analysis of sage data: an introduction kevin r. coombes section of bioinformatics
TRANSCRIPT

Analysis of SAGE Data:An Introduction
Kevin R. CoombesSection of Bioinformatics

Outline
• Description of SAGE method• Preliminary bioinformatics issues• Description of analysis methods
introduced in early paper• Review of literature: statistics and
SAGE

What is SAGE?
• Serial Analysis of Gene Expression• Method to quantify gene expression
levels in samples of cells• Open system
– Can potentially reveal expression levels of all genes: “unbiased” and “comprehensive”
– Microarrays are closed, since they only tell you about the genes spotted on the array
Ref: Velculescu et al., Science 1995; 270:484-487
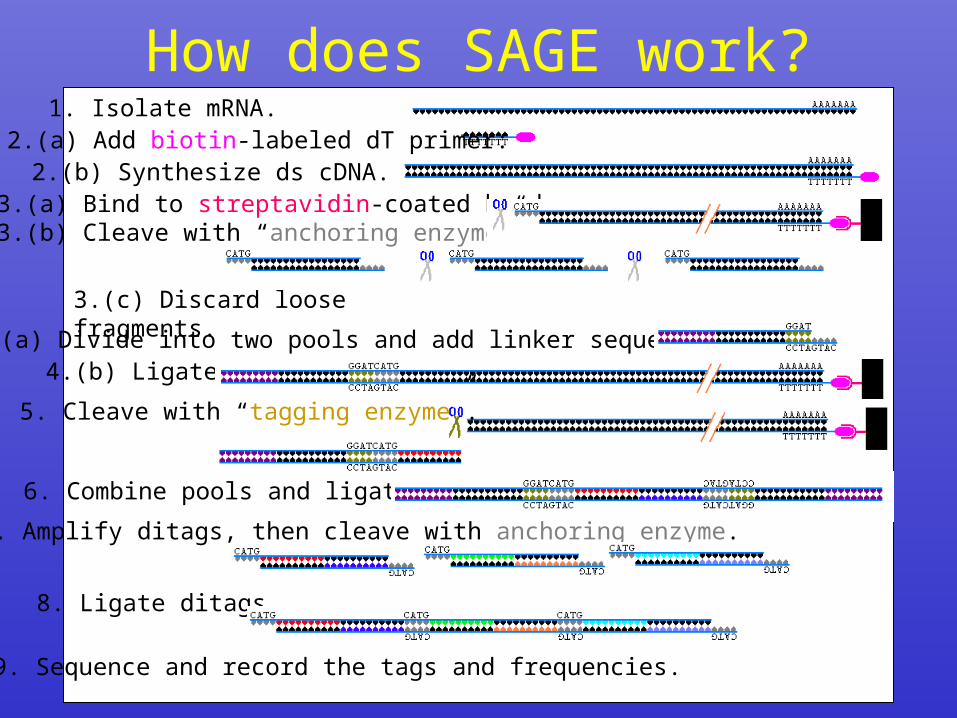
How does SAGE work?1. Isolate mRNA.
2.(b) Synthesize ds cDNA.2.(a) Add biotin-labeled dT primer:
4.(a) Divide into two pools and add linker sequences: 4.(b) Ligate.
3.(c) Discard loose fragments.
3.(a) Bind to streptavidin-coated beads.3.(b) Cleave with “anchoring enzyme”.
5. Cleave with “tagging enzyme”.
6. Combine pools and ligate.
7. Amplify ditags, then cleave with anchoring enzyme.
8. Ligate ditags.
9. Sequence and record the tags and frequencies.

