applying phylogenetic analysis to viral livestock diseases: moving beyond molecular typing
TRANSCRIPT

The Veterinary Journal 184 (2010) 130–137
Contents lists available at ScienceDirect
The Veterinary Journal
journal homepage: www.elsevier .com/locate / tv j l
Review
Applying phylogenetic analysis to viral livestock diseases: Moving beyondmolecular typing
Alex Olvera a,*, Núria Busquets a, Marti Cortey a, Nilsa de Deus a, Llilianne Ganges a,b, José Ignacio Núñez a,Bibiana Peralta a, Jennifer Toskano a, Roser Dolz a,*
a Centre de Recerca en Sanitat Animal (CReSA), UAB-IRTA, Campus de Bellaterra – Universitat Autònoma de Barcelona, 08193 Bellaterra, Barcelona, Spainb Departamento de Biotecnología, INIA, Madrid, Spain
a r t i c l e i n f o
Article history:Accepted 23 February 2009
Keywords:Phylogenetic analysisMolecular diagnosisVirusesLivestock diseases
1090-0233/$ - see front matter � 2009 Elsevier Ltd. Adoi:10.1016/j.tvjl.2009.02.015
* Corresponding authors. Tel.: +34 93 581 4567; faE-mail addresses: [email protected] (A.
uab.cat (R. Dolz).
a b s t r a c t
Changes in livestock production systems in recent years have altered the presentation of many diseasesresulting in the need for more sophisticated control measures. At the same time, new molecular assayshave been developed to support the diagnosis of animal viral disease. Nucleotide sequences generatedby these diagnostic techniques can be used in phylogenetic analysis to infer phenotypes by sequencehomology and to perform molecular epidemiology studies. In this review, some key elements of phylo-genetic analysis are highlighted, such as the selection of the appropriate neutral phylogenetic marker, theproper phylogenetic method and different techniques to test the reliability of the resulting tree. Examplesare given of current and future applications of phylogenetic reconstructions in viral livestock diseases.
� 2009 Elsevier Ltd. All rights reserved.
Introduction
The implementation of intensive production systems, morecomprehensive prevention programmes and enhanced biosecuritymeasures have contributed to the eradication of many conven-tional livestock diseases, such as classical swine fever (CSF) andfoot and mouth disease (FMD), in many regions of the world. How-ever, these factors have also modified the presentation of othercommon veterinary pathogens, such as avian infectious bronchitisand infectious bursal disease, increasing their economic impact(Lasher and Davis, 1997; Cavanagh, 2007). In addition, there isincreasing concern regarding zoonotic diseases, food safety andpublic health (Kahn, 2006). In the diagnosis of livestock disease,there is a need to establish the relationship of specific isolates toother strains of the same pathogen.
The extensive use of the polymerase chain reaction (PCR) in thedetection of infectious agents and the reduced cost of sequencinghave increasingly permitted the use of DNA sequences to confirmand more accurately diagnose disease (Zaidi et al., 2003). In com-parison with classical virological methods, molecular techniqueshave many advantages in that they are less expensive, more rapid,sensitive, have high resolution, do not require viable virus and offermore portability of the generated data (Foxman et al., 2005). Con-sequently, we can take advantage of phylogenetic analysis and use
ll rights reserved.
x: +34 93 581 4490.Olvera), Roser.Dolz@cresa.
nucleotide sequences to genotype isolates and compare them withpreviously characterised strains.
In this review we provide an overview of phylogenetic analysis,a powerful tool when applied to molecular epidemiology, but onethat can be easily misused and misinterpreted (Hall and Barlow,2006). We highlight important issues when constructing phyloge-nies, introduce the interpretation of phylogenetic trees and providesome general references for researchers who wish to pursue thesubject in more depth. Key points for performing phylogeneticanalysis are discussed and several examples of current or futureapplications of these tools in the diagnosis of livestock diseasesare included.
Phylogenetic inference
In this section, we describe some general concepts about con-struction and interpretation of phylogenetic trees. More detail isavailable in other reviews (Moreira and Philippe, 2000; Baldauf,2003; Hall and Barlow, 2006) and books (Nei and Kumar, 2000;Salemi and Vandamme, 2003; Felsenstein, 2004; Hall, 2007).
General concepts
The hierarchical genetic-evolutionary relationship amongst orwithin species is collectively referred to as a phylogeny. Phyloge-netic relationships are usually presented in the form of a tree, withthe branching pattern called a topology. The oldest point in a tree,located at its base, is the root and implies the order of branching inthe rest of the tree. The root is placed using an outgroup, usually a

1 For an extended list of available programmes with the implemented methods seehttp://evolution.genetics.washington.edu/phylip/software.html.
A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137 131
homologous sequence of a distantly related taxon, and its selectionis a critical issue in the sense of evolution. Alternatively, if a properoutgroup is not obtainable or we are not interested in the order ofdescent, an unrooted tree can be built. In addition, we have to keepin mind that a gene tree is not necessarily representative of thespecies tree, which should consider many other factors, as wellas the whole genome (Nei and Kumar, 2000).
Selection of the phylogenetic marker
Determination of the whole genome sequence is the most de-tailed genetic characterisation that can be done of a given patho-gen, but systematic and routine sequencing of large genomes isstill expensive. As a consequence, accurate selection of the optimalneutral phylogenetic marker for analysis is critical. The genome re-gion to be investigated depends mostly on the final objective whenconstructing phylogenetic trees. If correlation with a particularphenotype (such as protection, antigenicity or virulence) is desired,the region to be sequenced should contain sequences responsiblefor these functions and marker positions for each phenotype (Top-liff and Kelling, 1998; Xiao et al., 1998; Jensen et al., 2003). On theother hand, in epidemiological studies, nucleotide sequences thatreconstruct a tree similar to the species tree are preferred (Halland Barlow, 2006).
Ideally, a molecular marker should show sufficient variabilitywithout losing the strength of the phylogenetic signal due tosubstitution saturation (Xia et al., 2003) conditioned by thesubstitution rate, which is highly variable amongst virus families.Generally, the highest substitution rates are observed in RNAviruses (10�2–10�3 nucleotide substitutions/site/year) whereasDNA viruses have lower mutation rates (10�4 nucleotide substitu-tions/site/year for ssDNA viruses and 10�8–10�9 nucleotide substi-tutions/site/year for dsDNA viruses) (Duffy et al., 2008).
Constructing the data set
A molecular phylogeny is highly dependent on the alignment onwhich it is based (Aagesen, 2005). The first step in tree construc-tion is to build a data set, which could include sequences obtainedin the laboratory or retrieved from a database (e.g. GenBank, EMBL,DDBJ). The sequence set must be carefully checked and edited toavoid misinterpretations of sequence changes. Several pro-grammes and software packages are available for this purpose(e.g. BioEdit; Hall, 1999). Next, the sequences are aligned usingspecialised software (e.g. ClustalX; Thompson et al., 1997). Se-quences can be analysed at the nucleotide or amino acid levelbut, for closely related sequences, there will be more informationat the DNA level and for more distant relationships amino acid se-quences hold more information (Gibbs et al., 1995; Baldauf, 2003).
