analysis of cotton (gossypium hirsutum) root proteomes during a compatible interaction with the...
TRANSCRIPT

RESEARCH ARTICLE
Analysis of cotton (Gossypium hirsutum)
root proteomes during a compatible interaction with
the black root rot fungus Thielaviopsis basicola
Joëlle V. F. Coumans1, 2, Anne Poljak3, Mark J. Raftery3,David Backhouse4 and Lily Pereg-Gerk1, 2
1 Molecular and Cellular Biology, School of Science and Technology, University of New England,Armidale, NSW, Australia
2 Cotton Catchment Communities CRC, Narrabri, NSW, Australia3 Bioanalytical Mass Spectrometry Facility, The University of New South Wales, Sydney, NSW, Australia4 Botany, School of Environmental and Rural Science, University of New England, Armidale, NSW, Australia
A proteomic approach was used to uncover the inducible molecular defense mechanism of cot-ton root occurring during the compatible interaction with Thielaviopsis basicola. Microscopicobservation of cotton root inoculated with a suspension of conidia showed that this necrotrophichemibiotroph fungus interacts with the plant and completes its life cycle in our experimentalsystem. 2-DE analysis of root extracts taken after 1, 3, 5, and 7 days postinoculation and clusteranalysis of the protein expression levels showed four major profiles (constant, upregulated, oneslightly downregulated, and one dramatically downregulated). Spots significantly (p,0.05)upregulated were analyzed by LC-MS/MS and identified using MASCOT MS/MS ion searchsoftware and associated databases. These proteins included defense and stress related proteins,such as pathogenesis-related proteins and proteins likely to be involved in the oxidative burst,sugar, and nitrogen metabolism as well as amino acid and isoprenoid synthesis. While many ofthe identified proteins are common components of the defense response of most plants, a pro-teasome subunit and a protein reported to be induced only in cotton root following Meloidogyneincognita infection were also identified.
Received: 20 March, 2008Revised: 24 June, 2008
Accepted: 1 August, 2008
Keywords:
Differential 2-D SDS-PAGE / Gossypium hirsutum / Mass spectrometry / Proteomics /Thielaviopsis basicola
Proteomics 2009, 9, 335–349 335
1 Introduction
The soil-borne fungus Thielaviopsis basicola is a widespreadpathogen [1] that causes black root rot disease to a broadrange of important agricultural and horticultural crops [2, 3].
In Australia, black root rot has spread exponentially duringthe 1990s and has emerged as a major threat to the sustain-ability of the cotton industry [4]. The major effect of blackroot rot on cotton is to reduce early season growth, delayingcrop maturity, which leads to a yield diminution. Infectedplants generally appear stunted and chlorotic, but diagnosisof black root rot requires examination of the roots. T. basicolacauses a characteristic blackening of the roots due to thedestruction of the root cortex which contains the spores ofthe fungus. Studies of infection of tobacco and pansy roots [5,6] by T. basicola reported that penetration occurred directlyfrom swollen germ tube tips. The penetration hypha growsthrough the epidermal cell wall without any visible host cell
Correspondence: Dr. Joëlle Coumans, Molecular and Cellular Bi-ology, School of Science and Technology, University of New Eng-land, Armidale, NSW 2351, AustraliaE-mail: [email protected]: 161-2-6773-3267
Abbreviation: NI, non-inoculated
DOI 10.1002/pmic.200800251
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

336 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349
death. As infection and colonization of the host cell pro-gresses, evidence of necrosis becomes apparent in theinvaded cell. Subsequently, hyphae grow toward the rootcortex and move through the cortex to finally sporulate onnecrotic tissues. This behavior classifies T. basicola as anecrotrophic hemibiotroph [5, 6].
Plants respond to pathogen attack by constitutive andinduced responses. Constitutive defenses are the physicalstructures that prevent pathogen attachment and penetra-tion. The induced defenses comprise structural and chemicaldefenses that are activated only after pathogen attack. Thesecellular defenses are strictly orchestrated and follow a basicpathway through recognition and signal transduction to theexpression of proteins and molecules that will minimizepathogen infection. The ability of T. basicola to inducedefense responses in different plants has been reported. Inthe early sixties, Christou [7] observed the development ofyellow–brown granules in bean root cells as infection advan-ces; Hampton [8, 9] reported changes in phenolic com-pounds and an activity increase of polyphenol oxidases incarrot tissues infected with T. basicola. More recently, Punja[10] has shown an accumulation of polyphenol oxidase andphenolic compounds 5 days after fungal inoculation of beanleaves. He also reported the thickening of the cell wallaround the necrotic lesions as well as the production of cal-lose deposits. The presence of callose was also observed byMims [6] in pansy roots. This evidence suggests that infec-tion by T. basicola initiates radical changes in the proteinexpression pattern of the host plant. Nonessential proteinsmay be downregulated or switched off while proteins essen-tial to the defense and survival of the plant would be upreg-ulated or newly synthesized.
Currently nothing is known about the molecular back-ground of this plant-pathogen interaction. A better knowl-edge of the inducible molecular defense mechanism couldbe useful in designing protective strategies such as overexpression of defense genes in cotton plants. The two mostpromising approaches for understanding the full network ofmolecular responses are transcriptomic and proteomicanalyses. Transcriptomic analysis by microarray is anattractive analysis tool due to its relative simplicity, compre-hensive sampling capacity, and high throughput but it islimited to the analysis of gene expression. Moreover, it hasbeen shown that the level of gene expression does notnecessarily correlate with protein levels [11, 12]. Alter-natively, 2-DE technology combined with MS analysis allowsthe study of a complete set of proteins in a given sample, ata specified time, in a particular tissue but more importantlythe identification of the final product in the cellular re-sponse [13].
Previous work on responses of cotton to pathogen infec-tion across a wide range of genes has been done for the vas-cular wilt fungi Verticillium dahliae [14–16] and Fusariumoxysporum f. sp. vasinfectum [17], and the foliar bacteriumXanthomonas campestris pv. malvacearum [18]. These studiesall measured levels of gene expression. Analysis of protein
levels during infection has only been done for specific pro-teins such as d-cadinene synthase or Meloidogyne-associatedprotein [19, 20]. Of the previous work on gene expression,only Dowd et al. [17], Hill et al. [14], and Zuo et al. [16] inclu-ded root tissues in their experiments. The pathogens thatthey used were vascular wilts, which penetrate root cortexintercellularly and then colonize the xylem vessels [21]. Be-cause of their unusual mode of pathogenesis, these are notgood models for studying interactions with typical necro-trophic pathogens. Very little proteomic work has been doneso far on any roots of any plants infected with necrotrophicpathogens [22, 23].
In this work, we investigated protein expression changesat different time points during a compatible interaction be-tween a soilborne pathogen, T. basicola and its host plant,cotton. As far as we are aware, this study is the first publishedproteomic study on cotton roots and therefore providesinformation in an area so far not documented that could aidin the development of rational control strategies for mana-ging black root rot.
2 Materials and methods
2.1 Conidial inoculum, plant growth, and inoculation
system
T. basicola, isolate BRIP40192, recovered from a cotton fieldin Narrabri (NSW, Australia), was obtained from Jan Dean,Queensland Department of Primary Industries and Fish-eries. For conidial production, the isolate was grown on halfpotato dextrose agar (PDA) (19.5 g potato dextrose agar(Oxoid), 14.5 g Bacto-agar, distilled water 1 L) at 257C for5 days. Root-dip inoculation of seedlings was performed witha suspension of 106 conidia/mL in deionized water.
