april 19 (sat) -20 (sun), 2014 - jspn.jp · recombinant human erythropoietin therapy for a...
TRANSCRIPT

Sponsored by The Japanese Society of Pediatric NephrologyThe Korean Society of Pediatric NephrologyInternational Pediatric Nephrology Association
April 19 (Sat) - 20 (Sun), 2014Sysmex Hall, Kusunoki Campus Kobe University Graduate School of Medicine, Kobe, Japan

3
Dear Colleagues and Friends,
It is a great pleasure to have the 12th Japan-Korea Pediatric Nephrology Seminar in Kobe.
This seminar has assumed a crucial role to build a tight friendship among the pediatric
nephrologists in Japan, Korea and China, and to raise the scientific levels in each society.
Like in the previous 2 years, we will have an educational program, Continuous
Professional Development “Hereditary Diseases in Nephrology”, as a teaching course
supported by International Pediatric Nephrology Association.
We welcome you all for discussion and international scientific/cultural exchange in Kobe,
one of the most beautiful cities in Japan.
Sincerely yours,
Kazumoto Iijima, M.D., Ph.D.PresidentThe 12th Japan-Korea Pediatric Nephrology SeminarDepartment of PediatricsKobe University Graduate School of MedicineKobe, Japan
Invitation to the 12th Japan-Korea Pediatric Nephrology Seminar 2014
Invitation
Invitation
Organizing Committees
General Information
Program
Abstracts
Instruction for presenters
April 18th (Fri), 2014
Case ConferenceIrreversible severe kidney injury and anuria in a child with atypical hemolytic uremic syndrome under administration of eculizumab
Development of anti-Rituximab antibodies in children with nephrotic syndrome
Alport syndrome
Dense deposit disease with minor urinary abnormalities detected by school urinary screening
Nephronophthisis with heaptic and renal involvement in three siblings
Successful Treatment for Short Stature in a Pubertal Kidney Transplant Recipient Using a Combination of Gonadotropin Releasing Hormone Analog and Growth Hormone
Atypical Hemolytic Uremic Syndrome
Recombinant Human Erythropoietin Therapy for a Jehovah’s Witness Child with Severe Anemia due to Hemolytic Uremic Syndrome
A case with clinically diagnosed renal tubular dysgenesis characterized by pathological and genetic approach
Glomerulopathy with ANA, ANCA, and Antiphospholipid antibodies in Twins
Hereditary tubulopathies
Full-house pattern of glomerular immune deposits in patient who does not fulfilling SLE criteria
A Case of C3 Glomerulonephritis Diagnosed From School Urine Screening: Follow-up Clinical and Biopsy Findings Over a 5-year Observation Period
A case of intermittent hydronephrosis secondary to ureteropelvic junction obstruction
Epstein syndrome and MYH9 Disorder
Two young girls with Alport syndrome initially diagnosed as thin basement membrane nephropathy
Successful treatment of membranoproliferative glomerulonephritis concomitant with a splenorenal shunt
A case of lupus associated hemophagocytic lymphohistiocytosis
Congenital and infantile nephrotic syndrome
A Case of Familial Atypical HUS
Polypoid cystitis mimicking bladder tumor in a nephrotic patient treated with cyclosporine
Urate Transport Disorders
A Brother case of Hypotonia-Cystinuria Syndrome
Nephronophthisis with the mutation of NPHP4 gene in three siblings
Recurrent FSGS with mesangial IgA deposit after kidney transplantation in a child
A case with steroid resistant nephrotic syndrome successfully treated with carperitide
A novel CLCNKB gene mutation: severe hypomagnesemia and hypocalcemia
A case of TSC2/PKD1 contiguous gene deletion syndrome
Is a History of Previous Abdominal Surgery Contraindication for Peritoneal Dialysis?
A case of “not otherwise specified” variant of focal segmental glomerulosclerosis accompanied with crescent formation
New Guidelines for the Management and Investigation of Hemolytic Uremic Syndrome
Congenital nephrotic syndrome
Risk Factors and Management of Childhood Urinary Tract Infection
Treatment of atypical HUS
April 19th (Sat), 2014
Educational Topics
April 20th (Sun), 2014
Luncheon seminar
Continuous Professional Development
Posters
Poster Only
Contents
3
4
5
11
19
9
12
20-27
13-17
28-30
18
31
32-35
36-47
48-51
1.
1.
1.
13.
2.
2.
2.
14.
3.
3.
3.
15.
4.
4.
4.
16.
5.
5.
5.
6.
6.
6.
7.
7.
8.
8.
9.
10.
11.
12.
I.
II.
III.
Supported by Alexion Pharma, Japan
Theme: Hereditary Diseases in Nephrology

4 5
Kazumoto Iijima
Michio Nagata
Takashi Sekine
Shori Takahashi
Shoji Kagami
Ryugo Hiramoto
Kentaro Matsuoka
Kentaro Ogata
Shigeo Hara
Iekuni Ichikawa
Takashi Igarashi
Yuhei Ito
Department of Pediatrics, Nihon University Surugadai Hospital
1-8-13 Kanda-Surugadai, Chiyoda-Ku, Tokyo 101-8309, Japan
TEL
FAX
E-mail [email protected]
Kee Hwan Yoo
Kee Hyuck Kim
Il Soo Ha
Hae Il Cheong
Yong-Jin Kim
Kyoung Bun Lee
Jeong Hae Kie
Yong Choi
Chong Guk Lee
Seung Joo Lee
Yong Hoon Park
Department of Pediatrics, Seoul National University Children’s Hospital
28 Yongon-Dong, Chongro-Gu, Seoul 110-744, Korea
TEL
FAX
E-mail [email protected]
+ 81-3-3293-1711
+ 81-3-3293-1798
+ 82-2-2760-2810
+ 82-2-743-3455
Organizing Committee
Pathologists
Advisors
Office
Organizing Committee
Pathologists
Advisors
Office
General Information
Kobe University
University of Tsukuba
Toho University
Nihon University (Liaison officer)
Tokushima University
Matsudo City Hospital
National Center for Child Health and Development
Tachikawa Hospital
Kobe University
Tokai University
The University of Tokyo
Kurume University
Korea University
NHIC Ilsan Hospital
Seoul National University Children’s Hospital
Seoul National University Children’s Hospital (Liaison officer)
Yeungnam University
Seoul National University Hospital
National Health Insurance Corporation Ilsan Hospital
Seoul National University
Ilsan Paik Hospital
Ewha Womans University
Yeungnam University
JAPAN
KOREA
Registration for Korean and Chinese members staying at Chisun-Hotel
will be automatically processed at check in on April 18 (Friday).
Registration for Japanese members, registration desk is located in front of the Sysmex Hall (3F)
Registration fee: JPY10,000
During the seminar, on19 and 20, registration desk is located in front of the Sysmex Hall (3F).
We highly recommend you guys to use Limousine Bus bound for “Kobe Sannomiya” from Kansai International Airport (time required
65min/ fare: JPY1,900, Round trip fare: JPY3,000).
At “Sannomiya station” take a taxi and tell the driver to go to “Chisun Hotel” or show the card below.
The card says “Would you go to the Chisun Hotel?” The fare might be around JPY1,000.
Cloak may not be serviced
Pre-registered Korean and Chinese participants will stay in Chisun-Hotel with some Japanese members.
Please come directly to the Hotel and Check in on April 18.
April 19, Saturday – 20, SundaySysmex Hall, Kusunoki Campus Kobe University Graduate School of Medicine7-5-1 Kusunokicho, Chuo-ku, Kobe-shi, Hyogo 650-0017, JAPAN
Organizing CommitteesThe 12th Japan–Korea Pediatric Nephrology Seminar 2014
InformationInformation
Chisun Hotel
Congress Information
2-3-1 Nakamachi-dori, Chuo-ku, Kobe-shi, Hyogo 650-0027
TEL: 078-341-8111 FAX: 078-371-5577
http://www.solarehotels.com/en/hotel/kinki/chisunhotel-kobe.html
Dates
Venue
Adress
Registration
Tips! From Kansai Airport to Chisun-Hotel
Cloak
Access for Venue and Hotel from the Airport
神戸駅前のチサンホテルまでお願いします。

6 7
Limousine BusGetting to Kusunoki Campus
from Kansai International Airport
InformationInformation
Take JR ( Japan Railways) Kansai Airport Line from "Kansai Airport" station and get off at "Osaka" station (time required: 1 hour).
Change to JR Kobe Line and get off at "Kobe" station (time required: 25 min./fare: 1,660 yen).
When taking a train from "Kansai Airport," go up to the second floor of the passenger terminal building and go through the passage to
the concourse.
Train Route Finder >>> http://www.jorudan.co.jp/english/
For Kobe(Sannomiya)/Rokko Island,No.6 located outside the 1st floor ofKANSAI AIRPORT TERMINAL1.The buses to airport are arrivesat 4th floor of teriminal building.
For Kobe(Sannomiya)/Rokko Island, No.2 KANSAI AIRPORT TERMINAL2.
Please purchase ticketfrom ticket vending machine.
* Notice : Round Trip Ticket is valid 30 days from the date of purchase.* Due to traffic conditions, bus may delay. Please have a time to spare.
Please purchase ticketfrom ticket vending machine.
Please purchase ticketfrom ticket vending machine.
Adult ChildRound Trip
Ticket1Day RoundTrip Ticket
Commuter Ticket General information
TEL : 06-6844-11241 month 3 month
1,900yen 950yen 3,000yen 2,000yen 50,000yen 142,500yen
Take the Limousine Bus bound for "Kobe Sannomiya" from Kansai International Airport (time required: 65 min. /fare: 1,800 yen).
At "Sannomiya" station, change to Kobe Municipal Subway Seishin-Yamate Line and get off at "Okurayama" station (2nd stop, time
required: 3 min./fare: 200 yen).
When using a limousine bus, go out of the passenger terminal building to buy a ticket for "Kobe Sannomiya" at the ticket counter; board
the bus at No. 6 bus stop.
KATE Airport Limousine web site >>> http://www.kate.co.jp/pc/index.html
Take MK "Skygateshuttle" to your destination in Chuo-ku, Higashinada-ku, Nada-ku, or Hyogo-ku in Kobe city from Kansai
International Airport (fare: 2,300 yen, one-way/per person).
Reservation by phone or by web is required up to 2 days prior to the date of use.
When using the MK "Skygateshuttle" omnibus, go to the MK counter on the first floor of the passenger terminal.
MK "Skygateshuttle" web site >>> http://www.mktaxi-japan.com/#!kobe---kyoto/c13la
・
・
・
・
・
・
・
・
・
・
Fee Contact from Phone
By Railway
Bus Stop
Bus Stop
How to get ticket
How to get ticket
How to get ticket
By Bus
By MK "Skygateshuttle" Omnibus
KANSAI AIRPORT TERMINAL1.
KANSAI AIRPORT TERMINAL2.
SANNOMIYA
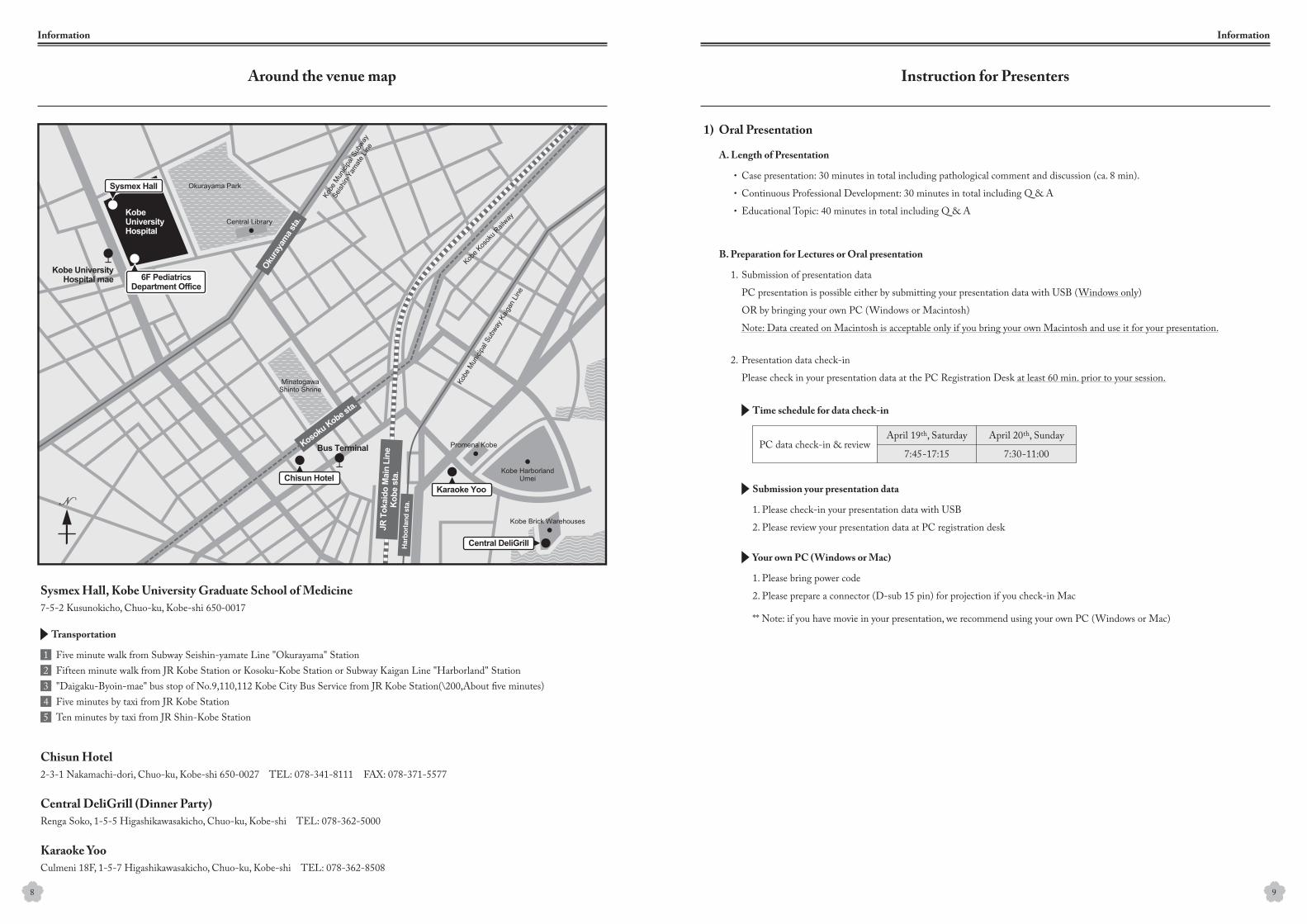
Sysmex Hall, Kobe University Graduate School of Medicine
Central DeliGrill (Dinner Party)
Chisun Hotel
Karaoke Yoo
7-5-2 Kusunokicho, Chuo-ku, Kobe-shi 650-0017
Five minute walk from Subway Seishin-yamate Line "Okurayama" StationFifteen minute walk from JR Kobe Station or Kosoku-Kobe Station or Subway Kaigan Line "Harborland" Station"Daigaku-Byoin-mae" bus stop of No.9,110,112 Kobe City Bus Service from JR Kobe Station(\200,About five minutes)Five minutes by taxi from JR Kobe StationTen minutes by taxi from JR Shin-Kobe Station
Renga Soko, 1-5-5 Higashikawasakicho, Chuo-ku, Kobe-shi TEL: 078-362-5000
2-3-1 Nakamachi-dori, Chuo-ku, Kobe-shi 650-0027 TEL: 078-341-8111 FAX: 078-371-5577
Culmeni 18F, 1-5-7 Higashikawasakicho, Chuo-ku, Kobe-shi TEL: 078-362-8508
8 9
12345
Transportation
Around the venue map
Information
Instruction for Presenters
Information
Case presentation: 30 minutes in total including pathological comment and discussion (ca. 8 min).
Continuous Professional Development: 30 minutes in total including Q & A
Educational Topic: 40 minutes in total including Q & A
1.
・
・
・
2.
Submission of presentation data
PC presentation is possible either by submitting your presentation data with USB (Windows only)
OR by bringing your own PC (Windows or Macintosh)
Note: Data created on Macintosh is acceptable only if you bring your own Macintosh and use it for your presentation.
Presentation data check-in
Please check in your presentation data at the PC Registration Desk at least 60 min. prior to your session.
1. Please check-in your presentation data with USB
2. Please review your presentation data at PC registration desk
1. Please bring power code
2. Please prepare a connector (D-sub 15 pin) for projection if you check-in Mac
** Note: if you have movie in your presentation, we recommend using your own PC (Windows or Mac)
1) Oral Presentation
A. Length of Presentation
B. Preparation for Lectures or Oral presentation
Time schedule for data check-in
Submission your presentation data
Your own PC (Windows or Mac)
April 20th, SundayApril 19th, Saturday
7:30-11:007:45-17:15PC data check-in & review

