between specific bases and block the synthesis of RNA, DNA, or both; cause DNA strand scission; and interfere with cell replication. All of the anticancer antibiotics now being used in clinical practice are products of various strains of the soil microbe Streptomyces . These include the anthracyclines, bleomycin, and mitomycin.
ANTHRACYCLINES
The anthracycline antibiotics, isolated from Streptomyces peucetius var caesius , are among the most widely used cytotoxic anticancer drugs. The structures of two congeners, doxorubicin and daunoru-bicin, are shown below. Several other anthracycline analogs have entered clinical practice, including idarubicin, epirubicin, and mitoxantrone. The anthracyclines exert their cytotoxic action through four major mechanisms: (1) inhibition of topoisomerase II; (2) high-affinity binding to DNA through intercalation, with consequent blockade of the synthesis of DNA and RNA, and DNA strand scission; (3) generation of semiquinone free radicals and oxygen free radicals through an iron-dependent, enzyme-mediated reductive process; and (4) binding to cellular membranes to alter fluidity and ion transport. While the precise mechanisms by which the anthracyclines exert their cytotoxic effects remain to be defined (and may depend upon the specific tumor type), it is now well-es-tablished that the free radical mechanism is the cause of the cardio-toxicity associated with the anthracyclines ( Table 54–4 ).
Doxorubicin
OHO
OHOOCH3 OH
R
OH
C CH2OH
O
R:
Daunorubicin
C CH3
O
R:
HO
NH2
O
CH3
In the clinical setting, anthracyclines are administered via the intravenous route. The anthracyclines are metabolized extensively in the liver, with reduction and hydrolysis of the ring substituents. The hydroxylated metabolite is an active species, whereas the agly-cone is inactive. Up to 50% of drug is eliminated in the feces via biliary excretion, and dose reduction is required in the setting of liver dysfunction. Although anthracyclines are usually administered on an every-3-week schedule, alternative schedules such as low-dose weekly or 72- to 96-hour continuous infusions have been shown to yield equivalent clinical efficacy with reduced toxicity.
Doxorubicin is one of the most important anticancer drugs in clinical practice, with major clinical activity in cancers of the breast, endometrium, ovary, testicle, thyroid, stomach, bladder, liver, and lung; in soft tissue sarcomas; and in several childhood cancers, including neuroblastoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. It also has clinical activity in hematologic malignancies, including acute lymphoblastic leukemia, multiple myeloma, and Hodgkin’s and non-Hodgkin’s lymphomas. It is gen-erally used in combination with other anticancer agents (eg, cyclo-phosphamide, cisplatin, and 5-FU), and clinical activity is improved with combination regimens as opposed to single-agent therapy.
Daunorubicin was the first agent in this class to be isolated, and it is still used in the treatment of acute myeloid leukemia. In contrast to doxorubicin, its efficacy in solid tumors is limited.
Idarubicin is a semisynthetic anthracycline glycoside analog of daunorubicin, and it is approved for use in combination with cytarabine for induction therapy of acute myeloid leukemia. When combined with cytarabine, idarubicin appears to be more active than daunorubicin in producing complete remissions and in improving survival in patients with acute myelogenous leukemia.
Epirubicin is an anthracycline analog whose mechanism of action and clinical pharmacology are identical to those of all other anthracyclines. It was initially approved for use as a compo-nent of adjuvant therapy in early-stage, node-positive breast cancer but is also used in the treatment of metastatic breast cancer and gastroesophageal cancer.
Mitoxantrone (dihydroxyanthracenedione) is an anthracene compound whose structure resembles the anthracycline ring. It binds to DNA to produce strand breakage and inhibits both DNA and RNA synthesis. It is currently used in the treatment of advanced, hormone-refractory prostate cancer and low-grade non-Hodgkin’s lymphoma. It is also indicated in breast cancer and in pediatric and adult acute myeloid leukemias. Myelosuppression with leukopenia is the dose-limiting toxicity, and mild nausea and vomiting, mucositis, and alopecia also occur. Although the drug is thought to be less cardiotoxic than doxorubicin, both acute and chronic cardiac toxicity are reported. A blue discoloration of the fingernails, sclera, and urine is observed 1–2 days after drug administration.
The main dose-limiting toxicity of all anthracyclines is myelo-suppression, with neutropenia more commonly observed than thrombocytopenia. In some cases, mucositis is dose-limiting. Two forms of cardiotoxicity are observed. The acute form occurs within the first 2–3 days and presents as arrhythmias and conduction abnormalities, other electrocardiographic changes, pericarditis, and myocarditis. This form is usually transient and in most cases
054-Katzung_Ch054_p949-976.indd 965 9/21/11 12:11:48 PM

966 SECTION VIII Chemotherapeutic Drugs
is asymptomatic. The chronic form results in a dose-dependent, dilated cardiomyopathy associated with heart failure. The chronic cardiac toxicity appears to result from increased production of free radicals within the myocardium. This effect is rarely seen at total doxorubicin dosages below 500–550 mg/m 2 . Use of lower weekly doses or continuous infusions of doxorubicin appear to reduce the incidence of cardiac toxicity. In addition, treatment with the iron-chelating agent dexrazoxane (ICRF-187) is currently approved to prevent or reduce anthracycline-induced cardiotoxicity in women with metastatic breast cancer who have received a total cumulative dose of doxorubicin of 300 mg/m 2 . The anthracyclines can also produce a “radiation recall reaction,” with erythema and desqua-mation of the skin observed at sites of prior radiation therapy.
MITOMYCIN
Mitomycin (mitomycin C) is an antibiotic isolated from Streptomyces caespitosus . It undergoes metabolic activation through an enzyme-mediated reduction to generate an alkylating agent that cross-links DNA. Hypoxic tumor stem cells of solid tumors exist in an environment conducive to reductive reactions and are more sensitive to the cytotoxic effects of mitomycin than normal cells and oxygenated tumor cells. It is active in all phases of the cell cycle, and is the best available drug for use in combination with radiation therapy to attack hypoxic tumor cells. Its main clinical use is in the treatment of squamous cell cancer of the anus in combination with 5-FU and radiation therapy. In addition, it is used in combination chemotherapy for squamous cell carcinoma of the cervix and for breast, gastric, and pancreatic cancer. One special application of mitomycin has been in the intravesical treat-ment of superficial bladder cancer. Because virtually none of the agent is absorbed systemically, there is little to no systemic toxicity when used in this setting.
The common toxicities of mitomycin are outlined in Table 54–4 . Hemolytic-uremic syndrome, manifested as microan-giopathic hemolytic anemia, thrombocytopenia, and renal failure, as well as occasional instances of interstitial pneumonitis have been reported.
BLEOMYCIN
Bleomycin is a small peptide that contains a DNA-binding region and an iron-binding domain at opposite ends of the molecule. It acts by binding to DNA, which results in single- and double-strand breaks following free radical formation, and inhibition of DNA biosynthesis. The fragmentation of DNA is due to oxida-tion of a DNA-bleomycin-Fe(II) complex and leads to chromo-somal aberrations. Bleomycin is a cell cycle-specific drug that causes accumulation of cells in the G 2 phase of the cell cycle.
Bleomycin is indicated for the treatment of Hodgkin’s and non-Hodgkin’s lymphomas, germ cell tumor, head and neck can-cer, and squamous cell cancer of the skin, cervix, and vulva. One advantage of this agent is that it can be given subcutaneously, intramuscularly, or intravenously. Elimination of bleomycin is
mainly via renal excretion, and dose modification is recommended in patients with renal dysfunction.
Pulmonary toxicity is dose-limiting for bleomycin and usually presents as pneumonitis with cough, dyspnea, dry inspiratory crackles on physical examination, and infiltrates on chest X-ray. The incidence of pulmonary toxicity is increased in patients older than 70 years of age, in those who receive cumulative doses greater than 400 units, in those with underlying pulmonary disease, and in those who have received prior mediastinal or chest irradiation. In rare cases, pulmonary toxicity can be fatal. Other toxicities are listed in Table 54–4 .
MISCELLANEOUS ANTICANCER DRUGS A large number of anticancer drugs that do not fit traditional categories have been approved for clinical use by the Food and Drug Administration (FDA); they are listed in Table 54–5 .
IMATINIB, DASATINIB, & NILOTINIB
Imatinib is an inhibitor of the tyrosine kinase domain of the Bcr-Abl oncoprotein and prevents phosphorylation of the kinase sub-strate by ATP. It is indicated for the treatment of chronic myelogenous leukemia (CML), a pluripotent hematopoietic stem cell disorder characterized by the t(9:22) Philadelphia chromo-somal translocation. This translocation results in the Bcr-Abl fusion protein, the causative agent in CML, and is present in up to 95% of patients with this disease. This agent also inhibits other receptor tyrosine kinases for platelet-derived growth factor recep-tor (PDGFR), stem cell factor, and c-kit.
Imatinib is well absorbed orally, and it is metabolized in the liver, with elimination of metabolites occurring mainly in feces via biliary excretion. This agent is approved for use as first-line ther-apy in chronic phase CML, in blast crisis, and as second-line therapy for chronic phase CML that has progressed on prior interferon-alfa therapy. Imatinib is also effective in the treatment of gastrointestinal stromal tumors expressing the c-kit tyrosine kinase. The main adverse effects are listed in Table 54–5 .
Dasatinib is an oral inhibitor of several tyrosine kinases, includ-ing Bcr-Abl, Src, c-kit, and PDGFR-α. It differs from imatinib in that it binds to the active and inactive conformations of the Abl kinase domain and overcomes imatinib resistance resulting from mutations in the Bcr-Abl kinase. It is approved for use in CML and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL) with resistance or intolerance to imatinib therapy.
Nilotinib is a second-generation phenylamino-pyrimidine mol-ecule that inhibits Bcr-Abl, c-kit, and PDGFR-β tyrosine kinases. It has a higher binding affinity (up to 20- to 50-fold) for the Abl kinase when compared with imatinib, and it overcomes imatinib resistance resulting from Bcr-Abl mutations. It was originally approved for chronic phase and accelerated phase CML with resis-tance or intolerance to prior therapy that included imatinib and was recently approved as first-line therapy of chronic phase CML.
054-Katzung_Ch054_p949-976.indd 966 9/21/11 12:11:48 PM
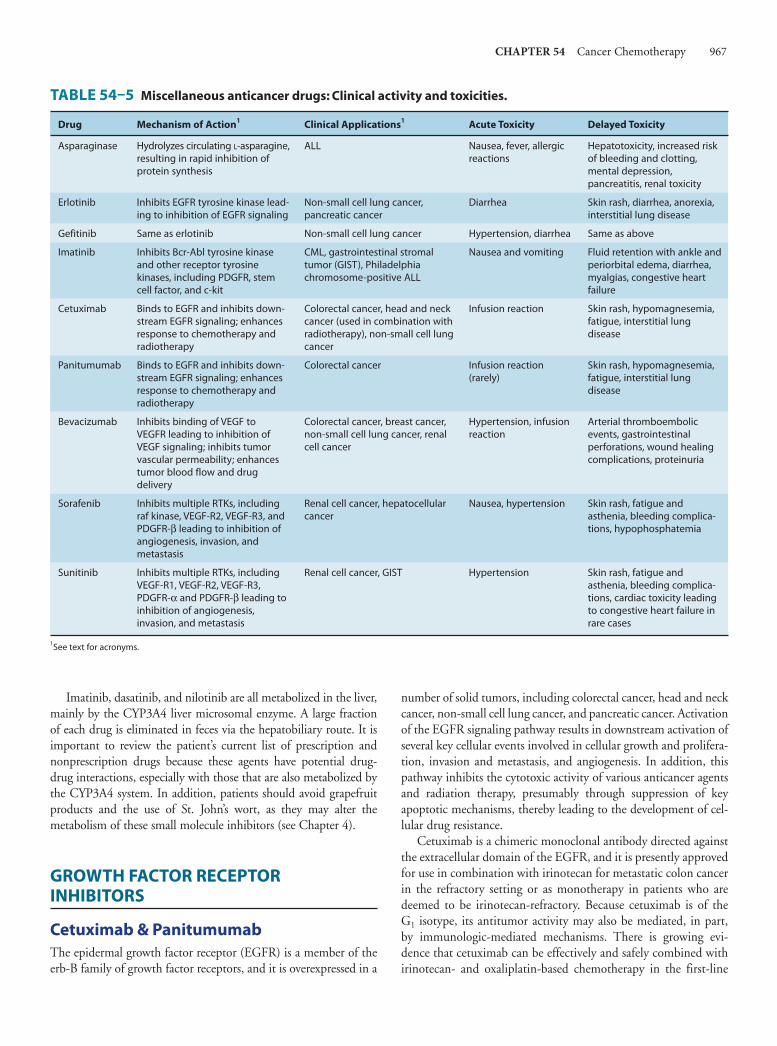
CHAPTER 54 Cancer Chemotherapy 967
Imatinib, dasatinib, and nilotinib are all metabolized in the liver, mainly by the CYP3A4 liver microsomal enzyme. A large fraction of each drug is eliminated in feces via the hepatobiliary route. It is important to review the patient’s current list of prescription and nonprescription drugs because these agents have potential drug-drug interactions, especially with those that are also metabolized by the CYP3A4 system. In addition, patients should avoid grapefruit products and the use of St. John’s wort, as they may alter the metabolism of these small molecule inhibitors (see Chapter 4 ).
GROWTH FACTOR RECEPTOR INHIBITORS
Cetuximab & Panitumumab The epidermal growth factor receptor (EGFR) is a member of the erb-B family of growth factor receptors, and it is overexpressed in a
number of solid tumors, including colorectal cancer, head and neck cancer, non-small cell lung cancer, and pancreatic cancer. Activation of the EGFR signaling pathway results in downstream activation of several key cellular events involved in cellular growth and prolifera-tion, invasion and metastasis, and angiogenesis. In addition, this pathway inhibits the cytotoxic activity of various anticancer agents and radiation therapy, presumably through suppression of key apoptotic mechanisms, thereby leading to the development of cel-lular drug resistance.
Cetuximab is a chimeric monoclonal antibody directed against the extracellular domain of the EGFR, and it is presently approved for use in combination with irinotecan for metastatic colon cancer in the refractory setting or as monotherapy in patients who are deemed to be irinotecan-refractory. Because cetuximab is of the G 1 isotype, its antitumor activity may also be mediated, in part, by immunologic-mediated mechanisms. There is growing evi-dence that cetuximab can be effectively and safely combined with irinotecan- and oxaliplatin-based chemotherapy in the first-line
TABLE 54–5 Miscellaneous anticancer drugs: Clinical activity and toxicities.
Drug Mechanism of Action1 Clinical Applications1 Acute Toxicity Delayed Toxicity
Asparaginase Hydrolyzes circulating L-asparagine, resulting in rapid inhibition of protein synthesis
ALL Nausea, fever, allergic reactions
Hepatotoxicity, increased risk of bleeding and clotting, mental depression, pancreatitis, renal toxicity
Erlotinib Inhibits EGFR tyrosine kinase lead-ing to inhibition of EGFR signaling
Non-small cell lung cancer, pancreatic cancer
Diarrhea Skin rash, diarrhea, anorexia, interstitial lung disease
Gefitinib Same as erlotinib Non-small cell lung cancer Hypertension, diarrhea Same as above
Imatinib Inhibits Bcr-Abl tyrosine kinase and other receptor tyrosine kinases, including PDGFR, stem cell factor, and c-kit
CML, gastrointestinal stromal tumor (GIST), Philadelphia chromosome-positive ALL
Nausea and vomiting Fluid retention with ankle and periorbital edema, diarrhea, myalgias, congestive heart failure
Cetuximab Binds to EGFR and inhibits down-stream EGFR signaling; enhances response to chemotherapy and radiotherapy
Colorectal cancer, head and neck cancer (used in combination with radiotherapy), non-small cell lung cancer
Infusion reaction Skin rash, hypomagnesemia, fatigue, interstitial lung disease
Panitumumab Binds to EGFR and inhibits down-stream EGFR signaling; enhances response to chemotherapy and radiotherapy
Colorectal cancer Infusion reaction (rarely)
Skin rash, hypomagnesemia, fatigue, interstitial lung disease
Bevacizumab Inhibits binding of VEGF to VEGFR leading to inhibition of VEGF signaling; inhibits tumor vascular permeability; enhances tumor blood flow and drug delivery
Colorectal cancer, breast cancer, non-small cell lung cancer, renal cell cancer
Hypertension, infusion reaction
Arterial thromboembolic events, gastrointestinal perforations, wound healing complications, proteinuria
Sorafenib Inhibits multiple RTKs, including raf kinase, VEGF-R2, VEGF-R3, and PDGFR-β leading to inhibition of angiogenesis, invasion, and metastasis
Renal cell cancer, hepatocellular cancer
Nausea, hypertension Skin rash, fatigue and asthenia, bleeding complica-tions, hypophosphatemia
Sunitinib Inhibits multiple RTKs, including VEGF-R1, VEGF-R2, VEGF-R3, PDGFR-α and PDGFR-β leading to inhibition of angiogenesis, invasion, and metastasis
Renal cell cancer, GIST Hypertension Skin rash, fatigue and asthenia, bleeding complica-tions, cardiac toxicity leading to congestive heart failure in rare cases
1See text for acronyms.
054-Katzung_Ch054_p949-976.indd 967 9/21/11 12:11:48 PM

968 SECTION VIII Chemotherapeutic Drugs
treatment of metastatic colorectal cancer as well. Of note, the efficacy of cetuximab is restricted to only those patients whose tumors express wild-type KRAS . Regimens combining cetuximab with cytotoxic chemotherapy may be of particular benefit in the neoadjuvant therapy of patients with liver-limited disease. Although this antibody was initially approved to be administered on a weekly schedule, pharmacokinetic studies have shown that an every-2-week schedule provides the same level of clinical activity as the weekly schedule. This agent is also approved for use in combination with radiation therapy in patients with locally advanced head and neck cancer. Cetuximab is well tolerated, with the main adverse effects being an acneiform skin rash, hypersensi-tivity infusion reaction, and hypomagnesemia.
Panitumumab is a fully human monoclonal antibody directed against the EGFR and works through inhibition of the EGFR signaling pathway. In contrast to cetuximab, this antibody is of the G 2 isotype, and as such, it would not be expected to exert any immunologic-mediated effects. Presently, panitumumab is approved for patients with refractory metastatic colorectal cancer who have been treated with all other active agents, and as with cetuximab, this antibody is only effective in patients whose tumors express wild-type KRAS . Recent clinical studies have shown that this anti-body is effectively and safely combined with oxaliplatin- and irino-tecan-based chemotherapy in the first- and second-line treatment of metastatic colorectal cancer. Acneiform skin rash and hypomag-nesemia are the two main adverse effects associated with its use. Because this is a fully human antibody, infusion-related reactions are rarely observed.
Gefitinib & Erlotinib Gefitinib and erlotinib are small molecule inhibitors of the tyrosine kinase domain associated with the EGFR, and both are used in the treatment of non-small cell lung cancer that is refractory to at least one prior chemotherapy regimen. Patients who are nonsmokers and who have a bronchoalveolar histologic subtype appear to be more responsive to these agents. In addition, erlotinib has been approved for use in combination with gemcitabine for the treat-ment of advanced pancreatic cancer. Both agents are metabolized in the liver by the CYP3A4 enzyme system, and elimination is mainly hepatic with excretion in feces. Caution must be taken when using these agents with drugs that are also metabolized by the liver CYP3A4 system, such as phenytoin and warfarin, and the use of grapefruit products should be avoided. An acneiform skin rash, diarrhea, and anorexia and fatigue are the most common adverse effects observed with these small molecules ( Table 54-5 ).
Bevacizumab, Sorafenib, Sunitinib, & Pazopanib The vascular endothelial growth factor (VEGF) is one of the most important angiogenic growth factors. The growth of both primary and metastatic tumors requires an intact vasculature. As a result, the VEGF-signaling pathway represents an attractive target for chemo-therapy. Several approaches have been taken to inhibit VEGF sig-naling; they include inhibition of VEGF interactions with its receptor by targeting either the VEGF ligand with antibodies or
soluble chimeric decoy receptors, or by direct inhibition of the VEGF receptor-associated tyrosine kinase activity by small mole-cule inhibitors.
Bevacizumab is a recombinant humanized monoclonal anti-body that targets all forms of VEGF-A. This antibody binds to and prevents VEGF-A from interacting with the target VEGF receptors. Bevacizumab can be safely and effectively combined with 5-FU-, irinotecan-, and oxaliplatin-based chemotherapy in the treatment of metastatic colorectal cancer. Bevacizumab is FDA approved as a first-line treatment for metastatic colorectal cancer in combination with any intravenous fluoropyrimidine-contain-ing regimen and is now also approved in combination with che-motherapy for metastatic non-small lung cancer and breast cancer. One potential advantage of this antibody is that it does not appear to exacerbate the toxicities typically observed with cytotoxic che-motherapy. The main safety concerns associated with bevacizumab include hypertension, an increased incidence of arterial throm-boembolic events (transient ischemic attack, stroke, angina, and myocardial infarction), wound healing complications, gastrointes-tinal perforations, and proteinuria.
Sorafenib is a small molecule that inhibits multiple receptor tyrosine kinases (RTKs), especially VEGF-R2 and VEGF-R3, platelet-derived growth factor-β (PDGFR-β), and raf kinase. It was initially approved for advanced renal cell cancer and is also approved for advanced hepatocellular cancer.
Sunitinib is similar to sorafenib in that it inhibits multiple RTKs, although the specific types are somewhat different. They include PDGFR-α and PDGFR-β, VEGF-R1, VEGF-R2, VEGF-R3, and c-kit. It is approved for the treatment of advanced renal cell cancer and for the treatment of gastrointestinal stromal tumors (GIST) after disease progression on or with intolerance to imatinib.
Pazopanib is a small molecule that inhibits multiple RTKs, espe-cially VEGF-R2 and VEGF-R3, PDGFR-β, and raf kinase. This oral agent is approved for the treatment of advanced renal cell cancer.
Sorafenib, sunitinib, and pazopanib are metabolized in the liver by the CYP3A4 system, and elimination is primarily hepatic with excretion in feces. Each of these agents has potential interac-tions with drugs that are also metabolized by the CYP3A4 system, especially warfarin. In addition, patients should avoid grapefruit products and the use of St. John’s wort, as they may alter the clinical activity of these agents. Hypertension, bleeding complica-tions, and fatigue are the most common adverse effects seen with both agents. With respect to sorafenib, skin rash and the hand-foot syndrome are observed in up to 30–50% of patients. For sunitinib, there is also an increased risk of cardiac dysfunction, which in some cases can lead to congestive heart failure .
ASPARAGINASE
Asparaginase ( L -asparagine amidohydrolase) is an enzyme used to treat childhood ALL. The drug is isolated and purified from Escherichia coli or Erwinia chrysanthemi for clinical use. It hydro-lyzes circulating L -asparagine to aspartic acid and ammonia. Because tumor cells in ALL lack asparagine synthetase, they require an exogenous source of L -asparagine. Thus, depletion of L -asparagine results in effective inhibition of protein synthesis. In
054-Katzung_Ch054_p949-976.indd 968 9/21/11 12:11:48 PM

CHAPTER 54 Cancer Chemotherapy 969
contrast, normal cells can synthesize L -asparagine and thus are less susceptible to the cytotoxic action of asparaginase. The main adverse effect of this agent is a hypersensitivity reaction manifested by fever, chills, nausea and vomiting, skin rash, and urticaria. Severe cases can present with bronchospasm, respiratory failure, and hypotension. Other side effects include an increased risk of both clotting and bleeding as a result of alterations in various clot-ting factors, pancreatitis, and neurologic toxicity with lethargy, confusion, hallucinations, and in severe cases, coma.
■ CLINICAL PHARMACOLOGY OF CANCER CHEMOTHERAPEUTIC DRUGS
A thorough knowledge of the kinetics of tumor cell proliferation along with an understanding of the pharmacology and mechanism of action of cancer chemotherapeutic agents is important in designing optimal regimens for patients with cancer. The strategy for developing drug regimens also requires knowledge of the spe-cific characteristics of individual tumors. For example, is there a high growth fraction? Is there a high spontaneous cell death rate? Are most of the cells in G 0 ? Is a significant fraction of the tumor composed of hypoxic stem cells? Are their normal counterparts under hormonal control? Similarly, an understanding of the phar-macology of specific drugs is important. Are the tumor cells sensi-tive to the drug? Is the drug cell cycle specific? Does the drug require activation in certain normal tissue such as the liver (cyclo-phosphamide), or is it activated in the tumor tissue itself (capecit-abine)? Knowledge of specific pathway abnormalities (eg, EGFR mutations, KRAS mutations) for intracellular signaling may prove important for the next generation of anticancer drugs.
For some tumor types, knowledge of receptor expression is important. In patients with breast cancer, analysis of the tumor for expression of estrogen or progesterone receptors is important in guiding therapy with selective estrogen receptor modulators. In addition, analysis of breast cancer for expression of the HER-2/ neu growth factor receptor can determine whether the human-ized monoclonal anti-HER-2/ neu antibody, trastuzumab, would be appropriate therapy. In the case of prostate cancer, chemical suppression of androgen secretion with gonadotropin-releasing hormone agonists or antagonists is important. The basic pharma-cology of hormonal therapy is discussed in Chapter 40 . The use of specific cytotoxic and biologic agents for each of the main cancers is discussed in this section.
THE LEUKEMIAS ACUTE LEUKEMIA
Childhood Leukemia Acute lymphoblastic leukemia (ALL) is the main form of leukemia in childhood, and it is the most common form of cancer in chil-dren. Children with this disease have a relatively good prognosis.
A subset of patients with neoplastic lymphocytes expressing surface antigenic features of T lymphocytes has a poor prognosis (see Chapter 55 ). A cytoplasmic enzyme expressed by normal thymo-cytes, terminal deoxycytidyl transferase (terminal transferase), is also expressed in many cases of ALL. T-cell ALL also expresses high levels of the enzyme adenosine deaminase (ADA). This led to inter-est in the use of the ADA inhibitor pentostatin (deoxycoformycin) for treatment of such T-cell cases. Until 1948, the median length of survival in ALL was 3 months. With the advent of methotrexate, the length of survival was greatly increased. Subsequently, cortico-steroids, 6-mercaptopurine, cyclophosphamide, vincristine, dauno-rubicin, and asparaginase have all been found to be active against this disease. A combination of vincristine and prednisone plus other agents is currently used to induce remission. Over 90% of children enter complete remission with this therapy with only minimal toxicity. However, circulating leukemic cells often migrate to sanctuary sites located in the brain and testes. The value of pro-phylactic intrathecal methotrexate therapy for prevention of central nervous system leukemia (a major mechanism of relapse) has been clearly demonstrated. Intrathecal therapy with methotrexate should therefore be considered as a standard component of the induction regimen for children with ALL.
Adult Leukemia Acute myelogenous leukemia (AML) is the most common leuke-mia in adults. The single most active agent for AML is cytarabine; however, it is best used in combination with an anthracycline, which leads to complete remissions in about 70% of patients. While there are several anthracyclines that can be effectively com-bined with cytarabine, idarubicin is preferred.
Patients often require intensive supportive care during the period of induction chemotherapy. Such care includes platelet transfusions to prevent bleeding, the granulocyte colony-stimulat-ing factor filgrastim to shorten periods of neutropenia, and anti-biotics to combat infections. Younger patients (eg, age < 55) who are in complete remission and have an HLA-matched donor are candidates for allogeneic bone marrow transplantation. The trans-plant procedure is preceded by high-dose chemotherapy and total body irradiation followed by immunosuppression. This approach may cure up to 35–40% of eligible patients. Patients over age 60 respond less well to chemotherapy, primarily because their toler-ance for aggressive therapy and resistance to infection are lower.
Once remission of AML is achieved, consolidation chemotherapy is required to maintain a durable remission and to induce cure.
CHRONIC MYELOGENOUS LEUKEMIA
Chronic myelogenous leukemia (CML) arises from a chromosomally abnormal hematopoietic stem cell in which a balanced translocation between the long arms of chromosomes 9 and 22, t(9:22), is observed in 90–95% of cases. This translocation results in constitu-tive expression of the Bcr-Abl fusion oncoprotein with a molecular weight of 210 kDa. The clinical symptoms and course are related to the white blood cell count and its rate of increase. Most patients with
054-Katzung_Ch054_p949-976.indd 969 9/21/11 12:11:49 PM

970 SECTION VIII Chemotherapeutic Drugs
white cell counts over 50,000/μL should be treated. The goals of treatment are to reduce the granulocytes to normal levels, to raise the hemoglobin concentration to normal, and to relieve disease-related symptoms. The tyrosine kinase inhibitor imatinib is considered as standard first-line therapy in previously untreated patients with chronic phase CML. Nearly all patients treated with imatinib exhibit a complete hematologic response, and up to 40–50% of patients show a complete cytogenetic response. As described previously, this drug is generally well tolerated and is associated with relatively minor adverse effects. Initially, dasatinib and nilotinib were approved for patients who were intolerant or resistant to imatinib; each shows clinical activity, but both are now also indicated as first-line treat-ment of chronic phase CML. In addition to these tyrosine kinase inhibitors, other treatment options include interferon-α, busulfan, other oral alkylating agents, and hydroxyurea.
CHRONIC LYMPHOCYTIC LEUKEMIA
Patients with early-stage chronic lymphocytic leukemia (CLL) have a relatively good prognosis, and therapy has not changed the course of the disease. However, in the setting of high-risk disease or in the presence of disease-related symptoms, treatment is indicated.
Chlorambucil and cyclophosphamide are the two most widely used alkylating agents for this disease. Chlorambucil is frequently combined with prednisone, although there is no clear evidence that the combination yields better response rates or survival compared with chlorambucil alone. In most cases, cyclophosphamide is com-bined with vincristine and prednisone (COP), or it can also be given with these same drugs along with doxorubicin (CHOP). Bendamustine is the newest alkylating agent to be approved for use in this disease, either as monotherapy or in combination with pred-nisone. The purine nucleoside analog fludarabine is also effective in treating CLL. This agent can be given alone, in combination with cyclophosphamide and with mitoxantrone and dexamethasone, or combined with the anti-CD20 antibody rituximab.
Monoclonal antibody-targeted therapies are being widely used in CLL, especially in relapsed or refractory disease. Rituximab is an anti-CD20 antibody that has documented clinical activity in this setting. This chimeric antibody appears to enhance the antitumor effects of cytotoxic chemotherapy and is also effective in settings in which resistance to chemotherapy has developed. Alemtuzumab is a humanized monoclonal antibody directed against the CD52 antigen and is approved for use in CLL that is refractory to alkylat-ing agent or fludarabine therapy. Response rates up to 30–35% are observed, with disease stabilization in another 30% of patients.
HODGKIN’S & NON-HODGKIN’S LYMPHOMAS HODGKIN’S LYMPHOMA
The treatment of Hodgkin’s lymphoma has undergone dramatic evolution over the last 30 years. This lymphoma is now widely rec-ognized as a B-cell neoplasm in which the malignant Reed-Sternberg
cells have rearranged VH genes. In addition, the Epstein-Barr virus genome has been identified in up to 80% of tumor specimens.
Complete staging evaluation is required before a definitive treatment plan can be made. For patients with stage I and stage IIA disease, there has been a significant change in the treatment approach. Initially, these patients were treated with extended-field radiation therapy. However, given the well-documented late effects of radiation therapy, which include hypothyroidism, an increased risk of secondary cancers, and coronary artery disease, combined-modality therapy with a brief course of combination chemotherapy and involved field radiation therapy is now the recommended approach. The main advance for patients with advanced stage III and IV Hodgkin’s lymphoma came with the development of MOPP (mechlorethamine, vincristine, procarba-zine, and prednisone) chemotherapy in the 1960s. This regimen resulted initially in high complete response rates, on the order of 80–90%, with cures in up to 60% of patients. More recently, the anthracycline-containing regimen termed ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) has been shown to be more effective and less toxic than MOPP, especially with regard to the incidence of infertility and secondary malignancies. In general, four cycles of ABVD are given to patients. An alternative regimen, termed Stanford V, utilizes a 12-week course of combination che-motherapy (doxorubicin, vinblastine, mechlorethamine, vincris-tine, bleomycin, etoposide, and prednisone), followed by involved radiation therapy.
With all of these regimens, over 80% of previously untreated patients with advanced Hodgkin’s lymphoma (stages III and IV) are expected to go into complete remission, with disappearance of all disease-related symptoms and objective evidence of disease. In general, approximately 50–60% of all patients with Hodgkin’s lymphoma are cured of their disease.
NON-HODGKIN’S LYMPHOMA
Non-Hodgkin’s lymphoma is a heterogeneous disease, and the clinical characteristics of non-Hodgkin’s lymphoma subsets are related to the underlying histopathologic features and the extent of disease involvement. In general, the nodular (or follicular) lym-phomas have a far better prognosis, with a median survival up to 7 years, compared with the diffuse lymphomas, which have a median survival of about 1–2 years.
Combination chemotherapy is the treatment standard for patients with diffuse non-Hodgkin’s lymphoma. The anthracy-cline-containing regimen CHOP (cyclophosphamide, doxorubi-cin, vincristine, and prednisone) has been considered the best treatment in terms of initial therapy. Randomized phase III clini-cal studies have now shown that the combination of CHOP with the anti-CD20 monoclonal antibody rituximab results in improved response rates, disease-free survival, and overall survival compared with CHOP chemotherapy alone.
The nodular follicular lymphomas are low-grade, relatively slow-growing tumors that tend to present in an advanced stage and are usually confined to lymph nodes, bone marrow, and spleen. This form of non-Hodgkin’s lymphomas, when presenting
054-Katzung_Ch054_p949-976.indd 970 9/21/11 12:11:49 PM

CHAPTER 54 Cancer Chemotherapy 971
at an advanced stage, is considered incurable, and treatment is generally palliative. To date, there is no evidence that immediate treatment with combination chemotherapy offers clinical benefit over close observation and “watchful waiting” with initiation of chemotherapy at the onset of disease symptoms.
MULTIPLE MYELOMA This plasma cell malignancy is one of the models of neoplastic disease in humans as it arises from a single tumor stem cell. Moreover, the tumor cells produce a marker protein (myeloma immunoglobulin) that allows the total body burden of tumor cells to be quantified. Multiple myeloma principally involves the bone marrow and bone, causing bone pain, lytic lesions, bone fractures, and anemia as well as an increased susceptibility to infection.
Most patients with multiple myeloma are symptomatic at the time of initial diagnosis and require treatment with cytotoxic che-motherapy. Treatment with the combination of the alkylating agent melphalan and prednisone (MP protocol) has been a stan-dard regimen for nearly 30 years. About 40% of patients respond to the MP combination, and the median remission is on the order of 2–2.5 years. Recently, combination regimens incorporating lenalidomide plus dexamethasone or the proteosome inhibitor bortezomib plus melphalan and prednisone have been shown to be more effective as first-line therapy.
In patients who are considered candidates for high-dose therapy with stem cell transplantation, melphalan and other alkylating agents are to be avoided, as they can affect the success of stem cell harvesting.
Thalidomide is a well-established agent for treating refractory or relapsed disease, and about 30% of patients will achieve a response to this therapy. More recently, thalidomide has been used in combi-nation with dexamethasone, and response rates on the order of 65% have been observed. Studies are now under way to directly compare the combination of vincristine, doxorubicin, and dexamethasone (VAD protocol) with the combination of thalidomide and dexa-methasone. In some patients, especially those with poor performance status, single-agent pulse dexamethasone administered on a weekly basis can be effective in palliating symptoms. Bortezomib was first approved for use in relapsing or refractory multiple myeloma and is now widely used in the first-line treatment of multiple myeloma. This agent is thought to exert its main cytotoxic effects through inhibition of the 26 S proteosome, which results in down-regulation of the nuclear factor kappa B (NF-κB) signaling pathway. Of note, inhibition of NF-κB has been shown to restore chemosensitivity. Based on this site of action, further efforts are focused on developing bortezomib in various combination regimens.
BREAST CANCER STAGE I & STAGE II DISEASE
The management of primary breast cancer has undergone a remarkable evolution as a result of major efforts at early diagnosis (through encouragement of self-examination as well as through
the use of cancer detection centers) and the implementation of combined modality approaches incorporating systemic chemo-therapy as an adjuvant to surgery and radiation therapy. Women with stage I disease (small primary tumors and negative axillary lymph node dissections) are currently treated with surgery alone, and they have an 80% chance of cure.
Women with node-positive disease have a high risk of both local and systemic recurrence. Thus, lymph node status directly indicates the risk of occult distant micrometastasis. In this situation, postop-erative use of systemic adjuvant chemotherapy with six cycles of cyclophosphamide, methotrexate, and fluorouracil (CMF proto-col) or of fluorouracil, doxorubicin, and cyclophosphamide (FAC) has been shown to significantly reduce the relapse rate and prolong survival. Alternative regimens with equivalent clinical benefit include four cycles of doxorubicin and cyclophosphamide and six cycles of fluorouracil, epirubicin, and cyclophosphamide (FEC). Each of these chemotherapy regimens has benefited women with stage II breast cancer with one to three involved lymph nodes. Women with four or more involved nodes have had limited benefit thus far from adjuvant chemotherapy. Long-term analysis has clearly shown improved survival rates in node-positive premeno-pausal women who have been treated aggressively with multiagent combination chemotherapy. The results from three randomized clinical trials clearly show that the addition of trastuzumab, a monoclonal antibody directed against the HER-2/ neu receptor, to anthracycline- and taxane-containing adjuvant chemotherapy ben-efits women with HER-2-overexpressing breast cancer with respect to disease-free and overall survival.
Breast cancer was the first neoplasm shown to be responsive to hormonal manipulation. Tamoxifen is beneficial in postmeno-pausal women when used alone or in combination with cytotoxic chemotherapy. The present recommendation is to administer tamoxifen for 5 years of continuous therapy after surgical resec-tion. Longer durations of tamoxifen therapy do not appear to add additional clinical benefit. Postmenopausal women who complete 5 years of tamoxifen therapy should be placed on an aromatase inhibitor such as anastrozole for at least 2.5 years, although the optimal duration is unknown. In women who have completed 2–3 years of tamoxifen therapy, treatment with an aromatase inhibitor for a total of 5 years of hormonal therapy is now recom-mended (see Chapter 40 ).
Results from several randomized trials for breast cancer have established that adjuvant chemotherapy for premenopausal women and adjuvant tamoxifen for postmenopausal women are of benefit to women with stage I (node-negative) breast cancer. While this group of patients has the lowest overall risk of recurrence after surgery alone (about 35–50% over 15 years), this risk can be fur-ther reduced with adjuvant therapy.
STAGE III & STAGE IV DISEASE
The approach to women with advanced breast cancer remains a major challenge, as current treatment options are only palliative. Combination chemotherapy, endocrine therapy, or a combination of both results in overall response rates of 40–50%, but only a
054-Katzung_Ch054_p949-976.indd 971 9/21/11 12:11:49 PM

972 SECTION VIII Chemotherapeutic Drugs
10–20% complete response rate. Breast cancers expressing estro-gen receptors (ER) or progesterone receptors (PR), retain the intrinsic hormonal sensitivities of the normal breast—including the growth-stimulatory response to ovarian, adrenal, and pituitary hormones. Patients who show improvement with hormonal abla-tive procedures also respond to the addition of tamoxifen. The aromatase inhibitors anastrozole and letrozole are now approved as first-line therapy in women with advanced breast cancer whose tumors are hormone-receptor positive. In addition, these agents and exemestane are approved as second-line therapy following treatment with tamoxifen.
Patients with significant involvement of the lung, liver, or brain and those with rapidly progressive disease rarely benefit from hor-monal maneuvers, and initial systemic chemotherapy is indicated in such cases. For the 25–30% of breast cancer patients whose tumors express the HER-2/ neu cell surface receptor, the human-ized monoclonal anti-HER-2/ neu antibody, trastuzumab, is avail-able for therapeutic use alone or in combination with cytotoxic chemotherapy.
About 50–60% of patients with metastatic disease respond to initial chemotherapy. A broad range of anticancer agents have activity in this disease, including the anthracyclines (doxorubicin, mitoxantrone, and epirubicin), the taxanes (docetaxel, paclitaxel, and albumin-bound paclitaxel) along with the microtubule inhib-itor ixabepilone, navelbine, capecitabine, gemcitabine, cyclophos-phamide, methotrexate, and cisplatin. The anthracyclines and the taxanes are two of the most active classes of cytotoxic drugs. Combination chemotherapy has been found to induce higher and more durable remissions in up to 50–80% of patients, and anthra-cycline-containing regimens are now considered the standard of care in first-line therapy. With most combination regimens, partial remissions have a median duration of about 10 months and com-plete remissions have a duration of about 15 months. Unfortunately, only 10–20% of patients achieve complete remissions with any of these regimens, and as noted, complete remissions are usually not long-lasting.
PROSTATE CANCER
Prostate cancer was the second cancer shown to be responsive to hormonal manipulation. The treatment of choice for patients with advanced prostate cancer is elimination of testosterone production by the testes through either surgical or chemical castration. Bilateral orchiectomy or estrogen therapy in the form of diethylstilbestrol was previously used as first-line therapy. Presently, the use of luteinizing hormone-releasing hormone (LHRH) agonists— including leuprolide and goserelin agonists, alone or in combina-tion with an antiandrogen (eg, flutamide, bicalutamide, or nilutamide)—is the preferred approach. There appears to be no survival advantage of total androgen blockade using a combination of LHRH agonist and antiandrogen agent compared with single-agent therapy. Hormonal treatment reduces symptoms—especially bone pain—in 70–80% of patients and may cause a significant reduction in the prostate-specific antigen (PSA) level, which is now widely accepted as a surrogate marker for response to treatment in
prostate cancer. Although initial hormonal manipulation is able to control symptoms for up to 2 years, patients usually develop pro-gressive disease. Second-line hormonal therapies include aminoglu-tethimide plus hydrocortisone, the antifungal agent ketoconazole plus hydrocortisone, or hydrocortisone alone.
Unfortunately, nearly all patients with advanced prostate cancer eventually become refractory to hormone therapy. A regimen of mitoxantrone and prednisone is approved in patients with hor-mone-refractory prostate cancer because it provides effective pallia-tion in those who experience significant bone pain. Estramustine is an antimicrotubule agent that produces an almost 20% response rate as a single agent. However, when used in combination with either etoposide or a taxane such as docetaxel or paclitaxel, response rates are more than doubled to 40–50%. The combination of doc-etaxel and prednisone was recently shown to confer survival advan-tage when compared with the mitoxantrone-prednisone regimen, and this combination has now become the standard of care for hormone-refractory prostate cancer.
GASTROINTESTINAL CANCERS Colorectal cancer (CRC) is the most common type of gastrointesti-nal malignancy. Nearly 145,000 new cases are diagnosed each year in the USA; worldwide, nearly one million cases are diagnosed each year. At the time of initial presentation, only about 40–45% of patients are potentially curable with surgery. Patients presenting with high-risk stage II disease and stage III disease are candidates for adjuvant chemotherapy with an oxaliplatin-based regimen in com-bination with 5-FU plus leucovorin (FOLFOX or FLOX) or with oral capecitabine (XELOX) and are generally treated for 6 months following surgical resection. Treatment with this combination regi-men reduces the recurrence rate after surgery by 35% and clearly improves overall patient survival compared with surgery alone.
Significant advances have been made over the past 10 years with respect to treatment of metastatic CRC. There are four active cytotoxic agents—5-FU, the oral fluoropyrimidine capecitabine, oxaliplatin, and irinotecan; and three active biologic agents—the anti-VEGF antibody bevacizumab and the anti-EGFR antibodies cetuximab and panitumumab. In general, a fluoropyrimidine with either intravenous 5-FU or oral capecitabine serves as the main foundation of cytotoxic chemotherapy regimens. Recent clinical studies have shown that FOLFOX/FOLFIRI regimens in combi-nation with the anti-VEGF antibody bevacizumab or with the anti-EGFR antibody cetuximab result in significantly improved clinical efficacy with no worsening of the toxicities normally observed with chemotherapy. In order for patients to derive maxi-mal benefit, they should be treated with each of these active agents in a continuum of care approach. Using this strategy, median survivals now are in the 24–28 month range, and in some cases, approach 3 years. One of the main challenges facing clinicians at present is to begin to identify which patients would benefit from these various cytotoxic and biologic agents as well as identify those who might experience increased toxicity.
The incidence of gastric cancer, esophageal cancer, and pancre-atic cancer is much lower than for CRC, but these malignancies tend to be more aggressive and result in greater tumor-related
054-Katzung_Ch054_p949-976.indd 972 9/21/11 12:11:49 PM

CHAPTER 54 Cancer Chemotherapy 973
symptoms. In most cases, they cannot be completely resected surgically, as most patients present with either locally advanced or metastatic disease at the time of their initial diagnosis. 5-FU-based chemotherapy, using either intravenous 5-FU or oral capecitabine, is generally considered the main backbone for regimens targeting gastroesophageal cancers. In addition, cisplatin-based regimens in combination with either irinotecan or one of the taxanes (pacli-taxel or docetaxel) also exhibit clinical activity. Response rates in the 40–50% range are now being reported. Recent studies have shown that the addition of the biologic agent trastuzumab to cis-platin-containing chemotherapy regimens provides significant clinical benefit in gastric cancer patients whose tumors overexpress the HER-2/ neu receptor. In addition, neoadjuvant approaches with combination chemotherapy and radiation therapy prior to surgery appear to have promise in selected patients.
Although gemcitabine is approved for use as a single agent in metastatic pancreatic cancer, the overall response rate is less than 10%, with complete responses being quite rare. Intense efforts con-tinue to be placed on incorporating gemcitabine into various com-bination regimens and on identifying novel agents that target signal transduction pathways thought to be critical for the growth of pan-creatic cancer. One such agent is the small molecule inhibitor erlo-tinib, which targets the EGFR-associated tyrosine kinase. This agent is now approved for use in combination with gemcitabine in locally advanced or metastatic pancreatic cancer although the improvement in clinical benefit is relatively small. There is also evidence to sup-port the use of adjuvant chemotherapy with either single-agent gemcitabine or 5-FU/leucovorin in patients with early-stage pancre-atic cancer who have undergone successful surgical resection.
LUNG CANCER Lung cancer is divided into two main histopathologic subtypes, non-small cell and small cell. Non-small cell lung cancer (NSCLC) makes up about 75–80% of all cases of lung cancer, and this group includes adenocarcinoma, squamous cell cancer, and large cell cancer, while small cell lung cancer (SCLC) makes up the remain-ing 20–25%. When NSCLC is diagnosed in an advanced stage with metastatic disease, the prognosis is extremely poor, with a median survival of about 8 months. It is clear that prevention (pri-marily through avoidance of cigarette smoking) and early detection remain the most important means of control. When diagnosed at an early stage, surgical resection results in patient cure. Moreover, recent studies have shown that adjuvant platinum-based chemo-therapy provides a survival benefit in patients with pathologic stage IB, II, and IIIA disease. However, in most cases, distant metastases have occurred at the time of diagnosis. In certain instances, radia-tion therapy can be offered for palliation of pain, airway obstruc-tion, or bleeding and to treat patients whose performance status would not allow for more aggressive treatments.
In patients with advanced disease, systemic chemotherapy is gen-erally recommended. Combination regimens that include a plati-num agent (“platinum doublets”) appear superior to non-platinum doublets, and either cisplatin or carboplatin are appropriate platinum agents for such regimens. For the second drug, paclitaxel and vino-relbine appear to have activity independent of histology, while the
antifolate pemetrexed should be used for non-squamous cell cancer, and gemcitabine for squamous cell cancer. For patients with good performance status and those with non-squamous histology, the combination of the anti-VEGF antibody bevacizumab with carbo-platin and paclitaxel is a standard treatment option. In patients deemed not to be appropriate candidates for bevacizumab therapy and those with squamous cell histology, a platinum-based chemo-therapy regimen in combination with the anti-EGFR antibody cetuximab is a reasonable treatment strategy. Maintenance chemo-therapy with pemetrexed is now used in patients with non-squamous NSCLC whose disease has not progressed after four cycles of plati-num-based first-line chemotherapy. Finally, first-line therapy with an EGFR tyrosine kinase inhibitor, such as erlotinib or gefitinib, sig-nificantly improves outcomes in NSCLC patients with sensitizing EGFR mutations.
Small cell lung cancer is the most aggressive form of lung cancer, and it is exquisitely sensitive, at least initially, to platinum-based combination regimens, including cisplatin and etoposide or cisplatin and irinotecan. When diagnosed at an early stage, this disease is potentially curable using a combined modality approach of chemo-therapy and radiation therapy. Unfortunately, drug resistance eventu-ally develops in nearly all patients with extensive disease. The topoisomerase I inhibitor topotecan is used as second-line mono-therapy in patients who have failed a platinum-based regimen.
OVARIAN CANCER In the majority of patients, ovarian cancer remains occult and becomes symptomatic only after it has already metastasized to the peritoneal cavity. At this stage, it usually presents with malignant ascites. It is important to accurately stage this cancer with laparos-copy, ultrasound, and CT scanning. Patients with stage I disease appear to benefit from whole-abdomen radiotherapy and may receive additional benefit from combination chemotherapy with cisplatin and cyclophosphamide.
Combination chemotherapy is the standard approach to stage III and stage IV disease. Randomized clinical studies have shown that the combination of paclitaxel and cisplatin provides survival benefit compared with the previous standard combination of cis-platin plus cyclophosphamide. More recently, carboplatin plus paclitaxel has become the treatment of choice. In patients who present with recurrent disease, the topoisomerase I inhibitor topo-tecan, the alkylating agent altretamine, and liposomal doxorubicin are used as single agent monotherapy.
TESTICULAR CANCER The introduction of platinum-based combination chemotherapy has made an impressive change in the treatment of patients with advanced testicular cancer. Presently, chemotherapy is recom-mended for patients with stage IIC or stage III seminomas and nonseminomatous disease. Over 90% of patients respond to che-motherapy and, depending upon the extent and severity of dis-ease, complete remissions are observed in up to 70–80% of patents. Over 50% of patients achieving complete remission are cured with chemotherapy. In patients with good risk features,
054-Katzung_Ch054_p949-976.indd 973 9/21/11 12:11:49 PM

974 SECTION VIII Chemotherapeutic Drugs
three cycles of cisplatin, etoposide, and bleomycin (PEB protocol) or four cycles of cisplatin and etoposide yield virtually identical results. In patients with high-risk disease, the combination of cis-platin, etoposide, and ifosfamide can be used as well as etoposide and bleomycin with high-dose cisplatin.
MALIGNANT MELANOMA Malignant melanoma is curable when it presents locally, with surgi-cal resection being the main treatment approach. However, once spread to metastatic sites has been diagnosed, it is one of the most difficult cancers to treat because it is a relatively drug-resistant tumor. While dacarbazine, temozolomide, and cisplatin are the most active cytotoxic agents for this disease, the overall response rates to these agents remain low. Biologic agents, including interferon-α and interleukin-2 (IL-2), have greater activity than traditional cytotoxic agents, and treatment with high-dose IL-2 has led to cures, albeit in a relatively small subset of patients. Significant efforts have focused on utilizing biologic therapy with combination chemotherapy in what have been labeled biochemotherapy regi-mens. Although overall response rates as well as complete response rates appear to be much higher with biochemotherapy regimens compared with chemotherapy alone, treatment toxicity also seems to be increased. As such, this approach remains investigational.
The BRAFV600E mutation has been identified in the large majority of melanomas. This mutation results in constitutive acti-vation of BRAF kinase, which then leads to activation of down-stream signaling pathways involved in cell growth and proliferation. In August 2011, the FDA approved a novel, oral, and highly selec-tive small molecule inhibitor of BRAFV600E (vemurafenib) with highly promising activity in metastatic melanoma. Further studies are ongoing to confirm its activity as a single agent as well as in combination with other cytotoxic and biologic agents.
BRAIN CANCER Chemotherapy has only limited efficacy in the treatment of malig-nant gliomas. In general, the nitrosoureas, because of their ability to cross the blood-brain barrier, are the most active agents in this
disease. Carmustine (BCNU) can be used as a single agent, or lomustine (CCNU) can be used in combination with procarbazine and vincristine (PCV regimen). In addition, the newer alkylating agent temozolomide is active when combined with radiotherapy and used in patients with newly diagnosed glioblastoma multi-forme (GBM) as well as in those with recurrent disease. The histo-pathologic subtype oligodendroglioma has been shown to be especially chemosensitive, and the PCV regimen is the treatment of choice for this disease. There is growing evidence that the anti-VEGF antibody bevacizumab alone or in combination with che-motherapy has promising activity in adult GBM, and bevacizumab was recently approved as a single agent for GBM in the setting of progressive disease following first-line chemotherapy. Of note, small molecule inhibitors of VEGFR-TKs are also undergoing active clinical development because they show interesting activity in adult brain tumors.
SECONDARY MALIGNANCIES & CANCER CHEMOTHERAPY The development of secondary malignancies is a late complication of the alkylating agents and the epipodophyllotoxin etoposide. The most frequent secondary malignancy is acute myelogenous leukemia (AML). In general, AML develops in up to 15% of patients with Hodgkin’s lymphoma who have received radiother-apy plus MOPP chemotherapy and in patients with multiple myeloma, ovarian carcinoma, or breast carcinoma treated with melphalan. The increased risk of AML is observed as early as 2–4 years after the initiation of chemotherapy and typically peaks at 5 and 9 years. With improvements in the clinical efficacy of various combination chemotherapy regimens resulting in prolonged sur-vival and in some cases actual cure of cancer, the issue of how second cancers may affect long-term survival assumes greater importance. There is already evidence that certain alkylating agents (eg, cyclophosphamide) may be less carcinogenic than oth-ers (eg, melphalan). In addition to AML, other secondary malig-nancies have been well-described, including non-Hodgkin’s lymphoma and bladder cancer, the latter most typically associated with cyclophosphamide therapy.
The reader is referred to the manufacturer’s literature for the most recent information on preparations available.
P R E P A R A T I O N S A V A I L A B L E
The reader is referred to the manufacturer’s literature for the most recent information on preparations available.
SUMMARY Anticancer Drugs
See Tables 54-2, -3, -4, -5
054-Katzung_Ch054_p949-976.indd 974 9/21/11 12:11:49 PM

CHAPTER 54 Cancer Chemotherapy 975
REFERENCES Books & Monographs
Abeloff MD et al: Clinical Oncology, 4th ed. Elsevier, 2008. Chabner BA, Longo DL: Cancer Chemotherapy and Biotherapy: Principles and
Practice , 4th ed. Lippincott Williams & Wilkins, 2006. Chu E, DeVita VT Jr: Cancer Chemotherapy Drug Manual 2011, 11th ed. Jones &
Bartlett, 2010. DeVita VT Jr, Hellman S, Rosenberg SA: Cancer: Principles and Practice of
Oncology, 9th ed. Lippincott Williams & Wilkins, 2011. Harris JR et al: Diseases of the Breast , 4th ed. Lippincott Williams & Wilkins,
2009. Hoppe R et al: Textbook of Radiation Oncology , 3rd ed. Elsevier, 2010. Hoskins WJ, Perez CA, Young RC: Principles and Practice of Gynecologic Oncology ,
4th ed. Lippincott Williams & Wilkins, 2004. Kantoff PW et al: Prostate Cancer: Principles and Practice . Lippincott Williams &
Wilkins, 2001.
Kelsen DP et al: Gastrointestinal Oncology: Principles and Practices , 2nd ed. Lippincott Williams & Wilkins, 2007.
Kufe D et al: Cancer Medicine , 7th ed. BC Decker, 2006. Mendelsohn J et al: The Molecular Basis of Cancer . WB Saunders, 2008. Pass HI et al: Principles and Practice of Lung Cancer: The Official Reference Text of
the International Association for the Study of Lung Cancer (IASLC), 4th ed. Lippincott Williams & Wilkins, 2010.
Pizzo PA, Poplack AG: Principles and Practice of Pediatric Oncology, 6th ed. Lippincott Williams & Wilkins, 2010.
Weinberg RA: Biology of Cancer . Taylor & Francis, 2006.
Articles & Reviews
DeVita VT, Chu E: The history of cancer chemotherapy. Cancer Res 2008;68:8643.
Redmond KM et al: Resistance mechanisms to cancer chemotherapy. Front Biosci 2008;13:5138.
C A S E S T U D Y A N S W E R
The 5-year survival rate for patients with high-risk stage III CRC is on the order of 25–30%. Because the patient has no symptoms after surgery and has no comorbid illnesses, he would be an appropriate candidate to receive aggressive adjuvant chemotherapy. The usual recommendation would be to administer 6 months of oxaliplatin-based chemo-therapy using either infusional 5-FU or oral capecitabine as the fluoropyrimidine base in combination with oxalipla-tin. Adjuvant chemotherapy is usually begun 4–6 weeks after surgery to allow sufficient time for surgical wound to heal.
Patients with partial or complete deficiency in the enzyme dihydropyrimidine dehydrogenase (DPD) experience an increased incidence of severe toxicity to fluoropyrimidines in the form of myelosuppression, gastrointestinal toxicity, and neurotoxicity. Although mutations in DPD can be identified in peripheral blood mononuclear cells, nearly 50% of patients who exhibit severe 5-FU toxicity do not have a defined muta-tion in the DPD gene. In addition, such mutations may not result in reduced expression of the DPD protein or in altered enzymatic activity. For this reason, genetic testing is not rec-ommended at this time as part of routine clinical practice.
054-Katzung_Ch054_p949-976.indd 975 9/21/11 12:11:49 PM

This page intentionally left blank

977
C H A P T E R
Agents that suppress the immune system play an important role in preventing the rejection of organ or tissue grafts and in the treatment of certain diseases that arise from dysregulation of the immune response. While precise details of the mechanisms of action of a number of these agents are still obscure, knowledge of the elements of the immune system is useful in understanding their effects. Agents that augment the immune response or selectively alter the balance of various components of the immune system are also becoming impor-tant in the management of certain diseases such as cancer, AIDS, and autoimmune or inflammatory diseases. A growing number of other conditions (infections, cardiovascular diseases, organ transplanta-tion) may also be candidates for immune manipulation.
■ ELEMENTS OF THE IMMUNE SYSTEM
NORMAL IMMUNE RESPONSES
The immune system has evolved to protect the host from invading pathogens and to eliminate disease. At its functioning best, the immune system is exquisitely responsive to invading pathogens while retaining the capacity to recognize self tissues and antigens to which it is tolerant. Protection from infection and disease is provided by the collaborative efforts of the innate and adaptive immune systems.
The Innate Immune System The innate immune system is the first line of defense against invading pathogens (eg, bacteria, viruses, fungi, parasites) and
consists of mechanical, biochemical, and cellular components. Mechanical components include skin/epidermis and mucus; bio-chemical components include antimicrobial peptides and proteins (eg, defensins), complement, enzymes (eg, lysozyme, acid hydro-lases), interferons, acidic pH, and free radicals (eg, hydrogen per-oxide, superoxide anions); cellular components include neutrophils, monocytes, macrophages, natural killer (NK), and natural killer-T (NKT) cells. Unlike adaptive immunity, the innate immune response exists prior to infection, is not enhanced by repeated infection, and is generally not antigen-specific. An intact skin or mucosa is the first barrier to infection. When this barrier is breached, an immediate innate immune response, referred to as “inflammation” is provoked that ultimately leads to destruction of the pathogen. The process of pathogen destruction can be accom-plished, for example, by biochemical components such as lysozyme (which breaks down the protective peptidoglycan cell wall) and the split products arising from complement activation. Complement components ( Figure 55–1 ) enhance macrophage and neutrophil phagocytosis by acting as opsonins (C3b) and chemoattractants (C3a, C5a), which recruit immune cells from the bloodstream to the site of infection. The activation of comple-ment eventually leads to pathogen lysis via the generation of a membrane attack complex that creates holes in the membrane and results in leakage of cellular components.
During the inflammatory response triggered by infection, neu-trophils and monocytes enter the tissue sites from peripheral circula-tion. This cellular influx is mediated by the action of chemoattractant cytokines (chemokines) (eg, interleukin-8 [IL-8; CXCL8], macrophage chemotactic protein-1 [MCP-1; CCL2],
55Immunopharmacology Douglas F. Lake, PhD, Adrienne D. Briggs, MD, & Emmanuel T. Akporiaye, PhD
C A S E S T U D Y
A 30-year-old woman has one living child, age 6. Her child and her husband are Rh positive and she is Rh o (D) and D u negative. She is now in her ninth month of pregnancy and is in the labor room having frequent contractions. Her Rh
antibody test taken earlier in the pregnancy was negative. What immunotherapy is appropriate for this patient? When and how should it be administered?
055-Katzung_Ch055_p977-1000.indd 977 9/23/11 5:16:50 PM

978 SECTION VIII Chemotherapeutic Drugs
and macrophage inflammatory protein-1α [MIP-1α; CCL3]) released from activated endothelial cells and immune cells (mostly tissue macrophages) at the inflammatory site. Egress of the immune cells from blood vessels into the inflammatory site is mediated by adhesive interactions between cell surface receptors (eg, L -selectin, integrins) on the immune cells and ligands (eg, sialyl-Lewis x, intercellular adhesion molecule-1 [ICAM-1]) on the activated endothelial cell surface. The tissue macrophages as well as dendritic cells express pattern recognition receptors (PRRs) that include Toll-like receptors (TLRs), nucleotide-binding oli-gomerization domain (NOD)-like receptors (NLRs), scavenger receptors, mannose receptors, and lipopolysaccharide (LPS)-binding protein, which recognize key evolutionarily conserved pathogen components referred to as pathogen-associated molecu-lar patterns (PAMPs). Examples of PAMPs include microbe-derived unmethylated CpG DNA, flagellin, double-stranded RNA, peptidoglycan, and LPS. The PRRs recognize PAMPs in various components of pathogens and stimulate the release of proinflammatory cytokines, chemokines, and interferons. If the innate immune response is successfully executed, the invading pathogen is ingested, degraded, and eliminated, and disease is either prevented or is of short duration.
In addition to monocytes and neutrophils, natural killer (NK), natural killer-T (NKT), and gamma-delta T (fc T) cells recruited to the inflammatory site contribute to the innate response by secret-ing interferon-gamma (IFN-γ) and IL-17, which activate resident tissue macrophages and dendritic cells and recruit neutrophils respectively to successfully eliminate invading pathogens. NK cells are so called because they are able to recognize and destroy virus-infected normal cells as well as tumor cells without prior stimula-tion. This activity is regulated by “killer cell immunoglobulin-like receptors” (KIRs) on the NK cell surface that are specific for major histocompatibility complex (MHC) class I molecules. When NK cells bind self MHC class I proteins (expressed on all nucleated cells), these receptors deliver inhibitory signals, preventing them from killing normal host cells. Tumor cells or virus-infected cells that have down-regulated MHC class I expression do not engage these KIRs, resulting in activation of NK cells and subsequent destruction of the target cell. NK cells kill target cells by releasing cytotoxic granules such as perforins and granzymes that induce programmed cell death.
NKT cells express T-cell receptors as well as receptors com-monly found on NK cells. NKT cells recognize microbial lipid antigens presented by a unique class of MHC-like molecules known as CD1 and have been implicated in host defense against microbial agents, autoimmune diseases, and tumors.
The Adaptive Immune System The adaptive immune system is mobilized by cues from the innate response when the innate processes are incapable of coping with an infection. The adaptive immune system has a number of char-acteristics that contribute to its success in eliminating pathogens. These include the ability to (1) respond to a variety of antigens, each in a specific manner; (2) discriminate between foreign (“non-self ”) antigens (pathogens) and self antigens of the host; and (3) respond to a previously encountered antigen in a learned way by initiating a vigorous memory response. This adaptive response culminates in the production of antibodies, which are the effec-tors of humoral immunity; and the activation of T lymphocytes, which are the effectors of cell-mediated immunity.
The induction of specific adaptive immunity requires the par-ticipation of professional antigen-presenting cells (APCs), which include dendritic cells (DCs), macrophages, and B lymphocytes. These cells play pivotal roles in the induction of an adaptive immune response because of their capacity to phagocytize particu-late antigens (eg, pathogens) or endocytose protein antigens, and enzymatically digest them to generate peptides, which are then loaded onto class I or class II MHC proteins and “presented” to the cell surface T-cell receptor (TCR) ( Figure 55–2 ). CD8 T cells recognize class I-MHC peptide complexes while CD4 T cells rec-ognize class II-MHC peptide complexes. At least two signals are necessary for the activation of T cells. The first signal is delivered following engagement of the TCR with peptide-bound MHC molecules. In the absence of a second signal, the T cells become unresponsive (anergic) or undergo apoptosis. The second signal involves ligation of costimulatory molecules (CD40, CD80 [also known as B7-1], and CD86 [also known as B7-2]) on the APC to
A C R O N Y M SADA Adenosine deaminase
ALG Antilymphocyte globulin
APC Antigen-presenting cell
ATG Antithymocyte globulin
CD Cluster of differentiation
CSF Colony-stimulating factor
CTL Cytotoxic T lymphocyte
DC Dendritic cell
DTH Delayed-type hypersensitivity
FKBP FK-binding protein
HAMA Human antimouse antibody
HLA Human leukocyte antigen
IFN Interferon
IGIV Immune globulin intravenous
IL Interleukin
LFA Leukocyte function-associated antigen
MAB Monoclonal antibody
MHC Major histocompatibility complex
NK cell Natural killer cell
SCID Severe combined immunodeficiency disease
TCR T-cell receptor
TGF-a Transforming growth factor-βTH1, TH2 T helper cell types 1 and 2
TNF Tumor necrosis factor
055-Katzung_Ch055_p977-1000.indd 978 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 979
their respective ligands (CD40L for CD40, CD28 for CD80 or CD86). Activation of T cells is regulated via a negative feedback loop involving another molecule known as T-lymphocyte-associated antigen 4 (CTLA-4). Following engagement of CD28 with CD80 or CD86, CTLA-4 in the cytoplasm is mobilized to the cell surface where, because of its higher affinity of binding to CD80 and CD86, it outcompetes or displaces CD28 resulting in suppression of T-cell activation and proliferation. This property of CTLA-4 has been exploited as a strategy for sustaining a desirable immune response such as that directed against cancer. A recombi-nant humanized antibody that binds CTLA-4 (ipilimumab) pre-vents its association with CD80/CD86. In so doing, the activated state of T cells is sustained. Recently completed vaccine trials in metastatic melanoma patients receiving anti-CTLA-4 antibody reported objective and durable clinical responses in a few patients. Unfortunately, these beneficial responses were associated with the development of autoimmune toxicity in some patients, raising concern about this approach.
T lymphocytes develop and learn to recognize self and non-self antigens in the thymus; those T cells that bind with high affinity to self antigens in the thymus undergo apoptosis (negative selection),
Antigen-presenting cell
CD40
T lymphocyte
T cell
TCRMHC
CD40L
ICAM-1 LFA-1
CD28CD80/86
CTLA-4
CD2LFA-3
Dendritic cell
1
2
Macrophage
Opsonization
Macrophage
Inflammatory site
Bacteria
Monocyte Bloodstream
Endothelial cell
2
1
3
2
1
3
C3a,C5a
C3a,C5a
C3a, C5aChemotaxis
Complement (C1–C9)
C5b, C6, C7, C8, C9
Bacterial lysis
Release of salts,proteins, water, etc
Bacterial destruction
C3breceptor
C3b
A
B
C
C3b
(MAC)
FIGURE 55–1 Role of complement in innate immunity. Complement is made up of nine proteins (C1–C9), which are split into fragments during activation. A: Complement components (C3a, C5a) attract phagocytes (1) to inflammatory sites (2), where they ingest and degrade pathogens (3). B: Complement components C5b, C6, C7, C8, and C9 associate to form a membrane attack complex (MAC) that lyses bacteria, causing their destruction. C: Complement component C3b is an opsonin that coats bacteria (1) and facilitates their ingestion (2) and digestion (3) by phagocytes.
FIGURE 55–2 T-cell activation by an antigen-presenting cell requires engagement of the T-cell receptor by the MHC-peptide complex (signal 1) and binding of the costimulatory molecules (CD80, CD86) on the dendritic cell to CD28 on the T cell (signal 2). The activation signals are strengthened by CD40/CD40L and ICAM-1/LFA-1 interac-tions. In a normal immune response, T-cell activation is regulated by T-cell-derived CTLA-4, which binds to CD80 or CD86 with higher affinity than CD28 and sends inhibitory signals to the nucleus of the T cell.
055-Katzung_Ch055_p977-1000.indd 979 9/23/11 5:16:51 PM
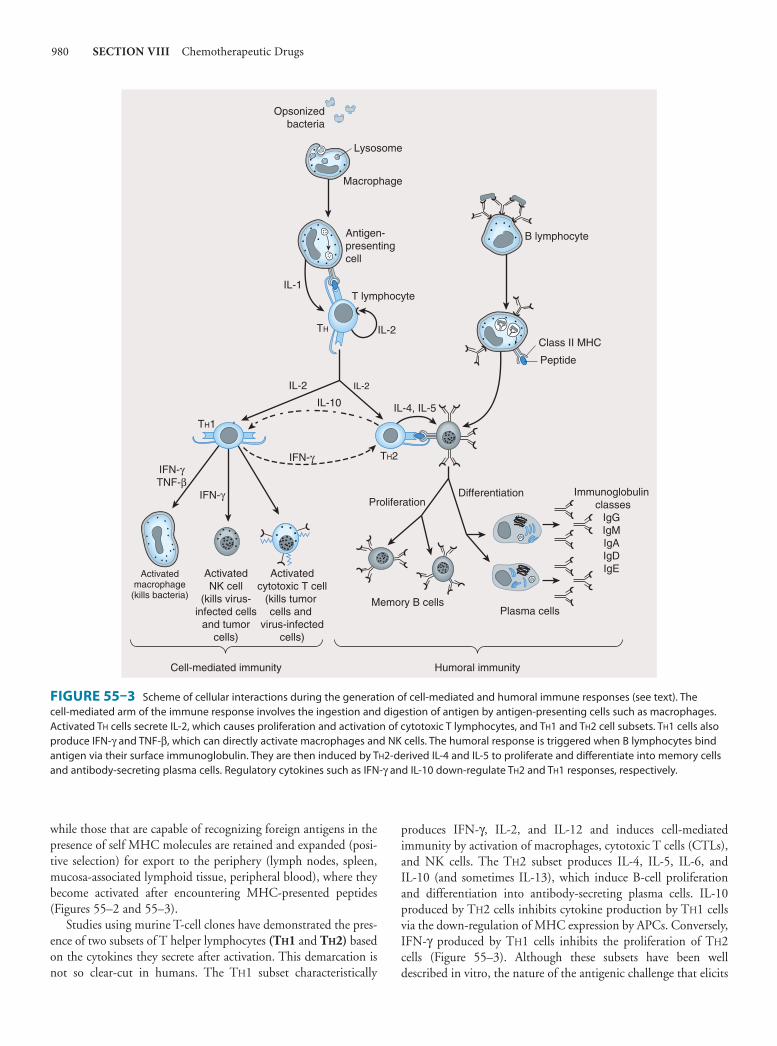
980 SECTION VIII Chemotherapeutic Drugs
while those that are capable of recognizing foreign antigens in the presence of self MHC molecules are retained and expanded (posi-tive selection) for export to the periphery (lymph nodes, spleen, mucosa-associated lymphoid tissue, peripheral blood), where they become activated after encountering MHC-presented peptides ( Figures 55–2 and 55–3 ).
Studies using murine T-cell clones have demonstrated the pres-ence of two subsets of T helper lymphocytes (T H 1 and T H 2) based on the cytokines they secrete after activation. This demarcation is not so clear-cut in humans. The T H 1 subset characteristically
produces IFN-γ, IL-2, and IL-12 and induces cell-mediated immunity by activation of macrophages, cytotoxic T cells (CTLs), and NK cells. The T H 2 subset produces IL-4, IL-5, IL-6, and IL-10 (and sometimes IL-13), which induce B-cell proliferation and differentiation into antibody-secreting plasma cells. IL-10 produced by T H 2 cells inhibits cytokine production by T H 1 cells via the down-regulation of MHC expression by APCs. Conversely, IFN-γ produced by T H 1 cells inhibits the proliferation of T H 2 cells ( Figure 55–3 ). Although these subsets have been well described in vitro, the nature of the antigenic challenge that elicits
Opsonizedbacteria
Macrophage
Lysosome
IL-2
IL-10
IFN-γ
IL-2
IL-4, IL-5
Activatedmacrophage
(kills bacteria)
ActivatedNK cell
(kills virus-infected cells
and tumorcells)
Activatedcytotoxic T cell
(kills tumor cells and
virus-infectedcells)
Cell-mediated immunity
Plasma cells
Humoral immunity
Memory B cells
B lymphocyte
Class II MHC
Peptide
Immunoglobulin classes
IgGIgMIgAIgDIgE
Antigen-presentingcell
IL-1
TH
TH1
TH2
IL-2
T lymphocyte
ProliferationIFN-γ
IFN-γTNF-β
Differentiation
FIGURE 55–3 Scheme of cellular interactions during the generation of cell-mediated and humoral immune responses (see text). The cell-mediated arm of the immune response involves the ingestion and digestion of antigen by antigen-presenting cells such as macrophages. Activated T H cells secrete IL-2, which causes proliferation and activation of cytotoxic T lymphocytes, and T H 1 and T H 2 cell subsets. T H 1 cells also produce IFN-γ and TNF-β, which can directly activate macrophages and NK cells. The humoral response is triggered when B lymphocytes bind antigen via their surface immunoglobulin. They are then induced by T H 2-derived IL-4 and IL-5 to proliferate and differentiate into memory cells and antibody-secreting plasma cells. Regulatory cytokines such as IFN-γ and IL-10 down-regulate T H 2 and T H 1 responses, respectively.
055-Katzung_Ch055_p977-1000.indd 980 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 981
a T H 1 or T H 2 phenotype is less clear. Extracellular bacteria typi-cally cause the elaboration of T H 2 cytokines, culminating in the production of neutralizing or opsonic antibodies. In contrast, intracellular organisms (eg, mycobacteria) elicit the production of T H 1 cytokines, which lead to the activation of effector cells such as macrophages. A less well-defined T-cell subset (T H 3) has been described that produces transforming growth factor-β (TGF-β), whose numerous functions include down-regulation of prolifera-tion and differentiation of T lymphocytes.
Recently, a subset of CD4 T cells that secrete IL-17 (T H 17) has been implicated in neutrophil recruitment to sites of inflamma-tion. A population of CD4 T cells that is essential for preventing autoimmunity and allergy as well as maintaining homeostasis and tolerance to self antigens is the regulatory (Treg) T cell. In the mouse, this cell population exists as natural Treg (nTreg), derived directly from the thymus, and induced (adaptive) Treg (iTreg), generated from naïve CD4 T cells in the periphery. Both popula-tions have also been shown to inhibit antitumor immune responses and are implicated in fostering tumor growth and progression. Recent attempts to distinguish both populations have resulted in the discovery of a transcription factor, Helios, in nTreg but not in iTreg cells.
CD8 T lymphocytes recognize endogenously processed pep-tides presented by virus-infected cells or tumor cells. These pep-tides are usually nine-amino-acid fragments derived from virus or protein tumor antigens in the cytoplasm and are loaded onto MHC class I molecules ( Figure 55–2 ) in the endoplasmic reticu-lum. In contrast, class II MHC molecules present peptides (usu-ally 11–22 amino acids) derived from extracellular (exogenous) pathogens to CD4 T helper cells. In some instances, exogenous antigens, upon ingestion by APCs, can be presented on class I MHC molecules to CD8 T cells. This phenomenon, referred to as “cross-presentation,” involves retro-translocation of antigens from the endosome to the cytosol for peptide generation in the prote-osome and is thought to be useful in generating effective immune responses against infected host cells that are incapable of priming T lymphocytes. Upon activation, CD8 T cells induce target cell death via lytic granule enzymes (“granzymes”), perforin, and the Fas-Fas ligand (Fas-FasL) apoptosis pathways.
B lymphocytes undergo selection in the bone marrow, during which self-reactive B lymphocytes are clonally deleted while B-cell clones specific for foreign antigens are retained and expanded. The repertoire of antigen specificities by T cells is genetically determined and arises from T-cell receptor gene rear-rangement while the specificities of B cells arise from immuno-globulin gene rearrangement; for both types of cells, these determinations occur prior to encounters with antigen. Upon an encounter with antigen, a mature B cell binds the antigen, inter-nalizes and processes it, and presents its peptide—bound to class II MHC—to CD4 helper cells, which in turn secrete IL-4 and IL-5. These interleukins stimulate B-cell proliferation and dif-ferentiation into memory B cells and antibody-secreting plasma cells. The primary antibody response consists mostly of IgM-class immunoglobulins. Subsequent antigenic stimulation results in a vigorous “booster” response accompanied by class (isotype) switching to produce IgG, IgA, and IgE antibodies with diverse
effector functions ( Figure 55–3 ). These antibodies also undergo affinity maturation, which allows them to bind more efficiently to the antigen. With the passage of time, this results in acceler-ated elimination of microorganisms in subsequent infections. Antibodies mediate their functions by acting as opsonins to enhance phagocytosis and cellular cytotoxicity and by activating complement to elicit an inflammatory response and induce bacte-rial lysis ( Figure 55–4 ).
ABNORMAL IMMUNE RESPONSES
Whereas the normally functioning immune response can success-fully neutralize toxins, inactivate viruses, destroy transformed cells, and eliminate pathogens, inappropriate responses can lead to extensive tissue damage (hypersensitivity) or reactivity against self antigens (autoimmunity); conversely, impaired reactivity to appro-priate targets (immunodeficiency) may occur and abrogate essen-tial defense mechanisms.
Hypersensitivity Hypersensitivity can be classified as antibody-mediated or cell-mediated. Three types of hypersensitivity are antibody-mediated (types I–III), while the fourth is cell-mediated (type IV). Hypersensitivity occurs in two phases: the sensitization phase and the effector phase. Sensitization occurs upon initial encounter with an antigen; the effector phase involves immunologic memory and results in tissue pathology upon a subsequent encounter with that antigen.
1. Type I— Immediate, or type I, hypersensitivity is IgE-mediated, with symptoms usually occurring within minutes following the patient’s reencounter with antigen. Type I hypersensitivity results from cross-linking of membrane-bound IgE on blood basophils or tissue mast cells by antigen. This cross-linking causes cells to degran-ulate, releasing substances such as histamine, leukotrienes, and eosinophil chemotactic factor, which induce anaphylaxis, asthma, hay fever, or urticaria (hives) in affected individuals ( Figure 55–5 ). A severe type I hypersensitivity reaction such as systemic anaphylaxis (eg, from insect envenomation, ingestion of certain foods, or drug hypersensitivity) requires immediate medical intervention.
2. Type II— Type II hypersensitivity results from the formation of antigen-antibody complexes between foreign antigen and IgM or IgG immunoglobulins. One example of this type of hypersen-sitivity is a blood transfusion reaction that can occur if blood is not cross-matched properly. Preformed antibodies bind to red blood cell membrane antigens that activate the complement cas-cade, generating a membrane attack complex that lyses the trans-fused red blood cells. In hemolytic disease of the newborn, anti-Rh IgG antibodies produced by an Rh-negative mother cross the placenta, bind to red blood cells of an Rh-positive fetus, and damage them. The disease is prevented in subsequent pregnancies by the administration of anti-Rh antibodies to the mother 24–48 hours after delivery (see Immunosuppressive Antibodies, below).
055-Katzung_Ch055_p977-1000.indd 981 9/23/11 5:16:51 PM

