atmospheric concentrations and chemistry of alkyllead compounds and environmental alkylation of lead
TRANSCRIPT

Environ. Sci. Technoi. 1987, 21, 260-266
Monarca, S.; Meier, J. R.; Bull, R. J. Water Res. 1983, 17,
Meier, J. R.; Ringhand, H. P.; Coleman, W. E.; Munch, J. W.; Streicher, R. P.; Kaylor, W. H.; Schenck, K. M. Mutat. Res. 1985, 157, 111-122. Ulitzur, S.; Weiser, I.; Yannai, S. Mutat. Res. 1980, 74, 113-124. Weiser, I.; Ulitzur, S.; Yannai, S. Mutat. Res. 1981, 91, 443-450. Mantura, R. F. L.; Riley, J. Anal. Chim. Acta 1975, 76, 97-100. De Serra, M. D.; Schnitzer, M. Can. J. Soil Sei. 1972,52, 365-374. Barton, D. H. R.; Schnitzer, M. Nature (London) 1963,198, 217-218. American Public Health Association Standard Methods for the Examination of Water and Wastewater, 15th ed.; American Public Health Association: New York, 1980.
1015-1026. (47) Masschelein, W. J. In Chlorine Dioxide; Ann Arbor Science:
(48) Wheeler, G. L.; Lott, P. F.; Yau, F. W. Microchem. J . 1978,
(49) Maron, D. M.; Ames, B. N. Mutat. Res. 1983,113,173-215. (50) Marnett, L. J.; Hurd, H. K.; Hollstein, M. C.; Levin, D. E.;
Esterbauer, E.; Ames, B. N. Mutat. Res. 1985,148,25-34. (51) Visser, S. A. Freshwater Biol. 1984, 14, 79-87. (52) Rav-Acha, Ch.; Blits, R., Environmental Health Laboratory,
Hebrew University, Jerusalem, unpublished results, 1985. (53) Reichert, J. K. Arch. Hyg. Bakteriol. 1968, 152, 265-276. (54) Trevors, J. T.; Bursuraba, J. Bull. Environ. Contam.
Ann Arbor, MI, 1979; p 129.
23, 160-164.
Toxicol. 1980, 25, 672-675.
Received for review February 4,1986. Revised manuscript re- ceived September 9,1986. Accepted October 21,1986. M.B.-A. was partially supported by a grant from the Israel Interior Ministry.
Atmospheric Concentrations and Chemistry of Alkyllead Compounds and Environmental Alkylation of Lead
C. Nicholas Hewltt
Department of Environmental Science, University of Lancaster, Lancaster LA 1 4YQ, U.K.
Roy M. Harrison"
Department of Chemistry, University of Essex, Coichester C04 3SQ, U.K.
The atmospheric chemistry of alkyllead compounds was investigated by determining the ratio of alkyllead to total lead in a variety of different air masses. Maritime air was found to contain significantly more alkyllead relative to total lead than continental or urban air. The presence of gas-phase trialkyllead in the rural atmosphere was con- firmed, with trialkyllead becoming progressively more im- portant relative to other lead species with increasing dis- tance from urban source areas. On the basis of estimates of the atmospheric lifethnes of the various lead species, it is concluded that these enhanced alkyllead ratios are explicable in terms of the atmospheric chemistry of the various species, with pollutant tetraalkyllead decomposing in the atmosphere to the relatively stable trialkyllead derivatives. It is therefore probably not necessary to in- voke the hypothesis of the natural alkylation of lead to explain these enhanced ratios. Notwithstanding this, ev- idence is also presented from experiments using intertidal sediments both with and without the addition of a labeled lead tracer that indicates that lead(I1) nitrate can be in- efficiently alkylated by a sediment system.
Introduction Evidence for the environmental formation of alkyllead
compounds from inorganic lead is, a t the present time, available from several sources, but despite extensive re- search is mainly circumstantial in nature. Experiments with environmental media (1, 2) and experiments with chemical systems (3) and environmental monitoring (4) all provide evidence that has been used to support the hy- pothesis that the alkylation of lead takes place in the environment.
We present here the results of a series of experiments and atmospheric monitoring that indicate that the at- mospheric chemistry of alkyllead is considerably more complex than was previously believed, with species other than tetraalkyllead (TAL) being present in the gas phase in both urban and rural air. Smog-chamber studies in-
dicate that these other alkyllead compounds, the ionic trialkyl and dialkyl species (TriAL and DiAL), are formed from TAL by reaction with hydroxyl (HO). They are more stable in the atmosphere than TAL, which casts doubts on the validity of the use of alkyllead-in-air data as an indicator of environmental alkylation. Notwithstanding this, we have also obtained experimental evidence using intertidal sediments both with and without the addition of a labeled lead tracer that indicates that lead(I1) nitrate can be inefficiently alkylated by a sediment system.
Prior to this study little was known of the atmospheric chemistry of alkyllead apart from the concentrations of TAL in the urban and rural atmosphere (5) and the rates of the main TAL decomposition reactions (6). No infor- mation was available concerning the nature or atmospheric concentrations of the products of these reactions, and it was assumed that these intermediate compounds (formed in the inevitable decay of TAL to inorganic Pb) were not significant in the atmosphere compared with TAL. Con- sequently, it was anticipated that measuring the ratios of alkyllead to total P b in different air masses, both in con- tinental air close to urban areas and in maritime air remote from anthropogenic lead sources, would be a suitable method for determining whether or not the natural al- kylation of lead takes place in the environment (4). This method assumes that both the gas-phase organic lead and the inorganic lead aerosol have similar atmospheric life- times, of the order of a few days. However, we show here that this is not the case.
The concentration of particulate lead found in the at mospheric aerosol in rural and maritime regions is now well established, with several recent papers containing extensive data sets of such measurements (7-10). Unfortunately, however, the same cannot be said of the organic lead species for which only a very few data are available for rural air (11-15), and none, to date, have been reported for true maritime air. We present here data on organic lead concentrations obtained at several rural sites in N.W.