Cotton seeds (Sicot 189, Narrabri, NSW, Australia) weresurface sterilized in a solution containing 1% NaOCl and10% ethanol for 15 min and then rinsed several times insterile distilled water. Seeds were then incubated in Petridishes containing yeast mannitol agar (1 g yeast extract, 10 gmannitol, 0.5 g K2HPO4, 0.2 g MgSO4?7H2O, 15 g agar, dis-tilled water 1 L) for 2 days at 257C. Germinated seedlings freefrom bacterial and fungal contamination were transferred toglass tubes containing 15 mL of hydroponic solution [24] andgrown for 6 days at 257C with a 12 h photoperiod(30 mmolm22/s21). Seedlings were then removed and root-dip-inoculated in either a T. basicola endoconidia suspensionor sterile water (non-inoculated (NI) sample) for 15 min andtransferred to Petri dishes containing a semi-solid hydro-ponic medium (0.5% agar). The seedlings were incubated at257C (12 h photoperiod), harvested at various times afterinoculation and rinsed in deionized water. The tap root andhypocotyl were cut, patted dry between paper towels, theirwet weights recorded and then stored at 2807C until proteinextraction.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 335–349 337
2.2 Protein extraction and quantification
For each sample, 15 to 30 treated or NI cotton roots werecombined and ground in a mortar prechilled with liquidnitrogen. The ground powder was collected, washed by cen-trifugation with ice-cold methanol until a clear supernatantwas obtained and washed one last time with ice-cold acetone.Finally, acetone residues were removed by drying undervacuum for 20 min. Proteins were extracted according to aphenol extraction procedure derived from that of Hurkmanand Tanaka [25]. In summary, plant powder was suspendedin the extraction buffer (30% sucrose, 0.1 M Tris-HCl pH 8,2 mM PMSF, 1% DTT, 100 mM KCl, 5 mM EDTA) and fur-ther disrupted with glass beads using a mini bead beaterapplied at four 30 s intervals interspersed with 5 min coolingon ice. An equal volume of phenol saturated with Tris-HCl(pH 8) was then added and the mixture vortexed for 2 minand centrifuged (10 0006g, 5 min). The phenolic phase wasremoved and re-extracted with the extraction buffer as above.Proteins were precipitated from the phenolic phase with fivevolumes of 0.1 M ammonium acetate in methanol overnightat 2207C and pelleted by centrifugation (10 0006g, 30 min).The resulting pellet was rinsed twice with ice-cold 0.1 Mammonium acetate in methanol, three times with ice-coldmethanol and once with ice-cold acetone/water (80:20 v/v).After air drying, the pellet was dissolved in IEF buffer (7 Murea, 2 M thiourea, 4% CHAPS, 1% DTT, 0.5% IPG bufferpH 4–7: GE Healthcare Life Sciences, Australia). Proteinconcentration was determined using the 2-D quant kit fromGE Healthcare Life Sciences and the amount of protein pergram of wet cotton root was calculated.
2.3 2-DE
Protein extract was loaded by cup loading onto a rehydrated18 cm IPG strip pH 4–7 (GE Healthcare Life Science) withprotein loadings of 300 mg (analytical gel) or 900 mg (pre-parative gel). IEF was carried out on the IPGphor II (GEHealthcare Life Science) at 207C with current limit 50 mA/strip to a total volt–hour product of 34 kVh (analytical gels) or45 kVh (preparative gels). Prior to the second dimensionanalysis, the individual strips were equilibrated according toGörg et al. [26]. For the second dimension separation, thestrips were positioned on lab cast 1 mm SDS polyacrylamidegels (12%) and sealed with 1% agarose. The gels were run on aPROTEAN II system (BioRad) at 10 mA/gel until the bromo-phenol blue dye front reached the anodic end of the SDS gel.
2.4 Protein visualization and image analysis
Proteins were visualized by Blue silver staining [27] for ana-lytical study and by CBB staining (50% methanol, 0.15%CBB R-250, 0.75% acetic acid) for preparative 2-DE. Stainedgels were recorded for image analysis using the Infinity im-aging system from Vilber Lourmat and analyzed withPDQuest advanced 2-D analysis software (BioRad). Three
biological samples and two technical replicates per biologicalsample for each of the inoculated period 1, 3, 5 and 7 dayswere obtained grouped and analyzed. NI plants were ana-lyzed after 1 and 7 days of incubation in the Petri dish. Be-cause no significant difference in their level of expressioncould be observed, samples were pooled as the NI group.Detection and matching of the protein spots was facilitatedwith the use of the software and re-evaluated by visualinspection, focusing on spots with altered expression. Nor-malization of the gels was performed using the local regres-sion method of the PDQuest software. Each sample set wasanalyzed independently. Spots showing at least a 1.75-foldincrease over those from NI samples at two or more timepoints and statistically different at a significance level ofp,0.05 based on t-tests analysis (assuming equal variance),were selected for LC-MS/MS analysis.
2.5 Cluster analysis
Analysis of the protein temporal expression pattern was per-formed using the Gene Cluster 3.0 and JavaTreeview soft-ware [28]. Centered correlation was used as a measure of thedistance between the different proteins while clustering wasperformed using the average linkage method. The relation-ship among the objects (protein spots) was visualized usingthe JavaTreeview software.
2.6 Protein identification and database search
Relevant protein spots from preparative gels were manuallyexcised, washed 20 min with 100 mM NH4HCO3 anddestained with 250 mL of 25 mM NH4HCO3 in ACN until theCBB disappeared. Protein spots were washed twice for10 min with 100% ACN and vacuum dried in a Speedvaccentrifugal dryer (Thermo, Australia). Proteins in the gelpieces were reduced for 1 h with 50 mL of 10 mM DTT at377C and alkylated for 1 h in 50 mL of 25 mM iodoacetamideat 377C. After three washes of 10 min with 200 mL of Milli-Qwater and one wash of 10 min with 200 mL of 100 mMNH4HCO3, the gel pieces were dehydrated twice for 10 minwith 100 mL of ACN and air dried for 5 min. Digestion of theproteins was carried out in 25 mM NH4HCO3 containing10 ng/mL of trypsin (Promega, Annandale, NSW, Australia)for 14 h at 377C. After digestion, the gel pieces were washedtwice for 15 min first with 50 mL of 0.1% formic acid, thenwith 0.1% formic acid/ACN (1:1). Combined extracts weredried under vacuum and peptides re-suspended in 20 mL of0.1% formic acid.
Digested peptides were separated by nano-LC using anUltimate HPLC and Famos autosampler system (LC-Pack-ings, Amsterdam, Netherlands). Samples (5 mL) were con-centrated and desalted onto a micro C18 precolumn(500 mm62 mm, Michrom Bioresources, Auburn, CA) withH2O/ACN (98:2, 0.05% HFBA) at 20 mL/min. After a 4 minwash the precolumn was switched (Switchos, LC Packings)into line with a fritless nano column (0.0756,100 mm2)
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

338 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349
manufactured according to Gatlin et al. [29]. Peptides wereeluted using a linear gradient of H2O/ACN (98:2, 0.1% for-mic acid) to H2O/ACN (55:45, 0.1% formic acid) at ,300 nL/min over 30 min. High voltage (2300 V) was applied to low avolume tee (Upchurch Scientific) and a column tip posi-tioned ,1 cm from the orifice of an API QStar Pulsar ihybrid tandem mass spectrometer (Applied Biosystems,Foster City, CA). Positive ions were generated by electrosprayand the QStar operated in information dependent acquisi-tion (IDA) mode. A TOF-MS survey scan was acquired (m/z 350–1700, 1 s). The two largest multiply charged ions(counts .15) were sequentially selected by Q1 for MS/MSanalysis. Nitrogen was used as collision gas and an optimumcollision energy chosen (based on charge state and mass).Tandem mass spectra were accumulated for 2.5 s (m/z 65–2000).
Peak lists were generated using MASCOT Distiller(Matrix Science, London, England) using the default parame-ters, and submitted to the database search program MASCOT(version 2.1 or 2.2, Matrix Science). Search parameters were:precursor and product ion tolerances 60.25 and 0.2 Da,respectively; Met(O) and Cys-carboxyamidomethylation spe-cified as variable modification, enzyme specificity was tryp-sin, one missed cleavage was possible and the NCBInr data-base (20070715) and EST-others (20060429) searched.
