analysis of potato virus x replicase and tgbp3 subcellular locations.pdf
TRANSCRIPT

Virology 393 (2009) 272–285
Contents lists available at ScienceDirect
Virology
j ourna l homepage: www.e lsev ie r.com/ locate /yv i ro
Analysis of potato virus X replicase and TGBp3 subcellular locations
Devinka Bamunusinghe a,1, Cynthia L. Hemenway b, Richard. S. Nelson c, Anton A. Sanderfoot d,Chang M. Ye a, Muniwarage A.T. Silva a, M. Payton e, Jeanmarie Verchot-Lubicz a,⁎a Department of Entomology and Plant Pathology, Noble Research Center, Oklahoma State University, Stillwater, OK 74078, USAb Department of Molecular and Structural Biochemistry, North Carolina State University, Raleigh, NC 27695-7622, USAc The Samuel Roberts Noble Foundation 2510 Sam Noble Pky., Ardmore, OK 73401, USAd Biology Department, University of Wisconsin-LaCrosse 1725 State St. La Crosse, WI 54601, USAe Department of Statistics, Oklahoma State University, Stillwater, OK 74078, USA
⁎ Corresponding author. Fax: +1 405 744 6039.E-mail addresses: [email protected] (D. Bamunusi
[email protected] (C.L. Hemenway), [email protected] (A.A. Sanderfoot), Verchot.lub(J. Verchot-Lubicz).
1 Current address for Devinka Bamunusinghe is UnivDepartment of Plant Pathology and Microbiology, 340Riverside, CA 92521.
0042-6822/$ – see front matter. Published by Elsevierdoi:10.1016/j.virol.2009.08.002
a b s t r a c t
a r t i c l e i n f oArticle history:Received 13 May 2009Returned to author for revision 9 July 2009Accepted 2 August 2009Available online 3 September 2009
Keywords:PotexvirusPotato virus XTriple gene blockReplicase
Potato virus X (PVX) infection leads to certain cytopathological modifications of the host endomembranesystem. The subcellular location of the PVX replicase was previously unknown while the PVX TGBp3 proteinwas previously reported to reside in the ER. Using PVX infectious clones expressing the green fluorescentprotein reporter, and antisera detecting the PVX replicase and host membrane markers, we examined thesubcellular distribution of the PVX replicase in relation to the TGBp3. Confocal and electron microscopicobservations revealed that the replicase localizes in membrane bound structures that derive from the ER. Asubset of TGBp3 resides in the ER at the same location as the replicase. Sucrose gradient fractionationshowed that the PVX replicase and TGBp3 proteins co-fractionate with ER marker proteins. This localizationrepresents a region where both proteins may be synthesized and/or function. There is no evidence toindicate that either PVX protein moves into the Golgi apparatus. Cerulenin, a drug that inhibits de novomembrane synthesis, also inhibited PVX replication. These combined data indicate that PVX replication relieson ER-derived membrane recruitment and membrane proliferation.
Published by Elsevier Inc.
Introduction
Successful virus infection is a consequence of specific interactionswith host subcellular machinery to enable essential events such asviral protein synthesis, replication, and cell-to-cell spread. The abilityof viruses to interact with host components underlies the appropriatesubcellular targeting of viral proteins and nucleic acids (Ahlquist,2006; Buck, 1999a; Hwang et al., 2008). It is often presumed that aviral movement protein (MP) carries infectious material intouninfected cells in the form of a ribonucleoprotein complex, althoughthe exact mechanism bywhich viral genomes are transferred from thesite of synthesis to the plasmodesmata has not been clearly shown(Haywood et al., 2002; Kiselyova et al., 2001; Lucas, 2006; Nelson,2005; Verchot-Lubicz et al., 2007).
For most positive strand RNA viruses, viral replicases causemembrane rearrangements to create a container or protectedenvironment for genome replication (Mackenzie, 2005; Salonen et
nghe),@noble.org (R.S. Nelson),[email protected]
ersity of California- Riverside,1, Watkins Drive, Boyce Hall,
Inc.
al., 2005; Sanfacon and Zhang, 2008; Schwartz et al., 2004; Villanuevaet al., 2005). There are wide ranging reports of plant-infecting RNAviruses associating with invaginations of the endoplasmic reticulum(ER), chloroplast outer membrane, vacuolar membranes, mitochon-dria, nuclear envelope, or peroxisomes (Goodin et al., 2007; Hwang etal., 2008; Jonczyk et al., 2007; Kim et al., 2006; Prod'homme et al.,2003; Rubino et al., 2001; Turner et al., 2004; Wei and Wang, 2008).Grapevine fanleaf virus (GFLV), Cowpea mosaic virus (CPMV), Redclover necrotic mosaic virus (RCNMV) and Tobacco mosaic virus (TMV)are a few recent examples of viruses whose replication requiresproliferation and/or invagination of ER membranes to sustain virusreplication (Carette et al., 2000; Heinlein et al., 1998; Mas and Beachy,1999; Ritzenthaler et al., 2002; Turner et al., 2004). CPMV in particularprovides an example of a virus that stimulates host cells to increasemembrane synthesis resulting in specific expansion of the ER with noobvious change in the Golgi apparatus (Carette et al., 2000), althoughlittle is known about how viruses are able to induce synthesis ofdistinct types of cellular membranes.
Tobacco mosaic virus (TMV) replicase is located on the ER or ERmodifications (Buck, 1999b; Ishikawa and Okada, 2004). For TMV,irregular shaped membrane bound structures containing the 183-and 126-kDa replication-associated proteins and viral genomes serveas a vehicle for trafficking virus within and between neighboringcells (Kawakami et al., 2004; Liu et al., 2005). A current model is thatthe TMV replication associated proteins and/or MP, through an

273D. Bamunusinghe et al. / Virology 393 (2009) 272–285
association with a membrane bound complex, traffics viral RNA to thecell wall near plasmodesmata, where it passes through the desmotu-bule (Asurmendi et al., 2004; Guenoune-Gelbart et al., 2008; Reicheland Beachy, 1998).
Potato virus X (PVX) is the type member of the Potexvirus genusand its genome contains five open reading frames (ORFs) (Huisman etal., 1988). The PVX replicase is a single 166 kDa protein whichcontains methyl transferase, helicase, and polymerase domainsrequired for virus replication (Davenport and Baulcombe, 1997). Aspecific PVX protein or protein domain that anchors the replicase tocellular membranes has not been identified. In addition, the site forPVX replication has not been described. The remaining four PVXproteins include the coat protein (CP) and triple gene block proteins(TGB) that are required for cell-to-cell movement and encapsidation(Huisman et al., 1988). The TGB proteins are derived from threepartially overlapping ORFs and are named TGBp1, TGBp2, and TGBp3(Beck et al., 1991; Verchot et al., 1998). TGBp1 is a multifunctionalprotein that suppresses gene silencing, has RNA binding and helicaseactivity, unwinds virion particles to promote translation of genomicRNA, and is responsible for increasing plasmodesmal permeabilityenabling viruses to pass from an infected cell into an uninfected cell(Bayne et al., 2005; Howard et al., 2004; Hsu et al., 2004; Kalinina etal., 2002; Kiselyova et al., 2003; Lough et al., 2000; Lough et al., 1998;Rodionova et al., 2003). While a typical plasmodesma restrictsdiffusion of molecules less than 1 kDa, TGBp1 causes expansion ofthe pore to enable transfer of larger macromolecules between cells.PVX TGBp2 and TGBp3 are low molecular weight (12 and 8 kDa,respectively) membrane-binding proteins (Ju et al., 2005, 2008;Krishnamurthy et al., 2003; Mitra et al., 2003). Since antibodies arenot available for immunodetection of TGBp2 or TGBp3, priorinvestigations employed noninvasive imaging of intrinsically fluores-cent proteins (GFP, YFP, CFP) fused to TGBp2 or TGBp3 to visualizeprotein accumulation in leaves and protoplasts (Samuels et al., 2007;Schepetilnikov et al., 2005; Zamyatnin et al., 2006). In addition,electron microscopic analysis of transgenic plants expressing GFPfused to TGBp2 or TGBp3 identified GFP–TGBp2 associating with theER and novel vesicles, whereas TGBp3–GFP fusions associated mainlywith ER strands. The ER lumenal binding protein BiP was identified inall TGBp2 and TGBp3 containing membrane compartments (Ju et al.,
Fig. 1. Infectious clones of PVX.GFP and PVX.TGBp3–GFP show green fluorescent foci. (A) Scprotein from a duplicated subgenomic RNA. PVX.TGBp3–GFP has GFP fused to the 3′ end of threspectively, were photographed at 3–5 dpi and were similar in dimension. Both viruses sp
2005; Krishnamurthy et al., 2003; Samuels et al., 2007). Importantly,we have noted the presence of spherical bodies containing TGBp3–GFP fusions in PVX infected cells and the nature of these TGBp3-containing structures remains unclear. These might be the samevesicles reported for TGBp2 or they represent another membranecompartment induced during PVX infection. More importantly,TGBp3–GFP is seen in spherical bodies only during virus infectionpointing to the possibility that another viral protein(s) are responsiblefor directing TGBp3–GFP from the polygonal ER into these structures.
The ER is vital for PVX cell-to-cell movement. Mutations blockingthe ability of TGBp2 or TGBp3 to bind to the ER inhibit virus cell-to-cell movement (Krishnamurthy et al., 2003). Recent attempts todissect the activities of TGBp2 led to the discovery of TGBp2-inducedvesicles that bud from the ER and are essential for virus traffickingbetween cells (Ju et al., 2005). The contents of these vesicles beyondthe presence of TGBp2 and BiP are unknown.
This study was undertaken to dissect the spatial relationships ofPVX replicase and the membrane bound TGBp3. We hypothesizethat, should PVX TGBp3 and the replicase lie in proximity, TGBp3may aid in the acquisition of newly synthesized viral genomes fromthe viral replicase. Considering that the ER and ER-derived vesiclesare required for virus movement, it is possible that the viral replicasemay also associate with the ER or ER-derived bodies, as was observedfor TMV. We used infectious clones of PVX expressing the reporterGFP, and antibodies to monitor the subcellular distribution of thePVX replicase and TGBp3 in relationship to host constituents duringinfection.
Results
Spherical membrane bound bodies contain PVX replicase and TGBp3 invirus infected leaves
In this investigation we explored the spatial relationship of thePVX replicase and TGBp3–GFP during PVX infection of N. benthamianaleaves. PVX.TGBp3–GFP is an infectious clone of PVX that contains GFPfused to the TGBp3 coding sequence in a manner that does not hindervirus infection (Fig. 1). We have previously reported that TGBp3–GFPfluorescence is seen in the tubular ER network with fluorescent
hematics of the viral genomes with GFP included. PVX.GFP has GFP expressed as a freee TGBp3 coding sequence. (B, C) Infection foci containing PVX-GFP and PVX.TGBp3–GFP,read systemically in N. benthamiana. Scale bar is 1 mm.