982 SECTION VIII Chemotherapeutic Drugs
Type II hypersensitivity can also be drug-induced and may occur during the administration of penicillin (for example) to allergic patients. In these patients, penicillin binds to red blood cells or other host tissue to form a neoantigen that evokes production of antibodies capable of inducing complement-mediated red cell lysis. In some circumstances, subsequent administration of the drug can lead to systemic anaphylaxis (type I hypersensitivity).
3. Type III— Type III hypersensitivity is due to the presence of elevated levels of antigen-antibody complexes in the circulation that ultimately deposit on basement membranes in tissues and vessels. Immune complex deposition activates complement to produce components with anaphylatoxic and chemotactic activi-ties (C5a, C3a, C4a) that increase vascular permeability and
recruit neutrophils to the site of complex deposition. Complex deposition and the action of lytic enzymes released by neutrophils can cause skin rashes, glomerulonephritis, and arthritis in these individuals. If patients have type III hypersensitivity against a particular antigen, clinical symptoms usually occur 3–4 days after exposure to the antigen.
4. Type IV: Delayed-type hypersensitivity— Unlike type I, II, and III hypersensitivities, delayed-type hypersensitivity (DTH) is cell-mediated, and responses occur 2–3 days after exposure to the sensitizing antigen. DTH is caused by antigen-specific DTH T H 1 cells and induces a local inflammatory response that causes tissue damage characterized by the influx of antigen- non specific inflammatory cells, especially macrophages. These cells are
–S-S––S-S–
Fc receptor
Fc region
Opsonization
Antigen-bindingregion
Hingeregion
Bacteria
C
B
A
Epitope
Macrophage
Bacterial lysis
Complement activation
–S-S––S-S–
Complementarity-determining
region (CDR)
CH1 VH
CH2
CH3
VLCL
FIGURE 55–4 Antibody has multiple functions. The prototypical antibody consists of two heavy (H) and two light (L) chains, each sub-divided into constant (C L , C H ) and variable (V L , V H ) domains. The structure is held together by intra- and interchain disulfide bridges. A: The complementarity-determining region (CDR) of the antigen-binding portion of the antibody engages the antigenic determinant (epitope) in a lock and key fashion. B: Antigen-antibody complexes activate complement to produce split complement components that cause bacterial lysis. C: The Fc portion of antibodies binds to Fc receptors on phagocytes (eg, macrophages, neutrophils) and facilitates uptake of bacteria (opsonization).
055-Katzung_Ch055_p977-1000.indd 982 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 983
recruited under the influence of T H 1-produced cytokines ( Figure 55–6 ), which chemoattract circulating monocytes and neutrophils, induce myelopoiesis, and activate macrophages. The activated macrophages are primarily responsible for the tissue damage associated with DTH. Although widely considered to be deleterious, DTH responses are very effective in eliminating infec-tions caused by intracellular pathogens such as Mycobacterium tuberculosis and Leishmania species. Clinical manifestations of DTH include tuberculin and contact hypersensitivities . Tuberculosis exposure is determined using a DTH skin test. Positive responses show erythema and induration caused by accu-mulation of macrophages and DTH T (T DTH ) cells at the site of the tuberculin injection. Poison ivy is the most common cause of contact hypersensitivity, in which pentadecacatechol, the lipo-philic chemical in poison ivy, modifies cellular tissue and results in a DTH T-cell response.
Autoimmunity Autoimmune disease arises when the body mounts an immune response against itself due to failure to distinguish self tissues and cells from foreign (nonself ) antigens or loss of tolerance to self. This phenomenon derives from the activation of self-reactive T and B lymphocytes that generate cell-mediated or humoral immune responses directed against self antigens. The pathologic conse-quences of this reactivity constitute several types of autoimmune diseases. Autoimmune diseases are highly complex due to MHC genetics, environmental conditions, infectious entities, and dys-functional immune regulation. Examples of such diseases include rheumatoid arthritis, systemic lupus erythematosus, multiple scle-rosis, and insulin-dependent diabetes mellitus (type 1 diabetes). In rheumatoid arthritis, IgM antibodies (rheumatoid factors) are pro-duced that react with the Fc portion of IgG and may form immune complexes that activate the complement cascade, causing chronic
Effects Smooth muscle contraction Vasodilation Increased vascular permeability Platelet aggregation Complement activation Mucus secretion
Mediators Histamine Serotonin Leukotrienes Prostaglandins Bradykinins Proteases Eosinophil chemotactic factor Neutrophil chemotactic factor
Clinical symptoms Asthma Hay fever Skin rashes Local anaphylaxis Systemic anaphylaxis
Naive B cell
+IL-4,-5T helper cell
IgE binds IgE Fcεreceptors on mastcells or basophils
IgE-secreting plasma cellIgE is specific for allergen Allergen cross-links IgE on mast
cell (or basophil) and triggers degranulation and release of pharmacologic mediators
Sensitization phase
Effector phase
FIGURE 55–5 Mechanism of type I hypersensitivity. Initial exposure to allergen (sensitization phase) leads to production of IgE by plasma cells differentiated from allergen-specific B cells (not shown). The secreted IgE binds IgE-specific receptors (FcεR) on blood basophils and tissue mast cells. Reexposure to allergen leads to cross-linking of membrane-bound IgE (effector phase). This cross-linking causes degranulation of cytoplasmic granules and release of mediators that induce vasodilation, smooth muscle contraction, and increased vascular permeability. These effects lead to the clinical symptoms characteristic of type I hypersensitivity.
055-Katzung_Ch055_p977-1000.indd 983 9/23/11 5:16:51 PM

984 SECTION VIII Chemotherapeutic Drugs
inflammation of the joints and kidneys. In systemic lupus erythe-matosus, antibodies are made against DNA, histones, red blood cells, platelets, and other cellular components. In multiple sclerosis and type 1 diabetes, cell-mediated autoimmune attack destroys myelin surrounding nerve cells and insulin-producing islet beta cells of the pancreas, respectively. In type 1 diabetes, activated CD4 T DTH cells that infiltrate the islets of Langerhans and recognize self islet beta cell peptides are thought to produce cytokines that stimu-late macrophages to produce lytic enzymes, which destroy islet beta cells. Autoantibodies directed against the islet beta cell antigens are produced, but do not contribute significantly to disease.
A number of mechanisms have been proposed to explain auto-immunity:
1. Exposure of antigens previously sequestered from the immune system (eg, lens protein, myelin basic protein) to self-reactive T lymphocytes.
2. Molecular mimicry by invading pathogens, in which immune responses are directed at antigenic determinants on pathogens that share identical or similar epitopes with normal host tissue. This phenomenon occurs in rheumatic fever following Streptococcus pyogenes infection, in which heart damage is thought to arise from an immune response directed against streptococcal antigens shared with heart muscle. The suggested viral etiology of autoimmune diseases has been ascribed to immune responses (both cell-mediated and humoral) directed against virus epitopes that mimic self antigens.
3. Inappropriate expression of class II MHC molecules on the membranes of cells that normally do not express class II MHC (eg, islet beta cells). Increased expression of MHC II may increase presentation of self peptides to T helper cells, which in
turn induce CTL, T DTH , and B-lymphocyte cells that react against self antigens.
Immunodeficiency Diseases Immunodeficiency diseases result from inadequate function in the immune system; the consequences include increased susceptibility to infections and prolonged duration and severity of disease. Immunodeficiency diseases are either congenital or arise from extrinsic factors such as bacterial or viral infections or drug treat-ment. Affected individuals frequently succumb to infections caused by opportunistic organisms of low pathogenicity for the immuno-competent host. Examples of congenitally acquired immunodefi-ciency diseases include X-linked agammaglobulinemia, DiGeorge’s syndrome, and severe combined immunodeficiency disease (SCID) due to adenosine deaminase (ADA) deficiency.
X-linked agammaglobulinemia is a disease affecting males that is characterized by a failure of immature B lymphocytes to mature into antibody-producing plasma cells. These individuals are susceptible to recurrent bacterial infections, although the cell-mediated responses directed against viruses and fungi are preserved. DiGeorge’s syndrome is due to failure of the thymus to develop, resulting in diminished T-cell responses (T DTH , CTL), while the humoral response remains functional, but does not benefit from T-cell help.
The ADA enzyme normally prevents the accumulation of toxic deoxy-ATP in cells. Deoxy-ATP is particularly toxic to lym-phocytes, and leads to death of T and B cells. Absence of the enzyme therefore results in SCID. Infusion of the purified enzyme ( pegademase, from bovine sources) and transfer of ADA
Sensitization phase: initial contact with antigen
Effector phase: secondary contact with antigen
Macrophage
Antigen-presentingcell (eg, macrophage,
Langerhans cell)
Class II MHC
TCR
CD4
IL-1
IL-1
IL-2 IL-2
IL-2
Memory TDTH
IL-2 receptor
Allergen
Proliferation and differentiation
T helpercell (TH1)
TDTH
Cytokines
Chemotaxisof macrophage
Activation of macrophages(increased phagocyticand microbicidal activities)
Induction of myelopoiesis of macrophage and neutrophil precursors
Chemotaxis and extravasation of macrophages
IL-8 MIF MCP
IFN-γ TNF-β
IL-3 GM-CSF
IL-8 TNF-α MIP
Effects
FIGURE 55–6 Mechanism of type IV hypersensitivity (DTH). In the sensitization phase, the processed allergen (eg, from poison oak) is presented to CD4 T H 1 cells by antigen-presenting cells in association with class II MHC. T cells are induced to express IL-2 receptors and are stim-ulated to proliferate and differentiate into memory T DTH cells. Secondary contact with antigen triggers the effector phase, in which memory T DTH cells release cytokines that attract and activate nonspecific inflammatory macrophages and neutrophils. These cells display increased phagocytic and microbicidal activities and release large quantities of lytic enzymes that cause extensive tissue damage.
055-Katzung_Ch055_p977-1000.indd 984 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 985
gene-modified lymphocytes have both been used successfully to treat this disease.
AIDS represents the classic example of immunodeficiency dis-ease caused by extrinsic viral infection, in this instance the human immunodeficiency virus (HIV). This virus exhibits a strong tro-pism for CD4 T helper cells; these become depleted, giving rise to increased frequency of opportunistic infections and malignancies in infected individuals. AIDS is also characterized by an imbalance in T H 1 and T H 2 cells, and the ratios of cells and their functions are skewed toward T H 2. This results in loss of cytotoxic T-lymphocyte activity, loss of delayed hypersensitivity, and hyper-gammaglobulinemia.
■ IMMUNOSUPPRESSIVE AGENTS
Immunosuppressive agents have proved very useful in minimizing the occurrence or impact of deleterious effects of exaggerated or inappropriate immune responses. Unfortunately, these agents also have the potential to cause disease and to increase the risk of infec-tion and malignancies.
GLUCOCORTICOIDS
Glucocorticoids (corticosteroids) were the first hormonal agents recognized as having lympholytic properties. Administration of any glucocorticoid reduces the size and lymphoid content of the lymph nodes and spleen, although it has no toxic effect on prolif-erating myeloid or erythroid stem cells in the bone marrow.
Glucocorticoids are thought to interfere with the cell cycle of activated lymphoid cells. The mechanism of their action is described in Chapter 39 . Glucocorticoids are quite cytotoxic to certain subsets of T cells, but their immunologic effects are prob-ably due to their ability to modify cellular functions rather than to direct cytotoxicity. Although cellular immunity is more affected than humoral immunity, the primary antibody response can be diminished, and with continued use, previously established anti-body responses are also decreased. Additionally, continuous administration of corticosteroid increases the fractional catabolic rate of IgG, the major class of antibody immunoglobulins, thus lowering the effective concentration of specific antibodies. Contact hypersensitivity mediated by T DTH cells, for example, is usually abrogated by glucocorticoid therapy.
Glucocorticoids are used in a wide variety of conditions ( Table 55–1 ). It is thought that the immunosuppressive and anti-
TABLE 55–1 Clinical uses of immunosuppressive agents.
Source Immunopharmacologic Agents Used Response
Autoimmune diseases
Idiopathic thrombocytopenic purpura (ITP)
Prednisone,1 vincristine, occasionally cyclophosphamide, mercaptopurine, or azathioprine; commonly high-dose gamma globulin, plasma immunoadsorption or plasma exchange
Usually good
Autoimmune hemolytic anemia Prednisone,1 cyclophosphamide, chlorambucil, mercaptopurine, azathioprine, high-dose gamma globulin
Usually good
Acute glomerulonephritis Prednisone,1 mercaptopurine, cyclophosphamide Usually good
Acquired factor XIII antibodies Cyclophosphamide plus factor XIII Usually good
Autoreactive tissue disorders (autoimmune diseases)2
Prednisone, cyclophosphamide, methotrexate, interferon-α and -β, azathioprine, cyclosporine, infliximab, etanercept, adalimumab
Often good, variable
Isoimmune disease
Hemolytic disease of the newborn Rho(D) immune globulin Excellent
Organ transplantation
Renal Cyclosporine, azathioprine, prednisone, ALG, OKT3, tacrolimus, basiliximab,3 daclizumab,3 sirolimus
Very good
Heart Cyclosporine, azathioprine, prednisone, ALG, OKT3, tacrolimus, basiliximab,3 daclizumab,3 sirolimus
Good
Liver Cyclosporine, prednisone, azathioprine, tacrolimus, sirolimus Fair
Bone marrow Cyclosporine, cyclophosphamide, prednisone, methotrexate, ALG Good
Prevention of cell proliferation
Coronary stents Sirolimus (impregnated stent) Good
Neovascular macular degeneration Ranibizumab (labeled), bevacizumab (off-label) Fair
1Drug of choice.2Including systemic lupus erythematosus, rheumatoid arthritis, scleroderma, dermatomyositis, mixed tissue disorder, multiple sclerosis, Wegener’s granulomatosis, chronic active hepatitis, lipoid nephrosis, and inflammatory bowel disease.3Basiliximab and daclizumab are approved for renal transplant only.
055-Katzung_Ch055_p977-1000.indd 985 9/23/11 5:16:51 PM

986 SECTION VIII Chemotherapeutic Drugs
inflammatory properties of corticosteroids account for their ben-eficial effects in diseases like idiopathic thrombocytopenic purpura and rheumatoid arthritis. Glucocorticoids modulate allergic reac-tions and are useful in the treatment of diseases like asthma or as premedication for other agents (eg, blood products, chemother-apy) that might cause undesirable immune responses. Gluco-corticoids are first-line immunosuppressive therapy for both solid organ and hematopoietic stem cell transplant recipients, with vari-able results. The toxicities of long-term glucocorticoid therapy can be severe and are discussed in Chapter 39 .
CALCINEURIN INHIBITORS
Cyclosporine Cyclosporine (cyclosporin A, CSA) is an immunosuppressive agent with efficacy in human organ transplantation, in the treat-ment of graft-versus-host disease after hematopoietic stem cell transplantation, and in the treatment of selected autoimmune disorders. Cyclosporine is a peptide antibiotic that appears to act at an early stage in the antigen receptor-induced differentiation of T cells and blocks their activation. Cyclosporine binds to cyclo-philin , a member of a class of intracellular proteins called immu-nophilins. Cyclosporine and cyclophilin form a complex that inhibits the cytoplasmic phosphatase, calcineurin, which is neces-sary for the activation of a T-cell-specific transcription factor. This transcription factor, NF-AT, is involved in the synthesis of inter-leukins (eg, IL-2) by activated T cells. In vitro studies have indi-cated that cyclosporine inhibits the gene transcription of IL-2, IL-3, IFN-γ, and other factors produced by antigen-stimulated T cells, but it does not block the effect of such factors on primed T cells nor does it block interaction with antigen.
Cyclosporine may be given intravenously or orally, though it is slowly and incompletely absorbed (20–50%). The absorbed drug is primarily metabolized by the P450 3A enzyme system in the liver with resultant multiple drug interactions. This propensity for drug interaction contributes to significant interpatient variability in bioavailability, such that cyclosporine requires individual patient dosage adjustments based on steady-state blood levels and the desired therapeutic ranges for the drug. Cyclosporine ophthal-mic solution is now available for severe dry eye syndrome, as well as ocular graft-versus-host disease. Inhaled cyclosporine is being investigated for use in lung transplantation.
Toxicities are numerous and include nephrotoxicity, hyperten-sion, hyperglycemia, liver dysfunction, hyperkalemia, altered mental status, seizures, and hirsutism. However, cyclosporine causes very little bone marrow toxicity. While an increased inci-dence of lymphoma and other cancers (Kaposi’s sarcoma, skin cancer) have been observed in transplant recipients receiving cyclosporine, other immunosuppressive agents may also predis-pose recipients to cancer. Some evidence suggests that tumors may arise after cyclosporine treatment because the drug induces TGF-β, which promotes tumor invasion and metastasis.
Cyclosporine may be used alone or in combination with other immunosuppressants, particularly glucocorticoids. It has been used
successfully as the sole immunosuppressant for cadaveric transplan-tation of the kidney, pancreas, and liver, and it has proved extremely useful in cardiac transplantation as well. In combination with methotrexate, cyclosporine is a standard prophylactic regimen to prevent graft-versus-host disease after allogeneic stem cell trans-plantation. Cyclosporine has also proved useful in a variety of autoimmune disorders, including uveitis, rheumatoid arthritis, psoriasis, and asthma. Its combination with newer agents is show-ing considerable efficacy in clinical and experimental settings where effective and less toxic immunosuppression is needed. Newer for-mulations of cyclosporine have been developed that are improving patient compliance (smaller, better tasting pills) and increasing bioavailability.
Tacrolimus Tacrolimus (FK 506) is an immunosuppressant macrolide antibi-otic produced by Streptomyces tsukubaensis. It is not chemically related to cyclosporine, but their mechanisms of action are similar. Both drugs bind to cytoplasmic peptidylprolyl isomerases that are abundant in all tissues. While cyclosporine binds to cyclophilin, tacrolimus binds to the immunophilin FK-binding protein (FKBP). Both complexes inhibit calcineurin, which is necessary for the activation of the T-cell-specific transcription factor NF-AT.
On a weight basis, tacrolimus is 10–100 times more potent than cyclosporine in inhibiting immune responses. Tacrolimus is utilized for the same indications as cyclosporine, particularly in organ and stem cell transplantation. Multicenter studies in the USA and in Europe indicate that both graft and patient survival are similar for the two drugs. Tacrolimus has proved to be effective therapy for preventing rejection in solid-organ transplant patients even after failure of standard rejection therapy, including anti-T-cell antibodies. It is now considered a standard prophylactic agent (usually in combination with methotrexate or mycophenolate mofetil) for graft-versus-host disease.
Tacrolimus can be administered orally or intravenously. The half-life of the intravenous form is approximately 9–12 hours. Like cyclosporine, tacrolimus is metabolized primarily by P450 enzymes in the liver, and there is potential for drug interactions. The dosage is determined by trough blood level at steady state. Its toxic effects are similar to those of cyclosporine and include neph-rotoxicity, neurotoxicity, hyperglycemia, hypertension, hyper-kalemia, and gastrointestinal complaints.
Because of the effectiveness of systemic tacrolimus in some dermatologic diseases, a topical preparation is now available. Tacrolimus ointment is currently used in the therapy of atopic dermatitis and psoriasis.
MAMMALIAN TARGET OF RAPAMYCIN (mTOR) INHIBITORS
The mTOR inhibitors include sirolimus (rapamycin) as well as its analogs (called “rapalogs”) such as everolimus and temsirolimus.
055-Katzung_Ch055_p977-1000.indd 986 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 987
Sirolimus is an immunosuppressant macrolide antibiotic produced by Streptomyces hygroscopicus and is structurally similar to tacroli-mus. Sirolimus binds the circulating immunophilin FK506-binding protein 12, resulting in an active complex that inhibits the kinase activity of mammalian target of rapamycin (mTOR). The mTOR is a key component of a complex intracellular signaling pathway involved in cellular processes such as cell growth and pro-liferation, angiogenesis, and metabolism. Thus, blockade of mTOR ultimately can, for example, lead to inhibition of interleukin-driven T-cell proliferation. The signaling pathways involved in mTor are an active area of investigation in immunotherapy and targeted cancer therapy. Sirolimus is available only as an oral drug. Its half-life is about 60 hours, while that of everolimus is about 43 hours. Both drugs are rapidly absorbed and elimination is similar to that of cyclosporine and tacrolimus, being substrates for both cytochrome P450 3A and P-glycoprotein. Hence, significant drug interactions can occur. For example, use with cyclosporine can increase the plasma levels of both sirolimus and everolimus such that drug levels need to be monitored. The target dose-range of these drugs will vary depending on clinical use.
Sirolimus has been used effectively alone and in combination with other immunosuppressants (corticosteroids, cyclosporine, tacrolimus, and mycophenolate mofetil) to prevent rejection of solid organ allografts. It is used as prophylaxis and as therapy for steroid-refractory acute and chronic graft-versus-host disease in hematopoietic stem cell transplant recipients. Topical sirolimus is also used in some dermatologic disorders and, in combination with cyclosporine, in the management of uveoretinitis. Recently, sirolimus-eluting coronary stents have been shown to reduce rest-enosis and other adverse cardiac events in patients with severe coronary artery disease. These benefits appear to be due to its antiproliferative effects. Everolimus is a rapalog with better bio-availability than sirolimus, but it appears that its clinical efficacy is similar to sirolimus in solid organ transplant recipients. Temsirolimus is not currently used as an immunosuppressant. Both temsirolimus and everolimus are used and being investigated as targeted therapy for various cancers.
Toxicities of the mTOR inhibitors can include profound myelosuppression (especially thrombocytopenia), hepatotoxicity, diarrhea, hypertriglyceridemia, pneumonitis, and headache. Because nephrotoxicity is of major concern when administering calcineurin inhibitors, sirolimus is frequently employed as first-line immunosuppressant therapy in both solid organ and stem cell transplantation because renal toxicity is usually not seen. However, increased use in stem cell transplantation regimens as graft-versus-host disease prophylaxis, particularly when combined with tacrolimus, has revealed an increased incidence of hemolyt-ic-uremic syndrome.
MYCOPHENOLATE MOFETIL
Mycophenolate mofetil (MMF) is a semisynthetic derivative of mycophenolic acid, isolated from the mold Penicillium glaucus . In vitro, it inhibits T- and B-lymphocyte responses, including mito-gen and mixed lymphocyte responses, probably by inhibition of
de novo synthesis of purines. Mycophenolate mofetil is hydrolyzed to mycophenolic acid, the active immunosuppressive moiety; it is synthesized and administered as MMF to enhance bioavailability.
Mycophenolate mofetil is available in both oral and intrave-nous forms. The oral form is rapidly metabolized to mycophenolic acid. Although the cytochrome P450 3A system is not involved, some drug interactions still occur. Plasma drug levels are fre-quently monitored, similar to the calcineurin inhibitors and PSIs.
Mycophenolate mofetil is used in solid organ transplant patients for refractory rejection and, in combination with predni-sone, as an alternative to cyclosporine or tacrolimus in patients who do not tolerate those drugs. Its antiproliferative properties make it the first-line drug for preventing or reducing chronic allograft vasculopathy in cardiac transplant recipients. Myco-phenolate mofetil is used as prophylaxis for and treatment of both acute and chronic graft-versus-host disease in hematopoietic stem cell transplant patients. Newer immunosuppressant applica-tions for MMF include lupus nephritis, rheumatoid arthritis, inflammatory bowel disease, and some dermatologic disorders.
Toxicities include gastrointestinal disturbances (nausea and vomiting, diarrhea, abdominal pain) headache, hypertension, and reversible myelosuppression (primarily neutropenia).
IMMUNOMODULATORY DERIVATIVES OF THALIDOMIDE (IMiDs)
Thalidomide is a sedative drug that was withdrawn from the market in the 1960s because of its disastrous teratogenic effects when used during pregnancy. Nevertheless, it has significant immunomodulatory actions and is currently in active use or in clinical trials for over 40 different illnesses. Thalidomide inhibits angiogenesis and has anti-inflammatory and immunomodulatory effects. It inhibits tumor necrosis factor-alpha (TNF-α), reduces phagocytosis by neutrophils, increases production of IL-10, alters adhesion molecule expression, and enhances cell-mediated immu-nity via interactions with T cells. The complex actions of thalido-mide continue to be studied as its clinical use evolves. Owing to thalidomide’s serious toxicity profile, considerable effort has been expended in the development of analogs. Immunomodulatory derivatives of thalidomide are termed IMiDs. Some IMiDs are much more potent than thalidomide in regulating cytokines and affecting T-cell proliferation.
Thalidomide is currently used in the treatment of multiple myeloma at initial diagnosis and for relapsed-refractory disease. Patients generally show signs of response within 2–3 months of starting the drug, with response rates from 20% to 70%. When combined with dexamethasone, the response rates in myeloma are 90% or more in some studies. Many patients have durable responses—up to 12–18 months in refractory disease and even longer in some patients treated at diagnosis. The success of thali-domide in myeloma has led to numerous clinical trials in other diseases such as myelodysplastic syndrome, acute myelogenous leukemia, and graft-versus-host disease, as well as in solid tumors like colon cancer, renal cell carcinoma, melanoma, and prostate
055-Katzung_Ch055_p977-1000.indd 987 9/23/11 5:16:51 PM

988 SECTION VIII Chemotherapeutic Drugs
cancer, with variable results to date. Thalidomide has been used for many years in the treatment of some manifestations of leprosy and has been reintroduced in the USA for erythema nodosum leprosum; it is also useful in management of the skin manifesta-tions of lupus erythematosus.
The adverse effect profile of thalidomide is extensive. The most important toxicity is teratogenesis. Because of this effect, thalido-mide prescription and use are closely regulated by the manufac-turer. Other adverse effects of thalidomide include peripheral neuropathy, constipation, rash, fatigue, hypothyroidism, and increased risk of deep vein thrombosis. Thrombosis is sufficiently frequent, particularly in the hematologic malignancy population, that most patients are placed on some type of anticoagulant when thalidomide treatment is initiated. Lenalidomide is an IMiD that in animal and in vitro studies has been shown to be similar to thalidomide in action, but with less toxicity, especially teratoge-nicity. Lenalidomide was approved by the Food and Drug Administration (FDA) in late 2005 as a consequence of trials that showed its effectiveness in the treatment of the myelodysplastic syndrome with the chromosome 5q31 deletion. Clinical trials using lenalidomide to treat newly diagnosed as well as relapsed or refractory multiple myeloma showed similar efficacy, leading to FDA approval for myeloma as well. Its side effect profile is similar to that of thalidomide, although with less teratogenic effect and fewer thromboembolic events. Pomalidomide (CC4047) is another oral IMiD that is being investigated for the treatment of multiple myeloma and myelodysplasia. The only IMiD currently used as an immunosuppressive medication (ie, in transplant recipients) is thalidomide.
CYTOTOXIC AGENTS
Azathioprine Azathioprine is a prodrug of mercaptopurine and, like mercap-topurine, functions as an antimetabolite (see Chapter 54 ). Although its action is presumably mediated by conversion to mer-captopurine and further metabolites, it has been more widely used than mercaptopurine for immunosuppression in humans. These agents represent prototypes of the antimetabolite group of cyto-toxic immunosuppressive drugs, and many other agents that kill proliferative cells appear to work at a similar level in the immune response.
Azathioprine is well absorbed from the gastrointestinal tract and is metabolized primarily to mercaptopurine. Xanthine oxidase splits much of the active material to 6-thiouric acid prior to excre-tion in the urine. After administration of azathioprine, small amounts of unchanged drug and mercaptopurine are also excreted by the kidney, and as much as a twofold increase in toxicity may occur in anephric or anuric patients. Since much of the drug’s inactivation depends on xanthine oxidase, patients who are also receiving allopurinol (see Chapters 36 and 54 ) for control of hyperuricemia should have the dose of azathioprine reduced to one-fourth to one-third the usual amount to prevent excessive toxicity.
Azathioprine and mercaptopurine appear to produce immuno-suppression by interfering with purine nucleic acid metabolism at steps that are required for the wave of lymphoid cell proliferation that follows antigenic stimulation. The purine analogs are thus cytotoxic agents that destroy stimulated lymphoid cells. Although continued messenger RNA synthesis is necessary for sustained antibody synthesis by plasma cells, these analogs appear to have less effect on this process than on nucleic acid synthesis in prolif-erating cells. Cellular immunity as well as primary and secondary serum antibody responses can be blocked by these cytotoxic agents.
Azathioprine and mercaptopurine appear to be of definite benefit in maintaining renal allografts and may be of value in transplantation of other tissues. These antimetabolites have been used with some success in the management of acute glomerulone-phritis and in the renal component of systemic lupus erythemato-sus. They have also proved useful in some cases of rheumatoid arthritis, Crohn’s disease, and multiple sclerosis. The drugs have been of occasional use in prednisone-resistant antibody-mediated idiopathic thrombocytopenic purpura and autoimmune hemo-lytic anemias.
The chief toxic effect of azathioprine and mercaptopurine is bone marrow suppression, usually manifested as leukopenia, although anemia and thrombocytopenia may occur. Skin rashes, fever, nausea and vomiting, and sometimes diarrhea occur, with the gastrointestinal symptoms seen mainly at higher dosages. Hepatic dysfunction, manifested by very high serum alkaline phosphatase levels and mild jaundice, occurs occasionally, particu-larly in patients with preexisting hepatic dysfunction.
Cyclophosphamide The alkylating agent cyclophosphamide is one of the most effica-cious immunosuppressive drugs available. Cyclophosphamide destroys proliferating lymphoid cells (see Chapter 54 ) but also appears to alkylate DNA and other molecules in some resting cells. It has been observed that very large doses (eg, > 120 mg/kg intravenously over several days) may induce an apparent specific tolerance to a new antigen if the drug is administered simultane-ously with, or shortly after, the antigen. In smaller doses, it has been effective against autoimmune disorders (including systemic lupus erythematosus) and in patients with acquired factor XIII antibodies and bleeding syndromes, autoimmune hemolytic ane-mia, antibody-induced pure red cell aplasia, and Wegener’s granu-lomatosis.
Treatment with large doses of cyclophosphamide carries con-siderable risk of pancytopenia and hemorrhagic cystitis and therefore is generally combined with stem cell rescue (trans-plant) procedures. Although cyclophosphamide appears to induce tolerance for marrow or immune cell grafting, its use does not prevent the subsequent graft-versus-host disease syn-drome, which may be serious or lethal if the donor is a poor histocompatibility match (despite the severe immunosuppres-sion induced by high doses of cyclophosphamide). Other adverse effects of cyclophosphamide include nausea, vomiting, cardiac toxicity, and electrolyte disturbances.
055-Katzung_Ch055_p977-1000.indd 988 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 989
Leflunomide Leflunomide is a prodrug of an inhibitor of pyrimidine synthesis (rather than purine synthesis). It is orally active, and the active metabolite has a long half-life of several weeks. Thus, the drug should be started with a loading dose, but it can be taken once daily after reaching steady state. It is approved only for rheuma-toid arthritis at present, although studies are underway combining leflunomide with mycophenolate mofetil for a variety of autoim-mune and inflammatory skin disorders, as well as preservation of allografts in solid organ transplantation. Leflunomide also appears (from murine data) to have antiviral activity.
Toxicities include elevation of liver enzymes with some risk of liver damage, renal impairment, and teratogenic effects. A low frequency of cardiovascular effects (angina, tachycardia) was reported in clinical trials of leflunomide.
Hydroxychloroquine Hydroxychloroquine is an antimalarial agent with immunosup-pressant properties. It is thought to suppress intracellular antigen processing and loading of peptides onto MHC class II molecules by increasing the pH of lysosomal and endosomal compartments, thereby decreasing T-cell activation.
Because of these immunosuppressant activities, hydroxychloro-quine is used to treat some autoimmune disorders, eg, rheumatoid arthritis and systemic lupus erythematosus. It has also been used to both treat and prevent graft-versus-host disease after allogeneic stem cell transplantation.
Other Cytotoxic Agents Other cytotoxic agents, including vincristine, methotrexate, and cytarabine (see Chapter 54 ), also have immunosuppressive properties. Methotrexate has been used extensively in rheuma-toid arthritis (see Chapter 36 ) and in the treatment of graft-versus-host disease. Although the other agents can be used for immunosuppression, their use has not been as widespread as the purine antagonists, and their indications for immunosuppres-sion are less certain. The use of methotrexate (which can be given orally) appears reasonable in patients with idiosyncratic reactions to purine antagonists. The antibiotic dactinomycin has also been used with some success at the time of impending renal transplant rejection. Vincristine appears to be quite useful in idiopathic thrombocytopenic purpura refractory to prednisone. The related vinca alkaloid vinblastine has been shown to pre-vent mast cell degranulation in vitro by binding to microtubule units within the cell and to prevent release of histamine and other vasoactive compounds.
Pentostatin is an adenosine deaminase inhibitor primarily used as an antineoplastic agent for lymphoid malignancies, and produces a profound lymphopenia. It is now frequently used for steroid-resistant graft-versus-host disease after allogeneic stem cell transplantation, as well as in preparative regimens prior to those transplants to provide severe immunosuppression to prevent allograft rejection.
IMMUNOSUPPRESSIVE ANTIBODIES
The development of hybridoma technology by Milstein and Kohler in 1975 revolutionized the antibody field and radically increased the purity and specificity of antibodies used in the clinic and for diagnostic tests in the laboratory. Hybridomas consist of antibody-forming cells fused to immortal plasmacytoma cells. Hybrid cells that are stable and produce the required antibody can be subcloned for mass culture for antibody production. Large-scale fermentation facilities are now used for this purpose in the pharmaceutical industry.
More recently, molecular biology has been used to develop monoclonal antibodies. Combinatorial libraries of cDNAs encod-ing immunoglobulin heavy and light chains expressed on bacterio-phage surfaces are screened against purified antigens. The result is an antibody fragment with specificity and high affinity for the antigen of interest. This technique has been used to develop anti-bodies specific for viruses (eg, HIV), bacterial proteins, tumor antigens, and even cytokines. Several antibodies developed in this manner are FDA-approved for use in humans.
Other genetic engineering techniques involve production of chimeric and humanized versions of murine monoclonal antibod-ies in order to reduce their antigenicity and increase the half-life of the antibody in the patient. Murine antibodies administered as such to human patients elicit production of human antimouse antibodies (HAMAs), which clear the original murine proteins very rapidly. Humanization involves replacing most of the murine antibody with equivalent human regions while keeping only the variable, antigen-specific regions intact. Chimeric mouse-human antibodies have similar properties with less complete replacement of the murine components. The current naming convention for these engineered substances uses the suffix “-umab” or “-zumab” for humanized antibodies, and “-imab” or “-ximab” for chimeric products. These procedures have been successful in reducing or preventing HAMA production for many of the antibodies dis-cussed below.
Antilymphocyte & Antithymocyte Antibodies Antisera directed against lymphocytes have been prepared spo-radically for over 100 years. With the advent of human organ transplantation as a therapeutic option, heterologous antilympho-cyte globulin (ALG) took on new importance. ALG and antithy-mocyte globulin (ATG) are now in clinical use in many medical centers, especially in transplantation programs. The antiserum is usually obtained by immunization of horses, sheep, or rabbits with human lymphoid cells.
ALG acts primarily on the small, long-lived peripheral lym-phocytes that circulate between the blood and lymph. With con-tinued administration, “thymus-dependent” (T) lymphocytes from lymphoid follicles are also depleted, as they normally par-ticipate in the recirculating pool. As a result of the destruction or inactivation of T cells, an impairment of delayed hypersensitivity and cellular immunity occurs while humoral antibody formation remains relatively intact. ALG and ATG are useful for suppressing
055-Katzung_Ch055_p977-1000.indd 989 9/23/11 5:16:51 PM

990 SECTION VIII Chemotherapeutic Drugs
certain major compartments (ie, T cells) of the immune system and play a definite role in the management of solid organ and bone marrow transplantation.
Monoclonal antibodies directed against specific cell surface and soluble proteins such as CD3, CD4, CD25, CD40, IL-2 receptor, and TNF-α (discussed below) much more selectively influence T-cell subset function. The high specificity of these antibodies improves selectivity and reduces toxicity of the therapy and alters the disease course in several different autoimmune disorders.
In the management of transplants, ALG and monoclonal anti-bodies can be used in the induction of immunosuppression, in the treatment of initial rejection, and in the treatment of steroid-resistant rejection. There has been some success in the use of ALG and ATG plus cyclosporine to prepare recipients for bone marrow transplantation. In this procedure, the recipient is treated with ALG or ATG in large doses for 7–10 days prior to transplantation of bone marrow cells from the donor. ALG appears to destroy the T cells in the donor marrow graft, and the probability of severe graft-versus-host syndrome is reduced.
The adverse effects of ALG are mostly those associated with injection of a foreign protein. Local pain and erythema often occur at the injection site (type III hypersensitivity). Since the humoral antibody mechanism remains active, skin-reactive and precipitating antibodies may be formed against the foreign IgG. Similar reactions occur with monoclonal antibodies of murine origin, and reactions thought to be caused by the release of cytok-ines by T cells and monocytes have also been described.
Anaphylactic and serum sickness reactions to ALG and murine monoclonal antibodies have been observed and usually require ces-sation of therapy. Complexes of host antibodies with horse ALG may precipitate and localize in the glomeruli of the kidneys. The incidence of lymphoma as well as other forms of cancer is increased in kidney transplant patients. It appears likely that part of the increased risk of cancer is related to the suppression of a normally potent defense system against oncogenic viruses or transformed cells. The preponderance of lymphoma in these cancer cases is thought to be related to the concurrence of chronic immune sup-pression with chronic low-level lymphocyte proliferation.
Muromonab-CD3 Monoclonal antibodies against T-cell surface proteins are increas-ingly being used in the clinic for autoimmune disorders and in transplantation settings. Clinical studies have shown that the murine monoclonal antibody muromonab-CD3 (OKT3) directed against the CD3 molecule on the surface of human T cells can be useful in the treatment of renal transplant rejection. In vitro, muromonab CD3 blocks killing by cytotoxic human T cells and several other T-cell functions. In a prospective randomized multi-center trial with cadaveric renal transplants, use of muromonab-CD3 (along with lower doses of steroids or other immunosuppressive drugs) proved more effective at reversing acute rejection than did conventional steroid treatment. Muromonab-CD3 is approved for the treatment of acute renal allograft rejection and steroid-resistant acute cardiac and hepatic transplant rejection. Many other mono-clonal antibodies directed against surface markers on lymphocytes
are approved for certain indications (see monoclonal antibody sec-tion below), while others are in various stages of development.
Immune Globulin Intravenous (IGIV) A different approach to immunomodulation is the intravenous use of polyclonal human immunoglobulin. This immunoglobulin preparation (usually IgG) is prepared from pools of thousands of healthy donors, and no single, specific antigen is the target of the “therapeutic antibody.” Rather, one expects that the pool of differ-ent antibodies will have a normalizing effect upon the patient’s immune networks.
IGIV in high doses (2 g/kg) has proved effective in a variety of different applications ranging from immunoglobulin deficiencies to autoimmune disorders to HIV disease to bone marrow trans-plantation. In patients with Kawasaki’s disease, it has been shown to be safe and effective, reducing systemic inflammation and pre-venting coronary artery aneurysms. It has also brought about good clinical responses in systemic lupus erythematosus and refractory idiopathic thrombocytopenic purpura. Possible mechanisms of action of IGIV include a reduction of T helper cells, increase of regulatory T cells, decreased spontaneous immunoglobulin pro-duction, Fc receptor blockade, increased antibody catabolism, and idiotypic-anti-idiotypic interactions with “pathologic antibodies.” Although its precise mechanism of action is still unknown, IGIV brings undeniable clinical benefit to many patients with a variety of immune syndromes.
Rh o (D) Immune Globulin Micro-Dose One of the earliest major advances in immunopharmacology was the development of a technique for preventing Rh hemolytic disease of the newborn. The technique is based on the observation that a primary antibody response to a foreign antigen can be blocked if specific antibody to that antigen is administered passively at the time of exposure to antigen. Rh o (D) immune globulin is a concen-trated (15%) solution of human IgG containing a higher titer of antibodies against the Rh o (D) antigen of the red cell.
Sensitization of Rh-negative mothers to the D antigen occurs usually at the time of birth of an Rh o (D)-positive or D u -positive infant, when fetal red cells leak into the mother’s bloodstream. Sensitization might also occur occasionally with miscarriages or ectopic pregnancies. In subsequent pregnancies, maternal anti-body against Rh-positive cells is transferred to the fetus during the third trimester, leading to the development of erythroblastosis fetalis (hemolytic disease of the newborn).
If an injection of Rh o (D) antibody is administered to the mother within 24–72 hours after the birth of an Rh-positive infant, the mother’s own antibody response to the foreign Rh o (D)-positive cells is suppressed because the infant’s red cells are cleared from circulation before the mother can generate a B-cell response against Rh o (D). Therefore she has no memory B cells that can activate upon subse-quent pregnancies with an Rh o (D)-positive fetus.
When the mother has been treated in this fashion, Rh hemo-lytic disease of the newborn has not been observed in subsequent pregnancies. For this prophylactic treatment to be successful, the
055-Katzung_Ch055_p977-1000.indd 990 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 991
mother must be Rh o (D)-negative and D u -negative and must not already be immunized to the Rh o (D) factor. Treatment is also often advised for Rh-negative mothers antepartum at 26–28 weeks’ gestation who have had miscarriages, ectopic pregnancies, or abortions, when the blood type of the fetus is unknown. Note: Rh o (D) immune globulin is administered to the mother and must not be given to the infant.
The usual dose of Rh o (D) immune globulin is 2 mL intramus-cularly, containing approximately 300 mcg anti-Rh o (D) IgG. Adverse reactions are infrequent and consist of local discomfort at the injection site or, rarely, a slight temperature elevation.
Hyperimmune Immunoglobulins Hyperimmune immunoglobulins are IGIV preparations made from pools of selected human or animal donors with high titers of antibodies against particular agents of interest such as viruses or toxins (see also Appendix I). Various hyperimmune IGIVs are available for treatment of respiratory syncytial virus, cytomegalo-virus, varicella zoster, human herpesvirus 3, hepatitis B virus, rabies, tetanus, and digoxin overdose. Intravenous administration of the hyperimmune globulins is a passive transfer of high titer antibodies that either reduces risk or reduces the severity of infec-tion. Rabies hyperimmune globulin is injected around the wound and given intravenously. Tetanus hyperimmune globulin is admin-istered intravenously when indicated for prophylaxis. Rattlesnake and coral snake hyperimmune globulins (antivenoms) are of equine or ovine origin and are effective for North and South American rattlesnakes and some coral snakes (but not Arizona coral snake). Equine and ovine antivenoms are available for rattle-snake envenomations, but only equine antivenom is available for coral snake bite. The ovine antivenom is a Fab preparation and is less immunogenic than whole equine IgG antivenoms, but retains the ability to neutralize the rattlesnake venom.
MONOCLONAL ANTIBODIES (MABs )
Recent advances in the ability to manipulate the genes of immu-noglobulins have resulted in development of a wide array of humanized and chimeric monoclonal antibodies directed against therapeutic targets. The only murine elements of humanized monoclonal antibodies are the complementarity-determining regions in the variable domains of immunoglobulin heavy and light chains. Complementarity-determining regions are primarily responsible for the antigen-binding capacity of antibodies. Chimeric antibodies typically contain antigen-binding murine variable regions and human constant regions. The following are brief descriptions of the engineered antibodies that have been approved by the FDA.
Antitumor MABs Alemtuzumab is a humanized IgG 1 with a kappa chain that binds to CD52 found on normal and malignant B and T lymphocytes, NK cells, monocytes, macrophages, and a small population of granulocytes. Currently, alemtuzumab is approved for the treatment
of B-cell chronic lymphocytic leukemia in patients who have been treated with alkylating agents and have failed fludarabine therapy. Alemtuzumab appears to deplete leukemic and normal cells by direct antibody-dependent lysis. Patients receiving this antibody become lymphopenic and may also become neutro-penic, anemic, and thrombocytopenic. As a result patients should be closely monitored for opportunistic infections and hemato-logic toxicity.
Bevacizumab is a humanized IgG 1 monoclonal antibody that binds to vascular endothelial growth factor (VEGF) and inhibits VEGF from binding to its receptor, especially on endothelial cells. It is an antiangiogenic drug that has been shown to inhibit growth of blood vessels (angiogenesis) in tumors. It is approved for first-line treatment of patients with metastatic colorectal cancer alone or in combination with 5-FU-based chemotherapy. It is also approved for treatment of non-small cell lung cancer, glioblastoma multiforme that has progressed after prior treatment, and meta-static kidney cancer when used with interferon-alpha. Since beva-cizumab is antiangiogenic, it should not be administered until patients heal from surgery. Patients taking the drug should be watched for hemorrhage, gastrointestinal perforations, and wound healing problems. Bevacizumab has also been used off label by intravitreal injection to slow progression of neovascular macular degeneration (see ranibizumab under Other MABs, below).
Cetuximab is a human-mouse chimeric monoclonal antibody that targets epidermal growth factor receptor (EGFR). Binding of cetuximab to EGFR inhibits tumor cell growth by a variety of mechanisms, including decreases in kinase activity, matrix metal-loproteinase activity, and growth factor production, and increased apoptosis. It is indicated for use in patients with EGFR-positive metastatic colorectal cancer and, along with radiation therapy, in patients with head and neck cancer. Cetuximab may be adminis-tered in combination with irinotecan or alone in patients who cannot tolerate irinotecan. HAMAs are generated by about 4% of patients being treated with cetuximab.
Ofatumumab is a human IgG 1 monoclonal antibody directed against a different epitope on CD20 than rituximab. It is approved for patients with chronic lymphocytic leukemia (CLL) who are refractory to fludarabine and alemtuzumab. Ofatumumab binds to all B cells including B-CLL. It is thought to lyse B-CLL cells in the presence of complement and to mediate antibody-dependent cellular cytotoxicity. There is a slight risk of hepatitis B virus reac-tivation in patients taking ofatumumab.
Panitumumab is a fully human IgG 2 kappa light chain mono-clonal antibody. It is approved for the treatment of EGFR-expressing metastatic colorectal carcinoma with disease progression on or following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens. Panitumumab binds to EGFR (similar to cetuximab), inhibiting epidermal growth factor from binding to its receptor, and prevents ligand-induced receptor autophosphorylation and activation of receptor-associated kinases. It inhibits cell growth, induces apoptosis, decreases vascular growth factor production, and suppresses internalization of the EGFR. Although some dermatologic and infusion-related toxici-ties have been observed following infusion of panitumumab, the distinct advantage over cetuximab is that it is fully human, and
055-Katzung_Ch055_p977-1000.indd 991 9/23/11 5:16:51 PM

992 SECTION VIII Chemotherapeutic Drugs
therefore does not elicit HAMAs. This is the first FDA-approved monoclonal antibody produced from transgenic mice expressing the human immunoglobulin gene loci.
Rituximab is a chimeric murine-human monoclonal IgG 1 (human Fc) that binds to the CD20 molecule on normal and malignant B lymphocytes and is approved for the therapy of patients with relapsed or refractory low-grade or follicular B-cell non-Hodgkin’s lymphoma and chronic lymphocytic leukemia. It is also approved for the treatment of rheumatoid arthritis in combination with methotrexate in patients for whom anti-TNF-α therapy has failed. The mechanism of action includes complement-mediated lysis, antibody-dependent cellular cyto-toxicity, and induction of apoptosis in the malignant lymphoma cells. In lymphoma this drug appears to be synergistic with che-motherapy (eg, fludarabine, CHOP, see Chapter 54 ). Recent reports indicate that rituximab may also be very useful in auto-immune diseases such as multiple sclerosis and systemic lupus erythematosus.
Trastuzumab is a recombinant DNA-derived, humanized monoclonal antibody that binds to the extracellular domain of the human epidermal growth factor receptor HER-2/ neu . This anti-body blocks the natural ligand from binding and down-regulates the receptor. Trastuzumab is approved for the treatment of HER-2 /neu -positive tumors in patients with breast cancer and patients with metastatic gastric or gastroesophageal junction ade-nocarcinoma. As a single agent it induces remission in about 15–20% of breast cancer patients; in combination with chemo-therapy, it increases response rates and duration as well as 1-year survival. Trastuzumab is under investigation for other tumors that express HER-2/neu (see Chapter 54 ).
MABs Used to Deliver Isotopes to Tumors Arcitumomab is a murine Fab fragment from an anti- carcinoembryonic antigen (CEA) antibody labeled with techne-tium 99m ( 99m Tc) that is used for imaging patients with metastatic colorectal carcinoma (immunoscintigraphy) to determine extent of disease. CEA is often upregulated on tumor in patients with gastrointestinal carcinomas. The use of the Fab fragment decreases the immunogenicity of the agent so that it can be given more than once; intact murine monoclonal antibodies would elicit stronger HAMA.
Capromab pendetide is a murine monoclonal antibody spe-cific for prostate specific membrane antigen. It is coupled to isoto-pic indium ( 111 In) and is used in immunoscintigraphy for patients with biopsy-confirmed prostate cancer and post-prostatectomy in patients with rising prostate specific antibody level to determine extent of disease.
Ibritumomab tiuxetan is an anti-CD20 murine monoclonal antibody labeled with isotopic yttrium ( 90 Y) or 111 In. The radia-tion of the isotope coupled to the antibody provides the major antitumor activity. Ibritumomab is approved for use in patients with relapsed or refractory low-grade, follicular, or B-cell non-Hodgkin’s lymphoma, including patients with rituximab-refrac-tory follicular disease. It is used in conjunction with rituximab in a two-step therapeutic regimen.
Tositumomab is another anti-CD20 monoclonal antibody and is complexed with iodine 131 ( 131 I). Tositumomab is used in two-step therapy in patients with CD20-positive, follicular non-Hodgkin’s lymphoma whose disease is refractory to rituximab and standard chemotherapy. Toxicities are similar to those for ibritu-momab and include severe cytopenias such as thrombocytopenia and neutropenia. Tositumomab should not be administered to patients with greater than 25% bone marrow involvement.
MABs Used as Immunosuppressants and Anti-Inflammatory Agents A. Anti-TNF-Alpha MABs Adalimumab, certolizumab pegol, etanercept, golimumab, and infliximab are antibodies that bind TNF-α, a proinflammatory cytokine that is important in rheumatoid arthritis and similar inflammatory diseases (see Chapter 36 ). Blocking TNF-α from binding to TNF receptors on inflammatory cells results in suppres-sion of downstream inflammatory cytokines such as IL-1 and IL-6 and adhesion molecules involved in leukocyte activation and migration. An increased risk of infection or reactivation of M tuber-culosis, hepatitis B virus, and invasive systemic fungi is common to each of these anti-TNF monoclonal antibodies. Patients may also be at increased risk for malignancies including lymphoma.
Adalimumab is a completely human IgG 1 approved for use in patients with rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, and plaque psoriasis. Like the other anti-TNF-α biologicals, adali-mumab blocks the interaction of TNF-α with TNF receptors on cell surfaces; it does not bind TNF-β. Adalimumab lyses cells expressing TNF-α in the presence of complement. Pharmacodynamic studies showed that administration of adalimumab reduced levels of C-reactive protein, erythrocyte sedimentation rate, serum IL-6, and matrix metalloproteinases MMP-1 and MMP-3.
Certolizumab pegol is a recombinant humanized Fab frag-ment that binds to TNF-α. It is coupled to a 40-kDa polyethylene glycol. It neutralizes the activity of membrane-associated and sol-uble TNF-α without lysing cells. Certolizumab is indicated for patients with Crohn’s disease and rheumatoid arthritis.
Etanercept is a dimeric fusion protein composed of human IgG 1 constant regions fused to the TNF receptor. Etanercept binds to both TNF-α and TNF-β and appears to have effects similar to those of adalimumab and infliximab, ie, inhibition of TNF-α-mediated inflammation, but its half-life is shorter due to its physical form (fusion protein) and the route of injection (subcutaneously, twice weekly). Etanercept is approved for adult rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, ankylosing spondylitis and psoriatic arthritis. It may be used in combination with meth-otrexate in some patients with arthritis.
Golimumab is a human IgG monoclonal antibody that also binds to soluble and membrane-associated TNF-α. It is an intact human IgG 1 and, like certolizumab pegol, it does not lyse cells expressing membrane-associated TNF-α. It is indicated for patients with rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It has the advantage of increased half-life such that subcu-taneous injections may be self-administered only once per month.
055-Katzung_Ch055_p977-1000.indd 992 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 993
Infliximab is a human-mouse chimeric IgG 1 monoclonal anti-body possessing human constant (Fc) regions and murine variable regions. It is administered intravenously but has the same anti-TNF-α activity as adalimumab and etanercept. Infliximab is cur-rently approved for use in Crohn’s disease, ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis, plaque psoriasis, and psoriatic arthritis.
B. Abatacept Abatacept is a recombinant fusion protein composed of the extra-cellular domain of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) fused to hinge, CH 2 , and CH 3 domains of human IgG 1 . CTLA-4 delivers an inhibitory signal to T cells. It binds more tightly to CD80/86 than CD28 ( Figure 55–7 ). This fusion protein blocks activation of T cells by binding to CD80 or CD86 so that CD28 on T cells cannot bind and stimulate the T cell and lead to cytokine release. Abatacept is approved for patients with rheumatoid arthritis and juvenile idiopathic arthritis. Patients should not take other anti-TNF drugs or anakinra while taking abatacept. As with anti-TNF monoclonal agents, patients should be screened and treated for latent tuberculosis infection before starting abatacept.
C. Alefacept Alefacept is an engineered protein consisting of the CD2-binding portion of leukocyte-function-associated antigen-3 (LFA-3) fused to a human IgG 1 Fc region (hinge, CH 2 , and CH 3 ). It is approved for the treatment of plaque psoriasis. It inhibits activation of T cells by binding to cell surface CD2, inhibiting the normal CD2/LFA-3
interaction ( Figure 55–7 ). Treatment of patients with alefacept also results in a dose-dependent reduction of the total number of circu-lating T cells, especially CD4 and CD8 memory effector subsets that predominate in psoriatic plaques. Peripheral T-cell counts of patients receiving alefacept must be monitored and the drug dis-continued if CD4 lymphocyte levels fall below 250 cells/μL.
D. Basiliximab and Daclizumab Basiliximab is a chimeric mouse-human IgG 1 that binds to CD25, the IL-2 receptor alpha chain on activated lymphocytes. Daclizumab is a humanized IgG 1 that also binds to the alpha subunit of the IL-2 receptor. Both agents function as IL-2 antago-nists, blocking IL-2 from binding to activated lymphocytes, and are therefore immunosuppressive. They are indicated for prophy-laxis of acute organ rejection in renal transplant patients and either may be used as part of an immunosuppressive regimen that also includes glucocorticoids and cyclosporine A.
E. Natalizumab Natalizumab is a humanized IgG4 monoclonal antibody that binds to the α4-subunit of α4β1 and α4β7 integrins expressed on the surfaces of all leukocytes except neutrophils, and inhibits the α4-mediated adhesion of leukocytes to their cognate receptor. It is indicated for patients with multiple sclerosis and Crohn’s disease who have not tolerated or had inadequate responses to conven-tional treatments.
F. Omalizumab Omalizumab is an anti-IgE recombinant humanized monoclonal antibody that is approved for the treatment of allergic asthma in adult and adolescent patients whose symptoms are refractory to inhaled corticosteroids (see Chapter 20 ). The antibody blocks the binding of IgE to the high-affinity Fc ε receptor on basophils and mast cells, which suppresses IgE-mediated release of type I allergy mediators such as histamine and leukotrienes. Total serum IgE levels may remain elevated in patients for up to 1 year after admin-istration of this antibody.
G. Tocilizumab Tocilizumab is recombinant humanized IgG 1 that binds to soluble and membrane-associated IL-6 receptors. It inhibits IL-6-mediated signaling on lymphocytes, suppressing inflammatory processes. It is indicated for treatment of patients with rheumatoid arthritis who are refractory to other anti-TNF-α biologicals. It may be used alone or in combination with methotrexate or other disease-modifying antirheumatic drugs. Patients taking tocilizumab have the same increased risk of infection as those taking anti-TNF-α monoclonal antibodies.
H. Ustekinumab Ustekinumab is a human IgG 1 monoclonal antibody that binds to the p40 subunit of IL-12 and IL-23 cytokines. It blocks IL-12 and IL-23 from binding to their receptors, therefore inhibiting recep-tor-mediated signaling in lymphocytes. Ustekinumab is indicated for patients with moderate to severe plaque psoriasis. The advantage
Antigen-presenting cell
CD40
T lymphocyte
T cell
TCRMHC
CD40L
Dendritic cell
BasiliximabandDaclizumab
ICAM-1 LFA-1
CD28CD80/86
CTLA-4
CD2
Alefacept
Abatacept
Ipilimumab
LFA-3
FIGURE 55–7 Actions of some monoclonal antibodies ( shown in red ). CTLA-4-lgFc fusion protein (CTLA-4-lg, abatacept) binds to CD80/86 on DC and inhibits T-cell costimulation. Alefacept inhibits activation of T cells by blocking the interaction of LFA-3 and CD2. Basiliximab and daclizumab block IL-2 from binding to the IL-2 receptor (CD25) on T cells, preventing activation; CD25 is also important for the survival of T regulatory cells. T-cell activation can be maintained or restored if CTLA-4 interaction with CD80/86 is blocked using an anti-CTLA-4 antibody (ipilimumab, currently in phase 3 clinical trials); ipilimumab inhibits CTLA-4 signaling and prolongs activation.
055-Katzung_Ch055_p977-1000.indd 993 9/23/11 5:16:51 PM

994 SECTION VIII Chemotherapeutic Drugs
SOLID ORGAN & BONE MARROW TRANSPLANTATION
In organ transplantation, tissue typing—based on donor and recipient histocompatibility matching with the human leukocyte antigen (HLA) haplotype system—is required. Close histocompat-ibility matching reduces the likelihood of graft rejection and may also reduce the requirements for intensive immunosuppressive therapy. Prior to transplant, patients may receive an immunosup-pressive regimen, including antithymocyte globulin, muromonab-CD3, daclizumab, or basiliximab. Four types of rejection can occur in a solid organ transplant recipient: hyperacute, accelerated, acute, and chronic. Hyperacute rejection is due to preformed antibodies against the donor organ, such as anti-blood group anti-bodies. Hyperacute rejection occurs within hours of the transplant and cannot be stopped with immunosuppressive drugs. It results in rapid necrosis and failure of the transplanted organ. Accelerated rejection is mediated by both antibodies and T cells, and it also cannot be stopped by immunosuppressive drugs. Acute rejection of an organ occurs within days to months and involves mainly cellular immunity. Reversal of acute rejection is usually possible with general immunosuppressive drugs such as azathioprine, myco-phenolate mofetil, cyclosporine, tacrolimus, glucocorticoids, cyclo- phosphamide, methotrexate, and sirolimus. Recently, biologic agents such as anti-CD3 monoclonal antibodies have been used to stem acute rejection. Chronic rejection usually occurs months or even years after transplantation. It is characterized by thickening and fibrosis of the vasculature of the transplanted organ, involving both cellular and humoral immunity. Chronic rejection is treated with the same drugs as those used for acute rejection.
Allogeneic hematopoietic stem cell transplantation is a well-established treatment for many malignant and nonmalignant dis-eases. An HLA-matched donor, usually a family member, is located, patients are conditioned with high-dose chemotherapy or radiation therapy, and then donor stem cells are infused. The con-ditioning regimen is used not only to kill cancer cells in the case of malignant disease, but also to totally suppress the immune system so that the patient does not reject the donor stem cells. As patients’ blood counts recover (after reduction by the conditioning regimen) they develop a new immune system that is created from the donor stem cells. Rejection of donor stem cells is uncommon, and can only be treated by infusion of more stem cells from the donor.
Graft-versus-host disease, however, is very common, occurring in the majority of patients who receive an allogeneic transplant. Graft-versus-host disease occurs as donor T cells fail to recognize the patient’s skin, liver, and gut (usually) as self and attack those tissues. Although patients are given immunosuppressive therapy (cyclosporine, methotrexate, and others) early in the transplant course to help prevent this development, it usually occurs despite these medications. Acute graft-versus-host disease occurs within the first 100 days, and is usually manifested as a skin rash, severe diar-rhea, or hepatotoxicity. Additional medications are added, invari-ably starting with high-dose corticosteroids, and adding drugs such as mycophenolate mofetil, sirolimus, tacrolimus, daclizumab, and others, with variable success rates. Patients generally progress to
of ustekinumab over anti-TNF-α drugs for psoriasis is faster and longer term improvement in symptoms along with very infre-quent dosing.
Other MABs Abciximab is a Fab fragment of a murine-human monoclonal antibody that binds to the integrin GPIIb/IIIa receptor on acti-vated platelets and inhibits fibrinogen, von Willebrand factor, and other adhesion molecules from binding to activated platelets, thus preventing their aggregation. It is indicated as an adjunct to per-cutaneous coronary intervention for the prevention of cardiac ischemic complications. See Chapter 34 for additional details.
Denosumab is a human IgG 2 monoclonal antibody specific for human RANKL (receptor activator of nuclear factor kappa-B ligand). By binding RANKL it inhibits the maturation of osteoclasts, the cells responsible for bone resorption. Denosumab is indicated for treatment of postmenopausal women with osteoporosis at high risk for fracture. Before starting denosumab, patients must be evaluated to be sure they are not hypocalcemic. During treatment, patients should receive supplements of calcium and vitamin D.
Eculizumab is a humanized IgG monoclonal antibody that binds the C5 complement component, inhibiting its cleavage into C5a and C5b thereby inhibiting the terminal pore-forming lytic activity of complement. Eculizumab is approved for patients with paroxysmal nocturnal hemoglobinuria (PNH) and dramatically reduces the need for red blood cell transfusions. It prevents PNH symptoms of anemia, fatigue, thrombosis, and hemoglobinemia by inhibiting intravascular hemolysis due to red cell lysis. Clinicians must be aware of increased risk of meningococcal infec-tion in patients receiving this anti-C5 monoclonal antibody.
Palivizumab is a monoclonal antibody that binds to the fusion protein of respiratory syncytial virus, preventing infection in sus-ceptible cells in the airways. It is used in neonates at risk for this viral infection and reduces the frequency of infection and hospi-talization by about 50% (see Chapter 49 ).
Ranibizumab is a recombinant human IgG 1 Fab that binds to VEGF-A. It also prevents new blood vessel formation by blocking VEGF from binding to its receptor. Ranibizumab is labeled for intravitreal injection in patients with neovascular age-related macu-lar degeneration and sudden blurring or vision loss secondary to macular edema following retinal vein occlusion. Pegaptanib is a pegylated oligonucleotide that binds extracellular VEGF and is also given by intravitreous injection to slow macular degeneration.
■ CLINICAL USES OF IMMUNOSUPPRESSIVE DRUGS
Immunosuppressive agents are commonly used in two clinical cir-cumstances: transplantation and autoimmune disorders. The agents used differ somewhat for the specific disorders treated (see specific agents and Table 55–1 ), as do administration schedules. Because autoimmune disorders are very complex, optimal treatment sched-ules have yet to be established in many clinical situations.
055-Katzung_Ch055_p977-1000.indd 994 9/23/11 5:16:51 PM

CHAPTER 55 Immunopharmacology 995
chronic graft-versus-host disease (after 100 days) and require ther-apy for variable periods thereafter. Unlike solid organ transplant patients, however, most stem cell transplant patients are able to eventually discontinue immunosuppressive drugs as graft-versus-host disease resolves (usually 1–2 years after their transplant).
AUTOIMMUNE DISORDERS
The effectiveness of immunosuppressive drugs in autoimmune disorders varies widely. Nonetheless, with immunosuppressive therapy, remissions can be obtained in many instances of autoim-mune hemolytic anemia, idiopathic thrombocytopenic purpura, type 1 diabetes, Hashimoto’s thyroiditis, and temporal arteritis. Improvement is also often seen in patients with systemic lupus erythematosus, acute glomerulonephritis, acquired factor VIII inhibitors (antibodies), rheumatoid arthritis, inflammatory myo-pathy, scleroderma, and certain other autoimmune states.
Immunosuppressive therapy is utilized in chronic severe asthma, where cyclosporine is often effective and sirolimus is another alternative. Omalizumab (anti-IgE antibody) has been approved for the treatment of severe asthma (see previous section). Tacrolimus is currently under clinical investigation for the man-agement of autoimmune chronic active hepatitis and of multiple sclerosis, where IFN-β has a definitive role.
IMMUNOMODULATION THERAPY
The development of agents that modulate the immune response rather than suppress it has become an important area of pharma-cology. The rationale underlying this approach is that such drugs may increase the immune responsiveness of patients who have either selective or generalized immunodeficiency. The major poten-tial uses are in immunodeficiency disorders, chronic infectious diseases, and cancer. The AIDS epidemic has greatly increased interest in developing more effective immunomodulating drugs.
Cytokines The cytokines are a large and heterogeneous group of proteins with diverse functions. Some are immunoregulatory proteins syn-thesized within lymphoreticular cells and play numerous interact-ing roles in the function of the immune system and in the control of hematopoiesis. The cytokines that have been clearly identified are summarized in Table 55–2 . In most instances, cytokines medi-ate their effects through receptors on relevant target cells and appear to act in a manner similar to the mechanism of action of hormones. In other instances, cytokines may have antiprolifera-tive, antimicrobial, and antitumor effects.
The first group of cytokines discovered, the interferons (IFNs), were followed by the colony-stimulating factors (CSFs, discussed in Chapter 33 ). The latter regulate the proliferation and differen-tiation of bone marrow progenitor cells. Most of the more recently discovered cytokines have been classified as interleukins (ILs) and numbered in the order of their discovery. Cytokines are produced using gene cloning techniques.
TABLE 55–2 The cytokines.
Cytokine Properties
Interferon-α (IFN-α) Antiviral, oncostatic, activates NK cells
Interferon-β (IFN-β) Antiviral, oncostatic, activates NK cells
Interferon-γ (IFN-γ) Antiviral, oncostatic, secreted by and acti-vates or up-regulates TH1 cells, NK cells, CTLs, and macrophages
Interleukin-1 (IL-1) T-cell activation, B-cell proliferation and differentiation
Interleukin-2 (IL-2) T-cell proliferation, TH1, NK, and LAK cell activation
Interleukin-3 (IL-3) Hematopoietic precursor proliferation and differentiation
Interleukin-4 (IL-4) TH2 and CTL activation, B-cell proliferation
Interleukin-5 (IL-5) Eosinophil proliferation, B-cell prolifera-tion and differentiation
Interleukin-6 (IL-6) HCF, TH2, CTL, and B-cell proliferation
Interleukin- 7 (IL-7) CTL, NK, LAK, and B-cell proliferation, thy-mic precursor stimulation
Interleukin-8 (IL-8) Neutrophil chemotaxis, proinflammatory
Interleukin-9 (IL-9) T-cell proliferation
Interleukin-10 (IL-10) TH1 suppression, CTL activation, B-cell proliferation
Interleukin-11 (IL-11) Megakaryocyte proliferation, B-cell differ-entiation
Interleukin-12 (IL-12) TH1 and CTL proliferation and activation
Interleukin-13 (IL-13) Macrophage function modulation, B cell proliferation
Interleukin-14 (IL-14) B-cell proliferation and differentiation
Interleukin-15 (IL-15) TH1, CTL, and NK/LAK activation, expan-sion of T-cell memory pools
Interleukin-16 (IL-16) T-lymphocyte chemotaxis, suppresses HIV replication
Interleukin-17 (IL-17) Stromal cell cytokine production
Interleukin-18 (IL-18) Induces TH1 responses
Interleukin-19 (IL-19) Proinflammatory
Interleukin-20 (IL-20) Promotes skin differentiation
Interleukin-21 (IL-21) Promotes proliferation of activated T cells, maturation of NK cells
Interleukin-22 (IL-22) Regulator of TH2 cells
Interleukin-23 (IL-23) Promotes proliferation of TH1 memory cells
Interleukin-24 (IL-24) Induces tumor apoptosis, induces TH1 responses
Interleukin-27 (IL-27) Stimulates naive CD4 cells to produce IFN-γ
Interleukin-28 and -29 (IL-28, IL-29)
Antiviral, interferon-like properties
Interleukin-30 (IL-30) p28 subunit of IL-27
Interleukin-31 (IL-31) Contributes to type I hypersensitivities and TH2 responses
(continued )
055-Katzung_Ch055_p977-1000.indd 995 9/23/11 5:16:52 PM

996 SECTION VIII Chemotherapeutic Drugs
Most cytokines (including TNF-α, IFN-γ, IL-2, granulocyte colony-stimulating factor [G-CSF], and granulocyte-macrophage colony-stimulating factor [GM-CSF]) have very short serum half-lives (minutes). The usual subcutaneous route of administration provides slower release into the circulation and a longer duration of action. Each cytokine has its own unique toxicity, but some toxicities are shared. For example, IFN-α, IFN-β, IFN-γ, IL-2, and TNF-α all induce fever, flu-like symptoms, anorexia, fatigue, and malaise.
Interferons are proteins that are currently grouped into three families: IFN-` , IFN-a , and IFN-f . The IFN-α and IFN-β fami-lies comprise type I IFNs, ie, acid-stable proteins that act on the same receptor on target cells. IFN-γ, a type II IFN, is acid-labile and acts on a separate receptor on target cells. Type I IFNs are usually induced by virus infections, with leukocytes producing IFN-α. Fibroblasts and epithelial cells produce IFN-β. IFN-γ is usually the product of activated T lymphocytes.
IFNs interact with cell receptors to produce a wide variety of effects that depend on the cell and IFN types. IFNs, particularly IFN-γ, display immune-enhancing properties, which include increased antigen presentation and macrophage, NK cell, and cytotoxic T-lymphocyte activation. IFNs also inhibit cell prolifera-tion. In this respect, IFN-α and IFN-β are more potent than IFN-γ. Another striking IFN action is increased expression of MHC molecules on cell surfaces. While all three types of IFN induce MHC class I molecules, only IFN-γ induces class II expres-sion. In glial cells, IFN-β antagonizes this effect and may, in fact, decrease antigen presentation within the nervous system.
IFN-α is approved for the treatment of several neoplasms, including hairy cell leukemia, chronic myelogenous leukemia,
TABLE 55–2 The cytokines.
Cytokine Properties
Interleukin-32 (IL-32) Involved in inflammation
Interleukin-34 (IL-34) Stimulates monocyte proliferation via the CSF-1 receptor (CSF-1R)
Interleukin-35 (IL-35) Induces regulatory T cells (iTR35)
Tumor necrosis factor-α (TNF-α)
Oncostatic, macrophage activation, proinflammatory
Tumor necrosis factor-β (TNF-β)
Oncostatic, proinflammatory, chemotactic
Granulocyte colony-stimulating factor
Granulocyte production
Granulocyte-macrophage colony-stimulating factor
Granulocyte, monocyte, eosinophil production
Macrophage colony-stimulating factor
Monocyte production, activation
Erythropoietin (epoetin, EPO)
Red blood cell production
Thrombopoietin (TPO) Platelet production
HCF, hematopoietic cofactor; LAK, lymphokine-activated killer cell.
Note: Many interleukin activities overlap and are influenced by each other.
malignant melanoma, and Kaposi’s sarcoma, and for use in hepati-tis B and C infections. It has also shown activity as an anticancer agent in renal cell carcinoma, carcinoid syndrome, and T-cell leu-kemia. IFN-β is approved for use in relapsing-type multiple sclero-sis. IFN-γ is approved for the treatment of chronic granulomatous disease and IL-2, for metastatic renal cell carcinoma and malignant melanoma. Clinical investigations of other cytokines, including IL-1, -3, -4, -6, -10, -11, and -12, are ongoing. Toxicities of IFNs, which include fever, chills, malaise, myalgias, myelosuppression, headache, and depression, can severely restrict their clinical use.
TNF-α has been extensively tested in the therapy of various malignancies, but results have been disappointing due to dose-limiting toxicities. One exception is the use of intra-arterial high-dose TNF-α for malignant melanoma and soft tissue sarcoma of the extremities. In these settings, response rates greater than 80% have been noted.
Cytokines have been under clinical investigation as adjuvants to vaccines, and IFNs and IL-2 have shown some positive effects in the response of human subjects to hepatitis B vaccine. Denileukin diftitox is IL-2 fused to diphtheria toxin, used for the treatment of patients with CD25+ cutaneous T-cell lymphomas. IL-12 and GM-CSF have also shown adjuvant effects with vac-cines. GM-CSF is of particular interest because it promotes recruitment of professional antigen-presenting cells such as the dendritic cells required for priming naive antigen-specific T-lymphocyte responses. There are some claims that GM-CSF can itself stimulate an antitumor immune response, resulting in tumor regression in melanoma and prostate cancer.
It is important to emphasize that cytokine interactions with target cells often result in the release of a cascade of different endogenous cytokines, which exert their effects sequentially or simultaneously. For example, IFN-γ exposure increases the num-ber of cell-surface receptors on target cells for TNF-α. Therapy with IL-2 induces the production of TNF-α, while therapy with IL-12 induces the production of IFN-γ.
Cytokine Inhibitors A more recent application of immunomodulation therapy involves the use of cytokine inhibitors for inflammatory diseases and septic shock, conditions in which cytokines such as IL-1 and TNF-α (see above) are involved in the pathogenesis. Drugs now in use or under investigation include anticytokine antibodies and soluble cytokine receptors. Anakinra is a recombinant form of the naturally occur-ring IL-1 receptor antagonist that prevents IL-1 from binding to its receptor, stemming the cascade of cytokines that would otherwise be released. Anakinra is approved for use in adult rheumatoid arthri-tis patients who have failed treatment with one or more disease-modifying antirheumatic drugs. Canakinumab is a recombinant human anti-IL-1β monoclonal antibody. It binds to human IL-1β and prevents it from binding to IL-1 receptors. Rilonacept is a dimeric fusion protein consisting of the ligand-binding domains of the extracellular portions of the human interleukin-1 receptor com-ponent (IL-1RI) and IL-1 receptor accessory protein (IL-1RAcP) fused to the Fc portion of human IgG1. These molecules are indi-cated for treatment of cryopyrin-associated periodic syndromes.
(Continued)
055-Katzung_Ch055_p977-1000.indd 996 9/23/11 5:16:52 PM

CHAPTER 55 Immunopharmacology 997
Patients must be carefully monitored for serious infections or malignancies if they are also taking an anti-TNF-α drug, have chronic infections, or are otherwise immunosuppressed.
■ IMMUNOLOGIC REACTIONS TO DRUGS & DRUG ALLERGY
The basic immune mechanism and the ways in which it can be sup-pressed or stimulated by drugs have been discussed in previous sections of this chapter. Drugs also activate the immune system in undesirable ways that are manifested as adverse drug reactions. These reactions are generally grouped in a broad classification as “drug allergy.” Indeed, many drug reactions such as those to penicil-lin, iodides, phenytoin, and sulfonamides are allergic in nature. These drug reactions are manifested as skin eruptions, edema, ana-phylactoid reactions, glomerulonephritis, fever, and eosinophilia.
Drug reactions mediated by immune responses can have sev-eral different mechanisms. Thus, any of the four major types of hypersensitivity discussed earlier in this chapter (pages 981-982) can be associated with allergic drug reactions:
• Type I: IgE-mediated acute allergic reactions to stings, pollens, and drugs, including anaphylaxis, urticaria, and angioedema. IgE is fixed to tissue mast cells and blood basophils, and after interaction with antigen the cells release potent mediators.
• Type II: Drugs often modify host proteins, thereby eliciting antibody responses to the modified protein. These allergic responses involve IgG or IgM in which the antibody becomes fixed to a host cell, which is then subject to complement-depen-dent lysis or to antibody-dependent cellular cytotoxicity.
• Type III: Drugs may cause serum sickness, which involves immune complexes containing IgG complexed with a foreign antigen and is a multisystem complement-dependent vasculitis that may also result in urticaria.
• Type IV: Cell-mediated allergy is the mechanism involved in al-lergic contact dermatitis from topically applied drugs or indura-tion of the skin at the site of an antigen injected intradermally.
In some drug reactions, several of these hypersensitivity responses may occur simultaneously. Some adverse reactions to drugs may be mistakenly classified as allergic or immune when they are actually genetic deficiency states or are idiosyncratic and not mediated by immune mechanisms (eg, hemolysis due to pri-maquine in glucose-6-phosphate dehydrogenase deficiency, or aplastic anemia caused by chloramphenicol).
IMMEDIATE (TYPE I) DRUG ALLERGY
Type I (immediate) sensitivity allergy to certain drugs occurs when the drug, not capable of inducing an immune response by itself, cova-lently links to a host carrier protein (hapten). When this happens, the immune system detects the drug-hapten conjugate as “modified self” and responds by generating IgE antibodies specific for the drug-hapten. It is not known why some people mount an IgE response to a drug, while others mount IgG responses. Under the influence of IL-4, IL-5, and IL-13 secreted by T H 2 cells, B cells specific for the drug
secrete IgE antibody. The mechanism for IgE-mediated immediate hypersensitivity is diagrammed in Figure 55–5 .
Fixation of the IgE antibody to high-affinity Fc receptors (FcεRs) on blood basophils or their tissue equivalent (mast cells) sets the stage for an acute allergic reaction. The most important sites for mast cell distribution are skin, nasal epithelium, lung, and gastrointestinal tract. When the offending drug is reintroduced into the body, it binds and cross-links basophil and mast cell-surface IgE to signal release of the mediators (eg, histamine, leu-kotrienes; see Chapters 16 and 18 ) from granules. Mediator release is associated with calcium influx and a fall in intracellular cAMP within the mast cell. Many of the drugs that block media-tor release appear to act through the cAMP mechanism (eg, cate-cholamines, glucocorticoids, theophylline), others block histamine release, and still others block histamine receptors. Other vasoac-tive substances such as kinins may also be generated during hista-mine release. These mediators initiate immediate vascular smooth muscle relaxation, increased vascular permeability, hypotension, edema, and bronchoconstriction.
Drug Treatment of Immediate Allergy One can test an individual for possible sensitivity to a drug by a simple scratch test, ie, by applying an extremely dilute solution of the drug to the skin and making a scratch with the tip of a needle. If allergy is present, an immediate (within 10–15 minutes) wheal (edema) and flare (increased blood flow) will occur. However, skin tests may be negative in spite of IgE hypersensitivity to a hapten or to a metabolic product of the drug, especially if the patient is taking steroids or antihistamines.
Drugs that modify allergic responses act at several links in this chain of events. Prednisone, often used in severe allergic reactions, is immunosuppressive; it blocks proliferation of the IgE-producing clones and inhibits IL-4 production by T helper cells in the IgE response, since glucocorticoids are generally toxic to lymphocytes. In the efferent limb of the allergic response, isoproterenol, epi-nephrine, and theophylline reduce the release of mediators from mast cells and basophils and produce bronchodilation. Epinephrine opposes histamine; it relaxes bronchiolar smooth muscle and con-tracts vascular muscle, relieving both bronchospasm and hypoten-sion. The antihistamines competitively inhibit histamine, which would otherwise produce bronchoconstriction and increased cap-illary permeability in the end organ. Glucocorticoids may also act to reduce tissue injury and edema in the inflamed tissue, as well as facilitating the actions of catecholamines in cells that may have become refractory to epinephrine or isoproterenol. Several agents directed toward the inhibition of leukotrienes may be useful in acute allergic and inflammatory disorders (see Chapter 20 ).
Desensitization to Drugs When reasonable alternatives are not available, certain drugs (eg, penicillin, insulin) must be used for life-threatening illnesses even in the presence of known allergic sensitivity. In such cases, desen-sitization (also called hyposensitization) can sometimes be accom-plished by starting with very small doses of the drug and gradually
055-Katzung_Ch055_p977-1000.indd 997 9/23/11 5:16:52 PM

998 SECTION VIII Chemotherapeutic Drugs
increasing the dose over a period of hours to the full therapeutic range (see Chapter 43 ). This practice is hazardous and must be performed under direct medical supervision, as anaphylaxis may occur before desensitization has been achieved. It is thought that slow and progressive administration of the drug gradually binds all available IgE on mast cells, triggering a gradual release of granules. Once all of the IgE on the mast cell surfaces has been bound and the cells have been degranulated, therapeutic doses of the offend-ing drug may be given with minimal further immune reaction. Therefore, a patient is only desensitized during administration of the drug.
AUTOIMMUNE (TYPE II) REACTIONS TO DRUGS
Certain autoimmune syndromes can be induced by drugs. Examples include systemic lupus erythematosus following hydral-azine or procainamide therapy, “lupoid hepatitis” due to cathartic sensitivity, autoimmune hemolytic anemia resulting from methyl-dopa administration, thrombocytopenic purpura due to quini-dine, and agranulocytosis due to a variety of drugs. As indicated in other chapters of this book, a number of drugs are associated with type I and type II reactions. In these drug-induced autoim-mune states, IgG antibodies bind to drug-modified tissue and are destroyed by the complement system or by phagocytic cells with Fc receptors. Fortunately, autoimmune reactions to drugs usually subside within several months after the offending drug is with-drawn. Immunosuppressive therapy is warranted only when the autoimmune response is unusually severe.
SERUM SICKNESS & VASCULITIC (TYPE III) REACTIONS
Immunologic reactions to drugs resulting in serum sickness are more common than immediate anaphylactic responses, but type II and type III hypersensitivities often overlap. The clinical features of serum sickness include urticarial and erythematous skin erup-tions, arthralgia or arthritis, lymphadenopathy, glomerulone-phritis, peripheral edema, and fever. The reactions generally last 6–12 days and usually subside once the offending drug is elimi-nated. Antibodies of the IgM or IgG class are usually involved. The mechanism of tissue injury is immune complex formation and deposition on basement membranes (eg, lung, kidney), fol-lowed by complement activation and infiltration of leukocytes, causing tissue destruction. Glucocorticoids are useful in attenuat-ing severe serum sickness reactions to drugs. In severe cases plas-mapheresis can be used to remove the offending drug and immune complexes from circulation.
Immune vasculitis can also be induced by drugs. The sulfon-amides, penicillin, thiouracil, anticonvulsants, and iodides have all been implicated in the initiation of hypersensitivity angiitis. Erythema multiforme is a relatively mild vasculitic skin disorder that may be secondary to drug hypersensitivity. Stevens-Johnson syndrome is probably a more severe form of this hypersensitivity reaction and consists of erythema multiforme, arthritis, nephritis, central nervous system abnormalities, and myocarditis. It has fre-quently been associated with sulfonamide therapy. Administration of nonhuman monoclonal or polyclonal antibodies such as rattle-snake antivenin may cause serum sickness.
055-Katzung_Ch055_p977-1000.indd 998 9/23/11 5:16:52 PM