Function characterization of the proteins identified wasdetermined by the Gene Ontology Tool (http://www.geneontology.org). Due probably to the lack of resolutionsometimes associated with 2-DE the MASCOT searches forsome protein spots occasionally gave several confident iden-tifications [30]. Only the top scoring proteins identifiedwithin each sample were reported in the results section.
3 Results
3.1 Black root rot development on cotton root in our
experimental system
A system for coordinated and reproducible infection of cot-ton root with T. basicola under controlled environmentalconditions was developed. Three days after inoculation,lesions characteristic of T. basicola infection were observedon root-dip inoculated seedlings transferred to semi-solidhydroponic medium. After 7 days, most of the tap root pre-sented the purplish-black discoloration characteristic of thedisease and the lateral root system was poorly developed(Fig. 1A). At this time, microscopic examination of the taproot confirmed that chlamydospores had formed on necrotictissues (Fig. 1B).
3.2 Changes of protein content in response to T.
basicola infection
Expression analysis of the protein profile was undertakenwith two purposes: (i) to observe the time course of the global
Figure 1. Infection of cotton seedlings with T. basicola. (A) Cottonseedling, grown aseptically in Petri dishes, after 7 days without(left) and with (right) root-dip inoculation with an endoconidiasuspension of T. basicola. (B) Section of cotton root colonized byT. basicola chlamydospores 7 days after root-dip inoculation.
protein changes occurring during T. basicola infection and(ii) to identify protein spots that were upregulated in re-sponse to infection. Therefore, cotton root protein samplesfrom NI, and day 1, 3, 5, and 7 postinoculation were analyzedby 2-DE in pH 4–7 and in the range of 15–150 kDa. Gelswere of high quality with reproducible protein patterns be-tween replicates of the same samples and between inde-pendent experiments. Moreover, we found that the betweengel variation was lower than the biological variation (averageCV 28.54% 6 19.08). Approximately 900 spots in gel imagesfrom NI and day 1 postinoculation samples were resolvedwhile on average 750, 700, and 650 spots were found in gelimages from day 3, 5, and 7 postinoculation, respectively.
To assess the global changes in the expression levelsoccurring during infection of the cotton root with T. basicola,gels from NI, day 1, 3, 5, and 7 postinoculation root werecompared and quantified using the PDQuest software. Theexpression profiles of the protein spots were further analyzedwith Gene Cluster 3.0 and JavaTreeview software [28]. Theresulting dendogram (Fig. 2) reveals four major proteinexpression profiles that were defined by taking groups ofclosely related expression patterns (correlation coefficient.0.8) and by calculating the average expression profile forthese proteins. About 90% of the proteins fell into these pro-files. Profile A and D contained approximately 18 and 14%,respectively, of the protein spots studied. They representprotein spots that showed a decrease in their expression levelduring infection with T. basicola. The main difference be-tween the expression patterns of the protein spots in profilesA and D was quantitative, with the expression levels of theprotein spots in profile A decreasing more strongly than inprofile D and becoming undetectable after either 3 or 5 dayspostinoculation. Profile C contained about 45% of proteinspots. These proteins exhibited relatively constant expressionlevels during the course of infection. Finally, profile B, which
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 335–349 339
Figure 2. Cluster representation and expression profiles of protein expression in infected cotton root with T. basicola. Dendogram andgrayscale image were produced as described in the text. The color scale ranges from white for low percentage of expression to black forhigh percentage of expression. Average expression profiles of the proteins included in the color bands are show to right hand of Fig 2.Profiles were defined by taking groups of closely related proteins (correlation coefficient .0.8) and by calculating the average expressionprofile for these proteins. About 90% of the proteins were used to define these profiles.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

340 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349
represented about 10% of the protein spots, contained pro-tein spots that displayed an increase in their expression levelfrom 1 to 7 days postinoculation with a peak of expression atday 5.
3.3 Identification of cotton root proteins upregulated
by T. basicola infection
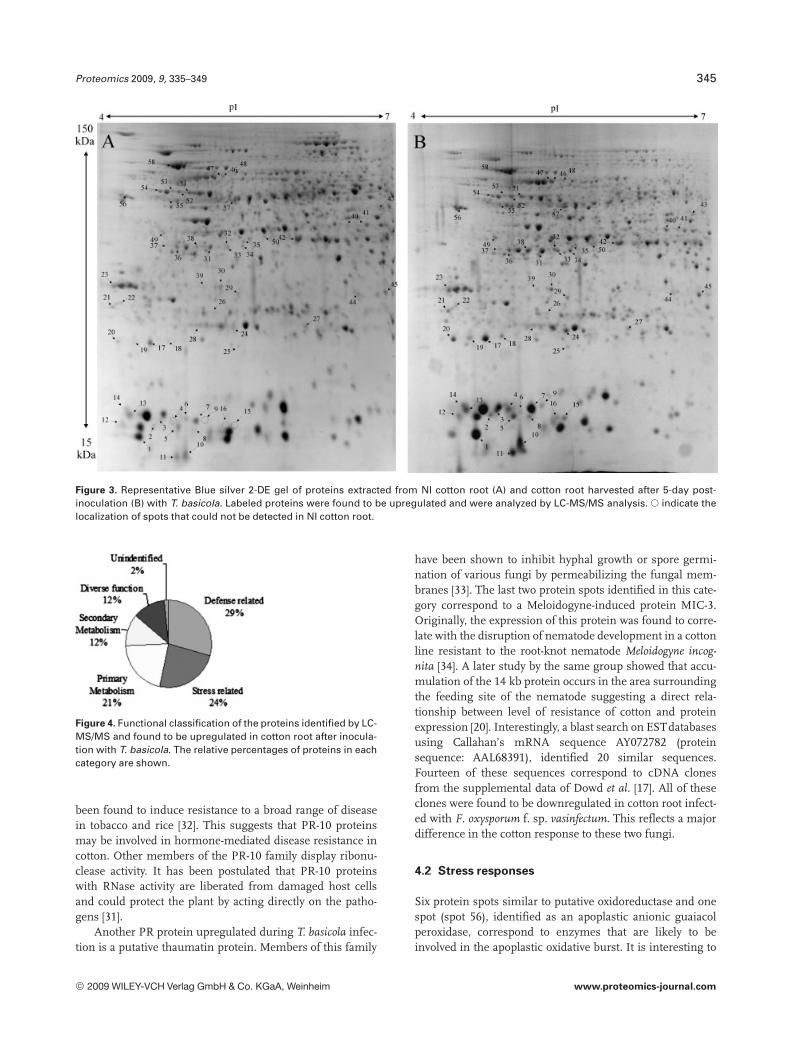
To reliably determine quantitative changes in proteinexpression and therefore overcome error imposed by techni-cal and biological variations, proteins were identified asupregulated if they were found to have an average expressionlevel at least 1.75 higher than those of NI samples over atleast two time points and at a statistical significance level ofp,0.05. We found that 97 proteins met these criteria. How-ever, we could only confidently manually excise (relativelyisolated and/or highly expressed) 58 spots from a preparative2-DE gel for further analyzed by LC-MS/MS analysis. All theMS/MS spectra were searched using the MASCOT softwareagainst the NCBInr database. Twenty one spots withsequence coverage between 17–99% were identified bymatching against a protein sequence from Gossypium spe-cies. Because the cotton genome is not yet fully sequenced,the other MS/MS spectra were searched against an ESTdatabase. Thirty four spots could be assigned confidently toan EST sequence from Gossypium species. The two remain-ing spots were identified by comparison with sequencesfrom different organisms. Table 1 provides the accessionnumbers corresponding to protein spots shown in Fig. 3, theexpression profile of these individual protein spots, theputative names of proteins, the organism from which theprotein has been identified, the MASCOTscore together withthe sequence coverage, the values for experimental and the-oretical pI and molecular mass as well as their putativefunction. The identified proteins could be classified into fivemajor groups according to their biological process as shownin Fig. 4: (i) defense related proteins, 17 spots: some of thesespots were identified as the same protein but displayed dif-ferent pI and MW values and might account for isoforms orpost-translationally modified forms of these proteins; (ii)stress response related proteins, 14 spots in total, with twospots (36 and 37), presenting a similar MW but a different pIand matching the same EST sequence containing a putativeconserved domain for an oxidoreductase; (iii) primary me-tabolism, 12 spots: four being associated with glycolysis/car-bohydrate degradation, four other spots are involved in ATPproduction, with spots 51, 52, and 53 matching the sameEST sequence, two spots corresponding to the same ESTsequence and containing a putative conserved domain for acysteine synthase, one spot associated with nitrogen metab-olism; and finally one spot associated with translation; (iv)secondary metabolism, seven spots all corresponding to pro-teins involved in the pathway leading to the formation ofisoprenoids, and finally (v) seven spots corresponding toproteins involved in diverse functions such as protein degra-
dation, ligand binding and protein stabilization. One spot (6)could not be identified unambiguously.