274 D. Bamunusinghe et al. / Virology 393 (2009) 272–285
spheres at the vertices of the connecting tubules (Ju et al., 2005, 2007;Krishnamurthy et al., 2003; Samuels et al., 2007). Immunofluores-cence and immunogold labeling in this study was carried out usinginfection foci that were similar in size to those presented in Fig. 1.
To clearly represent the polygonal nature of the ER network, N.benthamiana leaves were bombarded with plasmids expressing GFP-er, which has GFP fused to ER targeting and retention signals (Haseloffand Amos, 1995). Fig. 2A shows GFP-er fluorescence highlightsinterconnecting tubules that form a meshwork lattice around the cell.Furthermore, leaf epidermal cells bombarded with plasmids expres-sing TGBp3–GFP (Haseloff and Amos, 1995; Krishnamurthy et al.,2003) show a similar network, although the tubules appear shorter incomparison to GFP-er expressing cells, and there are cisternae at theintersection of connecting tubules (Fig. 2B). Intriguingly, the patternof spherical bodies seen in PVX.TGBp3–GFP infected cells is notexactly reproduced in cells expressing TGBp3–GFP alone (compareFigs. 2B and C), suggesting that there may be other PVX factors
Fig. 2. Localization of PVX replicase and TGBp3 proteins through fluorescence imaging of N.pRTL2-GFPer or -TGBp3–GFP, respectively. Arrows in panel B point to examples of ER cisternpanels A and B. Arrowheads point to vesicles. Bars represent 10 μm. (D–I) Immunolabelinggreen fluorescence due to TGB3–GFP fusion and red fluorescence indicating PVX replicafluorescence overlap. (D–F) Bars represent 10 μm. (G–I) Bars represent 200 μm. (J–L) Mofluorescence images, respectively show no indication of signal in mesophyll cell. Outline of
contributing to the build-up of these bodies. Since we were interestedto examine the membrane association of the PVX replicase, wehypothesized that the PVX replicase and TGBp3–GFP reside inproximity to facilitate transfer of newly synthesized genomic RNAsinto complexes with the viral movement proteins to facilitate viralegress.
Therefore we compared the spatial patterns of PVX replicase andTGBp3–GFP accumulation using immunofluorescence labeling andconfocal microscopy. PVX.TGBp3–GFP infected fluorescent foci wereharvested from N. benthamiana leaves at 7 dpi, when infection fociwere typically 10–15 cells in diameter. Transverse sections were fixedand subjected to immunolabeling using PVX replicase antiserafollowed by Alexafluor 633 conjugated secondary antisera. GFPfluorescence was not diminished by the immunolabeling processallowing comparison of the green and red fluorescence patterns inepidermal and mesophyll cells. Initial observations of TGBp3–GFPfluorescence confirmed the general integrity of the ER following
benthamiana leaf epidermal cells after infection. (A and B) Cells were bombarded withae. (C) PVX.TGBp3–GFP infected cell shows similar polygonal tubular network as seen inof PVX.TGBp3–GFP infected leaf epidermal cell (D–F) and mesophyll cells (G–I) showsse. (F, I) represent overlaid images show yellow highlighting where red and greenck inoculated leaf epidermal cell treated with replicase antisera. (J, K) Green and redmesophyll cell is shown in white. Bars represent 10 μm.

275D. Bamunusinghe et al. / Virology 393 (2009) 272–285
fixation and immunolabeling. In epidermal cells, the tubular networkis more condensed following fixation than the dispersed networkcommonly seen in living cells (compare Figs. 2B, C, and D). The greenfluorescent spheres remained associated with the ER network. Inmesophyll cells, which have a greater abundance of chloroplasts, theER is compressed by both the vacuole and chloroplasts making itdifficult to characterize ER architecture, although the spherical bodiesare easily identified. Immunolabeling of both epidermal (Figs. 2D–F)and mesophyll cells (Figs. 2G–I) shows spherical bodies co-labeledwith green and red fluorescence which appear yellow in the mergedimages (Figs. 2D–F). These data indicate that, although TGBp3–GFPbroadly associates with the ER network (Fig. 2F), the PVX replicaseand TGBp3 coincide in spherical ER-associated bodies.
PVX replicase antisera have been used extensively in pastbiochemical analysis of polymerase activity and its ability to reliablydetect the PVX replicase is well established (Braun and Hemenway,1992). However, for the purpose of this study, we included controls todemonstrate that red fluorescence is due to immuno-recognition ofthe PVX replicase. Transverse sections of mock inoculated leafsegments were subjected to the same immunolabeling procedures.We found no evidence of immunofluorescence labeling of mockinoculated host tissues (Figs. 2J, K).
PVX replicase localizes to spherical bodies along the ER networkin protoplasts
Given that leaf segments harvested at 7 dpi likely fail to representearly events during virus infection, and our inability to identifyinfected leaf cells at earlier times, we employed a protoplast systemfor studying the patterns of viral protein accumulation during the first48 h following inoculation. Protoplasts were prepared from BY-2suspension cells and inoculated with PVX–GFP (Figs. 3A–E) or PVX.TGBp3–GFP (Figs. 3H–M). PVX–GFP shows cytosolic and nuclearfluorescence that does not change over time. PVX replicase wasdetected in vesicles surrounding the nucleus and throughout theperiphery of the cell. At high magnification in Fig. 3D the GFPfluorescence has a lot of black holes in it. When we superimpose thered fluorescence image in Fig. 3E it is clear that these holes contain redfluorescence, indicating that GFP is excluded from the red spherescontaining the PVX replicase.
TGBp3–GFP fluorescence in fixed protoplasts during the earlyhours of infection is less intense than in leaf sections that wereinfected for seven days. Therefore, we employed dual-immunofluo-rescence labeling using PVX replicase polyclonal and GFP monoclonalantiserum. Alexafluor 633 conjugated secondary anti-goat sera wasused to detect replicase with peak emissions at 650 nm, whileAlexafluor 488 conjugated secondary anti-mouse sera was used todetect GFP, with peak emissions at 520 nm.
For PVX.TGBp3–GFP, green fluorescence was seen throughout theER network and in the nucleus (Fig. 3C) as reported previously (Ju etal., 2008; Samuels et al., 2007). We also reported that the ER changesshape during PVX infection of protoplasts. The ER is more densearound the nucleus in virus infected protoplasts and the strands areshorter (compare Figs. 3F and G with Figs. 3H and J) (Samuels et al.,2007). Red fluorescence identifies PVX replicase residing in sphericalbodies along the tubular network at 12 h post inoculation (Figs. 3I andJ). These data show the close association between the PVX replicaseand TGBp3, even at early stages of infection. Later at 48 h, the ERreticulum is less apparent and spherical bodies containing red andgreen fluorescence are seen in the cortical regions of the cell (Figs. 3K–M). Notably in protoplasts and in leaves the PVX replicase and TGBp3–GFP do not completely coincide. TGBp3–GFP and the replicase arelocated in the spherical bodies, but TGBp3 is also observed throughoutthe cortical ER.
To verify immunodetection of PVX replicase and TGBp3–GFP,mock-inoculated samples were treated with PVX replicase or GFP
antisera along with the appropriate secondary antisera. Theseprotoplasts failed to show evidence of green or red fluorescence(Figs. 3F–H). In addition, virus infected samples treated with bufferand secondary antisera did not exhibit red or green fluorescence (datanot shown).
PVX replicase related spherical bodies are membrane bound and containER markers
Although TGBp3–GFP and replicase have their own characteristicpatterns of subcellular accumulation, the combined leaf and proto-plast data indicate that the PVX replicase and TGBp3 do appear in thespherical bodies. To better visualize the precise membrane compart-ments containing TGBp3 and replicase we employed immunogoldlabeling and transmission electron microscopy. Thin sections of PVX.TGBp3–GFP-infected and healthy tobacco leaf segments were treatedwith GFP antisera alone, or both replicase and BiP antiserum. BiP is anER resident chaperone which is not present at high concentration inthe other areas of the endomembrane system and is typically used toconfirm the origin of ER-derived structures in virus infected cells(Fontes et al., 1991). For dual labeling experiments, secondaryantisera conjugated with 10 nm gold particles were used to detectBiP while secondary antiserum conjugated with 20 nm gold particleswere used to detect PVX replicase.
Immunogold labeling patterns were broadly analyzed to assess avariety of subcellular compartments. First, the distribution of goldparticles in major compartments of the cell was examined. Goldparticles were counted in 10 μm2
fields and were scored for theirassociation with the cytoplasm, vacuole, cell wall (Fig. 4A), chloro-plast (Figs. 4A, B, and C), and ER (Figs. 4D, E, and F). The averagenumbers of gold particles labeling specific structural components ofthe cell were reported in Table 1. Tabulated results were analyzedstatistically to provide an essential assessment of the subcellularlocations containing significant quantities of immunogold label.
Both PVX replicase (10 nm particles) and BiP (20 nm particles)associatedmainlywith strands of ER network (Table 1 and Figs. 4D–G).Moreover, the greatest amount of immunogold label detectingTGBp3–GFP was also along strands of ER (∼2 to 4 particles/10 μm2
area). Some replicase and TGBp3–GFP labeling was detected in thecytoplasm (∼1 particle/10 μm2), although the amount was not statis-tically significant.
PVX replicase, BiP and TGBp3–GFP labeling was not observedalong the cell wall at a significant levels (b1/10 μm2). There was nosignificant labeling in mock inoculated tissues (b1/10 μm2) probedwith antisera against PVX replicase or GFP, indicating the specificity ofthe respective antisera (Table 1 and data not shown).
A second analysis was conducted to quantify the extent ofimmunogold label associated with various organelles and vesicles.Table 2 presents the average numbers of gold particles associatingwithspecific vesicles or organelles: mitochondria (Fig. 4C), plasmodesmata(Fig. 4G), membrane bound bodies (MBB; see below) (Figs. 5A, B, andC), peroxisomes (Fig. 5D), coated vesicles (Fig. 5F; see below) and Golgi(Fig. 5G). Many of these structures were less abundant than thestructures noted in Table 1 orwere rarely seen, and thus the populationnumbers for each antiserum treatment vary in Table 2.
Two unique membrane bound compartments were identified inPVX.TGBp3–GFP infected samples that were not observed in healthyleaf segments. These structures may represent virus induced altera-tions of cellular membranes. First are the MBBs, which have anaverage diameter of 1048.14±228.72 nm (Figs. 5A–C) and containvirions, ribosomes, and membrane strands. Statistically significantamounts of immunogold label for PVX replicase, TGBp3–GFP and BiPwere identified in the MBBs (Table 2). These structures likely derivefrom the ER, since they contain significant quantities of BiP. Thesecond structures resembled coated vesicles and have an averagediameter of 375.95±82.04 nm (Fig. 5F). These vesicles are

276 D. Bamunusinghe et al. / Virology 393 (2009) 272–285

Fig. 4. Electron micrographs of thin sections through PVX.TGBp3–GFP infected leaf segments. (A–C) Low magnification images showing a region of neighboring cells, and majorstructures: Chloroplasts (Chl), mitochondria (M), cell wall (CW) and vacuole (Vac). (B and C) Samples were treated with PVX replicase and BiP antisera showing few or no goldparticles in these organelles. (D) PVX.TGBp3–GFP inoculated sample treated with PVX replicase and BiP antisera. Lines point to 10 nm gold particles which correspond to BiP antiseraand arrows point to 20 nm gold particles corresponding to PVX replicase. (F) Mock-inoculated samples show rough ER treated with PVX replicase antisera. No gold particles weredetected in mock samples. (G) Higher magnification of area represented by box in panel D shows 10 and 20 nm gold particles. Scale bars represent 500 nm.