10
Information
1.
2.
3.
4.
1.
2.
A poster board is provided to each speaker.
Poster Number and speaker name are prepared by secretariat.
Poster size: 1900mm high x 900 mm wide (please refer to the below)
* Please limit the size of your entire poster.
Pushpins to mount your poster are prepared by secretariat.
Schedule: 16:25 to 17:13 on April 19th Sat.
Each speaker has 8 min ; 5 min presentation + 3 min Q & A
Please be seated at the next chair’s seat which is located in the right side of the first line
of the lecture hall at least 10 minutes prior to the next session is begun.
2) Poster presentation
3) Instruction for Chairs
A. Time schedule for poster presentation is as follows.
B. Poster board
C. Oral presentation
April 19th, Saturday
7:45-11:30
Poster No, speaker name
Poster
Prepared by secretariat
200mm
900mm
2,100mm
15:55-16:25
16:25-17:13
17:13-18:00
Poster Mounting
Poster Viewing
Poster Presentation
Poster Removal

7:45 - 8:25
10:30 - 10:45
8:25 - 8:30
8:30 - 10:30
17:30 - 21:00
8:30 - 9:00
9:00 - 9:30
9:30 - 10:00
10:00 - 10:30
Registration
Coffee Break
Opening Remark
Case Conference I
Business Meeting
Case 1 Chairperson: Tae-Sun Ha (Chungbuk National University)
Case 2 Chairperson: Keisuke Sugimoto (Kinki University)
Case 3 Chairperson: Ji Hong Kim (Yonsei University College of Medicine)
Case 4 Chairperson: Ryugo Hiramoto (Matsudo City Hospital Children's Medical Center)
Irreversible severe kidney injury and anuria in a child with atypical hemolytic uremic syndrome under administration of eculizumab
Nephronophthisis with heaptic and renal involvement in three siblings
A case with clinically diagnosed renal tubular dysgenesis characterizedby pathological and genetic approach
A Case of C3 Glomerulonephritis Diagnosed From School Urine Screening:Follow-up Clinical and Biopsy Findings Over a 5-year Observation Period
Meeting Room at Pediatrics, Kobe University Graduate School of Medicine Kazumoto Iijima (Kobe University)
Yusuke Okuda1, Kenji Ishikura1,2, Chikako Terano1, Wataru Kubota1,Yasuhiro Yoshida1, Naoaki Mikami1, Shunsuke Shinozuka1, Ryoko Harada1,Riku Hamada1, Hiroshi Hataya1, Ryuji Fukuzawa3, Kentaro Ogata4, Masataka Honda1
Pathology commentary: Kentaro Ogata
Pathology commentary: Shigeo Hara
Jiwon M. Lee1, Hee Gyung Kang1,2, Il Soo Ha1, Kyoungbun Lee3,Kyung Chul Moon3, Yong Choi1, Hae Il Cheong1,2
Natsuki Matsunoshita1, Kandai Nozu1, Shigeo Hara2, Xue Jun Fu1, Hiroshi Kaito1,Takeshi Ninchoji1, Hironobu Kamiyoshi1, Hiromi Otsubo1, Kazumoto Iijima1
Myoung-Uk Kim1, Da-Eun Woo1, Yong-Jin Kim2, Yong-Hoon Park1
1. Department of Nephrology, Tokyo Metropolitan Children’s Medical Center2. Clinical Research Support Center, Tokyo Metropolitan Children’s Medical Center3. Department of Pathology and Laboratory Medicine, Tokyo Metropolitan Children’s Medical Center4. Division of Pathology, Federation of National Public Service Personnel Mutual Aid Associations, Tachikawa Hospital
1. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea 2. Research Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea3. Department of Pathology, Seoul National University Hospital, Seoul, Korea
1. Department of Pediatrics, Kobe University Graduate School of Medicine2. Department of Diagnostic Pathology, Kobe University Graduate School of Medicine
1. Department of Pediatrics, Yeungnam University College of Medicine, Daegu, Korea2. Department of Pathology, Yeungnam University College of Medicine, Daegu, Korea
7-5-2, Kusunokicho, Chuo-ku, Kobe-shi 650-0017
Registration for Korean and Chinese participants Automatically with hotel check in.
12 13
April 19th (Sat), 2014 [1]April 18th (Fri), 2014
ProgramProgram
Attendant: Core members of JK PN seminar with Chinese attendee.
Chisun Hotel 2-3-1 Nakamachi-dori, Chuo-ku, Kobe-shi 650-0027
Hana-Tamon (Sushi Bar)Dinner
TEL: 078-341-8111

10:45 - 11:25
15:15 - 15:55
11:25 - 12:05
15:55 - 16:25
16:25 - 17:13
18:30 -
12:15 - 12:55
15:00 - 15:15
13:00 - 15:00
13:00 - 13:30
13:30 - 14:00
14:00 - 14:30
14:30 - 15:00
Coffee Break
Case Conference II
Educational Topics I
Educational Topics III
Educational Topics II
Poster Viewing
Poster Presentation
Dinner Party
Luncheon seminar
Chairperson:Speaker:
Chairperson:Speaker:
Chairperson:Speaker:
Chairperson:
Chairperson:
Chairperson:Speaker:
Kyo Sun Kim (Konkuk University School of Medicine)
Jie Ding (Peking University First Hospital)
Shuichi Ito (National Center for Child Health and Development)
Kee Hyuck Kim (NHIS Ilsan Hospital)
Hiroshi Kaito (Kobe University Graduate School of Medicine)
Shori Takahashi (Nihon University)
Takashi Igarashi (National Center for Child Health and Development)
Seung Joo Lee (Ewha Womans University)
Jianhua Mao (Children's Hospital of Zhe Jiang University, IPNA speaker)
Hiroshi Hataya (Tokyo Metropolitan Children’s Medical Center)
Case 5 Chairperson: Jae Il Shin (Yonsei University College of Medicine)
Case 6 Chairperson: Akira Ashida (Osaka Medical College)
Case 7 Chairperson: Hyung Eun Yim (Korea University Medical Center)
Case 8 Chairperson: Yuko Shima (Wakayama Medical College)
Successful treatment of membranoproliferative glomerulonephritis concomitantwith a splenorenal shunt
New Guidelines for the Management and Investigation of Hemolytic Uremic Syndrome
Risk Factors and Management of Childhood Urinary Tract Infection
Congenital Nephrotic Syndrome
Session1: P1-P6 (3rd floor)
Session2: P7-P12 (4th floor)
Treatment of atypical HUS
A Case of Familial Atypical HUS
A Brother case of Hypotonia-Cystinuria Syndrome
Recurrent FSGS with mesangial IgA deposit after kidney transplantation in a child
Mari Okada1, Masaki Fuyama2, Ken Saida2, Hiroyuki Machida2, Mai Sato2,Akinori Miyazono4, Masao Ogura2, Koichi Kamei2, Kentaro Matsuoka3, Shuichi Ito2
Pathology commentary: Kentaro Matsuoka
-Supported by Alexion Pharma, Japan
Seong Heon Kim1, Su Young Kim1, Yong Choi2, Hae Il Cheong2
Shinichi Sakamoto1, Eturou Tokuhiro2, Yukio Naya3, Yasuhiro Shigeta4, Masaaki Fujimura5, Takeshi Ueda6,Kazuo Mikami5, Koichiro Akakura7, Motoyuki Masai4, Kuniyoshi Nozumi8, Tomohiko Ichikawa1
Jee Yeon Han1, Hye Ryun Yeh1, Min Jee Kim1, Eun Gu Kang1,Joo Hoon Lee1, Young Mi Cho2, Young Seo Park1
1. Division of Pediatrics, Musashino Red Cross Hospital2. Division of Nephrology and Rheumatology, National Center for Child Health and Development3. Division of Pathology, National Center for Child Health and Development4. Division of Pediatrics, Kagoshima University Medical and Dental Hospital
1.Department of Pediatrics, Pusan National University Children’s Hospital, Yangsan, Korea2.Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea
1. Department of Urology, Graduate School of Medicine, Chiba University3. Department of Urology, Teikyo University Chiba Medical Center5. Department of Urology, Saiseikai Narashino Hospital 7. Department of Urology, Tokyo Koseinenkin Hospital
2. Department of Pediatrics, Odawara City Hosptial 4. Mihama Hospital6. Department of Urology, Chiba Cancer Center8. Funabashi Clinic
1.Department of Pediatrics, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea2.Department of Pathology, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea
15
April 19th (Sat), 2014 [3]
Program
14
April 19th (Sat), 2014 [2]
Program
Central DeliGrillRenga Soko, 1-5-5 Higashikawasakicho, Chuo-ku, Kobe-shi
TEL: 078-362-5000

P-1:
P-7:
P-3:
P-9:
P-13:
P-4:
P-10:
P-14:
P-5:
P-11:
P-15:
P-6:
P-12:
P-16:
P-2:
P-8:
Yo Han Ahn1, Hee Gyung Kang1,2, Jiwon M Lee1, Hyun Jin Choi1, Il-Soo Ha1,3, Hae Il Cheong1,2,3
Min Jee Kim1, Jee Yeon Han1, Hye Ryun Yeh1, Eun Koo Kang1, Yoo Mi Choi3,Kyoung Hee Han4, Joo Hoon Lee1, Young Mi Cho2, Young Seo Park1, Hae II Cheong5
Hideki Matsumura1, Hyogo Nakakura1, Akihiko Shirasu1, Akira Ashida1, Motoshi Hattori2, Hiroshi Tamai1
Yuko Shima, Koichi Nakanishi, Taketsugu Hama, Masashi Sato, Hironobu Mukaiyama, Hiroko Togawa, Norishige Yoshikawa
Yo Han Ahn1, Jiwon M Lee1, Hyun Jin Choi1, Jae Il Shin2, Hyun Joo Jeong3, Hee Gyung Kang1,4,Il-Soo Ha1,5, Hae Il Cheong1,4,5, Yong Choi1, Kyoungbun Lee6, Kyul Chul Moon6
Sang Taek Lee1, Heeyeon Cho1, Young Seo Park2, Hae Il Cheong3
Ji Young Oh1, Hae Il Cheong2, Hyeon Joo Jeong3, Ji Hong Kim1, Jae Il Shin1, Se Jin Park4
Masano Akamatsu1, Akira Ashida1, Hideki Matsumura1, Tomoki Aomatsu1, Hyogo Nakakura1, Akihiko Shirasu1, Kenji Shimada2, Hiroshi Tamai1
Yusuke Kumagai1, Hiroaki Ueda1, Ichiro Kuki2, Shin Okazaki2, Rika Fujimaru1
Da Eun Woo, Sang Su Lee, Jae Min Lee, Yong Hoon Park Department of Pediatrics, Yeungnam University College of Medicine, Daegu, Korea
Jung Min Lee1, Jihei Cha1, Sun Hee Sung2, Seung Joo Lee1
Hee Sun Baek1, Seung Hyun Cho1, Cheol Woo Ko2, Min Hyun Cho1
Sun Ah Choi, Hye Won Park Department of Pediatrics, Seoul National University Bundang Hospital, Seongnam, Korea
Tomohiro Inoguchi1, Kazuya Matsumura1, Makoto Yoshida2, Yoko Ohwada3,Eiji Kikuchi4, Yasuaki Kobayashi2, Kaori Kameyama5, Midori Awazu1
Norio Omori1, Ryugo Hiramoto1, Tomohiko Yamamura1, Shinsuke Matsumoto1, Hironobu Eguchi1, Bunshiro Akikusa2
Hyung Eun Yim, Jee hoo Lee, Kee Hwan Yoo Department of Pediatrics, College of Medicine, Korea University, Seoul, Korea
1. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea2. Research Coordination Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea3. Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea
1. Department of Pediatrics, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea 2. Department of Pathology, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea3. Department of Pediatrics, Kyung Hee University Hospital, Seoul, Korea4. Department of Pediatrics, Jeju National University Hospital, Jeju, Korea5. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea
1. Department of Pediatrics, Osaka Medical College2. Department of Pediatric Nephrology, Tokyo Women’s Medical University
1. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea2. Department of Pediatrics, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, Korea3. Department of Pathology, Yonsei University College of Medicine, Seoul, Korea4. Research Coordination Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea5. Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea6. Department of Pathology, Seoul National University Hospital, Seoul, Korea
1. Department of Pediatrics, Samsung Medical Center, Seoul, Republic of Korea2. Department of Pediatrics, Asan Medical Center, Seoul, Republic of Korea3. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Republic of Korea
Department of Pediatrics, Wakayama medical university
1. Department of Pediatrics, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, Korea 2. Department of Pediatrics, Seoul National University Children's Hospital, Seoul, Korea.3. Department of Pathology, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea4. Department of Pediatrics, Ajou University School of Medicine, Suwon, Korea
1. Department of Pediatrics, Osaka Medical College2. Department of Urology, Osaka Medical Center and Research Institute for Maternal and Child Hearth
1. Department of Pediatrics, Osaka City General Hospital, Osaka, Japan2. Department of Pediatric neurology, Osaka City General Hospital, Osaka, Japan
1. Department of Pediatrics, Ewha Womans University School of Medicine2. Department of Pathology, Ewha Womans University School of Medicine
1. Department of Pediatrics, Kyungpook National University School of Medicine, Daegu, Korea 2. Department of Radiology, Kyungpook National University School of Medicine, Daegu, Korea
1. Department of Pediatrics, School of Medicine, Keio University3. Department of Pediatrics, Dokkyo Medical University School of Medicine5. Division of Diagnostic Pathology, Keio University Hospital
2. Department of Pediatrics, Ashikaga Red Cross Hospital4. Department of Urology, School of Medicine, Keio University
1. Department of Pediatrics, Matsudo City Hospital Children's Medical Center2. Department of Pathology, Matsudo City Hospital
16
Program
Development of anti-Rituximab antibodies in children with nephrotic syndrome
Nephronophthisis with the mutation of NPHP4 gene in three siblings
Glomerulopathy with ANA, ANCA, and Antiphospholipid antibodies in Twins
A novel CLCNKB gene mutation: severe hypomagnesemia and hypocalcemia
Dense deposit disease with minor urinary abnormalities detected by school urinary screening
A case of intermittent hydronephrosis secondary to ureteropelvic junction obstruction
A case of TSC2/PKD1 contiguous gene deletion syndrome
Recombinant Human Erythropoietin Therapy for a Jehovah’s Witness Child withSevere Anemia due to Hemolytic Uremic Syndrome
A case of lupus associated hemophagocytic lymphohistiocytosis
Is a History of Previous Abdominal Surgery Contraindication for Peritoneal Dialysis?
Full-house pattern of glomerular immune deposits in patient who does not fulfilling SLE criteria
Polypoid cystitis mimicking bladder tumor in a nephrotic patient treated with cyclosporine
A case of “not otherwise specified” variant of focal segmental glomerulosclerosis accompaniedwith crescent formation
Two young girls with Alport syndrome initially diagnosed as thin basement membrane nephropathy
Successful Treatment for Short Stature in a Pubertal Kidney Transplant RecipientUsing a Combination of Gonadotropin Releasing Hormone Analog and Growth Hormone
A case with steroid resistant nephrotic syndrome successfully treated with carperitide
17
Program
Poster with short oral presentation
Poster only (No presentation)

