assigning stereochemistry and determining enantiomeric ... · the use of an organic liquid crystal,...
TRANSCRIPT

Assigning Stereochemistry and Determining Enantiomeric Purity: New Liquid Crystals or Old Derivatives?
Eugene E. Kwan Supervised by Professor W. F. Reynolds
University of Toronto, Department of Chemistry 2003-2004
Cl
O
CF3
+OMe
Ph OH
d5-Py
CDCl3
O
F3C
OMe
Ph
O
Mosher Ester Synthesis ee determination
Abstract We examine the relative merits of three NMR methods of assigning stereochemistry and determining enantiomeric purity in terms of their practicality, efficacy, and accuracy. (+)- and (-)-menthol, secondary alcohols cheaply available in enantiomerically pure form, were used to compare two chiral auxiliaries for determining the absolute stereochemistry and enantiomeric excess of alcohols: the traditionally reliable Mosher reagent, 2-methoxy-2-phenyl-2-(trifluoromethyl)acetic acid chloride, to a recently reported silicon-based reagent, (trifluoromethyl)benzyl alcohol. Menthol’s structure and relative stereochemistry were first unambiguously assigned by standard 1D and 2D NMR techniques. Both the Mosher and silicon-based methods were able to then correctly assign the absolute stereochemistry of (+)- and (-)-menthol, however, the silicon-based method was found to be highly experimentally unreliable. Thus, assessment of the accuracy of determinations of enantiomeric purity was only possible for the Mosher method. Various mixtures of (+)- and (-)-menthol were derivitized using a purification-free NMR-tube protocol and analyzed by 1D proton NMR. While typical integration methods produced errors of around 10%, more sophisticated methods were able to reduce errors to within experimental error (~1%). The use of an organic liquid crystal, 4-n-pentyl-4’-cyanobiphenyl (5CB), was also examined as a method for determining relative stereochemistry. At temperatures slightly above its lyotropic phase transition, 5CB can dissolve and weakly align small organic molecules in magnetic fields. Dipolar “D” couplings, similar to NOE dipolar couplings, develop and can give stereochemical information. D-couplings were obtained for menthol and kauradienoic acid by determining 1JC-H via the coupled heteronuclear single quantum coherence (HSQC) method. Both phase-cycled and gradient-selected pulse sequences were investigated and the optimal experimental parameters determined for each.

2
Table of Contents
INTRODUCTION................................................................................................................................... 4
The Mosher Ester Method................................................................................................................. 5 The Silyl Diether Method .................................................................................................................. 6 Weakly Oriented Liquid Crystals...................................................................................................... 7 HSQC: Phase-Cycling vs. Gradient Selection.................................................................................. 9
RESULTS AND DISCUSSION ........................................................................................................... 10 FULL STRUCTURE ASSIGNMENT: MENTHOL......................................................................................... 11 MOSHER ESTERS.................................................................................................................................. 17
Determination of Absolute Configuration....................................................................................... 17 Determination of Enantiomeric Purity ........................................................................................... 18
THE SILYL DIETHER METHOD.............................................................................................................. 24 Determination of Configuration...................................................................................................... 24 Determination of Enantiomeric Purity ........................................................................................... 25
WEAKLY ORIENTED LIQUID CRYSTALS ............................................................................................... 25
CONCLUSIONS ................................................................................................................................... 28
ACKNOWLEDGEMENTS.................................................................................................................. 28
EXPERIMENTAL................................................................................................................................ 29 GENERAL ............................................................................................................................................. 29 NMR INTEGRATION OF MOSHER ESTERS............................................................................................. 30 CALIBRATION OF THE MOSHER ESTERS OF MENTHOL.......................................................................... 30 STRUCTURAL ASSIGNMENTS................................................................................................................ 32 SYNTHESIS ........................................................................................................................................... 33
TABLE OF FIGURES.......................................................................................................................... 43
LISTING OF TABLES......................................................................................................................... 43
REFERENCES...................................................................................................................................... 44

3
Abstract
We examine the relative merits of three NMR methods of assigning stereochemistry and
determining enantiomeric purity in terms of their practicality, efficacy, and accuracy. (+)- and (-)-
menthol, secondary alcohols cheaply available in enantiomerically pure form, were used to compare
two chiral auxiliaries for determining the absolute stereochemistry and enantiomeric excess of alcohols:
the traditionally reliable Mosher reagent, 2-methoxy-2-phenyl-2-(trifluoromethyl)acetic acid chloride,
to a recently reported silicon-based reagent, (trifluoromethyl)benzyl alcohol. Menthol’s structure and
relative stereochemistry were first unambiguously assigned by standard 1D and 2D NMR techniques.
Both the Mosher and silicon-based methods were able to then correctly assign the absolute
stereochemistry of (+)- and (-)-menthol, however, the silicon-based method was found to be highly
experimentally unreliable. Thus, assessment of the accuracy of determinations of enantiomeric purity
was only possible for the Mosher method. Various mixtures of (+)- and (-)-menthol were derivitized
using a purification-free NMR-tube protocol and analyzed by 1D proton NMR. While typical
integration methods produced errors of around 10%, more sophisticated methods were able to reduce
errors to within experimental error (~1%). The use of an organic liquid crystal, 4-n-pentyl-4’-
cyanobiphenyl (5CB), was also examined as a method for determining relative stereochemistry. At
temperatures slightly above its lyotropic phase transition, 5CB can dissolve and weakly align small
organic molecules in magnetic fields. Dipolar “D” couplings, similar to NOE dipolar couplings,
develop and can give stereochemical information. D-couplings were obtained for menthol and
kauradienoic acid by determining 1JC-H via the coupled heteronuclear single quantum coherence
(HSQC) method. Both phase-cycled and gradient-selected pulse sequences were investigated and the
optimal experimental parameters determined for each.

4
Introduction
Modern organic chemistry and pharmaceutical development heavily relies on the development
of asymmetric syntheses and methodologies. Researchers developing such protocols frequently seek to
rapidly determine enantiomeric excess (ee), sometimes in a high-throughput combinatorial format (1),
by monitoring the formation of a desired enantiomer by proton NMR (2). Many of the drug candidates
such protocols are meant to access are developed from natural product lead compounds with complex
structural elements, including multiple stereocenters. While modern 1D and 2D NMR experiments (3)
are sufficient to determine the overall structural connectivity and relative stereochemistry of these
natural products and their synthetic congeners, the determination of their absolute stereochemistry or
enantiomeric purity requires specialized supplemental techniques. For some particularly complex
natural products, such as azaspiracid (see Figure 1), even structural elucidation is difficult. This project
investigated the relative merits of three NMR methods of determining stereochemical configuration and
enantiomeric purity: Mosher Ester derivitzation of chiral alcohols, silyl diether derivitization of chiral
alcohols, and NMR in weakly oriented organic liquid crystals.
OH
O
O
O
O
O
H
H
H
OH
HOH
H
CO2H
Figure 1. The current proposed structure of azaspiracid.

5
The Mosher Ester Method
Developed by Harry Mosher (4) in the early 1970s, the “Mosher Ester Method” of determining
the absolute configuration and enantiomeric purity of chiral alcohols remains a highly influential
technique. In the technique, the chiral alcohol of interest is esterified with (+)- or (-)-2-methoxy-2-
phenyl-2-(trifluoromethyl)acetic acid chloride (MTPA-Cl):
OR
O
OMe
CF3MeO
pyridineOHR Cl
O
OMe
CF3Ph
+
chiral alcohol Mosher'sAcid Chloride
"Mosher Ester"
The NMR spectrum of the resulting diastereomer can be analyzed to reveal the absolute configuration
of the original chiral alcohol. Computational and spectroscopic studies (5) now verify Mosher’s
original conformational proposal (see Figure 2).
Figure 2. Conformation of Mosher Esters. Figure adapted from ref. (6).