Fig. 3. Localization of PVX replicase and TGBp3 proteins through fluorescence imaging of BY-2 protoplasts. (A, D) PVX–GFP infected protoplast shows green cytosolic and nuclearfluorescence at 24 h post inoculation. The vacuoles (v) and nucleus (n) are indicated. (B) Red fluorescence shows replicase in punctae throughout the protoplast. (C and E) Overlay ofred and green fluorescence shows red spherical bodies occur in sites that are devoid of green fluorescence. (F, G) GFP-er expressing protoplast show tubular network extending fromperinuclear region to the plasmamembrane. (H–J) PVX.TGBp3–GFP infected protoplast shows the tubular ER network at 12 h post inoculation. The tubular strands are shorter than incontrol samples although the polygonal pattern is maintained. Red fluorescent spherical bodies overlay the network. The yellow orange fluorescence highlights regions containingboth TGBp3–GFP and replicase. (K–M) PVX.TGBp3–GFP infected protoplast shows the tubular ER network at 44 h post inoculation (green tubular haze). Spherical bodies containTGBp3–GFP and replicase. (N–P)Mock inoculated protoplast treatedwith replicase antisera. (N) Transmitted light image of protoplast following immunolabeling procedure. (O andP)Green and red fluorescence images, respectively show no indication of signal. Bars in all panels represent 10 μm.
277D. Bamunusinghe et al. / Virology 393 (2009) 272–285
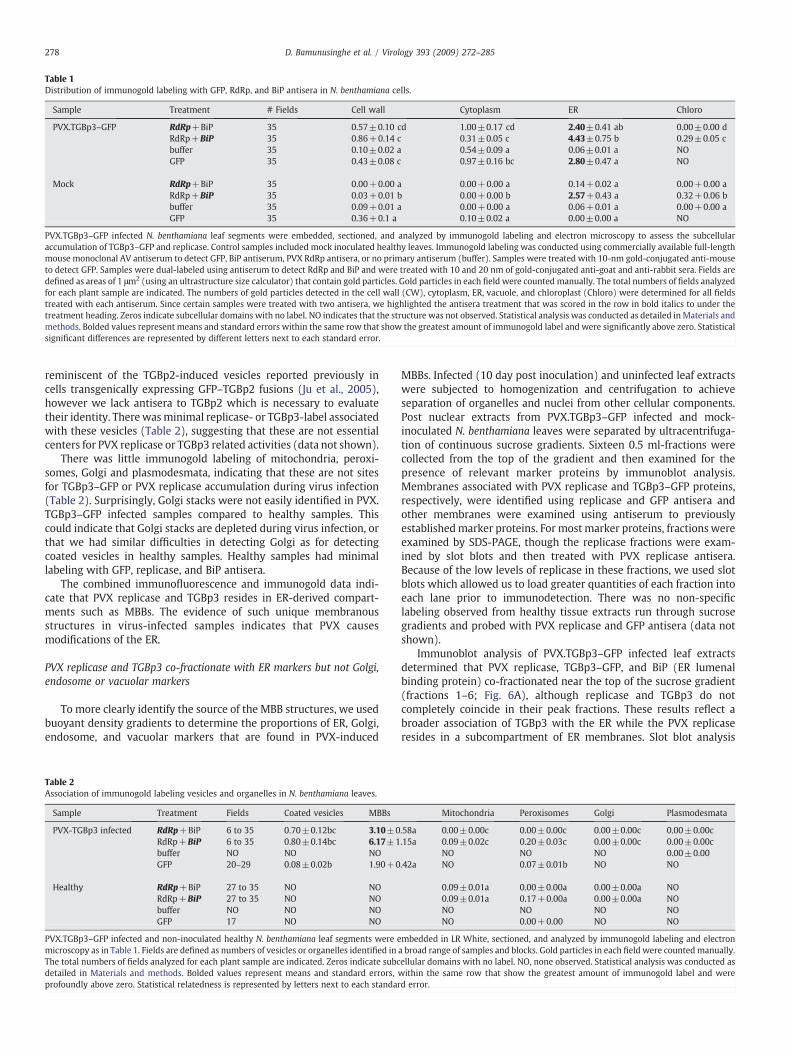
Table 1Distribution of immunogold labeling with GFP, RdRp, and BiP antisera in N. benthamiana cells.
Sample Treatment # Fields Cell wall Cytoplasm ER Chloro
PVX.TGBp3–GFP RdRp+BiP 35 0.57±0.10 cd 1.00±0.17 cd 2.40±0.41 ab 0.00±0.00 dRdRp+BiP 35 0.86+0.14 c 0.31±0.05 c 4.43±0.75 b 0.29±0.05 cbuffer 35 0.10±0.02 a 0.54±0.09 a 0.06±0.01 a NOGFP 35 0.43±0.08 c 0.97±0.16 bc 2.80±0.47 a NO
Mock RdRp+BiP 35 0.00+0.00 a 0.00+0.00 a 0.14+0.02 a 0.00+0.00 aRdRp+BiP 35 0.03+0.01 b 0.00+0.00 b 2.57+0.43 a 0.32+0.06 bbuffer 35 0.09+0.01 a 0.00+0.00 a 0.06+0.01 a 0.00+0.00 aGFP 35 0.36+0.1 a 0.10±0.02 a 0.00±0.00 a NO
PVX.TGBp3–GFP infected N. benthamiana leaf segments were embedded, sectioned, and analyzed by immunogold labeling and electron microscopy to assess the subcellularaccumulation of TGBp3–GFP and replicase. Control samples included mock inoculated healthy leaves. Immunogold labeling was conducted using commercially available full-lengthmouse monoclonal AV antiserum to detect GFP, BiP antiserum, PVX RdRp antisera, or no primary antiserum (buffer). Samples were treated with 10-nm gold-conjugated anti-mouseto detect GFP. Samples were dual-labeled using antiserum to detect RdRp and BiP and were treated with 10 and 20 nm of gold-conjugated anti-goat and anti-rabbit sera. Fields aredefined as areas of 1 μm2 (using an ultrastructure size calculator) that contain gold particles. Gold particles in each field were counted manually. The total numbers of fields analyzedfor each plant sample are indicated. The numbers of gold particles detected in the cell wall (CW), cytoplasm, ER, vacuole, and chloroplast (Chloro) were determined for all fieldstreated with each antiserum. Since certain samples were treated with two antisera, we highlighted the antisera treatment that was scored in the row in bold italics to under thetreatment heading. Zeros indicate subcellular domains with no label. NO indicates that the structure was not observed. Statistical analysis was conducted as detailed in Materials andmethods. Bolded values represent means and standard errors within the same row that show the greatest amount of immunogold label and were significantly above zero. Statisticalsignificant differences are represented by different letters next to each standard error.
278 D. Bamunusinghe et al. / Virology 393 (2009) 272–285
reminiscent of the TGBp2-induced vesicles reported previously incells transgenically expressing GFP–TGBp2 fusions (Ju et al., 2005),however we lack antisera to TGBp2 which is necessary to evaluatetheir identity. Therewasminimal replicase- or TGBp3-label associatedwith these vesicles (Table 2), suggesting that these are not essentialcenters for PVX replicase or TGBp3 related activities (data not shown).
There was little immunogold labeling of mitochondria, peroxi-somes, Golgi and plasmodesmata, indicating that these are not sitesfor TGBp3–GFP or PVX replicase accumulation during virus infection(Table 2). Surprisingly, Golgi stacks were not easily identified in PVX.TGBp3–GFP infected samples compared to healthy samples. Thiscould indicate that Golgi stacks are depleted during virus infection, orthat we had similar difficulties in detecting Golgi as for detectingcoated vesicles in healthy samples. Healthy samples had minimallabeling with GFP, replicase, and BiP antisera.
The combined immunofluorescence and immunogold data indi-cate that PVX replicase and TGBp3 resides in ER-derived compart-ments such as MBBs. The evidence of such unique membranousstructures in virus-infected samples indicates that PVX causesmodifications of the ER.
PVX replicase and TGBp3 co-fractionate with ER markers but not Golgi,endosome or vacuolar markers
To more clearly identify the source of the MBB structures, we usedbuoyant density gradients to determine the proportions of ER, Golgi,endosome, and vacuolar markers that are found in PVX-induced
Table 2Association of immunogold labeling vesicles and organelles in N. benthamiana leaves.
Sample Treatment Fields Coated vesicles MBBs
PVX-TGBp3 infected RdRp+BiP 6 to 35 0.70±0.12bc 3.10±0RdRp+BiP 6 to 35 0.80±0.14bc 6.17±1buffer NO NO NOGFP 20–29 0.08±0.02b 1.90+0
Healthy RdRp+BiP 27 to 35 NO NORdRp+BiP 27 to 35 NO NObuffer NO NO NOGFP 17 NO NO
PVX.TGBp3–GFP infected and non-inoculated healthy N. benthamiana leaf segments were emicroscopy as in Table 1. Fields are defined as numbers of vesicles or organelles identified inThe total numbers of fields analyzed for each plant sample are indicated. Zeros indicate subcdetailed in Materials and methods. Bolded values represent means and standard errors,profoundly above zero. Statistical relatedness is represented by letters next to each standar
MBBs. Infected (10 day post inoculation) and uninfected leaf extractswere subjected to homogenization and centrifugation to achieveseparation of organelles and nuclei from other cellular components.Post nuclear extracts from PVX.TGBp3–GFP infected and mock-inoculated N. benthamiana leaves were separated by ultracentrifuga-tion of continuous sucrose gradients. Sixteen 0.5 ml-fractions werecollected from the top of the gradient and then examined for thepresence of relevant marker proteins by immunoblot analysis.Membranes associated with PVX replicase and TGBp3–GFP proteins,respectively, were identified using replicase and GFP antisera andother membranes were examined using antiserum to previouslyestablishedmarker proteins. For most marker proteins, fractions wereexamined by SDS-PAGE, though the replicase fractions were exam-ined by slot blots and then treated with PVX replicase antisera.Because of the low levels of replicase in these fractions, we used slotblots which allowed us to load greater quantities of each fraction intoeach lane prior to immunodetection. There was no non-specificlabeling observed from healthy tissue extracts run through sucrosegradients and probed with PVX replicase and GFP antisera (data notshown).
