atresias, membranas, y duplicaciones - editorial …€¦ · contraste ingerido: el contraste en la...
TRANSCRIPT

Atresias, Membranas, y DuplicacionesJoel Shilyansky y Graeme Pitcher
CAPÍTULO 15
DEFINICIONES
La atresia intestinal (del griego que significa “no perfora-do”) denota una obliteración completa de la luz intestinal y se utiliza comúnmente para describir condiciones congé-nitas. Las telas o redes intestinales, son delgadas membranas intraluminales que se alinean con la mucosa, constituyen una forma de atresia. La red puede ser extendida para dar la apariencia de veleta. La estenosis es un estrechamiento de la luz intestinal. Las atresias intestinales y las redes pueden ser subclasificadas con base en la localización anatómica. Se pueden presentar en el esófago, estómago, duodeno, yeyu-no, íleon, colon o ano.1
La fisiopatología subyacente en todos los niños con atresia y redes es la obstrucción del flujo del contenido in-testinal. La forma de presentación puede ser aguda, con las características obvias de regurgitación, vómito, dolor abdo-minal y distensión abdominal. Los casos descuidados pue-den presentar descompensación sistémica y shock. La pre-sentación también puede ser sutil, especialmente cuando la obstrucción es incompleta como puede verse en la esteno-sis intestinal. Los pacientes pueden tener dolor abdominal crónico intermitente, vómito intermitente, y distensión. Pueden tener desde hace mucho tiempo compromiso nu-tricional o pérdida de peso, sin embargo, algunos niños se presentan con un peso normal, y la dieta se ajusta para mi-nimizar los síntomas. El diagnóstico precoz puede ser dado por una cuidadosa evaluación de los resultados clínicos, así como la historia prenatal y la interpretación posterior pre-cisa de los hallazgos radiológicos. La atención inicial debe incluir la reanimación adecuada e identificación de anoma-lías asociadas con la preparación del niño para el tratamien-to definitivo. Es fundamental que en las primeras etapas del trabajo en curso, sean identificadas y tratadas las condicio-nes que amenazan la vida del niño o lleven a la pérdida de la longitud intestinal, tales como enfermedad cardíaca, ma-
lrotación o perforación intestinal. La investigación secun-daria podría realizarse en forma segura. El tratamiento de la atresia intestinal es quirúrgico. El objetivo del tratamien-to quirúrgico es aliviar la obstrucción intestinal, mientras que se preserva la longitud intestinal y la función. Antes del advenimiento de la anestesia segura y el cuidado intensivo pediátrico, el 50% de los niños no sobrevivieron al trata-miento.1-3 La mortalidad disminuyó drásticamente a partir de la década de 1950, y actualmente la mortalidad aguda es menos de 4,8%. La mayoría de los pacientes pueden esperar una buena calidad de vida después de la corrección quirúr-gica, pero un peso significativo de la enfermedad recae so-bre quienes tienen inadecuada longitud intestinal.
ESÓFAGO
Atresia Esofágica Congénita
Definiciones y epidemiología
La atresia esofágica (AE) es una malformación congénita en la cual se interrumpe el esófago. La AE congénita se presen-ta aproximadamente en 1 de cada 3.000 nacidos vivos.4 La base embriológica de las anomalías del esófago aún es poco conocida. El esófago y la tráquea se separan 28-37 días des-pués de la fertilización. El fracaso del esbozo traqueal para separarse del intestino primordial anterior se cree que re-sulta en AE. La AE a menudo se asocia con otras anomalías congénitas, con mayor frecuencia cardiaca, y puede ser un componente complejo de anomalías vertebrales, anorrec-tales, cardíacas, traqueo-esofágica, renal y de extremidades (VACTERL) (Tabla 15-1).
