anita datta, md, frcpc, cscn diplomate (eeg)...
TRANSCRIPT

Anita Datta, MD, FRCPC, CSCN Diplomate (EEG)
Clinical Assistant Professor of Pediatrics
University of British Columbia
Pediatric Neurologist and Epileptologist

1. How genetic diagnosis can impact drug selection
– Sodium channelopathies
– KCNT1-related epilepsies
– KCNQ2-related epilepsies
– GRIN2A-related epilepsies
– Other
2. How genetic pathways diagnosis can impact medical and surgical management
– mTOR
3. Other forms of personalized medicine
– Adverse Reactions
– Ketogenic Diet
– CBD
– Rational Polypharmacy
4. Future of Precision Medicine

I have nothing to Disclose

• Affects 4% of the population
• 80% of epilepsy begins in children or adolescents
• Highest in the first year of life
• > 30 seizure types
• Many Epilepsy Syndromes

Idiopathic
Congential
Trauma
Vascular
Neoplastic
Infectious
Causes of Epilepsy
1975 Hauser

Genetics/WES:
DNA repair
Transcriptional regulation
Axon myelination
Protein translation/modn
Proxisomal function
Channelopathies
Synatpic dysfunction

• Structural/Metabolic – Better Imaging
modalities
– Understanding of mosaicism in brain tissue
– Focal/Lesional epilepsy can be genetic also!
• Inflammatory Causes and Neurotransmitters
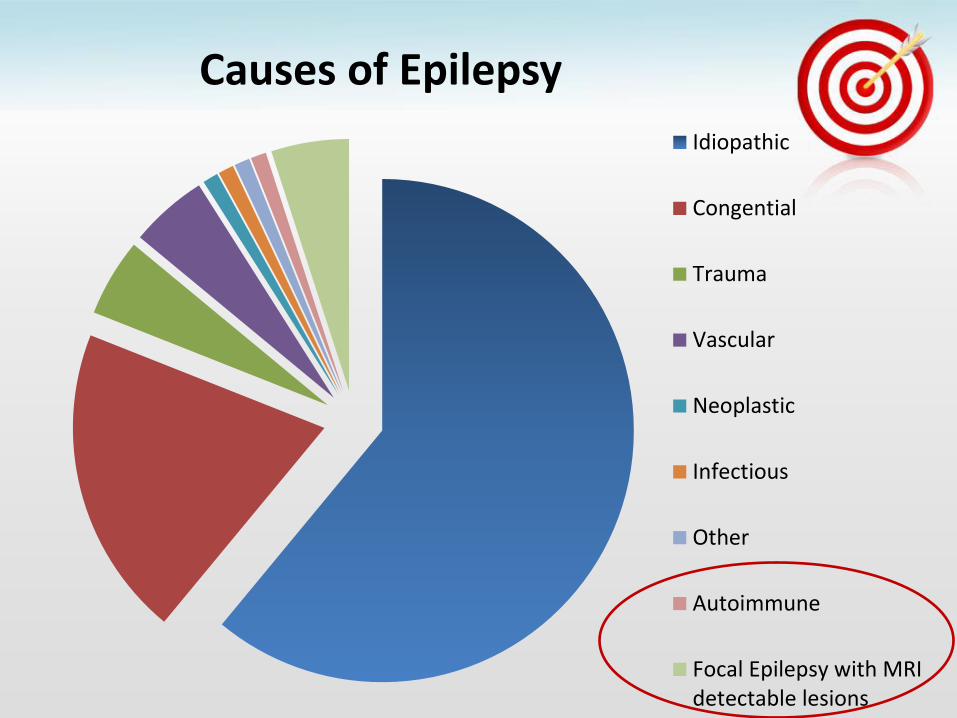
Idiopathic
Congential
Trauma
Vascular
Neoplastic
Infectious
Other
Autoimmune
Focal Epilepsy with MRIdetectable lesions
Causes of Epilepsy

Epilepsy genetic diagnosis
Precision Medicine

• De novo mutations often found
• Dravet syndrome: >80% have SCN1A mutations
• Many genes implicated
Phenotypic spectrum of genetic disease
Genetic heterogeneity of a phenotype

Dravet
MAE
Epilepsy of Infancy with Migrating Focal Seizures
Genetic Epilepsy with Febrile Seizures +
Ohtahara
Epilepsy of
Infancy
with Migrating
Focal Seizures
Benign Familial Neonatal-Infantile
Seizures
Genetic
Epilepsy
with Febrile
Seizures +
SCN1A
SCN1B
SCN2A
Sodium Channelopathies: Phenotypic Spectrum

• Establishing pathogenicity of variant
• De novo versus inherited mutation
• Mosaicism
• Comorbidities
• Large cohorts essential

• Genetic diagnosis Drug selection

• Different syndromic patterns – SCN1A – Dravet
– SCN2A – Ohtahara, Epilepsy of Infancy with Migrating Focal Seizures
– SCN9A – Mixed
• Does the specific sodium channel affect treatment?