8:30 - 12:00 Continuous Professional Development
Chairperson:
Chairperson:
Koichi Nakanishi (Wakayama Medical College)
Kee Hwan Yoo (Korea University Medical Center)
Hereditary Diseases in Nephrology
1. Alport syndrome (Kandai Nozu, Kobe University)
4. Epstein syndrome and MYH9 Disorder (Takashi Sekine, Toho University)
2. Atypical HUS (Hee Gyung Kang, Seoul National University)
5. Congenital and infantile nephrotic syndrome ( Joo Hoon Lee, University of Ulsan)
3. Hereditary Tubulopathies (Hae Il Cheong, Seoul National University)
6. Urate Transport Disorders (Hirotaka Matsuo, National Defence Medical College)
18
April 20th (Sun), 2014
Program
Coffee Break
Closing Address Kazumoto Iijima (Kobe University)
10:00 - 10:30

Irreversible severe kidney injury and anuria in a childwith atypical hemolytic uremic syndrome under administration of eculizumab
Nephronophthisis with heaptic and renal involvement in three siblings
Yusuke Okuda1, Kenji Ishikura1, 2, Chikako Terano1, Wataru Kubota1, Yasuhiro Yoshida1, Naoaki Mikami1,Shunsuke Shinozuka1, Ryoko Harada1, Riku Hamada1, Hiroshi Hataya1, Ryuji Fukuzawa3, Kentaro Ogata4, Masataka Honda1
Jiwon M. Lee1, Hee Gyung Kang1, 2, Il Soo Ha1, Kyoungbun Lee3, Kyung Chul Moon3, Yong Choi1, Hae Il Cheong1, 2
1. Department of Nephrology, Tokyo Metropolitan Children’s Medical Center2. Clinical Research Support Center, Tokyo Metropolitan Children’s Medical Center3. Department of Pathology and Laboratory Medicine, Tokyo Metropolitan Children’s Medical Center4. Division of Pathology, Federation of National Public Service Personnel Mutual Aid Associations, Tachikawa Hospital
1. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea2. Research Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea3. Department of Pathology, Seoul National University Hospital, Seoul, Korea
The efficacy and safety of eculizumab for atypical hemolytic uremic syndrome (aHUS) have been reported recently. We experienced a case with aHUS not recovered from anuria in spite of administration of eculizumab. Although gene mutations of complement system were not detected, pathological findings were compatible with aHUS, in terms of remarkable arteriolar and arterial change.A 3-month-old girl was transported to our hospital because of diarrhea without hemorrhage, vomiting, poor suckling, anemia, thrombocytopenia, and renal impairment. Laboratory data on admission were Hb 5.7 g/dL, Plt 34*103 /μL, Cr 4.63 mg/dL, LDH 780 U/L, C3 28 mg/dL, C4 17 mg/dL, and schizocytes were detected. She was diagnosed as aHUS due to negative stool culture, negative anti-lipopolysaccharide antibodies against the most frequent Shiga toxin-producing Escherichia coli, and normal ADAMTS13 activity. Intensive care was required and continuous hemodiafiltration (CHDF) was adopted from the day of admission because of anuria and severe hypertension. Plasma exchange was performed once at day 2 and eculizumab was administered three times (150 mg, 300 mg, and 300 mg at day 18, day 25, and day 32, respectively) as an induction. After the induction of eculizumab, although platelet counts and serum LDH level improved gradually, she was not recovered from anuria, and chronic peritoneal dialysis was adopted. No gene mutations of complement system such as factor H, I, B, MCP, THBD, or C3 were detected. Also, no mutations in diacylglycerol kinase epsilon were detected. Pathological findings of kidney biopsy at day 37 showed diffuse arteriolar and arterial luminal stenosis with remarkable thickness and sclerotic change of media and intima, which supports the diagnosis of aHUS. The findings would indicate poor renal prognosis. In addition, most glomeruli were in global sclerosis and collapse, and 80% of the tubular interstitial compartment showed atrophic change with infiltration of inflammatory cells, reflecting severe and irreversible kidney injury. IgG, IgA, IgM, C3, C4, C1q, fibrinogen was weakly and non-specifically positive.Diagnosis of aHUS without gene mutations depended on its clinical course, negation of typical HUS, and its pathological findings in this case. Pathological findings played an important role to support the diagnosis of aHUS and to predict prognosis. Although reasons for persisting anuria in spite of administration of eculizumab were unclear, fulminant type of aHUS, late administration of eculizumab, or other factors for kidney injury such as usage of multiple drugs in intensive care or long duration of CHDF could be considered as possibilities.
Nephronophthisis, a hereditary cystic kidney disease, is a major cause of childhood end stage renal disease (ESRD). To date, 13 genes are identified with regard to its pathogenesis. We introduce a familial case of NPHP13 with WDR19 mutations with peculiar hepatic and renal involvement.
NPHP13 is a recently identified disease. Various kinds of extrarenal manifestations including hepatic involvement have been documented in patients with NPHP13. In addition, renal biopsy may mimic immune complex-mediated glomerulopathy as shown in our cases.
1. Intrahepatic ductal dilatation (Caroli disease) is one of the major manifestations of NPHP13. 2. Thorough clinical examination and early suspicion is essential in prompt diagnosis.
A 6-year-old girl visited our clinic due to incidentally detected proteinuria. She had an older brother who presented with ESRD at age 6. Laboratory tests revealed normal serum creatinine (0.7 mg/dL), albumin, C3, and C4 levels. Albumin was 2+ at random urines, and 510mg in 24-hour urine. Utrasonography revealed echogenic both kidneys and, of note, focal intrahepatic dilatation in the left lobe of the liver. The latter finding was also found for her brother. A kidney biopsy showed severe glomerulosclerosis (65% global and 7% segmental) and tubulointerstitial changes. Immunofluorescent microscopy revealed mild mesangial deposit of IgA. On electron microscopy, tiny subendothelial deposits were noted. Three years later, she progressed to ESRD. Meanwhile, her younger sister was born, and at age 10, she showed elevation in the serum creatinine (1.21 mg/dL). Her kidney biopsy and ultrasonographic findings were nearly same as those of the patient. A targeted exome sequencing was performed for 96 ciliopathy-related genes under a suspicion of ciliopathy, and compound heterozygote mutations in WDR19 were detected in all three siblings. Two additional patients with similar kidney and liver involvement were detected by the targeted exome sequencing.
20
Background
Conclusion
Points of Discussion
Case
[Case 1]
Abstracts
21
[Case 2]
Abstracts

A case with clinically diagnosed renal tubular dysgenesis characterizedby pathological and genetic approach
A Case of C3 Glomerulonephritis Diagnosed From School Urine Screening: Follow-up Clinical and Biopsy Findings Over a 5-year Observation Period
Natsuki Matsunoshita1, Kandai Nozu1, Shigeo Hara2, Xue Jun Fu1, Hiroshi Kaito1,Takeshi Ninchoji1, Hironobu Kamiyoshi1, Hiromi Otsubo1, Kazumoto Iijima1
Myoung-Uk Kim1, Da-Eun Woo1, Yong-Jin Kim2, Yong-Hoon Park1
1. Department of Pediatrics, Kobe University Graduate School of Medicine2. Department of Diagnostic Pathology, Kobe University Graduate School of Medicine
1. Department of Pediatrics, Yeungnam University College of Medicine, Daegu, Korea2. Department of Pathology, Yeungnam University College of Medicine, Daegu, Korea
C3 glomerulonephiritis (C3GN) is a recently-described entity, defined as strong C3 deposition but without any immunoglobulins, C1q, or C4 on immunofluorescence (IF) microscopy. Proliferation either can or cannot be found on light microscopy (LM), and electron dense deposits in mesangium and/or subendothelium can be seen on electron microscopy (EM). However, treatment planning, prognosis, morphologic evolution are still unknown.We present a case of a 10-year old girl who visited for microscopic hematuria and proteinuria on school urine screening. She was asymptomatic with normal renal function. Persistent C3 hypocomplementemia led her to a kidney biopsy. First biopsy showed membranoproliferative glomerulonephritis (MPGN) pattern with only C3 deposition on IF. She was under supportive therapy including angiotensin-converting enzyme (ACE) inhibitor for over two years. During the period, she did not have symptoms nor proteinuria. But microscopic hematuria and C3 hypocomplementemia persisted. After 33 months of treatment, the patient quit medication on her own. Over one year later, she revisited clinic with nephrotic-range proteinuria (53.37 mg/m2/hr), microscopic hematuria and normal renal function at the age of 15. She denied of any symptoms during the entire period, and the only abnormality was persistent C3 hypocomplementemia. Kidney function was within normal range, without decline. A second kidney biopsy was taken, which showed MPGN pattern on LM, but only C3 deposition on IF as same as those of first biopsy. The pattern of proliferation, and location of dense deposits did not differ much from the first biopsy. Under the new entity, she was diagnosed as C3GN. Currently, she is treated with ACE inhibitor and angiotensin-receptor blocker. Proteinuria improved again, with no change in microscopic hematuria.The reclassification clarifies our patient’s stable renal function status better than conventional MPGN which would present with worse outcome. This is the first case report of C3GN with a follow-up biopsy. We were able to once again identify the usefulness of school urine screening.
Renal tubular dysgenesis (RTD) is a severe fetal disorder clinically characterized by oligohydramnios with normal sized kidney and Potter sequence without any renal dysplasia and urinary tract malformation. Most of the cases show early death from pulmonary hypoplasia, anuria and arterial hypotension. Pathological findings show absence or poor development of proximal tubules. In addition, renin is usually over expressed at the juxtaglomerular apparatus ( JGA) and arterial walls show marked thickening. RTD is genetically heterogeneous and linked to mutations in the four genes encoding the major components of the renin-angiotensin system (RAS): angiotensinogen (AGT), renin (REN), angiotensin converting enzyme (ACE) or angiotensin II receptor type I (AGTR1). Here, we report a clinically diagnosed RTD case accompanied by pathological and genetic approach.
Although the present case was clinically compatible with RTD, no unique pathological findings or disease causing mutations in the susceptible four genes were detected. One differential diagnosis is renal ischemic changes due to the renal artery constriction or stenosis, although no apparent arterial abnormalities were observed clinically. Another possible pathogenesis is unidentified congenital genetic disease in which clinical characteristics are quite similar to RTD; in this setting, pathological examination can be useful to make distinction from RTD. Accumulation of more similar cases and comprehensive analysis using next generation sequencing techniques is required to establish a genetic basis of the current case.
We report a fetus at 21 weeks of gestation. There was no family history of renal diseases or drug use of the mother during pregnancy. At 19 weeks of gestational age, the mother was detected oligohydramnios for the first time. Although ultrasonography and MRI imaging showed normal kidney size without any urinary tract malformation, no urine was detected in the fetus bladder. At 21 weeks of gestational age, no amniotic fluid was detected by ultrasonography and the pregnancy was terminated because of fetus having severe pulmonary hypoplasia. Macroscopically, fetal kidneys were normal in size without any urinary tract malformation. These findings completely matched RTD characteristics. However, histology showed intact number of proximal tubules and functionally developed glomeruli. Almost all tubules were atrophic and glomerular urinary spaces were narrow. Furthermore, JGA renin expression was not increased in immunostaining and arterial vessels did not show marked thickening. Genetic approach revealed no disease causing mutations in the four susceptible genes.
22
Introduction
Discussion
Case
[Case 3]
Abstracts
23
[Case 4]
Abstracts