260 Environ. Sci. Technoi., Vol. 21, No. 3, 1987 0013-936X/87/0921-0260$01.50/0 0 1987 American Chemical Society

England and N.W. Scotland at varying distances from motor-trafficked roads. By relating concentrations to wind direction sectors and backward air mass trajectory analysis, we believe that we are able to distinguish between “rural” and “maritime” air, that is, between air which has recently passed over land and that which has arrived at the sam- pling site after a long fetch over the sea. Rigorously clean sample collection and analytical techniques were used to minimize the possibilities of sample contamination. Nevertheless, our data do not, of course, represent the concentrations of organic lead to be found in the cleanest oceanic air such as might be expected in the mid South Pacific or other truly remote areas.
The data obtained at these sites are interpreted in light of experiments we have carried out on the photochemical decomposition and reaction rates of the alkyllead species. We also present the results obtained from respiration ex- periments using intertidal sediments, both with and without the addition of a labeled lead tracer.
Experimental Section Gas-Phase Alkyllead and Particulate Inorganic
h a d . Air was sampled at two sites near Lancaster in N.W. England, a t the Hazlerigg field station of the University of Lancaster in an elevated, rural, very well ventilated position with open aspects in all directions surrounded by pastures (grid reference SD 492573) and a t Cockerham (grid reference SD 451530). This latter site is 1 km west of the nearest road on a lightly trafficked agricultural lane, surrounded by meadows, and is -200 m from the River Lune estuary shore. Two sampling sites were used on the island of Harris in the Outer Hebrides, N.W. Scotland. One was at Husinish (grid reference NA 981118) on the extreme west coast of the island and was as remote as possible from vehicles. The sampling equipment was positioned -400 m upwind (west) of the nearest track and -15 km west of the nearest road carrying more than a few cars per day. The equipment was elevated 20 m above sea level and was positioned such that air arriving from the western half of the compass would do so without receiving any fresh anthropogenic inputs of lead after a long sea passage. Two locations were also used at Luskintyre (grid reference NG 087976) about 500 m from a minor road.
Samples were collected and analyzed with equipment and methods already described (12, 16). Particulate P b data were obtained by filtration, acid extraction, and at- omic absorption spectroscopy (AAS). Total gas-phase alkyllead (TAL plus any gas-phase ionic species present) was obtained by collection in iodine monochloride solution, a selective extraction procedure, and AAS while TAL was specifically determined by adsorption on a porous polymer followed by gas chromatography (GC)-AAS. Some sam- ples were also collected in IC1 solution following filtration of the air through 0.25 g of iron(I1) sulfate (which removes -50% of any gas-phase ionic TriAL salt present). The differences in concentrations obtained by simultaneous sampling with and without this filter could therefore be used to infer the relative proportions of TAL and ionic TriAL salt in the atmosphere.
Entirely separate particulate samples were simultane- ously obtained during the 1983 Harris sampling period by another worker using Fluoropore filters in Gelman holders (17). Particulate sodium analyses were carried out on both seta of filters by the same method (flame photometry with a Corning 400 instrument), and good correlation between the air concentrations was found. This provides confidence in the airflow rate measurements and hence in the derived air sample volumes. Blank filter papers, IC1 solutions, and adsorption tubes were transported, handled, stored, and
analyzed in the same way as the samples. No significant contamination of any of the blanks was observed, with minimal, acceptable, blank values being found. These were deducted from the sample concentrations in the normal manner.
Laboratory Experiments Using Sediments. Inter- tidal sediments were taken with plastic core tubes from the River Lune estuary, N.W. England (grid reference SD 456561). Composites of -9 kg were immediately placed in Pyrex containers and subsequently kept in the dark at room temperature. Charcoal-filtered air was passed through the head space above the sediment, filtered through a 0.45-pm Millipore membrane, and then bubbled through 0.1 M IC1 solution or through an adsorption tube packed with a porous polymer. Alkyllead evolved by the sediments was thus collected and either analyzed by di- thizone extraction-AAS (16) (giving a measure of the total alkyllead present) or by thermal desorption-GC-AAS (12) (which allows determination of the five TAL species in- dividually). Four sediment vessels and a fifth “blank” collector were used in parallel, utilizing the same filtered air supply.
Experiments were also carried out in which labeled lead(I1) nitrate solution containing 210Pb was added to sediments in the apparatus described above. A Millipore 0.45-pm filter in a stainless steel holder with a Teflon gasket was placed between each container and its collection bubbler in order to prevent the transfer of particulate or liquid 210Pb(N03)2 to the IC1 solution. This was an ad- ditional safeguard as the selective extraction procedure would, in any case, prevent interference from nonalkylated labeled lead. An activity of 9.678 MBq of 210Pb(N03)2 in 100 cm3 of deionized water was added to each 9-kg mass of sediment and well mixed. Gas-phase organic lead was collected and extracted as above and a 20-pL aliquot of the final acid solution used for lead analysis by flameless AAS. The zloPb activity of the bulk of the acid solution was then determined by y spectroscopy [using an Inter- technique IN90 with NaI(T1) detector] and the amount of zloPb present determined by comparison with standards prepared in the same medium.
A blank experiment was carried out by counting the y activity of the organic lead evolved from a set of sediments to which no labeled lead had been added. These sediments were quite productive, three out of four releasing alkyllead, but no 210Pb activity was detected from any.