4 Discussion
The absence of resistance and the exponential spread ofblack root rot disease on cotton (Australia), led us to under-take the present work. Previous microscopic studies havedescribed the events in the T. basicola–tobacco and pansy rootinteraction [5, 6] but to the best of our knowledge, this is thefirst study that aims to determine the nature of the bio-chemical response occurring in a host root during a com-patible interaction with this fungus. To reduce the effects ofchanges in protein expression attributable to factors otherthan infection, we have developed an environmentally con-trollable and reproducible experimental system that allowsthe development of black root rot symptoms observed in thefield. Moreover, the Petri dish bioassay showed that the fun-gus is capable of completing its biological cycle on the cottonroot as chlamydospores were observed on necrotic tissues.
To study changes in the pattern of protein expression incotton root in response to T. basicola infection, we used theBlue silver staining method to visualize and quantify around900 proteins on 2-DE gels with protein extracts from NI andT. basicola inoculated cotton root from days 1, 3, 5, and 7postinoculation. As a result, we found that the expressionlevel for almost half of the proteins (approximately 45%) wasnot altered and that more plant proteins were down-regulated, especially in the early stages of infection, thanupregulated (Fig. 2). This observation is similar to the studyof Dowd et al. [17] on cotton root infection by F. oxysporum f.sp. vasinfectum. Fifty eight protein spots significantly(p,0.05) upregulated over two time points were furtheridentified using LC-MS/MS analysis. We found that thesespots could be classified into five major categories accordingto their putative biological role (Fig. 4): defense, stress, pri-mary and secondary metabolism, and diverse function. Theirfunctional significance is discussed below.
4.1 Defense responses: pathogenesis-related genes