From ditags to counts
• Locate the punctuation “CATG”• Extract ditags of length 20-26 between the
punctuation• Discard duplicate ditags (including in
reverse direction) -- probably PCR artifacts• Take extreme 10 bases as the two tags,
reversing right-hand tag• Discard linker sequences• Count occurrences of each tag
SAGE software available at http://www.sagenet.org
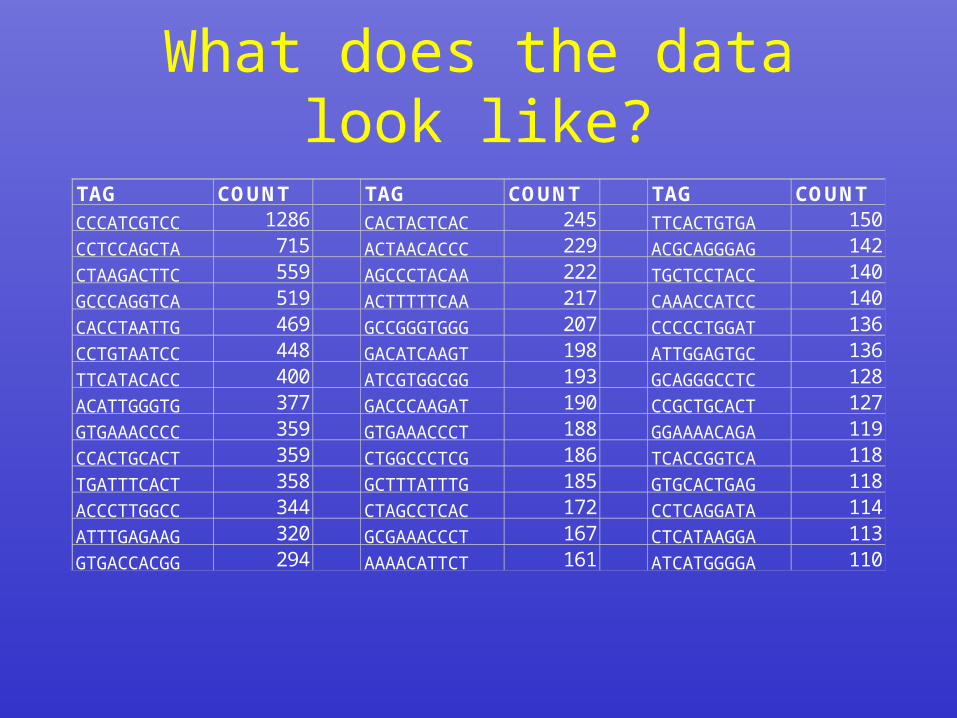
What does the data look like?
TAG COUNT TAG COUNT TAG COUNTCCCATCGTCC 1286 CACTACTCAC 245 TTCACTGTGA 150CCTCCAGCTA 715 ACTAACACCC 229 ACGCAGGGAG 142CTAAGACTTC 559 AGCCCTACAA 222 TGCTCCTACC 140GCCCAGGTCA 519 ACTTTTTCAA 217 CAAACCATCC 140CACCTAATTG 469 GCCGGGTGGG 207 CCCCCTGGAT 136CCTGTAATCC 448 GACATCAAGT 198 ATTGGAGTGC 136TTCATACACC 400 ATCGTGGCGG 193 GCAGGGCCTC 128ACATTGGGTG 377 GACCCAAGAT 190 CCGCTGCACT 127GTGAAACCCC 359 GTGAAACCCT 188 GGAAAACAGA 119CCACTGCACT 359 CTGGCCCTCG 186 TCACCGGTCA 118TGATTTCACT 358 GCTTTATTTG 185 GTGCACTGAG 118ACCCTTGGCC 344 CTAGCCTCAC 172 CCTCAGGATA 114ATTTGAGAAG 320 GCGAAACCCT 167 CTCATAAGGA 113GTGACCACGG 294 AAAACATTCT 161 ATCATGGGGA 110

From tags to genes• Collect sequence records from GenBank that
are represented in UniGene• Assign sequence orientation (by finding poly-
A tail or poly-A signal or from annotations)• Extract 10-bases 3’-adjacent to 3’-most
CATG• Assign UniGene identifier to each sequence
with a SAGE tag• Record (for each tag-gene pair)
– #sequences with this tag– #sequences in gene cluster with this tag
Maps available at http://www.ncbi.nlm.nih.gov/SAGE

From tags to genes
• Ideal situation:– one gene = one tag
• True situation– one gene = many tags (alternative
splicing; alternative polyadenylation)– one tag = many genes (conserved 3’
regions)