El hallazgo confirmatorio de AE es un esófago inte-rrumpido que termina en una bolsa ciega proximal, por lo general a nivel de la salida torácica.5 Además, se asocia una fístula traqueo-esofágica (FTE) con AE en más del 85% de

220 ■ Sección 3: Trastornos Estomacales e Intestinales
Anomalías Asociadas en Bebés con Atresia Esofágica
Anomalía Incidencia (%)
CardíacaVSDTetralogía de FallotASDCoartación aórtica Fisiología de ventrículo único
No cardíacaVACTERLCromosomal (18, 21)Atresia duodenal
3010542,52,5
331892,5
Los bebés pueden tener más de una anomalía asociada. 20
AE con/osin fístula
Fístula con/osin atresia
AE con/ fístula
proximal
AE con/fístuladistal
80-85% 5-7% 4-5% 2%FIGURA 15–1 ■ Clasificación de la atresia esofágica y fístula tra-queoesofágica.
Tabla 15–2. Tabla 15–1.
Hallazgos en la Historia, Examen Físico y Radiológicos del Paciente con Atresia Esofágica
HistoriaPolihidramniosIntolerancia a la alimentaciónPulverización con alimentaciónCianosis con la alimentaciónBabeo
FísicosInhabilidad para pasar la sonda o tubo nasogástricoBabeo
RadiológicosRXT: aire en la bolsa proximal contrasta con los tejidos blandos
mediastino.RXT: sonda naso/orogástrica no avanza en la bolsa del esófago
proximal.RXA: aire en el estómago, si está presente FTE. RXA: aire en el duodeno, pero no en el resto del tracto intestinal,
si se asocia con AD.Contraste ingerido: el contraste en la bolsa esofágica superior,
no en el estómago.
FTE = fístula traqueoesofágica; AD = atresia duodenal.
los infantes (Figura 15-1). En estos niños, el esófago distal tiene una conexión posterior fistulosa con la tráquea en o por encima de la carina. El aire que entra a la vía respira-toria puede pasar a través de la fístula al tracto intestinal. El contenido gástrico también por reflujo puede pasar a la tráquea a través de la fístula. La atresia pura del esófago distal corto sin comunicación con la tráquea representa un 5-7% de las anomalías del esófago. Se ven ocasionalmen-te otros subtipos como el tipo H, donde el esófago tiene la permeabilidad normal, pero se comunica con la tráquea a través de una FTE, y la variante aún más rara, una fístula entre la bolsa proximal y la tráquea.
Presentación clínica
El diagnóstico de la AE se sospecha en el período prenatal basado en la presencia de polihidramnios y un estómago pequeño, pero a menudo el diagnóstico no se realiza hasta después del nacimiento del niño. Los niños recién nacidos a menudo por filtración regurgitan o pueden experimentar episodios de cianosis debido a la aspiración de alimentos. Son los bebés donde típicamente se cae la baba, porque son
incapaces de tragar la saliva. Dado que el esófago se inte-rrumpe, no se puede pasar una sonda nasogástrica o tubo (Tabla 15-2).
