amorphous carbamazepine stabilization by the mesoporous silicate sba-15
TRANSCRIPT

Accepted Manuscript
Amorphous carbamazepine stabilization by the mesoporous silicate SBA-15
Valeria Ambrogi, Fabio Marmottini, Cinzia Pagano
PII: S1387-1811(13)00204-7
DOI: http://dx.doi.org/10.1016/j.micromeso.2013.04.008
Reference: MICMAT 6027
To appear in: Microporous and Mesoporous Materials
Received Date: 12 October 2012
Revised Date: 3 April 2013
Accepted Date: 5 April 2013
Please cite this article as: V. Ambrogi, F. Marmottini, C. Pagano, Amorphous carbamazepine stabilization by the
mesoporous silicate SBA-15, Microporous and Mesoporous Materials (2013), doi: http://dx.doi.org/10.1016/
j.micromeso.2013.04.008
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers
we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and
review of the resulting proof before it is published in its final form. Please note that during the production process
errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.

AMORPHOUS CARBAMAZEPINE STABILIZATION BY THE
MESOPOROUS SILICATE SBA-15
Valeria Ambrogia*, Fabio Marmottinib and Cinzia Paganoa
a Dipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, Via del Liceo 1, 06123
Perugia, Italy
b Dipartimento di Ingegneria Civile ed Ambientale, Università di Perugia, Via G. Duranti 93,
Perugia, Italy
* Corresponding author. Dipartimento Chimica e Tecnologia del Farmaco, Facoltà di Farmacia,
Università degli Studi di Perugia, 06123 Perugia, Italy,
Tel.: +39 0755855135; fax: +39 0755855135
E-mail address: [email protected] (V. Ambrogi)

Abstract
The poor solubility carbamazepine (CARBA) is the main responsible for its low and variable
bioavailability. This problem is strictly connected to the high energy required for CARBA crystal
lattice disruption that represents the rate determining step of the dissolution process. CARBA
dissolution enhancement can be achieved by converting the crystals ordered structure in the
amorphous solid state in which the molecules show a high mobility, requiring less energy for
intermolecular bond breaking. Unfortunately CARBA amorphous form is unstable and turns into
the crystalline one. The aim of this work was the improvement of CARBA dissolution by its
conversion in the amorphous form and its stabilization by inclusion in the mesoporous silica
material SBA-15. CARBA was loaded by a simple, safe and eco-friendly method obtaining a high
final drug loading (40% wt.). The obtained inclusion product showed improved drug dissolution
and good physical stability in comparison to crystalline CARBA. Interesting results were obtained
also for the physical mixture of SBA-15 and CARBA which showed an increase of drug
dissolution. Moisture effects on drug amorphous form stability are discussed as well.
Keywords: Carbamazepine; amorphous; SBA-15; dissolution; physical stability;

1. Introduction
Carbamazepine (CARBA) is a drug provided with antiepileptic and psychotropic properties and for
this reason used in the treatment of secondary generalised tonic-clonic and partial seizures and of
trigeminal neuralgia [1]. According to the Biopharmaceutics Classification System BCS [2]
CARBA is classified as class II drug [1, 3] as it is a poor soluble drug (water solubility: ~170 µg/ml
at 25°C) [4]. After oral administration, CARBA shows an erratic and unpredictable
pharmacokinetic profile mainly associated to the slow dissolution rate (due to poor solubility) thus,
high doses are required in order to obtain and to maintain the therapeutic effect. For example, the
suggested initial dose for epilepsy therapy is 100-200 mg once or twice daily, which gradually
increases by increments of 100 to 200 mg every 2 weeks to an usual maintenance dose of 0.8 - 1.2 g
daily in divided doses: up to 2 g daily may occasionally be necessary. Polymorphism is an
additional characteristic of CARBA. There are at least four anhydrous polymorphs of this molecule
and dihydrate as well as other solvates [5]. Among them the anhydrous form III (P-monoclinic
form) is thermodynamically the most stable at room temperature and for this reason it is the only
form used in marketed product formulation. It was evidenced that during the dissolution in aqueous
medium, CARBA anhydrous form converts into the dehydrate form [6] which shows a reduced
solubility. This process is responsible for the erratic dissolution and the low and variable CARBA
bioavailability [7, 8]. Thus, after oral administration, it is slowly and irregularly absorbed through
the GI tract and generally the plasmatic peak has a time lag of 4-8 hrs [2, 9, 10].
The rate determining step in CARBA dissolution is represented by the high energy required to
disrupt the highly ordered crystalline structure. Among the approaches that can be proposed for
CARBA dissolution enhancement the conversion of the crystalline phase into the amorphous form
can represent an interesting strategy. In fact, the conversion from an ordered to a disordered
structure allows to obtain an improvement of drug dissolution.
Previous studies demonstrated that CARBA inclusion into the ordered mesoporous silica (OMS)
material MCM-41 represents an interesting approach in the release enhancement of this drug [11].