SCN1A EE Dravet Avoid Sodium Channel blockers
– sodium channel blockers – Carbamazepine – Lamotrigine
– Topiramate – Stiripentol – Clobazam – valproate
Dalic et alMNCN 2014; Larsen et al Neurology 2015; Howell et al Neurology 2015

SCN1A EE Dravet Avoid Sodium Channel blockers
– sodium channel blockers – Carbamazepine – Lamotrigine
– Topiramate – Stiripentol – Clobazam – valproate
Dalic et alMNCN 2014; Larsen et al Neurology 2015; Howell et al Neurology 2015
SCN8A SCN2A EE Seizure control with sodium channel blockers
Carbemazepine Phenytoin Oxcarbazepine

• 1999: Studied as an add-on AED in exploratory observational trial
• Confirmed in two RCT’s as adjunctive therapy to CLB and VPA [19, 20].
• Reduced overall seizure rate by 70 %.
• Mechanism of action: – intrinsic GABAergic effect
– pharmacokinetic interaction by inhibiting cytochrome P450 enzymes [21]

• Amphetamine like drug launched for obesity in the 1990s.
• Withdrawn in 1997 for cardiac side effects
• Acts through inhibition of serotonin uptake and by release of serotonin due to a disruption of vesicular storage
• High density of serotonin receptors in structures critically involved in epilepsies, such as the hippocampus

• Reduced epileptiform activity in zebrafish larvae (antisense knockdown of scn1Lab
• Used as an add-on treatment in 12 patients with Dravet syndrome over 1–19 years. – 8 were seizure-free at their
last follow up visit.
– Patients were seizure-free for a mean of 6 (1–19) years.
– No serious adverse events occurred
• Clinical trials are under way Zhang, et al. PLoS ONE. 2015 Ceulemans et al, Epilepsia, 2012

• Clemizole, a US FDA–approved histamine antagonist, inhibited convulsive behaviors and electrographic seizures in the zebrafish.
• This drug is now a strong candidate for further preclinical evaluation

• KCNT1 encodes a weak voltage-dependent and intracellular sodium-activated potassium channel.
• Mutations in the KCNT1 gene: Missense:
Severe Autosomal dominant nocturnal frontal lobe epilepsy
De novo gain of function:
Epilepsy of infancy with migrating focal seizures
Milligan et al Ann Neurol 2014

• Patients with malignant migrating epilepsy have responded to doses ranging 34.4/kg/d - 60mg/kg/d.
• Bearden et al. treated a patient and reported a dramatic reduction in seizure frequency and developmental improvement
• Mikati et al. showed dramatic reduction in seizure frequency with the same mutation, while the other with a novel phenotype showed no improvement
Bearden et al, Ann Neurol. 2014 Mikati et al, Ann Neurol, 2015

• Cinchona plant
• Quinidine: FDA approved antiarrythmic 200-900 mg/day
• Broad spectrum K channel blocker (KCNT1)
• Also a treatment for Plasmodium falciparum malaria
• Crosses blood brain barrier
• Need evidence of efficacy and to minimize cardiac risk

• 1 week old male
• Diagnosed with malignant migrating partial epilepsy of infancy secondary to KCNT1 mutation (c.G1283A:p.R428Q)
• Following ineffective trials of 6 anti-seizure medications, this patient was trialled on oral quinidine.
• He was titrated up to a dose of 52mg/kg/d with careful cardiac monitoring.
• The parents noted no improvement in seizures.
– Prior to initiation of quinidine, this patient experienced 22 electrographic seizures over 24 hours. At target dose, this patient experienced greater than 70 seizures over 24 hours.