Selection of substitution model
Assuming that nucleotide or amino acid sequences have gradu-ally evolved from a common ancestor, the extent of nucleotide oramino acid substitutions can be estimated with several substitu-tion models (e.g. Jukes–Cantor, Kimura-2-parameter, Tamura–Nei). Phylogenetic algorithms make assumptions about the processof substitution and therefore may be dependent on the model ofnucleotide substitution used. ModelTest is an extended tool to se-lect the best-fit model out of 56 standard nucleotide substitutionmodels using several optimisation criteria (Posada and Crandall,1998).
However, codon-based models, which consider different evolu-tionary patterns amongst first, second and third codon positions,may be a more realistic approach when protein-coding sequencesare used, although computationally time-consuming. Codon-based
models are implemented in a growing number of analytical pack-ages (e.g. Mr. Bayes, BEAST) and their use with protein-coding se-quences is recommended (Shapiro et al., 2006).
Phylogenetic tree estimations
Once the data set is constructed and the substitution model hasbeen selected, there are four main groups of statistical methodsthat can be used for reconstructing phylogenetic trees from molec-ular data, namely, parsimony methods, distance methods, likeli-hood methods and Bayesian inference. In maximum parsimonymethods, the sum of the number of substitutions over all sites re-quired to explain each topology is computed. This sum is called thetree length. The maximum parsimony tree is simply the topologythat requires the smallest tree length. Parsimony methods oftenproduce several equally parsimonious trees that can be presentedin a single consensus tree.
A nucleotide substitution model is not needed for parsimonymethods, but is required for distance, likelihood and Bayesianmethods. In distance methods, evolutionary distances are com-puted for all pairs of taxa considered and a phylogenetic tree isconstructed by considering the relationships amongst these dis-tance values. Several of the most popular inference methods fallwithin this category, including the neighbour-joining method andthe unweighted pair group method with arithmetic mean (UP-GMA). However, under a wide variety of conditions, these methodsare outperformed by maximum likelihood and Bayesian methods,based on the likelihood function (i.e. the probability of observingthe data, in this case conditional on a tree) (Huelsenbeck et al.,2001).
In maximum likelihood methods, the likelihood of observing agiven set of sequence data for a specific substitution model is max-imised for each topology and the topology that gives the highestmaximum likelihood is chosen as the final tree. Unlike maximumlikelihood, Bayesian inference can incorporate prior informationabout phylogeny. Bayesian phylogeny estimation is based on theposterior probability distribution of trees, which are calculatedusing a Markov Chain Monte Carlo approximation. Because of theirflexibility, likelihood based methods (maximum likelihood andBayesian inference) have become the standard for many phyloge-netic analyses.
However, it is important to realise that every method has somestrengths and some weaknesses and no one method is superior. Onthe contrary, routine use of several phylogenetic methods based ondifferent optimisation criteria is a recommended strategy. Withsuch an approach, knowledge is gained from several viewpoints,allowing us to detect several inherent limitations of the algorithms(i.e. short and long-branch attraction in parsimony methods).Reporting coincident topologies using several criteria enhancesthe robustness of the data and improves the strength of the resultsobtained. It is noteworthy that the routine use of several analyticalpackages is necessary to perform all of the analyses describedabove (e.g. PAUP*, MEGA 4.0, PHYLIP, Mr. Bayes, BEAST).1
Evaluating the estimation
Once a phylogenetic tree or network is constructed, it is a com-mon and recommended strategy to evaluate the reliability of thetopology obtained under each approach. The bootstrap test is oneof the most commonly used techniques and assesses whether adata set is supporting any interior branch of the phylogeny. Ithas been assumed that bootstrapping indicates which percentage
:

132 A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137
of the subsampled trees supports the final topology, although thisassumption is not strictly correct (Efron et al., 1996). Nonetheless,bootstrapping is a simple, comprehensive and straightforwardmeasure of phylogenetic accuracy and values of 90% or higherare likely to indicate reliable groupings, although values of 70%can also yield useful information (Hillis and Bull, 1993).
In a consensus maximum parsimony tree, the reliability of thetree can be measured as the frequency of a given branching patternthat can be shown as a support percentage. Researchers areencouraged not only to use several phylogenetic methods and toevaluate branch confidences routinely, but also to test the consis-tency of the final tree obtained for each method with reliabilitytests used in other approaches (e.g. sum of branch lengths in dis-tance methods; tree length, consistency index and retention indexin parsimony; the Kishino and Hasegawa test for maximum likeli-hood) to score and evaluate which of the final trees gives the besttopology.
Phylogenetic trees versus networks
Phylogenetic analyses of livestock viral diseases are very oftenfocussed on differentiating strains of pathogen. Methods basedon classical trees, which were developed to estimate relationshipsamongst species, may not properly evaluate the exchange of genet-ic material amongst strains or individuals by recombination orreassortment. Both processes have long been recognised as a com-mon phenomena in virus evolution and as a primary cause ofincongruence amongst gene phylogenies (Worobey and Holmes,1999; Awadalla, 2003; Shackelton and Holmes, 2004). Therefore,it is highly recommended that potential recombination andreassortment events amongst sequences to be analysed aredetermined.
Numerous methods for detecting and analysing recombinationand reassortment signals are available.2 Phylogenetic networksthat examine for persistent ancestral nodes, multifurcations andreticulation could be a better representation of the viral evolution-ary process, and network algorithms for parsimony, distance, like-lihood and Bayesian approaches are also available (Posada andCrandall, 2001). Explicit models, such as hybridisation and recom-bination networks, can be used to provide an explicit description ofreticulate evolution (Huson and Bryant, 2006) that could havegreat applicability in the epidemiology of viral livestock diseases.
Applications of phylogenetic analysis in viral livestock diseases
Phylogenetic reconstructions have been used mainly to differ-entiate and classify isolates below the species level, mainly intogenotypes. Despite being designed to study evolutionary relation-ships amongst taxa, molecular phylogenetic analysis also providesa framework to show not only sequence similarity but also theclustering and relationships amongst different isolates. To link asequence to a particular phenotype, dendrograms should be usedto differentiate clusters that identify sequences with a homologousfunction that reflect a likely common ancestor. In epidemiologicalstudies, a stricter phylogenetic approach should be used to estab-lish the evolution of different sequences over time.
