asymmetric approaches towards ly290154 … · the work described in this thesis concerns the...
TRANSCRIPT

ASYMMETRIC APPROACHES TOWARDS LY290154
Antonio Garrido Montalban
A thesis submitted in partial fulfilment
of the requirements for the degree of
Doctor of Philosophy of the University of East Anglia
This copy of the thesis has been supplied on the conditions that anyone who consults it is understood to recognise that its copyright rests with the author and that no quotation from the thesis, nor any information derived therefrom, may be published without the author's prior written consent.
2
PREFACE
The research described in this thesis is, to the best of my knowledge, original
except where due reference is made, and has not previously been submitted for
any degree in this or any other university.
Antonio Garrido Montalban
Norwich, March 1995

3
ABSTRACT
The work described in this thesis concerns the development of asymmetric
approaches towards the synthesis of LY290154 35, a leukotriene antagonist.
The first chapter provides a general introduction to leukotrienes and leukotriene
antagonists, including racemic synthesis of 35 developed by Lilly.
In Chapter 2 a plausible synthetic route for the preparation of enantiomerically
enriched LY290154 from a corresponding optically active primary amine is
discussed. Chapter 3 describes the development and chapter 4 the synthesis of
the optically active amine 50b, which could in principle produce 35 in
enantiomerically enriched form.
In Chapter 5 the attempted construction of the key intermediate 48b, required to
achieve the asymmetric synthesis of LY290154 via the conversion of 3-
nitropyridinium salts into indoles is described. Unfortunately, the simple
unsubstituted pyridinium salt 153, essential for the formation of 48b, did not
undergo the reaction with imines derived from primary amines to yield the
corresponding indole derivatives.
Chapter 6 describes a new route for the preparation of 7-oxo-4,5,6,7-
tetrahydroindoles from reaction of primary amines with 7-oxo-4,5,6,7-
tetrahydrobenzofuran in a sealed tube at 150 °C. The aromatisation of 7-oxo-
4,5,6,7-tetrahydroindoles to the corresponding 7-hydroxyindoles could be achieved
via a novel selective halogenation/dehydrohalogenation sequence, which is
discussed in chapter 7.
In chapter 8 the results from attempted syntheses of the 7-oxotetrahydroindole
182, which would lead after aromatisation and alkylation to the required indole
derivative 48b from the amine 50b, are summarised. Unfortunately the amine 50b
could not be converted to 182 under the conditions used.
4
ABBREVIATIONS
Ac acetyl
anh anhydrous
aq aqueous
Ar aryl
BINAP 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
BDPP 2,4-bis(diphenylphosphino)pentane
bp boiling point
BPPM t-butyl-4-(diphenylphosphino)-2-
(diphenylphosphinomethy)-1-
pyrrolidinecarboxylate
br broad
t-BOC N-tert-butoxycarbonyl
Bu butyl
CI chemical ionisation
CHIRAPHOS 2,3-bis(diphenylphosphino)butane
conc concentrated
cycphos 1,2-bis(diphenylphosphino)-1-cyclohexylethane
Cys cysteine
d doublet (NMR)
d day(s)
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene
DDQ 2,3-dichloro-5,6-dicyano-1,4-benzoquinone
δ chemical shift (NMR)
DIBAL diisobutylaluminium hydride
DIOP 2,2-dimethyl-4,5-bis(diphenylphosphinomethyl)-
1,3-dioxolane

5
DIP-Cl diisopinocampheylchloroborane
DME 1,2-dimethoxyethane
DMF N,N-dimethylformamide
dmfdma N,N-dimethylformamide dimethyl acetal
DMSO dimethyl sulphoxide
DPPA diphenylphosphoryl azide
ee enantiomeric excess
e.g. for example
EI electron impact
eq equivalent(s)
Et ethyl
FTIR Fourier transform infrared
GGTP γ-glutamyl transpeptidase
Glu glutamic acid
Gly glycine
h hour(s)
5-HETE 5-hydroxy-6,8,11,14-eicosatetraenoic acid
5-HPET 5-hydroperoxy-6,8,11,14-eicosatetraenoic acid
Hz Hertz
IR infrared
J coupling constant (NMR)
LA Lewis acid
LDA lithium diisopropylamide
lit literature
LTA4 leukotriene A4
LTB4 leukotriene B4
LTC4 leukotriene C4
LTD4 leukotriene D4
LTE4 leukotriene E4
6
LTF4 leukotriene F4
m multiplet (NMR)
M molarity of solution
Me methyl
min minute(s)
mp melting point
Ms mesyl
MS mass spectrometry
NORPHOS 5,6-bis(diphenylphosphino)norbornene
NMR nuclear magnetic resonance
Ph phenyl
PPA polyphosphoric acid
ppm parts per million
PROPHOS 1,2-bis(diphenylphosphino)propane
PTC phase-transfer catalysis
q quartet (NMR)
quin quintet (NMR)
RAMP (R)-1-amino-2-methoxymethylpyrrolidine
Rf retention factor
rt room temperature
s singlet (NMR)
SAMP (S)-1-amino-2-methoxymethylpyrrolidine
SAR structure activity relationship
SRS slow reacting substance
t triplet (NMR)
TFAA trifluoroacetic anhydride
TFAE 2,2,2-trifluoro-(9-anthryl)ethanol
THF tetrahydrofuran
tlc thin layer chromatography

7
TMS tetramethylsilane
TMSCl trimethylsilyl chloride
trityl triphenylcarbenium
Ts tosyl
8
CONTENTS
1. Introduction....................................................................................................10
1.1 Slow Reacting Substance (SRS)...............................................................10
1.2 Leukotrienes..............................................................................................13
1.3 Leukotriene Antagonists............................................................................17
1.4 Development of LY290154....................................................................... 29
1.5 Racemic Synthesis of LY290154.............................................................. 33
2. Retrosynthetic Analysis of LY290154........................................................ 36
3. Preliminary Results .................................................................................... 39
4. Asymmetric Amine Synthesis...................................................................... 46
4.1 Results and Discussion............................................................................ 61
5. Synthesis of Indoles..................................................................................... 72
5.1 Bartoli Indole Synthesis............................................................................ 72
5.2 Conversion of 3-Nitropyridinium Salts into Indoles................................... 74
5.3 Results and Discussion............................................................................ 78
6. Synthesis of 4- and 7-Oxo-4,5,6,7-tetrahydroindoles................................ 85
6.1 Results and Discussion............................................................................ 94
7. Aromatisation.............................................................................................106
8. The final Battle towards LY290154.......................................................... 112

9
9. Conclusions............................................................................................... 124
10. Experimental.............................................................................................. 125
10.1 Solvents and Reagents........................................................................ 125
10.2 Purification and Characterisation Techniques...................................... 125
10.3 Experimental for Chapter 1, Section 1.5............................................... 126
10.4 Experimental for Chapter 3.................................................................. 132
10.5 Experimental for Chapter 4, Section 4.1............................................... 139
10.6 Experimental for Chapter 5, Section 5.3............................................... 152
10.7 Experimental for Chapter 6, Section 6.1............................................... 158
10.8 Experimental for Chapter 7.................................................................. 163
10.9 Experimental for Chapter 8.................................................................. 166
References........................................................................................................ 174
Appendix.......................................................................................................... 189
10
1. Introduction
1.1 Slow Reacting Substance (SRS)
In 1938 Feldberg and Kellaway introduced the term "slow reacting substance"
(SRS) for a material which was excreted from the lungs of guinea pigs after
treatment with cobra venom.1 Kellaway and Trethewie reported that SRS could
also be released by immunological challenge of sensitised lungs.2 The SRS
displayed smooth bronchial muscle contracting activity and subsequent studies
showed that it is an important mediator in asthma and other types of immediate
hypersensitivity reactions.3,4 Structural work on the SRS was initially severely
limited by the difficulty in obtaining sufficient quantities of pure substance.5 Murphy
et al., however, were able to biosynthesise relatively large amounts of SRS using
murine mastocytoma cells. Purification was achieved by high performance liquid
chromatography. The material obtained by this method produced a characteristic
contraction of guinea pig ileum which was reversed by FPL 55712.6 Experiments
with labelled precursors showed that arachidonic acid 1 and cysteine were
O
OH
1
incorporated into the products. Degradation of the SRS by desulfurisation on
Raney nickel afforded 5-hydroxyeicosanoic acid 2, which indicated that the
arachidonic acid derivative and cysteine were linked by a thioether bond. The
hydroxy group at C-5 in the fatty acid reinforced the hypothesis of a biogenetic

11
relationship between the arachidonic acid metabolites and the SRS. The positions
of the double bonds in the SRS were determined by ozonolysis and reduction of
the corresponding carbonyl derivatives. Isolation of 1-hexanol among the products
demonstrated that the C-14 double bond of arachidonic acid had been retained.
Murphy et al. also reported that arachidonic acid and related fatty acids containing
two cis double bonds at the C-5 and C-8 positions, separated by a methylene
group, are peroxygenated to give derivatives in which the C-5 double bond has
isomerised to C-6. Treatment of SRS with lipoxygenase resulted in isomerisation
of the C-14 double bond to form the 15-hydroperoxide of the conjugated tetraenoic
acid 3. The structural studies at this stage showed that SRS was a derivative of 5-
hydroxy-7,9,11,14-eicosatetraenoic acid with a cysteine-containing substituent
linked in a thioether-manner at C-6. That the cysteine is derivatised was suggested
by the failure to isolate alanine after desulfurisation. Additional studies involving
amino acid analyses of hydrolysed SRS demonstrated that in addition to cysteine,
glycine and glutamic acid were present in the ratio 1:1:1. The structure of the SRS
from murine mastocytoma cells was therefore 6-S-glutathionyl-5-hydroxy-
7,9,11,14-eicosatetraenoic acid, leukotriene C4 (LTC4) 47 (Scheme 1). The
stereochemistry proposed for LTC4 was confirmed and unambiguously assigned
from its total synthesis by Corey et al.8 The systematic name for LTC4 is thus
(5S,6R)-6-S-glutathionyl-5-hydroxy-(7E,9E,11Z,14Z)eicosatetraenoic acid. This
represented the first structure determination of an SRS.
12
Scheme 1
���
��
OH
O3/NaBH4
������
H2/Ni
Lipoxygenase
HCl
Cys
Glu
Gly
���
OH
OOH
OH
OOH
S
N
O O
OHNH2HN
O
OHO
H
����
R'
R"OOH
3
4
2

13
1.2 Leukotrienes
Polyunsaturated fatty acids play an important role as precursors of several
biologically active substances. Five main groups of metabolites, the
prostaglandins, the thromboxanes, the prostacyclins, the leukotrienes and the
lipoxins, are formed by oxygenation and further transformation of these compo-
unds, in particular arachidonic acid [(5Z,8Z,11Z,14Z)-eicosatetraenoic acid] 1, a
cascade, as it is often referred to.9-11 Arachidonic acid is formed in organisms from
linoleic and linolenic acids, which belong to the so-called essential fatty acids.
The leukotrienes exhibit three conjugated double bonds and were first detected in
the white blood cells (leukocytes). Lipoxygenase plays a central role in the
biosynthesis of the leukotrienes. This enzyme catalyses not only the formation of
5-hydroperoxy-6,8,11,14-eicosatetraenoic acid (5-HPET) 5 from arachidonic acid,
but also the subsequent conversion of 5-HPET to leukotriene A4 [LTA4, 5,6-epoxy-
(7E,9E,11Z,14Z)-eicosatetraenoic acid] 6. The expoxide 6 is formed from 5-HPET
via an intramolecular process, namely OH-elimination after abstraction of the
proton at C-10, which is activated by two allyl groups (Scheme 2). The structure of
LTA4 has been confirmed by chemical synthesis whereby the stereochemistry was
determined as the 5(S)-configuration.12 The enzyme glutathione peroxidase on the
other hand catalyses the conversion of the peroxide 5 to (S)-5-hydroxy-
(6E,8Z,11Z,14Z)-eicosatetraenoic acid (5-HETE) 7, a major metabolite isolated
OH
OOH
������
7
14
during the initial studies of the leukotriene biosynthesis.13 Leukotriene B4 [LTB4,
(5S,12R)-5,12-dihydroxy-(6Z,8E,10E,14Z)-eicosatetraenoic acid] 8 is formed en-
zymatically from compound 6. The configuration of the three conjugated double
bonds in LTB4 was also elucidated using a synthetic approach.14 The epoxide
function in LTA4 is also ring opened at the 6-position by the nucleophilic thio-
function of glutathione to form LTC4 4 (SRS). Leukotriene C4 is metabolised further
to leukotriene D4 [LTD4, (5S,6R)-6-S-cysteinylglycine-5-hydroxy-(7E,9E,11Z,14Z)-
eicosatetraenoic acid] 9 by enzymatic elimination of glutamic acid by g-glutamyl
transpeptidase (GGTP).15 The remaining peptide bond in leukotriene D4 is
hydrolysed by a renal dipeptidase to give leukotriene E4 [LTE4, (5S,6R)-6-S-
cysteinyl-5-hydroxy-(7E,9E,11Z,14Z)-eicosatetraenoic acid] 1016 (Scheme 3). Leu-
kotriene E4 can also act as an acceptor for γ-glutamic acid, thus forming the
glutamylcysteinyl derivative leukotriene F4 (LTF4) 11.17
OH
OOH
S
N OHOH
OO
O
NH2H
����
11
Following the structure determination of the SRS from mastocytoma cells6,7 and
the synthesis of LTC4, LTD4 and LTE4,7 all of these cysteine-containing
leukotrienes were identified in a variety of biological systems using comparison
with synthetic material. The SRS is therefore a mixture of leukotrienes containing
cysteine, e. g., the parent compound LTC4 and the metabolites LTD4 and LTE4.

15
The leukotrienes are not only formed from arachidonic acid, but in general from
C20-fatty acids possesing the same 5,8,11-double bond system with more or less
double bonds in the other half of the molecule. The index number, therefore, refers
to the unsaturation of the parent fatty acid from which they are derived. These
versatile, biologically active substances are also termed eicosanoids, after the
number of C atoms they contain.
Scheme 2
OOH
O�������
�����
Dehydrase��
Lipoxygenase��
O
OH
���������
������
���� O
OH
O
H
H
OH
������
1
5
6
16
Scheme 3
OH O
OH
HO
����
����
��Hydrolase ��
OH
OOH
S
N
O O
OHNH2HN
O
OHO
H
��
8
GlutathioneS-transferase
�� GGTP
OH
OOH
S
NH2N
O
H O
OH
��
9
Dipeptidase
OH
OOH
S
H2NO
OH
����
10
����
6
4

17
1.3 Leukotriene Antagonists
In an asthma attack the production of the SRS is triggered by pollen or other
allergens, and constriction of the air passages results. The major component of the
SRS in the human lung is leukotriene D4 9 and accounts for almost all of the
OH
OOH
S
H2NN
OHO
H O
����
9
biological activity.6,18 The leukotrienes exert their potent biological effects through
interaction with specific cellular receptors; in the human lung they are thought to
act on a common one. Over the past several years, much effort has been made to
discover leukotriene antagonists. Two major chemical approaches to the
development of such receptor antagonists have been pursued. The first involved
structural modification of the initial prototype leukotriene antagonist FPL 55712 12,
O
HO O OOH
O
O
OH
O
12
18
discovered at Fisons Corp. in 1977.19,20 Lack of bioavailability and a short half-life
have hindered clinical evaluation of this synthetic material. Propionic acid
derivatives at the 2-position of the chromone ring in compound 12 have been
reported to have longer biological half-lives though they were less potent in vitro.21
Structure activity relationships (SAR) of some derivatives of FPL 55712 have been
reported by Marshall et al.22 When the propylhydroxyacetophenone moiety was
separated from an acidic carboxyl group by one or two methylene groups, no
response could be found. Detectable leukotriene antagonist activity was observed
with three methylenes and maximum activity with the five methylene compound,
followed by a gradual diminishing of activity through 10 methylenes (Figure 1).
Figure 1
O
HO O COOHn
n = 1-10
Aromatic substitution changes were then studied, keeping the five-methylene chain
length and terminal carboxylic acid intact. The saturated n-propyl group was shown
to be better than allyl or hydrogen. There was no loss in activity when the acetyl
group was changed to propionyl, but greatly reduced potency when it was
substituted with a carbomethoxy group. Removal of the 3-hydroxy substituent
abolished activity. When acetyl and hydroxy groups were interchanged, profound
loss of potency was again observed (Figure 2). Next, a number of terminal groups

19
were investigated, without changing of the aromatic substitution pattern. While the
Figure 2
R' O
R
R"COOH
R = acetyl, propionyl, CH3OCO or hydroxy
R' = hydroxy, hydrogen or acetyl
R" = n-propyl, allyl or hydrogen
nitrile group had no in vitro activity, compounds in which the chain ended with
hydroxyl, dimethylamine, morpholine, or N-methylpiperazine were found to have
significant activity. Substitution of the carboxyl group by the bioisosteric tetrazole
resulted in substantial improvement in in vitro and in vivo LTD4 antagonist activity.
In contrast to the carboxylic acid series, in which maximum activity was obtained in
the compound with five methylenes in the chain, among tetrazoles the best activity
resulted in compounds with four, six and seven methylenes in the connecting chain
(Figure 3). An extensive pharmacological evaluation of one of these compounds,
Figure 3
HO O X
O
X = CN, OH, NMe2, N(CH2CH2)2O, N(CH2CH2)2NMe or 5-tetrazolyl
20
LY171883 13, has been reported and showed a greater stability and biological
HO O
O
NN
NN
H
13
activity than observed for FPL 55712 12.23 The second approach centred on
structures related to the leukotrienes themselves, since their elucidation in 1980.8
In recent years a number of specific receptor antagonists of LTD4 have been
synthesised. The compound SKF 104353 14, for example, was designed with the
S OH
OH
OOHO
����������
14
idea in mind, that shortening of the spatial separation between the eicosanoid
carboxyl and thioether functionality of the natural leukotrienes exhibited antagonist
properties.24 The (phenyloctyl)phenyl group was thought to mimic the unsaturated
planar triene moiety of LTD4. The compound 14, which posesses the same
stereochemistry as the agonist itself, showed high affinity for the receptor, while
the other enantiomer showed significantly reduced activity. SKF 104353 was
prepared from (phenyloctyl)benzaldehyde 15 as depicted in Scheme 4. Darzens

21
Scheme 4
other enantiomer
14 +
resolution���� OH
O
R
OH
SHO
O
NaOH����
18 17
+OMe
O
R
S
OH
OMe
O
OMe
O
R
OH
SMeO
O
HS(CH2)2CO2MeEt3N
������NaOMe
������
(CH2)7PhR =
1615
OMeO
O
R
NaOMe
ClCH2CO2Me���
O
R
22
condensation of compound 15 with methyl chloroacetate afforded the glycidic ester
16, which on reaction with methyl mercaptopropionate, gave a 1:1 mixture of the
regioisomers 17 and 18. Treatment of the mixture of regioisomers with methoxide
effected a retro-aldol degradation of the undesired isomer 18, affording compound
17 and recovered starting material. Ester hydrolysis of the intermediate 17 afforded
racemic 14, which was resolved to provide both enantiomers.
The discovery and preparation of the potent, specific and orally active leukotriene
D4 antagonist L-660,711 19, was originally carried out with a racemic mixture. As
classical resolution techniques failed to separate the racemate, an asymmetric
synthesis had to be developed in order to study the biological properties of each
enantiomer.25 Isophthalaldehyde was, therefore, reacted with one equivalent of
each of N,N-dimethyl-3-mercaptopropionamide and R-(-)-α-methoxyphenylthiola-
cetic acid 20 under acidic catalysis to provide a mixture of the diastereomeric
acylthioalkylthioacetals. Flash chromatography effected clean separation of the two
diastereoisomers, which were elaborated further individually. Reaction with sodium
methoxide and methyl acrylate gave the asymmetric dithioacetal 21. The
enantiomer 21 was then reacted with the ylid derived from (7-chloroquinolin-
2-yl)methyltriphenylphosphonium bromide (prepared from 7-chloroquinaldine by
photobromination and subsequent reaction with triphenylphosphine) to yield the
ester 22, which on hydrolysis provided the optically active product 19. The same
reaction sequence with the other diastereomer gave the opposite enantiomer of
the target molecule (Scheme 5).
Another orally active quinoline derivative, compound 23, has been reported by
White et al.26 The final step of the convergent synthesis was the coupling between
2-chloromethylquinoline 24 and the substituted phenol 25 (Scheme 6). The com-
pound 24 was conveniently prepared from quinaldine by conversion first to the N-
oxide, then reaction with benzenesulfonyl chloride. Synthesis of the phenol
derivative 25 began from m-hydroxyacetophenone via a kinetically controlled aldol

23
Scheme 5
OHCO
������
HSPh
O
MeO H
��������
HS(CH2)2CON(CH3)2
20OHC
S
S
Ph
O
HMeO
N
OH
�������
��
1. separation
2. NaOMe, COOMe
OHCS
SCON(CH3)2
COOMe����
S
SCON(CH3)2
COOMe
NCl
����
����
PPh3NCl
��
S
SCON(CH3)2
COOH
NCl
����
21
22
19
24
Scheme 6
OH
O
������
1. LDA
2. O
N
��H2O2
OH
O
NO
3. p-TsOH
����
H2, Pd/C
OH
OH
�� PhSO2Cl
NCl
���
K2CO3
NO
OH
23
24
25

25
condensation with n-butyraldehyde in the presence of LDA. Dehydration of the
crossed aldol product was carried out under acidic catalysis to give the α,β-
unsaturated kenone. Hydrogenation of the enone over palladium-on-carbon
yielded the desired alcohol 25.
Structure activity relationship studies are an important tool to improve the activity of
an initially discovered potent molecule. Very often, however, this approach is used
to modify an active compound towards reduced toxicity. Such an example is found
with L-695,499 26, which exhibited acute toxicity at high doses in mice. It was
N
S
COONa
OH
Et
Cl
����
������
26
noted that in comparison the ketone L-699,392 27 had a better overall biological
N
S
COONa
O
Et
Cl
��������
������
27
profile.27 The stereospecific synthesis of 27 is shown in Scheme 7. The aldehyde
28 was prepared by condensation of 7-chloroquinaldine with 1,3-benzenedicarb-
26
aldehyde in the presence of acetic anhydride.28 Treatment of 28 with
vinylmagnesium bromide gave the allylic alcohol 29, which was treated with methyl
2-bromobenzoate in the presence of palladium acetate to give the keto-ester
30. The ketone was reduced to the alcohol 31 with high ee using Corey's reagent
(see p. 43). The ester was transformed into the activated amide 32 using the
magnesium salt of N,O-dimethylhydroxylamine. The thiol chain was introduced by
first converting the alcohol to a better leaving group, followed by displacement with
the thiolate to give compound 33. The synthesis of L-699,392 27 was completed by
reacting 33 with methylmagnesium bromide to form the methyl ketone.
A "third-generation" of receptor antagonists, which bear little structural
resemblance to hydroxyacetophenones or leukotrienes has been described by
Matassa et al.29 This new family of indole and indazole benzoic acids and their N-
arylsulfonyl amide derivatives are potent and selective LTD4 antagonists. A
compound for clinical trials, ICI 204,219 34, resulted from this investigation. The
34
N
NO
O
H
OMe
NS
O O
OH
study showed that the LTD4 receptor is surprisingly tolerant to changes in the
electronic constitution of the template.

27
Scheme 7
��
NCl
O
O+
Ac2O
NCl
O
28
���
R
MgBr ROH
29
����
CO2MeBr Pd(OAc)2
RO CO2Me
30
������Corey's
reagent
RCO2MeOH
EtMgClMeHNOMe
ROH
O NOMe
31
32
���1. MsCl, Et3N
2.HS
COOH
Me����
NaH
����
R
O NOMe
S
Me COOH
��
�����
33
����
MeMgBr
27
28
This chapter covers the main approaches towards recently described leukotriene
antagonists and gives a general idea about how the activity and compatibility of a
compound, in connection with the human pathology, can be improved through SAR
studies. The significance of asymmetric synthesis is illustrated, since very often
only one enantiomer is able to interact in the desired manner with the "chiral"
environment of a biological system. Over the past few years, these efforts have
produced pharmacologically interesting molecules, some of which have been
important enough to warrant clinical evaluation.

29
1.4 Development of LY290154
In a search for novel potent leukotriene receptor antagonists, Eli Lilly developed a
highly active compound known as LY290154 35 for use to elucidate the role of the
SRS in human pathology and as a potential treatment for asthma. The structure of
NCl
N ON
NNN
NN N
N H
H
35
the LTD4 receptor site has not yet been determined. The design of binding
antagonists was, therefore, based on molecular modelling of both the natural
agonist and potential antagonists. The most important features of leukotriene D4 9
are probably an acidic group at one end, a lipophilic region at the other end and a
central planar triene-unit (Figure 4). A hypothetical receptor binding model was
designed based on these features. The low-energy conformations of potential
receptor antagonists were similarly derived and compared in size and shape with
the LTD4 structure. The initial structure activity exploration of a series of potential
antagonists led to the 2-thio-substituted quinoline compound LY231898 36.30 After
modelling of a number of possible groups the phenyltetrazole group was
selected as the part of the molecule that would fill the triene portion of the receptor.
It has approximately the same length as the triene of LTD4; each is similarly
unsaturated and flat. The phenyltetrazole also appeared to provide the appropriate
30
Figure 4
9
OH
OOH
S
H2NN
OHO
H O
����
lipophilic tail
planar region
steric determiner polar group
acid group
N S NN
NN
OHO
36
geometry for the acid and lipophilic groups. Quinoline, which is also part of the
lipophilic region of other receptor antagonists (see previous section), was shown to
increase activity. Compounds with aliphatic lipophilic substituents are essentially
inactive. Removal of the acidic group showed loss in activity, thus indicating the
significance of this substituent. Further optimisation of the compound 36 gave a
large increase in activity when the thiomethyl group was replaced by a
methyleneoxy group, possibly due in part to the shorter bond lengths associated
with the methyleneoxy linkage.31 No difference in activity was observed between
the methyleneoxy-linked compounds and those linked by a trans-olefin, a
phenomenon also found in other quinoline based series.32 Tetrazole analogues
exhibited somewhat better activity than their corresponding carboxylic acid

31
derivatives, a trend also observed in other SAR studies.22 Chlorine substitution of
the quinoline moiety at the 7-position resulted in increased activity. There is
possibly a lipophilic binding pocket at the receptor that acommodates this chlorine
atom. Other nitrogen-containing heterocycles may be substituted for quinoline
without loss of potency, e. g. benzothiazole.33 In summary, the SAR of a series of
highly potent quinoline-based, phenyltetrazole LTD4 receptor antagonists was
optimised and resulted in a 100-fold increase in activity over the parent compound
36, leading to the discovery of compound 37. Eli Lilly has since demonstrated that
NO
NN
NN
Cl
NN N
N Na
37
the incorporation of an indole moiety provided greater activity and further SAR
studies led subsequently to the development of LY290154 35. Compound 35
contains all of the important features of LTD4, namely, a lipophilic function, a
planar region, a steric determiner , an acid group, a polar group and an indole
linkage providing the correct steric orientation as shown in Figure 5.
32
Figure 5
NCl
N ON
NNN
NN N
N H
H
lipophilic tail
planarregion
steric
acid group
polar group
indole linkage
35
centre

33
1.5 Racemic Synthesis of LY290154
A racemic synthesis of LY290154 35 was achieved by Lilly and reproduced in our
laboratories using the same synthetic methodology (Scheme 8). The Finkelstein
reaction of 4-bromobutyronitrile 38 gave the corresponding iodo derivative 39 in
high yield. This was then converted into the organozinc reagent 40, which was
subsequently reacted with the aldehyde 28 to incorporate the pentanenitrile side
chain. Compound 28 was prepared from isophthalaldehyde and 7-chloroquinaldine
as described before (see p. 16). The resulting benzyl alcohol 41 was treated with
thionyl chloride to give the corresponding chloro compound 42 in essentially
quantitative yield. Coupling of the benzyl chloride 42 with 7-cyanomethoxyindole
45 in the presence of sodium hydride afforded the dinitrile 46 in very poor yield. By
far the major product from this reaction (in ~80% yield) is that derived from
dehydrohalogenation, namely the corresponding styrene . The low yielding nature
of this reaction is almost certainly a consequence of the severe steric congestion
present at the benzylic carbon in 46, and hence elimination is favoured rather than
substitution. The indole 45 was synthesised from 2-nitrophenol in three steps
according to the reported procedure.34 The cycloaddition reaction between the
dinitrile 46 and tributyltin azide proceeded smoothly to give LY290154 in modest
yield. It has recently become apparent that the yield of this last reaction can be
markedly increased just by using larger quantities of 46, and can be as high as
80%. The overall yield of racemic LY290154 35 following the Lilly procedure was
only 1% based on starting 7-chloroquinaldine.
34
Scheme 8
Br CN
���
3892%
i I CN
39
��
ii IZn CN
40
�� iii2862%
N
OHCN
Cl
41
N
ClCN
Cl
��� iv94%
42
NO2
OH
���
NO2
O
Ph
Ph
v48%
vi 54%
O
Ph
Ph
NH
��
���
ONC
NH vii
47%
��
N
N
Cl
CN
OCN
viii18%
��������
35ix
38%
43
44
45
46 (i) NaI, acetone; (ii) Zn, (CH2)2Br2, Me3SiCl, THF, ; (iii) THF, TiCl4; (iv) SOCl2; (v) Ph2CHBr, K2CO3, acetone; (vi) CH2=CHMgCl, THF; (vii) H2, Pd(OH)2/C, PhMe, MeOH, BrCH2CN, K2CO3, butanone; (viii) NaH, DMF; (ix) Bu3SnN3, DME.

35
One of the most difficult decisions affecting drug companies is whether to proceed
with the development of a chiral drug as a single enantiomer or as a racemate.
Traditionally, most synthetic drugs containing chiral centres have been marketed
as racemates, despite the fact that the biological activity for the two enantiomers
can be markedly different. One obvious example is thalidomide 47, that had to be
NN
O
O O
O
H
47
withdrawn from the market as a sedative because of association of one of the
enantiomers with teratogenic effects. The separation of compound 35 into
enantiomers using classical resolution techniques has not yet been successful.
The aim of this project, was therefore, to develop a homochiral synthesis of
LY290154. This would allow full evaluation of the in vivo and in vitro biological
activities of each enantiomer.
36
2. Retrosynthetic Analysis of LY290154
LY290154 35 can in principle be synthesised from a coupling reaction between an
appropriately functionalised indole 48 and a quinoline 49 as exemplified in Scheme
9 with the dinitrile precursor 46. The two fragments could be joined together via
Scheme 9
+N O
CN
CN YNClX
NCl
N O
CN
CN
48
49
46
a) X: CHO, Y: CH2PPh3; b) X: halogen, Y: CH2=CH2
either a Wittig reaction or a Heck procedure. Wittig chemistry of this general type,
using the ylid derived from (7-chloroquinolin-2-yl)methyltriphenylphosphonium

37
bromide has already been described for the synthesis of the LTD4 antagonist
L-660,711 19. The palladium catalysed Heck coupling reaction between the halo
compound 48b and the 2-vinylquinoline 49b would need to be investigated in more
detail.
The second disconnection is rather more complex since it requires a method for
the introduction of the neccessary chirality and another for the construction of the
indole ring system (Scheme 10). Asymmetric reduction of the corresponding oxime
Scheme 10
N O
CN
CN +
NH2
CN
RCN
O
O
or NMe
NO2
48
50
X: a) CHO, b) halogen
R: NOMe or O
X
X
X
+X -
ether could directly afford the amine 50. On the other hand, reduction of the
corresponding ketone would first give the optically enriched alcohol. Substitution of
the hydroxy group with an appropiate nucleophile (e. g. azide) and reduction would
38
than led to the key intermediate 50. Reaction of compound 50 with
7-oxotetrahydrobenzofuran or an N-methyl-3-nitropyridinium salt (for details on the
synthesis of indoles from 3-nitropyridinium salts see chapter 5) could result in
formation of the required indole moiety, in the former case after aromatisation. All
of these transformations would need to proceed without racemisation of the
products in order to fulfil the aim of this project.