Results and Discussion Rura l Air Sampling (Easterly Air Masses). Si-
multaneous sampling was carried out a t Hazlerigg and Cockerham in April 1984 during a 7-day period of well- established easterly (continental) airflow over the U.K. The results obtained are summarized in Table IA and shown in Figure 1. Mean particulate Pb concentrations of 115 and 101 ng of Pb m-3 were found at Hazlerigg and Cockerham (with ranges of 57-213 and 52-207 ng of Pb m-3, respectively), in close accord with values found pre- viously at these sites (18). Both sites had very similar mean Pb concentrations, indicating that local sources of particulate Pb were unimportant. This was also the case for the mean total organic lead and TAL concentrations, these being 2.4 and 3.3 ng of Pb m-3 for total organic lead (with ranges of 1.1-5.3 ng of Pb m-3 at Hazlerigg and 1.1-6.2 ng of Pb m-3 at Cockerham) and 0.7 and 0.8 ng of Pb mb3 for TAL (with ranges of 0.4-1.5 ng of Pb m-3 at Hazlerigg and 0.3-1.8 ng of Pb m-3 at Cockerham). The mean ratios of organic Pb/total Pb found at the two sites were also in close agreement, being 2.7 and 3.5% for total organic Pb/total Pb and 0.6 and 0.9% for TAL/total Pb
Environ. Sci. Technol., Vol. 21, No. 3, 1987 281
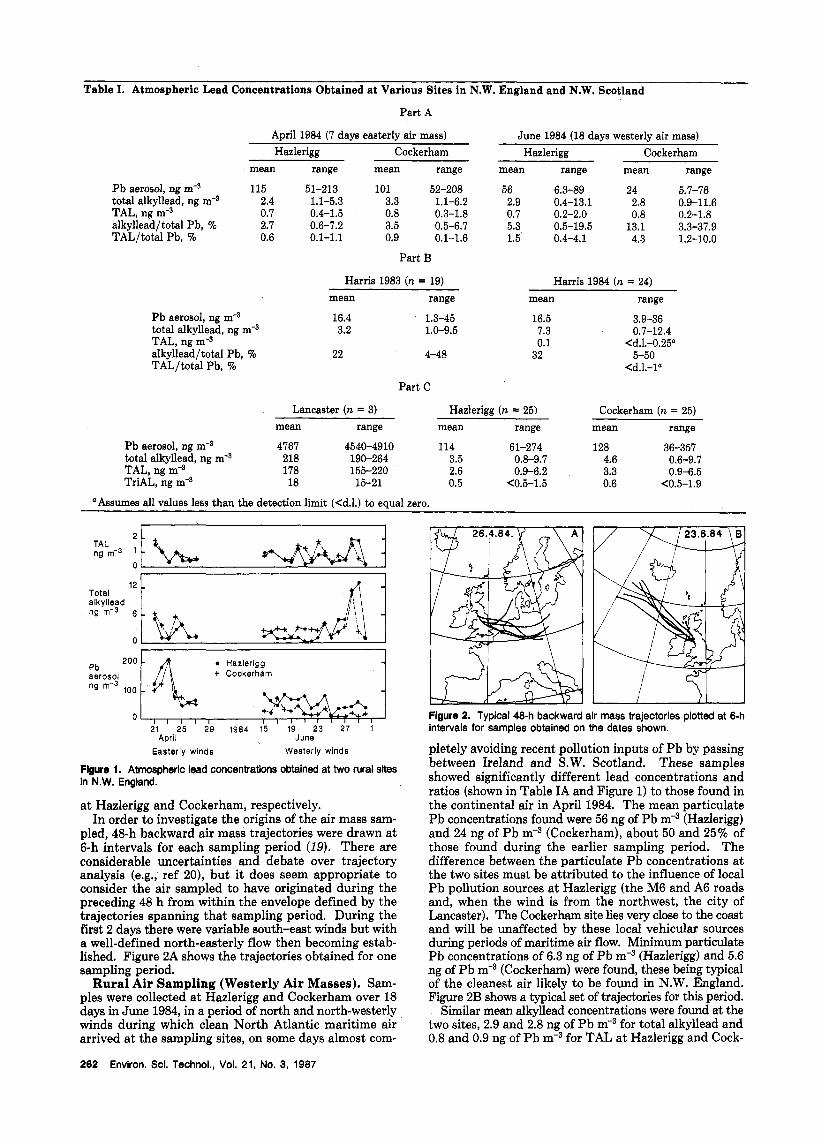
Table I. Atmospheric Lead Concentrations Obtained at Various Sites in N.W. England and N.W. Scotland
Part A
April 1984 (7 days easterly air mass) June 1984 (18 days westerly air mass) Hazlerigg Cockerham Hazlerigg Cockerham
mean range mean range mean range mean range
P b aerosol, ng m-3 115 51-213 101 52-208 56 6.3-89 24 5.7-76 total alkyllead, ng m-3 2.4 1.1-5.3 3.3 1.1-6.2 2.9 0.4-13.1 2.8 0.9-11.6 TAL, ng m-3 0.7 0.4-1.5 0.8 0.3-1.8 0.7 0.2-2.0 0.8 0.2-1.8 alkyllead/total Pb, % 2.7 0.6-7.2 3.5 0.5-6.7 5.3 0.5-19.5 13.1 3.3-37.9 TAL/total Pb, % 0.6 0.1-1.1 0.9 0.1-1.6 1.5 0.4-4.1 4.3 1.2-10.0
Part B
Harris 1983 (n = 19) Harris 1984 (n = 24) mean range mean range
Pb aerosol, ng m-3 16.4 1.3-45 16.5 3.9-36 total alkyllead, ng 3.2 1.0-9.5 7.3 0.7-12.4 TAL, ng m-s 0.1 <d.l.-0.25" alkyllead/total Pb, % 22 4-48 32 5-50 TAL/total Pb, % <d.l.-la
Part C
Lancaster (n = 3) Hazlerigg (n = 25) Cockerham (n = 25) mean range mean range mean range
Pb aerosol, ng rnb3 4767 4540-4910 114 61-274 128 36-357 total alkyllead, ng m-3 218 190-264 3.5 0.8-9.7 4.6 0.6-9.7 TAL, ng mq3 178 155-220 2.6 0.9-6.2 3.3 0.9-6.5 TriAL, ng m-3 18 15-21 0.5 <0.5-1.5 0.6 <0.5-1.9
"Assumes all values less than the detection limit (Cd.1.) to equal zero.
TAL ng m-3
0 -
Total 1
Hazlerigg + Cockerham
Pb aerosol ng m-3 ,oo -
0 1 1 1 1 I I O I I I I I
21 25 29 1984 15 19 23 27 1 April June
Flgure 1. Atmospheric lead concentrations obtained at two rural sltes In N.W. England.
a t Hazlerigg and Cockerham, respectively. In order to investigate the origins of the air mass sam-
pled, 48-h backward air mass trajectories were drawn at 6-h intervals for each sampling period (19). There are considerable uncertainties and debate over trajectory analysis (e.g., ref 20), but it does seem appropriate to consider the air sampled to have originated during the preceding 48 h from within the envelope defined by the trajectories spanning that sampling period. During the f i s t 2 days there were variable south-east winds but with a well-defined north-easterly flow then becoming estab- lished. Figure 2A shows the trajectories obtained for one sampling period.