Treatment 1 Wash-out Treatment 2 Wash-out
Sz freq measured Sz freq measured
Day 1 Day 2-4 Day 5-6 Day 7 Day 8-10 Day 11-12
Quinidine 3 days of quinidine 900mg/day versus placebo Decrease to 600 mg/day versus placebo Cardiac monitoring Video-EEG monitoring and precise seizure counts Saul Mullen, Patrick Carney

• Very large number of seizures (14/day)
• Exacerbation ¾
• No statistical effect
New research with Byostatin in the pipeline for the infantile encephalopathy

• A continuum of overlapping neonatal epileptic phenotypes: – KCNQ2-related benign familial neonatal epilepsy
KCNQ2-related epileptic encephalopathy [40].
• Encodes the potassium voltage-gated channel subfamily KQT member 2, also known as Kv7.2.
• KCNQ2-related epilepsies have recently been shown to respond well to drugs known to act on sodium channels including carbamazepine and phenytoin

• Selectively enhances the function of potassium channels formed by neuronal Kv7 subunits
• Significantly increases potassium currents in mutated KCNQ2 channels
• Retigabine: – blue skin discoloration
and retinal pigment abnormalities
– Urinary retention
Porter et al, Epilepsy Res, 2002


• Mutation in the GRIN2A gen encoding the GluN2A subunit of the NMDA receptors have been associated with: – epilepsy-aphasia spectrum – early onset epileptic encephalopathy
• Memantine (NMDAR antagonist) • in vitro: inhibit mutant channels with the mutation c.2434C9 A,
resulting in a leucine to methionine substitution at residue 812. • Decrease in seizure frequency and allowed for tapering of
conventional AEDs. • GluN2A mutation (N615K) was proven sensitive to another NMDAR
antagonist • Memantine is yet to be trialed clinically
Annals Clin Translat Neurol. 2014;
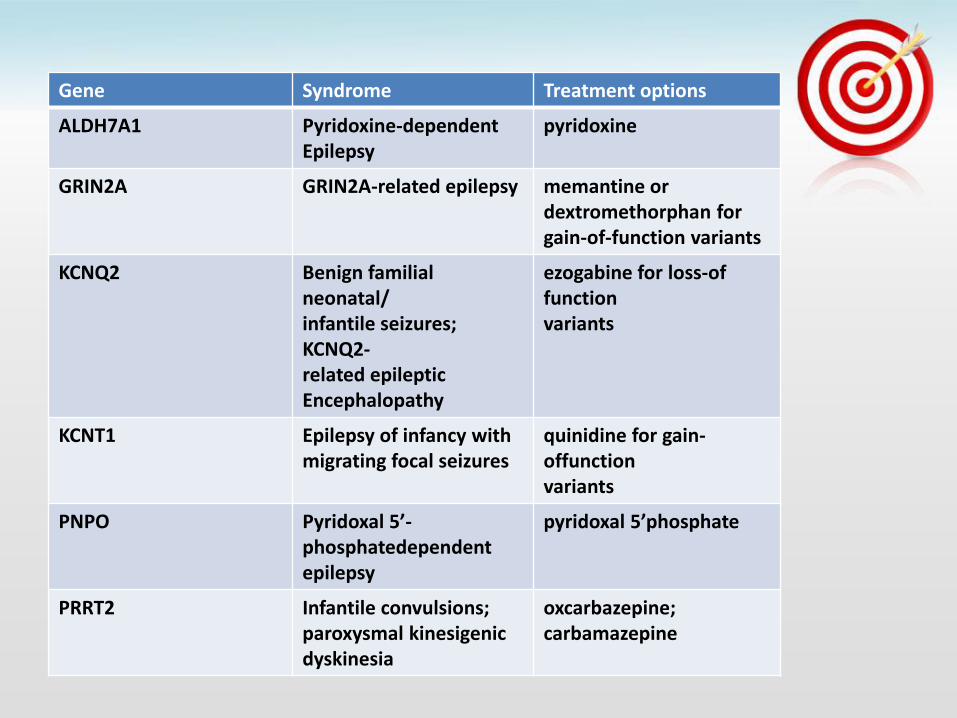
Gene Syndrome Treatment options
ALDH7A1 Pyridoxine-dependent Epilepsy
pyridoxine
GRIN2A GRIN2A-related epilepsy memantine or dextromethorphan for gain-of-function variants
KCNQ2 Benign familial neonatal/ infantile seizures; KCNQ2- related epileptic Encephalopathy
ezogabine for loss-of function variants
KCNT1 Epilepsy of infancy with migrating focal seizures
quinidine for gain-offunction variants
PNPO Pyridoxal 5’-phosphatedependent epilepsy
pyridoxal 5’phosphate
PRRT2 Infantile convulsions; paroxysmal kinesigenic dyskinesia
oxcarbazepine; carbamazepine