39
3. Preliminary Results
The racemic synthesis of the indole derivative 48a was required in order to study
the possibility of a Wittig reaction with the quinaldine derivative 49a. Coupling of
49a
NClPPh3
48a
N
CN
OCN
O
the chloride 51 with 7-cyanomethoxyindole (45) could result in formation of the
desired product 48a. Compound 51 should be readily accessible from the alcohol
51
ClCN
O OHCN
O
52
52, applying the Lilly methodology. 3-Bromobenzaldehyde was therefore reacted
with ethyleneglycol in the presence of p-toluenesulfonic acid (TsOH) to give the
acetal 53 in good yield, according to the published procedure35 (Scheme 11). This
acetal proved highly acid sensitive, and was cleaved under the most mild acidic
conditions. Lithiation of 53 with n-BuLi and quenching with N-formylpiperidine
gave the monoprotected isophthalaldehyde 54 in excellent yield. The reported
acidic work-up had to be avoided, because under these conditions complete
40
Scheme 11
O
Br BrO
O���
HOOH
TsOH
���n-BuLi
N
O
O
O
O
94% 93%53 54
hydrolysis of the acetal occurred.36 This two step route to 3-(1,3-dioxolan-2-
yl)benzaldehyde (54) was found to be superior to the one step acetalisation of
isophthalaldehyde using ethyleneglycol and TsOH in benzene, which required a
difficult chromatographic isolation of the product 54.37 Reaction of the aldehyde 54
with the zincate 40 in the presence of titanium tetrachloride, however, did not result
in formation of the expected alcohol 52 after hydrolysis, but gave the ether 55 in
low yield (Scheme 12). The organometallic reagent had reacted preferentially with
Scheme 12
����
IZn CN
40
TiCl4
OCN
OHO
22%
54
55
the acetal instead of the carbonyl group. The remaining product was shown to be
isophthalaldehyde, derived from hydrolysis of the acetal 54. When the reaction was
performed without Lewis acid catalyst only starting material could be recovered.
Treatment of 3-bromobenzaldehyde with the zincate 40, however, gave the
expected alcohol 56 in moderate yield (Scheme 13). The corresponding chloro

41
compound 57 was easily prepared, in almost quantitative yield, from 56 by simple
Scheme 13
BrO
���40TiCl4
Br CNOH
57%
56
stirring in thionyl chloride. Coupling of the indole 45 with the benzyl chloride 57 was
accomplished by preforming the sodium salt of 45 using sodium hydride, followed
by the addition of 57 (Scheme 14). The racemic dinitrile 48b was obtained in low
Scheme 14
40%
NaH���
98%57
Br CNCl
SOCl2���
56
BrN
CN
OCN
48b
45
yield, the major product being the olefin 58. Attempted coupling of the indoline 59
CNBr
58
42
with the benzyl chloride 57 under the same conditions, in the hope of obtaining a
better yield, led only to formation of 58. The indoline 59 was obtained by routine
reduction of the indole 45 with sodium cyanoborohydride in acetic acid (Scheme
15).38
Scheme 15
NHONC
����NaBH3CN
NHONC
90%45 59
AcOH
The failure to obtain the alcohol 52, required to exploit Wittig type chemistry, and
the successful preparation of the indole derivative 48b, indicated that the Heck
coupling was almost certainly going to be the method of choice for joining the
fragments 48 and 49 together. Nevertheless, reaction of 54 with 1,3-propanedithiol
in the presence of aluminium chloride, following the procedure described by Ong,39
yielded the corresponding monoprotected aldehyde 60 and the known bis-dithiane
61 in 8 and 28% yield respectively (Scheme 16). Several other spots were also
present in the reaction mixture, but the compounds could not be isolated. Again the
nucleophile reacted preferentially with the dioxolane instead of the aldehyde group.
None of the expected 1-(1,3-dioxolan-2-yl)-3-(1,3-dithiane-2yl)benzene (62) was
obtained. The same reaction, but with isophthalaldehyde and one equivalent of
the sulfur component, gave the desired product 60 in a slightly better yield (14%,
Scheme 16) together with the unwanted bis-dithiane 61 (14%). This reaction
proceeded much more cleanly than the previous one, and the only other
component in the product mixture was unreacted isophthalaldehyde. Reaction

43
Scheme 16
O
O
���HS SH
OO
O
54
S
SS
S
+
OS
S
60
AlCl3
61
S
SO
O
62
of compound 60 with the zincate 40 in the presence of titanium tetrachloride
afforded the alcohol 63 in low yield; most of the starting material was still present
in the product mixture (Scheme 17). This time no cleavage of the dithiane group
Scheme 17
63
S
S
OHCN
S
S
O
����40
60
TiCl4
22%
44
occurred, as had previously been observed with the dioxolane moiety. Now the
alcohol 52 could be accessible by conversion of the dithiane to the aldehyde
group, making the alternative Wittig reaction again a possibility.
The synthesis of the counterpart for the Heck coupling reaction, namely the
vinylquinoline 49b, was achieved by Dr. M. W. J. Urquhart at UEA using a novel
procedure.40 Thus, reaction of the anion derived from 7-chloroquinaldine with
diethyl chlorophosphonate gave a mixture of the ester 64 and unreacted quinaldine
(Scheme 18). The addition of a further equivalent of LDA to this mixture resulted in
Scheme 18
NCl
���1. LDA
NClP(OEt)2
O
2. (EtO)2POCl
71%
������
1. LDACH2O2.
NCl
58%49b
64
the disappearance of the quinaldine, and the ester 64 was isolated in good yield.
The ylid derived from 64 was then treated with paraformaldehyde to give the
desired vinylquinoline 49b in moderate yield. Work undertaken by Dr. R. A. Lewth-
waite at UEA showed that the vinylquinoline 49b could be successfully coupled
with the racemic dinitrile 48b in the presence of Pd(dppp)Cl2, to give the dinitrile

45
precursor 46 to LY290154 itself41 (Scheme 19). This important result made clear
Scheme 19
49b
60%46
NCl
N
CN
OCN
MeCNEt3NPd(dppp)Cl248b
������
BrN
CN
OCN
+ NCl
����������������������������������������������������������������������������������������������������������������������������������������������������
����
����
����
����
����
����
����
����
����
����
����
����
that the next goal in the asymmetric synthesis of the target compound 35 was to
achieve the homochiral preparation of the indole derivative 48b. This target would
be approach as outlined in the previous chapter (second disconnection). The
amine 50b had therefore to be synthesised. How we succeeded to produce this
crucial intermediate in a enantiomerically enriched form is discussed in the next
chapter.
46
4. Asymmetric Amine Synthesis
Extensive research during the past twenty five years has led to the discovery of a
number of catalysts based on transition metal complexes for the enantioselective
homogeneous hydrogenation of olefins and ketones. In contrast, the analogous
reduction of the C=N double bond, which represents a potentially important me-
thod for the synthesis of enantiomerically enriched amines, has received much less
attention. Most of the procedures reported so far involve the hydrogenation of
N-substituted imines under high pressure, using rhodium42-46 and iridium47,48
complexes of chiral bisphosphines to yield optically active secondary amines. The
proximity of an aromatic ring to the C=N bond appears to be essential for high
enantioselectivity. The RhI/(R)-cycphos (cycphos = 1,2-bis(diphenylphosphino)-1-
cyclohexylethane, 65) catalyst, for example, was shown to be inefficient for the
65
PPh2
PPh2
asymmetric hydrogenation of aliphatic imines.42,43 On the other hand, imines
derived from benzylamine and aniline were reduced with optical yields of up to
69% ee. The addition of halide ions, preferably iodide, can result in improved
enantioselectivity up to 91% ee.
The catalytic properties of water soluble complexes with sulfonated derivatives of
(1S,2S)-1,2-bis(diphenylphosphinomethyl)cyclobutane (66), (2S,4S)-2,4-bis(diphe-
nylphosphino)pentane (BDPP, 67), (2S,3S)-2,3-bis-(diphenylphosphino)butane
(CHIRAPHOS, 68) and (2R)-1,2-bis(diphenylphosphino)propane (PROPHOS, 69)

47
PPh2
PPh2
H
H
��
PPh2 PPh2
��������������
PPh2
PPh2
��������������
PPh2
PPh2
66 67 68 69
have been reported. Asymmetric hydrogenation of a benzylimine occurred in an
aqueous-organic two-phase solvent system using a rhodium complex of
tetrasulfonated 67 under high pressure, but only a 34% ee was obtained. The
same reaction, but with a mixture of the mono-, di- and trisulfonated ligand, gave
an optical yield of 58%.44 Other hydrogenations of N-benzylimines under similar
conditions, but using the monosulfonated derivative of 67, gave the corresponding
amines with 92-94% ee, whereas the disulfonated ligand showed no
enantioselectivity.45 The primary advantages of this catalytic system are the ease
of the workup and the ability to recover and reuse both the rhodium and the
optically active bisphosphine. Lensink and Vries reported the hydrogenation of
chiral imines derived from optically pure α-methylbenzylamines.46 With a rhodium-
BDPP 67 catalyst diastereoselectivity of up to 99.8% was obtained. The chiral
diphosphine ligand 2,2-dimethyl-4,5-bis(diphenylphosphinomethyl)-1,3-dioxolane
(DIOP, 70) was not as selective. Other 2-carbon bridged ligands like PROPHOS
O
O PPh2
PPh2
��������������
70
and CHIRAPHOS were not selective at all.
The results indicated that the selectivity of the hydrogenation of chiral imines is
mainly substrate controlled. Low enantioselectivities with catalysts prepared from
iridium(I) and 1,2-diphosphino ligands like CHIRAPHOS 68 and 5,6-bis(diphenyl-
48
phosphino)norbornene (NORPHOS, 71) have also been reported by Spindler et
71
Ph2P
Ph2P
��
al.47 With ligands which can form conformationally flexible six- or seven-membered
metallacycles, e. g. BDPP 67, DIOP 70 or t-butyl-4-(diphenylphosphino)-2-(diphe-
nylphosphinomethyl)-1-pyrrolidinecarboxylate (BPPM, 72) better results were
72
NBOC
Ph2PPPh2
����������������
����������
obtained. Effective asymmetric reductions of N-substituted prochiral imines using
iridium(III)/diphosphino complexes (diphosphines = BDPP 67, DIOP 70 and
NORPHOS 71) in moderate to good optical yields have been carried out.48
Interestingly, Noyori's 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (BINAP, 73) was
PPh2
PPh2
����
��
����������������
����������������
�����
73

49
ineffective in terms of both the rate and enantioselectivity of the hydrogenation.
Asymmetric hydrogenations of imines with a chiral titanium catalyst containing a
nonphosphine ligand have been studied by Willoughby and Buchwald.49-51 The
chiral auxiliary used was 2,2'-dihydroxy-1,1'-binaphthyl (74). While the reduction of
OHOH
����
��������������
��������������
������
74
acyclic N-substituted substrates proceeded with moderate to good enantioselec-
tivity, cyclic imines were transformed to the corresponding amines in excellent
optical yield. In the former case the enantiomeric excesses of the products with
large N-substituents correlated roughly with the anti/syn ratios of the imines. A
proposed model predicts that syn and anti imines react to give the amine with
opposite absolute configuration, thus lowering the optical purity. Furthermore, high
hydrogen pressures were required to achieve the maximum ees.
The chemical yields in the transition metal catalysed hydrogenations of imines are
generally high, making this process an attractive procedure for the synthesis of
homochiral amines. The major disadvantages are the necessity for expensive
chiral catalysts, the high pressures required, and the fact that only secondary ami-
nes can be prepared in this way.
A method for the preparation of optically active primary amines using a chiral
rhodium catalyst has been described by Burk and Feaster.52 In comparison with
other chiral diphosphines such as BDPP 67, CHIRAPHOS 68 or BINAP 73, Et-
DUPHOS 75 proved to be the most effective ligand. The synthesis consisted of the
50
P
P
EtEt
Et
Et
��������
����
��������������
������������
75
asymmetric hydrogenation of the C=N group of N-acylhydrazones of aryl alkyl
ketones and α-keto esters, followed by cleavage of the N-N bond with samarium
diiodide (Scheme 20). In contrast to the other procedures described so far, high
enantioselectivities were obtained at low hydrogen pressures. The reaction with
samarium diiodide occurred spontaneously with no loss of optical purity.
Scheme 20
R R'
NH2��
SmI2
��������
R R'
NNH
R"
O
H ����
Rh(I) 75��
��
R R'
NNH
R"
OR"CNHNH2
O
����
R R'
O
H2

51
In 1985, Itsuno reported the enantioselective reduction of ketones and oxime
ethers with reagents prepared from borane and chiral vicinal amino alcohols. The
best results for the synthesis of optically active primary amines were obtained from
borane reduction of acetophenone O-methyloximes in the presence of the amino
alcohol 76.53 High stereoselectivity (up to 99% ee) was always attained with the
H2N OH
PhPh
����������
76
reagent prepared from a 1:2 molar ratio of amino alcohol and borane, whereas the
reduction with a 1:1 molar ratio resulted in disappointingly low enantiomeric ex-
cesses. Reductions of oximes and their derivatives have also been carried out with
baker's yeast, but only modest ees were obtained.54
Corey et al. found that a fast reaction occurred between amino alcohol 76 and 2
equivalents of borane to give 2 equivalents of hydrogen gas and the
oxazaborolidine 7755 (Scheme 21). Solutions of 77 alone did not reduce ketones,
Scheme 21
7877
N O
PhPh
BHH3B
H
����������BH3���� ���
N O
PhPh
BH
H
��������BH3
������
76
however, but mixtures of 77 and borane effected complete reduction of
acetophenone with rates comparable to the Itsuno mixtures. 11B-NMR spectra of
52
mixtures of 77 and borane clearly indicated the formation of a 1:1 complex 78. It is
now possible to derive a reasonable mechanism for the enantioselective reduction
of oxime ethers, analogous to that postulated for the reduction of ketones, based
on the above results. It could occur by coordination of the electrophilic ring boron in
complex 78 with the C=N nitrogen and hydrogen transfer from the NBH3- unit to
the carbon via a six-membered cyclic transition state, as formulated in Scheme
22. Further reaction with BH3 and hydrolysis of the borane intermediates would
Scheme 22
N O
PhPh
BH2B
H
H
H
R'R
NOR"
��������
���� ��
���NOR"
H
H
H2BB
PhPh
ON
R'R
H
������
��
���
H
H
H3BB
PhPh
ON
����
BH3N
R'R
H
H2B OR"
��
+
���BH3
N
R'R
H
OR"B
H
H2B
HH
��
�����
����������NHBH2
R'R
H
���� R"OBH2+
��� H2ONH2
R'R
H���
��� + R"OH
yield the corresponding alcohol and the optically active primary amine. The highly
effective oxazaborolidine catalyst 79, known as Corey's reagent, for the
N OB
PhPh
HH3B
79

53
asymmetric reduction of prochiral ketones resulted from this study. For the
enantioselective reduction of N-substituted imines, however, Itsuno's reagent 78
was found to be superior.56 All the ketimines examined were reduced to the
corresponding amines in essentially quantitative yields. In the reduction of N-
phenyl aromatic ketimines, consistently high optical yields were obtained. In
contrast, the reduction of N-alkyl ketimines provided lower optical induction. This
observation is in accordance with the transition metal catalysed hydrogenation of
imines, where the proximity of an aromatic ring to the C=N bond was also found to
give higher enantioselectivities.
More recently, a different approach for the synthesis of optically active primary
amines has been described. The general procedure is to condense a prochiral ke-
tone with a chiral amino alcohol to give a chiral Schiff base (Scheme 23).
Scheme 23
8280, 81 [H]
80: R" = Ph, R''' = H81: R" = iPr, R''' = H82: R" = Me, R''' = Ph
R R'
NH2��
NaIO4��
R R'
N
R"OH
R'''
H ����
���
R R'
N
R"OH
R'''
R"
R'''H2N
OH����������
���
R R'
O
Reduction of the imine by an achiral catalyst such as Pd/C or PtO2, and oxidative
cleavage of the chiral auxiliary with periodate provides the corresponding amine.
Miao et al. obtained optical yields in the range of 40-60% using (R)-phenylglycinol
(80) and (R)-valinol (81),57 whereas Sreekumar and Pillai reported enantiomeric
54
excesses between 54 to 66% using norephedrine (82) as the chiral inducing
agent.58
A related procedure involves the diastereoselective addition of allylmetal
compounds to imines prepared by the condensation of aldehydes with esters of
(S)-valine.59 Excellent results were obtained with allyl bromide and zinc in the
presence of catalytic amounts of CeCl3 or SnCl2 (de up to 100%). Reduction of the
ester function in 83, followed by oxidative cleavage with periodic acid in the
presence of methylamine, afforded the primary homoallylic amine 84 86% optically
pure (Scheme 24). Further functionalisation of the C=C bond would allow the
Scheme 24
Ph NOR'
O
����M Ph NOR'
OH����
����
1.
2. H2O
LiAlH4
Ph NH
OH
�������H5IO6
MeNH2
Ph NH2����
83
84
synthesis of a variety of derivatives of such homochiral amines.
An approach for the synthesis of homoallylic amines based on SAMP/RAMP
hydrazones has been described by Enders et al.60 As shown in Scheme 25 , the
reaction of aldehydes with SAMP [(S)-1-amino-2-methoxymethylpyrrolidine (85)] or
RAMP [(R)-1-amino-2-methoxymethylpyrrolidine (86)] gave the corresponding

55
Scheme 25
R
O ������
NNH2
OMe
85: (S)86: (R)
R
NN
OMe
��
R
NN
OMe
MeO
O
���
R
NMeO
OH
1. M2. MeOCOCl
88
87
Li
NH3
hydrazones in high yields. Subsequent 1,2-addition of the in situ prepared
allylcerium reagent or allyl Grignard reagent to the CN-double bond of the
hydrazone 87 occurred with good yields and with high diastereoselectivities. The
intermediates were trapped with methyl chloroformate to obtain the carbamate
protected hydrazines 88. In contrast to the previous method, where no reaction
between allylcerium reagents and homochiral imines occurred, in this study they
were found to be superior to the corresponding Grignard derivatives. The
carbamate-protected hydrazines were cleaved by lithium in ammonia to the homo-
allylamines in good yields and without racemisation. This work represents a
continuation of the asymmetric reductive amination of ketones for the synthesis of
optically active primary amines from the corresponding SAMP/RAMP-hydrazones
described previously by the same author.61
56
A different borane reagent for the asymmetric reduction of imines has been
described by Kawate et al.62 These authors found that treatment of N-substituted
imines with chiral dialkoxyborane reagents in the presence of MgBr2.OEt2 gave the
corresponding amines in high chemical yields and moderate to good enantiose-
lectivities. Without MgBr2.OEt2 the reaction did not take place. The borane reagent
of choice found in this study was the tartaric acid derivative 89.
89
OB
O OMeOMe
H�������
Chiral sodium triacyloxyborohydrides 90, easily obtained from the reaction of
NaBH4 and N-acyl derivatives of optically active α-amino acis, are reported to be
90
NaBH
3
R N CO2
O
R'
R"
��
excellent reducing agents for cyclic imines, and optical yields of up to 95% have
been achieved. 63-65
The catalytic asymmetric hydrosilylation-hydrolysis procedure is an indirect method
of forming optically active secondary amines from N-substituted imines (Scheme
26). The reactions are carried out under mild conditions using a chiral
rhodium-phosphine catalyst such as [Rh(I)-DIOP] 91, with a best result of 65% ee
being obtained.66

57
Scheme 26
O
O
PRh
P
Cl
solvent
Ph
Ph Ph
Ph��������������
91
R R'
NHR"����H+ ���
R R'
NR" SiHPh2��
��
91
Ph2SiH2 ���
R R'
NR"
Chiral non-racemic sulfur reagents have found application in the asymmetric
synthesis of primary amines. Addition of the lithiated imine generated by the
reaction of methyllithium with benzonitrile to sulphinamide 92 resulted in clean
formation of the benzylidene sulphinamide 93 as a single diastereoisomer
(Scheme 27). Reduction of 93 with diisobutylaluminium hydride (DIBAL) gave the
diastereomeric products 94 and 95 in a 13:1 ratio. Treatment of this mixture with
methanolic trifluoroacetic acid, followed by the addition of hydrochloric acid,
resulted in formation of α-methylbenzylamine (96) with an optical purity of 86%.
The sulphinic acid 97 may be recycled after use and this method represents the
first example of a reagent of this type.67 More recently, Bolm and Felder have
reported the use of β-hydroxy sulfoximines in catalysed enantioselective amine
syntheses,68 as previously described for the asymmetric reduction of ketones.69,70
In the presence of (S)-98, reduction of ketimine derivatives with BH3.SMe2
occurred smoothly at ambient temperature. The N-substituent of a given ketimine
derivative had a major influence on the asymmetric induction. Ketoxime thioethers
58
Scheme 27
97
96
������
+
SO2H
N
OH
Ph NH2
����
2. HClCF3CO2H1. ���
���
9594
SN Ph
O
N
OH
H��
+
SN Ph
O
N
OH
H
��
����
DIBAL
������
93
SN Ph
O
N
OH
����
92
���+
Ph
NLi
NSO
O
������
���

59
98
PhS
Ph
N
O
OH H
Ph
����
gave the highest enantioselectivities (up to 70% ee). The more easily accessible
ketoxime O-ethers, however, gave significantly lower enantioselectivities.
Initial studies on the asymmetric addition of organolithium reagents to N-(4-
methoxybenzene)aldimines in the presence of bis-oxazolines 99 and (-)-sparteine
100 have been described by Denmark et al.71 Optical yields of the corresponding
N
O
N
O
R R
R' R'
NN
����
����������
99 100
secondary amines in the range of 30-91% were obtained for aromatic, olefinic and
aliphatic aldimines.
A totally different method, where the chirality is not introduced via the nitrogen-
containing precursor, is to start with an optically active alcohol. Chelucci et al., for
example, converted homochiral hydroxyalkylpyridines 101 into the azides 102 via
the non-isolated mesylates72 (Scheme 28). The azides 102 were reduced by
hydrogen on Pd/C to the corresponding aminopyridines 103 in good overall yields
and enantiomeric excesses in the range 66-92%.
60
Scheme 28
103
102101
H2Pd/C
NaN3
NR'
NH2
R
����
NR'
OMsR
�
NR'
N3
R
���
Et3N
MsCl ���
NR'
OHR
��

61
4.1 Results and Discussion
The mild conditions and readily available reagents for the reduction of oxime
ethers to optically active primary amines made the Itsuno procedure the initial
method of choice for the synthesis of the required enantiomerically enriched amine
50b*. The homochiral auxiliary 76 was synthesised by reaction of commercially
available L-valine methyl ester hydrochloride with an 8 fold excess of phenyl-
magnesium bromide (prepared in situ) according to the reported procedure53
(Scheme 29). For a model study, the O-methyl oxime 104 was obtained in high
Scheme 29
40%
OMe
O
H2N
��������
.HCl
���1. PhMgBr
2. NaOH OHH2N
PhPh
����������
76
yield as a mixture of stereoisomers from valerophenone and methoxylamine
hydrochloride in the presence of sodium acetate using the method of Karabatsos
and Hsi73 (Scheme 30). Reduction of compound 104 with a borane-THF complex
led to racemic 105 in modest yield.74 Asymmetric reduction of 104 with Itsuno's
reagent 78, generated in situ from BH3.THF and the optically active amino alcohol
76, at room temperature gave the enantiomerically enriched amine 105* in similar
yield. The enantiomeric excess was determined by proton NMR spectroscopy
using 2,2,2-trifluoro-(9-anthryl)ethanol (TFAE, 106) as the chiral solvating agent
and found to be 70%. The chiral auxiliary 76 was recovered without loss of optical
purity.
62
Scheme 30
104
47%
BH3.THF
NH2
O
�������OMe
N
105
.HCl
52%
ONB
Ph
Ph
H
H
H3B
����������
78
���MeONH2
91%
NaOAc.H2O
70% ee

63
OHF3C
106
The ketone 110, required to study the real system, was prepared in 35% overall
yield as shown in Scheme 31. 3-Bromobenzaldehyde was reacted with sodium
Scheme 31
��
98%
83%86%
50%110 109
108107
BrO
CN BrCN
NMe2
CN
2.1. LDA/TMEDA
Br(CH2)3CN��
BrNMe2
CN
��
Me2NH
NaCN
BrSO3Na
OH
���
NaHSO3Br
O
CuSO4.5H2O
hydrogen sulphite to give the adduct 107. The crude sulphonate 107 was
treated with dimethylamine and then sodium cyanide to give the aminonitrile 108
in high yield. Treatment of 108 with LDA, generated in situ from reaction of
diisopropylamine and n-butyllithium, followed by reaction with 4-bromobutyronitrile
gave compound 109 in essentially quantitative yield.75 The ketone 110 was
obtained by refluxing 109 in the presence of copper(II) sulphate pentahydrate. The
64
corresponding O-methyl oxime ether 111 was obtained in good yield following the
same method as before (Scheme 32). Borane reduction of 111, however, did not
Scheme 32
CNNH2
Br
CNN
Br
OMe
����
77%MeONH2
������
���
BH3 THF or
NH4OAc
NaBH3CN
13%
110
111
50b
78
NaOAc
.
lead to the required amine 50b with either BH3.THF or reagent 78; instead, a
product was obtained which could not be characterised from its proton NMR
spectrum. Competitive reduction of, and/or complexation of the boron reagent with,
the nitrile group in 111 was probably the reason for the disappointing result. The
racemic synthesis of the amine 50b could be achieved through reductive amination
of the ketone 110 with sodium cyanoborohydride, but only in a very poor yield.76
The same result was found with valerophenone, and the amine 105 was obtained
in only 11% yield after the mixture was left to react for 3 days. This particular
reaction was found to be very slow and most of the starting ketone could be
recovered.

65
Further attempts to reduce the functionalised O-methyl oxime ethers 113 and 114
with borane were also unsuccessful, presumably for the same types of reasons as
referred to above (Scheme 33). The oxime ethers 113 and 114 were best
Scheme 33
BH3.THFor 78���
���
113: R' = H, 39%114: R' = (CH2)3CN, 86%(CH2)3CN112: R' =
28: R' = H
R
R'R
NH2
N
R'R
OMe
pyridine
���
O
R'NCl MeONH2.HCl
synthesised under non-aqueous conditions, because of the insolubility of the
starting materials 28 and 112.74 The ketone 112 was obtained in moderate yield
from Swern oxidation of the benzyl alcohol 4177 (Scheme 34).
Because of the failure to obtain the enantiomerically enriched amine 50b* via the
asymmetric reduction of the corresponding O-methyl oxime ether 111 using
Itsuno's reagent 78, a different approach had to be considered.
Condensation of 3-bromobenzaldehyde with amino alcohol 76 in the presence of
anhydrous MgSO4 gave the corresponding homochiral imine 115 in essentially
quantitative yield 57 (Scheme 35). It was hoped that diastereoselective addition of
66
Scheme 34
60%11241
DMSO
TFAA����
OR CNR CN
OH
R = 7-chloro-2-vinylquinoline
Scheme 35
116
NBr
Ph
OHPh
H
CN
����
TiCl4
40IZn CN
������
98%
OBr ����76
NBr
Ph
OHPh
����
115
MgSO4
���

67
the zincate 40 in the presence of titanium tetrachloride would afford the optically
active amine 116. The chiral auxiliary would then be removed, giving the required
enantiomerically enriched amine 50b*. Unfortunately, no reaction between
compound 115 and the organometallic reagent 40 was observed (for a review on
organozinc reagents see ref78). A different approach was therefore pursued.
Thompson et al. reported a new method for the conversion of alcohols into their
corresponding azides with inversion of configuration using diphenylphosphoryl
azide (DPPA) and 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU).79 The method is
operationally simple and racemisation was typically less than 2%. Racemic benzyl
alcohol 56 was therefore converted to the azide 117 in good yield using the same
conditions (Scheme 36). Reduction of the azide 117, however, was not as
Scheme 36
5678%
DBU
Br CNN3
Br CNOH
117
���
DPPA
straightforward as expected (see Table 1). Stanovnik et al. found that the azido
group could be easily reduced by a diazo transfer reaction using acetylacetone in
the presence of triethylamine.80 The azide 117, however, did not react under the
conditions reported by Stanovnik (Entry 1). Attempts to hydrogenate 117
catalytically with Pd/C or Adam's catalyst led only to a mixture of products from
which the desired amine could not be isolated81,82 (Entries 2 and 3). Reduction
with sodium hydrogen telluride afforded the amine 50b, but only in a very poor
68
Table 1 Attempted reductions of azide 117 to amine 50b using different reducing
agents
50b
����
117
Br CNNH2
Br CNN3
Entry Reducing Agent Yield [%]
1 (CH3CO)2CH2 starting material
2 Pd/C/H2 "
3 PtO2/C mixture
4 NaBH4/Te 7
5 Ph3P/HCl 9
6 NaBH4/CuSO4 12
7 NaBH4/PTC 30
8 HS(CH2)3SH 60
yield83 (Entry 4). Most of the product was shown to be unreacted starting material.
When the azide 117 was reacted with triphenylphosphine spontaneous evolution of
nitrogen was observed, and tlc analysis showed that the reaction was complete
after one hour. The Staudinger product 118, however, was found to be very stable

69
BrN
CN
PPh
PhPh
118
towards hydrolysis (for reviews on azides and the Staudinger reaction see ref84
and ref85 respectively). Treatment of the iminophosphorane 118 with dilute
hydrochloric acid, for example, gave the required amine 50b in only 9% yield
(Entry 5). Reduction of 117 with sodium borohydride in the presence of copper(II)
sulphate, following the recent method described by Rao and Siva,86 gave again a
very low yield of the desired product (Entry 6). A better yield was obtained with
aqueous sodium borohydride under phase-transfer catalysis87 (PTC, Entry 7).
Finally, the amine 50b could be prepared in moderate yield by reaction of the
azide 117 with 1,3-propanedithiol in the presence of triethylamine88 (Entry 8).
The optically active series required the homochiral benzyl alcohol 56*. This
compound would be available from asymmetric reduction of the corresponding
ketone 110. The ketone 110 was obtained in a better overall yield than previously
described by oxidation of compound 56 under Swern conditions (Scheme 37).
Asymmetric reduction of 110 was best achieved with Itsuno's reagent 78 (see
Scheme 37
BrOH
CN
����DMSO
TFAA
BrO
CN
56 11084%
70
Table 2 Asymmetric reductions of ketone 110 to alcohol 56* using different chiral
reducing agents
O
CNBr CNBrOH
*
110 56*
Entry
Chiral reducing agent
Yield [%]
ee [%]
1
N COO-
O PhO3
NaBH
120
starting material
2
DIP-Cl 119
starting material
3
Corey's reagent 79
65
66
4
Itsuno's reagent 78
70
80
Table 2, Entry 4). Corey's reagent 79, however, was less effective89 (Entry 3). The
discovery of oxazaborolidines for the enantioselective reduction of prochiral
ketones has raised enormous interest in the synthetic community since the
pioneering work by Itsuno and Corey. Recently, new enantioselective catalysts of
this class have been developed using different optically active 1,2-amino alcohols
and borane reagents.90-101 Diaza analogues have also been reported.102 From the
many chiral borane reagents developed by Brown, commercially available (+)- or
(-)-diisopinocampheylchloroborane (DIP-Cl, 119) has been reported to be an

71
2
BCl
119
excellent reagent for the asymmetric reduction of aryl alkyl ketones.103-105
Reduction of aryl alkyl ketone 110 with this reagent, however, led only to recovery
of the starting material (Entry 2; for a review on asymmetric reductions with
organoborane reagents see ref106). The chiral sodium triacyloxyborohydride 120,
easily prepared from optically active N-benzyloxycarbonylproline and sodium
borohydride, which has been reported to reduce cyclic imines with excellent
enantioselectivities (see p. 47), did not react with compound 110 (Entry 1).
Conversion of the optically active alcohol 56* to its corresponding enantiomerically
enriched azide 117* was successfully achieved following the same method as for
the racemic series. Reduction of the azide 117* with 1,3-propanedithiol gave the
required amine 50b* in moderate yield without loss of optical activity. The
enantiomeric excesses of 117* and 50b* using TFAE as the chiral solvating agent
could not be determined because of lack of complexation.
With the chiral building block 50b* in hand, the remaining major challenge was to
construct the indole ring system in 48b. This is discussed in the following chapters.
72
5. Synthesis of Indoles
From the many methods which have been developed for the preparation of
indoles, the two that were of particular interest for the present project, namely the
Bartoli indole synthesis and the synthesis of indoles by the reaction of 3-
nitropyridinium salts with N-alkylketimines, are discussed in this chapter (for a
review on recent developments in indole synthesis see ref107).
5.1 Bartoli Indole Synthesis
In 1989 Bartoli and his co-workers discovered that reaction of three equivalents of
vinylmagnesium bromide with one equivalent of a nitroarene resulted in formation
of an indole.108 With less Grignard reagent no indoles could be isolated.
Satisfactory results were only obtained when the nitroarene was ortho substituted.
4-Chloro- and 4-bromonitrobenzene, for example, gave the corresponding indoles
in only 17 and 12% yield respectively. With 3-substituted arenes mixtures of the 4-
and 6-substituted indoles were obtained, but also in very low yields. Preliminary
mechanistic studies showed that nitroso arenes reacted with two equivalents of the
Grignard reagent to afford the corresponding indoles, suggesting that the first
stage of the reaction with nitroarenes involved reduction to nitrosoarenes by one
equivalent of the vinylmagnesium reagent.109,110 Direct evidence of nitrosoarene
involvement was obtained from GC-MS analysis of the reaction mixtures.
Nitrosoarene 121 could arise by attack of the carboanionic vinyl Grignard reagent
at the oxygen atoms of the nitro group, followed by elimination of the enolate of the
O-alkylated derivative (Scheme 38). The proposed mechanism explains the indole
formation via a 1,2-addition to the N=O double bond of 121 followed by an oxaza
[3,3] sigmatropic rearrangement of the N-aryl-O-vinylhydroxylamino magnesium

73
Scheme 38
OMgBr��
RN
O
��������
RN
O
OMgBr
���
�� ������
BrMgR
NO
O
121
MgBr
N
R H
H+
��
RN
O
MgBr
��� ��������
122
���
RNMgBr
O
������
���N
R
OMgBr
H
MgBr������
������
���
���
���
N
R
OMgBr
H
MgBr
123
124 125
salt 122. Attack of the aldehyde group by the negatively polarised nitrogen gives
the cyclised product 123. Aromatisation of the benzene ring, assisted by the third
equivalent of the vinyl Grignard reagent, leads to the intermediate 124, acidic
workup of which yields the corresponding indole 125.
In conclusion, the Bartoli indole synthesis represents one of the best methods for
the preparation of 7-substituted indoles, as exemplified in the Lilly synthesis of the
7-cyanomethoxyindole 45.34
74
5.2 Conversion of 3-Nitropyridinium Salts into Indoles
The mechanism of an earlier reported synthesis of indoles, by the reaction of 3-
nitropyridinium salts with N-alkyl-2-ketimines (Method A) or with mixtures of the
corresponding ketones and amines (Method B), has been discussed at length by
Yurovskaya et al.111 Table 3 summarises some of the results reported by the
Table 3 Conversion of 3-nitropyridinium salts into polyalkylindoles
MeNH2
NR
Me
RR
RR
2
4
5
6
7
RN
Me
7
NMe
R
RR
R NO2
2
4
5
6
��������
X
+ HXHNO2
Entry
R2
R4
R5
R6
R7
Yield [%] A B
1
H
H
H
H
H
0
2
Me
H
H
H
H
3
7
3
H
Me
H
H
H
11
5
4
H
H
Me
H
H
0
5
Me
Me
H
H
H
22
6
Me
H
H
Me
H
4
6
7
H
Me
H
Me
H
13
10

75
8
Me
Me
H
Me
H
62
9
Me
H
Me
Me
H
87
10
Me
Me
Me
Me
H
55
11
11
Me
Me
Me
Me
Me
57
52
Russian workers. The yield of the reaction generally increases with the number of
alkyl substituents, and can be up to 87% (Entry 9). With non-substituted 3-
nitropyridinium salts, however, no indole formation was observed (Entry 1).
Initially, a "molecular design" for the formation of the indole skeleton from the
structural fragments of the starting materials was described (Scheme 39).
Scheme 39
������
������
NR
R
RR
R NO2
2
4
5
6+
����
1R R
N
�����������
�������
72
34
5
6
��������
��������
NR
R
RR
RR
���������
�������������
������
1
2
4
5
6
7
234
5
6
Reaction of 1,2,4,6-tetramethyl-3-nitropyridinium iodide with N-cyclohexyl
isopropylimine showed that the nitrogen atom in the resulting indole originated
from the imine moiety and not from the pyridinium salt. The C2-C3 moiety of the
pyridinium cation is incorporated in the construction of the pyrrole ring, and the C4-
C5-C6 block in the formation of the benzene ring. The three carbon moiety of the
imine is inserted between the above mentioned fragments, that is, in the formation
of both rings of the indole molecule, with R7 occupying the 7 position. These rules
76
were established by the introduction of alkyl substituents into different positions of
the 3-nitropyridinium ring, and by using a mixture of acetone-d6 and methylamine.
The suggested mechanism of this rather complex process involves a presumably
stepwise 4,6-meta-bridging of the enamine form of the 2-ketimine to the 3-nitro-
pyridinium cation 126 to give the bicyclic compound 127, as shown in Scheme 40.
Ring opening of compound 127 in a retro-Michael type fashion generates the
intermediate 128 with a non-aromatic six-membered ring. Attack of the β-carbon of
the cyclic enamine 129 at the carbon adjacent to the nitro group eliminates nitrite
and forms the fused cyclopropane system 130. Opening of the three-membered
ring takes place with aromatisation of the six-membered ring to give 131.
Compound 131 then undergoes spontaneous cyclisation, leading to the indole 132.
Some experimental data obtained by Yurovskaya et al. elucidates certain mecha-
nistic features of the reaction. When mixtures of secondary amines and the
appropriate ketones were used, o-N,N-(dialkylaminobenzyl) ketones were isolated
among other products. The formation of such compounds, which may be regarded
as stable, non-cyclisable analogues of the indole precursors, provides evidence
that the elimination of the nitro group takes place prior to the pyrrolidine ring
closure and aromatisation steps. Moreover, in addition to 4,6-meta-bridging,
necessary for indole formation, p-nitroanilines, resulting from a possible 2,4-meta-
bridging, were in some cases obtained. Elimination of the nitro group as nitrite was
confirmed by acidic treatment of the reaction mixtures. The presence of primary
amines caused N2 evolution due to the decomposition of intermediate
alkyldiazonium salts.