CHAPTER 55 Immunopharmacology 999
Abatacept (Orencia)Parenteral: 250 mg/vial lyophilized powder
Abciximab (ReoPro)Parenteral: 2 mg/mL solution for IV injection
Adalimumab (Humira)Parenteral: 20, 40 mg/vial for SC injection
Alefacept (Amevive)Parenteral: 15 mg for IV injection
Alemtuzumab (Campath)Parenteral: 30 mg/mL vial for IV injection
Anakinra (Kineret)Parenteral: 100 mg/mL prefilled glass syringes for SC injection
Antithymocyte globulin (Thymoglobulin)Parenteral: 25 mg/vial for IV injection
Azathioprine (generic, Imuran)Oral: 50 mg tabletsParenteral: 100 mg/vial for IV injection
Basiliximab (Simulect)Parenteral: 20 mg powder; reconstitute for IV injection
Bevacizumab (Avastin)Parenteral: 25 mg/mL for injection
Canakinumab (Ilaris)Parenteral: 180 mg lyophilized powder for SC injection
Certolizumab (Cimzia)Parenteral: 200 mg powder for SC injection
Cetuximab (Erbitux)Parenteral: 2 mg/mL in 50 mL vials
Cyclosporine (Sandimmune, Neoral, SangCya)Oral: 25, 100 mg capsules; 100 mg/mL solutionParenteral: 50 mg/mL for IV administration
Daclizumab (Zenapax)Parenteral: 25 mg/5 mL vial for IV infusion
Denileukin dititox (Ontak)Parenteral: 150 mcg/mL for SC injection
Denosumab (Prolia)Parenteral: 60 mg/mL for SC injection
Etanercept (Enbrel)Parenteral: 25, 50 mg lyophilized powder for SC injection
Golimumab (Simponi)Parenteral: 50 mg/0.5 mL prefilled syringes for SC injection
Ibritumomab tiuxetan (Zevalin)Parenteral: 3.2 mg/2 mL for injection
Immune globulin intravenous [IGIV] (various)Parenteral: 5, 10% solutions; 2.5, 5, 6, 10, 12 g powder for injection
Infliximab (Remicade)Parenteral: 100 mg lyophilized powder for IV injection
Interferon alfa-2a (Roferon)Parenteral: 3, 6, 9 million IU
Interferon alfa-2b (Intron-A)Parenteral: 3–50 million units/vial
Interferon beta-1a (Avonex, Rebif)Parenteral: 22, 33, 44 mcg powder for IV injection
Interferon beta-1b (Betaseron, Extavia)Parenteral: 0.3 mg powder for SC injection
Interferon gamma-1b (Actimmune)Parenteral: 100 mcg vials
Interleukin-2 [IL-2, aldesleukin] (Proleukin)Parenteral: 22 million unit vials
Leflunomide (Arava)Oral: 10, 20, 100 mg tablets
Lenalidomide (Revlimid)Oral: 5, 10, 15, mg capsules
Lymphocyte immune globulin (Atgam)Parenteral: 50 mg/mL for injection (in 5 mL ampules)
Muromonab-CD3 [OKT3] (Orthoclone OKT3)Parenteral: 5 mg/5 mL ampule for injection
Mycophenolate mofetil (CellCept, Myfortic)Oral: 250 mg capsules; 500 mg tablets; 200 mg powder for suspen-
sion; 180, 360 mg delayed-release tabletsParenteral: 500 mg powder; reconstitute for injection
Natalizumab (Tysabri)Parenteral: 300 mg/15 mL for dilution and IV infusion
Ofatumumab (Arzerra)Parenteral: 100 mg/5 mL vial; dilute for IV infusion
Omalizumab (Xolair)Parenteral: 150 mg powder for SC injection
Panitumumab (Vectibix)Parenteral: single-use 20 mg/mL vials
Pegademase bovine (Adagen)Parenteral: 250 units/mL for IM injectionNote: Pegademase is bovine adenosine deaminase.
Pegaptanib (Macugen)Parenteral: 0.3 mL for intravitreal injection
Peginterferon alfa-2a (Pegasys)Parenteral: 180 mcg/mL
Peginterferon alfa-2b (PEG-Intron)Parenteral: 50, 80, 120, 150 mcg/0.5 mL
Ranibizumab (Lucentis)Parenteral: 10 mg/mL for intravitreal injection
Rho(D) immune globulin micro-dose (RhoGam, others)Parenteral: in single-dose and micro-dose vials
Rilonacept (Arcalyst)Parenteral: 220 mg lyophilized powder for SC injection
Rituximab (Rituxan)Parenteral: 10 mg/mL for IV infusion
Sirolimus (Rapamune)Oral: 1, 2 mg tablets; 1 mg/mL solution
Tacrolimus [FK 506] (Prograf)Oral: 0.5, 1, 5 mg capsulesParenteral: 5 mg/mLTopical (Protopic): 0.03%, 0.1% ointment
Thalidomide (Thalomid)Oral: 50, 100, 200 mg capsulesNote: Thalidomide is labeled for use only in erythema nodosum lep-
rosum in the USA.Tocilizumab (Actemra)
Parenteral: 20 mg/mL vials for dilution and IV infusionTrastuzumab (Herceptin)
Parenteral: 440 mg powder; reconstitute for IV infusionUstekinumab (Stelara)
Parenteral: 45 mg/0.5 mL, 90 mg/mL in prefilled syringes for SC injection
P R E P A R A T I O N S A V A I L A B L E *
Abatacept (Orencia)Parenteral: 250 mg/vial lyophilized powder
Abciximab (ReoPro)Parenteral: 2 mg/mL solution for IV injection
Adalimumab (Humira)Parenteral: 20, 40 mg/vial for SC injection
Alefacept (Amevive)Parenteral: 15 mg for IV injection
Alemtuzumab (Campath)Parenteral: 30 mg/mL vial for IV injection
Anakinra (Kineret)Parenteral: 100 mg/mL prefilled glass syringes for SC injection
Antithymocyte globulin (Thymoglobulin)Parenteral: 25 mg/vial for IV injection
Azathioprine (generic, Imuran)Oral: 50 mg tabletsParenteral: 100 mg/vial for IV injection
Basiliximab (Simulect)Parenteral: 20 mg powder; reconstitute for IV injection
Bevacizumab (Avastin)Parenteral: 25 mg/mL for injection
Canakinumab (Ilaris)Parenteral: 180 mg lyophilized powder for SC injection
Certolizumab (Cimzia)Parenteral: 200 mg powder for SC injection
Cetuximab (Erbitux)Parenteral: 2 mg/mL in 50 mL vials
Cyclosporine (Sandimmune, Neoral, SangCya)Oral: 25, 100 mg capsules; 100 mg/mL solutionParenteral: 50 mg/mL for IV administration
Daclizumab (Zenapax)Parenteral: 25 mg/5 mL vial for IV infusion
Denileukin dititox (Ontak)Parenteral: 150 mcg/mL for SC injection
Denosumab (Prolia)Parenteral: 60 mg/mL for SC injection
Etanercept (Enbrel)Parenteral: 25, 50 mg lyophilized powder for SC injection
Golimumab (Simponi)Parenteral: 50 mg/0.5 mL prefilled syringes for SC injection
Ibritumomab tiuxetan (Zevalin)Parenteral: 3.2 mg/2 mL for injection
Immune globulin intravenous [IGIV] (various)Parenteral: 5, 10% solutions; 2.5, 5, 6, 10, 12 g powder for injection
Infliximab (Remicade)Parenteral: 100 mg lyophilized powder for IV injection
Interferon alfa-2a (Roferon)Parenteral: 3, 6, 9 million IU
Interferon alfa-2b (Intron-A)Parenteral: 3–50 million units/vial
Interferon beta-1a (Avonex, Rebif)Parenteral: 22, 33, 44 mcg powder for IV injection
Interferon beta-1b (Betaseron, Extavia)Parenteral: 0.3 mg powder for SC injection
Interferon gamma-1b (Actimmune)Parenteral: 100 mcg vials
Interleukin-2 [IL-2, aldesleukin] (Proleukin)Parenteral: 22 million unit vials
Leflunomide (Arava)Oral: 10, 20, 100 mg tablets
Lenalidomide (Revlimid)Oral: 5, 10, 15, mg capsules
Lymphocyte immune globulin (Atgam)Parenteral: 50 mg/mL for injection (in 5 mL ampules)
Muromonab-CD3 [OKT3] (Orthoclone OKT3)Parenteral: 5 mg/5 mL ampule for injection
Mycophenolate mofetil (CellCept, Myfortic)Oral: 250 mg capsules; 500 mg tablets; 200 mg powder for suspen-
sion; 180, 360 mg delayed-release tabletsParenteral: 500 mg powder; reconstitute for injection
Natalizumab (Tysabri)Parenteral: 300 mg/15 mL for dilution and IV infusion
Ofatumumab (Arzerra)Parenteral: 100 mg/5 mL vial; dilute for IV infusion
Omalizumab (Xolair)Parenteral: 150 mg powder for SC injection
Panitumumab (Vectibix)Parenteral: single-use 20 mg/mL vials
Pegademase bovine (Adagen)Parenteral: 250 units/mL for IM injectionNote: Pegademase is bovine adenosine deaminase.
Pegaptanib (Macugen)Parenteral: 0.3 mL for intravitreal injection
Peginterferon alfa-2a (Pegasys)Parenteral: 180 mcg/mL
Peginterferon alfa-2b (PEG-Intron)Parenteral: 50, 80, 120, 150 mcg/0.5 mL
Ranibizumab (Lucentis)Parenteral: 10 mg/mL for intravitreal injection
Rho(D) immune globulin micro-dose (RhoGam, others)Parenteral: in single-dose and micro-dose vials
Rilonacept (Arcalyst)Parenteral: 220 mg lyophilized powder for SC injection
Rituximab (Rituxan)Parenteral: 10 mg/mL for IV infusion
Sirolimus (Rapamune)Oral: 1, 2 mg tablets; 1 mg/mL solution
Tacrolimus [FK 506] (Prograf)Oral: 0.5, 1, 5 mg capsulesParenteral: 5 mg/mLTopical (Protopic): 0.03%, 0.1% ointment
Thalidomide (Thalomid)Oral: 50, 100, 200 mg capsulesNote: Thalidomide is labeled for use only in erythema nodosum lep-
rosum in the USA.Tocilizumab (Actemra)
Parenteral: 20 mg/mL vials for dilution and IV infusionTrastuzumab (Herceptin)
Parenteral: 440 mg powder; reconstitute for IV infusionUstekinumab (Stelara)
Parenteral: 45 mg/0.5 mL, 90 mg/mL in prefilled syringes for SCinjection
∗Several drugs discussed in this chapter are available as orphan drugs but are not listed here. Other drugs not listed here will be found in other chapters (see Index).
055-Katzung_Ch055_p977-1000.indd 999 9/23/11 5:16:52 PM

1000 SECTION VIII Chemotherapeutic Drugs
C A S E S T U D Y A N S W E R
Within 24–72 hours postpartum, the woman should be given a 2-mL intramuscular injection of 300 mcg anti-Rh o (D) immune globulin. This will clear any fetal Rh-positive red
cells from her circulation so she does not generate anti-Rh o (D) B cells that might jeopardize any future pregnancy.
REFERENCES General Immunology
Bonneville M et al: γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat Rev Immunol 2010;10:467.
Kumar H, Kawai T, Akira S: Toll-like receptors and innate immunity. Biochem Biophys Res Comm 2009;388:621.
Levinson WE, Jawetz E: Medical Microbiology and Immunology, 7th ed. McGraw-Hill, 2008.
Murphy KM, Travers P, Walport M (editors): Janeway’s Immunobiology, 7th ed. Garland Science, 2008.
Thornton AM et al: Expression of helios, an ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol 2010;184:3433.
Hypersensitivity
Hausmann O: Drug hypersensitivity reactions involving skin. Handbk Exper Pharmacol 2010;196:29.
Phillips EJ: Pharmacogenetics of drug hypersensitivity. Pharmacogenomics 2010;11:973.
Autoimmunity
Bousvaros A: Use of immunomodulators and biologic therapies in children with inflammatory bowel disease. Expert Rev Clin Immunol 2010;6:659.
Carroll WM: Clinical trials of multiple sclerosis therapies: Improvements to dem-onstrate long-term patient benefit. Mult Scler 2009;15:951.
Kircik LH et al: How and when to use biologics in psoriasis. J Drugs Dermatol 2010;9:s106.
La Cava A: Anticytokine therapies in systemic lupus erythematosus. Immunotherapy 2010;2:575.
Ma MH et al: Remission in early rheumatoid arthritis. J Rheumatol 2010;37:1444.
Immunodeficiency Diseases
Wood PM: Primary antibody deficiency syndromes. Curr Opin Hematol 2010;17:356.
Immunosuppressive Agents
Braun J: Optimal administration and dosage of methotrexate. Clin Exp Rheumatol 2010;28:S46.
Galustian C: The anticancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother 2009;58:1033.
Li S, Gill N, Lentzch S: Recent advances of IMiDs in cancer therapy. Curr Opin Oncol 2010;22:579.
Ponticelli C: Calcineurin inhibitors in renal transplantation still needed but in reduced doses: A review. Transplant Proc 2010;42:2205.
Vicari-Christensen M: Tacrolimus: Review of pharmacokinetics, pharmacodynam-ics, and pharmacogenetics to facilitate practitioners’ understanding and offer strategies for educating patients and promoting adherence. Prog Transplant 2009;19:277.
Zhou H: Updates of mTOR inhibitors. Anticancer Agents Med Chem 2010;10:571.
Antilymphocyte Globulin & Monoclonal Antibodies
Cummings SR: Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009;361:756.
Gürcan HM: Information for healthcare providers on general features of IGIV with emphasis on differences between commercially available products. Autoimmunity Rev 2010;9:553.
Nelson AL: Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 2010;9:767.
Taylor PC: Pharmacology of TNF blockade in rheumatoid arthritis and other chronic inflammatory diseases. Curr Opin Pharmacol 2010;10:308.
Weiner LM: Monoclonal antibodies: Versatile platforms for cancer immunother-apy. Nat Rev Immunol 2010;10:317.
Cytokines
Foster GR: Pegylated interferons for the treatment of chronic hepatitis C: Pharmacological and clinical differences between peginterferon-alpha-2a and peginterferon-alpha-2b. Drugs 2010;70:147.
Gabay C: IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 2010;6:232.
Drug Allergy
Hamilton RG: Human IgE antibody serology: A primer for the practicing North American allergist/immunologist. J Allergy Clin Immunol 2010;126:33.
Khan DA: Drug allergy. J Allergy Clin Immunol 2010;125:S126.
055-Katzung_Ch055_p977-1000.indd 1000 9/23/11 5:16:52 PM

1001
C H A P T E R
56 Humans live in a chemical environment and inhale, ingest, or absorb from the skin many of these chemicals. Toxicology is con-cerned with the deleterious effects of these chemical agents on all living systems. In the biomedical area, however, the toxicologist is primarily concerned with adverse effects in humans resulting from exposure to drugs and other chemicals as well as the demonstra-tion of safety or hazard associated with their use.
Occupational Toxicology Occupational toxicology deals with the chemicals found in the workplace. The major emphasis of occupational toxicology is to identify the agents of concern, identify the acute and chronic diseases that they cause, define the conditions under which they may be used safely, and prevent absorption of harmful amounts of these chemicals. Occupational toxicologists may also define and carry out programs for the surveillance of exposed workers and the environment in which they work. Regulatory limits and voluntary guidelines have been elaborated to establish safe ambient air con-centrations for many chemicals found in the workplace.
Governmental and supragovernmental bodies throughout the world have generated workplace health and safety rules, including short-and long-term exposure limits for workers. These permissi-ble exposure limits (PELS) have the power of law. Copies of the United States Occupational Safety and Health Administration (OSHA) standards may be found on OSHA’s website at http://www.osha.gov . Copies of the United States Mine Safety and Health Administration (MSHA) standards may be found at http://www.msha.gov .
Voluntary organizations, such as the American Conference of Governmental Industrial Hygienists (ACGIH), periodically pre-pare lists of recommended threshold limit values (TLVs) for many chemicals. These guidelines are periodically updated, but regulatory imperatives in the United States are not updated except under certain extraordinary circumstances. These TLV guidelines are useful as reference points in the evaluation of potential work-place exposures. Copies of current TLV lists may be obtained from the ACGIH at http://www.acgih.org .
Environmental Toxicology Environmental toxicology deals with the potentially deleterious impact of chemicals, present as pollutants of the environment, on
SECTION IX TOXICOLOGY
Introduction to Toxicology: Occupational & EnvironmentalDaniel T. Teitelbaum, MD∗
∗The author thanks Gabriel L. Plaa, PhD, the previous author of this chapter, for his contributions.
056-Katzung_Ch056_p1001-1012.indd 1001 9/23/11 4:35:10 PM

1002 SECTION IX Toxicology
Risk is defined as the expected frequency of the occurrence of an undesirable effect arising from exposure to a chemical or physical agent. Estimation of risk makes use of dose-response data and extrapolation from the observed relationships to the expected responses at doses occurring in actual exposure situations. The quality and suitability of the biologic data used in such estimates are major limiting factors.
Routes of Exposure The route of entry for chemicals into the body differs in different exposure situations. In the industrial setting, inhalation is the major route of entry. The transdermal route is also quite impor-tant, but oral ingestion is a relatively minor route. Consequently, primary prevention should be designed to reduce or eliminate absorption by inhalation or by topical contact. Atmospheric pol-lutants gain entry by inhalation and by dermal contact. Water and soil pollutants are absorbed through inhalation, ingestion, and dermal contact.
Duration of Exposure Toxic reactions may differ qualitatively depending on the duration of the exposure. A single exposure—or multiple exposures occur-ring over a brief period from seconds to 1 or 2 days—represents acute exposure. Multiple exposures continuing over a longer period of time represent chronic exposure. In the occupational setting, both acute (eg, accidental discharge) and chronic (eg, repetitive handling of a chemical) exposures occur. Exposures to chemicals found in the environment such as air and water pollut-ants often cause chronic exposure, but sudden large chemical releases may result in acute massive population exposure with seri-ous or lethal consequences.
ENVIRONMENTAL CONSIDERATIONS
Certain chemical and physical characteristics are important for estimating the potential hazard involved for environmental toxicants. In addition to information regarding effects on differ-ent organisms, knowledge about the following properties is essential to predict the environmental impact: the degradability of the substance; its mobility through air, water, and soil; whether or not bioaccumulation occurs; and its transport and biomagnification through food chains. (See Box: Bioaccumulation & Biomagnification.) Chemicals that are poorly degraded (by abiotic or biotic pathways) exhibit environmental persistence and thus can accumulate. Typical examples of such chemicals include the persistent organic pollutants (POP) such as polychlorinated biphenyls and similar substances. Lipophilic substances such as the once-widespread organochlorine pesticides (eg, DDT) tend to bioaccumulate in body fat, resulting in tissue residues. Slowly released over time, these residues and their metabolites may have chronic adverse effects such as endocrine disruption. When the toxicant is incorporated into the food chain, biomagnification occurs as one species feeds on others and concentrates the chemi-cal. Humans stand at the apex of the food chain. They may be
living organisms. The term environment includes all the sur-roundings of an individual organism, but particularly the air, soil, and water. Although humans are considered a target species of particular interest, other species are of considerable importance as potential biologic targets.
Air pollution is a product of industrialization, technologic development, and increased urbanization. Humans may also be exposed to chemicals used in the agricultural environment as pesticides or in food processing that may persist as residues or ingredients in food products. Air contaminants are regulated in the United States by the Environmental Protection Agency (EPA) based on both health and esthetic considerations. Tables of regulated air contaminants and other regulatory issues that relate to air contaminants in the United States may be found at http://www.epa.gov . Many states also have individual air con-taminant regulations that may be more rigorous than those of the EPA. Many other nations and some supragovernmental orga-nizations regulate air contaminants.
The United Nations Food and Agriculture Organization and the World Health Organization (FAO/WHO) Joint Expert Commission on Food Additives adopted the term acceptable daily intake (ADI) to denote the daily intake of a chemical from food that, during an entire lifetime, appears to be without appreciable risk. These guidelines are reevaluated as new infor-mation becomes available. In the United States, the Food and Drug Administration (FDA) and the Department of Agriculture are responsible for the regulation of contaminants such as pes-ticides, drugs, and chemicals in foods. Major international problems have occurred because of traffic among nations in contaminated or adulterated foods from countries whose regu-lations and enforcement of pure food and drug laws are lax or nonexistent.
Ecotoxicology Ecotoxicology is concerned with the toxic effects of chemical and physical agents on populations and communities of living organ-isms within defined ecosystems; it includes the transfer pathways of those agents and their interactions with the environment. Traditional toxicology is concerned with toxic effects on individ-ual organisms; ecotoxicology is concerned with the impact on populations of living organisms or on ecosystems.
TOXICOLOGIC TERMS & DEFINITIONS
Hazard & Risk Hazard is the ability of a chemical agent to cause injury in a given situation or setting; the conditions of use and exposure are pri-mary considerations. To assess hazard, one needs to have know-ledge about both the inherent toxicity of the substance and the amounts to which individuals are liable to be exposed. Humans may be able to use potentially toxic substances when the necessary conditions minimizing absorption are established and respected. However, hazard is often a description based on subjective esti-mates rather than objective evaluation.
056-Katzung_Ch056_p1001-1012.indd 1002 9/23/11 4:35:11 PM

CHAPTER 56 Introduction to Toxicology: Occupational & Environmental 1003
exposed to highly concentrated pollutant loads as bioaccumula-tion and biomagnification occur. The pollutants that have the widest environmental impact are poorly degradable; are relatively mobile in air, water, and soil; exhibit bioaccumulation; and also exhibit biomagnification.
■ SPECIFIC CHEMICALS
AIR POLLUTANTS
Five major substances account for about 98% of air pollution: carbon monoxide (CO, about 52%), sulfur oxides (about 14%), hydrocarbons (about 14%), nitrogen oxides (about 14%), and particulate matter (about 4%). The sources of these chemicals include transportation, industry, generation of electric power, space heating, and refuse disposal. Sulfur dioxide and smoke resulting from incomplete combustion of coal have been associated with acute adverse effects, particu-larly among the elderly and individuals with preexisting car-diac or respiratory disease. Ambient air pollution has been implicated as a contributing factor in bronchitis, obstructive ventilatory disease, pulmonary emphysema, bronchial asthma, and lung cancer. EPA standards for these substances apply to the general environment, and OSHA standards apply to workplace exposure.
Carbon Monoxide Carbon monoxide (CO) is a colorless, tasteless, odorless, and nonirritating gas, a byproduct of incomplete combustion. The average concentration of CO in the atmosphere is about 0.1 ppm; in heavy traffic, the concentration may exceed 100 ppm. The recommended 2008 threshold limit values (TLV-TWA and TLV-STEL) are shown in Table 56–1 .
A. Mechanism of Action CO combines reversibly with the oxygen-binding sites of hemo-globin and has an affinity for hemoglobin that is about 220 times that of oxygen. The product formed—carboxyhemoglobin— cannot transport oxygen. Furthermore, the presence of carboxyhe-moglobin interferes with the dissociation of oxygen from the remaining oxyhemoglobin, thus reducing the transfer of oxygen to tissues. The brain and the heart are the organs most affected. Normal nonsmoking adults have carboxyhemoglobin levels of less than 1% saturation (1% of total hemoglobin is in the form of carboxyhemoglobin); this level has been attributed to the endog-enous formation of CO from heme catabolism. Smokers may exhibit 5–10% saturation, depending on their smoking habits. A person breathing air containing 0.1% CO (1000 ppm) would have a carboxyhemoglobin level of about 50%.
B. Clinical Effects The principal signs of CO intoxication are those of hypoxia and progress in the following sequence: (1) psychomotor impairment; (2) headache and tightness in the temporal area; (3) confusion and loss of visual acuity; (4) tachycardia, tachypnea, syncope, and coma; and (5) deep coma, convulsions, shock, and respiratory failure. There is great variability in individual responses to a given carboxy-hemoglobin concentration. Carboxyhemoglobin levels below 15% may produce headache and malaise; at 25% many workers com-plain of headache, fatigue, decreased attention span, and loss of fine motor coordination. Collapse and syncope may appear at around 40%; with levels above 60%, death may ensue as a result of irrevers-ible damage to the brain and myocardium. The clinical effects may be aggravated by heavy labor, high altitudes, and high ambient temperatures. Although CO intoxication is usually thought of as a form of acute toxicity, there is some evidence that chronic exposure to low levels may lead to undesirable effects, including the develop-ment of atherosclerotic coronary disease in cigarette smokers. The fetus may be quite susceptible to the effects of CO exposure.
Bioaccumulation & Biomagnification
If the intake of a long-lasting contaminant by an organism exceeds the latter’s ability to metabolize or excrete the substance, the chemical accumulates within the tissues of the organism. This is called bioaccumulation.
Although the concentration of a contaminant may be virtually undetectable in water, it may be magnified hundreds or thou-sands of times as the contaminant passes up the food chain. This is called biomagnification.
The biomagnification of polychlorinated biphenyls (PCBs) in the Great Lakes of North America is illustrated by the following residue values available from Environment Canada, a report pub-lished by the Canadian government, and other sources.
Thus, the biomagnification for this substance in the food chain, beginning with phytoplankton and ending with the herring
gull, is nearly 50,000-fold. Domestic animals and humans may eat fish from the Great Lakes, resulting in PCB residues in these spe-cies as well.
Source
PCB Concentration (ppm)1
Concentration Relative to Phytoplankton
Phytoplankton 0.0025 1
Zooplankton 0.123 49.2
Rainbow smelt 1.04 416
Lake trout 4.83 1,932
Herring gull 124 49,6001Sources: Environment Canada, The State of Canada’s Environment, 1991, Government of Canada, Ottawa; and other publications.
056-Katzung_Ch056_p1001-1012.indd 1003 9/23/11 4:35:11 PM

1004 SECTION IX Toxicology
C. Treatment In cases of acute intoxication, removal of the individual from the exposure source and maintenance of respiration are essential, fol-lowed by administration of oxygen—the specific antagonist to CO—within the limits of oxygen toxicity. With room air at 1 atm, the elimination half-time of CO is about 320 minutes; with 100% oxygen, the half-time is about 80 minutes; and with hyperbaric oxygen (2–3 atm), the half-time can be reduced to about 20 min-utes. If a hyperbaric oxygen chamber is readily available, it should be used in the treatment of CO poisoning for severely poisoned patients; however, there remain questions about its effectiveness. Progressive recovery from effectively treated CO poisoning, even of a severe degree, is often complete, although some patients dem-onstrate persistent impairment for a prolonged period of time.
Sulfur Dioxide Sulfur dioxide (SO 2 ) is a colorless, irritant gas generated primarily by the combustion of sulfur-containing fossil fuels. The 2008 TLVs are given in Table 56–1 .
A. Mechanism of Action On contact with moist membranes, SO 2 forms sulfurous acid, which is responsible for its severe irritant effects on the eyes, mucous mem-branes, and skin. Approximately 90% of inhaled SO 2 is absorbed in the upper respiratory tract, the site of its principal effect. The inhala-tion of SO 2 causes bronchial constriction; parasympathetic reflexes and altered smooth muscle tone appear to be involved. Exposure to 5 ppm SO 2 for 10 minutes leads to increased resistance to airflow in
most humans. Exposures of 5–10 ppm are reported to cause severe bronchospasm; 10–20% of the healthy young adult population is estimated to be reactive to even lower concentrations. The phenom-enon of adaptation to irritating concentrations has been reported in workers. However, current studies have not confirmed this phenom-enon. Asthmatic individuals are especially sensitive to SO 2 .
B. Clinical Effects and Treatment The signs and symptoms of intoxication include irritation of the eyes, nose, and throat and reflex bronchoconstriction. In asth-matic subjects, exposure to SO 2 may result in an acute asthmatic episode. If severe exposure has occurred, delayed-onset pulmonary edema may be observed. Cumulative effects from chronic low-level exposure to SO 2 are not striking, particularly in humans but these effects have been associated with aggravation of chronic cardiopulmonary disease. When combined exposure to high respi-rable particulate loads and SO 2 occurs, the mixed irritant load may increase the toxic respiratory response. Treatment is not spe-cific for SO 2 but depends on therapeutic maneuvers used in the treatment of irritation of the respiratory tract and asthma.
Nitrogen Oxides Nitrogen dioxide (NO 2 ) is a brownish irritant gas sometimes asso-ciated with fires. It is formed also from fresh silage; exposure of farmers to NO 2 in the confines of a silo can lead to silo-filler’s disease. The 2008 TLVs are shown in Table 56–1 .
A. Mechanism of Action NO 2 is a relatively insoluble deep lung irritant capable of produc-ing pulmonary edema. The type I cells of the alveoli appear to be the cells chiefly affected on acute exposure. At higher exposure, both type I and type II alveolar cells are damaged. Exposure to 25 ppm of NO 2 is irritating to some individuals; 50 ppm is mod-erately irritating to the eyes and nose. Exposure for 1 hour to 50 ppm can cause pulmonary edema and perhaps subacute or chronic pulmonary lesions; 100 ppm can cause pulmonary edema and death.
B. Clinical Effects and Treatment The signs and symptoms of acute exposure to NO 2 include irrita-tion of the eyes and nose, cough, mucoid or frothy sputum produc-tion, dyspnea, and chest pain. Pulmonary edema may appear within 1–2 hours. In some individuals, the clinical signs may sub-side in about 2 weeks; the patient may then pass into a second stage of abruptly increasing severity, including recurring pulmonary edema and fibrotic destruction of terminal bronchioles (bronchi-olitis obliterans). Chronic exposure of laboratory animals to 10–25 ppm NO 2 has resulted in emphysematous changes; thus, chronic effects in humans are of concern. There is no specific treatment for acute intoxication by NO 2 ; therapeutic measures for the manage-ment of deep lung irritation and noncardiogenic pulmonary edema are used. These measures include maintenance of gas exchange with adequate oxygenation and alveolar ventilation. Drug therapy may include bronchodilators, sedatives, and antibiotics.
TABLE 56–1 Threshold limit values (TLVs) of some common air pollutants and solvents.
TLV (ppm)
Compound TWA1 STEL2
Benzene 0.5 2.5
Carbon monoxide 25 NA
Carbon tetrachloride 5 10
Chloroform 10 NA
Nitrogen dioxide 3 5
Ozone 0.05 NA
Sulfur dioxide 2 5
Tetrachloroethylene 25 100
Toluene 50 NA
1,1,1-Trichloroethane 350 450
Trichloroethylene 50 1001TLV-TWA (time weighted average) is the concentration for a normal 8-hour workday or 40-hour workweek to which workers may be repeatedly exposed without adverse effects.2TLV-STEL (short-term exposure limit) is the maximum concentration that should not be exceeded at any time during a 15-minute exposure period.
NA, none assigned.
056-Katzung_Ch056_p1001-1012.indd 1004 9/23/11 4:35:11 PM

CHAPTER 56 Introduction to Toxicology: Occupational & Environmental 1005
Ozone Ozone (O 3 ) is a bluish irritant gas that occurs normally in the earth’s atmosphere, where it is an important absorbent of ultravio-let light. In the workplace, it can occur around high-voltage elec-trical equipment and around ozone-producing devices used for air and water purification. It is also an important oxidant found in polluted urban air. There is a near-linear gradient between expo-sure (1-hour level, 20–100 ppb) and response. See Table 56–1 for 2008 TLVs.
A. Clinical Effects and Treatment O 3 is an irritant of mucous membranes. Mild exposure produces upper respiratory tract irritation. Severe exposure can cause deep lung irritation, with pulmonary edema when inhaled at sufficient concentrations. Ozone penetration in the lung depends on tidal volume; consequently, exercise can increase the amount of ozone reaching the distal lung. Some of the effects of O 3 resemble those seen with radiation, suggesting that O 3 toxicity may result from the formation of reactive free radicals. The gas causes shallow, rapid breathing and a decrease in pulmonary compliance. Enhanced sensitivity of the lung to bronchoconstrictors is also observed.
Exposure around 0.1 ppm O 3 for 10–30 minutes causes irrita-tion and dryness of the throat; above 0.1 ppm, one finds changes in visual acuity, substernal pain, and dyspnea. Pulmonary function is impaired at concentrations exceeding 0.8 ppm. Airway hyper-responsiveness and airway inflammation have been observed in humans.
The response of the lung to O 3 is a dynamic one. The mor-phologic and biochemical changes are the result of both direct injury and secondary responses to the initial damage. Long-term exposure in animals results in morphologic and functional pul-monary changes. Chronic bronchitis, bronchiolitis, fibrosis, and emphysematous changes have been reported in a variety of spe-cies, including humans, exposed to concentrations above 1 ppm. There is no specific treatment for acute O 3 intoxication. Management depends on therapeutic measures used for deep lung irritation and noncardiogenic pulmonary edema (see Nitrogen Oxides, above).
SOLVENTS
Halogenated Aliphatic Hydrocarbons These agents once found wide use as industrial solvents, degreas-ing agents, and cleaning agents. The substances include carbon tetrachloride, chloroform, trichloroethylene, tetrachloroethylene (perchloroethylene), and 1,1,1-trichloroethane (methyl chloro-form). However, because of the likelihood that halogenated ali-phatic hydrocarbons are carcinogenic to humans, carbon tetrachloride and trichloroethylene have largely been removed from the workplace. Perchloroethylene and trichloroethane are still in use for dry cleaning and solvent degreasing, but it is likely that their use will be very limited in the future. Dry cleaning as an
occupation is listed as a class 2B carcinogenic activity by the International Agency for Research Against Cancer (IARC). Fluorinated aliphatics such as the freons and closely related com-pounds have also been used in the workplace and in consumer goods, but because of the severe environmental damage they cause, their use has been limited or eliminated by international treaty agreements. The common halogenated aliphatic solvents also create serious problems as persistent water pollutants. They are widely found in both groundwater and drinking water as a result of poor disposal practices.
See Table 56–1 for recommended TLVs.
A. Mechanism of Action and Clinical Effects In laboratory animals, the halogenated hydrocarbons cause central nervous system depression, liver injury, kidney injury, and some degree of cardiotoxicity. Several are also carcinogenic in animals and are considered probable carcinogens in humans. Trichloroethylene and tetrachloroethylene are listed as “reasonably anticipated to be a human carcinogen” by the US National Toxicology Program, and as class 2A probable human carcinogens by IARC. These sub-stances are depressants of the central nervous system in humans; chloroform is the most potent. Chronic exposure to tetrachloro-ethylene and possibly 1,1,1-trichloroethane can cause impaired memory and peripheral neuropathy. Hepatotoxicity is also a com-mon toxic effect that can occur in humans after acute or chronic exposures; carbon tetrachloride is the most potent of the series. Nephrotoxicity can occur in humans exposed to carbon tetrachlo-ride, chloroform, and trichloroethylene. With chloroform, carbon tetrachloride, trichloroethylene, and tetrachloroethylene, carcino-genicity has been observed in lifetime exposure studies performed in rats and mice and in some human epidemiologic studies. Reviews of the epidemiologic literature on the occupational expo-sure of workers to various halogenated aliphatic hydrocarbon sol-vents including trichloroethylene and tetrachloroethylene have found significant associations between exposure to the agent and renal, prostate, and testicular cancer. Other cancers have been found to be increased but their incidence has not reached statisti-cal significance.
B. Treatment There is no specific treatment for acute intoxication resulting from exposure to halogenated hydrocarbons. Management depends on the organ system involved.
Aromatic Hydrocarbons Benzene is used for its solvent properties and as an intermediate in the synthesis of other chemicals. The 2008 recommended TLVs are given in Table 56–1 . Benzene remains an important compo-nent of gasoline and may be found in premium gasolines at con-centrations as high as 2%. In cold climates such as Alaska, benzene concentrations in gasoline may reach 5%. The PEL promulgated by OSHA is 1 ppm in the air and a 5 ppm limit for skin exposure. The National Institute for Occupational Safety and Health (NIOSH) and others have recommended that the exposure limits
056-Katzung_Ch056_p1001-1012.indd 1005 9/23/11 4:35:11 PM

1006 SECTION IX Toxicology
for benzene be further reduced to 0.1 ppm because excess blood cancers occur at the current PEL. The acute toxic effect of benzene is depression of the central nervous system. Exposure to 7500 ppm for 30 minutes can be fatal. Exposure to concentrations larger than 3000 ppm may cause euphoria, nausea, locomotor problems, and coma; vertigo, drowsiness, headache, and nausea may occur at concentrations ranging from 250 to 500 ppm. No specific treat-ment exists for the acute toxic effect of benzene.
Chronic exposure to benzene can result in very serious toxic effects, the most significant of which is bone marrow injury. Aplastic anemia, leukopenia, pancytopenia, and thrombocytopenia occur at higher levels of exposure, as does leukemia. Chronic expo-sure to much lower levels has been associated with leukemia of several types as well as lymphomas, myeloma, and myelodysplastic syndrome. Recent studies have shown the occurrence of leukemia following exposures as low as 2 ppm-years. The pluri-potent bone marrow stem cells appear to be a target of benzene or its metabo-lites and other stem cells may also be targets. Epidemiologic data confirm a causal association between benzene exposure and an increased incidence of leukemia in workers. Most organizations now classify benzene as a known human carcinogen.
Toluene (methylbenzene) does not possess the myelotoxic properties of benzene, nor has it been associated with leukemia. It is, however, a central nervous system depressant and a skin and eye irritant. It is also fetotoxic. See Table 56–1 for the TLVs. Exposure to 800 ppm can lead to severe fatigue and ataxia; 10,000 ppm can produce rapid loss of consciousness. Chronic effects of long-term toluene exposure are unclear because human studies indicating behavioral effects usually concern exposures to several solvents. In limited occupational studies, however, metabolic interactions and modification of toluene’s effects have not been observed in work-ers also exposed to other solvents. Less refined grades of toluene contain benzene.
Xylene (dimethylbenzene) has been substituted for benzene in many solvent degreasing operations. Like toluene, the three
xylenes do not possess the myelotoxic properties of benzene, nor have they been associated with leukemia. Xylene is a central ner-vous system depressant and a skin irritant. Less refined grades of xylene contain benzene. Estimated TLV-TWA and TLV-STEL are 100 and 150 ppm, respectively.
PESTICIDES
Organochlorine Pesticides These agents are usually classified into four groups: DDT (chlo-rophenothane) and its analogs, benzene hexachlorides, cyclo-dienes, and toxaphenes ( Table 56–2 ). They are aryl, carbocyclic, or heterocyclic compounds containing chlorine substituents. The individual compounds differ widely in their biotransforma-tion and capacity for storage in tissues; toxicity and storage are not always correlated. They can be absorbed through the skin as well as by inhalation or oral ingestion. There are, however, important quantitative differences between the various deriva-tives; DDT in solution is poorly absorbed through the skin, whereas dieldrin absorption from the skin is very efficient. Organochlorine pesticides have largely been abandoned because they cause severe environmental damage. DDT continues to have very restricted use for domestic mosquito elimination in malaria-infested areas of Africa. This use is controversial, but it is very effective and is likely to remain in place for the foresee-able future. Organochlorine pesticide residues in humans, ani-mals, and the environment present long-term problems that are not yet fully understood.
A. Human Toxicology The acute toxic properties of all the organochlorine pesticides in humans are qualitatively similar. These agents interfere with inactivation of the sodium channel in excitable membranes and
TABLE 56–2 Organochlorine pesticides.
Chemical Class Compounds Toxicity Rating1 ADI
DDT and analogs Dichlorodiphenyltrichloroethane (DDT) 4 0.005
Methoxychlor 3 0.1
Tetrachlorodiphenylethane (TDE) 3 …
Benzene hexachlorides Benzene hexachloride (BHC; hexachlorocyclohexane) 4 0.008
Lindane 4 0.008
Cyclodienes Aldrin 5 0.0001
Chlordane 4 0.0005
Dieldrin 5 0.0001
Heptachlor 4 0.0001
Toxaphenes Toxaphene (camphechlor) 4 …
1Toxicity rating: Probable human oral lethal dosage for class 3 = 500−5000 mg/kg, class 4 = 50−500 mg/kg, and class 5 = 5−50 mg/kg. (See Gosselin et al, 1984.)
ADI, acceptable daily intake (mg/kg/d).
056-Katzung_Ch056_p1001-1012.indd 1006 9/23/11 4:35:11 PM

CHAPTER 56 Introduction to Toxicology: Occupational & Environmental 1007
cause rapid repetitive firing in most neurons. Calcium ion trans-port is inhibited. These events affect repolarization and enhance the excitability of neurons. The major effect is central nervous system stimulation. With DDT, tremor may be the first manifes-tation, possibly continuing to convulsions, whereas with the other compounds convulsions often appear as the first sign of intoxication. There is no specific treatment for the acute intoxi-cated state, and management is symptomatic.
The potential carcinogenic properties of organochlorine pesti-cides have been extensively studied, and results indicate that chronic administration to laboratory animals over long periods results in enhanced oncogenesis. Endocrine pathway disruption is the postulated mechanism. Extrapolation of the animal observa-tions to humans is controversial. However, several large epidemio-logic studies found no significant association between the risk of breast cancer and serum levels of DDE, the major metabolite of DDT. Similarly, the results of a case-control study conducted to investigate the relation between DDE and DDT breast adipose tissue levels and breast cancer risk did not support a positive asso-ciation. In contrast, recent work supports an association between prepubertal exposure to DDT and brain cancer. In addition, recent studies suggest that the risk of testicular cancer is increased in persons with elevated DDE levels. The risk of non-Hodgkin’s lymphoma (NHL) also seems to be increased in persons with elevated oxychlordane residues. Therefore, increased cancer risk in people exposed to the halogenated hydrocarbon pesticides is of concern.
B. Environmental Toxicology The organochlorine pesticides are considered persistent chemicals. Degradation is quite slow when compared with other pesticides, and bioaccumulation, particularly in aquatic ecosystems, is well documented. Their mobility in soil depends on the composition of the soil; the presence of organic matter favors the adsorption of these chemicals onto the soil particles, whereas adsorption is poor in sandy soils. Once adsorbed, they do not readily desorb. These compounds induce significant abnormalities in the endocrine bal-ance of sensitive animal and bird species, in addition to their adverse impact on humans. Their use is appropriately banned in most areas.
Organophosphorus Pesticides These agents, some of which are listed in Table 56–3 , are used to combat a large variety of pests. They are useful pesticides when in direct contact with insects or when used as plant systemics, where the agent is translocated within the plant and exerts its effects on insects that feed on the plant. These agents are based on com-pounds such as soman, sarin, and tabun, which were developed for use as war gases. Some of the less toxic organophosphorus com-pounds are used in human and veterinary medicine as local or systemic antiparasitics (see Chapters 7 and 53 ). The compounds are absorbed by the skin as well as by the respiratory and gastroin-testinal tracts. Biotransformation is rapid, particularly when com-pared with the rates observed with the chlorinated hydrocarbon
pesticides. Storm and collaborators reviewed current and suggested human inhalation occupational exposure limits for 30 organo-phosphate pesticides (see References).
A. Human Toxicology In mammals as well as insects, the major effect of these agents is inhibition of acetylcholinesterase through phosphorylation of the esteratic site. The signs and symptoms that characterize acute intoxication are due to inhibition of this enzyme and accumula-tion of acetylcholine; some of the agents also possess direct cholin-ergic activity. These effects and their treatment are described in Chapters 7 and 8 of this book. Altered neurologic and cognitive functions, as well as psychological symptoms of variable duration, have been associated with exposure to these pesticides. Furthermore, there is some indication of an association of low arylesterase activ-ity with neurologic symptom complexes in Gulf War veterans.
In addition to—and independently of—inhibition of acetyl-cholinesterase, some of these agents are capable of phosphorylat-ing another enzyme present in neural tissue, the so-called neuropathy target esterase . This results in progressive demyelina-tion of the longest nerves. Associated with paralysis and axonal degeneration, this lesion is sometimes called organophosphorus ester-induced delayed polyneuropathy (OPIDP). Delayed central and autonomic neuropathy may occur in some poisoned patients. Hens are particularly sensitive to these properties and have proved very useful for studying the pathogenesis of the lesion and for identifying potentially neurotoxic organophosphorus derivatives. In humans, neurotoxicity has been observed with triorthocresyl phosphate (TOCP), a noninsecticidal organophosphorus com-pound. It is also thought to occur with the pesticides dichlorvos, trichlorfon, leptophos, methamidophos, mipafox, trichloronat, and others. The polyneuropathy usually begins with burning and tingling sensations, particularly in the feet, with motor weakness a few days later. Sensory and motor difficulties may extend to the
TABLE 56–3 Organophosphorus pesticides.
Compound Toxicity Rating1 ADI
Azinphos-methyl 5 0.005
Chlorfenvinphos … 0.002
Diazinon 4 0.002
Dichlorvos … 0.004
Dimethoate 4 0.01
Fenitrothion … 0.005
Malathion 4 0.02
Parathion 6 0.005
Parathion-methyl 5 0.02
Trichlorfon 4 0.01
1Toxicity rating: Probable human oral lethal dosage for class 4 = 50-500 mg/kg, class 5 = 5-50 mg/kg, and class 6 = ≤ 5 mg/kg. (See Gosselin et al, 1984.)
ADI, acceptable daily intake (mg/kg/d).
056-Katzung_Ch056_p1001-1012.indd 1007 9/23/11 4:35:11 PM

1008 SECTION IX Toxicology
legs and hands. Gait is affected, and ataxia may be present. Central nervous system and autonomic changes may develop even later. There is no specific treatment for this form of delayed neurotoxic-ity. The long-term prognosis of neuropathy target esterase inhibi-tion is highly variable. Reports of this type of neuropathy (and other toxicities) in pesticide manufacturing workers and in agri-cultural pesticide applicators have been published.
B. Environmental Toxicology Organophosphorus pesticides are not considered to be persistent pesticides. They are relatively unstable and break down in the environment as a result of hydrolysis and photolysis. As a class they are considered to have a small impact on the environment in spite of their acute effects on organisms.
Carbamate Pesticides These compounds ( Table 56–4 ) inhibit acetylcholinesterase by carbamoylation of the esteratic site. Thus, they possess the toxic properties associated with inhibition of this enzyme as described for the organophosphorus pesticides. The effects and treatment are described in Chapters 7 and 8 . The clinical effects due to car-bamates are of shorter duration than those observed with organo-phosphorus compounds. The range between the doses that cause minor intoxication and those that result in lethality is larger with carbamates than with the organophosphorus agents. Spontaneous reactivation of cholinesterase is more rapid after inhibition by the carbamates. Although the clinical approach to carbamate poison-ing is similar to that for organophosphates, the use of pralidoxime is not recommended.
The carbamates are considered to be nonpersistent pesticides. They exert only a small impact on the environment.
Botanical Pesticides Pesticides derived from natural sources include nicotine, rote-none, and pyrethrum. Nicotine is obtained from the dried leaves of Nicotiana tabacum and N rustica . It is rapidly absorbed from mucosal surfaces; the free alkaloid, but not the salt, is readily absorbed from the skin. Nicotine reacts with the acetylcholine receptor of the postsynaptic membrane (sympathetic and para-sympathetic ganglia, neuromuscular junction), resulting in depo-larization of the membrane. Toxic doses cause stimulation rapidly followed by blockade of transmission. These actions are described in Chapter 7 . Treatment is directed toward maintenance of vital signs and suppression of convulsions.
Rotenone ( Figure 56–1 ) is obtained from Derris elliptica , D mallaccensis , Lonchocarpus utilis, and L urucu. The oral ingestion of rotenone produces gastrointestinal irritation. Conjunctivitis, dermatitis, pharyngitis, and rhinitis can also occur. Treatment is symptomatic.
Pyrethrum consists of six known insecticidal esters: pyrethrin I ( Figure 56–1 ), pyrethrin II, cinerin I, cinerin II, jasmolin I, and jasmolin II. Synthetic pyrethroids account for an increasing percent-age of worldwide pesticide usage. Pyrethrum may be absorbed after inhalation or ingestion; absorption from the skin is not significant. The esters are extensively biotransformed. Pyrethrum pesticides are not highly toxic to mammals. When absorbed in sufficient quanti-ties, the major site of toxic action is the central nervous system; excitation, convulsions, and tetanic paralysis can occur. Voltage-gated sodium, calcium, and chloride channels are considered tar-gets, as well as peripheral-type benzodiazepine receptors. Treatment of exposure is usually directed at management of symptoms. Anticonvulsants are not consistently effective. The chloride channel agonist, ivermectin, is of use, as are pentobarbital and mephenesin. The pyrethroids are highly irritating to the eyes, skin, and respira-tory tree. They may cause irritant asthma and, potentially, reactive airways dysfunction syndrome (RADS) and even anaphylaxis. The most common injuries reported in humans result from their aller-genic and irritant effects on the airways and skin. Cutaneous par-esthesias have been observed in workers spraying synthetic pyrethroids. The use of persistent synthetic pyrethroids for aircraft disinfection to comply with international rules regarding prevention of transfer of insect vectors has resulted in respiratory and skin problems, as well as some neurologic complaints in flight attendants and other aircraft workers. Severe occupational exposures to syn-thetic pyrethroids in China resulted in marked effects on the central nervous system, including convulsions.
HERBICIDES
Chlorophenoxy Herbicides 2,4-Dichlorophenoxyacetic acid (2,4-D), 2,4,5-trichlorophe-noxyacetic acid (2,4,5-T), and their salts and esters are com-pounds of interest as herbicides used for the destruction of weeds ( Figure 56–1 ). They have been assigned toxicity ratings of 4 or 3, respectively, which place the probable human lethal dosages at 50–500 or 500–5000 mg/kg, respectively.
TABLE 56–4 Carbamate pesticides.
Compound Toxicity Rating1 ADI
Aldicarb 6 0.005
Aminocarb 5 …
Carbaryl 4 0.01
Carbofuran 5 0.01
Dimetan 4 …
Dimetilan 4 …
Isolan 5 …
Methomyl 5 …
Propoxur 4 0.02
Pyramat 4 …
Pyrolan 5 …
Zectran 5 …
1Toxicity rating: Probable human oral lethal dosage for class 4 = 50−500 mg/kg, class 5 = 5−50 mg/kg, and class 6 = ≤ 5 mg/kg. (See Gosselin et al, 1984.)
ADI, acceptable daily intake (mg/kg/d).
056-Katzung_Ch056_p1001-1012.indd 1008 9/23/11 4:35:11 PM
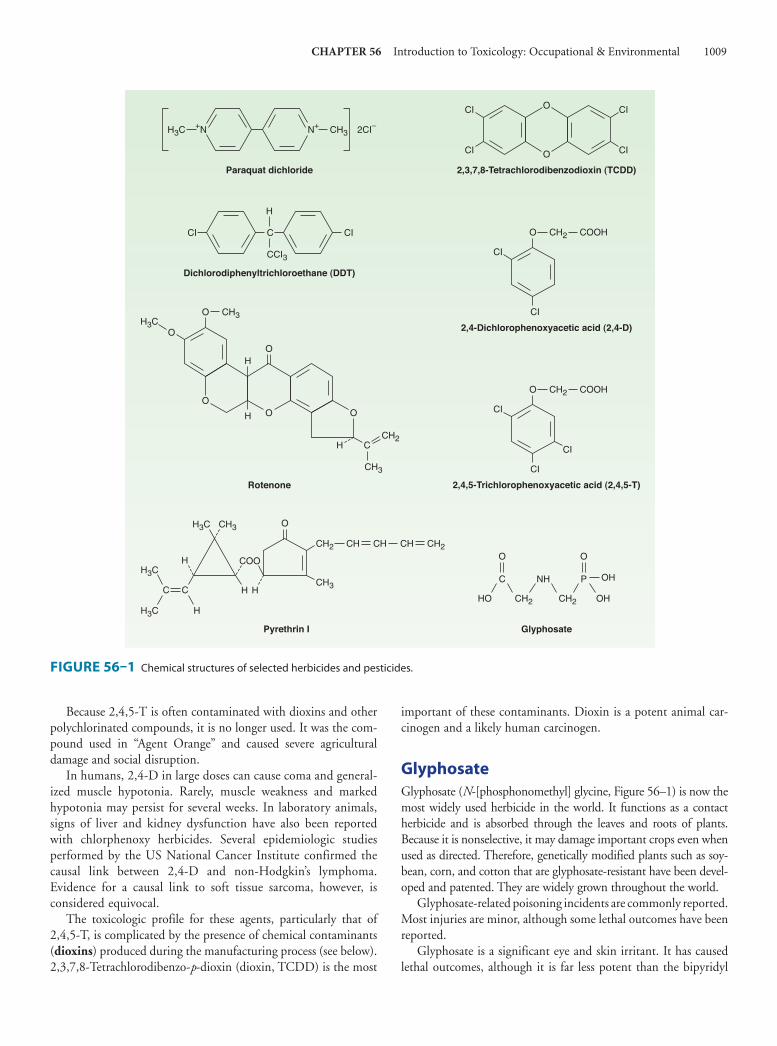
CHAPTER 56 Introduction to Toxicology: Occupational & Environmental 1009
Because 2,4,5-T is often contaminated with dioxins and other polychlorinated compounds, it is no longer used. It was the com-pound used in “Agent Orange” and caused severe agricultural damage and social disruption.
In humans, 2,4-D in large doses can cause coma and general-ized muscle hypotonia. Rarely, muscle weakness and marked hypotonia may persist for several weeks. In laboratory animals, signs of liver and kidney dysfunction have also been reported with chlorphenoxy herbicides. Several epidemiologic studies performed by the US National Cancer Institute confirmed the causal link between 2,4-D and non-Hodgkin’s lymphoma. Evidence for a causal link to soft tissue sarcoma, however, is considered equivocal.
The toxicologic profile for these agents, particularly that of 2,4,5-T, is complicated by the presence of chemical contaminants ( dioxins ) produced during the manufacturing process (see below). 2,3,7,8-Tetrachlorodibenzo- p -dioxin (dioxin, TCDD) is the most
important of these contaminants. Dioxin is a potent animal car-cinogen and a likely human carcinogen.
Glyphosate Glyphosate (N-[phosphonomethyl] glycine, Figure 56–1 ) is now the most widely used herbicide in the world. It functions as a contact herbicide and is absorbed through the leaves and roots of plants. Because it is nonselective, it may damage important crops even when used as directed. Therefore, genetically modified plants such as soy-bean, corn, and cotton that are glyphosate-resistant have been devel-oped and patented. They are widely grown throughout the world.
Glyphosate-related poisoning incidents are commonly reported. Most injuries are minor, although some lethal outcomes have been reported.
Glyphosate is a significant eye and skin irritant. It has caused lethal outcomes, although it is far less potent than the bipyridyl
FIGURE 56–1 Chemical structures of selected herbicides and pesticides.
CH3N+ 2CI–
Paraquat dichloride
H3C +N
CI
Dichlorodiphenyltrichloroethane (DDT)
CI C
CCI3
H
2,3,7,8-Tetrachlorodibenzodioxin (TCDD)
CI
CI
O
O
CI
CI
2,4-Dichlorophenoxyacetic acid (2,4-D)
CI
O CH2 COOH
CI
2,4,5-Trichlorophenoxyacetic acid (2,4,5-T)
CI
O CH2 COOH
CI
CI
O CH3
OH3C
OO O
HO
H
C
CH3
CH2
Rotenone
H
H
CH2 CH CH CH CH2
CH3
COO
H
CH3H3C
H
CC
H
H3C
H3C
Pyrethrin I
O
Glyphosate
HO OHCH2 CH2
OH
O
C PNH
O
056-Katzung_Ch056_p1001-1012.indd 1009 9/23/11 4:35:12 PM

1010 SECTION IX Toxicology
herbicides. Although the pure chemical seems to have little persis-tence and lower toxicity than other herbicides, the commercial formulations of glyphosate often contain surfactants and other active compounds that complicate the toxicity of the product. No specific treatment is available for glyphosate toxicity.
Bipyridyl Herbicides Paraquat is the most important agent of this class ( Figure 56–1 ). Its mechanism of action is said to be similar in plants and animals and involves single-electron reduction of the herbicide to free radical species. It has been given a toxicity rating of 4, which places the probable human lethal dosage at 50–500 mg/kg. Lethal human intoxications (accidental or suicidal) have been reported. Paraquat accumulates slowly in the lung by an active process and causes lung edema, alveolitis, and progressive fibrosis. It probably inhibits superoxide dismutase, resulting in intracellular free radical oxygen toxicity.
In humans, the first signs and symptoms after oral exposure are hematemesis and bloody stools. Within a few days, however, delayed toxicity occurs, with respiratory distress and the develop-ment of congestive hemorrhagic pulmonary edema accompanied by widespread cellular proliferation. Hepatic, renal, or myocardial involvement may also be evident. The interval between ingestion and death may be several weeks. Because of the delayed pulmo-nary toxicity, prompt removal of paraquat from the digestive tract is important. Gastric lavage, the use of cathartics, and the use of adsorbents to prevent further absorption have all been advocated; after absorption, treatment is successful in fewer than 50% of cases. Oxygen should be used cautiously to combat dyspnea or cyanosis, because it may aggravate the pulmonary lesions. Patients require prolonged observation, because the proliferative phase begins 1–2 weeks after ingestion. Management of severe paraquat poisoning is complex and largely symptomatic. Many approaches have been used, including immunosuppressive therapy to slow or stop the progressive pulmonary fibrosis. None of the currently proposed methods of treatment is universally successful.
ENVIRONMENTAL POLLUTANTS
Polychlorinated Biphenyls The polychlorinated biphenyls (PCBs, coplanar biphenyls) have been used in a large variety of applications as dielectric and heat trans-fer fluids, lubricating oils, plasticizers, wax extenders, and flame retar-dants. Unfortunately, PCBs persist in the environment. Their industrial use and manufacture in the USA were terminated by 1977. The products used commercially were actually mixtures of PCB iso-mers and homologs containing 12–68% chlorine. These chemicals are highly stable and highly lipophilic, poorly metabolized, and very resistant to environmental degradation; they bioaccumulate in food chains. Food is the major source of PCB residues in humans.
A serious exposure to PCBs—lasting several months—occurred in Japan in 1968 as a result of cooking oil contamination with PCB-containing transfer medium (Yusho disease). Possible effects on the fetus and on the development of the offspring of poisoned
women were reported. It is now known that the contaminated cooking oil contained not only PCBs but also polychlorinated dibenzofurans (PCDFs) and polychlorinated quaterphenyls (PCQs). Consequently, the effects that were initially attributed to the presence of PCBs are now thought to have been caused by a mixture of contaminants. Workers occupationally exposed to PCBs have exhibited the following clinical signs: dermatologic problems (chloracne, folliculitis, erythema, dryness, rash, hyper-keratosis, hyperpigmentation), some hepatic involvement, and elevated plasma triglycerides.
The effects of PCBs alone on reproduction and development, as well as their carcinogenic effects, have yet to be established in humans—whether workers or the general population—even though some subjects have been exposed to very high levels of PCBs. Repeated epidemiologic studies have found some increases in various cancers including melanoma, breast, pancreatic, and thyroid cancers, but the small number of cases and uncertain exposure status have left the carcinogenicity question unclear. In 1977, the IARC recommended that PCBs be regarded as likely carcinogenic to man, although the evidence for this classification was lacking. Some adverse behavioral effects in infants have been reported. An association between prenatal exposure to PCBs and deficits in childhood intellectual function was described for chil-dren born to mothers who had eaten large quantities of contami-nated fish. The polychlorinated dibenzo - p -dioxins (PCDDs), or dioxins , have been mentioned as a group of congeners of which the most important is 2,3,7,8-tetrachlorodibenzo - p -dioxin (TCDD). In addition, there is a larger group of dioxin-like com-pounds, including certain polychlorinated dibenzofurans (PCDFs) and coplanar biphenyls . While PCBs were used com-mercially, PCDDs and PCDFs are unwanted byproducts that appear in the environment and in manufactured products as con-taminants because of improperly controlled combustion processes. PCDD and PCDF contamination of the global environment is considered to represent a contemporary problem produced by human activities. Like PCBs, these chemicals are very stable and highly lipophilic. They are poorly metabolized and very resistant to environmental degradation.
In laboratory animals, TCDD administered in suitable doses has produced a wide variety of toxic effects, including a wasting syndrome (severe weight loss accompanied by reduction of muscle mass and adipose tissue), thymic atrophy, epidermal changes, hepatotoxicity, immunotoxicity, effects on reproduction and development, teratogenicity, and carcinogenicity. The effects observed in workers involved in the manufacture of 2,4,5-T (and therefore presumably exposed to TCDD) consisted of contact dermatitis and chloracne. In severely TCDD-intoxicated patients, discrete chloracne may be the only manifestation.
The presence of TCDD in 2,4,5-T is believed to be largely responsible for other human toxicities associated with the herbi-cide. There is epidemiologic evidence indicating an association between occupational exposure to the phenoxy herbicides and an excess incidence of non-Hodgkin’s lymphoma. The TCDD con-taminant in these herbicides seems to play a role in a number of cancers such as soft tissue sarcomas, lung cancer, Hodgkin’s lym-phomas, and others.
056-Katzung_Ch056_p1001-1012.indd 1010 9/23/11 4:35:12 PM

CHAPTER 56 Introduction to Toxicology: Occupational & Environmental 1011
Endocrine Disruptors The potential hazardous effects of some chemicals in the envi-ronment are receiving considerable attention because of their estrogen-like or antiandrogenic properties. Compounds that affect thyroid function are also of concern. Since 1998, the pro-cess of prioritization, screening, and testing of chemicals for such actions has been undergoing worldwide development. These chemicals mimic, enhance, or inhibit a hormonal action. They include a number of plant constituents (phytoestrogens) and some mycoestrogens as well as industrial chemicals, particularly persistent organochlorine agents such as DDT and PCBs. Some brominated flame retardants are now being investigated as pos-sible endocrine disrupters. Concerns exist because of their increasing contamination of the environment, the appearance of bioaccumulation, and their potential for toxicity. In vitro assays alone are unreliable for regulatory purposes, and animal studies are considered indispensable. Modified endocrine responses in some reptiles and marine invertebrates have been observed. In humans, however, a causal relation between exposure to a spe-cific environmental agent and an adverse health effect due to endocrine modulation has not been established. Epidemiologic studies of populations exposed to higher concentrations of endo-crine disrupting environmental chemicals are underway. There are indications that breast and other reproductive cancers are increased in these patients. Prudence dictates that exposure to environmental chemicals that disrupt endocrine function should be reduced.
Asbestos Asbestos in many of its forms has been widely used in industry for over 100 years. All forms of asbestos that have been used in industry have been shown to cause progressive lung disease that is characterized by a fibrotic process. Higher levels of exposure produce the process called asbestosis. Lung damage develops even at low concentrations of shorter fibers, whereas higher con-centrations of longer fibers are required to cause lung damage. Every form of asbestos, including chrysotile asbestos, causes an increase in lung cancer. Lung cancer occurs in people exposed at fiber concentrations well below concentrations that produce asbestosis. Cigarette smoking and exposure to radon daughters increase the incidence of asbestos-caused lung cancer in a syner-gistic fashion.
All forms of asbestos also cause mesothelioma of the pleura or peritoneum at very low doses. Other cancers including colon can-cer, laryngeal cancer, stomach cancer, and perhaps even lymphoma are increased in asbestos-exposed patients. The mechanism for asbestos-caused cancer is not yet delineated. Arguments that chrysotile asbestos does not cause mesothelioma are contradicted by many epidemiologic studies of worker populations. Recognition that all forms of asbestos are dangerous and carcinogenic has led many countries to ban all uses of asbestos. Countries such as Canada, Zimbabwe, and others that still produce asbestos argue that asbestos can be used safely with careful workplace environ-mental controls. However, studies of industrial practice make the “safe use” of asbestos highly improbable.
METALS
Occupational and environmental poisoning with metals, metal-loids, and metal compounds is a major health problem. Exposure in the workplace is found in many industries, and exposure in the home and elsewhere in the nonoccupational environment is wide-spread. The classic metal poisons (arsenic, lead, and mercury) continue to be widely used. (Treatment of their toxicities is dis-cussed in Chapter 57 .) Occupational exposure and poisoning due to beryllium, cadmium, manganese, and uranium are relatively new occupational problems, which present new and previously unaddressed problems.
Beryllium Beryllium (Be) is a light alkaline metal that confers special proper-ties on the alloys and ceramics in which it is incorporated. One attractive property of beryllium is its nonsparking quality, which makes it useful in such diverse applications as the manufacture of dental appliances and of nuclear weapons. Beryllium-copper alloys find use as components of computers, in the encasement of the first stage of nuclear weapons, in devices that require hardening such as missile ceramic nose cones, and in the space shuttle heat shield tiles. Because of the use of beryllium in dental appliances, dentists and dental appliance makers are often exposed to beryl-lium dust in toxic concentrations.
Beryllium is highly toxic by inhalation and is classified by IARC as a class 1, known human carcinogen. Inhalation of beryllium particles produces progressive pulmonary fibrosis and may lead to cancer. Skin disease also develops in workers over-exposed to beryllium. The pulmonary disease is called chronic beryllium disease (CBD) and is a chronic granulomatous pul-monary fibrosis. In the 5–15% of the population that is sensi-tive to beryllium, chronic beryllium disease is the result of activation of an autoimmune attack on the skin and lungs. The disease is progressive and may lead to severe disability and death. Although some treatment approaches to the manage-ment of chronic beryllium disease show promise, the prognosis is poor in most cases.
The current permissible exposure levels for beryllium of 0.01 mcg/m 3 averaged over a 30-day period or 2 mcg/m 3 over an 8-hour period are insufficiently protective to prevent chronic beryllium disease. Both NIOSH and the ACGIH have recom-mended that the PEL and TLV be reduced to 0.05 mcg/m 3 . These recommendations have not yet been implemented.
Environmental beryllium exposure is not generally thought to be a hazard to human health except in the vicinity of industrial sites where air, water and soil pollution have occurred.
Cadmium Cadmium (Cd) is a transition metal widely used in industry. Workers are exposed to cadmium in the manufacture of nickel cadmium batteries, pigments, low-melting-point eutectic materi-als; in solder; in television phosphors; and in plating operations. It is also used extensively in semiconductors and in plastics as a sta-bilizer. Cadmium smelting is often done from residual dust from
056-Katzung_Ch056_p1001-1012.indd 1011 9/23/11 4:35:12 PM

1012 SECTION IX Toxicology
lead smelting operations, and cadmium smelter workers often face both lead and cadmium toxicity.
Cadmium is toxic by inhalation and by ingestion. When met-als that have been plated with cadmium or welded with cadmium-containing materials are vaporized by the heat of torches or cutting implements, the fine dust and fumes released produce an acute respiratory disorder called cadmium fume fever . This disor-der, common in welders, is usually characterized by shaking chills, cough, fever, and malaise. Although it may produce pneumonia, it is usually transient. However, chronic exposure to cadmium dust produces a far more serious progressive pulmonary fibrosis. Cadmium also causes severe kidney damage, including renal fail-ure if exposure continues. Cadmium is a human carcinogen and is listed as a group 1, known human carcinogen by the IARC.
The current OSHA PEL for cadmium is 5 mcg/m 3 . This PEL, considered by OSHA to be the lowest feasible limit for the dust, is insufficiently protective of worker health.
REFERENCES Birnbaum LS, Staska DF: Brominated flame retardants: Cause for concern?
Environ Health Perspect 2004;112:9. Buckley A et al: Hyperbaric oxygen for carbon monoxide poisoning: A systematic
review and critical analysis of the evidence. Toxicol Rev 2005;24:75. Casals-Casas C, Desvergne B: Endocrine disruptors: from endocrine to metabolic
disruption. Annu Rev Physiol 2011;73:135. Crisp TM et al: Environmental endocrine disruption: An effects assessment and
analysis. Environ Health Perspect 1998;106(Suppl 1):11. Degen GH, Bolt HM: Endocrine disruptors: Update on xenoestrogens. Int Arch
Occup Environ Health 2000;73:433. Ecobichon DJ: Our changing perspectives on benefits and risks of pesticides: A
historical overview. Neurotoxicology 2000;21:211. Geusau A et al: Severe 2,3,7,8-tetrachlorodibenzo- p -dioxin (TCDD) intoxication:
Clinical and laboratory effects. Environ Health Perspect 2001;109:865. Gosselin RE, Smith RP, Hodge HC: Clinical Toxicology of Commercial Products , 5th
ed. Williams & Wilkins, 1984.
Haley RW et al: Association of low PON1 type Q (type A) arylesterase activity with neurologic symptom complexes in Gulf War veterans. Toxicol Appl Pharmacol 1999;157:227.
Hamm JI, Chen CY, Birnbaum LS: A mixture of dioxin, furans, and non-ortho PCBs based upon consensus toxic equivalency factors produces dioxin-like reproductive effects. Toxicol Sci 2003;74:182.
Jacobson JL, Jacobson SW: Association of prenatal exposure to an environmental contaminant with intellectual function in childhood. J Toxicol Clin Toxicol 2002;40:467.
Kao JW, Nanagas KA: Carbon monoxide poisoning. Emerg Med Clin North Am 2004;22:985.
Klaassen CD (editor): Casarett and Doull’s Toxicology , 7th ed. McGraw-Hill, 2007.
Lavin AL, Jacobson OF, DeSesso JM: An assessment of the carcinogenic potential of trichloroethylene in humans. Human Ecol Risk Assess 2000;6:575.
Lévesque B et al: Cancer risk associated with household exposure to chloroform. J Toxicol Environ Health A 2002;65:489.
Lotti M, Moretto A: Organophosphate-induced delayed polyneuropathy. Toxicol Rev 2005;24:37.
MacMahon B: Pesticide residues and breast cancer? J Natl Cancer Inst 1994;86:572.
Mundt KA, Birk T, Burch MT: Critical review of the epidemiological literature on occupational exposure to perchloroethylene and cancer. Int Arch Occup Environ Health 2003;76:473.
Olson KR et al (editors): Poisoning & Drug Overdose , 5th ed. McGraw-Hill, 2006.
Raub JA et al: Carbon monoxide poisoning—a public health perspective. Toxicology 2000;145:1.
Ray DE, Fry JR: A reassessment of the neurotoxicity of pyrethroid insecticides. Pharmacol Ther 2006;111:174.
Safe SH: Endocrine disruptors and human health—is there a problem? An update. Environ Health Perspect 2000;108:487.
Soderlund DM et al: Mechanisms of pyrethroid neurotoxicity: Implications for cumulative risk assessment. Toxicology 2002;171:3.
St Hilaire S et al: Estrogen receptor positive breast cancers and their association with environmental factors. Int J Health Geogr 2011;10:10.
Storm JE, Rozman KK, Doull J: Occupational exposure limits for 30 organophos-phate pesticides based on inhibition of red blood cell acetylcholinesterase. Toxicology 2000;150:1.
056-Katzung_Ch056_p1001-1012.indd 1012 9/23/11 4:35:12 PM

10131013
C H A P T E R
lines may increase the lead concentration of tap water. Environmental lead exposure, ubiquitous by virtue of the anthropo-genic distribution of lead to air, water, and food, has declined con-siderably in the last three decades as a result of the elimination of lead as an additive in gasoline, as well as diminished contact with lead-based paint and other lead-containing consumer products, such as lead solder in canned food. Although these public health measures, together with improved workplace conditions, have decreased the incidence of serious overt lead poisoning, there remains considerable concern over the effects of low-level lead exposure. Extensive evi-dence indicates that lead may have subtle subclinical adverse effects on neurocognitive function and on blood pressure at low blood lead concentrations formerly not recognized as harmful. Lead serves no useful purpose in the human body. In key target organs such as the developing central nervous system, no level of lead exposure has been shown to be without deleterious effects.
Pharmacokinetics Inorganic lead is slowly but consistently absorbed via the respiratory and gastrointestinal tracts. Inorganic lead is poorly absorbed through the skin. Absorption of lead dust via the respiratory tract is the most common cause of industrial poisoning. The intestinal tract is the primary route of entry in nonindustrial exposure ( Table 57–1 ). Absorption via the gastrointestinal tract varies with the nature of the lead compound, but in general, adults absorb about 10–15% of the ingested amount, whereas young children absorb up to 50%. Low dietary calcium, iron deficiency, and ingestion on an empty stomach all have been associated with increased lead absorption.
57
C A S E S T U D Y
A 48-year-old painter is referred for evaluation of recent onset of severe abdominal pains, headaches, and myalgias. For the last week, he has been removing old paint from an iron bridge using grinding tools and a blow torch. His
employer states that all the bridge workers are provided with the equivalent of “haz-mat” (hazardous materials) suits. What tests should be carried out? Assuming positive test results, what therapy would be appropriate?
Some metals such as iron are essential for life, whereas others such as lead are present in all organisms but serve no useful biologic purpose. Some of the oldest diseases of humans can be traced to heavy metal poisoning associated with metal mining, refining, and use. Even with the present recognition of the hazards of heavy metals, the incidence of intoxication remains significant, and the need for preventive strategies and effective therapy remains high. Toxic heavy metals interfere with the function of essential cations, cause enzyme inhibition, generate oxidative stress, and alter gene expression. As a result, multisystem signs and symptoms are a hallmark of heavy metal intoxication.
When intoxication occurs, chelator molecules (from chela “claw”), or their in vivo biotransformation products, may be used to bind the metal and facilitate its excretion from the body. Chelator drugs are discussed in the second part of this chapter.
■ TOXICOLOGY OF HEAVY METALS
LEAD
Lead poisoning is one of the oldest occupational and environmen-tal diseases in the world. Despite its recognized hazards, lead continues to have widespread commercial application, including production of storage batteries (nearly 90% of US consumption), ammunition, metal alloys, solder, glass, plastics, pigments, and ceramics. Corrosion of lead plumbing in older buildings or supply
Heavy Metal Intoxication & Chelators Michael J. Kosnett, MD, MPH
057-Katzung_Ch057_p1013-1026.indd 1013 9/21/11 12:13:12 PM

1014 SECTION IX Toxicology
Once absorbed from the respiratory or gastrointestinal tract, lead enters the bloodstream, where approximately 99% is bound to erythrocytes and 1% is present in the plasma. Lead is subse-quently distributed to soft tissues such as the bone marrow, brain, kidney, liver, muscle, and gonads; then to the subperiosteal surface of bone; and later to bone matrix. Lead also crosses the placenta and poses a potential hazard to the fetus. The kinetics of lead clearance from the body follows a multicompartment model, composed predominantly of the blood and soft tissues, with a half-life of 1–2 months; and the skeleton, with a half-life of years to decades. Approximately 70% of the lead that is eliminated appears in the urine, with lesser amounts excreted through the bile, skin, hair, nails, sweat, and breast milk. The fraction not undergoing prompt excretion, approximately half of the absorbed lead, may be incorporated into the skeleton, the repository of more than 90% of the body lead burden in most adults. In patients with high bone lead burdens, slow release from the skel-eton may elevate blood lead concentrations for years after expo-sure ceases, and pathologic high bone turnover states such as hyperthyroidism or prolonged immobilization may result in frank lead intoxication. Migration of retained lead bullet fragments into
a joint space or adjacent to bone has been associated with the development of lead poisoning signs and symptoms years or decades after an initial gunshot injury.
Pharmacodynamics Lead exerts multisystemic toxic effects that are mediated by mul-tiple modes of action, including inhibition of enzymatic function; interference with the action of essential cations, particularly cal-cium, iron, and zinc; generation of oxidative stress; changes in gene expression; alterations in cell signaling; and disruption of the integrity of membranes in cells and organelles.
A. Nervous System The developing central nervous system of the fetus and young child is the most sensitive target organ for lead’s toxic effect. Epidemiologic studies suggest that blood lead concentrations even less than 5 mcg/dL may result in subclinical deficits in neurocog-nitive function in lead-exposed young children, with no demon-strable threshold for a “no effect” level. The dose response between
TABLE 57–1 Toxicology of selected arsenic, lead, and mercury compounds.
Form Entering Body
Major Route of Absorption Distribution
Major Clinical Effects
Key Aspects of Mechanism
Metabolism and Elimination
Arsenic Inorganic arsenic salts
Gastrointestinal, respiratory (all mucosal surfaces)
Predominantly soft tissues (highest in liver, kidney). Avidly bound in skin, hair, nails
Cardiovascular: shock, arrhythmias. CNS: encephalopathy, peripheral neuropathy. Gastroenteritis; pan-cytopenia; cancer (many sites)
Inhibits enzymes; interferes with oxidative phosphorylation; alters cell signaling, gene expression
Methylation. Renal (major); sweat and feces (minor)
Lead Inorganic lead oxides and salts
Gastrointestinal, respiratory
Soft tissues; redistributed to skeleton (> 90% of adult body burden)
CNS deficits; peripheral neuropathy; ane-mia; nephropathy; hypertension; reproductive toxicity
Inhibits enzymes; interferes with essential cations; alters membrane structure
Renal (major); feces and breast milk (minor)
Organic (tetraethyl lead)
Skin, gastrointesti-nal, respiratory
Soft tissues, especially liver, CNS
Encephalopathy Hepatic dealkylation (fast) → trialkyme-tabolites (slow) → dissociation to lead
Urine and feces (major); sweat (minor)
Mercury Elemental mercury Respiratory tract Soft tissues, especially kidney, CNS
CNS: tremor, behavioral (erethism); gingivo- stomatitis; peripheral neuropa-thy; acrodynia; pneumonitis (high-dose)
Inhibits enzymes; alters membranes
Elemental Hg converted to Hg2+. Urine (major); feces (minor)
Inorganic: Hg+ (less toxic); Hg2+ (more toxic)
Gastrointestinal, skin (minor)
Soft tissues, especially kidney
Acute tubular necrosis; gastroen-teritis; CNS effects (rare)
Inhibits enzymes; alters membranes
Urine
Organic: alkyl, aryl Gastrointestinal, skin, respiratory (minor)
Soft tissues CNS effects, birth defects
Inhibits enzymes; alters microtubules, neuronal structure
Deacylation. Fecal (alkyl, major); urine (Hg2+ after deacyla-tion, minor)
057-Katzung_Ch057_p1013-1026.indd 1014 9/21/11 12:13:12 PM

CHAPTER 57 Heavy Metal Intoxication & Chelators 1015
low blood lead concentrations and cognitive function in young children is nonlinear, such that the decrement in intelligence associ-ated with an increase in blood lead from less than 1 to 10 mcg/dL (6.2 IQ points) exceeds that associated with a change from 10 to 30 mcg/dL (3.0 IQ points).
Adults are less sensitive to the central nervous system effects of lead, but long-term exposure to blood lead concentrations in the range of 10–30 mcg/dL may be associated with subtle, subclinical effects on neurocognitive function. At blood lead concentrations higher than 30 mcg/dL, behavioral and neurocognitive signs or symptoms may gradually emerge, including irritability, fatigue, decreased libido, anorexia, sleep disturbance, impaired visual-motor coordination, and slowed reaction time. Headache, arthral-gias, and myalgias are also common complaints. Tremor occurs but is less common. Lead encephalopathy, usually occurring at blood lead concentrations higher than 100 mcg/dL, is typically accompanied by increased intracranial pressure and may cause ataxia, stupor, coma, convulsions, and death. Recent epidemio-logical studies suggest that lead may accentuate an age-related decline in cognitive function in older adults. In experimental ani-mals, developmental lead exposure has been associated with increased expression of beta-amyloid, oxidative DNA damage, and Alzheimer’s-type pathology in the aging brain. There is wide inter-individual variation in the magnitude of lead exposure required to cause overt lead-related signs and symptoms.
Overt peripheral neuropathy may appear after chronic high-dose lead exposure, usually following months to years of blood lead concentrations higher than 100 mcg/dL. Predominantly motor in character, the neuropathy may present clinically with painless weakness of the extensors, particularly in the upper extremity, resulting in classic wrist-drop. Preclinical signs of lead-induced peripheral nerve dysfunction may be detectable by elec-trodiagnostic testing.
B. Blood Lead can induce an anemia that may be either normocytic or microcytic and hypochromic. Lead interferes with heme synthesis by blocking the incorporation of iron into protoporphyrin IX and by inhibiting the function of enzymes in the heme synthesis path-way, including aminolevulinic acid dehydratase and ferrochelatase. Within 2–8 weeks after an elevation in blood lead concentration (generally to 30–50 mcg/dL or greater), increases in heme precur-sors, notably free erythrocyte protoporphyrin or its zinc chelate, zinc protoporphyrin, may be detectable in whole blood. Lead also contributes to anemia by increasing erythrocyte membrane fragil-ity and decreasing red cell survival time. Frank hemolysis may occur with high exposure. Basophilic stippling on the peripheral blood smear, thought to be a consequence of lead inhibition of the enzyme 3’,5’-pyrimidine nucleotidase, is sometimes a suggestive—albeit insensitive and nonspecific—diagnostic clue to the presence of lead intoxication.
C. Kidneys Chronic high-dose lead exposure, usually associated with months to years of blood lead concentrations greater than 80 mcg/dL, may
result in renal interstitial fibrosis and nephrosclerosis. Lead neph-ropathy may have a latency period of years. Lead may alter uric acid excretion by the kidney, resulting in recurrent bouts of gouty arthritis (“saturnine gout”). Acute high-dose lead exposure some-times produces transient azotemia, possibly as a consequence of intrarenal vasoconstriction. Studies conducted in general popula-tion samples have documented an association between blood lead concentration and measures of renal function, including serum creatinine and creatinine clearance. The presence of other risk fac-tors for renal insufficiency, including hypertension and diabetes, may increase susceptibility to lead-induced renal dysfunction.
D. Reproductive Organs High-dose lead exposure is a recognized risk factor for stillbirth or spontaneous abortion. Epidemiologic studies of the impact of low-level lead exposure on reproductive outcome such as low birth weight, preterm delivery, or spontaneous abortion have yielded mixed results. However, a well-designed nested case-control study detected an odds ratio for spontaneous abortion of 1.8 (95% CI 1.1–3.1) for every 5 mcg/dL increase in maternal blood lead across an approximate range of 5–20 mcg/dL. Recent studies have linked prenatal exposure to low levels of lead (eg, maternal blood lead concentrations of 5–15 mcg/dL) to decrements in physical and cognitive development assessed during the neonatal period and early childhood. In males, blood lead concentrations higher than 40 mcg/dL have been associated with diminished or aberrant sperm production.
E. Gastrointestinal Tract Moderate lead poisoning may cause loss of appetite, constipation, and, less commonly, diarrhea. At high dosage, intermittent bouts of severe colicky abdominal pain (“lead colic”) may occur. The mecha-nism of lead colic is unclear but is believed to involve spasmodic contraction of the smooth muscles of the intestinal wall, mediated by alteration in synaptic transmission at the smooth muscle-neuro-muscular junction. In heavily exposed individuals with poor dental hygiene, the reaction of circulating lead with sulfur ions released by microbial action may produce dark deposits of lead sulfide at the gingival margin (“gingival lead lines”). Although frequently men-tioned as a diagnostic clue in the past, in recent times this has been a relatively rare sign of lead exposure.
F. Cardiovascular System Epidemiologic, experimental, and in vitro mechanistic data indi-cate that lead exposure elevates blood pressure in susceptible indi-viduals. In populations with environmental or occupational lead exposure, blood lead concentration is linked with increases in systolic and diastolic blood pressure. Studies of middle-aged and elderly men and women have identified relatively low levels of lead exposure sustained by the general population to be an indepen-dent risk factor for hypertension. In addition, epidemiologic stud-ies suggest that low to moderate levels of lead exposure are risk factors for increased cardiovascular mortality. Lead can also elevate blood pressure in experimental animals. The pressor effect of lead
057-Katzung_Ch057_p1013-1026.indd 1015 9/21/11 12:13:12 PM