Rural Air Sampling (Westerly Air Masses). Sam- ples were collected at Hazlerigg and Cockerham over 18 days in June 1984, in a period of north and north-westerly winds during which clean North Atlantic maritime air arrived at the sampling sites, on some days almost com-
262 Environ. Sci. Technol., Voi. 21, No. 3, 1987
Easterly winds Westerly winds
Flgure 2. Typical 48-h backward air mass trajectories plotted at 6-h intervals for samples obtained on the dates shown.
pletely avoiding recent pollution inputs of Pb by passing between Ireland and S.W. Scotland. These samples showed significantly different lead concentrations and ratios (shown in Table IA and Figure 1) to those found in the continental air in April 1984. The mean particulate Pb concentrations found were 56 ng of Pb m-3 (Hazlerigg) and 24 ng of Pb m-3 (Cockerham), about 50 and 25% of those found during the earlier sampling period. The difference between the particulate Pb concentrations at the two sites must be attributed to the influence of local Pb pollution sources a t Hazlerigg (the M6 and A6 roads and, when the wind is from the northwest, the city of Lancaster). The Cockerham site lies very close to the coast and will be unaffected by these local vehicular sources during periods of maritime air flow. Minimum particulate Pb concentrations of 6.3 ng of Pb mv3 (Hazlerigg) and 5.6 ng of Pb m-3 (Cockerham) were found, these being typical of the cleanest air likely to be found in N.W. England. Figure 2B shows a typical set of trajectories for this period.
Similar mean alkyllead concentrations were found at the two sites, 2.9 and 2.8 ng of Pb m-3 for total alkyllead and 0.8 and 0.9 ng of P b m-3 for TAL at Hazlerigg and Cock-
TAL ng Pb rn?
0
40
Pb aerosol ng Pb rn-320
0
h . Luskintyre
- + Husinish /A I‘ 1, -/‘\y 1
,+-- ’ +--+--+f
1 1 1 1 1 1 1 1 1 I l l I 1 1 1 1 1 I I I I
Flgure 3. Atmospheric lead concentratlons obtained at two rural sites in the Outer Hebrides. N.W. Scotland.
erham, respectively. However, as the particulate Pb con- centrations differed, so did the organic Pb/total Pb ratios. Total gas-phase alkyllead represented on average 5.3% of the total Pb at Hazlerigg but 13.1% at Cockerham, and the mean TAL/total P b ratios were 1.5 and 4.3%, re- spectively. These ratios are much higher than those found in the continental (easterly) air mass.
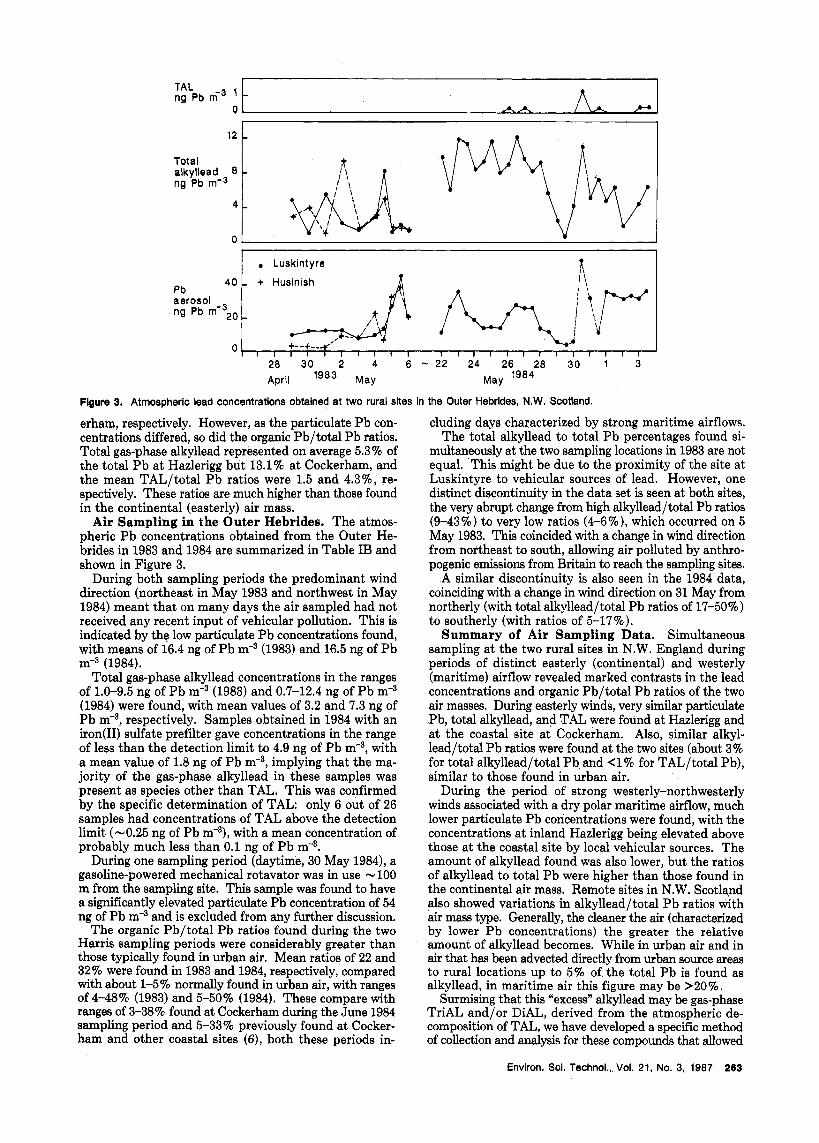
Air Sampling in the Outer Hebrides. The atmos- pheric P b concentrations obtained from the Outer He- brides in 1983 and 1984 are summarized in Table IB and shown in Figure 3.
During both sampling periods the predominant wind direction (northeast in May 1983 and northwest in May 1984) meant that on many days the air sampled had not received any recent input of vehicular pollution. This is indicated by the low particulate Pb concentrations found, with means of 16.4 ng of Pb m-3 (1983) and 16.5 ng of Pb m-3 (1984).
Total gas-phase alkyllead concentrations in the ranges of 1.0-9.5 ng of Pb m-3 (1983) and 0.7-12.4 ng of Pb m-3 (1984) were found, with mean values of 3.2 and 7.3 ng of P b m-3, respectively. Samples obtained in 1984 with an iron(I1) sulfate prefilter gave concentrations in the range of less than the detection limit to 4.9 ng of Pb m-3, with a mean value of 1.8 ng of Pb m-3, implying that the ma- jority of the gas-phase alkyllead in these samples was present as species other than TAL. This was confirmed by the specific determination of TAL: only 6 out of 26 samples had concentrations of TAL above the detection limit (-0.25 ng of Pb m-3), with a mean concentration of probably much less than 0.1 ng of P b m-3.