Species definition
Phylogeny has been demonstrated to be useful for differentiat-ing species according to taxonomy. The International Committeeon Taxonomy of Viruses (ICTV)3 lists the criteria to differentiate
2 See: http://www.bioinf.manchester.ac.uk/recombination/programs.shtml.3 See: http://www.ictvonline.org.
virus species, including demarcating new viral species; criteria canvary in different Families. Nucleotide or protein sequence phyloge-nies are usually included, since they provide sufficient power to dis-criminate intra- and inter-species relationships. Unfortunately, thelack of available genetic data often makes it difficult to define cut-off values suitable for discriminating species. Despite this, phyloge-netic concepts have been used to discover new viruses in differentGenera, such as Arenavirus (Charrel et al., 2008) and Flavivirus (Billoiret al., 2000; Gaunt et al., 2001). In the Pestivirus Genus, a threshold ofa 15% divergence in the 50 non-coding region was set to differentiatethe four species currently recognised by the ICTV: CSF virus (CSFV),bovine viral diarrhoea virus (BVDV) types 1 and 2 and border diseasevirus (BDV) (Sakoda et al., 1999). Once a species is established, spe-cies-specific PCR can be designed for routine diagnostic use (Patonet al., 2000).
Differentiation below the species level: Genotyping
The mandate of the ICTV does not include any consideration be-low the species level and the criteria for genotype definition havebeen left to the initiative of specialty groups (Fauquet and Stanley,2005). When defining genotypes for a particular virus on the basisof sequence similarities, a genetic distance threshold is usually set.Phylogenetic analysis provides a clear grouping of sequences, withstatistical support, and allows them to be classified in an intuitiveway. Clusters or genotypes can be used to extrapolate biologicalfeatures that are more difficult to determine, such as serotype (tosupport vaccine selection), zoonotic potential and virulence, or tosupport epidemiological studies at both local (reinfection, entryof a new strain or vaccine reversion) and global (relationship of aparticular isolate with others) levels.
Differences in virulence and diagnosis
Genotype differentiation has implications in the diagnosis ofviral livestock diseases, especially when strains with differentpathogenic potential co-circulate in the host population. In thisscenario, the identification of viral species has to be complementedwith the determination of the specific genotype present in theanimals.
Such is the case with infectious bursal disease virus (IBDV), amember of the Family Birnaviridae, which is distributed worldwideand causes an acute, highly contagious disease of chickens (vanden Berg et al., 1996). Traditionally, IBD control has relied on infec-tious attenuated vaccines, making it necessary to differentiate fieldand vaccine viruses when diagnosis is required (Rautenschleinet al., 2005). IBDV is classified into two serotypes, but only sero-type 1 includes pathogenic strains. This serotype can be further di-vided in subtypes based on its virulence and antigeniccharacteristics: attenuated vaccine strains, intermediate virulencevaccine strains, classical strains, highly virulent strains and anti-genic variant strains (Lukert and Saif, 1997). Genotyping basedon the VP2 gene correlates with viral subtypes, since this gene con-tains hypothetical virulence marker positions and the major epi-topes that induce viral neutralising antibodies (Brandt et al.,2001). If amino acid residues that are essential for virulence areknown, it is possible to deduce virulent phenotypes directly fromsequences (van den Berg et al., 2000).
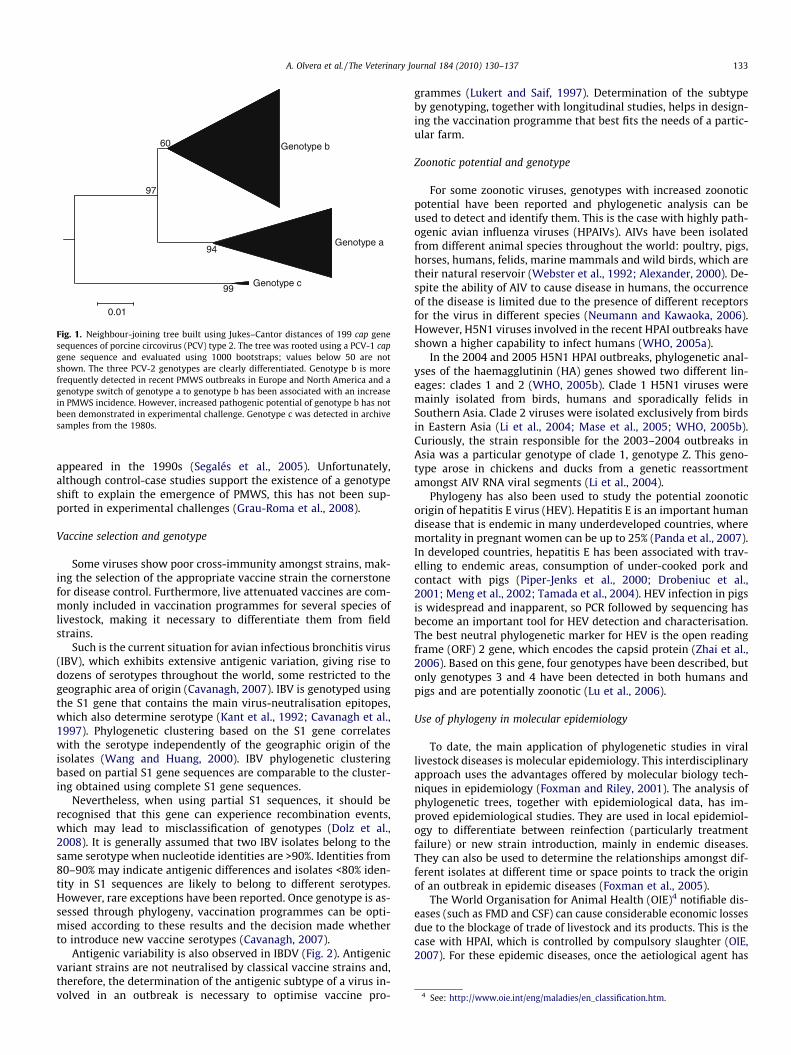
Another example of the potential diagnostic use of the associa-tion of an increased virulence with a particular genotype is porcinecircovirus type 2 (PCV-2). Although still under discussion, it wasrecently suggested that PCV-2 genotype b might be more fre-quently associated with post-weaning multisystemic wasting syn-drome (PMWS) (Segalés et al., 2008) (Fig. 1). One of the mainproblems of the study on PMWS was that, whereas PCV-2 is aubiquitous virus, PMWS has had an epizootic pattern since it first
Genotype b
Genotype a
Genotype c
60
94
97
99
0.01
Fig. 1. Neighbour-joining tree built using Jukes–Cantor distances of 199 cap genesequences of porcine circovirus (PCV) type 2. The tree was rooted using a PCV-1 capgene sequence and evaluated using 1000 bootstraps; values below 50 are notshown. The three PCV-2 genotypes are clearly differentiated. Genotype b is morefrequently detected in recent PMWS outbreaks in Europe and North America and agenotype switch of genotype a to genotype b has been associated with an increasein PMWS incidence. However, increased pathogenic potential of genotype b has notbeen demonstrated in experimental challenge. Genotype c was detected in archivesamples from the 1980s.