Immunoblot analysis of PVX.TGBp3–GFP infected leaf extractsdetermined that PVX replicase, TGBp3–GFP, and BiP (ER lumenalbinding protein) co-fractionated near the top of the sucrose gradient(fractions 1–6; Fig. 6A), although replicase and TGBp3 do notcompletely coincide in their peak fractions. These results reflect abroader association of TGBp3 with the ER while the PVX replicaseresides in a subcompartment of ER membranes. Slot blot analysis
Mitochondria Peroxisomes Golgi Plasmodesmata
.58a 0.00±0.00c 0.00±0.00c 0.00±0.00c 0.00±0.00c
.15a 0.09±0.02c 0.20±0.03c 0.00±0.00c 0.00±0.00cNO NO NO 0.00±0.00
.42a NO 0.07±0.01b NO NO
0.09±0.01a 0.00±0.00a 0.00±0.00a NO0.09±0.01a 0.17+0.00a 0.00±0.00a NONO NO NO NONO 0.00+0.00 NO NO
mbedded in LR White, sectioned, and analyzed by immunogold labeling and electrona broad range of samples and blocks. Gold particles in each field were countedmanually.ellular domains with no label. NO, none observed. Statistical analysis was conducted aswithin the same row that show the greatest amount of immunogold label and wered error.

Fig. 5. Electron micrographs of PVX.TGBp3–GFP and mock inoculated leaf segments. (A–C) MBBs found in PVX.TGBp3–GFP infected leaf tissues but are absent frommock-inoculatedsamples. These are PVX-induced structures which contain virions, ER membranes, and granules which resemble ribosomes. Bodies label with PVX replicase, BiP and GFP antisera.Images presented here show lines pointing to 10 nm gold particles which correspond to BiP antisera and arrows pointing to 20 nm gold particles corresponding to PVX replicase.Arrowheads point to virion particles. Boxed areas are shown as insets at higher magnification to show virions and gold particles (D) Example of peroxisomes. These failed to labelwith antisera. (E, F) Examples of coated vesicles which were more abundant in PVX.TGBp3–GFP infected samples but were rarely seen in mock inoculated samples. Statistically, theamount of replicase and GFP labeling these vesicles was not significant (Table 2). These resemble TGBp2 containing structures reported in Ju et al., (2005). (G) Example of Golgiapparatus. Scale bars represent 500 nm.
279D. Bamunusinghe et al. / Virology 393 (2009) 272–285
shows the greatest amount of PVX replicase in fraction 1 with lesseramounts in fractions 2–5. Western blots show peak levels of TGBp3–GFP in fractions 1, 2, 5, and 6 with lesser amounts in fractions 3–4.Peak levels of BiPwere seen in fractions 2 and 5. Identification of BiP inthe same fractions at TGBp3–GFP and replicase support immunogoldand immunofluorescence data indicating these proteins localize with
ER membranes, although there is not complete identity in theirfractionation patterns.
Further antisera were employed to characterize the distribution ofthe trans Golgi network, endosome, and vacuolar membranes acrossthe sucrose gradient. The additional antisera tested recognize: SYP61(trans Golgi network [TGN] and endosome); SYP41 (TGN); and SYP21

Fig. 6. (A) Presence of viral proteins in sucrose gradient fractions of membranous richextracts from PVX.TGBp3–GFP infected plants. Fractions were analyzed by slot orWestern blot using a variety of antisera. The identity of each antiserum is on the top leftof each panel. Fractions 1–16 are identified next to each lane. Fraction 1 is from the topof the gradient and fraction 16 is near the bottom. The bar on top of each Western blotindicates fractions 1–6 which contain PVX replicase, TGBp3–GFP and BiP. Theremaining fractions contain post-ER membranes. (B) Box plots representing qRT-PCRanalysis of host transcript levels in healthy and virus infected leaves. PVX-3D and PVX-9D indicate PVX.TGBp3–GFP infected leaves which were harvested at 3 and 9 day postinoculation. Box plots represent the range of values obtained for 20 samples at eachtime point and represent the variability of gene expression. The boundaries of each boxrepresent the 25th and 75th percentiles, and the horizontal line within the boxrepresents the median values (i.e. 50 percentile). The spacing of components within thebox indicates the degree of dispersal or skewness in the data. The lines at the top andbottom of the box (whiskers) represent the sample minimum and maximum. Longerlines at the top indicate a positive skewness. Outliers are indicated by “x”.
280 D. Bamunusinghe et al. / Virology 393 (2009) 272–285
(late endosome and vacuole), (Bar-Peled and Raikhel, 1997; Basshamet al., 2000; Sanderfoot et al., 1999). These Golgi, endosomal, andvacuolar proteins were mainly distributed across fractions 6–16.These data show that the ER resident proteins clearly separate fromlater components of the endomembrane network, and demonstratethat the PVX replicase and TGBp3–GFP do not associate with the Golgior post-Golgi networks.
We further investigated whether the changes in endomembranearchitecture, that are necessary for formation of MBBs, coincided withchanges in the expression of essential membrane resident proteins.We conducted qRT-PCR to determine if mRNAs representing BiP,SYP61, SYP41 and SYP21 are specifically upregulated in response toPVX infection. Total RNAwas extracted fromPVX.TGBp3–GFP-infectedor healthy leaves harvested at 3 and 9 day post inoculation. Initialexperiments using a small set of plants revealed that the mRNA levelsin PVX.TGBp3–GFP infected leaves, for some genes, ranged from 0.5 to5-fold increase in expression compared to healthy samples. Such widevariation could result if virus infection is asynchronous and geneexpression is altered for a short time and then subsides at some latertime. Therefore we undertook a more complex qRT-PCR analysis byisolating RNA from 20 plants representing healthy or PVX.TGBp3–GFPinoculated leaves. qRT-PCR provides an advantage over northernanalysis because it allows for simultaneous analysis of gene expressiontrends in larger populations than can be reasonably analyzed in a gel-based assay. Following qRT-PCR analysis the data were statisticallyanalyzed and presented in a box plot diagram (Fig. 6B). Box plots areemployed for nonparametric analysis of populations.
For healthy samples the qRT-PCR results show BiP, SYP61, SYP41and SYP21 values ranged from 0.25 to 1.8 with a median ofapproximately 1.0 (Fig. 6B). The boxes and whiskers were generallysmall indicating low dispersion of values among the 20 plantsanalyzed. Similarly, the median values for SYP61, SYP41, SYP21,using mRNA isolated from PVX.TGBp3–GFP infected leaves rangedfrom0.25 to 1.5. Thusmaximumandminimumvalueswere similar forhealthy and PVX.TGBp3–GFP infected samples. This is contrasted byqRT-PCR results detecting BiP expression in PVX.TGBp3–GFP infectedsamples at 3 and 9 dpi. The median value reflecting BiP geneexpression increased 2.25 fold. At 9 dpi, in particular, the range ofvalueswas positively skewed showing a 3.5 fold increase in expressionfor plants in the 75th percentile and maximum increase of 4.35 fold.These data indicate that PVX.TGBp3–GFP generally causes asmuch as a4.35-fold increase in BiP expression over a 9 day period, whileexpression of Golgi and post-Golgi resident proteins is unaltered.
Cerulenin treatment reduces PVX replication
Many positive strand RNA viruses induce proliferation of ERmembranes to sustain virus replication (Mackenzie, 2005; Salonen etal., 2005). Cerulenin treatment causes cessation of phospholipidbiosynthesis and has been employed to assess the role of membranesynthesis in the replication of Cowpea mosaic virus (CPMV), Bromemosaic virus (BMV), Grapevine fanleaf virus (GFLV) (Carette et al.,2000; Lee and Ahlquist, 2003; Ritzenthaler et al., 2002). These virusencoded replication complexes associate with the ER and studiesrevealed that virus-induced membrane biosynthesis plays an essen-tial role in the viral replication cycles.
For the purpose of the study, tobacco BY-2 protoplasts wereinfected with PVX–GFP and fluorometry was employed to monitorGFP expression as a measure of successful virus replication. Proto-plasts were treated with a range of cerulenin concentrations (0, 15,30, and 45 μM cerulenin) for 24 h and the percentage of fluorescentprotoplasts was recorded (Fig. 7). Seventy-eight percent of proto-plasts were infected with PVX–GFP with no cerulenin treatment.Infection decreased to 54% of cells treated with 15 μM cerulenin, and10–13% of cells treated with 30–45 μM cerulenin. The effects of highercerulenin concentrations on PVX–GFP accumulation was comparable

Fig. 7. Effect of cerulenin on PVX–GFP accumulation in infected protoplasts. Protoplastviability was measured immediately after electroporation with viral RNA using 0.1%FDA. Protoplasts were placed in culture medium containing 0, 15, 30, or 45 μMcerulenin. Protoplasts were incubated for 24 h and then assayed for the proportionexpressing GFP. GFP is derived from a viral subgenomic RNA and its expression isdependent upon successful virus infection. Controls were transfected with pRTL2-GFPplasmids which transcribe GFP in the nucleus. GFP expression is unaffected by cerulenintreatment. The numbers of GFP expressing protoplasts in each sample were counted.The results represent the proportion of viable protoplasts that express GFPfluorescence.
281D. Bamunusinghe et al. / Virology 393 (2009) 272–285
to related reports of GFLV and CPMV(Carette et al., 2000; Ritzenthaleret al., 2002) indicating a similar requirement for membrane synthesis.
To exclude the possibility that cerulenin decreased the viability ofprotoplasts and thereby hampered virus infection, the effect ofcerulenin on protoplasts transfected with pRTL2-GFP plasmids wasexamined. The percentage of protoplasts expressing GFP rangedbetween 40% and 50% in untreated and cerulenin treated samples(Fig. 7). Identical concentrations of cerulenin had no obvious impacton GFP expression from CaMV 35S promoter. The negative impact ofcerulenin on PVX-GFP infection indicates that formation of new ERmembranes is required for virus replication.
Discussion
In this report we have shown that the PVX replicase and TGBp3proteins associate with the ER during infection. Previous studies havereported the localization patterns of the TGB proteins when expressedectopically, alone or when co-expressed (Chang et al., 1997; Davies etal., 1993; Samuels et al., 2007), but not in relation to the replicase. Thesubcellular location of a potexvirus replicase previously has not beenreported. For other RNA viruses the replicase, or replication-associated proteins, have been shown to be essential for virusintracellular activities beyond virus accumulation (Guenoune-Gelbartet al., 2008; Liu et al., 2005). In particular the TMV 126 and/or 183 kDaproteins associate with virus accumulation and also function as asilencing suppressor, associate with microfilaments and are requiredfor intercellular virus movement (Ding et al., 2004; Liu et al., 2005;Hirashima and Watanabe, 2003). Some of these findings reliedstrongly on cell biological analyses. Considering the importance ofthe PVX replicase for virus accumulation and its potential to influenceother activities such as virus movement, it was important todetermine the subcellular location of this protein and its relationshipwith the TGB proteins during PVX infection to serve as a basis forfuture studies in those areas.