During one sampling period (daytime, 30 May 1984), a gasoline-powered mechanical rotavator was in use - 100 m from the sampling site. This sample was found to have a significantly elevated particulate Pb concentration of 54 ng of Pb rn9 and is excluded from any further discussion.
The organic Pb/total P b ratios found during the two Harris sampling periods were considerably greater than those typically found in urban air. Mean ratios of 22 and 32% were found in 1983 and 1984, respectively, compared with about 1-5% normally found in urban air, with ranges of 4-48% (1983) and 5-50% (1984). These compare with ranges of 3-38% found at Cockerham during the June 1984 sampling period and 5-33% previously found at Cocker- ham and other coastal sites (61, both these periods in-
cluding days characterized by strong maritime airflows. The total alkyllead to total Pb percentages found si-
multaneously at the two sampling locations in 1983 are not equal. This might be due to the proximity of the site at Luskintyre to vehicular sources of lead. However, one distinct discontinuity in the data set is seen at both sites, the very abrupt change from high alkyllead/total Pb ratios (9-43%) to very low ratios (4-6%), which occurred on 5 May 1983. This coincided with a change in wind direction from northeast to south, allowing air polluted by anthro- pogenic emissions from Britain to reach the sampling sites.
A similar discontinuity is also seen in the 1984 data, coinciding with a change in wind direction on 31 May from northerly (with total alkyllead/total Pb ratios of 17-5070) to southerly (with ratios of 5-17%).
Summary of Air Sampling Data. Simultaneous sampling at the two rural sites in N.W. England during periods of distinct easterly (continental) and westerly (maritime) airflow revealed marked contrasts in the lead concentrations and organic Pb/total Pb ratios of the two air masses. During easterly winds, very similar particulate Pb, total alkyllead, and TAL were found at Hazlerigg and at the coastal site a t Cockerham. Also, similar alkyl- lead/total Pb ratios were found at the two sites (about 3% for total alkyllead/total P b and <1% for TAL/total Pb), similar to those found in urban air.
During the period of strong westerly-northwesterly winds associated with a dry polar maritime airflow, much lower particulate Pb concentrations were found, with the concentrations at inland Hazlerigg being elevated above those at the coastal site by local vehicular sources. The amount of alkyllead found was also lower, but the ratios of alkyllead to total Pb were higher than those found in the continental air mass. Remote sites in N.W. Scotland also showed variations in alkyllead/total Pb ratios with air mass type. Generally, the cleaner the air (characterized by lower P b concentrations) the greater the relative amount of alkyllead becomes. While in urban air and in air that has been advected directly from urban source areas to rural locations up to 5% of the total Pb is found as alkyllead, in maritime air this figure may be >20%.
Surmising that this “excess” alkyllead may be gas-phase TriAL and/or DiAL, derived from the atmospheric de- composition of TAL, we have developed a specific method of collection and analysis for these compounds that allowed
Environ. Sci. Technol.,. Vol. 21, No. 3, 1987 263

us to confirm, for the first time, that gas-phase tri- methyllead, at least, is present in the atmosphere (21). Air sampling using this method was carried out at an urban curb site at Lancaster City Hall and at the two rural sites a t Hazlerigg and Cockerham. The results obtained are shown in Table IC. Gas-phase ionic TriML represents, in these samples, between 8 p d 14% of the total alkyllead. It is thus a significant component of the alkyllead content of the atmosphere, which is overlooked if a TAL-specific analytical method only is used.
Formation of Gas-Phase Ionic Alkyllead. In order to confirm that the gas-phase TriML compounds observed in the atmosphere are due to the atmospheric decompo- sition of TAL, we have carried out a series of smog-cham- ber experiments. This has allowed the identification of the intermediate lead compounds formed by the reactions of TAL with the hydroxyl radical in the gas-phase. Es- timates have been made of the rates of reaction of these compounds with HO at ambient temperatures and pres- sure and therefore of their atmospheric lifetimes (22).
After reaction with hydroxyl, it was found that tetra- methyllead (TML) and tetraethyllead (TEL) produced a suite of lead products that were present in both the gas and the aerosol phases. Both TriAL and DiAL products were found, together with inorganic Pb. The rate constants measured for the reaction of HO with the TriAL com- pounds were (2.2 f 0.2) X lo5 and (8.1 f 1.0) X lo5 ppm-l h-' for TriML and TriEL, respectively. These values are approximately 3 times less than the corresponding TAL- HO rate constants. If reaction with HO is the most im- portant decomposition route for the TriAL compounds in the atmosphere (as in the case for TML and TEL and for a very wide range of other compounds), then these data suggest that the atmospheric lifetimes of TriML and TriEL will be about 3 times greater than those of TML and TEL. By use of a global mean hydroxyl concentration of 1 X lo6 HO (23), mean atmospheric half-lives of about 5 and 1.5 days are indicated for TriML and TriEL, respectively. Trimethyllead in particular is therefore quite persistent in the troposphere and may be advected to remote areas distant from urban sources, forming mean- while in the atmosphere from TML.
These reaction rates may adequately explain the en- hancement of alkyllead to total Pb ratios observed in the maritime atmosphere. Trimethyllead, at least, may persist in the atmosphere longer than the Pb aerosol, assuming a mean lifetime for the latter of 1-3 days in the marine boundary layer (24-26). Thus, anthropogenic TAL will degrade to the relatively stable TriAL and DiAL while the Pb aerosol is depleted by deposition processes, so pro- gressively increasing the amount of alkyllead relative to total P b over time. It may therefore be unnecessary to invoke the natural alkylation of lead as a mechanism to explain the observed variations in lead concentrations and ratios in the atmosphere.
Experiments with Labeled and Unlabeled Sedi- ments. Forty sediment samples were investigated for their ability to release gaseous alkyllead in the laboratory over periods of 5-120 days. Spasmodic and variable alkyllead release was found from approximately half of the samples studied with a maximum release rate of 0.085 ng (Pb) kg-' h-l. The limit of detection was 2 ng (Pb) with IC1 or 0.2 ng (Pb) with GC-AAS. There was no apparent correlation between the alkyllead release rate and the total Pb con- tents of the sediments (100-198 mg kg-l with a mean of 137 mg kg-'1.