4 See: http://www.oie.int/eng/maladies/en_classification.htm.
A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137 133
appeared in the 1990s (Segalés et al., 2005). Unfortunately,although control-case studies support the existence of a genotypeshift to explain the emergence of PMWS, this has not been sup-ported in experimental challenges (Grau-Roma et al., 2008).
Vaccine selection and genotype
Some viruses show poor cross-immunity amongst strains, mak-ing the selection of the appropriate vaccine strain the cornerstonefor disease control. Furthermore, live attenuated vaccines are com-monly included in vaccination programmes for several species oflivestock, making it necessary to differentiate them from fieldstrains.
Such is the current situation for avian infectious bronchitis virus(IBV), which exhibits extensive antigenic variation, giving rise todozens of serotypes throughout the world, some restricted to thegeographic area of origin (Cavanagh, 2007). IBV is genotyped usingthe S1 gene that contains the main virus-neutralisation epitopes,which also determine serotype (Kant et al., 1992; Cavanagh et al.,1997). Phylogenetic clustering based on the S1 gene correlateswith the serotype independently of the geographic origin of theisolates (Wang and Huang, 2000). IBV phylogenetic clusteringbased on partial S1 gene sequences are comparable to the cluster-ing obtained using complete S1 gene sequences.
Nevertheless, when using partial S1 sequences, it should berecognised that this gene can experience recombination events,which may lead to misclassification of genotypes (Dolz et al.,2008). It is generally assumed that two IBV isolates belong to thesame serotype when nucleotide identities are >90%. Identities from80–90% may indicate antigenic differences and isolates <80% iden-tity in S1 sequences are likely to belong to different serotypes.However, rare exceptions have been reported. Once genotype is as-sessed through phylogeny, vaccination programmes can be opti-mised according to these results and the decision made whetherto introduce new vaccine serotypes (Cavanagh, 2007).
Antigenic variability is also observed in IBDV (Fig. 2). Antigenicvariant strains are not neutralised by classical vaccine strains and,therefore, the determination of the antigenic subtype of a virus in-volved in an outbreak is necessary to optimise vaccine pro-
grammes (Lukert and Saif, 1997). Determination of the subtypeby genotyping, together with longitudinal studies, helps in design-ing the vaccination programme that best fits the needs of a partic-ular farm.
Zoonotic potential and genotype
For some zoonotic viruses, genotypes with increased zoonoticpotential have been reported and phylogenetic analysis can beused to detect and identify them. This is the case with highly path-ogenic avian influenza viruses (HPAIVs). AIVs have been isolatedfrom different animal species throughout the world: poultry, pigs,horses, humans, felids, marine mammals and wild birds, which aretheir natural reservoir (Webster et al., 1992; Alexander, 2000). De-spite the ability of AIV to cause disease in humans, the occurrenceof the disease is limited due to the presence of different receptorsfor the virus in different species (Neumann and Kawaoka, 2006).However, H5N1 viruses involved in the recent HPAI outbreaks haveshown a higher capability to infect humans (WHO, 2005a).
In the 2004 and 2005 H5N1 HPAI outbreaks, phylogenetic anal-yses of the haemagglutinin (HA) genes showed two different lin-eages: clades 1 and 2 (WHO, 2005b). Clade 1 H5N1 viruses weremainly isolated from birds, humans and sporadically felids inSouthern Asia. Clade 2 viruses were isolated exclusively from birdsin Eastern Asia (Li et al., 2004; Mase et al., 2005; WHO, 2005b).Curiously, the strain responsible for the 2003–2004 outbreaks inAsia was a particular genotype of clade 1, genotype Z. This geno-type arose in chickens and ducks from a genetic reassortmentamongst AIV RNA viral segments (Li et al., 2004).
Phylogeny has also been used to study the potential zoonoticorigin of hepatitis E virus (HEV). Hepatitis E is an important humandisease that is endemic in many underdeveloped countries, wheremortality in pregnant women can be up to 25% (Panda et al., 2007).In developed countries, hepatitis E has been associated with trav-elling to endemic areas, consumption of under-cooked pork andcontact with pigs (Piper-Jenks et al., 2000; Drobeniuc et al.,2001; Meng et al., 2002; Tamada et al., 2004). HEV infection in pigsis widespread and inapparent, so PCR followed by sequencing hasbecome an important tool for HEV detection and characterisation.The best neutral phylogenetic marker for HEV is the open readingframe (ORF) 2 gene, which encodes the capsid protein (Zhai et al.,2006). Based on this gene, four genotypes have been described, butonly genotypes 3 and 4 have been detected in both humans andpigs and are potentially zoonotic (Lu et al., 2006).
Use of phylogeny in molecular epidemiology
To date, the main application of phylogenetic studies in virallivestock diseases is molecular epidemiology. This interdisciplinaryapproach uses the advantages offered by molecular biology tech-niques in epidemiology (Foxman and Riley, 2001). The analysis ofphylogenetic trees, together with epidemiological data, has im-proved epidemiological studies. They are used in local epidemiol-ogy to differentiate between reinfection (particularly treatmentfailure) or new strain introduction, mainly in endemic diseases.They can also be used to determine the relationships amongst dif-ferent isolates at different time or space points to track the originof an outbreak in epidemic diseases (Foxman et al., 2005).
The World Organisation for Animal Health (OIE)4 notifiable dis-eases (such as FMD and CSF) can cause considerable economic lossesdue to the blockage of trade of livestock and its products. This is thecase with HPAI, which is controlled by compulsory slaughter (OIE,2007). For these epidemic diseases, once the aetiological agent has

VARIANT-A
GZ29112
GLS
VARIANT-E
DEL-E
U.S. variant strains
BURSINE2
BURSINEPLUS
Spain-62-08
Intermediate virulence
classical strains
STC
BURSAVAC
Cu-1wt
52/70
Classical strains
Int/228E
Spain-34-08 Low attenuated classical strains
D78
HARBIN
CEF94
PBG98
P2
Spain-33-08HIPRAGUMBO
CU-1
Attenuated classical strains
849VB
Spain-33-026145
UK661
Spain-61-02HK46
D6948
5939
NETHERLAND
VG-248
OKYM
Very virulent strains
100
100
73
78
99
99
72
96
99
99
98
100
74
0.005
Fig. 2. Neighbour-joining unrooted tree built using gamma Tamura–Nei substitution rates (gamma parameter = 1). The tree was evaluated using 1000 bootstraps; valuesbelow 70 are not shown. A fragment of 400 bp of the IBDV VP2 gene was amplified and sequenced from different Spanish samples and aligned with reference strains from theGenBank database. Spanish isolates are highlighted in italics. Two field sequences (Spain-62-08 and Spain-33-08) were grouped with vaccine strains, which are usuallyclustered as attenuated, intermediate virulence or low attenuated strains. Two other sequences (Spain-61-02 and Spain-33-02) were clustered with very virulent strains,indicating that they are true field isolates.