6
Mosher proposed that the most populated solution-phase conformer places the carbinyl proton,
ester carbonyl, and trifluoromethyl moieties in the same plane (6). In this conformation, the L2 signal
of the (R)-MTPA ester should appear upfield relative to the L2 signal of the (S)-MTPA ester. Thus, a
pair of NMR-indistinguishable enantiomers is transformed into NMR-distinguishable diastereomers. A
given mixture of enantiomers of a chiral alcohol can then be analyzed by comparing the relative
integrals of their corresponding diastereomers.
The Silyl Diether Method
The derivitization of chiral alcohols with chiral silanes is a modern variant of the Mosher Ester
technique. In the silyl diether methodology proposed by Williamson et al., (7), chiral alcohols are
derivitized with (R)- or (S)-(trifluoromethyl)benzyl alcohol (see Figure 3). Treatment with
tetrabutylammonium fluoride affords the original chiral alcohol.
Ph OH
CF3
Et3NMe2SiCl2
Ph O
CF3
SiCl
OSi
O Ph
CF3
REt3N
OHR
Figure 3. Silyl diether derivitization of chiral alcohols.
Several advantages over the Mosher technique have been reported for this methodology: ease of
use; ease of spectroscopic interpretation; scope which includes hindered or elimination-prone alcohols;
and ease of recovery of the starting chiral alcohol. A similar conformational model (see Figure 4) has

7
been proposed for these silyl diethers in which the key identifying interaction is that between the
carbinyl proton of the (trifluoromethyl)benzyl ether and the oxygen of the derivitized alcohol. In this
manner, the (R) L1 protons should be upfield relative to the (S) L1 protons. Similarly, the (R) L2
protons should be upfield relative to the (S) L2 protons.
Figure 4. Proposed conformational model for silyl diethers. Figure reproduced from (7).
Weakly Oriented Liquid Crystals
The use of liquid crystals in determining structure and stereochemistry is a relatively new
development which has been made possible by the development of organic liquid crystals (8). Small
organic molecules can be dissolved in such liquid crystals and develop detectable dipolar “D”
couplings. Structural and stereochemical information may be inferred because these D couplings have
an inherent angular and spatial dependence:
Dij α (3 cos2 θ - 1)(1 – r3).
These couplings are comparable to the dipolar couplings used in NOE experiments. Once the
couplings in a particular molecule are established, they can be fitted to an overall molecular axis and
geometry with a computer, a technique which has been used in protein structure determinations (9).
The 1/r3 spatial dependence means that this technique should be more sensitive than 1/r6-dependent
NOE experiments. Since previous work has focused on polypeptides such as poly-γ-benzyl-L-

8
glutamate (10), an incredibly viscous liquid, we chose to work instead on the relatively fluid liquid
crystal 4’-n-pentyl-biphenyl-4-carbonitrile (5CB) (11) (see Figure 5).
NC (CH2)4 CH3
5CB - used in perdeuterated form
Figure 5. The liquid crystal 4’-n-pentyl-biphenyl-4-carbonitrile.
A common pulse-sequence for obtaining carbon-proton correlations is the heteronuclear single
quantum coherence (HSQC) experiment. The use of this inverse-detection method reliably affords not
only carbon-proton correlation information, but also stereochemical and DEPT information.
Information appears in a standard F1 (carbon), F2 (proton) format, with cross-peaks representing
carbon-proton correlations. In its coupled form (cHSQC), no decoupling is used, such that each
carbon-proton correlation peak appears as a doublet, rather than a singlet. This is the result of a
geminal C-H coupling. Thus, 1JC-H values can be easily and rapidly obtained.
Should the molecule under study be weakly aligned in a liquid crystal, then the observed
doublet separations will incorporate both 1JC-H and D couplings. At high temperatures, the liquid
crystal is in its disordered state and will be unable to align organic molecules in a magnetic field.
cHSQC measurements will therefore reveal only the 1JC-H values. At temperatures slightly above the
liquid crystal’s lyotropic phase transition temperature, significant alignment will be possible, and the
doublet splitting in the cHSQC spectrum will be a sum of 1JC-H and D couplings. By taking the
difference between the high and low temperature doublet splittings, the D-alignment of a molecule can
be established. Computer programs, such as those already in use for protein structure determinations,

9
can then be used in conjunction with other information already determined from standard 1D and 2D
NMR experiments to produce a consistent structure.
HSQC: Phase-Cycling vs. Gradient Selection
Both the HSQC and cHSQC experiments are now routinely available in sensitivity-enhanced
phase-cycled and gradient-selected versions (12, 13). These modified sequences specifically enhance
the detection sensitivity for methine groups (by factors of 2 and 2, respectively). The gradient-
selected version is, in principle, particularly advantageous because sampling of both coherence
pathways allows the normal loss in detection sensitivity for gradient-selection for methylene and
methyl groups, a factor of 2 , to be recovered. However, studies have shown that phase-cycling may
be preferable to gradient-selection under conditions of very low sample mass (14). The purpose of
phase-cycling and gradient-selection in the HSQC sequence is to suppress the resonances of protons
directly bonded to 12C. Since the natural abundance of 12C is 99%, it is often difficult to remove these
resonances entirely and they appear as “t1 ridges”. The intensity of these undesirable ridges is
proportional to the intensity of the proton signal causing the ridge, so methyl signals can be particularly
overcome. HSQC spectra also have other random noise whose intensity is independent of signal
strength. Therefore, under low sample-mass conditions, the intensity of the t1 ridges may be small
compared to the other random noise. Thus, it was of interest to compare the performance of the
cHSQC sequence under both gradient-selected (hereafter, cgHSQC) and phase-cycled (hereafter,
cHSQC) modes.

10
Results and Discussion
To test the relative merits of the various methods described above, we employed the
stereochemically rigid compound menthol (1, see Figure 6) as a test compound. It is cheaply available
in both enantiomerically pure forms and racemic form.
HO
1
Figure 6. Menthol.
The structural connectivity and relative stereochemistry of 1 were fully assigned using traditional 1D
and 2D NMR methods. The absolute configuration of both enantiomers was then verified using the
Mosher and silyl diether techniques. Mixtures of the enantiomers of menthol were derivitized using
MTPA-Cl to test the validity of the Mosher method in determining enantiomeric purity. Finally, the
1JC-H values in menthol were determined in deuterochloroform and 5CB with coupled HSQC
experiments.

11
Full Structure Assignment: Menthol
To illustrate the exact procedures that would be used in determining the absolute
stereochemistry of an unknown compound, all the proton and carbon NMR peaks, as well as the
relative stereochemistry, in menthol were unambiguously assigned. A high-resolution 500 MHz proton
spectrum (see Figure 7) and a 125 MHz carbon spectrum of rac-menthol in deuterochloroform were
obtained.
1.45 1.40 1.35 1.30 1.25 1.20 1.15 1.10 1.05 1.00 0.95 0.90 0.85 0.80Chemical Shift (ppm)
3.181.043.251.501.000.921.02
3.450 3.425 3.400Chemical Shift (ppm)
1.02
2.2 2.1 2.0 1.9 1.8 1.7 1.6Chemical Shift (ppm)
1.121.051.061.01
Figure 7. 500 MHz 1H NMR spectrum of menthol in CDCl3.
An extensive simplex-based lock autoshim on Z1-Z5 was used to reduce the TMS FWHM to
0.55 Hz. The Levenberg-Marquardt peak-fitting algorithm was used to fit each multiplet to determine
coupling constants more accurately. A carbon-decoupled gradient-selected heteronuclear single

12
quantum coherence (gHSQC) experiment (with 2X 16-coefficient linear prediction and 4X zero-filling)
was used to establish one-bond carbon-proton connectivity in the molecule. It was initially assumed
that menthol would take on a stable chair conformation with the isopropyl group equatorial (see Figure
9). To determine if particular protons were in equatorial or axial positions, gHSQC F2 slices (proton
transects) were obtained. Because F2 resolution is limited, equatorial methylene protons show only as
doublets (only the geminal coupling appears) and axial methylene protons show as quartets (both the
geminal coupling and large diaxial coupling appear) (see Figure 8). These initial results are shown in
Table 1.
13C δ (ppm)
Type 1H δ (ppm) Relative Stereochemistry
1 16.1 Methyl 0.81 (3H, d, J=6.97 Hz) 2 21.0 Methyl 0.94 (3H, m) 3 22.2 Methyl 0.91 (3H, d, J=6.63Hz) 4 23.2 Methylene 0.99a (1H, m)
1.61 (1H, dq, J=12.66, 3.24 Hz) axial equatorial
5 25.9 Methine 2.17 (1H, m) 6 31.6 Methine 1.43 (1H, m) 7 34.5 Methylene 0.86 (1H, td, J=12.66, 3.31Hz)
1.66 (1H, m) axial equatorial
8 45.1 Methylene 0.99b (1H, m) 1.97 (1H, m)
axial equatorial
9 50.2 Methine 1.11 (1H, m) 10 71.6 Methine 3.41 (1H, td, J=10.43, 4.27 Hz)
Table 1: Initial gathered information from proton, carbon, and gHSQC spectra.

13
1.8 1.7 1.6 1.5 1.4 1.3 1.2 1.1 1.0 0.9 0.8 0.7 0.6Chemical Shift (ppm)
equatorialproton
axialprotongeminal coupling:
"doublet"
geminal and vicinalcouplings present:
"quartet"
Figure 8. Distinguishing axial and equatorial protons. Above, an F2 slice from a gHSQC spectrum.
To establish the overall structural connectivity and analyze the proton couplings, gradient-
selected heteronuclear multiple-bond correlation (gHMBC) and gradient-selected proton-proton
correlation spectroscopy (gCOSY) experiments were run (2X linear prediction with 16 coefficients and
4X zero-filling for both and standard COSY symmetrization). The gHMBC experiment gives C-H
correlations over more than one bond in an F1, F2 format. The gCOSY experiment gives through-bond
H-H coupling information in a diagonal, off-diagonal format. The correlations results are summarized
in Table 2.