77
Scheme 40
132
131 130
129128
127
126
MeNH2-
RR
RR
NR
R
1
2
4
5
6
7
RR
RR
NR
R
MeHN
H
1
2
4
5
6
7
������
���
���
RHNR
R
RR
MeN
R 1
4
5
6
7
��
���
RHNR
R
R
RMeN
HR H
1
45
6
7
������
���
RHNR
R
R
R
NMeN
R
OHO
1
2
45
6
7
�����
����
��� ���
NR
R
R
R
NMeHN
OO
HR
R1
45
6
7
����
�� -H
1R H
NMe
RR
NO2N
R
R
26
5R
7
4
����
�� ��
1R H
26
5R
N
R
RNO2
R RMeN
7
4
����
���
��
NMe
R
RR
R NO2
2
4
5
6
N
R
H1R
7
�� ������
����
�������
1R H
NMe
R
NO2N
H
R
RR
2
5R
4
������ ���
�����
������
41R H
26
5R
N
R
NO2
R RMeN
HR
7
67
������
�� �����
2 �����
���
����
2
���
RHNR
R
R
R
NMeN
R
OOH
1
2
45
6
7
����
-H
NO2-
��
2
���
���
���
78
5.3 Results and Discussion
Reaction of the optically active imine 133 with a 3-nitropyridinium salt, using the
method described by Yurovskaya et al., could in principle result in formation of the
required enantiomerically enriched indole derivative 48b* (Scheme 41). The imine
Scheme 41
Br CNNH2��
��
50b*
OR7
Br CNN
R
����
7���
��
N
NO2
MeX
133
BrN
CN
R��
7
48b*
133 would be synthesised from condensation of the optically active amine 50b*
with an appropriate ketone.
The synthetic strategy outlined in Scheme 41 requires use of an unsubstituted 3-
nitropyridinium salt. It was therefore necessary first to validate the general results
claimed by the Russian chemists, because of their failure to observed any indole

79
formation from such pyridinium cations and, if successful, to then reexamine the
reactions with the simple 1-methyl-3-nitropyridinium salt.
Nitration of 2,4,6-trimethylpyridine (2,4,6-collidine) 134, following the method
described by Plazek, gave the corresponding 3-nitro derivative 135 in 26%
yield.112 Reaction of compound 135 in a sealed tube with an excess of methyl
iodide, gave the 3-nitropyridinium salt 136 in good yield113 (Scheme 42). The
reactions of com-
Scheme 42
N
Me
MeMe N
Me
MeMe
NO2
���
N
Me
MeMe
NO2
MeI
MeI
������oleum
KNO326% 71%
134 135136
pound 136 with various imines under different conditions are summarised in Table
4. Reaction with N-methyl-2-propylimine (137), prepared in 76% yield by
condensation of acetone with methylamine,114 gave as expected 1,2,4,6-
tetramethylindole (138), but in a much lower yield than reported by the Russian
workers (Entry 3). Treatment of compound 136 with N-methyl-2-butylimine (142),
synthesised by the same method, also resulted in a lower yield of the desired
product 143 (Entry 7). When 136 was treated with mixtures of methylamine and the
corresponding ketones (Method B) the yields were even lower, an observation that
was in general also made by the Russian chemists. Reaction of 137 with
compound 136 under acidic conditions resulted in decreased yield (Entry 1 and 2).
The desired catalytic effect, by shifting the equilibrium of the initial intermediates,
as proposed in the reaction mechanism, towards product formation, was not
observed. When the reaction was carried out in the presence of sodium 3-
nitrobenzenesulfonate (139) or 2,4-dinitrobenzenesulfonic acid (140) (10 mol%
80
respectively), however, the expected catalytic effect was obtained (Entries 4 and
5). These reagents probably assisted in the aromatisation of the proposed
intermediate 130. The best result in terms of catalysis was achieved with the
sodium salt of 2,4-dinitrobenzenesulfonic acid (141) (Entry 6). No changes in yield
SO3Na
NO2
SO3NaNO2
NO2
NO2
NO2
SO3H
139140 141
were observed by using larger amounts (2 equivalents) of the catalysts. Similar
improved results were also obtained from reaction of 136 with the imine 142 in the
presence of the catalyst 139 (Entry 8).
Attempts to introduce larger groups than methyl into the 7-position of the product
failed. Reaction of compound 136 with N-methyl-4-methyl-2-pentylimine (144,
prepared following the same method as previously described) or N-cyclohexyl-1-
phenyl-2-propylimine (145) did not lead to indole formation even in the presence
of the catalysts 139 or 141 (Entries 9-13). Compound 145 was prepared following
a literature procedure.115 When 136 was treated with a mixture of hydroxyacetone
and methylamine again no product was obtained (Entry 14). On the other hand,
from imines that contained a large substituent only on nitrogen, the corresponding
indoles could be isolated in moderate yields using method A or B (Entries 15 and
17 respectively). Treatment of compound 136 with N-cyclohexyl-2-propylimine
(146)116 or N-benzyl-2-propylimine (148)117 in the presence of sodium 3-
nitrobenzenesulfonate (139), however, did not significantly improve the yields
(Entries 16 and 18 respectively). When 136 was reacted with a mixture of acetone
and (+)-α-methylbenzylamine the corresponding optically active indole 150 was

81
Table 4 Reactions of 1,2,4,6-tetramethyl-3-nitropyridinium iodide 136 with different
imines
���
NMe
NO2
Me
MeMe
RN
R1
7
NRR
Me
Me
Me 17
136
Entry
Imine
R1
R7
Catalyst
Indole
Yield [%] A B
1
137
Me
H SO3H
Me 138
13
-
2
137
Me
H
EtCO2H
138
29
-
3
137
Me
H
-
138
38
4
4
137
Me
H
139
138
57
-
5
137
Me
H
140
138
60
-
6
137
Me
H
141
138
92
-
7
142
Me
Me
-
143
32
17
8
142
Me
Me
139
143
47
-
9
144
Me
iPr
-
-
0
-
10
144
Me
iPr
139
-
0
0
82
11
144
Me
iPr
141
-
0
-
12
145
C6H11
Ph
-
-
0
-
13
145
C6H11
Ph
139
-
0
-
14
-
Me
OH
-
-
-
0
15
146
C6H11
H
147
59
71
16
146
C6H11
H
139
147
65
-
17
148
PhCH2
H
-
149
58
48
18
148
PhCH2
H
139
149
62
-
19
-
PhCHCH3
H
-
150
-
40
20
-
PhCH(CH2)3CH3
H
-
-
-
0
obtained in 40% yield (Entry 19). From reaction of the pyridinium salt 136 with α-
butylbenzylamine (105) and acetone a product was obtained in low yield, the
structure of which could not be assigned to the expected indole from its proton
NMR spectrum (Entry 20). The mass fragmentation, however, showed a molecular
ion that could correspond to the desired product.

83
Having established the general accuracy of the Russian work with 1,2,4,6-
tetramethyl-3-nitropyridinium iodide (136), and shown that the transformation can
be effectively catalysed with nitrobenzenesulfonic acids, we next turned attention
to the reactivity of the simple unsubstituted pyridinium salt (153), essential for the
formation of 48b*.
Oxidation of 3-aminopyridine (151) with hydrogen peroxide in oleum, gave 3-
nitropyridine (152) in low yield.118 Compound 152 was methylated in high yield to
give 1-methyl-3-nitropyridinium iodide (153), following the same procedure as
before (Scheme 43). Table 5 summarises the results obtained from reaction of
Scheme 43
���
MeI
93%15%H2O2
����oleum
N
NO2
MeI
N
NO2
N
NH2
151 152153
the 3-nitropyridinium cation 153 with different imines. Unfortunately, compound 153
did not undergo the reaction to yield the corresponding indoles, not even in the
presence of the catalyst 139 (Entries 2 and 4). These findings are thus in full
accordance with those reported by Yurovskaya et al.
The above results clearly indicate that although an optically active product and, in
some cases, improved yields using various catalysts could be obtained, this
synthesis was not going to be the method for the construction of the required 7-
substituted indole derivative 48b*. The reaction is limited to the preparation of
polyalkylindoles.
84
Table 5 Reactions of 1-methyl-3-nitropyridinium iodide 153 with different imines
���
NMe
NO2 RN
Me
7
N
R Me7
153
Entry
Imine
R7
Catalyst
Yield [%] A B
1
137
H
-
0
0
2
137
H
139
0
-
3
142
Me
-
0
-
4
142
Me
139
0
-

85
6. Synthesis of 4- and 7-Oxo-4,5,6,7-tetrahydroindoles
The first generally useful method for the preparation of 4-oxo-4,5,6,7-tetrahydro-
indoles was described by Stetter and Lauterbach in 1962.119 In this procedure, 1,3-
cyclohexanediones were alkylated with α-halo ketones to give the 4-oxo-4,5,6,7-
tetrahydrobenzofuran derivatives 154, which on treatment with ammonia or primary
amines, in a steel bomb at 150 °C, gave the corresponding tetrahydroindoles 155
in high yields (Scheme 44). In contrast, reaction of 1,3-cyclohexanedione with
Scheme 44
155
154
R"'NH2
OX
R'R"
H+
��������
2)
1)
O
NR'
R'
R"
R"'
O
O
R'
R"R'
O
OHR
����
ethyl bromopyruvate gave 4-oxotetrahydrobenzofuran-3-carboxylic acid 156, from
initial aldol condensation. Reaction of 156 with ammonia resulted in formation of
86
the decarboxylated parent 4-oxotetrahydroindole 157 in moderate yield (Scheme
45). Matsumoto and Watanabe synthesised the indole 157 in 96% yield by heating
Scheme 45
NH3
������
����
158
O
O
2) H+
1) ClO
������
156
NH3
O
OH
O
O
CO2H
O
NH
1)
2)���
H+
OBr
CO2Et
157
4-oxo-4,5,6,7-tetrahydrobenzofuran (158) in the presence of ammonia under
similar conditions as described above.120 The furan 158 was obtained in good yield
from initial aldol condensation of 1,3-cyclohexanedione with chloroacetaldehyde in
the presence of sodium bicarbonate (Scheme 45). Analogous reaction of 158 with
a variety of primary amines afforded the corresponding 1-substituted 4-
oxotetrahydroindoles in high yields.
Torii et al. described the synthesis of novel cyclohexanone derivatives via an
electrochemical C-C coupling reaction.121,122 When a basic mixture of, for
example, 1,3-cyclohexanedione, ethyl vinyl ether and ethanol was electrolysed at
room temperature under a constant current, a 3:4 mixture of the products 159 and

87
160 was obtained (Scheme 46). Both compounds could be easily converted to 4-
Scheme 46
O
OH
���OEt
EtOH
O
OOEt +
O
OH
OEt
OEt
159 160
oxotetrahydroindoles in moderate to good yields by reaction with ammonia or the
corresponding primary amines in a sealed tube at 150 °C.
When 1,3-cyclohexanediones are condensed with aminoacetaldehyde dimethyl
acetals the corresponding enamines 161 are obtained, which can be cyclised with
acid to 4-oxo-4,5,6,7-tetrahydroindoles in moderate yields123 (Scheme 47). The
use of 1,2-cyclohexanediones can result in formation of the corresponding 7-
oxotetrahydroindoles. 1-Methyl-7-oxo-4,5,6,7-tetrahydroindole (162), for example,
was obtained using this general method, but only in very low yield124 (Scheme 48).
Recently a modification of this method, for the synthesis of 3-substituted 4-
oxotetrahydroindoles, has been reported by Edstrom.125 Reaction of 1,3-
cyclohexanedione with the sodium salt derived from sarcosine (N-methylglycine)
afforded the condensation adduct 163, which was isolated as its crude acid.
Compound 163 was heated in acetic anhydride to give 3-acetoxy-4-oxo-4,5,6,7-
tetrahydroindole (164) in 80% yield (Scheme 49).
88
Scheme 47
161
H+
R'NH OMe
OMe O
N
OMeMeO
R'R
R
����
O
NRR R'
O
OHRR
��
Scheme 48
Me
NH OMe
OMe
N
OMeMeO
MeO
����
NMeO
OHO
�� CH3COOH
16213%

89
Scheme 49
164
163
O
OH
��������
MeN CO2NaH
O
N
CO2H
Me
������
Ac2O
O
NH
OAc
A different approach for the synthesis of 4- and 7-oxotetrahydroindoles, starting
from pyrrole, was found by Julia and Pascal.126 When the pyrrole N-magnesium
derivative 165 was reacted with 4-chlorobutyronitrile, a mixture of the 2- and 3-
substituted pyrrole isomers was obtained (Scheme 50). Conversion of the nitrile to
the carboxylic acid group and subsequent ring closure gave the corresponding 4-
and 7-oxotetrahydroindoles 157 and 166.
In another procedure, Friedel-Crafts acylation of the trisubstituted pyrrole 167 gave
the derivative 168 (Scheme 51). Reduction of the carbonyl group in 168 was found
to proceed best via hydrogenation with Pd/C. The diester 169 was cyclised in
polyphosphoric acid (PPA) to give the 7-oxotetrahydroindole 170 in moderate
yield.127
90
Scheme 50
H2O2)
165 ������
+
���
NMgX
Cl CN1)
NH
CN
NH
CN
NH
CO2H
NH
CO2H
+
N
O
H
NHO
����
+
157 166

91
Scheme 51
N
O H
Me
Et
������
NEt
Me
EtO2C
H
MeO2C
NEt
Me
EtO2C
H
MeO2C
O
���
N
Me
EtO2C
H
Et
PPA
��
H2Pd/C
MeO2C Cl
O
AlCl3
167 168
169170
A more elegant method involves the regioselective AlCl3-catalysed 3-acylation of
1-(phenylsulfonyl)pyrrole (171)128 (Scheme 52). Clemmensen reduction of 172
gave 173 in high yield. Treatment of 173 with oxalyl chloride gave 174, which
cyclised even in the absence of any catalyst to give the 7-oxotetrahydroindole 175.
The production of 175 was found to be more efficient when 174 was reacted with
trifluoroacetic anhydride. Subsequent hydrolysis gave 166 in high yield.
Reaction of 2,3-epoxy-3-methylcyclohexanones 176 with benzylamine has been
reported to yield 2-amino-3-methylcyclohex-2-enones 177, which on treatment with
dimethylformamide dimethyl acetal (dmfdma) cyclised to the corresponding 1-
benzyl-7-oxotetrahydroindoles 178 in high yields129 (Scheme 53). Benedetti et al.
prepared 7-oxotetrahydroindoles 180 from reaction of N-substituted enamines 179
92
Scheme 52
NaOH
(COCl)2
Zn/HCl������
AlCl3
NSO2Ph
O
O
O
���
+NSO2Ph
O
O
HO
NSO2Ph
O
HO����
NSO2Ph
O
Cl
��
NSO2PhO
������
NHO
171 172
173174
175 166
(CF3CO)2O

93
Scheme 53
������
O
RR
���
O
OR
R
H2O2 ����
ONHBn
RR
O
NBn
RR
dmfdma
K2CO3
BnNH2
176 177
178
(analogous to 177) with nitroolefins130 (Scheme 54). The reaction was found to be
Scheme 54
3
2
1O
NR
R
R
O
O ���
ONHR
RNH21
1 NO2
R
R2
3
179 180
���
very substrate dependent. In fact, when R1 was n-butyl, the reaction proceeded
rapidly and almost quantitatively.
94
6.1 Results and Discussion
The shortest and most effective method for the synthesis of 1-substituted 4-
oxotetrahydroindoles is that described by Matsumoto and Watanabe, from reaction
of an aqueous ethanolic solution of 4-oxo-4,5,6,7-tetrahydrobenzofuran (158) with
primary amines in a sealed tube at 150 °C for twelve hours.120 In principle, it
should be possible to perform the analogous reaction with the 7-oxo isomer 181,
thus opening a new and better route for the synthesis of 1-substituted 7-
oxotetrahydroindoles. Reaction of the optically active amine 50b* with 181 could
therefore give the 7-oxotetrahydroindole derivative 182*, and this in turn could be
converted into the required enantiomerically enriched 1,7-disubstituted indole 48b*,
after aromatisation and alkylation (Scheme 55).
Scheme 55
48b*
182*
181
50b*
BrN
CN
OCN
����
BrN
CN
O������
O
O
���BrNH2
CN

95
Initial studies towards the synthesis of 7-oxo-4,5,6,7-tetrahydrobenzofuran (181)
were carried out by Justin Cowell.131 The best result was obtained from reaction of
1,2-cyclohexanedione with chloroacetaldehyde in the presence of sodium
bicarbonate, as previously reported for the synthesis of the 4-oxo isomer 158
(Scheme 56). The modest yield stems probably from the lower acidity of the
Scheme 56
181
O
O
����
OOH
ClO
1)
2) H+
40%
hydrogens at C-3 in 1,2-cyclohexanedione compared to the highly activated
methylene hydrogens at C-2 in 1,3-cyclohexanedione. A longer-winded Wittig
approach gave 181 in only 26% overall yield (Scheme 57). Thus, reaction of
commercially available (but expensive) furan-3-carbaldehyde with the ylid derived
from 3-chloropropanoic acid gave the 4-(3-furyl)butenoic acid (183) in 36% yield.
Compound 183 was hydrogenated at atmospheric pressure over a palladised
charcoal catalyst to yield the butanoic acid 184. The saturated acid proved to be
fairly unstable and was thus reacted in crude form with oxalyl chloride to produce
4-(3-furyl)butanoyl chloride (185). The desired 7-oxotetrahydroindole 181 was
obtained from intramolecular tin(IV) chloride catalysed Friedel-Crafts reaction of
185 as reported by Walsh and Stone.132 A possible alternative Paal-Knorr
approach had to be abandoned because of failure to obtain the required tricarbonyl
intermediate 186.
96
Scheme 57
185 184
183
O
O
����
Ph3P OH
OCl
OHO
O
NaH
������
Pd/CH2
OHO
O�������� (COCl)2
OCl
O
����
SnCl4
181
186
O
OOH

97
The next step was to find out if condensation of the furan 181 with primary amines
would give the corresponding 7-oxotetrahydroindoles, and if chirality would be
retained during the course of the reaction. Homochiral α-methylbenzylamine was
chosen as the condensation partner for the initial model studies, because it is
benzylic and one of the cheapest optically pure amines available. Cowell showed
that treatment of 181 with an excess of α-methylbenzylamine under an inert
atmosphere and different conditions such as use of a solvent (MeOH), addition of
protic acid catalysts (HCl, acetic acid) and variation of temperature and reaction
time, always gave complex mixtures from which the desired product could not be
isolated. Three other products were instead obtained, which were found to be 7-
(N-α-methylbenzylimino)-4,5,6,7-tetrahydrobenzofuran (187), acetophenone and
N-(α-methylbenzyl)-α-methylbenzylimine (188) (Scheme 58). From reaction with
Scheme 58
Ph NH2
181188
187
Ph O+Ph N Ph+
N
O
Ph
O
O
����
the less sterically demanding benzylamine, however, the desired reaction took
place to some extent to give 1-benzyl-7-(N-benzylimino)-4,5,6,7-tetrahydroindole
(189), in addition to the expected two other products (Scheme 59). A large excess
of benzylamine was required in order to consume all the starting furan 181. The
desired 7-oxotetrahydroindole 190 was obtained from hydrolysis of 189 with
saturated ammonium chloride, but only in 10% yield. Attempts to increased the
nucleophilicity of α-methylbenzylamine by using its lithium salt, prepared in situ
98
Scheme 59
���PhCH2NH2
O
O
N
N
Ph
Ph
+ Ph N Ph + Ph O
����
aq NH4Cl
N
PhO
189181
190
from reaction with LDA, led only to the recovery of starting material. In this case
the lithium species acted as a base rather than a nucleophile and simply
deprotonated 181, as indicated from reaction with methyl iodide where the only
product isolated was shown to be 6-methyl-7-oxo,4,5,6,7-tetrahydrobenzofuran
(191).
191
O
O
From the above results obtained by Cowell, it seemed that the drastic conditions
previously reported by Matsumoto and Watanabe were going to be necessary. An
aqueous ethanolic solution of 7-oxo-4,5,6,7-tetrahydrobenzofuran (181) and
homochiral α-methylbenzylamine was therefore heated in a sealed tube for twelve
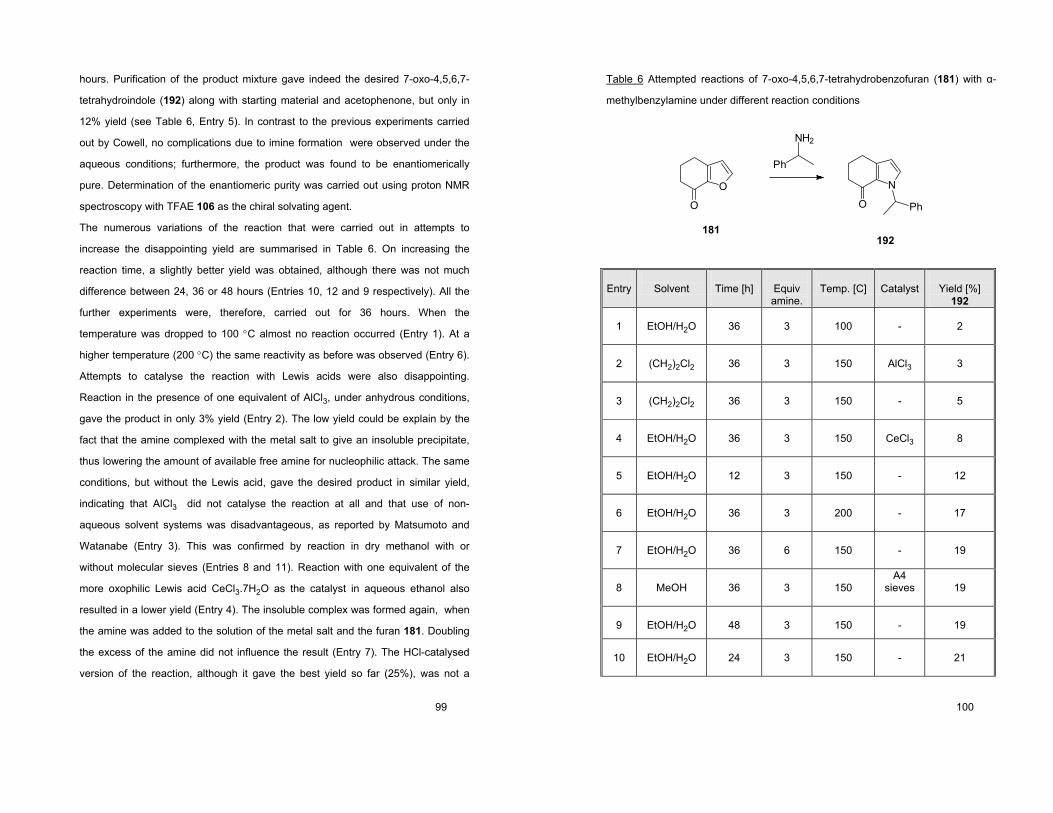
99
hours. Purification of the product mixture gave indeed the desired 7-oxo-4,5,6,7-
tetrahydroindole (192) along with starting material and acetophenone, but only in
12% yield (see Table 6, Entry 5). In contrast to the previous experiments carried
out by Cowell, no complications due to imine formation were observed under the
aqueous conditions; furthermore, the product was found to be enantiomerically
pure. Determination of the enantiomeric purity was carried out using proton NMR
spectroscopy with TFAE 106 as the chiral solvating agent.
The numerous variations of the reaction that were carried out in attempts to
increase the disappointing yield are summarised in Table 6. On increasing the
reaction time, a slightly better yield was obtained, although there was not much
difference between 24, 36 or 48 hours (Entries 10, 12 and 9 respectively). All the
further experiments were, therefore, carried out for 36 hours. When the
temperature was dropped to 100 °C almost no reaction occurred (Entry 1). At a
higher temperature (200 °C) the same reactivity as before was observed (Entry 6).
Attempts to catalyse the reaction with Lewis acids were also disappointing.
Reaction in the presence of one equivalent of AlCl3, under anhydrous conditions,
gave the product in only 3% yield (Entry 2). The low yield could be explain by the
fact that the amine complexed with the metal salt to give an insoluble precipitate,
thus lowering the amount of available free amine for nucleophilic attack. The same
conditions, but without the Lewis acid, gave the desired product in similar yield,
indicating that AlCl3 did not catalyse the reaction at all and that use of non-
aqueous solvent systems was disadvantageous, as reported by Matsumoto and
Watanabe (Entry 3). This was confirmed by reaction in dry methanol with or
without molecular sieves (Entries 8 and 11). Reaction with one equivalent of the
more oxophilic Lewis acid CeCl3.7H2O as the catalyst in aqueous ethanol also
resulted in a lower yield (Entry 4). The insoluble complex was formed again, when
the amine was added to the solution of the metal salt and the furan 181. Doubling
the excess of the amine did not influence the result (Entry 7). The HCl-catalysed
version of the reaction, although it gave the best yield so far (25%), was not a
100
Table 6 Attempted reactions of 7-oxo-4,5,6,7-tetrahydrobenzofuran (181) with α-
methylbenzylamine under different reaction conditions
Ph
NH2
O
O
181����
O
N
Ph
192
Entry
Solvent
Time [h]
Equiv amine.
Temp. [C]
Catalyst
Yield [%]
192
1
EtOH/H2O
36
3
100
-
2
2
(CH2)2Cl2
36
3
150
AlCl3
3
3
(CH2)2Cl2
36
3
150
-
5
4
EtOH/H2O
36
3
150
CeCl3
8
5
EtOH/H2O
12
3
150
-
12
6
EtOH/H2O
36
3
200
-
17
7
EtOH/H2O
36
6
150
-
19
8
MeOH
36
3
150
A4 sieves
19
9
EtOH/H2O
48
3
150
-
19
10
EtOH/H2O
24
3
150
-
21

101
11
MeOH
36
3
150
-
22
12
EtOH/H2O
36
3
150
-
23
13
EtOH/H2O
36
3
150
HCl
25
significant improvement (Entry 13). Despite all the attempts, no real success was
achieved in terms of optimisation.
The furan 181 was then reacted with ammonia and a variety of simple primary
amines in a sealed tube at 150 °C for 36 h. The yields of 7-oxotetrahydroindoles
were, as expected, not very high and in general much lower than those obtained
with the 4-oxo isomer 158 (See Table 7). The synthesis of the corresponding 4-
oxotetrahydroindole derivatives was carried out by Sven Baum,133 except for
compound 196. When both 7- and 4-oxotetrahydrobenzofuran were treated with
the more complex amine 199, the products obtained were those derived from
HCl.H2N Ph
CO2Me
199
hydrolysis and decarboxylation of the ester group, namely the corresponding N-
benzyloxotetrahydroindoles 190 and 198. This could be potentially a problem,
since ultimately the functionalised amine 50b would need to survive the harsh
conditions, in order to yield the required 7-oxotetrahydroindole 182. As previously
mentioned, besides formation of the 7-oxotetrahydroindoles the formation of the
corresponding carbonyl compounds derived from the benzylic amines used was
also observed (Entries 1, 3 and 4). In the case where the amine was not benzylic,
no carbonyl compounds were obtained (Entries 2, 5, 6 and 7). In the 4-oxo series
102
Table 7 Reactions of 7-oxo-4,5,6,7-tetrahydrobenzofuran (181) with various
primary amines
O
NR
���RNH2
O
O EtOH/H2Osealed tube
36 h181
Entry R Product Yield [%] Product
*
Yield [%]*
1 PhCHC4H9 193 17 196 8
2 CH3 162 22 197 89
3 PhCHCH3 192 23 83
4 PhCH2 190 30 198 83
5 C6H11 194 34 36
6 H 166 36 157 91
7 PhCH2CH2 195 46 71
*Yields obtained from reaction of 4-oxo-4,5,6,7-tetrahydrobenzofuran (158) with the correspon- ding amines under the same conditions, except that the reaction mixture was left to react for twelve hours.
the corresponding carbonyl compounds were never formed, whether the amine
was benzylic or not. With this information it is now possible to propose a
reasonable mechanism for the oxidation of benzylic amines to the corresponding
carbonyl derivatives during the course of the reaction, as shown in Scheme 60.
Nucleophilic attack of the amine at the 2-position of 181 would result in formation

103
Scheme 60
202
201
200
181
Ph
O+
OOH
NH2
������
�� ���
OOH
N PhH
OH
��������
����
��������
����
HOHO
OH
N Ph
����
OOH
N Ph
��������
����
������
������������ O
O
NPh
H H����
������
������
������ ����
H2N PhO
O
OOH
N PhH
104
of the enamine 200. A sequence of tautomeric equilibrium reactions would give the
imine 201 as the key intermediate, the driving force being the conjugation to the
phenyl ring. This compound would then be readily hydrolysed by the water present
to form the corresponding carbonyl derivative of the amine and compound 202.
The amine 202, however, could never be isolated since it is probably far too
reactive under the conditions to survive unchanged. Furthermore, the reactivity of
the 7-oxotetrahydrobenzofuran 181 is certainly lower than that of the 4-oxo isomer
158. The two intermediates involved in the cyclisation reaction are quite different.
Initial nucleophilic attack of the amine to 7-oxotetrahydrobenzofuran 181 forms
203, where the equilibrium lies preferentially to the left, thus preventing ring closure
203O
O
NHR
���� ���
OOH
NHR
and decreasing reactivity. In the case of 4-oxotetrahydrobenzofuran 158, the initial
intermediate from attack of the amine is 204 with the tautomer on the right being
204
O
NHR
OH
��� ���
O
NHR
O
the major component, therefore, increasing reactivity and product formation. All of
these observations account for the lower yields obtained in the reaction of 181
with primary amines compared with 158.

105
Although the yields of 7-oxotetrahydroindoles were not as high as desired, the
synthesis of a series of 1-substituted 7-oxotetrahydroindoles could be achieved
successfully with this novel route. The fact that the reaction is enantiospecific fulfils
all the conditions needed for the synthesis of the required enantiomerically
enriched 1-substituted 7-oxo derivative 182*. But first, it was necessary to
investigate the aromatisation of such systems into 7-substituted indoles and to see
if this transformation would proceed without racemisation. How we developed a
novel procedure for this purpose is described in the next chapter.
106
7. Aromatisation
The classical method for dehydrogenation of 4-oxo-4,5,6,7-tetrahydroindoles to the
corresponding 4-hydroxyindoles, involves heating with palladium on charcoal in an
aromatic hydrocarbon solvent such as cumene or mesitylene.134 4-Hydroxyindole
was thus prepared from 157 in good yield when mesitylene was used.135
Treatment of a 7-oxotetrahydroindole under similar conditions also gave the co-
rresponding aromatised product.127 When Baum attempted to aromatise the 4-
oxotetrahydroindoles 157 and 197 using this general method, however, only
starting materials were recovered133 (Scheme 61). Different concentrations of
Scheme 61
RN
O
Pd/C
157: R = H197: R = Me
NR
OH
������
Pd/C systems (5 and 10%) as well as different solvent mixtures (cumene/mesi-
tylene) and reaction times were tried, but in no case was the desired result
obtained. As catalytic dehydrogenation is quite sensitive to the catalyst, it is
probable that our particular sample of catalyst was inferior in activity.
Baum further showed that treatment of 197 with dichlorodicyanobenzoquinone
(DDQ) yielded a polymeric black residue. Reaction of the corresponding silyl enol
ether 205, prepared from reaction of 197 with LDA and trimethylsilyl chloride
(TMSCl) following a procedure described by Fleming and Paterson,136 with DDQ
or trityl tetrafluoroborate137 in the presence of collidine gave a similar result

107
(Scheme 62). Remers et al. reported that 4-hydroxyindoles were found to be
Scheme 62
197 205
DDQ
N
OTMS
Me
2)TMSCl
1) LDA ����
N
O
MeNMe
OH
���
unstable to DDQ unless they contained an additional electron-withdrawing group at
C-5.138 It seems therefore likely that dehydrogenation occurred, but the products
were unstable in the presence of DDQ. On the other hand, when a solution of 7-
oxotetrahydrobenzofuran (181) in dichloromethane was treated with DDQ at room
temperature or under reflux conditions, no decomposition took place and all the
starting material was recovered. The corresponding silyl enol ether 206, prepared
using the same method as before, could be aromatised to 7-hydroxybenzofuran
(207) with Pd(OAc)2139,140 (Scheme 63).
Scheme 63
����
O
OH
O
O
����1) LDA
2)TMSCl O
OTMS
206181
Pd(OAc)2
207
108
Reaction of the 4-oxotetrahydroindole 197 with LDA at -40 °C and subsequent
quenching with phenylselenium chloride following the general procedure described
by Williams and Nishitani,141 gave the corresponding α-phenylseleno derivative
208 (Scheme 64). Oxidative elimination of 208 with hydrogen peroxide, however,
Scheme 64
197 208
N
O
Me
PhSe
2) PhSeCl
1) LDA���
N
O
Me
NMe
OH
����
H2O2
������
N
O
Me
PhSeO
gave only a multispot tlc as shown by Baum.133 Probably the same reasons as
referred to above (instability of the 4-hydroxyindole under the oxidative conditions)
were responsible for the observed result.
When a solution of the 7-oxotetrahydroindole 192 in benzene was treated with
commercially availalbe MnO2 at room temperature or under reflux conditions no
reaction occurred.142,143 Treatment of 192 with ferric hexafluorophosphate in

109
dichloromethane at room temperature and under nitrogen, led also only to recovery
of the starting material.144,145
Kotnis reported that 1,3-cyclohexanediones can be aromatised to the corres-
ponding resorcinols in high yields with iodine and methanol.146 Attempted aroma-
tisation of the homochiral 7-oxotetrahydroindole 192 following this method,
however, gave only a mixture of the 6- and 3-iodo isomers 209 and 210, which
could be separated by column chromatography (Scheme 65). The 6-iodo derivative
Scheme 65
21020919260%
I2
LDA
��
10%
O
N
Ph
I
+
O
N
Ph
II2
MeOH���
O
N
Ph
209, which was shown to be a diastereomeric mixture from its proton NMR
spectrum, gave a single spot on tlc and could not be separated into the single
diastereomers. This was not a problem, since base catalysed dihydroiodination of
the mixture 209 would produce the same 7-hydroxyindole. Treatment of 192 with
LDA, prepared in situ from reaction of diisopropylamine and n-butyllithium, followed
by addition of iodine gave 209 in 15% yield; none of the 3-iodo isomer or 7-
hydroxyindole was formed. This particular reaction was found to proceed very
slowly, most of the starting material being recovered unchanged. When 209 was
heated in dichloromethane in the presence of triethylamine, however, the desired
110
7-hydroxyindole 211 was obtained, but only in low yield, the starting material being
the major component of the reaction mixture. Treatment of 209 with neat 1,8-
diazabyciclo[5.4.0]-undec-7-ene (DBU) at 80 °C resulted in consumption of all the
starting material, but still the product 211 was isolated in low yield. Reaction with
DBU under the same conditions as above, followed by in situ alkylation with
bromoacetonitrile resulted in formation of the corresponding 7-cyanomethoxyindole
212 but only in 20% yield. The 7-hydroxyindole 211 was found to decompose
readily, as did also the 6-iodo precursor 209, and this was probably the reason for
the low yields obtained. Finally, the reaction was carried out at room temperature.
After 15 min all the starting material 209 had disappeared by tlc and the reaction
mixture was worked up. Subsequent alkylation of the crude isolated 7-
hydroxyindole 211 with bromoacetonitrile in the presence of potassium carbonate
gave the desired 7-cyanomethoxyindole 212 in 48% overall yield (Scheme 66). A
very good result, however, was the fact that aromatisation and introduction of the
required O-nitrile chain proceeded without loss of optical activity ( α D20 132.5).
Aromatisation of several 4-oxotetrahydro- into the corresponding 4-hydroxyindoles,
including an optically active one, using this procedure was achieved by Baum.133
The yields were in general higher than obtained with 192 (up to 83%), since no
pyrrole ring iodination and instability problems were encountered with either the 5-
iodo-4-oxo-tetrahydro- or the 4-hydroxyindoles. This represents a novel and milder
route for the synthesis of 4- and 7-substituted indoles, via a selective
halogenation/dehydrohalogenation sequence of the 4- and 7-oxo derivatives,
compared with other known methods based on the same stragegy.147-149