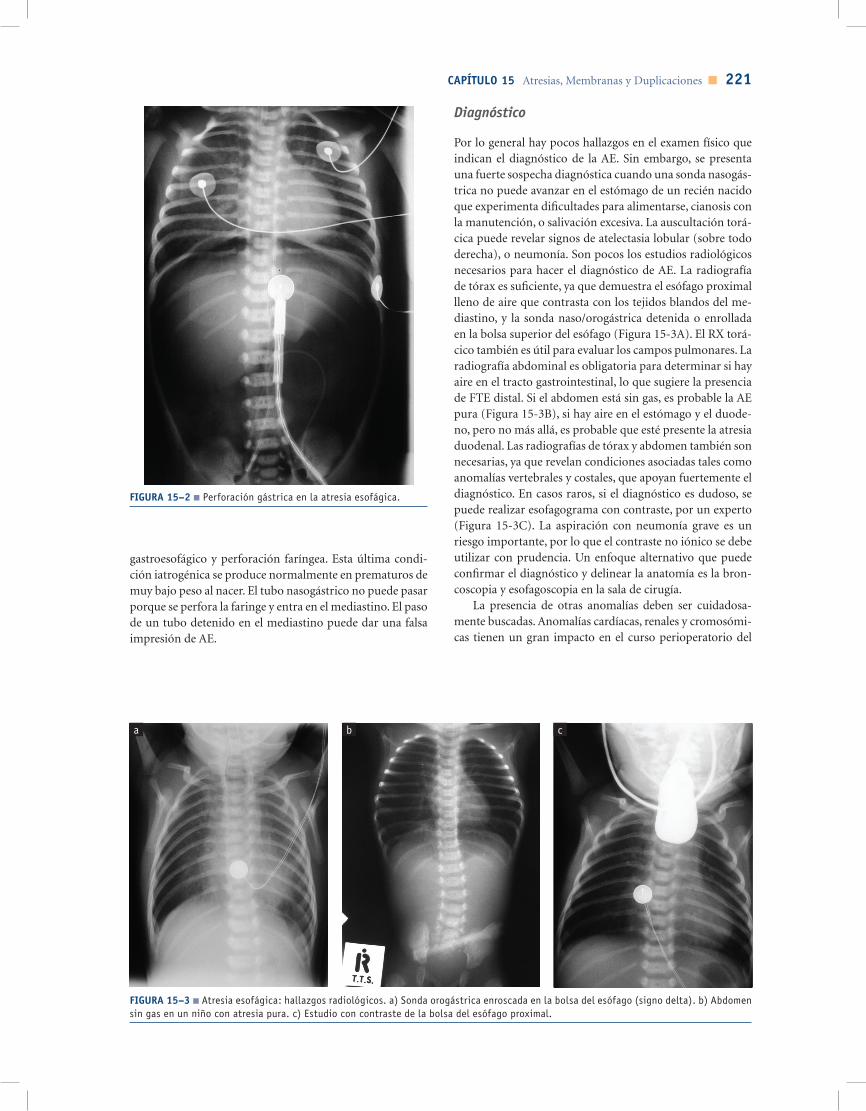
El curso clínico de los niños con AE puede ser compli-cado debido a broncoaspiración, neumonía y compromiso respiratorio. Puede ocurrir neumonía por aspiración debi-do al reflujo del contenido gástrico a través de la FTE, así como aspiración de la excesiva saliva de la bolsa superior. La aspiración puede conducir a insuficiencia respiratoria o neumonía, que requieren ventilación mecánica. Las com-plicaciones específicas pueden producirse en pacientes con ventilación mecánica, especialmente si la fístula es grande o si se requieren altas presiones de ventilación. El paso del aire a través de la fístula puede causar dilatación gástrica e in-testinal con ferulización diafragmática, haciendo ineficiente la ventilación. En los casos en que hay obstrucción intesti-nal distal (como atresia duodenal asociada), el estómago se distiende enormemente, dando lugar a compromiso respi-ratorio y ruptura, incluso gástrica, que se presenta con un neumoperitoneo muy grande y el deterioro clínico agudo (Figura 15-2). Estos pacientes a veces requieren salvamento con la interrupción de una fístula emergente ya sea median-te la colocación de un catéter balón o cirugía.
Diagnóstico diferencial
El diagnóstico diferencial de AE es corto. Las condiciones a considerar incluyen fisura laringo-traqueo-esofágica, age-nesia traqueal, estenosis esofágica, enfermedad por reflujo
CAPÍTULO 15 Atresias, Membranas y Duplicaciones ■ 221
FIGURA 15–2 ■ Perforación gástrica en la atresia esofágica.
gastroesofágico y perforación faríngea. Esta última condi-ción iatrogénica se produce normalmente en prematuros de muy bajo peso al nacer. El tubo nasogástrico no puede pasar porque se perfora la faringe y entra en el mediastino. El paso de un tubo detenido en el mediastino puede dar una falsa impresión de AE.