Successful treatment of membranoproliferative glomerulonephritis concomitantwith a splenorenal shunt
A Case of Familial Atypical HUS
Mari Okada1, Masaki Fuyama2, Ken Saida2, Hiroyuki Machida2, Mai Sato2,Akinori Miyazono4, Masao Ogura2, Koichi Kamei2, Kentaro Matsuoka3, Shuichi Ito2
Seong Heon Kim1, Su Young Kim1, Yong Choi2, Hae Il Cheong2
1. Division of Pediatrics, Musashino Red Cross Hospital2. Division of Nephrology and Rheumatology, National Center for Child Health and Development3. Division of Pathology, National Center for Child Health and Development4. Division of Pediatrics, Kagoshima University Medical and Dental Hospital
1. Department of Pediatrics, Pusan National University Children’s Hospital, Yangsan, Korea2. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea
Secondary membranoproliferative glomerulonephritis (MPGN) is induced by various disorders. Hepatic diseases such as viral infection and liver cirrhosis are well-known causes of MPGN. However, MPGN triggered by splenorenal shunt is rarely reported. We encountered a patient with MPGN triggered by splenorenal shunt due to portal hypertension.A 10-year-old boy with 21 trisomy and postoperative biliary atresia showed proteinuria and hematuria at his school urinary screening 8 months before admission. His clinical findings at admission were proteinuria (urinary total protein/Creatinine, 4–5), hematuria (urinary red blood cells, 30–49/HPF), hypoalbuminemia (serum albumin level, 2.1 g/dl), and renal insufficiency (serum Cr level, 0.48 mg/dl; cystatin C level, 1.29 mg/l). Hepatic enzymes were increased (aspartate aminotransferase, 200 IU/l; alanine aminotransferase, 106 IU/l), but hepatitis B and C were negative. Cryoglobulinemia was ruled out. A renal biopsy showed diffuse mesangial proliferation, segmental endocapillary proliferation, and some double contours. Immunofluorescence microscopy showed diffuse deposition of immunoglobulin G, A, and M, and complement (C3, C4, and C1q). Electron microscopy revealed subendothelial deposition. Abdominal ultrasonography showed splenomegaly and splenorenal shunt. We diagnosed our patient with secondary MPGN caused by splenorenal shunt. He was initially treated with methylprednisolone pulse therapy (MPT), followed by prednisolone, mizoribine, and candesartan. He responded well to these therapies, and his urinary findings completely recovered. We performed a second biopsy 6 months later. However, light microscopy findings were unchanged. Electron microscopy showed a marked decrease in dense deposits compared with the previous biopsy. Few studies have reported glomerulonephritis concomitant with splenorenal shunt, and its incidence is quite low. A review of the literature showed 209 patients with this condition, but their renal prognosis was rarely described. Most of the cases progressed to end-stage renal disease.Nevertheless two cases decreased their urinary protein by MPT, there aren’t any cases which were reported to have a complete remission. The liver and spleen play an important role in removing circulating immune complexes. Interestingly, Soma et al. reported three patients who developed de novo MPGN type I after surgical creation of a portosystemic shunt to treat non-cirrhotic portal hypertension. They hypothesized that MPGN might be induced by reduced clearance of immune complexes through the liver. Primary treatment of MPGN concomitant with splenorenal shunt could be an early cure for primary disease, but liver transplantation was too early for our patient because he had not yet developed severe liver failure or severe portal hypertension. In our patient, multiple treatments, including steroids and immunosuppressive agents, were effective. Our experience suggests that it is worth attempting combined therapy with multiple therapies, including steroids and immunosuppressive agents.
The hemolytic uremic syndrome is characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury. A rare form known as atypical HUS which may be familial or sporadic has generally a poor prognosis. Here we present a case of familial atypical HUS which caused by membrane cofactor protein(MCP) mutation.
1. Onset age of MCP mutation 2. Effect of plasma therapy in MCP mutation
A 9 year-old girl without recent history of diarrheal disease was referred to our hospital for the evaluation of fever, thrombocytopenia, gross hematuria persisted for 3days and acute kidney injury. She had suffered from upper respiratory tract infection 4 days before that gradually worsened. Her initial laboratory findings were as follows : WBC 5,540/uL, Hb 8.9g/dL, plt 10k/uL, BUN 38mg/dL, creatinine 0.9mg/dL, LDH 3827 IU/L, C3 102mg/dL, C4 17.5mg/dL, PT INR 1.08, aPTT 42sec. Peripheral blood smear(PBS) showed typical microangiopathic hemolytic anemia and hematuria with nephrotic range proteinuria was found on urinalysis. Under the diagnosis of atypical HUS, we had treated her with several transfusions of fresh frozen plasma and supportive therapies and she improved. She had been treated for two or three times of clinical events like this in other hospital. She had 6 year-old brother who had suffered from suspected atypical HUS and his clinical manifestation were very similar to hers. The other 19 month -old sister had gross hematuria on diaper at about 11 months of age whose laboratory results revealed anemia with a few fragmented RBC on PBS and thrombocytopenia which improved rapidly. Suspecting familial atypical HUS, genetic tests were performed which revealed a compound heterozygote mutation of MCP gene previously reported by Maga et al. in all three children. As far as we know, this is the first case of familial atypical HUS confirmed by MCP mutation in Korea.
24
Introduction
Discussion point
Case
[Case 5]
Abstracts
25
[Case 6]
Abstracts

A Brother case of Hypotonia-Cystinuria Syndrome
Shinichi Sakamoto1, Eturou Tokuhiro2, Yukio Naya3, Yasuhiro Shigeta4, Masaaki Fujimura5, Takeshi Ueda6,Kazuo Mikami5, Koichiro Akakura7, Motoyuki Masai4, Kuniyoshi Nozumi8, Tomohiko Ichikawa1
1. Department of Urology, Graduate School of Medicine, Chiba University2. Department of Pediatrics, Odawara City Hosptial3. Department of Urology, Teikyo University Chiba Medical Center4. Mihama Hospital5. Department of Urology, Saiseikai Narashino Hospital6. Department of Urology, Chiba Cancer Center7. Department of Urology, Tokyo Koseinenkin Hospital8. Funabashi Clinic
Hypotonia–cystinuria syndrome (HCS) is an autosomal recessive disorder caused by combined deletions of SLC3A1(rBAT) and PREPL. Clinical features include cystinuria, neonatal hypotonia with spontaneous improvement, poor feeding in neonates, hyperphagia in childhood, growth hormone deficiency, and variable cognitive problems.Only 14 families with 6 different deletions have been reported, previously. Patients are often initially misdiagnosed, while correct diagnosis enables therapeutic interventions.We report a brother case of HCS, further characterizing the clinical and molecular genetics spectrum of HCS. As far as we searched, this is a first case of HCS in Japan.
Array CGH on genomic DNA obtained from blood sample were performed in both patients and parents using whole Genome DNA array ver5. Direct sequences were performed in SLC3A1(rBAT) and SLC7A9(BAT1).
Similar to reported cases, genomic deletion of SLC3A1, PREPL and C2/f34 were observed as a result of micro-chromosomal deletion of 2p21. Our data may suggest existence of potential cases of HCS among Japanese cystinuria patients.
As a result of array CGH, in both patients, genomic deletion of SLC3A1(rBAT) at chromosome 2p21(0.66, 0.73) were identified. No genomic deletion were observed in SLC7A9(BAT1)(. 1.00, 1.00). In addition to SLC3A1, deletion of PREPL and C2/f34, both exist at chromosome 2p21, were also identified. In direct sequence, complete deletion in exon 10 were observed in SLC3A1(rBAT1). Only a polymorphism; L223M were observed In SLC7A9(BAT1).
Genetic analysis of 31 and 29 years old cystinuria brothers were requested to our institute on purpose of further genetic investigation of cystinuria. Patients were born after a normal pregnancy to consanguineous parents. Hypotonia was observed in mother. Both patients have symptoms of neonatal hypotonia, growth retardation. They were also noted to have a long face, a slight ptosis with high and narrow palate. Both patients have history for recurrent urinary stones and performed multiple extracorporeal shock wave lithotripsies(ESWL) and endoscopic operations. Currently they were medicated with thiols.
26
Background
Method
Conclusion
Result
Case
[Case 7]
Abstracts
Recurrent FSGS with mesangial IgA deposit after kidney transplantation in a child
Jee Yeon Han1, Hye Ryun Yeh1, Min Jee Kim1, Eun Gu Kang1, Joo Hoon Lee1, Young Mi Cho2, Young Seo Park1
1. Department of Pediatrics, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea2. Department of Pathology, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea
Focal segmental glomerulosclerosis (FSGS) is a major cause of end-stage renal disease (ESRD) in children accounting for 11% of ESRD cases. The recurrence of FSGS occurs in 20~40% of allografts with risk of graft failure. In most cases of recurrent FSGS, pathologic finding shows glomerular foot process effacement consistent with FSGS without immunoglobulin deposit. We report a case of recurrent FSGS with mesangial Ig A deposit after kidney transplantation(KT).
Recurrence of FSGS is frequent in pediatric renal transplant recipients. However, recurrent FSGS combined with immunoglobin deposit is rare. We reported our experience of recurrent FSGS with mesangial Ig A deposit.
1. Which is the cause of proteinuria, IgA nephropathy or FSGS?2. Did Ig A nephropathy (HS nephritis?) occur as a “de novo nephropathy” or as a “recurrent coexisting disease”
A 17-year-old boy was admitted for renal biopsy due to recurrent proteinuria after cadaveric KT 21 months ago. Ten years ago, generalized edma developed 1 month after purpura on both lower extremities. He had been managed under the diagnosis of Henoch-Schonlein nephritis with steroid, cyclophoshamide and cyclosporine. Renal biopsy performed 8 years ago due to persistent proteinuria showed diffuse proliferative glomerulonephritis with weak Ig A deposit. Cyclophosphamide and cyclosporin was added after biopsy and mPD pulse therapy was repeated 10 times but proteinuria persisted. Repeated renal biopsy result was FSGS with Ig M deposit. His renal function decreased to ESRD 7 years ago. He received KT after the 5-year duration of peritonel dialysis and hemodialysis. He showed heavy proteinuria immediately after KT and received plasmapheresis at day 9 post-transplant under the impression of recurrent FSGS and then, proteinuria decreased. We changed mycophenolate mofetil to cyclophosphamide. He has taken tacrolimus, deflazacort and cyclophosphamide for immunosuppression and amlodipine and enalapril for control of hypertension. The cycle of plasmapheresis was performed 3 more times because of increased proteinuria to above 1,500 mg/day and after plasmapheresis proteinuria decreased to below 250 mg/day. The laboratory findings at admission were as follows: Hb 13.1 g/dL, WBC 5,600 x 103/uL, platelet 201K/uL, serum BUN 20 mg/dL, creatinine 1.1 mg/dL, protein 4.8 g/dL, albumin 2.4 g/dL, urinalysis: S.G. 1.017, pH 6.5, albumin 3+, occult blood -, urine albumin/creatinine 5.67mg/mg, 24 hour urine protein 4682 mg. The renal biopsy results showed focal global and segmental sclerosis, focal fibrous crescent formation and mild chronic tubulointerstitial change. The immunofluorescence study revealed predominant mesangial Ig A immunopositivity, which was supported by mesangial electron dense deposit on the EM study. The donor kidney specimen harvested on the zero-day did not show significant immunopositivity for IgG, IgM and IgA. The result of BK virus immunostaining was negative.
Background
Conclusion
Point of discussion
Case
27
[Case 8]
Abstracts

New Guidelines for the Management and Investigation of Hemolytic Uremic Syndrome
Takashi Igarashi, Shuichi Ito, Mayumi Sako
National Center for Child Health and DevelopmentStudy subgroup for establishing guidelines for the diagnosis and therapy of hemolytic uremic syndromeStudy group for pathological factors in severe form of enterohemorrhagic Escherichia coli infections and the generalization of therapy
The Japanese Society of Pediatric Nephrology ( JSPN) published the previous guidelines for the diagnosis and treatment of HUS following the Shiga toxin producing Escherichia coli (STEC) infection in 2000 after the outbreak of STEC infection among schoolchildren in Sakai city in Osaka. Since then, there has been considerable advancement in the understanding and treatment of acute encephalopathy - one of the most serious complications in HUS. Furthermore, the etiology, conditions and treatments of atypical HUS have been elucidated. Therefore, a set of comprehensive guidelines for HUS that reflects recent clinical evidence is now necessary. The aim of this set of guidelines is to provide a tool for daily medical practice and to contribute to the standardization and accessibility of HUS-related medical care, as well as to improve level of safety for HUS patients. In addition, the reliable and contemporary guidelines must be made by the procedures proposed by the Medical Information Network Distribution Service (Minds) of the Japan Council for Quality Health Care.The Ministry of Health, Labor and Welfare supported the three-year (from 2012 to 2014) grant for us to make the guidelines and to investigate the long-term prognosis of the HUS patients. We formed the guidelines writing committee (GWC) consisted by the members from JSPN, The Japanese Society of Nephrology ( JSN), The Japanese Society of Child Neurology, Japanese Society for Pediatric Infectious Diseases and The Japanese Association for Infectious Diseases.The GWC members set the keywords in conjunction with the clinical question and critically reviewed relevant literatures published between 1992 and August 31, 2012, through the use of major databases (PubMed and the Japana Centra Revuo Medicina) in cooperation with The Japan Medical Library Association. As there is a lack of high quality publications on HUS, publications with low quality evidences or without retrieval target period were also carefully reviewed. The level of evidence (from Level I to Level VI) or the grade of recommendation for treatment or procedure (from Grade A to Grade D) was determined based on the level of evidence, as well as on the quality and clinical significance of the evidence. The independent assessment committee consisting of three representatives from JSPN and one representative from Child Support Whole Country Network of Intractable Diseases reviewed the present guidelines. The final draft of the guidelines, together with a request for public comments, was published on the websites of Japan Pediatric Society, JSN and JSPN. The GWC then took on board the comments and suggestions by the public to revise and finalize the present set of guidelines as Japanese in 2013 and as English in 2014.
28
Educational Topics I
Abstracts
Congenital nephrotic syndrome
Mao Jianhua Department of Nephrology, The Children Hospital of Zhejiang University School of Medicine
1. Definition of congenital nephrotic syndromeWith massive proteinuria, severe hypoalbuminemia and edema, the clinical onset within the first 3 months of life.
2. Classification of congenital nephrotic syndromeNon-genetic CNS: Congenital infections: HBV, HCV, HIV, EBV, Syphilis, CMV, Parvovirus B19, Varicella-Zoster Virus, Toxoplasmosis, Malaria...Others: Maternal SLE, steroid–chlorpheniramine treatment, RVT, Alloimmunization against maternal neutral neuropeptidase, HUS… Genetic CNS: Syndromic: Denys-Drash syndrome, Frasier syndrome, WAGR syndrome, Pierson syndrome, Nail-patella syndrome, Fabry disease, Schimke immuno-osseous dysplasia, mitochondrial cytopathy, Galloway Mowat syndrome, CNS + ventriculomegalyIsolated: NPHS1, NPHS2, WT1, LAMB2, TRPC6, PLCE1, COQ2, PDSS2, NEPH1...
3. Differentiation of genetic CNSBy isolated or systemic syndrome, the age of proteinuria onset, pathology, inheritance model, and molecular diagnosis.
4. Treatment strategy for CNSAlbumin infusionsMedication: ACE-inhibitor, indomethacin, Thyroxin supplementation, AnticoagulationNutrition: Hypercaloric diet, Protein and lipid supplementation, Vitamins, Calcium and magnesium supplementationNephrectomyKidney transplantationImmunosuppressive reagents?
29
Educational Topics II
Abstracts