However, in this case the maximum drug loading obtained was 15 wt. %. On the basis of a previous
work [12], the problem of the low CARBA loading could be solved by using a OMS with larger
pores than MCM-41. In 1998, a new family of highly OMS was synthesized in an acid medium by
the use of commercially available non-ionic triblock copolymers (EOnPOmEOn) with large
polyethyleneoxide (EO)n and polypropyleneoxide (PO)m blocks [13]. Different materials with a
diversity of periodic arrangements have been prepared and denoted as SBA materials (the acronym
for Santa Barbara Acids). Among them, SBA-15 immediately attracted a lot of attention for the
application in drug delivery. SBA-15 is a combined micro- and mesoporous material with
hexagonally ordered tuneable uniform mesopores (4-14 nm) [14]. SBA-15 silica exhibits attractive
features, including large mesopore size and volume, high specific surface area. OMS materials
possess a network of channels of well defined size that make them suitable candidates for hosting
and delivering a variety of molecules of pharmaceutical interests [15, 16]. The suitability in the use
of OMS for drug hosting is due to the high surface area which allows a high drug loading and a
wide contact area between solid particles and biological fluids; the unidirectional and size
uniformity of mesopores shuns any tortuosity and narrowing that could slow the diffusion of the
adsorbed drug [17]; the light interactions between silicate silanols and the adsorbed molecules break
easily in the presence of water; moreover, once adsorbed, the drug molecules are confined in a
nanosized space that prevents their re-crystallization [18]; additionally OMS results non-toxic and
safe when used for oral administration [15].
In this research work, with the aim of improving CARBA release after oral administration, the drug
was converted into the amorphous form by its inclusion in the mesoporous silica material SBA-15
by a safe and eco-friendly method. Moreover the physical stability of the amorphous CARBA form
was evaluated.
2. Experimental details
2.1. Materials

CARBA, sodium metasilicate (Na2SiO3) and tetraethyl orthosilicate (TEOS) were purchased from
Sigma-Aldrich GmbH (Steinheim, Germany). The surfactant Pluronic® (P123, EO20PO70EO20) was
kindly provided from BASF Corporation 3000 Continental Drive (North Mount Olive, New Jersey,
USA). Commercially available carbamazepine IR tablets (Tegretol® 200 mg) were purchased in
pharmacy. Deionized water was obtained by reverse osmosis process with a MilliQ system
(Millipore, Roma, Italy). Other reagents and solvents were of reagent grade and were used without
further purification.
2.2. SBA-15 synthesis and loading
SBA-15 was synthesized following the method reported by Fulvio et al [19]. Four grams of P123
was added to 144 ml of an aqueous solution of HCl 1.7 M and stirred for 4 hrs at 40°C.
Successively, TEOS was added dropwise (ratio TEOS:P123=2) and then stirred for 2 hrs.
Afterwards, the mixture was transferred to Teflon-lined sealed container and kept at 100°C for 24
hrs. The final product was filtered, washed (water and ethanol, EtOH) and finally calcined at 540°C
for 24 hrs. CARBA was loaded aiming at a theoretical drug content of 40 wt.%. The drug solution
(400 mg for CARBA) was prepared in absolute ethanol (EtOH, 30 ml) and then added to SBA-15
(600 mg) dispersed in the same solvent (~5 ml). At this step, in order to remove air from matrix
pore and to make easy drug entrance, the vacuum was applied at the dispersion until bubble airs
were removed from the sample. Afterward, the mixture was kept under stirring for 30 minutes.
Finally the solvent was removed by using a rotary evaporator. Then the sample was kept under
vacuum at room temperature.
2.3. Thermogravimetric analysis (TGA)
TGA was carried out by a thermoanalyzer (Stanton-Redcroft STA 781, England) operating at
heating rate of 10°C min-1 and air flow of 30 ml/min.