14
Cross Peaks (ppm) 13C δ (ppm)
1H δ (ppm) gCOSY gHMBC
1 16.1 0.81 (CH3) 0.94, 2.17 21.0, 25.9, 50.2 2 21.0 0.94 (CH3) 0.81, 0.99, 1.11, 1.61, 2.17 16.1, 25.9, 50.2 3 22.2 0.91 (CH3) 0.99, 1.43, 1.66 31.6, 34.5, 45.1 4 23.2 0.99a (CH2, ax.)
1.61 (CH2, eq.) 0.86, 0.94, 0.99, 1.11, 2.17, 3.41
31.6, 34.5, 50.2, 71.6
5 25.9 2.17 (CH) 0.81, 0.94, 1.11, 1.61, 3.41 16.1, 21.0, 23.2, 50.2, 71.6 6 31.6 1.43 (CH) 0.86, 0.91, 0.99, 1.66, 1.97 22.2, 34.5, 45.1, 71.6 7 34.5 0.86 (CH2, ax.)
1.66 (CH2, eq.) 0.99, 1.43, 1.61, 1.66 0.86, 0.91, 0.99, 1.43
21.0, 25.9, 50.2 34.5, 45.1
8 45.1 0.99b (CH2, ax.) 1.97 (CH2, eq.)
0.99, 1.43, 3.41
31.6, 34.5, 50.2, 71.6
9 50.2 1.11 (CH) 0.94, 0.99, 1.61, 2.17, 3.41 25.9, 31.6, 34.5, 45.1, 50.2, 71.6
10 71.6 3.41 (CH) 0.99, 1.11, 1.30, 1.61, 1.97
22.2, 23.2, 34.5, 45.1
Table 2. gHSQC, gCOSY, and gHMBC correlations in menthol.
The gHMBC cross-peaks suggest that carbon peaks 16.1 and 21.0 belong to the isopropyl. The
remaining carbon methyl peak at 22.2 is therefore easy to assign. Based on chemical shift arguments,
the carbon peak at 71.6 and the proton peaks at 1.30 and 3.41 are assigned to the CHOH (see Figure 9).
H
O
H
CH3
H
CH3H3C
H
H
0.91
22.2
16.1
0.81
21.0
0.94
1.30
71.6
3.41
Figure 9. Initial assignments of menthol.

15
The common gHMBC cross-peaks for protons 0.81 and 0.94 are 25.9 and 50.2. Carbon peak
25.9 probably corresponds to CH(CH3)2 because of its lower chemical shift. Also, it only shows one
gHMBC cross-peak to one ring-methylene (23.2) whereas carbon peak 50.2 shows gHMBC cross-
peaks to three ring-methylenes (31.6, 34.5, and 45.1). Thus, carbon peak 50.2 can be assigned to the
methine adjacent to the isopropyl group and the remaining methine peak (31.6) to the CHCH3 (see
Figure 10).
H
O
H
CH3
H
CH3H3C
H
H
0.91
22.2
16.1
0.81
21.0
0.94
1.30
71.6
3.4131.6
1.43
50.2
1.11
25.9
2.17
Figure 10. Second round of assignments for menthol.
To establish the assignments for the remaining ring-methylenes, note that proton 2.17 shows a
cross-peak only to ring-methylene 23.2. Thus, using the gHSQC axial/equatorial information, we may
assign carbon 23.2 to the ring-methylene adjacent to the methine containing the isopropyl group.
Proton 3.41 shows gCOSY couplings to protons 0.99 and 1.97 but not protons 0.86 and 1.66.
Therefore, the ring-methylene between the methine containing the methyl group and the CHOH can be
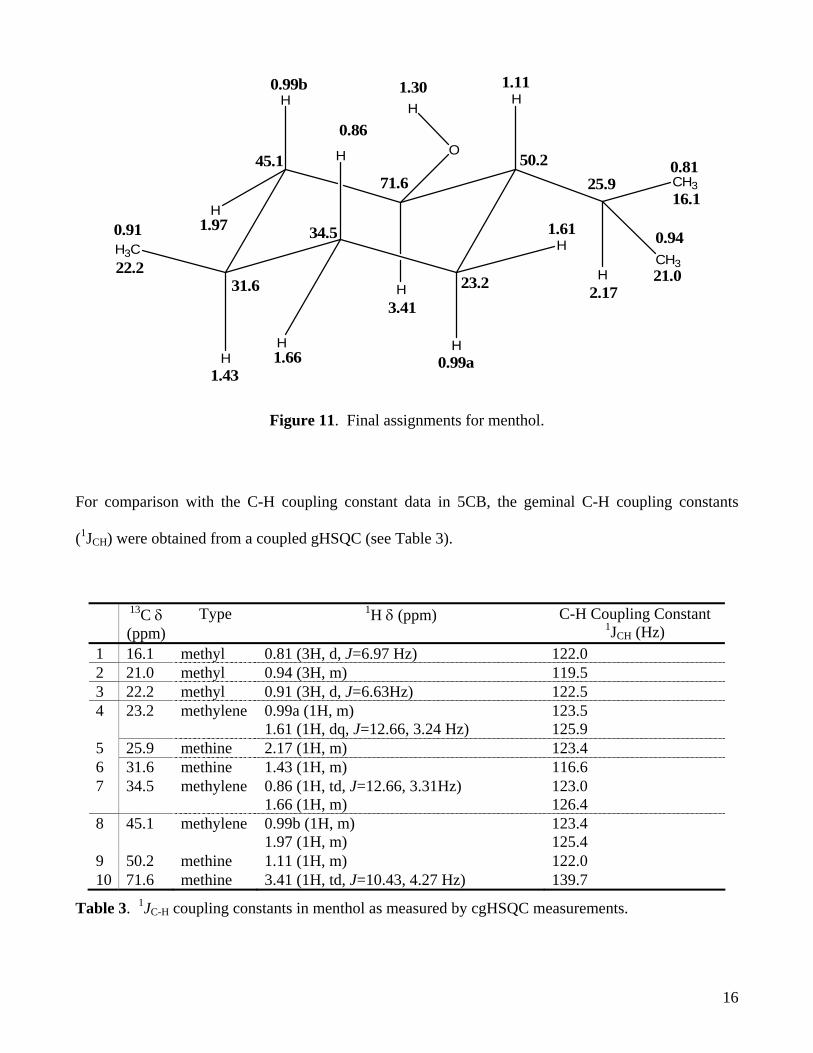
assigned to carbon 45.1. The final assignments are shown in Figure 11.
16
H
O
H
CH3
HCH3
H3C
H
H
0.91
22.2
16.1
0.81
21.0
0.94
1.30
71.6
3.4131.6
1.43
50.2
1.11
25.9
2.17
H
H
H
H H
H
0.99b
1.97
45.1
0.86
34.5
1.66
23.2
1.61
0.99a
Figure 11. Final assignments for menthol.
For comparison with the C-H coupling constant data in 5CB, the geminal C-H coupling constants
(1JCH) were obtained from a coupled gHSQC (see Table 3).
13C δ (ppm)
Type 1H δ (ppm) C-H Coupling Constant 1JCH (Hz)
1 16.1 methyl 0.81 (3H, d, J=6.97 Hz) 122.0 2 21.0 methyl 0.94 (3H, m) 119.5 3 22.2 methyl 0.91 (3H, d, J=6.63Hz) 122.5 4 23.2 methylene 0.99a (1H, m)
1.61 (1H, dq, J=12.66, 3.24 Hz) 123.5 125.9
5 25.9 methine 2.17 (1H, m) 123.4 6 31.6 methine 1.43 (1H, m) 116.6 7 34.5 methylene 0.86 (1H, td, J=12.66, 3.31Hz)
1.66 (1H, m) 123.0 126.4
8 45.1 methylene 0.99b (1H, m) 1.97 (1H, m)
123.4 125.4
9 50.2 methine 1.11 (1H, m) 122.0 10 71.6 methine 3.41 (1H, td, J=10.43, 4.27 Hz) 139.7
Table 3. 1JC-H coupling constants in menthol as measured by cgHSQC measurements.