134 A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137
been confirmed, phylogenetic studies can be used to determine theorigin/source of the strain responsible of introducing the diseasewithin a country, as well as the evolution patterns of virus subtypesin the field (Beck and Strohmaier, 1987; Lowings et al., 1996; Moen-nig et al., 2003).
Classically, FMD virus (FMDV) has been classified into sero-types. Global phylogenetic comparison of complete sequences ofmany FMDV lineages and their geographic location has allowedfurther classification of FMDV strains from the same serotype intonew genotypes, known as ‘topotypes’ (Samuel and Knowles, 2001).For example, the PanAsia strain, responsible for a pandemic in Asiathat extended to different parts of Africa and reached Europe in2001, was clustered in the Middle East-South Asia topotype (Gib-bens et al., 2001). The use of phylogenetic studies has clarified
the origin of many FMD outbreaks caused by the introduction ofthe virus from different geographical regions. For example, theintroduction of European FMDV strains in India in the 1960s wasdetermined when C-Bombay/64 strain and European isolates circu-lating at those dates were branched together. Importation of ani-mals for cross-breeding with local cattle or pigs was the mostlikely source of infection (Hemadri et al., 2003).
In other cases, the analysis of the phylogenetic relationships be-tween field isolates and vaccine strains has identified vaccines asthe possible origin of an outbreak, as occurred during the 1980sin Europe, where FMDV isolates from 1987 and 1988 were groupedwith vaccine strain O1 Kaufbeuren Ger/66 (Sáiz et al., 1993). InArgentina in 1983 and 1984, serotype C isolates showed a closephylogenetic relationship with C3/Resende/Bra/55, a strain
North American
duck/NZL76H1N3
duck/Bavaria77H1N1
duck/MiyagiH1N1
duck/Australia80H1N1
duck/Australia80H1N1(2)
duck/HokkaidoH1N1
pintail/Shimane98H1N9
duck/Italy07H1N1
duck/Italy06H1N1
goose/Italy03H1N1
Italy
Eurasian teal/Spain07H1N1
mallard duck/Spain07H1N1
Eurasian teal/Spain/1404/07H1N1
Spain 2007
Eurasian
98
100
98
90
55
60
9999
9999
96
100
990.02
Eurasian teal/Spain07H1N1
mallard duck/Spain07H1N1
Eurasian teal/Spain/1404/07H1N1
Spain 2007
100
100
99
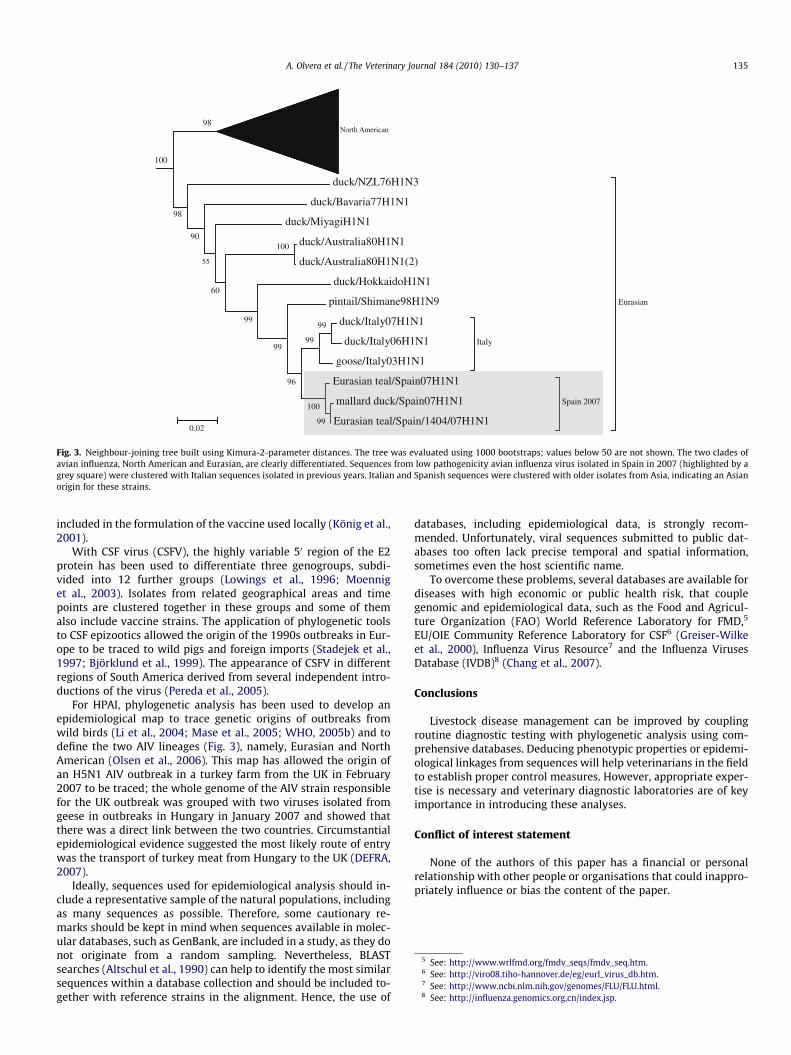
Fig. 3. Neighbour-joining tree built using Kimura-2-parameter distances. The tree was evaluated using 1000 bootstraps; values below 50 are not shown. The two clades ofavian influenza, North American and Eurasian, are clearly differentiated. Sequences from low pathogenicity avian influenza virus isolated in Spain in 2007 (highlighted by agrey square) were clustered with Italian sequences isolated in previous years. Italian and Spanish sequences were clustered with older isolates from Asia, indicating an Asianorigin for these strains.
5 See: http://www.wrlfmd.org/fmdv_seqs/fmdv_seq.htm.6 See: http://viro08.tiho-hannover.de/eg/eurl_virus_db.htm.7 See: http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html.8 See: http://influenza.genomics.org.cn/index.jsp.
A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137 135
included in the formulation of the vaccine used locally (König et al.,2001).
With CSF virus (CSFV), the highly variable 50 region of the E2protein has been used to differentiate three genogroups, subdi-vided into 12 further groups (Lowings et al., 1996; Moenniget al., 2003). Isolates from related geographical areas and timepoints are clustered together in these groups and some of themalso include vaccine strains. The application of phylogenetic toolsto CSF epizootics allowed the origin of the 1990s outbreaks in Eur-ope to be traced to wild pigs and foreign imports (Stadejek et al.,1997; Björklund et al., 1999). The appearance of CSFV in differentregions of South America derived from several independent intro-ductions of the virus (Pereda et al., 2005).