2.4. X-ray powder diffraction (XRPD)
The X-Ray powder diffraction (XRPD) patterns were performed by a diffractometer (PW 1710
Philips, Almelo, The Netherland), using the Ni-filtered Cu Kα radiation, that works at 40 KV, 30
mA with goniometer PW 1820 and graphite’s monocromater for diffracted ray. Diffractograms
were registered with step scanning method (step size 2θ = 0.03°) and were elaborated by PC-APD
program.
2.5. Differential scanning calorimetry (DSC)
DSC analyses were performed using an automatic thermal analyser (Mettler Toledo DSC821e) and
indium standard for temperature calibrations. Holed aluminium pans were employed in the
experiments for all samples and an empty pan, prepared in the same way, was used as a reference.
Samples of 3-6 mg were weighted directly into the aluminium pans and the thermal analyses of
CARBA were conducted at heating rate of 10°C/min, from 20 to 300°C.
2.6. Fourier transform infrared spectroscopy (FT-IR)
FT-IR spectra were recorded in KBr dispersion (Jasco model FT/IR-410, 420 Herschel series –
Jasco Corporation Tokyo, Japan) using the EasiDiffTM Diffuse Reflectance Accessory.
2.7. Surface analysis
Nitrogen adsorption-desorption isotherms at 77 K was determined using a computer controlled
Micromeritics (Norcross, GA, USA) ASAP 2010 apparatus. Prior to adsorption measurements, the
SBA-15 samples and loaded samples were outgassed under vacuum at room temperature overnight.
The specific surface area was determined by application of the Brunauer, Emmett and Teller

(B.E.T.) theory [20] to the isotherm, while the mesopore size and volume characterization was
performed following the BJH method [21].
2.8. In vitro release studies
CARBA in vitro release studies were performed by using the paddle apparatus Farmacopea
Ufficiale Italiana XII Edizione (F.U. XII Ed.) working at 60 rpm and at the temperature of 37.0°C ±
0.5. The tests were carried out in 1000 ml of pH 1.2 ± 0.1 gastric juice (USP XX) without pepsin,
working in sink conditions (sample containing 20 mg of free drug). Four ml of dissolution medium
were removed from the vessel at appropriate intervals and replaced by fresh dissolution medium.
Samples were filtered through a cellulose membrane (Filter paper Whatman 41, Whatman GmbH,
Dassel, Germany). Drug concentration was determined by using a UV-Vis spectrophotometer
(Agilent mod. 8453). Drug calibration curve was previously prepared in acidic fluid at pH 1.2 (λmax
= 285.0 nm, r = 0.9996). All experiments were performed in triplicate, each result represents an
average of three measurements and the error was expressed as standard deviation (SD).
CARBA release from the inclusion product SBA-15-CARBA was also compared to that from the
commercial formulation Tegretol®. This experiment was performed in the same conditions above
described but in a pH 1.2 ± 0.1 gastric fluid (USP XX) with 0.5% of SLS as dissolution medium in
order to guarantee sink conditions [11, 22, 23]. The test was carried out working with an amount of
sample containing 200 mg of CARBA, which is the dose present in the commercial formulation.
Drug concentration was determined by UV spectrophotometry (λmax = 285.0 nm).
2.9. Physical stability studies
Stability accelerated tests were performed following the European Agency for the Evaluation of
Medicinal Products [24] guidelines for solid dosage forms which prescribes to keep the samples at
40°C ± 2 and 75% ± 5 RH for 6 months. Each sample was put into a glass vial and then