30
Educational Topics III
Abstracts
Risk Factors and Management of Childhood Urinary Tract Infection
Seung Joo Lee Department of Pediatrics, Ewha Womans University School of Medicine
Childhood urinary tract infection(UTI) is the most common serious bacterial infection in the pediatric population. The incidence is especially high in febrile infants (5-25%), and the recurrence is common after the first UTI (12-50%). The consequences of childhood UTI is the development of renal scar, a cause of substantial long-term morbidity such as childhood hypertension and chronic kidney disease. To prevent UTI and subsequent renal scar, risk factors should be searched and well managed. Many risk factors have been known and are listed in the recent edition of Nelson Textbook (2011). However there has been some significant development in understanding of the risk factors in the recent years. Primary vesicoureteral reflux(pVUR) had been considered as a major risk factor since the relationship among pVUR, recurrent pyelonephritis and renal scarring (reflux nephropathy) was suggested, and several guidelines for diagnosing and managing pVUR had developedand used for the last three decades. However, the recent systemic review could not conclude that pVUR was a prerequisite for UTI and renal scar and have challenged the relationship. Although original guidelines were recently revised to a less aggressive approach, a more restrictive revision will be required. Uncircumcised male is a definite risk factor with good evidences. The low rate in neonatal circumcision is significantly related to the higher incidence and male preponderance of infantile UTI. Since the target of bacterial colonization in uncircumcised male is the prepuce, a real risk factor can be switched from uncircumcised male to physiologic phimosis. Although the traditional approach for physiologic phimosis is neonatal circumcision, recent AAP policy (2011) still did not recommend it as a routine procedure because of the possible surgical complication. Non-surgical alternative is topical steroid application for 2-4 weeks which is provn to be easy, safe and effective. Therefore, topical steroid can be a reasonable first-line management instead of neonatal circumcision, in order to reduce infantile UTI. Inadequate genital hygiene is a generally acceptable risk factor, which is not listed in Nelson textbook and easily neglected. Good genital (preputial and perineal) hygiene is so natural that the clinical studies are almost absent. Practically good genital hygiene has not well performed, proper education should be emphasized. Labial adhesion is an anatomic risk factor that induce urinary stasis although the prevalence is not known. Meticulous perineal examination is mandatory in children with UTI because labial adhesion is easily manageable with topical steroid or estrogen. Voiding dysfunction(VD) is a common pediatric problem inducing urinary stasis. The pathogenesis is not well known but was suggested to relate with later toilet training. Early toilet training is recommended to prevent VD. For treatment of VD, timed frequent voiding, behavioral modification and anticholinergics is recommended. Deficit of urogenital microflora is a newly suggested risk factor for UTI. Since UTI was proved as an ascending infection of own fecal uropathogens, the role of urogenital microflora was suggested. Therefore urogenital lactobacilli (probiotics) have been introduced as a novel approach to prevent UTI in women. Bacterial adhesiveness is a prerequisite for UTI development. Cranberry was proved to inhibit bacterial adhesiveness and reduce recurrent UTI in women. Given that lactobacilli and cranberries are harmless, readily available and may be beneficial, they should be considered even in children with UTI.
Treatment of atypical HUS
Hiroshi Hataya Department of General Pediatrics, Tokyo Metropolitan Children’s Medical CenterDepartment of Nephrology, Tokyo Metropolitan Children’s Medical Center
Atypical hemolytic uremic syndrome (atypical HUS) is a heterogeneous disorder. It is defined as 1) HUS without Shiga toxin-producing Escherichia coli (STEC) infection in the broad sense or 2) HUS caused by dysregulation of the alternative complement activation pathway in the narrow sense. The former definition is used in “Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society”, but in this seminar the term “atypical HUS” represents HUS primarily caused by complement dysregulation.Complement dysregulation accounts for most of the non-STEC cases of HUS. Atypical HUS has an incidence of only 7 per million child population per year and accounts for 5% of the total cases of HUS in European countries. These patients have a poorer outcome and prognosis than patients with typical HUS. Plasma exchange and infusion is the only therapy for patients with atypical HUS up until a few years ago. Recently, the revolutionary development in the treatment of atypical HUS occurred. Several case reports have demonstrated that eculizumab (Soliris®, Alexion Pharmaceuticals), a humanized monoclonal antibody against C5, is effective in the treatment of atypical HUS. This complement blockade binds to C5 and prevents the production of the terminal complement components C5a and the membrane attack complex (MAC) composed of C5b-9, the final effector pathway of complement activation. Although data are observational, the efficacy of eculizumab demonstrated improvement in renal function and hematological parameters in patients who were treated with ongoing plasma therapy. Because host defense against encapsulated organisms is dependent on the ability to form MAC, vaccination against Neisseria meningitides is required. Patients should receive vaccinations for Neisseria meningitides and for Streptococcus pneumonia and Haemophilus influenza type b.In this seminar I will review the treatment of atypical HUS and report our cases which we experienced last year.
31
Luncheon seminar
Abstracts

32
Continuous Professional DevelopmentTheme: Hereditary Diseases in Nephrology
Abstracts
1. Alport syndrome
2. Atypical Hemolytic Uremic Syndrome
3. Hereditary tubulopathies
Kandai Nozu Department of Pediatrics, Kobe University Graduate School of Medicine
Hee Gyung Kang Pediatric Nephrology, Pediatrics, Seoul National University Children’s Hospital
Hae Il Cheong
Alport syndrome is a hereditary disorder that generally runs a progressive course. It usually presents in children as hematuria and proteinuria associated with neurosensory deafness and progresses to end-stage renal disease (ESRD). Alport syndrome is genetically heterogeneous and is mainly transmitted as an X-linked trait (XLAS: 85%). Autosomal recessive Alport syndrome (ARAS) and autosomal dominant modes of inheritance are much less prevalent.In XLAS, disease-causing mutations in COL4A5 result in abnormal α5(IV) expression and typically in complete absence of α5(IV) in the glomerular basement membrane (GBM) and Bowman’s capsule (BC). However, a previous review suggested that 20% of male XLAS sufferers showed complete or partial staining for this collagen chain, though the genetic and clinical backgrounds of male XLAS patients presenting with such atypical immunohistological findings have not yet been elucidated. We retrospectively studied 52 genetically diagnosed male X-linked Alport syndrome patients to evaluate differences in clinical characteristics and renal outcomes between α5(IV)-positive (n = 15, 29%) and α5(IV)-negative (n = 37, 71%) patients. Thirteen patients in the α5(IV)-positive group had non-truncating mutations (nine missense mutations, three in-frame deletions, and one splice-site mutation resulting in small in-frame deletions of transcripts), while the remaining two showed somatic mutations with mosaicism. Missense mutations in the α5(IV)-positive group were more likely to be located before exon 25 compared with missense mutations in the α5(IV)-negative group. Furthermore, urinary protein levels were significantly lower and age at onset of end-stage renal disease was significantly higher in the positive group than in the negative group. These results help to clarify the milder clinical manifestations and molecular characteristics of male X-linked Alport syndrome patients expressing the α5(IV) chain. On the other hand, autosomal recessive and autosomal dominant forms of Alport syndrome are caused by mutations in the COL4A3 or COL4A4 genes, which encode type IV collagen α3 (α3(IV)) or α4 (α4(IV)) chains. With type IV collagen tissue staining, typically the glomerular basement membrane lacks the α3(IV), α4(IV) and α5(IV) chains, but the α5(IV) chain persists in Bowman’s capsule, distal tubules and the epidermal basement membrane. We retrospectively analyzed 30 patients in 24 pedigrees of genetically diagnosed ARAS. Interestingly, 20% of the ARAS patients showed normal expression of α5 in kidney tissue. The median age of developing ESRD was 21 years and the median age of developing hearing loss was 20 years. Three of 30 patients showed ocular lesions (10%). No genotype-phenotype correlation could be seen in our study in ARAS. Although immunohistochemical analysis of α5 can provide diagnostic information, normal distribution does not exclude diagnosis of both XLAS and ARAS.
Hereditary atypical hemolytic uremic syndrome (aHUS) has been known since 1978, often in association with hypocomplementemia. Alternative pathway complement activation from deficiency of complement factor H (CFH) was hypothesized, and linkage study identified CFH mutation in familial cases of aHUS. Hot spot for CFH mutation (short consensus repeat (SCR) 16~20) is located in C-terminal of the protein recognizing host cell. Subsequently, mutations affecting other regulators of complement activation, membrane cofactor protein (MCP, CD46), complement factor I (CFI), and thrombomodulin, were found to cause aHUS. Cobalamin C defect, prothrombotic variation of the prothrombin gene, mutations of C4b-binding protein or clusterin, methylmalonic aciduria and homocystinuria, and partial ADAMTS13 deficiency were also reported as genetic causes of aHUS. At the moment, frequency of genetic causes of aHUS is as follows; CFH deficiency ~30%, MCP defect ~12%, CFI deficiency 5-10%, thrombomodulin defect 3~5%, gain of function mutations of complement factor B (CFB, 2-10%) and C3 (< 5%). Anti-factor H antibody interfering CFH function, commonly associated with large deletion of CFHRs, ‘deficiency of complement factor H-related genes (CFHR) plasma proteins and CFH autoantibody positive (DEAP)-HUS’, accounts for 5~30% of aHUS. Recently, defect of diacylglycerol kinase ε (DGKE) was discovered to cause one third of infantile aHUS.
The kidney is one of the most highly differentiated organs in the body. A mature kidney is composed of approximately 30 different cell types, along the nephron. This diverse cellularity makes the nephron to be capable of modulating a variety of complex physiologic processes, including endocrine functions, regulation of blood pressure, solute and water transport, acid-base balance, mineral homeostasis, and removal of drug metabolites. Each segment of the renal tubules plays different function in such complex physiologic processes, and accordingly, the tubular epithelial cells vary dramatically in morphology and cell types from segment to segment. In addition, each mono-layered lining epithelial cell is so called polarized, i.e., the cell membrane can be functionally demarcated into two discrete domains by the tight junction: the apical membrane facing the tubular lumen and the basolateral membrane facing the interstitium. The asymmetric assignment of membrane proteins, especially solute transporters and/or channels, provides the machinery for directional movement of fluid and solutes by the nephron.Such cellular diversity and complex functions of the renal tubule make it prone to numerous genetic abnormalities resulting in various kinds of hereditary tubulopathies. The phenotypic expression of hereditary tubulopathies depends on the mode of interference, the normal physiology of the segment affected, and whether the abnormality is caused by loss of function or, less commonly, gain of function. In this lecture, I will address an introductory overview of the current knowledge about genotypes, phenotypes, and genotype-phenotype correlation of some representative disorders affecting the length of the nephron, highlighting the molecular defects responsible for underlying pathophysiology. The most focusing diseases will be as follows; 1) disorders involving the proximal tubules including proximal renal tubular acidosis and Dent disease, 2) disorders involving the loop of Henle including Bartter syndrome, 3) disorders involving the distal convoluted tubules including Gitelman syndrome, and 4) disorders involving the collecting ducts including distal renal tubular acidosis, and nephrogenic diabetes insipidus. On the basis of sufficient understanding of the molecular defects responsible for underlying pathophysiology, novel biomarkers and therapeutic candidate substances can be developed in the future for these rare and difficult diseases.
According to the causative genetic problem, onset age of aHUS varies. Those with two defective alleles of DGKE or CFH uniformly present aHUS during infancy, while majority of patients with one defective allele of CFI, C3, or CFB present during infancy and others present throughout their childhood. DEAP-HUS and MCP defect commonly present in older children. Obtaining genetic diagnosis is necessary to predict the prognosis and establish treatment strategy, because most of the hereditary aHUS patients experience recurrent episodes of aHUS and eventually progress to end-stage renal disease, and recurrence after kidney allograft transplantation (CFH, CFI defect). On the other hand, patients with DGKE or MCP defect have negligible risk of recurrence after transplantation. While plasma therapy is a mainstream of treatment for aHUS from compliment dys-regulation, it is not required for MCP mutation because 90% would recover with or without plasma treatment. For hereditary aHUS, genetic study is necessary to select an allograft kidney donor, since carriers of certain mutations are at risk of aHUS or other renal disease such as type II membranoproliferative glomerulonephritis (in CFH defect). For CFH defect, liver transplantation should be considered as well, because the liver is the main source of circulating CFH.
33
Abstracts
Department of Pediatrics, Seoul National University Children’s HospitalResearch Coordination Center for Rare Diseases, Seoul National University HospitalKidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea

34
Abstracts
35
Abstracts
4. Epstein syndrome and MYH9 Disorder
6. Urate Transport Disorders
5. Congenital and infantile nephrotic syndrome
Takashi Sekine Department of Pediatrics, Toho University School of Medicine
Hirotaka Matsuo, MD, PhD. Department of Integrative Physiology and Bio-Nano Medicine, National Defense Medical College, Japan
Joo Hoon Lee Department of Pediatrics, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea
Hae Il Cheong Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea
Epstein syndrome is an autosomal dominant disorder characterized by macrocytic thrombocytopenia, progressive renal disease, and hearing disability. During 2000 to 2001, it was revealed that four distinct disorders including May-Hegglin anomaly and Epstein disease are caused by mutations in MYH9 gene encoding non-muscle myosin heavy chain IIA (myosin-IIA). In contrast to patients with May-Hegglin anomaly manifesting only macrocytic thrombocytopenia, those with Epstein syndrome develop progressive renal disease and hearing disability. This difference depends on the mutational sites in MYH9 gene. Especially, patients with R702 and S96 mutations in MYH9 develop end stage renal failure before 20-years of age; renal histology of Epstein syndrome is FSGS. Interestingly, SNPs in MYH9 gene have also been implicated to cause idiopathic FSGS from several GWAS studies. Thus, analyses of Epstein syndrome and myosin-IIA would give some clues to unveil the pathogenesis of FSGS.We have been studying Epstein syndrome from clinical and pathophysiological aspects. We identified distinct localization of myosin-IIA in podocyte from other podocyte-related proteins, and its decreased expression both in Epstein syndrome and idiopathic FSGS. In PAN nephrotic rats, the same phenomenon was observed. In vitro study, we showed that myosin-IIA is related to the formation of the specific morphology of podocyte. At this moment, we are considering that myosin-IIA is one of critical molecules on pathophysiological basis, not only Epstein syndrome, but also idiopathic FSGS.
Two pathophysiological conditions are known as urate transport disorders; renal hypouricemia (RHUC) and hyperuricemia/gout. RHUC is a common inherited disease characterized by low serum uric acid (SUA) levels, and is associated with severe complications such as urolithiasis and exercise-induced acute renal failure. We have previously reported that urate transporter 1 (URAT1/SLC22A12) and glucose transporter 9 (GLUT9/SLC2A9) are causative genes for renal hypouricemia type 1 (RHUC1) and renal hypouricemia type 2 (RHUC2), respectively. These findings enable us to propose a physiological model of the renal urate reabsorption and can be promising therapeutic targets for hyperuricemia, gout and associated diseases, such as cardiovascular diseases, cerebrovascular diseases and renal failure. In addition to the outpatients of hospitals, patients are collected out of more than 5000 examinees of medical checkups. Moreover,
Nephrotic syndrome that presents at birth or within the first three months of life is defined as congenital nephrotic syndrome. Later onset, between three months and one year of age, is called infantile nephrotic syndrome. Most children with congenital or infantile nephrotic syndrome have a genetic basis for the renal disease and a poor outcome. Mutations of the following genes are responsible for the majority of cases of congenital and infantile nephrotic syndrome: NPHS1, which encodes nephrin (a key component of the podocyte slit diaphragm) and is responsible for the Finnish-type congenital nephrotic syndrome. NPHS2, which encodes podocin (a protein that interacts with nephrin at the slit diaphragm) and is responsible for familial focal segmental glomerulosclerosis. WT1, which encodes the transcription tumor suppressor (a protein involved in kidney and gonad development) and is responsible for the Denys-Drash syndrome. LAMB2, which encodes laminin beta 2 (a component of the glomerular basement membrane) and is responsible for the Pierson syndrome. PLCE1 gene, which encodes phospholipase C epsilon, is responsible for the early onset of isolated diffuse mesangial sclerosis. Other etiologies of congenital or infantile nephrotic syndrome include secondary causes such as infections (eg, syphilis or toxoplasmosis), toxins such as mercury exposure, and genetic disorders. Because most cases of congenital and infantile nephrotic syndrome are caused by genetic mutations and fail to respond to immunosuppressive therapy, we suggest genetic screening be performed before starting such treatment.
we send questionnaires to the physicians of related medical society in Japan as a field survey. In this presentation, I show the latest result of these studies with the findings of the candidate genes for RHUC3 by the next-generation sequencers.While RHUC is relatively rare in general clinics, gout and hyperuricemia which show elevated SUA are common diseases and the numbers of patients are still increasing. From the function-based genetic analysis, we previously reported that ABCG2/BCRP is the major causative gene of gout and hyperuricemia through the findings that ABCG2 is a high-capacity urate exporter and that its common dysfunctional variants strongly associate with the risk of gout/hyperuricemia (Odds ratio is more than 3.0 (25.8 at maximum)). Interestingly, the common variants of ABCG2, non-functional Q126X and half-functional Q141K, are independent genetics risks and therefore enable us to estimate the level of ABCG2 function. Subsequent study revealed that ABCG2 dysfunction associates with the young-onset gout. Furthermore, we observed paradoxically findings that dysfunctional functions of urate exporter ABCG2 increase the urinary excretion of urate. Together with high ABCG2 expression in extra-renal tissues and experiments with Abcg2 knockout mice, these results suggest that common dysfunctional variants of ABCG2 decrease extra-renal urate excretion, especially in intestines, and cause hyperuricemia. This novel mechanism would have been mistaken for urate “overproduction.” Thus, we proposes a novel classification of gout/hyperuricemia that “overproduction type” in the current concept of hyperuricemia should be renamed “renal overload type,” which is caused by two different mechanisms, “extra-renal urate underexcretion” and genuine “urate overproduction.”Our approaches on urate transporters will provide a more accurate diagnosis and more effective treatment for RHUC, hyperuricemia and gout.