Not surprisingly, a number of the upregulated proteins cor-respond to pathogenesis-related proteins (PR). The majorityof them belong to the PR-10 family. PR-10 proteins havebeen shown to be induced in response to pathogen attacks,including fungal pathogens. In cotton, Dowd et al. [17]reported that PR-10-related genes were the most commonPR genes induced in hypocotyls infected with F. oxysporum f.sp. vasinfectum. A gene for a putative PR-10 has also beenreported to be induced in bacterial-blight resistant cottonafter inoculation with X. campestris pv. malvacearum [18].While the specific function of PR-10 proteins is stillunknown, it has been proposed that in plants some PR-10proteins have a steroid carrier function [31]. Brassinolide isone of the main plant steroids that promotes growth and has
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
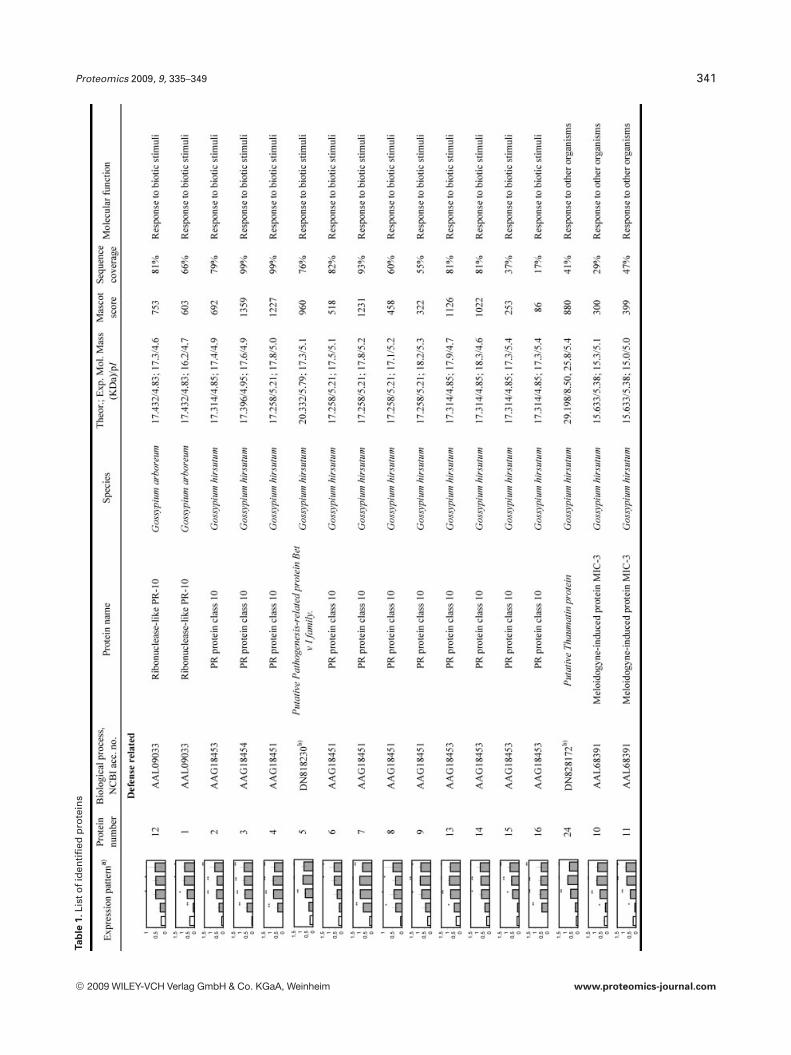
Proteomics 2009, 9, 335–349 341Ta
ble
1.Li
sto
fid
enti
fied
pro
tein
s
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
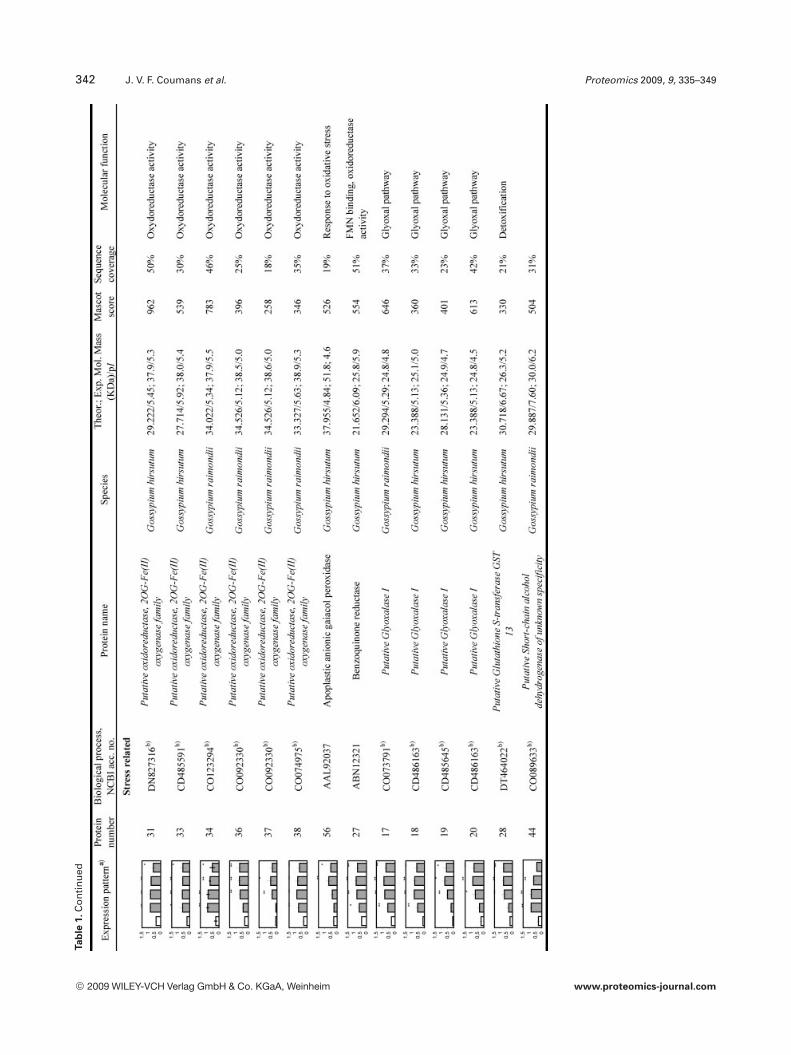
342 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349Ta
ble
1.C
on
tin
ued
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
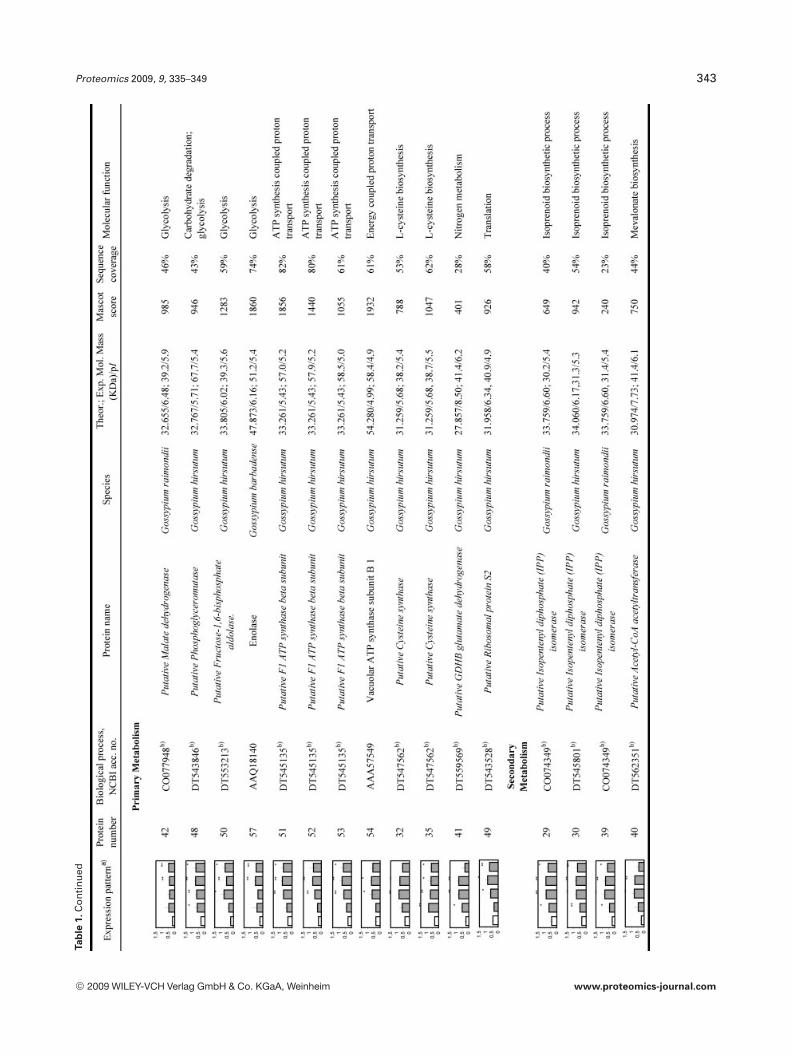
Proteomics 2009, 9, 335–349 343Ta
ble
1.C
on
tin
ued
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
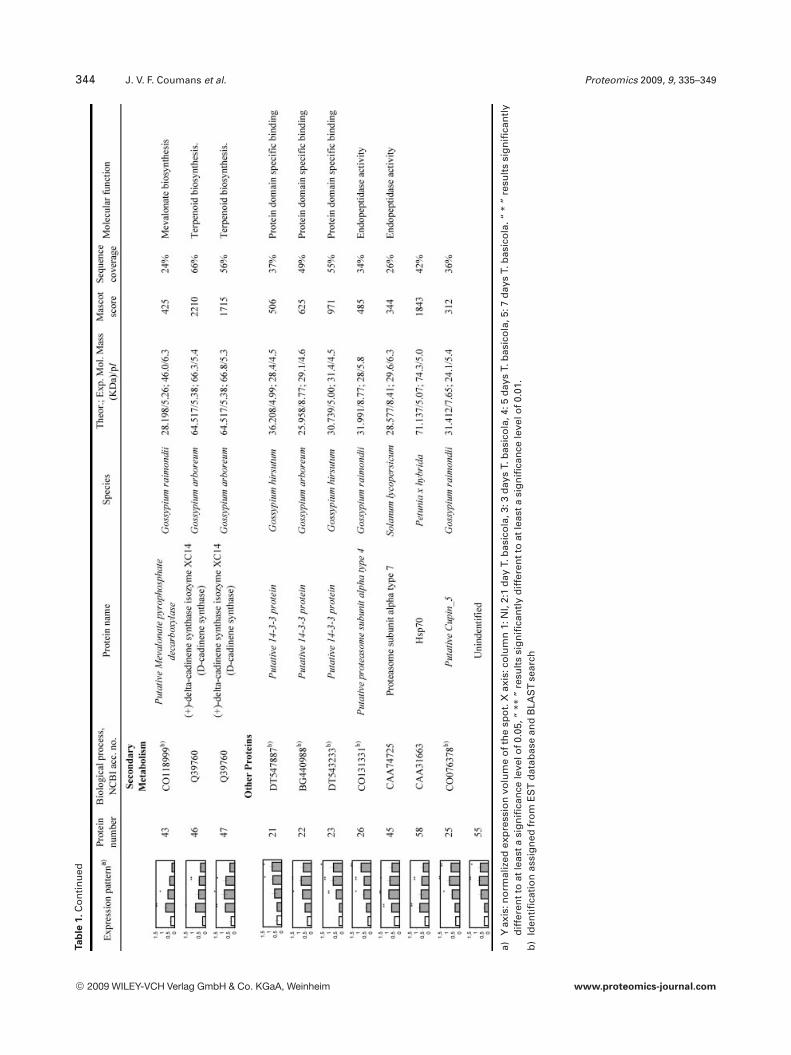
344 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349Ta
ble
1.C
on
tin
ued
a)Y
axis
:no
rmal
ized
exp
ress
ion
volu
me
oft
he
spo
t.X
axis
:co
lum
n1:
NI,
2:1
day
T.b
asic
ola
,3:3
day
sT.
bas
ico
la,4
:5d
ays
T.b
asic
ola
,5:7
day
sT.
bas
ico
la.“
*”
resu
lts
sig
nif
ican
tly
dif
fere
ntt
oat
leas
tasi
gn
ific
ance
leve
lof0
.05,
“**
”re
sult
ssi
gn
ific
antl
yd
iffe
ren
tto
atle
asta
sig
nif
ican
cele
velo
f0.0
1.b
)Id
enti
fica
tio
nas
sig
ned
fro
mE
ST
dat
abas
ean
dB
LAS
Tse
arch
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com
Proteomics 2009, 9, 335–349 345
Figure 3. Representative Blue silver 2-DE gel of proteins extracted from NI cotton root (A) and cotton root harvested after 5-day post-inoculation (B) with T. basicola. Labeled proteins were found to be upregulated and were analyzed by LC-MS/MS analysis. s indicate thelocalization of spots that could not be detected in NI cotton root.