thermostated at 40°C ± 2 and 75% RH ± 5%. Samples were analyzed by XRPD and DSC at
established times (1, 2 and 6 months) to evaluate if drug crystal growth occurred during storage
time.
3. Results and Discussions
Drug dissolution enhancement can be achieved by the conversion of the ordered crystalline
structure into the amorphous form. However, the latter is characterized from a high free energy
meaning that the conversion in a less soluble polymorphic form can take place in a short time
giving rise to a product with different plasmatic profiles in comparison to the pristine. Thus, the use
of the amorphous form requires a suitable system able to stabilize it for a reasonable time. A
valuable approach in this field is represented by the use of ordered mesoporous materials
characterized by the high specific surface area and pores in which the drug is adsorbed and stored.
Previous studies demonstrated that the realization of inorganic-organic hybrids obtained by
CARBA inclusion into the inorganic matrix MCM-41 represents an interesting approach in its
release enhancement [11]. However, the limit of this product is the low drug loading that resulted
ca. 15 wt.%. This aspect represents a limit in the use of these systems for CARBA oral
administration as a high amount of the inclusion product MCM-41-CARBA (1.33 g) should be
required in order to administer the same dose of the commercial IR tablets (Tegretol®, 200 mg). The
cause of the low loading could be associated to the loading procedure, which consisted on the
adsorption process. This is a low yield process as it consists on the formation, on the particles
surface, of a monolayer of both solvent and drug molecules. It is noteworthy that several factors can
influence the drug loading into OMS such as pore size, surface functionality, morphology, drug
molecule size and solvent employed. Taking into account these considerations the use of SBA-15,
an OMS with larger pores than MCM-41, represented a valid strategy in CARBA loading
enhancement. CARBA inclusion into SBA-15 has already been prepared by Van Speybroeck et al.

[25] by the incipient wetness procedure, using methylene chloride as solvent for drug solubilisation
reaching a final loading of 20 wt.%. The low drug loading limits the possible application of such
system as alternative to conventional formulations for CARBA oral administration. In this paper a
different method for SBA-15-CARBA preparation is described. This loading procedure is simple,
scalable and implies the use of non toxic solvent giving a final product with high drug content.
In this work SBA-15-CARBA was prepared by the solvent evaporation method properly modified
[12] and four products were prepared with increasing loadings (30, 35, 40 and 50 wt.%) with the
aim to obtain a final product with the highest loading and the proper physicochemical properties.
Through X-ray powder diffraction (XRPD) analysis it was possible to evaluate the presence or not
of CARBA crystals in the final inclusion products. From the sample diffractograms analysis (data
not reported) resulted that the inclusion products showing the highest drug content, free from
crystals, was that with a theoretical loading of 40% wt. In fact, the XRPD pattern of such product
(SBA-15-CARBA) shows only the typical reflections of pristine SBA-15 and no peaks attributable
to crystalline CARBA, meaning that the inclusion procedure preserves the matrix original structure
and that, once included, the drug is not organized as crystals (Fig. 1). SBA-15 typical XRPD
diffractogram reveals the 2-D hexagonally structured pores (p6mm space group) at low angles,
while crystalline CARBA XRPD pattern shows the typical reflections of the polymorphic form III
[5].
The experimental drug loading was measured by TGA resulting of 40.0 ± 0.5 wt.% (data not
reported).
The lack of crystals in the inclusion product was evidenced by DSC analysis too. Crystalline
CARBA thermal profile shows a little endothermic peak at 175.5°C, due the melting of polymorph
III, that soon re-crystallizes in polymorph I and then melts at 192.1°C [5, 26]. This behaviour was
not observed for the inclusion product (Fig. 2).
The interactions established between CARBA and SBA-15 were studied by FT-IR analysis.
CARBA FT-IR spectrum shows a characteristic peak at 3466 cm-1 (–NH2 stretching), two bands at

3341 and 3283 cm-1 (-CONH stretching in solid state), 1678 cm-1 (–CO-R stretching), 1595 and
1605 cm-1 (primary amide in solid state, –NH2 bending), and 1384 cm-1(-C–N- amide stretching) [5,
27]. From SBA-15 spectrum it must be highlighted the presence of a peak at 3740 cm-1 attributable
to isolated terminal silanol groups. The inclusion product spectrum revealed a remarkably decreased
peak at 3740 cm-1 and the presence of a big and large band between 2800 and 3500 cm-1, in
addition the intensities of CARBA amino group at 3466 cm-1 and the two bands at 3341 and 3283
cm-1 (-CONH stretching in solid state) decreased significantly. These observations allow to
hypothesize the formation of a hydrogen bond between the amino group of CARBA and the silanol
groups of the mesoporous silica. It must be highlighted that the physical blend of CARBA and
SBA-15 shows, in respect to SBA-15, the decreased peak at 3740 cm-1 meaning that probably
CARBA in solid form is able to interact with terminal silanols (Fig. 3).
The study of the change of the specific surface area, pore diameter and volume of SBA-15 before
and after loading furnished important information about the space occupied from drug molecules.
This aspect was evaluated by the nitrogen adsorption–desorption isotherms recorded for SBA-15
before and after loading (Fig. 4). From these data results that the adsorbed nitrogen decreases from
precursor (SBA-15) to the corresponding inclusion product (SBA-15-CARBA). Comparing the
calculated B.E.T. specific surface area and the BJH pore volume of the precursor and SBA-15-
CARBA resulted that the B.E.T. specific surface area changes from 784 m2/g to 195 m2/g after
CARBA inclusion as well as the BJH pore volume greatly decreases from 0.95 cm3/g to 0.31 cm3/g.
It is possible to relate the volume of the included CARBA with pore volume decrease. In a previous
paper [11] the CARBA specific volume was determined and resulted 0.61 cm3/g. Thus the volume
occupied from CARBA molecules in 1g of SBA-15-CARBA is 0.61 × 0.40 = 0.24 cm3. As 1g of
SBA-15-CARBA contains 0.60 g of the SBA-15, a pore volume of 0.60 × 0.95 = 0.57 cm3 is
available for CARBA inclusion. By comparison this value with the measured pore volume for SBA-
15-CARBA sample of 0.31 cm3/g, it is possible to calculate that the pore volume occupied from
CARBA molecules in this sample is 0.57 – 0.31 = 0.26 cm3/g.