Most positive strand RNA viruses infecting plants and mammalsreplicate in association with cellular membranes (Salonen et al.,2005). Viruses often induce membrane modifications such asproliferation or invaginations to create available membrane scaffoldsfor assembly of the replication machinery (Ahlquist, 2006; Netherton
et al., 2007; Suhy et al., 2000; Villanueva et al., 2005). Recent cellbiological investigations with RCNMV and GFLV revealed extensiveredistribution of the ER as a result of virus infection (Ritzenthaler etal., 2002; Turner et al., 2004). Both viruses cause accumulation of ERaggregates or vesicles in the perinuclear region. In this study,immunolabeling of leaf cells and BY-2 protoplasts showed PVXreplicase and TGBp3–GFP both localized to spherical bodies occurringalong the ER network. In protoplasts, MBBs containing PVX replicaseand TGBp3–GFP were observed between 12 and 48 h indicating thatformation of these structures is an early event. The MBBs observed inleaf cells at 7 dpi appeared larger than in protoplasts, which couldresult from increasing modifications and distortion of the ER overtime, due to the presence and accumulation of viral products.Fractionation experiments provide further indication that both thePVX replicase and TGBp3–GFP proteins coincide with the ER markerBiP, and are excluded from Golgi and endosomal vesicles, pointing tothe ER as the primary site for PVX replication.
Localization of a viral MP and replicase in the same membranebound complexes was first reported for TMV (Heinlein et al., 1998;Kahn et al., 1998). The TMV MP is an integral membrane protein thatcauses distortions in the ER network (Reichel and Beachy, 1998;Reichel et al., 1999). Thus, the data reported in this study provide anexample of another virus whose replicase and MP appear in the samemembrane bound complexes. These similarities between TMV andPVX suggest that certain interactions between plant viral replicases,or replication-associated proteins, and MPs could be conserved.Although it is logical that this localization represents a region whereboth proteins are synthesized and/or function, evidence presentedhere showing the proximity of the PVX replicase and the TGBp3movement protein in particular subcellular sites provides the basis forfuture work to examine whether the PVX replicase and TGBp3 act inconcert to promote virus infection and movement.
In leaf cells and protoplasts, the primary site for TGBp3–GFPaccumulation is the cortical ER (Ju et al., 2005; Krishnamurthy et al.,2003) and the same spherical bodies containing PVX replicase. Thus,TGBp3–GFP associates broadly with the ER while the PVX replicase isrestricted to a subcompartment containing ER membranes. Given thatearly studies revealed that deleting TGBp3 from the viral genome doesnot hamper virus replication, TGBp3 is not likely to function as themembrane anchor for the PVX replicase (Beck et al., 1991; Verchot etal., 1998). Typical membrane-anchoring proteins are essential andknockout mutations would eliminate virus replication. Mutationalanalysis has shown that TGBp3 binding to the ER is essential for virusintercellular transport although there has been little progress towardclarifying the role for the ER in modulating virus cell-to-cellmovement. Thus, the exact role of TGBp3 in the ER is unknown,although we have recently proposed that TGBp3 could play a role inmodulating viral protein turnover (Ju et al., 2008). Moreover, wechose to examine the amount of TGBp3–GFP in the cytoplasm in thisstudy based on our recently reported results (Ju et al., 2008) whichsuggested that TGBp3–GFP is turned over by ER-associated degrada-tion pathways (ERAD). TGBp3–GFP is dislodged from the ER forcytosolic degradation by the 26S proteasome (Brandizzi et al., 2003; Juet al., 2008; Meusser et al., 2005). Given the role of TGBp3 in viralprotein turnover and virus movement, it is possible that TGBp3 mayfunction to target the PVX replicase for degradation and acquiringviral RNAs for transfer to plasmodesmata. Further research is neededto determine if the stability of the PVX replicase is altered by thepresence of TGBp3.
In this study, electron microscopy of virus-infected tissuesidentified MBBs, which were filled with ER strands and virions.Immunolabeling detected PVX replicase, TGBp3 and the ER markerprotein, BiP, in these bodies. Since no other ER associated bodies weredetected in virus infected cells, it is reasonable to consider these MBBsto be the spherical bodies seen using confocal microscopy. Virusinfected cells also showed an abundance of coated vesicles that were

282 D. Bamunusinghe et al. / Virology 393 (2009) 272–285
not detected in healthy tissues. The identity of these vesicles was notfurther examined in this study because they failed to showimmunoreactivity with GFP or replicase antisera. However, theyhave some resemblance to TGBp2 induced vesicles reported in arelated study (Ju et al., 2005). The TGBp2-related vesicles are similarin size and show the same granular appearance along the outermembrane. GFLV polyprotein precursors were reported to associatewith membranous vesicles that resembled the COP-coated vesicleswhich traffic from the ER to the Golgi apparatus. Researchersproposed that GFLV might employ the COPII budding machinery torecruit membrane scaffolds for the viral replicase (Ritzenthaler et al.,2002). The COP vesiculation machinery has been shown to beessential for poliovirus replication (Belov et al., 2005; Cuconati etal., 1998; Fogg et al., 2003). While we found no evidence that coatedvesicles (Fig. 5E and F) are COP-related structures their abundance inPVX.TGBp3–GFP infected cells warrants further investigations.
Through treatment with the lipid biosynthesis antagonist, cerule-nin, we determined that membrane synthesis was necessary for PVX.TGBp3–GFP replication (Fig. 7). Cerulenin was also shown to inhibitreplication of GFLV, CPMV, and poliovirus (Carette et al., 2000; Fogg etal., 2003; Ritzenthaler et al., 2002). Thus, PVX belongs to a class ofviruses that require de novo membrane synthesis to support virusreplication. The moderate increase in BiP expression seen in PVXinfected cells may correlate with expansion of the ER or may reflect amoderate level of stress exerted upon the ER as the result of virusinfection. Viral pathogenesis can sometimes trigger the unfoldedprotein response (UPR), leading to increased cellular synthesis of ERresident chaperones necessary to restore ER homeostasis. BiP is one ERresident chaperone that is known to play a vital role in UPR (Kamauchiet al., 2005; Urade, 2007). Similar events have been reported to occurduring mammalian virus infection. Specifically, flaviviral nonstructur-al proteins trigger UPR signaling, which, for viruses such as Hepatitis Cvirus, leads to restoration of ER homeostasis necessary for a persistentvirus infection (Tardif et al., 2005). Increased expression of ER residentchaperones ensures proper protein folding and processing in the ER, asa result of the increaseddemandon the cellularmachinery. In addition,misfolded proteins can be relocated to the cytoplasm for proteasomaldegradation. The role for TGBp3 or PVX replicase in regulatingaccumulation of ER resident chaperones has not yet been examined,although we have reported TGBp3 accumulation is monitored by the26S proteasome (Ju et al., 2008).
Taken together, the data demonstrate that the ER-derived MBBscontain both the PVX replicase and TGBp3 proteins. PVX replicase andTGBp3 do not exactly coincide throughout the ER or in membranefractions. While the top fractions 1–5 contain BiP, which is a marker forER membranes, replicase was predominantly in fraction 1 while peaklevels of TGBp3 were in fraction 5. These results are reflective ofevidence that the replicasedoes not occur throughout the cortical ER butare restricted to a subcompartment of the ER, such as MBBs. Particlesresembling PVX virions were also seen inside MBBs, although furtherstudies are needed to learn howmany PVX-encoded proteins associatewith these structures. Importantly, this is thefirst report to demonstratea spatial relationship between the PVX TGBp3 and replicase. Given theclose relationship of the TMV replicase and movement protein, it isworth considering that interactions between the PVX replicase andTGBp3 may serve analogous functions. However, further dynamicinvestigations are necessary to reveal whether TGBp3 is similar to theTMVmovement protein and drives trafficking ofmembrane bound viralreplication complexes toward the plasmodesmata.
Materials and methods
Infectious clones, in vitro transcription, and plant inoculations
PVX–GFP and PVX.TGBp3–GFP are infectious clones of PVXdescribed in prior studies (Fig. 1; Krishnamurthy et al., 2003; Mitra
et al., 2003; Verchot et al., 1998). Infectious clones weremaintained inEscherichia coli strain JM109 (Sambrook et al., 1989). One microgramof SpeI linearized DNA was transcribed using the mMESSAGEmMACHINE™ kit (Ambion Inc., Austin, TX). The leaves of N.benthamianawere dustedwith cellite powder and then rub inoculatedwith 5 μg of transcripts. Mock inoculated plants were treated similarlywith 5 μL of ddH2O.
BY-2 protoplast preparation and transfection
Protoplasts were prepared using tobacco BY-2 suspension cells asdescribed previously (Ju et al., 2005; Nagata et al., 1992). For RNAtransfection, 500 μL of protoplasts was mixed with 5–10 μg oftranscripts and placed in 0.4 cm gap cuvettes on ice. Electroporationwas carried out at 0.25 kV, 100 Ω and 125 μF using a BioRad GenePulser (Bio-Rad Laboratories, Hercules, CA). Protoplasts were trans-ferred to 6-well cell culture plates containing BY-2 culture media plus0.45 M mannitol and 1% agarose (w/v; pH 5.7). Samples weremaintained at 27–28 °C in dark.
Immunofluorescence labeling of virus infected leaf segmentsand protoplasts
Leaf segments containing fluorescent infection sites were fixed ineppendorf tubes with a solution of 3.7% (v/v) formaldehyde, 5% (v/v)dimethyl sulfoxide, and PHEM buffer (60 mM PIPES, 25 mM HEPES,5 mM EGTA, 2 mM MgCl2, pH 6.9) for 2 h (Liu et al., 2005). Fixedsegments were placed on microscope slides and digested with cellwall degrading enzymes (1% Onozuka R10 cellulase, 0.1% pectolyase[Kikkoman, Tokyo, JP] and 0.1% bovine serum albumin [BSA; fractionV] in PHEM buffer) for 2 h. Following washes with PHEM (3 min/wash) the segments were incubated with 1% (v/v) Triton X-100 for20 min. Ice cold methanol was added, incubated for 10 min at roomtemperature, and then washed thrice for 5 min with PHEM.
Protoplast samples were collected by centrifugation at 59×g for5min, washed twicewith PHEM buffer, and fixedwith a solution of 2%[v/v] paraformaldehyde and 0.5% [v/v] DMSO for 15 min at roomtemperature. Samples were transferred onto a cover glass atop a thinfilm of 0.7% Bacto-agar and 200 μL of 1% Triton X-100 (in PHEM) wasadded for 10 min. Following three washes with PHEM, NaBH4 wasadded to the cover glass, incubated for 10min, and thenwashed thricewith PHEM. Ice cold methanol was added, incubated for 10 min atroom temperature, and then washed thrice for 5 min with PHEM.
For immunolabeling, leaf segments and protoplasts were incubat-ed overnight in a moisture chamber at 4 °C with PVX replicase goatantisera (diluted 1:100 or 1:500) in a moisture chamber at 4 °C.Samples were washed five times for 5 min with PHEM and thenincubated with Alexa Fluor 633 conjugated secondary antisera (1:100dil) (Molecular Probes, Inc., Eugene, OR) for 2 h in a moisturechamber at room temperature. Cover glasses were washed five timesfor 5 min with PHEM and then mounted on a slide with commerciallyavailable Vectashield™ mounting media (Vector Laboratories, Inc.,Burlingame, CA).