17
Mosher Esters
The Mosher method was easy to carry out in good yield and was highly reliable. None of the
over thirty attempts to derivitized alcohols failed. (+)- and (-)-menthol were successfully derivitized as
all four diastereomers (see compounds 2-5). The related primary alcohols (R)- and (S)-2,2-dimethyl-
1,3-dioxolane-4-methanol were also successfully derivitized as all four diastereomers (see compounds
12-15).
Determination of Absolute Configuration
The absolute configuration of menthol was verified using the Mosher conformational model.
The diastereomeric (-)-menthol, (S,+)-MTPA (2) and (+)-menthol, (S,+)-MTPA (3) Mosher esters show
the expected chemical shift differences (see Figure 12).
4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0Chemical Shift (ppm)
(-)-menthol, (+)MTPA
(+)-menthol, (+)MTPA
Figure 12. Chemical shift differences between the diastereomeric Mosher esters of menthol. (500
MHz in CDCl3)

18
Similarly, the diastereomeric (S)-dioxolane, (S,+)-MTPA (12) and (R)-dioxolane, (S,+)-MTPA (14)
Mosher esters also showed the expected chemical shift differences (see Figure 13).
(R)-dioxolane, (S, +)-MTPA
(S)-dioxolane, (S, +)-MTPA
4.4 4.3 4.2 4.1 4.0 3.9 3.8 3.7 3.6 3.5Chemical Shift (ppm)
Figure 13. Chemical shift differences between the diastereomeric Mosher esters of 2,2-dimethyl-1,3-
dioxolane-4-methanol. (500 MHz, CDCl3)
As predicted, the chemical shift differences between the diastereomeric primary dioxolane
Mosher esters are less than those for the diastereomeric menthol Mosher esters.
Determination of Enantiomeric Purity
To assess the accuracy of Mosher Ester determinations of enantiomeric purity,
deuterochloroform solutions with varying mass ratios of (+)-menthol and (-)-menthol were Mosher-
derivitized. The proton NMR signal of the sensitive CHOH proton adjacent to the Mosher Ester in the

19
product should, in principle, integrate to the corresponding mass ratio of the solution; however, initial
attempts gave a large dispersion about the expected correlation on the order of ten percent. One major
source of error was undoubtedly related to a “kinetic resolution” effect in which the (+)-menthol, (+)
Mosher Ester / (-)-menthol, (-) Mosher Ester formation reactions approach equilibrium faster than the
corresponding (-)-menthol, (+) Mosher Ester / (+)-menthol, (-) Mosher Ester formation reactions. This
necessitates a reaction time of at least 48 hours. A molar excess of at least 2:1 of Mosher’s acid
chloride must be used to counteract slow hydrolysis of the derivitizing agent.
Equilibration time errors will insufficient to explain all of the deviations, however. The
remaining deviations were attributed to errors in integration. The diastereomeric CHOH protons are
sufficiently close in the proton NMR spectrum to overlap slightly; moreover, any spinning side-bands
or 13C satellites may cause significant errors. Several measures were implemented to correct for these
issues. First, simplex lock-autoshimming on Z1-Z5 was employed to narrow the spectral line widths as
much as possible. Manual corrections on the low-order XY shims currents were also made to reduce
spinning sidebands. Next, each diastereomeric multiplet, including spinning side-bands and 13C
satellites, was fitted (see Figure 14) to a linear combination of Gauss-Lorentz lineshapes using the
Levenberg-Marquardt algorithm.

20
3.56 3.55 3.54 3.53 3.52 3.51 3.50 3.49 3.48 3.47 3.46Chemical Shift (ppm)
3.480
3.484
3.488
3.492
3.536
3.5403.545
3.549 residuals
peak fits
Figure 14. The carbinyl proton in the diastereomeric Mosher esters of menthol. The peaks have been
fitted to Gauss-Lorentz lineshapes. (300 MHz, CDCl3)
The corrected areas were then used. Finally, the peak positions of each diastereomeric multiplet were
accurately estimated for the most enantioenriched mixtures by carrying over the peak positions from
less enantiorich mixtures since errors in the peak-fitted areas for the minor diastereomer may arise
when the peaks have low S/N. To further reduce peak-position errors and improve digitization, 4X
zero-filling was employed as a form of Fourier interpolation. To reduce T1 relaxation time errors, a
generous delay was used between scans.
To correct for baseline errors, a number of acquisition and data processing protocols were used.
The standard 1H pulse sequence incorporates a pre-acquisition delay time between the pulse which
necessitates a “zero-order phase correction”. Recall that a time series f(t), when shifted in time by ∆t,
develops a phase-shift in the frequency domain of e-iw∆t, where w is the conjugate frequency variable.

21
During this delay time, the magnetization vectors evolve at different rates, such that the vectors with
the largest chemical shifts evolve the furthest. Thus, a frequency-dependent phase-shift is introduced.
If the delay time is small, then a combination of zero- and first-order phase corrections will be
sufficient to properly align each peak into a purely absorptive mode. The Varian ‘calfa’ macro was
used to empirically set the left phase correction to zero, eliminating a rolling baseline, at the time of
acquisition. To remove the DC component, a drift-correction was applied. The ACD automated phase
correction macro, used in baseline-optimized mode, was used to uniformly correct the phase for all
spectra. Finally, a fourth-order polynomial baseline correction was applied. The algorithm splits the
frequency spectrum into 64 parts and fits a fourth-order polynomial to the segments that contain only
noise. The resulting polynomial was subtracted from the entire spectrum.
The resulting calibration curve gave an excellent correlation (R=0.998) between the actual mass
ratio of (+)-menthol:(-)-menthol and the integrated mass ratio (see Figure 15).

Figure 15. Excellent correlation between Mosher-estimated mass fraction of (+)-menthol and actual mass fraction.
22

A Wald f-test showed the calibration curve was statistically indistinguishable from the expected
1:1 correspondence at the 95% confidence level. While the average error was approximately 3%, the
average error for highly enantioenriched mixtures (er > 95%) was approximately 1%, which is the
expected accuracy limit for the procedure, given that the MTPA-Cl reagents were approximately 99%
enantiomerically pure (according to Aldrich). The large errors for the marginally enantioenriched
mixtures were attributed to mass errors resulting from a faulty balance, rather than NMR integration
error. Thus, the Mosher Ester method, when applied with the appropriate rigor, is a very reliable way
of determining the enantiomeric purity of chiral alcohols.
23

24
The Silyl Diether Method
The silyl diether derivitization was very hard to implement. Despite many repeated attempts,
derivitization of menthol was largely unsuccessful. Reactions were very moisture sensitive, producing
complicated and unidentifiable mixtures. The purification step was particularly difficult, producing
broad, inseparable bands on silica. As such, this method is clearly inferior to the Mosher method.
Determination of Configuration
As with the Mosher esters, the silyl diether method showed the expected chemical shift
differences between the (+)-menthol, (R)-(trifluoromethyl)benzyl (PhTFE) alcohol silyl diether (10)
and the (-)-menthol, (R)-PhTFE alcohol silyl diether (11) (see Figure 16). However, the shifts were not
as large.
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0Chemical Shift (ppm)
(+)-menthol, (R)-PhTFE
(-)-menthol, (R)-PhTFE
Figure 16. Chemical shift differences between the silyl diethers of menthol. (400 MHz, CDCl3)

25
Determination of Enantiomeric Purity
Due to the unreliability of the method, it was impossible to synthesize sufficient quantities of
silyl diethers to effect a calibration curve. However, due to the moisture-sensitivity of the reaction, it is
unlikely that the reaction would be suitable for the same purification-free NMR tube procedure
undertaken for the Mosher method.
Weakly Oriented Liquid Crystals
To evaluate the liquid crystal methodology, menthol and kauradienoic acid (16, see Figure 17)
were chosen as test compounds. Geminal C-H coupling constants (1JC-H) were obtained at non-oriented
(high) and weakly-oriented (low) temperatures by use of the coupled HSQC sequence (see Figure 18).
The sequence was run in both gradient-selected (cgHSQC) and phase-cycled (cHSQC) modes. The
observed couplings at both temperatures and the inferred D-couplings are shown in . To determine the
correct temperatures for the experiment, various coupled HSQC experiments were run and the 1JC-H and
peak widths were noted. As temperature was increased, 1JC-H reached a maximum as liquid crystal
orientation was lost. Good non-oriented temperature values were found to be between 45°C (for 1) and
33°C (for 16). As temperature was decreased, the difference between the high temperature 1JC-H values
and the observed 1JC-H increased, indicating increasing D coupling. However, as the liquid crystal
aligned, the solution viscosity increase significantly, leading to increased line-broadening. Thus, the
optimal low temperatures were found to be approximately 24.5°C for both 1 and 16. The cgHSQC
sequence was found to be preferable to the cHSQC sequence, as sufficient S/N was present to give
significant cHSQC t1 ridges.
To obtain the best S/N and resolution for the D-coupling measurements, a number of post-
acquisition strategies were used. In the detected proton axis (F2), a line-broadening filter was used.
Since F2 can show proton multiplet structure, as well as geminal C-H coupling structure, a large

26
amount of line broadening (>5 Hz) was used. This had the effect of reducing dq or dt splitting to d
splittings. To reduce digitization errors, the t2 domain was zero-filled to 32K points. In the indirectly
detected carbon axis (F1), a matched line-broadening filter and 4X 8-coefficient linear prediction were
used. The D-couplings were measured from peak-to-peak for each doublet. While the D-couplings
were disappointingly small in menthol, they were fairly large in kauradienoic acid, suggesting a
possible aligning interaction between the carboxylic acid group (C-18) and 5CB.
HCO2H
1
3
1918
20 11 13
14
15
17
5
16
Figure 17. Kauradienoic acid, a secondary metabolite of Montanoa Tomentosa (15).
2.0 1.5 1.0 0.5F2 Chemical Shift (ppm)
20
25
30
35
40
45
50
F1 C
hem
i cal
Shi
ft (p
pm)
Figure 18. Coupled cgHSQC of menthol in 5CB at 25.4°C.