1016 SECTION IX Toxicology
may be mediated by an interaction with calcium mediated con-traction of vascular smooth muscle, as well as generation of oxida-tive stress and an associated interference in nitric oxide signaling pathways.
Major Forms of Lead Intoxication A. Inorganic Lead Poisoning ( Table 57–1 ) 1. Acute— Acute inorganic lead poisoning is uncommon today. It usually results from industrial inhalation of large quantities of lead oxide fumes or, in small children, from ingestion of a large oral dose of lead in the form of lead-based paint chips; small objects, eg, toys coated or fabricated from lead; or contaminated food or drink. The onset of severe symptoms usually requires several days or weeks of recurrent exposure and manifests as signs and symptoms of encephalopathy or colic. Evidence of hemolytic anemia (or ane-mia with basophilic stippling if exposure has been subacute) and elevated hepatic aminotransferases may be present.
The diagnosis of acute inorganic lead poisoning may be diffi-cult, and depending on the presenting symptoms, the condition has sometimes been mistaken for appendicitis, peptic ulcer, biliary colic, pancreatitis, or infectious meningitis. Subacute presenta-tion, featuring headache, fatigue, intermittent abdominal cramps, myalgias, and arthralgias, has often been mistaken for a flu-like viral illness. When there has been recent ingestion of lead-contain-ing paint chips, glazes, or weights, radiopacities may be visible on abdominal radiographs.
2. Chronic— The patient with chronic lead intoxication usually presents with multisystemic findings, including complaints of anorexia, fatigue, and malaise; neurologic complaints, including headache, difficulty in concentrating, and irritability or depressed mood; weakness, arthralgias, or myalgias; and gastrointestinal symptoms. Lead poisoning should be strongly suspected in any patient presenting with headache, abdominal pain, and anemia; and less commonly with motor neuropathy, gout, and renal insuf-ficiency. Chronic lead intoxication should be considered in any child with neurocognitive deficits, growth retardation, or develop-mental delay. It is important to recognize that adverse effects of lead that are of considerable public health significance, such as subclinical decrements in neurodevelopment in children and hypertension in adults, are usually nonspecific and may not come to medical attention.
The diagnosis of lead intoxication is best confirmed by measur-ing lead in whole blood. Although this test reflects lead currently circulating in blood and soft tissues and is not a reliable marker of either recent or cumulative lead exposure, most patients with lead-related disease have blood lead concentrations higher than the normal range. Average background blood lead concentrations in North America and Europe have declined by 90% in recent decades, and the geometric mean blood lead concentration in the United States in 2005–2006 was estimated to be 1.29 mcg/dL. Though predominantly a research tool, the concentration of lead in bone assessed by noninvasive K X-ray fluorescence measurement of lead has been correlated with long-term cumulative lead expo-sure, and its relationship to numerous lead-related disorders is a
subject of ongoing investigation. Measurement of lead excretion in the urine after a single dose of a chelating agent (sometimes called a “chelation challenge test”) primarily reflects the lead content of soft tissues and may not be a reliable marker of long-term lead exposure, remote past exposure, or skeletal lead burden. Because of the lag time associated with lead-induced elevations in circulating heme precursors, the finding of a blood lead concentration of 30 mcg/dL or more with no concurrent increase in zinc protopor-phyrin suggests that the lead exposure was of recent onset.
B. Organolead Poisoning Poisoning from organolead compounds is now very rare, in large part because of the worldwide phase-out of tetraethyl and tetra-methyl lead as antiknock additives in gasoline. However, organo-lead compounds such as lead stearate or lead naphthenate are still used in certain commercial processes. Because of their volatility or lipid solubility, organolead compounds tend to be well absorbed through either the respiratory tract or the skin. Organolead com-pounds predominantly target the central nervous system, producing dose-dependent effects that may include neurocognitive deficits, insomnia, delirium, hallucinations, tremor, convulsions, and death.
Treatment A. Inorganic Lead Poisoning Treatment of inorganic lead poisoning involves immediate termi-nation of exposure, supportive care, and the judicious use of chela-tion therapy. (Chelation is discussed later in this chapter.) Lead encephalopathy is a medical emergency that requires intensive supportive care. Cerebral edema may improve with corticosteroids and mannitol, and anticonvulsants may be required to treat sei-zures. Radiopacities on abdominal radiographs may suggest the presence of retained lead objects requiring gastrointestinal decon-tamination. Adequate urine flow should be maintained, but over-hydration should be avoided. Intravenous edetate calcium disodium (CaNa 2 EDTA) is administered at a dosage of 1000–1500 mg/m 2 /d (approximately 30–50 mg/kg/d) by continuous infusion for up to 5 days. Some clinicians advocate that chelation treatment for lead encephalopathy be initiated with an intramus-cular injection of dimercaprol, followed in 4 hours by concurrent administration of dimercaprol and EDTA. Parenteral chelation is limited to 5 or fewer days, at which time oral treatment with another chelator, succimer, may be instituted. In symptomatic lead intoxication without encephalopathy, treatment may some-times be initiated with succimer. The end point for chelation is usually resolution of symptoms or return of the blood lead con-centration to the premorbid range. In patients with chronic expo-sure, cessation of chelation may be followed by an upward rebound in blood lead concentration as the lead re-equilibrates from bone lead stores.
Although most clinicians support chelation for symptomatic patients with elevated blood lead concentrations, the decision to chelate asymptomatic subjects is more controversial. Since 1991, the Centers for Disease Control and Prevention (CDC) has rec-ommended chelation for all children with blood lead concentra-tions of 45 mcg/dL or greater. However, a recent randomized,
057-Katzung_Ch057_p1013-1026.indd 1016 9/21/11 12:13:12 PM

CHAPTER 57 Heavy Metal Intoxication & Chelators 1017
double-blind, placebo-controlled clinical trial of succimer in chil-dren with blood lead concentrations between 25 mcg/dL and 44 mcg/dL found no benefit on neurocognitive function or long-term blood lead reduction. Prophylactic use of chelating agents in the workplace should never be a substitute for reduction or pre-vention of excessive exposure.
Management of elevated blood lead levels in children and adults should include a conscientious effort to identify and reduce all potential sources of future lead exposure. Many local, state, or national governmental agencies maintain lead poisoning preven-tion programs that can assist in case management. Blood lead screening of family members or coworkers of a lead poisoning patient is often indicated to assess the scope of the exposure. Although the CDC blood lead level of concern for childhood lead poisoning of 10 mcg/dL has not been revised since 1991, the adverse impact of lower levels on children is widely acknowledged, and primary prevention of lead exposure is receiving increased emphasis. Although the US Occupational Safety and Health Administration (OSHA) lead regulations introduced in the late 1970s mandate that workers be removed from lead exposure for blood lead levels higher than 50–60 mcg/dL, an expert panel in 2007 recommended that removal be initiated for a single blood lead level greater than 30 mcg/dL, or when two successive blood lead levels measured over a 4-week interval are 20 mcg/dL or more. The longer-term goal should be for workers to maintain blood lead levels at lower than 10 mcg/dL, and for pregnant women to avoid occupational or avocational exposure that would result in blood lead levels higher than 5 mcg/dL. Environmental Protection Agency (EPA) regulations effective since 2010 require that contractors who perform renovation, repair, and painting projects that disturb lead-based paint in pre-1978 residences and child-occupied facilities must be certified and must follow specific work practices to prevent lead contamination.
B. Organic Lead Poisoning Initial treatment consists of decontaminating the skin and pre-venting further exposure. Treatment of seizures requires appropri-ate use of anticonvulsants. Empiric chelation may be attempted if high blood lead concentrations are present.
Arsenic Arsenic is a naturally occurring element in the earth’s crust with a long history of use as a constituent of commercial and industrial products, as a component in pharmaceuticals, and as an agent of deliberate poisoning. Recent commercial applications of arsenic include its use in the manufacture of semiconductors, wood pre-servatives for industrial applications (eg, marine timbers or utility poles), nonferrous alloys, glass, gel-based insecticidal ant baits, and veterinary pharmaceuticals. In some regions of the world, ground-water may contain high levels of arsenic that has leached from natural mineral deposits. Arsenic in drinking water in the Ganges delta of India and Bangladesh is now recognized as one of the world’s most pressing environmental health problems. Arsine, an arsenous hydride (AsH 3 ) gas with potent hemolytic effects, is manu-factured predominantly for use in the semiconductor industry but
may also be generated accidentally when arsenic-containing ores come in contact with acidic solutions.
It is of historical interest that Fowler’s solution, which contains 1% potassium arsenite, was widely used as a medicine for many conditions from the eighteenth century through the mid- twentieth century. Organic arsenicals were the first pharmaceutical antimi-crobials ∗ and were widely used for the first half of the twentieth century until supplanted by sulfonamides and other more effective and less toxic agents.
Other organoarsenicals, most notably lewisite (dichloro-[2-chlorovinyl]arsine), were developed in the early twentieth cen-tury as chemical warfare agents. Arsenic trioxide was reintroduced into the United States Pharmacopeia in 2000 as an orphan drug for the treatment of relapsed acute promyelocytic leukemia and is find-ing expanded use in experimental cancer treatment protocols (see Chapter 54 ). Melarsoprol, another trivalent arsenical, is used in the treatment of advanced African trypanosomiasis (see Chapter 52 ).
Pharmacokinetics Soluble arsenic compounds are well absorbed through the respira-tory and gastrointestinal tracts ( Table 57–1 ). Percutaneous absorp-tion is limited but may be clinically significant after heavy exposure to concentrated arsenic reagents. Most of the absorbed inorganic arsenic undergoes methylation, mainly in the liver, to monomethyl-arsonic acid and dimethylarsinic acid, which are excreted, along with residual inorganic arsenic, in the urine. When chronic daily absorption is less than 1000 mcg of soluble inorganic arsenic, approximately two thirds of the absorbed dose is excreted in the urine within 2–3 days. After massive ingestions, the elimination half-life is prolonged. Inhalation of arsenic compounds of low solu-bility may result in prolonged retention in the lung and may not be reflected by urinary arsenic excretion. Arsenic binds to sulfhy-dryl groups present in keratinized tissue, and following cessation of exposure, hair, nails, and skin may contain elevated levels after urine values have returned to normal. However, arsenic in hair and nails as a result of external deposition may be indistinguishable from that incorporated after internal absorption.
Pharmacodynamics Arsenic compounds are thought to exert their toxic effects by several modes of action. Interference with enzyme function may result from sulfhydryl group binding by trivalent arsenic or by substitution for phosphate. Inorganic arsenic or its metabolites may induce oxidative stress, alter gene expression, and interfere with cell signal transduction. Although on a molar basis, inorganic trivalent arsenic (As 3+ , arsenite) is generally two to ten times more acutely toxic than inorganic pentavalent arsenic (As 5+ , arsenate), in vivo interconversion is known to occur, and the full spectrum of arsenic toxicity has occurred after sufficient exposure to either form. Recent studies suggest that the trivalent form of the methy-lated metabolites (eg, monomethylarsonous acid [MMA III ])
∗ Paul Ehrlich’s “magic bullet” for syphilis (arsphenamine, Salvarsan) was an arsenical.
057-Katzung_Ch057_p1013-1026.indd 1017 9/21/11 12:13:12 PM

1018 SECTION IX Toxicology
may be more toxic than the inorganic parent compounds. Reduced efficiency in the methylation of MMA to DMA, result-ing in an elevated percentage of MMA in the urine, has been associated with an increased risk of chronic adverse effects. Arsenic methylation requires S-adenosylmethionine, a universal methyl donor in the body, and arsenic-associated perturbations in one-carbon metabolism may underlie some arsenic-induced epigenetic effects such as altered gene expression.
Arsine gas is oxidized in vivo and exerts a potent hemolytic effect associated with alteration of ion flux across the erythrocyte membrane; it also disrupts cellular respiration in other tissues. Arsenic is a recognized human carcinogen and has been associated with cancer of the lung, skin, and bladder. Marine organisms may contain large amounts of a well-absorbed trimethylated organo-arsenic, arsenobetaine, as well as a variety of arsenosugars and arsenolipids. Arsenobetaine exerts no known toxic effects when ingested by mammals and is excreted in the urine unchanged; arsenosugars are partially metabolized to dimethylarsinic acid. Thio-dimethylarsinic acid has recently been identified as a com-mon but minor human arsenic metabolite of uncertain toxico-logical significance.
Major Forms of Arsenic Intoxication A. Acute Inorganic Arsenic Poisoning Within minutes to hours after exposure to high doses (tens to hundreds of milligrams) of soluble inorganic arsenic compounds, many systems are affected. Initial gastrointestinal signs and symp-toms include nausea, vomiting, diarrhea, and abdominal pain. Diffuse capillary leak, combined with gastrointestinal fluid loss, may result in hypotension, shock, and death. Cardiopulmonary toxicity, including congestive cardiomyopathy, cardiogenic or noncardiogenic pulmonary edema, and ventricular arrhythmias, may occur promptly or after a delay of several days. Pancytopenia usually develops within 1 week, and basophilic stippling of eryth-rocytes may be present soon after. Central nervous system effects, including delirium, encephalopathy, and coma, may occur within the first few days of intoxication. An ascending sensorimotor peripheral neuropathy may begin to develop after a delay of 2–6 weeks. This neuropathy may ultimately involve the proximal musculature and result in neuromuscular respiratory failure. Months after an acute poisoning, transverse white striae (Aldrich-Mees lines) may be visible in the nails.
Acute inorganic arsenic poisoning should be considered in an individual presenting with abrupt onset of gastroenteritis in com-bination with hypotension and metabolic acidosis. Suspicion should be further heightened when these initial findings are fol-lowed by cardiac dysfunction, pancytopenia, and peripheral neu-ropathy. The diagnosis may be confirmed by demonstration of elevated amounts of inorganic arsenic and its metabolites in the urine (typically in the range of several thousand micrograms in the first 2–3 days after acute symptomatic poisoning). Arsenic disap-pears rapidly from the blood, and except in anuric patients, blood arsenic levels should not be used for diagnostic purposes. Treatment is based on appropriate gut decontamination, intensive
supportive care, and prompt chelation with unithiol, 3–5 mg/kg intravenously every 4–6 hours, or dimercaprol, 3–5 mg/kg intra-muscularly every 4–6 hours. In animal studies, the efficacy of chelation has been highest when it is administered within minutes to hours after arsenic exposure; therefore, if diagnostic suspi-cion is high, treatment should not be withheld for the several days to weeks often required to obtain laboratory confirmation.
Succimer has also been effective in animal models and has a higher therapeutic index than dimercaprol. However, because it is available in the United States only for oral administration, its use may not be advisable in the initial treatment of acute arsenic poi-soning, when severe gastroenteritis and splanchnic edema may limit absorption by this route.
B. Chronic Inorganic Arsenic Poisoning Chronic inorganic arsenic poisoning also results in multisystemic signs and symptoms. Overt noncarcinogenic effects may be evi-dent after chronic absorption of more than 0.01 mg/kg/d (∼ 500–1000 mcg/d in adults). The time to appearance of symp-toms varies with dose and interindividual tolerance. Constitutional symptoms of fatigue, weight loss, and weakness may be present, along with anemia, nonspecific gastrointestinal complaints, and a sensorimotor peripheral neuropathy, particularly featuring a stocking glove pattern of dysesthesia. Skin changes—among the most characteristic effects—typically develop after years of expo-sure and include a “raindrop” pattern of hyperpigmentation, and hyperkeratoses involving the hands and feet ( Figure 57–1 ). Peripheral vascular disease and noncirrhotic portal hypertension may also occur. Epidemiologic studies suggest a possible link to hypertension, diabetes, chronic nonmalignant respiratory disease, and adverse reproductive outcomes. Cancer of the lung, skin, bladder, and possibly other sites, may appear years after exposure to doses of arsenic that are not high enough to elicit other acute or chronic effects.
Administration of arsenite in cancer chemotherapy regimens, often at a daily dose of 10–20 mg for weeks to a few months, has been associated with prolongation of the QT interval on the elec-trocardiogram and occasionally has resulted in malignant ventric-ular arrhythmias such as torsades de pointes.
The diagnosis of chronic arsenic poisoning involves inte-gration of the clinical findings with confirmation of exposure. The urine concentration of the sum of inorganic arsenic and its primary metabolites MMA and DMA is less than 20 mcg/L in the general population. High urine levels associated with overt adverse effects may return to normal within days to weeks after exposure ceases. Because it may contain large amounts of nontoxic organoarsenic, all seafood should be avoided for at least 3 days before submission of a urine sample for diagnostic purposes. The arsenic content of hair and nails (normally less than 1 ppm) may sometimes reveal past elevated exposure, but results should be interpreted cautiously in view of the potential for external contamination.
Management of chronic arsenic poisoning consists primarily of termination of exposure and nonspecific supportive care. Although
057-Katzung_Ch057_p1013-1026.indd 1018 9/21/11 12:13:13 PM

CHAPTER 57 Heavy Metal Intoxication & Chelators 1019
empiric short-term oral chelation with unithiol or succimer for symptomatic individuals with elevated urine arsenic concentra-tions may be considered, it has no proven benefit beyond removal from exposure alone. Preliminary studies suggest that dietary supplementation of folate—thought to be a cofactor in arsenic methylation—might be of value in arsenic-exposed individuals, particularly men, who are also deficient in folate.
C. Arsine Gas Poisoning Arsine gas poisoning produces a distinctive pattern of intoxication dominated by profound hemolytic effects. After a latent period that may range from 2 hours to 24 hours postinhalation (depend-ing on the magnitude of exposure), massive intravascular hemoly-sis may occur. Initial symptoms may include malaise, headache, dyspnea, weakness, nausea, vomiting, abdominal pain, jaundice, and hemoglobinuria. Oliguric renal failure, a consequence of hemoglobin deposition in the renal tubules, often appears within 1–3 days. In massive exposures, lethal effects on cellular respira-tion may occur before renal failure develops. Urinary arsenic levels are elevated but are seldom available to confirm the diagnosis dur-ing the critical period of illness. Intensive supportive care— including exchange transfusion, vigorous hydration, and, in the case of acute renal failure, hemodialysis—is the mainstay of therapy.
Currently available chelating agents have not been demonstrated to be of clinical value in arsine poisoning.
MERCURY
Metallic mercury as “quicksilver”—the only metal that is liquid under ordinary conditions—has attracted scholarly and scientific interest from antiquity. The mining of mercury was early recog-nized as being hazardous to health. As industrial use of mercury became common during the last 200 years, new forms of toxicity were recognized that were found to be associated with various transformations of the metal. In the early 1950s, a mysterious epidemic of birth defects and neurologic disease occurred in the Japanese fishing village of Minamata. The causative agent was determined to be methylmercury in contaminated seafood, traced to industrial discharges into the bay from a nearby factory. In addition to elemental mercury and alkylmercury (including meth-ylmercury), other key mercurials include inorganic mercury salts and aryl mercury compounds, each of which exerts a relatively unique pattern of clinical toxicity.
Mercury is mined predominantly as HgS in cinnabar ore and is then converted commercially to a variety of chemical forms. Key industrial and commercial applications of mercury are found in the electrolytic production of chlorine and caustic soda; the manufacture of electrical equipment, thermometers, and other instruments; fluorescent lamps; and dental amalgam. The wide-spread use of elemental mercury in artisanal gold production is a growing problem in many developing countries. Mercury use in pharmaceuticals and in biocides has declined substantially in recent years, but occasional use in antiseptics and folk medicines is still encountered. Thimerosal, an organomercurial preservative that is metabolized in part to ethylmercury, has been removed from almost all the vaccines in which it was formerly present. Environmental exposure to mercury from the burning of fossil fuels, or the bioaccumulation of methylmercury in fish, remains a concern in some regions of the world. Low-level exposure to mer-cury released from dental amalgam fillings occurs, but systemic toxicity from this source has not been established.
Pharmacokinetics The absorption of mercury varies considerably depending on the chemical form of the metal. Elemental mercury is quite volatile and can be absorbed from the lungs ( Table 57–1 ). It is poorly absorbed from the intact gastrointestinal tract. Inhaled mercury is the primary source of occupational exposure. Organic short-chain alkylmercury compounds are volatile and potentially harmful by inhalation as well as by ingestion. Percutaneous absorption of metallic mercury and inorganic mercury can be of clinical concern following massive acute or long-term chronic exposure. Alkylmercury compounds appear to be well absorbed through the skin, and acute contact with a few drops of dimethylmercury has resulted in severe, delayed toxicity. After absorption, mercury is distributed to the tissues within a few hours, with the highest concentration occurring in the kidney. Inorganic mercury is
FIGURE 57–1 Dermatologic lesions associated with chronic inges-tion of arsenic in drinking water. (Photo courtesy of Dipankar Chakraborti, PhD.)
057-Katzung_Ch057_p1013-1026.indd 1019 9/21/11 12:13:13 PM

1020 SECTION IX Toxicology
excreted through the urine and feces. Excretion of inorganic mer-cury follows a multicompartment model: most is excreted within weeks to months, but a fraction may be retained in the kidneys and brain for years. After inhalation of elemental mercury vapor, urinary mercury levels decline with a half-life of approximately 1–3 months. Methylmercury, which has a blood and whole body half-life of approximately 50 days, undergoes biliary excretion and enterohepatic circulation, with more than two thirds eventually excreted in the feces. Mercury binds to sulfhydryl groups in kera-tinized tissue, and as with lead and arsenic, traces appear in the hair and nails.
Major Forms of Mercury Intoxication Mercury interacts with sulfhydryl groups in vivo, inhibiting enzymes and altering cell membranes. The pattern of clinical intoxication from mercury depends to a great extent on the chem-ical form of the metal and the route and severity of exposure.
A. Acute Acute inhalation of elemental mercury vapors may cause chemical pneumonitis and noncardiogenic pulmonary edema. Acute gingivo-stomatitis may occur, and neurologic sequelae (see following text) may also ensue. Acute ingestion of inorganic mercury salts, such as mercuric chloride, can result in a corrosive, potentially life-threatening hemorrhagic gastroenteritis followed within hours to days by acute tubular necrosis and oliguric renal failure.
B. Chronic Chronic poisoning from inhalation of mercury vapor results in a classic triad of tremor, neuropsychiatric disturbance, and gingivo-stomatitis. The tremor usually begins as a fine intention tremor of the hands, but the face may also be involved, and progression to choreiform movements of the limbs may occur. Neuropsychiatric manifestations, including memory loss, fatigue, insomnia, and anorexia, are common. There may be an insidious change in mood to shyness, withdrawal, and depression along with explosive anger or blushing (a behavioral pattern referred to as erethism ). Recent studies suggest that low-dose exposure may produce subclinical neurologic effects. Gingivostomatitis, sometimes accompanied by loosening of the teeth, may be reported after high-dose exposure. Evidence of peripheral nerve damage may be detected on elec-trodiagnostic testing, but overt peripheral neuropathy is rare. Acrodynia is an uncommon idiosyncratic reaction to subacute or chronic mercury exposure and occurs mainly in children. It is characterized by painful erythema of the extremities and may be associated with hypertension, diaphoresis, anorexia, insomnia, irritability or apathy, and a miliary rash. Chronic exposure to inorganic mercury salts, sometimes via topical application in cos-metic creams, has been associated with neurological symptoms and renal toxicity in case reports and case series.
Methylmercury intoxication affects mainly the central nervous system and results in paresthesias, ataxia, hearing impairment, dysarthria, and progressive constriction of the visual fields. Signs and symptoms of methylmercury intoxication may first appear
several weeks or months after exposure begins. Methylmercury is a reproductive toxin. High-dose prenatal exposure to methylmer-cury may produce mental retardation and a cerebral palsy-like syndrome in the offspring. Low-level prenatal exposures to meth-ylmercury have been associated with a risk of subclinical neurode-velopmental deficits.
A 2004 report by the Institute of Medicine’s Immunization Safety Review Committee concluded that available evidence favored rejection of a causal relation between thimerosal-containing vac-cines and autism. In like manner, a recent retrospective cohort study conducted by the CDC did not support a causal association between early prenatal or postnatal exposure to mercury from thimerosal-containing vaccines and neuropsychological functioning later in childhood.
Dimethylmercury is a rarely encountered but extremely neurotoxic form of organomercury that may be lethal in small quantities.
The diagnosis of mercury intoxication involves integration of the history and physical findings with confirmatory laboratory testing or other evidence of exposure. In the absence of occu-pational exposure, the urine mercury concentration is usually less than 5 mcg/L, and whole blood mercury is less than 5 mcg/L. In 1990, the Biological Exposure Index (BEI) Committee of the American Conference of Governmental Industrial Hygienists (ACGIH) recommended that workplace exposures should result in urinary mercury concentrations less than 35 mcg per gram of creatinine and end-of-work-week whole blood mercury concen-trations less than 15 mcg/L. To minimize the risk of developmen-tal neurotoxicity from methylmercury, the US Environmental Protection Agency and the Food and Drug Administration (FDA) have advised pregnant women, women who might become preg-nant, nursing mothers, and young children to avoid consumption of fish with high mercury levels (eg, swordfish) and to limit con-sumption of fish with lower levels of mercury to no more than 12 ounces (340 g, or two average meals) per week.
Treatment A. Acute Exposure In addition to intensive supportive care, prompt chelation with oral or intravenous unithiol, intramuscular dimercaprol, or oral succimer may be of value in diminishing nephrotoxicity after acute exposure to inorganic mercury salts. Vigorous hydration may help to maintain urine output, but if acute renal failure ensues, days to weeks of hemodialysis or hemodiafiltration in conjunction with chelation may be necessary. Because the efficacy of chelation declines with time since exposure, treatment should not be delayed until the onset of oliguria or other major systemic effects.
B. Chronic Exposure Unithiol and succimer increase urine mercury excretion following acute or chronic elemental mercury inhalation, but the impact of such treatment on clinical outcome is unknown. Dimercaprol has been shown to redistribute mercury to the central nervous system
057-Katzung_Ch057_p1013-1026.indd 1020 9/21/11 12:13:14 PM

CHAPTER 57 Heavy Metal Intoxication & Chelators 1021
from other tissue sites, and since the brain is a key target organ, dimercaprol should not be used in treatment of exposure to ele-mental or organic mercury. Limited data suggest that succimer, unithiol, and N -acetyl-L-cysteine (NAC) may enhance body clear-ance of methylmercury.
■ PHARMACOLOGY OF CHELATORS
Chelating agents are drugs used to prevent or reverse the toxic effects of a heavy metal on an enzyme or other cellular target, or to accelerate the elimination of the metal from the body. By form-ing a complex with the heavy metal, the chelating agent renders the metal unavailable for toxic interactions with functional groups of enzymes or other proteins, coenzymes, cellular nucleophiles, and membranes. Chelating agents contain one or more coordinat-ing atoms, usually oxygen, sulfur, or nitrogen, which donate a pair of electrons to a cationic metal ion to form one or more coordi-nate-covalent bonds. Depending on the number of metal-ligand bonds, the complex may be referred to as mono-, bi-, or polyden-tate. Figure 57–2 depicts the hexadentate chelate formed by inter-action of edetate (ethylenediaminetetraacetate) with a metal atom, such as lead.
In some cases, the metal-mobilizing effect of a therapeutic chelating agent may not only enhance that metal’s excretion—a desired effect—but may also redistribute some of the metal to other vital organs. This has been demonstrated for dimercaprol, which redistributes mercury and arsenic to the brain while also enhancing urinary mercury and arsenic excretion. Although several chelating agents have the capacity to mobilize cadmium, their tendency to redistribute cadmium to the kidney and increase nephrotoxicity has negated their therapeutic value in cadmium intoxication.
In addition to removing the target metal that is exerting toxic effects on the body, some chelating agents may enhance excretion of essential cations, such as zinc in the case of calcium EDTA and diethylenetriaminepentaacetic acid (DTPA), and zinc and copper in the case of succimer. No clinical significance of this effect has been demonstrated, although some animal data suggest the possibility of adverse developmental impact. If prolonged chelation during the prenatal period or early childhood period is necessary, judicious supplementation of the diet with zinc might be considered.
The longer the half-life of a metal in a particular organ, the less effectively it will be removed by chelation. For example, in the case of lead chelation with calcium EDTA or succimer, or of pluto-nium chelation with DTPA, the metal is more effectively removed from soft tissues than from bone, where incorporation into bone matrix results in prolonged retention.
In most cases, the capacity of chelating agents to prevent or reduce the adverse effects of toxic metals appears to be greatest when such agents are administered very soon after an acute metal exposure. Use of chelating agents days to weeks after an acute metal exposure ends—or their use in the treatment of chronic metal intoxication—may still be associated with increased metal excretion. However, at that point, the capacity of such enhanced
excretion to mitigate the pathologic effect of the metal exposure may be reduced.
The most important chelating agents currently in use in the USA are described below.
DIMERCAPROL (2,3-DIMERCAPTOPROPANOL, BAL)
Dimercaprol ( Figure 57–3 ), an oily, colorless liquid with a strong mercaptan-like odor, was developed in Great Britain during
FIGURE 57–2 Salt and chelate formation with edetate (ethyl-enediaminetetraacetate, EDTA). A: In a solution of calcium disodium salt of EDTA, the sodium and hydrogen ions are chemically and bio-logically available. B: In solutions of calcium disodium edetate, cal-cium is bound by coordinate-covalent bonds with nitrogens as well as by the usual ionic bonds. C: In the lead–edetate chelate, lead is incorporated into five heterocyclic rings. (Modified and reproduced, with
permission, from Meyers FH, Jawetz E, Goldfien A: Review of Medical Pharmacology,
7th ed. Originally published by Lange Medical Publications. McGraw-Hill, 1980.)
HO C
O
CH2 CH2
CH2
CH2
CH2
CH2CH2CH2
N
CNa O
O
N
C
C OH
NaO
O
OA
Ca
OC
H2C
N
CNa O
O
CO
N
C H2
C H2
C H2
C
O
O Na
B
Pb
O
N
C
OO
O
N
O
C O
O
O O
057-Katzung_Ch057_p1013-1026.indd 1021 9/21/11 12:13:14 PM

1022 SECTION IX Toxicology
World War II as a therapeutic antidote against poisoning by the arsenic-containing warfare agent lewisite. It thus became known as British anti-Lewisite, or BAL. Because aqueous solutions of di-mercaprol are unstable and oxidize readily, it is dispensed in 10% solution in peanut oil and must be administered by intra-muscular injection, which is often painful.
In animal models, dimercaprol prevents and reverses arsenic-induced inhibition of sulfhydryl-containing enzymes and, if given soon after exposure, may protect against the lethal effects of inor-ganic and organic arsenicals. Human data indicate that it can increase the rate of excretion of arsenic and lead and may offer therapeutic benefit in the treatment of acute intoxication by arse-nic, lead, and mercury.
Indications & Toxicity Dimercaprol is FDA-approved as single-agent treatment of acute poisoning by arsenic and inorganic mercury and for the treatment of severe lead poisoning when used in conjunction with edetate calcium disodium (EDTA; see below). Although studies of its metabolism in humans are limited, intramuscularly administered dimercaprol appears to be readily absorbed, metabolized, and excreted by the kidney within 4–8 hours. Animal models indicate that it may also undergo biliary excretion, but the role of this excretory route in humans and other details of its biotransforma-tion are uncertain.
When used in therapeutic doses, dimercaprol is associated with a high incidence of adverse effects, including hypertension, tachy-cardia, nausea, vomiting, lacrimation, salivation, fever (particu-larly in children), and pain at the injection site. Its use has also been associated with thrombocytopenia and increased prothrom-bin time—factors that may limit intramuscular injection because of the risk of hematoma formation at the injection site. Despite its protective effects in acutely intoxicated animals, dimercaprol may redistribute arsenic and mercury to the central nervous system, and it is not advocated for treatment of chronic poisoning. Water-soluble analogs of dimercaprol—unithiol and succimer—have higher therapeutic indices and have replaced dimercaprol in many settings.
SUCCIMER (DIMERCAPTOSUCCINIC ACID, DMSA)
Succimer is a water-soluble analog of dimercaprol, and like that agent it has been shown in animal studies to prevent and reverse metal-induced inhibition of sulfhydryl-containing enzymes and to protect against the acute lethal effects of arsenic. In humans, treat-ment with succimer is associated with an increase in urinary lead excretion and a decrease in blood lead concentration. It may also decrease the mercury content of the kidney, a key target organ of inorganic mercury salts. In the USA, succimer is formulated exclu-sively for oral use, but intravenous formulations have been used successfully elsewhere. It is absorbed rapidly but somewhat vari-ably after oral administration. Peak blood levels of succimer occur at approximately 3 hours. The drug binds in vivo to the amino acid cysteine to form 1:1 and 1:2 mixed disulfides, possibly in the kidney, and it may be these complexes that are the active chelating moieties. Experimental data suggest that multidrug-resistance protein 2 (Mrp2), one of a group of transporter proteins involved in the cellular excretion of xenobiotics, facilitates the renal excre-tion of mercury compounds that are bound to the transformed succimer and to unithiol. The elimination half-time of trans-formed succimer is approximately 2–4 hours.
Indications & Toxicity Succimer is currently FDA-approved for the treatment of children with blood lead concentrations greater than 45 mcg/dL, but it is also commonly used in adults. The typical dosage is 10 mg/kg orally three times a day. Oral administration of succimer is compa-rable to parenteral EDTA in reducing blood lead concentration and has supplanted EDTA in outpatient treatment of patients who are capable of absorbing the oral drug. However, despite the dem-onstrated capacity of both succimer and EDTA to enhance lead elimination, their value in reversing established lead toxicity or in otherwise improving therapeutic outcome has yet to be established by a placebo-controlled clinical trial. In a recent study in lead-ex-posed juvenile rats, high-dose succimer did reduce lead-induced neurocognitive impairment when administered to animals with moderate- and high-dose lead exposure. Conversely, when admin-istered to the control group that was not lead exposed, succimer
FIGURE 57–3 Chemical structures of several chelators. Ferroxamine (ferrioxamine) without the chelated iron is deferoxam-ine. It is represented here to show the functional groups; the iron is actually held in a caged system. The structures of the in vivo metal-chelator complexes for dimercaprol, succimer, penicillamine, and uni-thiol (see text) are not known and may involve the formation of mixed disulfides with amino acids. (Modified and reproduced, with permis-
sion from Meyers FH, Jawetz E, and Goldfien A: Review of Medical Pharmacology,
7th ed. Originally published by Lange Medical Publications. McGraw-Hill, 1980.)
SHCH
COOH
COOH
OH
O
N
O
C
CONH
(CH2)5(CH2)5(CH2)5 (CH2)2(CH2)2
CONH
O
N
O
C
O
N
O
C
CH3
H2N
Fe3+
Ferroxamine
H2C
H2C
SH
HC SH
Dimercaprol(2, 3-dimercaptopropanol)
H3C C
NH2
Penicillamine
CH3
CH C OH
SH
O
SHCH
Succimer(DMSA)
057-Katzung_Ch057_p1013-1026.indd 1022 9/21/11 12:13:14 PM

CHAPTER 57 Heavy Metal Intoxication & Chelators 1023
was associated with a decrement in neurocognitive performance. Based on its protective effects against arsenic in animals and its ability to mobilize mercury from the kidney, succimer has also been used in the treatment of arsenic and mercury poisoning.
In limited clinical trials, succimer has been well tolerated. It has a negligible impact on body stores of calcium, iron, and magne-sium. It induces a mild increase in urinary excretion of zinc and, less consistently, copper. This effect on trace metal balance has not been associated with overt adverse effects, but its long-term impact on neurodevelopment is uncertain. Gastrointestinal dis-turbances, including anorexia, nausea, vomiting, and diarrhea, are the most common side effects, occurring in less than 10% of patients. Rashes, sometimes requiring discontinuation of the medication, have been reported in less than 5% of patients. Mild, reversible increases in liver aminotransferases have been noted in 6–10% of patients, and isolated cases of mild to moderate neutro-penia have been reported.
EDETATE CALCIUM DISODIUM (ETHYLENEDIAMINETETRAACETIC ACID, EDTA)
Ethylenediaminetetraacetic acid ( Figure 57–2 ) is an efficient che-lator of many divalent and trivalent metals in vitro. To prevent potentially life-threatening depletion of calcium, the drug should be administered only as the calcium disodium salt.
EDTA penetrates cell membranes relatively poorly and there-fore chelates extracellular metal ions much more effectively than intracellular ions.
The highly polar ionic character of EDTA limits its oral absorption. Moreover, oral administration may increase lead absorption from the gut. Consequently, EDTA should be admin-istered by intravenous infusion. In patients with normal renal function, EDTA is rapidly excreted by glomerular filtration, with 50% of an injected dose appearing in the urine within 1 hour. EDTA mobilizes lead from soft tissues, causing a marked increase in urinary lead excretion and a corresponding decline in blood lead concentration. In patients with renal insufficiency, excretion of the drug—and its metal-mobilizing effects—may be delayed.
Indications & Toxicity Edetate calcium disodium is indicated chiefly for the chelation of lead, but it may also have usefulness in poisoning by zinc, manga-nese, and certain heavy radionuclides. In spite of repeated claims in the alternative medicine literature, EDTA has no demonstrated use-fulness in the treatment of atherosclerotic cardiovascular disease.
Because the drug and the mobilized metals are excreted via the urine, the drug is relatively contraindicated in anuric patients. In such instances, the use of low doses of EDTA in combination with high-flux hemodialysis or hemofiltration has been described. Nephrotoxicity from EDTA has been reported, but in most cases can be prevented by maintenance of adequate urine flow, avoid-ance of excessive doses, and limitation of a treatment course to
5 or fewer consecutive days. EDTA may result in temporary zinc depletion that is of uncertain clinical significance. Analogs of EDTA, the calcium and zinc disodium salts of DTPA, pentetate, have been used for removal (“decorporation”) of certain tran-suranic, rare earth, and transition metal radioisotopes, and in 2004 were approved by the FDA for treatment of contamination with plutonium, americium, and curium.
UNITHIOL (DIMERCAPTOPROPANESULFONIC ACID, DMPS)
Unithiol, a dimercapto chelating agent that is a water-soluble analog of dimercaprol, has been available in the official formular-ies of Russia and other former Soviet countries since 1958 and in Germany since 1976. It has been legally available from com-pounding pharmacies in the USA since 1999. Unithiol can be administered orally and intravenously. Bioavailability by the oral route is approximately 50%, with peak blood levels occurring in approximately 3.7 hours. Over 80% of an intravenous dose is excreted in the urine, mainly as cyclic DMPS sulfides. The elimi-nation half-time of total unithiol (parent drug and its transforma-tion products) is approximately 20 hours. Unithiol exhibits protective effects against the toxic action of mercury and arsenic in animal models, and it increases the excretion of mercury, arse-nic, and lead in humans. Animal studies and a few case reports suggest that unithiol may also have usefulness in the treatment of poisoning by bismuth compounds.
CH2
Unithiol
SH
CH
SH
CH2
SO2H
Indications & Toxicity Unithiol has no FDA-approved indications, but experimental studies and its pharmacologic and pharmacodynamic profile suggest that intravenous unithiol offers advantages over intra-muscular dimercaprol or oral succimer in the initial treatment of severe acute poisoning by inorganic mercury or arsenic. Aqueous preparations of unithiol (usually 50 mg/mL in sterile water) can be administered at a dosage of 3–5 mg/kg every 4 hours by slow intravenous infusion over 20 minutes. If a few days of treatment are accompanied by stabilization of the patient’s cardiovascular and gastrointestinal status, it may be possible to change to oral administration of 4–8 mg/kg every 6–8 hours. Oral unithiol may also be considered as an alterna-tive to oral succimer in the treatment of lead intoxication.
Unithiol has been reported to have a low overall incidence of adverse effects (< 4%). Self-limited dermatologic reactions (drug exanthems or urticaria) are the most commonly reported adverse effects, although isolated cases of major allergic reactions, includ-ing erythema multiforme and Stevens-Johnson syndrome, have
057-Katzung_Ch057_p1013-1026.indd 1023 9/21/11 12:13:14 PM

1024 SECTION IX Toxicology
been reported. Because rapid intravenous infusion may cause vasodilation and hypotension, unithiol should be infused slowly over an interval of 15–20 minutes.
PENICILLAMINE (D-DIMETHLCYSTEINE)
Penicillamine ( Figure 57–3 ) is a white crystalline, water-soluble derivative of penicillin. D-Penicillamine is less toxic than the L isomer and consequently is the preferred therapeutic form. Penicillamine is readily absorbed from the gut and is resistant to metabolic degradation.
Indications & Toxicity Penicillamine is used chiefly for treatment of poisoning with cop-per or to prevent copper accumulation, as in Wilson’s disease (hepatolenticular degeneration). It is also used occasionally in the treatment of severe rheumatoid arthritis (see Chapter 36 ). Its abil-ity to increase urinary excretion of lead and mercury had occa-sioned its use in outpatient treatment for intoxication with these metals, but succimer, with its stronger metal-mobilizing capacity and lower adverse-effect profile, has generally replaced pen-icillamine for these purposes.
Adverse effects have been seen in up to one third of patients receiving penicillamine. Hypersensitivity reactions include rash, pruritus, and drug fever, and the drug should be used with extreme caution, if at all, in patients with a history of penicillin allergy. Nephrotoxicity with proteinuria has also been reported, and protracted use of the drug may result in renal insufficiency. Pancytopenia has been associated with prolonged drug intake. Pyridoxine deficiency is a frequent toxic effect of other forms of the drug but is rarely seen with the D form. An acetylated deriva-tive, N -acetylpenicillamine, has been used experimentally in mer-cury poisoning and may have superior metal-mobilizing capacity, but it is not commercially available.
DEFEROXAMINE
Deferoxamine is isolated from Streptomyces pilosus. It binds iron avidly ( Figure 57–3 ) but binds essential trace metals poorly. Furthermore, though competing for loosely bound iron in iron-carrying proteins (hemosiderin and ferritin), it fails to compete for biologically chelated iron, as in microsomal and mitochondrial cytochromes and hemoproteins. Consequently, it is the parenteral chelator of choice for iron poisoning (see Chapters 33 and 58 ). Deferoxamine plus hemodialysis may also be useful in the treat-ment of aluminum toxicity in renal failure. Deferoxamine is poorly absorbed when administered orally and may increase iron absorption when given by this route. It should therefore be admin-istered intramuscularly or, preferably, intravenously. It is believed to be metabolized, but the pathways are unknown. The iron- chelator complex is excreted in the urine, often turning the urine an orange-red color.
Rapid intravenous administration may result in hypotension. Adverse idiosyncratic responses such as flushing, abdominal
discomfort, and rash have also been observed. Pulmonary compli-cations (eg, acute respiratory distress syndrome) have been reported in some patients undergoing deferoxamine infusions last-ing longer than 24 hours, and neurotoxicity and increased suscep-tibility to certain infections (eg, with Yersinia enterocolitica) have been described after long-term therapy of iron overload conditions (eg, thalassemia major).
DEFERASIROX
Deferasirox is a tridentate chelator with a high affinity for iron and low affinity for other metals, eg, zinc and copper. It is orally active and well absorbed. In the circulation, it binds iron, and the com-plex is excreted in the bile. Deferasirox was approved by the FDA in 2005 for the oral treatment of iron overload caused by blood transfusions, a problem in the treatment of thalassemia and myelodysplastic syndrome. More than five years of clinical experi-ence suggest that daily long-term usage is generally well tolerated, with the most common adverse effects consisting of mild to mod-erate gastrointestinal disturbances (< 15% of patients) and skin rash (≈ 5% of patients).
PRUSSIAN BLUE (FERRIC HEXACYANOFERRATE)
Ferric hexacyanoferrate (insoluble Prussian blue) is a hydrated crystalline compound in which Fe 2+ and Fe 3+ atoms are coordi-nated with cyanide groups in a cubic lattice structure. Although used as a dark blue commercial pigment for nearly 300 years, it was only three decades ago that its potential usefulness as a phar-maceutical chelator was recognized. Primarily by ion exchange, and secondarily by mechanical trapping or adsorption, the com-pound has high affinity for certain univalent cations, particularly cesium and thallium. Used as an oral drug, insoluble Prussian blue undergoes minimal gastrointestinal absorption (< 1%). Because the complexes it forms with cesium or thallium are nonabsorb-able, oral administration of the chelator diminishes intestinal absorption or interrupts enterohepatic and enteroenteric circula-tion of these cations, thereby accelerating their elimination in the feces. In clinical case series, the use of Prussian blue has been asso-ciated with a decline in the biologic half-life (ie, in vivo retention) of radioactive cesium and thallium.
Indications & Toxicity In 2003, the FDA approved Prussian blue for the treatment of contamination with radioactive cesium ( 137 Cs) and intoxication with thallium salts. Approval was prompted by concern over potential widespread human contamination with radioactive cesium caused by terrorist use of a radioactive dispersal device (“dirty bomb”). The drug is part of the Strategic National Stockpile of pharmaceuticals and medical material maintained by the CDC ( http://www.bt.cdc.gov/stockpile/#material ). ( Note : Although soluble forms of Prussian blue, such as potassium
057-Katzung_Ch057_p1013-1026.indd 1024 9/21/11 12:13:14 PM

CHAPTER 57 Heavy Metal Intoxication & Chelators 1025
ferric hexacyanoferrate, may have better utility in thallium poi-soning, only the insoluble form is currently available as a phar-maceutical.)
After exposure to 137 Cs or thallium salts, the approved adult dosage is 3 g orally three times a day; the corresponding pediatric dosage (2–12 years of age) is 1 g orally three times a day. Serial monitoring of urine and fecal radioactivity ( 137 Cs) and urinary
thallium concentrations can guide the recommended duration of therapy. Adjunctive supportive care for possible acute radiation illness ( 137 Cs) or systemic thallium toxicity should be instituted as needed.
Prussian blue has not been associated with significant adverse effects. Constipation, which may occur in some cases, should be treated with laxatives or increased dietary fiber.
P R E P A R A T I O N S A V A I L A B L E
Deferasirox (Exjade)Oral: 125, 250, 500 mg tablets
Deferoxamine (Desferal)Parenteral: Powder to reconstitute, 500, 2000 mg/vial
Dimercaprol (BAL in Oil)Parenteral: 100 mg/mL for IM injection
Edetate calcium [calcium EDTA] (Calcium Disodium Versenate)Parenteral: 200 mg/mL for injection
Penicillamine (Cuprimine, Depen)Oral: 125, 250 mg capsules; 250 mg tablets
Pentetate Calcium Trisodium ([calcium DTPA] and Pentetate Zinc Trisodium [zinc DTPA])
Parenteral: 200 mg/mL for injectionPrussian Blue (Radiogardase)
Oral: 500 mg capsulesSuccimer (Chemet)
Oral: 100 mg capsulesUnithiol (Dimaval)
Bulk powder available for compounding as oral capsules, or for infusion (50 mg/mL)
Deferasirox (Exjade)Oral: 125, 250, 500 mg tablets
Deferoxamine (Desferal)Parenteral: Powder to reconstitute, 500, 2000 mg/vial
Dimercaprol (BAL in Oil)Parenteral: 100 mg/mL for IM injection
Edetate calcium [calcium EDTA] (Calcium Disodium Versenate)Parenteral: 200 mg/mL for injection
Penicillamine (Cuprimine, Depen)Oral: 125, 250 mg capsules; 250 mg tablets
Pentetate Calcium Trisodium ([calcium DTPA] and Pentetate Zinc Trisodium [zinc DTPA])
Parenteral: 200 mg/mL for injectionPrussian Blue (Radiogardase)
Oral: 500 mg capsulesSuccimer (Chemet)
Oral: 100 mg capsulesUnithiol (Dimaval)
Bulk powder available for compounding as oral capsules, or for infusion (50 mg/mL)
REFERENCES Lead
Brubaker CJ et al: The influence of age of lead exposure on adult gray matter volume. Neurotoxicology 2010;31:259.
Carlisle JC et al: A blood lead benchmark for assessing risks from childhood lead exposure. J Environ Sci Health Part A 2009;44:1200.
Centers for Disease Control and Prevention (CDC): Guidelines for the Identification and Management of Lead Exposure in Pregnant and Lactating Women. CDC, 2010. http://www.cdc.gov/nceh/lead/publications/LeadandPregnancy2010.pdf
Kosnett MJ et al: Recommendations for medical management of adult lead expo-sure. Environ Health Perspect 2007;115:463.
Lanphear BP et al: Low-level environmental lead exposure and children’s intellec-tual development: An international pooled analysis. Environ Health Perspect 2005;113:894.
U.S. Environmental Protection Agency. Air Quality Criteria for Lead (Final). EPA: Washington, DC, 2006. http://cfpub.epa.gov/ncea/cfm/recordisplay.cfm?deid=158823
Weisskopf MG et al: A prospective study of bone lead concentration and death from all causes, cardiovascular diseases, and cancer in the Department of Veterans Affairs Normative Aging Study. Circulation 2009;120:1056.
Weuve J et al: Cumulative exposure to lead in relation to cognitive function in older women. Environ Health Perspect 2009;117:574.
Arsenic
Caldwell KL et al: Levels of urinary total and speciated arsenic in the US popula-tion: National Health and Nutrition Examination Survey 2003–2004. J Exp Sci Environ Epid 2009;19:59.
Chen CJ et al: Arsenic and diabetes and hypertension in human populations. Toxicol Appl Pharmacol 2007;222:298.
Gamble MV: Folate and arsenic metabolism: A double-blind, placebo-controlled folic acid supplementation trial in Bangladesh. Am J Clin Nutr 2006;84:1093.
Lindberg AL et al: The risk of arsenic induced skin lesions in Bangladeshi men and women is affected by arsenic metabolism and the age at first exposure. Tox Appl Pharmacol 2008;230:9.
Parvez F et al: A prospective study of respiratory symptoms associated with chronic arsenic exposure in Bangladesh: findings from the Health Effects of Arsenic Longitudinal Study (HEALS). Thorax 2010;65:528.
Vahter M: Effects of arsenic on maternal and fetal health. Annu Rev Nutr 2009;29:381.
Mercury
Bellinger DC et al: Dental amalgam restorations and children’s neuropsychological function: The New England Children’s Amalgam Trial. Environ Health Perspect 2007;115:440.
Caldwell KL et al: Total blood mercury concentrations in the U.S. population: 1999–2006. Int J Hyg Environ Health 2009;212:588.
Clarkson TW, Magos L: The toxicology of mercury and its chemical compounds. Crit Rev Toxicol 2006;36:609.
EPA website: What You Need to Know about Mercury in Fish and Shellfish. http://water.epa.gov/scitech/swguidance/fishshellfish/outreach/advice_index.cfm
Hertz-Picciotto I et al: Blood mercury concentrations in CHARGE study children with and without autism. Environ Health Perspect 2010;118:161.
Lederman SA et al: Relation between cord blood mercury levels and early child development in a World Trade Center cohort. Environ Health Perspect 2008;116:1085.
057-Katzung_Ch057_p1013-1026.indd 1025 9/21/11 12:13:14 PM

1026 SECTION IX Toxicology
Yorifuji T et al: Long-term exposure to methylmercury and neurologic signs in Minamata and neighboring communities. Epidemiology 2008;19:3.
Chelating Agents
Agarwal MB: Deferasirox: Oral, once daily iron chelator—an expert opinion. Indian J Pediatr 2010;77:185.
Bradberry S, Vale A: A comparison of sodium calcium edetate (edetate calcium disodium) and succimer (DMSA) in the treatment of inorganic lead poison-ing. Clin Toxicol 2009;47:841.
Dargan PI et al: Case report: Severe mercuric sulphate poisoning treated with 2,3-dimercaptopropane-1-sulphonate and haemodiafiltration. Crit Care 2003;7:R1.
Gong Z et al: Determination of arsenic metabolic complex excreted in human urine after administration of sodium 2,3-dimercapto-1-propane sulfonate. Chem Res Toxicol 2002;15:1318.
Kosnett MJ: Chelation for heavy metals (arsenic, lead, and mercury): Protective or perilous? Clin Pharmacol Ther 2010;88:412.
Stangle DE et al: Succimer chelation improves learning, attention, and arousal regulation in lead-exposed rats but produces lasting cognitive impairment in the absence of lead exposure. Environ Health Perspect 2007;115:201.
Thompson DF, Called ED: Soluble or insoluble prussian blue for radiocesium and thallium poisoning? Ann Pharmacother 2004;38:1509.
C A S E S T U D Y A N S W E R
This scenario is highly suspicious for acute lead intoxica-tion. Lead-based paints have been commonly used as anticorrosion coatings on iron and steel structures, and grinding and torch cutting can result in high-dose expo-sure to inhaled lead dust and fumes. Measurement of a whole blood lead concentration would be a key diagnostic test. If an elevated blood lead concentration is confirmed, the primary therapeutic intervention will be removal of
the individual from further work exposure until blood lead concentration has declined and symptoms resolved. If the blood lead concentration is in excess of 80 mcg/dL (∼ 4 μmol/L), treatment with a chelating agent, such as oral succimer or parenteral edetate calcium disodium, should be strongly considered. Upon return to work, use of proper respiratory protection and adherence to protec-tive work practices are essential.
057-Katzung_Ch057_p1013-1026.indd 1026 9/21/11 12:13:15 PM

1027
SPECIAL ASPECTS OF TOXICOKINETICS
Volume of Distribution The volume of distribution (V d ) is defined as the apparent volume into which a substance is distributed in the body (see Chapter 3 ). A large V d implies that the drug is not readily accessible to mea-sures aimed at purifying the blood, such as hemodialysis. Examples of drugs with large volumes of distribution (> 5 L/kg) include antidepressants, antipsychotics, antimalarials, opioids, propra-nolol, and verapamil. Drugs with a relatively small V d (< 1 L/kg) include salicylate, ethanol, phenobarbital, lithium, valproic acid, and phenytoin (see Table 3–1 ).
Clearance Clearance is a measure of the volume of plasma that is cleared of drug per unit time (see Chapter 3 ). The total clearance for most drugs is the sum of clearances via excretion by the kidneys and metabolism by the liver. In planning a detoxification strategy, it is important to know the contribution of each organ to total clearance. For example, if a drug is 95% cleared by liver metabolism and only 5% cleared by renal excretion, even a dramatic increase in urinary concentration of the drug will have little effect on overall elimination.
Overdosage of a drug can alter the usual pharmacokinetic pro-cesses, and this must be considered when applying kinetics to
C H A P T E R
Over 1 million cases of acute poisoning occur in the USA each year, although only a small number are fatal. Most deaths are due to intentional suicidal overdose by an adolescent or adult. Childhood deaths due to accidental ingestion of a drug or toxic household product have been markedly reduced in the last 40 years as a result of safety packaging and effective poisoning prevention education.
Even with a serious exposure, poisoning is rarely fatal if the victim receives prompt medical attention and good supportive care. Careful management of respiratory failure, hypotension, seizures, and thermoregulatory disturbances has resulted in improved survival of patients who reach the hospital alive.
This chapter reviews the basic principles of poisoning, initial management, and specialized treatment of poisoning, including methods of increasing the elimination of drugs and toxins.
■ TOXICOKINETICS & TOXICODYNAMICS
The term toxicokinetics denotes the absorption, distribution, excre-tion, and metabolism of toxins, toxic doses of therapeutic agents, and their metabolites. The term toxicodynamics is used to denote the injurious effects of these substances on body functions. Although many similarities exist between the pharmacokinetics and toxicoki-netics of most substances, there are also important differences. The same caution applies to pharmacodynamics and toxicodynamics.
58Management of the Poisoned Patient Kent R. Olson, MD
administers a drug intravenously, followed by another substance via a nasogastric tube. The patient is admitted to the intensive care unit for continued supportive care and recovers the next morning. What drug might be used intravenously to prevent further seizures? What substance is commonly used to adsorb drugs still present in the gastrointestinal tract?
C A S E S T U D Y
A 62-year-old woman with a history of depression is found in her apartment in a lethargic state. An empty bottle of bupro-pion is on the bedside table. In the emergency department, she is unresponsive to verbal and painful stimuli. She has a brief generalized seizure, followed by a respiratory arrest. The emergency physician performs endotracheal intubation and
058-Katzung_Ch058_p1027-1038.indd 1027 9/21/11 12:13:35 PM

1028 SECTION IX Toxicology
poisoned patients. For example, dissolution of tablets or gastric emptying time may be slowed so that absorption and peak toxic effects are delayed. Drugs may injure the epithelial barrier of the gastrointestinal tract and thereby increase absorption. If the capac-ity of the liver to metabolize a drug is exceeded, the first-pass effect will be reduced and more drug will be delivered to the circulation. With a dramatic increase in the concentration of drug in the blood, protein-binding capacity may be exceeded, resulting in an increased fraction of free drug and greater toxic effect. At normal dosage, most drugs are eliminated at a rate proportional to the plasma concentration (first-order kinetics). If the plasma concen-tration is very high and normal metabolism is saturated, the rate of elimination may become fixed (zero-order kinetics). This change in kinetics may markedly prolong the apparent serum half-life and increase toxicity.
SPECIAL ASPECTS OF TOXICODYNAMICS
The general dose-response principles described in Chapter 2 are relevant when estimating the potential severity of an intoxication. When considering quantal dose-response data, both the therapeutic index and the overlap of therapeutic and toxic response curves must be considered. For instance, two drugs may have the same therapeu-tic index but unequal safe dosing ranges if the slopes of their dose-response curves are not the same. For some drugs, eg, sedative-hypnotics, the major toxic effect is a direct extension of the therapeutic action, as shown by their graded dose-response curve (see Figure 22–1 ). In the case of a drug with a linear dose-response curve (drug A), lethal effects may occur at 10 times the normal therapeutic dose. In contrast, a drug with a curve that reaches a plateau (drug B) may not be lethal at 100 times the normal dose.
For many drugs, at least part of the toxic effect may be different from the therapeutic action. For example, intoxication with drugs that have atropine-like effects (eg, tricyclic antidepressants) reduces sweating, making it more difficult to dissipate heat. In tricyclic antidepressant intoxication, there may also be increased muscular activity or seizures; the body’s production of heat is thus enhanced, and lethal hyperpyrexia may result. Overdoses of drugs that depress the cardiovascular system, eg, β blockers or calcium channel blockers, can profoundly alter not only cardiac function but all functions that are dependent on blood flow. These include renal and hepatic elimination of the toxin and that of any other drugs that may be given.
■ APPROACH TO THE POISONED PATIENT
HOW DOES THE POISONED PATIENT DIE?
An understanding of common mechanisms of death due to poisoning can help prepare the caregiver to treat patients effectively. Many toxins depress the central nervous system (CNS), resulting in obtundation or coma. Comatose patients frequently lose their airway protective
reflexes and their respiratory drive. Thus, they may die as a result of airway obstruction by the flaccid tongue, aspiration of gastric contents into the tracheobronchial tree, or respiratory arrest. These are the most common causes of death due to overdoses of narcotics and sedative-hypnotic drugs (eg, barbiturates and alcohol).
Cardiovascular toxicity is also frequently encountered in poison-ing. Hypotension may be due to depression of cardiac contractility; hypovolemia resulting from vomiting, diarrhea, or fluid sequestra-tion; peripheral vascular collapse due to blockade of α-adrenoceptor-mediated vascular tone; or cardiac arrhythmias. Hypothermia or hyperthermia due to exposure as well as the temperature-dysregulat-ing effects of many drugs can also produce hypotension. Lethal arrhythmias such as ventricular tachycardia and fibrillation can occur with overdoses of many cardioactive drugs such as ephedrine, amphetamines, cocaine, digitalis, and theophylline; and drugs not usually considered cardioactive, such as tricyclic antidepressants, antihistamines, and some opioid analogs.
Cellular hypoxia may occur in spite of adequate ventilation and oxygen administration when poisoning is due to cyanide, hydrogen sulfide, carbon monoxide, and other poisons that interfere with transport or utilization of oxygen. Such patients may not be cyan-otic, but cellular hypoxia is evident by the development of tachy-cardia, hypotension, severe lactic acidosis, and signs of ischemia on the electrocardiogram.
Seizures, muscular hyperactivity, and rigidity may result in death. Seizures may cause pulmonary aspiration, hypoxia, and brain damage. Hyperthermia may result from sustained muscular hyperactivity and can lead to muscle breakdown and myoglobinu-ria, renal failure, lactic acidosis, and hyperkalemia. Drugs and poisons that often cause seizures include antidepressants, isoniazid (INH), diphenhydramine, cocaine, and amphetamines.
Other organ system damage may occur after poisoning and is sometimes delayed in onset. Paraquat attacks lung tissue, resulting in pulmonary fibrosis, beginning several days after ingestion. Massive hepatic necrosis due to poisoning by acetaminophen or certain mushrooms results in hepatic encephalopathy and death 48–72 hours or longer after ingestion.
Finally, some patients may die before hospitalization because the behavioral effects of the ingested drug may result in traumatic injury. Intoxication with alcohol and other sedative-hypnotic drugs is a common contributing factor to motor vehicle accidents. Patients under the influence of hallucinogens such as phencycli-dine (PCP) or lysergic acid diethylamide (LSD) may suffer trauma when they become combative or fall from a height.
■ INITIAL MANAGEMENT OF THE POISONED PATIENT
The initial management of a patient with coma, seizures, or oth-erwise altered mental status should follow the same approach regardless of the poison involved: supportive measures are the basics ( “ABCDs” ) of poisoning treatment.
First, the airway should be cleared of vomitus or any other obstruction and an oral airway or endotracheal tube inserted if
058-Katzung_Ch058_p1027-1038.indd 1028 9/21/11 12:13:36 PM

CHAPTER 58 Management of the Poisoned Patient 1029
needed. For many patients, simple positioning in the lateral, left-side-down position is sufficient to move the flaccid tongue out of the airway. Breathing should be assessed by observation and pulse oxi-metry and, if in doubt, by measuring arterial blood gases. Patients with respiratory insufficiency should be intubated and mechanically ventilated. The circulation should be assessed by continuous moni-toring of pulse rate, blood pressure, urinary output, and evaluation of peripheral perfusion. An intravenous line should be placed and blood drawn for serum glucose and other routine determinations.
At this point, every patient with altered mental status should receive a challenge with concentrated dextrose , unless a rapid bedside blood glucose test demonstrates that the patient is not hypoglycemic. Adults are given 25 g (50 mL of 50% dextrose solution) intravenously, children 0.5 g/kg (2 mL/kg of 25% dex-trose). Hypoglycemic patients may appear to be intoxicated, and there is no rapid and reliable way to distinguish them from poi-soned patients. Alcoholic or malnourished patients should also receive 100 mg of thiamine intramuscularly or in the intravenous infusion solution at this time to prevent Wernicke’s syndrome.
The opioid antagonist naloxone may be given in a dose of 0.4–2 mg intravenously. Naloxone reverses respiratory and CNS depression due to all varieties of opioid drugs (see Chapter 31 ). It is useful to remember that these drugs cause death primarily by respiratory depression; therefore, if airway and breathing assistance have already been instituted, naloxone may not be necessary. Larger doses of naloxone may be needed for patients with overdose involv-ing propoxyphene, codeine, and some other opioids. The benzodi-azepine antagonist flumazenil (see Chapter 22 ) may be of value in patients with suspected benzodiazepine overdose, but it should not be used if there is a history of tricyclic antidepressant overdose or a seizure disorder, as it can induce convulsions in such patients.
History & Physical Examination Once the essential initial ABCD interventions have been instituted, one can begin a more detailed evaluation to make a specific diagno-sis. This includes gathering any available history and performing a toxicologically oriented physical examination. Other causes of coma or seizures such as head trauma, meningitis, or metabolic abnor-malities should be sought and treated. Some common intoxications are described under Common Toxic Syndromes.
A. History Oral statements about the amount and even the type of drug ingested in toxic emergencies may be unreliable. Even so, family members, police, and fire department or paramedical personnel should be asked to describe the environment in which the toxic emergency occurred and should bring to the emergency depart-ment any syringes, empty bottles, household products, or over-the-counter medications in the immediate vicinity of the possibly poisoned patient.
B. Physical Examination A brief examination should be performed, emphasizing those areas most likely to give clues to the toxicologic diagnosis. These include vital signs, eyes and mouth, skin, abdomen, and nervous system.
1. Vital signs— Careful evaluation of vital signs (blood pressure, pulse, respirations, and temperature) is essential in all toxicologic emergencies. Hypertension and tachycardia are typical with amphetamines, cocaine, and antimuscarinic (anticholinergic) drugs. Hypotension and bradycardia are characteristic features of overdose with calcium channel blockers, β blockers, clonidine, and sedative hypnotics. Hypotension with tachycardia is common with tricyclic antidepressants, trazodone, quetiapine, vasodilators, and β agonists. Rapid respirations are typical of salicylates, carbon monoxide, and other toxins that produce metabolic acidosis or cellular asphyxia. Hyperthermia may be associated with sympath-omimetics, anticholinergics, salicylates, and drugs producing sei-zures or muscular rigidity. Hypothermia can be caused by any CNS-depressant drug, especially when accompanied by exposure to a cold environment.
2. Eyes— The eyes are a valuable source of toxicologic information. Constriction of the pupils (miosis) is typical of opioids, clonidine, phenothiazines, and cholinesterase inhibitors (eg, organophosphate insecticides), and deep coma due to sedative drugs. Dilation of the pupils (mydriasis) is common with amphetamines, cocaine, LSD, and atropine and other anticholinergic drugs. Horizontal nystagmus is characteristic of intoxication with phenytoin, alcohol, barbiturates, and other sedative drugs. The presence of both vertical and horizon-tal nystagmus is strongly suggestive of phencyclidine poisoning. Ptosis and ophthalmoplegia are characteristic features of botulism.
3. Mouth— The mouth may show signs of burns due to corro-sive substances, or soot from smoke inhalation. Typical odors of alcohol, hydrocarbon solvents, or ammonia may be noted. Poisoning due to cyanide can be recognized by some examiners as an odor like bitter almonds.
4. Skin— The skin often appears flushed, hot, and dry in poison-ing with atropine and other antimuscarinics. Excessive sweating occurs with organophosphates, nicotine, and sympathomimetic drugs. Cyanosis may be caused by hypoxemia or by methemoglo-binemia. Icterus may suggest hepatic necrosis due to acetamino-phen or Amanita phalloides mushroom poisoning.
5. Abdomen— Abdominal examination may reveal ileus, which is typical of poisoning with antimuscarinic, opioid, and sedative drugs. Hyperactive bowel sounds, abdominal cramping, and diar-rhea are common in poisoning with organophosphates, iron, arsenic, theophylline, A phalloides , and A muscaria .
6. Nervous system— A careful neurologic examination is essen-tial. Focal seizures or motor deficits suggest a structural lesion (eg, intracranial hemorrhage due to trauma) rather than toxic or meta-bolic encephalopathy. Nystagmus, dysarthria, and ataxia are typi-cal of phenytoin, carbamazepine, alcohol, and other sedative intoxication. Twitching and muscular hyperactivity are common with atropine and other anticholinergic agents, and cocaine and other sympathomimetic drugs. Muscular rigidity can be caused by haloperidol and other antipsychotic agents, and by strychnine or by tetanus. Generalized hypertonicity of muscles and lower extremity clonus are typical of serotonin syndrome. Seizures are
058-Katzung_Ch058_p1027-1038.indd 1029 9/21/11 12:13:36 PM

1030 SECTION IX Toxicology
often caused by overdose with antidepressants (especially tricyclic antidepressants and bupropion [as in the case study]), cocaine, amphetamines, theophylline, isoniazid, and diphenhydramine. Flaccid coma with absent reflexes and even an isoelectric electro-encephalogram may be seen with deep coma due to sedative-hypnotic or other CNS depressant intoxication and may be mistaken for brain death.
Laboratory & Imaging Procedures A. Arterial Blood Gases Hypoventilation results in an elevated PCO2 (hypercapnia) and a low PO2 (hypoxia). The PO2 may also be low in a patient with aspiration pneumonia or drug-induced pulmonary edema. Poor tissue oxygenation due to hypoxia, hypotension, or cyanide poisoning will result in metabolic acidosis. The PO2 measures only oxygen dissolved in the plasma and not total blood oxygen content or oxyhemoglobin saturation and may appear normal in patients with severe carbon monoxide poisoning. Pulse oxi-metry may also give falsely normal results in carbon monoxide intoxication.
B. Electrolytes Sodium, potassium, chloride, and bicarbonate should be mea-sured. The anion gap is then calculated by subtracting the mea-sured anions from cations:
Anion gap = (Na+ + K+) – (HCO3– + Cl–)
Normally, the sum of the cations exceeds the sum of the anions by no more than 12–16 mEq/L (or 8–12 mEq/L if the formula used for estimating the anion gap omits the potassium level). A larger-than expected anion gap is caused by the presence of unmeasured anions (lactate, etc) accompanying metabolic acidosis. This may occur with numerous conditions, such as diabetic ketoacidosis, renal failure, or shock-induced lactic acidosis. Drugs that may induce an elevated anion gap metabolic acidosis ( Table 58–1 ) include aspirin, metformin, methanol, ethylene glycol, isoniazid, and iron.
Alterations in the serum potassium level are hazardous because they can result in cardiac arrhythmias. Drugs that may cause hyper-kalemia despite normal renal function include potassium itself, β
blockers, digitalis glycosides, potassium-sparing diuretics, and fluo-ride. Drugs associated with hypokalemia include barium, β ago-nists, caffeine, theophylline, and thiazide and loop diuretics.
C. Renal Function Tests Some toxins have direct nephrotoxic effects; in other cases, renal failure is due to shock or myoglobinuria. Blood urea nitrogen and creatinine levels should be measured and urinalysis performed. Elevated serum creatine kinase (CK) and myoglobin in the urine suggest muscle necrosis due to seizures or muscular rigidity. Oxalate crystals in large numbers in the urine suggest ethylene glycol poisoning.
D. Serum Osmolality The calculated serum osmolality is dependent mainly on the serum sodium and glucose and the blood urea nitrogen and can be estimated from the following formula:
2 × Na+ (mEq/L) + +Glucose (mg/dL)
18BUN (mg/dL)
3
This calculated value is normally 280–290 mOsm/L. Ethanol and other alcohols may contribute significantly to the measured serum osmolality but, since they are not included in the calcula-tion, cause an osmol gap:
Osmolargap
Measuredosmolality
Calculatedosmolality–=
Table 58–2 lists the concentration and expected contribution to the serum osmolality in ethanol, methanol, ethylene glycol, and isopropanol poisonings.
E. Electrocardiogram Widening of the QRS complex duration (to more than 100 milli-seconds) is typical of tricyclic antidepressant and quinidine over-doses ( Figure 58–1 ). The QT c interval may be prolonged (to more than 440 milliseconds) in many poisonings, including quinidine, antidepressants and antipsychotics, lithium, and arsenic (see also http://www.torsades.org/ ). Variable atrioventricular (AV) block and
TABLE 58–2 Some substances that can cause an osmol gap.
Substance1Serum Level (mg/dL)
Corresponding Osmol Gap (mOsm/kg)
Ethanol 350 75
Methanol 80 25
Ethylene glycol 200 35
Isopropanol 350 60
1Other substances that can increase the osmol gap include propylene glycol and other glycols, acetone, mannitol, and magnesium.
TABLE 58–1 Examples of drug-induced anion gap acidosis.
Type of Elevation of the Anion Gap Agents
Organic acid metabolites
Methanol, ethylene glycol, diethylene glycol
Lactic acidosis Cyanide, carbon monoxide, ibuprofen, isoniazid, metformin, salicylates, valproic acid; any drug-induced seizures, hypoxia, or hypotension
Note: The normal anion gap calculated from (Na+ + K+) − (HCO3− + Cl−) is 12–16 mEq/L;
calculated from (Na+) − (HCO3− + Cl−), it is 8–12 mEq/L.
058-Katzung_Ch058_p1027-1038.indd 1030 9/21/11 12:13:36 PM

CHAPTER 58 Management of the Poisoned Patient 1031
a variety of atrial and ventricular arrhythmias are common with poisoning by digoxin and other cardiac glycosides. Hypoxemia due to carbon monoxide poisoning may result in ischemic changes on the electrocardiogram.
F. Imaging Findings A plain film of the abdomen may be useful because some tablets, particularly iron and potassium, may be radiopaque. Chest radio-graphs may reveal aspiration pneumonia, hydrocarbon pneumo-nia, or pulmonary edema. When head trauma is suspected, a computed tomography (CT) scan is recommended.
Toxicology Screening Tests It is a common misconception that a broad toxicology “screen” is the best way to diagnose and manage an acute poisoning. Unfortunately, comprehensive toxicology screening is time- consuming and expensive and results of tests may not be available for days. Moreover, many highly toxic drugs such as calcium chan-nel blockers, β blockers, and isoniazid are not included in the screening process. The clinical examination of the patient and selected routine laboratory tests are usually sufficient to generate a tentative diagnosis and an appropriate treatment plan. Although screening tests may be helpful in confirming a suspected intoxica-tion or for ruling out intoxication as a cause of apparent brain death, they should not delay needed treatment.
When a specific antidote or other treatment is under consid-eration, quantitative laboratory testing may be indicated. For example, determination of the acetaminophen level is useful in assessing the need for antidotal therapy with acetylcysteine. Serum levels of salicylate (aspirin), ethylene glycol, methanol, theophylline, carbamazepine, lithium, valproic acid, and other drugs and poisons may indicate the need for hemodialysis ( Table 58–3 ).
Decontamination Decontamination procedures should be undertaken simultane-ously with initial stabilization, diagnostic assessment, and labora-tory evaluation. Decontamination involves removing toxins from the skin or gastrointestinal tract.
A. Skin Contaminated clothing should be completely removed and double-bagged to prevent illness in health care providers and for possible laboratory analysis. Wash contaminated skin with soap and water.
B. Gastrointestinal Tract Controversy remains regarding the efficacy of gut emptying by emesis or gastric lavage, especially when treatment is initiated more than 1 hour after ingestion. For most ingestions, clinical toxicologists recommend simple administration of activated char-coal to bind ingested poisons in the gut before they can be absorbed (as in the case study). In unusual circumstances, induced emesis or gastric lavage may also be used.
1. Emesis— Emesis can be induced with ipecac syrup (never extract of ipecac), and this method was previously used to treat some childhood ingestions at home under telephone supervision of a physician or poison control center personnel. However, the risks involved with inappropriate use outweighed the unproven
TABLE 58–3 Hemodialysis in drug overdose and poisoning.1
Hemodialysis may be indicated depending on the severity of poisoning or the blood concentration:
Carbamazepine
Ethylene glycol
Lithium
Methanol
Metformin
Phenobarbital
Salicylate
Theophylline
Valproic acid
Hemodialysis is ineffective or is not useful:
Amphetamines
Antidepressants
Antipsychotic drugs
Benzodiazepines
Calcium channel blockers
Digoxin
Metoprolol and propranolol
Opioids
1This listing is not comprehensive.
A
B
C
FIGURE 58–1 Changes in the electrocardiogram in tricyclic anti-depressant overdosage. A : Slowed intraventricular conduction results in prolonged QRS interval (0.18 s; normal, 0.08 s). B and C: Supraventricular tachycardia with progressive widening of QRS com-plexes mimics ventricular tachycardia. (Reproduced, with permission, from
Benowitz NL, Goldschlager N: Cardiac disturbances in the toxicologic patient. In:
Haddad LM, Winchester JF [editors]. Clinical Management of Poisoning and Drug
Overdose. WB Saunders, 1983.)
058-Katzung_Ch058_p1027-1038.indd 1031 9/21/11 12:13:36 PM

1032 SECTION IX Toxicology
benefits, and this treatment is rarely used in the home or hospital. Ipecac should not be used if the suspected intoxicant is a corrosive agent, a petroleum distillate, or a rapid-acting convulsant. Previously popular methods of inducing emesis such as fingertip stimulation of the pharynx, salt water, and apomorphine are inef-fective or dangerous and should not be used.
2. Gastric lavage— If the patient is awake or if the airway is protected by an endotracheal tube, gastric lavage may be per-formed using an orogastric or nasogastric tube—as large a tube as possible. Lavage solutions (usually 0.9% saline) should be at body temperature to prevent hypothermia.
3. Activated charcoal— Owing to its large surface area, acti-vated charcoal can adsorb many drugs and poisons. It is most effective if given in a ratio of at least 10:1 of charcoal to estimated dose of toxin by weight. Charcoal does not bind iron, lithium, or potassium, and it binds alcohols and cyanide only poorly. It does not appear to be useful in poisoning due to corrosive mineral acids and alkali. Studies suggest that oral activated charcoal given alone may be just as effective as gut emptying (eg, ipecac-induced emesis or gastric lavage) followed by charcoal. Repeated doses of oral activated charcoal may enhance systemic elimination of some drugs (including carbamazepine, dapsone, and theophylline) by a mechanism referred to as “gut dialysis,” although the clinical ben-efit is unproved.
4. Cathartics— Administration of a cathartic (laxative) agent may hasten removal of toxins from the gastrointestinal tract and reduce absorption, although no controlled studies have been done. Whole bowel irrigation with a balanced polyethylene glycol- electrolyte solution (GoLYTELY, CoLyte) can enhance gut decon-tamination after ingestion of iron tablets, enteric-coated medicines, illicit drug-filled packets, and foreign bodies. The solution is administered orally at 1–2 L/h (500 mL/h in children) for several hours until the rectal effluent is clear.
Specific Antidotes There is a popular misconception that there is an antidote for every poison. Actually, selective antidotes are available for only a few classes of toxins. The major antidotes and their characteristics are listed in Table 58–4 .
Methods of Enhancing Elimination of Toxins After appropriate diagnostic and decontamination procedures and administration of antidotes, it is important to consider whether measures for enhancing elimination, such as hemodialysis or uri-nary alkalinization, can improve the clinical outcome. Table 58–3 lists intoxications for which dialysis may be beneficial.
A. Dialysis Procedures 1. Peritoneal dialysis— Although it is a relatively simple and available technique, peritoneal dialysis is inefficient in removing most drugs.
2. Hemodialysis— Hemodialysis is more efficient than perito-neal dialysis and has been well studied. It assists in correction of fluid and electrolyte imbalance and may also enhance removal of toxic metabolites (eg, formic acid in methanol poisoning; oxalic and glycolic acids in ethylene glycol poisoning). The efficiency of both peritoneal dialysis and hemodialysis is a function of the molecular weight, water solubility, protein binding, endogenous clearance, and distribution in the body of the specific toxin. Hemodialysis is especially useful in overdose cases in which the precipitating drug can be removed and fluid and electrolyte imbal-ances are present and can be corrected (eg, salicylate intoxication).
B. Forced Diuresis and Urinary pH Manipulation Previously popular but of unproved value, forced diuresis may cause volume overload and electrolyte abnormalities and is not recom-mended. Renal elimination of a few toxins can be enhanced by alteration of urinary pH. For example, urinary alkalinization is use-ful in cases of salicylate overdose. Acidification may increase the urine concentration of drugs such as phencyclidine and amphet-amines but is not advised because it may worsen renal complications from rhabdomyolysis, which often accompanies the intoxication.
■ COMMON TOXIC SYNDROMES
ACETAMINOPHEN
Acetaminophen is one of the drugs commonly involved in suicide attempts and accidental poisonings, both as the sole agent and in combination with other drugs. Acute ingestion of more than 150–200 mg/kg (children) or 7 g total (adults) is considered potentially toxic. A highly toxic metabolite is produced in the liver (see Figure 4–5 ).
Initially, the patient is asymptomatic or has mild gastrointestinal upset (nausea, vomiting). After 24–36 hours, evidence of liver injury appears, with elevated aminotransferase levels and hypoprothrom-binemia. In severe cases, fulminant liver failure occurs, leading to hepatic encephalopathy and death. Renal failure may also occur.
The severity of poisoning is estimated from a serum acetamin-ophen concentration measurement. If the level is greater than 150–200 mg/L approximately 4 hours after ingestion, the patient is at risk for liver injury. (Chronic alcoholics or patients taking drugs that enhance P450 production of toxic metabolites are at risk with lower levels.) The antidote acetylcysteine acts as a gluta-thione substitute, binding the toxic metabolite as it is produced. It is most effective when given early and should be started within 8–10 hours if possible. Liver transplantation may be required for patients with fulminant hepatic failure.
AMPHETAMINES & OTHER STIMULANTS
Stimulant drugs commonly abused in the USA include metham-phetamine (“crank,” “crystal”), methylenedioxymethamphetamine (MDMA, “ecstasy”), and cocaine (“crack”) as well as pharmaceu-ticals such as pseudoephedrine (Sudafed) and ephedrine (as such
058-Katzung_Ch058_p1027-1038.indd 1032 9/21/11 12:13:36 PM