111
Scheme 66
48%212
K2CO3
���
O
N
Ph
CN
Br CN
211
OH
N
Ph
DBU����
209
O
N
Ph
I
112
8. The final Battle towards LY290154
Having established a route for the synthesis of homochiral 1-substituted 7-
oxotetrahydroindoles and shown that aromatisation of the latter proceeds without
loss of optical activity, we finally turned our attention to the preparation of the
enantiomerically enriched 7-cyanomethoxyindole derivative 48b*, required as the
key intermediate for the asymmetric synthesis of LY290154 35. An aqueous
ethanolic solution of racemic 50b was therefore reacted with 7-oxotetrahydro-
benzofuran (181) in a sealed tube at 150 °C in the hope of producing the desired
7-oxotetrahydroindole derivative 182, which would be converted to 48b after
aromatisation and O-alkylation as described in the previous chapter. The same
reaction sequence with the optically active amine 50b* would thus give 48b*.
Unfortunately the desired transformation did not take place; instead the lactam 213
was obtained in modest yield (Scheme 67).
The intramolecular cyclisation of 50b could in principle be avoided by converting
the nitrile into the tetrazoyl group,150-152 which is present in the target molecule 35.
When a dimethoxyethane solution of the amine 50b was refluxed with tributyltin
azide, prepared from reaction of sodium azide with tributyltin chloride, an oil was
obtained, the structure of which could not be assigned unambiguously to the
desired aminotetrazole 214 on the basis of its proton NMR or mass spectra
(Scheme 68). Reaction without solvent gave the same result. On the other hand,
reaction of the ketone 110 and the oxime ether 111 using the same conditions as
above resulted in formation of the corresponding tetrazoles 215 and 216
respectively (Scheme 69). The reaction was found to give better yields when no
solvent was employed. The aminotetrazole 214 could now be approached via
reduction of the oxime ether 216 (Scheme 70). Unfortunately, all the attempts to
reduce 216 with a borane-THF complex, following the same method as described
in Chapter 4.1, failed. Instead, a solid was isolated which could not be identified
from its proton NMR spectrum.
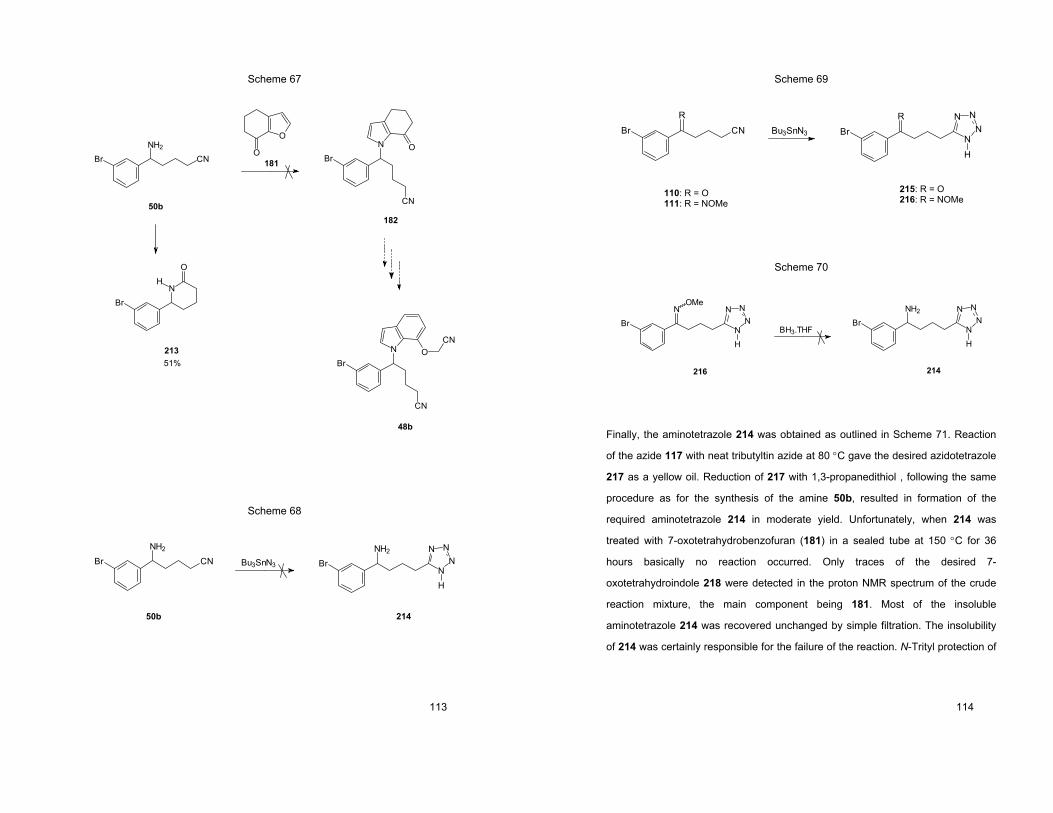
113
Scheme 67
48b
��
BrN
CN
OCN
����
��
182
BrN
CN
O
��
50b
Br CNNH2
O
O
BrN
OH
���181
21351%
Scheme 68
21450b
Br CNNH2
����Bu3SnN3 BrN
NNN
H
NH2
114
Scheme 69
Br CNR ���
���Bu3SnN3 BrR
NN
NN
H
110: R = O111: R = NOMe
215: R = O216: R = NOMe
Scheme 70
214
BrN
NNN
H
NH2
BrN
NN
NN
H
OMe
216
BH3.THF
������
Finally, the aminotetrazole 214 was obtained as outlined in Scheme 71. Reaction
of the azide 117 with neat tributyltin azide at 80 °C gave the desired azidotetrazole
217 as a yellow oil. Reduction of 217 with 1,3-propanedithiol , following the same
procedure as for the synthesis of the amine 50b, resulted in formation of the
required aminotetrazole 214 in moderate yield. Unfortunately, when 214 was
treated with 7-oxotetrahydrobenzofuran (181) in a sealed tube at 150 °C for 36
hours basically no reaction occurred. Only traces of the desired 7-
oxotetrahydroindole 218 were detected in the proton NMR spectrum of the crude
reaction mixture, the main component being 181. Most of the insoluble
aminotetrazole 214 was recovered unchanged by simple filtration. The insolubility
of 214 was certainly responsible for the failure of the reaction. N-Trityl protection of

115
Scheme 71
181
214
117
Bu3SnN3
BrN
NNN
H
NH2
��
HS SH
217
BrN
NNN
H
N3
BrN3
CN ���
43%
55%
O
O���
BrN
NN N
N H
O
218
���������
BrN
NN N
N H
OCN
116
the tetrazole moiety could result in increased solubility. Treatment of a suspension
of 214 in dichloromethane at room temperature with pyridine followed by trityl
chloride resulted in formation of the N-protected derivative 219153 (Scheme 72).
Scheme 72
214
BrN
NNN
H
NH2 ���Ph3CCl
pyridine
BrN
NNN
CPh3
NH2
219
Sealed tube reaction of 219, however, was not attempted because the trityl group
dropped off very easily under aqueous conditions.
The results obtained so far indicated that the required 7-oxotetrahydroindole
derivative 182 was not going to be produced from sealed tube reaction of the
amine 50b with 7-oxotetrahydrobenzofuran (181). A different approach for the
construction of 182 had therefore to be considered. Based on the work reported by
Kasum et al.129 (see Chapter 6), commercially available 3-methyl-2-cyclohexene-
1-one was treated with basic hydrogen peroxide to give the 2,3-epoxy ketone 220
in moderate yield154 (Scheme 73). Reaction of 220 with benzylamine in aqueous
methanol under reflux conditions gave the expected enamine 221, but in a much
lower yield than previously reported by the Australian chemists. When 221 was
treated with N,N-dimethylformamide dimethyl acetal (dmfdma) at 150 °C the
desired 1-benzyl-7-oxotetrahydroindole 190 could be isolated, but again in a much
lower yield than obtained by Kasum et al.
.

117
Scheme 73
19018%
22113%
22047%
O
����
O
O
���
ONHBn
N
O Bn
BnNH2
Me2NCH(OMe)2���� 150 ºC
H2O2
K2CO3
Having shown that 1-substituted 7-oxotetrahydroindoles could be prepared via this
general method, the remaining question was: would it also be stereospecific?
Reaction of 220 with homochiral α-methylbenzylamine using the same conditions
as before gave the expected enamine 222, but again in very low yield (see Table
8, Entry 3). A good result, however, was the fact that the product was shown to be
optically active. The yield of the enamine formation had therefore to be improved.
The results obtained from reaction of 222 with α-methylbenzylamine under
different reaction conditions are summarised in Table 8. Since at room temperature
no product formation was observed, all the experiments were carried out under
reflux conditions. An attempt to catalyse the reaction with the Lewis acid
CeCl3.7H2O155 (10 mol%) failed; instead a multispot tlc was obtained from which
the desired product could not be isolated (Entry 1). Reaction in the presence of
118
Table 8 Attempted reactions of 2,3-epoxy-3-methylcyclohexanone (220) with
α-methylbenzylamine under different reaction conditions
222
Ph NH2
220
O
O ���
ONH
Ph
Entry
Solvent Catalyst Yield [%]
1
MeOH/H2O CeCl3.7H2O 0
2
MeOH/H2O HCl 0
3
MeOH/H2O - 20
4
CH3CN/H2O
- 21
HCl gave a similar result (Entry 2). Using aqueous acetonitrile instead of aqueous
methanol as the solvent gave the same yield as before (Entry 4). When the
reaction was carried out with one equivalent CeCl3.7H2O in refluxing aqueous
methanol, a totally unexpected result was obtained. None of the desired enamine
222 was formed; instead, the cyclic 2-chloro-2-ene-1-one 223 and the toluidine
derivative 224 were isolated through column chromatography (Scheme 74). At
room temperature no reaction occurred. Compound 224 showed no optical activity,
indicating that racemisation of the benzylic position of the amine occurred during

119
Scheme 74
9%35%
+
OCl����
����
O
OCeCl3.7H2O
MeOH/H2Oreflux
Ph NH2
NH
Ph
220 223 224
the transformation. When the reaction was repeated in the absence of the amine
the expected chloro derivative 223 was obtained in high yield. Use of CeCl3 under
anhydrous conditions, however, gave the 2-chloro-3-hydroxy-1-one 225 in good
yield (Scheme 75). Simple refluxing of 225 in aqueous methanol did not produce
Scheme 75
1
OCl
OHMeCN
CeCl3 ����
O
O
220 22574%
����MeOH/H2O
OCl
HCl
223
223. In the presence of acid, however, the enone 223 was obtained quantitatively.
Based on the observation that CeCl3.7H2O is somehow involved in the elimination
step to give 223 from 225, it is now possible to postulate a reasonable mechanism
for the formation of racemic 224. The mechanism proposed by the Australian
chemists for the synthesis of the enamines 227 is shown in Scheme 76. The first
120
Scheme 76
O
NHR���
NRHO
OH
���
������
���
O
NHRHO
H2O��� ���
NR
O
226 227
step is straightforward imine formation from reaction of the 2,3-epoxy ketone with
the amine, after which reaction with water ring opens the epoxide via the
aziridine intermediate 226. Compound 226 is then converted into the enamine 227
after opening of the three membered ring followed by elimination of water. It is
likely that in the CeCl3.7H2O catalysed reaction of 220 with homochiral α-
methylbenzylamine, the initial step is also the formation of imine 228 (Scheme 77),
Scheme 77
N PhH
���
O HH���
��� �����
������
N Ph
H
���
���
O HH
���
O HH
��
������
���
N Ph
H
����������
��������
N Ph
OH
H
LA
���������
O
N PhH
LA
���
����
��
N Ph
O
H
228 229
230224

121
which, assisted by the Lewis acid (LA), tautomerises to the conjugated, more
stable imine 229, with destruction of the stereogenic centre. Ring opening of the
epoxide and elimination of water produces the intermediate 230, which aromatises
spontaneously to give the racemic product 224.
Not having been able to improve the yield for the formation of the enamine 222, we
next turned attention to the synthesis of the corresponding 7-oxotetrahydroindole.
Reaction of optically active 222 with neat dmfdma gave 192, but as expected in
very low yield (Scheme 78). The reaction, however, was shown to be
Scheme 78
222
���
ONH
Ph
dmfdma
O
N
Ph
19214%
stereospecific, the product having the same optical rotation as that obtained via
sealed tube methodology. When the reaction was carried out in refluxing DMF, the
same yield was obtained. Refluxing of the more sterically hindered N,N-
dimethylformamide diisopropyl or di-tert-butyl acetals with 222 in methanol in the
presence of one equivalent of triethylamine, however, led only to recovery of the
starting material.
Since, again, attempts to improved the yield of 192 were unsuccessful, it was
decided to investigated the synthesis of the required 7-oxotetrahydroindole 182
using this methodology without optimisation of the reaction. The amine 50b was
therefore reacted with the α,β-epoxy ketone 220 in refluxing aqueous acetonitrile to
give a yellow oil in low yield, the proton NMR spectrum of which showed it to be a
122
complex mixture (Scheme 79). The mass fragmentation, however, showed a
Scheme 79
BrNH2
CN �������� Br
N
CN
O
HO
O
MeCN/H2O
������
dmfdma
BrN
CN
O
182
220
50b231
BrNH2
CN �������� Br
N
CN
O
HO
O
MeCN/H2O
������
dmfdma
BrN
CN
O
182
220
50b231
peak that could correspond to the desired enamine 231. Attempts to purify the
material through column chromatography were unsuccessful, and hence the crude
product was treated with dmfdma in the hope of producing some of the required 7-
oxotetrahydroindole 182. Unfortunately, a dark brown oil was obtained from which
the desired product could not be isolated. Instead two fractions were obtained after

123
column chromatography, which were shown by proton NMR spectroscopy to be
complex mixtures.
Due to the failure to produce the optically active key intermediate 48b*, required to
achieved the asymmetric synthesis of LY290154 35, from the amine 50b* via its
corresponding 7-oxotetrahydroindole derivative 182*, it was clear that this
particular synthetic strategy was not going to be the method to provide the desired
target molecule in enantiomerically enriched form.
124
9. Conclusions
The work described in this thesis demonstrates that a variety of 1-substituted 7-
oxotetrahydroindoles are accessible from reaction of primary amines with 7-
oxotetrahydrobenzofuran (181), using the same method as reported by Matsumoto
and Watanabe for the synthesis of the 4-oxo derivatives (see Chapter 6.1).
Aromatisation to give the corresponding 4- and 7-hydroxyindoles can be achieved
via a new procedure based on an iodination/dihydroiodination sequence (see
Chapter 7). Furthermore, when homochiral amines are employed the
corresponding optically active 4- and 7-hydroxyindoles are obtained.
Unfortunately, construction of the 7-cyanomethoxyindole derivative 48b, the key
intermediate for the asymmetric synthesis of LY290154 35, using this methodology
was unsuccessful (see Chapter 8). The reason was the failure to obtain the
precursor 182 from reaction of the amine 50b, which was prepared in racemic and
enantiomerically enriched form (see Chapter 4.1), with 181. The explanation as to
why 50b did not react in the desired manner with 181, was that it cyclised
intramolecularly under the harsh conditions employed to give the lactam 212.
Since other methods also failed (see Chapter 5 and 8), further attempts to
synthesise the required enantiomerically enriched LY290154 precursor 48b* from
amine 50b* had to be abandoned.

125
10. Experimental
10.1 Solvents and Reagents
Commercially available materials were used without further pufirication unless
stated otherwise. Reference to the supplier is made when appropiate.
All solvents were of reagent or analytical grade and were used as supplied
commercially unless specified as dry, in which case they were dried and distilled
before use under oxygen-free nitrogen as follows: acetonitrile and dichloromethane
from calcium hydride and stored over A4 molecular sieves; DMF from and stored
over magnesium sulphate; toluene from sodium, and acetone from and stored over
potassium carbonate; diethyl ether and THF from sodium/benzophenone.
Triethylamine was distilled from sodium hydroxide.
Air- and moisture-sensitive reagents were handled under oxygen-free nitrogen in
flame-dried glassware. Sensitive liquids were transferred with thoroughly dried (in
a desiccator at room temperature in vacuo) gas-tight Hamilton syringes and added
through a rubber septum. Brine refers to a saturated aqueous solution of sodium
chloride.
10.2 Purification and Characterisation Techniques
Melting points were determined on a Kofler hot-stage microscope apparatus and
are uncorrected. Infrared spectra were recorded on a Perkin-Elmer 1720X FTIR
spectrometer using the standard nujol mull technique between sodium chloride
plates. 1H-NMR and 13C-NMR spectra were recorded using Jeol PMX60, EX90Q,
EX270 and GX400 NMR spectrometers. Tetramethylsilane (TMS) was used as the
internal standard in all NMR spectra run in CDCl3, DMSO-d6, acetone-d6 or MeOD,
126
and the chemical shifts (δ) are quoted in parts per million (ppm) from TMS (TMS=0
ppm). Low resolution EI mass spectra (Kratos MS25 mass spectrometer with an
ionisation potential of 70 eV at 200 °C) and microanalyses (Carlo Erba 1106
Elemental Analyser) were performed as a service by Mr. A. W. R. Saunders at the
University of East Anglia. High resolution mass spectra were recorded at the
SERC Mass Spectrometry Service Centre at the University College of Swansea.
Chemical Ionisation (CI) mass spectra and proton NMR chiral shift experiments
were carried out by Dr. J. Gilmore at Lilly Research Centre Limited in Surrey.
Optical rotations were determined on a Jasco DIP-370 digital polarimeter.
Column chromatography at atmospheric pressure was performed using Merck
7734 silica gel (200-600 microns). Flash column chromatography was performed
on Sorbsil C60 silica gel (40-60 microns). Thin layer chromatography (tlc) was
performed on Merck 60F254 backed silica plates, compounds being visualised by
UV irradiation (254 nm) or exposure to iodine.
10.3 Experimental for Chapter 1, Section 1.5
3-(2(E)-(7-Chloroquinolin-2-yl)ethenyl)benzenecarbaldehyde (28)
A magnetically stirred suspension of 7-chloroquinaldine (49.4 g, 0.28 mol),
isophthalaldehyde (55.7 g, 0.41 mol), dry toluene (320 ml) and acetic anhydride
(80 ml) was refluxed overnight under a nitrogen atmosphere. After cooling to rt the
solid present was separated by filtration and washed with a small amount of
toluene. The dried material was then slurried in boiling dichloromethane (1000 ml)
and the mixture filtered hot to separate the product from the less soluble
bisadduct. The filtrate was acidified with 2 M HCl to give the product as the
hydrochloride salt, which was readily removed from unreacted isophthalaldehyde
by filtration. The salt was then poured into a mixture of CH2Cl2 (1000 ml) and 2 M
NaOH (500 ml) and the resulting two-phase suspension heated under stirring until

127
most of the material was dissolved. The layers were separated and insoluble
residues removed by filtration. The organic phase was dried over anh MgSO4 and
concentrated under vacuo to yield the crude product as a yellow solid.
Recrystallisation from ethyl acetate and petroleum ether (40-60 °C) afforded the
pure compound as a yellow solid (30. 5 g, 37%), mp 136-142 °C, lit28 mp 156-7 °C;
1H-NMR (60 MHz, CDCl3, δH): 7.26-8.26 (11H, m, 11 x ArH), 10.14 (1H, s, CHO);
IR (nujol): 1688 (CHO),1640 (C=C), 1598 (C=C), 1590, 1498, 870, 785, 686 cm-1;
m/z (%): 293/295 (72/25, M+), 292 (100, M+-H), 264/266 (22/8, M+-CHO), 228 (10,
M+-CHO-Cl), 163 (13, 7-chloroquinoline); Anal calcd for C18H12ClNO (%): C, 73.60;
H, 4.12; N, 4.77; Cl, 12.07; Found (%): C, 73.43; H, 3.85; N, 4.65; Cl, 12.35; tlc
(SiO2, cyclohexane/EtOAc 1:1, Rf): 0.49.
4-Iodobutyronitrile (39)
A magnetically stirred mixture of 4-bromobutyronitrile (25.0 g, 0.17 mol) and
sodium iodide (38.2 g, 0.26 mol) in dry acetone (170 ml) in a flask closed with a
calcium chloride tube was refluxed overnight. After cooling, the inorganics were
filtered off and the organic yellow solution concentrated in a rotary evaporator.
Further inorganic material which precipitated was removed by filtration. The
residual yellow liquid was taken up in diethyl ether (50 ml), and the resulting
solution was washed with aq sodium thiosulphate (3 x 30 ml) and brine (3 x 30 ml).
The organic layer was dried over anh MgSO4 and the solvent evaporated under
reduced pressure. Vacuum distillation of the crude product gave the title compound
as a colourless liquid (23.5 g, 71%), bp 74 °C/0.8 mm; 1H-NMR (60 MHz, CDCl3,
δH): 1.96-2.70 (4H, m, 2 x CH2), 3.32 (2H, t, 3J 6.0 Hz, CH2I); IR (neat): 2250
(CN), 1430, 1230 cm-1; m/z (%): 195 (53, M+), 127 (10, I), 68 (100, M+-I), 41 (64,
M+-I-CN-H); Anal calcd for C4H6IN (%): C, 24.64; H, 3.10; N, 7.18; Found (%): C,
24.80; H, 3.01; N, 7.19.
128
5-Hydroxy-5-{3-[2-(7-chloroquinolin-2-yl)ethenyl]phenyl}valeronitrile (41)
A mixture of zinc dust (2.75, 42.0 mmol) in dry THF (4 ml) containing
dibromoethane (150 ml, 1.6 mmol) was refluxed under nitrogen for 1 min. After
cooling to rt, trimethylsilyl chloride (160 ml, 1.3 mmol) was added dropwise and the
resulting mixture was stirred for 15 min. Neat iodobutyronitrile (7.8 g, 40.0 mmol)
was added dropwise over 15 min (exothermic) and the mixture was stirred
overnight at 50 °C. The crude zincate 40 was obtained as a viscous orange oil and
subsequently used in the next step after being cooled to rt.
The aldehyde 28 (7.84 g, 26.7 mmol) was dissolved in dry THF (40 ml) and the
solution stirred under nitrogen as the zincate 40, diluted in dry THF (5 ml), was
added rapidly. After cooling in an ice-bath, a 1.0 M solution of titanium tetrachloride
in CH2Cl2 (26.7 ml, 26.7 mmol) was added dropwise over 15 min, keeping the
temperature between 0-10 °C. After stirring for 30 min at 0 °C, the dark brown
solution was further stirred for 5 h at rt under a nitrogen atmosphere. Water (100
ml) was added, and the solution was made basic with aq sodium carbonate,
whereby a deep blue colour arose. The aqueous mixture was extracted with
dichloromethane (3 x 80 ml), when the deep blue colour disappeared. The organic
extracts were washed with brine (3 x 30 ml), dried over anh MgSO4 and
concentrated under vacuo to yield a brown oil. The pure product was obtained after
flash column chromatography on silica, eluting with 25-60% ethyl acetate in light
petroleum ether (40-60 °C), as a pale yellow oil that solidified upon standing (6.0 g,
62%), mp 93-96 °C; 1H-NMR (60 MHz, DMSO-d6, δH): 1.52-1.82 (4H, m, 2 x CH2),
2.40-2.76 (2H, m, CH2CN), 4.54-4.92 (1H, m, OCHCH2), 5.40 (1H, d, 3J 4.8 Hz,
D2O exch, OH), 7.26-8.20 (10H, m, 10 x ArH), 8.44 (1H, d, 3J 8.4 Hz, quinoline 4-
H); IR (nujol): 3260 (OH), 2246 (CN), 1608 (C=C), 1592, 1499, 860, 797, 698 cm-1;
m/z (%): 362/364 (8.7/2.8, M+), 294 (12, M+- (CH2)3CN), 292 (14, M+-(CH2)3CN-
2H), 264 (6.0, M+-(CH2)3CN-2H-CO), 163 (29, 7-chloroquinoline); Anal calcd for
C22H19ClN2O (%): C, 72.80; H, 5.28; N, 7.72; Cl, 9.77; Found (%): C, 72.74; H,
5.19; N, 7.52; Cl, 9.68; tlc (SiO2, cyclohexane/EtOAc 1:1, Rf): 0.16.

129
5- Chloro-5-{3-[2-(7-chloroquinolin-2-yl)ethenyl]phenyl}valeronitrile (42)
The alcohol 41 (5.70 g, 15.8 mmol) was placed in a round bottomed flask and
cooled in an ice-bath. Thionyl chloride (15 ml) was added dropwise (cautiously,
exothermic) and the mixture was stirred for 1 h at 0 °C. It was then warmed to rt
and stirred for a further 1 h. The excess SOCl2 was evaporated under reduced
pressure. The brown residue obtained was poured into water (50 ml) and the two
phase mixture extracted with dichloromethane (3 x 20 ml). The organic extracts
were combined and washed with aq sodium bicarbonate (3 x 20 ml), brine (3 x 20
ml), dried over anh MgSO4, and concentrated in a rotary evaporator to yield a
brown oil after being dried under vacuo (4.57 g, 76%). The crude product was
used in the next step without further purification. 1H-NMR (400 MHz, CDCl3, δH):
1.70-1.80 (1H, m, CHCHHCH2), 1.90-2.00 (1H, m, CHCHHCH2), 2.20-2.35 (2H, m,
CH2CH2CH2), 2.42 (2H, t, 3J 8.0 Hz, CH2CH2CN), 4.91 (1H, dd, 3J 8.4 and 8.6 Hz,
ClCHCH2), 7.30-8.16 (11H, m, 11 x ArH); IR (neat): 2950, 2250 (CN), 1640 (C=C),
1600 (C=C), 1500, 865, 775, 700 cm-1; m/z (%): 281 (14, M+), 279 (16, M+-2H),
343 (88, M+-2H-Cl), 302 (25, M+-3H-Cl-CH2CN), 290 (16, M+- 3H-Cl-(CH2)2CN),
264 (14, M+-Cl-CH-(CH2)3CN), 36/38 (100/34, Cl); tlc (SiO2, cyclohexane/EtOAc
1:1, Rf): 0.58.
2-Benzhydroloxynitrobenzene (43)
A magnetically stirred suspension of 2-nitrophenol (20.9 g, 0.15 mol), anhydrous
K2CO3 (69 g, 0.5 mol) and benzhydryl bromide (39.5 g, 16 mol) in acetone (400 ml)
was refluxed under nitrogen for 5 h. The cooled reaction mixture was filtered and
concentrated under reduced pressure to give an orange/brown solid. This residue
was taken up in diethyl ether (100 ml) and solids removed by filtration. The filtrate
was again concentrated to dryness and the residue triturated with petroleum ether
(60-80 °C, 200 ml). The product was obtained as brown crystals after filtration and
being dried under vacuo at rt (30.6 g, 67%), mp 92-102 °C, lit34 mp 96-98 °C. 1H-
130
NMR (60 MHz, CDCl3, δH): 6.44 (1H, s, OCHPh2), 6.96-8.04 (14H, m, 14 x ArH);
tlc (SiO2, 30% EtOAc in hexane, Rf): 0.53.
7-Benzhydroloxyindole (44)
Following the procedure reported by Dobson et al.,34 to a magnetically stirred
solution of 43 (9.15 g, 30.0 mmol) in dry THF (200 ml) 1.7 M vinylmagnesium
chloride in THF (62.5 ml, 0.11 mol) was added dropwise over 30 min at -40 °C
under nitrogen. After stirring for a further 40 min at -40 °C the reaction solution was
poured into aqueous ammonium chloride (200 ml) and the two phase mixture
extracted with diethyl ether (3 x 100 ml). The combined organic phases were dried
over anh MgSO4 and concentrated under reduced pressure to yield a yellow oil.
Flash column chromatography on silica with 12.5% ethyl acetate in light petroleum
ether (40-60 °C) as eluent afforded the desired product as a pale yellow oil (4.1 g,
46%); 1H-NMR (60 MHz, CDCl3, δH): 6.36 (1H, s, OCHPh2), 6.44-7.60 (15H, m, 15
x ArH), 8.32 (1H, br s, NH); IR (neat): 3436 (NH), 3063, 1578, 1494, 1248, 1065
cm-1; tlc (SiO2, CH2Cl2, Rf): 0.76.
7-Cyanomethoxyindole (45)
According to the same paper as above, a solution of 44 (17.8 g, 59.5 mmol) in
methanol (150 ml)/toluene (150 ml) was hydrogenated at 50 psi for 1 h at rt in the
presence of 1.5 g Pd(OH)2 (Pearlman's catalyst). The reaction mixture was filtered
through Celite and the filtrate concentrated in vacuo to give a brown oil. The crude
7-hydroxyindole [1H-NMR (60 MHz, CDCl3, δH): 5.00 (1H, br s, OH), 6.40-7.60
(5H, m, 5 x ArH), 8.46 (1H, br s, NH); tlc (SiO2, CH2Cl2, Rf): 0.1] was taken up in
butanone (200 ml) and degassed with N2. Anhydrous potassium carbonate (21.0 g,
0.15 mol) and bromoacetonitrile (6.5 ml,93.3 mmol) were added, and the reaction
mixture refluxed under nitrogen for 1h. The cooled brown solution was slowly
poured into 2 M hydrochloric acid (200 ml) and the resulting mixture extracted with
CH2Cl2 (3 x 100 ml).. The combined organic extracts were dried over anh MgSO4

131
and concentrated under reduced pressure to yield a brown oil. Flash column
chromatography on silica with 12.5% ethyl acetate in light petroleum ether (40-60
°C) as eluent afforded the desired product as a brown solid (7.3 g, 72%), mp 64 °
C, lit34 mp 63-5 °C; 1H-NMR (60 MHz, CDCl3, δH): 4.80 (2H, s, OCH2CN), 6.36-
7.40 (5H, m, 5 x ArH), 8.40 (1H, br s, NH); IR (nujol): 3369 (NH), 1578, 1462,
1377, 1347, 1247 cm-1; tlc (SiO2, CH2Cl2, Rf): 0.49.
7-Chloro-2-(2-{3-[1-(7-cyanomethoxyindol-1-yl)-4-cyanobutyl]phenyl}ethenyl)-
quinoline (46)
7-Cyanomethoxyindole (45, 2.01 g, 11.7 mmol) was dissolved in dry DMF (55 ml)
and the solution stirred as sodium hydride (60% dispersion in oil, 0.47 g, 11.7
mmol) was added in portions over 10 min under nitrogen. The resulting suspension
was stirred for 30 min at rt. The crude benzyl chloride 42 (4.47 g, 11.7 mmol),
dissolved in dry DMF (10 ml), was added dropwise over 10 min. The resulting
mixture was stirred for 20 h in an oil-bath at 60 °C under nitrogen, then diluted with
ethyl acetate (100 ml) and the solution washed with water (3 x 50 ml) and brine (3
x 50 ml). The organic layer was dried over anh MgSO4 and concentrated in a
rotary evaporator to give a dark brown oil. The residue was flash column
chromatographed on silica with 20-40% ethyl acetate in light petroleum ether (40-
60 °C) as eluent to yield the title compound as a pale yellow oil after being dried
under vacuo (1.09 g, 18%). A satisfactory elemental analysis could not be
obtained. 1H-NMR (270 MHz, CDCl3, δH): 1.76 (2H, m, CH2CH2CH2), 2.42 (2H, m,
CHCH2CH2), 2.42 (2H, m, CH2CN), 4.86 (2H, s, OCH2CN), 6.31 (1H, t, 3J 5.6 Hz,
CH2CHN), 6.61 (1H, d, 3J 3.3 Hz, indole 3-H), 6.69 (1H, d, 3J 7.6 Hz, indole 6-H),
7.04 (1H, t, 3J 7.9. Hz, 5'-H), 7.17 (1H, d, 3J 7.6 Hz, indole 4-H), 7.25-7.73 (10H,
m, 10 x ArH), 8.09 (2H, m, quinoline 4-H and 8-H); IR (nujol): 2245 (CN), 1607
(C=C), 1575, 1485, 888, 781, 722 cm1; m/z (%): 347 (0.71, M+-(7-cyanomethoxyin-
dole)), 333 (0.85, M+-(7-cyanomethoxyindole)-N), 292 (0.92, M+-(7-cyanomethoxy-
132
indole)-(CH2)2CN-H), 278 (2.0, M+-(7-cyanomethoxyindole)-(CH2)3CN-H), 264
(2.7, M+-(7-cyanomethoxyindole)-CH(CH2)3CN-2H); tlc (SiO2, cyclohexane
/EtOAc 1:1, Rf): 0.40.
7-Chloro-2-(2-{3-[1-(7-{1H-tetrazol-5-ylmethoxy}indol-1-yl)-4-(1H-tetrazol-5-yl)-
butyl]phenyl}ethenyl)quinoline (35)
A magnetically stirred solution of the dinitrile 46 (0.75 g, 1.45 mmol) and tributyltin
azide (1.45 g, 4.36 mmol) in dimethoxyethane ( 5 ml) was heated at 150 °C for 5 h,
allowing the solvent to evaporate. After cooling of the reaction mixture to rt, the
crude product was taken up in methanol (15 ml) containing acetic acid (1 ml) and
the solution left overnight. The orange microcrystalline product which had
separated was filtered off, washed with dichloromethane (3 x 1 ml) and dried under
vacuo (0.87 g, 40%). A satisfactory elemental analysis could not be obtained for
this compound. 1H-NMR (270 MHz, MeOD, δH): 1.80 (2H, m, CH2CH2CH2), 2.11
(1H, m, CHCHHCH2), 2.29 (1H, m, CHCHHCH2), 2.90 (2H, t, 3J 7.3 Hz,
CH2CH2Tet), 5.47 (2H, s, OCH2Tet), 6.40 (1H, t, 3J 8.1 Hz, NCHCH2), 6.49 (1H, d,
3J 3.3 Hz, indole 3-H), 6.83 (1H, d, 3J 7.3 Hz, indole 6-H), 6.92 (2H, m, 2 x ArH),
7.16 (4H, m, 4 x ArH), 7.44 (4H, m, 4 x ArH), 7.76 (1H, d, 3J 8.4 Hz, 1 x ArH), 7.87
(1H, d, 3J 8.9 Hz, quinoline 3-H), 7.90 (1H, d, 4J 2.0 Hz, quinoline 8-H), 8.17 (1H,
d, 3J 8.9 Hz, quinoline 4-H); IR (nujol): 3406 (br NH), 1635 (C=C), 1608 (C=C),
1573, 1488, 845, 783, 722 cm-1.
10.4 Experimental for Chapter 3
3-(1,3-Dioxolan-2-yl)bromobenzene (53)
Following the same procedure as for the preparation of the o-isomer,35 a
magnetically stirred solution of 3-bromobenzaldehyde (10.0 g, 54.1 mmol), ethane-
1,2-diol (9.40 g, 0.15 mol) and 4-toluenesulfonic acid monohydrate (0.20 g, 1.08