27
13C δ (ppm)
1H δ (ppm) Isotropic Splitting (45°C, Hz)
Aligned Splitting (45°C, Hz)
D coupling (Hz)
1 16.1 0.81 (CH3) 123.7 120.6 -3.1 2 21.0 0.94 (CH3) 123.7 121.0 -2.7 3 22.2 0.91 (CH3) 123.8 123.4 -0.4 4 23.2 0.99a (CH2, ax.)
1.61 (CH2, eq.) 127.5 121.2
124.9 121.2
-2.6 ~0
5 25.9 2.17 (CH) 122.6 128.1 +5.5 6 31.6 1.43 (CH) 119.4 126.6 +7.2 7 34.5 0.86 (CH2, ax.)
1.66 (CH2, eq.) 127.7 124.6
127.3 127.8
-0.4 +3.2
8 45.1 0.99b (CH2, ax.) 1.97 (CH2, eq.)
123.2 127.0
127.2 125.3
+4.0 -1.7
9 50.2 1.11 (CH) 121.9 125.0 +3.1 10 71.6 3.41 (CH) 138.2 143.8 +5.6
Table 4a. D-couplings of menthol (1) in 5CB as obtained from cgHSQC experiments.
1H δ (ppm) 13C δ (ppm) α β
Isotropic Splitting (45°C, Hz)
Aligned Splitting (45°C, Hz)
D coupling (Hz)
1 40.75 1.93 1.24 unclear 2 20.16 1.88 1.50 unclear 3 38.31 2.16 1.02 137, unclear 134, unclear -3, unclear 5 46.56 - 1.67 120 113 -7 6 18.48 2.47 1.68 unclear 7 29.66 1.97 1.46 unclear 11 114.90 5.24b 156 163 +7 12 37.93 2.43 1.99 unclear, 136 unclear, 125 -11 13 41.24 2.77 - 135 130 -5 14 44.94 1.60 1.50 127, 133 133, 133 +6, ~0 15 50.32 2.20 2.62 129, 137 128, 140 -1, +3 17 105.48 4.91b,c 4.80 157, 157 148, 164 -9, +7 19 28.23 - 1.24 123 121 -2 20 23.62 1.02 - 126 123 -3
Table 5a. D-couplings of kauradienoic acid (16) in 5CB as obtained by cgHSQC measurements. C-H correlations are from ref. (15). Methylene proton-carbon couplings are indicated with the α coupling followed by the β coupling.
aProton and carbon chemical shifts are referenced to TMS in their respective 1D spectra; referencing within cgHSQC/5CB experiments is ambiguous. bThe α/β nomenclature does not apply to olefinic hydrogens. cThis refers to the proton cis to C-13.

28
Conclusions
For routine determinations of absolute configuration or enantiomeric purity, the Mosher method
is superior to the silyl diether method in terms of its ease of use and reliability. Determinations of
enantiomeric purity with the Mosher method can be made in an NMR-tube without purification and can
be made accurate to within 1%, which is within the experimental error constraint imposed by the purity
of MTPA-Cl. The liquid crystal method shows much promise and may be useful for certain
compounds with orienting groups. The cgHSQC sequence is ideal for extracting the D-couplings.
Acknowledgements
NSERC is acknowledged for its financial support. Professor Batey and his group members are
acknowledged for graciously providing laboratory space and equipment. Tim Burrow is acknowledged
for his NMR support. Professor Reynolds is especially acknowledged for his support and his work on
this project.

29
Experimental
General
All reagents were obtained from commercial suppliers (e.g., Aldrich) and were used without
further purification. All reactions were carried out under an inert atmosphere of dry nitrogen.
Deuterated solvents were used as received. Preparative thin-layer chromatography was performed on
Aldrich glass-backed, inorganically bound 250 µm silica gel plates with F254 indicator. Analytical
thin-layer chromatography was performed on pre-coated aluminum-backed silica gel plates, with UV
(254 nm) visualization. Staining was performed with ninhydrin or potassium permanganate. Solvent
ratios for Rf values are reported as v/v.
NMR spectra were recorded on Varian Mercury-300, Unity-400, and Inova-500 instruments
with chloroform/TMS as internal reference standard. 2X zero-filling was used on all one-dimensional
spectra. Resolved multiplets were fitted to Gauss-Lorentz lineshapes with the Levenberg-Marquardt
algorithm. Automated coupling constant analysis which accounted for the peak-fitting results was
carried out using ACD/SpecManager 7. Phase artifacts were corrected with 4-point 8-coefficient
backward linear prediction. 2X 8-coefficient forward linear prediction and 2X zero-filling was
performed on all magnitude-mode two-dimensional spectra. 4X 8-coefficient forward and 2X zero-
filling was performed on all phase-sensitive two-dimensional spectra. Peak multiplicities are reported
using the abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; J, coupling
constant (Hz). Low and high resolution electron impact (EI) mass spectra were obtained on a
Micromass 70-S-250 mass spectrometer.

30
NMR Integration of Mosher Esters
The proton NMR resonance of the carbinyl proton (CHO-Mosher Ester) was acquired at room
temperature using the standard phase-cycled one-dimensional proton acquisition sequence with 32
scans and a long relaxation delay time of 3.0 s. Gradient shimming or coarse manual adjustment of the
axial shims was supplemented by a simplex-based lock autoshim of Z1-Z5. The low-order XY shims
were corrected manually Spectra were recorded at a 20 Hz spinning rate. The pre-acquisition dead-
time was pre-set to render a null zero-order phase correction using the Varian ‘calfa’ macro. The FIDs
were 4X zero-filled to enhance digitization and apodized using a 0.2 Hz line broadening function. The
FIDs were processed with the FFT algorithm and first-order phase corrections were carried out using
the baseline-optimized phase correction feature of ACD/SpecManager 7. A fourth-order polynomial
was subtracted from each spectrum to correct the baseline further. The spectra were referenced to
TMS. The peak positions of each diastereomeric multiplet, including any spinning sidebands or 13C
satellites, were found by manual peak picking when S/N allowed. When this was impossible, peak
positions from similar spectra were used. The peaks were fitted to Gauss-Lorentz lineshapes using the
Levenberg-Marquardt algorithm. Automated integration which accounted for the peak-fitting data was
used to determine the diastereomeric ratios.
Calibration of the Mosher Esters of Menthol
Calibration of the technique was carried out by analyzing, via the 1D NMR technique outlined
above, mixtures of (+)-menthol and (-)-menthol. (+)-menthol (99%) and (-)-menthol (99%) were used
as received from Aldrich. The appropriate amount of each enantiomer was weighed out to make a total
mass of approximately 40 mg. Masses were weighed to an accuracy of 0.01 mg. 3 mL of
deuterochloroform (without TMS) was added to each mixture to afford solutions with enantiomeric
ratios of (+)-menthol:(-)-menthol ranging from >99.9:0.01 to <0.01:99.9.

31
225 µL of each solution was added to a clean, dry 5 mm NMR tube under nitrogen. d5-pyridine
(11 µL, 130 µmol) (ampoules used as received from Aldrich), followed by (+)- or (-)-2-methoxy-2-
phenyl-2-(trifluoromethyl)acetic acid chloride (2 µL, 11 µmol) were added using a dry microsyringe.
Deuterochloroform (with 0.01% v/v TMS) was added to make a sample height of approximately 4.5
cm. The mixtures were allowed to react for 48 hours and 1D proton NMR spectra were taken as
described above.