For HPAI, phylogenetic analysis has been used to develop anepidemiological map to trace genetic origins of outbreaks fromwild birds (Li et al., 2004; Mase et al., 2005; WHO, 2005b) and todefine the two AIV lineages (Fig. 3), namely, Eurasian and NorthAmerican (Olsen et al., 2006). This map has allowed the origin ofan H5N1 AIV outbreak in a turkey farm from the UK in February2007 to be traced; the whole genome of the AIV strain responsiblefor the UK outbreak was grouped with two viruses isolated fromgeese in outbreaks in Hungary in January 2007 and showed thatthere was a direct link between the two countries. Circumstantialepidemiological evidence suggested the most likely route of entrywas the transport of turkey meat from Hungary to the UK (DEFRA,2007).
Ideally, sequences used for epidemiological analysis should in-clude a representative sample of the natural populations, includingas many sequences as possible. Therefore, some cautionary re-marks should be kept in mind when sequences available in molec-ular databases, such as GenBank, are included in a study, as they donot originate from a random sampling. Nevertheless, BLASTsearches (Altschul et al., 1990) can help to identify the most similarsequences within a database collection and should be included to-gether with reference strains in the alignment. Hence, the use of
databases, including epidemiological data, is strongly recom-mended. Unfortunately, viral sequences submitted to public dat-abases too often lack precise temporal and spatial information,sometimes even the host scientific name.
To overcome these problems, several databases are available fordiseases with high economic or public health risk, that couplegenomic and epidemiological data, such as the Food and Agricul-ture Organization (FAO) World Reference Laboratory for FMD,5
EU/OIE Community Reference Laboratory for CSF6 (Greiser-Wilkeet al., 2000), Influenza Virus Resource7 and the Influenza VirusesDatabase (IVDB)8 (Chang et al., 2007).
Conclusions
Livestock disease management can be improved by couplingroutine diagnostic testing with phylogenetic analysis using com-prehensive databases. Deducing phenotypic properties or epidemi-ological linkages from sequences will help veterinarians in the fieldto establish proper control measures. However, appropriate exper-tise is necessary and veterinary diagnostic laboratories are of keyimportance in introducing these analyses.
Conflict of interest statement
None of the authors of this paper has a financial or personalrelationship with other people or organisations that could inappro-priately influence or bias the content of the paper.

136 A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137
Acknowledgements
The authors would like to thank Joaquim Segalès and VirginiaAragón for helpful comments and suggestions.
References
Aagesen, L., 2005. Direct optimization, affine gap costs, and node stability.Molecular Phylogenetic Evolution 36, 641–653.
Alexander, D.J., 2000. A review of avian influenza in different bird species.Veterinary Microbiology 74, 3–13.
Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J., 1990. Basic localalignment search tool. Journal of Molecular Biology 215, 403–410.
Awadalla, P., 2003. The evolutionary genomics of pathogen recombination. NatureReviews Genetics 4, 50–60.
Baldauf, S.L., 2003. Phylogeny for the faint of heart: a tutorial. Trends in Genetics 19,345–351.
Beck, E., Strohmaier, K., 1987. Subtyping of European foot-and-mouth disease virusstrains by nucleotide sequence determination. Journal of Virology 61, 1621–1629.
Billoir, F., de Chesse, R., Tolou, H., de Micco, P., Gould, E.A., de Lamballerie, X., 2000.Phylogeny of the genus Flavivirus using complete coding sequences ofarthropod-borne viruses and viruses with no known vector. Journal ofGeneral Virology 81, 781–790.
Björklund, H., Lowings, P., Stadejek, T., Vilcek, S., Greiser-Wilke, I., Paton, D., Belák,S., 1999. Phylogenetic comparison and molecular epidemiology of classicalswine fever virus. Virus Genes 19, 189–195.
Brandt, M., Yao, K., Liu, M., Heckert, R.A., Vakharia, V.N., 2001. Moleculardeterminants of virulence, cell tropism, and pathogenic phenotype ofinfectious bursal disease virus. Journal of Virology 75, 11974–11982.
Cavanagh, D., Elus, M.M., Cook, J.K., 1997. Relationship between sequence variationin the S1 spike protein of infectious bronchitis virus and the extent of cross-protection in vivo. Avian Pathology 26, 63–74.
Cavanagh, D., 2007. Coronavirus avian infectious bronchitis virus. VeterinaryResearch 38, 281–297.
Chang, S., Zhang, J., Liao, X., Zhu, X., Wang, D., Zhu, J., Feng, T., Zhu, B., Gao, G.F.,Wang, J., Yang, H., Yu, J., Wang, J., 2007. Influenza virus database (IVDB): anintegrated information resource and analysis platform for influenza virusresearch. Nucleic Acids Research 35, 376–380.
Charrel, R.N., de Lamballerie, X., Emonet, S., 2008. Phylogeny of the genusArenavirus. Current Opinion in Microbiology 11, 362–368.
Department for Environment, Food and Rural Affairs (DEFRA). Avian influenzareports published, June 2007. <http://www.defra.gov.uk/news/latest/2007/animal-0216.htm> (accessed 12.12.08).
Dolz, R., Pujols, J., Ordóñez, G., Porta, R., Majó, N., 2008. Molecular epidemiology andevolution of avian infectious bronchitis virus in Spain over a fourteen-yearperiod. Virology 25, 50–59.
Duffy, S., Shackelton, L.A., Holmes, E.C., 2008. Rates of evolutionary change inviruses: patterns and determinants. Nature Reviews Genetics 9, 267–276.
Drobeniuc, J., Favorov, M.O., Shapiro, C.N., Bell, B.P., Mast, E.E., Dadu, A., Culver, D.,Iarovoi, P., Robertson, B.H., Margolis, H.S., 2001. Hepatitis E virus antibodyprevalence among persons who work with swine. Journal of Infectious Diseases184, 1594–1597.
Efron, B., Halloran, E., Holmes, S., 1996. Bootstrap confidence levels for phylogenetictrees. Proceedings of the Natural Academy of Sciences of the USA 93, 7085–7090.
Fauquet, C.M., Stanley, J., 2005. Revising the way we conceive and name virusesbelow the species level: a review of Geminivirus taxonomy calls for newstandardized isolate descriptors. Archives Virology 150, 2151–2179.
Felsenstein, J., 2004. Inferring Phylogenies. Sinauer Associates, Sunderland,Massachussets, USA.
Foxman, B., Riley, L., 2001. Molecular epidemiology: focus on infection. AmericanJournal of Epidemiology 153, 1135–1141.
Foxman, B., Zhang, L., Koopman, J.S., Manning, S.D., Marrs, C.F., 2005. Choosing anappropriate bacterial typing technique for epidemiologic studies. EpidemiologyPerspectives and Innovation 2, 10.
Gaunt, M.W., Sall, A.A., de Lamballerie, X., Falconar, A.K., Dzhivanian, T.I., Gould, E.A.,2001. Phylogenetic relationships of flaviviruses correlate with theirepidemiology, disease association and biogeography. Journal of GeneralVirology 82, 1867–1876.