This value is near to that of 0.24 cm3/g calculated from the chemical composition indicating that
most of the CARBA is inserted in the SBA-15 mesopores.
At last from the comparison between the pore size distribution of the precursor and the inclusion
compound (Fig. 5) it is possible to note a diminution in the mean pore diameter probably caused by
the adsorption on the pore walls.
3.1. CARBA in vitro release studies
CARBA in vitro release was evaluated by the paddle apparatus (F.U. XII Ed.) by using as
dissolution medium acidic fluid at pH 1.2 (USP XX).
The profiles reported in Fig. 6 highlight that the amount of drug released from SBA-15-CARBA is
higher than the two controls represented from crystalline CARBA and CARBA/SBA-15 physical
mixture. After 10 min a drug release of 70% was achieved in respect to ~50% and ~20% of the
physical mixture and crystalline CARBA respectively. The rapid drug release from the inclusion
product is related to lack of crystalline CARBA form and to the high pore size that allows a rapid
penetration of the dissolution medium that guarantees the breaking of the hydrogen bonds among
CARBA molecules and SBA-15 silanols. Interesting results have been obtained from the physical
mixture between CARBA and SBA-15 as well. In fact, an improvement of CARBA release is
observed from the physical mixture too. This effect could be due to the partial CARBA
amorphization observed in DSC thermal profile. Whereas crystalline CARBA showed two
endothermic peaks, relative to the melting of two different polymorphs, at 175.5 °C with a ΔH = -
19.0 J/g of drug and at 190.5 °C with a ΔH = -105.5 J/g of drug. In the physical mixture, CARBA
melting peaks had a ΔH = -1.5 J/g and ΔH = -22.1 J/g of drug respectively. The enthalpy change of
both peaks is proof for CARBA crystallinity decrease obtained by simple mechanical mixing.
Spontaneous amorphization has been shown to convert several crystalline compounds to the