133
mmol) in benzene (80 ml) was heated under reflux for 3.5 h, using a Dean & Stark
trap to removed the water formed during the reaction. After cooling to rt the
reaction mixture was diluted with water (50 ml) and the organic layer separated.
The water phase was back extracted with ethyl acetate (3 x 20 ml) and the
combined organic extracts washed with brine (3 x 40 ml) and dried over anh
MgSO4. Evaporation of the solvent under reduced pressure gave an oily residue
(11.6 g, 94%). Vacuum distillation of the crude material yielded the title compound
as a colourless oil (11.4 g, 92%), bp 180 °C/0.35 mm, lit156 bp 98-101 °C/0.01 mm;
1H-NMR (60 MHz, CDCl3, δH): 4.04 (4H, s, 2 x CH2), 5.76 (1H, s, CH), 7.18-7.70
(4H, m, 4 x ArH); IR (neat): 2887, 1573, 1474, 1212, 1103, 1080, 883, 786, 696
cm-1; tlc (SiO2, 5% EtOAc in hexane, Rf): 0.21.
3-(1,3-Dioxolan-2-yl)benzaldehyde (54)
According to the method described for the o-isomer,36 to a magnetically stirred
solution of 3-(1,3-dioxolan-2-yl)bromobenzene (53) (6.00 g, 26.2 mmol) in dry
tetrahydrofuran (50 ml) at -74 °C under a nitrogen atmosphere was added
dropwise a 1.6 M solution of n-butyllithium in hexane (17.2 ml, 27.5 mmol)
maintaining the addition temperature below -60 °C. The resulting pale yellow
solution was stirred below -70 °C for 30 min, during which time the lithium salt
separated, then treated dropwise with a solution of N-formylpiperidine (3.26 g, 28.8
mmol) in dry THF (8 ml) keeping the internal temperature below -60 °C. The
reaction mixture was stirred below -40 °C for 1 h, then quenched with aq NH4Cl
(50 ml), and the resulting mixture was extracted with ethyl acetate (3 x 30 ml). The
combined organic extracts were washed with aq NH4Cl (3 x 30 ml), brine (3 x 30
ml), dried over anh MgSO4 and concentrated in a rotary evaporator to yield a
yellow oil (4.46 g, 96%). Bulb to bulb distillation gave the product as a colourless
oil (4.35 g, 93%), bp 210-2 °C/1.2 mm, lit156 bp 95-9 °C/0.01 mm; 1H-NMR (60
MHz, CDCl3, δH): 4.04 (4H, s, 2 x CH2), 5.82 (1H, s, CH), 7.36-8.06 (4H, m, 4 x
134
ArH), 9.98 (1H, s, CHO); IR (neat): 2889, 1700 (CHO), 1609, 1590, 1451, 1151,
1097, 907, 795, 694 cm-1; tlc (SiO2, 20% EtOAc in hexane, Rf): 0.32.
Attempted synthesis of 5-(3-formylphenyl)-5-hydroxypentanenitrile (52)
The same method was followed as for the preparation of the benzyl alcohol 41.
Thus, the aldehyde 54 (4.0 g, 22.4 mmol) was reacted with the zincate 40 (33.6
mmol) in the presence of TiCl4 (22.4 ml, 22.4 mmol). After the usual work up, two
products were isolated after flash column chromatography on silica with 25-60%
EtOAc in light petroleum ether (40-60 °C). Spectroscopic analysis showed them to
be isophthalaldehyde (0.85 g, 28%) and the aldehyde 55 (1.15 g, 22%).
Compound 55 was isolated as a yellow oil from which a satisfactory elemental
analysis could not be obtained due to decomposition of the material. 1H-NMR (90
MHz, CDCl3, δH): 1.87 (4H, m, 2 x CH2), 2.42 (2H, t, 3J 6.5 Hz, CH2CN), 3.07 (1H,
s, D2O exch, OH), 3.47 and 3.73 (4H, AB t, OCH2CH2O), 4.47 (1H, t, 3J 6.5 Hz,
OCHCH2), 7.24-8.00 (4H, m, 4 x ArH), 10.04 (1H, s, CHO); IR (neat): 3477 (OH),
2934, 2246 (CN), 1695 (CHO), 1603, 1454, 1240, 1112, 1058, 890, 803, 699 cm-1;
m/z (%): 247 (5.6, M+), 202 (2.4, M+-(CH2)2OH), 186 (M+-O(CH2)2OH), 179 (94,
M+-(CH2)3CN), 135 (100, M+-(CH2)3CN-(CH2)2OH), 117 (31, M+-(CH2)3CN-
O(CH2)2OH); tlc (SiO2, EtOAc/light petroleum ether [40-60 °C] 1:1, Rf): 0.18.
5-(3-Bromophenyl)-5-hydroxypentanenitrile (56)
Following the same procedure as above, reaction of the zincate 40 (40.0 mmol)
with 3-bromobenzaldehyde (4.95 g, 26.7 mmol) in the presence of titanium
tetrachloride (26.7 ml, 26.7 mmol) yielded the title compound as a yellow oil (3.87
g, 57%) after flash column chromatography on silica with 25% EtOAc in light
petroleum ether (40-60 °C) as eluent. 1H-NMR (270 MHz, CDCl3, δH): 1.77 (4H, m,
2 x CH2), 2.35 (2H, t, 3J 7.1 Hz, CH2CN), 2.96 (1H, s, D2O exch, OH), 4.63 (1H, t,
3J 7.1 Hz, OCHCH2), 7.20-7.60 (4H, m, 4 x ArH); 13C-NMR (22.4 MHz, CDCl3, δC):
17.1 (CH2CN), 21.8 (CH2CH2), 37.5 (CHCH2), 72.5 (CH), 119.6 (CN), 122.9 (3'-C),

135
124.3 (6'-C), 128.9 (5'-C), 130.7 (2'-C), 131.1 (4'-C), 146.4 (1'-C); IR (neat): 3435
(OH), 2939, 2247 (CN), 1595, 1475, 1098, 886, 786, 698; m/z (%): 253/255
(13.8/13.2, M+-H), 185/187 (100/90, M+-H-(CH2)3CN), 157/159 (19/14,
bromophenyl), 77 (60, phenyl); Anal calcd for C11H12BrNO (%): C, 51.99; H, 4.76;
N, 5.51; Found (%): C, 51.79; H, 4.86; N, 5.69; tlc (SiO2, EtOAc/light petroleum
ether [40-60 °C] 1:1, Rf): 0.49.
5-(3-Bromophenyl)-5-chloropentanenitrile (57)
The same procedure as for the synthesis of the benzyl chloride 42 was followed.
Thus, reaction of the benzyl alcohol 56 (1,93 g, 7.59 mmol) with thionyl chloride (5
ml) gave the title compound as a brown oil (1.85 g, 89%). Column chromatography
on silica with 25% EtOAc in light petroleum ether (40-60 °C) yielded the product as
a yellow oil. A satisfactory elemental analysis was not obtained. 1H-NMR (270
MHz, CDCl3, δH): 1.61-1.81 (1H, m, CHCHH), 1.81-2.0 (1H, m, CHCHH), 2.08-
2.28 (2H, m, CH2CH2CH2), 2.40 (2H, t, 3J 6.9 Hz, CH2CN), 4.81 (1H, dd, 3J 6.9
and 7.1 Hz, ClCHCH2), 7.15-7.40 (2H, m, 2 x ArH), 7.45 (1H, m, 1 x ArH), 7.55
(1H, m, 1 x ArH); IR (neat): 2941, 2247 (CN), 1592, 1477, 879, 788, 695; m/z (%):
271/273 (11/15, M+-H), 236/238 (31/30, M+-Cl), 157 (26, M+-Cl-Br), 116 (100, M+-
Cl-Br-CH2CN-H); tlc (SiO2, EtOAc/light petroleum ether [40-60 °C] 1:1, Rf): 0.70.
N-(1-[3-Bromophenyl]-4-cyanobutyl)-7-cyanomethoxyindole (48b)
According to the procedure for compound 46, the indole 45 (1.0 g, 5.80 mmol) was
treated successively with NaH (0.23 g, 5.80 mmol) and the benzyl chloride 57 (1.5
g, 5.50 mmol) to give a dark brown oil. Column chromatography on silica with 25%
EtOAc in light petroleum ether (40-60 °C) as eluent yielded the product as a yellow
oil (0.90 g, 40%). 1H-NMR (270 MHz, CDCl3, δH): 1.66 (2H, m, CH2CH2CH2), 2.34
(2H, t, 3J 6.9 Hz, CH2CN), 2.35 (2H, m, CHCH2), 4.78 (2H, s, OCH2CN), 6.18 (1H,
dd, 3J 6.6 and 8.9 Hz, NCHCH2), 6.59 (1H, d, 3J 3.3 Hz, 3-H), 6.65 (1H, d, 3J 7.6
Hz, 6-H), 7.01 (1H, t, 3J 7.7 Hz, 5'-H), 7.12 (2H, m, 4-H and 6'-H), 7.20 (1H, d, 3J
136
3.3 Hz, 2-H), 7.30 (3H, m, 2'-, 4'- and 5'-H); 13C-NMR (67.8 MHz, CDCl3, δC): 16.8
(CH2CN), 22.4 (CH2CH2), 34.5 (CHCH2), 53.6 (OCH2CN), 59.7 (CH), 103.7 (3-C),
104.4 (6-C), 114.9 (OCH2CN), 116.3 (4-C), 119.1 (CN), 120.0 (5-C), 122.8 (3'-C),
125.0 (2-C), 125.3 (6'-C), 125.7 (3a-C), 129.3 (5'-C), 130.4 (4'-C), 130.8 (2'-C),
131.5 (1'-C), 143. 9 (7a-C), 144.0 (7-C); IR (neat): 2934, 2247 (CN), 1594, 1572;
m/z (%): 407/409 (1.88/1.86, M+-H), 236/238 (1.7/1.5, M+-[7-cy-
anomethoxyindole]), 43 (100); Anal calcd for C21H18BrN3O (%): C, 61.78; H, 4.44;
N, 10.29; Found (%): C, 61.60; H, 4.34; N, 10.29; tlc (SiO2, EtOAc/light petroleum
ether [40-60 °C] 1:1, Rf): 0.45.
7-Cyanomethoxyindoline (59)
A magnetically stirred solution of the indole 45 (4.0 g, 23.2 mmol) in glacial acetic
acid (200 ml) at 10 °C under a nitrogen atmosphere was treated portionwise with
sodium cyanoborohydride (7.30 g, 0.12 mol). The resulting solution was stirred at rt
for 1 h, quenched with water (50 ml) and the AcOH removed under reduced
pressure. The brown residue was basified with 2 M NaOH and the resulting
solution extracted with ethyl acetate (3 x 30 ml). The combined organic extracts
were dried over anh MgSO4 and concentrated to give a yellow/brown oil. The crude
product was passed through silica on a short column using 30-50% EtOAc in light
petroleum ether (40-60 °C) as eluent to afford the title compound as an orange oil
(3.6 g, 90%). 1H-NMR (60 MHz, CDCl3, δH): 3.12 (2H, m, 3-CH2), 3.56 (2H, m, 2-
CH2), 4.70 (2H, s, OCH2CN), 6.60-7.00 (3H, m, 3 x ArH); IR (neat): 3375 (NH),
2850, 1620, 1593, 1490, 1292, 945, 747 cm-1; tlc (SiO2, EtOAc/light petroleum
ether [40-60 °C] 1:1, Rf): 0.26.
Attempted synthesis of 1-(1,3-dioxolan-2yl)-3-(1,3-dithiane-2-yl)benzene (62)
A solution of the aldehyde 54 (1.0 g, 5.61 mmol) and 1,3-propanedithiol (0.79 g,
7.28 mmol) in 1,2-dichloroethane (8 ml) was stirred at rt under nitrogen. AlCl3 (0.25

137
g, 1.90 mmol) was added in small portions (exothermic) and the reaction mixture
stirred for a further 15 min. The turbid liquid was poured into water (30 ml) and the
resulting two-phase mixture extracted with dichloromethane (3 x 20 ml). The
combined organic layers were washed with brine (3 x 20 ml), dried over anh
MgSO4 and concentrated in a rotary evaporator to give a yellow oil that solidified
upon standing. Column chromatography on silica with CH2Cl2 as eluent yielded
two products. The first compound was obtained as a white solid and shown to be
the known bisdithioacetal 61 (0.50 g, 28%), mp 133 °C, lit157 mp 129.0-129.5 °C,
1H-NMR (270 MHz, CDCl3, δH): 1.76-1.99 (2H, m, 2 x SCH2CHHCH2S), 2.03-2.20
(2H, m, 2 x SCH2CHHCH2S), 2.77-2.90 (4H, m, 4 x SCHH), 2.90-3.10 (4H, m, 4 x
SCHH), 5.07 (2H, s, 2 x CH), 7.17-7.29 (1H, m, 5-H), 7.30 (2H, d, 3J 7.9 Hz, 4-
and 6-H), 7.51 (1H, s, 2-H); 13C-NMR (22.4 MHz, CDCl3, δC): 25.1
(SCH2CH2CH2S), 31.9 (SCH2), 51.2 (CH), 127.5 (5-C), 127.8 (4-C), 129.0 (2-C),
139.6 (1-C); IR (nujol): 909, 750, 707 cm-1; m/z (%): 314 (30, M+), 196 (100, M+-
dithiane), 122 (83, [PhCHS]+), 106 (25, [S(CH2)3S]+), 91 (37, tropylium), 77 (30,
phenyl); Anal calcd for C14H18S4 (%): C, 53.46; H, 5.77; S, 40.77; Found (%): C,
53.70; H, 5.54; S, 40.66; tlc (SiO2, CH2Cl2, Rf): 0.54. The second compound was
isolated as a yellow oil that solidified upon standing and was shown to be 3-(1,3-
dithiane-2-yl)benzaldehyde (60) (0.10 g, 8%), mp 57 °C; 1H-NMR (270 MHz,
CDCl3, δH): 1.83-2.03 (1H, m, SCH2CHHCH2S), 2.07-2.23 (1H, m,
SCH2CHHCH2S), 2.85-2.98 (2H, m, 2 x SCHH), 2.98-3.13 (2H, m, 2 x SCHH),
5.23 (1H, s, CH), 7.45 (1H, t, 3J 9.0 Hz, 5-H), 7.73 (1H, d, 3J 9.0 Hz, 4-H), 7.83
(1H, d, 3J 9.0 Hz, 6-H), 8.0 (1H, s, 2-H), 10.00 (1H, s, CHO); 13C-NMR (67.8 MHz,
CDCl3, δC): 24.9 (SCH2CH2), 31.9 (SCH2), 50.6 (CH), 129.39 (6-C), 129.44 (5-C),
129.5 (2-C), 133.8 (4-C), 136.8 (1-C), 140.3 (3-C), 191.8 (CHO); IR (nujol): 1688
(CHO), 1600, 874, 751, 689 cm-1; m/z (%): 224 (100, M+), 150 (43, M+-(CH2)3S),
105 (24, M+-dithiane); Anal calcd for C11H12OS2 (%): C, 58.89; H, 5.39; S, 28.58;
Found (%): C, 58.61; H, 5.22; S, 28.47; tlc (SiO2, CH2Cl2, Rf): 0.41.
138
3-(1,3-Dithiane-2-yl)benzaldehyde (60)
Following the same procedure as above, reaction of isophthalaldehyde (1.0 g, 7.46
mmol) with 1,3-propanedithiol (0.81 g, 7.46 mmol) gave the desired product as a
yellow solid (0.24 g, 14%). Compound 61 was also isolated from this reaction in
14% yield (0.32 g). The remainder of the reaction mixture was shown to be starting
material.
5-(3-[1,3-Dithiane-2-yl]phenyl)-5-hydroxypentanenitrile (63)
Analogous to the procedure used for compound 41, 3-(1,3-dithiane-2-
yl)benzaldehyde (60) (0.15 g, 0.67 mmol) was reacted with the zincate 40 (8.0
mmol) in the presence of TiCl4 (0.67 ml, 0.67 mmol). Column chromatography on
silica with 10% EtOAc in light petroleum ether (40-60 °C) as eluent gave the
desired product as a yellow oil (43.7 mg, 22%). A satisfactory elemental analysis
for this compound could not be obtained. 1H-NMR (270 MHz, CDCl3, δH): 1.62-
2.26 (6H, m, 6 x aliph), 2.37 (2H, t, 3J 7.7 Hz, CH2CN), 2.86-2.98 (2H, m, 2 x
SCHH), 3.00-3.14 (2H, m, 2 x SCHH), 4.71 (1H, dd, 3J 6.8 and 7.4 Hz, OCHCH2),
5.17 (1H, s, SCHS), 7.25-7.48 (4H, m, 4 x ArH); 13C-NMR (67.8 MHz, CDCl3, δC):
17.1 (CH2CN), 21.9 (CHCH2CH2), 25.1 (SCH2CH2), 32.1 (SCH2), 37.6 (CHCH2),
51.4 (SCH), 73.4 (OCH), 119.6 (CN), 125.1 (6'-C), 125.8 (5'-C), 127.3 (2'-C), 129.1
(4'-C), 139.6 (3'-C), 144.7 (1'-C); IR (neat): 3438 (OH), 2932, 2245 (CN), 1604,
705, 760, 907 cm-1; m/z (%): 279 (6.7, M+-N), 224 (1.4, M+-H-(CH2)3CN), 183 (3.8,
M+-(CH2)3CN)-(CH2)3), 167 (11, M+-(CH2)3CN)-(CH2)3-O); tlc (SiO2, EtOAc/light
petroleum ether [40-60 °C] 1:1, Rf): 0.46.

139
10.5 Experimental for Chapter 4, Section 4.1
S-(-)-2-Amino-3-methyl-1,1-diphenylbutanol (76)
According to the procedure reported by Itsuno et al.,53 in a round bottomed flask
fitted with an addition funnel, magnetic stirrer bar and reflux condenser carrying a
calcium chloride tube, were place magnesium tunnings (6.70 g, 0.28 mol), dry THF
(50 ml) and a crystal of iodine. A solution of bromobenzene (39.3 g, 0.25 mol) in
dry THF (50 ml) was added dropwise until the formation of the Grignard started.
The remaining bromobenzene solution was added at such a rate as to maintain a
gentle reflux. After the addition was complete, the reaction mixture was refluxed for
a further 30 min, then cooled to 0 °C and L-valine methyl ester hydrochloride (5.25
g, 31.3 mmol) added slowly with a spatula, keeping the temperature between 0-10
°C. After 5 h stirring at rt, the reaction mixture was poured into ice (200 ml) and the
resulting suspension was extracted with ethyl acetate (3 x 100 ml). The combined
organics were dried over anh MgSO4 and concentrated in a rotary evaporator to
give a yellow oil that solidified upon standing. The crude product was dissolved in
dichloromethane (20 ml), and 2 M HCl added to precipitate the product as the
hydrochloride salt; this was collected under suction and washed with CH2Cl2 (3 x 5
ml). The free base was released by treatment of the hydrochloride salt with a
boiling mixture of 2 M NaOH (50 ml) and dichloromethane (50 ml). The layers were
separated and the organic solvent evaporated under reduce pressure to yield the
title compound as a white solid after being dried in an oven at 80 °C (3.23 g, 40%),
mp 96 °C, α D23 -127.8 (c 0.006262 gml-1, CHCl3), lit53 mp 95-6 °C, α D
25 -127.7 (c
0.00639 gml-1, CHCl3); 1H-NMR (90 MHz, CDCl3, δH): 0.89 (3H, d, 3J 3.8 Hz,
CH3), 0.97 (3H, d, 3J 3.8 Hz, CH3), 1.80 (1H, m, (CH3)2CH), 3.86 (1H, d, 3J 2.5 Hz,
CHCHNH2), 7.11-7.77 (10H, m, 10 x ArH); IR (nujol): 3338 (br, NH2, OH), 1593,
1489, 693, 743 cm-1; m/z (%): 238 (0.67, M+-OH), 222 (4.4, M+-OH-NH2), 72 (100,
phenyl); Anal calcd for C17H21NO (%): C, 79.96; H, 8.29; N, 5.49; Found (%): C,
79.64; H, 8.30; N, 5.59; tlc (SiO2, EtOAc, Rf): 0.44.
140
1-O-Methyloximino-1-phenylpentan (104)
Ethanol was added dropwise to a mixture of valerophenone (4.86 g, 30.0 mmol),
methoxylamine hydrochloride (2.92 g, 33.0 mmol) and sodium acetate trihydrate
(4.50 g, 33.0 mmol) in water (30 ml) until the solution cleared. The reaction mixture
was refluxed for 5 h and after cooling to rt extracted with diethyl ether (3 x 20 ml).
The combined organic extracts were washed with brine (3 x 20 ml), 5% aq
NaHCO3 (3 x 20 ml) and dried over anh MgSO4. The solvent was removed in a
rotary evaporator to leave a yellow liquid. Bulb to bulb distillation of the crude
product yielded the pure material as a mixture of stereoisomers (5.08 g, 89%), bp
60 °C/0.02 mm; 1H-NMR (90 MHz, CDCl3, δH): 0.80-1.00 (3H, m, CH3), 1.20-1.60
(4H, m, 2 x CH2), 2.74 (2H, t, 3J 7.8 Hz, N=CCH2), 3.80 and 3.97 (3H, s, OCH3),
7.26-7.43 (3H, m, 3 x ArH), 7.51-7.64 (2H, m 2 x ArH); IR (neat): 2957, 1596
(C=N), 1466, 1054 (OMe), 765, 696; m/z (%): 191 (26, M+), 176 (2.8, M+-CH3), 162
(12, M+-CH3CH2), 149 (100, M+-CH3CH2CH), 119 (47, M+-CH3CH2CH2CH2-CH3),
104 (96, M+-CH3CH2CH2CH2-OCH3), 91 (22, M+-CH3CH2CH2CH2-OCH3-N), 77
(39, phenyl); Anal calcd for C12H17NO (%): C, 75.36; H, 8.96; N, 7.32; Found (%):
C, 75.48; H, 8.91; N, 7.36; tlc (SiO2, 10% EtOAc in light petroleum ether [40-60 °
C], Rf): 0.39 and 0.54.
1-Phenylpentylamine (105)
A) A solution of valerophenone (100.0 mg, 0.62 mmol) in methanol (1.4 ml) was
stirred with 4A molecular sieves (75.5 mg) as ammonium acetate was added (0.48
g, 6.2 mmol). After the addition of sodium cyanoborohydride (39.0 mg, 0.62 mmol),
the reaction mixture was stirred at rt for 3 d under a nitrogen atmosphere. The
resulting suspension was filtered through Celite, and the inorganics washed with
CH2Cl2 (10 ml). The organic filtrate was washed with water (3 x 5 ml), dried over
anh MgSO4 and concentrated in a rotary evaporator to yield a yellow liquid. The
crude mixture was acidified with 2 M HCl (5 ml) and the solution extracted with
dichloromethane (1 x 5 ml) . Basification of the aqueous layer with 2 M NaOH and

141
extraction with CH2Cl2 (3 x 5 ml) yielded the product as a clear liquid after being
dried under reduced pressure (10.7 mg, 11%).
B) According to the general procedure described by Feuer and Braunstein,74 to a
solution of the oxime ether 104 (1.35 g, 7.06 mmol) in dry THF (6 ml) at 0 °C under
nitrogen, BH3.THF (1.0 M in THF, 21.2 ml, 10.6 mmol) was added dropwise by
syringe at such a rate that the temperature did not exceed 10 °C. The reaction
mixture was refluxed for 2 h and then cooled to 0 °C. Water (4 ml) was added
cautiously followed by aq KOH (20%, 4 ml) and the resulting mixture was refluxed
for 1 h. The cooled mixture was then extracted with diethyl ether (3 x 20 ml). The
combined organics were washed with brine (3 x 20 ml), dried over anh MgSO4 and
concentrated under reduced pressure to yield a pale yellow liquid. Bulb to bulb
distillation afforded the title compound as a colourless liquid (0.70 g, 61%), bp 90
°C/2 mm, lit158 bp 120 °C/20 mm. The product could also be successfully purified
by flash column chromatography on silica with EtOAc as eluent. A satisfactory
elemental analysis for carbon was not obtained. 1H-NMR (90 MHz, CDCl3, δH):
0.88 (3H, t, 3J 7.1 Hz, CH3), 1.28 (4H, m, 2 x CH2), 1.66 (2H, m, NCHCH2), 3.85
(1H, t, J 7.1 Hz, NCHCH2), 7.30 (5H, m, 5 x ArH); 13C-NMR (22.4 MHz, CDCl3,
dC): 14.0 (CH3), 22.7 (CH2CH3), 28.8 (CH2CH2) , 39.4 (CHCH2), 56.3 (CHNH2),
126.3 (4'-C), 126.8 (2'-C), 128.4 (3'-C), 147.0 (1'-C); IR (neat): 3386 (NH2), 3333
(NH2), 2957, 1602, 1453, 757, 696 cm-1; m/z (%): 165 (1.0, M++2H), 106 (100,
M+-(CH2)3CH3), 77 (12, phenyl); tlc (SiO2, 10% EtOAc in light petroleum ether [40-
60 °C], Rf): 0.10.
(-)-1-Phenylpentylamine (105*)
A solution of BH3.THF (1.0 M solution in THF, 13.08 ml, 13.08 mmol) was added
dropwise to a stirred solution of the S-(-)-aminoalcohol 76 (1.67 g, 6.54 mmol) in
dry THF (6.5 ml) at 0°C under a nitrogen atmosphere. The resulting mixture was
stirred at the same temperature for 8 h, then a solution of the oxime ether 104 (1.0
142
g, 5.23 mmol) in dry THF (3 ml) was added by syringe, keeping the internal
temperature between 0 and 10 °C. The reaction mixture was stirred at rt for 24 h
and then decomposed by the addition of 2 M HCl (10 ml). Evaporation of the THF
deposited the hydrochloride salt of the chiral auxiliary 76 as a white solid, which
was collected under suction and washed with water (10 ml). The aqueous acidic
filtrate was basified with 2 M NaOH and the solution extracted with diethyl ether (3
x 20 ml). The combined organic extracts were dried over anh MgSO4 and
evaporated under reduced pressure to yield a pale yellow liquid. The product was
obtained as a colourless liquid after being purified as above (0.44 g, 52%), α D25
-11.41 (c 0.008410 gml-1, CHCl3) 70% ee, lit159 α D22 -13.9 (neat) 83% ee. The
enantiomeric excess was determined by proton NMR spectroscopy with 10
equivalents of 2,2,2-trifluoro-1-(9-anthryl)ethanol (TFAE, 106) as the chiral
solvating agent at 60 °C. All the other spectroscopic data were identical to those of
the racemate 105.
5-(3-Bromophenyl)-5-oxopentanenitrile (110)
A) A solution of NaHSO3 (5.62 g, 54.0 mmol) in water (4 ml) was added to 3-
bromobenzaldehyde (10.0 g, 54.0 mmol). The reaction mixture spontaneously
warmed up with formation of a white solid. The precipitate of (107) was filtered off
under suction, washed with petroleum ether (40-60 °C, 10 ml), then ethanol (10 ml)
and subsequently used in the next step; mp 165 °C; IR (nujol): 3249 (OH), 1572,
1198, 1069, 880, 762, 698 cm-1; m/z (%): 264 (0.84, M+-Na-2H), 183 (94, M+-
SO3Na-2H), 155/157 (47/46, bromophenyl), 77 (29, phenyl), 64 (29, SO2).
The crude sulfonate 107 was suspended in water (7 ml) and an aq solution of
dimethylamine (25%, 11.2 ml, 58.3 mmol) added dropwise. The white suspension
was stirred at rt for 1.5 h, then a solution of NaCN (2.53 g, 56.2 mmol) in water (5
ml) was added dropwise at 0 °C. The reaction mixture was stirred overnight at rt
and extracted with diethyl ether (3 x 20 ml). The combined organic extracts were
washed with aq NaHSO3 (3 x 20 ml), brine (3 x 20 ml) and dried over anh MgSO4.

143
Evaporation of the solvent under reduced pressure yielded a clear oil (108) after
being dried under vacuo (10.8 g, 83%). 1H-NMR (270 MHz, CDCl3, δH): 2.33 (6H,
s, 2 x CH3), 4.83 (1H, s, CH), 7.29 (1H, t, 3J 8.3 Hz, 5-H), 7.50 (2H, m, 4- and 6-H),
7.70 (1H, s, 2-H); 13C-NMR (67.8 MHz, CDCl3, δC): 42.0 (CH3), 62.7 (CH), 114.7
(CN), 123.2 (3'-C), 126.6 (6'-C), 130.6 (4'-C), 131.0 (5'-C), 132.4 (2'-C), 136.3 (1'-
C).
To a solution of diisopropylamine (7.0 ml, 49.6 mmol) in dry THF (30 ml) containing
TMEDA (14.0 ml, 90.0 mmol), n-BuLi (2.5 M solution in hexane, 20.0 ml, 49.6
mmol) was added dropwise keeping the temperature below -10 °C under a
nitrogen atmosphere. The reaction mixture was stirred for 30 min and allowed to
warm up to 0 °C, then cooled down to -78 °C and a solution of the aminonitrile 108
(9.18 g, 38.4 mmol) in dry THF (10 ml) added drowise under nitrogen. After 10 min
neat bromobutyronitrile (3.8 ml, 38.4 mmol) was added slowly and the reaction
mixture was stirred for a further 1 h at -78 °C. The yellow solution was warmed up
to rt and poured into aq NH4Cl (50 ml). The resulting two phase mixture was
extracted with diethyl ether (3 x 30 ml), and the combined organics were dried over
anh MgSO4 and concentrated in a rotary evaporator to give a yellow oil. Column
chromatography on silica with 20% EtOAc in light petroleum ether (40-60 °C)
afforded compound 109 as a yellow oil (11.5 g, 98%). A satisfactory elemental
analysis for nitrogen could not be obtained. 1H-NMR (270 MHz, CDCl3, δH): 1.20-
1.30 (1H, m, NCCCHH), 1.50-1.70 (1H, m, NCCCHH), 2.20-2.40 (10H, m, 10 x
aliph H), 7.30 (1H, t, 3J 7.4 Hz, 5-H), 7.50 (2H, m, 4- and 6-H), 7.70 (1H, s, 2-H);
13C-NMR (67.8 MHz, CDCl3, δC): 16.9 (CH2CN), 20.7 (CH2CH2CH2), 39.0
(Me2NCCH2), 40.9 (NCH3), 70.9 (Me2NC), 116.9 (CCN), 118.6 (CH2CN), 123.2
(3'-C), 125.2 (6'-C), 129.4 (4'-C), 130.5 (5'-C), 132.2 (2'-C), 140.2 (1'-C); IR (neat):
2962, 2246 (CN), 1594, 1473, 892, 788, 698 cm-1; m/z (%): 305/307 (0.37/0.34,
M+-H), 278/280 (14/12, M+-CN-2H), 238/240 (100/97, M+-(CH2)3CN), 159 (15, M+-
(CH2)3CN-Br), 116 (44, M+-(CH2)3CN-Br-NMe2), 44 (21, NMe2).
144
To a solution of 109 (11.5 g, 37.6 mmol) in ethanol (190 ml) was added
CuSO4.5H2O (24.0 g, 96.1 mmol) and the reaction mixture was refluxed for 2 h.
After being cooled to rt, the inorganics were filtered through Celite and washed
with ethanol (3 x 10 ml). The filtrate was diluted with diethyl ether (200 ml) and the
organic solution was washed with brine (3 x 80 ml). The organic phase was dried
over anh MgSO4 and concentrated in a rotary evaporator to give a brown residue.
Column chromatography on silica with 20% EtOAc in light petroleum ether (40-60
°C) as eluent yielded the title compound 110 as a yellow solid (4.80 g, 50%).
B) According to the procedure described for the synthesis of compound 112, the
benzyl alcohol 56 (1.0 g, 3.93 mmol) was oxidised with DMSO (0.57 ml, 7.87
mmol) in the presence of TFAA (0.86 ml, 5.90 mmol), to yield the product (0.83 g,
84%) as a yellow solid, mp 68 °C, after column chromatography on silica with 25%
EtOAc in light petroleum ether (40-60 °C) as eluent. 1H-NMR (270 MHz, CDCl3,
δH): 2.12 (2H, quin, 3J 5.9 Hz, CH2CH2CH2), 2.53 (2H, t, 3J 5.9 Hz, CH2CN), 3.16
(2H, t, 3J 5.9 Hz, COCH2), 7.38 (1H, t, 3J 7.9 Hz, 5-H), 7.72 (1H, ddd, 3J 7.9 Hz
and 4J 1.3 and 1.3 Hz, 4-H), 7.89 (1H, tt, 3J 7.9 Hz and 4J 1.3 Hz, 6-H), 8.10 (1H, t,
4J 1.3 Hz, 2-H); 13C-NMR (22.4 MHz, CDCl3, δC): 16.6 (CH2CN), 19.7 (CH2CH2),
36.5 (COCH2), 119.2 (CN), 123.2 (3'-C), 126.5 (6'-C), 130.4 (5'-C), 131.1 (2'-C),
136.6 (4'-C), 138.3 (1'-C), 196.7 (CO); IR (nujol): 2246 (CN), 1689 (CO), 1565,
786, 683 cm-1; m/z (%): 251/253 (13.8/13.2, M+-H), 183/185 (100/98, M+-H-
(CH2)3CN), 76 (17, phenyl); Anal calcd for C11H10BrNO (%): C, 52.41; H, 4.00; N,
5.56; Found (%): C, 52.47; H, 3.84; N, 5.44; tlc (SiO2, EtOAc/light petroleum ether
[4-60 °C] 1:1, Rf): 0.55. The product can also be recrystallised from ethanol, but
large quantities are required.