36
[Poster 1]
Abstracts
Development of anti-Rituximab antibodies in children with nephrotic syndrome
37
[Poster 2]
Abstracts
Yo Han Ahn1, Hee Gyung Kang1, 2, Jiwon M Lee1, Hyun Jin Choi1, Il-Soo Ha1, 3, Hae Il Cheong1, 2, 3
1. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, 2. Research Coordination Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea3. Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea
Rituximab is actively used as a rescue therapy for nephrotic syndrome (NS). The development of anti-drug antibodies, including anti-rituximab antibodies (ARA) and human anti-chimeric antibodies (HACA), has been reported with rituximab treatment in various diseases. Here, we report two pediatric patients with NS who developed ARA.
We report the development of HACA in two patients with NS who did not achieve B cell depletion after repeated administration of rituximab. This report suggests that additional studies are needed to determine the incidence of anti-rituximab antibodies in patients with NS and its clinical significance.
Rituximab was given as a rescue therapy for the two patients with steroid- dependent NS. Both patients had been treated with oral glucocorticosteroid, methylprednisolone, and calcineurin inhibitors, but experienced frequent relapses. With the rituximab treatment, the patients remained in remission for several months. After the B cell count recovered, the patients received a second course of rituximab and experienced a hypersensitivity reaction during the drug infusion. The CD19 cell counts rose despite treatment with rituximab. The ARA titers were monitored before and after treatment with rituximab, and development of ARA after the second course of rituximab was confirmed.
Background
Conclusions
Case
Successful Treatment for Short Stature in a Pubertal Kidney Transplant Recipient Using a Combination of Gonadotropin Releasing Hormone Analog and Growth Hormone
Hideki Matsumura1, Hyogo Nakakura1, Akihiko Shirasu1, Akira Ashida1, Motoshi Hattori2, Hiroshi Tamai1
1. Department of Pediatrics, Osaka Medical College2. Department of Pediatric Nephrology, Tokyo Women’s Medical University
Despite various developments in the management of CKD, including the use of recombinant human growth hormone (rhGH), achieving an adequate final body height remains a challenge for such children, especially around the time of puberty. Here, we report a pubertal kidney transplant recipient with short stature who was treated with a combination of gonadotropin releasing hormone analog (GnRHa) and rhGH.
Despite undergoing kidney transplantation at puberty, our patient’s height SDS was effectively improved using rhGH and GnRHa combination therapy. No adverse events have been observed, and her allograft function has remained good for five years after transplantation. Combination therapy with rhGH and GnRHa is safe and effective for treatment of short stature in pubertal kidney transplant recipients, and can be a good therapeutic option i .
We treated an 18-year-old girl with ESRD due to Senior-Loken syndrome with the features of familial juvenile nephronophthisis and retinitis pigmentosa. Peritoneal dialysis had been started at the age of 8 years. Her height standard deviation score (SDS) had deteriorated, becoming -3.5 at the age of 12 years 5 months, and we started rhGH therapy. Although her height velocity improved from 0.5 cm/year to 6 cm/year within one year after starting rhGH therapy, her height SDS did not improve. At the start of rhGH therapy, the patient has been at Tanner stage II; therefore we decided to add GnRHa therapy in order to prolong the pre-pubertal growth phase and obtain catch-up growth. At the age of 13 years 9 months, 4 months after initiation of the combined therapy, the patient underwent living-donor kidney transplantation. Immunosuppressive agents included cyclosporine, mycophenolate mofetil, and methylprednisolone. The methylprednisolone was reduced quickly to 0.3 mg/kg/day at 4 weeks after transplantation, and to 4 mg on alternate days from 4 months. At the age of 15 years 8 months, her height SDS had improved to -2.3, and we terminated the GnRHa therapy. At the age of 16 years 6 months, the patient had her first menstrual period, and as her height SDS had reached -2.1, we completed the rhGH therapy. Finally, by the age of 18 years, her height SDS had improved to -1.8. Since then, her transplanted kidney function has been stable and her menstruation cycles have been regular.
Background
Conclusion
Case report

38
[Poster 3]
Abstracts
Glomerulopathy with ANA, ANCA, and Antiphospholipid antibodies in Twins
39
[Poster 4]
Abstracts
Yo Han Ahn1, Jiwon M Lee1, Hyun Jin Choi1, Jae Il Shin2, Hyun Joo Jeong3, Hee Gyung Kang1, 4,Il-Soo Ha1, 5, Hae Il Cheong1, 4, 5, Yong Choi1, Kyoungbun Lee6, Kyul Chul Moon6
1. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea2. Department of Pediatrics, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, Korea3. Department of Pathology, Yonsei University College of Medicine, Seoul, Korea4. Research Coordination Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea5. Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea6. Department of Pathology, Seoul National University Hospital, Seoul, Korea
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies directed against nuclear components, antinuclear antibodies (ANAs). Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) are a group of uncommon diseases characterized by small-sized vessel vasculitis associated with ANCAs positivity. These two diseases are only rarely associated.
What is the diagnosis of case 1 characterized by ANA, MPO-ANCA, and anti-phospholipid antibodies positivity? 2. Does the case 2 have the same disease?3. What would be optimal treatments for the cases?
Case 1, a 15-year-old girl, was admitted for fever, cough, hemoptysis, and rapidly progressive glomerulonephritis. She was born on the first baby of monozygotic twins at gestational age of 32 weeks with birth weight of 2 kg. Laboratory findings revealed severe anemia (Hb 5.9 g/dL), thrombocytopenia (104,000/μℓ), and elevated serum creatinine (2.7 mg/dL). A renal biopsy disclosed focal crescentic and necrotizing glomerulonephritis with IgG, IgA, C3, and C1q deposits. MPO-ANCA was positive. Her ANA and anti-DNA antibody were positive with hypocomplementemia. Lupus anticoagulant, anti-cardiolipin antibody (IgG), and anti-β2-glycoprotein 1 antibodies (IgG and IgM) were also positive. After administration of methylprednisolone and cyclophosphamide, her complement and creatinine levels became normal, ANA and anti-DNA antibody converted negative whereas MPO-ANCA and antiphospholipid antibodies were still positive. Two months later, she was admitted for acute respiratory distress syndrome with the laboratory findings as follows: WBC 4,410/μℓ, platelet 61,000/μℓ, creatinine 1.72 mg/dL, C3 44 mg/dL, C4 6 mg/dL, anti-DNA Ab (-), ANA (-), and MPO-ANCA (+). Case 2, an older twin sister of case 1, was found to have asymptomatic proteinuria and microscopic hematuria at a school urinary screening. She had a history of idiopathic thrombocytopenic purpura due to anti-platelet antibody. Serum C3 and C4 levels were normal. ANA and anti-DNA antibody were negative, whereas MPO-ANCA and antiphospholipid antibodies were positive. A kidney biopsy revealed 3 glomeruli suggesting immune complex mediated glomerulonephritis with mesangial deposits of IgG and C3. A second renal biopsy performed 3 months after discontinuation of steroid treatment disclosed a focal endocapillary proliferation with segmental sclerosis without immune complex and complement deposits.
Background
Discussion points
Case
A case of intermittent hydronephrosis secondary to ureteropelvic junction obstruction
Masano Akamatsu1, Akira Ashida1, Hideki Matsumura1, Tomoki Aomatsu1,Hyogo Nakakura1, Akihiko Shirasu1, Kenji Shimada2, Hiroshi Tamai1
1. Department of Pediatrics, Osaka Medical College2. Department of Urology, Osaka Medical Center and Research Institute for Maternal and Child Hearth
Intermittent uretero-pelvic junction (UPJ) obstruction is usually acute and self-limiting. It causes distinct clinical symptoms that include severe episodic abdominal pain, nausea and vomiting associated with intermittent hydronephrosis due to rapid distension of the pelvis and stretching of the renal capsule. The diagnosis of intermittent hydronephrosis due to UPJ obstruction can be confirmed if hydronephrosis is present when the child is symptomatic, and resolves when the child is well. Therefore, definitive diagnosis of intermittent hydronephrosis can be difficult.
For diagnosis of intermittent hydronephrosis in patients with antenatal hydronephrosis, it is necessary to compare the size of the affected pelvis during attacks with that during convalescence, and to demonstrate aggravation of hydronephrosis in the acute phase. For this purpose, although ultrasonography is very useful, we believe that the timing of diagnostic studies is actually more important than the imaging modality employed.
We describe an 8-year-old boy who was referred to our hospital for management of persistent left-sided antenatal hydronephrosis (SFU grade I to II). The patient had normal renal function and no serological abnormalities. Ultrasonography demonstrated left-sided pelvic dilatation and no stones. At the age of two months, voiding cystourethrography had shown no vesicoureteral reflux. At the age of two years, diuretic renography with MAG3 had shown wash out delay from the affected side, and accelerated response to diuretics (Lasix®). Therefore, follow-up had been continued using serial ultrasonography, urinalysis, and blood examinations. Although renal function had been stable, renal sonography had shown gradually increasing hydronephrosis (SFU grade II or III) from about five years of age, and the patient had complained of left-sided intermittent abdominal pain since about six years of age. Periodic renal sonography had shown no apparent change in the degree of hydronephrosis during period when abdominal pain was absent. At the age of eight, the patient developed severe left-sided abdominal pain while playing soccer, and brought to our hospital. At that time, the left pelvis was twice as large as that during convalescence. Therefore, the patient was finally diagnosed as having intermittent hydronephrosis with severe pelvic dilatation, and surgical treatment is now being planned.
Background
Conclusion
Patient presentation

40
[Poster 5]
Abstracts
A case of lupus associated hemophagocytic lymphohistiocytosis
41
[Poster 6]
Abstracts
Jung Min Lee1, Jihei Cha1, Sun Hee Sung2, Seung Joo Lee1
1. Department of Pediatrics, Ewha Womans University School of Medicine2. Department of Pathology, Ewha Womans University School of Medicine
Hemophagocytic lymphohistiocytosis (HLH) is a rare but potentially lethal disease. HLH syndrome is classified as genetic (primary) HLH or acquired (secondary) HLH. Secondary HLH includes infection-associated hemophagocytic syndrome (IAHS), autoimmune-associated hemophagocytic syndrome (AAHS), malignancy-associated HLH. Here. we report a rare case of HLH developed in a patient with lupus nephritis.
The proper treatment of lupus associated HLH is not well established. It consists of intensive immunosuppressants, IVIG and rituximab but sometimes is not successful. HLH 2004 protocol was originally developed for primary HLH and has been used in secondary HLH, This is the first successful lupus associated HLH to be treated with HLH 2004 protocol.
A 14-year-old girl presented with fever, edema and weight gain (7kg in 2 months). She looked pale and edematous. Laboratory values were as follows: hemoglobin 7.7 g/dl, hematocrit 23.6%, WBC 4800/mm2, Platelet 139k/mm2, BUN/Serum creatinine 19/0.9 mg/dl, total protein/albumin 6.9/2.9 g/dl, cholesterol 150 mg/dl. Urine protein (3+), occult blood (4+), RBC many/HPF, 24 hour urine protein 1.6 g/m2/day. C3 23.4 mg/dl, C4 3.3 mg/dl, Antinuclear antibody (3+), Anti-dsDNA 1986.5 IU, Anti SSA(-), Anti-SSB(-), Renal biopsy revealed lupus nephritis ISN/RPS stage IV-GA. She was treated with intensive induction therapy with intravenous pulse methylprednisolone (1 g/day) for 3 days followed by mycophenolate mophetil (MMF, 480 mg/m2) and rituximab (500 mg). Since switching to maintenance therapy with oral deflazacort (72mg/day) and MMF, her lupus activity was slightly wax and waned. 31 months later, fever with mild upper respiratory symptoms developed but high fever (39-40°C) persisted for 10 days and was followed by severe generalized weakness. She was re-admitted underr the impression of lupus flare. Laboratory findings were as follows: hemoglobin 9.6 g/dl, hematocrit 27.8%, WBC 810/mm2 (ANC 520), Platelet 46k, BUN/Serum creatinine 10/ 0.7 mg/dl, total protein/albumin 6.3/4.2 g/dl, triglyceride 154 mg/dl and Ferritin 9746 mg/dl. Urine protein (2+), C3 33.5 mg/dl, C4 11.2mg/dl. Antinuclear antibody (4+), Anti-dsDNA 139 IU, Anti RNP(+), Anti-sm(+), Bone marrow biopsy showed increased hemophagocytosis which was compatible with HLH. Under the diagnosis of lupus associated HLH, she was treated with HLH 2004 protocol (intravenous VP-16 (etoposide) and oral dexamethasone, cyclosporine) and was improved .
Introduction
Discussion
Case
Polypoid cystitis mimicking bladder tumor in a nephrotic patient treated with cyclosporine
Tomohiro Inoguchi1, Kazuya Matsumura1, Makoto Yoshida2, Yoko Ohwada3,Eiji Kikuchi4, Yasuaki Kobayashi2, Kaori Kameyama5, Midori Awazu1
1. Department of Pediatrics, School of Medicine, Keio University2. Department of Pediatrics, Ashikaga Red Cross Hospital3. Department of Pediatrics, Dokkyo Medical University School of Medicine4. Department of Urology, School of Medicine, Keio University5. Division of Diagnostic Pathology, Keio University Hospital
We report a patient with frequent relapsing nephrotic syndrome (NS) who was treated with cyclosporine A (CyA) for a total of approximately 4 years and developed polypoid cystitis mimicking bladder tumor.
Urothelial carcinoma and papillomatous lesions have been associated with CyA, which is due to viral infection resulting from immunosuppression. Although viral infection was not documented in our patient, CyA is likely to be involved in the development of polypoid cyctitis.
The patient is a 20-year-old male with frequently relapsing NS. He developed NS at age 12. Renal and bladder ultrasonography was normal then. Because of frequent relapses, a renal biopsy was performed at age 14, which showed minimal change disease. He was treated with CyA for 2 years with trough levels varying from 41 to 105 ng/mL. At age 19, because of 8 relapses after the discontinuation of CyA, he was restarted on CyA. Twenty months after the readministration of CyA, he had several episodes of gross hematuria with no pain or fever. He denied edema, oliguria, or a recent history of upper respiratory infection. Physical examination was unremarkable. Urinalysis showed protein +/-, blood 2+, RBC >100/HPF, WBC <2/HPF, and no casts. Urine ß2 microglobulin and NAG per creatinine were 168 μg/L and 2.1, respectively. Blood test showed urea nitrogen 11.1, creatinine 0.83 mg/dL, normal electrolytes, total protein 7.4, albumin 3.9 g/dL, IgG 1207, IgA 166, IgM 160, C3 100, C4 26 mg/dL, and ASO 125 IU/mL. IgM antibodies to CMV and EBV VCA were negative. Urine culture grew no bacteria, and no virus was isolated from the urine. Ultrasonography revealed normal kidneys and a mass in the bladder, 7 mm in diameter. Urine cytology was benign. Lymphocyte subset showed slightly decreased CD4/CD8 ratio of 0.76. Lymphocyte blastoid transformation test revealed normal T cell reproductive activity. Magnetic resonance imaging confirmed a mass suggestive of inverted papilloma. The patient was referred to urology, and cystoscopy revealed a pedunculated tumor on the anterior wall of the bladder. He had no history of bladder catheterization or exposure to organic solvent. He claimed use of condom for casual sexual contact. Transurethral resection of bladder tumor was performed. The specimen was polypoid mass showing mildly hyperplastic urothelium overlying an edematous congested stroma. There was mild infiltration of inflammatory cells. Atypia of the epithelial cells was absent. Real time PCR for 36 types of human papilloma virus on the specimen was negative.
Introduction
Conclusion
Case