32
Structural Assignments
Rac-menthol (1): colorless prisms; mp 34-36 ºC; 1H and 13C NMR (CDCl3) in Table 6; m/z (EI) 138
(M-H2O, 35), 123 (44), 109 (11), 96 (27), 95 (88), 82 (38), 81 (88), 71 (100), 69 (34), 67 (36), 57 (20),
56 (15), 55 (47), 53 (12). Found: [M-H2O] 138.1413. C10H18 requires m/z 138.1408.
H
O
H
CH3
HCH3
H3C
H
H
H
H
H
H H
H
1
2
3
4
53'
6 1'2'
2''
position 13Ca 1Hb gCOSY gHMBC OH - 1.30 (br s) H-1’ C-1, C-2, C-6 2’ 16.1 0.81 (d, 6.97) H-2’’, H-1’ C-2’’, C-1’, C-2 2’’ 21.0 0.94 (m) H-2’, H-2, H-3eq, H-1’ C-2’, C-1’, C-2 3’ 22.2 0.91 (d, 6.63) H-5, H-4eq C-5, C-4, C-6 3 23.2 0.99 (m)
1.61 (dq, 12.66, 3.24) H-4ax, H-2’’, H-2, H-1’, H-1
C-5, C-4, C-2, C-1
1’ 25.9 2.17 (m) H-2’, H-2’’, H-2, H-3eq, H-1 C-2’, C-2’’, C-3, C-2, C-1
5 31.6 1.43 (m) H-4ax, H-3’, H-4eq, H-6eq C-3’, C-4, C-6, C-1 4 34.5 0.86 (td, 12.66, 3.31)
1.66 (m) H-5, H-3eq, H-4eq H-4ax, H-3’, H-5
C-2’’, C-1’, C-2 C-4, C-6
6 45.1 0.99 (m) 1.97 (m)
H-5, H-1
C-5, C-4, C-2, C-1
2 50.2 1.11 (m) H-2’’, H-3eq, H-1’, H-1 C-1’, C-5, C-4, C-6, C-2, C-1
1 71.6 3.41 (td, 10.43, 4.27) H-2, 1.30, H-3eq, H-6eq
C-3’, C-3, C-4, C-6
Table 6. 1H and 13C NMR (CDCl3) Spectral Data and Assignments for rac-menthol (1).
a Assignments made using gHSQC, gHMBC, and gCOSY techniques. b Multiplicities and coupling constants in Hz in parentheses. For the methylenes, the axial protons are given first, followed by the equatorial protons.
33
Kauradienoic Acid (16): Please see reference 15.
Synthesis
(1S,2R,5S,2'S)-3',3',3'-Trifluoro-2'-methoxy-2'-phenylpropionic acid 2-isopropyl-5-methyl-
cyclohexyl ester (2):
O
CH3
CH3 CH3
O
OMe
PhF3C
pyridineCDCl3
OH
CH3
CH3 CH3
Cl
O
OMe
PhF3C
+
2
Pyridine (20 µL, 247 µmol) was added to a solution of (-)-menthol (1.0 mg, 6.0 µmol) in 400
µL deuterochloroform. (+)-2-methoxy-2-phenyl-2-(trifluoromethyl)acetic acid chloride (2.3 µL, 12
µmol) was then added and the solution stirred for two days at room temperature. The solvent was
removed in vacuo and the resulting clear oil was purified by preparative TLC (20% EtOAc/Hexanes) to
yield 2 as a clear oil in moderate yield: TLC: Rf = 0.70 (20% EtOAc/Hexanes); 1H NMR (300 MHz,
CDCl3) δ 0.78 (3H, d, J=6.98 Hz), 0.88 (3H, d, J=7.02 Hz), 0.92 (3H, d, J=6.50 Hz), 1.03 (1H, m),
1.03 (1H, m), 1.46 (1H, m), 1.71 (1H, m), 1.89 (1H, m, J=13.98, 6.98, 6.98, 3.08 Hz), 2.09 (1H, m,
J=11.99, 3.98, 3.88, 1.98 Hz), 3.54 (3H, q, J=1.25 Hz, OCH3), 4.89 (1H, td, J=10.92, 4.42 Hz,
CHOC(O)), 7.40 (3H, m), 7.53 (2 H, m); 13C NMR (75 MHz, CDCl3) δ 15.6, 20.8, 22.0, 22.8, 25.8,
31.4, 34.0, 40.0, 46.6, 55.4, 77.5, 127.5, 128.3, 129.5, 132.4; m/z (EI) 373 (M+H, 1%), 189 (23), 139
(53), 105 (13), 97 (22), 83 (100), 69 (37), 57 (32), 55 (40). Found: [M+H], 373.2004. C20H28O3F3
requires m/z 373.1991.

34
(1S,2R,5S,2'R)-3',3',3'-Trifluoro-2'-methoxy-2'-phenylpropionic acid 2-isopropyl-5-methyl-
cyclohexyl ester (3):
O
CH3
CH3 CH3
O
OMe
PhF3C
pyridineCDCl3
OH
CH3
CH3 CH3
Cl
O
OMe
PhF3C
+
3
Pyridine (60 µL, 741 µmol) was added to a solution of (+)-menthol (3.0 mg, 19.2 µmol) in 1
mL deuterochloroform. (+)-2-methoxy-2-phenyl-2-(trifluoromethyl)acetic acid chloride (7.0 µL, 36
µmol) was then added and the solution stirred for two days at room temperature. The solvent was
removed in vacuo and the resulting clear oil was purified by preparative TLC (20% EtOAc/Hexanes) to
yield 3 (5.5 mg, 77%) as a clear oil: TLC: Rf = 0.85 (20% EtOAc/Hexanes); 1H NMR (500 MHz,
CDCl3) δ 0.60 (3H, d, J=6.95 Hz), 0.68 (3H, d, J=7.02 Hz), 0.80 (1H, m), 0.87 (3H, d, J=6.56 Hz),
0.97 (1H, m), 1.04 (1H, q, J=11.69 Hz), 1.34 (1H, m), 1.47 (1H, m), 1.50 (1H, m), 1.60 (1H, m), 1.63
(1H, m), 2.06 (1H, m), 3.51 (3H, q, J=1.34 Hz), 4.83 (1H, td, J=10.97, 4.36 Hz), 7.32 (3H, m), 7.47
(2H, m); 13C NMR (100 MHz, CDCl3) 15.5, 20.6, 22.0, 22.8, 25.3, 31.5, 34.0, 40.5, 46.7, 55.5, 77.1,
127.1, 128.2, 129.4, 132.7, 166.1; m/z (EI) 373 (M+H, 1%), 189 (25), 139 (65), 105 (16), 97 (26), 95
(10), 84 (11), 83 (100), 81 (18), 69 (39), 57 (36), 55 (41). Found: [M+H], 373.1971. C20H28O3F3
requires m/z 373.1991. See the structural assignments section.

35
(1R,2S,5R,2'S)-3',3',3'-Trifluoro-2'-methoxy-2'-phenylpropionic acid 2-isopropyl-5-methyl-
cyclohexyl ester (4):
O
CH3
CH3 CH3
O
OMe
CF3MeO
pyridineCDCl3
OH
CH3
CH3 CH3
Cl
O
OMe
CF3Ph
+
4
Pyridine (60 µL, 741 µmol) was added to a solution of (-)-menthol (3.0 mg, 19.2 µmol) in 1 mL
deuterochloroform. (-)-2-methoxy-2-phenyl-2-(trifluoromethyl)acetic acid chloride (7.0 µL, 36 µmol)
was then added and the solution stirred for two days at room temperature. The solvent was removed in
vacuo and the resulting clear oil was purified by preparative TLC (20% EtOAc/Hexanes) to yield 4 (4.0
mg, 56%) as a clear oil: TLC: Rf = 0.60 (20% EtOAc/Hexanes); 1H NMR (300 MHz, CDCl3) δ 0.60
(3H, d, J=6.88 Hz), 0.68 (3H, d, J=7.03 Hz), 0.80 (1H, m), 0.87 (3H, d, J=6.59 Hz), 0.97 (1H, m), 1.04
(1H, m), 1.34 (1 H, m), 1.47 (1H, m), 1.50 (1H, m), 1.59 (1H, m), 1.63 (1H, m), 2.06 (1H, m), 3.51
(3H, q, J=1.34 Hz), 4.83 (1H, td, J=10.93, 4.33 Hz), 7.32 (3H, m), 7.48 (2 H, m); m/z (EI) 373 (M+H,
2%), 189 (26), 140 (11), 139 (73), 105 (15), 97 (26), 95 (10), 84 (11), 83 (100), 81 (20), 69 (39), 57
(35), 55 (43). Found: [M+H], 373.1990. C20H28O3F3 requires m/z 373.1991.
36
(1R,2S,5R,2'S)-3',3',3'-Trifluoro-2'-methoxy-2'-phenylpropionic acid 2-isopropyl-5-methyl-
cyclohexyl ester (5):
O
CH3
CH3 CH3
O
OMe
CF3MeO
pyridineCDCl3
OH
CH3
CH3 CH3
Cl
O
OMe
CF3Ph
+
5
Pyridine (60 µL, 741 µmol) was added to a solution of (+)-menthol (3.0 mg, 19.2 µmol) in 1
mL deuterochloroform. (-)-2-methoxy-2-phenyl-2-(trifluoromethyl)acetic acid chloride (7.0 µL, 36
µmol) was then added and the solution stirred for two days at room temperature. The solvent was
removed in vacuo and the resulting clear oil was purified by preparative TLC (20% EtOAc/Hexanes) to
yield 5 (6.5 mg, 92%) as a clear oil: TLC: Rf = 0.80 (20% EtOAc/Hexanes); 1H NMR (300 MHz,
CDCl3) δ 0.78 (3H, d, J=6.98 Hz), 0.88 (3H, d, J=7.02 Hz), 0.92 (3H, d, J=6.52 Hz), 1.03 (1H, m),
1.03 (1H, m), 1.46 (1H, m), 1.71 (1H, m), 1.89 (1H, m), 2.09 (1H, m), 3.54 (3H, q, J=1.25 Hz), 4.89
(1H, td, J=10.92, 4.39 Hz), 7.41 (3H, m), 7.53 (2H, m); m/z (EI) 373 (M+H, 5%), 189 (28), 139 (91),
105 (15), 97 (25), 83 (100), 69 (39), 57 (37), 55 (42). Found: [M+H], 373.1999. C20H28O3F3 requires
m/z 373.1991.