amorphous form [28] and a corresponding increase in the release profile of the relative formulations
in aqueous media has been shown [29].
3.2. CARBA physical stability testing
Physical stability studies were performed with the aim to evaluate the moisture effects on the
amorphous state maintenance and on the amorphization process. In particular effects of moisture
during storage remain unclear. In fact it has been reported that in the presence of moisture the
amorphous nature can be retained and in other cases the amorphous content also increased [28]. On
the contrary other studies reported a detrimental effect of moisture on drug release. Thus the effect
of moisture on physical stability of SBA-15-CARBA and SBA-15/CARBA physical mixture was
evaluated.
SBA-15-CARBA and CARBA/SBA-15 physical mixture were submitted to stressed conditions of
temperature and humidity (40°C, RH 75%) as prescribed from EMEA in the guidelines for
accelerated stability testing [24]. As expected thermal analysis performed after 2 months of storage
showed a change of the CARBA melting ΔH from -1.48 J/g and -22.1 J/g of CARBA in the
physical mixture sample before storage to -3.83 J/g and -27.7 J/g of drug in the sample after two
months storage (Fig. 7). Considering the increase of ΔH value to polymorph III at 175,5 °C after
storage, on the basis of literature data [30], we can conclude that the stressed conditions of humidity
and temperature enhance the growth of this form from about 15% to ~35% [30].
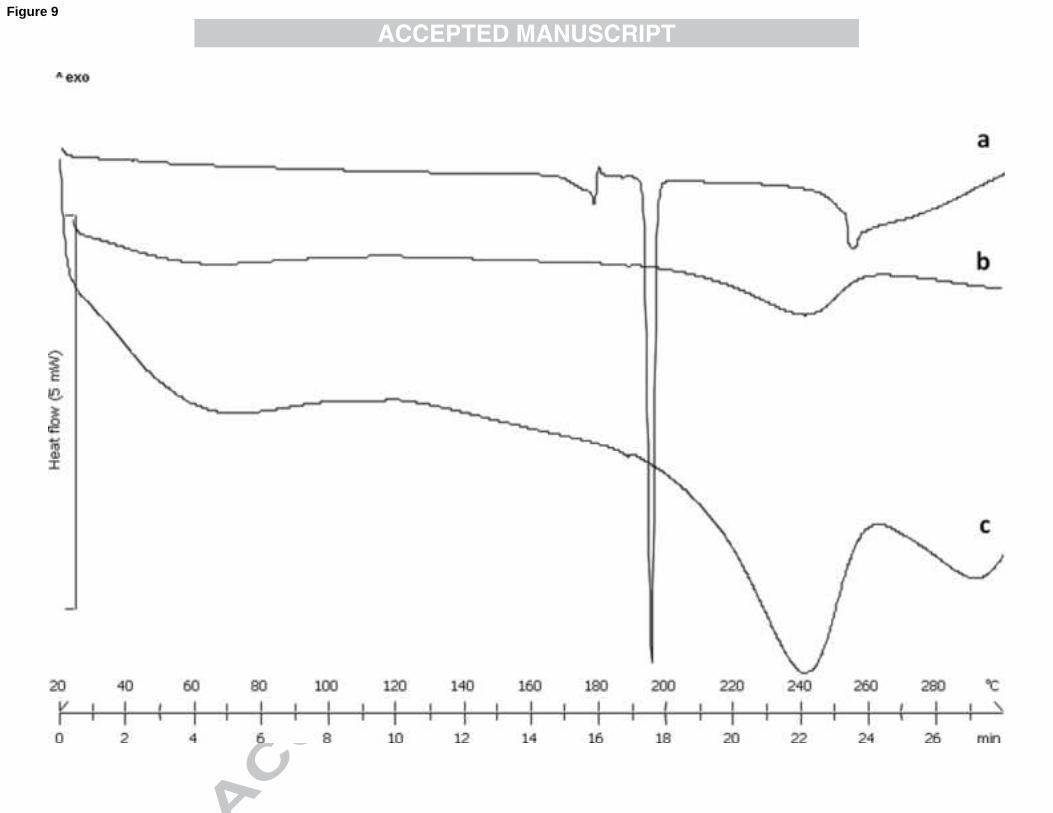
SBA-15-CARBA physical stability was verified also after 6 months of storage by XRPD and DSC.
Both XRPD patterns (Fig. 8) and DSC profiles (Fig. 9) confirmed that drug physical status does not
change. In the XRPD diffractogram is clearly evidenced the lack of crystalline CARBA typical
reflexes moreover, the thermal profile does not show the peak relative to CARBA recrystallization
and melting. These results suggest that the inclusion of CARBA into SBA-15 is able to stabilize it
from physical status modifications.

3.3. SBA-15-CARBA dissolution vs marketed product
The stabilization of CARBA amorphous by the inclusion into SBA-15 pores represents an
interesting approach for the realization of new formulations intended for CARBA immediate
release. This aspect is important in particular for the treatment of diseases requiring an immediate
pharmacological effect such as trigeminal neuralgia, characterized by episodes of intense pain. A
conventional formulation containing CARBA in crystalline form could not act promptly to reduce
the symptoms as the low solubility reduces the total amount of drug absorbed and available at the
action site.
In order to evaluate the real benefits that the use of SBA-15-CARBA can offer, the release profiles
of the inclusion product have been compared to that obtained from the commercial formulation
containing crystalline CARBA (Tegretol® 200 mg tablets). The dissolution profile of the inclusion
product SBA-15-CARBA was studied at the dose of 200 mg in order to compare the tested
compound to the commercial IR tablets of Tegretol®. CARBA release for the inclusion product
SBA-15-CARBA (Fig. 10) was faster than that from Tegretol® reaching the complete release after
50 minutes vs 180 minutes of the commercial product. After 10 minutes the drug release was of
78% and 43% for SBA-15-CARBA and the commercial formulation respectively.
4. Conclusions
The use of the mesoporous materials as stabilizer for CARBA amorphous form represents an
interesting approach in the dissolution enhancement.
In this work the preparation of the inclusion product was performed by the solvent evaporation
method avoiding the use of toxic solvents and obtaining a high final drug loading.
From the release studies it was demonstrated that the conversion of CARBA in the amorphous form
allows to obtain an improvement of its dissolution. Moreover it was demonstrated that CARBA
inclusion into SBA-15 pores is able to stabilize the amorphous form of the drug. CARBA