Nephronophthisis with the mutation of NPHP4 gene in three siblings A case with steroid resistant nephrotic syndrome successfully treated with carperitide
Min Jee Kim1, Jee Yeon Han1, Hye Ryun Yeh1, Eun Koo Kang1, Yoo Mi Choi3,Kyoung Hee Han4, Joo Hoon Lee1, Young Mi Cho2, Young Seo Park1, Hae II Cheong5
1. Department of Pediatrics, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea2. Department of Pathology, Asan Medical Center, Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea3. Department of Pediatrics, Kyung Hee University Hospital, Seoul, Korea4. Department of Pediatrics, Jeju National University Hospital, Jeju, Korea5. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Korea
Nephronophthisis(NPHP) is an autosomal recessive tubulointerstitial nephropathy and one of the most frequent genetic disorders causing end-stage renal disease (ESRD), Eleven different genes have been identified by positional cloning(NPHP1-11, NPHP1L) for NPHP. Among them, NPHP4 mutation was identified on chromosome 1p36, which encoded nephrocystin-4. Mutation in NPHP4 accounts for about 2% of NPHP cases and can results in isolated NPHP, NPHP with oculomotor apraxia or Senior-Löken syndrome. We present three siblings with nephronophthisis, who have mutation in NPHP4 gene. Case 1An eight-years-old boy was transferred to our hospital due to azotemia. Elevated creatinine level was found incidentally on laboratory tests due to chronic fatigue. He had pale appearance and short stature (under 3 percentile) on physical examination. He was diagnosed with retinal dysplasia at 3-years of age. He had two elder brothers, who had progressed to ESRD. His mother’s sister also had ESRDs. The laboratory findings were as follows: Hb 8.0 g/dL, WBC 6,900 x 103/uL, platelet 208K/uL, serum BUN 157mg/dL, creatinine 10.8 mg/dL, protein 7.0 g/dL, albumin 3.6 g/dL, IgG/A/M 1600/126/81.6 mg/dL, C3/C4 86.6/19.9 mg/dL, HBs-Ag/Ab (-/-), HCV Ab (-), anti-nuclear antibody: reactive nuclear dot, anti-ds-DNA 3.3 IU/mL, urinalysis: S.G. 1.015, pH 5.0, albumin -, glucose -, occult blood -, RBC 0-2/HPF, WBC many/HPF, 24-hour urine protein 70.0 mg/m2/day. Histopathological findings on renal biopsy showed global glomerulosclerosis, increased mesangial matrix and cells, marked tubular atrophy and interstitial chronic inflammatory cell infiltration. He was already progressed to end-stage renal disease(ESRD) and started dialysis at that time of transfer. Case 2A twelve-years-old boy, who is a second elder brother of above case, also had severe azotemia andunderwent laboratory tests and renal biopsy. He was also diagnosed with cataract at 5-years of age. The results of laboratory tests and renal biopsy were similar to his younger brother. He was progressed to ESRD and needed dialysis eventually at 14-years of old. Case 3A fifteen-years-old boy, who is the eldest brother of above cases, also had similar results of laboratory tests and renal biopsy. He had no ocular abnormality. He was also progressed to ESRD at the age of 20-years old. Based on the clinical and pathological finding, the diagnosis of nephronophthisis was suspected and the mutation of NPHP4 was identified in all three siblings, finally. We report these three sibiling, as the cases of nephronophthisis with mutation of NPHP4 gene, which progressed to ESRD.
A 2-year-old boy was admitted to a regional hospital with complaints of body weight gain and an eyelid edema. Dipstick analysis of his urine revealed 4+ protein, - blood. Serum albumin was 0.9 g/dL, serum creatinine was 0.32 mg/dL, and complement level was normal. He was diagnosed as having idiopathic nephrotic syndrome. Oral prednisolone 2 mg/kg/day by three divided dose was initiated, however, his urinalysis showed no improvement and serum albumin was 0.5 g/dL after 4-week prednisolone administration. Then he was diagnosed as steroid-resistant nephrotic syndrome and transferred to our hospital. Kidney biopsy revealed focal segmental glomerulosclerosis (tip variant). Because his diuretic was hardly obtained even by administration of large doses of furosemide, he was suffered from deterioration of renal function, hypertension, edema, and posterior reversible encephalopathy syndrome (PRES). The combination of infused carperitide and furosemide promoted diuretic and improved his general condition.After a total ten courses of intravenous methylprednisolone according to Mendoza’s protocol with oral cyclosporine and mizoribine, his renal function recovered but he still showed nephrotic status. Then oral cyclophosphamide was added to the treatment of cyclosporine and mizoribine. After 3-month administration of oral cyclophosphamide he achieved complete remission and maintained remission until the discontinuation of treatments. He experienced steroid dependent relapses after stopping of oral cyclosporine and mizoribine. He received oral cyclosporine again and maintains complete remission.Carperitide (hANP) is one of a cardiac hormone secreted from the atrium, which ① extends afferent arteries and deflates efferent arteries with increasing glomerular filtration rate, ② increases medullary blood flow and inhibits the reabsorption of sodium and water by reducing the osmotic gradient, ③ inhibits the secretion of ADH, and ④ suppresses RAAS. And it exerts a diuretic effect depending on the fluid balance. Carperitide showed marked response in fluid overload state in this case.
42
[Poster 7]
Abstracts
43
[Poster 8]
Abstracts
Yuko Shima, Koichi Nakanishi, Taketsugu Hama, Masashi Sato, Hironobu Mukaiyama, Hiroko Togawa, Norishige Yoshikawa
Department of Pediatrics, Wakayama Medical University

A novel CLCNKB gene mutation: severe hypomagnesemia and hypocalcemia A case of TSC2/PKD1 contiguous gene deletion syndrome
Sang Taek Lee1, Heeyeon Cho1, Young Seo Park2, Hae Il Cheong3 Yusuke Kumagai1, Hiroaki Ueda1, Ichiro Kuki2, Shin Okazaki2, Rika Fujimaru1
1. Department of Pediatrics, Samsung Medical Center, Seoul, Republic of Korea2. Department of Pediatrics, Asan Medical Center, Seoul, Republic of Korea3. Department of Pediatrics, Seoul National University Children’s Hospital, Seoul, Republic of Korea
1. Department of Pediatrics, Osaka City General Hospital, Osaka, Japan2. Department of Pediatric neurology, Osaka City General Hospital, Osaka, Japan
Bartter syndrome (BS) is an autosomal recessive inherited renal tubular disorder characterized with low or normal blood pressure, hypokalemic metabolic alkalosis and hyperreninemic hyperaldosteronism and classified into 5 genotypes according to underlying mutant genes. Type III BS is caused by loss-of-function mutations in the CLCNKB gene encoding for basolateral ClC-Kb and the most common genotype in Korean patients with BS. Type III BS manifests with hypokalemia, hypercalciuria, or nephrocalcinosis. CLCNKB gene mutation also cause Gitelman syndrome (GS) which is distinguished from other hypokalemic tubulopathies by the presence of both hypomagnesemia and hypocalciuria. We describe the pediatric patient with severe hypomagnesemia, hypocalcemia with hypercalciuria and hypokalemic periodic paralysis caused by CLCNKB gene mutation difficult to categorise as type III BS or GS. A 10-year-old boy was referred to our consultation due to persistent hypokalemia and hypomagnesemia. He had the birth history of maternal polyhydramnios and normal full term spontaneous delivery. He had the past medical history of muscle cramps and rigidity six years ago. At that time, the laboratory findings in other hospital showed hypokalemia, hypocalcemia with mild hypercalciuria, severe hypomagnesemia, and nephrocalcinosis. At that time, the levels of renin and aldosterone were elevated. He had been treated with potassium and magnesium oral supplements, amiloride, and spironolactone for 6 years. He showed persistent hypomagnesemia in spite of magnesium oral supplement and was given intermittent magnesium intravenous supplement. His height and body weight were all more than the 50th percentile and physical examination was normal. After referral, the investigations showed normal renal function, low serum potassium and magnesium, elevated urinary potassium and hypercalciuria. Kidney ultrasonography showed bilateral medullary nephrocalcinosis or renal stone. These findings were not fully compatible with any kind of tubulopathy, however, the presumed diagnosis includes type III BS, type V BS, GS, or familial hypomagnesemia with hypercalciuria and nephrocalcinosis. The genetic study revealed no mutation in CLDN16 gene. Genetic tests for SLC12A3 gene mutation present in GS came negative. Genetic test for CASR gene present in type V BS was negative. CLCNKB gene mutation analysis revealed a heterozygous c.139G>A in exon 13 [p.Gly(GGG)465Glu(GAG)]. The patient is now being treated with potassium and magnesium oral supplements, and indomethacin without muscle weakness or muscle cramps. Even though this change in CLCNKB gene is not known mutation, the clinical findings and in silico prediction are suggestive of causative mutation.
44
[Poster 9]
Abstracts
45
[Poster 10]
Abstracts
Tuberous sclerosis complex (TSC) is caused by mutations in either of two genes, TSC1 and TSC2. Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in either PKD1 or PKD2. TSC is characterized by developing benign hamartomas in various organs. Although renal cysts develop as a kidney complication of TSC, the onset of the disorder commonly occurs in the second decade. Clinically, a small percentage of TSC patents develop severe infantile polycystic kidney disease, which is believed to be caused by deletion of the contiguous TSC2 and PKD1 gene on chromosome 16. We report a TSC patient, who presented with multiple large bilateral renal cysts by fetal ultrasound.
PKDTS had been identified in our patient with TSC and early-onset severe ADPKD by array-CGH. Early detection of this syndrome may help to prevent complications such as renal hypertension and intracranial aneurysms rupture.
Our patient, a boy, was born in our hospital. In fetal life, ultrasonography and magnetic resonance imaging (MRI) demonstrated cardiac tumors, nodular lesions around the lateral ventricle and multiple renal cysts. In family history, his mother was revealed several kidney cysts, but with normal function. Physical examination at birth was remarkable for bilateral nephromegaly. Abdominal ultrasonography showed bilaterally enlarged kidneys with multiple cysts resembling those seen in ADPKD. Echocardiography showed multiple cardiac rhabdomyomas. A brain MRI revealed typical signs of TSC, such as multiple subependymal nodules and cortical tubers. He fulfilled the definitive criteria for TSC in infancy, and at the same had been diagnosed as having severe polycystic kidney disease. At 8 months old, he presented with recurrent episodes of convulsions. Electroencephalography showed hypsarrhythmia. A diagnosis of West syndrome with TSC was made. About 2 years later, he developed blood pressure elevation requiring antihypertensive treatment. At the age of 3 years, an abdomen MRI showed the increase in both the number and size of renal cysts, but the extrarenal signs for ADPKD such as liver cysts had not been found. His renal function was still normal. Our findings were highly suspicious of TSC2/PKD1 contiguous gene deletion syndrome (PKDTS). Array comparative genomic hybridization (array-CGH) for the 16p13.3 locus detected reduction by half of the genomic copy number of TSC2 and PKD1 and large heterozygous deletions. Recently, a mammalian target of rapamycin inhibitor was started as an investigational new drug, because his seizures were not well controlled by several different types of antiepileptic agents.
Objective
Conclusions
Case report

46
[Poster 11]
Abstracts
Is a History of Previous Abdominal Surgery Contraindication for Peritoneal Dialysis?
47
[Poster 12]
Abstracts
Hee Sun Baek1, Seung Hyun Cho1, Cheol Woo Ko2, Min Hyun Cho1
1. Department of Pediatrics, Kyungpook National University School of Medicine, Daegu, Korea2. Department of Radiology, Kyungpook National University School of Medicine, Daegu, Korea
An 18-year-old girl undergoing hemodialysis (HD) with end-stage renal disease (ESRD) was admitted for insertion of PD catheter. When she was 3-year-old (15 years ago), she was diagnosed with chronic renal failure caused by neurogenic bladder and reflux nephropathy. A percutaneous vesicostomy was performed. Nine years ago she received a renal transplant from a living-related donor, her father. At that time, renal transplantation was performed via transperitoneal approach. The artery and vein of this graft were anastomosed to the abdominal aorta and inferior vena cava, respectively. The right native kidney was removed. However, seven years after kidney transplantation (when she was 16-year-old), HD was inevitably restarted because of loss of graft function with chronic allograft dysfunction. It was actually thought that PD would be a contraindication in this patient due to her history of a previous abdominal surgery (renal transplantation via transperitoneal approach). When she became a university student (18-year-old), it was thought that PD would be more suitable to her life style than HD, but more information about the status of peritoneal cavity such as the extent of peritoneal adhesion was necessary for PD catheter insertion. At that time, she had severe growth retardation (Her body weight was about 31 kilogram).
1. Do you think that the history of previous abdominal surgery is contraindication of PD? 2. If you regard the history of previous abdominal surgery as the contraindication of PD, how do you perform dialysis in children with the history of abdominal surgery, especially very small children?
Case report
Point of Discussion
A case of “not otherwise specified” variant of focal segmental glomerulosclerosis accompanied with crescent formation
Norio Omori1, Ryugo Hiramoto1, Tomohiko Yamamura1, Shinsuke Matsumoto1, Hironobu Eguchi1, Bunshiro Akikusa2
1. Department of Pediatrics, Matsudo City Hospital Children's Medical Center2. Department of Pathology, Matsudo City Hospital
Primary focal segmental glomerulosclerosis (FSGS) has recently been divided into five subtypes based on the Columbia classification. We report a case of steroid-resistant nephrotic syndrome presenting “not otherwise specified (NOS)” FSGS accompanied with crescent formation.
Large sized crescents accounted for approximately 10% out of 43 glomeruli. The etiology of crescent formation, however, is unclear. Although the Columbia classification of histological variants of FSGS has been constituted to unify the complexity of diagnosing FSGS, it has been controversial regarding prognostic and therapeutic utility. The further studies are inevitable to clarify the clinical features and outcome of FSGS variants of pediatric patients on the basis of the Columbia classification.
In this case, combination therapy of CyA, MPT and PSL has been effective. Because the cause of crescent formation is not clearly understood and the prognosis of FSGS NOS variant is uncertain, close and careful follow up towards this patient is essential.
An otherwise healthy 15-year-old male with proteinuria, hematuria and edema was diagnosed with nephrotic syndrome in December, 2010. Initial data were as follows: total protein 4.1g/dL; albumin 1.6g/dL; total cholesterol 392 mg/dL; serum creatinine 0.54mg/dL; spot urine protein 4g/dL; urine RBC 10-19/HPF. He responded well to initial prednisolone (PSL) therapy of 80mg/day with complete resolution of proteinuria on the twelfth day. However, when the dose of PSL was tapered to 10mg/day, the first relapse occurred. The dose of PSL then increased up to 60 mg/day. This time, he did not respond to PSL treatment for four weeks, and was diagnosed with steroid-resistant nephrotic syndrome in April, 2011. The first renal biopsy revealed minor glomerular abnormalities on light microscope and pauci-immune on immunofluorescence (IF), which lead to the diagnosis of minimal change nephrotic syndrome. Combination therapy of cyclosporine (CyA), methylprednisolone pulses (MPT), and oral PSL (1mg/kg/every other day) was continued. After five series of MPT, complete remission was finally obtained. While PSL therapy was discontinued one year after starting the combination therapy, CyA was continued to keep complete remission. The second renal biopsy performed two years later indicated FSGS accompanied with crescent formation (2 fibro-cellular crescents and 2 fibrous crescents). Tip lesions and perihilar sclerosis without foam cells were identified. Because perihilar sclerosis was not more than 50% of segmental lesions, NOS variant was confirmed. There were no episodes of gross hematuria and renal impairment throughout the clinical course. Both PR3-ANCA and MPO-ANCA were negative.
Introduction
Discussion
Conclusion
Case report