Sequencing Errors
• Estimated sequencing error rate:– 0.7% per base (range 0.2% - 1%)
• Affect– ditags in a SAGE experiment
• can improve by using phred scores and discarding ambiguous sequences
– tag-gene mappings from GenBank• RNA better than EST

Reliable tag-gene assignments
Sequence Type Poly-A signal Annotation
mRNA/cDNA NA NA
EST Yes 3’
EST Yes None
EST Yes 5’
EST No 3’

SAGE and cancer
• Ten SAGE libraries, two each from– normal colon– colon tumors– colon cancer cell lines– pancreatic tumors– pancreatic cell lines
• Pooled each pair
Ref: Zhang et al., Science 1997; 276:1268-1272

Variability in SAGE libraries
NC1
NC2
0 2 4 6 8 10
02
46
810

Distribution of tags
• 303,706 total tags• 48,471 distinct tags• Distribution
– 85.9% seen up to 5 times (25% of mass)
– 12.7% between 5 and 50 times (30%)– 0.1% between 50 and 500 times
(26%)– 0.1% more than 500 times (19%)Ref: Zhang et al., Science 1997; 276:1268-1272

How many tags were missed?
• They simulated to find 92% chance of detecting tags at 3 copies/cell
• Using binomial approximation– Get 95% chance for 3 copies/cell– Only get 63% chance for 1 copy/cell
• Most of what they saw occurred at 1-5 copies per cell

Differential Expression
• Found 289 tags differentially expressed between normal colon and colon cancer (181 decreased; 108 increased)
• Method: Monte Carlo simulation.– 100000 sims per transcript for relative
likelihood of seeing observed difference– Used observed distribution of transcripts
to simulate 40 experiments.
Ref: Zhang et al., Science 1997; 276:1268-1272

Sensitivity
• Claim: 95% chance of detecting 6-fold difference
• Method: Monte Carlo– 200 simulations, assuming
abundance of 0.0001 in first sample and 0.0006 in second sample
Ref: Zhang et al., Science 1997; 276:1268-1272

Weaknesses in Analysis
• Failed to account for intrinsic variability in samples (which changes depending on abundance) in assessing significance
• Monte Carlo used observed distribution, which is definitely not true distribution.
• Sensitivity only measured at one abundance level.

Alternative Analytic Methods
• Audic and Claverie, Genome Res 1997; 7:986-995
• Chen et al., J Exp Med 1998; 9:1657-1668• Kal et al., Mol Biol of Cell 1999; 10:1859-1872• Michiels et al., Physiol Genomics 1999; 1:83-91• Stollberg et al., Genome Res 2000; 10:1241-
1248• Man et al., Bioinformatics 2000; 16:953-959

Audic and Claverie
• Main goal: confidence limits for differential expression
• Use Poisson approximation for number of times x you see the same tag.
• Put a uniform prior on the Poisson parameter; get posterior probability of see tag y times in new experimentp(y | x) = (x + y)! / [x! y! 2^(x + y +1)]
• Generalizes to unequal sample sizes

Chen et al.
• Assume– equal sample sizes– tag has concentration X, Y in two samples
• Look at W = X/(X+Y)• Use a symmetric Beta prior distribution with a
peak near 0.5 (since most genes don’t change)
• Use Bayes theorem to compute posterior probability of threefold difference in expression

Unequal sample sizes
• This analysis generalizes easily to the case of unequal size SAGE libraries– Lal et al., Cancer Res 1999; 59:5403-5407
• This method is used at the NCBI SAGEmap web site for online differential expression queries– http://www.ncbi.nlm.nih.gov/SAGE

Kal et al.
• Assume the proportion of times you see a tag has binomial distribution
• Replace with a normal approximation to compute confidence limits
• Used at http://www.cmbi.kun.nl/usage• Equivalent to chi-square test on 2x2
table: Library 1 Library 2
Gene ofInterest
x1 x2
All OtherGenes
N1 N2

Michiels et al.
• First perform overall chi-square test to decide if the two SAGE libraries being compared are different.
• Get significance by Monte Carlo simulation
• Perform gene-by-gene chi-square tests and use them to rank genes in order of “most likely to be different”