Fixation and LR-White or Spurr's resin embedding for electronmicroscopic analysis
Virus-infected and mock-inoculated leaf segments were harvestedat 5 dpi and subjected to chemical fixation and embedding in Spurr'sresin. Stepwise chemical fixation was carried out as described(Dunoyer et al., 2002a, 2002b). Leaf segments were pre-incubatedwith 1% glutaraldehyde in 25 mM potassium phosphate buffer (pH7.4) for 1 h in a bench top vacuum chamber and then immersed in aprimary fixative [2% (v/v) glutaraldehyde, 0.1 ml of saturated picricacid in 25 mM potassium phosphate buffer (pH 7.4)] for 15 min atroom temperature. Segments were incubated at 4 °C for 16 h, washed

Table 3Primers for qRT-PCR.
Genes Accession Primers Primer sequences
18S rRNA AJ236016 Forward 5′-ATGGCCGTTCTTAGTTGGTGGAGC-3′Reverse 5′-AGTTAGCAGGCTGAGGTCTCGAAC-3′
BiP FJ463755 Forward 5′-AGCTTTGAGCAGTCAACACCAAGT-3′Reverse 5′-AAAACGTGCCCGAGTAAGTGGTTC-3′
SYP21 L41651 Forward 5′- CCGATTTACGCGACAAGTTGCACA-3′Reverse 5′- ACTCGATGATCTGTTTCGCTGGCT-3′
SYP41 NM_001036873 Forward 5′- TTCAGTGAACTGCAGACGACCACT-3′Reverse 5′- TAGAACGACCCGGCATGAATCCAA-3′
SYP61 NM_001036025 Forward 5′- ACTTGTGGAAGCATTGAGTGGCAG-3′Reverse 5′- TTCACATTCCGCACCTGTGTCCTA-3′
283D. Bamunusinghe et al. / Virology 393 (2009) 272–285
four times (5 min per wash) in 25 mM PIPES buffer (pH 7.0) and thentransferred into secondary fixatives [2% (w/v) OsO4 and 0.5% (w/v)potassium ferrocyanide in 25 mM PIPES buffer (pH 7.0)] for 2 h atroom temperature. Samples were washed twice with 25 mM PIPESbuffer (pH 7.0) for 15 min and then twice with ddH2O for another15 min. Samples were transferred to 2% (w/v) aqueous uranyl acetatefor 16 h at 4 °C and washed twice with ddH2O for 15 min. Sampleswere dehydrated in an acetone series (10%, 20%, 40%, 60%, 80%, and100%) and infiltrated with acetone/Spurr's (1:1) or LR-White resinovernight and then embedded in Spurr's or LR-White resin.
Ultra thin sections (700 nm) were cut using a diamond knife on aSorvall MT 6000 ultramicrotome. Sectionsweremounted on formvar-coated nickel grids (Electron Microscopy Science, Hatfield, PA) andused for immunogold labeling.
Cerulenin treatment
Following electroporation, protoplast viability was measuredusing 0.1% fluorescein diacetate (FDA) in 1 mL of 0.05 M phosphatebuffer. Cerulenin (Sigma-Aldrich Co., St. Louis, MO) was dissolved indimethyl sulfoxide and added to the protoplast culture medium to afinal concentration of 0, 15, 30, and 45 μM (Carette et al., 2000;Ritzenthaler et al., 2002). Protoplasts were incubated for 24 h andharvested. A haemocytometer was employed to calculate theproportion of GFP expressing protoplasts. The proportions of viableprotoplasts that were virus infected were recorded.
Immunogold labeling of LR-White or Spurr's resin-embeddedplant material
Immunogold labeling of tissues were conducted using monoclonalGFP (BD Living Colors™; Clontech Laboratories, Mountain View, CA)and PVX replicase polyclonal goat antisera as previously described(Mitra et al., 2003). Grids were incubated in blocking solutionconsisting of PBS, pH 7.5 (130 mM NaCl, 7.0 mM Na2HPO4, 3.0 mMNaH2PO4) plus 2% BSA (w/v) for 15 min, and then was incubated with5% normal sheep sera (Sigma-Aldrich Co.) in PBS plus 2% BSA for15 min. Then samples were incubated with GFP monoclonal antiseradiluted 1:500 in PBS plus 2% BSA (w/v), PVX replicase goat antiseradiluted 1:500 in PBS plus 0.1% Tween (v/v), or buffer containing noprimary antisera for 2 h. Grids were then washed five times for 5 minwith PBS and then with PBS plus 2% fish gelatin (v/v) for 15 min. Gridswere then incubated for 1 h with either 10 or 20 nm gold-conjugatedgoat antisera (EY Labs, San Mateo, CA) diluted 1:10 in PBS plus 2% fishgelatin.
Some grids were dual labeled with antiserum to detect replicaseand GFP and a longer procedure was followed (Mitra et al., 2003).Grids were first incubated with replicase goat antisera diluted 1:500in PBS plus 0.1% Tween (v/v) for 2 h and then with the 20 nm goldconjugated rabbit anti-goat serum diluted 1:10 in PBS plus 2% fishgelatin for 2 h as described above. Samples were washed twice for10 min with PBS and then incubated for 30 min with GFP monoclonalantisera diluted 1:500 in PBS. Grids were washed five times for 5 minwith PBS, then for 15 min with PBS with 2% fish gelatin, and wereincubated 30 min with 10 nm gold-conjugated goat anti-mouseserum (EY Labs) diluted 1:10 in PBS plus 2% fish gelatin. Grids werewashed three times for 5 min with ddH2O, and stained with a solutionof 2.5% uranyl acetate and 70% methanol (v/v) for 30 min, and thenwith a solution of 2% Reynold's lead citrate pH 12.0 (in ddH2O) for20 min. Samples were washed with mildly warm ddH2O three timesfor 5 min and then dried. Resin embedded sections were consecu-tively labeled with PVX replicase goat primary, anti-goat secondaryand GRP78 (BiP) rabbit primary and anti-rabbit secondary antisera(ABR-Affinity Bioreagents Inc., Rockford, IL) using established proto-cols (Ju et al., 2005). Grids were stained with uranyl acetate andReynolds lead citrate.
Laser scanning confocal microscopy and transmissionelectron microscopy
A Leica DMRE microscope with Leica TCS SP2 confocal imagingsystem was used. Ar/Kr lasers were used for detecting GFP (488 nmexcitation) and He/Ne lasers for detecting Alexa Fluor 633 fluores-cence. Electron microscopy was carried out using a JEOL JEM-2100Scanning Transmission Electron Microscope System with an EDAXGenesis 2000 EDS system (JEOL Ltd., Tokyo, Japan). Electronmicroscopic images were taken, and number of gold particles labelingspecific structural components of the cell was scored in 10 μm2
fields(using an ultrastructure size calculator) or in each organelle. Goldparticles in each field/organelle were counted manually. Average andstandard error was calculated and tabulated. Images were compiledusing Adobe Photoshop CS software (Adobe Systems, San Jose, CA).
Sucrose gradient fraction of plant extracts containing PVX replicase
Forty grams of PVX.TGBp3–GFP infected N. benthamiana leaveswas collected from plants at 14 dpi and then homogenized in 100 mlof ice-cold buffer A [50 mM Tris–HCl, pH 8.2; 15 mM MgCl2, 120 mMKCl, 20% glycerol, 1 mM DTT, 5 mM EDTA and 10 μL/mL of ProteaseInhibitor Cocktail for plant cell and tissue extracts (Sigma-Aldrich Co.)(Hu et al., 2007; Plante et al., 2000). The crude homogenate wasfiltered through gauze and miracloth (Calbiochem, La Jolla, CA) into afresh tube and centrifuged at 500×g for 10 min at 4 °C. Thesupernatant was removed and centrifuged at 30,000×g for 30 minusing a SW 32.1 ultracentrifuge rotor and OptimaTM L-XP Seriespreparative ultracentrifuge (Beckman Coulter, Inc., Fullerton, CA). Theresulting pellet was resuspended in 6ml buffer B (50mMTris–HCl, pH8.2; 15 mM MgCl2, 5% glycerol, 1 mM DTT, 5 mM EDTA and 10 μL/mLof Protease Inhibitor Cocktail) and divided into 3 ml samples whichwere loaded onto 7.5 mL, 20–60% continuous sucrose gradients. Aftercentrifugation at 189,000×g for 1 h at 4 °C, 0.5 mL fractions werecollected from the top of each gradient. Approximately 16 to 18fractions were collected per gradient (Plante et al., 2000).
Gradient fractions were concentrated five-fold as described(Wessel and Flugge, 1984). A 0.2 mL aliquot of each fraction wascombined with 0.8 mL of methanol, followed by vortexing and lowspeed centrifugation at 9000×g for 10 s. Samples were extractedusing sequential addition of 0.2mL of chloroform and 0.6mL of ddH2Ofollowed again by centrifugation at 9000×g for 1 min. Methanol(0.6 mL) was added to the organic and inter-phase. Proteins werepelleted by centrifugation at 9000×g for 2 min, dried under a streamof air, and then boiled with 40 μL protein dissociation buffer(Sambrook et al., 1989) before loading onto 12.5% SDS-polyacryl-amide gels. Vertical transfer of denatured proteins onto nitrocellulosemembranes (Perkin Elmer, Boston, MA) was conducted using aBioRad Trans-Blot apparatus (Bio-Rad Laboratories). To detect PVXreplicase, 150 μL fractions plus 50 μL of protein dissociation bufferwere applied directly to a nitrocellulose membrane using a Bio-Dot SFapparatus (Bio-Rad Laboratories).

284 D. Bamunusinghe et al. / Virology 393 (2009) 272–285
Membranes were probed with PVX replicase goat polyclonal orcommercially available
GFP monoclonal or BiP (GRP 78) rabbit polyclonal antiserum. Inaddition, membranes were probed with Arabidopsis thaliana (At)SYP61, AtSYP21, and AtSYP41 rabbit polyclonal antiserum (Bar-Peledand Raikhel, 1997; Bassham et al., 2000; Sanderfoot and Raikhel,1999) to determine if these cellular proteins reside in the samefractions as the PVX proteins. Blots were developed using theWestern Lightning Chemiluminescence reagent plus developer(Perkin Elmer), and then exposed to Kodak Bio Max Light Film(Kodak, Rochester, NY).
Density values were calculated using the FluorChem software. Foreach autoradiograph, the background was automatically subtractedby determining the average of the 10 lowest pixel values surroundingeach individual band. For a more rigorous quantification of the data,the density values for an empty lane was calculated and subtractedfrom each density value recorded for lanes 1–16. The final values arepresented as relative density values (RDV) and were plotted usingMicrosoft Excel 2003.