Gene Syndrome Treatment Options
Metabotropic glutamate receptor 5
Fragile X MGlurR5 antagonist
Ring chromosome 20
Voltage-gated potassium channel
ezogabine
ATP1A Hemiplegic migraine flunarizine
Nicotinic receptor ADFNLE carbemazepine
SCN8A SCN8A-related epilepsy phenytoin; high-dose carbamazepine
SLC2A1 Glucose transporter deficiency
ketogenic diet
SCN1A Dravet syndrome; SCN1Arelated epilepsy
avoid sodium channel blockers
SCN2A SCN2A-related epilepsy phenytoin; high-dose carbamazepine

Focal epilepsy originating in different brain regions
in different family members
Ingrid E. Scheffer, FRACP, Hilary A. Philips, BSc, Catherine E. O’Brien, MA, Michael M. Saling, PhD, Jacqueline A. Wrennall, MSc, Robyn H. Wallace, BSc(Hons), John C. Mulley, PhD, and Samuel F.
Berkovic, FRACP
Annals of Neurology, 1998

Di EP D C
5 • Mutations in DEPDC5 cause familial focal
epilepsy with variable foci
• 7/8 families had DEPDC5 mutations
• Penetrance mean 66%

• 60% all epilepsy is focal
• Many cases are non-lesional
• In the past, it was believed that these were unlikely genetic
• DEPDC5 mutations in 12% families with >/= 2 individuals with non-lesional focal epilepsy

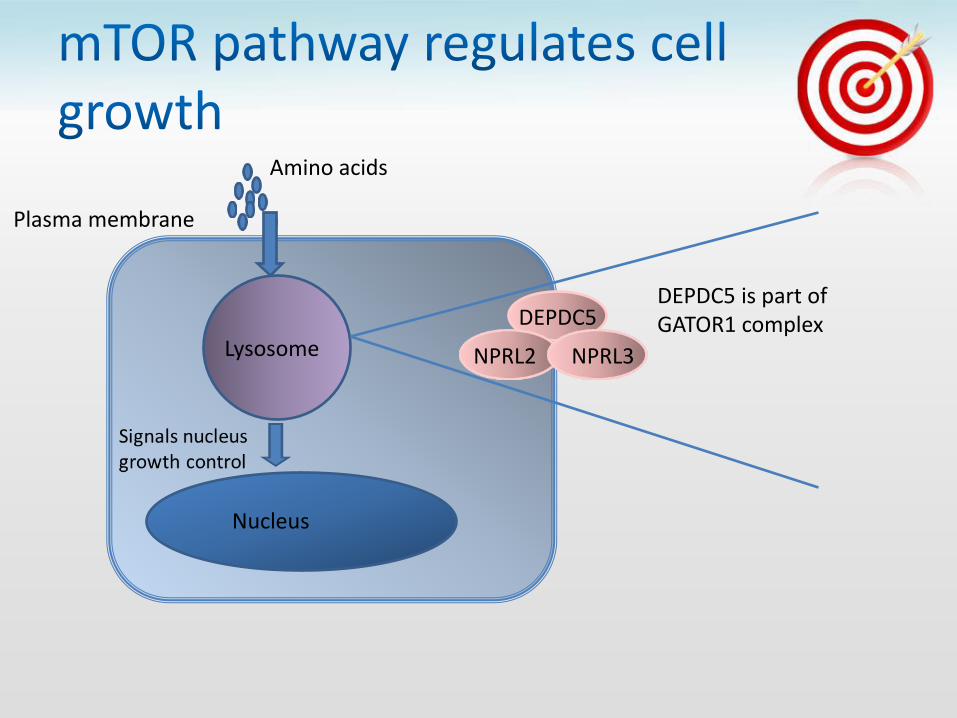
Lysosome
Nucleus
Amino acids
Plasma membrane

Lysosome
Nucleus
Amino acids
Plasma membrane
Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
DEPDC5 is part of GATOR1 complex

Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
DEPDC5 is part of GATOR1 complex
Growth repression

Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
DEPDC5 is part of GATOR1 complex
Signals nucleus growth control
Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
DEPDC5 is part of GATOR1 complex
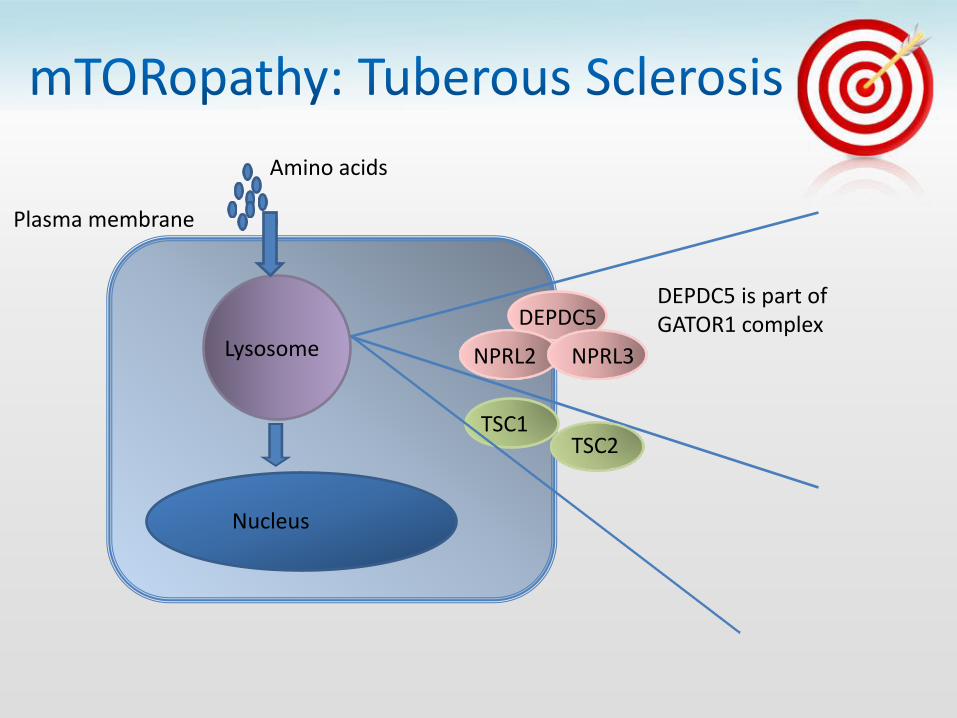
TSC1 TSC2

Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
DEPDC5 is part of GATOR1 complex
TSC1 TSC2

• Nocturnal Frontal Lobe Epilepsy
– FCD IIA in 2 brothers
– Dyspmorphic cytomegalic neurons
• Also:
– FCD IIB
– Hemimegencephaly
Epilepsy Surgery Candidates?

Growth factor
Receptor PIK3CA P13K
AKT3
TSC1
TSC2
Rheb mTORC1
Cell
DEPDC5
Nprl2 Nprl3
RagA
RagC
Hemimegencephaly
Hemimegencephaly
Hemimegencephaly Focal cortical dysplasia
Hemimegencephaly Focal cortical dysplasia
Hemimegencephaly Focal cortical dysplasia
Ricos et al Ann Neurol 2015 Poduri et al 2012 Lee et al 2012

DEPDC5
Nprl2 Nprl3
GATOR 1
mTORC1
Cell growth
RagA
RagC
TSC1
TSC2
Rheb

DEPDC5
Nprl2 Nprl3
GATOR 1
mTORC1
Cell growth
RagA
RagC
TSC1
TSC2
Rheb
GATOR 1 epilepsies Focal epilepsy often mild Onset child-adolescent Intellectual disability rare ASD rare Familial or de novo mutations Variable penetrance Dysplastic lesions FCD I, IIA, IIB
Tuberous Sclerosis Focal epilepsy often severe Onset infant-child Intellectual disability common ASD common Familial or de novo mutations Multiple tubers with FCD IIB

Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
TSC1 TSC2
mTOR inhibitors Everolimus in Tuberous Sclerosis
Reduction of size of SEGA’s EXIST-3 trial
mTOR inhibitors

Lysosome
Nucleus
Amino acids
Plasma membrane
DEPDC5
NPRL3 NPRL2
TSC1 TSC2
mTOR inhibitors Everolimus in Tuberous Sclerosis
Reduction of size of SEGA’s EXIST 3:
mTOR inhibitors
Review MRI for FCD Surgical candidate?