One set of sediments was modified by autoclaving. These sediments were initially active with three out of four
264 Envlron. Scl. Technol., Vol. 21, No. 3, 1987
producing TML (identified by GC-AAS). After 20 days the sediments were sterilized by autoclaving to prevent any further biological activity taking place, and the respiration experiment was then continued under sterile conditions. No alkyllead was evolved during the subsequent 20 days. This result might be taken to imply that the initial release of alkyllead was due to biological activity since alkyllead release stopped when the sediment was sterilized. How- ever, the limitations of autoclaving as a technique for distinguishing between biological and chemical alkylation mechanisms are severe (27), and the results of this type of experiment must be treated with caution.
Only TML was found to be released by the sediments, except in three samples when TEL was also initially re- leased. In these cases TEL production ceased after 10 days, but TML release continued. If the alkyllead released by these samples was due to alkyllead already present in the sediments as a result of pollution (by road run-off, by gasoline spillage, or, unlikely, by airborne contamination), it might be expected that the much less volatile TEL would persist longer in the sediments than TML and so be re- leased more slowly over a longer period of time. This is the reverse of the observed pattern. However with the presently achievable detection limits for the various al- kyllead species in sediments, we are not able to exclude the hypothesis that the production of alkyllead by the sediments was due solely to the release of alkyllead already present from the pollution sources. Therefore in order to indicate whether a true alkylation process was occurring during the period of the experiment, an experiment was devised with a radioactive tracer as the methyl acceptor.
For each sediment sample the total alkyllead and the 210Pb-labeled alkyllead released in two 14-day periods were determined. After 28 days a fresh set of four samples were labeled with 210Pb, and the experiment was repeated. The results obtained are shown in Table 11.
The release rates of total alkyllead were consistent with those previously found for unmodified sediments, with conversion efficiencies (defined as the mass of alkyllead as Pb emitted in 14 days divided by the total Pb content of the sediment) in the range (0.1-0.5) X These efficiencies must be considered to be minimum estimates since the rigorous chemical extraction procedure employed may include a fraction of the element that is unavailable far transformation (e.g., Pb held in the silicate lattice of the sediment). Z1oPb-labeled alkyllead was not detected from any sample during the first 14-day period. This may imply that inorganic Pb salts are not immediately available for alkylation on addition to a sediment system. However, during the second 14-day period 210Pb-labeled alkyllead was detected from every sample. The conversion effi- ciencies found for the zloPb (defined as the activity of 210Pb-labeled alkyllead emitted in 14 days divided by the total 210Pb activity of the sediment) were in the range (0.9-2.6) x similar to those found for total Pb. The process involving labeled Pb seems to be slightly more efficient, which may indicate that added lead(I1) nitrate in aqueous solution is, after an initial period of physico- chemical change, more readily alkylated than complexes or adsorbed P b already present in the sediment.
The addition of 10 MBq of 210Pb was accompanied by the addition of only 7.2 pg of stable Pb2+; this compares with about 0.9 g of inorganic Pb already present in the 9 kg of sediment. The labeled lead did not therefore sig- nificantly alter the total Pb content of the sediment. However, it may not have been acting as a true tracer for all the Pb species in the sediment but only for those with which the Pb2+ ions were able to exchange on the time
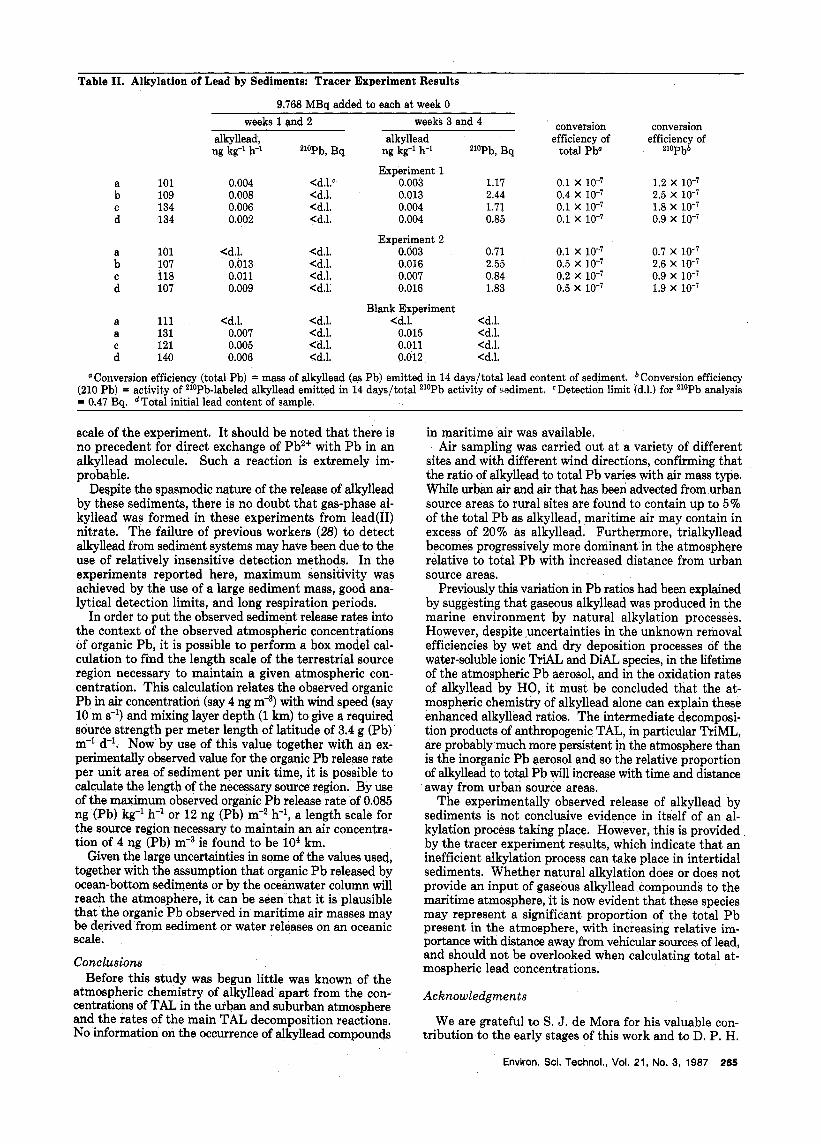
Table 11. Alkylation of Lead by Sediments: Tracer Experiment Results
9.768 MBa added to each at week 0
conversion conversion alkyllead, alkyllead efficiency of efficiency of
weeks 1 and 2 weeks 3 and 4
ng kg-l h-' 210Pb, Bq ng kg-' h-' zlOPb, Bq total Pb" 2lOpbb
Experiment 1 0.1 x 10-7 1.2 x 10-7
C 134 0.006 <d.l. 0.004 1.71 0.1 x 10-7 1.8 x 10-7 d 134 0.002 <d.l. 0.004 0.85 0.1 x 10-7 0.9 x 10-7
a 101 <d.l. <d.l. 0.003 0.71 0.1 x 10-7 0.7 x 10-7 b 107 0.013 <d.l. 0.016 2.55 0.5 x 10-7 2.6 x 10-7 C 118 0.011 <d.l. 0.007 0.84 0.2 x 10-7 0.9 x 10-7 d 107 0.009 <d.l. 0.016 1.83 0.5 x 10-7 1.9 x 10-7
a 101 0.004 <d.l.' 0.003 1.17 b 109 0.008 <d.l. 0.013 2.44 0.4 X lo-' 2.5 X
Experiment 2
Blank Experiment a 111 <d.l. <d.l. <d.l. <d.l. a 131 0.007 <d.l. 0.015 <d.l. C 121 0.005 <d.l. 0.011 <del. d 140 0.006 <d.l. 0.012 <d.l.