Diagnóstico
Por lo general hay pocos hallazgos en el examen físico que indican el diagnóstico de la AE. Sin embargo, se presenta una fuerte sospecha diagnóstica cuando una sonda nasogás-trica no puede avanzar en el estómago de un recién nacido que experimenta dificultades para alimentarse, cianosis con la manutención, o salivación excesiva. La auscultación torá-cica puede revelar signos de atelectasia lobular (sobre todo derecha), o neumonía. Son pocos los estudios radiológicos necesarios para hacer el diagnóstico de AE. La radiografía de tórax es suficiente, ya que demuestra el esófago proximal lleno de aire que contrasta con los tejidos blandos del me-diastino, y la sonda naso/orogástrica detenida o enrollada en la bolsa superior del esófago (Figura 15-3A). El RX torá-cico también es útil para evaluar los campos pulmonares. La radiografía abdominal es obligatoria para determinar si hay aire en el tracto gastrointestinal, lo que sugiere la presencia de FTE distal. Si el abdomen está sin gas, es probable la AE pura (Figura 15-3B), si hay aire en el estómago y el duode-no, pero no más allá, es probable que esté presente la atresia duodenal. Las radiografías de tórax y abdomen también son necesarias, ya que revelan condiciones asociadas tales como anomalías vertebrales y costales, que apoyan fuertemente el diagnóstico. En casos raros, si el diagnóstico es dudoso, se puede realizar esofagograma con contraste, por un experto (Figura 15-3C). La aspiración con neumonía grave es un riesgo importante, por lo que el contraste no iónico se debe utilizar con prudencia. Un enfoque alternativo que puede confirmar el diagnóstico y delinear la anatomía es la bron-coscopia y esofagoscopia en la sala de cirugía.
La presencia de otras anomalías deben ser cuidadosa-mente buscadas. Anomalías cardíacas, renales y cromosómi-cas tienen un gran impacto en el curso perioperatorio del
FIGURA 15–3 ■ Atresia esofágica: hallazgos radiológicos. a) Sonda orogástrica enroscada en la bolsa del esófago (signo delta). b) Abdomen sin gas en un niño con atresia pura. c) Estudio con contraste de la bolsa del esófago proximal.
a b c

222 ■ Sección 3: Trastornos Estomacales e Intestinales
Tasas de Supervivencia de Bebés con Atresia Esofágica20
Grupo Riesgo Peso(g)
Anomalías cardiacas
Supervivencia(%)
IIIIIIIV
BajoModeradoModerado
altoAlto
>2000<2000<2000<2000
––++
100817227
Tabla 15–3.
lactante, la morbilidad y la mortalidad. Habitualmente se realiza un ecocardiograma para evaluar las anomalías car-díacas y para determinar la ubicación del arco aórtico. Debe ser establecida la permeabilidad del ano y excluir atresia duodenal y buscar otras características de la asociación VACTERL. Con tal fin, los estudios de ecografía abdominal se realizan de forma rutinaria para evaluar anomalías rena-les, de la columna vertebral para descartar médula anclada, y de la cabeza para evaluar patología intracraneal. Los estudios deben realizarse con rapidez, pero no es necesario efectuar-los de forma urgente, hasta cuando el niño esté estable.