Figure 4. Functional classification of the proteins identified by LC-MS/MS and found to be upregulated in cotton root after inocula-tion with T. basicola. The relative percentages of proteins in eachcategory are shown.
been found to induce resistance to a broad range of diseasein tobacco and rice [32]. This suggests that PR-10 proteinsmay be involved in hormone-mediated disease resistance incotton. Other members of the PR-10 family display ribonu-clease activity. It has been postulated that PR-10 proteinswith RNase activity are liberated from damaged host cellsand could protect the plant by acting directly on the patho-gens [31].
Another PR protein upregulated during T. basicola infec-tion is a putative thaumatin protein. Members of this family
have been shown to inhibit hyphal growth or spore germi-nation of various fungi by permeabilizing the fungal mem-branes [33]. The last two protein spots identified in this cate-gory correspond to a Meloidogyne-induced protein MIC-3.Originally, the expression of this protein was found to corre-late with the disruption of nematode development in a cottonline resistant to the root-knot nematode Meloidogyne incog-nita [34]. A later study by the same group showed that accu-mulation of the 14 kb protein occurs in the area surroundingthe feeding site of the nematode suggesting a direct rela-tionship between level of resistance of cotton and proteinexpression [20]. Interestingly, a blast search on ESTdatabasesusing Callahan’s mRNA sequence AY072782 (proteinsequence: AAL68391), identified 20 similar sequences.Fourteen of these sequences correspond to cDNA clonesfrom the supplemental data of Dowd et al. [17]. All of theseclones were found to be downregulated in cotton root infect-ed with F. oxysporum f. sp. vasinfectum. This reflects a majordifference in the cotton response to these two fungi.
4.2 Stress responses
Six protein spots similar to putative oxidoreductase and onespot (spot 56), identified as an apoplastic anionic guaiacolperoxidase, correspond to enzymes that are likely to beinvolved in the apoplastic oxidative burst. It is interesting to
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

346 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349
note that the temporal expression pattern between thesespots was different, with spots 31 and 34 being highlyinduced at day 1 and subsequently decreasing, while spots36, 37, and 56 showed a peak of expression at day 5. Thesecontrasting temporal expression patterns may reflect thedifferent functional roles of these putative oxidoreductases.For example we could speculate that spot 31 and 34 may beinvolved in the initial oxidative burst while the others are apart of a more general stress response. Peroxidases areknown to be induced by several stresses [35]. In response topathogen attack, it has been proposed that the production ofROS such as superoxide anions and hydrogen peroxide cre-ates an unfriendly environment that could eventually reducepathogen growth as observed in cotton infected with X. cam-pestris pv. malvacearum [36]. Moreover, peroxidases have alsobeen associated with oxidative H2O2-mediated cross-linkingof cell wall proteins such as formation of lignin that reinforcethe cell wall thereby reducing the spread of the pathogen aswell as with cell wall deposition of phenolic compounds [35].A putative benzoquinone reductase was also found to beupregulated. This protein is known to play a key role in lig-nin degradation by wood-rotting fungi via quinone inter-mediates [37]. Four spots corresponding to putative glyox-alase I were upregulated. Interestingly, glyoxalase I wasreported to increase in response to various environmentalstress conditions in several plants and to be involved in thedetoxification of cytotoxic compounds such as methylglyoxal[38]. GST, a key defense enzyme against xenobiotic toxicitywas also found to be upregulated. These enzymes are knownto be induced by biotic stress or by treatment stimulating theplant defense reaction [39]. While the exact function of thisprotein is a matter of speculation, it may protect the plantfrom the oxidative damage occurring during T. basicolainfection. Finally, a putative short-chain alcohol dehy-drogenase was identified. In cotton, alcohol dehydrogenasesare known to be induced in roots following anaerobic stress[40].
4.3 Metabolism: primary
Another major group of proteins found to be responsive to T.basicola infection are those involved in primary metabolism.Upregulation of enzymes (fructose-1, 6-bisphosphate aldo-lase, phosphoglyceromutase, enolase) that catalyze reversiblereactions required for both glycolysis and gluconeogenesis aswell as upregulation of a putative malate dehydrogenase, anenzyme of the citric acid cycle but also of the glyoxylate cyclesuggested an important role for sugar metabolism in thedefense response. Interestingly, recent publications havecorrelated the sucrose levels found in plants with the level ofresistance to a broad range of pathogens [41, 42] and Gomez-Ariza et al. [41] have also shown that rice plants pretreatedwith sucrose had increased resistance to Magnaporthe oryzaeinfection. Moreover, some spots were identified as putativeATP synthase suggesting the plants need for energy. Finally,an increase in the level of expression of a putative cysteine
synthase, a glutamate dehydrogenase and a putative riboso-mal protein suggest an alteration of amino acid synthesisand nitrogen metabolism. It has been hypothesized thatduring a plant-fungus interaction, the fungus is in a nitrogenstarvation state during in planta growth and has to acquirenitrogen from the plant [43]. In tomato leaves, Solomon andOliver [44] reported that the levels of most amino acids andnitrogen increase during infection with Clasdosporium ful-vum except for cysteine and tryptophan, which could not bedetected. It is possible that the increase of glutamate dehy-drogenase and cysteine synthase reflect the needed to com-pensate for the consumption of plant cysteine and nitrogenby the fungi and provides a possible explanation for the earlyinduction (day 1) of a putative cysteine synthase.
4.4 Metabolism: secondary
All protein spots identified in this group are enzymes leadingto the formation of isoprenoids which include gossypol andrelated phytoalexins. Production of gossypol has beenthought to contribute to the resistance of cotton to Fusariumwilt [45], has been shown to be induced in cotton followinginfection with V. dahliae and to occur early in a resistant cul-tivar compared to a susceptible cultivar [19]. Dowd et al. [17]also reported the induction of phytoalexin synthesis genesduring cotton infection with F. oxysporum f. sp. vasinfectum.However, their induction by F. oxysporum f. sp. vasinfectumoccurred late during infection and therefore could not pro-vide an effective resistance mechanism. In this study,expression levels of these enzymes was highest at day 1 sug-gesting that gossypol production is not likely to be efficientin conferring resistance to T. basicola.
4.5 Other proteins
Three protein spots corresponding to a putative 14-3-3 pro-tein were induced following infection with T. basicola. In1999, Hill et al. [14] reported the transient expression of a 14-3-3 like gene in cotton roots inoculated with V. dahliae. It isknown that the 14-3-3 protein family is involved in variousstress and disease responses. However, the understanding ofthe significance of 14-3-3 proteins in the defense response isstill in its infancy. A common feature of the 14-3-3 proteinfamily is the interaction with other proteins as binding part-ners. It appears that they play a significant role in signaltransduction through their ability to bind phosphorylatedproteins regulating their activity and or localization [46].They are involved in the hypersensitive response throughregulation of the proton pump [47] and they control keyenzymes of the nitrogen assimilation pathway as well asenzymes involved in carbohydrate metabolism [48]. From anutrient starvation study, it was suggested that they mayparticipate in nutrient sensing binding and stabilizing targetproteins [49].