"Conversion efficiency (total Pb) = mass of alkyllead (as Pb) emitted in 14 days/total lead content of sediment. bConversion efficiency (210 Pb) = activity of 210Pb-labeled alkyllead emitted in 14 days/total zloPb activity of sediment. CDetection limit (d.1.) for 210Pb analysis = 0.47 Bq. dTotal initial lead content of sample.
scale of the experiment. It should be noted that there is no precedent for direct exchange of Pb2+ with P b in an alkyllead molecule. Such a reaction is extremely im- probable.
Despite the spasmodic nature of the release of alkyllead by these sediments, there is no doubt that gas-phase al- kyllead was formed in these experiments from lead(I1) nitrate. The failure of previous workers (28) to detect alkyllead from sediment systems may have been due to the use of relatively insensitive detection methods. In the experiments reported here, maximum sensitivity was achieved by the use of a large sediment mass, good ana- lytical detection limits, and long respiration periods.
In order to put the observed sediment release rates into the context of the observed atmospheric concentrations of organic Pb, it is possible to perform a box model cal- culation to find the length scale of the terrestrial source region necessary to maintain a given atmospheric con- centration. This calculation relates the observed organic Pb in air concentration (say 4 ng m-3) with wind speed (say 10 m s-l) and mixing layer depth (1 km) to give a required source strength per meter length of latitude of 3.4 g (Pb) m-l d-l. Now by use of this value together with an ex- perimentally observed value for the organic Pb release rate per unit area of sediment per unit time, it is possible to calculate the length of the necessary source region. By use of the maximum observed organic Pb release rate of 0.085 ng (Pb) kg-l h-l or 12 ng (Pb) m-2 h-l, a length scale for the source region necessary to maintain an air concentra- tion of 4 ng (Pb) m-3 is found to be lo4 km.
Given the large uncertainties in some of the values used, together with the assumption that organic Pb released by ocean-bottom sediments or by the oceanwater column will reach the atmosphere, it can be seen that it is plausible that the organic Pb observed in maritime air masses may be derived from sediment or water releases on an oceanic scale.
Conclusions Before this study was begun little was known of the
atmospheric chemistry of alkyllead apart from the con- centrations of TAL in the urban and suburban atmosphere and the rates of the main TAL decomposition reactions. No information on the occurrence of alkyllead compounds
in maritime air was available. Air sampling was carried out a t a variety of different
sites and with different wind directions, confirming that the ratio of alkyllead to total Pb varies with air mass type. While urban air and air that has been advected from urban source areas to rural sites are found to contain up to 5% of the total Pb as alkyllead, maritime air may contain in excess of 20% as alkyllead. Furthermore, trialkyllead becomes progressively more dominant in the atmosphere relative to total Pb with increased distance from urban source areas.
Previously this variation in Pb ratios had been explained by suggesting that gaseous alkyllead was produced in the marine environment by natural alkylation processes. However, despite uncertainties in the unknown removal efficiencies by wet and dry deposition processes of the water-soluble ionic TriAL and DiAL species, in the lifetime of the atmospheric Pb aerosol, and in the oxidation rates of alkyllead by HO, it must be concluded that the at- mospheric chemistry of alkyllead alone can explain these enhanced alkyllead ratios. The intermediate decomposi- tion products of anthropogenic TAL, in particular TriML, are probably much more persistent in the atmosphere than is the inorganic Pb aerosol and so the relative proportion of alkyllead to total Pb will increase with time and distance away from urban source areas.
The experimentally observed release of alkyllead by sediments is not conclusive evidence in itself of an al- kylation process taking place. However, this is provided by the tracer experiment results, which indicate that an inefficient alkylation process can take place in intertidal sediments. Whether natural alkylation does or does not provide an input of gaseous alkyllead compounds to the maritime atmosphere, it is now evident that these species may represent a significant proportion of the total Pb present in the atmosphere, with increasing relative im- portance with distance away from vehicular sources of lead, and should not be overlooked when calculating total at- mospheric lead concentrations.
Acknowledgments
We are grateful to S. J. de Mora for his valuable con- tribution to the early stages of this work and to D. P. H.
Environ. Sci. Technol., Vol. 21, No. 3, 1987 265

Envlron. Sci. Technol, 1987, 21, 266-272
Laxen for discussions. We also thank the staff of the Associated Octel Co., Ellesmere Port, U.K., for their dis- cussions, practical assistance, interest, and encouragement.
Registry No. Pb, 7439-92-1.
Literature Cited (1) Wong, P. T. S.; Chau, Y. K.; Luxon, P. L. Nature (London)
(2) Thompson, J. A. J.; Crerar, J. A. Mar. Pollut. Bull. 1980, 1975,253, 263-264.