Statistical analysis
ANOVA procedures with PC SAS version 8.2 (SAS Institute) andPROC GLM were used to evaluate differences in location in thenumber of gold particles observed in the electron micrographs thatwere reported in Tables 1 and 2. The analysis was performed for eachcombination of virus-infected or healthy plus antiserum. Due toproblems in homogeneity of variance and distributional assumptions,a square-root transformation was used prior to conducting theANOVAs. When the ANOVA was significant, pairwise comparisons ofthe locations were made with a PDIFF option in an LSMEANSstatement. A significance level of 0.05 was used for all comparisons.
Quantitative RT-PCR (qRT-PCR)
N. benthamiana leaves were inoculated with 100 μg/ml purifiedPVX.TGBp3–GFP. Total RNA was extracted from inoculated leaves of20 plants at 3 and 9 day post inoculation with SV Total RNA IsolationKit (Promega Corp., Madison, WI). First strand cDNA synthesis wascarried out using Superscript reverse transcriptase III (InvitrogenCorp., Carlsbad, CA) and hexamer random primers. Primers of SYP21,SYP41 and SYP61 are designed based on N. benthamiana homologousESTs of the Arabidopsis syntaxins (Table 3). 18S rRNA serves as theendogenous control. Twenty five nanograms cDNA of each sampleand 100 to 900 nM primers were used to perform qPCR with powerSYBR Green II and ABI7500 system (Applied Biosystem). Twenty-fivemicroliters PCR reactions were incubated at 95 °C 10 min to activateTaq polymerase, followed by 40 cycles of 95 °C for 15 s and 60 °C for60 s. Efficiencies of all primers are verified by normal RT-PCR and gelelectrophoresis prior to qRT-PCR. Duplicate PCR reactions for eachsample were carried out and averaged. The comparative CT methodemploys the formula 2 -dd CT where the values of the endogenouscontrol and calibrator (constant quantity of healthy sampletemplate) and are subtracted from the target sample value toprovide the ddCT value. The 2 -dd CT represents the fold of RNAaccumulation. The raw qRT-PCR data were treated in Excel, and boxplots were drawn in GenStat 11th edition (The Numerical Algo-rithms Group Ltd, Oxford, UK).
References
Ahlquist, P., 2006. Parallels among positive-strand RNA viruses, reverse-transcribingviruses and double-stranded RNA viruses. Nat. Rev. Microbiol. 4 (5), 371–382.
Asurmendi, S., Berg, R.H., Koo, J.C., Beachy, R.N., 2004. Coat protein regulates formationof replication complexes during Tobacco mosaic virus infection. Proc. Natl. Acad. Sci.U. S. A. 101 (5), 1415–1420.
Bar-Peled, M., Raikhel, N.V., 1997. An efficient method for cloning in-frame fusionprotein genes. Anal. Biochem. 250 (2), 262–264.
Bassham, D.C., Sanderfoot, A.A., Kovaleva, V., Zheng, H., Raikhel, N.V., 2000. AtVPS45complex formation at the trans-Golgi network. Mol. Biol. Cell 11 (7), 2251–2265.
Bayne, E.H., Rakitina, D.V., Morozov, S.Y., Baulcombe, D.C., 2005. Cell-to-cell movementof potato potexvirus X is dependent on suppression of RNA silencing. Plant J. 44 (3),471–482.
Beck, D.L., Guilford, P.J., Voot, D.M., Andersen, M.T., Forster, R.L., 1991. Triple gene blockproteins of white clover mosaic potexvirus are required for transport. Virology 183(2), 695–702.
Belov, G.A., Fogg, M.H., Ehrenfeld, E., 2005. Poliovirus proteins induce membraneassociation of GTPase ADP-ribosylation factor. J. Virol. 79 (11), 7207–7216.
Brandizzi, F., Hanton, S., DaSilva, L.L., Boevink, P., Evans, D., Oparka, K., Denecke, J.,Hawes, C., 2003. ER quality control can lead to retrograde transport from the ERlumen to the cytosol and the nucleoplasm in plants. Plant J. 34 (3), 269–281.
Braun, C.J., Hemenway, C.L., 1992. Expression of amino-terminal portions or full-lengthviral replicase genes in transgenic plants confers resistance to potato virus Xinfection. Plant Cell 4 (6), 735–744.
Buck, K.W., 1999a. Replication of tobacco mosaic virus RNA. Philos. Trans. R. Soc. Lond.,B Biol. Sci. 354 (1383), 613–627.
Buck, K.W., 1999b. Replication of tobacco mosaic virus RNA. Philos. Trans. R. Soc. Lond.B. Biol. Sci. 354 (1383), 613–627.
Carette, J.E., Stuiver, M., Van Lent, J., Wellink, J., Van Kammen, A., 2000. Cowpea mosaicvirus infection induces a massive proliferation of endoplasmic reticulum but notGolgi membranes and is dependent on de novo membrane synthesis. J. Virol. 74(14), 6556–6563.
Chang, B.Y., Lin, N.S., Liou, D.Y., Chen, J.P., Liou, G.G., Hsu, Y.H., 1997. Subcellularlocalization of the 28 kDa protein of the triple-gene-block of bamboo mosaicpotexvirus. J. Gen. Virol. 78 (Pt. 5), 1175–1179.
Cuconati, A., Molla, A., Wimmer, E., 1998. Brefeldin A inhibits cell-free, de novosynthesis of poliovirus. J. Virol. 72 (8), 6456–6464.
Davenport, G.F., Baulcombe, D.C., 1997. Mutation of the GKS motif of the RNA-dependent RNA polymerase from potato virus X disables or eliminates virusreplication. J. Gen. Virol. 78 (Pt. 6), 1247–1251.
Davies, C., Hills, G., Baulcombe, D.C., 1993. Sub-cellular localization of the 25-kDaprotein encoded in the triple gene block of potato virus X. Virology 197 (1),166–175.
Ding, X.S., Liu, J., Cheng, N.H., Folimonov, A., Hou, Y.M., Bao, Y., Katagi, C., Carter, S.A.,Nelson, R.S., 2004. The Tobacco mosaic virus 126-kDa protein associated with virusreplication and movement suppresses RNA silencing. Mol. Plant-Microbe Interact.17 (6), 583–592.
Dunoyer, P., Pfeffer, S., Fritsch, C., Hemmer, O., Voinnet, O., Richards, K.E., 2002a.Identification, subcellular localization and some properties of a cysteine-richsuppressor of gene silencing encoded by Peanut clump virus. Plant J. 29 (5), 555–567.
Dunoyer, P., Ritzenthaler, C., Hemmer, O., Michler, P., Fritsch, C., 2002b. Intracellularlocalization of the Peanut clump virus replication complex in tobacco BY-2protoplasts containing green fluorescent protein-labeled endoplasmic reticulumor Golgi apparatus. J. Virol. 76 (2), 865–874.
Fogg, M.H., Teterina, N.L., Ehrenfeld, E., 2003. Membrane requirements for uridylylationof the poliovirus VPg protein and viral RNA synthesis in vitro. J. Virol. 77 (21),11408–11416.
Fontes, E.B., Shank, B.B., Wrobel, R.L., Moose, S.P., GR, O.B., Wurtzel, E.T., Boston, R.S.,1991. Characterization of an immunoglobulin binding protein homolog in themaize floury-2 endosperm mutant. Plant Cell 3 (5), 483–496.
Goodin, M.M., Chakrabarty, R., Yelton, S., Martin, K., Clark, A., Brooks, R., 2007.Membrane and protein dynamics in live plant nuclei infected with Sonchus yellownet virus, a plant-adapted rhabdovirus. J. Gen. Virol. 88 (Pt. 6), 1810–1820.
Guenoune-Gelbart, D., Elbaum, M., Sagi, G., Levy, A., Epel, B.L., 2008. Tobacco mosaicvirus (TMV) replicase andmovement protein function synergistically in facilitatingTMV spread by lateral diffusion in the plasmodesmal desmotubule of Nicotianabenthamiana. Mol. Plant-Microb. Interact. 21 (3), 335–345.
Haseloff, J., Amos, B., 1995. GFP in plants. Trends Genet 11 (8), 328–329.Haywood, V., Kragler, F., Lucas, W.J., 2002. Plasmodesmata: pathways for protein and
ribonucleoprotein signaling. Plant Cell 14 Suppl, S303–S325.Heinlein, M., Padgett, H.S., Gens, J.S., Pickard, B.G., Casper, S.J., Epel, B.L., Beachy, R.N.,
1998. Changing patterns of localization of the tobacco mosaic virus movementprotein and replicase to the endoplasmic reticulum and microtubules duringinfection. Plant Cell 10 (7), 1107–1120.
Hirashima, K., Watanabe, Y., 2003. RNA helicase domain of tobamovirus replicaseexecutes intercellular movement possibly through collaboration with its non-conserved region. J. Virol. 77, 12357–12362.
Howard, A.R., Heppler, M.L., Ju, H.-J., Krishnamurthy, K., Payton, M.E., Verchot-Lubicz, J.,2004. Potato virus X TGBp1 induces plasmodesmata gating and moves betweencells in several host species whereas CP moves only in N. benthamiana leaves.Virology 328, 185–197.
Hsu, H.T., Hsu, Y.H., Bi, I.P., Lin, N.S., Chang, B.Y., 2004. Biological functions of thecytoplasmic TGBp1 inclusions of bamboo mosaic potexvirus. Arch. Virol. 149 (5),1027–1035.
Hu, B., Pillai-Nair, N., Hemenway, C., 2007. Long-distance RNA-RNA interactionsbetween terminal elements and the same subset of internal elements on the potatovirus X genome mediate minus- and plus-strand RNA synthesis. RNA 13, 267–280.
Huisman, M.J., Linthorst, H.J., Bol, J.F., Cornelissen, J.C., 1988. The complete nucleotidesequence of potato virus X and its homologies at the amino acid level with variousplus-stranded RNA viruses. J. Gen. Virol. 69 (Pt. 8), 1789–1798.
Hwang, Y.T., McCartney, A.W., Gidda, S.K., Mullen, R.T., 2008. Localization of theCarnation Italian ringspot virus replication protein p36 to the mitochondrial outer

285D. Bamunusinghe et al. / Virology 393 (2009) 272–285
membrane is mediated by an internal targeting signal and the TOM complex. BMCCell Biol 9, 54.
Ishikawa, M., Okada, Y., 2004. Replication of tobamovirus RNA. Proc. Jpn. Acad. Ser. B2004, 215–224.
Jonczyk, M., Pathak, K.B., Sharma, M., Nagy, P.D., 2007. Exploiting alternative subcellularlocation for replication: tombusvirus replication switches to the endoplasmicreticulum in the absence of peroxisomes. Virology 362 (2), 320–330.
Ju, H.J., Samuels, T.D., Wang, Y.S., Blancaflor, E., Payton, M., Mitra, R., Krishnamurthy, K.,Nelson, R.S., Verchot-Lubicz, J., 2005. The potato virus X TGBp2 movement proteinassociates with endoplasmic reticulum-derived vesicles during virus infection.Plant Physiol. 138 (4), 1877–1895.
Ju, H.J., Brown, J.E., Ye, C.M., Verchot-Lubicz, J., 2007. Mutations in the central domain ofpotato virus X TGBp2 eliminate granular vesicles and virus cell-to-cell trafficking.J. Virol. 81 (4), 1899–1911.