Two other spots which are upregulated correspond toproteasome a subunits (a4 and a7). Previous studies have
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 335–349 347
already suggested the involvement of the proteasome inplant defense [50], while others have shown that some pro-teasome subunits are upregulated after elicitation of theplant defense reaction by cryptogein suggesting a possibleinitiating role in systematic acquired resistance [51, 52].Other spots with altered expression levels in cotton root fol-lowing T. basicola exposure were identified as Hsp70, a puta-tive cupin-5. Hsp70 proteins function as molecular chaper-ones, interacting with other proteins to facilitate proteinfolding. They also prevent irreversible aggregation of dena-turated protein and maintain the conformation of proteinsduring translocation. This protective role has been shown toconfer stress tolerance (e.g., heat resistance) [53]. Plant Hsp70 are also induced by other abiotic and biotic signals such asviruses [54]. Recently, Sung and Guy [55] have shown thatover-expression of Hsp 70 in Arabidopsis altered the growthand development of the plant indicating potential modifica-tion in signal transduction or nutrient utilization. Cupin-5belongs to a protein super-family with multiple functionsand therefore it is difficult in this case to postulate its invol-vement in the cotton defense response.
There were several proteins found to be upregulated inthis study whose gene expression was also upregulated inmost other studies on cotton. Among these were PR-10 pro-teins, thaumatin-like proteins, GSTs, d-cadinene synthase,alcohol dehydrogenase-like proteins, and oxidoreductases.These could be considered part of the general reaction ofcotton tissues to infection. In contrast to shoot tissuesinfected with V. dahliae, F. oxysporum f. sp. vasinfectum or X.campestris pv. malvacearum [15, 17, 18] no chitinases or b-1,3-glucanases were identified among proteins upregulated inroots infected with T. basicola. They were also not foundamong genes upregulated in cotton roots after infection withF. oxysporum f. sp. vasinfectum [17]. These proteins are acommon component of the defense response of most plants,and it would be interesting to determine whether their lackof induction is a common feature of infection of cotton rootsby pathogens, or is specific to the two pathogens (T. basicolaand F. oxysporum f. sp. vasinfectum) that have been investi-gated.
In summary, our research has demonstrated that analy-sis of the cotton proteome has provided insight into the plantresponse to a compatible interaction with the fungus T. basi-cola. We have shown that more proteins (around 30%) arerepressed than induced (around 10%) and could identifyknown cotton defense responses such as induction of PRproteins and formation of isoprenoids. We identified Meloi-dogyne-induced protein MIC-3 proteins that so far were onlyknow to be induced in M. incognita infected cotton andreported a stress response that may be secondary to infectionas proteins identified are also known to be induced by otherstress conditions. We also show that an alteration of carbo-hydrate and nitrogen metabolism occurs and that the pro-teasome may be important in the cotton defense response tothis pathogen. Finally, this research is the first report of someof the biochemical changes associated with the black root rot
disease and has laid the foundation for further studiesincluding proteome sub-fractionation, with the aim ofunderstanding this host–pathogen interaction in order todeveloped better control strategies for T. basicola infection.
J. V. F. Coumans was supported by grants from the Uni-versity of New England, Cotton Research and Development Cor-poration and Cotton Cooperative Research Centres. Mass spec-trometric results were obtained at the Bioanalytical Mass Spec-trometry Facility within the Analytical Centre of the University ofNew South Wales. This work was undertaken using infra-structure provided by NSW Government co-investment in theNational Collaborative Research Infrastructure Scheme(NCRIS). Subsidized access to this facility is gratefully acknowl-edged.
The authors have declared no conflict of interest.
5 References
[1] Yarwood, C. E., The occurrence of Chalara elegans. Mycolo-gia 1981, 73, 524–530.
[2] Johnson, J., Host plants of Thielavia basicola. J. Agri. Res.1916, 7, 289–300.
[3] Otani, Y., Studies on the black root rot disease caused byThielaviopsis basicola (Berk. & Br.) Ferraris. Bull. OkayamaTob. Exp. Stn. 1962, 23, 1–118.
[4] Nehl, D. B., Allen, S. J., Mondal, A. H., Lonergan, P. A., Blackroot rot: A pandemic in Australian cotton. Australas. PlantPathol. 2004, 33, 87–95.
[5] Hood, M. E., Shew, H. D., Initial cellular interactions betweenThielaviopsis basicola and tobacco root hairs. Phytopathol-ogy 1997, 87, 228–235.
[6] Mims, C. W., Copes, W. E., Richardson, E. A., Ultrastructureof the penetration and infection of pansy roots by Thiela-viopsis basicola. Phytopathology 2000, 90, 843–850.
[7] Christou, T., Penetration and host parasite relationships ofThielaviopsis basicola in the bean plant. Phytopathology1962, 52, 194–198.
[8] Hampton, R. E., Changes in phenolic compounds in carrotroot tissue infected with Thielaviopsis basicola. Phyto-pathology 1962, 52, 413–415.
[9] Hampton, R. E., Activity of some soluble oxidases in carrotslices infected with Thielaviopsis basicola. Phytopathology1963, 53, 497–499.
[10] Punja, Z. K., Virulence of Chalara elegans on bean leaves,and host-tissue responses to infection. Can. J. Plant Pathol.2004, 26, 52–62.
[11] Coumans, J. V. F., Yeoh, T., Seeto, R. K., Keogh, A. et al., Var-iations in the relative mRNA levels of actins and myosinheavy chains do not produce corresponding differences intheir proteins in the adult human heart. J. Mol. Cell. Cardiol.1997, 29, 895–905.
[12] Gygi, S. P., Rochon, Y., Franza, B. R., Aebersold, R., Correla-tion between protein and mRNA abundance in yeast. Mol.Cell. Biol. 1999, 19, 1720–1730.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

348 J. V. F. Coumans et al. Proteomics 2009, 9, 335–349
[13] Görg, A., Weiss, W., Dunn, M. J., Current two-dimensionalelectrophoresis technology for proteomics. Proteomics2004, 4, 3665–3685.
[14] Hill, M. K., Lyon, K. J., Lyon, B. R., Identification of diseaseresponse genes expressed in Gossypium hirsutum uponinfection with the wilt pathogen Verticillium dahliae. PlantMol. Biol. 1999, 40, 289–296.
[15] McFadden, H. G., Chapple, R., de Feyter, R., Dennis, E.,Expression of pathogenesis-related genes in cotton stems inresponse to infection by Verticillium dahliae. Physiol. Mol.Plant Pathol. 2001, 58, 119–131.
[16] Zuo, K., Wang, J., Wu, W., Chai, Y. et al., Identification andcharacterization of differentially expressed ESTs of Gossy-pium barbadense infected by Verticillium dahliae with sup-pression subtractive hybridization. Mol. Biol. 2005, 39, 191–199.
[17] Dowd, C., Wilson, I. W., McFadden, H., Gene expressionprofile changes in cotton root and hypocotyl tissues in re-sponse to infection with Fusarium oxysporum f. sp. vasin-fectum. Mol. Plant Microbe Interact. 2004, 17, 654–667.
[18] Patil, M. A., Pierce, M. L., Phillips, A. L., Venters, B. J.,Essenberg, M., Identification of genes up-regulated in bac-terial-blight-resistant upland cotton in response to inocula-tion with Xanthomonas campestris pv. malvacearum. Phy-siol. Mol. Plant Pathol. 2005, 67, 319–335.
[19] Bianchini, G. M., Stipanovic, R. D., Bell, A. A., Induction ofdelta-cadinene synthase and sesquiterpenoid phytoalexinsin cotton by Verticillium dahliae. J. Agric. Food Chem. 1999,47, 4403–4406.
[20] Callahan, F. E., Zhang, X. D., Ma, D. P., Jenkins, J. N. et al.,Comparison of MIC-3 protein accumulation in response toroot-knot nematode infection in cotton lines displaying arange of resistance levels. J. Cotton Sci. 2004, 8, 186–190.
[21] Di Pietro, A., Madrid, M., Caracuel, Z., Delgado-Jarana, J.,Roncero, M., Fusarium oxysporum: Exploring the moleculararsenal of a vascular wilt fungus. Mol. Plant Pathol. 2003, 4,315–325.
[22] Jorrin, J. V., Maldonado, A. M., Castillejo, M. A., Plant pro-teome analysis: A 2006 update. Proteomics 2007, 7, 2947–2962.