11. 251-253. Rhode, S. F.; Weber, J. H. Environ. Technol. Lett. 1984,
Harrison, R. M.; Laxen, D. P. H. Nature (London) 1978,
De Jonghe, W. R. A.; Adams, F. C. Talanta 1982, 29,
Harrison, R. M.; Laxen, D. P. H. Environ. Sci. Technol.
Chester, R.; Sharples, E. J.; Murphy, K.; Saydam, A. C.; Sanders, G. S. Mar. Chem. 1983,13, 57-72. Duce, R. A.; Arimoto, R.; Ray, B. J.; Unni, C. K.; Harder, P. J. J. Geophys. Res., C: Oceans Atmos. 1983, 88(9),
Lannefors, H.; Hanson, H. C.; Granat, L. Atmos. Environ.
Patterson, C. C.; Settle, D.; Schank, B.; Burnett, M. In Marine Pollutant Transfer; Windom, H. L.; Duce, R. A., Ed.; Lexington Books: Lexington, MA, 1976. Harrison, R. M.; Radojevic, M.; Hewitt, C. N. Sci. Total Enuiron. 1985, 44, 235-244. Hewitt, C. N.; Harrison, R. M. Anal. Chim. Acta 1985,167,
Nielsen, T.; Egsgaard, H.; Larsen, E.; Schroll, G. Anal. Chim. Acta 1981, 124, 1-13.
5, 63-68.
275, 738-740.
1057-1067.
1978,12, 1384-1392.
5321-5342.
1983, 17, 87-101.
227-287.
(14) De Jonghe, W. R. A.; Chakraborti, D.; Adams, F. C. Environ.
(15) Jiang, S.; Ma, C.; Ge, J.; Li, M.; Adams, F. C.; Winchester,
(16) Birch, J.; Harrison, R. M.; Laxen, D. P. H. Sci. Total En-
(17) Sturges, W. Ph.D. Thesis, University of Lancaster, U.K.,
(18) Harrison, R. M.; Williams, C. R. Atmos. Environ. 1982, 16,
(19) Sykes, R. I.; Hatton, L. Atmos. Environ. 1976,10,925-934. (20) Smith, F. B.; Hunt, R. D. Atmos. Environ. 1978,12,461-477. (21) Hewitt, C. N.; Harrison, R. M. Proceedings o f the Inter-
national Conference on Heavy Metals in the Environment, Athens, 1985; CEP Consultants: Edinburgh, 1985; pp
(22) Hewitt, C. N.; Harrison, R. M. Environ. Sci. Technol. 1986,
(23) Hewitt, C. N.; Harrison, R. M. Atmos. Environ. 1985,19,
(24) Moore, H. E.; Poet, S. E.; Martell, E. A. Enuiron. Sci.
(25) Junge, C. E. Air Chemistry and Radioactivity; Academic:
(26) Hewitt, C. N. Ph.D. Thesis, University of Lancaster, U.K.,
(27) Jarvie, A. W. P.; Markall, R. N.; Potter, H. R. Nature
(28) Reisinger, K.; Stoeppler, M.; Nurnberg, H. W. Nature
Sci. Technol. 1981, 15, 1217-1222.
J. W. Atmos. Environ. 1984, 28, 2553-2556.
uiron. 1980, 14, 31-42.
1984.
2669-2681.
171-173.
20, 797-802.
545-554.
Technol. 1976, 10, 586-591.
New York, 1963.
1985.
(London) 1985,255, 217-218.
(London) 1981,291, 228-229.
Received for review April 29, 1986. Accepted October 21, 1986. Financial support was provided by the Natural Environment Research Council, U.K., with assistance from The Associated Octel Co., Ltd., U.K.
Oxidative Degradation of Organic Acid Conjugated with Sulfite Oxidation in Flue Gas Desulfurization: Products, Kinetics, and Mechanism7
Y. Joseph Lee and Gary T. Rochelle"
Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712
H Organic acid degradation conjugated with sulfite oxi- dation has been studied under flue gas desulfurization (FGD) conditions. The oxidative degradation constantklz is defined as the ratio of organic acid degradation rate and sulfite oxidation rate times the ratio of the concentration of dissolved S(1V) and organic acid. It is not significantly affected by pH or dissolved oxygen in the absence of manganese or iron. However, i t lz is increased by certain transition metals such as Fe, Co, and Ni and is decreased by Mn and halides. Lower dissolved S(IV) magnifies these effects. A free radical mechanism was proposed to describe the kinetics. Hydroxy and sulfonated carboxylic acids degrade approximately 3 times slower than saturated di- carboxylic acids, while maleic acid, an unsaturated di- carboxylic acid, degraded an order of magnitude faster. A wide spectrum of degradation products of adipic acid were found, including car<bon dioxide-the major product- smaller dicarboxylic acids, monocarboxylic acids, other carbonyl comr>ounds, and hydrocarbons.
Introduction Currently, limestone slurry scrubbing is the dominant
commercial technology for flue gas desulfurization (FGD).
Presented at 189th National Meeting of the American Chemical Society, Miami Beach, March 1985.
The performance of limestone scrubbing is chemically limited by two pH extremes: (a) low pH near the gas/ liquid interface, which decreases the solubility and ab- sorption rate of SO,; (b) high pH near the liquid/solid interface, which decreases the solubility and dissolution rate of limestone (1). Organic acids that buffer between pH 3.0 and pH 5.5 enhance SOz removal efficiency and limestone utilization at concentrations of 5-10 mM (2).
Adipic acid was the first organic acid buffer additive successfully and generally applied to FGD processes up to commercial scale (3, 4). It has been replaced com- mercially by dibasic waste acid (DBA), a waste from adipic acid and cyclohexanone production, containing primarily adipic, glutaric, and succinic acids. DBA was found to be as effective as adipic acid (2, 3) . Other potential alter- natives include hydroxy carboxylic acids and sulfonated carboxylic acids (4). They are of interest because of re- duced volatility and potentially lower degradation rates.
In addition to the expected loss of organic acid additive by entrainment of solution in waste solids, loss by chemical degradation and coprecipitation is also observed (2). Chemical degradation, which is conjugated with sulfite oxidation (5)) is the most important mechanism of buffer loss under forced oxidation conditions (2, 3) .
Assuming that both oxidation and degradation are free radical reactions proceeding by a common radical, Rochelle
286 Environ. Scl. Technol., Vol. 21, No. 3, 1987 0013-936X/87/0921-0266$01.50/0 0 1987 American Chemical Soclety