145
5-(3-Bromophenyl)-5-O-methyloximinopentanenitrile (111)
According to the preparation of compound 104, reaction of the ketone 110 (0.50 g,
1.98 mmol) with methoxylamine hydrochloride (0.17 g, 2.05 mmol) gave the
desired product after column chromatography on silica with 25% EtOAc in light
petroleum ether (40-60 °C) as eluent, as a pale yellow oil (0.43 g, 77%). 1H-NMR
(90 MHz, CDCl3, δH): 2.95 (2H, m, CH2CH2CH2), 2.38 ( 2H, t, 3J 7.9 Hz, CH2CN),
2.92 (2H, t, 3J 7.9 Hz, MeONCCH2), 3.86 and 4.00 (3H, s, OCH3), 7.25-7.62 (3H,
m, 4'-, 5'- and 6'-H), 7.83 (1H, t, 4J 2.6 Hz, 2'-H); 13C-NMR (22.4 MHz, CDCl3, δC):
17.0 (CH2CN), 22.4 (CH2CH2), 25.1 (MeONCCH2), 62.2 (OCH3), 119.0 (CN),
122.9 (3'-C), 124.6 (6'-C), 129.1 (5'-C), 130.1 (2'-C), 132.3 (4'-C), 137.0 (1'-C),
154.9 (CNOMe); IR (neat): 2938, 2247 (CN), 1589 (C=N), 1458, 1051 (OMe), 903,
787, 693 cm-1; m/z (%): 280/282 (88/87, M+-H), 249/251 (29/28, M+-OMe-H),
197/199 (57/51, M+-OMe-(CH2)2CN), 182/184 (100/97, M+-OMe-(CH2)3CN),
155/157 (58/57, bromophenyl), 102 (81, M+-OMe-(CH2)3CN-Br), 76 (67, phenyl);
Anal calcd for C12H13BrN2O (%): C, 51.27; H, 4.66; N, 9.96; Found (%): C, 51.38;
H, 4.53; N, 9.95; tlc (SiO2, EtOAc in light petroleum ether [40-60 °C] 1:1, Rf): 0.57
and 0.66.
5-{3-[2-(7-Chloroquinolin-2-yl)ethenyl]phenyl}-5-oxopentanenitrile (112)
Using the method described by Swern et al.,77 to a magnetically stirred solution of
dry DMSO (1.60 ml, 21.5 mmol) in dry CH2Cl2 (12 ml) at -60 °C under nitrogen,
trifluoroacetic anhydride (2.30 ml, 16.2 mmol) was added dropwise, keeping the
temperature between -50 to -60 °C. After 10 min below -60 °C, a solution of the
benzyl alcohol 41 (3.90 g, 10.8 mmol) in dichloromethane (20 ml) was added
dropwise, maintaining the temperature below -50 °C. The reaction mixture was
stirred below -60 °C for 30 min, followed by addition of triethylamine (4.30 ml, 30.8
mmol), keeping the temperature between -50 to -60 °C. After warming up to rt, the
reaction mixture was washed with water (3 x 20 ml) and the aqueous layer back
extracted with CH2Cl2 (3 x 10 ml). The combined organics were dried over anh
146
MgSO4 and concentrated under vacuo to give a yellow residue. Flash column
chromatography of the crude material on silica with 25-40% EtOAc in light
petroleum ether (40-60 °C) as eluent, yielded the pure product as a yellow solid
(2.31 g, 60%), mp 128 °C. 1H-NMR (90 MHz, CDCl3, δH): 2.0-2.40 (2H, m,
CH2CH2CH2), 2.57 (2H, t, 3J 5.6 Hz, CH2CN), 3.23 (2H, t, 3J 7.0 Hz, COCH2), 7.30
(1H, d, 3J 4.2 Hz, quinoline 3-H), 7.36-8.30 (10H, m, 10 x ArH); 13C-NMR (22.4
MHz, CDCl3, δC): 16.7 (CH2CN), 19.8 (CH2CH2CH2), 36.6 (COCH2), 119.3 (CN),
119.7 (quinoline 3-C), 125.9 (quinoline 4a-C), 127.0 ,127.5, 128.1, 128.3, 128.8,
129.4, 129.9, 132.0, 133.9, 135.8 (quinoline 7-C), 136.4 (quinoline 4-C), 137.1,
149.3 (quinoline 8a-C), 156.9 (quinoline 2-C), 198.3 (CO); IR (nujol): 2245 (CN),
1679 (CO), 1591, 1495 cm-1; m/z (%): 360 (0.64, M+), 195 (51, M+-[7-
chloroquinoline]), 126 (100, M+-[7-chloroquinoline]-(CH2)3CN); Anal calcd for
C22H17ClN2O (%): C, 73.23; H, 4.75; N, 7.76; Found (%): C, 73.30; H, 4.54; N,
7.65; tlc (SiO2, EtOAc/light petroleum ether [40-60 °C] 1:1, Rf): 0.44.
3-(2-(7-Chloroquinolin-2-yl)ethenyl)-benzaldehyde-O-methyloximinobenzene (113)
Following the same procedure as for compound 114, the aldehyde 28 (1.0 g, 3.40
mmol) was reacted with methoxylamine hydrochloride (0.29 g, 3.52 mmol) to yield
the product, after the usual workup, as a yellow solid. Recrystallisation from
EtOH/H2O (1:1) afforded the pure compound (0.43 g, 39%), mp 104 °C. 1H-NMR
(60 MHz, CDCl3, δH): 4.06 (3H, s, OCH3), 7.20-8.32 (12H, m, 11 x ArH and 1 x
CHN); IR (nujol): 1608 (C=C), 1592 (C=N), 1497, 1065 (OMe), 832, 787, 690 cm-1;
m/z (%): 322/324 (24/8, M+), 289 (100, M+-OMe-2H), 264/266 (11/3, M+-
CHNOMe), 228 (5.7, M+-CHNOMe-Cl); Anal calcd for C19H15ClN2O (%): C, 70.70;
H, 4.68; N, 8.68; Found (%): C, 70.98; H, 4.50; N, 8.33; tlc (SiO2, 30% EtOAc in
light petroleum ether [40-60 °C], Rf): 0.18 and 0.50.

147
5-{3-[2-(7-Chloroquinolin-2-yl)ethenyl]phenyl}-5-O-methyloximinopentanenitrile
(114)
A solution of the ketone 112 (1.0 g, 2.77 mmol) and methoxylamine hydrochloride
(0.24 g, 2.87 mmol) in pyridine (10 ml) and ethanol (10 ml) was refluxed for 4 h.
After cooling to rt, the solvent was removed under reduce pressure in a rotary
evaporator and the residue taken up in CH2Cl2 (50 ml). The organic phase was
washed with water (3 x 20 ml) and concentrated under vacuo to give a brown oil.
Column chromatography on silica with 50% EtOAc in light petroleum ether (40-60
°C) as eluent yielded the desired product as a pale yellow oil after being dried
under reduced pressure with a vacuum pump (0.93 g, 86%). A satisfactory
elemental analysis was not obtained. 1H-NMR (270 MHz, CDCl3, δH): 1.78 (2H, m,
CH2CH2CH2), 2.24 (2H, m, CH2CN), 2.80 (NCCH2), 3.73 and 3.89 (3H, s, OCH3),
7.67-8.00 (11H, m, 11 x ArH); IR (neat): 2936, 2247 (CN), 1611 (C=C), 1596
(C=N), 1496, 1050 (OMe), 844, 705 cm1; m/z (%): 105 (35, [PhCH2N]+), 43 (100,
[CHNO]+); tlc (SiO2, EtOAc/light petroleum ether [40-60 °C] 1:1, Rf): 0.50 and 0.61.
2-(3-Bromobenzylideneamino)-3-methyl-1,1-diphenylbutanol (115)
A suspension of 3-bromobenzaldehyde (0.50 g, 2.70 mmol), the S-aminoalcohol
76 (0.69 g, 2.70 mmol) and anh MgSO4 (0.65 g, 5.40 mmol) in CHCl3 (10 ml) was
refluxed for 48 h under a nitrogen atmosphere. After cooling to rt, the reaction
mixture was filtered through Celite and the filtrate concentrated in a rotary
evaporator to give a yellow oil after being dried under vacuo (1.12 g, 98%). 1H-
NMR (90 MHz, CDCl3, δH): 0.50 (3H, d, 3J 5.6 Hz, CH3), 1.03 (3H, d, 3J 5.6 Hz,
CH3), 1.86 (1H, m, (CH3)2CH), 2.60 (1H, br s, D2O exch, OH), 3.92 (1H, d, 3J 5.6
Hz, CHCH), 7.00-8.00 (15H, m, 14 x ArH and CH=N); IR (neat): 3338 (OH), 2959,
1653 (C=N), 1597, 1491, 881, 785, 701; m/z (%): 329/331 (1.20/1.18, M+-Ph-Me-
H), 184/186 (93/91, [BrPhCHNH]+), 155/157 (45/45, bromophenyl), 91 (54,
tropylium); tlc (SiO2, EtOAc, Rf): 0.50.
148
(-)-5-(3-Bromophenyl)-5-hydroxypentanenitrile (56*)
According to the procedure for compound 105*, reaction of the ketone 110 (100.0
mg, 0.40 mmol) with BH3.THF (0.80 ml, 0.80 mmol) and the chiral auxiliary 76
(102.1 mg, 0.40 mmol) gave the title compound, after the usual workup, as a
yellow oil (74.2 mg, 74%), α D25 -14.87 (c 0.007420 gml-1, CHCl3), 80% ee (see
Table 2, Entry 4). The enantiomeric excess was determined at rt as described
previously. The spectroscopic data for this product were in accordance with those
of the racemic material 56.
(+)-5-(3-Bromophenyl)-5-hydroxypentanenitrile (56*)
Asymmetric reduction of compound 110 (0.20 g, 0.79 mmol) as described above,
except that commercially available S-(-)-a,a-diphenyl-2-pyrrolidinemethanol (0.20
g, 0.79 mmol) was used as the chiral auxiliary, gave the title compound as a yellow
oil (0.13 g, 65%), α D25 +12.27 (c 0.007660 gml-1, CHCl3), 66% ee (see Table 2,
Entry 3). The enantiomeric excess was determined as above. The spectroscopic
data corresponded to those of the racemate 56.
5-Azido-5-(3-bromophenyl)pentanenitrile (117)
The benzyl alcohol 56 (2.33 g, 9.17 mmol) and diphenylphosphoryl azide (2.50 ml,
11.14 mmol) were dissolved in dry toluene (30 ml). The resulting reaction mixture
was cooled to 0 °C, neat DBU (1.70 ml, 11.14 mmol) was added and the mixture
was stirred for 2 h at the same temperature under a nitrogen atmosphere. After
being stirred overnight at rt, the resulting two-phase mixture was washed with
water (3 x 20 ml) and 5% HCl (3 x 20 ml). The organic layer was dried over anh
MgSO4 and concentrated under reduced pressure to give a brown oil. Column
chromatography of the crude product on silica with CH2Cl2 as eluent yielded the
title compound as a yellow oil (2.0 g, 78%). 1H-NMR (270 MHz, CDCl3, δH): 1.56-
1.95 (4H, m, 2 x CH2), 2.37 (2H, t, 3J 6.7 Hz, CH2CN), 4.46 (1H, dd, 3J 5.6 and 7.6
Hz, N3CHCH2), 7.24-7.32 (2H, m , 5'- and 6'-H), 7.45-7.51 (2H, m, 2'- and 4'-H);

149
13C-NMR (67.8 MHz, CDCl3, δC): 16.9 (CH2CN), 22.1 (CH2CH2), 35.2 (CHCH2),
64.8 (CH), 119.0 (CN), 123.1 (3'-C), 125.4 (6'-C), 129.8 (5'-C), 130.6 (2'-C), 131.7
(4'-C), 141.2 (1'-C); IR (neat): 2941, 2247 (CN), 2104 (N3), 1595, 1476, 884, 788,
698 cm-1; m/z (%): 278/280 (4.23/4.14, M+-H), 250/252 (11.7/9.5, M+-N2-H),
236/238 (16.3/15.7, M+-N3-H), 223/225 (86/81, M+-N4), 197 (97, M+-Br-H), 182/184
(92/89, M+-N3-(CH2)2CN-H), 155/157 (51/50, bromophenyl), 116 (100, M+-Br-N3-
CH2CN-H); Anal calcd for C11H11BrN4 (%): C, 47.33; H, 3.97; N, 20.07; Found (%):
C, 47.32; H, 3.91; N, 19.72; tlc (SiO2, CH2Cl2, Rf): 0.59.
(-)-5-Azido-5-(3-bromophenyl)pentanenitrile (117*)
The title compound was obtained from the (+)-alcohol 56* according to the above
procedure, α D25 -49.12 (c 0.006393 gml-1, CHCl3). The spectroscopic data for this
compound corresponded to those of the racemate 117. The enantiomeric excess
could not be determined, as there was no complexation with TFAE 106.
5-Amino-5-(3-bromophenyl)pentanenitrile (50b)
A) Reduction of ketone 110 (100.0 mg, 0.40 mmol), following method A) as
described for the synthesis of compound 105, gave the desired product as a pale
yellow oil (13.5 mg, 14%). Attempts to purify the crude material by column
chromatography on silica or neutral alumina led to decomposition.
B) A mixture of powdered tellurium (0.11 g, 0.90 mmol), sodium borohydride (81.0
mg, 2.14 mmol) and dry ethanol (1.7 ml) was refluxed under a nitrogen
atmosphere until the tellurium disappeared. After cooling to rt, a solution of the
azide 117 (100.0 mg, 0.36 mmol) in dry diethyl ether (1 ml) was added dropwise
and the reaction mixture was stirred overnight. The suspension was filtered
through Celite, the inorganics washed with ethanol (3 x 5 ml), and the filtrate
concentrated to yield a brown oil. Acidic workup of this residue, as described for
150
the amine 105, yielded the title compund as a pale yellow oil (6.4 mg, 7%, see
Table 1, Entry 4).
C) The azide 117 (100.0 mg, 0.36 mmol) was mixed with triphenylphosphine
(94.42 mg, 0.36 mmol), which resulted in spontaneous evolution of nitrogen. THF
(3 ml) and water (1 ml) were added to the resulting suspension and the two-phase
mixture was stirred overnight at rt under a nitrogen atmosphere. The product was
obtained as a pale yellow oil following the same acidic workup as above (8.2 mg,
9%, see Table 1, Entry 5).
D) A magnetically stirred solution of CuSO4.5H2O (0.90 mg, 3.6.10-3 mmol) in
methanol (1 ml) was cooled in an ice bath and treated portionwise with NaBH4
(13.62 mg, 0.36 mmol). A solution of the azide 117 (100.0 mg, 0.36 mmol) in
methanol (0.5 ml) was added dropwise to the resulting black suspension and the
mixture was stirred overnight under N2. After the usual acidic workup the product
was obtained as a pale yellow oil after being dried under vacuo (10.7 mg, 12%,
see Table 1, Entry 6).
E) To a magnetically stirred solution of the azide 117 (0.20 g, 0.72 mmol) and
hexadecyltributylphosphonium bromide (36.55 mg, 0.072 mmol) in toluene (1 ml),
a solution of NaBH4 (81.71 mg, 2.16 mmol) in water (2 ml) was added dropwise at
80 °C. The reaction mixture was stirred at the same temperature for a further 2 h.
After cooling to rt the layers were separated and the organic phase exposed to the
usual acidic work up. The product was obtained as a pale yellow oil (54.41 mg,
30%, see Table 1, Entry 7).
F) A magnetically stirred solution of the azide 117 (2.0 g, 7.16 mmol) in dry MeOH
(18 ml), was treated with a 3-fold excess of 1,3-propanedithiol (2.2 ml, 21.48 mmol)
under a nitrogen atmosphere. Triethylamine ( 2.75 ml, 21.48 mmol) was added by

151
syringe and the reaction mixture was stirred overnight under N2 at rt. The
precipitated 1,2-dithiolane160 was filtered under suction and washed with CH2Cl2 (3
x 5 ml). The filtrate was diluted with CH2Cl2 (20 ml) and the resulting solution was
washed with water (3 x 10 ml) and dried over anh MgSO4. The solvent was
removed under reduced pressure in a rotary evaporator to yield a yellow liquid.
The usual acidic treatment of the crude material, gave the product as a pale
yellow oil (1.09 g, 60%, see Table 1, Entry 8). A satisfactory elemental analysis
could not be obtained. 1H-NMR (270 MHz, CDCl3, δH): 1.50-1.90 (4H, m, 2 x CH2),
2.34 (2H, t, 3J 6.8 Hz, CH2CN), 3.90 (1H, t, 3J 6.8 Hz, H2NCHCH2), 7.23 (2H, m,
4'- and 6'-H), 7.39 (1H, m, 5'-H), 7.48 (1H, s, 2'-H); 13C-NMR (67.8 MHz, CDCl3,
δC): 17.3 (CH2CN), 22.3 (CH2CH2), 38.0 (CHCH2), 55.3 (CH), 119.5 (CN), 123.0
(3'-C), 125.0, 129.5, 130.03, 130.04, 148.0 (1'-C); IR (neat): 3377 (NH2), 3310
(NH2), 2933, 2246 (CN), 1593, 1474, 886, 785, 698 cm-1; m/z (%): 252/254
(4.7/4.0, M+), 223/225 (2.4/2.4, M+-NH2-N+H), 184/186 (100/93, M+-(CH2)3CN),
105 (8, M+-Br-(CH2)3CN+H), 77 (20, phenyl), [Found: m/z (EI) 252.0260; Calc for
C11H13BrN2: 252.0262]; tlc (SiO2, CH2Cl2, Rf): 0.08.
(-)-5-Amino-5-(3-bromophenyl)pentanenitrile (50b*)
Reduction of the (-)-azide 117* following the above method F) gave the desired
product in 58% yield, α D25 -10.69 (c 0.004210 gml-1, CHCl3). The spectroscopic
data for this compound corresponded to those of the racemate 50b. The
enantiomeric excess could not be determined, as there was no complexation with
TFAE 106.
152
10.6 Experimental for Chapter 5, Section 5.3
2,4,6-Trimethyl-3-nitropyridine (135)
In accordance with the method described by Plazek,112 2,4,6-trimethylpyridine
(16.4 ml, 0.12 mol) was dropped slowly into 70 ml of fuming sulphuric acid (20%
SO3), and the resulting mixture was heated to 100C. During the next 30 min
potassium nitrate (30.0 g, 0.30 mol) was added portionwise, and the solution
further heated at the same temperature for 5 h. The reaction mixture was cooled,
poured slowly into ice (1000 ml), and the resulting mixture carefully made alkaline
with a mixture of sodium and potassium hydroxide pellets. The inorganics were
filtered under suction, and the aqueous layer extracted with diethyl ether (3 x 200
ml). The combined organic layers were dried over potassium hydroxide, and
concentrated in a rotary evaporator to give a brown residue. Distillation of the
crude material yielded the product as a white crystalline solid (5.35 g, 26%), bp
200C, mp 37 C, lit112 bp 228-30 C, mp 38 C. 1H-NMR (60 MHz, CDCl3, δH): 2.30
(3H, s, 4-CH3), 2.56 (6H, s, 2-CH3 and 6-CH3), 6.96 (1H, s, 5-H); IR (nujol): 1604,
1528 (NO2), 1457 (NO2), 1322, 934, 834 cm-1; tlc (SiO2, EtOAc, Rf): 0.61.
1,2,4,6-Tetramethyl-3-nitropyridinium iodide (136)
A solution of the 3-nitropyridine 135 (2.16 g, 13.0 mmol) in MeI (7 ml) was heated
for 4 h in a sealed tube at 100C. After cooling of the reaction mixture to rt, the
precipitated product was collected under suction, washed with ethanol (5 ml), and
dried in an oven at 70C to give the crude material as orange crystals ( 3.20 g,
80%). Recrystallisation from ethanol afforded the pure product as orange plates
(2.86 g, 71%), mp 240 C, lit113 mp 210-11 C. 1H-NMR (60 MHz, DMSO-d6, δH):
2.52 (3H, s, 6-CH3), 2.74 (3H, s, 4-CH3), 2.84 (3H, s, 2-CH3), 4.12 (3H, s, NCH3),
8.18 (1H, s, 5-H); IR (nujol): 1640, 1550 (NO2), 1020, 850 cm-1; m/z (%): 180
(12.5, M+-H-I), 166 (31.2, M+-CH3-I), 120 (100, M+-H-I-CH3-NO2); Anal calcd for
C9H13IN2O2 (%): C, 35.08; H, 4.25; N, 9.09; Found (%): C, 35.32; H, 4.05; N, 8.90.

153
N-Methyl-2-propylimine (137)
A 25-30% aqueous solution of methylamine (66.8 ml, 0.50 mol) was
added dropwise to magnetically stirred acetone (18.4 ml, 0.25 mol) at 0C under a
nitrogen atmosphere. Then solid KOH was poured into the reaction mixture until
the bottom of the flask was covered, keeping the same temperature. The flask was
filled with nitrogen, securely stoppered, and allowed to stand at rt for 48 h with
occasional agitation. After cooling in an ice bath the flask was opened, the layers
separated, and the organic phase distilled under a nitrogen atmosphere to yield the
desired product as a clear liquid (13.6g, 76%), bp 67-8 C, nD20 1.4021, lit114 bp
65-6 C, nD20 1.4023. 1H-NMR (60 MHz, CDCl3, δH): 1.88 (3H, s, CH3), 2.04 (3H, s,
CH3), 3.12 (3H, s, NCH3); IR (neat): 1675 cm-1 (C=N).
N-Methyl-2-butylimine (142)
Following the same procedure as above, butanone (44.7 ml, 0.50 mol) was reacted
with aqueous methylamine (133.6 ml, 1.0 mol) to give the title compound after
distillation as a colourless liquid (14.2 g, 43%), bp 78-80 C, nD20 1.404; 1H-NMR
(270 MHz, CDCl3, δH): 0.84 (3H, t, 3J 8.3 Hz, CH3CH2), 1.81 (3H, s, CH3C=N),
2.24 (2H, q, 3J 8.3 Hz, CH3CH2C=N), 3.08 (3H, s, NCH3); IR (neat): 1669 cm-1
(C=N).
N-Methyl-4-methyl-2-pentylimine (144)
Following the same procedure as for the preparation of compound 137, 4-methyl-
2-pentanone (15.6 ml, 0.13 mol) was reacted with aqueous methylamine (33.4 ml,
0.25 mol) in the presence of solid KOH to yield the desired product after distillation
as a colourless liquid (9.02 g, 65%), bp 98 C; 1H-NMR (60 MHz, CDCl3, δH): 0.94
(6H, d, 3J 7.2 Hz, (CH3)2CH), 1.82 (3H, s, CH3C=N), 2.00-2.28 (3H, m,
(CH3)2CHCH2C=N), 3.12 (3H, s, NCH3); IR (neat): 1665 cm-1 (C=N).
154
N-Cyclohexyl-1-phenyl-2-propylimine (145)
To a magnetically stirred solution of cyclohexylamine (12.6 ml, 0.11 mol) in 30 ml
of dry diethyl ether, 20 g of anh Na2SO4 and phenylacetone ( 13.4 ml, 0.10 mol)
were added at -20 °C. The reaction mixture was allowed to stand at rt for 40 h, the
inorganics were filtered off, and the solvent was evaporated under reduced
pressure. The residue was distilled under vacuo to yield the product as a
colourless liquid (0.60 g, 2.8%), bp 75-80 C/0.3 mm, lit115 bp 155-160 C/15 mm.
The product was found to polymerise readily during distillation. 1H-NMR (60 MHz,
CDCl3, δH): 0.92-2.02 (10H, m, 5 x CH2), 1.70 (3H, s, CH3C=N), 3.15-3.27 (1H, m,
CHN), 3.52 (2H, s, PhCH2C=N ), 7.26 (5H, s, 5 x ArH); IR (neat): 1660 cm-1 (C=N).
N-Cyclohexyl-2-propylimine (146)
The same procedure as above was followed. Reaction of cyclohexylamine (12.6
ml, 0.11 mol) with dry acetone (7.3 ml, 0.10 mol) in the presence of anh Na2SO4
gave the desired product after distillation under reduced pressure as a colourless
liquid (4.80 g, 35%), bp 28 C/0.3 mm, lit116 bp 181 or 67-9 C/17 mm. 1H-NMR (270
MHz, CDCl3, δH): 0.96-1.82 (10H, m, 5 x CH2), 1.84 (3H, s, CH3C=N), 1.99 (3H, s,
CH3C=N), 3.15-3.27 (1H, m, CHN); IR (neat): 1665 cm-1 (C=N).
N-Benzyl-2-propylimine (148)
Following the same procedure, benzylamine (12.0 ml, 0.11 mol) was condensed
with acetone (7.3 ml, 0.10 mol) to give the title compound after vacuum distillation
as a clear liquid (3.80 g, 23%), bp 52 C/0.2 mm, lit117 bp 107 C/13 mm. 1H-NMR
(60 MHz, CDCl3, δH): 1.88 (3H, s, CH3C=N); 2.04 (3H, s, CH3C=N); 4.40 (2H, s,
PhCH2N); 7.24 (5H, s, 5 x ArH); IR (neat): 1663 cm-1 (C=N).
Note: GC and spectroscopic analyses of the prepared imines showed them always
to be contaminated with the corresponding ketone, presumably due to the easy
hydrolysis of the Schiff bases.

155
3-Nitropyridine (152)
Based on the procedure described by Schickh et al.,118 to a magnetically stirred
solution of fuming sulphuric acid (100 ml, 20% SO3) and 30% aq H2O2 (50 ml), a
solution of 3-aminopyridine (5.0 g, 52.6 mmol) in conc sulphuric acid (14 ml) was
added dropwise at 0 C. The reaction mixture was kept at 0 C for 5 h, for 4 d at rt,
and then poured slowly onto ice (1000 ml). The acidic aqueous solution was
carefully made alkaline with NaOH pellets, and the resulting mixture extracted with
diethyl ether (3 x 200 ml). The combined organic extracts were dried over anh
MgSO4 and the solvent evaporated under reduced pressure to leave a brown
residue. The crude material was purified by column chromatography on silica gel
with ethyl acetate as eluent to afford the product as a yellow solid (1.0 g, 15%), mp
37 C, lit118 mp 35-6 C. 1H-NMR (60 MHz, CDCl3, δH): 7.52 (1H, dd, 3J 4.8 and 8.4
Hz, 5-H), 8.48 (1H, dd, 4J 2.4 and 3J 8.4 Hz, 4-H), 8.90 (1H, d, 3J 4.8 Hz, 6-H),
9.40 (1H, d, 4J 2.4 Hz, 2-H); tlc (SiO2, EtOAc, Rf): 0.58.
1-Methyl-3-nitropyridinium iodide (153)
The same procedure as for compound 136 was followed. Reaction of 152 (0.98 g,
7.9 mmol) with MeI (5 ml) gave the title compound after recrystallisation from EtOH
as yellow crystals (1.96 g, 93%), mp 208 C, lit161 mp 212-15 C; 1H-NMR (60 MHz,
DMSO-d6, δH): 4.52 (3H, s, NCH3), 8.24-8.64 (1H, m, 5-H), 9.12-9.52 (2H, m, 4-H
and 6-H), 10.10 (1H, s, 2-H); IR (nujol): 1647, 1588, 1548 (NO2), 1356 (NO2),
1030, 729, 689 cm-1; m/z (%): 143 (100, M+-I+3H), 139 (4.1, M+-I), 127 (25, M+-I-
CH3+3H), 124 (6.3, M+-I-CH3); Anal calcd for C6H7IN2O2 (%): C, 27.09; H, 2.65; N,
10.53; Found (%): C, 27.09; H, 2.47; N, 10.27.
General Procedures for the Synthesis of Polyalkylindoles111
A) The 3-nitropyridinium salt (100.0 mg) was dissolved in 1.2 ml of dry DMF. To
this orange solution 3 mol equivalents of the corresponding N-methylketimine was
156
added at rt, which resulted in spontaneous darkening of the solution (dark brown).
The reaction mixture was kept at rt for 3 d then poured onto ice (10 ml); when the
product precipitated out of solution it was collected under suction, otherwise it was
extracted with CH2Cl2 (3 x 10 ml). The combined organic layers were washed with
brine (3 x10 ml), dried over anh MgSO4 and concentrated under vacuo to leave a
brown residue. The crude material was column chromatographed on silica with
cyclohexane/ethyl acetate (2.5:1) as eluent to yield the pure product.
B) A mixture of the 3-nitropyridinium salt (100.0 mg), 3 mol equivalents of the
corresponding amine, and 15 mol equivalents of the appropriate ketone (15.0
mmol) was kept at rt for 4 d. Again spontaneous darken of the reaction mixture
was observed. The 3-nitropyridium salt, contrary to the previous procedure,
dissolved only very slowly, and needed the whole reaction time to completely
disappear. The same work up method as described above was then followed.
Modifications of the above procedures and yields of the corresponding
polyalkylindoles obtained are summarised in Table 4.
1,2,4,6-Tetramethylindole (138)
The product was obtained as a pale yellow solid, mp 81 °C, lit162 mp 82-3 °C;
1H-NMR (60 MHz, CDCl3, δH): 2.46 (9H, m, 2-, 4- and 6-CH3), 3.62 (3H, s, NCH3),
6.22 (1H, s, 3-H), 6.72 (1H, s, 5-H), 6.92 (1H, s, 7-H); m/z (%): 173 (100,M+), 158
(33, M+-CH3); tlc (SiO2, cyclohexane/EtOAc 2.5:1, Rf): 0.27.
1,2,4,6,7-Pentamethylindole (143)
The product was obtained as a pale yellow solid, mp 132 °C, lit163 mp 132-34 °C;
1H-NMR (60 MHz, CDCl3, δH): 2.36 (9H, m, 4-, 6- and 7-CH3), 2.62 (3H, s, 2-CH3),
3.90 (3H, s, NCH3), 6.18 (1H, s, 3-H), 6.70 (1H, s, 5-H); IR (nujol): 1591, 1557,

157
1329, 1303, 1208, 1060, 1020, 824, 772, 739 cm-1; m/z (%): 187 (100, M+), 172
(64, M+-CH3); Anal calcd for C13H17N (%): C, 83.37; H, 9.15; N, 7.48; Found (%):
C, 83.14; H, 9.27; N, 7.14; tlc (SiO2, cyclohexane/EtOAc 2.5:1, Rf): 0.26.
1-Cyclohexyl-2,4,6-trimethylindole (147)
The product was obtained as a yellow oil, lit164 mp 53-5 °C; 1H-NMR (270 MHz,
CDCl3, δH): 1.20-2.38 (10H, m, 5 x CH2), 2.44 (9H, m, 2-, 4- and 6-CH3), 4.09 (1H,
m, CHN ), 6.17 (1H, s, 3-H), 6.74 (1H, s, 5-H), 7.18 (1H, s, 7-H); IR (neat): 2932,
2359, 1653, 1609, 1558, 1447, 1400, 1381, 1333, 1313, 1286, 1233, 1054, 827,
772, 736 cm-1; m/z(%): 241 (100, M+), 198 (3.5, M+-(CH3)2CH), 159 (57, M+-
cyclohexyl+H), 158 (38, M+-cyclohexyl); tlc (SiO2, cyclohexane/EtOAc 2.5:1, Rf):
0.64.
1-Benzyl-2,4,6-trimethylindole (149)
The product was obtained as a yellow oil, 1H-NMR (270 MHz, CDCl3, δH): 2.34
(3H, s, 6-CH3), 2.38 (3H, s, 4-CH3), 2.50 (3H, s, 2-CH3), 5.26 (2H, s, PhCH2N),
6.28 (1H, s, 3-H), 6.73 (1H, s, 5-H), 6.85 (1H, s, 7-H), 6.98 (2H, dd, 4J 1.8 Hz and
3J 8.0 Hz, 2'- and 6'-H), 7.05-7.48 (3H, m, 3'-, 4'- and 5'-H); 13C-NMR (67.8 MHz,
CDCl3, δC): 12.8 (6-CH3), 18.6 (4-CH3), 21.8 (2-CH3), 46.4 (CH2), 98.7 (3-C),
106.9 (7-C), 121.7 (5-C), 126.0 (4'-C), 126.4 (3a-C), 126.7 (6-C), 127.1 (3'-C),
128.7 (2'-C), 130.6 (4-C), 135.3 (7a-C), 137.3 (2-C), 138.1 (1'-C); IR (neat): 2923,
2359, 1716, 1662, 1604, 1551, 1527, 1495, 1453, 1400, 1381, 1332, 1295, 1241,
1207, 1072, 1028, 826, 775, 731, 697 cm-1; m/z (%): 249 (65, M+), 173 (100, M+-
phenyl), 91 (87, tropylium) [Found: m/z (EI) 249.1517; Calc for C18H19N:
249.1517]; tlc (SiO2, cyclohexane/EtOAc 2.5:1, Rf): 0.57.
(+)-1-(a-Methylbenzyl)-2,4,6-trimethylindole (150)165
The product was obtained as a yellow oil, α D25 18.6 (c 0.00335 gml-1, CHCl3); 1H-
NMR (270 MHz, CDCl3, δH): 1.94 (3H, d, 3J 6.7 Hz, CH3CHN), 2.30 (6H, s, 4- and
158
6-CH3), 2.48 (3H, s, 2-CH3), 5.73 (1H, q, 3J 6.7 Hz, CH3CHN), 6.25 (1H, s, 3-H),
6.68 (1H, s, 5-H), 6.73 (1H, s, 7-H), 7.15 (2H, d, 3J 7.3 Hz, 2'- and 6'-H), 7.20-7.35
(3H, m, 3'-, 4'- and 5'H); 13C-NMR (67.8 MHz, CDCl3, δC): 14.1 (6-CH3), 18.6 (4-
CH3), 18.7 (CH3), 21.8 (2-CH3), 52.3 (CH), 99.4 (3-C), 108.5 (7-C), 121.3 (5-C),
126.2 (4'-C), 126.8 (3a-C), 127.0 (6-C), 128.5 (3'-C), 128.6 (2'-C), 130.0 (4-C),
135.4 (7a-C), 136.2 (2-C), 141.7 (1'-C); IR (neat): 2916, 2361, 1714, 1655, 1605,
1550, 1527, 1495, 1448, 1393, 1376, 1333, 1278, 1237, 1057, 1027, 832, 773,
740, 698 cm-1; m/z (%): 263 (55, M+), 159 (100, M+-phenethyl), 105 (56,
phenethyl), 77 (11, phenyl); tlc (SiO2, 30% EtOAc in light petroleum ether (40-60 °
C), Rf): 0.60.
10.7 Experimental for Chapter 6, Section 6.1
7-Oxo-4,5,6,7-tetrahydrobenzofuran (181)
An aqueous solution of chloroacetaldehyde (50%, 14.0 ml, 0.11 mol) and sodium
hydrogen carbonate (10.0 g, 0.12 mol) were added to water (80 ml) at 0-5 °C. To
this mixture, a solution of 1,2-cyclohexanedione (11.2 g, 0.10 mol) in water (90 ml)
was added dropwise under stirring, keeping the same temperature as above. The
resulting yellow solution was stirred overnight at rt to give a red solution, which was
diluted with EtOAc (100 ml). The reaction mixture was acidified with concentrated
sulphuric acid (pH 1) and stirred for a further 1 h at rt. The layers were separated,
the organic phase washed with aqueous sodium carbonate solution (3 x 50 ml) and
dried over anh MgSO4. Evaporation of the solvent under reduced pressure yielded
a brown oil. Flash column chromatography of the crude material on silica with 10%
EtOAc in light petroleum ether (40-60 °C) as eluent yielded the title compound as a
yellow oil that solidified upon standing (2.40 g, 18%), mp 60 °C, lit132 mp 58-61 °C.
1H-NMR (270 MHz, CDCl3, δH): 2.16 (2H, m, 5-CH2), 2.57 (2H, t, 3J 6.14 Hz, 4-
CH2), 2.78 (2H, t, 3J 6.14 Hz, 6-CH2), 6.43 (1H, d, 3J 2.45 Hz, 3-CH), 7.58 (1H, d,