[23] Salekdeh, G. H., Komatsu, S., Crop proteomics: Aim at sus-tainable agriculture of tomorrow. Proteomics 2007, 7, 2976–2996.
[24] Zeman, A. M., Tchan, Y. T., Elmerich, C., Kennedy, I. R.,Nitrogenase activity in wheat seedlings bearing para-nodules induced by 2,4-dichlorophenoxyacetic acid (2,4-D)and inoculated with Azospirillum. Res. Microbiol. 1992, 143,847–855.
[25] Hurkman, W. J., Tanaka, C. K., Solubilisation of plant men-brane proteins for analysis by two-dimensional gel electro-phoresis. Plant Physiol. 1986, 81, 802–806.
[26] Görg, A., Obermaier, C., Boguth, G., Harder, A. et al., Thecurrent state of two-dimensional electrophoresis withimmobilized pH gradients. Electrophoresis 2000, 21, 1037–1053.
[27] Candiano, G., Bruschi, M., Musante, L., Santucci, L. et al.,Blue silver: A very sensitive colloidal Coomassie G-250staining for proteome analysis. Electrophoresis 2004, 25,1327–1333.
[28] de Hoon, M. J., Imoto, S., Nolan, J., Miyano, S., Open sourceclustering software. Bioinformatics 2004, 20, 1453–1454.
[29] Gatlin, C. L., Kleemann, G. R., Hays, L. G., Link, A. J., Yates, J.R., IIIrd, Protein identification at the low femtomole levelfrom silver-stained gels using a new fritless electrosprayinterface for liquid chromatography-microspray and nano-spray mass spectrometry. Anal. Biochem. 1998, 263, 93–101.
[30] Lim, H., Eng, J., Yates, J. R., IIIrd, Tollaksen, S. L. et al., Iden-tification of 2D-gel proteins: A comparison of MALDI/TOFpeptide mass mapping to mu LC-ESI tandem mass spec-trometry. J. Am. Soc. Mass Spectrom. 2003, 14, 957–970.
[31] Liu, J. J., Ekramoddoullah, A. K. M., The family 10 of plantpathogenesis-related proteins: Their structure, regulation,and function in response to biotic and abiotic stresses. Phy-siol. Mol. Plant Pathol. 2006, 68, 3–13.
[32] Nakashita, H., Yasuda, M., Nitta, T., Asami, T. et al., Brassi-nosteroid functions in a broad range of disease resistance intobacco and rice. Plant J. 2003, 33, 887–898.
[33] Anzlovar, S., Dermastia, M., The comparative analysis ofosmotins and osmotin-like PR-5 proteins. Plant Biol. 2003, 5,116–124.
[34] Callahan, F. E., Jenkins, J. N., Creech, R. G., Lawrence, G. W.,Changes in cotton root proteins correlated with resistance toroot knot nematode development. J. Cotton Sci. 1997, 1, 38–47.
[35] Passardi, F., Cosio, C., Penel, C., Dunand, C., Peroxidaseshave more functions than a Swiss army knife. Plant Cell Rep.2005, 24, 255–265.
[36] Delannoy, E., Jalloul, A., Assigbetse, K., Marmey, P. et al.,Activity of class III peroxidases in the defense of cotton tobacterial blight. Mol. Plant Microbe Interact. 2003, 16, 1030–1038.
[37] Akileswaran, L., Brock, B., Cereghino, J., Gold, M., 1,4-Ben-zoquinone reductase from Phanerochaete chrysosporium:cDNA cloning and regulation of expression. Appl. Environ.Microbiol. 1999, 65, 415–421.
[38] Yadav, S. K., Singla-Pareek, S. L., Ray, M., Reddy, M. K.,Sopory, S. K., Methylglyoxal levels in plants under salinitystress are dependent on glyoxalase I and glutathione. Bio-chem. Biophys. Res. Commun. 2005, 337, 61–67.
[39] Dixon, D. P., Lapthorn, A., Edwards, R., Plant glutathionetransferases. Genome Biol. 2002, 3, reviews 3004 ? 1 –reviews 3004 ? 10.
[40] Millar, A. A., Olive, M. R., Dennis, E. S., The expression andanaerobic induction of alcohol dehydrogenase in cotton.Biochem. Genet. 1994, 32, 279–300.
[41] Gomez-Ariza, J., Campo, S., Rufat, M., Estopa, M. et al.,Sucrose-mediated priming of plant defense responses andbroad-spectrum disease resistance by overexpression of themaize pathogenesis-related PRms protein in rice plants.Mol. Plant Microbe Interact. 2007, 20, 832–842.
[42] Murillo, I., Roca, R., Bortolotti, C., Segundo, B. S., Engineer-ing photoassimilate partitioning in tobacco plants improvesgrowth and productivity and provides pathogen resistance.Plant J. 2003, 36, 330–341.
[43] Divon, H. H., Fluhr, R., Nutrition acquisition strategies duringfungal infection of plants. FEMS Microbiol. Lett. 2007, 266,65–74.
[44] Solomon, P. S., Oliver, R. P., The nitrogen content of thetomato leaf apoplast increases during infection by Clados-porium fulvum. Planta 2001, 213, 241–249.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com

Proteomics 2009, 9, 335–349 349
[45] Zhang, J., Mace, M., Stipanovic, R., Bell, A., Production andfungitoxicity of the terpenoid phytoalexins in cotton inocu-lated with Fusarium-oxysporum f. sp vasinfectum. J. Phyto-pathol. 1993, 139, 247–252.
[46] Ferl, R. J., 14-3-3 proteins: Regulation of signal-inducedevents. Physiol. Plant 2004, 120, 173–178.
[47] Zhou, F., Andersen, C. H., Burhenne, K., Fischer, P. H. et al.,Proton extrusion is an essential signalling component in theHR of epidermal single cells in the barley-powdery mildewinteraction. Plant J. 2000, 23, 245–254.
[48] Comparot, S., Lingiah, G., Martin, T., Function and specific-ity of 14-3-3 proteins in the regulation of carbohydrate andnitrogen metabolism. J. Exp. Bot. 2003, 54, 595–604.
[49] Cotelle, V., Meek, S. E., Provan, F., Milne, F. C. et al., 14-3-3sRegulate global cleavage of their diverse binding partners insugar-starved Arabidopsis cells. EMBO J. 2000, 19, 2869–2876.
[50] Becker, J., Kempf, R., Jeblick, W., Kauss, H., Induction ofcompetence for elicitation of defense responses in cucum-ber hypocotyls requires proteasome activity. Plant J. 2000,21, 311–316.
[51] Suty, L., Lequeu, J., Lancon, A., Etienne, P. et al., Preferentialinduction of 20S proteasome subunits during elicitation ofplant defense reactions: Towards the characterization of“plant defense proteasomes”. Int. J. Biochem. Cell Biol.2003, 35, 637–650.
[52] Dahan, J., Etienne, P., Petitot, A. S., Houot, V. et al., Crypto-gein affects expression of alpha3, alpha6 and beta1 20Sproteasome subunits encoding genes in tobacco. J. Exp.Bot. 2001, 52, 1947–1948.
[53] Sung, D.-Y., Kaplan, F., Guy, C. L., Plant Hsp70 molecularchaperones: Protein structure, gene family, expression andfunction. Physiol. Plant 2001, 113, 443–451.
[54] Aranda, M. A., Escaler, M., Wang, D., Maule, A. J., Inductionof HSP70 and polyubiquitin expression associated withplant virus replication. Proc. Natl. Acad. Sci. 1996, 93, 15289–15293.
[55] Sung, D.-Y., Guy, C. L., Physiological and molecular assess-ment of altered expression of Hsc70-1 in Arabidopsis. Evi-dence for pleiotropic consequences. Plant Physiol. 2003,132, 979–987.
© 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com