CHAPTER 58 Management of the Poisoned Patient 1033
and in the herbal agent Ma-huang ) (see Chapter 32 ). Caffeine is often added to dietary supplements sold as “metabolic enhancers” or “fat-burners.” Newer synthetic analogs of amphetamines such as 3,4-methylenedioxypyrovalerone (MDPV) and various deriva-tives of methcathinone are becoming popular drugs of abuse, often sold on the street as “bath salts” with names like “Ivory Wave,” “Bounce,” “Bubbles,” “Mad Cow,” and “Meow Meow.”
At the doses usually used by stimulant abusers, euphoria and wakefulness are accompanied by a sense of power and well-being. At higher doses, restlessness, agitation, and acute psychosis may
occur, accompanied by hypertension and tachycardia. Prolonged muscular hyperactivity or seizures may contribute to hyperthermia and rhabdomyolysis. Body temperatures as high as 42°C (107.6°F) have been recorded. Hyperthermia can cause brain damage, hypotension, coagulopathy, and renal failure.
Treatment for stimulant toxicity includes general supportive measures as outlined earlier. There is no specific antidote. Seizures and hyperthermia are the most dangerous manifestations and must be treated aggressively. Seizures are usually managed with intravenous benzodiazepines (eg, lorazepam). Temperature is
TABLE 58–4 Examples of specific antidotes.
Antidote Poison(s) Comments
Acetylcysteine (Acetadote, Mucomyst)
Acetaminophen Best results if given within 8–10 hours of overdose. Follow liver function tests and acetaminophen blood levels. Acetadote is given intravenously; Mucomyst is given orally.
Atropine Anticholinesterase intoxication: organophosphates, carbamates
An initial dose of 1–2 mg (for children, 0.05 mg/kg) is given IV, and if there is no response, the dose is doubled every 10–15 minutes, with decreased wheezing and pulmonary secretions as therapeutic end points.
Bicarbonate, sodium Membrane-depressant cardiotoxic drugs (tricyclic antidepressants, quinidine, etc)
1–2 mEq/kg IV bolus usually reverses cardiotoxic effects (wide QRS, hypotension). Give cautiously in heart failure (avoid sodium overload).
Calcium Fluoride; calcium channel blockers Large doses may be needed in severe calcium channel blocker overdose. Start with 15 mg/kg IV.
Deferoxamine Iron salts If poisoning is severe, give 15 mg/kg/h IV. 100 mg of deferoxamine binds 8.5 mg of iron.
Digoxin antibodies Digoxin and related cardiac glycosides
One vial binds 0.5 mg digoxin; indications include serious arrhythmias, hyper-kalemia.
Esmolol Theophylline, caffeine, metaproterenol
Short-acting β blocker. Infuse 25–50 mcg/kg/min IV.
Ethanol Methanol, ethylene glycol A loading dose is calculated so as to give a blood level of at least 100 mg/dL (42 g/70 kg in adults). Fomepizole (see below) is easier to use.
Flumazenil Benzodiazepines Adult dose is 0.2 mg IV, repeated as necessary to a maximum of 3 mg. Do not give to patients with seizures, benzodiazepine dependence, or tricyclic overdose.
Fomepizole Methanol, ethylene glycol More convenient than ethanol. Give 15 mg/kg; repeat every 12 hours.
Glucagon β blockers 5–10 mg IV bolus may reverse hypotension and bradycardia.
Hydroxocobalamin Cyanide Adult dose is 5 g IV over 15 minutes. Converts cyanide to cyanocobalamin (vitamin B12).
Naloxone Narcotic drugs, other opioid derivatives
A specific antagonist of opioids; give 0.4–2 mg initially by IV, IM, or SC injection. Larger doses may be needed to reverse the effects of overdose with propoxy-phene, codeine, or fentanyl derivatives. Duration of action (2–3 hours) may be significantly shorter than that of the opioid being antagonized.
Oxygen Carbon monoxide Give 100% by high-flow nonrebreathing mask; use of hyperbaric chamber is con-troversial but often recommended for severe poisoning.
Physostigmine Suggested for delirium caused by anticholinergic agents
Adult dose is 0.5–1 mg IV slowly. The effects are transient (30–60 minutes), and the lowest effective dose may be repeated when symptoms return. May cause bradycardia, increased bronchial secretions, seizures. Have atropine ready to reverse excess effects. Do not use for tricyclic antidepressant overdose.
Pralidoxime (2-PAM) Organophosphate (OP) cholinesterase inhibitors
Adult dose is 1 g IV, which should be repeated every 3–4 hours as needed or preferably as a constant infusion of 250–400 mg/h. Pediatric dose is approxi-mately 250 mg. No proved benefit in carbamate poisoning; uncertain benefit in established OP poisoning.
058-Katzung_Ch058_p1027-1038.indd 1033 9/21/11 12:13:36 PM

1034 SECTION IX Toxicology
reduced by removing clothing, spraying with tepid water, and encouraging evaporative cooling with fanning. For very high body temperatures (eg, > 40–41°C [104–105.8°F]), neuromuscular paralysis is used to abolish muscle activity quickly.
ANTICHOLINERGIC AGENTS
A large number of prescription and nonprescription drugs, as well as a variety of plants and mushrooms, can inhibit the effects of acetylcholine at muscarinic receptors. Some drugs used for other purposes (eg, antihistamines) also have anticholinergic effects, in addition to other potentially toxic actions. For example, antihista-mines such as diphenhydramine can cause seizures; tricyclic anti-depressants, which have anticholinergic, quinidine-like, and α-blocking effects, can cause severe cardiovascular toxicity.
The classic anticholinergic (technically, “antimuscarinic”) syn-drome is remembered as “red as a beet” (skin flushed), “hot as a hare” (hyperthermia), “dry as a bone” (dry mucous membranes, no sweating), “blind as a bat” (blurred vision, cycloplegia), and “mad as a hatter” (confusion, delirium). Patients usually have sinus tachy-cardia, and the pupils are usually dilated (see Chapter 8 ). Agitated delirium or coma may be present. Muscle twitching is common, but seizures are unusual unless the patient has ingested an antihis-tamine or a tricyclic antidepressant. Urinary retention is common, especially in older men.
Treatment for anticholinergic syndrome is largely supportive. Agitated patients may require sedation with a benzodiazepine or an antipsychotic agent (eg, haloperidol). The specific antidote for peripheral and central anticholinergic syndrome is physostigmine, which has a prompt and dramatic effect and is especially useful for patients who are very agitated. Physostigmine is given in small intravenous doses (0.5–1 mg) with careful monitoring, because it can cause bradycardia and seizures if given too rapidly. Physostigmine should not be given to a patient with suspected tricyclic antidepressant overdose because it can aggravate cardio-toxicity, resulting in heart block or asystole. Catheterization may be needed to prevent excessive distention of the bladder.
ANTIDEPRESSANTS
Tricyclic antidepressants (eg, amitriptyline, desipramine, doxe-pin, many others; see Chapter 30 ) are among the most common prescription drugs involved in life-threatening drug overdose. Ingestion of more than 1 g of a tricyclic (or about 15–20 mg/kg) is considered potentially lethal.
Tricyclic antidepressants are competitive antagonists at musca-rinic cholinergic receptors, and anticholinergic findings (tachycar-dia, dilated pupils, dry mouth) are common even at moderate doses. Some tricyclics are also strong α blockers, which can lead to vasodilation. Centrally mediated agitation and seizures may be followed by depression and hypotension. Most important is the fact that tricyclics have quinidine-like cardiac depressant effects on the sodium channel that cause slowed conduction with a wide QRS interval and depressed cardiac contractility. This cardiac
toxicity may result in serious arrhythmias ( Figure 58–1 ), including ventricular conduction block and ventricular tachycardia.
Treatment of tricyclic antidepressant overdose includes general supportive care as outlined earlier. Endotracheal intubation and assisted ventilation may be needed. Intravenous fluids are given for hypotension, and dopamine or norepinephrine is added if necessary. Many toxicologists recommend norepinephrine as the initial drug of choice for tricyclic-induced hypotension. The anti-dote for quinidine-like cardiac toxicity (manifested by a wide QRS complex) is sodium bicarbonate: a bolus of 50–100 mEq (or 1–2 mEq/kg) provides a rapid increase in extracellular sodium that helps overcome sodium channel blockade. Do not use physo-stigmine ! Although this agent does effectively reverse anticholin-ergic symptoms, it can aggravate depression of cardiac conduction and cause seizures.
Monoamine oxidase inhibitors (eg, tranylcypromine, phenel-zine) are older antidepressants that are occasionally used for resis-tant depression. They can cause severe hypertensive reactions when interacting foods or drugs are taken (see Chapters 9 and 30 ), and they can interact with the selective serotonin reuptake inhibi-tors (SSRIs).
Newer antidepressants (eg, fluoxetine, paroxetine, citalopram, venlafaxine) are mostly SSRIs and are generally safer than the tri-cyclic antidepressants and monoamine oxidase inhibitors, although they can cause seizures. Bupropion (not an SSRI) has caused sei-zures even in therapeutic doses. Some antidepressants have been associated with QT prolongation and torsades de pointes arrhyth-mia. SSRIs may interact with each other or especially with mono-amine oxidase inhibitors to cause the serotonin syndrome, characterized by agitation, muscle hyperactivity, and hyperthermia (see Chapter 16 ).
ANTIPSYCHOTICS
Antipsychotic drugs include the older phenothiazines and butyro-phenones, as well as newer atypical drugs. All of these can cause CNS depression, seizures, and hypotension. Some can cause QT prolongation. The potent dopamine D 2 blockers are also associ-ated with parkinsonian movement disorders (dystonic reactions) and in rare cases with the neuroleptic malignant syndrome, char-acterized by “lead-pipe” rigidity, hyperthermia, and autonomic instability (see Chapters 16 and 29 ).
ASPIRIN (SALICYLATE)
Salicylate poisoning (see Chapter 36 ) is a much less common cause of childhood poisoning deaths since the introduction of child-resistant containers and the reduced use of children’s aspirin. It still accounts for numerous suicidal and accidental poisonings. Acute ingestion of more than 200 mg/kg is likely to produce intoxication. Poisoning can also result from chronic overmedica-tion; this occurs most commonly in elderly patients using salicy-lates for chronic pain who become confused about their dosing. Poisoning causes uncoupling of oxidative phosphorylation and disruption of normal cellular metabolism.
058-Katzung_Ch058_p1027-1038.indd 1034 9/21/11 12:13:36 PM

CHAPTER 58 Management of the Poisoned Patient 1035
The first sign of salicylate toxicity is often hyperventilation and respiratory alkalosis due to medullary stimulation. Metabolic aci-dosis follows, and an increased anion gap results from accumula-tion of lactate as well as excretion of bicarbonate by the kidney to compensate for respiratory alkalosis. Arterial blood gas testing often reveals a mixed respiratory alkalosis and metabolic acidosis. Body temperature may be elevated owing to uncoupling of oxida-tive phosphorylation. Severe hyperthermia may occur in serious cases. Vomiting and hyperpnea as well as hyperthermia contribute to fluid loss and dehydration. With very severe poisoning, pro-found metabolic acidosis, seizures, coma, pulmonary edema, and cardiovascular collapse may occur. Absorption of salicylate and signs of toxicity may be delayed after very large overdoses or inges-tion of enteric coated tablets.
General supportive care is essential. After massive aspirin inges-tions (eg, more than 100 tablets), aggressive gut decontamination is advisable, including gastric lavage, repeated doses of activated charcoal, and consideration of whole bowel irrigation. Intravenous fluids are used to replace fluid losses caused by tachypnea, vomit-ing, and fever. For moderate intoxications, intravenous sodium bicarbonate is given to alkalinize the urine and promote salicylate excretion by trapping the salicylate in its ionized, polar form. For severe poisoning (eg, patients with severe acidosis, coma, and serum salicylate level > 100 mg/dL), emergency hemodialysis is performed to remove the salicylate more quickly and restore acid-base balance and fluid status.
BETA BLOCKERS
In overdose, β blockers inhibit both β 1 and β 2 adrenoceptors; selectivity, if any, is lost at high dosage. The most toxic β blocker is propranolol. As little as two to three times the therapeutic dose can cause serious toxicity. This may be because propranolol in high doses may cause sodium channel blocking effects similar to those seen with quinidine-like drugs, and it is lipophilic, allowing it to enter the CNS (see Chapter 10 ).
Bradycardia and hypotension are the most common manifesta-tions of toxicity. Agents with partial agonist activity (eg, pindolol) can cause tachycardia and hypertension. Seizures and cardiac conduction block (wide QRS complex) may be seen with propranolol overdose.
General supportive care should be provided as outlined earlier. The usual measures used to raise the blood pressure and heart rate, such as intravenous fluids, β-agonist drugs, and atropine, are gener-ally ineffective. Glucagon is a useful antidote that—like β agonists—acts on cardiac cells to raise intracellular cAMP but does so independent of β adrenoceptors. It can improve heart rate and blood pressure when given in high doses (5–20 mg intravenously).
CALCIUM CHANNEL BLOCKERS
Calcium antagonists can cause serious toxicity or death with rela-tively small overdoses. These channel blockers depress sinus node automaticity and slow AV node conduction (see Chapter 12 ). They also reduce cardiac output and blood pressure. Serious
hypotension is mainly seen with nifedipine and related dihydro-pyridines, but in severe overdose all of the listed cardiovascular effects can occur with any of the calcium channel blockers.
Treatment requires general supportive care. Since most ingested calcium antagonists are in sustained-release form, it may be pos-sible to expel them before they are completely absorbed; initiate whole bowel irrigation and oral activated charcoal as soon as pos-sible, before calcium antagonist-induced ileus intervenes. Calcium, given intravenously in doses of 2–10 g, is a useful antidote for depressed cardiac contractility but less effective for nodal block or peripheral vascular collapse. Other treatments reported to be help-ful in managing hypotension associated with calcium channel blocker poisoning include glucagon and high-dose insulin (0.5–1 unit/kg/h) plus glucose supplementation to maintain eugly-cemia. Recently case reports have suggested benefit from adminis-tration of lipid emulsion (Intralipid, normally used as an intravenous dietary fat supplement) for severe verapamil overdose.
CARBON MONOXIDE & OTHER TOXIC GASES
Carbon monoxide (CO) is a colorless, odorless gas that is ubiqui-tous because it is created whenever carbon-containing materials are burned. Carbon monoxide poisoning is the leading cause of death due to poisoning in the USA. Most cases occur in victims of fires, but accidental and suicidal exposures are also common. The diagnosis and treatment of carbon monoxide poisoning are described in Chapter 56 . Many other toxic gases are produced in fires or released in industrial accidents ( Table 58–5 ).
CHOLINESTERASE INHIBITORS
Organophosphate and carbamate cholinesterase inhibitors (see Chapter 7 ) are widely used to kill insects and other pests. Most cases of serious organophosphate or carbamate poisoning result from intentional ingestion by a suicidal person, but poisoning has also occurred at work (pesticide application or packaging) or, rarely, as a result of food contamination or terrorist attack (eg, release of the chemical warfare nerve agent sarin in the Tokyo subway system in 1995).
Stimulation of muscarinic receptors causes abdominal cramps, diarrhea, excessive salivation, sweating, urinary frequency, and increased bronchial secretions (see Chapters 6 and 7 ). Stimulation of nicotinic receptors causes generalized ganglionic activation, which can lead to hypertension and either tachycardia or brady-cardia. Muscle twitching and fasciculations may progress to weak-ness and respiratory muscle paralysis. CNS effects include agitation, confusion, and seizures. The mnemonic DUMBELS (diarrhea, urination, miosis and muscle weakness, bronchospasm, excitation, lacrimation, and seizures, sweating, and salivation) helps recall the common findings. Blood testing may be used to document depressed activity of red blood cell (acetylcholinest-erase) and plasma (butyrylcholinesterase) enzymes, which provide an indirect estimate of synaptic cholinesterase activity.
058-Katzung_Ch058_p1027-1038.indd 1035 9/21/11 12:13:36 PM
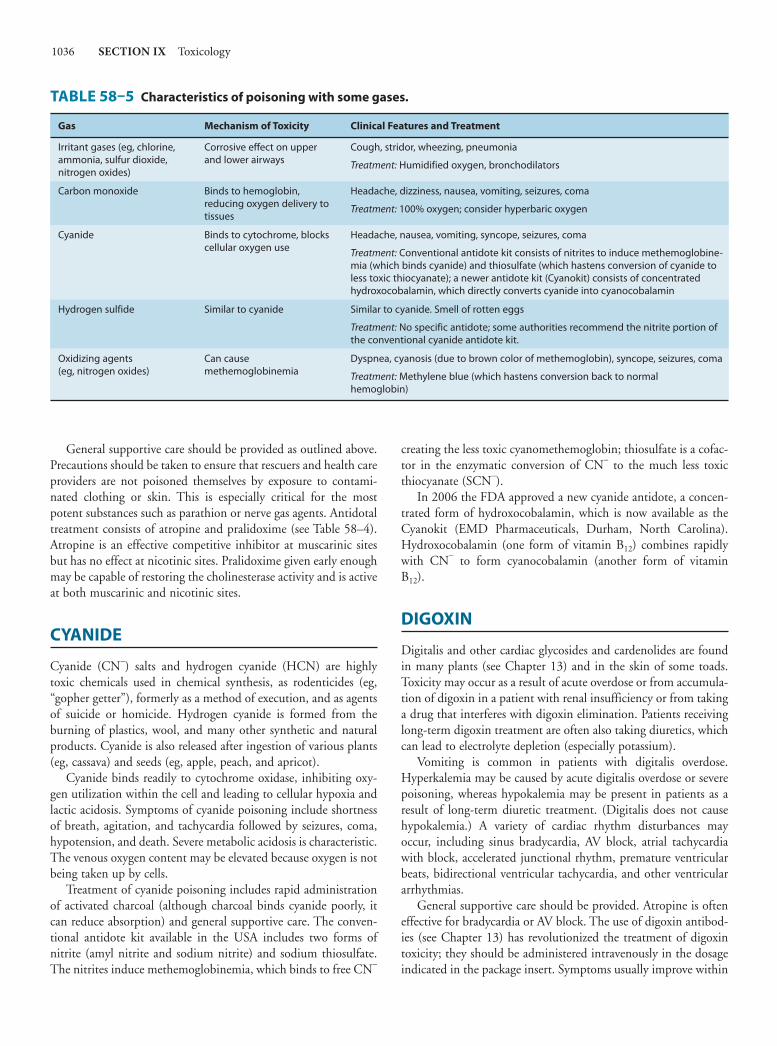
1036 SECTION IX Toxicology
General supportive care should be provided as outlined above. Precautions should be taken to ensure that rescuers and health care providers are not poisoned themselves by exposure to contami-nated clothing or skin. This is especially critical for the most potent substances such as parathion or nerve gas agents. Antidotal treatment consists of atropine and pralidoxime (see Table 58–4 ). Atropine is an effective competitive inhibitor at muscarinic sites but has no effect at nicotinic sites. Pralidoxime given early enough may be capable of restoring the cholinesterase activity and is active at both muscarinic and nicotinic sites.
CYANIDE
Cyanide (CN − ) salts and hydrogen cyanide (HCN) are highly toxic chemicals used in chemical synthesis, as rodenticides (eg, “gopher getter”), formerly as a method of execution, and as agents of suicide or homicide. Hydrogen cyanide is formed from the burning of plastics, wool, and many other synthetic and natural products. Cyanide is also released after ingestion of various plants (eg, cassava) and seeds (eg, apple, peach, and apricot).
Cyanide binds readily to cytochrome oxidase, inhibiting oxy-gen utilization within the cell and leading to cellular hypoxia and lactic acidosis. Symptoms of cyanide poisoning include shortness of breath, agitation, and tachycardia followed by seizures, coma, hypotension, and death. Severe metabolic acidosis is characteristic. The venous oxygen content may be elevated because oxygen is not being taken up by cells.
Treatment of cyanide poisoning includes rapid administration of activated charcoal (although charcoal binds cyanide poorly, it can reduce absorption) and general supportive care. The conven-tional antidote kit available in the USA includes two forms of nitrite (amyl nitrite and sodium nitrite) and sodium thiosulfate. The nitrites induce methemoglobinemia, which binds to free CN −
creating the less toxic cyanomethemoglobin; thiosulfate is a cofac-tor in the enzymatic conversion of CN − to the much less toxic thiocyanate (SCN − ).
In 2006 the FDA approved a new cyanide antidote, a concen-trated form of hydroxocobalamin, which is now available as the Cyanokit (EMD Pharmaceuticals, Durham, North Carolina). Hydroxocobalamin (one form of vitamin B 12 ) combines rapidly with CN − to form cyanocobalamin (another form of vitamin B 12 ).
DIGOXIN
Digitalis and other cardiac glycosides and cardenolides are found in many plants (see Chapter 13 ) and in the skin of some toads. Toxicity may occur as a result of acute overdose or from accumula-tion of digoxin in a patient with renal insufficiency or from taking a drug that interferes with digoxin elimination. Patients receiving long-term digoxin treatment are often also taking diuretics, which can lead to electrolyte depletion (especially potassium).
Vomiting is common in patients with digitalis overdose. Hyperkalemia may be caused by acute digitalis overdose or severe poisoning, whereas hypokalemia may be present in patients as a result of long-term diuretic treatment. (Digitalis does not cause hypokalemia.) A variety of cardiac rhythm disturbances may occur, including sinus bradycardia, AV block, atrial tachycardia with block, accelerated junctional rhythm, premature ventricular beats, bidirectional ventricular tachycardia, and other ventricular arrhythmias.
General supportive care should be provided. Atropine is often effective for bradycardia or AV block. The use of digoxin antibod-ies (see Chapter 13 ) has revolutionized the treatment of digoxin toxicity; they should be administered intravenously in the dosage indicated in the package insert. Symptoms usually improve within
TABLE 58–5 Characteristics of poisoning with some gases.
Gas Mechanism of Toxicity Clinical Features and Treatment
Irritant gases (eg, chlorine, ammonia, sulfur dioxide, nitrogen oxides)
Corrosive effect on upper and lower airways
Cough, stridor, wheezing, pneumonia
Treatment: Humidified oxygen, bronchodilators
Carbon monoxide Binds to hemoglobin, reducing oxygen delivery to tissues
Headache, dizziness, nausea, vomiting, seizures, coma
Treatment: 100% oxygen; consider hyperbaric oxygen
Cyanide Binds to cytochrome, blocks cellular oxygen use
Headache, nausea, vomiting, syncope, seizures, coma
Treatment: Conventional antidote kit consists of nitrites to induce methemoglobine-mia (which binds cyanide) and thiosulfate (which hastens conversion of cyanide to less toxic thiocyanate); a newer antidote kit (Cyanokit) consists of concentrated hydroxocobalamin, which directly converts cyanide into cyanocobalamin
Hydrogen sulfide Similar to cyanide Similar to cyanide. Smell of rotten eggs
Treatment: No specific antidote; some authorities recommend the nitrite portion of the conventional cyanide antidote kit.
Oxidizing agents (eg, nitrogen oxides)
Can cause methemoglobinemia
Dyspnea, cyanosis (due to brown color of methemoglobin), syncope, seizures, coma
Treatment: Methylene blue (which hastens conversion back to normal hemoglobin)
058-Katzung_Ch058_p1027-1038.indd 1036 9/21/11 12:13:37 PM

CHAPTER 58 Management of the Poisoned Patient 1037
30–60 minutes after antibody administration. Digoxin antibodies may also be tried in cases of poisoning by other cardiac glycosides (eg, digitoxin, oleander), although larger doses may be needed due to incomplete cross-reactivity.
ETHANOL & SEDATIVE-HYPNOTIC DRUGS
Overdosage with ethanol and sedative-hypnotic drugs (eg, benzo-diazepines, barbiturates, γ-hydroxybutyrate [GHB], carisoprodol [Soma]; see Chapters 22 and 23 ) occurs frequently because of their common availability and use.
Patients with ethanol or other sedative-hypnotic overdose may be euphoric and rowdy (“drunk”) or in a state of stupor or coma (“dead drunk”). Comatose patients often have depressed respira-tory drive. Depression of protective airway reflexes may result in pulmonary aspiration of gastric contents leading to pneumonia. Hypothermia may be present because of environmental exposure and depressed shivering. Ethanol blood levels greater than 300 mg/dL usually cause deep coma, but regular users are often tol-erant to the effects of ethanol and may be ambulatory despite even higher levels. Patients with GHB overdose are often deeply comatose for 3–4 hours and then awaken fully in a matter of minutes.
General supportive care should be provided. With careful attention to protecting the airway (including endotracheal intuba-tion) and assisting ventilation, most patients recover as the drug effects wear off. Hypotension usually responds to intravenous fluids, body warming if cold, and, if needed, dopamine. Patients with isolated benzodiazepine overdose may awaken after intrave-nous flumazenil, a benzodiazepine antagonist. However, this drug is not widely used as empiric therapy for drug overdose because it may precipitate seizures in patients who are addicted to benzodi-azepines or who have ingested a convulsant drug (eg, a tricyclic antidepressant). There are no antidotes for ethanol, barbiturates, or most other sedative-hypnotics.
ETHYLENE GLYCOL & METHANOL
Ethylene glycol and methanol are alcohols that are important toxins because of their metabolism to highly toxic organic acids (see Chapter 23 ). They are capable of causing CNS depression and a drunken state similar to ethanol overdose. In addition, their products of metabolism—formic acid (from methanol) or hippuric, oxalic, and glycolic acids (from ethylene glycol)—cause a severe metabolic acidosis and can lead to coma and blindness (in the case of formic acid) or renal failure (from oxalic acid and glycolic acid). Initially, the patient appears drunk, but after a delay of up to several hours, a severe anion gap metabolic acidosis becomes apparent, accompanied by hyperventilation and altered mental status. Patients with meth-anol poisoning may have visual disturbances ranging from blurred vision to blindness.
Metabolism of ethylene glycol and methanol to their toxic prod-ucts can be blocked by inhibiting the enzyme alcohol dehydrogenase
with a competing drug, such as fomepizole (4-methylpyrazole). Ethanol is also an effective antidote, but it can be difficult to achieve a safe and effective blood level.
IRON & OTHER METALS
Iron is widely used in over-the-counter vitamin preparations and is a leading cause of childhood poisoning deaths. As few as 10–12 prenatal multivitamins with iron may cause serious illness in a small child. Poisoning with other metals (lead, mercury, arsenic) is also important, especially in industry. See Chapters 33, 56, and 57 for detailed discussions of poisoning by iron and other metals.
OPIOIDS
Opioids (opium, morphine, heroin, meperidine, methadone, etc) are common drugs of abuse (see Chapters 31 and 32 ), and over-dose is a common result of using the poorly standardized prepara-tions sold on the street. See Chapter 31 for a detailed discussion of opioid overdose and its treatment.
RATTLESNAKE ENVENOMATION
In the USA, rattlesnakes are the most common venomous reptiles. Bites are rarely fatal, and 20% do not involve envenomation. However, about 60% of bites cause significant morbidity due to the destructive digestive enzymes found in the venom. Evidence of rattlesnake envenomation includes severe pain, swelling, bruising, hemorrhagic bleb formation, and obvious fang marks. Systemic effects include nausea, vomiting, muscle fasciculations, tingling and metallic taste in the mouth, shock, and systemic coagulopathy with prolonged clotting time and reduced platelet count.
Studies have shown that emergency field remedies such as inci-sion and suction, tourniquets, and ice packs are far more damag-ing than useful. Avoidance of unnecessary motion, on the other hand, does help to limit the spread of the venom. Definitive therapy relies on intravenous antivenom (also known as antivenin) and this should be started as soon as possible.
THEOPHYLLINE
Although it has been largely replaced by inhaled β agonists, theo-phylline continues to be used for the treatment of bronchospasm by some patients with asthma and bronchitis (see Chapter 20 ). A dose of 20–30 tablets can cause serious or fatal poisoning. Chronic or subacute theophylline poisoning can also occur as a result of accidental overmedication or use of a drug that interferes with theophylline metabolism (eg, cimetidine, ciprofloxacin, erythro-mycin; see Chapter 4 ).
In addition to sinus tachycardia and tremor, vomiting is com-mon after overdose. Hypotension, tachycardia, hypokalemia, and hyperglycemia may occur, probably owing to β 2 -adrenergic activa-tion. The cause of this activation is not fully understood, but the
058-Katzung_Ch058_p1027-1038.indd 1037 9/21/11 12:13:37 PM

1038 SECTION IX Toxicology
effects can be ameliorated by β blockers (see below). Cardiac arrhythmias include atrial tachycardias, premature ventricular contractions, and ventricular tachycardia. In severe poisoning (eg, acute overdose with serum level > 100 mg/L), seizures often occur and are usually resistant to common anticonvulsants. Toxicity may be delayed in onset for many hours after ingestion of sustained-release tablet formulations.
General supportive care should be provided. Aggressive gut decontamination should be carried out using repeated doses of activated charcoal and whole bowel irrigation. Propranolol or other β blockers (eg, esmolol) are useful antidotes for β-mediated hypotension and tachycardia. Phenobarbital is preferred over
phenytoin for convulsions; most anticonvulsants are ineffective. Hemodialysis is indicated for serum concentrations greater than 100 mg/L and for intractable seizures in patients with lower levels.
REFERENCES Dart RD (editor): Medical Toxicology , 3rd ed. Lippincott Williams & Wilkins,
2004. Goldfrank LR et al (editors): Goldfrank’s Toxicologic Emergencies , 9th ed. McGraw-
Hill, 2010. Olson KR et al (editors): Poisoning & Drug Overdose , 6th ed. McGraw-Hill,
2011. POISINDEX . (Revised Quarterly.) Thomson/Micromedex.
C A S E S T U D Y A N S W E R
Overdose of bupropion can cause seizures that are often recurrent or prolonged. Drug-induced seizures are treated with an intravenous benzodiazepine such as lorazepam or diazepam. If this is not effective, phenobarbital or another
more efficacious central nervous system depressant may be used. To prevent ingested drugs and poisons from being absorbed systemically, a slurry of activated charcoal is often given orally or by nasogastric tube.
058-Katzung_Ch058_p1027-1038.indd 1038 9/21/11 12:13:37 PM

11711171
AAbacavir, 863f, 869, 870t, 872, 888
drug interactions with, 874tAbatacept, 643, 656, 993, 993f, 999Abbokinase, 617. See also UrokinaseAbbreviated new drug application
(ANDA), 75–76Abciximab, 613, 994, 617, 999Abdomen, poisoning effects on, 1029Abelcet
properties of, 850tAbilify, 519. See also AripiprazoleAbiraterone, 710, 738Abortion
eicosanoids and, 324Abreva, 888. See also DocosanolAbscess(es)
amebic liverchloroquine for, 917
Absence seizures, 421Absorption of drugs. See also specific drugs
bioavailability effects on, 43in the elderly, 1052in infants and children, 1044, 1044tpercutaneous, 1061, 1062fpharmacokinetic mechanisms of, 1149as pharmacokinetic variable, 47–48
Abstinence syndrome, 549Abuse
alcoholprevalence of, 389prevention of
drugs in, 400drugs of, 565–580. See also specific drugs
and Drug abuseAcamprosate
in alcoholism management, 397Acarbose, 766
for diabetes mellitus, 759Acceptable daily intake (ADI), 1002Accolate, 328, 355. See also ZafirlukastAccumulation factors, 43Accumulation of drugs, 42–43, 42fAccupril, 190, 225. See also QuinaprilACE inhibitors. See Angiotensin-converting
enzyme (ACE) inhibitorsAcebutolol
as antihypertensive drug, 179preparations available, 167, 189, 249
Aceon, 190, 225. See also PerindoprilAcephen, 656. See also Acetaminophen
Acetaldehydemetabolism of, 390–391, 390f
Acetaminophenhalf-life of
in neonates and adults, 1045tionization constants of, 11tmetabolism of
to hepatotoxic metabolites, 60–61, 61f
oral absorption ofin neonates vs. older children and
adults, 1044toverdose of, 1032pharmacokinetic parameters for, 39t
Acetazolamide, 257, 270for epilepsy, 420ionization constants of, 11t
5-Acetylaminosalicylic acidstructure of, 1101f
Acetylation, 60tAcetylcholine (ACh), 99f, 124f,
467f, 112activation of endothelial cell muscarinic
receptors by, 103, 103fin ANS, ENS, and NANC neurons, 80f,
81f, 83f, 84tas CNS neurotransmitter, 366t, 369effects of, 92tproperties of, 99, 100t
Acetylcholine-blocking drugs. See Antimuscarinic drugs
Acetylcholinesterase (AChE), 84Acetylcysteine
as poisoning antidote, 1033tAcetylsalicylic acid, 656. See also AspirinN-Acetyltransferases (NATs), 60Acid(s). See also specific types
secretion ofphysiology of, 1081–1082, 1082f
weakdefined, 10ionization of, 10–12, 11t, 12f, 12t
Acid-peptic diseasesdrugs used for, 1081–1091
Acid reducersingredients in, 1118t
Acid secretionphysiology of, 1081–1082, 1082f
Acidityintragastric
drugs for reducing, 1081–1089
Acidosisanion gap
drug-induced, 1030, 1030tmetabolic. See Metabolic acidosis
Aciphex, 1111. See also RabeprazoleAcitretin
for psoriasis, 1071Acne preparations, 1069–1071
ingredients in, 1118ttopical antibiotics, 1064–1065
Aconiterisks associated with, 1127t
Acromegaly, 665dopamine agonists for, 673
Actemra, 656, 999. See also TocilizumabACTH. See also Adrenocorticotropic
hormone (ACTH)Acthar Gel, 678. See also CorticotropinActhrel, 678. See also Corticorelin ovine
triflutateActigall, 1112. See also UrsodiolActimmune, 999 Action potential(s)
resting potential effects on, 231–233, 232f, 233f, 233t
Activase, 617. See also Alteplase recombinant
Activated charcoalin decontamination of poisoned patient,
1032Activated partial thromboplastin time
(aPTT), 605Active cell membrane, 230–231, 231fActivin, 726Actonel, 786. See also Risedronate sodiumActos, 766. See also PioglitazoneAcute coronary syndrome, 193
clinical pharmacology of, 207Acute heart failure
management of, 222sympathomimetic drug effects on,
144Acute mountain sickness
carbonic anhydrase inhibitors for, 257–258
Acute pulmonary edemaopioid analgesics for, 553
Acute renal failureloop diuretics for, 259potassium-sparing drugs
and, 263
IndexNote: Page numbers in boldface type indicate major discussions. A t following a page number indicates tabular material, and an ffollowing a page number indicates a figure.
Katzung_Index_p1171-1234.indd 1171 9/24/11 12:03:33 PM

1172 Index
Acyclovir, 863f, 888dosage of, 865tfor HSV and VZV infections, 862–864,
863f, 864fpharmacokinetic and pharmacodynamic
parameters for, 39ttopical, 1067
Adagen, 999. See also Pegademase bovineAdalat, 190, 209. See also NifedipineAdalimumab, 647–648, 647f, 992
for inflammatory bowel disease, 1104–1106, 1105t
preparations available, 656, 999, 1112for psoriasis, 1072
Adapalenefor acne, 1070
Adcirca, 209. See also TadalafilAddiction
animal models of, 566clinical pharmacology of, 578defined, 565dopamine hypothesis of, 568dopamine level effects of, 565–566,
567f, 569f, 569tsynaptic plasticity and, 571
Addison’s diseasesynthetic corticosteroids for, 703–704
Adefovir, 863f, 888for HBV infection, 883t, 884
Adenocard, 250. See also AdenosineAdenosine
for cardiac arrhythmias, 245–246inhibitory effects of, 92tmembrane actions of, 237tpharmacologic properties of, 238tin renal tubule transport, 256
Adenosine A1-receptor antagonists, 258Adenosine diphosphate (ADP)
in coagulation, 601, 602fAdenosine triphosphate (ATP)
in ANS, ENS, and NANC neurons, 80f, 81f, 83f, 84t
S-Adenosyl-L-methionine (SAMe), 60ADH. See Antidiuretic hormone (ADH)ADHD
sympathomimetic drugs for, 146Adherence
in clinical trials, 73ADI. See Acceptable daily intake (ADI)Adipose tissue
insulin effects on, 746, 747tAdjuvant chemotherapy, 950–951Adrenal androgens, 709Adrenal cortex
angiotensin II effects on, 298Adrenal steroid inhibitors, 712Adrenal suppression
synthetic corticosteroids and, 707Adrenalin, 148, 355.
See also Epinephrine
Adrenergic nervescotransmitters in, 87
Adrenergic-neuron-blocking drugsas antihypertensive drugs, 175t,
177–178Adrenergic receptors, 87, 89tAdrenergic transmission, 84–87, 85f, 86fAdrenoceptor(s), 87, 89t
agonists of, 129–149.See also Sympathomimetic drugs
antagonists of, 151–168alpha-receptor antagonists, 151–157as antihypertensive drugs, 178beta-receptor antagonists. See also Beta
receptors, antagonists ofbasic pharmacology of, 157–161
beta. See Beta adrenoceptorsAdrenoceptor-blocking actions
of H1-receptor antagonists, 278Adrenocortical agents
antagonists of, 709–711.See also specific drugs
structures of, 709fAdrenocortical hypo- and hyperfunction
synthetic corticosteroids for, 704Adrenocortical insufficiency
synthetic corticosteroids for, 703–704Adrenocorticosteroids, 697–709. See also
specific drugsACTH vs., 707mineralocorticoids, 708–709synthetic corticosteroids, 703–708
Adrenocorticotropic hormone (ACTH), 661, 661t
adrenocorticosteroids vs., 707biosynthesis of
major pathways in, 698, 698fdiagnostic uses of, 662t
Adrenomedullin, 308Advair Diskus, 355. See also Salmeterol/
fluticasoneAdverse drug reactions (ADRs).
See also specific drugsduring drug development, 76–77in the elderly, 1057–1058
Advicor, 633Advil, 655. See also IbuprofenAeroBid, 355. See also FlunisolideAerosol corticosteroids, 355Aerospan, 355. See also FlunisolideAfrican trypanosomiasis
drugs used for, 930–934, 930t, 932t
Afrin, 148. See also OxymetazolineAftate, 858. See also TolnaftateAfterdepolarization(s)
delayed, 233f, 234Afterload
in cardiac performance, 215, 215f
AgammaglobulinemiaX-linked, 984–985
Ageas factor in drug dosage in infants and
children, 1047, 1049, 1049tas factor in drug metabolism, 66
Aggrastat, 617. See also TirofibanAging. See also Elderly
androgen production effects of, 736behavioral and lifestyle changes, 1053hepatic clearance of drugs and, 1052tpharmacologic changes associated with,
1051–1053, 1052f, 1052tpractical aspects of, 1058
phosphorylated enzyme complex of, 107physiologic function effects of, 1051,
1052fAgomelatine, 283Agonist(s), 6–8, 7f, 8f
binding molecule inhibitors, 6defined, 4full, 7, 8f, 19, 20finverse, 8, 8fopioids with, 552partial, 6–7, 8f, 19–20, 20freceptor binding of
concentration-effect curves and, 16, 17f
receptors in mediation of, 15AIDS, 985. See also HIV
antimicrobial drugs in patients with, 909antiretroviral drugs in, 869
Air pollutants, 1002–1005, 1005tTLVs of, 1005t
Airwayslipoxygenase effects on, 323prostaglandins and thromboxanes effects
on, 320Akathisia
tardive, 496Akinesia
end-of-dose, 487Akineton, 499. See also BiperidineAlbendazole
adverse effects of, 939clinical uses of, 938–939contraindications to, 939for helminthic infections, 937–939, 938tpharmacology of, 937precautions with, 939
Albenza, 935, 946. See also AlbendazoleAlbumin
concentration ofin drug concentration measurements,
49Albuterol, 343f, 355
for asthma, 344ionization constants of, 11t
Albuterol/ipratropium, 355Alcaine, 463. See also Proparacaine
Katzung_Index_p1171-1234.indd 1172 9/24/11 12:03:33 PM

Index 1173
Alcohol(s), 389–401. See also Ethanolabuse of. See also Alcohol abuse;
Alcoholismanimal studies of, 392as antiseptics/disinfectants, 893t, 894benzyl, 1068clinical pharmacology of, 395–397drug interactions with, 395, 1150tdurations of action of, 106tneurotoxicity of, 393in OTC products, 1123ttherapeutic uses for, 106ttolerance to, 393
Alcohol abuse, 569t, 575acute intoxication
management of, 395prevalence of, 389prevention of
drugs in, 400treatment for, 575
Alcohol dehydrogenase pathway, 390, 390f
Alcoholismprevalence of, 389treatment of, 396–397
Aldactazidedosage of, 262t
Aldactone, 225, 270. See also Spironolactone
dosage of, 262tAldara, 889. See also ImiquimodAldehydes
as antiseptics/disinfectants, 893t, 896Aldicarb
toxicity rating for, 1008tAldosterone, 697, 708
antagonists of, 225sites of action of, 252f
Aldosteronismsynthetic corticosteroids for,
704–705Aldrin
toxicity rating for, 1006tAlefacept, 993
for psoriasis, 1071Alemtuzumab, 991Alendronate
for osteoporosis, 775, 783Alendronate sodium, 786Aleve, 656. See also NaproxenAlfenta, 562. See also AlfentanilAlfentanil, 557
pharmacokinetics of, 545tAlferon N, 889. See also Interferon alfa-n3Alfuzosin, 155Alinia, 935. See also NitazoxanideAliskiren, 300
in heart failure management, 219Alitretinoin, 1077
dermatologic uses of, 1078t
Alkaloid(s)belladonna, 127vinca
for cancer, 962–964, 963tAlkalosis
metaboliccarbonic anhydrase inhibitors for,
257, 257thypokalemic
thiazide diuretics and, 261Alkylamine(s), 279tAlkylamine derivative, 278fAlkylating agents. See also specific drugs
adverse effects of, 955, 955f, 956tfor cancer, 953–958
Alkylsulfonate, 954fAllegra, 292. See also FexofenadineAllergic contact dermatitis
topical dermatologic medications and, 1063t
Allergic drug reactionsto antipsychotic drugs, 512autoimmune (type II), 998to cephalosporins, 800to H1-receptor antagonists, 279–280immediate (type I), 997
desensitization to drugs, 997–998treatment of, 997
immunologic, 997–998to insulin, 753to loop diuretics, 260serum sickness due to, 998to thiazide diuretics, 261vascular (type III), 998
Allergic rhinoconjunctivitis, 349Allergy preparations
ingredients in, 1118tAllied health professionals
prescribing authority of, 1144–1145, 1144tAllopurinol, 653–654, 962f
for angina pectoris, 206drug interactions with, 654, 1150tionization constants of, 11t
Allosteric modulators, 19Allylamines
topical, 856, 1065Almotriptan
pharmacokinetics of, 286tAloe
in constipation management, 1093Alosetron
for irritable bowel syndrome, 1096Aloxil, 1111. See also PalonosetronAlpha1-acid glycoprotein
concentration ofin drug concentration measurements,
49Alpha adrenoceptors, 87, 89t
antagonists ofas antihypertensive drugs, 180
Alpha1 adrenoceptorsantagonists of, 189
Alpha agonistsfor open-angle glaucoma, 161t
Alpha2 antagonistsapplications of, 157
Alpha-glucosidase inhibitorsfor diabetes mellitus, 759–760, 759tstructures of, 759t
Alpha receptor(s), 131, 131t, 132f, 137tAlpha1 receptor(s)
activation ofcardiovascular effects of, 137, 138t,
139f, 140fAlpha2 receptor(s)
activation ofcardiovascular effects of, 137
Alpha receptor antagonists, 151–157Alpha1 responses
activation of, 130, 130fAlpha2-selective agonists, 141Alphagan, 148. See also BrimonidineAlphanate, 617. See also Antihemophilic
factorAlphaNine SD, 617. See also Factor IX
complex, humanAlprazolam
dosages of, 383thepatic clearance of
aging effects on, 1052tpharmacokinetic properties of, 377tfor tremors, 494
Alprenololionization constants of, 11t
Alprostadil, 315, 326for erectile dysfunction, 199
Altace, 190, 225. See also RamiprilAlteplase
pharmacology of, 612Alteplase recombinant, 617AlternaGEL, 1111. See also Aluminum
hydroxide gelAltretamine
clinical activity and toxicities of, 956tAluminum hydroxide
as antacid, 1083Aluminum hydroxide gel, 1111Aluminum hydroxide/magnesium
hydroxide, 1111Alupent, 355. See also MetaproterenolAlveolar ventilation
of inhaled anesthetics, 430–432, 432fAlvimopan, 560
in constipation management, 1094preparations available, 562, 1112
Alzheimer’s diseaseantipsychotic drugs for, 509in the elderly
drugs used for, 1054–1055, 1055f, 1055t
Katzung_Index_p1171-1234.indd 1173 9/24/11 12:03:33 PM

1174 Index
Amantadinefor influenza, 887for parkinsonism, 491
Amaryl, 766. See also GlimepirideAmbenonium
duration of action of, 106ttherapeutic uses for, 106t
Ambien, 387. See also ZolpidemAmBisome
properties of, 850tAmbrisentan, 305Amebiasis
drugs used for, 927–930, 928t, 929f
types of, 927Amebic colitis
drugs used for, 927Amebic liver abscess
chloroquine for, 917Amerge, 292. See also NaratriptanAmevive, 999. See also AlefaceptAmicar, 617. See also Aminocaproic acidAmidate, 446. See also EtomidateAmide(s), local anesthetics,
structures of, 451tAmikacin, 826
dosage of, 840tpharmacokinetic parameters for,
39tfor tuberculosis, 840t, 844
Amikin, 828. See also AmikacinAmiloride, 261–263
ionization constants of, 11tAmine(s)
biogenicdrugs binding to transporters of
abuse of, 576–578, 577fprimary, secondary, etc 11
Amino acidsas CNS neurotransmitters, 366–369,
366t–367t, 368fAmino group
sympathomimetic drug substitution on, 135
p-Aminobenzoic acid (PABA), 832f
Aminocaproic acid, 604for bleeding disorders, 604f, 616
Aminocarbtoxicity rating for, 1008t
Aminoglutethimide, 709–710Aminoglycosides, 821–827Aminolevulinic acids, 1076Aminopenicillins
clinical uses of, 796Aminophylline
for asthma, 345Aminopterin
fetal effects of, 1042t4-Aminoquinolones, 918f
Aminosalicylate(s)for inflammatory bowel disease. See
5-Aminosalicyclic acid (5-ASA)
5-Aminosalicylic acid (5-ASA)adverse effects of, 1102–1103Azo compounds, 1101–1102,
1101fchemistry and formulations of,
1101–1102, 1101fclinical uses of, 1102dosage of, 840tfor inflammatory bowel disease,
1101–1103, 1101f, 1102fmesalamine compounds, 1102,
1102fmetabolism of, 1101–1102, 1101f,
1102fpharmacodynamics of, 1102pharmacokinetics of, 1102for tuberculosis, 840t, 844
Amiodaronefor cardiac arrhythmias, 242–243drug interactions with, 610tionization constants of, 11tthyrotoxicosis due to, 693–694
Amlodipineclinical pharmacology of, 202tclinical uses of, 205pharmacokinetics/dosage of, 175t
Amitiza, 1112. See also LubiprostoneAmitriptyline
dosage of, 535tpharmacodynamics of, 530tpharmacokinetics of, 529t
Amnesiaanesthesia and, 435
Amobarbital, 387Amodiaquine
clinical uses of, 917tfor malaria, 920
Amoxapine, 528f, 539pharmacology of, 527–530, 530t
Amoxicillindosage of, 795tpharmacokinetic parameters for,
39tAmoxicillin/potassium clavulanate, 795t,
807Amoxil, 807. See also AmoxicillinAmphetamines
abuse of, 569t, 576–577, 577feffects, 136f, 142fetal effects of, 1042tionization constants of, 11toverdose of, 1032–1034
Amphotecproperties of, 850t
Amphotericinclinical uses of, 934
Amphotericin (Cont.):pharmacokinetic parameters for,
39tAmphotericin B, 850f, 858
for systemic infections, 849–852topical, 1066
Ampicillinclinical uses of, 796ionization constants of, 11tduring lactation, 1048toral absorption of
in neonates vs. older children and adults, 1044t
pharmacokinetic parameters for, 39t
Ampicillin/sulbactam sodium, 807Amrix, 481. See also CyclobenzaprineAmVaz, 209Amyl nitrite, 197Amylin, 743, 763Amylin analogs
for diabetes mellitus, 760Amyloid β synthesis
inhibitors offor Alzheimer’s disease, 1055t
Amytal, 387. See also AmobarbitalAnabolic steroids, 734–737Anacetrapib, 631Anafranil, 539. See also ClomipramineAnakinra, 996, 999Analgesia/analgesics
in the elderly, 1054ingredients in, 1119topioid, 446, 543–564. See also Opioid
analgesicspatient-controlled, 554
Anaphylactic shocksympathomimetic drugs for, 145
Anaphylaxissympathomimetic drugs for, 145
Anaprox, 656. See also NaproxenAnaspaz, 127, 1111. See also HyoscyamineAnastrozole, 733, 740Ancef, 807. See also CefazolinAncobon, 858. See also FlucytosineANDA, 75–76Androgen(s), 726, 734–737. See also
Testosteroneadrenal, 709fetal effects of, 1042t
Androgen suppression, 737Androl-50, 740. See also OxymetholoneAnectine, 481. See also SuccinylcholineAnemia(s). See also specific types
agents used in, 581–599.See also specific drugs
folic acid/folates, 588–590, 589firon, 582–586vitamin B12, 584t, 586–588
androgens in, 736
Katzung_Index_p1171-1234.indd 1174 9/24/11 12:03:33 PM

Index 1175
Anemia(s). (Cont.):defined, 582iron deficiency
treatment of, 585–586, 586tmicrocytic hypochromic, 582nutritional
features of, 584, 584tpernicious, 588
Anesthesia/anestheticsgeneral, 429–447. See also specific drugs
and General anestheticsinhaled, 430–438. See also Inhaled
anestheticsintravenous, 438–446. See also specific
drugs and Intravenous anesthetics
local, 449–464.See also specific drugs and Local anesthesia/anesthetics
H1-receptor antagonists in, 279monitored care, 429, 430neuromuscular blocking drugs with, 475opioid analgesics in, 553–554premedication before clonidine as, 146sedative-hypnotic drugs as, 380sites of action of, 435
Angina pectorisdrugs used for. See also specific drugs
pharmacology of, 193–208vasodilators, 193–210.See also specific
types and drugsAngiomax, 617. See also BivalirudinAngiotensin, 295–300
agents blocking production or action of, 172
inhibitors ofas antihypertensive drugs, 183–185
mechanism and sites of action of, 183–184
Angiotensin-converting enzyme (ACE) inhibitors, 299
as antihypertensive drugs, 184–185fetal effects of, 1042tin heart failure management, 219, 220pharmacokinetics/dosage of, 175t, 185
Angiotensin I, 296f, 297Angiotensin II
actions of, 298excitatory effects of, 92t
Angiotensin receptor(s)mechanism of action of, 299
Angiotensin receptor blockers (ARBs), 299–300
as antihypertensive drugs, 185in heart failure management, 219, 220
Angiotensinase, 298Angiotensinogen, 296f, 297Anidulafungin, 855, 858Anion(s)
overdose ofloop diuretics for, 259
Anion gap acidosisdrug-induced, 1030, 1030t
Anion inhibitors, 689Anisindione, 617Anistreplase, 612ANOs, 2ANS. See Autonomic nervous system (ANS)Ansaid, 655. See also FlurbiprofenAntabuse, 400. See also DisulfiramAntacids
for acid-peptic diseases, 1082–1083drug interactions with, 1150t
Antagonismamong antimicrobial drugs, 910drug
mechanisms of, 20neutral, 7physiologic, 20
Antagonist(s), 6, 8fadrenoceptor, 151–168.See also
Adrenoceptor(s), antagonists ofchemical, 20
defined, 4competitive, 18–19, 18fdefined, 4drugs acting as, 15–16irreversible, 18f, 19opioids with, 552pure, 15–16receptors in mediation of, 15
Antagonist-precipitated withdrawal, 555Antara, 633. See also FenofibrateAnterior pituitary hormones, 660–673Anthracyclines
for cancer, 965–966Anthraquinone derivatives
in constipation management, 1093Anti-amyloid antibodies
for Alzheimer’s disease, 1055tAnti-amyloid vaccines
for Alzheimer’s disease, 1055tAntiandrogens, 737–739
receptor inhibitors, 738–739steroid synthesis inhibitors, 737
Antiarrhythmic drugs, 227–250. See also specific drugs
for atrial fibrillation, 246basic pharmacology of, 235–237benefits of, 247beta-adrenoceptor antagonists, 242calcium channel blockers, 245in the elderly, 1056mechanisms of action of, 235–237,
236fneuromuscular blocking drugs with,
475pharmacologic properties of, 238tprolonging effective refractory period,
242–245risks associated with, 247
Antibacterial drugstopical, 1063–1065
Antibiotics. See specific drugs and Antimicrobial drugs
Antibody, 978antithymocyte, 989–990anti-amyloid
for Alzheimer’s disease, 1055tantilymphocyte, 989–990digitalis, 225digoxin
as poisoning antidote, 1033tfunctions of, 981, 982fimmunosuppressive, 989–991monoclonal. See Monoclonal antibodies
(MABs)Anticholinergic drugs
for irritable bowel syndrome, 1096for nausea and vomiting, 1100overdose of, 1034
Anticholinergic syndrome, 1034Anticholinoceptor actions
of H1-receptor antagonists, 278Anticoagulants, 601–618. See also specific
drugscoumarin, 608–611direct thrombin inhibitors, 607–608indirect thrombin inhibitors, 604–607,
605foral
drug interactions with, 1151t–1152t
oral direct factor XA inhibitors, 607warfarin, 608–611
Anticonvulsantssedative-hypnotic drugs as, 381
Antidepressant(s), 521–541. See also specific drugs
drug interactions with, 536–537, 537t, 1152t
in the elderly, 1054heterocyclic
drug interactions with, 1152toverdose of, 536, 1034SNRIs. See Serotonin-norepinephrine
reuptake inhibitors (SNRIs)SSRIs. See Selective serotonin reuptake
inhibitors (SSRIs)subgroups of, 525–528, 526f–528fsummary table, 538tetracyclic. See Tetracyclic antidepressantstricyclic. See Tricyclic antidepressants
(TCAs)unicyclic. See Unicyclic antidepressants
Antidepressant–CYP450 drug interactions, 537t
Antidiabetic drugsoral, 753–762.See also specific drugs and
Diabetes mellitus, oral antidiabetic drugs for
Katzung_Index_p1171-1234.indd 1175 9/24/11 12:03:33 PM

1176 Index
Antidiarrheal agents, 1094–1095. See also specific drugs
ingredients in, 1119tAntidiuretic hormone (ADH), 674–675
agonists of, 264antagonists of, 264–265elevated
causes of, 264–265Antiemetic drugs, 1097–1100.
See also specific drugs5-HT3-receptor antagonists, 1097–1098neurokinin receptor antagonists,
1099–1100pathophysiology of, 1097–1098, 1098fphenothiazines, 1100substituted benzamides, 1100
Antiepileptic drugs (AEDs), 403–427. See also Antiseizure drugs
Antifolate drugs, 831–834, 958–960, 959t. See also specific drugs
for cancer, 958–960, 959tAntifungal drugs, 849–859, 1065–1067.
See also specific drugs and indications
drug interactions with, 1152ttopical, 856, 1065–1066
ingredients in, 1120tvaginal preparations
ingredients in, 1120tAntigen-presenting cells (APCs), 978Antihelminthic drugs
clinical pharmacology of, 937–947ingredients in, 1119t
Antihemophilic factor, 617Antihepatitis drugs, 882–886.
See also specific drugsAntihistamines. See also H1-receptor
antagonists; H2-receptor antagonists
dermatologic uses of, 1078tin OTC products, 1123tpreparations available, 292
Antihypertensive drugs, 169–191. See also specific drugs
in the elderly, 1056Anti-IgE monoclonal antibodies
for asthma, 350, 352Anti-inflammatory drugs, 1072–1074.
See also specific drugsantimicrobial drugs, 1056–1057MABs as, 992–994, 993ftopical
ingredients in, 1120tAnti-influenza drugs, 886–887Anti-inhibitor coagulant complex, 617Anti-integrin therapy
for inflammatory bowel disease, 1106Antilipid drugs
for Alzheimer’s disease, 1055tAntilymphocyte antibodies, 989–990
Antimalarial drugs, 915–927. See also specific drugs
dermatologic uses of, 1078tAntimetabolites. See also specific drugs
for cancer, 958–962Antimicrobial drugs, 891–899.
See also specific drugsbacteriostatic vs. bactericidal, 907, 907tclinical uses of, 901–913combinations of, 908–910in the elderly, 1056introduction to, 789for malaria, 926–927neuromuscular blocking drugs with, 475prophylaxis with, 910–913,
911t, 912ttopical
for acne, 1064–1065toxicity of
management of, 908–909, 908tAntiminth, 946. See also Pyrantel pamoateAntimuscarinic drugs, 115–124
clinical uses of, 347, 492intoxication by
cholinomimetic drugs in management of, 109
for parkinsonism, 491–492Antimycobacterial drugs, 839–848
for leprosy, 845–846for tuberculosis, 839–845.See also specific
drugs and Tuberculosis, drugs used for
Antineoplastic agents, 1077Antioxidants
for Alzheimer’s disease, 1055tAntiplatelet drugs
basic pharmacology of, 612–614Antiprotozoal drugs, 914–936. See also
specific drugs and indicationsAntipruritic drugs, 1076–1077Antipsychotic drugs, 501–520. See also
specific drugsin the elderly, 1054overdose of, 513, 517, 1034
Antipyreticsingredients in, 1119t
Antiretroviral drugs, 869–882. See also specific drugs and indications
Antiseborrhea agents, 1077–1078, 1078tingredients in, 1120t
Antiseizure drugs, 403–427. See also specific drugs
Antisense oligonucleotides (ANOs), 2Antisepsis, 89tAntiseptic drugs, 896–897Antispasmodics
for irritable bowel syndrome, 1096Antithrombin, 603–604Antithrombin III, 617Antithymocyte antibodies, 989–990
Antithymocyte globulin, 999Antithyroid drugs, 681–696.
See also specific drugsAntitrichogenic agents, 1077Antitumor antibiotics
for cancer, 965–966Anti-tumor necrosis factor (TNF) therapy
for inflammatory bowel disease, 1104–1106, 1105t
Antitussives, 559ingredients in, 1121t
Antivert, 292. See also MeclizineAntiviral drugs, 861–890. See also specific
drugs and indicationsanti-influenza agents, 886–887for CMV infections, 866–869for HBV and HCV infections,
882–886for HSV and VZV infections, 862–866topical, 1067
Antizol, 400. See also FomepizoleAnturane, 656. See also SulfinpyrazoneAnxiety
performancebeta-receptor antagonists for, 164
Anxiety disordersantidepressants for, 532–533sedative-hypnotic drugs for, 382, 382t
Anzemet, 1111. See also DolasetronApixaban, 607Apocrine sweat glands
sympathomimetic drug effects on, 139Apokyn, 499. See also ApomorphineApolipoprotein (apo) B-100, 619Apomorphine
for parkinsonism, 491Apraclonidine
for ophthalmic disorders, 145preparations available, 148
Aprepitantfor nausea and vomiting, 1099
Apresoline, 189. See also HydralazineApriso, 1102Aprotinin
for bleeding disorders, 617Aptivus, 889. See also TipranaviraPTT, 605Aquachloral Supprettes, 387. See also
Chloral hydrateAquaretics, 263–265Aquatensen, 270. See also MethyclothiazideAqueous diffusion, 9, 9fArachidonic acid, 313, 314f–316f
metabolism ofdietary manipulation of, 328
release and metabolism of, 313, 314fAralen, 935. See also ChloroquineAramine, 148. See also MetaraminolAranesp, 598. See also Darbepoetin alfaArava, 656, 999. See also Leflunomide
Katzung_Index_p1171-1234.indd 1176 9/24/11 12:03:33 PM

Index 1177
ARBs. See Angiotensin receptor blockers (ARBs)
Arcalyst, 999. See also RilonaceptArcitumomab, 992Area under the curve (AUC), 41Aredia, 786. See also Pamidronate sodiumArfonad, 127. See also TrimethaphanArformoterol, 355Argatroban, 608, 617Arginine vasopressin, 673fAricept, 112. See also DonepezilArimidex, 740. See also AnastrozoleAripiprazole, 504f, 509–510, 519Aristospan, 712. See also Triamcinolone
hexacetonideArixtra, 606. See also FondaparinuxArmodafinil, 148Armour, 695. See also Thyroid desiccatedAromasin, 740. See also ExemestaneAromatic (4)-hydroxylation, 64Aromatic hydrocarbons
TLVs of, 1005ttoxic exposure to, 1005–1006
Arrestin(s), 133–134Arrhythmia(s), 227–250. See also Cardiac
arrhythmiasalcohol and, 394
Arsenic, 1014t, 1017–1019Arsine gas poisoning, 1019Artane, 499. See also TrihexyphenidylArtemether, 920Artemether/lumefantrine, 935Artemisinin(s), 919t, 920–921Arterial blood gases
in poisoning assessment, 1030Arterial thrombosis, 614Artesunate, 920Arthritis
rheumatoideicosanoids and, 328
Articaine, 451t, 459–460, 463Arzerra, 999. See also Ofatumumab5-ASA. See 5-Aminosalicyclic acid (5-ASA)Asacol, 1102, 1102fAsbestos
as environmental pollutant, 1011
Ascariasisalbendazole for, 938–939
Asenapine, 519Asmanex Twisthaler, 355. See also
MometasoneAsparaginase
for cancer, 967–969Aspirin, 638–640
as antiplatelet agent, 612ionization constants of, 11tduring lactation, 1048tin OTC products, 1123toverdose of, 1034–1035
Aspirin (Cont.):pharmacokinetics of, 39t, 638,
638t, 639fresistance to, 613
Association-dissociation process, 10Asthma
acutemanagement of, 352–353
drugs used for, 339–357.See also specific drugs
sympathomimetic drugs, 145, 341–344, 343f
Atacand, 190, 225. See also CandesartanAtarax, 387. See also HydroxyzineAtazanavir, 870t, 878
drug interactions with, 874tAtenolol, 158f, 157–163,
167, 189as antihypertensive drug, 175t, 179pharmacokinetic parameters for, 39t
Atgam, 999. See also Lymphocyte immune globulin
Athetosis, 483, 495Athletes
androgen and anabolic steroid abuse in, 736
Ativan, 387, 400, 427, 446. See also Lorazepam
Atomoxetinesympathomimetic drug effects, 143
Atonic seizures, 421Atorvastatin, 633Atovaquone, 918f, 935
for malaria, 924–925Atovaquone-proguanil
clinical uses of, 917tATP
inhibitory effects of, 92tAtracurium
pharmacology of, 467–468, 470tAtrial fibrillation
antiarrhythmic drug-use principles applied to, 246
Atropinepharmacology of, 115–124ionization constants of, 11tas poisoning antidote, 1033tpreparations available, 127, 1111
Atropine fever, 120Atrovent, 127, 355. See also
IpratropiumAttention deficit hyperactivity disorder
(ADHD)sympathomimetic drugs for, 146
AUC (area under the curve), 41Augmentin, 807. See also Amoxicillin/
potassium clavulanateAuranofin, 656Aurolate, 656. See also Gold sodium
thiomalate
Aurothioglucose, 656Autacoid(s), 273
renalin renal tubule transport, 255–256
Autoclaving, 897Autoimmune diseases
immunosuppressive drugs for, 985t, 995Autoimmunity, 983–984Autologous stem cell transplantation
myeloid growth factors in, 593, 593fAutomatisms, 421Autonomic drugs, 79–168Autonomic junctions
anatomy of, 82Autonomic nervous system (ANS), 79–96
activity ofsystemic effects of, 90t
anatomy of, 80–82, 80f, 81fantipsychotic drugs effects on, 507t, 508,
512functional organization of, 88–93neurotransmitter chemistry of, 82–87
Autonomic pharmacologyintroduction to, 79–96. See also
Autonomic nervous system (ANS)
Autonomic receptors, 87, 89ttypes of, 89t
Autonomic synapsesanatomy of, 82
Autonomic transmissiondrug effects of, 93, 94t
Autoplexfor bleeding disorders, 616
Autoplex T, 617. See also Anti-inhibitor coagulant complex
Autoreceptor(s), 92, 92tAutosomal dominant hypophosphatemia,
783Avandamet, 766. See also Rosiglitazone plus
metforminAvandaryl, 766. See also Rosiglitazone plus
glimepirideAvandia, 766. See also RosiglitazoneAvapro, 190, 225. See also IrbesartanAvastin, 999. See also BevacizumabAvelox, 838. See also MoxifloxacinAvodart, 740. See also DutasterideAvonex, 999 Axert, 292. See also AlmotriptanAxid, 1111. See also NizatidineAygestin, 740. See also Norethindrone
acetateAzactam, 807. See also AztreonamAzathioprine, 643–644
adverse effects of, 644, 1104clinical uses of, 1103drug interactions with, 1104as immunosuppressive drug, 985t, 988indications for, 644
Katzung_Index_p1171-1234.indd 1177 9/24/11 12:03:34 PM

1178 Index
Azathioprine (Cont.):for inflammatory bowel disease, 1103–1104mechanism of action of, 643, 1103
Azelaic acidfor acne, 1070–1071
Azelastine, 292Azidothymidine (AZT). See ZidovudineAzilect, 499. See also RasagilineAzilsartan, 190Azinphos-methyl
toxicity rating for, 1007tAzithromycin, 814, 819Azmacort, 355. See also TriamcinoloneAzo compounds, 1101–1102, 1101fAzole(s)
for systemic infections, 853–855. See also specific drugs
topical, 856, 1065–66Azopt, 270. See also BrinzolamideAztreonam, 801, 807Azulfidine, 656, 1112. See also Sulfasalazine
BB-48, 620B-100, 620Babesia spp.
drugs used for, 931tBabesiosis
quinine for, 922Bacitracin, 805
topical, 1063Baclofen, 477, 477f, 478f, 481
in addiction management, 578Bacterial cell wall synthesis, 792, 792fBactrim, 837. See also Trimethoprim-
sulfamethoxazole (TMP-SMZ)Bactroban, 898. See also MupirocinBAL in Oil, 1025. See also DimercaprolBalanced anesthesia, 429, 438Balantidium coli
drugs used for, 931tBallismus, 495Balsalazide, 1112Balsalazide sodium, 1101fBanthine, 127. See also MethanthelineBanzel, 427. See also RufinamideBaraclude, 889. See also EntecavirBarbiturate(s), 377, 387, 441
abuse of, 575biotransformation of, 377chemistry of, 373–374clinical uses of, 442dosage of, 442drug interactions with, 1153tfetal effects of, 1042thepatic clearance of
aging effects on, 1052tas intravenous anesthetic, 441–442neuropharmacology of, 379tolerance and dependence of, 381
Barbiturate nucleus, 375fBaroreceptor
renalin renin release, 296, 297f
Baroreflexpostural, 171, 171f
Basal gangliacircuitry of, 483, 484f
Base(s)weak
ionization of, 10–12, 11t, 12f, 12t
Basiliximab, 993, 999BCHE, 65BDNF. See Brain-derived neurotrophic
factor (BDNF)Becaplermin
dermatologic uses of, 1078tBeclomethasone, 355Bedulin VH, 617. See also Factor IX
complex, humanBehavior(s)
antipsychotic drug effects on, 511levodopa effects on, 487
Behavioral effectsof aging, 1053
Belladonna alkaloids, 127, 1111Benadryl, 292. See also DiphenhydramineBenazepril
pharmacokinetics/dosage of, 175tpreparations available, 190, 225
Bendamustinefor cancer, 957clinical activity and toxicities of, 956t
Bendroflumethiazide, 270dosage of, 261t
Benefix, 617. See also Factor IX complex, human
Benicar, 190, 225. See also OlmesartanBentyl, 127, 1111. See also DicyclomineBenylin DM, 563. See also
DextromethorphanBenzalkonium, 898Benzalkonium chloride, 896Benzamides
for nausea and vomiting, 1100Benzathine penicillin
clinical uses of, 795pharmacokinetics of, 794
BenzeneTLVs of, 1005ttoxic exposure to, 1005–1006
Benzene hexachloridestoxicity rating for, 1006t
Benzisoxazoleadvantages and disadvantages of,
509tchemical structure effects on potency
and toxicity of, 505tBenznidazole, clinical uses of, 934
Benzocaine, 451t, 460, 463Benzodiazepine(s), 373–387
abuse of, 569t, 575for alcohol abuse, 575clinical uses of, 443dosage of, 443for epilepsy, 419–420as intravenous anesthetic, 442–443inverse agonists of, 379for nausea and vomiting, 1100pharmacodynamics of, 378–381, 378f,
442Benzodiazepine antagonists, 381–382Benzoic acid, 897Benzomorphans
with mixed receptor actions, 559Benzoyl peroxide
for acne, 1070preparations available, 898
Benztropine, 117f , 492t, 499Benzyl alcohol, 1068o-Benzyl-p-chlorophenol, 895Beryllium
as environmental pollutant, 1011Beta adrenoceptor(s), 87, 89t
agonists ofin heart failure management, 218
antagonists offor angina pectoris, 205as antihypertensive drugs, 178–180for cardiac arrhythmias, 242cardiac effects of, 242in heart failure management, 219,
221for open-angle glaucoma, 161toverdose of, 1035
glucagon for, 763preparations available, 189, 225, 249
rapid desensitization, resensitization, and down-regulation of, 26, 27f
Beta blockers. See Beta adrenoceptor(s), antagonists of
Beta carbonsympathomimetic drug substitution on,
136Beta-lactam compounds, 790–797
carbapenems, 801–802cephalosporins, 797–801cephamycins, 797–801monobactams, 801penicillins, 790–797. See also Penicillin(s)
Beta-lactamase assayfor infections with known etiology, 903
Beta-lactamase inhibitors, 801Beta receptor(s), 131–132, 131t, 132f, 137t
activation ofcardiovascular effects of, 137–138,
138t, 139fBeta-receptor antagonists
basic pharmacology of, 157–161
Katzung_Index_p1171-1234.indd 1178 9/24/11 12:03:34 PM

Index 1179
Beta-receptor antagonists (Cont.):clinical pharmacology of, 162–164, 163fdrug interactions with, 1153tspecific agents, 159t, 161–162for variceal hemorrhage, 1108
Beta-selective agonists, 133t, 141Betadine, 898. See also Povidone-iodineBetagan Liquifilm, 167. See also
LevobunololBetamethasone
clinical uses and dosage of, 703tBetamethasone sodium phosphate, 712Betapace, 167, 249. See also SotalolBetaseron, 999 Betaxolol, 159t, 161, 167, 189
as antihypertensive drug, 179Bethanechol, 99f, 112
properties of, 99, 100tBevacizumab, 991, 999
for cancer, 968clinical activity and toxicities of, 967t
Bexarotene, 1077dermatologic uses of, 1078t
Bezold-Jarisch reflex, 283Bias
in clinical trials, 72–73Biaxin, 819. See also ClarithromycinBicalutamide, 739, 740Bicarbonate
as poisoning antidote, 1033tBicillin, 807. See also Penicillin G
benzathineBiguanides,
for diabetes mellitus, 757Bile acid–binding resins
chemistry and pharmacokinetics of, 629for diabetes mellitus, 760drug interactions with, 1153tfor hyperlipidemias, 629–630mechanism of action of, 624f, 629–630therapeutic uses and dosage of, 630toxicity of, 630
Bile salt–binding resinsfor diarrhea, 1095
Biliary tractopioid analgesics effects on, 552
Biltricide, 946. See also PraziquantelBimatoprost, for glaucoma, 328, 1077Bioaccumulation
defined, 1003Bioavailability, 39t–40t, 43–44, 43f, 43t.
See also specific drugsabsorption and, 43, 43f, 44defined, 43first-pass elimination and, 43–44
Bioclate, 617. See also Antihemophilic factor
Biogenic aminesdrugs binding to transporters of
abuse of, 576–578, 577f
Biologic agentsfor psoriasis, 1071–1072
Biological License Application, 75Biomagnification
defined, 1003Biotransformation of drugs, 53–68. See also
Drug metabolism; Metabolismphase I reactions, 53–54, 54fphase II reactions, 54, 54f, 59–60, 59f,
60tBiperiden, for parkinsonism, 492tBiperidine, 499Biphenyl(s)
coplanar, 1010polychlorinated
as environmental pollutants, 1010Bipolar (affective) disorder
antipsychotic drugs for, 508, 513–517lithium for, 513–518.See also Lithium
Bipyridines, in heart failure management, 218
Bipyridyl herbicides, 1010Bisacodyl, 1112Bismuth compounds
for acid-peptic diseases, 1090–1091Bisoprolol
as antihypertensive drug, 179preparations available, 167, 189, 225properties of, 159t
Bisphosphonatesin bone mineral homeostasis, 776
Bithionolfor helminthic infections, 938t, 939
Bitolterol, 355Bivalirudin, 607–608, 617Black cohosh
risks associated with, 1127tBlack widow spider bite
passive immunization for, 1168tBlackwater fever, 922Bleeding
heparin and, 605–606mucosal
stress-relatedproton pump inhibitors in
prevention of, 1088uterine
estrogen and, 722Bleeding disorders
aprotinin in, 617drugs used for, 614–617, 615tfibrinolytic inhibitors in, 604f, 616plasma fractions in, 615–616, 615trecombinant factor VIIA in, 603f, 616serine protease inhibitors in, 617vitamin K for, 614–615
Bleomycinfor cancer, 966clinical activity and toxicities of, 963t
Blocadren, 189. See also Timolol
Blood alcohol concentration (BAC)clinical effects of
in nontolerant persons, 391, 391tBlood cells
lipoxygenase and, 323Blood clotting
initiation of, 602–604, 603fBlood coagulation, 601–602
disorders ofdrugs used for, 601–618.See also
specific drugs and Anticoagulantsestrogen effects on, 720
Blood coagulation cascade, 602, 603f, 603tBlood gases
arterialin poisoning assessment, 1030
Blood pressureregulation of, 169–171, 170f, 170t, 171f
Blood schizonticides, 915Blood vessels
autonomic nerve activity effects on, 90tBMI
calculation of, 664Boceprevir
for HCV infection, 886Body mass index (BMI)
calculation of, 664Bond(s)drug-receptor, 4Bone(s)
Paget’s disease of, 784PTH, FGF23, and vitamin D effects on,
774ttetracyclines effects on, 812
Bone marrow transplantationimmunosuppressive drugs in, 994–995passive immunization for, 1168t
Bone metabolismprostaglandins effects on, 322
Bone mineral homeostasis, 769–785FGF23 in, 770, 770f, 771f, 773hormonal interactions controlling, 771fhormonal regulators of
principal, 770–774secondary, 774–775
mechanisms contributing to, 770fnonhormonal agents affecting, 776–778,
776fparathyroid hormone and, 770vitamin D and, 772
Bone mineral–regulating hormonesdisorders involving, 780–785
Boniva, 786. See also Ibandronate sodiumBorage
risks associated with, 1127tBortezomib
for multiple myeloma, 971Bosentan, 305, 305f, 255
in heart failure management, 219Botanical pesticides, 1008, 1009fBotanical substances, 1126–1134
Katzung_Index_p1171-1234.indd 1179 9/24/11 12:03:34 PM

1180 Index
Botox, 481. See also Botulinum toxin, type ABotulinum toxin, 84, 479, 481Botulism
passive immunization for, 1168tBrain
cellular organization of, 364–366, 365fhierarchical systems in, 365, 365fnonspecific/diffuse neuronal systems
in, 365–366Brain cancer
chemotherapeutic drugs for, 974Brain-derived neurotrophic factor (BDNF)
in major depression, 522–523, 522tBrain natriuretic peptide (BNP)
in heart failure management, 219Bravelle, 678. See also UrofollitropinBreast(s)
oral contraceptives effects on, 727Breast cancer
chemotherapeutic drugs for, 971–972Brethine, 355. See also TerbutalineBreviBloc, 167, 189, 249. See also EsmololBrevital, 446. See also MethohexitalBrimonidine, for ophthalmic disorders,
145, 148Brinzolamide, 270Bromfenac, 655Bromocriptine, 672
for parkinsonism, 489, 499Bromocriptine mesylate, 678Brompheniramine
dosage of, 279t, 292Bronchiolar smooth muscle
autonomic nerve activity effects on, 90thistamine effects on, 276
Bronchodilationsympathomimetic drugs and,
137t, 138Bronchodilators
for asthma, 351Brovano, 355. See also ArformoterolBrovex, 292. See also BrompheniramineBrugia spp.
diethylcarbamazine citrate for, 940Bucindolol, 162Bucladin-S Softabs, 292. See also BuclizineBuclizine, 292Budesonide
for inflammatory bowel disease, 1103preparations available, 355, 1112
Bulk-forming laxatives, 1093, 1112Bumetanide
dosage of, 259t, 270Bumex, 270. See also BumetanideBupivacaine, 451t, 460, 463
ionization constants of, 11tpharmacokinetic properties of, 452ttoxicity of
reversal of, 458Buprenex, 562. See also Buprenorphine
Buprenorphine, 558–559, 562pharmacokinetics of, 545t
Bupropion, 527–531, 539dosage of, 535tin nicotine addiction management, 575overdose of, 1034
Burimamide, 276–277BuSpar, 387. See also BuspironeBuspirone, 283–285, 374, 376, 387Busulfan
clinical activity and toxicities of, 956tfetal effects of, 1042t
Butenafinepreparations available, 858from prescription to OTC status,
1116ttopical, 1065
Butoconazole, 858from prescription to OTC status,
1116tButorphanol, 559, 562
pharmacokinetics of, 545tButoxamine, 162Butyrophenone(s)antipsychotics, 503f,
504–505, 505tadvantages and disadvantages of, 509tchemical structure effects on potency
and toxicity of, 505tfor nausea and vomiting, 1100
Butyrylcholinesterase (BCHE), 65Byetta, 766. See also ExenatideBystolic, 167, 189, 225. See also Nebivolol
CCabazitaxel
for cancer, 963t, 964Cabergoline, 672, 678Calcineurin inhibitors
as immunosuppressive drugs, 985t, 986Calcitonin
in bone mineral homeostasis, 774–775Cadmium
as environmental pollutant, 1011–1012
Cadmium fume fever, 1012Cafergot, 292. See also Ergotamine
mixturesCaffeine, 344–345
during lactation, 1048tin OTC products, 1123t
Cal-Sup, 786. See also Calcium carbonateCalan, 190, 209, 249. See also VerapamilCalciferol, 786. See also ErgocalciferolCalcimimetics
in bone mineral homeostasis, 777Calcionate, 786. See also
Calcium glubionateCalcipotriene
for psoriasis, 1071Calciquid, 786. See also Calcium glubionate
Calcitoninfor hypercalcemia, 778, 786
Calcitonin gene–related peptide (CGRP), 308
in ANS, ENS, and NANC neurons, 81f, 84t
in migraine, 285Calcitonin-Salmon, 786Calcitriol, 786
for psoriasis, 1071Calcium
abnormal levels ofin bone mineral homeostasis, 778–780
contractile proteins sensitivity to, 212digitalis with, 218for hypocalcemia, 779–780as poisoning antidote, 1033tas second messengers, 28–29, 28fstored in and released from sarcoplasmic
reticulum, 212trigger
amount of, 212Calcium salts, 786Calcium carbonate as antacid, 1083Calcium channel(s)
voltage-activatedproperties of, 201t
Calcium channel blockers. See also specific drugs
for angina pectoris, 201–205as antihypertensive drug, 183for cardiac arrhythmias, 245in control of smooth muscle contraction,
194, 195fdrug interactions with, 1154toverdose of, 1035
Calcium Disodium Versenate, 1025. See also Edetate calcium
Calcium ion, 25cAMP, 25Campath, 999. See also AlemtuzumabCampral, 400. See also Acamprosate
calciumCamptothecins
for cancer, 963t, 964–965Canakinumab, 996, 999Canasa, 1102, 1102fCancer(s). See also specific types and sites
alcohol effects on, 395causes of, 949–950cell cycle and, 951, 952festrogen and, 722garlic effects on, 1129insulin and, 753oral contraceptives and, 730prostaglandins effects on, 322treatment modalities for, 950–951.
See also Cancer chemotherapyCancer chemotherapy, 949–976. See also
specific drugs and types of cancer
Katzung_Index_p1171-1234.indd 1180 9/24/11 12:03:34 PM

Index 1181
Cancer chemotherapy (Cont.):alkylating agents, 953–958antimetabolites, 958–962antitumor antibiotics, 965–966cell cycle kinetics and, 951–953, 951f,
952f, 952tcell cycle–nonspecific drugs, 951, 952tcell cycle–specific drugs, 951, 952tdrug combinations in, 951–953drug resistance in, 953milk thistle effects on, 1132nausea and vomiting due to
5-HT3-receptor antagonists for, 1099Cancidas, 858. See also CaspofunginCandesartan, 190, 225Cannabinoids
abuse of, 569t, 572–573for nausea and vomiting, 1100
Cantil, 127. See also MepenzolateCapacity-limited elimination, 39t–40t,
41–42Capacity-limited protein binding
in drug concentration measurements, 49Capastat Sulfate, 848. See also CapreomycinCapecitabine
for cancer, 960clinical activity and toxicities of, 959t
Capoten, 190, 225. See also CaptoprilCapreomycin
for tuberculosis, 840t, 844Capromab pendetide, 992Captopril, 190, 225, 299
as antihypertensive drug, 184–185dosage of, 175t, 185pharmacokinetics of, 39t, 175t, 185
Carafate, 1111. See also SucralfateCarbachol, 99f, 112
properties of, 99, 100tCarbamate(s)
durations of action of, 106ttherapeutic uses for, 106t
Carbamate pesticides, 1008, 1008tCarbamazepine, 409–410, 427, 517, 519
for bipolar disorder, 517drug interactions with, 410, 1154tfetal effects of, 1042tpharmacokinetics of, 39t, 410for seizures, 409–410therapeutic levels/dosage of, 410
Carbapenem(s), 791f, 801–802, 807–808
pharmacokinetics and dosage of, 798t, 801–802
Carbaryl, structure of, 105ftoxicity rating for, 1008t
Carbenicillinclinical uses of, 796
Carbenoxolonehepatic clearance of
aging effects on, 1052t
Carbidopa, 485, 499Carbidopa/levodopa, 499Carbidopa/levodopa/entacapone, 499Carbimazole
structure of, 688fCarbinoxamine, 292
dosage of, 279tCarbocaine, 463. See also MepivacaineCarbofuran
toxicity rating for, 1008tCarbohydrate(s)
impaired tolerance forthiazide diuretics and, 261
metabolism oforal contraceptives effects on, 728
Carbon monoxide, TLVs of, 1005tCarbon monoxide poisoning, 1003–1004,
1035, 1036tCarbon tetrachloride, TLVs of, 1005tCarbonic anhydrase inhibitors, 256–258,
257tfor open-angle glaucoma, 161t
Carboplatinclinical activity and toxicities of, 956t
Carboprost, 328Carboprost tromethamine, 324, 325fCardene, 190, 209. See also NicardipineCardiac arrhythmias
beta-receptor antagonists for, 162, 163f
drugs used for, 227–250.See also Antiarrhythmic drugs
mechanisms of, 233–235, 233f–236f
disturbances of impulse conduction, 234–235, 234f–236f
disturbances of impulse formation, 233f, 234
nonpharmacologic therapy of, 228Cardiac disease. See Heart diseaseCardiac muscle
calcium channel blockers effects on, 203–204
Cardiac outputin heart failure, 212–213inhaled anesthetics effects on, 433
Cardiac resynchronization therapyin chronic heart failure management,
221Cardiac rhythm
normalelectrophysiology of, 227–233
Cardiac stress testdobutamine in, 144
Cardiogenic shocksympathomimetic drugs for, 144
Cardiomyopathyalcohol and, 393–394
Cardioquin, 249. See also Quinidine polygalacturonate
Cardiovascular disordersantimuscarinic drugs for, 121beta-receptor antagonists for, 163sympathomimetic drugs for, 144–145
Cardiovascular drugsin the elderly, 1056
Cardiovascular system, see specific drugsCardizem, 189, 209, 250. See also
DiltiazemCardura, 167, 189. See also DoxazosinCarisoprodol, 481Carmustine, 954f
clinical activity and toxicities of, 956tCarperitide
in heart failure management, 219Carteolol, 159t, 161
as antihypertensive drug, 179Cartrol, 189. See also CarteololCarvedilol, 159t, 162
as antihypertensive drug, 179preparations available, 167, 189, 225racemate of, 5, 5t
Cascarain constipation management, 1093
Cascara sagrada, 1112Casodex, 740. See also BicalutamideCaspofungin, 855, 858Cataflam, 655. See also DiclofenacCatapres, 189. See also ClonidineCatechol, 136fCatechol-O-methyltransferase (COMT)
in catecholamine metabolism, 87, 88fCatechol-O-methyltransferase (COMT)
inhibitorsfor parkinsonism, 490–491
Catecholamine(s)biosynthesis of, 84–86, 86fendogenous, 138t, 140–141
Catecholamine reuptake inhibitorssympathomimetic drug effects on, 143
Catharticsactivated, 1032in constipation management, 1093
Causal prophylactic drugs, 915Ceclor, 807. See also CefaclorCedax, 807. See also CeftibutenCefaclor, 807, 797fCefadroxil, 797f, 807
pharmacokinetics and dosage of, 797, 798t
Cefazolin, 797f, 807pharmacokinetics and dosage of, 798t,
799Cefdinir, 797f , 807Cefditoren, 807Cefepime, 797f, 807
pharmacokinetics and dosage of, 798t, 800
Cefixime, 807Cefizox, 807. See also Ceftizoxime
Katzung_Index_p1171-1234.indd 1181 9/24/11 12:03:34 PM