159
3J 2.45 Hz, 2-CH); 13C-NMR (67.8 MHz, CDCl3, δC): 23.0 (5-C), 24.4 (4-C), 38.3
(6-C), 111.6 (3-C), 140.0 (7a-C), 147.4 (2-C), 147.7 (3a-C), 186.3 (CO); IR (neat):
3122, 2941, 1663 (CO), 1434, 1108, 889, 810 cm-1; m/z (%): 136 (100, M+), 121
(21, M++H-O); Anal calcd for C8H8O2 (%): C, 70.57; H, 5.92; Found (%): C, 70.60;
H, 5.86; tlc (SiO2, 50% EtOAc in light petroleum ether (40-60 °C), Rf): 0.36.
General Procedure for the Synthesis of N-Substituted 4- and 7-Oxo-4,5,6,7-
tetrahydroindoles
A solution of 4- or 7-oxo-4,5,6,7-tetrahydrobenzofuran (100.0 mg, 0.73 mmol) and
three equivalents of the corresponding amine in 20% aqueous ethanol (~2 ml) was
heated in a sealed tube at 150 °C for 12 or 36 h respectively. The reaction mixture
was poured into water (10 ml) and the resulting solution extracted with
dichloromethane (3 x 10 ml). The combined organic extracts were dried over anh
MgSO4, concentrated in a rotary evaporator and the brown residue
chromatographed on silica gel to give the corresponding 4- or 7-oxo-4,5,6,7-
tetrahydroindole.
1-Methyl-7-oxo-4,5,6,7-tetrahydroindole (162)
Column chromatography on silica with 5% EtOAc in light petroleum ether (40-60
°C) as eluent afforded the title compound as a yellow oil (23.9 mg, 22%). 1H-NMR
(270 MHz, CDCl3, δH): 2.06 (2H, m, 5-CH2), 2.47 (2H, t, 3J 5.94 Hz, 4-CH2), 2.73
(2H, t, 3J 5.94 Hz, 6-CH2), 3.91 (3H, s, NCH3), 5.96 (1H, d, 3J 2.18 Hz, 3-H), 6.72
(1H, d, 3J 2.18 Hz, 2-H); 13C-NMR (67.8 MHz, CDCl3, δC): 24.0 (5-C), 25.3 (4-C),
36.6 (6-C), 39.3 (NCH3), 106.6 (3-C), 130.3 (2-C), 131.1 (7a-C), 137.7 (3a-C),
189.3 (CO); IR (neat): 2920, 1650 (CO), 1510, 1440, 1410, 1210, 1005, 760 cm-1;
m/z (%): 149 (100, M+), 134 (8.1, M+-CH3); tlc (SiO2, 5% EtOAc in light petroleum
160
ether (40-60 °C), Rf): 0.07. The 1H-NMR and the IR spectra were in agreement
with the published spectroscopic data.124
7-Oxo-4,5,6,7-tetrahydroindole (166)
Column chromatography on silica with 30% EtOAc in light petroleum ether (40-60
°C) as eluent yielded the title compound as a white solid (35.5 mg, 36%), mp 92-5
°C, lit126 mp 95 °C. 1H-NMR (270 MHz, CDCl3, δH): 2.11 (2H, m, 5-CH2), 2.52
(2H, t, 3J 5.70 Hz, 4-CH2), 2.77 (2H, t, 3J 5.7 Hz, 6-CH2), 6.10 (1H, d, 3J 2.28 Hz,
3-H), 7.07 (1H, d, 3J 2.28 Hz, 2-H), 10.78 (1H, br s, D2O exch, NH); 13C-NMR
(22.4 MHz, CDCl3, δC): 23.5 (5-C), 25.4 (4-C), 37.9 (6-C), 108.6 (3-C), 126.2 (2-
C), 128.2 (7a-C), 137.6 (3a-C), 189.0 (CO); IR (nujol): 3270 (NH), 1680 (CO) cm-1;
m/z (%): 135 (100, M+), 118 (10, M+-O-H), 79 (64, cyclohexyl); tlc (SiO2, 50%
EtOAc in light petroleum ether (40-60 °C), Rf): 0.36.
1-Benzyl-7-oxo-4,5,6,7-tetrahydroindole (190)
Column chromatography on silica with 5% EtOAc in light petroleum ether (40-60
°C) as eluent afforded the title compound as a yellow oil (47.9 mg, 30%). 1H-NMR
(270 MHz, CDCl3, δH): 2.06 (2H, m, 5-CH2), 2.47 (2H, t, 3J 6.16 Hz, 4-CH2), 2.74
(2H, t, 3J 6.16 Hz, 6-CH2), 5.52 (2H, s, PhCH2N), 6.01 (1H, d, 3J 2.16 Hz, 3-H),
6.81 (1H, d, 3J 2.16 Hz, 2-H), 7.14-7.35 (5H, m, 5 x ArH); 13C-NMR (67.8 MHz,
CDCl3, δC): 24.0 (5-C), 25.0 (4-C), 39.4 (6-C), 52.0 (PhCH2N), 107.4 (3-C), 126.5
(7a-C), 127.5 (4'-C), 128.6 (3'-C), 129.5 (2'-C), 138.1 (1'-C), 189.0 (CO); IR (neat):
2933, 1646 (CO), 1499, 1411, 1304, 1213, 1005, 759, 732, 708 cm-1; m/z (%): 225
(100, M+), 91 (65, tropylium); tlc (SiO2, 5% EtOAc in light petroleum ether (40-60
°C), Rf): 0.09. The 1H-NMR and the IR spectra were in agreement with the
published spectroscopic data.129

161
(+)-1-(α-Methylbenzyl)-7-oxo-4,5,6,7-tetrahydroindole (192)
Column chromatography on silica with 5% EtOAc in light petroleum ether (40-60
°C) as eluent afforded the title compound as a yellow oil. Modifications of the
general procedure and yields are summarise in Table 6. α D20 140.7 (c 0.006610
gml-1, CHCl3). 1H-NMR (270 MHz, CDCl3, δH): 1.75 (3H, d, 3J 7.07 Hz, CH3CHN),
2.03 (2H, m, 5-CH2), 2.45 (2H, t, 3J 6.71 Hz, 4-CH2), 2.73 (2H, t, 3J 6.36 Hz, 6-
CH2), 6.01 (1H, d, 3J 2.83 Hz, 3-H), 6.60 (1H, q, 3J 7.07 Hz, CH3CHN), 6.94 (1H,
d, 3J 2.83 Hz, 2-H), 7.18-7.35 (5H, m, 5 x ArH); 13C-NMR (22.4 MHz, CDCl3, δC):
21.6 (CH3), 24.0 (5-C), 25.0 (4-C), 39.6 (6-C), 55.5 (NCH), 107.3 (3-C), 126.0 (2-
C), 126.5 (2'-C), 127.3 (3'-C), 128.5 (4'-C), 138.1 (3a-C), 142.6 (1'-C), 188.8 (CO);
IR (neat): 2934, 1645 (CO), 1495, 1411, 1285, 1205, 1003, 759 cm-1; m/z (%): 239
(75, M+), 224 (4.3, Me+-CH3), 135(100, M++H-phenethyl), 105 (73, phenethyl);
Anal calcd for C16H17NO (%): C, 80.30; H, 7.16; N, 5.85; Found (%): C, 80.50; H,
7.19; N, 6.15; tlc (SiO2, 5% EtOAc in light petroleum ether (40-60 °C), Rf): 0.09.
1-(α-Butylbenzyl)-7-oxo-4,5,6,7-tetrahydroindole (193)
Column chromatography on silica with 5% EtOAc in light petroleum ether (40-60
°C) as eluent afforded the title compound as a yellow oil (30.7 mg, 17%). 1H-NMR
(270 MHz, CDCl3, δH): 0.87 (3H, t, 3J 7.38 Hz, CH3), 1.20-1-41 (4H, m, 2 x CH2),
1.97-2.17 (4H, m, 2 x CH2), 2.45 (2H, tt, 2J 2.11 Hz and 3J 6.33 Hz, 4-CH2), 2.72
(2H, tt, 2J 1.99 Hz and 3J 6.33 Hz, 6-CH2), 6.02 (1H, d, 3J 2.51 Hz, 3-H), 6.50 (1H,
t, 3J 7.85 Hz, NCH), 7.03 (1H, d, 3J 2.51 Hz, 2-H), 7.28 (5H, m, 5 x ArH); 13C-NMR
(67.8 MHz, CDCl3, δC): 13.9 (CH3), 22.4 (CH3CH2), 24.1 (5-C), 24.9 (4-C), 28.6
(CH2CH2), 35.4 (CHCH2), 39.8 (6-C), 59.9 (NCH), 107.5 (3-C), 125.9 (2-C), 126.7
(7a-C), 127.0 (2'-C), 127.4 (3'-C), 128.5 (4'-C), 137.9 (3a-C), 141.9 (1'-C), 189.1
(CO); IR (neat): 2931, 1646 (CO), 1491, 1411, 1282, 1202, 1009, 757 cm-1; m/z
(%): 281 (61, M+), 146 (50, phenylpentyl), 117 (45, phenylpropyl), 104 (22,
phenethyl), 91 (100, tropylium), 77 (13, phenyl); tlc (SiO2, 5% EtOAc in light
petroleum ether (40-60 °C), Rf): 0.12.
162
1-Cyclohexyl-7-oxo-4,5,6,7-tetrahydroindole (194)
Column chromatography on silica with 5% EtOAc in light petroleum ether (40-60
°C) as eluent afforded the title compound as a green oil that solidified upon
standing (54.6 mg, 34%), mp 58-60 °C. 1H-NMR (270 MHz, CDCl3, δH): 1.49 (4H,
m, 2 x CH2), 1.17-1.43 (4H, m, 2 x CH2), 2.00-2.14 (4H, m, 2 x CH2), 2.47 (2H, t,
3J 6.00 Hz, 4-CH2), 2.74 (2H, t, 3J 6.00 Hz, 6-CH2), 4.95-5.10 (1H, m, NCH), 5.99
(1H, d, 3J 2.91 Hz, 3-H), 7.04 (1H, d, 3J 2.91 Hz, 2-H); 13C-NMR (67.8 MHz,
CDCl3, δC): 24.1 (5-C), 25.0 (4-C), 25.6 (4'-C), 25.8 (3'-C), 34.4 (2'-C), 39.9 (6-C),
56.5 (1'-C), 106.9 (3-C), 125.0 (2-C), 126.1 (7a-C), 137.8 (3a-C), 188.8 (CO); IR
(nujol): 1646 (CO), 1494, 1430, 1412, 1205, 1004, 753 cm-1; m/z (%): 217 (63,
M+), 135 (100, M+-cyclohexyl); tlc (SiO2, 5% EtOAc in light petroleum ether (40-60
°C), Rf): 0.13. A satisfactorily elemental analysis could not be obtained.
1-Phenethyl-7-oxo-4,5,6,7-tetrahydroindole (195)
Column chromatography on silica with 5% EtOAc in light petroleum ether (40-60
°C) as eluent afforded the title compound as a yellow oil (80.2 mg, 46%). 1H-NMR
(270 MHz, CDCl3, δH): 2.07 (2H, m, 5-CH2), 2.49 (2H, t, 3J 6.75 Hz, 4-CH2), 2.73
(2H, t, 3J 6.75 Hz, 6-CH2), 3.01 (2H, t, 3J 7.40 Hz, CH2Ph), 4.44 (2H, t, 3J 7.40 Hz,
NCH2), 5.89 (1H, d, 3J 2.16 Hz, 3-H), 6.55 (1H, d, 3J 2.16 Hz, 2-H), 7.09-7.31 (5H,
m, 5 x ArH); 13C-NMR (67.8 MHz, CDCl3, δC): 24.0 (5-C), 25.2 (4-C), 38.1 (6-C),
39.4 (CH2Ph), 51.0 (NCH2), 106.5 (3-C), 126.2 (7a-C), 126.4 (2-C), 128.4 (4'-C),
129.0 (3'-C), 129.7 (2'-C), 138.3 (3a-C), 138.5 (1'-C), 188.6 (CO); IR (neat): 2936,
1645 (CO), 1499, 1411, 1309, 1211, 1004, 754 cm-1; m/z (%): 239 (78, M+), 148
(100, M+-benzyl), 135 (96, M+-phenethyl+H), 92 (17, tropylium), 77 (14, phenyl);
Anal calcd for C16H17NO (%): C, 80.30; H, 7.16; N, 5.85; Found (%): C, 80.00; H,
7.12; N, 6.17; tlc (SiO2, 5% EtOAc in light petroleum ether (40-60 °C), Rf): 0.09.

163
1-(α-Butylbenzyl)-4-oxo-4,5,6,7-tetrahydroindole (196)
Column chromatography on silica with 5% and then 50% EtOAc in light petroleum
ether (40-60 °C) as eluent afforded the title compound as a yellow oil (14.4 mg,
8%). 1H-NMR (270 MHz, CDCl3, δH): 0.91 (3H, t, 3J 7.34 Hz, CH3), 1.24-1.44 (4H,
m, 2 x CH2), 2.02-2.24 (4H, m, 2 x CH2), 2.40-2.80 (4H, m, 2 x CH2), 5.35 (1H, t,
3J 7.71 Hz, CHN), 6.62 (1H, d, 3J 3.44 Hz, 3-H), 6.79 (1H, d, 3J 3.44 Hz, 2-H),
7.08-7.40 (5H, m, 5 x ArH); 13C-NMR (67.8 MHz, CDCl3, δC): 13.9 (CH3), 22.1
(CH3CH2), 22.4 (5-C), 23.6 (4-C), 28.8 (CH2CH2), 35.2 (CHCH2), 37.8 (6-C), 60.6
(NCH), 105.7 (3-C), 119.5 (2-C), 120.9 (7a-C), 126.3 (2'-C), 127.8 (3'-C), 128.9 (4'-
C), 141.0 (3a-C), 143.7 (1'-C), 194.4 (CO); IR (neat): 2931, 1653 (CO), 1496,
1413, 1111 700 cm-1; m/z (%): 281 (17, M+), 147 (16, phenylpentyl), 117 (23
phenylpropyl), 104 (11, phenylethyl), 91 (100, tropylium), 77 (18, phenyl); tlc (SiO2,
50% EtOAc in light petroleum ether (40-60 °C), Rf): 0.49.
10.8 Experimental for Chapter 7
7-Hydroxybenzofuran (207)
To a magnetically stirred solution of diisopropylamine (0.35 ml, 2.50 mmol) in dry
THF (10 ml), n-butyllithium (2,5 M solution in hexane, 1.10 ml, 2.75 mmol) was
added at -40 °C under a nitrogen atmosphere. After stirring at the same
temperature for 1 h, a solution of 7-oxo-4,5,6,7-tetrahydrobenzofuran (181, 0.34 g,
2.50 mmol) in dry THF (5 ml) was added dropwise keeping the temperature below
-20 °C. The deep yellow solution was stirred for a further 1 h, then
chlorotrimethylsilane (0.32 ml, 2.50 mmol) was added slowly below -20 °C. The
reaction mixture was allowed to warm up to room temperature, and after stirring for
1 h the solvent was distilled under reduced pressure. Dry hexane (20 ml) was
added and the insoluble material removed by filtration. The organic filtrate was
concentrated under vacuo to yield a yellow oil (206) that was subsequently used in
164
the next step without further purification [tlc (SiO2, 30% EtOAc in light petroleum
ether (40-60 °C), Rf): 0.88].
The crude silyl enol ether 206 was taken up in dry acetonitrile (10 ml) and stirred in
the presence of Pd(OAc)2 (0.56 g, 2.50 mmol) for 4 h at rt. The inorganics were
filtered through Celite, washed with CH2Cl2 (5 ml), and the filtrate concentrated
under reduce pressure to yield a yellow oil. Column chromatography of this residue
on silica with 30% EtOAc in light petroleum ether (40-60 °C) as eluent gave the title
compound as a white solid, mp 42-45 °C, lit166 mp 43 °C ; 1H-NMR (60 MHz,
CDCl3, δH): 6.80-7.20 (5H, m, 5 x ArH); IR (neat): 3400 (br, OH), 2959, 1625,
1595, 1476, 1251, 1075, 843; tlc (SiO2, 30% EtOAc in light petroleum ether (40-60
°C), Rf): 0.34.
(+)-6-Iodo-7-oxo-1-(a-methylbenzyl)-4,5,6,7-tetrahydroindole (209)
A) The title compound was prepared according to the above procedure by reaction
of the 7-oxotetrahydroindole 192 (0.44 g, 1.84 mmol) with LDA [n-butyllithium (0.88
ml, 2.2 mmol), diisopropylamine (0.28 ml, 2.20 mmol)] and iodine (0.51 g, 2.02
mmol). Column chromatography on silica with 5% EtOAc in light petroleum ether
(40-60 °C) as eluent gave the product as a yellow oil (99.0 mg, 15%).
B) Following the method described by Kotnis,146 iodine crystals (0.48 g, 1.9 mmol)
were added to a solution of 192 (0.23 g, 0.95 mmol) in methanol (3 ml). The
reaction mixture was refluxed and stirred for 4 h under N2. After cooling of the dark
brown solution to rt the solvent was removed under vacuo, the dark brown residue
thus obtained was dissolved in CH2Cl2 (10 ml), and the mixture was washed with a
saturated solution of sodium bicarbonate (3 x 10 ml), sodium thiosulfate (3 x 10
ml), 5% sodium hydroxide (3 x 10 ml) and water (3x 10 ml) before being dried over
anh MgSO4. The solvent was distilled in a rotary evaporator to give a brown oil.
Column chromatography as above afforded the desired product as a
diastereomeric mixture (0.21 g, 60%), α D22 151.2 (c 0.008030 gml-1, CHCl3). 1H-

165
NMR (270 MHz, CDCl3, δH): 1.77 (3H, t, 3J 7.10 Hz, CHCH3), 2.00-2.29 (2H, m, 5-
CH2), 2.50-2.77 (2H, m, 4-CH2), 4.81 (1H, t, 3J 3.59 Hz, CHI), 6.05 and 6.06 (1H,
d, 3J 2.63 Hz, 3-H), 6.54 (1H, q, 3J 7.10 Hz, CH3CH), 6.98 and 7.05 (1H, d, 3J 2.63
Hz, 2-H), 7.14-7.35 (5H, m, 5 x Ar'H); IR (neat): 2930, 1646 (CO), 1495, 1425,
1285, 765, 701 cm-1; m/z (%): 365 (1.5, M+), 238 (100, M+-I), 105 (56,
phenylethyl), 77 (13, phenyl), [Found: m/z (EI) 365.0277; Calc for C16H16INO:
365.0277]; tlc (SiO2, CH2Cl2, Rf): 0.55.
A second fraction was isolated in low yield (35.0 mg, 10%) after column
chromatography and identified as 3-iodo-7-oxo-1-(a-methylbenzyl)-4,5,6,7-
tetrahydroindole (210): 1H-NMR (270 MHz, CDCl3, δH): 1.75 (3H, d, 3J 7.43 Hz,
CHCH3), 2.06 (2H, m, 5-CH2), 2.48 (2H, t, 3J 6.75 Hz, 4-CH2), 2.60 (2H, t, 3J 6.62
Hz, 6-CH2), 6.60 (1H, q, 3J 7.43 Hz, CHCH3), 7.00 (1H, s, 2-H), 7.20-7.38 (5H, m,
5 x Ar'H); 13C-NMR (67.8 MHz, CDCl3, δC): 21.7 (CH3), 24.6 (5-C), 24.9 (4-C),
39.2 (6-C), 56.1 (CH), 63.1 (3-C), 126.6 (2'-C), 127.2 (7a-C), 127.7 (3'-C), 128.7
(4'-C), 130.0 (2-C), 140.5 (3a-C), 141.7 (1'-C), 188.5 (CO); IR (neat): 2930, 1653
(CO), 1452, 1375, 1074, 697 cm-1; m/z (%): 365 (64, M+), 261 (100, M++H-
phenylethyl), 105 (92, phenylethyl), 77 (23, phenyl); tlc (SiO2, CH2Cl2, Rf): 0.34.
(+)-7-Cyanomethoxy-1-(a-methylbenzyl)indole (212)
A mixture of compound 209 (95.9 mg, 0.26 mmol) and neat DBU (2 ml) was stirred
for 15 min at rt under a nitrogen atmosphere. The dark brown solution was poured
into water (10 ml) and the solution was extracted with CH2Cl2 (3 x 10 ml). The
combined organic layers were washed with saturated aqueous NH4Cl solution (3 x
10 ml), dried over anh MgSO4 and concentrated in a rotary evaporator to give a
brown oil (211). This residue was subsequently used in the next step without
further purification. 1H-NMR (270 MHz, CDCl3, δH): 1.85 (3H, d, 3J 7.11 Hz,
CH3CH), 6.46 (1H, d, 3J 3.41 Hz, 3-H), 6.60 (1H, dd, 4J 1.14 Hz and 3J 7.82 Hz, 6-
H), 6.68 (1H, q, 3J 7.11 Hz, CH3CH), 6.86 (1H, t, 3J 7.82 Hz, 5-H), 7.08-7.31 (7H,
m, 7 x ArH); tlc (SiO2, CH2Cl2, Rf): 0.40.
166
A solution of the crude 7-hydroxyindole 211 in butanone (2 ml) was degassed with
N2. Anhydrous K2CO3 (90.0 mg, 0.65 mmol) and bromoacetonitrile (0.03 ml, 0.43
mmol) were added and the mixture refluxed for 1h under N2. The cooled reaction
mixture was poured into 2 M HCl (10 ml) and the resulting solution was extracted
with dichloromethane (3 x 10 ml). The combined organic extracts were dried over
anh MgSO4 and concentrated in vacuo to give a brown oil. Column
chromatography of this residue on silica with 5% EtOAc in light petroleum ether
(40-60 °C) as eluent afforded the title compound as a yellow oil (35.1 mg, 48%),
α D20 132.5 (c 0.001940 gml-1, CHCl3). 1H-NMR (270 MHz, CDCl3, δH): 1.90 (3H,
d, 3J 6.97 Hz, CH3CH), 4.69 (2H, d, 2J 6.75 Hz, OCH2), 6.31 (1H, q, 3J 6.97 Hz,
CH3CH), 6.55 (1H, d, 3J 3.22 Hz, 3-H), 6.64 (1H, d, 3J 7.72 Hz, 6-H), 7.00 (1H, t,
3J 7.72 Hz, 5-H), 7.04-7.09 (2H, m, 2 x Ar'H), 7.20-7.32 (4H, m, 3-H and 3 x Ar'H),
7.35 (1H, dd, 4J 1.07 Hz and 3J 7.72 Hz , 4-H); 13C-NMR (67.8 MHz, CDCl3, δC):
22.5 (CH3), 54.0 (OCH2), 56.8 (CHN), 102.2 (3-C), 104.6, 115.0 (CN), 116.3,
119.7, 125.7, 126.0 (3a-C), 126.1, 127.2, 128.6, 131.9 (7a-C), 144.1 (1'-C), 144.8
(7-C); IR (neat): 2930, 1573, 1453, 1233, 721, 699 cm-1; m/z (%): 276 (30, M+),
172 (17, M+-phenylethyl+H), 132 (20, M+-phenylethyl-CH2CN+H), 105 (100,
phenylethyl), 77 (14, phenyl), [Found: m/z (EI) 276.1263; Calc for C18H16N2O:
276.1263]; tlc (SiO2, CH2Cl2, Rf): 0.59.
10.9 Experimental for Chapter 8
Attempted preparation of 5-(3-bromophenyl)-5-(7-oxo-4,5,6,7-tetrahydroindol-1-yl)-
pentanenitrile (182)
Reaction of the benzofuran 181 with the benzylamine 50b in a sealed tube at 150
°C following the general procedure described previously for the synthesis of 4- and
7-oxotetrahydroindoles gave, after column chromatography on silica with 50%
EtOAc in light pet ether (40-60 °C) as eluent, exclusively the lactam 213 as a white

167
solid (0.28 g, 51%), mp 104 °C. 1H-NMR (270 MHz, CDCl3, δH): 1.51-1.90 (3H, m,
3 x aliph H), 1.99-2.13 (1H, m, 1 x aliph H), 2.35 (2H, m, CH2CO), 4.67 (1H, dd, 3J
7.41 and 7.80 Hz, CH2CHN), 7.22-7.49 (5H, m 5 x ArH); 13C-NMR (67.8 MHz,
CDCl3, δC): 19.0, 31.1, 31.7 (CH2CO), 56.6 (CHN), 122.7 (3-C), 124.8, 129.1,
130.3, 130.7, 145.1 (1-C), 172.8 (HNCO); IR (nujol): 3193 (NH), 3071 (NH), 1649
(CO), 1593, 1400, 880, 789, 698 cm-1; m/z (%): 253/255 (42/39, M+), 174 (100,
M+-Br); Anal calcd for C11H12BrNO (%): C, 51.99; H, 4.76; N, 5.51, Br, 31.44;
Found (%): C, 52.09; H, 4.52; N, 5.35; Br, 31.27; tlc (SiO2, 50% EtOAc in light
petroleum ether (40-60 °C), Rf): 0.09.
1-(3-Bromophenyl)-4-(1H-tetrazol-5-yl)butan-1-one (215)
According to the procedure for the bis-tetrazole 35, reaction of 110 (100.0 mg, 0.40
mmol) with Bu3SnN3 (132.8 mg, 0.40 mmol) gave a brown oil. After treatment of
the crude material with glacial acetic acid in methanol for several days, the desired
product separated as a yellow solid (64.3 mg, 55%), mp 122 °C. 1H-NMR (270
MHz, DMSO-d6, δH): 2.05 (2H, m, CH2CH2CH2), 2.95 (2H, t, 3J 6.75 Hz, CH2Tet),
3.16 (2H, t, 3J 6.75 Hz, COCH2), 7.50 (1H, t, 3J 8.10 Hz, 5'-H), 7.85 (1H, d, 3J 8.10
Hz, 4'-H), 7.96 (1H, d, 3J 8.10 Hz, 6'-H), 8.08 (1H, s, 2'-H); 13C-NMR (67.8 MHz,
DMSO-d6, δC): 37.0, 122.1 (3'-C), 126.8 (6'-C), 130.3 (5'-C), 130.9 (2'-C), 135.7
(4'-C), 138.5 (1'-C), 155.6 (tetrazole-C), 198.2 (CO); IR (neat): 2955, 1692 (CO),
1590, 1464, 878, 779, 680 cm-1; m/z (CI, %): 295/297 (100/95, M+); tlc (SiO2, 50%
EtOAc in light petroleum ether (40-60 °C), Rf): 0.15. A satisfactory elemental
analysis for nitrogen was not obtained.
1-(3-Bromophenyl)-4-(1H-tetrazol-5-yl)butan-1-one-O-methyloxime (216)
Following the same procedure as above, reaction of the oxime ether 111 (1.75 g,
6.22 mmol) with tributyltin azide (2.30 g, 6.85 mmol) with or without solvent gave a
brown oil. Column chromatography of the residue on silica with 10-40% EtOAc in
light petroleum ether (40-60 °C) as eluent yielded the title compound as a white
168
solid (1.31 g, 65%), mp 115-7 °C. 1H-NMR (270 MHz, acetone-d6, δH): 2.05 (2H,
m, CH2CH2CH2), 2.89 (2H, t, 3J 7.20 Hz, CH2Tet), 3.04 (2H, t, 3J 7.20 Hz,
CNCH2), 3.94 (3H, s, OCH3). 7.37 (1H, t, 3J 7.20 Hz, 5'-H), 7.59 (1H, d, 3J 7.20
Hz, 4'-H), 7.71 (1H, d, 3J 7.20 Hz, 6'-H), 7.91 (1H, s, 2'-H); 13C-NMR (67.8 MHz,
acetone-d6, δC): 23.9, 25.0, 25.8, 62.4 (OCH3), 123.1 (3'-C), 126.0 (6'-C), 129.7
(5'-C), 131.3 (2'-C), 132.9 (4'-C), 138.5 (1'-C), 156.7 (tetrazole-C); IR (nujol): 1732,
1587, 1459, 889, 787, 685 cm-1; m/z (CI, %): 324/326 (100/100, M+); tlc (SiO2,
50% EtOAc in light pet ether (40-60 °C), Rf): 0.24. A satisfactory elemental
analysis for carbon could not be obtained, presumably due to the tin residues in
the product.
1-(3-Bromophenyl)-4-(1H-tetrazol-5-yl)butyl azide (217)
The same procedure as above gave the title compound from reaction of the azide
31 (1.00 g, 3.50 mmol) with Bu3SnN3 (1.20 g, 3.60 mmol). Column
chromatography on silica with 50% EtOAc in light petroleum ether (40-60 °C) as
eluent afforded the product as a yellow oil (0.50 g, 43%). 1H-NMR (270 MHz,
CDCl3, δH): 1.77-2.10 (4H, m, 2 x CH2), 3.11 (2H, t, 3J 7.00 Hz, CH2Tet), 4.48 (1H,
t, 3J 7.00 Hz, N3CHCH2). 7.20 (2H, m, 2 x Ar'H), 7.40 (2H, m,2 x Ar'H), 9.13 (1H,
br s, NH); 13C-NMR (67.8 MHz, CDCl3, δC): 23.1, 24.2, 35.4, 65.1 (CHN3), 122.9
(3'-C), 125.5 (6'-C), 129.8 (5'-C), 130.5 (2'-C), 131.6 (4'-C), 141.5 (1'-C), 156.5
(tetrazole-C); IR (neat): 2916, 2100 (N3), 1569, 1476 cm-1; m/z (CI, %): 322 (81,
M+), 294 (65, M+-N2); tlc (SiO2, 50% EtOAc in light petroleum ether (40-60 °C), Rf):
0.12. A satisfactory elemental analysis could not be obtained. This type of
compound lose nitrogen gradually, so that when analysed the result is high in
carbon and low in nitrogen.

169
1-(3-Bromophenyl)-4-(1H-tetrazol-5-yl)butylamine (214)
Following the same procedure as for the synthesis of the amine 50b, reduction of
the azide 217 (0.31 g, 0.95 mmol) with 1,3-propanedithiol (0.30 ml, 2.85 mmol) in
the presence of Et3N (0.40 ml, 2.85 mmol) gave the desired product as a white
solid (0.16 g, 55%), mp 216-20 °C, after the neutralised aqueous solution was kept
overnight in a refrigerator. 1H-NMR (270 MHz, DMSO-d6, δH): 1.40-1.93 (4H, m, 2
x CH2), 2.70 (2H, t, 3J 7.12 Hz, CH2Tet), 4.13 (1H, t, 3J 7.12 Hz, H2NCH). 7.31-
7.46 (2H, m, 4'- and 5'-H), 7.55 (1H, d, 3J 7.12 Hz, 6'-H), 7.68 (1H, s, 2'-H); 13C-
NMR (67.8 MHz, DMSO-d6, δC): 24.0, 24.4, 35.3, 53.7 (H2NCH), 121.7 (3'-C),
126.1 (6'-C), 129.8 (5'-C), 130.65 (2'-C), 130.71 (4'-C), 143.2 (1'-C), 158.9
(tetrazole-C); IR (nujol): 3308 (NH), 1650, 1571, 1402, 896, 787, 697 cm-1; m/z (CI,
%): 296/298 (67/65, M+), 279/281 (42/40, M+-NH2-H), 184/186 (100/96, 3-
bromobenzylamine-H); tlc (SiO2, 50% EtOAc in light petroleum ether (40-60 °C),
Rf): 0. The product is not uv active but colours red on exposure to ninhydrin. A
satisfactory elemental analysis could not be obtained.
2,3-Epoxy-3-methylcyclohexanone (220)
The title compound was prepared following the procedure described by Yamazaki
et al.154 for the synthesis of 2,3-epoxy-3-methylcyclopentanone. 3-Methyl-2-
cyclohexene-1-one (10.0 g, 90.9 mmol) was therefore dissolved in MeOH (37 ml).
To this magnetically stirred light yellow solution aq H2O2 (30%, 30.5 g, 269 mmol)
was added dropwise at 0 °C. Then aq K2CO3 solution (5%, 19.2 g, 6.95 mmol) was
added slowly at the same temperature to the reaction mixture, whereby the colour
disappeared . The clear solution was stirred for 5 h at rt and then extracted with
ethyl acetate (3 x 30 ml). The combined organic extracts were washed with brine
(3 x 20 ml) and concentrated to give a yellow liquid. Column chromatography on
silica with 25% EtOAc in light petroleum ether (40-60 °C) as eluent afforded the
pure product as a colourless liquid (5.35 g, 47%), bp 85 °C/15, lit167 bp 85 °C/15.
1H-NMR (270 MHz, CDCl3, δH): 1.45 (3H, s, CH3), 1.60-2.20 (6H, m, 3 x CH2),
170
3.09 (1H, s, CH); 13C-NMR (67.8 MHz, CDCl3, δC): 17.2 (5-C), 22.2 (CH3), 28.4 (4-
C), 35.7 (6-C), 62.0 (2-C), 62.4 (3-C), 206.7 (CO); IR (neat): 2948, 1700 (CO),
1400, 1287, 1088, 809 cm-1; m/z (%): 126 (81, M+), 111 (8.1, M+-CH3); tlc (SiO2,
50% EtOAc in light petroleum ether (40-60 °C), Rf): 0.55. The product is not uv
active but colours blue when treated with an ammonium molybdate solution in 2N
H2SO4.
2-Benzylamino-3-methyl-2-cyclohexen-1-one (221)
A solution of the epoxy ketone 220 (0.30 g, 2.38 mmol) and benzylamine (0.31 ml,
2.86 mmol) in MeOH/H2O (1.5 ml and 0.5 ml respectively) was refluxed for 4 h
under a nitrogen atmosphere. After cooling to rt, the reaction mixture was extracted
with dichloromethane (3 x 10 ml). The combined organic layers were washed with
brine (3 x 10 ml), dried over anh MgSO4 and concentrated in a rotary evaporator to
yield a red oil. The residue was column chromatographed on silica or alumina with
CH2Cl2 as eluent to yield the product as a brown oil (60.0 mg, 13%). 1H-NMR (270
MHz, CDCl3, δH): 1.84 (2H, m, 5-CH2), 1.96 (3H, s, CH3), 2.35 (4H, m, 2 x CH2),
3.98 (2H, s, CH2Ph), 7.27 (5H, m, 5 x Ar'H); 13C-NMR (67.8 MHz, CDCl3, δC): 20.3
(CH3), 21.9 (5-C), 32.2 (4-C), 37.0 (6-C), 52.4 (CH2Ph), 127.0 (2'-C), 127.7 (3'-C),
128.4 (4'-C), 139.0 (2-C), 139.8 (3-C), 140.2 (1'-C), 196.1 (CO); IR (neat): 3322
(NH), 2929, 1664 (CO), 1635, 1453, 739, 699 cm-1; m/z (%): 215 (70, M+), 200
(6.9, M+-CH3), 138 (8.6, M+-Ph), 106 (64, [PhCH2NH]+), 91 (100, tropylium); tlc
(SiO2, CH2Cl2, Rf): 0.17. The spectroscopic data were in agreement with those
reported.129

171
3-Methyl-2-(a-methylbenzylamino)-2-cyclohexen-1-one (222)
According to the above procedure, reaction of 220 (0.30 g, 2.38 mmol) with R(+)-α-
methylbenzylamine (0.37 ml, 2.86 mmol) gave the title compound as a brown oil,
after being column chromatographed on silica with CH2Cl2 as eluent. Different
reaction conditions and yields are summarise in Table 8. 1H-NMR (270 MHz,
CDCl3, δH): 1.43 (3H, d, 3J 6.75 Hz, CHCH3), 1.49-1.64 (1H, m, 5-CHH), 1.73-1.88
(1H, m, 5-CHH), 1.90 (3H, s, CH3), 2.19-2.36 (4H, m, 2 x CH2), 4.16 (1H, q, 3J
6.75 Hz, CHPh), 7.15-7.25 (5H, m, 5 x Ar'H); 13C-NMR (67.8 MHz, CDCl3, δC):
20.5 (CHCH3), 21.9 (5-C), 23.9 (CH3), 32.3 (4-C), 36.9 (6-C), 56.6 (CHPh), 126.0
(2'-C), 126.8 (3'-C), 128.2 (4'-C), 138.8 (2-C), 145.3 (1'-C), 196.3 (CO); IR (neat):
3330 (NH), 2929, 1663 (CO), 1635, 1452, 763, 701 cm-1; m/z (%): 229 (49, M+),
214 (48, M+-CH3), 125 (34, M+-phenylethyl), 105 (100, [PhCH2CH]+), 77 (14,
phenyl), [Found: m/z (EI) 229.1467; Calc for C15H19NO: 229.1467]; tlc (SiO2,
CH2Cl2, Rf): 0.15.
Reaction of 220 in the presence of 1 equivalent CeCl3.7H2O
The same reaction as above was repeated with the exception that 1 equivalent of
CeCl3.7H2O (0.90 g, 2.38 mmol) was added. Directly after the clear solution
started refluxing a white precipitate was formed. After cooling to rt the reaction
mixture was filtered through Celite and the inorganics washed with CH2Cl2 (3 x 5
ml). After the usual workup a yellow oil was obtained. Column chromatography on
silica with CH2Cl2 as eluent yielded two new products which were identified as: (1)
2-chloro-3-methyl-2-cyclohexene-1-one (223, 121.5 mg, 35%), 1H-NMR (270 MHz,
CDCl3, δH): 2.02 (2H, q, 3J 6.75 Hz, 5-CH2), 2.14 (3H, s, CH3), 2.55 (4H, quin, 3J
6.75 Hz, 2 x CH2); 13C-NMR (67.8 MHz, CDCl3, δC): 21.6 (5-C), 22.4 (CH3), 33.3
(4-C), 37.8 (6-C), 128.9 (2-C), 156.9 (3-C), 190.9 (CO); IR (neat): 2929, 1685
(CO), 1611 (C=C), 1278, 810 cm-1; m/z (%): 144/146 (67//22, M+), 116/118
(100/32), 102/102 (16/5), 88/90 (36/12); tlc (SiO2, CH2Cl2, Rf): 0.39. A satisfactory
172
elemental analysis could not be obtained. (2) N-(α-Methylbenzyll)-3-toluidine (224,
42.7 mg, 9%), 1H-NMR (270 MHz, CDCl3, δH): 1.49 (3H, d, 3J 6.75 Hz, CHCH3),
2.20 (3H, s, ArCH3), 4.48 (1H, q, 3J 6.75 Hz, NCHCH3), 6.30 (1H, dd, 4J 2.14 Hz
and 3J 7.86 Hz, 4-H), 6.36 (1H, s, 2-H), 6.47 (1H, d, 3J 7.86 Hz, 6-H), 6.98 (1H, t,
3J 7.86 Hz, 5-H) 7.18-7.40 (5H, m, 5 x Ar'H); 13C-NMR (67.8 MHz, CDCl3, δC):
21.6 (CHCH3), 24.9 (ArCH3), 53.4 (CHN), 110.4 (6-C), 114.2 (2-C), 118.3 (4-C),
125.9 (2'-C), 126.8 (3'-C), 128.6 (4'-C), 129.0 (5-C), 138.8 (5-C), 145.3 (1'-C),
147.3 (1-C); IR (neat): 3412 (NH), 2966, 1607, 1490, 1325, 767, 700 cm-1; m/z
(%): 211 (63, M+), 196 (100, M+-CH3), 180 (2.1, M+-2 x CH3-H), 134 (11, M+-Ph),
107 (56, M+- phenylethyl-H), 91 (20, tropylium), 77 (21, phenyl); Anal calcd for
C15H17N (%): C, 85.26; H, 8.11; N, 6.63; Found (%): C, 85.17; H, 8.14; N, 6.73; tlc
(SiO2, CH2Cl2, Rf): 0.65.
2-Chloro-3-hydroxy-3-methylcyclohexanone (225)
To a solution of 220 (100.0 mg, 0.79 mmol) in dry acetonitrile (2 ml) anhydrous
CeCl3 (0.19 g, 0.79 mmol) was added. The resulting suspension was refluxed for 6
h under a nitrogen atmosphere. After being cooled to rt, the reaction mixture was
filtered through Celite and the inorganics washed with CH2Cl2 (3 x 5 ml). The
organic filtrate was concentrated under reduce pressure to give the title compound
as a yellow oil (95.2 mg, 74%) after being dried under vacuo. 1H-NMR (270 MHz,
CDCl3, δH): 1.31. (3H, s, CH3), 1.60-2.80 (6H, m, 3 x CH2), 4.26 (1H, s, CHCl);
13C-NMR (67.8 MHz, CDCl3, δC): 20.4 (5-C), 24.1 (CH3), 35.0 (4-C), 37.4 (6-C),
70.8 (2-C), 76.4 (3-C), 203.0 (CO); IR (neat): 3444 (br, OH), 2950, 1729 (C=O),
1378, 1140, 949.