Gibbens, J.C., Sharpe, C.E., Wilesmith, J.W., Mansley, L.M., Michalopoulou, E., Ryan,J.B., Hudson, M., 2001. Descriptive epidemiology of the 2001 foot-and-mouthdisease epidemic in Great Britain: the first five months. Veterinary Record 15,729–743.
Gibbs, A., Calisher, C.H., García-Arenal, F., 1995. Molecular Basis of Virus Evolution,first ed. Cambridge University Press, Cambridge, UK.
Grau-Roma, L., Crisci, E., Sibila, M., López-Soria, S., Nofrarias, M., Cortey, M., Fraile, L.,Olvera, A., Segalés, J., 2008. A proposal on porcine circovirus type 2 (PCV2)genotype definition and their relation with postweaning multisystemic wastingsyndrome (PMWS) occurrence. Veterinary Microbiology 128, 23–35.
Greiser-Wilke, I., Zimmermann, B., Fritzemeier, J., Floegel, G., Moennig, V., 2000.Structure and presentation of a World Wide Web database of CSF virus isolatesheld at the EU reference laboratory. Veterinary Microbiology 73, 131–136.
Hall, T.A., 1999. BioEdit: a user-friendly biological sequence alignment editor andanalysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41,95–98.
Hall, B.G., Barlow, M., 2006. Phylogenetic analysis as a tool in molecularepidemiology of infectious diseases. Annals of Epidemiology 16, 157–169.
Hall, B.G., 2007. Phylogenetic Trees Made Easy: A How-to Manual. SinauerAssociates, Massachussets, USA.
Hemadri, D., Sanyal, A., Tosh, C., Venkataramanan, R., Pattnaik, B., 2003. Serotype Cfoot-and-mouth disease virus isolates from India belong to a separate so far notdescribed lineage. Veterinary Microbiology 20, 25–35.
Hillis, D.M., Bull, J.J., 1993. An empirical test of bootstrapping as a method forassessing confidence in phylogenetic analysis. Systematic Biology 42, 182–192.
Huelsenbeck, J.H., Ronquist, F., Nielsen, R., Bollback, J., 2001. Bayesian inference ofphylogeny and its impact on evolutionary biology. Science 294, 2310–2314.
Huson, D.H., Bryant, D., 2006. Application of phylogenetic networks in evolutionarystudies. Molecular Biology and Evolution 23, 254–267.
Jensen, M.A., Li, F.S., van’t Wout, A.B., Nickle, D.C., Shriner, D., He, H.X., McLaughlin,S., Shankarappa, R., Margolick, J.B., Mullins, J.I., 2003. Improved coreceptorusage prediction and genotypic monitoring of R5-to-X4 transition by motifanalysis of human immunodeficiency virus type 1 env V3 loop sequences.Journal of Virology 77, 13376–13388.
Kahn, L.H., 2006. Confronting zoonoses, linking human and veterinary medicine.Emerging Infectious Diseases 12, 556–561.
Kant, A., Koch, G., van Roozelaar, D.J., Kusters, J.G., Poelwijk, F.A., van der Zeijst, B.A.,1992. Location of antigenic sites defined by neutralizing monoclonal antibodieson the S1 avian infectious bronchitis virus glycopolypeptide. Journal of GeneralVirology 73, 591–596.
König, G., Blanco, C., Knowles, N.J., Palma, E.L., Maradei, E., Piccone, M.E., 2001.Phylogenetic analysis of foot-and-mouth disease viruses isolated in Argentina.Virus Genes 23, 175–181.
Lasher, H.N., Davis, V.S., 1997. History of infectious bursal disease in the USA – thefirst two decades. Avian Diseases 41, 11–19.
Li, K.S., Guan, Y., Wang, J., Smith, G.J., Xu, K.M., Duan, L., Rahardjo, A.P.,Puthavathana, P., Buranathai, C., Nguyen, T.D., Estoepangestie, A.T., Chaisingh,A., Auewarakul, P., Long, H.T., Hanh, N.T., Webby, R.J., Poon, L.L., Chen, H.,Shortridge, K.F., Yuen, K.Y., Webster, R.G., Peiris, J.S., 2004. Genesis of a highlypathogenic and potentially pandemic H5N1 influenza virus in eastern Asia.Nature 430, 209–213.
Lowings, P., Ibata, G., Needham, J., Paton, D., 1996. Classical swine fever virusdiversity and evolution. Journal of General Virology 77, 1311–1321.
Lu, L., Li, C., Hagedorn, C.H., 2006. Phylogenetic analysis of global hepatitis E virussequences: genetic diversity, subtypes and zoonosis. Reviews in MedicalVirology 16, 5–36.
Lukert, P.D., Saif, Y.M., 1997. Infectious bursal disease. In: Calnek, B.W., Barnes, H.J.,Beard, C.W., McDougald, L.R. (Eds.), Disease of Poultry. Iowa State UniversityPress, Ames, Iowa, USA, pp. 721–738.
Mase, M., Tsukamoto, K., Imada, T., Imai, K., Tanimura, N., Nakamura, K., Yamamoto,Y., Hitomi, T., Kira, T., Nakai, T., Kiso, M., Horimoto, T., Kawaoka, Y., Yamaguchi,S., 2005. Characterization of H5N1 influenza A viruses isolated during the 2003–2004 influenza outbreaks in Japan. Virology 332, 167–176.
Meng, X.J., Wiseman, B., Elvinger, F., Guenette, D.K., Toth, T.E., Engle, R.E., Emerson,S.U., Purcell, R.H., 2002. Prevalence of antibodies to hepatitis E virus inveterinarians working with swine and in normal blood donors in the UnitedStates and other countries. Journal of Clinical Microbiology 40, 117–122.
Moennig, V., Floegel-Niesmann, G., Greiser-Wilke, I., 2003. Clinical signs andepidemiology of classical swine fever: a review of new knowledge. TheVeterinary Journal 165, 11–20.
Moreira, D., Philippe, H., 2000. Molecular phylogeny: pitfalls and progress.International Microbiology 3, 9–16.
Nei, M., Kumar, S., 2000. Molecular Evolution and Phylogenetics. Oxford UniversityPress, New York, USA.
Neumann, G., Kawaoka, Y., 2006. Host range restriction and pathogenicity in thecontext of influenza pandemic. Emerging Infectious Diseases 12, 881–886.
Olsen, B., Munster, V.J., Wallensten, A., Waldenstrom, J., Osterhaus, A.D.M.E.,Fouchier, R.A., 2006. Global patterns of influenza A virus in wild birds. Science312, 384–388.
Organisation Mondiale de la Santé Animale (OIE), 2007. Chapter 2.7.12. of theTerrestrial Animal Health Code. <http://www.oie.int/eng/normes/mcode/en_chapitre_2.7.12.htm> (accessed 12.06.08).