Growth factor
Receptor PIK3CA P13K
AKT3
TSC1
TSC2
Rheb mTORC1
Cell
DEPDC5
Nprl2 Nprl3
RagA
RagC
Hemimegencephaly
Hemimegencephaly
Hemimegencephaly Focal cortical dysplasia
Hemimegencephaly Focal cortical dysplasia
Hemimegencephaly Focal cortical dysplasia
Ricos et al Ann Neurol 2015 Poduri et al 2012 Lee et al 2012

• The phenotypic spectrum of PIK3CA-related segmental overgrowth includes: – bilateral dysplastic megalencephaly
– hemimegalencephaly
– focal cortical dysplasia
• Acute 1 hr-suppression of PI3K signaling despite the ongoing presence of dysplasia has dramatic anti-epileptic benefit.
• PI3K inhibitors are promising for new effective anti-epileptic therapy
Roy et al. eLife 2015

6 day old female with treatment TRE
(trial of 9 anti-seizure medications and the ketogenic diet
While awaiting epilepsy surgery, rapamycin was initiated.

• After 1 week: more than 50% reduction in seizure frequency and the duration of seizures decreased.
• At 2 weeks: the parents felt that for the first time, she was making developmental gains. She also appeared brighter and more interactive.
• Hemispherectomy was deferred by two months, allowing her to gain more weight for decreased risk of complications from the surgery.

• EXIST III Trial for TSC
Reports of seizure control:
• Focal Cortical Dysplasia (esp FCD II)
• Polyhydramnios Megencephaly and Symptomatic Epilepsy
• Hypothalamic Hamartoma
Lee et al, Nature genetics. 2012; D'Gama et al, Annals of Neurology, 2015; French et al, Lancet 2016

Genetic Pathway diagnosis
Medical and surgical management

• Predicting adverse affects
• Ketogenic diet
• CBD
• Rational polypharmacy?
• Future

• HLA‐B*1502 allele: highly associated with CBZ SJS in Han Chinese , as well as phenytoin, and lamotrigine induced SJS
• Identification of cellular and molecular biomarkers including RNA, microRNA, protein and metabolites from blood and CSF is needed.
• May allow:
– early prediction of disease following a brain insult or a first seizure,
– measurement of progression
– stratification of drug resistance based on drug responsiveness.

- First-line treatment for:
GLUT1 deficiency PDH deficiency
- Metabolic disorders:
Phosphofructokinase deficiency. Glycogen storage disease type V. Mitochondrial respiratory chain complex disorders.
- Epileptic syndromes:
Myoclonic astatic epilepsy Seizures caused by tuberous sclerosis complex West syndrome refractory to vigabatrin or adrenocorticotropic hormone (ACTH) Dravet syndrome Angelman
- Symptomatic epilepsies:
Lafora body disease. Seizures caused by Rett syndrome Landau-Kleffner syndrome Sub acute sclerosing panencephalitis Febrile infection-related epilepsy syndrome Refractory status epilepticus
Alberti et al. Arch Argent Pediatr 2016

• CBD may reduce neuronal excitability and neuronal transmission by:
– TRP channels
– G-coupled protein receptor protein 55 (GPR55)
– voltage-dependent anion-selective channel protein 1 (VDAC1)
• Several publications of parental reports on the efficacy of CBD for children with Dravet syndrome, Lennox Gastaut Syndrome or Inafantile Spasms.
– Overall responder rate: (33-89%)
– Seizure freedom: (11-14%)
– Much higher responder rate of 89% for the LGS patients
• RCT’s are required to evaluate efficacy, dosing and safety
• Such studies are currently underway for patients with Dravet syndrome and LGS Devinsky et al, Epilepsia 2014, Hussain et al, Epilepsy
Behav: E&B. 2015

• Combination of drugs with different, rather than the same, mechanisms of action.
• Does it work in clinical practice?
• Perampanel:
– Selective noncompetitive AMPA-receptor antagonist
– May be effective in patients with epilepsy and movement disorders
– Examined in Parkinson’s Disease
– Progressive myoclonic epilepsies
Rascol et al, Clin Neuropharmacol. 2012; Goldsmith et al, Epilepsy Behav. 2016

• Large scale (national & international) cohort and registration studies for epilepsy
• Fast, convenient and economic genetic testing
• Enhanced bioinformatics capacity for data analysis to improve the accuracy of risk assessment and genetic counseling
• Better/More functional studies for pathogenic variation
• Develop more specific / “tailored” AEDs therapy, even gene therapy through specific genetic markers or targets, or based on certain pathogenic pathway

EpiPM Consortium, Lancet, 2015