Ju, H.J., Ye, C.M., Verchot-Lubicz, J., 2008. Mutational analysis of potato virus X TGBp3links subcellular accumulation to protein turnover during virus infection. Virology375, 103–117.
Kahn, T.W., Lapidot, M., Heinlein, M., Reichel, C., Cooper, B., Gafny, R., Beachy, R.N., 1998.Domains of the TMVmovement protein involved in subcellular localization. Plant J.15 (1), 15–25.
Kalinina, N.O., Rakitina, D.V., Solovyev, A.G., Schiemann, J., Morozov, S.Y., 2002. RNAhelicase activity of the plant virus movement proteins encoded by the first gene ofthe triple gene block. Virology 296 (2), 321–329.
Kamauchi, S., Nakatani, H., Nakano, C., Urade, R., 2005. Gene expression in response toendoplasmic reticulum stress in Arabidopsis thaliana. FEBS J. 272 (13), 3461–3476.
Kawakami, S., Watanabe, Y., Beachy, R.N., 2004. Tobacco mosaic virus infection spreadscell to cell as intact replication complexes. Proc. Natl. Acad. Sci. U. S. A. 101 (16),6291–6296.
Kim, M.J., Kim, H.R., Paek, K.H., 2006. Arabidopsis tonoplast proteins TIP1 and TIP2interact with the cucumber mosaic virus 1a replication protein. J. Gen. Virol. 87(Pt. 11), 3425–3431.
Kiselyova, O.I., Yaminsky, I.V., Karger, E.M., Frolova, O.Y., Dorokhov, Y.L., Atabekov, J.G.,2001. Visualization by atomic force microscopy of tobacco mosaic virus movementprotein-RNA complexes formed in vitro. J. Gen. Virol. 82 (Pt. 6), 1503–1508.
Kiselyova, O.I., Yaminsky, I.V., Karpova, O.V., Rodionova, N.P., Kozlovsky, S.V.,Arkhipenko, M.V., Atabekov, J.G., 2003. AFM study of potato virus X disassemblyinduced by movement protein. J. Mol. Biol. 332 (2), 321–325.
Krishnamurthy, K., Heppler, M., Mitra, R., Blancaflor, E., Payton, M., Nelson, R.S.,Verchot-Lubicz, J., 2003. The potato virus X TGBp3 protein associates with the ERnetwork for virus cell-to-cell movement. Virology 309 (1), 135–151.
Lee, W.M., Ahlquist, P., 2003. Membrane synthesis, specific lipid requirements, andlocalized lipid composition changes associated with a positive-strand RNA virusRNA replication protein. J. Virol. 77 (23), 12819–12828.
Liu, J.Z., Blancaflor, E.B., Nelson, R.S., 2005. The tobacco mosaic virus 126-kilodaltonprotein, a constituent of the virus replication complex, alone or within the complexaligns with and traffics along microfilaments. Plant Physiol. 138, 1853–1865.
Lough, T.J., Shash, K., Xoconostle-Cázares, B., Hofstra, K.R., Beck, D.L., Balmori, E., Forster,R.L.S., Lucas, W.J., 1998. Molecular dissection of the mechanism by whichpotexvirus triple gene block proteins mediate cell-to-cell transport of infectiousRNA. Mol. Plant-Microb. Interact. 11, 801–814.
Lough, T.J., Netzler, N.E., Emerson, S.J., Sutherland, P., Carr, F., Beck, D.L., Lucas, W.J.,Forster, R.L., 2000. Cell-to-cell movement of potexviruses: evidence for aribonucleoprotein complex involving the coat protein and first triple gene blockprotein. Mol. Plant-Microb. Interact. 13 (9), 962–974.
Lucas, W.J., 2006. Plant viral movement proteins: agents for cell-to-cell trafficking ofviral genomes. Virology 344 (1), 169–184.
Mackenzie, J., 2005. Wrapping things up about virus RNA replication. Traffic 6 (11),967–977.
Mas, P., Beachy, R.N., 1999. Replication of tobacco mosaic virus on endoplasmicreticulum and role of the cytoskeleton and virus movement protein in intracellulardistribution of viral RNA. J. Cell Biol. 147 (5), 945–958.
Meusser, B., Hirsch, C., Jarosch, E., Sommer, T., 2005. ERAD: the long road to destruction.Nat. Cell Biol. 7 (8), 766–772.
Mitra, R., Krishnamurthy, K., Blancaflor, E., Payton, M., Nelson, R.S., Verchot-Lubicz, J.,2003. The potato virus X TGBp2 protein association with the endoplasmicreticulum plays a role in but is not sufficient for viral cell-to-cell movement.Virology 312 (1), 35–48.
Nagata, T., Nemoto, Y., Hasezawa, S., 1992. Tobacco BY-2 cell line as the “Hela” cell in thecell biology of higher plants. Internat. Rev. Cytol. 132, 1–31.
Nelson, R., 2005. Movement of viruses to and through plasmodesmata. In: Oparka, K.(Ed.), Plasmodesmata. InBlackwell Publishing Ltd, Oxford, pp. 188–209.
Netherton, C., Moffat, K., Brooks, E., Wileman, T., 2007. A guide to viral inclusions,membrane rearrangements, factories, and viroplasm produced during virusreplication. Adv. Virus Res. 70, 101–182.
Plante, C.A., Kim, K.H., Pillai-Nair, N., Osman, T.A., Buck, K.W., Hemenway, C.L., 2000.Soluble, template-dependent extracts from Nicotiana benthamiana plants infectedwith potato virus X transcribe both plus- and minus-strand RNA templates.Virology 275 (2), 444–451.
Prod'homme, D., Jakubiec, A., Tournier, V., Drugeon, G., Jupin, I., 2003. Targeting of theturnip yellow mosaic virus 66 K replication protein to the chloroplast envelope ismediated by the 140 K protein. J. Virol. 77 (17), 9124–9135.
Reichel, C., Beachy, R.N., 1998. Tobacco mosaic virus infection induces severemorphological changes of the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S.A. 95 (19), 11169–11174.
Reichel, C., Mas, P., Beachy, R.N., 1999. The role of the ER and cytoskeleton in plant viraltrafficking. Trends Plant Sci. 4 (11), 458–462.
Ritzenthaler, C., Laporte, C., Gaire, F., Dunoyer, P., Schmitt, C., Duval, S., Piequet, A.,Loudes, A.M., Rohfritsch, O., Stussi-Garaud, C., Pfeiffer, P., 2002. Grapevine fanleafvirus replication occurs on endoplasmic reticulum-derived membranes. J. Virol. 76(17), 8808–8819.
Rodionova, N.P., Karpova, O.V., Kozlovsky, S.V., Zayakina, O.V., Arkhipenko, M.V.,Atabekov, J.G., 2003. Linear remodeling of helical virus by movement proteinbinding. J. Mol. Biol. 333 (3), 565–572.
Rubino, L., Weber-Lotfi, F., Dietrich, A., Stussi-Garaud, C., Russo, M., 2001. The openreading frame 1-encoded (‘36 K’ protein) of Carnation Italian ringspot viruslocalizes to mitochondria. J. Gen. Virol. 82 (Pt. 1), 29–34.
Salonen, A., Ahola, T., Kaariainen, L., 2005. Viral RNA replication in association withcellular membranes. Curr. Top. Microbiol. Immunol. 285, 139–173.
Sambrook, J., Fritsch, E.F., Maniatis, T., 1989. Molecular Cloning: A Laboratory Manual,2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY.
Samuels, T.D., Ye, C.-M., Ju, H.-J., Motes, C.M., Blancaflor, E.B., Verchot-Lubicz, J., 2007.Potato virus X TGBp1 protein accumulates independently of TGBp2 and TGBp3 topromote virus cell-to-cell movement. Virol. 367, 375–389.
Sanderfoot, A.A., Raikhel, N.V., 1999. The specificity of vesicle trafficking: coat proteinsand SNAREs. Plant Cell 11 (4), 629–642.
Sanderfoot, A.A., Kovaleva, V., Zheng, H., Raikhel, N.V., 1999. The t-SNARE AtVAM3presides on the prevacuolar compartment in Arabidopsis root cells. Plant Physiol.121 (3), 929–938.
Sanfacon, H., Zhang, G., 2008. Analysis of interactions between viral replicase proteinsand plant intracellular membranes. Methods Mol. Biol. 451, 361–375.
Schepetilnikov, M.V., Manske, U., Solovyev, A.G., Zamyatnin Jr., A.A., Schiemann, J.,Morozov, S.Y., 2005. The hydrophobic segment of potato virus X TGBp3 is a majordeterminant of the protein intracellular trafficking. J. Gen. Virol. 86 (Pt. 8),2379–2391.
Schwartz, M., Chen, J., Lee, W.M., Janda, M., Ahlquist, P., 2004. Alternate, virus-inducedmembrane rearrangements support positive-strand RNA virus genome replication.Proc. Natl. Acad. Sci. U. S. A. 101 (31), 11263–11268.
Suhy, D.A., Giddings Jr., T.H., Kirkegaard, K., 2000. Remodeling the endoplasmicreticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74 (19), 8953–8965.
Tardif, K.D., Waris, G., Siddiqui, A., 2005. Hepatitis C virus, ER stress, and oxidativestress. Trends Microbiol. 13 (4), 159–163.
Turner, K.A., Sit, T.L., Callaway, A.S., Allen, N.S., Lommel, S.A., 2004. Red clover necroticmosaic virus replication proteins accumulate at the endoplasmic reticulum.Virology 320 (2), 276–290.
Urade, R., 2007. Cellular response to unfolded proteins in the endoplasmic reticulum ofplants. FEBS J. 274 (5), 1152–1171.
Verchot-Lubicz, J., Ye, C.-M., Bamunusinghe, D., 2007. Molecular biology of potex-viruses: recent advances. J. Gen. Virol. 88, 1643–1655.
Verchot, J., Angell, S.M., Baulcombe, D.C., 1998. In vivo translation of the triplegene block of potato virus X requires two subgenomic mRNAs. J. Virol. 72 (10),8316–8320.
Villanueva, R.A., Rouille, Y., Dubuisson, J., 2005. Interactions between virus proteins andhost cell membranes during the viral life cycle. Int. Rev. Cytol. 245, 171–244.
Wei, T., Wang, A., 2008. Biogenesis of cytoplasmic membranous vesicles for plantpotyvirus replication occurs at endoplasmic reticulum exit sites in a COPI- andCOPII-dependent manner. J. Virol. 82 (24), 12252–12264.
Wessel, D., Flugge, U.I., 1984. Amethod for the quantitative recovery of protein in dilutesolution in the presence of detergents and lipids. Anal. Biochem. 138 (1), 141–143.
Zamyatnin Jr., A.A., Solovyev, A.G., Bozhkov, P.V., Valkonen, J.P., Morozov, S.Y.,Savenkov, E.I., 2006. Assessment of the integral membrane protein topology inliving cells. Plant J. 46 (1), 145–154.