Panda, S.K., Thakral, D., Rehman, S., 2007. Hepatitis E virus. Reviews in MedicalVirology 17, 151–180.
Paton, D.J., McGoldrick, A., Bensaude, E., Belak, S., Mittelholzer, C., Koenen, F.,Vanderhallen, H., Greiser-Wilke, I., Scheibner, H., Stadejek, T., Hofmann, M.,Thuer, B., 2000. Classical swine fever virus: a second ring test to evaluate RT-PCR detection methods. Veterinary Microbiology 77, 71–81.
Pereda, A.J., Greiser-Wilke, I., Schmitt, B., Rincon, M.A., Mogollon, J.D., Sabogal, Z.Y.,Lora, A.M., Sanguinetti, H., Piccone, M.E., 2005. Phylogenetic analysis of classicalswine fever virus (CSFV) field isolates from outbreaks in South and CentralAmerica. Virus Research 110, 111–118.
Piper-Jenks, N., Horowitz, H.W., Schwartz, E., 2000. Risk of hepatitis E infection totravelers. Journal of Travel Medicine 7, 194–199.
Posada, D., Crandall, K., 1998. ModelTest: testing the model of DNA substitution.Bioinformatics 14, 817–818.
Posada, D., Crandall, K., 2001. Intraspecific gene genealogies: trees grafting intonetworks. Trends in Ecology and Evolution 16, 37–45.

A. Olvera et al. / The Veterinary Journal 184 (2010) 130–137 137
Rautenschlein, S., Kraemer, C., Vanmarcke, J., Montiel, E., 2005. Protective efficacy ofintermediate and intermediate plus infectious bursal disease virus (IBDV)vaccines against very virulent IBDV in commercial broilers. Avian Diseases 49,231–237.
Sakoda, Y., Ozawa, S., Damrongwatanapokin, S., Sato, M., Ishikawa, K., Fukusho, A.,1999. Genetic heterogeneity of porcine and ruminant pestiviruses mainlyisolated in Japan. Veterinary Microbiology 65, 75–86.
Sáiz, J.C., Sobrino, F., Dopazo, J., 1993. Molecular epidemiology of foot-and-mouthdisease virus type O. Journal of General Virology 74, 2281–2285.
Salemi, M., Vandamme, AM., 2003. The Phylogenetic Handbook: A PracticalApproach to DNA and Protein Phylogeny. Cambridge University Press,Cambridge, UK.
Samuel, A.R., Knowles, N.J., 2001. Foot-and-mouth disease type O viruses exhibitgenetically and geographically distinct evolutionary lineages (topotypes).Journal of General Virology 82, 609–621.
Segalés, J., Allan, G.M., Domingo, M., 2005. Porcine circovirus diseases. AnimalHealth Research Reviews 6, 119–142.
Segalés, J., Olvera, A., Grau-Roma, L., Charreyre, C., Nauwynck, H., Larsen, L., Dupont,K., McCullough, K., Ellis, J., Krakowka, S., Mankertz, A., Fredholm, M., Fossum, C.,Timmusk, S., Stockhofe-Zurwieden, N., Beattie, V., Armstrong, D., Grassland, B.,Baekbo, P., Allan, G., 2008. PCV-2 genotype definition and nomenclature.Veterinary Record 162, 867–868.
Shapiro, B., Rambaut, A., Drummond, A.J., 2006. Choosing appropriate substitutionmodels for the phylogenetic analysis of protein-coding genes. MolecularBiology Evolution 23, 7–9.
Shackelton, L.A., Holmes, E.C., 2004. The evolution of large DNA viruses: combininggenomic information of viruses and their hosts. Trends in Microbiology 12,458–465.
Stadejek, T., Vilcek, S., Lowings, J.P., Ballagi-Pordány, A., Paton, D.J., Belák, S., 1997.Genetic heterogeneity of classical swine fever virus in Central Europe. VirusResearch 52, 195–204.
Tamada, Y., Yano, K., Yatsuhashi, H., Inoue, O., Mawatari, F., Ishibashi, H., 2004.Consumption of wild boar linked to cases of hepatitis E. Journal of Hepatology40, 869–870.
Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G., 1997. TheClustalX windows interface: flexible strategies for multiple sequence alignmentaided by quality analysis tools. Nucleic Acids Research 24, 4876–4882.
Topliff, C.L., Kelling, C.L., 1998. Virulence markers in the 50 untranslated region ofgenotype 2 bovine viral diarrhea virus isolates. Virology 250, 164–172.
van den Berg, T.P., Gonze, M., Morales, D., Meulemans, G., 1996. Acute infectiousbursal disease in poultry: immunological and molecular basis of antigenicity ofhighly virulent strain. Avian Pathology 25, 751–768.
van den Berg, T.P., Eterradossi, N., Toquin, D., Meulemans, G., 2000. Infectious bursaldisease (Gumboro disease). Revue Scientifique et Technique 19, 509–543.
Wang, C.H., Huang, Y.C., 2000. Relationship between serotypes and genotypes basedon the hypervariable region of the S1 gene of infectious bronchitis virus.Archives of Virology 145, 291–300.
Webster, R.G., Bean, W.J., Gorman, O.T., Chambers, T.M., Kawaoka, Y., 1992.Evolution and ecology of influenza A viruses. Microbiology Reviews 56, 152–179.
World Health Organization (WHO), August 2005a. Confirmed human cases of avianinfluenza A (H5N1) since 28 January 2004. <http://www.who.int/csr/disease/avian_influenza/country/cases_table_2005_01_26/en/> (accessed 10.12.08).
World Health Organization (WHO), 2005b. Global Influenza Program SurveillanceNetwork, evolution of H5N1 avian influenza viruses in Asia. Emerging InfectiousDiseases 11, 1515–1521.
Worobey, M., Holmes, E.C., 1999. Evolutionary aspects of recombination in RNAviruses. Journal of General Virology 80, 2535–2543.
Xia, X., Xie, Z., Salemi, M., Chen, L., Wang, Y., 2003. An index of substitutionsaturation and its application. Molecular Phylogenetics and Evolution 26, 1–7.
Xiao, L., Owen, S.M., Goldman, I., Lal, A.A., deJong, J.J., Goudsmit, J., Lal, R.B., 1998.CCR5 coreceptor usage of non-syncytium-inducing primary HIV-1 isindependent of phylogenetically distinct global HIV-1 isolates: delineation ofconsensus motif in the V3 domain that predicts CCR-5 usage. Virology 240, 83–92.
Zaidi, N., Konstantinou, K., Zervos, M., 2003. The role of molecular biology andnucleic acid technology in the study of human infection and epidemiology.Archives of Pathology Laboratory Medicine 127, 1098–1105.
Zhai, L., Dai, X., Meng, J., 2006. Hepatitis E virus genotyping based on full-lengthgenome and partial genomic regions. Virus Research 120, 57–69.