Dense deposit disease with minor urinary abnormalities detectedby school urinary screening
Ji Young Oh1, Hae Il Cheong2, Hyeon Joo Jeong3, Ji Hong Kim1, Jae Il Shin1, Se Jin Park4
1. Department of Pediatrics, Severance Children’s Hospital, Yonsei University College of Medicine, Seoul, Korea2. Department of Pediatrics, Seoul National University Children's Hospital, Seoul, Korea.3. Department of Pathology, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea4. Department of Pediatrics, Ajou University School of Medicine, Suwon, Korea
Dense deposit disease (DDD) is a rare glomerulonephritis that is clinically characterized by hematuria, proteinuria, acute nephritic syndrome, or nephrotic syndrome. DDD is also pathologically described as an intense deposition of complement 3 (C3) in a ribbon-like pattern along the glomerular basement membranes, tubular basement membrane, and the wall of Bowman’s capsule. Dysregulation of complement alternative pathway is one of the important pathomechanisms in DDD. It is known that about 50% of the patients with DDD proceed to end-stage renal failure within 10 years of onset, but there is no universally effective treatment for children with DDD.We herein report an 11-year-old girl who was transferred to our hospital for evaluation of hematuria and proteinuria discovered by school urinary screening. When she was found to be positive for antinuclear antibody and have low C3 levels, lupus nephritis was suspected at a regional hospital. On physical examination, her blood pressure was within normal limit and she had no generalized edema. Her blood urea nitrogen and serum creatinine were 10.0 mg/dL and 0.57 mg/dL, respectively. However, the levels of C3 and CH50 were decreased to 14 mg/dL and 11.5 mg/dL. Urinary laboratory findings disclosed persistent microscopic hematuria and proteinuria. In electron microscopy, histopathological findings of the renal biopsy specimen confirmed the ribbon-like thickenings of the glomerular basement membranes by intramembranous electron-dense deposits. An analysis of the complement alternative pathway revealed normal levels of complement factor H (CFH), together with a focus on genetic variations in CFH gene cluster and CFH autoantibodies in the patient. We identified no homolozygous deletions in CFH-related genes 1 and 3 (delCFHR1-CFHR3). CFH autoantibodies were not detected in the patient. At 3-year follow-up, laboratory findings were not specific except persistent microscopic hematuria and proteinuria, and low C3 levels.
48
Poster Only[Poster 13]
Abstracts
Recombinant Human Erythropoietin Therapy for a Jehovah’s Witness Child with Severe Anemia due to Hemolytic Uremic Syndrome
Hemolytic uremic syndrome (HUS) is a common cause of acute renal injury in children. It is characterized by microangiopathic hemolytic anemia, thrombocytopenia and acute renal injury. Because of hemolysis of RBC, the patient with HUS can become anemic status, rapidly and profoundly. Therefore, when the patient’s Hb level is lower than 6g/dL, packed RBC transfusion should be considered to avoid cardiac or pulmonary complications.Most of the Jehovah’s Witnesses refuse whole blood or blood components transfusion even in the face of life-threatening medical conditions according to their religious conviction.Erythropoietin is a glycopeptide that enhancing endogenous erythropoiesis by regulating proliferation, differentiation and maturation of RBC precursors in the bone marrow. Since recombinant human erythropoietin (rHuEPO) became available, several cases have been reported in which rHuEPO was successfully administered to patients refusing blood transfusion. But the optimal dose and duration of treatment with rHuEPO is not established.We present a case of a 2-year old Jehovah’s Witness boy with diarrhea-associated HUS. At the time of admission, he presented with fever, abdominal pain and bloody diarrhea. The laboratory examination revealed hemolytic anemia, thrombocytopenia and increased serum BUN and creatinine level. After all, the Hb level decreased to 3.6g/dL, and oliguric status persisted. We decided to start renal replacement therapy and packed RBC transfusion, but his parents refused transfusion owing to their belief. Thus, we treated him with rHuEPO (300IU/kg/day), in the first 5 consecutive days, daily folic acid, vitamin B12 and intravenous iron. The Hb level increased steadily, to 7.4g/dL after 10 days of treatment and the renal function was improved without any complications. We report the first case of successful rHuEPO treatment for a Jehovah’s Witness child with severe anemia due to HUS.
49
Poster Only[Poster 14]
Abstracts
What is the appropriate treatment for the patient only with minor urinary abnormalities?What do you think will be the prognosis of the patient with DDD in this case?
Points of discussion
Da Eun Woo, Sang Su Lee, Jae Min Lee, Yong Hoon Park Department of Pediatrics, Yeungnam University College of Medicine, Daegu, Korea

50
Poster Only[Poster 15]
Abstracts
Full-house pattern of glomerular immune deposits in patientwho does not fulfilling SLE criteria
Sun Ah Choi, Hye Won Park Department of Pediatrics, Seoul National University Bundang Hospital, Seongnam, Korea
Immune complex-mediated glomerulopathy can be caused by autoimmune diseases, most commonly with systemic lupus erythematosus, as well as other conditions like posthepatic cirrhosis, diabetic nephropathy, and chronic infections. We report a case of immune complex disease in the mesangial and subendothelial , subepithelial deposits with IgG, IgA, IgM, C3, and C1q in a patient with proteinuria, thrombocytopenia, and a positive lupus anticoagulant test. In 18 months follow up, without taking immunosuppressant, he doesn’t show any clinical manifestations of lupus.
X-linked Alport syndrome accounts for 85% of all cases and arises from mutations in the COL4A5 gene. Although the clinical course and glmomerular basement membeane (GBM) ultrastructural changes in females with X-linked Alport syndrome can sometimes mimic thin basement membrane nephropathy (TBMN), they tend to be more progressive than usually seen in TBMN. The possibility of autosomal recessive Alport syndrome is often overlooked, but its identification is also important. Autosomal recessive inheritance may be suspected where a female individual develops renal failure, especially at a young age and no pathogenic variant in COL4A5 gene is identified. Here, we report two young girls with X-linked and autosomal recessive Alport syndrome, respectively, initially diagnosed as TBMN.
In the literature reviews, patients with full house immunofluorescence and tubuloreticular inclusions are likely to have positive serology and clinical symptoms of SLE during follow up. In our case, patient had 3 of the criteria for SLE thus failing to meet the diagnosis based on the American College of Rheumatology definition. Without taking any immunosuppressant, although the pathologic findings suggest lupus nephropathy, the patient showed no flare up, nor clinical manifestations related to lupus. In the absence of SLE, other causes of immune complex mediated glomerulonephritis have been excluded. In 18 months follow up, this patient has seronegative and free of extrarenal symptoms of lupus but he needs regular follow up for the delayed onset of frank SLE.
An 8-year-old girl was admitted for persistent gross hematuria since infancy. She has had recurrent gross hematuria since 15 months old and first visited our hospital when she was 21 months old. At that time, imaging studies were not specific except mild hydronephrosis of left kidney on sonography. Mild proteinuria and hematuria were detected (protein to creatinine ratio 0.68, RBC > 60/HPF). Blood tests were within normal range. After 1 month kidney biopsy was performed because of persistent gross hematuria. Biopsy findings were consistent with TBMN, showing diffuse thinning of GBMs (average 145 nm) and focal effacement of foot processes. She was discharged with oral corticosteroids and lost to follow-up for 4 years. At this time, the patient revisited the clinic complaining of still existing gross hematuria. Laboratory tests were performed; leukocyte count, 14,200/uL; Hb, 9.6 mg/dL; platelets, 405,000/uL; BUN, 37.1 mg/dL; creatinine, 1.36 mg/dL; protein, 7.2 g/dL; albumin, 4.4 g/dL; cholesterol, 181 mg/dL. The IgA, C3, C4, anti-dsDNA antibody, HBs antigen, and anti-neutrophil cytoplasmic antibody were all not specific. Urinalysis showed mild proteinuria and hematuria (protein 1+, RBC > 61/HPF). The 24-hour urine study showed proteinuria (267 mg/day (10.61 mg/m2/hr)) and decreased GFR (56.8 mL/m2/min). Kidney re-biopsy showed the findings of Alport syndrome with mild chronic tubulointerstitial changes. There were some sclerotic glomeruli, focal tubular atrophy, and patchy areas of mild interstitial fibrosis. Segmental multilamellation of GBM and focal effacement of foot process were seen on electron microscopy. Gene study of whole sequencing of COL4A5 showed no pathogenic mutation.
1. How can we interpret the results of gene study in two cases? 2. How genetic mutations may determine clinical features of Alport syndrome and TBMN?3. What is the association of scrub typhus with renal manifestations in case 1?4. Can routine genetic evaluation be warranted in pediatric patients with TBMN?
A 14-year-old boy presented with a complaint of both eyelid swelling and foamy urine. He was previously healthy patient without remarkable past medical history. Family history related to renal diseases was denied. Physical examinations showed no remarkable findings except both eyelid swelling and pretibial pitting edema. Initial blood pressure was 137/63mmHg. Laboratory investigations revealed thrombocytopenia (platelet 84,000/μL), hypoalbuminemia (serum albumin 2.0g/dL) and proteinuria with microscopic hematuria (spot urine protein/creatinine ratio 8.98, urine alb 3+, RBC 30-49/HPF, dRBC 90%). Mild left pleural effusion was noted in simple chest radiography. In renal ultrasonography, the echogenicity of renal parenchyme was slightly increased without focal lesion. Renal biopsy revealed overall hypercellularity of glomerulus involving mesangial cell proliferation. There were diffuse thickening of capillary wall and tram track appearance of capillary loop suggesting membranoproliferative glomerulonephritis. In electron microscopy, there were no cytoplasmic tubuloreticular inclusions. Large subepithelial, mesangial, and moderate subendothelial deposits were seen in electron and immunofluorescence microscopy. Immunofluorescence microscopy also showed full-house pattern of IgG, IgA, IgM, C3 and C1q in glomerular immune deposits. As these pathologic findings were suggestive of lupus as an underlying cause, we evaluated for the clinical and immunological evidence of SLE. Anti-nuclear antibody was negative, whereas anti-cardiolipin IgG and lupus anticoagulant were positive. Positive anti-ds DNA and low C3, C4 level were detected when proteinuria was detected. Serologic evaluation was negative for hepatitis A, B, C and syphilis. There was no evidence of chronic and systemic infection which could be expected as a cause of immune complex mediated nephropathy. Proteinuria, thrombocytopenia and positive lupus anticoagulant were recognized as positive findings according to the American College of Rheumatology definition. He was treated with oral prednisolone (1mg/kg per day) and cyclosporine (3mg/kg per day). As proteinuria gradually improved, the prednisolone dosage was tapered every other week and discontinued after 7 months. The cyclosporine was tapered off after 16 months. In one year follow up, anti-nuclear antibody and anti-Smith antibody were still negative. In 18 months follow up, without taking any immunosuppressant, proteinuria has been resolved and complement levels are lower margin of normal range. Although the pathologic findings suggest lupus nephropathy, clinical manifestations related to lupus has not been reported. Serial monitoring of ANA and clinical symptoms of SLE are necessary as appearance of autoantibodies and/or clinical symptoms may be delayed for several years in those who have full house nephropathy.
A 7-year- old girl was admitted for evaluation of recurrent coke-colored urine. She had been treated with scrub typus and gross hematuria at other hospital 1 month ago. She had no family history of any kind of kidney disease. The vital sign and physical examination were not specific. The results of serologic tests revealed: leukocytes, 6,800/uL; Hb, 10.7 mg/dL; platelets, 363,000/uL; c-reactive protein, < 0.1 mg/L; BUN, 20.8 mg/dL; creatinine, 0.41 mg/dL; protein, 7.6 g/dL; albumin, 4.3 g/dL; cholesterol, 143 mg/dL; tsutsugamushi antibody IgG, positive. The C3, C4, anti-dsDNA antibody, and anti-neutrophil cytoplasmic antibody were all normal. Urinalysis showed only microscopic hematuria (RBC 30-60/HPF) and urine culture result was negative. The 24-hour urine collection study showed no specific findings (protein 3.94 mg/m2/hr, calcium 2.7 mg/kg/day, urate 12.3 mg/kg/day). Abdominal sonography showed equivocally increased cortical echogenicity in upper pole of right kidney, but DMSA scan was non-specific. Gross hematuria was improved during admission; however, microscopic hematuria persisted. Isolated hematuria was diagnosed and she was discharged. About a month later the patient was re-admitted for kidney biopsy due to prolonged intermittent gross hematuria. Renal biopsy findings were of TBMN. The GBMs were thinned ranging from 125 nm to 175 nm, averaging 150 nm. There was focal lamination in many segments, but the foot processes of visceral epithelial cells were relatively preserved. Gene study of whole sequencing of COL4A5 revealed a heterozygous c.1852G>C in exon 25 p.Gly(GGG)618Arg(CGG) that implies the diagnosis of X-linked Alport syndrome. Her mother did not have this mutation.
Introduction
Background
Conclusion
Case 2
Points of Discussion
AbstractCase 1
Two young girls with Alport syndrome initially diagnosed as thin basement membrane nephropathy
51
Poster Only[Poster 16]
Abstracts
Hyung Eun Yim, Jee hoo Lee, Kee Hwan Yoo Department of Pediatrics, College of Medicine, Korea University, Seoul, Korea