37
(1S, 2R, 2’S, 5S)-(2,2,2-Trifluoro-1-[1-(2-isopropyl-5-methyl-cyclohexyloxy)-1-methyl-ethoxy]-
ethyl)-benzene (10):
Ph OH
CF3
Et3NMe2SiCl2
Ph O
CF3
SiCl
(+)-mentholEt3N
OSi
O Ph
CF3
10
Dichlorodimethylsilane (180 µL, 150 µmol) was added dropwise to a solution of R-
(trifluoromethyl)benzyl alcohol (10 µL, 75 µmol) in 750 µL deuterochloroform. Following the slow
addition of triethylamine (20 µL, 150 µmol) the solution was stirred at room temperature for ten
minutes. The solvent was then removed from the mixture in vacuo to afford a white residue. The
residue was dissolved in 650 µL deuterochloroform and (+)-menthol (3.0 mg, 19 µmol) was added.
Following the slow addition of triethylamine (10 µL, 75 µmol), the mixture was stirred at room
temperature for twenty minutes. The dark yellow solution was eluted through a silica plug (50%
EtOAc/Hexanes) followed by a small amount of deuterochloroform. Solvent was removed from the
resulting liquid in vacuo and the resulting clear oil was purified by preparative TLC (20%
EtOAc/Hexanes) to yield 10 (0.6 mg, 8%) as a clear oil: TLC: Rf = 0.75 (30% EtOAc/Hexanes); 1H
NMR (300 MHz, CDCl3) δ 0.07 (3H, m), 0.16 (3H, m), 0.83 (3H, m), 0.86 (3H, m), 0.91 (1H, m), 0.99
(1H, m), 1.11 (1H, m), 1.25 (1H, m), 1.31 (1H, m), 1.58 (1H, m), 1.58 (1H, m), 1.86 (1H, m), 2.02 (1H,
m), 3.46 (1H, td, J=10.32, 4.39 Hz), 5.07 (1H, q, J=6.44 Hz), 7.37 (3H, m), 7.45 (2H, m); 13C NMR

38
(500 MHza, CDCl3) δ 15.6, 20.7, 21.9, 23.0, 25.8, 29.6, 31.5, 34.1, 40.2, 46.7, 55.3, 77.6, 127.7, 128.4,
129.6; m/z (EI) 388 (M+, 10%), 377 (12), 304 (25), 303 (100), 159 (13), 149 (18), 147 (21), 139 (19),
138 (61), 137 (51), 123 (16), 110 (14), 109 (71), 96 (11), 95 (28), 91 (13), 86 (10), 84 (15), 83 (26), 82
(21), 81 (40), 75 (17), 73 (11), 69 (22), 67 (11), 57 (20), 55 (27). Found: [M+H], 388.2046.
C20H31O2F3Si requires m/z 388.2045.
(1R, 2S, 2’S, 5R)-(2,2,2-Trifluoro-1-[1-(2-isopropyl-5-methyl-cyclohexyloxy)-1-methyl-ethoxy]-
ethyl)-benzene (11):
Ph OH
CF3
Et3NMe2SiCl2
Ph O
CF3
SiCl
(-)-mentholEt3N
OSi
O Ph
CF3
11
Dichlorodimethylsilane (180 µL, 150 µmol) was added dropwise to a solution of R-
(trifluoromethyl)benzyl alcohol (10 µL, 75 µmol) in 750 µL deuterochloroform. Following the slow
addition of triethylamine (20 µL, 150 µmol) the solution was stirred at room temperature for ten
minutes. The solvent was then removed from the mixture in vacuo to afford a white residue. The
residue was dissolved in 650 µL deuterochloroform and (-)-menthol (3.0 mg, 19 µmol) was added.
Following the slow addition of triethylamine (10 µL, 75 µmol), the mixture was stirred at room
a Obtained from (inverse-detection) gHSQC measurements.

39
temperature for twenty minutes. The dark yellow solution was eluted through a silica plug (50%
EtOAc/Hexanes) followed by a small amount of deuterochloroform. Solvent was removed from the
resulting liquid in vacuo and the resulting clear oil was purified by preparative TLC (10%
EtOAc/Hexanes) to yield 11 in very poor yield as a clear oil: TLC: Rf = 0.70 (10% EtOAc/Hexanes);
1H NMR (400 MHz, CDCl3) δ -0.03-0.01 (3H, m), 0.04-0.12 (3H, m), 0.14-0.19 (3H, m), 0.69-0.76
(3H, m), 0.84-0.87 (1H, m), 0.89 (3H, d, J=6.78 Hz), 0.91-1.08 (3H, m), 1.56-1.69 (2H, m), 3.53 (1H,
td, J=10.05, 4.41 Hz), 5.01-5.12 (1H, m), 7.34-7.40 (3H, m), 7.42-7.48 (2H, m).
(2R,4S)-3,3,3-Trifluoro-2-methoxy-2-phenyl-propionic acid 2,2-dimethyl-[1,3]dioxolan-4-
ylmethyl ester (12):
O
O
HO
pyridineCDCl3
Cl
O
OMe
PhF3C
+
O
O
O
O
OMe
PhF3C
12
(S)-(+)-2,2-dimethyl-1,3-dioxolane-4-methanol (2.4 µL, 19.2 µmol) was added to a solution of
d5-pyridine (40 µL, 500 µmol) in 600 µL deuterochloroform in a 5 mm NMR tube. (+)-2-methoxy-2-
phenyl-2-(trifluoromethyl)acetic acid chloride (7.2 µL, 38.4 µmol) was added and the mixture was
allowed to react for 48 hours. The solvent was then removed in vacuo and the crude mixture was
purified by preparative TLC (20% EtOAc/Hexanes) to give 12 in good yield. TLC: Rf = 0.50 (20%
EtOAc/Hexanes); 1H NMR (500 MHz, CDCl3) δ 1.34 (3H, d, J=0.69 Hz, O2C(CH3)2), 1.38 (3H, d,

40
J=0.69 Hz, O2C(CH3)2), 1.54 (1H, s), 3.56 (1H, q, J=1.14 Hz, OCH3), 3.73 (1H, dd, J=8.60, 5.53 Hz),
4.04 (1H, dd, J=8.60, 6.26 Hz), 4.28 (1H, dd, J=10.98, 5.25 Hz), 4.35 (1H, dt, J=11.35, 5.58 Hz), 4.42
(1H, dd, J=10.86, 5.25 Hz), 7.40 (3H, m), 7.53 (2H, m); m/z (EI) 333 (M+H, 84%), 334 (11), 189
(100), 105 (21), 101 (16), 91 (11), 77 (12), 59 (16), 57 (13). Found: [M+H], 333.0954. C15H16O5F3
requires m/z 333.0950.
(2S,4S)-3,3,3-Trifluoro-2-methoxy-2-phenyl-propionic acid 2,2-dimethyl-[1,3]dioxolan-4-ylmethyl
ester (13):
O
O
HO
pyridineCDCl3
Cl
O
OMe
CF3Ph
+
O
O
O
O
OMe
CF3
Ph
13
(S)-(+)-2,2-dimethyl-1,3-dioxolane-4-methanol (2.4 µL, 19.2 µmol) was added to a solution of
d5-pyridine (40 µL, 500 µmol) in 600 µL deuterochloroform in a 5 mm NMR tube. (-)-2-methoxy-2-
phenyl-2-(trifluoromethyl)acetic acid chloride (7.2 µL, 38.4 µmol) was added and the mixture was
allowed to react for 48 hours. The solvent was then removed in vacuo and the crude mixture was
purified by preparative TLC (20% EtOAc/Hexanes) to give 13 in good yield. TLC: Rf = 0.50 (20%
EtOAc/Hexanes); 1H NMR (500 MHz, CDCl3) δ 1.34 (3H, m), 1.38 (3H, m), 1.54 (1H, m), 3.56 (3H,
q, J=1.26 Hz), 3.73 (1H, m), 4.03 (1H, m), 4.35 (1H, m), 4.41 (1H, m), 7.42 (3H, m), 7.53 (2H, m); 13C
NMR (100 MHz, CDCl3) δ 25.0, 26.2, 55.1, 65.3, 65.8, 72.5, 109.6, 121.5, 124.3, 126.8, 127.0, 128.1,

41
128.2, 129.4, 131.7, 166.1; m/z (EI) 333 (M+H, 95%), 334 (20), 189 (100), 105 (18), 101 (26), 77 (10),
72 (11), 59 (20), 57 (21). Found: [M+H], 333.0954. C15H16O5F3 requires m/z 333.0950.
(2R,4R)-3,3,3-Trifluoro-2-methoxy-2-phenyl-propionic acid 2,2-dimethyl-[1,3]dioxolan-4-
ylmethyl ester (14):
O
O
HO
pyridineCDCl3
Cl
O
OMe
PhF3C
+
O
O
O
O
OMe
PhF3C
14
(R)-(+)-2,2-dimethyl-1,3-dioxolane-4-methanol (2.4 µL, 19.2 µmol) was added to a solution of
d5-pyridine (40 µL, 500 µmol) in 600 µL deuterochloroform in a 5 mm NMR tube. (+)-2-methoxy-2-
phenyl-2-(trifluoromethyl)acetic acid chloride (7.2 µL, 38.4 µmol) was added and the mixture was
allowed to react for 48 hours. The solvent was then removed in vacuo and the crude mixture was
purified by preparative TLC (20% EtOAc/Hexanes) to give 14 in good yield. TLC: Rf = 0.50 (20%
EtOAc/Hexanes); 1H NMR (500 MHz, CDCl3) δ ppm 1.34 (3H, s), 1.38 (3H, s), 3.56 (3H, m), 3.73
(1H, dd, J=8.48, 5.31 Hz), 4.02 (1H, dd, J=8.60, 5.80 Hz), 4.34 (1H, m), 4.37 (1H, m), 4.39 (1H, m),
7.41 (3H, m), 7.53 (2H, m); 13C NMR (125 MHz, CDCl3) δ 13.9, 22.5, 25.1, 26.4, 31.4, 62.8, 65.8,
66.0, 72.7, 125.1, 127.1, 128.0, 128.3, 128.8, 129.5, 166.6; m/z (EI) 333 (M+H, 100%), 334 (17), 189
(95), 105 (12), 101 (14). Found: [M+H], 333.0954. C15H16O5F3 requires m/z 333.0950.