amorphization by inclusion into SBA-15 is able to produce promising results in terms of dissolution
improvement compared to the commercial formulation actually available on the market.
Acknowledgments
The authors are grateful to Mr. Marco Marani for technical assistance.

References
[1] K. Parfitt, Martindale, The complete drug reference, 32nd ed., Pharmaceutical Press, London,
1999, pp. 339-343.
[2] J.M. Custodio, C.Y. Wu, L.Z. Benet, Adv. Drug Deliv. Rev. 60 (2008) 717-733.
[3] M. Lindenberg, S. Kopp, J.B. Dressman, Eur. J. Pharm. Biopharm. 58 (2004) 265-278.
[4] E. Gavini, A.B. Hegge, G. Rassu, V. Sanna, C. Testa, G. Pirisino, J. Karlsen, P. Giunchedi, Int.
J. Pharm. 307 (2006) 9-15.
[5] C. Rustichelli, G. Gamberini, V. Ferioli, M.C. Gamberini, R. Ficarra, S. Tommasini, J. Pharm.
Biomed. Anal. 23 (2000) 41-54.
[6] F. Tian, J.A. Zeitler, C.J. Strachan, D.J. Saville, K.C. Gordon, T. Rades, J. Pharm. Biomed.
Anal. 40 (2006) 271-280.
[7] Y. Kobayashi, S. Ito, S. Itai, K. Yamamoto, Int. J. Pharm. 193 (2000) 137-146.
[8] S. Šehić, G. Betza, Š. Hadžidedić, S.K. El-Arini, H. Leuenberger, Int. J. Pharm. 386 (2010) 77-
90.
[9] L.S. Koester, J.B. Bertuol, K.R. Groch, C.R. Xavier, R. Moellerke, P. Mayorga, T. Dalla Costa,
V.L. Bassani, Eur. J. Pharm. Sci. 22 (2004) 201-207.
[10] R. Levy, A.J. Wilensky, G.D. Anderson in: W.E. Evans, J.J. Jusko (Eds.), third ed., Applied
Therapeutics, Vancouver, (1992) (Chapter 26), p. 1.
[11] V. Ambrogi, L. Perioli, F. Marmottini, O. Accorsi, C. Pagano, M. Ricci, C. Rossi, Micropor.
Mesopor. Mater. 113 (2008) 445-452.
[12] V. Ambrogi, L. Perioli, C. Pagano, F. Marmottini, M. Ricci, A. Sagnella, C. Rossi, Eur. J.
Pharm. Sci. 46 (2012) 43-48.
[13] D. Zhao, J. Feng, Q. Huo, N. Melosh, G.H. Fredricksin, B.F. Chmelka, G.D. Stucky, Science
279 (1998) 548-552.
[14] E.B. Celera, M. Kruk, Y. Zuzek, M. Jaroniec, J. Mater. Chem. 16 (2006) 2824-2833.