1182 Index
Cefmetazole, 807Cefotan, 807. See also CefotetanCefotaxime, 797f, 807
pharmacokinetics and dosage of, 798t, 800
Cefotetan, 797f, 807 pharmacokinetics and dosage of, 798t
Cefoxitin, 797f, 807 pharmacokinetics and dosage of,
798tCefpodoxime, 797fCefprozil, 797f, 807Ceftaroline, 797f, 800Ceftaroline fosamil
pharmacokinetics and dosage of, 798t, 800
Ceftazidime, 797f, 807pharmacokinetics and dosage of, 798t,
800Ceftibuten, 797f, 807Ceftin, 807. See also
CefuroximeCeftizoxime, 797f, 807Ceftriaxone, 797f, 807
pharmacokinetics and dosage of, 798t, 800
Cefuroxime, 797f, 807 pharmacokinetics and dosage of, 798t
Cefzil, 807. See also CefprozilCelebrex, 655. See also CelecoxibCelecoxib, 638t, 640, 655Celestone, 712. See also BetamethasoneCelestone Phosphate, 712. See also
Betamethasone sodium phosphate
Celexa, 539. See also CitalopramCeliprolol, properties of, 159tCelivarone, 242Cell cycle kinetics
in cancer chemotherapy, 951–953, 951f, 952f, 952t
Cell growthangiotensin II effects on, 298
Cell-mediated immunity, 978Cell proliferation
prevention ofimmunosuppressive drugs for, 985t
CellCept, 999. See also Mycophenolate mofetil
Celontin, 427. See also MethsuximideCenestin, 740. See also Esterified estrogensCentral integration
in functional organization of autonomic activity, 81f, 89
Central nervous system (CNS)alcohol effects on, 391, 391tangiotensin II effects on, 298antimuscarinic drug effects on, 116, 118antipsychotic drugs effects on, 507t, 508barbiturates effects on, 441benzodiazepines effects on, 442
Central nervous system (Cont.):dexmedetomidine effects on, 445digitalis effects on, 217direct-acting cholinomimetic drug effects
on, 102t, 104disorders of
antimuscarinic drugs for, 120–121cholinomimetic drugs for, 109sympathomimetic drugs for, 146
drugs acting in, 359–580.See also Central nervous system (CNS) drugs
ergot alkaloids effects on, 288–289etomidate effects on, 443ganglion-blocking drug effects on, 124ginkgo effects on, 1130indirect-acting cholinomimetic drug
effects on, 107ketamine effects on, 444local anesthetics effects on,
457–458methylxanthines effects on, 345morphine effects on, 549–551, 551tnitric oxide effects on, 336oral contraceptives effects on, 727propofol effects on, 440prostaglandins and thromboxanes effects
on, 321sympathomimetic drug effects on, 140
Central nervous system (CNS) drugs, 359–580. See also specific types and drugs
in the elderly, 1053–1055pharmacology of, 359–371
in cellular organization of brain, 364–366, 365f
central neurotransmitters in, 366–371, 366t–367t
identification of, 364ion channels in, 360–361, 362fmethods for study of, 359–360, 361fneurotransmitter receptors in,
360–361, 362fsynapses and synaptic potentials in,
361–363, 362f, 363fsites of action of, 363–364, 364f
Central neurotransmittersidentification of
CNS drugs and, 364Central precocious puberty
gonadotropins in, 670–671Cephalexin, dosage of, 797, 798t
pharmacodynamics of, 797, 798tCephalosporin(s), 797–801. See also specific
drugsactive against methicillin-resistant
staphylococci, 800adverse effects of, 800–801chemistry of, 797, 797ftoxicity of, 800–801
Cephalothin, pharmacokinetics 39tCephamycins, 797–801Cephradine, pharmacokinetics and dosage
of, 797Cephulac, 1112. See also LactuloseCerebral vasospasm
following subarachnoid hemorrhagecalcium channel blockers effects on, 204
Cerebrospinal fluid (CSF)antimicrobial drugs in, 908, 909t
Cerebrovascular diseaseoral contraceptives and, 729
Cerebyx, 427. See also FosphenytoinCertolizumab, 648
for inflammatory bowel disease, 1104–1106, 1105t
preparations available, 656, 999, 1112Certolizumab pegol, 992Cervidil, 328. See also DinoprostoneCesamet, 1112. See also NabiloneCestodes
drugs used for, 938tCetirizine, 292
dosage of, 279tfrom prescription to OTC status, 1116t
Cetrorelix, 671, 678Cetrotide, 678. See also Cetrorelix acetateCetuximab, 991
for cancer, 967–968Cevimeline, 112cGMP
as second messengers, 29Channel(s)
ligand-gated, 23–24, 24f, 362fvoltage-gated, 23–24, 24f, 362f
Chantix, 112. See also VareniclineChaparral
risks associated with, 1127tCharcoal
activatedin decontamination of poisoned
patient, 1032ChAT, 82, 83fChelators
pharmacology of, 1021–1025. See also specific drugs
Chemet, 1025. See also SuccimerChemical(s)
toxic exposure to, 1003–1005, 1005tChemical antagonists, 20
defined, 4Chemical contraception
in men, 739Chemoattractant cytokines, 977–978Chemokines, 977–978Chemoreceptor reflex, 283Chemotherapeutic drugs, 789–1000.
See also specific drugs, types of cancer, and Cancer chemotherapy
Katzung_Index_p1171-1234.indd 1182 9/24/11 12:03:34 PM

Index 1183
Children. See Pediatric patientsChirality, 4–5Chirocaine, 463. See also LevobupivacaineChlor-Trimeton, 292. See also
ChlorpheniramineChloral hydrate, 375f, 387
during lactation, 1048tChlorambucil
clinical activity and toxicities of, 956tChloramine T
as antiseptic/disinfectant, 895Chloramphenicol, 816–817, 819
drug interactions with, 1154tduring lactation, 1048tpharmacokinetics of, 39t, 816
Chlordanetoxicity rating for, 1006t
Chlordiazepoxidedosages of, 383thepatic clearance of
aging effects on, 1052tionization constants of, 11tpharmacokinetics of, 39t, 377t
Chlorfenvinphostoxicity rating for, 1007t
Chlorhexidineas antiseptic/disinfectant, 893t, 894–895
Chloride channel activatorin constipation management, 1094for irritable bowel syndrome, 1097
Chloride channel GABA receptor complex, 378f, 380
Chlorineas antiseptic/disinfectant, 893t, 895
Chlorine dioxideas antiseptic/disinfectant, 893t, 895
Chlormethiazolehepatic clearance of
aging effects on, 1052tChloroform
TLVs of, 1005tChloromycetin, 819. See also
ChloramphenicolChlorophenoxy herbicides, 1008–1009,
1009fChloroprocaine, 460, 463Chloroquine, 644, 916–920, 917t, 918f,
919t, 920, 935ionization constants of, 11tfor malaria, 916–920, 917t, 918f, 919tpharmacokinetics of, 39t, 644, 916, 918f
Chlorothiazide, 261t, 270ionization constants of, 11tduring lactation, 1048t
Chlorotrianisene, 718f, 719tChlorpheniramine, 278f, 279t, 292
ionization constants of, 11tChlorpromazine, 155, 503f, 519
advantages and disadvantages of, 509t–510t
Chlorpromazine (Cont.):ionization constants of, 11tduring lactation, 1048t
Chlorpropamide, 755t, 766for diabetes mellitus, 754, 755tfetal effects of, 1042tionization constants of, 11tpharmacokinetic parameters for, 39t
Chlorthalidone, 261t, 270Chlorzoxazone, 481Cholecalciferol, 786Cholecystokinin (CCK)
in ANS, ENS, and NANC neurons, 84t
Cholesterol, 619Cholesteryl ester(s), 619Cholesteryl ester transfer protein (CETP)
inhibitorsfor hyperlipidemias, 631
Cholestyramine, 629–630, 633for diarrhea, 1095
Choline acetyltransferase (ChAT), 82, 83f
Choline estersproperties of, 99, 100t
Choline salicylate, 655Choline transporter, 82, 83fCholinergic fibers, 80f, 82Cholinergic nerves
cotransmitters in, 87Cholinergic poisoning
antimuscarinic drugs for, 122–123Cholinergic receptors, 87, 89tCholinergic transmission, 82–84, 83f, 84tCholinesterase inhibitors, 105f, 110–112
for Alzheimer’s disease, 1055toverdose of, 1035–1036
Cholinesterase regenerator(s), 127Cholinesterase regenerator compounds
antimuscarinic drugs for, 123Cholinoceptor, 84
characteristics of, 98tsubtypes of, 98t
Cholinoceptor-activating drugs, 97–113. See also Cholinomimetic drugs
Cholinoceptor-blocking drugs, 115–128. See also Cholinoreceptor antagonists
Cholinomimetic drugs, 97–113clinical pharmacology of, 108–110clinical uses of, 108–110direct-acting, 99–105in gastrointestinal motility stimulation,
1092groups of, 97, 98findirect-acting, 105–107mode of action of, 99for open-angle glaucoma, 161tspectrum of action of, 97–98, 98ttoxicity of, 109–110
Cholinoreceptor antagonists, 115–128ganglion-blocking drugs, 124–125muscarinic receptor–blocking drugs,
115–120. See also Antimuscarinic drugs
Choreadescribed, 483forms of, 495
Choriogonadotropin alfa, 667preparations available, 678
Chorionic gonadotropin, 678Chronic kidney disease,
vitamin D for, 781–782Chronic lymphocytic leukemia (CLL)
passive immunization for, 1168tChronic obstructive pulmonary disease
(COPD)antimuscarinic drugs for, 121treatment of, 353
Chronic orthostatic hypotensionsympathomimetic drugs for, 144
Chronotropic effectpositive, 138
Chronulac, 1112. See also LactuloseChrousos syndrome
synthetic corticosteroids for, 704Chylomicron(s), 620Chylomicronemia
primary, 623drugs used for, 622t
Cialis, 209. See also TadalafilCiclesonide, 348Ciclopirox olamine
topical, 1065Cidex, 898. See also GlutaraldehydeCidex OPA, 898. See also Ortho-
phthalaldehyde (OPA)Cidofovir, 863f, 867t, 888
for CMV infections, 868–869Cigarette smoking
coronary disease related to, 619Cilastatin, 802Cilostazol, 209, 617
as antiplatelet agent, 614for intermittent claudication, 207
Cimetidine, 1083t, 1111drug interactions with, 610t, 1155tpharmacokinetics, 39tfrom prescription to OTC status,
1116tCimzia, 999, 1112. See also CertolizumabCinacalcet, 780, 786Cinchonism, 922Cipro, 837. See also CiprofloxacinCiprofloxacin
clinical uses of, 836ionization constants of, 11tpharmacokinetics of, 39t, 836, 836t
Circus movement, 234, 234fCirrhosis, hepatic, diuretics for, 267
Katzung_Index_p1171-1234.indd 1183 9/24/11 12:03:34 PM

1184 Index
Cisapride, 286in constipation management, 1094
Cisatracurium, 468–470, 481Cisplatin
clinical activity and toxicities of, 956tdrug interactions with, 1155t
Citalopramdosage of, 535tpharmacokinetics of, 529t
Citanest, 463. See also PrilocaineCitracal, 786. See also Calcium citrateCitrucel, 1112. See also MethylcelluloseCladribine
for cancer, 962clinical activity and toxicities of, 959t
Clarinex, 292. See also DesloratadineClarithromycin, 814, 819Claritin, 292. See also LoratadineClassic angina, 193Claudication, intermittent treatment of,
207Clavulanic acid, 801fClearance, 38–42, 39t–41t, 1027–1028
capacity-limited elimination and, 39t–40t, 41–42
defined, 38drug accumulation and
time course of, 42–43, 42fin drug concentration measurements,
49, 50in the elderly, 1052f, 1052t, 1053flow-dependent elimination and,
39t–40t, 42in infants and children, 1045–1046models of, 38, 41fpharmacokinetic mechanisms of, 1161as pharmacokinetic variable, 48
Clemastine, 292from prescription to OTC status, 1116t
Cleocin, 819, 935. See also Clindamycin; Lincomycin
Clevidipine, 189, 209Cleviprex, 189, 209. See also ClevidipineClidinium, 127Clindamycin, 815, 815f
for acne, 1064Clinical trials, 72–77
controlled, 2types of evidence in, 73
Clinoril, 656. See also SulindacClobazam, for epilepsy, 420
hepatic clearance ofaging effects on, 1052t
Clofazimine, dosage of, 840tfor leprosy, 846
Clomid, 740. See also ClomipheneClomiphene, 731, 732f, 733–734, 740Clomipramine, 527f, 539
dosage of, 535tfetal effects of, 1042t
Clomipramine (Cont.):pharmacodynamics of, 530tpharmacokinetics of, 529t
Clonazepam, 387, 427for epilepsy, 420
Clonidine, 176,176f, 189for hypertension, 146, 176–177ionization constants of, 11tpharmacokinetics of, 39t, 175t, 177as premedication before anesthesia, 146for tics, 495
Clonorchiasispraziquantel for, 944
Clopidogrel, 617as antiplatelet agent, 613
Clorazepate, 387dosages of, 383tpharmacokinetic properties of, 377t
Clorazepate dipotassiumfor epilepsy, 420, 427
Clotrimazole, 853f, 858from prescription to OTC status, 1116t
Cloxacillin, dosage of, 795tClozapine, 504f, 519
advantages and disadvantages of, 509tdosage of, 510t
Clozaril, 519. See also ClozapineCMV infections. See Cytomegalovirus
(CMV) infectionsCNS. See Central nervous system (CNS)Co-Lyte, 1112. See also Polyethylene glycol
electrolyte solutionCoagulation
disorders ofdrugs used for, 601–618, 615t.
See also specific drugs and Anticoagulants
mechanisms of, 601–602, 602fCoagulation factor VIIa recombinant, 617Coartem, 935. See also Artemether/
lumefantrinefor malaria, 919t
Cocaine, 451t, 460, 463abuse of, 569t, 576fetal effects of, 1042tionization constants of, 11tsympathomimetic drug effects of, 143
Cockcroft-Gault formula, 1053Codeine, 558, 559, 562, 563
ionization constants of, 11tduring lactation, 1048tpharmacokinetics of, 545t
Codeine/acetaminophen, 562Codeine/aspirin, 562Coenzyme Q10, 1134–1135Cogentin, 499. See also BenztropineCognex, 112. See also TacrineCognitive remediation
antipsychotic drugs in, 513Colace, 1112. See also Docusate
Colazal, 1112. See also BalsalazideColchicine, 656
drug interactions with, 1155tColcrys, 656. See also Colchicine“Cold” preparations
ingredients in, 1118tColesevelam, 629–630, 633
for diabetes mellitus, 760for diarrhea, 1095
Colestid, 633. See also ColestipolColestipol, 629–630
for diarrhea, 1095Colistimethate sodium, 898Colitis
amebicdrugs used for, 927
Collecting tubule systemin renal tubule transport, 252t,
254–255, 254fColorectal cancer
chemotherapeutic drugs for, 972Coltsfoot
risks associated with, 1127tColy-mycin M, 898. See also Colistimethate
sodiumComa
hyperosmolarnonketotic, 744
myxedema, 691Combantrin, 946. See also Pyrantel
pamoateCombivent, 355. See also Albuterol/
ipratropiumComfrey
risks associated with, 1127tCompazine, 1112. See also ProchlorperazineComplement
in innate immunity, 979fComplex partial seizures, 421Compliance
in clinical trials, 73in prescribing drugs, 1143–1144
Compound(s)with estrogenic activity, 718, 718f
Compound F, 698–702. See also CortisolCOMT inhibitors
for parkinsonism, 490–491Comtan, 499. See also EntacaponeConcentration, target, 45Concentration-dependent killing, 823, 907Concentration-effect curves
receptor binding of agonists and, 16, 17f
Concentration gradientas factor in pharmacologic response to
dermatologic drugs, 1061Congenital adrenal hyperplasia
synthetic corticosteroids for, 704Congestive heart failure
sympathomimetic drugs in, 225
Katzung_Index_p1171-1234.indd 1184 9/24/11 12:03:34 PM

Index 1185
Conivaptan, 264, 270, 675, 678in acute heart failure management, 222
Conjugated estrogensdosage of, 731t
Conscious sedation, 430Consciousness
anesthesia and, 435Constitutive activity, 7Contact dermatitis
topical dermatologic medications and, 1063t
topical dermatologic medications and, 1063t
Contact hypersensitivity, 983Contact urticaria
topical dermatologic medications and, 1063t
Context-sensitive half-time, 440Continuous subcutaneous insulin infusion
(CSII) devices, 750–751Contraception
chemicalin men, 739
hormonal, 725t, 726–731“morning after,” 731
Contraceptivesemergency
ingredients in, 1121timplantable, 725foral, 725fpostcoital, 731, 731t
Contractile proteinssensitivity to calcium, 212
Contractilityin cardiac performance, 215, 215f
Controlled clinical trial, 2Controlled ovarian hyperstimulation, 667,
668fgonadotropins in, 670
Controlled substancesclassification of, 1145–1146, 1145t
Converting enzyme, 297–298, 298fConvulsion(s)
neuromuscular blocking drugs for, 476
COPD. See Chronic obstructive pulmonary disease (COPD)
Coplanar biphenyls, 1010Cordarone, 249. See also AmiodaroneCoreg, 167, 189, 225. See also CarvedilolCorgard, 167, 189. See also NadololCorlopam, 148, 189. See also FenoldopamCoronary artery disease
alcohol and, 394myxedema and, 690–691smoking and, 619
Coronary blood flowdeterminants of, 194
Cortef, 712. See also HydrocortisoneCortenema, 1112. See also Hydrocortisone
Cortexcircuitry of, 483, 484f
Corticorelin ovine triflutate, 678Corticosteroids, 355, 697–709
aerosolfor asthma, 347–348, 351–352clinical uses of, 347–348, 703–706dermatologic uses of, 1078tin the elderly, 1057mechanism of actions of, 347for nausea and vomiting, 1099synthetic, 703–708topical, 1072–1074, 1073t, 1074t
Corticotropin, 678Corticotropin-releasing hormone (CRH),
661, 661fclinical uses of, 661t
Cortifoam, 1112. See also HydrocortisoneCortisol, 697, 698–702Cortisone, 712
clinical uses and dosage of, 703tCortone Acetate, 712. See also CortisoneCortrosyn, 678. See also CosyntropinCorvert, 249. See also DronedaroneCosyntropin, 678Cotransmitter(s)
in cholinergic and adrenergic nerves, 84t, 87
Cotransmitter substances, 82Cough suppression. See also Antitussives
opioid analgesics in, 551, 553Coumadin, 617. See also WarfarinCoumarin anticoagulants, 608–611Coupling
receptor-effectorspare receptors and, 16–18, 17f
COX-2 inhibitorsselective, 640
COX inhibitorsnonselective, 640–642
Cozaar, 190, 225. See also LosartanCreon, 1111. See also PancrelipaseCrestor, 633. See also RosuvastatinCRH. See Corticotropin-releasing hormone
(CRH)Crixivan, 889. See also IndinavirCromolyn
for asthma, 348–349, 352ionization constants of, 11tfrom prescription to OTC status, 1116t
Crotamiton, 1068Cryoablation
for cardiac arrhythmias, 228Cryoprecipitate
for bleeding disorders, 616Cryptosporidium spp.
drugs used for, 931tCubicin, 808. See also DaptomycinCuprimine, 499, 656, 1025. See also
Penicillamine
Cushing’s syndromesynthetic corticosteroids for, 704
Cyanidepoisoning by, 1036, 1036t
Cyanocobalamin, 586Cyclic adenosine monophosphate (cAMP)
as second messenger, 25, 26, 28, 28fCyclic guanosine monophosphate (cGMP)
as second messenger, 29Cyclizine, 292
dosage of, 279tionization constants of, 11t
Cyclobenzaprine, 481Cyclodienes
toxicity rating for, 1006tCyclogyl, 127. See also CyclopentolateCyclooxygenase(s), 314–316, 315fCyclopentolate, 127Cyclophilin, 986Cyclophosphamide, 644
clinical activity and toxicities of, 956tfetal effects of, 1042tas immunosuppressive drugs, 985t,
988–989metabolism of, 955fstructure of, 954f
Cycloplegia, 118, 119fCycloserine, 805, 808, 848
dosage of, 840tfor tuberculosis, 840t, 844
Cyclospasm, 93, 95fCyclospora cayetanensis
drugs used for, 931tCyclosporine, 644, 999
dermatologic uses of, 1078tdrug interactions with, 1156tas immunosuppressive drug,
985t, 986pharmacokinetics of, 39t, 644
Cyklokapron, 617. See also Tranexamic acidCymbalta, 539. See also DuloxetineCYP. See Cytochrome P450 (CYP)Cyproheptadine, 279, 286
dosage of, 279tpreparations available. See also Cyclizine
Cyproterone, 738Cyproterone acetate, 738Cystospaz-M, 127. See also HyoscyamineCytarabine
for cancer, 960–961clinical activity and toxicities of, 959tfetal effects of, 1042tas immunosuppressive drug, 989
Cytisinein nicotine addiction management, 574
Cytochrome P450 (CYP), 55–64in enzyme induction, 57–59, 58tin enzyme inhibition, 58t, 59
Cytochrome P450 (CYP)–antidepressant drug interactions, 537t
Katzung_Index_p1171-1234.indd 1185 9/24/11 12:03:34 PM

1186 Index
Cytochrome P450 (CYP) cyclein drug oxidation, 55, 55f
Cytochrome P450 (CYP)–dependent oxidations, 56t–57t
Cytochrome P450 (CYP)–independent oxidations, 57t
Cytochrome P450–derived metabolitesprostaglandins and thromboxanes effects
on, 322–323Cytokine(s)
chemoattractant, 977–978in immunomodulation therapy,
995–996, 995t–996tCytokine inhibitors
in immunomodulation therapy, 996–997
Cytokine receptors, 23, 24fCytomegalovirus (CMV) infections
drugs used for, 866–869passive immunization for, 1168t
Cytomel, 695. See also LiothyronineCytosine arabinoside, 960fCytosine deoxyriboside, 960fCytotec, 328, 1111. See also MisoprostolCytotoxic agents
as immunosuppressive drugs, 985t, 988–989
Cytovene, 889. See also Ganciclovir
DDabigatran etexilate mesylate, 608Dacarbazine
for cancer, 957clinical activity and toxicities of, 956t
Daclizumab, 999DAG. See Diacylglycerol (DAG)Dalbavancin, 803Dalcetrapib, 631Dalmane, 387. See also FlurazepamDalteparin, 617Danaparoid, 617Danazol, 732–733Danocrine, 740. See also DanazolDantrium, 481. See also DantroleneDantrolene, 479Dapsone, 846
for acne, 1064–1065dermatologic uses of, 1078tfor leprosy, 846
Daptomycin, 803–804, 808Daranide, 270. See also DichlorphenamideDaraprim, 837, 935. See also
PyrimethamineDarbepoetin alfa, 598Darifenacin
for urinary disorders, 122, 122tDarunavir, 870t, 879
drug interactions with, 874tDasatinib
for cancer, 966–967, 967t
Daunorubicinfor cancer, 963t, 965clinical activity and toxicities of, 963t
Daypro, 656. See also OxaprozinDDT
toxicity rating for, 1006tDealkylation
oxidative, 56tDeamination, 56tDebrisoquin-sparteine oxidation, 63Decadron, 712. See also DexamethasoneDecadron-LA, 712. See also Dexamethasone
acetateDecadron Phosphate, 712. See also
Dexamethasone sodium phosphate
Declomycin, 270, 819. See also Demeclocycline
Decongestantsingredients in, 1121t
Decontaminationdefined, 894tof poisoned patients, 1031–1032
Deep sedation, 430Deferasirox, 598, 1024, 1025
for iron poisoning, 586Deferoxamine, 1024
for iron poisoning, 586as poisoning antidote, 1033t
Degarelix, 671, 678Degree of spareness, receptors, 18Dehydration
osmotic diuretics and, 264Dehydroemetine
for amebiasis, 930Delatestryl, 740. See also Testosterone
enanthate in oilDelavirdine, 870t, 876–877, 888Delayed afterdepolarizations (DADs), 216,
218f, 233f, 234Delirium tremens, 393Delsym, 563. See also DextromethorphanDelta-Cortef, 712. See also PrednisoloneDelta-D, 786. See also CholecalciferolDemadex, 270. See also TorsemideDemecarium
duration of action of, 106tpreparations available, 112therapeutic uses for, 106t
Demeclocycline, 812, 270, 819Demerol, 562. See also MeperidineDemser, 167. See also MetyrosineDenavir, 889. See also PenciclovirDenervation supersensitivity, 92Denileukin diftitox, 999
dermatologic uses of, 1078tDenosumab, 771, 786, 994, 999
in bone mineral homeostasis, 776–777
for osteoporosis, 775, 783
Deoxycorticosterone, 708–709Deoxycytidine analogs
for cancer, 960–961Depade, 561. See also NaltrexoneDepakene, 427, 519. See also Valproic acid/
valproateDepakote, 519. See also DivalproexDepen, 499, 656, 1025. See also
PenicillamineDependence
addiction vs., 565clinical pharmacology of, 578defined, 565
Depo-Estradiol, 740. See also Estradiol cypionate in oil
Depo-testosterone, 740. See also Testosterone cypionate
Depolarization(s)delayed after, 216, 218f, 233fearly after, 233fpremature, 216, 218f
Depo-Medrol, 712. See also Methylprednisolone acetate
Depressionantidepressants for. See Antidepressant(s)major, 521–522. See also Major
depressive disorderoral contraceptives and, 730pathophysiology of
hypotheses integration in, 525neuroendocrine factors in, 524–525
psychoticantipsychotic drugs for, 509
recurrent endogenouslithium for, 516
St. John’s wort for, 1132–1133unipolar
lithium for, 516Dermatitis
contactallergic, photoallergic
topical dermatologic medications and, 1063t
Dermatologic drugs, 1061–1079for acne, 1069–1071affecting pigmentation, 1068–1069anti-inflammatory drugs, 1072–1074antibacterial drugs, 1063–1065antifungal drugs, 1065–1067antineoplastic agents, 1077antipruritic agents, 1076–1077antiseborrhea agents, 1077–1078, 1078tantiviral drugs, 1067ectoparasiticides, 1068immunomodulators, 1067keratolytic and destructive agents,
1074–1076for psoriasis, 1071–1072reactions to, 1061–1063, 1063tsunscreens, 1069
Katzung_Index_p1171-1234.indd 1186 9/24/11 12:03:34 PM

Index 1187
Dermatologic drugs (Cont.):trichogenic and antitrichogenic agents,
1077vehicles for, 1061–1062
as factor in pharmacologic response to dermatologic drugs, 1061
Dermopathythyroid disease–related, 693
Desensitizationheterologous, of receptors, 133homologous, of receptors, 133to drugs, 997–998
Desferal, 598, 1025. See also DeferoxamineDesflurane, 432f, 446
pharmacologic properties of, 433tDesoxycorticosterone acetate
clinical uses and dosage of, 703tDesipramine
chemistry of, 526, 527fdosage of, 535tpharmacodynamics of, 530t
Desirudin, 617Desloratadine, 292Desmethyldiazepam, 374f
hepatic clearance ofaging effects on, 1052t
Desmopressin, 616, 674, 678Desogestrel, 719f
properties of, 723tDesoxyn, 148. See also MethamphetamineDesulfuration, 56t–57tDesvenlafaxine
dosage of, 535tDesyrel, 539. See also TrazodoneDetrol, 127. See also TolterodineDevelopment, fetal
critical periods of, 1041fDexamethasone, 712
clinical uses and dosage of, 703tDexamethasone suppression test, 705Dexchlorpheniramine, 292Dexedrine, 148. See also DextroamphetamineDexfenfluramine, 285Dexilant, 1111. See also DexlansoprazoleDexlansoprazole
pharmacokinetics of, 1086tDexmedetomidine, 141, 148, 446
as intravenous anesthetic, 445–446for sedation, 146
Dexmethylphenidate, 148Dexrazoxane
in cancer therapy, 966Dextroamphetamine, 148Dextromethorphan, 559Diaβeta, 766. See also GlyburideDiabetes insipidus
diuretics for, 268nephrogenic
ADH antagonists and, 265lithium effects on, 516–517
Diabetes mellitusdefined, 743drugs used for, 764–765glucagon in, 743, 762–763. See also
Glucagonglycemic control in
benefits of, 751, 752insulin for, 743, 744–745. See also
Insulinoral antidiabetic drugs for, 753–762.
See also specific drugsalpha-glucosidase inhibitors,
759–760, 759tamylin analog, 760biguanides, 757bile acid sequestrants, 760combination therapy with, 761–762DPP-4 inhibitors, 761GLP-1 receptor antagonists, 760–761insulin secretagogues, 754–756meglitinide, 756, 756tD-phenylalanine derivative, 756, 756tsulfonylureas, 754–756, 755tthiazolidinediones, 757–759
type 1 (insulin-dependent), 743–744combination therapy for, 762
type 2 (noninsulin-dependent), 744combination therapy for, 761–762
type 3, 744type 4 (gestational), 744
Diabetic ketoacidosis, 744insulin for, 751–752
Diabinese, 766. See also ChlorpropamideDiacetylmonoxime, 123f
for cholinergic poisoning, 123Diacomit, 427. See also StiripentolDiacylglycerol (DAG), 131, 131t, 132f,
137tas second messengers, 28–29, 28f
Dialysisperitoneal
in decontamination of poisoned patient, 1032
Diamox, 270. See also AcetazolamideDiarrhea
drugs used for, 1094–1095.See also spe-cific agents and Antidiarrheal agents
opioid analgesics for, 553Diastat, 481. See also DiazepamDiastolic heart failure, 211Diazepam, 476
dosages of, 383tfor epilepsy, 420fetal effects of, 1042thalf-life of
in neonates and adults, 1045thepatic clearance of
aging effects on, 1052tionization constants of, 11t
Diazepam (Cont.):during lactation, 1048toral absorption of
in neonates vs. older children and adults, 1044t
pharmacokinetics of, 39t, 377tpharmacologic properties of, 378, 439tpreparations available, 387, 400, 427,
446, 481Diazinon
toxicity rating for, 1007tDiazoxide, 182f, 189
as antihypertensive drug, 182–183Dibenamine
cardiovascular effects of, 153fDibenzo-p-dioxins
polychlorinated, 1010Dibenzodiazepine
advantages and disadvantages of, 509tchemical structure effects on potency
and toxicity of, 505tDibenzofurans
polychlorinated, 1010Dibenzothiazepine
advantages and disadvantages of, 509tchemical structure effects on potency
and toxicity of, 505tDibenzoxazepine
advantages and disadvantages of, 509tDibenzyline, 167. See also
PhenoxybenzamineDibucaine, 463Dichlorodiphenyltrichloroethane (DDT),
1009ftoxicity rating for, 1006t
2,4-Dichlorophenoxyacetic acid, 1008–1009
Dichlorphenamide, 270dosage of, 257t
Dichlorvostoxicity rating for, 1007t
Diclofenac, 640properties of, 638t
Dicloxacillindosage of, 795t
Dicumarol, structure of, 609fduring lactation, 1048t
Dicyclomine, 117f, 127, 1111for irritable bowel syndrome, 1096
Didanosine, 863f, 870t, 872, 888drug interactions with, 874t
Didronel, 786. See also Etidronate disodium
Dieldrintoxicity rating for, 1006t
Dientamoeba fragilisdrugs used for, 931t
Dietin drug metabolism, 65–66in hyperlipidemia, 625
Katzung_Index_p1171-1234.indd 1187 9/24/11 12:03:35 PM

1188 Index
Dietary Supplement and Non-Prescription Drug Consumer Protection Act, 1126
Dietary Supplement Health and Education Act (DSHEA), 74t, 1125–1126
Dietary supplements, 1125–1138. See also specific types
Diethylcarbamazine, for helminthic infections, 938t, 939–940
Diethylstilbestrol, 718f, 740dosage of, 731tfetal effects of, 1042t
Difenoxin, 558, 1112Diffuse neuronal systems
of CNS, 365–366Diffusible second messenger, 361, 362fDiffusion
aqueous, 9, 9flipid, 9, 9f
Diflucan, 858. See also FluconazoleDiflunisal, 641, 655
properties of, 638tDigestive enzymes, 1111Digibind, 225 DigiFab, 225Digitalis
in heart failure management, 215–218, 221
Digitalis antibody, 225Digitalis glycosides
drug interactions with, 1156tDigoxin
in chronic heart failure management, 215–218, 221
half-life ofin neonates and adults, 1045t
during lactation, 1048toral absorption of
in neonates vs. older children and adults, 1044t
overdose of, 1036–1037pharmacokinetics, 39t
Digoxin antibodies, as poisoning antidote, 1033t
Digoxin immune fab, 225Dihydroartemisinin, 920Dihydrocarbostyril
advantages and disadvantages of, 509t
chemical structure effects on potency and toxicity of, 505t
Dihydrocodeine, 558Dihydroergotamine, 155, 292Dihydroindolone
advantages and disadvantages of, 509tchemical structure effects on potency
and toxicity of, 505tDihydropyridine(s), 202tDihydroxyphenylalanine, 485fDiiodotyrosine (DIT), 682
Dilacor, 250. See also DiltiazemDilantin, 427. See also PhenytoinDilaudid, 562. See also HydromorphoneDiloxanide furoate
for amebiasis, 929–930Diltiazem
for angina, 202tfor cardiac arrhythmias, 245clinical pharmacology of, 202tmembrane actions of, 237tpharmacokinetics/dosage of, 175tpharmacologic properties of, 238tpreparations available, 189,
209, 250Dimaval, 1025. See also UnithiolDimenhydrinate, 278f, 292
dosage of, 279tfor nausea and vomiting, 1100
Dimercaprol, 1021–1022, 1022f, 1025indications and toxicity of, 1022
Dimercaptopropanesulfonic acid. See Unithiol
2,3-Dimercaptopropanol. See Dimercaprol
Dimercaptosuccinic acid. See SuccimerDimetan
toxicity rating for, 1008tDimethisterone, 719f
properties of, 723tDimethoate
toxicity rating for, 1007tD-Dimethylcysteine. See PenicillamineDimetilan
toxicity rating for, 1008tDinoprostone, 324, 325f, 328Diovan, 190, 225. See also ValsartanDioxins, 1009Dipentum, 1112. See also OlsalazineDipeptidyl peptidase-4 (DPP-4) inhibitors
for diabetes mellitus, 761Diphenhydramine, 278f, 292
dosage of, 279tionization constants of, 11tfor nausea and vomiting, 1100
Diphenoxylate, 558, 1112for diarrhea, 1094–1095ionization constants of, 11t
Diphenylmethane derivativesin constipation management, 1093
Diphtheriapassive immunization for, 1168t
Diphtheria tetanus acellular pertussis (DTaP) vaccine, 1164t
Diphyllobothriasispraziquantel for, 944
Diphyllobothrium latumniclosamide for, 942
Diprivan, 446. See also PropofolDipyridamole, 617
as antiplatelet agent, 613–614
Direct thrombin inhibitors, 607–608. See also specific drugs
oral, 608parenteral, 607–608
Direct vasodilators, 172Disalcid, 656. See also SalsalateDisease-modifying antirheumatic
drugs (DMARDs), 643–650, 656
in inflammation reduction, 636Disinfectants, 893–896, 898Disinfection
defined, 894tDisopyramide
for cardiac arrhythmias, 239–240Disseminated intravascular coagulation
(DIC), 604Dissociation constants (KD)
of enantiomers, 5, 5tDistal convoluted tubule
in renal tubule transport, 252t, 254, 254f
Distribution of drugs. See Volume of distribution
Disulfiramin alcoholism management, 397, 400drug interactions with, 610t, 1156t
Ditropan, 127. See also OxybutyninDiurese, 270. See also TrichlormethiazideDiuresis forced for poisoned patient, 1032
salinefor hypercalcemia, 778
Diuretic(s), 251–271. See also specific drugsadenosine A1-receptor antagonists, 258as antihypertensive drugs, 172–174,
174fbasic pharmacology of, 256–265clinical pharmacology of, 265–268for diabetes insipidus, 268for edematous states, 265–267in heart failure management, 219,
265–266hemodynamic effects of, 173toxicity of, 174, 258–263use of, 173–174
Diuril, 270. See also ChlorothiazideDivalproex, 519DMARDs. See Disease-modifying
antirheumatic drugs (DMARDs)
DNA gyrase inhibitors, 834–836. See also specific drugs
Doan’s Pills, 655. See also Magnesium salicylate
Dobutamine, 141, 142f, 148, 225in cardiac stress test, 144in heart failure management, 218
Dobutrex, 148. See also DobutamineDocetaxel
for cancer, 963t, 964
Katzung_Index_p1171-1234.indd 1188 9/24/11 12:03:35 PM

Index 1189
Docosanoldosage of, 865tfor HSV and VZV infections, 866
Docusate, 1093, 1112Dofetilide
for cardiac arrhythmias, 237–244Dolasetron, 1097f, 1111
for nausea and vomiting, 1098–1099Dolobid, 655. See also DiflunisalDolophine, 562. See also MethadoneDomperidone
in gastrointestinal motility stimulation, 1092
Donepezil, 112for Alzheimer’s disease, 1055
Dopamine, 136f, 138t, 141, 148, 225, 661, 661t
addictive drugs effects on, 565–566, 567f, 569f, 569t
in ANS, ENS, and NANC neurons, 84tas CNS neurotransmitter, 366t, 370inhibitory effects of, 92t
Dopamine agonists, 143–144, 278, 488–490, 672–673
for parkinsonism, 488–490, 488fDopamine hypothesis
of addiction, 568of schizophrenia, 502
Dopamine receptor(s), 87, 89t, 131, 132, 137t
activation ofcardiovascular effects of, 138
antagonists of, 506antipsychotic drugs and, 506–507,
506fDopamine transporter, 576Dopaminergic systems
antipsychotic drugs and, 506Dopar, 499. See also LevodopaDoral, 387. See also QuazepamDoribax, 807. See also DoripenemDoripenem
pharmacokinetics and dosage of, 798t, 801–802
Dorzolamide, 270Dose, 16–18
reducing interval between cancer treatment cycles, 953
Dose axis, 30, 30fDose-effect curves
graded, 16–18quantal, 31–32, 31f
Dose escalation, 953Dose-response curves, 16–18
for sedative-hypnotic drugs, 374fshape of, 30f, 31
Dose-response relationship, 30–34, 30f, 31fDosing schedule
as factor in pharmacologic response to dermatologic drugs, 1061
Dostinex, 678. See also CabergolineDouble-blind design, 72Down-regulation
receptor, 23Doxazosin, 154, 167, 189Doxepin, 1076
dosage of, 535tpharmacodynamics of, 530t
Doxercalciferol, 786Doxorubicin
for cancer, 963t, 965Doxycycline, 810–813, 819, 935
antiparasitic uses of, 917tfor helminthic infections, 938t, 940in malaria prevention in travelers, 919t
DPP-4 inhibitors. See Dipeptidyl peptidase-4 (DPP-4) inhibitors
Dramamine, 292. See also DimenhydrinateDrisdol, 786. See also ErgocalciferolDromotropic effect, positive, 138Dronabinol, 1111 abuse of, 572Dronedarone, 242, 249
for cardiac arrhythmias, 243membrane actions of, 237t
Droperidol, 446for nausea and vomiting, 1100
Drospirenone, 711Drospirenone/ethinyl estradiol
dermatologic uses of, 1078tDrug(s). See also specific types, indications,
and agentsof abuse, 565–580. See also specific drugs
and Drug abusewith actions on smooth muscle,
273–357autonomic, 79–168autonomic transmission effects of, 93,
94tbiotransformation of, 53–68. See also
Biotransformation of drugsCNS-related, 359–580. See also Central
nervous system (CNS) drugsdevelopment of, 69–77. See also Drug
developmentdiscovery of, 70–71, 70fefficacy of, 30f, 31excretion of. See Clearanceionization constants of, 11tlabeled and off-label uses of
legal issues related to, 1146legislation pertaining to
in USA, 73, 74tlipid:aqueous partition coefficient of,
9, 9fnature of, 3–6, 5f, 5torphan
development of, 77permeation of
mechanisms of, 9–10, 9f
Drug(s) (Cont.):physical nature of, 4potency of, 30–31, 30frational design of, 6reactivity of, 4shape of, 4–5, 5f, 5tsize of, 4
Drug abuse, 565–580. See also specific drugs and Addiction
nonaddictive, 571tolerance in, 566–567withdrawal in, 567
Drug accumulation, 42–43, 42fDrug action
duration of, 8signaling mechanisms and, 21–29,
21f–28f, 26ttime course of, 37–51
Drug antagonismmechanisms of, 7–8, 20
Drug concentrationdrug response and
relationship between, 16–20, 17f, 18f, 20f
measurements ofinterpretation of, 49–50
Drug conjugates, 59, 60tDrug development, 69–77
clinical trials in, 72–77. See also Clinical trials
orphan drugs in, 77pharmaceutical industry in, 69–70preclinical safety and toxicity testing in,
71–72, 71tfor rare diseases, 77receptor classes and, 22f, 29–30translational research in, 76
Drug dispositionbiotransformation of drugs in, 53–54, 54f
Drug distributionmodels of, 38, 41fphysical volumes of body compartments
in, 38, 41tDrug dose
clinical response andrelationship between, 30–34, 30f, 31f.
See also Dose-response relationship
individualization ofpharmacokinetics and
pharmacodynamics in, 47–49rational
time course of drug action related to, 37–51
regimen fortarget concentration approach to
designing of, 45–47, 47fDrug-drug interactions
during drug metabolism, 66–67, 66t, 67t
Katzung_Index_p1171-1234.indd 1189 9/24/11 12:03:35 PM

1190 Index
Drug groups, 12–13Drug holidays, 488Drug interactions, 1149–1162,
1150t–1161t. See also specific drugs
combined toxicity, 1161–1162pharmacodynamic mechanisms of, 1161pharmacokinetic mechanisms of, 1149,
1161predictability of, 1149–1162Drug metabolism, 61–68diseases affecting, 67–68, 67tin the elderly, 1052–1053, 1052tfetal
during pregnancy, 1040in infants and children, 1045, 1045tpharmacokinetic mechanisms of, 1149,
1161placental
during pregnancy, 1040to toxic products, 60–61, 61f
Drug overdose. See also specific drugshemodialysis in, 1031, 1031t, 1032
Drug penetrationregional variation in
as factor in pharmacologic response to dermatologic drugs, 1061
Drug receptor(s), 2. See specific types and Receptor(s)
Drug-receptor bonds, 4Drug resistance
in cancer chemotherapy, 953Drug response
drug concentration andrelationship between, 16–20, 17f, 18f,
20fidiosyncratic, 32variations in, 32–33
Drug screening, 70–71Drug tolerance, 32, 66, 566–567Drug “trapping”
through pH-partitioning phenomenon, 11, 12t
Drug withdrawal, 567Drug-body interactions, 6–12, 7f–9f,
10t–12t, 12fDrug-receptor interactions
types of, 6, 7fDSHEA. See Dietary Supplement Health
and Education Act (DSHEA)Duetact, 766. See also Pioglitazone plus
glimepirideDulcolax, 1112. See also BisacodylDuloxetine, 526f, 539
dosage of, 535tpharmacokinetics of, 528, 529tsympathomimetic drug effects on, 143
Duocaine, 463. See also Lidocaine and bupivacaine
DuoNeb, 355. See also Albuterol/ipratropium
Duricef, 807. See also CefadroxilDutasteride, 738, 740Dyazide
dosage of, 262tDyclone, 463. See also DyclonineDyclonine, 463Dylix, 355. See also DyphyllineDynaCirc, 190, 209. See also IsradipineDynorphin(s), 543–544, 544t
CREB-mediated up-regulationduring withdrawal from dependence,
568, 570fDyphylline, 355Dyrenium, 270. See also Triamterene
dosage of, 262tDysbetalipoproteinemia
familial, 623–624drugs used for, 622t
Dyskinesia(s)dopamine receptor agonists and, 489drug-induced, 495–496levodopa and, 487tardive, 496
antipsychotic drugs and, 511Dyslipidemia(s)
drugs used in, 619–633.See also specific disorders, e.g., Hyperlipidemia(s)
Dysmenorrheaeicosanoids and, 326
Dysmorphogenesis, 1041–1043antipsychotic drugs and, 512
Dyspepsianonulcer. See Nonulcer dyspepsia
Dystonia(s), 495described, 483tardive, 496
EE-prescribing, 1143Early afterdepolarizations (EADs), 233f,
234Easprin, 655. See also AspirinEating disorders
antidepressants for, 533Ecallantide, 302Echinacea, 1126–1128Echinocandins, 849
for systemic infections, 855Echothiophate, 106f, 112
durations of action of, 106ttherapeutic uses for, 106t
Econazole, 858Ecotoxicology, 1002Ecstasy
abuse of, 569t, 577–578Ectoparasiticides, 1068Eculizumab, 994Edarbi, 190. See also AzilsartanEdecrin, 270. See also Ethacrynic acid
Edemaidiopathic
diuretics for, 267lithium and, 517pulmonary, acute,
opioid analgesics for, 553Edematous states
diuretics for, 265–267Edetate calcium disodium, 1021f, 1023
indications and toxicity of, 1023Edrophonium, 105–107, 112
durations of action of, 106tEfavirenz, 870t, 877, 888
drug interactions with, 874tEffector(s)
of G proteins, 25, 26tEffector mechanism, 6Effector pathways
beneficial and toxic drug effects related to, 34
Effexor, 539. See also VenlafaxineEfficacy
intrinsic, 7Effient, 617. See also PrasugrelEffort angina, 193
clinical pharmacology of, 206–207, 206f, 207t
nitrate effects in, 200Eflornithine, 935, 1077
clinical uses of, 933–934EGF receptor
mechanism of activation of, 22, 23fEicosanoid(s). See also Leukotriene(s);
Prostaglandin(s)basic pharmacology of, 317–324clinical pharmacology of, 324–328
Eicosanoid receptors, 319–320, 319tElavil, 539. See also AmitriptylineEldepryl, 499. See also SelegilineElderly. See also Aging
ADRs in, 1057–1058anti-inflammatory drugs in, 1056–1057antimicrobial drugs in, 1056cardiovascular drugs in, 1056CNS drugs in, 1053–1055.See also
Central nervous system (CNS) drugs, in the elderly
ophthalmic drugs in, 1057Electrocardiography (ECG)
in poisoning assessment, 1030–1031, 1031f
Electroencephalography (EEG)antipsychotic drugs effects on, 508
Electrolyte(s)lithium effects on, 514in poisoning assessment, 1030, 1030t
Electrolyte balancealcohol effects on, 394
Electrostatic bonding, 4Elestat, 292. See also Epinastine
Katzung_Index_p1171-1234.indd 1190 9/24/11 12:03:35 PM

Index 1191
Eletriptan, 286t, 292Eligard, 678. See also Leuprolide acetateElimination of drugs. See also Clearance
capacity-limited, 39t–40t, 41–42flow-dependent, 39t–40t, 42models of, 38, 41f
Elixirsin pediatric patients, 1046
Elixophyllin, 355. See also TheophyllineEltrombopag, 595, 598Emadine, 292. See also EmedastineEmedastine, 292Emend, 1111. See also AprepitantEmergency contraceptives
ingredients in, 1121tEmesis
in decontamination of poisoned patient, 1031–1032
Emetinefor amebiasis, 930
EMLA (eutectic mixture of local anesthetics), 461
Empiric therapy, antimicrobials, 902–906
Empirin Compound, 562. See also Codeine/aspirin
Emtricitabine, 863f, 870t, 872–874, 888Enablex, 127. See also DarofemacomEnalapril, 190, 225, 299
as antihypertensive drug, 184, 190pharmacokinetics, 39t
Enantiomers, 5, 5f, 5tEnbrel, 656, 999. See also EtanerceptEnd-of-dose akinesia, 487Endocannabinoids
as CNS neurotransmitter, 367t, 371Endocrine disorders
diagnosis ofglucagon in, 763
Endocrine disruptorsas environmental pollutants, 1011
Endocrine drugs, 659–787. See also specific types, drugs, and indications
Endocrine glandskinins effects on, 301
Endocrine pancreas, 743–744Endocrine system
alcohol effects on, 394antipsychotic drugs effects on, 507t, 508,
512beta-receptor antagonists effects on, 160etomidate effects on, 444garlic effects on, 1129oral contraceptives effects on, 727properties in common with nervous
system, 80Endocytosis, 9–10, 9fEndogenous catecholamines, 138t,
140–141Endogenous opioid peptides, 543–544, 544t
Endometriosisgonadotropins in, 670
Endomorphin-1, 544Endomorphin-2, 544Endoperoxides
structures of, 918fEndorphin(s), 543–544, 544tEndothelial-derived relaxing factor
(EDRF), 103, 331Endothelin(s), 304–306Endothelium-derived hyperpolarizing
factors, 317Endotracheal intubation, 476Enduron, 270. See also MethyclothiazideEntecavir, 863fEnflurane, 432f, 446
pharmacologic properties of, 433tEnfuvirtide, 870t, 881, 888Enjuvia, 740. See also Esterified estrogensEnkephalin(s), 543–544, 544t
in ANS, ENS, and NANC neurons, 84tinhibitory effects of, 92t
Enoxaparin, 617ENS. See Enteric nervous system (ENS)Entacapone
for parkinsonism, 488f, 490, 499Entecavir
for HBV infection, 883t, 884, 889Entereg, 562, 1112. See also AlvimopanEnteric infections
proton pump inhibitors and, 1088–1089Enteric nervous system (ENS), 81–82,
81f, 84tphysiology of, 1091–1092, 1092ftransmitter substances found in, 84t
Enteric oxaluria, 785Enterochromaffin-like (ECL) cells, 81f,
1082, 1082fEnteroglucagon, 762Entocort, 1112. See also BudesonideEntry inhibitors, 870t–871t, 881–882.
See also specific drugsEnvironmental factors
cancer related to, 949in drug metabolism, 65–66
Environmental pollutants, 1010–1011asbestos, 1011endocrine disruptors, 1011metals, 1011–1012polychlorinated biphenyls, 1010
Environmental toxicology, 1001–1003Enzyme(s), 16, 1111
converting, 297–298, 298fdigestivehuman liver P450, 55–59. See also
Human liver P450 enzymeslithium effects on, 514, 515ttransmembrane
ligand-regulated, 22–23, 23fZiegler’s, 65
Enzyme inductionP450 in, 57–59, 58t
Enzyme inhibition, 58t, 59Ephedra
risks associated with, 1127tEphedrine, 136f, 148, 355
for asthma, 343–344ionization constants of, 11tsympathomimetic drug effects of, 136f,
141–142Epidermal growth factor (EGF) receptor
mechanism of activation of, 22, 23fEpilepsy
drug development for, 403–404, 405f, 406f
drugs used for, 419–420, 422–423prevalence of, 403VNS for, 404
Epinastine, 292Epinephrine, 138t, 140–141
for anaphylactic shock, 145for asthma, 343cardiovascular responses to, 138texcitatory effects of, 92tionization constants of, 11t
Epinephrine reversal, 152Epipodophyllotoxins
for cancer, 963t, 964Epirubicin
for cancer, 965Eplerenone, 711, 225, 270Epoetin alfa, 598Epoetin beta, 598Epogen. See also Epoetin alfaEpoprostenol, 328, 325fEpoxidation, 56tEpoxide hydrolases, 60Epoxygenase products, 317Eprosartan, 190, 225Epsom Salt, 1112. See also Magnesium
hydroxideEptifibatide, 613, 617Equalactin, 1112. See also PolycarbophilEquanil, 387. See also MeprobamateEraxis, 858. See also AnidulafunginErbitux, 999. See also CetuximabErectile dysfunction
drugs used for, 199alpha-receptor antagonists, 156–157
Erethism, 1020Ergocalciferol, 786Ergomar, 292. See also Ergotamine tartrateErgonovine, 292Ergosterol, 850Ergot alkaloids, 287–290Ergot poisoning, 287Ergotamine, 155
ionization constants of, 11tErgotamine mixtures, 292Ergotamine tartrate, 292
Katzung_Index_p1171-1234.indd 1191 9/24/11 12:03:35 PM

1192 Index
Ergotism, 287Ergotrate, 292. See also ErgonovineErlotinib
for cancer, 968clinical activity and toxicities of, 967t
Epogen, 598Ertapenem
pharmacokinetics and dosage of, 798t, 801–802
Erythromycin, 813–814for acne, 1064in gastrointestinal motility stimulation,
1092pharmacokinetics of, 39t, 813
Erythropoietin, 591–592Escitalopram, 529t
dosage of, 535t, 539Eserine, 112. See also PhysostigmineEsidrix, 270. See also HydrochlorothiazideEskalith. See also Lithium carbonateEslicarbazepine, 427
for seizures, 411Esmolol, 162, 167, 189, 249
as antihypertensive drug, 180cardiac effects of, 242membrane actions of, 237tpharmacologic properties of, 238tas poisoning antidote, 1033tproperties of, 159t
Esomeprazole, 1111pharmacokinetics of, 1086t
Essential tremor, 493–494Estazolam, preparations available, 387Ester(s),
choline, properties of, 99, 100tEsterified estrogens, 740Estrace, 740. See also EstradiolEstraderm, 740. See also Estradiol transdermalEstradiol, 716f, 717–718, 717fEstradiol cypionate
dosage of, 719tEstriol, 716f, 717–718, 717fEstrogen(s), 716–723, 740
blood coagulation effects of, 720in bone mineral homeostasis, 775drug interactions with, 1157t
Estrogen inhibitors, 731–734Estrogen replacement therapy
for osteoporosis, 775Estrogen response elements (EREs), 719Estrone, 716f, 717–718, 717f, 740Estropipate
dosage of, 719tEszopiclone, 374
biotransformation of, 378pharmacokinetic properties of, 377t
Etanercept, 647, 656, 992, 999for psoriasis, 1071–1072
Ethacrynic acid, 259f, 270dosage of, 259t
Ethambutol, for tuberculosis, 840t, 842Ethanol. See also Alcohol(s)
as antiseptic/disinfectant, 893t, 894pharmacology of, 390–397fetal effects of, 1042tduring lactation, 1048tmetabolism of, 390, 390foverdose of, 1037pharmacokinetics of, 390–391, 390fas poisoning antidote, 1033t
Ethanolamines (antihistamines)dosage of, 279t
Ethinyl estradiol, dosage of, 719t, 731t17-Ethinyl testosterone derivatives
properties of, 723tEthionamide, 843f, 848
dosage of, 840tfor tuberculosis, 840t, 843–844
Ethosuximidefor seizures, 417–418, 422–423
Ethotoinfor seizures, 409
Ethrane, 446. See also EnfluraneEthylene glycol
in alcoholism, 398poisoning by, 1037
Ethylene oxide, 897Ethylenediaminetetraacetic acid. See
Edetate calcium disodiumEthyleneimonium intermediate,
152, 152fEthynodiol diacetate
properties of, 723tEtidocaine, 460Etidronate, 776fEtidronate disodium, 786Etodolac, 641, 655
properties of, 638tEtomidate, 438f, 446
as intravenous anesthetic, 439t, 443–444Etoposide
clinical activity and toxicities of, 963tEtravirine, 870t, 877, 889
drug interactions with, 874tEtretinate
fetal effects of, 1042tEulexin, 740. See also FlutamideEuphoria
opioid analgesics and, 550Evista, 740. See also RaloxifeneEvoxac, 112. See also CevimelineExcitatory postsynaptic potential (EPSP),
92, 93f, 362–363Exelderm, 858. See also SulconazoleExelon, 112. See also RivastigmineExemestane, 733, 740Exenatide
for diabetes mellitus, 760Exjade, 598, 1025. See also Deferasirox
Exocrine glandskinins effects on, 301
Exocytosis, 9–10, 9fEx-Lax, 1112. See also SennaExpectorants
ingredients in, 1121tExperimental pharmacology, 2Experimental physiology, 2Extavia, 999 Extensive metabolizers, 64Extracellular volume expansion
osmotic diuretics and, 264Extraction ratio, 44Extrinsic factor, 586Eye(s). See also Ophthalmic disorders;
Ophthalmologic disordersanterior chamber of
structures of, 95fantimuscarinic drug effects on, 118, 119fantipsychotic drugs effects on, 512autonomic nerve activity effects on, 90tbeta-receptor antagonists effects on, 160,
161tcholinomimetic drug effects on
direct-acting, 102, 102tindirect-acting, 107
disorders ofcholinomimetic drugs for, 108
ganglion-blocking drug effects on, 125pharmacology of, 93, 95fpoisoning effects on, 1029prostaglandins effects on, 322serotonin effects on, 284sympathomimetic drug effects on, 138
Ezetimibefor hyperlipidemias, 630–631
Ezogabine (retigabine), 415
FFactive, 838. See also GemifloxacinFactor IX complex, human, 617Factrel, 678. See also Gonadorelin
hydrochlorideFadrozole, 733Famciclovir, 865t
for HSV and VZV infections, 866topical, 1067
Familial combined hyperlipoproteinemia, 623, 624
drugs used for, 622tFamilial dysbetalipoproteinemia,
623–624drugs used for, 622t
Familial hypercholesterolemia, 624, 624fdrugs used for, 622t
Familial hypertriglyceridemia, 623drugs used for, 622t
Familial ligand-defective apo Bdrugs used for, 622t
Familial ligand-defective apo B-100, 624
Katzung_Index_p1171-1234.indd 1192 9/24/11 12:03:35 PM

Index 1193
Famotidine, 1083from prescription to OTC status, 1116t
Famvir, 889. See also EtravirineFansidar, 837, 925–926, 935. See also
Pyrimethamine/sulfadoxineFareston, 740. See also ToremifeneFaslodex, 740. See also FulvestrantFasudil
for angina pectoris, 206Fatty acids
omega-3, 633FDA
in clinical trials, 73–74, 74treview of OTC drug ingredients by,
1115–1124Febuxostat
for gout, 654preparations available, 656
Federal Food, Drug, and Cosmetic Act of 1938, 73, 74t
FEIBA (factor eight inhibitor bypassing activity)
for bleeding disorders, 616Feiba VH Immuno, 617. See also Anti-inhibitor
coagulant complexFelbamate, for seizures, 412–413, 427Felbatol, 427. See also FelbamateFeldene, 656. See also PiroxicamFelodipine
clinical pharmacology of, 202t, 190, 209Female infertility
GnRH agonists for, 669–670Female maturation
estrogen effects on, 719–720Female reproductive system
eicosanoid effects on, 324–326melatonin effects on, 1136prostaglandins and thromboxanes effects
on, 321Femara, 740. See also LetrozoleFenitrothion
toxicity rating for, 1007tFenofibrate, 628f, 633
in drug combinations, 631for hyperlipidemias, 628–629
Fenoldopamas antihypertensive drug, 183, 189sympathomimetic drug effects of, 144, 148
Fenoprofenproperties of, 638t
Fentanyl, 557,557f, 562, 563pharmacokinetics of, 545t
Ferric hexacyanoferrate. See Prussian blueFerrioxamine, 1022fFesoterodine
for urinary disorders, 122, 122tFetal alcohol syndrome, 394Fetal drug metabolism
during pregnancy, 1040Fetal hydantoin syndrome, 424
Fetusadverse drug effects on, 1042t–1043tlung maturation in
corticosteroids in stimulation of, 705predictable toxic drug actions in, 1041teratogenic drug action effects on,
1042t–1043ttherapeutic drug actions in
during pregnancy, 1040–1041Fever
atropine, 120Blackwater, 922cadmium fume, 1012prostaglandins and, 321
Fexmid, 481. See also CyclobenzaprineFexofenadine dosage of, 279tFGF23. See Fibroblast growth factor 23
(FGF23)Fiber-Lax, 1112. See also PolycarbophilFiberCon, 1112. See also PolycarbophilFibrates
in drug combinations, 631for hyperlipidemias, 628–629, 629f
Fibric acid derivatives. See FibratesFibrinolysis, 604, 604fFibrinolytic drugs
basic pharmacology of, 611–612Fibrinolytic inhibitors
for bleeding disorders, 604f, 616Fibroblast growth factor 23 (FGF23)
in bone mineral homeostasis, 770–774Fibroids
uterinegonadotropins in, 670
Fick’s law of diffusion, 10Fidaxomicin, 892Filgrastim, 598Finasteride, 738, 1077Firmagon, 678. See also DegarelixFirst-pass effect, 44, 54
alternative routes of administration and, 44
First-pass eliminationbioavailability and, 43–44
FK-binding protein (FKBP), 986Flagyl, 898, 935. See also MetronidazoleFlavin monooxygenase, 65Flavoxate, 127Flecainide
for cardiac arrhythmias, 237t, 241Fleets Phospho-soda, 1112. See also Sodium
phosphateFlexeril, 481. See also CyclobenzaprineFlexible regulation, 29Flolan, 328. See also EpoprostenolFlomax, 167. See also TamsulosinFlorinef Acetate, 712. See also
Fludrocortisone acetateFlovent, 355. See also FluticasoneFlow-dependent elimination, 39t–40t, 42
Floxin, 838. See also OfloxacinFluconazole
for skin infections, 1066for systemic infections, 854–855
Flucytosinefor systemic infections, 852–853
Fludarabinefor cancer, 962clinical activity and toxicities of, 959t
Fludrocortisone, 709clinical uses and dosage of,
703t, 712Fluke(s)
drugs used for, 938tFlumadine, 889. See also RimantadineFlumazenil, 379, 381–382
as poisoning antidote, 1033tFlunarizine
for migraine, 285Flunisolide, 355Fluoride
in bone mineral homeostasis, 777Fluoropyrimidines
for cancer, 960Fluoroquinolones, 834–836
for tuberculosis, 840t, 844–845Fluorouracil, 10765-Fluorouracil
for cancer, 960Fluothane, 446. See also HalothaneFluoxetine, 528–529, 539
dosage of, 535tpharmacodynamics of, 39t, 530t
Fluoxymesterone, dosage of, 736tFluphenazine
advantages and disadvantages of, 509t
dosage of, 510tionization constants of, 11t
Fluphenazine decanoate, 519Fluprednisolone
clinical uses and dosage of, 703tFlurazepam
hepatic clearance ofaging effects on, 1052t
pharmacokinetic properties of, 377tFlurbiprofen,637f, 641, 655
properties of, 638tFlutamide, 738, 740Fluticasone, 355Fluvastatin, 633Fluvoxamine
dosage of, 535tpharmacodynamics of, 530tpharmacokinetics of, 529t
Focalin, 148. See also DexmethylphenidateFolate antagonists, 918fFolate synthesis
inhibitors offor malaria, 925–926
Katzung_Index_p1171-1234.indd 1193 9/24/11 12:03:35 PM

1194 Index
Folic acid/folates, 589f, 598, 958f, for anemia, 588–590, 589fdeficiency of
anemia due to, 584tsupplementation with, 589
Follicle-stimulating hormone (FSH), 666–669, 715
Follistim, 678. See also Follitropin betaFollitropin, 667, 678Fomepizole, 398, 398f, 400
as poisoning antidote, 1033tFondaparinux, 605f, 606, 617Food and Drug Administration (FDA)
in clinical trials, 73–74, 74treview of OTC drug ingredients by,
1115–1124Foradil, 355. See also FormoterolForane, 446. See also IsofluraneFormaldehyde
as antiseptic/disinfectant, 893t, 896Formalin, 896Formoterol
for asthma, 344, 355Fortaz, 807. See also CeftazidimeForteo, 786. See also TeriparatideFosamax, 786. See also Alendronate sodiumFosamprenavir, 870t, 879, 889
drug interactions with, 874tFosaprepitant, 307
for nausea and vomiting, 1099Foscarnet
for CMV infections, 868, 889dosage of, 863f, 865t, 867t, 889
Foscavir, 889. See also FoscarnetFosfomycin, 805, 808Fosinopril, 190, 225Fosphenytoin, 407, 427Fospropofol, 441, 446Fragmin, 617. See also DalteparinFrova, 292. See also FrovatriptanFrovatriptan
pharmacokinetics of, 286tFull agonists, 19, 20fFulvestrant, 733, 740Fulvicin P/G, 858. See also GriseofulvinFungizone
properties of, 850tFuracin, 898. See also NitrofurazoneFurosemide, 259f, 270
in chronic heart failure management, 220dosage of, 259tionization constants of, 11tpharmacokinetics, 39t
Fuzeon, 888. See also Enfuvirtide
GGi0-coupled receptors, drugs of abuse and,
572–573G-CSF. See Granulocyte colony-stimulating
factor (G-CSF)
G protein(s), 25–26, 25f, 26f, 26tG protein–coupled receptor(s) (GPCRs),
25, 26fG protein–coupled receptor kinase (GRK),
133GABA (gamma-aminobutyric acid)
in ANS, ENS, and NANC neurons, 84tas CNS neurotransmitter, 366t, 369
GABA (gamma-aminobutyric acid) receptorheterogeneity and pharmacologic
selectivity of, 379GABAA receptor
molecular pharmacology of, 378–379, 378f
GABAB agonist, 477Gabapentin, 413, 478
for seizures, 413for tremors, 494
Gabapentin/pregabalinfor chronic pain, 550
Gabitril, 427. See also TiagabineGablofen, 481. See also BaclofenGalanin
in ANS, ENS, and NANC neurons, 84t
Galantamine, 112for Alzheimer’s disease, 1055
Gallaminetissue effects of, 474t
Gallium nitratefor hypercalcemia, 778–779, 786
Gallstones, 1112bile acid therapy for, 1107
Gametocides, 915Gamma-delta T cells, 978Gamma-hydroxybutyric acid
abuse of, 569t, 573, 574fGanciclovir, 863f, 889
for CMV infections, 866–867dosage of, 867t
Ganglionprevertebral, 81
Ganglion-blocking drugs, 124–125as antihypertensive drug, 177
Ganirelix, 671, 678Ganite, 786. See also Gallium nitrateGantacurium
pharmacokinetics of, 468Garamycin, 828. See also GentamicinGarlic, 1128–1129Gas(es)
irritant poisoning by, 1036ttoxic poisoning by, 1035, 1036t
Gastric aciditydecreased
proton pump inhibitors and, 1089Gastric emptying
impaireddomperidone for, 1092metoclopramide for, 1092
Gastric lavagein decontamination of poisoned patient,
1032Gastrin, 743, 1082fGastrinoma
proton pump inhibitors for, 1088Gastritis
stress-relatedprevention of bleeding from
H2-receptor antagonists in, 1085Gastroesophageal reflux disease (GERD)
domperidone for, 1092H2-receptor antagonists for, 1083t, 1084metoclopramide for, 1092proton pump inhibitors for, 1087
Gastrointestinal cancers. See also specific types
chemotherapeutic drugs for, 972–973Gastrointestinal diseases
drugs used for, 1081–1114. See also specific drugs and diseases
acid-peptic disease, 1081–1091antidiarrheal agents, 1094–1095antiemetic drugs, 1097–1100antimuscarinic drugs, 121–122, 122tcholinomimetic drugs, 108gastrointestinal motility stimulation,
1091–1092, 1091finflammatory bowel disease,
1100–1106, 1101f, 1102f, 1105t
irritable bowel syndrome, 1095–1097, 1097f
laxatives, 1092–1094oral contraceptives and, 730
Gastrointestinal tractalcohol effects on, 392–393antimuscarinic drug effects on, 120, 120fautonomic nerve activity effects on, 90tcholinomimetic drug effects on
direct-acting, 102t, 104indirect-acting, 107
decontamination of poisoned patient, 1031–1032
disorders ofcholinomimetic drugs for, 108
dopamine receptor agonists effects on, 489
drug absorption effects onin infants and children, 1044, 1044t
eicosanoid effects on, 327ganglion-blocking drug effects on, 125lead poisoning effects on, 1015levodopa effects on, 487lipoxygenase effects on, 323methylxanthines effects on, 346opioid analgesics effects on, 551t, 552prostaglandins and thromboxanes effects
on, 320serotonin effects on, 284
Katzung_Index_p1171-1234.indd 1194 9/24/11 12:03:35 PM

Index 1195
Gastrointestinal tract (Cont.):smooth muscle of
histamine effects on, 276stimulation of motility of
drugs used for, 1091–1092, 1091ftetracyclines effects on, 812
Gatifloxacin, 835pharmacokinetics of, 836t
Gaviscon, 1111. See also Aluminum hydroxide/magnesium hydroxide
Gefitinibfor cancer, 967–968
Gelusil, 1111. See also Aluminum hydroxide/ magnesium hydroxide
Gemcitabinefor cancer, 959–961
Gemfibrozilfor hyperlipidemias, 628–629
Gemifloxacin, 835pharmacokinetics of, 836t
Genderin drug metabolism, 66
Gene therapy, 3for parkinsonism, 492
General anesthetics, 429–447. See also specific drugs
in current clinical practice, 446inhaled, 430–438intravenous, 438–446mechanism of action of, 429–430
Generalized seizuresdrugs used for, 417–418, 422–423
Generalized tonic-clonic seizures, 421drugs used for, 407–417, 422
Generic prescriptions, 1147Generic products, 75–76Genetic(s)
pharmacology of, 3Genetic factors
in drug metabolism, 61–65, 62t–63t, 64fphase I enzyme polymorphisms,
61–65, 62t–63t, 64fphase II enzyme polymorphisms, 65
Genitourinary smooth muscleautonomic nerve activity effects on, 90t
Genitourinary systemantimuscarinic drug effects on, 120, 120fdirect-acting cholinomimetic drug effects
on, 102t, 104disorders of
sympathomimetic drugs for, 145–146ganglion-blocking drug effects on, 125sympathomimetic drug effects on, 139
Gentamicin, 825–826pharmacokinetics, 39ttopical, 825, 1064
Gentamicin, netilmicin, 822fGeocillin, 807. See also CarbenicillinGeodon, 519. See also Ziprasidone
GERD. See Gastroesophageal reflux disease (GERD)
Geriatric patients. See Aging; ElderlyGermander
risks associated with, 1127tGHRH. See Growth hormone–releasing
hormone (GHRH)Giardia lamblia
drugs used for, 931tGiardiasis
drugs used for, 928Gigantism, 665Gilles de la Tourette’s syndrome, 495Ginkgo, 1129–1130Ginseng, 1130–1131Gland-derived extracts
risks associated with, 1127tGlaucoma
drugs used for, 160, 161t, 1057beta-receptor antagonists,
163–164carbonic anhydrase inhibitors, 257,
257teicosanoid effects on, 328treatment of, 160, 161t, 1057
Glicentin, 762Glimepiride, 755t, 766
for diabetes mellitus, 756Glipizide, 755t, 766
for diabetes mellitus, 755–756Glipizide plus metformin, 766Globulin
antithymocyte, 999GLP-1 receptor antagonists. See
Glucagon-like polypeptide-1 (GLP-1) receptor agonists
Glucagon, 743, 762–763“gut,” 762as poisoning antidote, 1033t
Glucagon-like polypeptide-1 (GLP-1) receptor agonists
for diabetes mellitus, 760–761, 766
Glucocorticoid(s), 697, 700f, 712antagonists of, 709–711anti-inflammatory effects of, 636in bone mineral homeostasis, 775clinical uses of, 703–706for diagnostic purposes, 705for gout, 655for hypercalcemia, 779as immunosuppressive drugs, 985–986,
985tfor inflammatory bowel disease,
1103mechanism of action of, 21–22, 22fpharmacodynamics of, 699–701pharmacokinetics of, 698, 703
Glucocorticoid receptor elements (GREs), 699
Glucophage, 766. See also MetforminGlucosamine, 1135Glucose
in acute alcohol intoxication management, 395
Glucose transporters, 746, 746tGlucotrol, 766. See also GlipizideGlucovance, 766. See also Glyburide plus
metforminGlucuronidation, 60tGlutamate, 365
as CNS neurotransmitter, 366t, 367–369, 368f
Glutamate hypothesisof schizophrenia, 502–503
Glutaraldehyde, 893t, 896, 898Glutathione conjugation, 60tGlutethimide, structure of, 375fGlyburide
for diabetes mellitus, 754–755, 766Glyburide plus metformin, 766Glycemic control
benefits of, 751, 752Glycemic memory, 752Glycerin suppository, 1093Glycine, 365, 478
as CNS neurotransmitter, 366t, 369
Glycine conjugation, 60tGlycol poisoning, 398, 1037Glycopeptide(s), 802–803
pharmacokinetics and dosage of, 798t, 802–803
Glycoprotein IIB/IIIA receptorsblockade of, 613
Glycopyrrolate, 117f, 127, 1111Glycosides
digitalis, 215–218drug interactions with, 1156t
Glynase PresTab, 766. See also GlyburideGlyphosphate, 1009–1010Glyset, 766. See also MiglitolGM-CSF. See Granulocyte-macrophage
colony-stimulating factor (GM-CSF)
GnRH. See Gonadotropin-releasing hormone (GnRH)
Goiter(s)nontoxic, 694toxic multinodular, 692toxic uninodular, 692
Gold sodium thiomalate, 656Goldman-Hodgkin-Katz equation, 230Golimumab, 648–649, 992–993
adverse effects of, 649indications for, 649mechanism of action of, 648–649pharmacokinetics of, 649
GoLYTELY, 1112. See also Polyethylene glycol electrolyte solution
Katzung_Index_p1171-1234.indd 1195 9/24/11 12:03:35 PM

1196 Index
Gonadal hormones, 715–741inhibitors of, 731–734, 737–739
Gonadarche, 715Gonadorelin, 669–671Gonadorelin hydrochloride, 678Gonadotropin(s), 666–669. See also specific
typesGonadotropin antagonists, 678Gonadotropin-releasing hormone (GnRH),
661, 661t, 669–671, 715, 733
receptor agonists, 671–672Gonal-f, 678. See also Follitropin alfaGoserelin acetate, 678Gossypol, 739Gout
drugs used for, 656Graft-versus-host disease, 994–995Gramicidin
topical, 1063Grand mal seizures, 421
drugs used for, 404–417, 422Granisetron
for nausea and vomiting, 1098–1099, 1111
Granulocyte colony-stimulating factor (G-CSF), 591t, 593–594, 593f
Granulocyte-macrophage colony-stimulating factor (GM-CSF), 591t, 593–594, 593f
Graves’ disease, 691–692Gray baby syndrome, 817Grifulvin, 858. See also GriseofulvinGrisactin, 858. See also GriseofulvinGriseofulvin
for mucocutaneous infections, 855–856
oral, 1066–1067Growth factor(s)
megakaryocyte, 594–595Growth factor receptor inhibitors
for cancer, 967–968Growth hormone, 660, 661t, 662–665,
678Growth hormone antagonists, 665–666,
665f, 678Growth hormone–releasing hormone
(GHRH), 661, 661tGrowth stimulators
androgens as, 736Guanabenz, 176, 189Guanadrel, 189Guanethidine
as antihypertensive drug, 175t, 177Guanfacine, 176, 189Guanine, 962fGuanylate cyclase C agonists
in constipation management, 1094Gynazole-1, 858. See also Butoconazole
Gynecologic disordersandrogens in, 736
Gynecomastiapotassium-sparing drugs and, 263
HH2 antagonists
ingredients in, 1118tH2-histamine receptor blockers, 1111H1-receptor antagonists, 277–280, 279t
for nausea and vomiting, 1100H2-receptor antagonists, 280–281, 292
for acid-peptic diseases, 1083–1085, 1083t, 1084f
serotonin-blocking actions of, 278–279H3-receptor antagonists, 281H4-receptor antagonists, 281Haemophilus influenzae type b conjugate
vaccine, 1164tHair growth stimulants
ingredients in, 1121tHalazepam
dosages of, 383tHalcion, 387. See also TriazolamHaldol, 519. See also HaloperidolHalf-life, 39t–40t, 42, 42f
as pharmacokinetic variable, 48HalfLytely, 1112. See also Polyethylene
glycol electrolyte solutionHalofantrine
clinical uses of, 917tfor malaria, 927structure of, 918f
Halogen(s)as antiseptics/disinfectants, 893t, 895
Halogenated aliphatic hydrocarbonsTLVs of, 1005ttoxic exposure to, 1005
Haloperidol, 155advantages and disadvantages of, 509tdosage of, 510tfor Huntington’s disease, 495for tics, 495
Halothanepharmacologic properties of, 433t, 446
Halothane hepatitis, 437Hand hygiene, 894Haplotype(s), 134Hashish
abuse of, 572Hazard
defined, 1002HDLs. See High-density lipoproteins
(HDLs)Headache(s)
migraine5-HT1D/1B agonists for, 285–286, 285f
Heartalcohol effects on, 391antipsychotic drugs effects on, 512
Heart (Cont.):autonomic nerve activity effects on, 90tcontractility of
control of, 212, 213fdigitalis effects on, 216–217, 217f, 217tglucagon effects on, 763lipoxygenase effects on, 323lithium effects on, 517performance of
pathophysiology of, 215, 215fHeart disease
alcohol and, 394cholinomimetic drugs for, 109ischemic
beta-receptor antagonists for, 162, 163f
coenzyme Q10 for, 1134Heart failure
acutemanagement of, 222sympathomimetic drug effects on, 144
alcohol and, 393–394cardiac resynchronization therapy for,
221chronic
classification of, 220tmanagement of, 220–221, 220t
coenzyme Q10 for, 1134diastolic, 211
management of, 221–222drugs used in, 211–226, 212t.See also
specific drugsACE inhibitors, 219ARBs, 219basic pharmacology of, 215–219beta-adrenoceptor antagonists, 219beta-adrenoceptor stimulants, 218beta-receptor antagonists, 162–163bipyridines, 218digitalis, 215–218diuretics, 219, 265–266positive inotropic drugs, 212t, 218vasodilators, 219without positive inotropic effects,
212t, 218–219“high-output” failure, 214pathophysiology of, 212, 214–215, 214fsystolic, 211
Heart musclecalcium channel blockers effects on,
203–204Heart rate
in cardiac performance, 215Heart rhythm
normalelectrophysiology of, 227–233
Heavy metalsas antiseptics/disinfectants, 897toxicology of, 1013–1321. See also
specific types
Katzung_Index_p1171-1234.indd 1196 9/24/11 12:03:35 PM