42
(2S,4R)-3,3,3-Trifluoro-2-methoxy-2-phenyl-propionic acid 2,2-dimethyl-[1,3]dioxolan-4-
ylmethyl ester (15):
O
O
HO
pyridineCDCl3
Cl
O
OMe
CF3Ph
+
O
O
O
O
OMe
CF3
Ph
15
(R)-(+)-2,2-dimethyl-1,3-dioxolane-4-methanol (2.4 µL, 19.2 µmol) was added to a solution of
d5-pyridine (40 µL, 500 µmol) in 600 µL deuterochloroform in a 5 mm NMR tube. (-)-2-methoxy-2-
phenyl-2-(trifluoromethyl)acetic acid chloride (7.2 µL, 38.4 µmol) was added and the mixture was
allowed to react for 48 hours. The solvent was then removed in vacuo and the crude mixture was
purified by preparative TLC (20% EtOAc/Hexanes) to give 15 in good yield. TLC: Rf = 0.50 (20%
EtOAc/Hexanes); 1H NMR (500 MHz, CDCl3) δ 1.34 (3H, s), 1.38 (3H, s), 3.56 (3H, q, J=1.18 Hz),
3.73 (1H, dd, J=8.60, 5.53 Hz), 4.04 (1H, dd, J=8.60, 6.26 Hz), 4.28 (1H, dd, J=10.98, 5.25 Hz), 4.35
(1H, dt, J=11.25, 5.58 Hz), 4.42 (1H, dd, J=10.98, 5.37 Hz), 7.41 (3H, m), 7.53 (2H, m); m/z (EI) 333
(M+H, 100), 334 (21), 189 (100), 105 (19), 101 (28), 77 (12), 72 (12), 59 (21), 57 (21). Found:
[M+H], 333.0953 C15H16O5F3 requires m/z 333.0950.

43
Table of Figures FIGURE 1. THE CURRENT PROPOSED STRUCTURE OF AZASPIRACID. ........................................................... 4 FIGURE 2. CONFORMATION OF MOSHER ESTERS ....................................................................................... 5 FIGURE 3. SILYL DIETHER DERIVITIZATION OF CHIRAL ALCOHOLS. ........................................................... 6 FIGURE 4. PROPOSED CONFORMATIONAL MODEL FOR SILYL DIETHERS ..................................................... 7 FIGURE 5. THE LIQUID CRYSTAL 4’-N-PENTYL-BIPHENYL-4-CARBONITRILE............................................... 8 FIGURE 6. MENTHOL. ............................................................................................................................... 10 FIGURE 7. 500 MHZ 1H NMR SPECTRUM OF MENTHOL IN CDCL3. ......................................................... 11 FIGURE 8. DISTINGUISHING AXIAL AND EQUATORIAL PROTONS ............................................................... 13 FIGURE 9. INITIAL ASSIGNMENTS OF MENTHOL. ...................................................................................... 14 FIGURE 10. SECOND ROUND OF ASSIGNMENTS FOR MENTHOL. ................................................................ 15 FIGURE 11. FINAL ASSIGNMENTS FOR MENTHOL. .................................................................................... 16 FIGURE 12. CHEMICAL SHIFT DIFFERENCES BETWEEN THE DIASTEREOMERIC MOSHER ESTERS OF
MENTHOL ......................................................................................................................................... 17 FIGURE 13. CHEMICAL SHIFT DIFFERENCES BETWEEN THE DIASTEREOMERIC MOSHER ESTERS OF 2,2-
DIMETHYL-1,3-DIOXOLANE-4-METHANOL ....................................................................................... 18 FIGURE 14. THE CARBINYL PROTON IN THE DIASTEREOMERIC MOSHER ESTERS OF MENTHOL. THE PEAKS
HAVE BEEN FITTED TO GAUSS-LORENTZ LINESHAPES. (300 MHZ, CDCL3)..................................... 20 FIGURE 15. EXCELLENT CORRELATION BETWEEN MOSHER-ESTIMATED MASS FRACTION OF (+)-MENTHOL
AND ACTUAL MASS FRACTION. ......................................................................................................... 22 FIGURE 16. CHEMICAL SHIFT DIFFERENCES BETWEEN THE SILYL DIETHERS OF MENTHOL ....................... 24 FIGURE 17. KAURADIENOIC ACID, A SECONDARY METABOLITE OF MONTANOA TOMENTOSA.................... 26 FIGURE 18. COUPLED CGHSQC OF MENTHOL IN 5CB AT 25.4°C............................................................ 26
Listing of Tables TABLE 1: INITIAL GATHERED INFORMATION FROM PROTON, CARBON, AND GHSQC SPECTRA. ................ 12 TABLE 2. GHSQC, GCOSY, AND GHMBC CORRELATIONS IN MENTHOL................................................. 14 TABLE 3. 1JC-H COUPLING CONSTANTS IN MENTHOL AS MEASURED BY CGHSQC MEASUREMENTS. ........ 16 TABLE 4. D-COUPLINGS OF MENTHOL (1) IN 5CB AS OBTAINED FROM CGHSQC EXPERIMENTS.............. 27 TABLE 5A. D-COUPLINGS OF KAURADIENOIC ACID (16) IN 5CB AS OBTAINED BY CGHSQC
MEASUREMENTS. C-H CORRELATIONS ARE FROM REF. (15). METHYLENE PROTON-CARBON COUPLINGS ARE INDICATED WITH THE α COUPLING FOLLOWED BY THE β COUPLING. ...................... 27
TABLE 6. 1H AND 13C NMR (CDCL3) SPECTRAL DATA AND ASSIGNMENTS FOR RAC-MENTHOL. ............ 32

44
references
References
1. Reetz, M.T. Angew. Chem. Int’l. Ed. 2001, 40, 284-310.
2. Finn, M.G. Chirality. 2002, 14, 534-540.
3. Reynolds, W.F.; Enriquez, R.G. J. Nat. Prod. 2002, 65, 221-244.
4. (i) Dale, J.A.; Mosher, H.S. J. Am. Chem. Soc. 1973, 95, 512. (ii) Sullivan, G.R.; Dale, J.A.;
Mosher, H.S. J. Org. Chem. 1973, 38, 2143.
5. (i) Merckx, E.M.; Vanhoeck, L.; Lepoivre, J.A.; Alderweireldt, F.C.; Van Der Veken, B.J.;
Tollenaere, J.P.; Raymaekers, L.A. Spectros. Int’l J. 1983, 2, 30. (ii) Doesburg, J.M.; Petit, G.H.;
Merckx, E.M. Acta Crystallogr. 1982, B38, 1181.
6. Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113(11), 4093.
7. Williamson, R.T.; Sosa, A.C.B.; Mitra, A.; Seaton, P.J.; Weibel, D.B.; Schroeder, F.C.; Meinwald,
J.; Koehn, F.E. Org. Lett. 2003, 5(10), 1745-1748.
8. (i) Meier, S.; Haussinger, D.; Grzesiek, S. J. Biomol. NMR 2002, 24, 351-356. (ii) Tycko,
R.; Blanco, F. J.; Ishii, Y. J. Am. Chem. Soc. 2000, 122, 9340-9341.
9. Louhivuori, M.; Paakkonen, K.; Fredriksson, K.; Permi, P.; Lounila, J.; Annilla, A. J. Am. Chem.
Soc. 2003, 125, 15647-15650.
10. Parenty A.; Campagne J.-M.; Aroulanda C.; Lesot P. Org. Lett. 2002, 4, 1663–1666.
11. Auger, C.; Lesage, A.; Caldarelli, S.; Hodgkinson, P.; Emsley, L. J. Am. Chem. Soc. 1997, 119,
12000-12001.
12. Palmer, A.G.; Cavanaugh, J.; Wright, P.E.; Rance, M. J. Magn. Reson. 1991, 93, 151-170.
13. Kay, L.E.; Keifer, P.; Saarinen, T. J. Am. Chem. Soc. 1992, 114, 10663-10665.
14. Reynolds, W.F.; Enriquez, R.G. Magn. Reson. Chem. 2001, 39, 531-538.
15. Enriquez,R.G.; Barajas, J.; Ortiz, B.; Lough, A.J.; Reynolds, W.F.; Yu, M.; Leon, I.; Gnecco, D.
Can. J. Chem. 1997, 75, 342-347.