[15] J. Hirvonen, T. Laaksonen, L. Peltonen, H. Santos, V.-P. Lehto, T. Heikkilä, J. Riikonen, E.
Mäkilä, J. Salonen, Dosis 24 (2008) 129-149.
[16] T. Heikkil�, J. Salonen, J. Tuura, N. Kumar, T. Salmi, D.Y. Murzin, M. S. Hamdy, G. Mul, L.
Laitinen, A.M. Kaukonen, J. Hirvonen, V.-P. Lehto, Drug Deliv. 14 (2007) 337-347.
[17] A. Silvestre-Albero, E.O. Jardim, E. Bruijn, V. Meynen, P. Cool, A. Sepùlveda-Escribano, J.
Silvestre-Albero, F. Rodrìguez-Reinoso, Langmuir 25 (2009) 939-943.
[18] M. Sliwinska-Bartkowiak, G. Dudziak, R. Gras, R. Sikorski, R. Radhakrishnan, K.E. Gubbins,
Colloids Surf. A Physicochem. Eng. Asp. 187-188 (2001) 523-529.
[19] P.F. Fulvio, S. Pikus, M. Jaroniec, J. Colloid Interface Sci. 287 (2005) 717-720.
[20] S. Brunauer, P.H. Emmet, E. Teller, J. Am. Chem. Soc. 60 (1938) 309-319.
[21] E.P. Barrett, L.G. Joyner, P.P. Halenda, J. Am. Chem. Soc. 73 (1951) 373-380.
[22] V.P. Shah, A. Noory, C. Noory, B. McCullough, S. Clarke, R. Everett, H. Naviasky, B.N.
Srinivasan, D. Fortman, J.P. Skelly, Int. J. Pharm. 125 (1995) 99-106.
[23] Food and Drug Administration Center for Drug Evaluation and Research (CDER) Guidance
for Industry Dissolution Testing of Immediate Release Solid Oral Dosage Forms, August, 1997.
[24] EMEA, Guideline on stability testing: stability testing of existing active substances and related
finished products, CPMP/QWP/122/02, rev 1. (2003).
[25] M. Van Speybroeck, V. Barillaro, T.D. Thi, R. Mellaerts, J. Martens, J. van Humbeeck, J.
Vermant, P. Annaert, G. van Den Mooter, P. Augustijns, J. Pharm. Sci. 98 (2009) 2648-2658.
[26] C. McGregor, M.H. Saunders, G. Buckton, R.D. Saklatvala, Thermochim. Acta 417 (2004)
231-237.
[27] J.G. Panchal, R.V. Patel, S.K. Menon, J. Incl. Phenom. Macrocycl. Chem. 67 (2010) 201-208.
[28] K.K. Qian, R.H. Bogner, J. Pharm. Sci. 101 (2012) 444-463.
[29] T. Konno, Chem. Pharm. Bull. 38 (1990) 2003-2007.
[30] R. Bettini, L. Bonassi, V. Castoro, A. Rossi, L. Zema, A. Gazzaniga, F. Giordano. Eur. J.
Pharm. Sci. 13 (2001) 281-286.

Research Highlights
CARBA has been included into mesoporous SBA‐15 with a high drug loading.
CARBA amorphous form is stabilized into SBA‐15 pores.
CARBA release from SBA‐15 is enhanced in comparison to the crystalline form.

Figures captions
Fig. 1. XRPD spectra of: crystalline CARBA (a); SBA-15 (b); SBA-15/CARBA physical mixture
(c); SBA-15-CARBA (d).
Fig. 2. Thermal profiles of: crystalline CARBA (a); SBA-15 (b); SBA-15/CARBA physical
mixture (c); SBA-15- CARBA (d).
Fig. 3. FT-IR spectra of: crystalline CARBA (a); SBA-15 (b); SBA-15/CARBA physical mixture
(c); SBA-15-CARBA (d).
Fig. 4 . Nitrogen adsorption–desorption isotherms obtained with the samples SBA-15 and SBA-15-CARBA.
Fig. 5. Pore size distribution calculated for SBA-15 and SBA-15-CARBA.
Fig. 7. Thermal profiles of: crystalline CARBA (a); SBA-15 (b); SBA-15/CARBA physical
mixture (c); SBA-15/CARBA physical mixture after 2 months at 40°C RH 75% (d).
Fig. 8. XRPD spectra of: crystalline CARBA (a); SBA-15-CARBA after preparation (b) and SBA-
15-CARBA after storage for 6 months at 40°C RH 75% (c).
Fig. 9. Thermal profiles of: crystalline CARBA (a); SBA-15-CARBA after preparation (b) and
SBA-15-CARBA after storage for 6 months at 40°C RH 75% (c).
Fig. 6. In vitro release profiles at pH 1.2, 37.0°C ± 0.5 of crystalline CARBA ( ), SBA-
15/CARBA physical mixture ( ) and SBA-15-CARBA ( ), n=3, mean ± SD.
Fig. 10. In vitro release profiles at pH 1.2, 37.0°C ± 0.5 of CARBA from the inclusion product
SBA-15-CARBA ( ) and from the commercial formulation Tegretol® ( ). n=3, mean ± SD.










*Graphical Abstract (for review)