Index 1197
Heavy metals, toxicology of (Cont.):lead, 1013–1017mercury, 1014t, 1019–1021
Hectorol, 786. See also DoxercalciferolHelicobacter pylori ulcers
proton pump inhibitors for, 1087–1088Helixate, 617. See also Antihemophilic
factorHelminthic infections
chemotherapy of, 937–947. See also specific drugs
Hemabate, 328. See also CarboprostHematopoiesis
sulfonamides effects on, 833Hematopoietic growth factors, 590–595.
See also specific typeserythropoietin, 591–592megakaryocyte growth factors, 594–595myeloid growth factors, 592–594
Hemicholiniums, 82, 83fHemochromatosis
causes of, 586Hemodialysis
in drug overdose and poisoning, 1031, 1031t, 1032
Hemofil M, 617. See also Antihemophilic factor
Hemorrhagepostpartum
ergot alkaloids for, 290subarachnoid
cerebral vasospasm and infarct following
calcium channel blockers effects on, 204
varicealdrugs used for, 1107–1108
Henderson-Hasselbalch equation, 10, 449Heparin, 604–607, 605fHeparin-induced thrombocytopenia, 606Hepatic cirrhosis
diuretics for, 267Hepatic injury
milk thistle effects on, 1131Hepatitis
halothane, 437isoniazid-induced, 841viral
drugs used for, 882–886. See also specific drugs
Hepatitis Apassive immunization for, 1168t
Hepatitis A vaccine, 1164tHepatitis B
passive immunization for, 1168tHepatitis B vaccine, 1164tHepatitis B virus (HBV) infection
drugs used for, 883–885, 883tHepatitis C virus (HCV) infection
drugs used for, 883t, 885–886
Hepoxillin(s), 317Hepsera, 888. See also AdefovirHeptachlor
toxicity rating for, 1006tHerbal medications, 1125–1138. See also
specific typesHerbicides, 1008–1010
bipyridyl, 1010chlorophenoxy, 1008–1009, 1009fglyphosphate, 1009–1010
Herceptin, 999. See also TrastuzumabHereditary vitamin D–resistant rickets,
783–784Heroin, 557
fetal effects of, 1042tduring lactation, 1048t
Herpes simplex virus (HSV) infectiondrugs used for, 862–866
Heterocyclic antidepressantsdrug interactions with, 1152t
Heterodimers, 25Heterologous desensitization, 133Heteroreceptor(s), 92, 92tHetrazan, 946. See also DiethylcarbamazineHexachlorocyclohexane, 1068Hexachlorophene, 895, 898Hexamethonium, 124Hibiclens, 898. See also Chlorhexidine
gluconateHibistat, 898. See also Chlorhexidine
gluconateHigh-density lipoproteins (HDLs),
619–621deficiency of, 625
“High-output” heart failure, 214Hirudin, 607Histamine, 274–277
basic pharmacology of, 274–277as CNS neurotransmitter, 367tinhibitory effects of, 92t“triple response” of, 276
Histamine antagonists, 277. See also specific types
Histamine receptorssubtypes of, 275, 275t
Histamine receptor antagonists, 277–281. See also specific antagonists, e.g., H1-receptor antagonists
Histex, 292. See also CarbinoxamineHistrelin acetate, 678HIV
life cycle of, 873fHIV infection, 985
in childrenIGIV passive immunization, 1168t
Hivid, 889. See also ZalcitabineHMG-CoA reductase inhibitors,
625–627, 626fin drug combinations, 631drug interactions with, 1157t
Hodgkin’s lymphomachemotherapeutic drugs for, 970
Homatropine, 127Homeostasis
bone mineral, 769–785Homodimers, 25Homologous desensitization, 133Hookworm infections
albendazole for, 938–939Hormonal contraception, 725t, 726–731.
See also specific types and Oral contraceptives
Hormonal therapypostmenopausal
estrogen in, 720–722Hormone(s). See also specific types
adrenal, 697–713bone mineral–regulating
disorders involving, 780–785defined, 4gonadal, 750–741
inhibitors of, 750–741hypothalamic-pituitary, 659–679ovarian, 726thyroid, 684–688. See also Thyroid
hormonesHormone secretion
sympathomimetic drug effects on, 139–140
HSV infection. See Herpes simplex virus (HSV) infection
5-HT agonists, 2925-HT1D/1B agonists
for migraine, 285–286, 285f5-HT2 antagonists, 286, 526–531, 539
adverse effects of, 5365-HT3-receptor antagonists, 1092–1098,
11115-HT4-receptor agonists, 1092
in constipation management, 1094for irritable bowel syndrome, 1096–1097
Human chorionic gonadotropin (hCG), 666–669
Human developmentcritical periods of
schematic diagram of, 1041fHuman liver P450 enzymes, 55–59
described, 55–57enzyme induction, 57–59, 58tenzyme inhibition, 58t, 59
Human menopausal gonadotropins (hMGs), 666
Human papillomavirus (HPV) vaccine, 1164t
Human placenta derivativesrisks associated with, 1127t
Humatin, 828, 935. See also ParomomycinHumira, 656, 999, 1112. See also
AdalimumabHumoral immunity, 978
Katzung_Index_p1171-1234.indd 1197 9/24/11 12:03:36 PM

1198 Index
Humorsol, 112. See also DemecariumHuntington’s disease, 494–495, 494fHydatid disease
albendazole for, 939praziquantel for, 944
Hydeltrasol, 712. See also Prednisolone sodium phosphate
Hydralazine, 181, 181f, 189as antihypertensive drug, 181in chronic heart failure management, 220dosage of, 175t, 181ionization constants of, 11tpharmacokinetics of, 39t, 175t, 181
Hydrazineschemistry of, 527–528
Hydro-Diuril, 270. See also Hydrochlorothiazide
Hydrocarbonsaromatic
TLVs of, 1005ttoxic exposure to, 1005
halogenated aliphaticTLVs of, 1005ttoxic exposure to, 1005–1006, 1005t
Hydrochlorothiazide, 260f, 270dosage of, 261tpharmacokinetics/dosage of, 175t
Hydrocodone, 558pharmacokinetics of, 545tmixtures, 562
Hydrocortisone, 698–702. See also Cortisolpreparations available, 712, 1112
Hydroflumethiazide, 270Hydrogen peroxide
as antiseptic/disinfectant, 896–897Hydrogen sulfide
poisoning by, 1036tHydrolyses, 57tHydromorphone, 556, 562, 563
pharmacokinetics of, 545tHydromox, 270. See also QuinethazoneHydrophobic bonds, 4Hydroquinone, 1068–1069Hygroton, 270. See also ChlorthalidoneHydroxocobalamin, 586
as poisoning antidote, 1033tHydroxyamphetamine, 148Hydroxychloroquine, 644
as immunosuppressive drug, 989Hydroxyprogesterone
structure of, 719fHydroxyprogesterone caproate
properties of, 723t5-Hydroxytryptamine, 281–287. See also
SerotoninHydroxyurea, 582, 961fHydroxyzine, 292, 387
dosage of, 279tHygiene
hand, 894
Hylorel, 189. See also GuanadrelHymenolepis nana
praziquantel for, 944Hyoscine
for nausea and vomiting, 1100Hyoscyamine, 127, 1111
for irritable bowel syndrome, 1096Hypercalcemia, 778–779
diuretics for, 267–268Hypercalciuria
idiopathic, 784Hyperchloremic metabolic acidosis
carbonic anhydrase inhibitors and, 257t, 258
potassium-sparing drugs and, 263Hypercholesterolemia(s)
familial, 624, 624fdrugs used for, 622t
primary, 624–625Hyperhidrosis
antimuscarinic drugs for, 123Hyperimmune immunoglobulins, 991Hyperkalemia
heart rhythm and, 230loop diuretics for, 259neuromuscular blocking drugs and, 474osmotic diuretics and, 264potassium-sparing drugs and, 263
Hyperlipidemia(s), 619drugs used for, 625–631thiazide diuretics and, 261
Hyperlipoproteinemia(s), 619dietary management of, 625drugs used for, 622tfamilial combined, 623, 624
drugs used for, 622tlipoprotein(a), 624
drugs used for, 622tpathophysiology of, 620–625secondary, 625
causes of, 622, 623tHypernatremia
osmotic diuretics and, 264Hyperosmolar coma
nonketotic, 744Hyperosmolar hyperglycemic syndrome
insulin for, 752Hyperparathyroidism
primary, 780Hyperphosphatemia, 780Hyperplasia
congenital adrenalsynthetic corticosteroids for, 704
prostatic, 738–739Hyperprolactinemia
dopamine agonists for, 672–673, 672f
ergot alkaloids for, 290Hypersecretory conditions
proton pump inhibitors for, 1088
Hypersensitivity, 981–983, 983f, 984fcontact, 983defined, 32tuberculin, 983type I, 981, 983ftype II, 981–982type III, 982type IV (delayed-type), 982–983, 984f
Hyperstat IV, 189. See also DiazoxideHypertension, 169–171
alcohol and, 394alpha-receptor antagonists for, 156causes of, 170classification of, 170tcoenzyme Q10 for, 1134diagnosis of, 169–170, 170tdiuretics for, 172–174, 174f, 267outpatient therapy in, 186prevalence of, 169pulmonary
eicosanoids and, 326resistant
polypharmacy for, 173treatment of, 171–187
Hypertensive emergencies, 187alpha-receptor antagonists for, 156
Hyperthermia, 284, 284t, 479. See Malignant hyperthermia
Hyperthyroidism, 691–694beta-receptor antagonists for, 164
Hypertriglyceridemia(s)familial, 623
drugs used for, 622tprimary, 622–624
Hyperuricemiagout and, 651–655loop diuretics and, 260thiazide diuretics and, 261
Hypnosissedative-hypnotic drugs causing, 380
Hypocalcemia, 259, 779–780calcium for, 779–780vitamin D for, 780
Hypoglycemiaglucagon for, 763insulin and, 753
Hypogonadismprimary
estrogens for, 720Hypokalemia, 230Hypokalemic metabolic acidosis
loop diuretics and, 257t, 260Hypokalemic metabolic alkalosis
thiazide diuretics and, 261Hypomagnesemia
loop diuretics and, 260Hyponatremia
osmotic diuretics and, 264thiazide diuretics and, 261
Hypoparathyroidism, 780–781
Katzung_Index_p1171-1234.indd 1198 9/24/11 12:03:36 PM

Index 1199
Hypophosphatemia, 780autosomal dominant, 783X-linked, 783
Hypotension, sympathomimetic drugs for, 144
Hypothalamic hormones, 659–679clinical uses of, 661treceptors of, 660–673
Hypothalamic-pituitary endocrine system, 659, 660f
Hypothalamic-pituitary-thyroid axis, 684fHypothyroidism, 687t, 690–691Hypoxanthine, 962fHytrin, 167, 189. See also Terazosin
IIbandronate
for osteoporosis, 775, 783, 786Ibritumomab tiuxetan, 992, 999Ibuprofen, 641, 655
ionization constants of, 11tfrom prescription to OTC status, 1116tproperties of, 638t
Ibutilidefor cardiac arrhythmias, 244–245membrane actions of, 237tpharmacologic properties of, 238t
Icatibant, 302Idarubicin
for cancer, 963t, 965clinical activity and toxicities of, 963t
Idiopathic edemadiuretics for, 267
Idiopathic hypercalciuria, 784Idiopathic short stature
growth hormone for, 663, 663tIdiopathic thrombocytopenic purpura (ITP)
passive immunization for, 1168tIDLs. See Intermediate-density lipoproteins
(IDLs)Idrocilamide, 478IGIV. See Immune globulin intravenous
(IGIV)Ilaris, 999. See also CanakinumabIloprost, 325f, 326, 328Imatinib
for cancer, 966–967, 967tImidazoline receptor, 176IMiDs. See Immunomodulatory derivatives
of thalidomide (IMiDs)Imipenem
pharmacokinetics and dosage of, 798t, 801–802
Imipenem/cilastatin, 807Imipramine, 525–530, 539
hepatic clearance ofaging effects on, 1052t
ionization constants of, 11tpharmacokinetics of, 39t, 529tfor urinary disorders, 122, 122t
Imiquimod, 888, 1067, 889Imitrex, 292. See also SumatriptanImmobility
anesthesia and, 435Immune globulin intravenous (IGIV), 990,
999, 1163, 1168tImmune insulin resistance, 753Immune modulation
echinacea in, 1126–1128Immune response(s), 635–636
abnormal, 981–985, 982f–984fnormal, 977–981, 979f, 980f
Immune systemadaptive, 978–981, 979f, 980falcohol effects on, 394–395eicosanoid effects on, 327–328elements of, 977–985innate, 977–978, 979fisoniazid effects on, 841
Immunitycell-mediated, 978humoral, 978innate
complement in, 979fprostaglandins and, 322
Immunization(s), 1163–1170. See also specific vaccines
active, 1163, 1164t–1167tmaterials used for, 1164t–1166t
passive, 1163, 1167, 1168t–1169tmaterials used for, 1168t–1169t
routine childhood schedule, 1167tfor travel, 1170untoward reactions to
legal liability for, 1167, 1170Immunodeficiency diseases, 984–985
primarypassive immunization for, 1168t
Immunoglobulin(s)hyperimmune, 991
Immunologic contact urticariatopical dermatologic medications and,
1063tImmunologic reactions to drugs and drug
allergy, 997–998Immunomodulation therapy
immunosuppressive drugs in, 995–997, 995t–996t
Immunomodulators, 1067Immunomodulatory derivatives of
thalidomide (IMiDs)as immunosuppressive drugs, 987–988
Immunopharmacology, 977–1000Immunosuppressive antibodies, 989–991Immunosuppressive drugs, 985–994.
See also specific drugsin autoimmune diseases, 995
Imodium, 1112. See also LoperamideImplantable cardioverter-defibrillator (ICD)
for cardiac arrhythmias, 228
Impulse conductiondisturbances of, 234–235, 234f–236f
Impulse formationdisturbances of, 233f, 234
Imuran, 999Inamrinone, 225Inapsine, 446. See also DroperidolIncertohypothalamic pathway, 506Increlex, 678. See also MecaserminIND (Investigational new drug), 74–76Indapamide, 260f, 270
dosage of, 261tInderal, 167, 189, 249. See also PropranololIndinavir, 871t, 879
drug interactions with, 874tIndirect thrombin inhibitors, 604–607,
605f. See also specific drugsheparin, 604–607, 605f
Indocin, 655. See also IndomethacinIndomethacin, 641, 638t
pharmacokinetic, 39tIndoramin, 155Infant(s)
drug therapy in, 1044–1046Infantile spasms, 421
drugs used for, 423Infarction(s)
following subarachnoid hemorrhagecalcium channel blockers effects on,
204Infection(s). See also specific types
eicosanoids and, 328Infergen, 889Infertility
femaleGnRH agonists for, 669–670
maleGnRH agonists for, 670gonadotropins for, 668
Inflammationechinacea for, 1127–1128eicosanoids and, 328kinins role in, 302lipoxygenase and, 323nitric oxide and, 335–336prostaglandins and, 322systemic
treatment of, 636Inflammatory bowel disease
described, 1100–1101drugs used for, 1100–1106, 1101f,
1102f, 1105tpancreatic enzyme supplementation,
1106–1107for variceal hemorrhage, 1107–1108
Infliximab, 649, 993for inflammatory bowel disease,
1104–1106, 1105tfor rheumatoid arthritis, 649for psoriasis, 1072
Katzung_Index_p1171-1234.indd 1199 9/24/11 12:03:36 PM

1200 Index
Influenzadrugs used for, 886–887
Influenza vaccine, 1164t–1165tInhalant(s)
abuse of, 576Inhaled anesthetics, 430–438. See also
specific drugsInhibin, 726Inhibitory postsynaptic potential (IPSP),
92, 93f, 363, 363fInnohep, 617. See also TinzaparinINOmax, 337. See also Nitric oxideInositol 1,4,5-trisphosphate (IP3), 28,
131, 131t, 132f, 137t
as second messenger, 28–29, 28fInotropic drugs
positivein the elderly, 1056in heart failure management, 212t,
218Inotropic effect
positive, 138Insomnia
melatonin for, 1136Inspired concentration of inhaled
anesthetics, 430–432, 432fInspra, 225, 262t, 270.
See also EplerenoneInsulin, 743, 744–753
targets foreffects of, 747, 747f
treatment with, 751Insulin aspart, 747, 748t, 749Insulin delivery systems, 748t, 750–751
insulin pumps, 750–751portable pen injectors, 748t, 750
Insulin detemir, 748t, 750Insulin glargine, 748t, 749–750Insulin glulisine, 747, 748t, 749Insulin-like growth factor-I (IGF-I), 661,
661fInsulin lispro, 747, 748t, 749Insulin pumps, 750–751Insulin receptor, 746–747, 746f, 746tInsulin resistance
immune, 753Insulin secretagogues, 754–756. See also
specific types, eg, SulfonylureasIntegrase strand transfer inhibitors, 871t,
882Integrilin, 617. See also EptifibatideIntention tremor, 494Interferon alfa-2a
for HBV infection, 883tInterferon alfa-2b
for HBV infection, 883tInterferon(s), 882–887, 995
dermatologic uses of, 1078tproperties of, 995t
Interleukin(s)properties of, 995t–996t
Interleukin-1 inhibitorsfor gout, 655
Interleukin-2, 999Interleukin-11
agents mimicking actions of, 591t, 594–595
clinical uses of, 591t, 594–595Intermediate-density lipoproteins (IDLs),
619Intermittent claudication
treatment of, 207Intestinal osteodystrophy, 782Intestine
actions of PTH, FGF23, and vitamin D on, 774t
disorders of, 1091–1114Intracellular signaling pathways
in renin release, 296, 297fIntracellular sodium
concentration of, 212Intracranial pressure
reduction ofosmotic diuretics for, 264
Intragastric acidityreducing of
drugs used for, 1081–1089Intragastric pressure
increasedneuromuscular blocking drugs and, 474
Intraocular pressureincreased
neuromuscular blocking drugs and, 474
reduction ofosmotic diuretics for, 264
Intravenous anesthetics, 438–446. See also specific drugs
pharmacologic properties of, 439tIntrinsic efficacy, 7Intrinsic factor, 586Intron-A, 889, 999. See also Interferon
alfa-2bIntropin, 148, 225. See also DopamineInvanz, 807. See also ErtapenemInvega, 519. See also PaliperidoneInverse agonists, 8, 8fInversine, 127, 189. See also MecamylamineIodide(s)
antithyroid activity of, 689fetal effects of, 1042tmetabolism of, 681
Iodide organification, 682Iodine
as antiseptic/disinfectant, 895during lactation, 1048tradioactive
antithyroid activity of, 689for Graves’ disease, 692
Iodophor(s)as antiseptic/disinfectant, 895
Iodoquinol, 929f, 935for amebiasis, 929–930
Iodotope, 695. See also Radioactive iodine sodium
Ion channel(s)characterization of
tools for, 360–360tCNS drugs and, 360–361novel analgesic targets and, 550
Ion channel blockersin chronic heart failure
management, 221Ion transport
lithium effects on, 514Ionization
of weak acids and bases, 10–12, 11t, 12f, 12t
Ionization constantof common drugs, 11t
Ionotropic receptors, 360–361drugs mediating their effects via
abuse of, 573–576Iopidine, 148. See also ApraclonidineIpratropium, 127, 347, 355
for COPD, 121, 353Iprivask, 617. See also DesirudinIPSP. See Inhibitory postsynaptic potential
(IPSP)Irbesartan, 190, 225Irinotecan
clinical activity and toxicities of, 963tIron, 582–586, 598
drug interactions with, 1157tpoisoning by, 1037
Iron deficiency anemiatreatment of, 585–586, 586t
Iron dextran, 585Iron-sucrose complex, 585–586Irritable bowel syndrome
drugs used for, 1095–1097, 1097fIrritant(s)
gasespoisoning by, 1036t
topical dermatologic medications as, 1063t
Ischemic heart diseasebeta-receptor antagonists for, 162, 163fcoenzyme Q10 for, 1134
Isentress, 889. See also RaltegravirIslet amyloid polypeptide (IAPP), 743, 763Ismelin, 189. See also GuanethidineIsmo, 209. See also Isosorbide mononitrateIsocarboxazid
dosage of, 535tIsoeicosanoids, 317Isoetharine, 355Isoflurane, 432f, 446
pharmacologic properties of, 432f, 433t
Katzung_Index_p1171-1234.indd 1200 9/24/11 12:03:36 PM

Index 1201
Isoimmune diseaseimmunosuppressive drugs for, 985t
Isolantoxicity rating for, 1008t
Isoniazid, 840–841, 848during lactation, 1048tfor tuberculosis, 840–841, 840t
Isophane insulin, 748t, 749Isopropyl alcohol
as antiseptic/disinfectant, 893t, 894
Isoproterenol, 136f, 138t, 139f, 141, 148, 343f, 355
for asthma, 343cardiovascular responses to, 138tionization constants of, 11t
Isoptin, 190, 209, 249. See also VerapamilIsopto Carpine, 112. See also PilocarpineIsopto Homatropine, 127. See also
HomatropineIsordil, 209. See also Isosorbide dinitrateIsosorbide dinitrate, 196, 209Isosorbide mononitrate, 197, 209Isospora belli
drugs used for, 931tIsotretinoin
for acne, 1070fetal effects of, 1042t
Isradipineclinical pharmacology of, 202t
Istaroximein heart failure management, 218
Isuprel, 148, 355. See also IsoproterenolItraconazole, 853f, 858
pharmacologic properties of, 854tfor systemic infections, 854
Ivermectin, 940–941for helminthic infections, 938t,
940–941
JJanumet, 766. See also Sitagliptin plus
metforminJanus-kinase (JAK) family, 23, 24fJanuvia, 766. See also SitagliptinJerking
myotonic, 421Jet lag
melatonin for, 1135–1136Jin Bu Huan
risks associated with, 1127t
KKaletra, 889. See also Lopinavir/ritonavirKallikrein(s), 300Kallikrein-kinin system
drugs affecting, 302–303Kanamycin, 822f, 826–827, 828
for tuberculosis, 840t, 844Kantrex, 828. See also Kanamycin
Kava-kavarisks associated with, 1127t
Kawasaki diseasepassive immunization for, 1168t
Kefauver-Harris Amendments of 1962, 74, 74t
Keflex, 807. See also CephalexinKefurox, 807. See also CefuroximeKefzol, 807. See also CefazolinKemadrin, 499. See also ProcyclidineKenalog, 712. See also Triamcinolone Keppra, 427. See also LevetiracetamKeratolytic agents, 1074–1076Kerlone, 189. See also BetaxololKetalar, 446. See also KetamineKetamine, 438f, 444–445, 446
abuse of, 569t, 575–576, 577ffor chronic pain, 550as intravenous anesthetic, 444–445pharmacologic properties of, 439t
Ketanserin, 286Ketek, 819. See also TelithromycinKetoacidosis
diabetic, 744insulin for, 751–752
Ketoconazole, 709f, 710, 712, 737, 853f, 858
oral, 1066pharmacologic properties of, 854tfrom prescription to OTC status, 1116tfor systemic infections, 854
Ketolides, 814–815, 819Ketoprofen, 641–642, 655
properties of, 638tKetorolac, 642, 655
properties of, 638tKetotifen, 292Ketotifen fumarate
from prescription to OTC status, 1116t
Kidney(s). See also under Renalactions of PTH, FGF23, and vitamin D
on, 774tangiotensin II effects on, 298inhaled anesthetics effects on, 436–437lead poisoning effects on, 1015lipoxygenase effects on, 323lithium effects on, 516–517methylxanthines effects on, 346opioid analgesics effects on, 552prostaglandins and thromboxanes effects
on, 320–321Kidney disease
chronic, 781–782vitamin D for, 781–782
diuretics for, 266–267Kidney stones
carbonic anhydrase inhibitors and, 258potassium-sparing drugs and, 263
Kineret, 999. See also Anakinra
Kinin(s), 300–303Kinin receptors
mechanisms of action of, 302Kininogen(s), 301Klonopin, 387, 427. See also ClonazepamKnockin mice, 3Knockout mice, 3
adrenoceptor subtypes and, 133Koate-HP, 617. See also Antihemophilic
factorKogenate, 617. See also Antihemophilic
factorKombiglyze, 766. See also Saxagliptin plus
metforminKonyne 80, 617. See also Factor IX
complex, humanKrystexxa, 656. See also PegloticaseKytril, 1111. See also Granisetron
LLabetalol, 155, 158f, 159t, 161–162, 167,
189as antihypertensive drug, 179for hypertensive emergencies, 156pharmacokinetic and pharmacodynamic
parameters for, 39tLabor, obstetrical
facilitation ofeicosanoids in, 325–326
prematuresuppression of
sympathomimetic drugs for, 145Lacosamide
for seizures, 413–414Lactation
drug use during, 1046–1047, 1048tmilk thistle effects on, 1132physiologic
dopamine agonists for, 673Lactation stimulation
postpartumdomperidone for, 1092
Lactulosein constipation management, 1093
Lamictal, 427, 519. See also LamotrigineLamisil, 858. See also TerbinafineLamivudine, 863f, 871t, 874, 889
for HBV infection, 883t, 884–885during pregnancy, 876, 876t
Lamotrigine, 414f, 427, 519for bipolar disorder, 517for seizures, 414
Lamprene, 848. See also ClofazimineLanoxicaps, 225. See also DigoxinLanoxin, 225. See also DigoxinLanreotide, 666, 678Lansoprazole, 1086f, 1111
pharmacokinetics of, 1086tfrom prescription to OTC status, 1116t
Lariam, 935. See also Mefloquine
Katzung_Index_p1171-1234.indd 1201 9/24/11 12:03:36 PM

1202 Index
Lasix, 270. See also FurosemideLatanoprost, 315, 328Laxative(s), 1092–1094. See also specific
typesingredients in, 1122t
LDLs. See Low-density lipoprotein(s) (LDLs)
Leadtoxicology of, 1013–1017
Lead compounds, 71toxicology of, 1014t
Lead poisoning, 1013–1017Learning
maladaptiveaddiction as, 568–571
Leflunomide, 645, 656, 999as immunosuppressive drug, 989, 999
Legal issuesimmunization-related, 1167, 1170in prescribing drugs, 1144–1147, 1144t,
1145tLeiomyomata
uterinegonadotropins in, 670
Leishmaniasisdrugs used for, 931t, 933–934pentamidine for, 932
Lenalidomide, 988, 999for multiple myeloma, 971
Lepirudin, 607, 617Leprosy
drugs used for, 845–846, 848Lescol, 633. See also FluvastatinLethal dose
median, 31–32, 31f, 71–72minimum, 71
Letrozole, 733, 740Leu-enkephalin, 543–544, 544tLeukemias
chemotherapeutic drugs for, 969–970
chronic lymphocyticpassive immunization for, 1168t
Leukine, 598. See also SargramostimLeukotriene antagonists
for asthma, 352Leukotriene inhibitors, 349–350Leuprolide acetate, 678Levalbuterol, 355Levaquin, 838. See also LevofloxacinLevatol, 167, 189. See also PenbutololLevetiracetam, 414f, 427
for seizures, 414–415Levitra, 209. See also VardenafilLevo-Dromoran, 562. See also LevorphanolLevo-T, 695. See also LevothyroxineLevobunolol, 161, 167Levobupivacaine, 460, 463Levocabastine, 292Levocetirizine, 292
Levodopadrug interactions with, 488, 1157tionization constants of, 11tfor parkinsonism, 484–488, 485f, 486fsympathomimetic drug effects of, 143
Levofloxacin, 835f, 838clinical uses of, 836dosage of, 840t, 838pharmacokinetics of, 836t
Levolet, 695. See also LevothyroxineLevomethadyl acetate, 562Levonorgestrel, 740
from prescription to OTC status, 1116tLevophed, 148. See also NorepinephrineLevopropoxyphene, 559Levorphanol, 557, 562
pharmacokinetics of, 545tLevosimendan, 212
in heart failure management, 218Levothroid, 695. See also LevothyroxineLevothyroxine, 687–688, 695Levoxyl, 695. See also LevothyroxineLevsin, 127, 1111. See also HyoscyamineLexapro, 539. See also EscitalopramLexiva, 889. See also FosamprenavirLialda, 1102, 1102fLibrium, 387, 400. See also
ChlordiazepoxideLidocaine, 240f, 249, 451t, 460, 463
for cardiac arrhythmias, 237–241cardiac effects of, 237t, 240, 240ffor chronic pain, 550ionization constants of, 11tpharmacokinetics of, 39t, 452t
Lidocaine mixtures, 463Lifestyle changes
aging and, 1053Ligand-gated channels, 23–24, 24f,
360–361Linaclotide
in constipation management, 1094Linagliptin
for diabetes mellitus, 761, 766Lincomycin, 819Lindane, 1068
toxicity rating for, 1006tLinezolid, 817, 819
for tuberculosis, 840t, 845Lioresal, 481. See also BaclofenLiothyronine, 687–688, 695Liotrix, 695Lipid diffusion, 9, 9fLipid metabolism
oral contraceptives effects on, 728Lipid resuscitation
local anesthetics and, 459Lipid-soluble drugs
intracellular receptors for, 21–22, 21fduring pregnancy, 1039
Lipid:aqueous partition coefficient, 9, 9f
Lipodystrophyinsulin and, 753
Lipopeptidespharmacokinetics and dosage of, 798t,
802–803Lipoprotein(s), 619. See also specific types,
e.g., High-density lipoproteins (HDLs)
disorders of, 621–622, 622t, 623tmetabolism of, 620–621, 621fstructure of, 630synthesis and catabolism of, 620, 621f
Lipoprotein lipase, 620Lipoprotein(a), 619, 620Lipoprotein(a) hyperlipoproteinemia, 624
drugs used for, 622tLipoxin(s), 317Lipoxygenase(s)
products of, 316–317, 316fprostaglandins and thromboxanes effects
on, 322–323Liquaemin, 617. See also Heparin sodiumLiraglutide
for diabetes mellitus, 760–761, 766Lisinopril
pharmacokinetics/dosage of, 175t, 190, 225
Lithiumfor bipolar disorder, 513–517drug interactions with, 516, 1158tfetal effects of, 1042tduring lactation, 1048tpharmacokinetics of, 39t, 514, 514t
Lithium carbonate, 519Liver
alcohol effects on, 392–393inhaled anesthetics effects on, 437insulin effects on, 746, 747toral contraceptives effects on, 727–728
Liver abcess, amebic, chloroquine for, 917Liver impairment
antimicrobial drugs in patients with, 908t, 909
Livostin, 292. See also LevocabastineLoa loa
diethylcarbamazine citrate for, 940Loading dose, 46–47, 47fLobeline, 100fLocal anesthesia/anesthetics, 449–464.
See also specific drugsH1-receptor antagonists in, 279neuromuscular blocking drugs with, 475neuronal factors affecting, 455in OTC products, 1123ttransient neurologic symptoms of, 459
Local circuit neurons, 365, 365fLodine, 655. See also EtodolacLodosyn, 499. See also CarbidopaLofibra, 633. See also FenofibrateLog-kill hypothesis, 951, 951f
Katzung_Index_p1171-1234.indd 1202 9/24/11 12:03:36 PM

Index 1203
Lomefloxacinpharmacokinetics of, 836t, 838
Lomotil, 1112. See also DiphenoxylateLomustine
clinical activity and toxicities of, 956tLong-term potentiation (LTP), 369, 571Loniten, 189. See also MinoxidilLoop diuretics, 258–260
sites of action of, 252fthiazide diuretics and, 265toxicity of, 260
Loop of Henlein renal tubule transport, 252t,
253–254, 254fsites of action of, 252t
Loperamide, 558for diarrhea, 1094, 1112
Lopid, 633. See also GemfibrozilLopinavir, 871t, 879–880Lopinavir/ritonavir, 889
drug interactions with, 874tduring pregnancy, 876, 876t
Lopressor, 167, 189, 225. See also Metoprolol
Lorabid, 807. See also LoracarbefLoracarbef, 807Loratadine
dosage of, 279t, 292from prescription to OTC status,
1116tLorazepam
dosages of, 383tfor epilepsy, 420pharmacology of, 377t, 439tpreparations available, 387, 400, 427,
446Lorcaserin
in obesity management, 664, 665tLortab, 562. See also Hydrocodone/acet-
aminophenLosartan
pharmacokinetics/dosage of, 175t, 185, 190, 225
Lotensin, 190, 225. See also BenazeprilLotrimin, 858. See also ClotrimazoleLotrimin Ultra, 858. See also ButenafineLotronex, 1111. See also AlosetronLovastatin, 626f, 633Lovaza, 633. See also Omega-3 fatty acidsLovenox, 617. See also EnoxaparinLow-density lipoproteins (LDLs), 619, 6205-LOX-activating protein (FLAP), 316,
316fLoxapine
advantages and disadvantages of, 509tdosage of, 510t, 519
Loxitane, 519. See also LoxapineLozol, 270. See also IndapamideLSD
abuse of, 569t, 573
LTP. See Long-term potentiation (LTP)Lubiprostone
in constipation management, 1094
for irritable bowel syndrome, 1097, 1112
Lucentis, 999. See also RanibizumabLufyllin, 355. See also DyphyllineLugol’s Solution, 898. See also Iodine Lumefantrine, 918f
clinical uses of, 917tfor malaria, 927
Lumigan, 328. See also BimatoprostLuminal Sodium, 387, 427. See also
PhenobarbitalLunesta, 387. See also EszopicloneLung(s)
maturation of, fetal, corticosteroids and, 705–706, 706t
Lung cancerchemotherapeutic drugs for, 973
Lupron, 678. See also Leuprolide acetateLusedra, 446. See also FospropofolLuteinizing hormone (LH),
666–669, 715Lutropin alfa, 667, 678Luveris, 678. See also Lutropin alfaLymphocyte(s)
T, 978–981, 979f, 980fLymphocyte immune globulin, 999Lymphoma(s)
Hodgkin’schemotherapeutic drugs for, 970
non-Hodgkin’schemotherapeutic drugs for,
970–971Lynestrenol, 723tLyrica, 427. See also PregabalinLysodren, 712. See also Mitotane
MMa-huang
risks associated with, 1127tMaalox, 1111 MABs. See Monoclonal antibodies (MABs)Macrodantin, 898. See also
NitrofurantoinMacrolides, 813–815. See also specific drugs
drug interactions with, 1158tin gastrointestinal motility stimulation,
1092Macrophage colony-stimulating factor
properties of, 996tMacugen, 999. See also PegaptanibMacula densa
in renin release, 296, 297fMacular degeneration
drugs used in, 1057Mafenide, 837Magan, 655. See also Magnesium salicylate
Magnesiumfor cardiac arrhythmias, 246digitalis with, 218
Magnesium citratein constipation management, 1093
Magnesium hydroxideas antacid, 1083in constipation, 1093, 1112
Magnesium salicylate, 655Magnesium sulfate, 250Maintenance dose, 46Major depressive disorder. See also
Depressionantidepressants for, 521–541. See also
Antidepressants(s)BDNF in, 522–523, 522fcharacteristics of, 521monoamine(s) and, 523, 524fmonoamine hypothesis of, 522, 523,
523fneurotransmitters and, 523, 524fneurotrophic hypothesis of, 522–523,
522fpathophysiology of, 522
neuroendocrine factors in, 524–525prevalence of, 521
Maladaptive learningaddiction as, 568–571
Malaoxon, 106fMalaria, 915–927
chemoprophylaxis of, 922, 924drugs used for, 915–927. See also specific
drugs and Antimalarial drugsfalciparum
chloroquine-resistanttreatment of, 926
treatment of, 920, 920t, 922ovale
treatment of, 924parasites carrying
life cycle of, 915, 916fprevention of
drugs used for, 919tvivax
treatment of, 924Malarone, 935. See also
Atovaquone-proguanilfor malaria, 924in malaria prevention in travelers, 919t
Malathion, 1068structure of, 106ftoxicity rating for, 1007t
Male chemical contraception, 739Male infertility
GnRH agonists for, 670gonadotropins for, 668
Male reproductive systemeicosanoid effects on, 326prostaglandins and thromboxanes effects
on, 321
Katzung_Index_p1171-1234.indd 1203 9/24/11 12:03:36 PM

1204 Index
Malignancy(ies)secondary
chemotherapeutic drugs for, 974Malignant hyperthermia, 284, 284t, 475
inhaled anesthetics and, 437treatment of
dantrolene in, 479Malignant melanoma
chemotherapeutic drugs for, 974Mammalian target of rapamycin (mTOR)
inhibitors, 985t, 986–987Mannitol, 270MAOIs. See Monoamine oxidase inhibitors
(MAOIs)Maprotiline, 527–530Maraviroc, 871t, 881–882, 889
drug interactions with, 874tMarcaine, 463. See also BupivacaineMarezine. See also CyclizineMarijuana
abuse of, 572Marinol, 1111. See also DronabinolMarplan, 539. See also IsocarboxazidMateria medica
defined, 2Maternal drug actions
during pregnancy, 1040Maturation
femaleestrogen effects on, 719–720
male, 735Mavik, 190, 225. See also TrandolaprilMaxair, 355. See also PirbuterolMaxaquin, 838. See also LomefloxacinMaximum effect
as pharmacodynamic variable, 48Maxipime, 807. See also CefepimeMaxzide, 262tMean arterial pressure, 89Measles
passive immunization for, 1168tMeasles-mumps-rubella (MMR) vaccine,
1165tMebaral, 387, 427. See also MephobarbitalMebendazole, 941–942, 946
for helminthic infections, 938t, 941–942Met-enkephalin, 543–544, 544tMecamylamine, 124, 127, 189Mecasermin, 665, 678Mechlorethamine, 954f
clinical activity and toxicities of, 956tMeclizine
dosage of, 279t, 292for nausea and vomiting, 1100
Meclofenamate sodium, 656Meclofenamic acid, 637fMectizan, 946. See also IvermectinMed Watch program, 1146Median effective dose (ED50), 31–32, 31fMedian lethal dose (LD50), 31–32, 31f, 71–72
Median toxic dose (TD50), 31–32Medicare Modernization Act of 2003, 1146Medication Therapy Management (MTM)
program, 1146Medrol, 712. See also MethylprednisoloneMedrol Enpack, 1112. See also
MethylprednisoloneMedroxalol, 162Medroxyprogesterone, 719f
properties of, 723tMedroxyprogesterone acetate, 725, 740Medullary-periventricular pathway, 506Mefenamic acid, 656Mefloquine
clinical uses of, 917tfor malaria, 919t, 923in malaria prevention in travelers, 919t
Mefoxin, 807. See also CefoxitinMegace, 740. See also Megestrol acetateMegakaryocyte growth factors, 591t,
594–595Megestrol acetate, 723t, 740Meglitinide
for diabetes mellitus, 756, 756t, 766Melagatran, 607Melanoma(s)
malignantchemotherapeutic drugs for, 974
Melarsoprolclinical uses of, 933
Melatonin, 283, 1135–1136Melatonin receptor agonists, 292Mellaril, 519. See also ThioridazineMeloxicam, 640, 656
properties of, 638tMelphalan, 954f
clinical activity and toxicities of, 956tMemantine
for Alzheimer’s disease, 1055Membrane-delimited pathways, 361, 362fMembrane electrical activity
ionic basis of, 229–230, 229fMenest, 740. See also Esterified estrogens;
EstroneMeningococcal conjugate vaccine, 1165tMenopause, 715, 716fMenopur, 678. See also MenotropinsMenotropins, 666, 678Menstrual cycle, 715, 716fMental disturbances
dopamine receptor agonists and, 489–490
Mentax, 858. See also ButenafineMepenzolate, 127Meperidine,40t, 557, 562
abuse of, 572cardiovascular system effects of, 551–552hepatic clearance of
aging effects on, 1052tpharmacokinetics of, 40t, 545t
Mephenytoinfor seizures, 409, 427
Mephobarbital, 387, 427Mepivacaine, 451t, 460–461, 463
pharmacokinetic properties of, 452tMeprednisone
clinical uses and dosage of, 703tMeprobamate, 375fMepron, 935. See also AtovaquoneMequinol, 1068–10696-Mercaptopurine (6-MP)
for inflammatory bowel disease, 1103–1104
mechanism of action of, 961, 961fMercury poisoning, 1014t, 1019–1021Meropenem
pharmacokinetics and dosage of, 798t, 801–802
Merrem, 807. See also MeropenemMersol, 898. See also ThimerosalMesalamine, 1112Mesalamine compounds, 1102, 1102fMesantoin, 427. See also MephenytoinMescaline
abuse of, 569t, 573Mesolimbic-mesocortical pathway, 506Mestinon, 112. See also PyridostigmineMestranol, 718fMetabolic acidosis
hyperchloremiccarbonic anhydrase inhibitors and,
257t, 258potassium-sparing drugs and, 263
hypokalemicloop diuretics and, 257t, 260
Metabolic alkalosiscarbonic anhydrase inhibitors for, 257,
257thypokalemic
thiazide diuretics and, 261Metabolic functions
autonomic nerve activity effects on, 90tMetabolic syndrome, 664Metabolism
antipsychotic drugs effects on, 512beta-receptor antagonists effects on,
160bone
prostaglandins effects on, 322estrogen effects on, 720ginkgo effects on, 1130glucagon effects on, 762–763histamine effects on, 276oral contraceptive effects on, 728sympathomimetic drug effects on, 139synthetic corticosteroids effects on, 706
Metabolizers, drugextensive, 64poor, 63 ultrarapid, 63–64
Katzung_Index_p1171-1234.indd 1204 9/24/11 12:03:36 PM

Index 1205
Metabotropic receptors, 361Metaglip, 766. See also Glipizide plus
metforminMetal(s)
as environmental pollutant, 1011–1012
Metalloprotein(s)nitric oxide interactions with, 331–332
Metamucil, 1112. See also PsylliumMetaproterenol
for asthma, 344, 355Metaraminol, 148
ionization constants of, 11tMetaxalone, 481Metformin, 757, 766Methacholine
properties of, 99, 100tMethadone, 557, 562
fetal effects of, 1042tionization constants of, 11tduring lactation, 1048tpharmacokinetics of, 545t
Methallenestrildosage of, 719t
Methamphetamine, 148ionization constants of, 11tsympathomimetic drug effects of, 142
Methanolpharmacology of, 397–398,
398f, 400Methanol poisoning, 1037Methantheline, 127Methazolamide, 270
dosage of, 257tMethenamine, 898
as urinary antiseptic, 893Methergine, 292. See also MethylergonovineMethimazole
for Graves’ disease, 691–692, 695Methocarbamol, 481Methohexital, 446
pharmacologic properties of, 439tMethomyl
toxicity rating for, 1008tMethotrexate, 645, 958
adverse effects of, 645, 1104for cancer, 958clinical activity and toxicities of, 959tclinical uses of, 1104fetal effects of, 1043tas immunosuppressive drug, 989indications for, 645for inflammatory bowel disease, 1104ionization constants of, 11tmechanism of action of, 645pharmacodynamics of, 40t, 1104pharmacokinetics of, 40t, 645, 1104
Methoxamine, 136f, 141for acute hypotension, 144
Methoxsalen, 1069
Methoxychlortoxicity rating for, 1006t
Methscopolamine, 127Methsuximide, 427
for seizures, 418N-Methyl-D-aspartate glutamate
antagonistsfor Alzheimer’s disease, 1055t
Methylation, 60tMethylcellulose, 1093, 1112Methyclothiazide, 270
dosage of, 261tMethyldopa
as antihypertensive drug, 175–176, 175t
ionization constants of, 11tpharmacokinetics/dosage of, 175t, 176
Methylene-dioxymethamphetamine (MDMA)
abuse of, 569f, 577–578Methylergonovine, 292Methylnaltrexone, 560, 562Methylnaltrexone bromide
in constipation management, 1094, 1112
Methylphenidate, 142for ADHD, 146
Methylprednisoloneclinical uses and dosage of, 703tpreparations available, 712, 1112
Methyltestosterone, 735t, 740in replacement therapy
dosage of, 736tMethylthiouracil
fetal effects of, 1043tMethylxanthine(s)
for asthma, 344–346Meticorten, 712. See also PrednisoneMetipranolol, 167Metoclopramide
in gastrointestinal motility stimulation, 1092
for nausea and vomiting, 1100Metolazone
dosage of, 261tMetoprolol, 159t, 161
as antihypertensive drug, 175t, 179ionization constants of, 11tpharmacokinetics, 40tpreparations available, 167, 189, 225
Metrifonate, 942for helminthic infections, 938t, 942
Metronidazole, 891–892for acne, 1064adverse effects of, 929for amebiasis, 927–929, 928tdrug interactions with, 610tfetal effects of, 1043tpharmacokinetics of, 40t, 928preparations available, 898, 935
Metyrapone, 710Metyrosine, 85
for pheochromocytoma, 156, 167Mevacor, 633. See also LovastatinMevalonate, 626fMexiletine
for cardiac arrhythmias, 241for chronic pain, 550membrane actions of, 237t
Mexitil, 249. See also MexiletineMibefradil, 203Micafungin, 855, 858Micardis, 190, 225. See also TelmisartanMicatin, 858. See also MiconazoleMichaelis-Menten elimination, 42Miconazole, 858
from prescription to OTC status, 1116tMicrocytic hypochromic anemia, 582Micronase, 766. See also GlyburideMicronized estradiol
dosage of, 719tMicroorganisms
chemical and physical killing ofterms related to, 894t
MicroRNAs (miRNAs), 2Microsomal ethanol oxidizing system
(MEOS), 390Microsomal mixed function
oxidase systemphase I reactions and, 54–55, 55f,
56t–57tMicrosporidia
drugs used for, 931tMidamor, 270. See also Amiloride
dosage of, 262tMidazolam, 438f
pharmacokinetics, 40tpreparations available, 387, 446
Midodrine, 141, 148Mifeprex, 712, 740. See also
MifepristoneMifepristone (RU-486), 710–711,
731–732Miglitol
for diabetes mellitus, 759, 766Migraine(s)
beta-receptor antagonists for, 164ergot alkaloids for, 2905-HT1D/1B agonists for, 285–286, 285f
Milk of magnesia, 1112in constipation management, 1093
Milk thistle, 1131–1132Milnacipran, 525–529
dosage of, 535t, 539sympathomimetic drug effects of, 143
Milophene, 740. See also ClomipheneMilrinone, 225Miltefosine
clinical uses of, 934Miltown, 387. See also Meprobamate
Katzung_Index_p1171-1234.indd 1205 9/24/11 12:03:36 PM

1206 Index
Mineral oilas stool softener, 1093
Mineralocorticoids, 708–709. See also specific drugs
aldosterone, 708antagonists of, 711fludrocortisone, 709
Minimal bactericidal concentration (MBC), 903
Minimum inhibitory concentration (MIC), 792, 903
Minimum lethal dose, 71Minipress, 167, 189. See also PrazosinMinirin, 678. See also Desmopressin acetateMinocin, 819. See also MinocyclineMinocycline, 812, 819Minoxidil
as antihypertensive drug, 181, 181f, 189
pharmacokinetics/dosage of, 175t, 181from prescription to OTC status, 1116t
Mintezol, 946. See also ThiabendazoleMiochol-E, 112. See also Acetylcholine
(ACh)Miosis
opioid analgesics and, 551, 551tMiradon, 617. See also AnisindioneMirapex, 499. See also PramipexoleMircera, 598. See also Epoetin betamiRNAs, 2Mirtazapine, 527, 530
dosage of, 535tMisoprostol, 315, 327, 1090
fetal effects of, 1043tpreparations available, 328, 1111
Mithracin, 786. See also PlicamycinMitomycin
for cancer, 966clinical activity and toxicities of, 963t
Mitotane, 711Mitoxantrone
for cancer, 965Mitrolan, 1112. See also PolycarbophilMivacron, 481. See also MivacuriumMivacurium
pharmacodynamics of, 470tpharmacokinetics of, 468, 470ttissue effects of, 474t
Mixed function oxidases, 55Moban, 519. See also MolindoneMobic, 656. See also MeloxicamMobidin, 655. See also Magnesium
salicylateModafinil
for narcolepsy, 146, 148sympathomimetic drug effects of,
142–143Moduretic, dosage of, 262tMoexipril
preparations available, 190, 225
Molecular weightof drugs, 4
Molecule(s)special carrier, 9, 9ftransport, 9, 10t
Molindonedosage of, 510t, 519
Mometasone, 355Monistat 1, 858. See also TioconazoleMonoamine(s)
as CNS neurotransmitters, 370major depression and, 523, 524f
Monoamine hypothesisof major depression, 522, 523, 523f
Monoamine oxidase (MAO)metabolism of catecholamines by,
87, 88fMonoamine oxidase inhibitors (MAOIs)
for depression, 527–528, 528foverdose of, 1034for parkinsonism, 490
Monoamine transporterspharmacologic targeting of,
134, 135fMonobactam(s), 801, 807–808Monobenzone, 1068–1069Monoclate, 617. See also Antihemophilic
factorMonoclonal antibodies (MABs), 991–994,
993factions of, 993, 993fanti-IgE
for asthma, 350, 352antitumor, 991–992to deliver isotopes to tumors, 992described, 989–990as immunosuppressants and
anti-inflammatory agents, 992–994, 993f
Monoiodotyrosine (MIT), 682Mononine, 617. See also Factor IX
complex, humanMonooxygenase(s), 55Monopril, 190, 225. See also FosinoprilMontelukast, 328, 349, 349f, 355Monurol, 808. See also FosfomycinMood stabilizers, 519Moricizine
for cardiac arrhythmias, 242“Morning after” contraception, 731Morphinans
with mixed receptor actions, 559strong, 557
Morphine, 556. See also Opioid analgesics
ionization constants of, 11tmechanism of action of, 546–549,
547f–549fmetabolism of, 545–546organ system effects of, 549–552
Morphine (Cont.):pharmacodynamics of, 40t, 546–552pharmacokinetic and pharmacodynamic
parameters for, 40tpharmacokinetics of, 40t, 545tsource of, 543
Motility disordersdrugs used for, 1111
Motion sicknessantimuscarinic drugs for, 121H1-receptor antagonists for, 280
Motofen, 1112. See also DifenoxinMotrin, 655. See also IbuprofenMountain sickness
carbonic anhydrase inhibitors for, 257–258
Mouthpoisoning effects on, 1029
Movement disorders, 493–497, 498, 499drugs used for, 483–500. See also specific
drugs and Parkinsonism, drugs used for
MoviPrep, 1112. See also Polyethylene glycol electrolyte solution
Moxifloxacin, 835, 835f, 838pharmacokinetics of, 836t
Mozobil, 598. See also Plerixafor6-MP, 961. See also 6-MercaptopurinemTOR inhibitors. See Mammalian target of
rapamycin (mTOR) inhibitorsMucocutaneous infections
oral systemic antifungal drugs for, 855–856
Mucosal bleedingstress-related, 1088
Mucosal protective agents, 1111Multidrug resistance type 1 (MDR1)
transporter, 9, 10tMultidrug resistance–associated protein
(MRP) transporter, 9, 10tMultiple myeloma
chemotherapeutic drugs for, 971Mupirocin, 892, 898
topical, 1063Muromonab-CD3, 990, 999Muscarine, 100fMuscarinic agonists, 99–110Muscarinic antagonists, 115–124
for asthma, 351Muscarinic receptors, 87, 89tMuscarinic signaling, 101fMuscle
insulin effects on, 746, 747tMuscle pain
neuromuscular blocking drugs and, 474–475
Muscle relaxants, 465–482. See also specific drugs
neuromuscular blocking drugs, 465–476nondepolarizing vs. depolarizing, 471t
Katzung_Index_p1171-1234.indd 1206 9/24/11 12:03:36 PM

Index 1207
Muscle relaxants (Cont.):sedative-hypnotic drugs, 381spasmolytic drugs, 476–479tizanidine, 146
Muscle spasmacute local
drugs for, 479Mushroom poisoning
antimuscarinic drugs for, 123Myambutol, 848. See also EthambutolMycamine, 858. See also MicafunginMycelex-3, 858. See also ButoconazoleMycifradin, 828. See also NeomycinMycobacteria
atypicaldrugs active against,
845, 846tMycobacterial infections
drugs used for, 845, 846tisoniazid, 840–841rifampin, 841–842streptomycin, 824, 843
Mycobutin, 848. See also RifabutinMycophenolate mofetil, 645–646
dermatologic uses of, 1078tfetal effects of, 1043tas immunosuppressive drug, 985t, 987,
999Mycostatin, 858. See also NystatinMydriacyl Ophthalmic, 127. See also
TropicamideMydriasis, 118, 119fMyeloid growth factors, 592–594Myeloma multiple,
chemotherapeutic drugs for, 971Myenteric plexus, 81f, 82Myfortic, 999. See also Mycophenolate
mofetilMykrox, 270. See also MetolazoneMylanta, 1111 Myobloc, 481. See also Botulinum toxin
type BMyocardial infarction
acutethrombolytic drugs for,
611, 612oral contraceptives and, 729
Myocardial oxygen demanddeterminants of, 194, 194t
Myocardial oxygen supplydeterminants of, 194
Myoclonic jerking, 421Myopathy
coenzyme Q10 in, 1134Mysoline, 427. See also PrimidoneMytelase, 112. See also AmbenoniumMyxedema
coronary artery disease and, 690–691
Myxedema coma, 691
NNa+/K+-ATPase
activity of, 212Nabilone
abuse of, 572–573for nausea and vomiting, 1100, 1112
Nabumetone, 642Nadolol, 161
as antihypertensive drug, 179properties of, 159t
NADPH-cytochrome P450 oxidoreductase, 55
Nafarelin acetate, 678Nafcillin
adverse effects of, 796–797dosage of, 795tpharmacokinetics of, 794–795
Naftifine, 858topical, 1065
Naftin, 858. See also NaftifineNalbuphine, 558, 562
pharmacokinetics of, 545tNalfon, 655. See also FenoprofenNalidixic acid, 835fNalmefene, 559–560Naloxone, 559–560
clinical uses of, 560for opioid abuse, 572pharmacokinetics of, 560as poisoning antidote, 1033t
Naltrexone, 559–560in addiction management, 578in alcoholism management, 396–397
NANC neuronstransmitter substances found in, 83f, 84t,
87–88Nandrolone decanoate, 735t, 740Naphazoline, 148Naphazoline/pheniramine
from prescription to OTC status, 1116tNaprosyn, 656. See also NaproxenNaproxen, 638t, 642
preparations available, 656from prescription to OTC status, 1116t
Naqua, 270. See also TrichlormethiazideNaratriptan
pharmacokinetics of, 286tNarcan, 562. See also NaloxoneNarcolepsy
modafinil for, 146Nardil, 539. See also PhenelzineNaropin, 463. See also RopivacaineNatacyn, 858. See also NatamycinNatalizumab, 993, 999Natamycin, 858Nateglinide
for diabetes mellitus, 756, 766National Cholesterol Education Program:
Adult Treatment Guidelines (2011), 622t
National Institute for Occupational Safety and Health (NIOSH), 1005–1006
National Research Council (NRC) Wound Classification Criteria, 911
Natrecor, 225, 270. See also NesiritideNatriuretic peptides, 303–304Natural estrogens, 716f, 717–718, 717fNatural killer (NK) cells, 978Natural killer–T (NKT) cells, 978Natural products
for cancer, 962–965, 963tclinical activity and toxicities of, 963t
Natural progestins, 723Natural toxins, 360, 360tNature-Throid, 695. See also Thyroid
desiccatedNaturetin, 270. See also
BendroflumethiazideNausea
H1-receptor antagonists for, 278, 279tNausea and vomiting
chemotherapy-induced5-HT3-receptor antagonists for, 1099
drugs used for, 1097–1100. See also specific drugs and Antiemetic drugs
opioid analgesics and, 551, 551tpathogenesis of
neurologic pathways involved in, 1097, 1098f
postoperative and postradiation5-HT3-receptor antagonists for, 1099
of pregnancyH1-receptor antagonists for, 280
Navane, 519. See also ThiothixeneNDA, 75–76Nebcin, 828. See also TobramycinNebivolol, 161
as antihypertensive drug, 179–180pharmacokinetics/dosage of, 175tpreparations available, 167,
189, 225properties of, 159t
Nedocromilfor asthma, 348–349, 352
Nefazodonechemistry of, 526, 527fdosage of, 535tpharmacodynamics of, 530t, 531pharmacokinetics of, 529, 529t
Nelfinavir, 871t, 880, 889drug interactions with, 874t
Nematodesdrugs used for, 938t
Nembutal, 427. See also PentobarbitalNembutal Sodium, 387.
See also PentobarbitalNeo-Synephrine, 148.
See also Oxymetazoline
Katzung_Index_p1171-1234.indd 1207 9/24/11 12:03:36 PM

1208 Index
Neoadjuvant chemotherapy, 950Neoendorphin(s)
alpha and beta, 544Neomycin, 826–827, 828
topical, 827, 1064Neonate(s)
Graves’ disease in, 693half-lives of drugs in, 1045toral drug absorption in, 1044, 1044tspecial pharmacodynamic features in,
1046Neoplasm(s), 949
thyroid, 694Neoral, 999. See also CyclosporineNeostigmine, 105–106, 112Nephrogenic diabetes insipidus
ADH antagonists and, 265lithium effects on, 516–517
Nephrolithiasisdiuretics for, 267
Nephronsegments of, 252t
Nephrotic syndrome, 784Neptazane, 270. See also MethazolamideNernst equation, 230Nerve growth factor
for Alzheimer’s disease, 1055tNervous system. See also Autonomic
nervous system (ANS); Central nervous system (CNS); Enteric nervous system; Peripheral nervous system
alcohol effects on, 393antipsychotic drugs effects on, 511histamine effects on, 275inhaled anesthetics effects on, 434–436lead poisoning effects on, 1014–1015lithium effects on, 516local anesthetics effects on, 458–459poisoning effects on, 1029–1030properties in common, endocrine
system, 80serotonin effects on, 282–283
Nesacaine, 463. See also ChloroprocaineNesiritide
in heart failure management, 219preparations available, 225, 270
Netilmicin, 826Neulasta, 598. See also PegfilgrastimNeumega, 598. See also OprelvekinNeupogen, 598. See also FilgrastimNeural injury
local anesthetics and, 458–459Neurocysticercosis
albendazole for, 939praziquantel for, 944
Neuroendocrine systemin depression, 524–525opioid analgesics effects on, 551t, 552
Neuroendocrine tumorsdiarrhea due to
octreotide for, 1095Neurokinin A, 307Neurokinin B, 307Neurokinin receptor antagonists
for nausea and vomiting, 1099–1100Neuroleptanesthesia, 509Neuroleptic(s), 501Neuroleptic malignant syndrome, 284,
284t, 496antipsychotic drugs and, 512–513
Neurologic diseasesbeta-receptor antagonists for, 164
Neuromuscular blocking drugs, 465–476. See also specific drugs
depolarizing, 468–469, 470f, 470t, 473
neuromuscular transmission ofassessment of, 471f, 472–473
nondepolarizing, 467–468, 468f, 470t
Neuromuscular junctioncholinomimetic drug effects on
direct-acting, 102t, 104–105indirect-acting, 107
disorders ofcholinomimetic drugs for, 108–109
Neuromuscular responseaging effects on, 475disease effects on, 475
Neuron(s)local circuit, 365, 365fNANC
transmitter substances found in, 83f, 84t, 87–88
in parkinsonism, 483, 485fprojection, 365, 365frelay, 365, 365f
Neurontin, 427, 481. See also GabapentinNeuropathy target esterase, 1007Neuropeptide Y, 308–309
in ANS, ENS, and NANC neurons, 84tinhibitory effects of, 92t
Neuroprotective therapyfor parkinsonism, 492
Neurotensin, 307–308Neurotransmission
prostaglandins effects on, 321Neurotransmitter(s)
centralidentification of
CNS drugs and, 364major depression and, 523, 524f
Neurotransmitter receptorsCNS drugs and, 360–361, 362f
Neurotransmitter uptake carriers, 87Neurotrophic hypothesis
of major depression, 522–523, 522fNeutral antagonism, 7
Neutropenia, 582chemotherapy-induced
G-CSF for, 593, 593fNevirapine, 871t, 877–878, 889
drug interactions with, 874tduring pregnancy, 876, 876t
New Drug Application (NDA), 75–76Nexium, 1111. See also EsomeprazoleNiacin
for hyperlipidemias, 627–628Nicardipine
clinical pharmacology of, 202torgan system effects of, 204
Niclosamide, for helminthic infections, 938t, 942–943
Nicotineabuse of, 569t, 573–575
treatment of, 574–575ionization constants of, 11tas pesticide, 1008structure of, 100ftoxicity of
acute, 109–110chronic, 110
Nicotine polacrilex gumfrom prescription to OTC status, 1116t
Nicotine transdermal systemfrom prescription to OTC status,
1116tNicotinic acetylcholine receptor (nAChR),
24, 24fNicotinic acid, 627–628. See also NiacinNicotinic receptors, 87, 89tNicotinic signaling, 101fNicotinic stimulants
direct-actingtoxicity of, 109–110
Nifedipine, 190, 203f, 209clinical pharmacology of, 202tdosage of, 175tpharmacokinetics of, 40t, 175t
Nifurtimoxclinical uses of, 934
Nigrostriatal pathway, 506Nilandron, 740. See also NilutamideNilotinib
for cancer, 966–967, 967tNilutamide, 739, 740Nimbex, 481. See also CisatracuriumNirvanol, 409Nisoldipine, 209
clinical pharmacology of, 202tNitazoxanide, 933Nitrates/nitrites
for angina pectoris, 195–201carcinogenicity of, 200, 200tin chronic heart failure management,
220Nitrazepam
for epilepsy, 420
Katzung_Index_p1171-1234.indd 1208 9/24/11 12:03:37 PM

Index 1209
Nitrendipineclinical pharmacology of, 202t
Nitric oxide, 331–337in ANS, ENS, and NANC neurons, 84tas CNS neurotransmitter, 370–371
Nitric oxide donors, 334Nitric oxide gas inhalation, 334Nitric oxide synthase (NOS)
isoforms ofproperties of, 331, 332t
Nitro-vasodilatorsfor angina pectoris, 201
Nitrofurantoin, 898as urinary antiseptic, 892–893
Nitrofurazone, 898Nitrogen dioxide
TLVs of, 1005tNitrogen oxides, 332, 333t
toxic exposure to, 1004–1005Nitroglycerin, 195–201Nitropress, 189. See also NitroprussideNitroprusside, 189. See also Sodium
nitroprussideNitrosourea(s)
for cancer, 955, 957Nitrous oxide
pharmacologic properties of, 433tNizatidine, 1111
from prescription to OTC status, 1116tvs. other H2-receptor antagonists, 1083t
Nizoral, 712, 858. See also KetoconazoleNNRTIs. See Nonnucleoside reverse tran-
scriptase inhibitors (NNRTIs)No-effect dose, 71Nolahist, 292. See also PhenindamineNolvadex, 740. See also TamoxifenNon-Hodgkin’s lymphoma
chemotherapeutic drugs for, 970–971Non-immunologic contact urticaria
topical dermatologic medications and, 1063t
Nonadrenal disorderssynthetic corticosteroids for, 705–706,
706tNonadrenergic, noncholinergic (NANC)
neuronstransmitter substances found in, 83f, 84t,
87–88Nonketotic hyperosmolar coma, 744Nonnucleoside reverse transcriptase inhibi-
tors (NNRTIs), 870t–871t, 876–878
Nonsteroidal anti-inflammatory drugs (NSAIDs), 636–643, 1076. See also specific drugs
for Alzheimer’s disease, 1055tdrug interactions with, 1159tin the elderly, 1057ulcers due to
proton pump inhibitors for, 1088
Nontoxic goiter, 694Nontuberculous infections
streptomycin for, 824Nonulcer dyspepsia
domperidone for, 1092H2-receptor antagonists for, 1085metoclopramide for, 1092proton pump inhibitors for, 1088
NOP (nociceptin receptor), 544Noradrenergic fibers, 80f, 82Norco, 562. See also Hydrocodone/
acetaminophenNorcuron, 481. See also VecuroniumNorepinephrine, 136f, 138t, 141, 148
for acute hypotension, 144in ANS, ENS, and NANC neurons, 84tas CNS neurotransmitter, 367t, 370dose-response curves to, 152, 153finhibitory effects of, 92tionization constants of, 11t
Norepinephrine transporter (NET), 85–87, 134
Norethindrone, properties of, 719f, 723tNorethindrone acetate, 740
properties of, 723tNorethynodrel
properties of, 723tNorflex, 481. See also OrphenadrineNorfloxacin, 835, 836t, 838 Norgestrel
dosage of, 731tL-Norgestrel
dosage of, 731tproperties of, 723t
Normodyne, 167, 189. See also LabetalolNoroxin, 838. See also NorfloxacinNorpace, 249. See also DisopyramideNorplant, 740. See also LevonorgestrelNorpramin, 539. See also Desipramine19-Nortestosterone derivatives
properties of, 723tNortriptyline, 527f, 539
dosage of, 535thepatic clearance of
aging effects on, 1052tpharmacodynamics of, 40t, 530tpharmacokinetics of, 40t
Norvasc, 189, 209. See also AmlodipineNorvir, 889. See also RitonavirNotice of Claimed Investigational
Exemption for a New Drug (IND), 74–76
Novo-Seven, 617. See also Coagulation factor VIIa recombinant
Novocain, 463. See also ProcaineNovothyrox, 695. See also LevothyroxineNoxafil, 858. See also PosaconazoleNPH (neutral protamine Hagedorn), 748t,
749Nplate, 598. See also Romiplostim
NRC. See National Research Council (NRC)
NRTIs. See Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs)
NSAIDs. See Nonsteroidal anti-inflamma-tory drugs (NSAIDs)
Nubain, 562. See also NalbuphineNucleoside/nucleotide reverse transcriptase
inhibitors (NRTIs), 869–876, 870t–872t. See also specific drugs
during pregnancy, 876, 876tNucleus accumbens, 565–566Nucynta, 562. See also TapentadolNumorphan, 562. See also OxymorphoneNupercainal, 463. See also DibucaineNuprin, 655. See also IbuprofenNurse practitioners
prescribing authority of, 1144–1145, 1144t
Nutritionproton pump inhibitors effects on, 1088
Nutritional supplementspurified, 1134–1136
coenzyme Q10, 1134–1135glucosamine, 1135melatonin, 1135–1136
Nuvigil, 148. See also ArmodafinilNystatin, 858
topical, 856, 1066
OObesity
management of, 664, 665tObsessive compulsive disorder (OCD)
antidepressants for, 532Occlusion
as factor in pharmacologic response to dermatologic drugs, 1061
Octreotide, 665–666, 665f, 678adverse effects of, 1095clinical uses of, 1095for diarrhea, 1095for variceal hemorrhage,
1107–1108Ofatumumab, 991, 999Ofloxacin, 838
pharmacokinetics of, 836tOgen, 740. See also EstropipateOlanzapine, 504f, 519
advantages and disadvantages of, 509t
dosage of, 510tfor Huntington’s disease, 495
Oligonucleotide(s)antisense, 2
Olmesartan, 190, 225Olopatadine, 292Olsalazine, 1101f, 1112
Katzung_Index_p1171-1234.indd 1209 9/24/11 12:03:37 PM

1210 Index
Omalizumab, 993for asthma, 350preparations available, 355, 999
Omecantiv mecarbil, 212Omega-3 fatty acids, marine, 633Omeprazole
pharmacokinetics of, 1086tfrom prescription to OTC status, 1116t
Omnicef, 807. See also CefdinirOn-off phenomenon, 487Onchocerciasis
ivermectin for, 941Oncogenes
cancer related to, 950Ondansetron, 287, 1097f, 1111
for nausea and vomiting, 1098–1099Onglyza, 766. See also SaxagliptinOntak, 999. See also Denileukin diftitoxOphthalmologic disorders. See also Eye(s)
antimuscarinic drugs for, 121, 121t in the elderly
drugs for, 1057sympathomimetic drugs for, 145
Ophthalmopathythyroid disease–related, 693
Opioid(s), 572abuse of, 569t, 572
treatment of, 572overdose of, 1037
Opioid agonistsfor diarrhea, 1094–1095
Opioid analgesics, 446, 543–564, 545t, 549–550, 553. See also specific drugs
with agonist and antagonist actionseffects of, 552
antitussives, 559basic pharmacology of, 543–552classification of, 543, 544tclinical pharmacology of, 552–560CNS effects of, 549–551, 551tcontraindications to, 556dependence on, 549, 551t, 555mechanism of action of, 546–549,
547f–549fsites of action of, 548, 548fsource of, 543specific agents, 556–560supraspinal actions of, 548tolerance to, 549, 551t, 554–555toxicity of, 554–556, 554t
Opioid antagonists, 559–560, 562Opioid peptides
in ANS, ENS, and NANC neurons, 84tas CNS neurotransmitter, 367tendogenous, 543–544, 544t
Opioid receptor antagonistsin constipation management, 1094
Opisthorchiasispraziquantel for, 944
Opium, 543Oprelvekin, 594, 598Optipranolol, 167. See also MetipranololOptometrists
prescribing authority of, 1144–1145, 1144t
Oral contraceptives, 725t, 726–731during lactation, 1048t
Oral direct factor XA inhibitors, 607Orap, 519. See also PimozideOrencia, 656, 999. See also AbataceptOrgan transplant rejection
cell-mediatedeicosanoids and, 327
Organ transplantationimmunosuppressive drugs in, 985t,
994–995Organic nitrates, 334Organic nitrites, 334Organic solvents
fetal effects of, 1043tOrganochlorine pesticides, 1006–1007
toxicity rating for, 1006tOrganolead poisoning, 1014t, 1016Organon, 678. See also Ganirelix acetateOrganophosphate(s)
for cholinergic poisoning, 123durations of action of, 106ttherapeutic uses for, 106t
Organophosphorus pesticides, 1007–1008toxicity rating for, 1007t
Orgaran, 617. See also DanaparoidOrinase, 766. See also TolbutamideOrlaam, 562. See also Levomethadyl acetateOrlistat
in obesity management, 664, 665tfrom prescription to OTC status, 1116t
Ornidyl, 935. See also EflornithineOrphan drugs
development of, 77Orphan receptors, 16Orphanin FQ, 544Orphanin opioid-receptor-like subtype I,
544Orphenadrine
for parkinsonism, 492t, 499Ortho-phthalaldehyde, 896, 898Orthoclone OKT3, 999. See also
Muromonab-CD3Orudis, 655. See also KetoprofenOs-Cal, 786. See also Calcium carbonateOseltamivir
for influenza, 886–887, 889Osmitrol, 270. See also MannitolOsmol gap
causes of, 1030, 1030tOsmoPrep, 1112. See also Sodium
phosphateOsmotic agents
sites of action of, 252f
Osmotic diuretics, 263–264Osmotic laxatives, 1093Osteoarthritis
in the elderly, 1056–1057Osteodystrophy
intestinal, 782Osteoporosis, 782–783, 782f
androgens in, 736OTC drugs. See Over-the-counter (OTC)
drugsOtotoxicity
aminoglycosides and, 824loop diuretics and, 260
Otrivin, 148. See also XylometazolineOvarian cancer
chemotherapeutic drugs for, 973Ovarian hormones, 726Ovarian hyperstimulation syndrome, 667,
668fgonadotropins and, 668–669
Ovary, 715–731functions of, 715–716
disturbances in, 716mechanism of action of, 718–719oral contraceptives effects on, 727
Over-the-counter (OTC) drugs, 1115–1124
ingredients into be avoided in selected patients,
1117, 1123tFDA review of, 1115–1124hidden, 1117, 1123tof known efficacy, 1117,
1118t–1122toveruse/misuse of, 1117, 1123t, 1124switched from prescription to,
1115–1117, 1116tOvidrel, 678. See also Choriogonadotropin
alfaOvulation induction
drugs for, 733–734gonadotropins for, 667–668, 668f
Oxacillindosage of, 795t
Oxaliplatinclinical activity and toxicities of, 956t
Oxaluria enteric, 785Oxamniquine, 943
for helminthic infections, 938t, 943
Oxandrin, 740. See also OxandroloneOxandrolone, 735t, 740Oxantel pamoate, 946Oxaprozin, 642, 656
properties of, 638tOxazepam
dosages of, 383tpharmacokinetic properties of, 377t
Oxazolidinediones for seizures, 419
Katzung_Index_p1171-1234.indd 1210 9/24/11 12:03:37 PM

Index 1211
Oxazolidinones, for infection, 817
Oxcarbazepinefor seizures, 410–411, 427
Oxiconazole, 858Oxidase(s)
mixed function, 55N-Oxidation, 56tS-Oxidation, 56tOxidative dealkylation, 56tOxidizing agents
poisoning by, 1036tOxistat, 858. See also OxiconazoleOxybutynin
for urinary disorders, 122, 122t, 127Oxycodone, 558
pharmacokinetics of, 545tOxycodone mixtures, 562Oxygen
as poisoning antidote, 1033tOxymetazoline, 141, 148Oxymetholone, 735t, 740Oxymorphone, 556
pharmacokinetics of, 545tOxytocin, 673–674Oxytocin antagonists, 674Ozone
TLVs of, 1005ttoxic exposure to, 1004–1005
PP-glycoprotein transporter, 9, 10tPaclitaxel
for cancer, 963t, 964clinical activity and toxicities of, 963t
Paget’s disease of bone, 784Pain. See also Analgesia/Analgesics
acetaminophen and, 650chronic
opioid analgesics for, 550kinins role in, 302muscle
neuromuscular blocking drugs and, 474–475
Pain disordersantidepressants for, 533
Paliperidone, 519Palivizumab, 888, 889, 994Palonosetron
for nausea and vomiting, 1098–1099Palosuran, 309Pamelor, 539. See also NortriptylinePamidronate, 776f, 786Pamine, 127. See also MethscopolaminePanadol, 656. See also AcetaminophenPancreas
endocrine, 743–744Pancrease, 1111. See also PancrelipasePancreatic cancer
chemotherapeutic drugs for, 973
Pancreatic enzyme supplementation, 1106–1107
Pancreatic islet cells, 744tPancreatic peptide, 743Pancreatin, 1106–1107Pancrelipase, 1106–1107, 1111Pancuronium
pharmacokinetic and pharmacodynamic properties of, 470t
tissue effects of, 474tPanitumumab, 991–992, 999
for cancer, 967–968Pantoprazole,1086f, 1111
pharmacokinetics of, 1086tPapaverine
for erectile dysfunction, 156–157Parabens, 897Paragonimiasis
praziquantel for, 944Paraldehyde, 387Paramethasone
clinical uses and dosage of, 703tParaoxon
structure of, 106fParaquat, 1009f, 1010Parathion, 106f
toxicity rating for, 1007tParathion-methyl
toxicity rating for, 1007tParathyroid hormone (PTH)
in bone mineral homeostasis, 769–771, 770f, 771f
vitamin D and FGF23 interactions with, 773–774
Paravertebral chains, 81Paremyd, 148. See also
HydroxyamphetamineParicalcitol, 786Parkinsonism
drug-induced, 493drugs used for, 483–500. See also
specific drugsgene therapy for, 492neurons involved in, 483, 485fneuroprotective therapy for, 492nonmotor manifestations of
therapy for, 492pathogenesis of, 484surgical procedures for, 492
Parlodel, 499, 678. See also Bromocriptine
Parnate, 539. See also TranylcypromineParomomycin
clinical uses of, 934preparations available, 828, 935structure of, 929f
Paromomycin sulfatefor amebiasis, 930
Paroxetine, 526–530, 535t, 539Partial agonists, 19–20, 20f
Partial seizuresclinical pharmacology of, 420–421
Paser, 848. See also 5-Aminosalicylic acidPasteurization
defined, 894tPatanol, 292. See also OlopatadinePatent ductus arteriosus
eicosanoids and, 326Pathilon, 127. See also TridihexethylPatient-controlled analgesia (PCA), 554Paxil, 539. See also ParoxetinePazopanib
for cancer, 968Pediatric patients
clinical pharmacology of, 1039–1049immunization for, 1167tpassive immunization for, 1168t
Pediculicidesingredients in, 1122t
PEG-Intron, 889, 999. See also Peginterferon alfa-2b
Pegademase, 985, 999Peganone, 427. See also EthotoinPegaptanib, 994, 999Pegasys, 889, 999. See also Peginterferon
alfa-2aPegfilgrastim, 598Peginterferon alfa-2a
for HBV infection, 883tpreparations available, 889, 999
Peginterferon alfa-2bfor HBV infection, 883tpreparations available,
889, 999Pegloticase
for gout, 654–655Pegvisomant, 665, 666Pen-Vee K, 807. See also Penicillin VPenbutolol, 159t
as antihypertensive drug, 179Penciclovir
dosage of, 865tfor HSV and VZV infections, 866topical, 1067
Penicillamine, 1024fetal effects of, 1043tindications and toxicity of, 1024ionization constants of, 11tfor Wilson’s disease, 496–497
Penicillin(s), 790–797. See also specific typesadverse effects of, 796–797clinical uses of, 795–796during lactation, 1048tmechanism of action of, 792,
792f–794funits and formulations of, 791–792
Penicillin G, 790Penicillin G benzathine, 807Penicillin G procaine, 807
clinical uses of, 795
Katzung_Index_p1171-1234.indd 1211 9/24/11 12:03:37 PM

1212 Index
Penicillin V, 807dosage of, 795t
Pennyroyalrisks associated with, 1127t
Pentacarinat, 935. See also PentamidinePentam 300, 935. See also PentamidinePentamidine, 929f, 930–932, 935
for trypanosomiasis, 930–932Pentasa, 1102, 1102fPentazocine, 545t, 559
ionization constants of, 11tPentetate calcium trisodium
preparations available, 1025Pentetate Zinc Trisodium, 1025. See also
Pentetate calcium trisodiumPentids, 807. See also Penicillin GPentobarbital, 375f, 387, 427
ionization constants of, 11tPentostatin
as immunosuppressive drug, 989Pentothal, 446. See also ThiopentalPentoxifylline, 209, 346, 355
for intermittent claudication, 207Pepcid, 1111. See also FamotidinePeptic ulcer disease
H2-receptor antagonists for, 1084–1085proton pump inhibitors for, 1087–1088
Peptide(s)as CNS neurotransmitters, 370natriuretic, 303–304. See also Natriuretic
peptidesopioid
in ANS, ENS, and NANC neurons, 84t
as CNS neurotransmitter, 367tendogenous, 543–544, 544t
pancreatic, 743in renal tubule transport, 256vasoactive, 295–312. See also specific
drugs and Vasoactive peptidesvasoactive intestinal
in ANS, ENS, and NANC neurons, 84t
Pepto-Bismol, 1112. See also Bismuth subsalicylate
Peracetic acid, 896–897Peramivir
for influenza, 887Percocet, 562. See also Oxycodone mixturesPercodan, 562. See also Oxycodone
mixturesPerformance anxiety
beta-receptor antagonists for, 164Pergolide
for parkinsonism, 489, 499Perinatal pharmacology, 1039–1045Perindopril, 190, 225Peripheral artery disease
drugs used for, 209treatment of, 207
Peripheral blood stem cells, 592Peripheral nervous system
direct-acting cholinomimetic drug effects on, 102t, 104
nitric oxide effects on, 336opioid analgesics effects on, 551–552prostaglandins and thromboxanes effects
on, 321Peripheral tissues
muscarinic receptor subgroups important in, 118t
Peripheral vascular diseasealpha-receptor antagonists for, 156eicosanoids and, 326
Peritoneal dialysisin decontamination of poisoned patient,
1032Permapen, 807. See also Penicillin G
benzathinePermax, 499. See also PergolidePermeation
drug, 9–10, 9fPermethrin, 1068Pernicious anemia, 588Peroxisome proliferator–activated
receptor-gamma (PPAR-γ)in diabetes mellitus, 757–758
Peroxygen compoundsas antiseptics/disinfectants, 896–897
Perphenazine, 503fdosage of, 510t, 519for Huntington’s disease, 495
Persantine, 617. See also DipyridamolePesticide(s)
botanical, 1008, 1009fcarbamate, 1008, 1008torganochlorine, 1006–1007organophosphorus, 1007–1008toxic exposure to, 1006–1008,
1006t–1008tPetit mal seizures, 421Pfizerpen, 807. See also Penicillin GpFOX inhibitors
for angina pectoris, 205pH
of drugsduring pregnancy, 1040
pH-partitioning phenomenonbody fluids with potential for drug
“trapping” through, 11, 12tPharmaceutical industry
described, 69–70pharmacology and, 3
Pharmacistsprescribing authority of, 1144–1145,
1144tPharmacodynamics
components in, 38fdefined, 6in dose individualization, 47–49
Pharmacodynamics (Cont.):drug receptors and, 15–35principles of, 6–8, 7f, 8fprocesses involved in, 2f, 6variables related to, 48–49
Pharmacogenetic testingin drug therapy, 65
Pharmacogenomicsdefined, 2, 3
Pharmacokinetics, 37–48bioavailability and, 39t–40t, 43–44, 43f,
43tclearance and, 38–42, 39t–41tcomponents in, 38, 38fdefined, 6in dose individualization, 47–49drug accumulation and, 42–43, 42fdrug effects, 44–45, 45f. See also Drug
effects, time course ofextraction ratio and, 44first-pass effect and, 44half-life and, 39t–40t, 42, 42fprimary processes of, 38, 38fprinciples of, 8–12, 9f, 10t–12t, 12fvariables related to, 47–48volume of distribution and, 38,
39t–41tPharmacology
areas of study in, 2fautonomic
introduction to, 79–96. See also Autonomic nervous system (ANS)
basic principles of, 1–77defined, 1dermatologic, 1061–1079experimental, 2general principles of, 3–12genetics and, 3history of, 1–3medical
defined, 1, 2fpediatric, 1039–1045perinatal, 1039–1045pharmaceutical industry and, 3sources of information, 13transport molecules in, 9, 10t
Phase I reactions, 56t–57t. See also Biotransformation of drugs, phase I reactions
Phase II reactions, 59–60, 59f, 60tPhenacemide
for seizures, 409Phenanthrene analgesics
mild to moderate agonists, 558with mixed receptor actions, 558–559strong, 556–557
Phencyclidine (PCP)abuse of, 569t, 575–576, 577ffetal effects of, 1043t
Katzung_Index_p1171-1234.indd 1212 9/24/11 12:03:37 PM