Stollberg et al.
• Assume binomial distributions • Model the binomial parameters as a
sum of two exponentials – fit to the Zhang step function data
• Simulate from this model, adding– sequencing errors– nonuniqueness of tags– nonrandomness of DNA sequences

Stollberg et al.
• Key finding:– Naively using observed data to fit
model parameters cannot recover the observed data by simulation
– Maximum likelihood estimate of parameters that recover the observed data give very different looking parameters
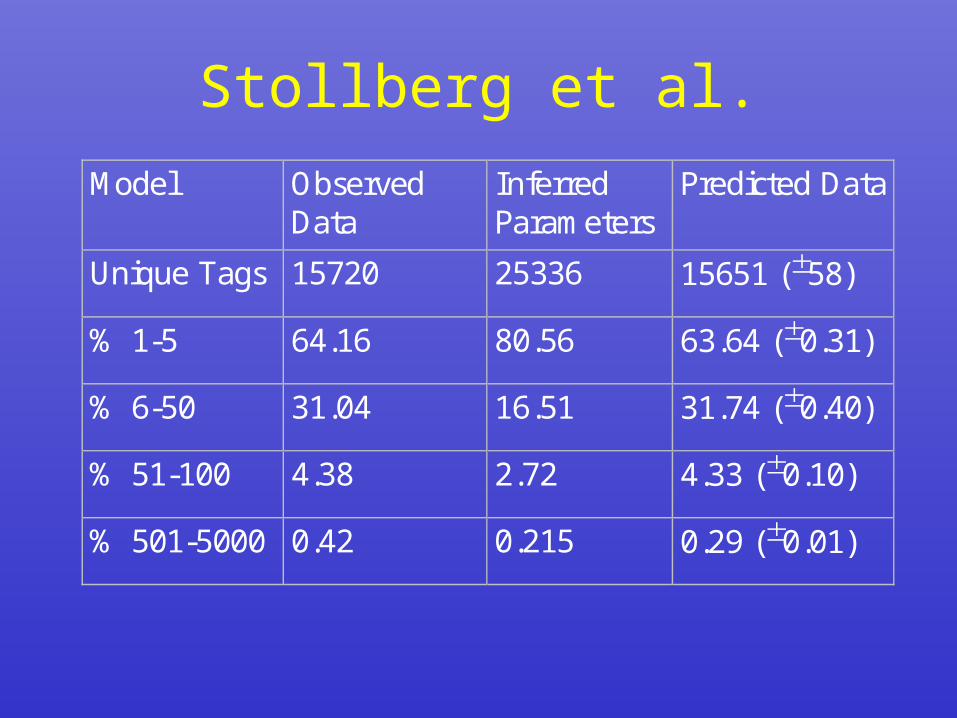
Stollberg et al.Model Observed
DataInferredParameters
Predicted Data
Unique Tags 15720 25336 15651 (58)
% 1-5 64.16 80.56 63.64 (0.31)
% 6-50 31.04 16.51 31.74 (0.40)
% 51-100 4.38 2.72 4.33 (0.10)
% 501-5000 0.42 0.215 0.29 (0.01)

Man et al.
• Compares specificity and sensitivity of different tests for differential expression– Audic and Claverie– Kal– Fisher’s exact test
• Monte Carlo simulation of experiments• Findings
– Similar power at high abundance– Kal has highest power at low abundance

Questions
• Sample size computations:– How many tags should we sequence if we
want to see tags of a given frequency?– How many tags should we sequence if we
want to see a given percentage of tags?
• How many tags are expressed in a sample?
• Best method for identifying differential expression?

Additional SAGE references
• Review– Madden et al., Drug Disc Today 2000; 5:415-425
• Online Tools– Lash et al., Genome Res 2000; 10:1051-1060– van Kampen et al., Bioinformatics 2000; 16:899-905
• Comparison of SAGE and Affymetrix– Ishii et al., Genomics 2000; 68:136-143
• Combine SAGE and custom microarrays– Nacht et al., Cancer Res 1999; 59:5464-5470
• Mapping SAGE data onto genome– Caron et al., Science 2001; 291:1289-1292
• Data mining the public SAGE libraries– Argani et al., Cancer Res 2001; 61:4320-4324