Tratamiento
El primer paso del tratamiento es la reanimación y apoyo cardiopulmonar. Para reducir el riesgo de broncoaspira-ción de secreciones orales, se debe colocar un tubo de as-piración orogástrica estilo Replogle (un tubo de ventilación de doble luz con aberturas de succión cerca de la punta) en la bolsa del esófago proximal y se conecta a succión para evacuar la saliva. La cabecera de la cama tiene que ser elevada para reducir el reflujo gastroesofágico. Se pre-fiere la ventilación espontánea, sin un tubo endotraqueal. Si es necesaria la ventilación mecánica o de control de la vía aérea, se deben emplear estrategias que minimicen la presión del ventilador. Una vez que el trabajo se haya com-pletado, se realiza la reparación quirúrgica. El objetivo de la cirugía es doble: dividir y ligar la FTE para evitar com-promiso pulmonar por la aspiración y distensión abdomi-nal, y para reconstruir el esófago. La mayoría de los niños con AE y FTE pueden someterse a reparación quirúrgi-ca exitosa del esófago durante los primeros días de vida. Los bebés que desarrollan distensión abdominal clínica-mente significativa o no se pueden aspirar las secreciones orales requieren intervención quirúrgica urgente. Los bebés inestables con compromiso pulmonar severo o cardiopatía cianótica, y en los cuales el esófago no puede ser reconstrui-do pueden beneficiarse de la reparación esofágica retarda-da. Estos bebés pueden requerir la colocación del tubo de gastrostomía para descomprimir el estómago y facilitar el acceso a la alimentación enteral. Los pacientes con enfer-medad cardíaca no cianótica se pueden tratar después de la reparación de AE y FTE.
La reparación exitosa del esófago depende de la longi-tud de la fisura entre la bolsa proximal dilatada del esófago y el esófago distal. En los niños con AE y FTE, el esófago usualmente se puede reparar todo. La AE aislada o pura puede estar asociada con un estómago muy pequeño y una distancia muy larga entre los extremos proximal y distal. Los niños con AE de distancia larga se manejan con repara-ción retardada la cual se realiza usualmente a los 2-3 meses de edad. Se han reportado métodos innovadores para colo-cación de tracción en los extremos del esófago con el fin de fomentar el crecimiento y permitir la anastomosis primaria, sin embargo, la aceptación general y el éxito a largo plazo ha sido difícil de lograr. Si la anastomosis primaria no es posi-ble se puede lograr la reconexión del esófago, por “tracción” del estómago hacia el mediastino. La reconstrucción tem-
prana del esófago, en los primeros meses de vida, incluso si se requiere un procedimiento de ascenso gástrico, se ha visto favorecida recientemente con la esperanza de evitar la aversión alimentaria.
Resultados
La mortalidad después de la reparación esofágica depende del peso al nacer y la presencia de enfermedades cardíacas congénitas importantes. Los bebés con anomalías cardíacas que pesan menos de 2.000 g tienen una tasa aproximada de supervivencia del 40% (Tabla 15-3). Los bebés de alto riesgo pueden considerarse para la reparación esofágica retrasada. La reconstrucción de la AE puede ser complicada por fuga anastomótica, fístula traqueo-esofágica recurrente, sepsis y estenosis. Las complicaciones tempranas se deben a errores técnicos, brecha larga entre los extremos del esófago, ten-sión en la anastomosis, e isquemia. La estenosis tardía puede ser el resultado de la enfermedad de reflujo gastroesofágico. Los pacientes se quejan a menudo de dificultad para comer y que la comida se pegue en el esófago. La anastomosis de la estenosis suele responder al tratamiento que incluye el con-trol de reflujo ácido y dilataciones. La dilatación con balón bajo el control directo endoscópico o fluoroscópico parece segura. Algunos bebés con el tiempo pueden requerir rem-plazo del esófago con el estómago, el colon o yeyuno como un conducto. Además de las complicaciones relacionadas con la cirugía esofágica, los niños pueden sufrir compromi-so de la vía aérea debido a traqueomalacia.
Estenosis Esofágica CongénitaPresentación y diagnóstico
Embriológicamente, la estenosis esofágica congénita está relacionada con la AE, y generalmente se presenta después del nacimiento con dificultad para tragar sólidos y comi-da atrancada en el esófago.5 Los estudios de contraste de-muestran un segmento corto y estrecho en el tercio medio o distal del esófago. La tomografía computarizada (TC) suele demostrar anillos cartilaginosos ectópicos. El diagnóstico diferencial incluye estenosis esofágica secundaria a cirugía previa, esofagitis resultante de enfermedad por reflujo gas-troesofágico y esofagitis eosinofílica.