Index 1213
Phenelzinedosage of, 535tpharmacokinetics of, 529t
Phenergan, 292, 1112. See also Promethazine
Phenindamine, 292Phenindione, 609fPhenmetrazine, 142Phenobarbital
clinical uses of, 411dosages of, 383t, 411half-life of
in neonates and adults, 1045tionization constants of, 11tduring lactation, 1048tmechanism of action of, 411oral absorption of
in neonates vs. older children and adults, 1044t
pharmacokinetics of, 40t, 411for seizures, 411therapeutic levels/dosage of, 411
Phenolicsas antiseptics/disinfectants, 893t, 895
Phenothiazine(s)advantages and disadvantages of, 509tfor nausea and vomiting, 1100potency and toxicity of
chemical structure effects on, 505tPhenothiazine derivatives
of antipsychotic drugs, 503–504, 503f, 505t
Phenoxybenzamine, 154, 286mechanism of action of, 151–152for pheochromocytoma, 155–156, 156f
Phensuximidefor seizures, 418
Phentolamine, 154cardiovascular effects of, 152, 153ffor erectile dysfunction, 156–157, 199
D-Phenylalanine derivativefor diabetes mellitus, 756, 756t
Phenylbutazonedrug interactions with, 610thepatic clearance of
aging effects on, 1052tPhenylephrine, 133t, 136f, 141, 148
for acute hypotension, 144cardiovascular responses to, 138tionization constants of, 11t
Phenylethylamine, 134, 136fPhenylethylmalonamide (PEMA)
structure of, 412fPhenylheptylamine analgesics
mild to moderate agonists, 558strong, 557
O-Phenylphenol, 895Phenylpiperidine analgesics
mild to moderate, 558strong, 557
Phenylpropanolaminesympathomimetic drug effects of, 142
Phenytoin, 407–409, 427drug interactions with, 409, 1159tfetal effects of, 1043thalf-life of
in neonates and adults, 1045tionization constants of, 11tduring lactation, 1048toral absorption of
in neonates vs. older children and adults, 1044t
pharmacokinetics of, 40t, 408, 408fPheochromocytoma, 155–156, 156fpHisoHex, 898. See also HexachlorophenePhosLo, 786. See also Calcium acetatePhosphate, 786
for hypercalcemia, 7793′-Phosphoadenosine 5′-phosphosulfate
(PAPS), 60Phosphodiesterase(s)
in heart failure management, 218Phosphoinositide(s), 25
as second messengers, 28–29, 28fPhospholine, 112. See also EchothiophatePhosphorus
for hypocalcemia, 780Phosphorylation
second messenger signaling in, 29Photoallergic contact dermatitis
topical dermatologic medications and, 1063t
Photoirritationtopical dermatologic medications and,
1063tPhysician’s assistants
prescribing authority of, 1144–1145, 1144tPhysiologic antagonism, 20Physostigmine, 105–106, 112
ionization constants of, 11tas poisoning antidote, 1033t
Phytonadione, 609fPigmentation
drugs affecting, 1068–1069Pilocarpine, 100f, 112
ionization constants of, 11tPimecrolimus, 1067Pimozide, 504, 519
drug interactions with, 1159tfor tics, 495
Pin-rid, 946. See also Pyrantel pamoatePin-X, 946. See also Pyrantel pamoatePindolol, 159t, 167, 189
as antihypertensive drug, 179ionization constants of, 11t
Pinworm infectionsalbendazole for, 938–939
Pioglitazone, 758t, 786for diabetes mellitus, 758
Pioglitazone mixtures, 766
Pipecuronium, 469fPiperacillin, 791f, 807
dosage of, 795tPiperacillin and tazobactam sodium, 807Piperaquine, 918f
clinical uses of, 917tfor malaria, 920
Piperazine, 943for helminthic infections,
938t, 943Piperazine antihistamines, 279tPiperidine antipsychotics,
advantages and disadvantages of, 509tPiperidine antihistamines, 278fPipracil, 807. See also PiperacillinPirbuterol, 355
for asthma, 344Pirenzepine, 117fPiroxicam, 637f, 642, 656
preparations availableproperties of, 638t
Pitavastatin, 633Pitocin, 678. See also OxytocinPitressin, 678. See also VasopressinPituitary hormones, 659–679
anterior, 660–673posterior, 673–675
Placental drug metabolismduring pregnancy, 1040
Placental transportersof drugs, 1040
Plant(s)medical use of
history of, 1125Plant systemic pesticides, 1007Plasma fractions
for bleeding disorders, 615–616, 615tPlasma protein binding
importance of, 49Platelet(s)
nitroglycerin effects on, 198prostaglandins and thromboxanes effects
on, 320Platelet-rich thrombi, 601–602Platinum analogs
for cancer, 957–958Plavix, 617. See also ClopidogrelPlendil, 190, 209. See also FelodipinePlerixafor
G-CSF withclinical uses for, 594
preparations available, 598Pletal, 209, 617. See also CilostazolPlexus
myenteric, 81fsubmucous, 81f, 82
Plicamycinin bone mineral homeostasis, 777for hypercalcemia, 779preparations available, 786
Katzung_Index_p1171-1234.indd 1213 9/24/11 12:03:37 PM

1214 Index
PLX4032, 974Pneumococcal conjugate vaccine,
1165tPneumococcal polysaccharide vaccine,
1165tPneumocystis spp.
P. cariniidrugs used for, 931t
P. jirovecidrugs used for, 924, 931t
Pneumocystosisdrugs used for, 924, 926
pentamidine, 932Podofilox, 1075–1076Podophyllum resin, 1075–1076Poison ivy
contact hypersensitivity due to, 983Poisoning(s). See also specific types
arsenic, 1014t, 1017–1019arsine gas, 1019cholinergic
antimuscarinic drugs for, 122–123death due to, 1028ergot, 287heavy metals, 1013–1021.See also specific
types and Heavy metalshemodialysis in, 1031, 1031t, 1032lead, 1013–1017management of, 1027–1038mercury, 1014t, 1019–1021mushroom
antimuscarinic drugs for, 123organolead, 1014t, 1016sulfur dioxide, 1004
Poisons, 4Poke root
risks associated with, 1127tPoliovirus vaccine, 1165tPollutant(s)
air, 1002–1005, 1005tTLVs of, 1005t
environmental, 1010–1011.See also specific types and Environmental pollutants
Polycarbophil, 1093, 1112Polychlorinated biphenyls
as environmental pollutants, 1010
Polychlorinated dibenzo-p-dioxins (PCDDs), 1010
Polychlorinated dibenzofurans (PCDFs), 1010
Polyethylene glycolbalanced
in constipation management, 1093from prescription to OTC status,
1116tPolyethylene glycol electrolyte solution,
1112Polymyxin(s), 892
Polymyxin B, 892, 898topical, 1063–1064
Polymyxin E, 892Polythiazide, 270
dosage of, 261tPolyvinyl pyrrolidone (PVP)
as antiseptic/disinfectant, 895Ponstel, 656. See also Mefenamic acidPontocaine, 463. See also TetracainePortable pen injectors
in insulin delivery, 748t, 750Posaconazole, 858
pharmacologic properties of, 854tfor systemic infections, 855
Positive chronotropic effect, 138Positive dromotropic effect, 138Positive inotropic drugs
in the elderly, 1056in heart failure management, 212t, 218
Positive inotropic effect, 138Postantibiotic effects, 823Postantibiotic leukocyte enhancement, 907Postcoital contraceptives, 731, 731tPostganglionic sympathetic nerve terminal
blockers, 189Postmenopausal hormonal therapy
estrogen in, 720–722Postpartum hemorrhage
ergot alkaloids for, 290Postsynaptic density, 369Postsynaptic regulation
functional organization of, 92–93, 93fPost-traumatic stress disorder (PTSD)
antidepressants for, 533Postural baroreflex, 87, 171, 171fPosture, 786. See also Tricalcium phosphatePotassium
for cardiac arrhythmias, 246digitalis with, 218effects of, 230
Potassium iodidepreparations available, 695
Potassium-sparing diuretics, 261–265drug interactions with, 1159t
Povidone-iodineas antiseptic/disinfectant, 895preparations available, 898
Prader-Willi syndromegrowth hormone for, 663, 663t
Pralatrexatefor cancer, 959–960
Pralidoxime, 123f, 127for cholinergic poisoning, 123as poisoning antidote, 1033t
Pramipexole, 489f, 499for parkinsonism, 489for restless legs syndrome, 496
Pramlintide, 760, 763, 766for diabetes mellitus
combination therapy with, 762
Pramoxine, 1077local anesthetic, 463
Prandin, 766. See also RepaglinidePrasugrel, 617
as antiplatelet agent, 613Pravachol, 633. See also PitavastatinPraziquantel, 943–945, 946
for helminthic infections, 938t, 943–945
Prazosin, 152f, 154, 167,189as antihypertensive drug, 180pharmacokinetics of, 40t, 175t, 180
Precedex, 148, 446. See also Dexmedetomidine
Precocious pubertygonadotropins in, 670–671
Precose, 766. See also AcarbosePrednisolone
clinical uses and dosage of, 703t, 712Prednisone
clinical uses and dosage of, 703t, 712during lactation, 1048t
Pregabalin, 413f, 427mechanism of action of, 413for seizures, 413
Pregnancydrug therapy during, 1039–1043
antipsychotic drugs, 512antiretroviral drugs, 876, 876tlithium, 517opioid analgesics, 556pharmacodynamics of, 1040–1043pharmacokinetics of, 1039–1040
hypothyroidism during, 691multiple
gonadotropins and, 668–669nausea and vomiting of
H1-receptor antagonists for, 280thyrotoxicosis during, 693
Pregnyl, 678. See also Chorionic gonadotropin
Preloadin cardiac performance, 215, 215f
Prelone, 712. See also PrednisolonePremarin, 740. See also Conjugated
estrogensPremature depolarizations, 216, 218fPremature labor
suppression ofsympathomimetic drugs for, 145
Premenstrual dysphoric disorderantidepressants for, 533
Pemetrexedfor cancer, 959
Prenalterol, 141, 142fPrepidil, 328. See also DinoprostonePrescribing, 1139–1148. See also
Prescription(s)compliance issues in, 1143–1144cost factors in, 1147–1148
Katzung_Index_p1171-1234.indd 1214 9/24/11 12:03:37 PM

Index 1215
Prescribing (Cont.):E-prescribing, 1143errors in, 1141–1143generic, 1147legal issues in, 1144–1147, 1144t, 1145trational, 1139–1141, 1140fsocioeconomic factors in, 1147–1148
Prescription(s), 1139–1148. See also Prescribing
abbreviations for, 1142tcosts of, 1147–1148elements of, 1140–1141, 1140fgeneric, 1147inappropriate, 1143writing of, 1139–1141, 1140f
poor, 1142–1143Prescription drug(s)
dietary supplements vs., 1125Prescription Drug User Fee Act (PDUFA)
of 1992, 76Preservatives
as antiseptics/disinfectants, 897Presynaptic inhibition, 363Presynaptic regulation
in functional organization of autonomic activity, 91–92, 92t
Prevacid, 1111. See also LansoprazolePrevertebral ganglia, 81Prezista, 888. See also DarunavirPrialt, 562. See also ZiconotidePriftin, 848. See also RifapentinePrilocaine, 461
pharmacokinetic properties of, 452tpreparations available, 463
Prilosec, 1111. See also OmeprazolePrimacor, 225. See also MilrinonePrimaquine, 919t, 923–924
for malaria, 919t, 923–924in malaria prevention in travelers, 919t
Primary amine, 11Primary chylomicronemia, 623
drugs used for, 622tPrimary generalized glucocorticoid
resistancesynthetic corticosteroids for, 704
Primary hyperparathyroidism, 780Primary immunodeficiency disorders
passive immunization for, 1168tPrimary induction chemotherapy, 950Primaxin, 807. See also Imipenem/cilastatinPrimidone
for seizures, 411–412for tremors, 494
Prinivil, 190, 225. See also LisinoprilPrinzmetal angina, 193Priscoline, 167. See also TolazolinePristiq, 539. See also DesvenlafaxinePrivine, 148. See also NaphazolinePro-Banthine, 127. See also PropanthelineProAmatine, 148. See also Midodrine
Probenecid, 656drug interactions with, 1160t
Procainamidefor cardiac arrhythmias, 238–239ionization constants of, 11tpharmacokinetics of, 40t, 239pharmacologic properties of, 238t
Procaine, 451t, 463ionization constants of, 11t
Procaine penicillin, 794, 807Procarbazine
for cancer, 957clinical activity and toxicities of, 956t
Procardia, 190, 209. See also NifedipineProchlorperazine
preparations available, 1112Procrit, 598. See also Epoetin alfaProctoFoam-HC, 1112. See also
HydrocortisoneProcyclidine
for parkinsonism, 492tProdrug, 8Profasi, 678. See also Chorionic
gonadotropinProfenal, 656. See also SuprofenProfilnine, 617Profilnine SD, 617. See also Factor IX
complex, humanProgabide, 478Progesterone, 719f, 723, 740
effects of, 724Progesterone inhibitors, 731–734Progestin(s), 723–726
contraception with, 725t, 730–731preparations available, 740
Prograf, 999. See also TacrolimusProguanil, 925, 918fProinsulin, 744fProjection neurons, 365, 365fProkinetic agents, 1091Prolactin, 660, 672Prolactin antagonists
preparations available, 678Proleukin, 999. See also Interleukin-2Prolia, 786, 999. See also DenosumabProlixin, 519. See also FluphenazineProloprim, 837. See also TrimethoprimPromacta, 598. See also EltrombopagPromethazine, 279, 1100
ionization constants of, 11tPronestyl, 249. See also ProcainamidePropafenone
for cardiac arrhythmias, 242membrane actions of, 237tpharmacologic properties of,
238tPropantheline, 117f, 127Proparacaine, 463Prophylactic drugs, in malaria, causal,
915
Propiverinefor urinary disorders, 122, 122t
Proplex SX-T, 617. See also Factor IX complex, human
Proplex T, 617. See also Factor IX complex, human
Propofol, 438–441as intravenous anesthetic, 438–441
Propoxurtoxicity rating for, 1008t
Propoxyphenemild to moderate agonists, 558
Propranolol, 161, 178–179, 238tas antihypertensive drug, 178cardiac effects of, 242dosage of, 175t, 179hepatic clearance of
aging effects on, 1052tionization constants of, 11tduring lactation, 1048tmembrane actions of, 237tproperties of, 159tstructure of, 158ffor tremors, 493
Propylene glycol, 1075Propylthiouracil, 688f, 695
fetal effects of, 1043tfor Graves’ disease, 691–692ionization constants of, 11tduring lactation, 1048t
Prorenin receptors, 300ProSom, 387. See also EstazolamProstacyclin, 315Prostaglandin(s). See also Eicosanoid(s)
for open-angle glaucoma, 161torgan system effects of, 320–322PGE2, 315PGF2α, 315PGF2α, 325fin renal tubule transport, 256
Prostaglandin analogsfor acid-peptic diseases, 1090
Prostaglandin E1, E2
inhibitory effects of, 92tProstaglandin endoperoxide synthases
products of, 314–316, 315fProstanoid(s)
biosynthesis of, 315fProstanoid receptors
signaling pathways of, 318fProstate cancer
advancedGnRH receptor agonists in,
671chemotherapeutic drugs for,
972gonadotropins in, 670
Prostigmin, 112. See also NeostigmineProstin E2, 328. See also DinoprostoneProtamine sulfate, 607, 617
Katzung_Index_p1171-1234.indd 1215 9/24/11 12:03:37 PM

1216 Index
Protease inhibitors, 870t–871t, 878–881. See also specific drugs
during pregnancy, 876, 876tProtein(s)
contractilesensitivity to calcium, 212
FK-binding, 986G. See G proteinsregulatory, 16structural, 16synaptosomal nerve–associated, 82–83transport, 16
Protein anabolic agentsandrogens and anabolic steroids as, 736
Protein bindingcapacity-limited
in drug concentration measurements, 49of drugs
during pregnancy, 1040plasma
importance of, 49Protein C, 604Protein S, 604Proton pump inhibitors
for acid-peptic diseases, 1085–1089, 1086f, 1086t
ingredients in, 1118tProtonix, 1111. See also PantoprazoleProtopam, 127. See also PralidoximeProtozoal infections
drugs used for, 915–936. See also specific drugs and indications
Protriptyline, 527f, 539dosage of, 535tpharmacodynamics of, 530t
Proventil, 355. See also AlbuterolProvera, 740. See also Medroxyprogesterone
acetateProvigil, 148. See also ModafinilProximal tubule
in renal tubule transport, 251–253, 252f, 252t, 253f
Proximal tubule diureticscombination therapy, 265
Prozac, 539. See also FluoxetinePrucalopride
in constipation management, 1094Pruritus
opioid analgesics and, 552treatment of, 1076
Prussian blue, 1024indications and toxicity of, 1024–1025preparations available, 1025
Pseudoephedrine, 142, 148ionization constants of, 11t
Pseudovitamin D deficiency rickets, 783–784Psilocybin
abuse of, 569t, 573Psoriasis
drugs used for, 1071–1072
Psychosis(es)nature of, 502
Psychotic depressionantipsychotic drugs for, 509
Psyllium, 1093preparations available, 1112
PTH. See Parathyroid hormone (PTH)PTSD
antidepressants for, 533Puberty
precociouscentral
gonadotropins in, 670–671Pulmicort, 355. See also BudesonidePulmonary edema
acutediuretics for, 265–266opioid analgesics for, 553
Pulmonary hypertensioneicosanoids and, 326
Pure Food and Drug Act of 1906, 73, 74t
Purified nutritional supplements, 1134–1136. See also Nutritional supplements, purified
Purine analogsfor inflammatory bowel disease,
1103–1104Purine antagonists
for cancer, 961–962Pyramat
toxicity rating for, 1008tPyrantel pamoate, 945, 946
for helminthic infections, 938t, 945
Pyrazinamidefor tuberculosis, 840t, 842–843
Pyrethrin I, 1009fPyrethrum, 1008Pyridostigmine
durations of action of, 106tpharmacokinetic and pharmacodynamic
parameters for, 40ttherapeutic uses for, 106t
Pyridoxine, 840fPyrimethamine, 833, 925–926
ionization constants of, 11tmechanism of action of, 833
Pyrimethamine/sulfadoxineclinical uses of, 917tpreparations available, 837, 925–926,
935Pyrimidines
pharmacokinetics of, 832tPyrithione zinc
in OTC products, 1120tPyrolan
toxicity rating for, 1008tPyrophosphate, 776f
QQinghaosu, 919t, 920–921. See also
Artemisinin(s)Quantrel, 946. See also Oxantel pamoateQuarzan, 127. See also ClidiniumQuaternary amines, 11Quaternary ammonium compounds
as antiseptics/disinfectants, 893t, 895–896
Quazepam, 387Quelicin, 481. See also SuccinylcholineQuestran, 633. See also CholestyramineQuetiapine, 504f, 519
advantages and disadvantages of, 509tdosage of, 510t
Quinagolide, 672Quinapril, 190, 225Quinestrol, 718f
dosage of, 719tQuinethazone, 270
dosage of, 261tQuinidine, 238, 921–922
for cardiac arrhythmias, 238chemistry of, 918f, 921–922drug interactions with, 1160thepatic clearance of
aging effects on, 1052tionization constants of, 11tfor malaria, 921–922pharmacokinetics of, 40t, 239, 921–922
Quinine, 918f, 921–922, 935for babesiosis, 922hepatic clearance of
aging effects on, 1052tfor malaria, 921–922
Quinoline methanols, 918fQuinolones
antibacterial activity of, 835described, 834, 834fdrug interactions with, 1160t
Quinupristin-dalfopristin, 815–816, 819QVAR, 355. See also Beclomethasone
RRabbit syndrome, 496Rabeprazole, 1086f, 111
pharmacokinetics of, 1086tRabies vaccine, 1165t
passive immunization for, 1169tRadical cure, malaria, 915Radioactive iodine
antithyroid activity of, 689for Graves’ disease, 692
Radiofrequency catheter ablationfor cardiac arrhythmias, 228
Radiogardase, 1025. See also Prussian blueRaloxifene, 731, 740
for osteoporosis, 783Raltegravir, 871t, 882, 889Ramelteon, 283, 374, 376, 387
Katzung_Index_p1171-1234.indd 1216 9/24/11 12:03:37 PM

Index 1217
Ramipril, 190, 225Ranexa, 209. See also RanolazineRanibizumab, 994, 999Ranitidine, 1083t, 1111
pharmacokinetics, 40tfrom prescription to OTC status, 1116t
RANK ligand (RANKL), 771Ranolazine, 209
for angina pectoris, 205Rapaflo, 167. See also SilodosinRapamune, 999. See also SirolimusRare diseases
drugs fordevelopment of, 77
Rasagiline, 499for parkinsonism, 488f, 490
Rattlesnake envenomation, 1037Razadyne, 112. See also GalantamineRebetron, 889. See also Interferon alfa-2bRebif, 999 Reboxetine
sympathomimetic drug effects on, 143Receptor(s). See also specific types
beneficial and toxic drug effects related to, 34
classes ofdrug development and, 22f, 29–30
consequences of, 15cytokine, 23, 24fdefined, 4, 15EGF
mechanism of action of, 22, 23fof G proteins, 25, 26tinert binding sites vs., 8intracellular
for lipid-soluble agents, 21–22, 21fmacromolecular nature of, 16nomenclature of, 6“orphan,” 16pharmacodynamics and, 15–35“spare,” 17–18, 17f
Receptor down-regulation, 23Receptor-effector coupling
spare receptors and, 16–18, 17fReceptor-effector mechanism
beneficial and toxic drug effects related to, 33–34
Receptor recycling, 549Receptor regulation, 25, 133–134Receptor selectivity, 133, 133t
stereo (chiral), 4–5Receptor tyrosine kinase(s)
inhibitors of, 22–23, 23fReceptor tyrosine kinase signaling pathway,
22–23, 23fReceptor uncoupling, opioid, 549Recombinant activated factor VII
for bleeding disorders, 616Recombinant factor VIIA
for bleeding disorders, 603f, 616
Recombinate, 617. See also Antihemophilic factor
Rectal suppositoriesopioid analgesic, 554
Recurrent endogenous depressionlithium for, 516
Red thrombi, 602Reductions, metabolic, 57tReentry
defined, 234, 234fReflex(es). See specific typesRefludan, 617. See also LepirudinRefractory period, 231Reglan, 1112. See also MetoclopramideRegonol, 112. See also PyridostigmineRegulation of receptors
flexible, 29Regulatory proteins, 16Relaxin, 726Relay neurons, 365, 365fRelcovaptan, 303Relenza, 889. See also ZanamivirRelistor, 562, 1112. See also
MethylnaltrexoneRelpax, 292. See also EletriptanRemeron, 539. See also MirtazapineRemicade, 656, 999, 1112. See also
InfliximabRemifentanil, 557, 562
pharmacokinetics of, 545tReminyl, 112. See also GalantamineRemodulin, 328. See also TreprostinilRenagel, 786. See also Sevelamer carbonateRenal autacoids
in renal tubule transport, 255–256Renal baroreceptor
in renin release, 296, 297fRenal failure
acuteloop diuretics for, 259potassium-sparing drugs and, 263
ADH antagonists and, 265diuretics for, 266–267
Renal function testsin poisoning assessment, 1030
Renal impairmentantimicrobial drugs in patients with,
908t, 909Renal potassium wasting
carbonic anhydrase inhibitors and, 258Renal stones. See Kidney stonesRenal system
eicosanoid effects on, 326lipoxygenase effects on, 323
Renal tubuletransport mechanisms in, 251–256
adenosine, 256collecting tubule system, 252t,
254–255, 254fdistal convoluted tubule, 252t, 254, 254f
Renal tubule, transport mechanisms in (Cont.):loop of Henle, 252tpeptides, 256prostaglandins, 256proximal tubule, 251–253, 252f, 252t,
253frenal autacoids, 255–256
Renese, 270. See also PolythiazideRenin, 295–297, 296f, 297f
release ofcontrol of, 296–297, 297fdrugs blocking, 299pharmacologic alteration of, 297
Renin-angiotensin systemchemistry of, 296finhibition of, 299–300
Renin inhibitors, 190, 300Renvela, 786. See also Sevelamer carbonateReoPro, 617, 999. See also AbciximabRepaglinide
for diabetes mellitus, 756, 756tRepaglinide plus metformin, 766Repinotan, 282Reproduction
inhaled anesthetics effects on, 437–438Reproductive smooth muscle
prostaglandins and thromboxanes effects on, 320
Reproductive systemfemale
eicosanoid effects on, 324–326hormones and, 715–734melatonin effects on, 1136
lead poisoning effects on, 1015male
eicosanoid effects on, 326hormones and, 734–739melatonin effects on, 1136
prostaglandins and thromboxanes effects on, 321
Repronex, 678. See also MenotropinsRequip, 499. See also RopiniroleRescriptor, 888. See also DelavirdineResearch
translational, 70in drug development, 76
Reserpineas antihypertensive drug, 178for Huntington’s disease, 494mechanism and sites of action of 83f, 178
Resin(s)bile acid–binding. See Bile acid–binding
resinsbile salt–binding
for diarrhea, 1095podophyllum, 1075–1076
Respirationsedative-hypnotic drugs effects on, 381
Respiratory depressionopioid analgesics in, 551
Katzung_Index_p1171-1234.indd 1217 9/24/11 12:03:37 PM

1218 Index
Respiratory disordersantimuscarinic drugs for, 121nitric oxide effects on, 336
Respiratory infectionsproton pump inhibitors and, 1088–1089
Respiratory syncytial viruspassive immunization for, 1169t
Respiratory systemantimuscarinic drug effects on, 119–120barbiturates effects on, 442benzodiazepines effects on, 443beta-receptor antagonists effects on, 158,
160cholinomimetic drug effects on
direct-acting, 102t, 103indirect-acting, 107
dexmedetomidine effects on, 445eicosanoid effects on, 327etomidate effects on, 444inhaled anesthetics effects on, 436ketamine effects on, 445propofol effects on, 440serotonin effects on, 283
Response elements, 21Rest tremor, 494Resting potential
effect on action potentials, 231–233, 232f, 233f, 233t
Restless legs syndrome, 496Restoril, 387. See also TemazepamResuscitation
lipidlocal anesthetics and, 459
Retapamulintopical, 1063
Retavase, 617. See also ReteplaseReteplase
pharmacology of, 612preparations available, 617
Retigabine (ezogabine), 415Retinoic acid, derivatives of,
for acne, 1069–1070Retrograde signaling, 363Retrovir, 889. See also ZidovudineRevatio, 209. See also SildenafilReverse potential, 230Revex, 562. See also NalmefeneReVia, 400, 562. See also NaltrexoneRevlimid, 999. See also LenalidomideRevonto, 481. See also DantroleneRexolate, 656. See also Sodium thiosalicylateReyataz, 888. See also AtazanavirRheumatoid arthritis
eicosanoids and, 328Rheumatrex, 656. See also MethotrexateRhinoconjunctivitis
allergic, 349RHo(D) immune globulin micro-dose,
990–991preparations available, 999
RhoGam, 999. See also RHo(D) immune globulin micro-dose
Ribavirin, 888for HCV infection, 886
Ricketspseudovitamin D deficiency, 783–784vitamin D–dependent
types I and II, 783–784vitamin D–resistant
hereditary, 783–784Ridaura, 656. See also AuranofinRifabutin
dosage of, 840tfor tuberculosis, 840t, 845
Rifadin, 848. See also RifampinRifampin, 840t, 841–842, 848
drug interactions with, 1160tfor leprosy, 846
Rifapentine, 848dosage of, 840tfor tuberculosis, 840t, 845
Rifaximin, 845Rigidity
truncalopioid analgesics and, 551, 551t
Rilonacept, 996, 999Rilutek, 481. See also RiluzoleRiluzole, 478, 481Rimactane, 848. See also RifampinRimantadine, 889
for influenza, 887Rimonabant
in addiction management, 578Risedronate
for osteoporosis, 775, 783Risk
defined, 1002Risperidone, 504f, 519
advantages and disadvantages of, 509tdosage of, 510t
Ritalin, 148. See also MethylphenidateRitanserin, 286–287Ritodrine, 142f
in premature labor suppression, 145Ritonavir, 871t, 880, 889
drug interactions with, 874tRituxan, 656, 999. See also RituximabRituximab, 646, 656, 992, 999Rivaroxaban, 607Rivastigmine
for Alzheimer’s disease, 1055, 112Rizatriptan, 292
pharmacokinetics of, 286tRobaxin, 481. See also MethocarbamolRobinul, 127, 1111. See also
GlycopyrrolateRocephin, 807. See also CeftriaxoneRocuronium, 469f, 481
pharmacokinetic and pharmacodynamic properties of, 470t
Rocuronium (Cont.):tissue effects of, 474t
Roferon, 999. See also Interferon alfa-2aRoferon-A, 889. See also Interferon alfa-2aRomidepsin, 1077
dermatologic uses of, 1078tRomiplostim
clinical uses of, 591t, 594–595Ropinirole, 489f, 499
for parkinsonism, 487for restless legs syndrome, 496
Ropivacaine, 451t, 461, 463pharmacokinetic properties of, 452t
Rosiglitazone, 758t, 766for diabetes mellitus, 758
Rosiglitazone mixtures, 766Rosuvastatin, 633Rotavirus vaccine, 1165tRotenone, 1009f
as pesticide, 1008Rotigotine
for parkinsonism, 489Roundworms
drugs used for, 938tRoutes of administration. See also specific types
alternativefirst-pass effect and, 44
Rowasa, 1102, 1102fRoyal jelly
risks associated with, 1127tRozerem, 387. See also RamelteonRU-486. See Mifepristone (RU-486)Rubella
passive immunization for, 1169tRufen, 655. See also IbuprofenRufinamide, 415f, 427
clinical uses of, 415mechanism of action of, 415for seizures, 415
Ryanodine receptor (RyR) channel, 479Rythmol, 249. See also Propafenone
SSafety tests
in drug development, 71–72, 71tSalicylate(s)
drug interactions with, 1160thalf-life of
in neonates and adults, 1045tmetabolism of, 638, 639fnonacetylated, 640in OTC products, 1123toverdose of, 1034–1035properties of, 638tstructure of, 639f
Salicylic acid, 1074–1075ionization constants of, 11tpharmacokinetic and pharmacodynamic
parameters for, 40tstructure of, 1074f
Katzung_Index_p1171-1234.indd 1218 9/24/11 12:03:38 PM

Index 1219
Salicylsalicylic acid, 656Saline diuresis
for hypercalcemia, 778Salivary glands
sympathomimetic drug effects on, 139Salmeterol, 343f, 355
for asthma, 344Salmeterol/fluticasone, 355Salsalate
preparations available, 656Salt(s)
nonabsorbablein constipation management, 1093
Saluron, 270. See also HydroflumethiazideSamsca, 270, 678. See also TolvaptanSanctura, 127. See also TrospiumSandimmune, 999. See also CyclosporineSandostatin, 678. See also Octreotide acetateSangCya, 999. See also CyclosporineSanitization
defined, 894tSaphris, 519. See also AsenapineSaquinavir, 871t, 880, 889
drug interactions with, 874tSaralasin, 299Sarcomere(s)
cardiac muscle, 213fSarcoplasmic reticulum
calcium stored in and released from, 212Sargramostim
preparations available, 598Sassafras
risks associated with, 1127tSavella, 539. See also MilnacipranSaw palmetto, 1133–1134Saxagliptin
for diabetes mellitus, 761Saxagliptin plus metformin, 766Scavenger receptors, 619Schistosomiasis
praziquantel for, 944Schizoaffective disorder
antipsychotic drugs for, 508lithium for, 516
Schizonticidesblood, 915tissue, 915
Schizophreniaantipsychotic drugs for, 508basic pharmacology of, 503–508, 509t,
510tclinical pharmacology of, 508–513dopamine hypothesis of, 502glutamate hypothesis of, 503lithium for, 516nature of, 502serotonin hypothesis of, 502
Scopolamine, 116, 127, 1111ionization constants of, 11torgan system effects of, 116, 118
Screeningdrug, 70–71
Secobarbital, 375f, 387Seconal, 387. See also SecobarbitalSecond messengers, 25–26, 25f, 26f, 26t
calcium, 28–29, 28fcAMP, 26, 28, 28fcGMP, 29DAG, 28–29, 28fdescribed, 25lithium effects on, 514–515, 515f, 514tphosphoinositides, 28–29, 28fwell-established, 26, 28–29, 28f
Secondarily generalized seizure attack, 421Secondary amine, 11Secretory glands
direct-acting cholinomimetic drug effects on, 102t, 104
Secretory tissuesmooth muscle of, 276
Sectral, 167, 189, 249. See also AcebutololSedation
conscious, 430deep, 430dexmedetomidine for, 146H1-receptor antagonists for, 278, 279tmonitored anesthesia care and, 430opioid analgesics in, 550–551
Sedative-hypnotic drugs, 373–388. See also specific drugs
in the elderly, 1053–1054overdose of, 1037
Seizure(s)absence, 421antipsychotic drugs and, 511atonic, 421classification of, 404t, 420–421complex partial, 421described, 403drugs used for, 403–427. See also
Antiseizure drugsgeneralized
clinical pharmacology of, 421drugs used for, 417–418, 422–423
generalized tonic-clonic, 421drugs used for, 407–417, 422
grand mal, 421drugs used for, 407–417, 422
partialclinical pharmacology of, 420–421
petit mal, 421types of, 404t
Selective estrogen receptor modulators (SERMs), 731
in bone mineral homeostasis, 775Selective serotonin reuptake inhibitors
(SSRIs)adverse effects of, 535chemistry of, 525, 526fdosage of, 535t
Selective serotonin reuptake inhibitors (Cont.):
drug interactions with, 536, 537t, 1161tfetal effects of, 1043tpharmacodynamics of, 530t, 531pharmacokinetics of, 528, 529t
Selegilinedosage of, 535tfor parkinsonism, 488f, 490, 499pharmacokinetics of, 529t, 530
Selenium sulfatein OTC products, 1120t
Selzentry, 889. See also MaravirocSemustine, 954fSenile cerebral insufficiency
ergot alkaloids for, 290Senna
in constipation management, 1093, 1112
Senokot, 1112. See also SennaSensipar, 786. See also CinacalcetSensitivity
as pharmacodynamic variable, 48–49Sensorcaine, 463. See also BupivacaineSeptic shock
nitric oxide in, 335, 335tSeptocaine, 463. See also ArticaineSeptra, 837. See also Trimethoprim-
sulfamethoxazole (TMP-SMZ)Serevent, 355. See also SalmeterolSerine protease inhibitors
for bleeding disorders, 617Seromycin Pulvules, 808, 848. See also
CycloserineSerophene, 740. See also ClomipheneSeroquel, 519. See also QuetiapineSerotonin, 281–287
in ANS, ENS, and NANC neurons, 84tbasic pharmacology of, 281–284chemistry of, 281, 281fclinical pharmacology of, 284–286as CNS neurotransmitter, 366t–377t,
370in coagulation, 601, 602finhibitory effects of, 92tmechanisms of action of, 282, 282tpharmacodynamics of, 282–284, 282t,
284tpharmacokinetics of, 281–282tissue and organ system effects of,
282–284, 284tSerotonin agonists, 284–285Serotonin antagonists, 286Serotonin-blocking actions
of H1-receptor antagonists, 278–279Serotonin hypothesis
of schizophrenia, 502Serotonin-norepinephrine reuptake
inhibitors (SNRIs)adverse effects of, 535
Katzung_Index_p1171-1234.indd 1219 9/24/11 12:03:38 PM

1220 Index
Serotonin-norepinephrine reuptake inhibitors (Cont.):
chemistry of, 525–526, 527fdosage of, 535tdrug interactions with, 536–537pharmacodynamics of, 530t, 531pharmacokinetics of, 528–529, 529t
Serotonin receptor(s)subtypes of, 282t
Serotonin syndrome, 284, 284t, 537, 1034
Serotonin transporter, 577Serotonin-receptor antagonists, 286–287Sertoli cells, 734Sertraline, 526f, 539
dosage of, 535tpharmacodynamics of, 530tpharmacokinetics of, 529t
Serum gastrinincreased
proton pump inhibitors and, 1089
Serum osmolalityin poisoning assessment, 1030, 1031t
Serum sicknessimmunologic reactions to drugs and,
998Serutan, 1112. See also PsylliumSevelamer, 780, 786Seven-transmembrane (7-TM) receptors,
25, 26fSevoflurane, 432f, 446
pharmacologic properties of, 433tShivering
reducingopioid analgesics for, 553
Shockanaphylactic
sympathomimetic drugs for, 145cardiogenic
sympathomimetic drugs for, 144septic
nitric oxide in, 335, 335tsympathomimetic drugs for, 144
Short staturegrowth hormone for children with,
662–663, 663tidiopathic
growth hormone for, 663, 663t
SIADH secretionADH antagonists for, 264
Sibutraminein obesity management, 664, 665tsympathomimetic drug
effects on, 143Sickle cell disease, 582Signal transducers and activators of
transcription (STATs), 23, 24f
Signaling mechanismscytokine receptors, 23, 24fdrug action and, 21–29, 21f–28f, 26tG proteins, 25–26, 25f, 26f, 26tinterplay among, 29intracellular receptors for lipid-soluble
agents, 21–22, 21fligand-gated channels, 23–24, 24fligand-regulated transmembrane
enzymes, 22–23, 23fphosphorylation and, 29receptor regulation, 25–26, 27fsecond messengers, 25–26, 25f, 26f, 26t,
28–29, 28ftransmembrane, 21, 21fvoltage-gated channels, 23–24, 24f
Sildenafilfor erectile dysfunction, 199preparations available, 209
Silodosin, 155, 167Silvadene, 837. See also Silver sulfadiazineSilver nitrate, 897, 898Silver sulfadiazine, 897, 837Simcor, 633Simponi, 656, 999. See also GolimumabSimulect, 999. See also BasiliximabSimvastatin, 633Sinecatechins, 1076Sinemet, 499. See also Carbidopa/levodopaSinequan, 539. See also DoxepinSingle-blind design, 72Singulair, 328, 355. See also MontelukastsiRNAs, 2Sirolimus
as immunosuppressive drug, 985t, 986–987
Sisomicin, 825Sitagliptin
for diabetes mellitus, 761Sitagliptin plus metformin, 766Skelaxin, 481. See also MetaxaloneSkeletal muscle
calcium channel blockers effects on, 204dantrolene effects on, 479methylxanthines effects on, 346serotonin effects on, 284
Skeletal muscle paralysis, 472Skeletal muscle relaxants, 465–482. See also
Muscle relaxantsSkelid, 786. See also Tiludronate disodiumSkin
autonomic nerve activity effects on, 90tdecontamination of poisoned patient,
1031–1032isoniazid effects on, 841oral contraceptives effects on, 728poisoning effects on, 1029
Skin diseasesdrugs used for, 1061–1079.See also
Dermatologic drugs
Sleepprostaglandins and, 321sedative-hypnotics and, 382
Sleep aidsingredients in, 1122t
Sleep disorderssedative-hypnotic drugs for, 382–383
Slo-Phyllin, 355. See also TheophyllineSlow acetylator phenotype, 65Slow-reacting substance of anaphylaxis
(SRS-A), 317Small interfering RNAs (siRNAs), 2Smoking
cigarettecoronary disease related to, 619
fetal effects of, 1043tSmoking cessation
aids foringredients in, 1122t
antidepressants in, 533varenicline in, 110
Smooth musclealcohol effects on, 391bronchiolar
autonomic nerve activity effects on, 90t
histamine effects on, 276calcium channel blockers effects on, 203contraction of
calcium channel blockers in control of, 194, 195f
digitalis effects on, 217drugs with important actions on,
273–357genitourinary
autonomic nerve activity effects on, 90t
glucagon effects on, 763lipoxygenase effects on, 323methylxanthines effects on, 346nitrates/nitrites in
mechanism of action of, 196f, 197reproductive
prostaglandins and thromboxanes effects on, 320
uterineergot alkaloids effects on, 289inhaled anesthetics effects on, 437
vascularergot alkaloids effects on, 288t,
289, 289fnitroglycerin effects on, 196f, 197
vascularnitrates/nitrites effects on, 196f, 197nitroglycerin effects on, 196f, 197prostaglandins and thromboxanes
effects on, 320Snake bite
passive immunization for, 1169tSNAPs, 82–83
Katzung_Index_p1171-1234.indd 1220 9/24/11 12:03:38 PM

Index 1221
SNRIs. See Serotonin-norepinephrine reuptake inhibitors (SNRIs)
Social anxiety disorderantidepressants for, 532
Socioeconomic factorsin prescribing drugs, 1147
Sodiumintracellular
concentration of, 212in OTC products, 1123t
Sodium balancedrugs altering, 172–174, 174f
Sodium bicarbonateas antacid, 1083
Sodium-calcium exchangeractivity of, 212
Sodium channel blockers, 238–242. See also specific drugs
preparations available, 209, 249Sodium ferric gluconate complex, 585–586Sodium fluoride, 786Sodium Iodide I 131, 695. See also
Radioactive iodine sodiumSodium nitroprusside, 182, 334. See also
Nitroprussideas antihypertensive drug, 182pharmacokinetics/dosage of, 182
Sodium phosphatein constipation management, 1093
Sodium removalin chronic heart failure management,
220Sodium salicylate, 656Sodium stibogluconate,929f, 933Sodium sulfacetamide
for acne, 1064Sodium thiosalicylate, 656Sodium valproate
for seizures, 418therapeutic levels/dosage of, 419
Solganal, 656. See also AurothioglucoseSolid organ transplantation
immunosuppressive drugs in, 994–995
Solifenacinfor urinary disorders, 122, 122t
Solu-Cortef, 712. See also Hydrocortisone sodium succinate
Solu-Medrol, 712. See also Methylprednisolone sodium succinate
Solventsorganic
fetal effects of, 1043tTLVs of, 1005ttoxic exposure to, 1005–1006,
1005tSoma, 481. See also CarisoprodolSoman, 106fSomatomedin C, 662
Somatostatin, 661, 661f, 743for diarrhea, 1095for variceal hemorrhage, 1107–1108
Somatostatin analogs, 665–666, 665fSomatotropin
structure of, 662Somatropin, 678Somatuline Depot, 678. See also Lanreotide
acetateSomavert, 678. See also PegvisomantSonata, 387. See also ZaleplonSorafenib
for cancer, 968clinical activity and toxicities of, 967t
Sorbitolin constipation management, 1093
Sotalolfor cardiac arrhythmias, 244cardiac effects of, 242membrane actions of, 237tpharmacologic properties of, 238tproperties of, 159t
Spare receptors, 17–18, 17freceptor-effector coupling and,
16–18, 17fSpasm(s)
acute localdrugs for, 479
infantile, 421drugs used for, 423
Spasmex, 127. See also TrospiumSpasmolytic drugs, 476–479, 481. See also
specific drugsSpecial carrier molecules, 9, 9fSpectazole, 858. See also EconazoleSpectinomycin, 827
structure of, 827fSpectracef, 807. See also CefditorenSpiriva, 127, 355. See also TiotropiumSpironolactone, 261–263, 711, 739
during lactation, 1048tSpontaneously resolving hyperthyroidism,
692Sporanox, 858. See also ItraconazoleSRS-A, 317SRX251, 303St. Anthony’s fire, 287St. John’s wort, 1132–1133Stadol, 562. See also ButorphanolStage fright
beta-receptor antagonists for, 164Stalevo, 499. See also Carbidopa/levodopa/
entacaponefor parkinsonism, 491
Staphylococcus(i)methicillin-resistant
cephalosporins active against, 800Starlix, 766. See also NateglinideState-dependent drug action,
235, 236f
Statin-induced myopathyprevention of
coenzyme Q10 in, 1134STATs (signal transducers and activators of
transcription), 23, 24fStatus epilepticus
drugs used for, 423Stavudine, 863f, 871t, 874–875, 889Stedesa, 427. See also EslicarbazepineStelara, 999. See also UstekinumabSterilants
as antiseptics/disinfectants, 897preparations available, 898
Sterilization, 894tStereoisomerism, 4–5Steroid(s)
anabolic, 734–737. See also Anabolic steroids
Steroid synthesis inhibitors, 709–711, 737Sterol(s)
intestinal absorption ofinhibitors of
for hyperlipidemias, 630–631Stilphostrol, 740. See also DiethylstilbestrolStimate, 678. See also Desmopressin acetateStimulant(s)
overdose of, 1032–1034Stimulant laxatives, 1093Stiripentol, 427
for seizures, 415Stool softeners, 1093STOP-NIDDM trial, 752, 759Streptase, 617. See also StreptokinaseStreptogramins, 815–816Streptokinase, 604, 604f, 617
pharmacology of, 611Streptomycin, 822f, 824–825, 828, 848
for tuberculosis, 840t, 843Stress
bleeding due toproton pump inhibitors in prevention
of, 1088gastritis due to
prevention of bleeding fromH2-receptor antagonists in, 1085
Stress testcardiac
dobutamine in, 144Stretch reflex arc
structures involved in, 476, 477fStromectol, 946. See also IvermectinStrongyloidiasis
ivermectin for, 941Strontium ranelate
in bone mineral homeostasis, 777–778
for osteoporosis, 783Structural proteins, 16Structure-activity relationship
of drug congeners, 22f, 29–30
Katzung_Index_p1171-1234.indd 1221 9/24/11 12:03:38 PM

1222 Index
Strychnine, 366tionization constants of, 11t
Subacute thyroiditis, 692Subarachnoid hemorrhage
cerebral vasospasm and infarct followingcalcium channel blockers effects on,
204Submucous plexus, 81f, 82Substance P, 307
in ANS, ENS, and NANC neurons, 84tSubstituted benzamides
for nausea and vomiting, 1100Succimer, 1022–1023, 1025
indications and toxicity of, 1022–1023Succinylcholine, 466, 467f, 481
pharmacokinetic and pharmacodynamic properties of, 470t
tissue effects of, 474tSucralfate
for acid-peptic diseases, 1089–1090Sudafed, 148. See also PseudoephedrineSufenta, 562. See also SufentanilSufentanil, 557, 562
pharmacokinetics of, 545tSugar(s)
nonabsorbablein constipation management, 1093
Suicidality, 536antiseizure drugs and, 424
Suicide inhibitors, of enzymes, 59Sular, 190, 209. See also NisoldipineSulbactam, 801fSulconazole, 858Sulfacetamide sodium, 837Sulfacytine
pharmacokinetics of, 832tSulfadiazine
ionization constants of, 11tpharmacokinetics of, 832t
Sulfadoxine, 918f, 925pharmacokinetics of, 832t
Sulfadoxine/pyrimethamine. See Pyrimethamine/sulfadoxine
Sulfamethizole, 832tSulfamethoxazole
pharmacokinetics of, 40t, 832tSulfamylon, 837. See also MafenideSulfanilamide, 832fSulfapyridine, 1101f
ionization constants of, 11tpharmacokinetics of, 832t
Sulfasalazine, 646, 1101Sulfathalidine, 832fSulfation, 60tSulfinpyrazone, 656
drug interactions with, 610tSulfisoxazole, 832f, 837
pharmacokinetics of, 832tSulfonamides, 831–833, 837
antimicrobial activity of, 831, 832f
Sulfonamides (Cont.):oral absorption of
in neonates vs. older children and adults, 1044t
Sulfonylureas, 754–756, 755t, 766Sulfur, for scabies, 1068Sulfur dioxide
TLVs of, 1005tSulfur dioxide poisoning, 1004Sulindac, 642, 656
properties of, 638tSulthiame, 420Sumatriptan, 285f, 292
for migraine, 285pharmacokinetics of, 286t
Sunitinibfor cancer, 967t, 968
Sunscreens, 1069Superoxidized water
as antiseptic/disinfectant, 896Supersensitivity
denervation, 92Supprelin LA, 678. See also Histrelin
acetateSuprane, 446. See also DesfluraneSuprax, 807. See also CefiximeSuprofen, 656Suramin
clinical uses of, 933Surface area
as factor in drug dosage in infants and children, 1047, 1049, 1049t
Surgical relaxationneuromuscular blocking drugs for, 476
Surmontil, 539. See also TrimipramineSusceptibility testing
of pathogens to antimicrobial drugs, 903Suspensions
in pediatric patients, 1046Sustiva, 888. See also EfavirenzSweat glands
antimuscarinic drug effects on, 120Symlin, 766. See also PramlintideSymmetrel, 499, 888. See also AmantadineSympathetic nervous system
function ofdrugs altering, 174–175
in renin release, 296, 297fSympathomimetic drugs, 129–149, 355
actions ofmolecular pharmacology underlying,
130–134alpha receptors, 131, 131t, 132f, 137tamphetamine-like, 136f, 142–143, 143tfor anaphylaxis, 145for asthma, 341–344, 343fbeta receptors, 131–132, beta2-selective drugs, 343–344, 343fcardiovascular applications of, 144–145
Sympathomimetic drugs (Cont.):cardiovascular system effects of, 136–138,
137t, 138t, 139f, 140fcatecholamine reuptake inhibitors,
143CNS applications of, 146in congestive heart failure, 225described, 129–130desensitization of, 133direct-acting, 133t, 136f, 141dopamine agonists, 143–144dopamine receptors, 131, 132, 137tgenitourinary applications of, 145–146indirect-acting, 136f, 142–143, 143tmixed-acting, 136f, 141–142noncardiac effects of, 138–140norepinephrine transporter of, 134ophthalmic applications of, 145organ system effects of, 136–140in OTC products, 1123tpulmonary applications of, 145receptors
regulation of, 133–134selectivity of, 133, 133tsubtypes of, 131
distribution of, 136, 137tphysiologic functions of, 133polymorphisms of, 134
therapeutic uses for, 144–146toxicities of, 344types of, 131
Sympathoplegic drugs, 172, 189as antihypertensive drugs, 174–175centrally acting, 175–177
mechanisms and sites of action of, 175–176
Synagis, 889. See also PalivizumabSynapse(s)
CNS drugs and, 361–363, 362f, 363fSynaptic plasticity
addiction and, 571Synaptic potentials
CNS drugs and, 361–363, 362f, 363fSynaptobrevin, 83Synaptosomal nerve–associated proteins
(SNAPs), 82–83Synaptotagmin, 84Synarel, 678. See also Nafarelin acetateSyndrome of inappropriate ADH (SIADH)
secretionADH antagonists for, 264
Synercid, 819. See also Quinupristin-dalfopristin
Synergismamong antimicrobial drugs, 909–910
Synergistic killing, 823Synergy studies
for infections with known etiology, 903Syntaxin, 83Synthetic estrogens, 718, 718f
Katzung_Index_p1171-1234.indd 1222 9/24/11 12:03:38 PM

Index 1223
Synthetic progestins, 723physiologic effects of, 724, 725t
Synthetic steroidswith androgenic and anabolic action,
735, 735tSynthroid, 695. See also LevothyroxineSyprine, 499. See also TrientineSystemic infections
antifungal drugs for, 849–855Systolic failure, 211
TT lymphocytes, 978–981, 979f, 980ft-PA. See Tissue plasminogen activator
(t-PA)Tachykinins
as CNS neurotransmitters, 367tTachyphylaxis, 32Tacrine
for Alzheimer’s disease, 1054–1055, 112Tacrolimus, 999, 1067
as immunosuppressive drug, 985t, 986Tadalafil
for erectile dysfunction, 199Taenia spp.
niclosamide for, 942Taeniasis
praziquantel for, 944Tagamet, 1111. See also CimetidineTalwin, 562. See also PentazocineTambocor, 249. See also FlecainideTamiflu, 889. See also OseltamivirTamoxifen, 731, 740
fetal effects of, 1043tTamsulosin, 152f, 154–155, 167Tapazole, 695. See also MethimazoleTapentadol, 559, 562Tapeworms
drugs used for, 938tTar compounds, 1074Tardive akathisia, 496Tardive dyskinesia, 496
antipsychotic drugs and, 511Tardive dystonia, 496Target concentration, 45Target concentration intervention, 47–49
in designing rational dosage regimen, 45–47, 47f
loading dose, 46–47, 47fmaintenance dose, 46
Target concentration strategy, 48Tasmar, 499. See also TolcaponeTavist, 292. See also ClemastineTaxanes
for cancer, 963t, 964Tazarotene
for acne, 1070for psoriasis, 1071
Tazidime, 807. See also CeftazidimeTazobactam, 801f
TCAs. See Tricyclic antidepressants (TCAs)Teeth
fluoride effects on, 777tetracyclines effects on, 812
Teflaro, 807. See also Ceftaroline fosamilTegaserod, 286
in constipation management, 1094, 1097f, 1112
Tegretol, 427, 519. See also CarbamazepineTeicoplanin, 803Tekturna, 190. See also AliskirenTelaprevir
for HCV infection, 886Telavancin, 798t, 803, 808
pharmacokinetics and dosage of, 798t, 803Telbivudine
for HBV infection, 883t, 885Telithromycin, 814–815, 819Telmisartan, 190, 225Temazepam
pharmacokinetic properties of, 377tTemozolomide
clinical activity and toxicities of, 956tTemperature
hyperthermia, 284topioid analgesics effects on, 551, 551t
Tempra, 656. See also AcetaminophenTenecteplase
pharmacology of, 612Tenex, 189. See also GuanfacineTenofovir, 863f, 871t, 875, 889
drug interactions with, 874tfor HBV infection, 883t, 885
Tenormin, 167, 189. See also AtenololTensilon, 112. See also EdrophoniumTeratogen(s)
defined, 1042FDA risk categories for, 1042, 1043trisks associated with
counseling women about, 1042–1043Teratogenic drug actions
during pregnancy, 1041–1043, 1041f, 1042t–1043t
Terazol 3, 858. See also TerconazoleTerazol 7, 858. See also TerconazoleTerazosin, 154, 189Terbinafine
for mucocutaneous infections, 856oral, 1067from prescription to OTC status, 1116ttopical, 1065
Terbutalinefor asthma, 344ionization constants of, 11tpharmacokinetic parameters for, 40tin premature labor suppression, 145structure of, 142f, 343f
Terconazole, 858Teriparatide, 771
for osteoporosis, 775, 783
Terlipressin, 303for variceal hemorrhage, 1108
Tertiary amine, 11p-Tertiary amylphenol, 895Testicular cancer
chemotherapeutic drugs for, 973–974Testis(es), 734–739Testopel, 740. See also Testosterone pelletsTestosterone, 734–737. See also
Androgen(s)preparations available, 735t, 740
Tetanuspassive immunization for, 1169t
Tetanus-diphtheria-pertussis vaccine, 1166tTetanus-diphtheria vaccine, 1165tTetrabenazine
for Huntington’s disease, 494–495preparations available, 499
Tetracaine, 463, 451t2,3,7,8-Tetrachlorodibenzo-p-dioxin
(TCDD), 1010, 1009fTetrachlorodiphenylethane (TDE)
toxicity rating for, 1006tTetrachloroethylene
TLVs of, 1005tTetracyclic antidepressants
dosage of, 535tdrug interactions with, 537pharmacodynamics of, 530t, 531pharmacokinetics of, 529–530, 529t
Tetracycline(s), 810–813fetal effects of, 1043tduring lactation, 1048t
Tetraethylammonium, 124Tetrahydrocannabinol
abuse of, 572for chronic pain, 550
Tetrahydrozolinepreparations available, 148
Tetraiodothyronine (T4), 681Tetrathiomolybdate
for Wilson’s disease, 497Teveten, 190, 225. See also EprosartanTezosentan
in heart failure management, 219Thalamus
circuitry of, 483, 484fThalidomide
dermatologic uses of, 1078tfetal effects of, 1043timmunomodulatory derivatives of,
987–988as immunosuppressive drug, 987–988
Thalitone, 270. See also ChlorthalidoneThalomid, 999. See also ThalidomideTheo-24, 355. See also TheophyllineTheo-Dur, 355. See also TheophyllineTheobromine, 345fTheophylline, 344–346, 355
drug interactions with, 1161t
Katzung_Index_p1171-1234.indd 1223 9/24/11 12:03:38 PM

1224 Index
Theophylline (Cont.):half-life of
in neonates and adults, 1045thepatic clearance of
aging effects on, 1052tionization constants of, 11tduring lactation, 1048toverdose of, 1037–1038pharmacokinetic and pharmacodynamic
parameters for, 40tTherapeutic index, 32Thiabendazole, 945
for helminthic infections, 938t, 945Thiamine
in acute alcohol intoxication management, 395
Thiazide diuretics, 260–261in bone mineral homeostasis, 777
Thiazolidinedione(s)combination therapy with
preparations available, 766for diabetes mellitus, 757–759
Thiazolidinedione derivatives, 766Thienobenzodiazepine antipsychotics
advantages and disadvantages of, 509tchemical structure effects on potency
and toxicity of, 505tThimerosal, 897, 898Thioamides
antithyroid activity of, 688–6896-Thioguanine
clinical activity and toxicities of, 959tmechanism of action of, 961, 961f
Thiol(s)nitric oxide interactions with, 332–333
Thiopentalpharmacologic properties of, 439t
6-Thiopurinesfor cancer, 961–962, 961f
Thioridazineadvantages and disadvantages of, 509tdosage of, 510tionization constants of, 11t
Thiotepaclinical activity and toxicities of, 956tstructure of, 954f
Thiothixeneadvantages and disadvantages of, 509tdosage of, 510t
Thioxanthene antipsychotics, 503f, 504advantages and disadvantages of, 509tchemical structure effects on potency
and toxicity of, 505tThorazine, 519. See also ChlorpromazineThreshold limit values (TLVs), 1001
of air pollutants and solvents, 1005tThrombate III, 617. See also Antithrombin
IIIThrombin inhibitors
direct, 607–608. See also specific drugs
Thrombocytopenia, 582heparin-induced, 606
Thrombolytic drugsfor acute myocardial infarction, 611, 612
Thrombopoietinproperties of, 996t
Thrombosis, treatment ofarterial, 614venous, 614
Thromboxane(s), 315. See also Eicosanoid(s)
organ system effects of, 320–322Thromboxane A2, 315, 320, 601, 612Thrombus(i)
platelet-rich, 601–602red, 602white, 601–602
Thymoglobulin, 999. See also Antithymocyte globulin
Thyrogen, 678, 695. See also ThyrotropinThyroid. See also Antithyroid drugs
autoregulation of, 684drug effects on, 684, 685tfunction of
lithium effects on, 516physiology of, 681–684
Thyroid desiccated, 695Thyroid disease, 695Thyroid drugs, 681–696. See also specific
drugs and Antithyroid drugsThyroid function tests
values for, 683–684, 683tThyroid hormones, 684–688Thyroid neoplasms, 694Thyroid-pituitary relationships, 683–684,
684fThyroid stimulators
abnormal, 684Thyroid storm, 692–693Thyroidectomy
for Graves’ disease, 692Thyroiditis
subacute, 692Thyrolar, 695. See also LiotrixThyrotoxicosis
amiodarone-induced, 693–694manifestations of, 687tduring pregnancy, 693treatment of, 691–694
Thyrotropin, 695Thyrotropin alfa, 678Thyrotropin-releasing hormone (TRH),
661, 661tclinical uses of, 661tdiagnostic uses of, 662t
Thyroxine, 681during lactation, 1048tperipheral metabolism of, 682, 682f
Tiagabine, 415f, 416for seizures, 415–416
Ticar, 807. See also TicarcillinTicarcillin
dosage of, 795t, 807Ticarcillin/clavulanate potassium, 807Ticlid, 617. See also TiclopidineTiclopidine
as antiplatelet agent, 612–613, 617Tics, 495
described, 483treatment of, 495
Tigan, 1112. See also TrimethobenzamideTigecycline, 810f , 812Tikosyn, 249. See also DofetilideTiludronate disodium, 786Time-dependent killing, 907Timentin, 807. See also Ticarcillin/
clavulanate potassiumTimolol, 159t, 161, 167, 189Tinactin, 858. See also TolnaftateTindamax, 935. See also TinidazoleTinidazole, 892
adverse effects of, 929for amebiasis, 927–929, 928t
Tinzaparin, 617Tioconazole, 858
from prescription to OTC status, 1116tTiotropium, 117f ,127, 347,355
for COPD, 121Tipranavir, 871t, 880–881, 889
drug interactions with, 874tTirofiban, 613, 617Tirosint, 695. See also LevothyroxineTissue factor-VIIa complex, 602–604, 603fTissue plasminogen activators (t-PAs), 604,
604fpharmacology of, 612
Tissue schizonticides, 915Tizanidine, 141, 148, 477–478, 481
as muscle relaxant, 146TLVs. See Threshold limit values (TLVs)TMP-SMZ. See also Trimethoprim-
sulfamethoxazole (TMP-SMZ)TNKase, 617. See also TenecteplaseTobramycin, 822f, 826, 828
pharmacokinetic and pharmacodynamic parameters for, 40t
Tocainidepharmacokinetic and pharmacodynamic
parameters for, 40tTocilizumab, 646–647, 993Tofranil, 539. See also ImipramineTolazamide
for diabetes mellitus, 754, 755tTolazoline
cardiovascular effects of, 153fTolbutamide
for diabetes mellitus, 754, 755thepatic clearance of
aging effects on, 1052tionization constants of, 11t
Katzung_Index_p1171-1234.indd 1224 9/24/11 12:03:38 PM

Index 1225
Tolbutamide (Cont.):during lactation, 1048tpharmacokinetic and pharmacodynamic
parameters for, 40tTolcapone
for parkinsonism, 488f, 490, 499Tolectin, 656. See also TolmetinTolerance, 66Tolinase, 766. See also TolazamideTolmetin, 642Tolnaftate, 858
topical, 1066Tolterodine
for urinary disorders, 122, 122tToluene, 1006
TLVs of, 1005tTolvaptan, 264, 675
in heart failure, 222preparations available, 270, 678
Tonic-clonic seizuresgeneralized, 421
drugs used for, 407–417, 422Topamax, 427, 519. See also TopiramateTopiramate, 416f, 427, 519
for seizures, 416for tremors, 494
Topotecanclinical activity and toxicities of, 963t
Toprol, 167. See also MetoprololToprol XL, 225. See also MetoprololToradol, 655. See also KetorolacTorcetrapib, 631Toremifene, 731, 740Tornalate, 355. See also BitolterolTorsemide
dosage of, 259t, 270Tositumomab, 992Tourette’s syndrome, 495
antipsychotic drugs for, 509Toviaz, 127. See also FesoterodineToxaphenes
toxicity rating for, 1006tToxic dose
median (TD50), 31–32Toxic multinodular goiter, 692Toxic products
metabolism of drugs to, 60–61, 61f
Toxic syndromes, 1032–1038Toxic uninodular goiter, 692Toxicodynamics, 1027–1028Toxicokinetics, 1027–1028Toxicology, 1001–1038
chemicals, 1003–1005, 1005tdefined, 1, 2fenvironmental, 1001–1003herbicides, 1008–1010occupational, 1001pesticides, 1006–1008, 1006t–1008tscreening tests, 1031, 1031t
Toxicology (Cont.):solvents, 1005–1006, 1005tterminology related to, 1002
Toxin(s)defined, 4exposure to
duration of, 1002routes of, 1002
ion channel effects, 360, 360tToxoplasma gondii
drugs used for, 932tToxoplasmosis
drugs used for, 926Tracheal intubation
neuromuscular blocking drugs for, 476Tracleer, 225. See also BosentanTrademark, 76Tradjenta, 766. See also LinagliptinTramadol, 559, 562Trandate, 167, 189. See also LabetalolTrandolapril, 190, 225Tranexamic acid
for bleeding disorders, 616Transderm Scop, 127, 1111, 1112. See also
ScopolamineTransdermal patch
opioid analgesic, 554Transferrin, 583f, 584Transient neurologic symptoms
of local anesthetics, 459Translational research, 70
in drug development, 76Transmembrane enzymes
ligand-regulated, 22–23, 23fTransmembrane signaling mechanisms,
21, 21fTransplantation
autologous stem cellmyeloid growth factors in,
593, 593fbone marrow
immunosuppressive drugs in, 994–995
organimmunosuppressive drugs in, 985t
solid organimmunosuppressive drugs in,
994–995Transport molecules, 9, 10tTransport proteins, 16Transporter(s)
drug, 9, 10tTranxene, 387, 427. See also ClorazepateTranylcypromine, 528f, 539
chemistry of, 527–528dosage of, 535tpharmacokinetics of, 530
Trastuzumab, 992preparations available, 999
Travatan, 328. See also Travoprost
Travelersimmunizations recommended for, 1170malaria prevention in
drugs used for, 919tTravoprost, 328Trazodone, 155, 529
chemistry of, 526, 527fdosage of, 535t
Trecator-SC, 848. See also EthionamideTrelstar, 678. See also Triptorelin pamoateTrematodes
drugs used for, 938tTremor(s), 493–494
beta-receptor antagonists for, 164described, 483, 493essential, 493–494intention, 494lithium and, 516rest, 494
Trental, 209, 355. See also PentoxifyllineTreprostinil, 326, 328Treprostinil sodium, 325fTRH. See Thyrotropin-releasing hormone
(TRH)Triamcinolone, 700f, 712
clinical uses and dosage of, 703tTriamterene, 270Triazolam, 374f
pharmacokinetic properties of, 377tTricalcium phosphate
preparations available, 786Trichlorfon, 942
for helminthic infections, 938t, 942toxicity rating for, 1007t
Trichlormethiazidedosage of, 261t
1,1,1-TrichloroethaneTLVs of, 1005t
TrichloroethyleneTLVs of, 1005t
2,4,5-Trichlorophenoxyacetic acid, 1008–1009
structure of, 1009fTrichogenic agents, 1077Trichomonas vaginalis
drugs used for, 932tTrichomoniasis
drugs used for, 928Trichuriasis
albendazole for, 938–939Trichlormethiazide, 270Tricor, 633. See also FenofibrateTricyclic antidepressants (TCAs) 526,
527f, 539adverse effects of, 535dosage of, 535tdrug interactions with, 536–537, 1152tfetal effects of, 1042toverdose of, 1034pharmacodynamics of, 530t, 531
Katzung_Index_p1171-1234.indd 1225 9/24/11 12:03:38 PM

1226 Index
Tridihexethyl, 127Tridione, 427. See also TrimethadioneTrientine
for Wilson’s disease, 497, 499Triethylenemelamine, 954fTrifluoperazine, 503f
dosage of, 510tTrifluridine
dosage of, 865t, 889for HSV and VZV infections, 866
Trihexyphenidylfor parkinsonism, 492t, 499
Triiodothyronine (T3), 681Trileptal, 427. See also OxcarbazepineTrilostane, 710Trimethadione
fetal effects of, 1043tfor seizures, 419
Trimethaphan, 127Trimethobenzamide
for nausea and vomiting, 1100Trimethoprim, 833–834, 837
pharmacokinetics of, 40t, 832t, 834Trimethoprim-sulfamethoxazole
(TMP-SMZ), 833–834drug interactions with, 610t
Trimipraminepharmacodynamics of, 530t
Trimipramine maleatedosage of, 535t
Trimpex, 837. See also TrimethoprimTriorthocresyl phosphate (TOCP), 110,
1007–1008Trioxsalen, 1069“Triple response”
of histamine, 276Triprolidine, 292Triptan(s), 285–286Triptorelin pamoate, 678Tronothane, 463. See also PramoxineTropicamide, 117f, 127Trospium, 127
for urinary disorders, 122, 122tTRPV1, 550Truncal rigidity
opioid analgesics and, 551, 551tTrusopt, 270. See also DorzolamideTrypanosoma cruzi
drugs used for, 932tTrypanosomiasis
Africandrugs used for, 930–934, 930f, 932t
L-Tryptophanstructure of, 281f
Tuberculin hypersensitivity, 983Tuberculosis
drugs used for, 839–845.See also specific drugs
adverse reactions to, 843amikacin, 840t, 844
Tuberculosis, drugs used for (Cont.):aminosalicylic acid, 840t, 844capreomycin, 840t, 844cycloserine, 840t, 844dosages of, 840tduration of therapy, 840tethambutol, 840t, 842ethionamide, 840t, 843–844fluoroquinolones, 840t, 844–845isoniazid, 840–841, 840tkanamycin, 840t, 844linezolid, 840t, 845preparations available, 848pyrazinamide, 840t, 842–843rifabutin, 840t, 845rifampin, 840t, 841–842rifapentine, 840t, 845second-line drugs, 840t, 843–845streptomycin, 840t, 843
Tuberoinfundibular system, 506Tubocurarine, 466–470, 468f, 481
pharmacokinetics of, 40t, 470ttissue effects of, 474t
Tumor(s)neuroendocrine
diarrhea due tooctreotide for, 1095
Tumor necrosis factor-α (TNF-α)properties of, 996t
Tumor necrosis factor-α (TNF-α) antagonists, 647–649, 647f. See also specific drugs
for psoriasis, 1071–1072Tumor suppressor genes
cancer related to, 950Tums, 786, 1111. See also Calcium
carbonateTurner syndrome
growth hormone for, 663, 663tTygacil, 819. See also TetracyclineTylenol, 656. See also AcetaminophenTylenol with Codeine, 562. (Oxycodone/
acetaminophen)Tylox, 562. (Oxycodone/acetaminophen)Typhoid vaccine, 1166tTyramine, 143, 143tTyrosine hydroxylase inhibitors, 167Tyrosine nitration
nitric oxide interactions with, 333, 333t
Tysabri, 999. See also NatalizumabTyzeka, 889. See also Telbivudine
UUlaritide, 256
in heart failure management, 219Ulcer(s)
H. pyloriproton pump inhibitors for,
1087–1088
Ulcer(s) (Cont.):NSAID–associated
proton pump inhibitors for, 1088peptic. See Peptic ulcer(s)
Uloric, 656. See also FebuxostatUltane, 446. See also SevofluraneUltiva, 562. See also RemifentanilUltram, 562. See also TramadolUltrarapid metabolism, 63–64Unasyn, 807. (Ampicillin/sulbactam)Unicyclic antidepressants
effects of, 531, 536chemistry of, 527, 528fdosage of, 535t
Uniphyl, 355. See also TheophyllineUnipolar depression
lithium for, 516United Kingdom Prospective Diabetes
Study (UKPDS), 752United Nations Food and Agriculture
Organization and the World Health Organization (FAO/WHO) Joint Expert Commission on Food Additives, 1002
Unithiol, 1023–1024Unithroid, 695. See also LevothyroxineUnivasc, 190, 225. See also MoexiprilUnstable angina, 193
clinical pharmacology of, 207nitrate effects in, 200
Uracil, 960fUrantide, 309Urapidil, 155Urea as drug, 1075Urecholine, 112. See also BethanecholUricosuric drugs
for gout, 651f, 652–653Uridine 5′-diphosphate
(UDP)-glucuronosyl transferases (UGTs), 59–60, 60f
Urinary alkalinizationcarbonic anhydrase inhibitors for, 257,
257tUrinary antiseptics, 892–893Urinary disorders
antimuscarinic drugs for, 122, 122t
Urinary obstructionalpha-receptor antagonists for, 156
Urinary pH manipulationin decontamination of poisoned patient,
1032Urinary tract
disorders ofcholinomimetic drugs for, 108
indirect-acting cholinomimetic drug effects on, 107
sulfonamides effects on, 833
Katzung_Index_p1171-1234.indd 1226 9/24/11 12:03:38 PM

Index 1227
Urine volumeincrease in
osmotic diuretics for, 263–264Urispas, 127. See also FlavoxateUrofollitropin, 666–667, 678Urokinase, 604, 604f
pharmacology of, 611Urotensin, 309Ursodiol
for gallstones, 1107Uroxatral, 167. See also AlfuzosinUrticaria
contactimmunologic
topical dermatologic medications and, 1063t
non-immunologictopical dermatologic medications
and, 1063tUse-dependent drug action, 235, 236fUstekinumab, 993–994, 999
for psoriasis, 1072Uterine bleeding
estrogen and, 722Uterine fibroids
gonadotropins in, 670Uterine leiomyomata
gonadotropins in, 670Uterus
ergot alkaloids effects on, 289inhaled anesthetics effects on, 437opioid analgesics effects on, 552oral contraceptives effects on, 727
VV-Cillin, 807. See also Penicillin VVaccine(s), 1163–1170. See also specific
types and Immunization(s)Vaccinia
passive immunization for, 1169tVagistat-1, 858. See also TioconazoleVagus nerve stimulation (VNS)
for seizures, 404Valacyclovir, 863f, 889
dosage of, 865tfor HSV and VZV infections, 864topical, 1067
Valcyte, 889. See also ValganciclovirValganciclovir, 863f, 889
for CMV infections, 867–868Valium, 387, 400, 427. See also DiazepamValproic acid/valproate, 418,
427, 519for bipolar disorder, 517fetal effects of, 1043tpharmacokinetics of, 40t, 418–419for seizures, 418
Valsartanpreparations available, 190, 225
Valtrex, 889. See also Valacyclovir
VAMPs, 82Vancocin, 808. See also VancomycinVancoled, 808. See also VancomycinVancomycin, 802–803, 808
dosage of, 798t, 802mechanisms of action of, 802pharmacokinetics of, 40t, 798t, 802
Vantas, 678. See also Histrelin acetateVantin, 807. See also CefotaximeVaprisol, 270, 678. See also ConivaptanVardenafil
for erectile dysfunction, 199Varenicline
in nicotine addiction, 574–575in smoking cessation, 110, 112
Variant angina, 193diagnosis of
ergot alkaloids in, 290nitrate effects in, 200
Variceal hemorrhagedrugs used for, 1107–1108
Varicella vaccine, 1166tVaricella-zoster virus (VZV) infection
drugs used for, 862–866passive immunization for, 1169t
Vascular disordersoral contraceptives and, 729peripheral
alpha-receptor antagonists for, 156Vascular smooth muscle
ergot alkaloids effects on, 288t, 289, 289fnitrates/nitrites effects on, 196f, 197nitroglycerin effects on, 196f, 197peptides and, 295–312prostaglandins and thromboxanes effects
on, 320Vascular tone
determinants of, 194–195, 194tVasoactive intestinal peptide (VIP),
306–307in ANS, ENS, and NANC neurons, 84t
Vasoactive peptides, 295–312. See also specific drugs
adrenomedullin, 308angiotensin, 295–300CGRP, 308endothelins, 304–306kinins, 300–303natriuretic peptides, 303–304neuropeptide Y, 308–309neurotensin, 307–308substance P, 307urotensin, 309vasopeptidase inhibitors, 304vasopressin, 303VIP, 306–307
Vasoconstrictioninduction of
sympathomimetic drugs for, 144–145
Vasodilator(s). See also specific drugsfor angina pectoris, 193–210as antihypertensive drugs, 180–183calcium channel blockers, 183diazoxide, 182–183direct
described, 172fenoldopam, 183in heart failure management, 219, 220hydralazine, 181mechanism and sites of action of, 174f,
180, 181tminoxidil, 181sodium nitroprusside, 182
Vasopeptidase inhibitors, 304Vasopressin, 303, 674–675
clinical pharmacology of, 674–675contraindications to, 675for variceal hemorrhage, 1108
Vasopressin agonists, 678Vasopressin antagonists, 303, 675, 678Vasopressin receptors, 303Vasospasm(s)
cerebralfollowing subarachnoid hemorrhage
calcium channel blockers effects on, 204
Vasospastic angina, 193clinical pharmacology of, 207
Vasotec, 190, 225. See also EnalaprilVectibix, 999. See also PanitumumabVecuronium
pharmacokinetic and pharmacodynamic properties of, 469f, 470t, 474t
Venlafaxine, 526f, 539dosage of, 535tpharmacodynamics of, 530t, 531pharmacokinetics of, 528, 529t
Venous thromboembolic diseaseoral contraceptives and, 729
Venous thrombosis, 614Ventavis, 328. See also IloprostVentilation
alveolarof inhaled anesthetics, 430–432, 432f
control ofneuromuscular blocking drugs for,
476Ventolin, 355. See also AlbuterolVentral tegmental area (VTA), 565Verapamil
for angina, 201–205for cardiac arrhythmias, 245clinical pharmacology of, 202tdosage of, 175t, 245for hypertension, 183membrane actions of, 237tfor migraine, 285pharmacokinetics of, 40t, 175t, 245pharmacologic properties of, 238t
Katzung_Index_p1171-1234.indd 1227 9/24/11 12:03:38 PM

1228 Index
Verapamil (Cont.):therapeutic use of, 245toxicity of, 245
Vermox, 946. See also MebendazoleVernakalant
for cardiac arrhythmias, 243–244Versed, 387, 446. See also MidazolamVery-low-density lipoproteins (VLDLs),
619, 620, 621fVesamicol, 82Vesicare, 127. See also SolifenacinVesicle-associated membrane proteins
(VAMPs), 82Vesicle-associated transporter, 82, 83fVesicular glutamate transporter, 367Vesicular monoamine transporter, 85Vesicular proteoglycan, 82Vestibular disturbances
H1-receptor antagonists for, 280Vfend, 858. See also VoriconazoleViagra, 209. See also SildenafilVibativ, 808. See also TelavancinVibramycin, 819, 935. See also DoxycyclineVicodin, 562. See also Hydrocodone/
acetaminophenVicoprofen, 562. See also Hydrocodone/
ibuprofenVictoza, 766. See also LiraglutideVigabatrin
for seizures, 416–417Viibryd, 539. See also VilazodoneVilazodone, 539Vimpat, 427. See also LacosamideVinblastine
for cancer, 962, 963tas immunosuppressive drug, 989
Vinca alkaloidsfor cancer, 962–964, 963t
Vincristinefor cancer, 962–964, 963tas immunosuppressive drug, 989
Vinorelbinefor cancer, 963t, 964
VIP. See Vasoactive intestinal peptide (VIP)Viracept, 889. See also NelfinavirViral infections
drugs used for, 861–890. See also specific drugs, indications, and Antiviral drugs
Viral replication, 861–862Viramune, 889. See also NevirapineViread, 889. See also TenofovirViroptic, 889. See also TrifluridineVirus(es)
cancer related to, 949–950described, 861
Visicol, 1112. See also Sodium phosphateVisine, 148. See also TetrahydrozolineVisken, 167, 189. See also PindololVistaril, 292, 387. See also Hydroxyzine
Vistide, 888. See also CidofovirVitamin B12
for anemia, 584t, 586–588Vitamin D
in bone mineral homeostasis, 770, 770f, 771f, 772–773, 772f
for chronic kidney disease, 781–782deficiency of
nutritional, 781for hypocalcemia, 780metabolites and analogs of, 772–773,
773tPTH and FGF23 interactions with,
773–774Vitamin D–dependent rickets
hereditary, 783–784types I and II, 783–784
Vitamin Kfor bleeding disorders, 614–615
Vitamin K1, 609fVitamin K cycle, 609, 609fVivactil, 539. See also ProtriptylineVLDLs. See Very-low-density lipoproteins
(VLDLs)VNS. See Vagus nerve stimulation (VNS)Voltage-gated channels, 23–24, 24fVolume of distribution, 38, 39t–41t, 1027
defined, 38in drug concentration measurements, 50in the elderly, 1052, 1052tin infants and children, 1044–1045pharmacokinetic mechanisms of, 1149as pharmacokinetic variable, 48
Vomitingnausea and. See Nausea and vomitingprevention of
domperidone in, 1092metoclopramide in, 1092
Voriconazole, 853f, 858for systemic infections, 855
Vorinostat, 1077dermatologic uses of, 1078t
Vytorinpreparations available, 633
VZV infection. See Varicella-zoster virus (VZV) infection
WWarfarin, 608–611, 609f, 617
fetal effects of, 1043tionization constants of, 11tduring lactation, 1048tpharmacokinetics of, 40t,
608–609, 609fWater
superoxidizedas antiseptic/disinfectant, 896
Water balance. See also Diureticsdrugs altering, 172–174, 174f
Water conjugation, 60t
Water excretionagents altering, 263–265. See also
specific diureticsWearing-off reactions, 487Weight
as factor in drug dosage in infants and children, 1047, 1049, 1049t
Weight loss aidsingredients in, 1122t
WelChol, 633. See also ColesevelamWellbutrin, 539. See also BupropionWernicke-Korsakoff syndrome, 393Westhroid, 695. See also Thyroid desiccatedWhite thrombi, 601–602Whole bowel irrigation
for iron poisoning, 586Wilson’s disease, 496–497Withdrawal syndrome, 549, 565Wound(s)
classification criteria forof NRC, 911
Wuchereria bancroftidiethylcarbamazine citrate for, 940
Wytensin, 189. See also Guanabenz
XX-linked agammaglobulinemia, 984–985X-linked hypophosphatemia, 783Xalatan, 328. See also LatanoprostXanax, 387. See also AlprazolamXanthine, 345fXenazine, 499. See also TetrabenazineXenobiotics, 4, 53Xenopex, 355. See also LevalbuterolXibrom, 655. See also BromfenacXifaxan, 848. See also RifaximinXolair, 355, 999. See also OmalizumabXylocaine, 249, 463. See also LidocaineXylometazoline, 141, 148Xyzal, 292. See also Levocetirizine
YYellow fever vaccine, 1166tYodoxin, 935. See also IodoquinolYohimbine, 155
ZZaditor, 292. See also KetotifenZafirlukast, 328, 349, 355Zalcitabine, 863f, 872t, 875, 889Zaleplon
biotransformation of, 374, 375f, 378, 387
pharmacokinetic properties of, 377tZanaflex, 148, 481. See also TizanidineZanamivir
for influenza, 886–887Zantac, 1111. See also RanitidineZarontin, 427. See also EthosuximideZaroxolyn, 270. See also Metolazone
Katzung_Index_p1171-1234.indd 1228 9/24/11 12:03:39 PM

Index 1229
Zebeta, 167, 189, 225. See also BisoprololZectran
toxicity rating for, 1008tZefazone, 807. See also CefmetazoleZegerid, 1111. See also OmeprazoleZelnorm, 1112. See also TegaserodZemplar, 786. See also ParicalcitolZemuron, 481. See also RocuroniumZenapax, 999. See also DaclizumabZenpep, 1111. See also PancrelipaseZephiran, 898. See also BenzalkoniumZestril, 190, 225. See also LisinoprilZetia, 633. See also EzetimibeZevalin, 999. See also Ibritumomab tiuxetanZiconotide, 562
for chronic pain, 550, 562Zidovudine, 863f, 872t, 875–876,
889pharmacokinetic parameters for, 40tduring pregnancy, 876, 876t
Ziegler’s enzyme, 65Zileuton, 349, 328, 355Zinacef, 807. See also CefuroximeZinc
for Wilson’s disease, 497Ziprasidone, 504f, 519
advantages and disadvantages of, 509t
dosage of, 510tZithromax, 819. See also
AzithromycinZocor, 633. See also SimvastatinZofran, 1111. See also OndansetronZoladex, 678. See also Goserelin acetateZoledronate, zoledronic acid
for osteoporosis, 775, 783
Zolmitriptanpharmacokinetics of, 286t
Zoloft, 539. See also Sertraline
Zolpidem, 374biotransformation of, 377–378pharmacokinetic properties of,
377tZometa, 786. See also Zoledronic acidZomig, 292. See also ZolmitriptanZonegran, 427. See also ZonisamideZonisamide
for seizures, 417, 427Zoster vaccine, 1166tZosyn, 807. See also Piperacillin
and tazobactam sodium
Zovirax, 888. See also AcyclovirZyflo, 328, 355. See also ZileutonZyloprim, 656. See also AllopurinolZymine, 292. See also TriprolidineZyprexa, 519. See also OlanzapineZyrtec, 292. See also CetirizineZyvox, 819. See also Linezolid
Katzung_Index_p1171-1234.indd 1229 9/24/11 12:03:39 PM