173
1-Benzyl-7-oxo-4,5,6,7-tetrahydroindole (190)
A mixture of 2-benzylamino-3-methyl-2-cyclohexen-1-one (221) (50.0 mg, 0.25
mmol) and dmfdma (0.20 ml, 0.80 mmol) was heated at 150 °C overnight under a
nitrogen atmosphere. Removal of the excess acetal under vacuum left a red oil.
Purification as described previously gave the title compound in 18% yield (9.8 mg).
(+)-1-(α-Methylbenzyl)-7-oxo-4,5,6,7-tetrahydroindole (192)
Reaction of 3-methyl-2-(a-methylbenzylamino)-2-cyclohexen-1-one (222) (50.0 mg,
0.22 mmol) with dmfdma (0.20 ml, 0.80 mmol) following the same procedure as
above gave the title compound as a yellow oil (7.4 mg, 14%). The spectroscopic
data were identical with those reported previously.
174
References
1. W. Feldberg and C. H. Kellaway, J. Physiol. (London), 1938, 94, 187.
2. C. H. Kellaway and E. R. Trethewie, Q. J. Exp. Physiol., 1940, 30, 121.
3. R. P. Orange and K. F. Austen, Adv. Immunol., 1969, 10, 105.
4. K. F. Austen, J. Immunol., 1978, 121, 793.
5. R. P. Orange, R. C. Murphy, M. L. Karnovsky and K. F. Austen, J. Immunol.,
1973, 110, 760.
6. R. C. Murphy, S. Hammarström and B. Samuelsson, Proc. Natl. Acad. Sci.
USA, 1979, 76, 4275.
7. S. Hammarström, R. C. Murphy, B. Samuelsson, D. A. Clark, C. Mioskowski
and E. J. Corey, Biochem. Biophys. Res. Commun., 1979, 91, 1266.
8. S. Hammarström, B. Samuelsson, D. A. Clark, G. Goto, A. Marfat, C.
Mioskowski and E. J. Corey, Biochem. Biophys. Res. Commun., 1980, 92,
946.
9. B. Samuelsson, Angew. Chem., Int. Ed. Engl., 1982, 21, 902.
10. B. Samuelsson, Angew. Chem., Int. Ed. Engl., 1983, 22, 805.

175
11. B. Samuelsson, S.-E. Dahlen, J. A. Lindgren, C. A. Rouzer and C. N. Serhan,
Science, 1987, 237, 1171.
12. O. Radmark, C. Malmsten, B. Samuelsson, D. A. Clark, G. Giichi, A. Marfat
and E. J. Corey, Biochem. Biophys. Res. Commun., 1980, 92, 954.
13. P. Borgeat, M. Hamberg and B. Samuelsson, J. Biol. Chem., 1976, 251,
7816.
14. E. J. Corey, A. Marfat, G. Goto and F. Brion, J. Am. Chem. Soc., 1980, 102,
7984.
15. L. Örning, S. Hammarström and B. Samuelsson, Proc. Natl. Acad. Sci. USA,
1980, 77, 2014.
16. K. Bernström, S. Hammarström, J. Biol. Chem., 1981, 256, 9579.
17. M. E. Anderson, R. D. Allison and A. Meister, Proc. Natl. Acad. Sci. USA,
1982, 79, 1088.
18. R. A. Lewis, K. F. Austen. J. M. Drazen, D. A. Clark, A. Marfat and E. J.
Corey, Proc. Natl. Acac. Sci. USA, 1983, 77, 3710.
19. J. Augstein, J. B. Farmer, T. B. Lee, P. Sheard and M. L. Tattersall, Nature
(London) New Biol., 1973, 245, 215.
20. R. A. Appleton, J. R. Bantick, T. R. Chamberlain, D. N. Hardern, T. B. Lee
and A. D. Pratt, J. Med. Chem., 1977, 20, 371.
176
21. P. Sheard, M. C. Holyroyde, A. M. Ghelani, J. R. Bantick, and T. B. Lee, Adv.
Prostaglandin, Thromboxane Leukotriene Res., 1982, 9, 229.
22. W. S. Marshall, T. Goodson, G. J. Cullinan, D. Swanson-Bean, K. D. Haisch,
L. E. Rinkema and J. H. Fleisch, J. Med. Chem., 1987, 30, 682.
23. J. H. Fleisch, L. E. Rinkema, K. D. Haisch, D. Swanson-Bean, T. Goodson,
P. P. K. Ho and W. S. Marshall, J. Pharmacol. Exp. Ther., 1985, 223, 148.
24. J. G. Gleason, R. F. Hall, C. D. Perchonock, K. F. Erhard, J. S. Frazee, T. W.
Ku, K. Kondrad, M. E. McCarthy, S. Mong, S. T. Crooke, G. Chi-Rosso, M. A.
Wasserman, T. J. Thorpy, R. M. Muccitelli, D. W. Hay, S. S. Tucker and L.
Vickery-Clark, J. Med. Chem., 1987, 30, 959.
25. R. N. Young, J. Y. Gauthier, M. Therien and R. Zamboni, Heterocycles, 1989,
28, 967.
26. J. D. White, K. M. Yager and F. Stappenbeck, J. Org. Chem., 1993, 58,
3466.
27. M. Labelle, M. Belley, E. Champion, R. Gordon, K. Hoogsteen, T. R. Jones,
Y. Leblanc, A. Lord, M. McAuliffe, C. McFarlane, P. Masson, K. M. Metters,
D. Nicoll-Griffith, N. Ouiment, H. Piechuta, C. Rochette, N. Sawyer, Y. B.
Xiang, J. Yergey, A. W. Ford-Hutchinson, C. B. Pickett, R. J. Zamboni and R.
N. Yound, Biorg. Med. Chem. Lett., 1994, 3, 463.
28. J. M. McNamara, J. L. Leazer, M. Bhupathy, J. S. Amato, R. A. Reamer, P. J.
Reider and E. J. J. Grabowski, J. Org. Chem., 1989, 54, 3718.

177
29. V. G. Matassa, T. P. Maduskuie Jr., H. S. Shapiro, B. Hesp, D. W. Snyder,
D. Aharony, R. D. Krell and R. A. Keith, J. Med. Chem., 1990, 33, 1781.
30. R. W. Harper, D. K. Herron, N. G. Bollinger, J. S. Sawyer, R. F. Baldwin, C.
R. Roman, L. E. Rinkema and J. H. Fleisch, J. Med. Chem., 1992, 35, 1191.
31. J. S. Sawyer, R. F. Baldwin, L. E. Rinkema, C. R. Roman, and J. H. Fleisch,
J. Med. Chem., 1992, 35, 1200.
32. F.-C. Huang, R. A. Galemmo Jr., W. H. Johnson Jr., G. B. Poli, M. M.
Morrissette, J. J. Mencel, J. D. Warus, H. F. Campbell, G. W. Nuss, G. W.
Carnathan and R. G. Van Inwegen, J. Med. Chem., 1990, 33, 1194.
33. J. H. Musser, A. F. Kreft, R. H. W. Bender, D. M. Kubrak, D. Grimes, R. P.
Carlson, J. M. Hand and J. Chang, J. Med. Chem., 1990, 33, 240.
34. D. Dobson, A. Todd and J. Gilmore, Synth. Commun., 1991, 21, 611.
35. M. S. Newman and L. F. Lee, J. Org. Chem., 1975, 40, 2650.
36. G. D. Hartman, B. T. Phillips and W. Halczenko, J. Org. Chem., 1985, 50,
2423.
37. R. N. Young, J. Y. Gauthier, R. Zamboni and M. L. Michel, Eur. Pat. 399,818,
1990; Chem. Abstr., 1991, 115, 256016g.
38. Y. Kumar and L. Florvall, Synth. Commun., 1983, 13, 489.
39. B. S. Ong, Tetrahedron Lett., 1980, 21, 4225.
178
40. M. W. J. Urquhart, Research Report, University of East Anglia, 1994.
41. R. A. Lewthwaite, Research Report, University of East Anglia, 1994.
42. G.-J. Kang, W. R. Cullen, M. D. Fryzukl, B. R. James and J. P. Kutney, J.
Chem. Soc., Chem. Commun., 1988, 1466.
43. A. G. Becalski, W. R. Cullen, M. D. Fryzuk, B. R. James, G.-J. Kang and S.
J. Rettig, Inorg. Chem., 1991, 30, 5002.
44. Y. Amrani, L. Lecomte, D. Sinou, J. Bakos, I. Toth and B. Heil,
Organometallics, 1989, 8, 542.
45. C. Lensink and J. G. de Vries, Tetrahedron: Asymmetry, 1992, 3, 235.
46. C. Lensink and J. G. de Vries, Tetrahedron: Asymmetry, 1993, 4, 215.
47. F. Spindler, B. Pugin and H.-U. Blaser, Angew. Chem., Int. Ed. Engl., 1990,
29, 558.
48. Y. Ng Cheong Chan and J. A. Osborn, J. Am. Chem. Soc., 1990, 112, 9400.
49. C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1992, 114, 7562.
50. C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116 8952.
51. C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 11703.

179
52. M. J. Burk and J. E. Feaster, J. Am. Chem. Soc., 1992, 114, 6266. See also
C. Bolm, Angew. Chem., Int. Ed. Engl., 1993, 32, 232.
53. S. Itsuno, M. Nakano, K. Miyazaki, H. Masuda, K. Ito, A. Hirao and S.
Nakahama, J. Chem. Soc., Perkin Trans. 1, 1985, 2039.
54. D. E. Gibbs and D. Barnes, Tetrahedron Lett., 1990, 31, 5555.
55. E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109,
5551.
56. B. T. Cho and Y. S. Chun, J. Chem. Soc., Perkin Trans. 1, 1990, 3200.
57. C. K. Miao, R. Sorcek and P.-J. Jones, Tetrahedron Lett., 1993, 34, 2259.
58. R. Sreekumar and C. N. Pillai, Tetrahedron: Asymmetry, 1993, 4, 2095.
59. T. Basile, A. Bocoum, D. Savoia and A. Umani-Ronchi, J. Org. Chem., 1994,
59, 7766.
60. D. Enders, J. Schankat and M. Klatt, Synlett, 1994, 795.
61. D. Enders in Current Trends in Organic Synthesis, H. Nozaki (Ed.),
Pergamon Press, Oxford, 1983; p. 151.
62. T. Kawate, M. Nakagawa, T. Kakikawa and T. Hino, Tetrahedron:
Asymmetry, 1992, 3, 227.
63. K. Yamada, M. Takeda and T. Iwakuma, Tetrahedron Lett., 1981, 22, 3869.
180
64. K. Yamada, M. Takeda and T. Iwakuma, J. Chem. Soc., Perkin Trans. 1,
1983, 265.
65. S. Atarashi, H. Tsurumi, T. Fujiwara and I. Hayakawa, J. Heterocyclic Chem.,
1991, 28, 329.
66. N. Langlois, T.-P. Dang and B. Kagan, Tetrahedron Lett., 1973, 4865.
67. D. R. J. Hose, T. Raynham and M. Wills, Tetrahedron: Asymmetry, 1993, 4,
2159.
68. C. Bolm and M. Felder, Synlett, 1994, 655.
69. C. Bolm and M. Felder, Tetrahedron Lett., 1993, 34, 6041.
70. C. Bolm, A. Seger and M. Felder, Tetrahedron Lett., 1993, 34, 8079.
71. S. E. Denmark, N. Nakajima and L. J.-C. Nicaise, J. Am. Chem. Soc., 1994,
16, 8797.
72. G. Chelucci, M. A. Cabras and A. Saba, Tetrahedron: Asymmetry, 1994, 5,
1973.
73. G. J. Karabatsos and N. Hsi, Tetrahedron, 1967, 23, 1079.
74. H. Feuer and D. M. Braunstein, J. Org. Chem., 1969, 34, 1817.
75. M. Zervos and L. Wartski, Tetrahedron Lett., 1984, 25, 4641.

181
76. D. M. Ryckman and R. V. Stevens, J. Org. Chem., 1987, 52, 4274.
77. S. L. Huang, K. Omura and D. Swern, J. Org. Chem., 1976, 41, 3329.
78. P. Knochel and R. D. Singer, Chem. Rev., 1993, 93, 2117.
79. A. S. Thompson, G. R. Humphrey, A. M. DeMarco, D. J. Mathre and E. J. J.
Grabowski, J. Org. Chem., 1993, 58, 5886.
80. B. Stanovnik, M. Tisler, S. Polanc and J. Zitnik, Synthesis, 1977, 491.
81. D. S. Matterson and E. C. Beedle, Tetrahedron Lett., 1987, 28, 4499.
82. W. S. Mungall, G. L. Greene, G. A. Heavner and R. L. Letsinger, J. Org.
Chem., 1975, 40, 1659.
83. H. Suzuki and K. Takaoka, Chem Lett., 1984, 1733.
84. E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 298.
85. Y. G. Gololobov and L. F. Kasukhin, Tetrahedron, 1992, 48, 1353.
86. H. S. P. Rao and P. Siva, Synth. Commun., 1994, 24, 549.
87. F. Rolla, J. Org. Chem., 1982, 47, 4327.
88. H. Bayley, D. N. Standring and J. R. Knowles, Tetrahedron Lett., 1978, 3633.
182
89. E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen and V. K. Singh, J. Am.
Chem. Soc., 1987, 109, 7925.
90. G. J. Quallich and T. M. Woodall, Synlett, 1993, 929.
91. G. J. Quallich, J. F. Blake and T. M. Woodall, J. Am. Chem. Soc., 1994, 116,
8516.
92. R. Berenguer, J. Garcia and J. Vilarrasa, Tetrahedron: Asymmetry, 1994, 5,
165.
93. T. Mehler, W. Behnen, J. Wilken and J. Martens, Tetrahedron: Asymmetry,
1994, 5, 185.
94. G. B. Stone, Tetrahedron: Asymmetry, 1994, 5, 465.
95. J. Yaozhong, Q. Yong and M. Aiqiao, Tetrahedron: Asymmetry, 1994, 5,
1211.
96. Y. Hong, Y. Gao, X. Nie and C. M. Zepp, Tetrahedron Lett., 1994, 35, 6631.
97. T. Gajda, Tetrahedron: Asymmetry, 1994, 5, 1965.
98. Y.-J. Shi, D. Cai, U.-H. Dolling, A. W. Douglas, D. M. Tschaen and T. R.
Verhoeven, Tetrahedron Lett., 1994, 35, 6409.
99. P. Delair, C. Einhorn, J. Einhorn and J. L. Luche, Tetrahedron, 1995, 51, 165.

183
100. J. G. H. Willems, F. J. Dommerholt, J. B. Hammink, A. M. Vaarhorst, L Thijs
and B. Zwanenburg, Tetrahedron Lett., 1995, 36, 603.
101. B. Di Simone, D. Savoia, E. Tagliavini and A. Umani-Ronchi, Tetrahedron:
Asymmetry, 1995, 6, 301.
102. O. Froelich, M. Bonin, J.-C. Quirion and H.-P. Husson, Tetrahedron:
Asymmetry, 1993, 4, 2335.
103. H. C. Brown, J. Chandrasekharan and P. V. Ramachandran, J. Am. Chem.
Soc., 1988, 110, 1539.
104. H. C. Brown, M. Srebnik and P. V. Ramachandran, J. Org. Chem., 1989, 54,
1577.
105. P. V. Ramachandran, B. Gong and H. C. Brown, Tetrahedron: Asymmetry,
1993, 4, 2399.
106. M. M. Midland, Chem. Rev., 1989, 89, 1553.
107. G. W. Gribble, Contemp. Org. Synth., 1994, 1, 145.
108. G. Bartoli, G. Palmieri, M. Bosco and R. Dalpozzo, Tetrahedron Lett., 1989,
30, 2129.
109. M. Bosco, R. Dalpozzo, G. Bartoli, G. Palmieri and M. Patrini, J. Chem. Soc.,
Perkin Trans. 2, 1991, 657.
184
110. G. Bartoli, M. Bosco, R. Dalpozzo, G. Palmieri and E. Marcantoni, J. Chem.
Soc., Perkin Trans. 1, 1991, 2757.
111. M. A. Yurovskaya, A. Z. Afanasyev, F. V. Maximova and Y. G. Bundel,
Tetrahedron, 1993, 49, 4945.
112. E. Plazek, Chem. Ber., 1939, 72, 577.
113. P. J. Van Rijn, Rec. Trav. Chim., 1926, 45, 267.
114. American Home Products Corp., Brit. Pat. 702,985, 1954: Chem. Abstr.,
1955, 49, 5515g.
115. D. Savoia, C. Trombini and A. Umani-Ronchi, J. Org. Chem., 1978, 43, 2907.
116. D. G. Norton, V. E. Haury, F. C. Davis, L. J. Mitchell and S. A. Ballard, J.
Org. Chem., 1954, 19, 1054; T. Takeshima, M. Muraoka, H. Asaba and M.
Tokoyama, Bull. Chem. Soc. Jpn., 1968, 41, 506.
117. R. Kuhn and H. Schretzmann, Chem. Ber., 1957, 90, 557.
118. O. v. Schickh, A. Binz and A. Schulz, Chem. Ber., 1936, 12, 2593.
119. H. Stetter and R. Lauterbach, Liebigs Ann. Chem., 1962, 655, 20.
120. M. Matsumoto and N. Watanabe, Heterocycles, 1984, 22, 2313.
121. S. Torii, K. Uneyama, T. Onishi, Y. Fujita, M. Ishiguro and T. Nishida, Chem.
Lett., 1980, 1603.

185
122. S. Torii, K. Uneyama, T. Onishi, Y. Fujita, M. Ishiguro and T. Nishida, Eur.
Pat. 41,656, 1981; Chem. Abstr., 1982, 96, 181142y.
123. J. M. Bobbitt, C. L. Kulkarni, C. P. Dutta, H. Kofod and K. Ng Chiong, J. Org.
Chem., 1978, 43, 3541.
124. S. Massa, G. Stefancich, M. Artico, F. Corelli and R. Silvestri, Farmaco Ed.
Sci., 1987, 42, 567.
125. E. D. Edstrom, Synlett, 1995, 49.
126. M. Julia and Y. R. Pascal, Chim. Ther., 1970, 4, 279.
127. J. G. Berger, S. R. Teller and I. J. Pachter, J. Org. Chem., 1970, 35, 3122.
128. M. Kakushima, P. Hamel, R. Frenette and J. Rokach, J. Org. Chem., 1983,
48, 3214.
129. B. Kasum, R. H. Prager and C. Tsopelas, Aust. J. Chem., 1990, 43, 355.
130. F. Benedetti, F. Berti, P. Nitti, G. Pitacco and E. Valentin, Gazz. Chim. Ital.,
1990, 120, 25.
131. J. Cowell, MSc Thesis, University of East Anglia, 1995.
132. E. J. Walsh Jr. and G. B. Stone, Tetrahedron Lett., 1986, 27, 1127.
133. S. Baum, Third Year Project Report, University of East Anglia, 1994.
186
134. W. A. Remers and M. J. Weiss, J. Org. Chem., 1971, 36, 1241.
135. H. Plieninger and K. Klinga, Chem. Ber., 1968, 101, 2605.
136. I. Fleming and I. Paterson, Synthesis, 1979, 736.
137. M. E. Jung and Y.-G. Pan, J. Org. Chem., 1977, 42, 3961.
138. W. A. Remers, R. H. Roth, G. J. Gibs and M. J. Weiss, J. Org. Chem., 1971,
36, 1232.
139. J. Enda and I. Kuwajima, J. Am. Chem. Soc., 1985, 107,5495.
140. S. Danishefsky, S. Chackalamannil, P. Harrison, M. Silvestri and P. Cole, J.
Am. Chem. Soc., 1985, 107, 2474.
141. D. R. Williams and K. Nishitani, Tetrahedron. Lett., 1980, 21, 4417.
142. A. J. Fatiadi, Synthesis, 1976, 133.
143. B. Hughes and H. Suschitzky, J. Chem. Soc., 1965, 875.
144. H.-J. Knölker; M. Bauermeister and J.-B. Pannek, Tetrahedron, 1993, 49,
841.
145. H.-J. Knölker, Synlett, 1992, 371.
146. A. S. Kotnis, Tetrahedron Lett., 1991, 32, 3441.

187
147. M. Matsumoto, Y. Ishida and N. Watanabe, Heterocycles, 1985, 23, 165.
148. Y. Murakami, M. Tani, T. Ariyasu, C. Hishiyama, T. Watanabe and Y.
Yokoyama, Heterocycles, 1988, 27, 1855.
149. K. G. Estep, Synth. Commun., 1995, 25, 507.
150. J. V. Duncia, M. E. Pierce and J. B. Santella III, J. Org. Chem., 1991, 56,
2395.
151. S. J. Wittenberger and B. G. Donner, J. Org. Chem., 1993, 58, 4139.
152. B. E. Huff and M. A. Staszak, Tetrahedron Lett., 1993, 34, 8011.
153. J. E. Baldwin, A. C. Spivey, C. J. Schofield and J. B. Sweeney, Tetrahedron,
1993, 49, 6309.
154. T. Yamazaki, M. Nakai, Y. Kuroki and M. Mishimura, Jpn. Pat. 77,111,546,
1977; Chem. Abstr., 1978, 88, 50632v.
155. G. Rücker, H. Hörstner and W. Cajewski, Synth. Commun., 1980, 10, 623.
156. T. Marx and E. Breitmaier, Liebigs Ann. Chem., 1992, 183.
157. T. Hylton and V. Boeckelheide, J. Am. Chem. Soc., 1968, 90, 6887.
158. T. Asai, T. Aoyama, T. Shioiri, Synthesis, 1980, 811,
188
159. D. Enders, H. Schubert and C Nübling, Angew. Chem., Int. Ed. Engl., 1986,
25, 1109.
160. J. A. Baretrop, P. M. Hayes and M. Calvin, J. Am. Chem. Soc., 1954, 76,
4348.
161. G. Pfleiderer, E. Sann and A. Stock, Chem. Ber., 1960, 93, 3083; B. Barlin
and J. A. Benhow, J. Chem. Soc., Perkin Trans. 2, 1974, 7, 790.
162. M. A. Yurovskaya, A.Z. Afanasyev, V. Chertkov, E. M. Gizatullina and Y. G.
Bundel, Khim. Geterotsikl. Soedin., 1987, 1625; Chem. Abstr., 1988, 109,
128791v.
163. S. P. Gromov and Y. G. Bundel, Dokl. Acad. Nauk SSSR., 1985, 281, 585;
Chem. Abstr., 1986, 104, 50748m.
164. A. Z. Afanasyev, M. A. Yurovskaya and Y. G. Bundel, Khim. Geterotsikl.
Soedin., 1987, 134; Chem. Abstr., 1987, 107, 197998p.
165. S. P. Gromov, M. Bchaumik and Y. G. Bundel, Khim. Geterotsikl. Soedin.,
1985, 522; Chem. Abstr., 1985, 103, 87738p.
166. S. Tanaka, J. Chem. Soc. Jpn., 1952, 73, 282.
167. G. Magnussen and S. Thoren, J. Org. Chem., 1973, 38, 1380.

189
Appendix
O
OH
1
OH
OH O
2
R'
OOHR"
3
OH
OOH
S
N OHN
O O
NH2H
O OH
H
O
��
4
190
OH
OOOH���
5
OH
OO�������
��6
OH
OOH���
7
OH
OH OH O����
����
8
OH
OOH
S
H2NN
OHO
OH
��
9

191
OH
OOH
S
H2NO
OH
��
10
OH
OOH
S
N OHOH
OO
O
NH2H
����
11
O
HO O OOH
O
O
OH
O
12
192
HO O
O
NN
NN
H
13
S OH
OH
OOHO
����������
14
O
15
OMe
OO
16

193
OMe
O
OH
S
O
OMe17
OMe
O
S
OH
OMe
O
18
19
S
SCON(CH3)2
COOH
NCl
����
20
HSPh
O
MeO H
������
21
OHCS
SCON(CH3)2
COOMe����
194
22
S
SCON(CH3)2
COOMe
NCl
����
23
NO
OH
24
NCl
25
OH
OH
N
S
COONa
OH
Et
Cl
����
������������
26

195
N
S
COONa
O
Et
Cl
����
������������
27
NCl
O
28
29
NCl
OH
30
O CO2Me
NCl
196
CO2MeOH
NCl
31
OHNOOMe
NCl
32
NCl
O NOMe
S
Me COOH
����
����������
33
34
N
NO
O
H
OMe
NS
O O
OH

197
NCl
N ON
NNN
NN N
N H
H
35
N S NN
NN
OHO
36
NO
NN
NN
Cl
NN N
N Na
37
Br CN
38
I CN
39
IZn CN
40
198
NCl
OHCN
41
NCl
ClCN
42
O
Ph
PhNO2
43
O
Ph
Ph
NH
44
ONC
NH
45
NCl
N O
CN
CN
46

199
NN
O
O O
O
H
47
N O
CN
CN
O
48a
BrN O
CN
CN
48b
NClPPh3
49a
NCl
49b
NH2
CNO
50a
BrNH2
CN
50b
200
ClCN
O
51
OHCN
O
52
O
O Br
53
O
O
O
54
OCN
OOH
55
CNOH
Br
56
CNCl
Br
57
CNBr
58
ONC
NH
59
S
S
O
60
S
S S
S
61

201
S
SO
O
62
S
S
OHCN
63
NClP(OEt)2
O
64 65
PPh2
PPh2
PPh2
PPh2
H
H
����
PPh2 PPh2
�������������� PPh2
PPh2
��������������
PPh2
PPh2
66 67 68 69
O
O PPh2
PPh2
��������������
70 71
Ph2P
Ph2P
��
72
NBOC
Ph2PPPh2��������
����������
202
PPh2
PPh2
����
��������������
������������
73
OHOH
����
��������������
������������
74
P
P
EtEt
Et
Et
��������
����
��������������
������������
75
H2N OH
PhPh
����������
76
N O
PhPh
BH
H
����������
77
N O
PhPh
BHH3B
H
����������
78
N OB
PhPh
HH3B
79
Ph
HH2N
OH
����
80
HH2N
OH
�����
81
PhH2N
OH��������
82
83
Ph NOR'
OH
����
84
Ph NH2����
NNH2
OMe
��������������
85

203
NNH2
OMe
86
R
NN
OMe
87
R
NN
OMe
MeO
O
88
89
OB
O OMeOMe
H
����������������
90
NaBH
3
R N CO2
O
R'
R"
����
O
O
PRh
P
Cl
solvent
Ph
Ph Ph
Ph�������
91
NSO
O
������
��
92
SN Ph
O
N
OH
��
93
SN Ph
O
N
OH
H
�� ����
94
SN Ph
O
N
OH
H
��95
204
Ph NH2
����
96
SO2H
N
OH
97 98
PhS
Ph
N
O
OH H
Ph
����
N
O
N
O
R R
R' R'
NN
����
��������������
99 100
NR'
OHR
����
101
NR'
N3
R
102
NR'
NH2
R
103 104
OMeN
NH2
105
OHF3C
106
BrSO3Na
OH
107

205
BrNMe2
CN
108
BrCN
NMe2
CN
109
BrO
CN
110
BrN
CN
OMe
111
O
NClCN
112
N
NCl
OMe
113
206
N
NCl
OMe
CN
114
115
NBr
Ph
OHPh
����
��
NBr
Ph
OHPh
H
CN
����
116
117
Br CNN3
BrN
CN
PPh
PhPh
118
2
BCl
119
NaBH
3
N COO-
O PhO
120

207
121
RN
O
RN
O
MgBr
122 123
N
R
OMgBr
124
N
R
OMgBr
MgBr
125
N
R H
126
6
5
4
2NMe
R
RR
R NO2
127
4
7R
5
6 2NMe
RR
NO2N
R
RHR1
2
128
7
6
54
1RN
R
R
R
R
NO2
MeHN
R
RHNR
R
R
R
NMeN
R
OHO
1
2
45
6
7
129
2
RHNR
R
R
RMeN
R1
45
6
7
130
208
2
131
7
6
5
4
1RHN
RR
RR
MeN
R
132
7
6
5
4
2
1R
R
RR
NR
R
133
7
����Br CN
N
R
N
134
N
NO2
135
N
NO2
Me
136
NMe
137
NMe
Me
Me
Me
138
SO3Na
NO2
SO3NaNO2
NO2
NO2
NO2
SO3H
139140 141

209
NMe
142
NMe
Me
Me
MeMe
143
NMe
144
N
Ph
145
N
146 147
NMe
Me
Me
N Ph
148
NMe
Me
MePh
149
NMe
Me
MePh
150 151
N
NH2
152
N
NO2
153
N
NO2
MeI
O
O
R'
R"R'
154
210
O
NR'
R'
R"
R"'
155
O
O
CO2H
156 157
O
NH
O
O
158
159
O
OOEt
160
O
OH
OEt
OEt
O
N
OMeMeO
R'R
R
161
162
NMeO
O
N
CO2H
Me
163
O
NH
OAc
164
NMgX
165
166
NHO
167
N
Me
EtO2C
H
Et
168
NEt
Me
EtO2C
H
MeO2C
O

211
169
NEt
Me
EtO2C
H
MeO2C
170
N
O H
Me
Et
171
NSO2Ph
172
NSO2Ph
O
O
HO
173
NSO2Ph
O
HO
174
NSO2Ph
O
Cl
175
NSO2PhO
176
O
OR
R
177
ONHBn
RR
178
O
NBn
RR
179
1
ONHR
212
180
3
2
1O
NR
R
R
O
O181
N
CN
OBr
182
OHO
O
183
OHO
O
184
OCl
O
185
186
O
OOH
N
O
Ph187
Ph N Ph
188 189
N
N
Ph
Ph
190
N
PhO
191
O
O
N
O Ph
192
N
O Ph
193

213
N
O
194
N
OPh
195 196
N
Ph
O
NMe
O
197
N
Ph
O
198
HCl.H2N Ph
CO2Me
199
OOH
N PhH
200
OOH
N Ph
201
OOH
NH2
202
OOH
NHR
203
O
NHR
O
204
214
N
OTMS
Me
205 206
O
OTMS
207
O
OH
N
O
Me
PhSe
208
O
N
Ph
I
209
O
N
Ph
I
210
OH
N
Ph
211
O
N
Ph
CN
212 213
BrN
OH
BrN
NNN
H
NH2
214
BrO
NN
NN
H
215

215
BrN
NN
NN
H
OMe
216
BrN
NNN
H
N3
217
218
BrN
NN N
N H
O
219
BrN
NNN
CPh3
NH2
O
O
220
ONHBn
221 222
ONH
Ph
223
OCl
216
224
NH
Ph
225
OCl
OH
226
NRHO
OH
227
ONHR
228
N Ph
O
229
O
N Ph
230
N Ph
BrN
CN
O
H
231
BrN
CN
O
H
231